Isoform Variable Action among Thyroid Hormone Receptor Mutants Provides Insight into Pituitary Resistance to Thyroid Hormone J. D. Safer, M. F. Langlois, R. Cohen, T. Monden, D. John-Hope, J. Madura, A. N. Hollenberg, and F. E. Wondisford Thyroid Unit Department of Medicine Beth Israel Hospital and Harvard Medical School Boston, Massachusetts 02215 Resistance to thyroid hormone (RTH) is due to mu- tations in the b-isoform of the thyroid hormone receptor (TR-b). The mutant TR interferes with the action of normal TR to cause the clinical syndrome. Selective pituitary resistance to thyroid hormone (PRTH) results in inappropriate TSH secretion and peripheral sensitivity to elevated thyroid hormone levels. Association of the PRTH phenotype with in vitro behavior of the mutant TR has proved elusive. Alternative exon utilization results in two TR-b iso- forms, TR-b1 and TR-b2, which differ only in their amino termini. Although the TR-b1 isoform is ubiq- uitous, the TR-b2 isoform is found predominantly in the anterior pituitary and brain. To date, in vitro evaluation of RTH mutations has focused on the TR-b1 isoform. Site-directed mutagenesis was used to create several PRTH (R338L, R338W, V349M, R429Q, I431T) and generalized RTH (D337T, P453H) mutations in both TR-b isoforms. The ability of mutant TRs to act as dominant neg- ative inhibitors of wild type TR-b function on pos- itive and negative thyroid hormone response ele- ments (pTREs and nTREs, respectively) was evaluated in transient transfection assays. PRTH mutants had no significant dominant negative ac- tivity as TR-b1 isoforms on pTREs found in periph- eral tissues or on nTREs found on genes regulating TSH synthesis. PRTH mutants, in contrast, had strong dominant negative activity on these same nTREs as TR-b2 isoforms. Cotransfected retinoid X receptor-a was required for negative T 3 regula- tion via the TR-b1 isoform but was not necessary for negative regulation via the TR-b2 isoform in CV-1 cells. The differing need for retinoid X recep- tor cotransfection demonstrates two distinct neg- ative T 3 -regulatory pathways, one mediated by the TR-b1 and the other mediated by TR-b2. The selective effect of PRTH mutations on the TR-b2 isoform found in the hypothalamus and pituitary vs. the TR-b1 isoform found in peripheral tissues sug- gests a molecular mechanism for the PRTH disor- der. (Molecular Endocrinology 11: 16–26, 1997) INTRODUCTION Resistance to thyroid hormone (RTH) is the result of mutations in the carboxyl terminus of the b-thyroid hormone receptor (TR-b) (1–4). Individuals with the disorder require greater thyroid hormone (T 3 ) concen- trations to achieve T 3 -dependent actions. RTH is a dominant disorder in which most individuals are het- erozygous for the mutant allele. In a phenomenon called dominant negative activity, the mutant allele interferes with the activity of the normal allele (5–8). Clinically, thyroid hormone resistance can be di- vided into two entities: generalized resistance to thy- roid hormone (GRTH) and central or pituitary resis- tance to thyroid hormone (PRTH) (4). In both syn- dromes there is thyroid hormone resistance at the level of the pituitary and hypothalamus causing inappropri- ate TSH secretion and, in turn, elevated thyroid hor- mone levels. In GRTH there is also peripheral resis- tance, often resulting in a hypothyroid- or euthyroid- appearing patient. In PRTH, however, peripheral sensitivity to thyroid hormone is preserved and the individual suffers thyrotoxic symptomatology from the elevated levels of circulating T 3 (4a). A molecular mechanism to explain these two clinical phenotypes has proved elusive, and many authors have concluded that they are part of a spectrum of the same disorder (9–11). Both GRTH and PRTH mutations congregate in two major hot spots in the ligand-binding domain of TR-b (Fig. 1) (12–13). GRTH mutations have also been re- ported in the hinge region, suggesting a third hot spot for mutations at the TR-b locus (11, 14, 15). Alternative 0888-8809/97/$3.00/0 Molecular Endocrinology Copyright © 1997 by The Endocrine Society 16

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Isoform Variable Action amongThyroid Hormone Receptor MutantsProvides Insight into PituitaryResistance to Thyroid Hormone
J. D. Safer, M. F. Langlois, R. Cohen, T. Monden,D. John-Hope, J. Madura, A. N. Hollenberg, andF. E. Wondisford
Thyroid UnitDepartment of MedicineBeth Israel Hospital and Harvard Medical SchoolBoston, Massachusetts 02215
Resistance to thyroid hormone (RTH) is due to mu-tations in the b-isoform of the thyroid hormonereceptor (TR-b). The mutant TR interferes with theaction of normal TR to cause the clinical syndrome.Selective pituitary resistance to thyroid hormone(PRTH) results in inappropriate TSH secretion andperipheral sensitivity to elevated thyroid hormonelevels. Association of the PRTH phenotype with invitro behavior of the mutant TR has proved elusive.Alternative exon utilization results in two TR-b iso-forms, TR-b1 and TR-b2, which differ only in theiramino termini. Although the TR-b1 isoform is ubiq-uitous, the TR-b2 isoform is found predominantlyin the anterior pituitary and brain. To date, in vitroevaluation of RTH mutations has focused on theTR-b1 isoform. Site-directed mutagenesis wasused to create several PRTH (R338L, R338W,V349M, R429Q, I431T) and generalized RTH(D337T, P453H) mutations in both TR-b isoforms.The ability of mutant TRs to act as dominant neg-ative inhibitors of wild type TR-b function on pos-itive and negative thyroid hormone response ele-ments (pTREs and nTREs, respectively) wasevaluated in transient transfection assays. PRTHmutants had no significant dominant negative ac-tivity as TR-b1 isoforms on pTREs found in periph-eral tissues or on nTREs found on genes regulatingTSH synthesis. PRTH mutants, in contrast, hadstrong dominant negative activity on these samenTREs as TR-b2 isoforms. Cotransfected retinoidX receptor-a was required for negative T3 regula-tion via the TR-b1 isoform but was not necessaryfor negative regulation via the TR-b2 isoform inCV-1 cells. The differing need for retinoid X recep-tor cotransfection demonstrates two distinct neg-ative T3-regulatory pathways, one mediated by theTR-b1 and the other mediated by TR-b2. The
selective effect of PRTH mutations on the TR-b2isoform found in the hypothalamus and pituitary vs.the TR-b1 isoform found in peripheral tissues sug-gests a molecular mechanism for the PRTH disor-der. (Molecular Endocrinology 11: 16–26, 1997)
INTRODUCTION
Resistance to thyroid hormone (RTH) is the result ofmutations in the carboxyl terminus of the b-thyroidhormone receptor (TR-b) (1–4). Individuals with thedisorder require greater thyroid hormone (T3) concen-trations to achieve T3-dependent actions. RTH is adominant disorder in which most individuals are het-erozygous for the mutant allele. In a phenomenoncalled dominant negative activity, the mutant alleleinterferes with the activity of the normal allele (5–8).Clinically, thyroid hormone resistance can be di-
vided into two entities: generalized resistance to thy-roid hormone (GRTH) and central or pituitary resis-tance to thyroid hormone (PRTH) (4). In both syn-dromes there is thyroid hormone resistance at the levelof the pituitary and hypothalamus causing inappropri-ate TSH secretion and, in turn, elevated thyroid hor-mone levels. In GRTH there is also peripheral resis-tance, often resulting in a hypothyroid- or euthyroid-appearing patient. In PRTH, however, peripheralsensitivity to thyroid hormone is preserved and theindividual suffers thyrotoxic symptomatology from theelevated levels of circulating T3 (4a). A molecularmechanism to explain these two clinical phenotypeshas proved elusive, and many authors have concludedthat they are part of a spectrum of the same disorder(9–11).Both GRTH and PRTH mutations congregate in two
major hot spots in the ligand-binding domain of TR-b(Fig. 1) (12–13). GRTH mutations have also been re-ported in the hinge region, suggesting a third hot spotfor mutations at the TR-b locus (11, 14, 15). Alternative
0888-8809/97/$3.00/0Molecular EndocrinologyCopyright © 1997 by The Endocrine Society
16

promoter utilization at the TR-b locus yields two iso-forms of the TR-b: TR-b1, which is ubiquitously ex-pressed, and TR-b2, which is expressed almost ex-clusively in the anterior pituitary, hypothalamus, andelsewhere in the brain (16–20). While the TR-b iso-forms are identical in their DNA- and ligand-bindingdomains, they differ significantly in their N-terminalA/B domains. A functional difference between the twoTR-b isoforms in thyroid hormone regulation wouldprovide a molecular mechanism for tissue-specificregulation of gene expression (21). In this study, there-fore, the role of the TR-b2 isoform in mediating thePRTH syndrome was evaluated.
RESULTS
TR-b Mutations Used in This Study
As shown in Fig. 1, seven TR-b mutations were eval-uated in this study. Two of the mutations (D337T andP453H) significantly reduce T3 binding and causeGRTH (3, 22). One of the mutations (R338W) preservesT3 binding and is reported to cause both GRTH andPRTH, although reports of this mutation causing PRTHpredominate (11, 23, 24). The latter studies have in-cluded specific measurements of thyroid hormone ac-tion in the periphery. The remaining four mutations(R338L, V349M, R429Q, and I431T) are reported tocause PRTH (11, 25). Patients with R338L (25a) andR429Q (25) mutations have had detailed measure-ments of thyroid hormone action in the periphery, in-dicating they indeed have PRTH. Measurements ofperipheral thyroid hormone action in patients withV349M and I431T mutations have not been described,making the diagnosis of PRTH less certain in thesepatients. With the exception of I431T, TRs containingthese mutations bind well to T3. These mutations were
introduced into either a TR-b1 or TR-b2 cDNA, usingsite-directed mutagenesis, and inserted into a viralexpression vector (pSG5) for use in transient transfec-tion studies. All transfections in this study were per-formed in a TR-deficient cell line (CV-1 cells), in theabsence of a suitable pituitary cell line, which is alsoTR deficient.
Certain PRTH Mutants Have Weak DominantNegative Activity on Positive Thyroid HormoneResponse Elements
We first sought to determine the function of each ofthese mutations on thyroid hormone-regulated genesexpressed in peripheral tissues. In peripheral tissuessuch as the kidney, heart, and liver, TR-b1 and TR-a1mRNAs are readily detected by Northern blot analysisand together represent 82–90% of total T3 bindingactivity (25b). In contrast, TR-b2 mRNA is not detectedby Northern analysis, and TR-b2 protein represents asmall fraction of T3-binding capacity in these sametissues (10–18%). Because TR-b1 is the major TR-bisoform expressed in peripheral tissues, each mutationwas only tested in the context of the TR-b1 isoform. Asa model for thyroid hormone action in peripheral tis-sues, two copies of positive thyroid hormone responseelements (pTREs) were fused upstream of a heterolo-gous promoter luciferase construct (pTK109-Luc) foruse in transient transfection assays. Shown in Fig. 2Aare the dominant negative activities of each mutant ona well characterized pTRE found in a number of thyroidhormone-regulated genes (Direct repeat with a 4-bpgap [Dr14]). Dominant negative activity is plotted with100% meaning complete interference with WT TR-b1T3-mediated stimulation. The I431T, V349M, P453H,and D337T mutations all had significant dominant neg-ative activity on this element at 2.5 nM T3. Essentiallyequivalent results were obtained at 10 nM T3 (data not
Fig. 1. Schematic Representation of the Location of TR-b Mutations Used in This StudyMutations cluster in two hot spots in the TR-b C terminus and surround a region with nine heptad repeats.
Pituitary Resistance to Thyroid Hormone 17

shown). In contrast, the PRTH mutants, R338L,R338W, and R429Q, were unable to block wild type(WT) TR-b1 function on this element at 2.5 nM (Fig. 2A)or 10 nM T3 (data not shown). Similar results wereobtained with another naturally occurring pTRE [in-verted palindrome (F2)] except that the putative PRTHmutant I431T was without effect, and the GRTH mu-tant P453H had less dominant negative activity (Fig.2B). Others have also reported the lack of dominantnegative activity of PRTH mutants as TR-b1 isoformson pTREs (27, 28).We next tested the effect of these mutants on the
palindromic element, an artificial pTRE. GRTHmutantshave significant dominant negative activity (.75%) onthe element at either 2.5 nM T3 (Fig. 2C) or 10 nM T3(Fig. 2D). The I431T and V349M mutants had between50% and 70% dominant negative activity on the WTTR at either 2.5 or 10 nM T3. Finally, R429Q, R338W,and R338L had weak dominant negative activity onthis element (,40%) at 2.5 nM T3 (Fig. 2C) and evenless activity (,30%) at 10 nM T3 (Fig. 2D). Taken to-
gether, the I431T and V349M putative PRTHmutationsfunction like GRTH mutants on pTREs found in theperiphery. In contrast, three PRTH mutations (R429Q,R338W, and R338L) have weak or no dominant neg-ative activity on these same elements.
The Dominant Negative Activity of PRTH Mutantson Negative Thyroid Hormone ResponseElements Depends on the TR-b Isoform
Dominant negative activity on genes negatively regu-lated by thyroid hormone was next evaluated. In con-trast to peripheral tissues, TR-b2 is a major isoform inthe anterior pituitary and hypothalamus. We thereforetested the function of both TR-b isoforms on genesexpressed in the anterior pituitary and hypothalamusthat are responsible for control of thyroid hormonesynthesis. The human TRH and common a-subunitgene promoters were inserted into a luciferase re-porter construct and used in transient transfectionassays of CV-1 cells. The TSH-b promoter was not
Fig. 2. Dominant Negative Activity of Mutant TR-b1 Expression Vectors on a 2xDr14 Reporter Construct (A), a 2xF2 ReporterConstruct (B), and a 2xPal Reporter Construct (C and D)A 10-fold excess of mutant to WT TR-b1 expression vector (1 mg to 100 ng) was transfected with the indicated reporter
construct in CV-1 cells and compared with transfection of the WT TR-b1 expression vector alone (100 ng). Dominant negativeactivity is plotted on the x-axis, where 100% represents complete blockade and 0% represents no change in T3 stimulation [at2.5 nM T3 (panels A, B, and C) and 10 nM T3 (panel D)] relative to the WT TR-b1 transfection alone.
MOL ENDO · 1997 Vol 11 No. 118

used in this study because of its very low expressionin this cell line. On the TRH promoter, the D337Tmutation had complete dominant negative activity inthe context of the TR-b1 isoform (Fig. 3). Dominantnegative activity is plotted with 100% meaning com-plete interference with WT TR-b1 T3-mediated inhibi-tion. The P453H, V349M, and I431T mutations hadapproximately 50% dominant negative activity asTR-b1 isoforms. In contrast the PRTH mutants,R429Q, R338W, and R338L had little or no activity onthe same reporter as TR-b1 isoforms even at 10-foldexcess of mutant vs. WT TR-b1 expression vector.Interestingly, these same PRTH mutants, which had
weak dominant negative activity as TR-b1 isoforms onthe TRH gene, had strong dominant negative activityon the TRH gene as TR-b2 isoforms at 2.5 nM T3 (Fig.3). The PRTH mutants, R429Q, R338W, and R338L, asTR-b2 isoforms were converted into strong dominantnegative inhibitors on the TRH gene; and the dominantnegative activity of the I431T and V349M mutants wasgreater as TR-b2 vs. TR-b1 isoforms. This was nottrue, however, of the GRTH mutants that had essen-tially similar dominant negative activity on the TRHpromoter as either TR-b1 or TR-b2 isoforms, suggest-ing that the isoform differences on the TRH promoterwere due to specific TR-b mutations.Identical experiments were performed using the com-
mon a-subunit gene promoter as the reporter constructand a 10-fold excess of mutant vs. WT TR-b1 or TR-b2expression vector (Fig. 4). Note that the PRTH mutants,R429Q, R338W, and R338L, had no significant dominantnegative activity on this promoter in the context of theTR-b1 isoform and near 100% activity as TR-b2 iso-
forms. The putative PRTH mutants, I431T and V349M,competed as effectively as either TR-b1 or TR-b2 iso-forms, similar to data obtained with the TRH promoter(Fig. 3). Likewise, the GRTHmutants were effective com-petitors in either isoform. These results indicate that theR429Q, R338W, and R338L mutations display consis-tent and dramatic TR-b isoform differences on two neg-atively regulated genes involved in the central control ofthyroid hormone synthesis.
PRTH Mutants Have Significant DominantNegative Effects on the TRH Gene over a WideRange of T3 Concentrations
To determine whether changes in T3 concentrationwould alter the dominant negative effect of theTR-b2 isoform on the TRH gene, T3 concentrationswere varied from 1–10 nM in transfection experi-ments in which a 10-fold excess of mutant vs. WTTR-b2 expression vector was used. Similar to dataobtained in Fig. 3, both GRTH and PRTH mutantshad significant dominant negative activity as TR-b2isoforms at all concentrations of T3 tested (Fig. 5).Interestingly, however, the dominant negative activ-ity for all of the PRTH mutations (except for I431Tand V349M) and one of the GRTH mutants (P453H)was reduced at higher T3 concentration from ap-proximately 80% to 100% at 1 nM T3 to 40% to 60%at 10 nM T3. Other investigators have noted thatsupraphysiological T3 concentrations can reducedominant negative activity of RTH mutations pre-sumably by increasing T3 binding to less avid recep-tors (26). Parallel experiments were also performed
Fig. 3. Dominant Negative Activity of Mutant TR-b Isoform Expression Vectors on a TRH Reporter ConstructA 10-fold excess of mutant to WT TR-b isoform expression vector (1 mg to 100 ng) was transfected with the indicated reporter
construct in CV-1 cells and compared with transfection of the WT TR-b1 or WT TR-b2 expression vector alone (100 ng). Dominantnegative activity is plotted on the x-axis, where 100% represents complete blockade and 0% represents no change in T3 inhibition(at 2.5 nM T3) relative to the WT TR-b1 or WT TR-b2 transfection alone.
Pituitary Resistance to Thyroid Hormone 19

using the TR-b1 isoform and varying the T3 concen-tration from 1–10 nM. At 10 nM T3, none of themutations in this study had dominant negative ac-tivity on the TRH promoter except for D337T, whichretained 100% activity (data not shown). Thus,PRTH mutations in the TR-b2 isoform display sig-nificant dominant negative activity on the TRH pro-moter over a wide range of T3 concentrations.
PRTH Mutants in the TR-b2 Isoform Inhibit theFunction of Both WT TR-b1 and TR-b2 on theTRH and Common a-Subunit Genes
Since all of the transfections on the TRH and commona-subunit genes had been performed at a 10-fold ex-cess of the mutant vs. WT TR-b, experiments wererepeated at a 1:1 ratio of WT to mutant TRb expression
Fig. 5. Dominant Negative Activity of Mutant TR-b2 Expression Vectors on a TRH Reporter Construct at Various T3ConcentrationsExperiment identical to that described in Fig. 3 except that it was performed at the indicated T3 concentration.
Fig. 4. Dominant Negative Activity of Mutant TR-b Isoform Expression Vectors on the Common Glycoprotein a-Subunit ReporterConstructA 10-fold excess of mutant to WT TR-b isoform expression vector (1 mg to 100 ng) was transfected with the indicated reporter
construct in CV-1 cells and compared with transfection of the WT TR-b1 or WT TR-b2 expression vector alone (100 ng). Dominantnegative activity is plotted on the x-axis, where 100% represents complete blockade and 0% represents no change in T3 inhibition(at 2.5 nM T3) relative to the WT TR-b1 or WT TR-b2 transfection alone.
MOL ENDO · 1997 Vol 11 No. 120
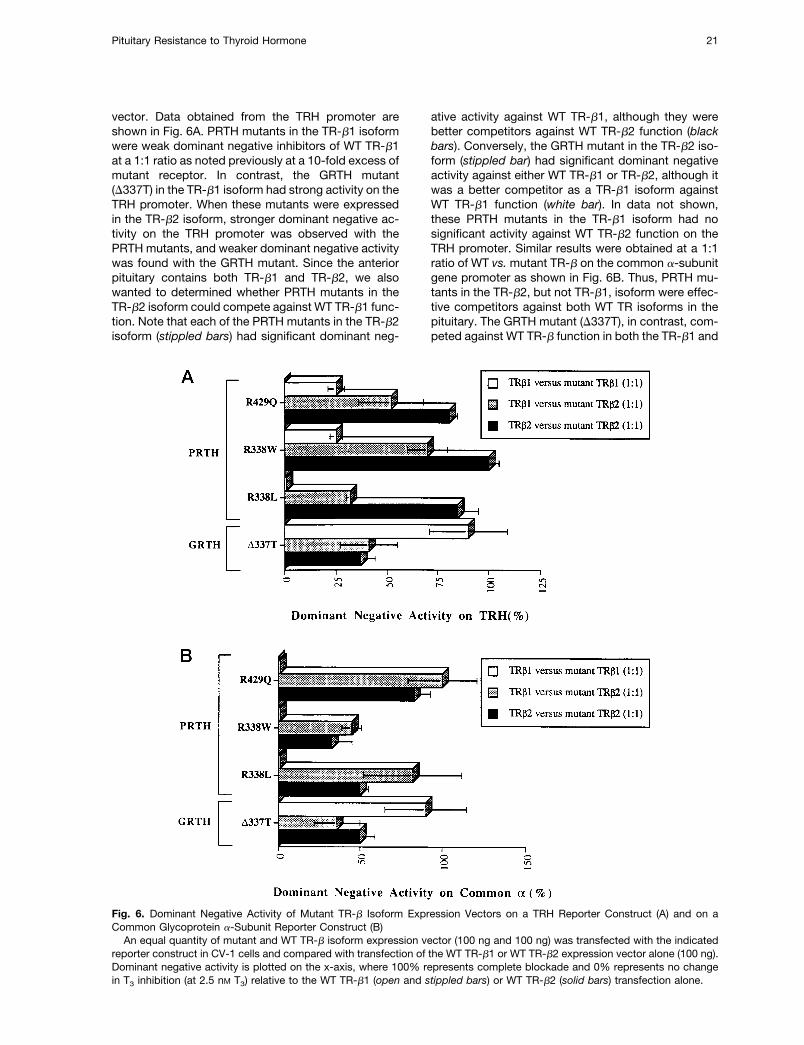
vector. Data obtained from the TRH promoter areshown in Fig. 6A. PRTH mutants in the TR-b1 isoformwere weak dominant negative inhibitors of WT TR-b1at a 1:1 ratio as noted previously at a 10-fold excess ofmutant receptor. In contrast, the GRTH mutant(D337T) in the TR-b1 isoform had strong activity on theTRH promoter. When these mutants were expressedin the TR-b2 isoform, stronger dominant negative ac-tivity on the TRH promoter was observed with thePRTH mutants, and weaker dominant negative activitywas found with the GRTH mutant. Since the anteriorpituitary contains both TR-b1 and TR-b2, we alsowanted to determined whether PRTH mutants in theTR-b2 isoform could compete against WT TR-b1 func-tion. Note that each of the PRTH mutants in the TR-b2isoform (stippled bars) had significant dominant neg-
ative activity against WT TR-b1, although they werebetter competitors against WT TR-b2 function (blackbars). Conversely, the GRTH mutant in the TR-b2 iso-form (stippled bar) had significant dominant negativeactivity against either WT TR-b1 or TR-b2, although itwas a better competitor as a TR-b1 isoform againstWT TR-b1 function (white bar). In data not shown,these PRTH mutants in the TR-b1 isoform had nosignificant activity against WT TR-b2 function on theTRH promoter. Similar results were obtained at a 1:1ratio of WT vs.mutant TR-b on the common a-subunitgene promoter as shown in Fig. 6B. Thus, PRTH mu-tants in the TR-b2, but not TR-b1, isoform were effec-tive competitors against both WT TR isoforms in thepituitary. The GRTH mutant (D337T), in contrast, com-peted against WT TR-b function in both the TR-b1 and
Fig. 6. Dominant Negative Activity of Mutant TR-b Isoform Expression Vectors on a TRH Reporter Construct (A) and on aCommon Glycoprotein a-Subunit Reporter Construct (B)An equal quantity of mutant and WT TR-b isoform expression vector (100 ng and 100 ng) was transfected with the indicated
reporter construct in CV-1 cells and compared with transfection of the WT TR-b1 or WT TR-b2 expression vector alone (100 ng).Dominant negative activity is plotted on the x-axis, where 100% represents complete blockade and 0% represents no changein T3 inhibition (at 2.5 nM T3) relative to the WT TR-b1 (open and stippled bars) or WT TR-b2 (solid bars) transfection alone.
Pituitary Resistance to Thyroid Hormone 21
TR-b2 isoform but was distinctly superior as a com-petitor in the TR-b1 isoform.
Pathways for Negative Regulation by ThyroidHormone Are TR-b Isoform-Specific
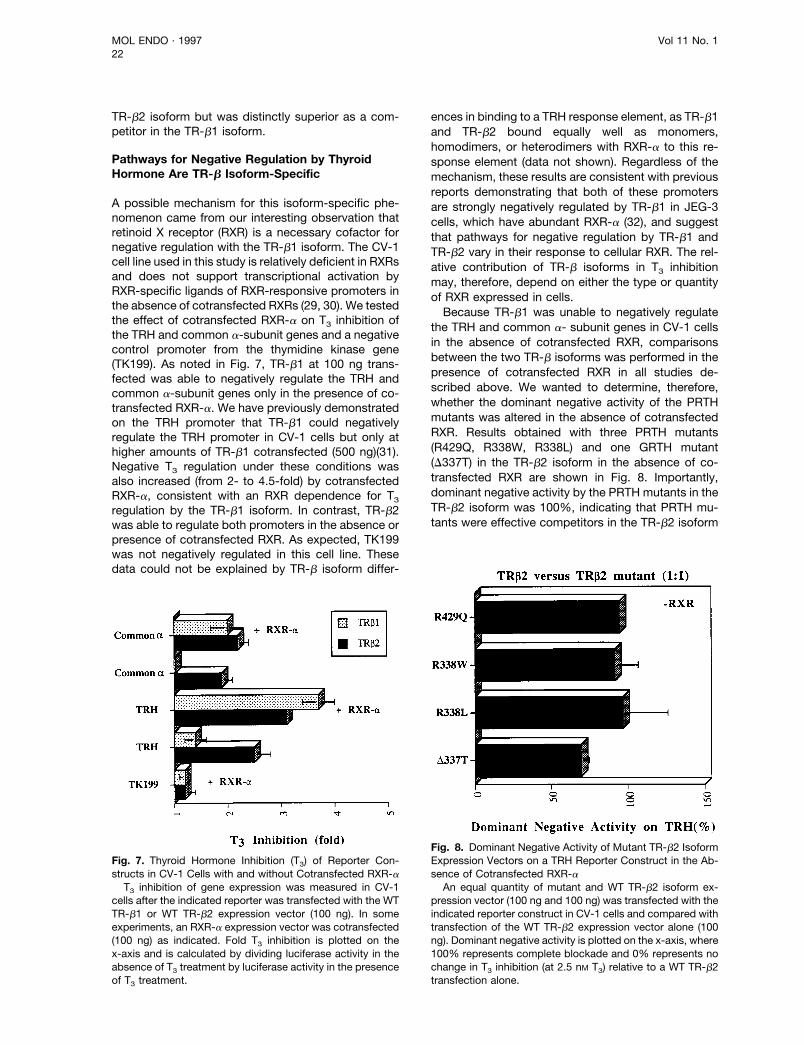
A possible mechanism for this isoform-specific phe-nomenon came from our interesting observation thatretinoid X receptor (RXR) is a necessary cofactor fornegative regulation with the TR-b1 isoform. The CV-1cell line used in this study is relatively deficient in RXRsand does not support transcriptional activation byRXR-specific ligands of RXR-responsive promoters inthe absence of cotransfected RXRs (29, 30). We testedthe effect of cotransfected RXR-a on T3 inhibition ofthe TRH and common a-subunit genes and a negativecontrol promoter from the thymidine kinase gene(TK199). As noted in Fig. 7, TR-b1 at 100 ng trans-fected was able to negatively regulate the TRH andcommon a-subunit genes only in the presence of co-transfected RXR-a. We have previously demonstratedon the TRH promoter that TR-b1 could negativelyregulate the TRH promoter in CV-1 cells but only athigher amounts of TR-b1 cotransfected (500 ng)(31).Negative T3 regulation under these conditions wasalso increased (from 2- to 4.5-fold) by cotransfectedRXR-a, consistent with an RXR dependence for T3regulation by the TR-b1 isoform. In contrast, TR-b2was able to regulate both promoters in the absence orpresence of cotransfected RXR. As expected, TK199was not negatively regulated in this cell line. Thesedata could not be explained by TR-b isoform differ-
ences in binding to a TRH response element, as TR-b1and TR-b2 bound equally well as monomers,homodimers, or heterodimers with RXR-a to this re-sponse element (data not shown). Regardless of themechanism, these results are consistent with previousreports demonstrating that both of these promotersare strongly negatively regulated by TR-b1 in JEG-3cells, which have abundant RXR-a (32), and suggestthat pathways for negative regulation by TR-b1 andTR-b2 vary in their response to cellular RXR. The rel-ative contribution of TR-b isoforms in T3 inhibitionmay, therefore, depend on either the type or quantityof RXR expressed in cells.Because TR-b1 was unable to negatively regulate
the TRH and common a- subunit genes in CV-1 cellsin the absence of cotransfected RXR, comparisonsbetween the two TR-b isoforms was performed in thepresence of cotransfected RXR in all studies de-scribed above. We wanted to determine, therefore,whether the dominant negative activity of the PRTHmutants was altered in the absence of cotransfectedRXR. Results obtained with three PRTH mutants(R429Q, R338W, R338L) and one GRTH mutant(D337T) in the TR-b2 isoform in the absence of co-transfected RXR are shown in Fig. 8. Importantly,dominant negative activity by the PRTH mutants in theTR-b2 isoform was 100%, indicating that PRTH mu-tants were effective competitors in the TR-b2 isoform
Fig. 7. Thyroid Hormone Inhibition (T3) of Reporter Con-structs in CV-1 Cells with and without Cotransfected RXR-aT3 inhibition of gene expression was measured in CV-1
cells after the indicated reporter was transfected with the WTTR-b1 or WT TR-b2 expression vector (100 ng). In someexperiments, an RXR-a expression vector was cotransfected(100 ng) as indicated. Fold T3 inhibition is plotted on thex-axis and is calculated by dividing luciferase activity in theabsence of T3 treatment by luciferase activity in the presenceof T3 treatment.
Fig. 8. Dominant Negative Activity of Mutant TR-b2 IsoformExpression Vectors on a TRH Reporter Construct in the Ab-sence of Cotransfected RXR-aAn equal quantity of mutant and WT TR-b2 isoform ex-
pression vector (100 ng and 100 ng) was transfected with theindicated reporter construct in CV-1 cells and compared withtransfection of the WT TR-b2 expression vector alone (100ng). Dominant negative activity is plotted on the x-axis, where100% represents complete blockade and 0% represents nochange in T3 inhibition (at 2.5 nM T3) relative to a WT TR-b2transfection alone.
MOL ENDO · 1997 Vol 11 No. 122

in either the presence or absence of cotransfectedRXR.
DISCUSSION
The data presented here represent a plausible expla-nation for PRTH and its molecular distinction formGRTH. All RTH mutations described to date, exceptfor deletion of the TR-b locus, are point mutationswithin a common region of the receptor (4). Thus, boththe TR-b1 and TR-b2 isoforms found in the anteriorpituitary and other regions of the brain would carry thismutation. Previous studies have extensively exploredthe function of the TR-b1 isoform on a number ofnatural or artificial thyroid hormone response elements(5, 6, 8, 26, 27, 33–35) and concluded that most PRTHmutants would be expected to cause a mild form ofRTH, which could not be distinguished from GRTH inin vitro studies. The role of the TR-b2 isoform in thegenesis of PRTH is less well known even though it ishighly expressed in the anterior pituitary. We proposethat PRTH mutants function as dominant inhibitors ofthyroid hormone action in the TR-b2 isoform, but notthe TR-b1 isoform, and RTH caused by these muta-tions is therefore limited to tissues that express thisisoform, most notably the anterior pituitary andhypothalamus.In order for PRTH to represent a distinct clinical and
biochemical entity, PRTH mutants should have thefollowing two in vitro characteristics: 1) lack of domi-nant negative activity on genes expressed in periph-eral tissues; and 2) significant dominant negative ac-tivity on genes involved in regulating TSH secretion.Three of the PRTHmutants have these characteristics:R338L, R338W, and R429Q. Each has weak dominantnegative activity on positive TREs found in peripheraltissues via the TR-b1 isoform expressed in those tis-sues. This is in part due to the fact that these muta-tions do not interfere with T3 binding. Two PRTH mu-tations (V349M and I431T), in contrast, behavedbiochemically like GRTHmutations on these elements.In addition, R338L, R338W, and R429Q are all strongdominant inhibitors on the TRH and common a-sub-unit genes in the TR-b2, but not the TR-b1, isoform.These PRTH mutants should, however, lack dominantnegative activity on other genes negatively regulatedby T3 in the periphery (e.g. myosin heavy chain b),since TR-b2 is not expressed in peripheral tissues.Thus, TR-b isoforms mediate negative T3 regulationvia different mechanisms. The TR-b1 isoform acts byan RXR-dependent pathway in which the PRTH mu-tants have little effect in antagonizing normal T3 inhi-bition by TR-b1. On the other hand, the TR-b2 isoformacts through a distinct pathway, which is not depen-dent on cotransfected RXR, such that PRTH mutants,as TR-b2 isoforms, exert significant dominant negativeactivity against normal T3 inhibition by either isoform.GRTH mutants, by contrast, interfere with WT TR
function as either TR-b1 or TR-b2 isoforms. Both
D337T and P453H have significant dominant negativeactivity against pTREs as TR-b1 isoforms and negativeTREs via either isoform. On certain TREs (F2, andTRH), D337T was a stronger dominant inhibitor thanP453H, consistent with previous reports. In addition,D337T was a superior competitor on negative TREs(TRH and common a-subunit, Fig. 6) as a TR-b1 vs.TR-b2 isoform, in contradistinction to PRTH mutants.These data suggest that GRTH mutants can act eitherthrough the TR-b1 or TR-b2 pathway to cause centralresistance to thyroid hormone.Attempts have been made to correlate phenotype
with location of mutation and/or T3-binding capacity ofthe mutant receptor (5, 7, 36). Previous authors havebeen unable to use these receptor characteristics todistinguish RTH syndromes. Our data confirm andextend these observations. Mutations found in eithermajor hot spot caused clinical PRTH and have similarbiochemical characteristics (R338L and R338W vs.R429Q) in our study. In addition, each of these PRTHmutations preserved near-normal T3 binding as did theV349M mutation, which biochemically resembles aGRTH mutation. Our data suggest that tissue-specificexpression of TR-b isoforms, coupled with a distincteffect of PRTH mutants on T3 inhibition via the TR-b2isoform, explains PRTH syndrome.How PRTH mutants selectively alter the function of
the TR-b2 isoform is unclear. Electrophoretic mobilityshift assay studies reveal that R338L, R338W, andR429Q are all homodimer deficient but heterodimerizewith RXR normally on both TRH and DR14 elementsas either isoform, while V349M and I431T both homo-and heterodimerize on these elements (data notshown and Ref. 25). Perhaps the TR homodimer iscritical for T3 inhibition and mutations that disrupt ho-modimerization cause PRTH. This is unlikely becauseTR homodimers are not thought to be important fornegative T3 regulation of the common a-subunit gene.In addition, it would not explain why PRTH mutants,only as TR-b2 isoforms, act as dominant negativeinhibitors on the TRH and common a-subunit genes.Alternatively, the homodimer defect may be a markerfor a conformation change in the receptor that causesPRTH via the TR-b2 isoform. Finally, the homodimerdefect may be unrelated to the pathogenesis of thePRTH syndrome.In summary, certain TR-b mutations cause PRTH by
converting the centrally expressed TRb2 isoform into adominant inhibitor of genes that regulate TSH secre-tion. Importantly, these mutations do not significantlyalter T3 binding or the function of the TR-b1 isoform onthyroid hormone-responsive genes, thus limiting resis-tance to only those tissues that express the TR-b2.We, therefore, propose the following model (Fig. 9) toexplain our findings and to provide a molecular frame-work for future study of GRTH and PRTH syndromes.Two discrete pathways mediate negative regulation ofgene expression by thyroid hormone in the anteriorpituitary and hypothalamus. PRTH mutants interferewith the TR-b2 pathway to cause selective resistance
Pituitary Resistance to Thyroid Hormone 23
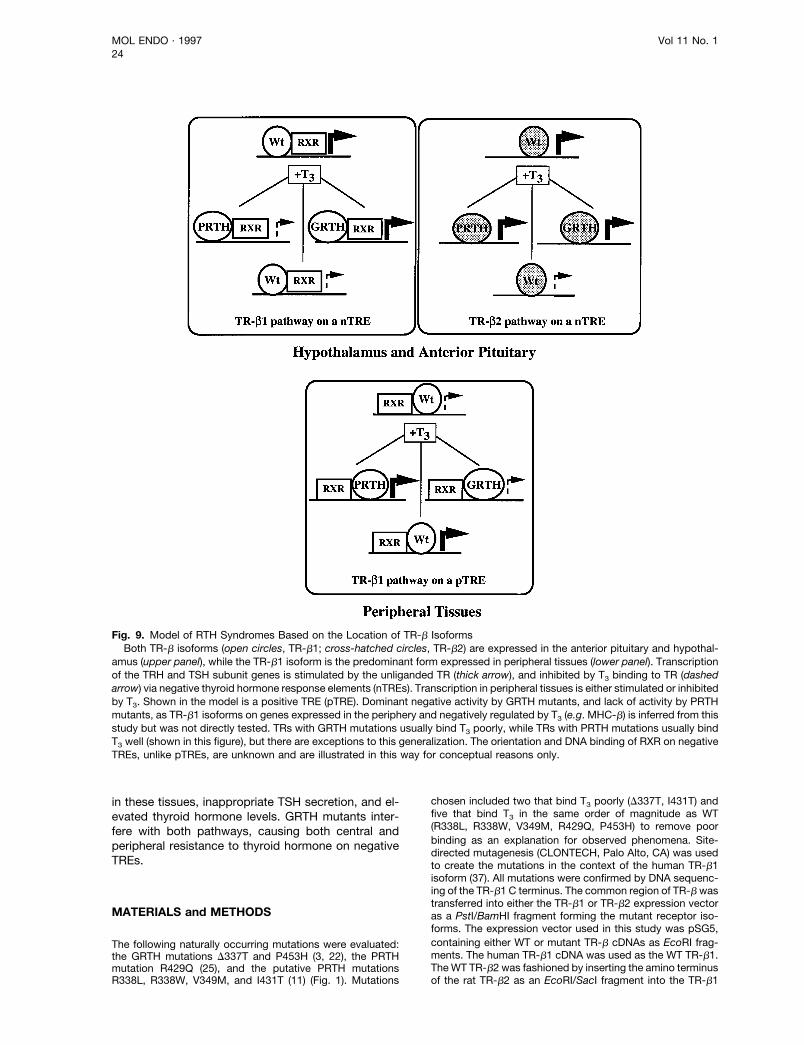
in these tissues, inappropriate TSH secretion, and el-evated thyroid hormone levels. GRTH mutants inter-fere with both pathways, causing both central andperipheral resistance to thyroid hormone on negativeTREs.
MATERIALS and METHODS
The following naturally occurring mutations were evaluated:the GRTH mutations D337T and P453H (3, 22), the PRTHmutation R429Q (25), and the putative PRTH mutationsR338L, R338W, V349M, and I431T (11) (Fig. 1). Mutations
chosen included two that bind T3 poorly (D337T, I431T) andfive that bind T3 in the same order of magnitude as WT(R338L, R338W, V349M, R429Q, P453H) to remove poorbinding as an explanation for observed phenomena. Site-directed mutagenesis (CLONTECH, Palo Alto, CA) was usedto create the mutations in the context of the human TR-b1isoform (37). All mutations were confirmed by DNA sequenc-ing of the TR-b1 C terminus. The common region of TR-b wastransferred into either the TR-b1 or TR-b2 expression vectoras a PstI/BamHI fragment forming the mutant receptor iso-forms. The expression vector used in this study was pSG5,containing either WT or mutant TR-b cDNAs as EcoRI frag-ments. The human TR-b1 cDNA was used as the WT TR-b1.TheWT TR-b2 was fashioned by inserting the amino terminusof the rat TR-b2 as an EcoRI/SacI fragment into the TR-b1
Fig. 9. Model of RTH Syndromes Based on the Location of TR-b IsoformsBoth TR-b isoforms (open circles, TR-b1; cross-hatched circles, TR-b2) are expressed in the anterior pituitary and hypothal-
amus (upper panel), while the TR-b1 isoform is the predominant form expressed in peripheral tissues (lower panel). Transcriptionof the TRH and TSH subunit genes is stimulated by the unliganded TR (thick arrow), and inhibited by T3 binding to TR (dashedarrow) via negative thyroid hormone response elements (nTREs). Transcription in peripheral tissues is either stimulated or inhibitedby T3. Shown in the model is a positive TRE (pTRE). Dominant negative activity by GRTH mutants, and lack of activity by PRTHmutants, as TR-b1 isoforms on genes expressed in the periphery and negatively regulated by T3 (e.g. MHC-b) is inferred from thisstudy but was not directly tested. TRs with GRTH mutations usually bind T3 poorly, while TRs with PRTH mutations usually bindT3 well (shown in this figure), but there are exceptions to this generalization. The orientation and DNA binding of RXR on negativeTREs, unlike pTREs, are unknown and are illustrated in this way for conceptual reasons only.
MOL ENDO · 1997 Vol 11 No. 124

cDNA in place of the amino terminus of the TR-b1. Thehuman RXR-a as an EcoRI fragment was inserted into thesame expression vector (38). Expression vector plasmidpreparations used in this study were carefully quantitated byagarose gel electrophoresis. To confirm the integrity andquality of each expression vector, plasmid DNA preparation,in vitro translation with [35S]methionine, was performed usingT7 polymerase, and the products were analyzed bySDS-PAGE.Reporter constructs included two copies of idealized
pTREs: palindrome (Pal, Ref. 11), inverted palindrome (chick-en lysozyme F2, Ref. 39), and direct repeat with a 4-bpinterval (DR14, Ref. 11). The negative response elementsincluded the 59-flanking sequences for the TRH and the com-mon glycoprotein a-subunit gene (5, 31). In the case of thepositive reporters, constructs contained the pTRE elementfused upstream of a2109 thymidine kinase promoter and theluciferase gene to measure activity. The negative reporterconstructs included their own promoters and were also fusedupstream of the luciferase gene. The luciferase reporter genewas derived from pSV0 and contains two transcriptional stopsequences upstream of the promoter to prevent read-through transcription. The reporter was documented to haveneither positive nor negative thyroid hormone responses inthe absence of response elements.Transfections were performed in CV-1 cells, which are
relatively RXR deficient (29, 30), and thereby provided anopportunity to examine the importance of limiting concentra-tions of RXR. Each transfection included a WT TR-b expres-sion vector (100 ng), a mutant or WT TR-b expression vector(100 ng or 1 mg), an RXR-a expression vector except whereindicated (100 ng), and a response element reporter con-struct (10 mg). Cell cultures were transfected using a calcium-phosphate precipitation method, and precipitate was appliedfor 16 h in DMEM containing 10% FBS, glutamine, and ap-propriate antimicrobials. The next day, medium was removedand DMEM containing both anion exchange and charcoal-stripped FBS (10%) was added 6 T3. Most of the experi-ments used 2.5 nM T3, although other concentrations wereused as noted. Data were pooled from at least three inde-pendent experiments and displayed as mean 6 SE.Gel-shift studies were performed with proteins derived
from a coupled in vitro transcription/translation reaction inreticulocyte lysate. Three to four microliters of either TR-b1,TR-b2, or RXR-a lysate were used in binding studies on eithera radiolabeled Dr14 or TRH element according to publishedmethods (31).
Acknowledgments
We would like to thank Dr. Steve Usala for certain mutantTRs, Dr. Larry Jameson for the common a-glycoprotein sub-unit gene promoter, Dr. Mitchell Lazar for the rat TR-b2cDNA, and Dr. Ronald Evans for the RXR-a cDNA.
Received August 2, 1996. Re-revision recieved October 9,1996. Accepted October 10, 1996.Address requests for reprints to: Fredric E. Wondisford,
M.D., Thyroid Unit, Beth Israel Hospital, Research North,Room 330C, 99 Brookline Avenue, Boston, Massachusetts02215.This work was supported by NIH Grants DK-02423 (to
J.D.S), DK-02354 (to A.N.H.), and DK-43653 and DK-49126(to F.E.W.) and the McLaughlin Foundation (to M.F.L.).
REFERENCES
1. Usala SJ, Bale AE, Gesundheit N, Weinberger C, LashRW, Wondisford FE, McBride OW, Weintraub BD 1988
Tight linkage between the syndrome of generalized thy-roid hormone resistance and the human c-erbA betagene. Mol Endocrinol 2:1217–1220
2. Sakurai A, Takeda K, Ain K, Ceccarelli P, Nakai A, SeinoS, Bell GI, Refetoff S, Degroot LJ 1989 Generalized re-sistance to thyroid hormone associated with a mutationin the ligand-binding domain of the human thyroid hor-mone receptor b. Proc Natl Acad Sci USA 86:8977–8981
3. Usala SJ, Tennyson GE, Bale AE, Lash RW, GesundheitN, Wondisford FE, Accili D, Hauser P, Weintraub BD1990 A base mutation of the c-erbAb thyroid hormonereceptor in a kindred with generalized thyroid hormoneresistance. J Clin Invest 85:93–100
4. Refetoff S, Weiss RE, Usala SJ 1993 The syndromes ofresistance to thyroid hormone. Endocr Rev 14:348–399
4a.Gershengorn MC, Weintraub BD 1975 Thyrotropin-in-duced hyperthyroidism caused by selective pituitaryresistance to thyroid hormone. A new syndrome ofinappropriate secretion of TSH. J Clin Invest56:633–642
5. Chatterjee VK, Nagaya T, Madison LD, Datta S, Rentou-mis A, Jameson JL 1991 Thyroid hormone resistancesyndrome. Inhibition of normal receptor function by mu-tant thyroid hormone receptors. J Clin Invest87:1977–1984
6. Yen PM, Darling DS, Carter RL, Forgione M, Umeda PK,Chin WW 1992 Triiodothyronine (T3) decreases binding toDNA by T3-receptor homodimers but not receptor-aux-iliary protein heterodimers. J Biol Chem 267:3565–3568
7. Nagaya T, Eberhardt NL, Jameson JL 1993 Thyroid hor-mone resistance syndrome: correlation of dominant neg-ative activity an location of mutations. J Clin EndocrinolMetab 77:982–990
8. Zavacki AM, Harney JW, Brent GA, Larsen PR 1993Dominant negative inhibition by mutant thyroid hormonereceptors is thyroid hormone response element and re-ceptor isoform specific. Mol Endocrinol 7:1319–1330
9. Beck-Peccoz P, Chatterjee VK, Chin WW, DeGroot LJ,Jameson JL, Nakamura H, Refetoff S, Usala SJ, Wein-traub BD 1994 Nomenclature of thyroid hormone recep-tor beta gene mutations in resistance to thyroid hormone.First workshop on thyroid hormone resistance, July 10–11, 1993, Cambridge, UK. J Endocrinol Invest17:283–287
10. Beck-Peccoz P, Chatterjee VK 1994 The variable clinicalphenotype in thyroid hormone resistance syndrome.Thyroid 4:225–232
11. Adams M, Matthews C, Collingwood, TN, Tone Y, Beck-Peccoz P, Chatterjee VK 1994 Genetic analysis of 29kindreds with generalized and pituitary resistance to thy-roid hormone. J Clin Invest 94:506–515
12. Parrilla R, Mixson AJ, McPherson JA, McClaskey JH,Weintraub BD 1991 Characterization of seven novel mu-tations of the c-erbAb gene in unrelated kindreds withgeneralized thyroid hormone resistance. Evidence fortwo “hot spot” regions of the ligand binding domain.J Clin Invest 88:2123–2130
13. Takeda K, Weiss RE, Refetoff S 1992 Rapid localisationof mutations in the thyroid hormone receptor b gene bydenaturing gradient gel electrophoresis in 18 familieswith thyroid hormone resistance. J Clin Endocrinol Metab74:712–719
14. Behr M, Loos U 1992 A point mutation (Ala 229 to Thr) inthe hinge domain of the c-erbAb thyroid hormone recep-tor gene in a family with generalized thyroid hormoneresistance. Mol Endocrinol 6:1119–1126
15. Onigata K, Yagi H, Sakuri A, Nagashima T, Nomura Y,Nagashima K, Hashizume K, Morikawa A 1995 A novelpoint mutation (R243Q) in exon 7 of the c-erbA betathyroid hormone receptor gene in a family with resistanceto thyroid hormone. Thyroid 5:355–358
16. Hodin RA, Lazar MA, Wintman BI, Darling DS, Koenig RJ,Larsen PR, Moore DD, Chin WW 1989 Identification of a
Pituitary Resistance to Thyroid Hormone 25

thyroid hormone receptor that is pituitary specific. Sci-ence 244:76–79
17. Wood WM, Ocran KW, Gordon DF, Ridgway EC 1991Isolation and characterization of mouse complementaryDNAs encoding alpha and beta thyroid hormone recep-tors from thyrotrope cells: the mouse pituitary specificbeta 2 isoform differs at the amino terminus from thecorresponding species from rat pituitary tumor cells. MolEndocrinol 5:1049–1061
18. Lazar M 1993 Thyroid hormone receptors: multipleforms, multiple possibilities. Endocr Rev 14:184–193
19. Cook CB, Kakucska I, Lechan RM, Koenig RJ 1992 Ex-pression of thyroid hormone receptor b2 in rat hypothal-amus. Mol Cell Endocrinol 130:1077–1079
20. Li M, Boyages SC 1996 Detection of extended distribu-tion of b2-thyroid hormone receptor messenger ribonu-cleic acid (RNA) in adult rat brain using complementaryRNA in Situ hybridization histochemistry. Endocrinology137:1272–1275
21. Ng L, Forrest D, Haugen BR, Wood WM, Curran T 1995N-terminal variants of thyroid hormone receptor b: dif-ferential function and potential contribution to syndromeof resistance to thyroid hormone. Mol Endocrinol9:1202–1213
22. Usala SJ, Menke JB, Watson TL, Wondisford FE,Weintraub BD, Berard J, Bradley WEC, Ono S, MuellerOT, Bercu BB 1991 A homozygous deletion in the c-erbAb thyroid hormone receptor gene in a patient withgeneralized thyroid hormone resistance: isolation andcharacterization of the mutant receptor. Mol Endocrinol5:327–335
23. Mixson AJ, Renault JC, Ransom S, Bodenner DL,Weintraub BD 1993 Identification of a novel mutation inthe gene encoding the beta-triiodothyronine receptor in apatient with apparent selective pituitary resistance tothyroid hormone. Clin Endocrinol (Oxf) 38:227–234
24. Sasaki S, Nakamura H, Tagami T, Miyoshi Y, Nogimori T,Mitsma T, Imura H 1993 Pituitary resistance to thyroidhormone associated with a base mutation in the hor-mone binding domain of the human 3,5,39-triiodothyro-nine receptor beta. J Clin Endocrinol Metab76:1254–1258
25. Flynn TR, Hollenberg AN, Cohen O, Menke JB, Usala SJ,Tollin S, Hegarty MK, Wondisford FE 1994 A novel C-terminal domain in the thyroid hormone receptor selec-tively mediates thyroid hormone inhibition. J Biol Chem269:32713–32716
25a.Crino A, Borrelli P, Salvatroi R, Cortelazzi, Roncoroni R,Beck-Peccoz P 1992 Anti-iodothyronine autoantibodiesin a girl with hyperthyroidism due to pituitary resistanceto thyroid hormones. J Endocrinol Invest 15:113–120
25b.Schwartz HL, Lazar MA, Oppenheimer JH 1994 Wide-spread distribution of immunoreactive thyroid hormoneb2 receptor (Trb2) in the nuclei of extrapituitary rat tis-sues. J Biol Chem 269:24777–24782
26. Meier CA, Dickstein BM, Ashizawa K, McClaskey JH,Muchmore P, Ransom SC, Menke JB, Hao EH, Usala SJ,Bercu BB, Cheng SY, Weintraub BD 1992 Variable tran-scriptional activity and ligand binding of mutant b13,5,39-triiodothyronine receptors from four families withgeneralized resistance to thyroid hormones. Mol Endo-
crinol 6:248–25827. Collinwood TN, Adams M, Tone Y, Chatterjee VKK 1994
Spectrum of transcriptional, dimerization, and dominantnegative properties of twenty different mutant thyroidhormone b receptors in thyroid hormone resistance syn-drome. Mol Endocrinol 8:1262–1277
28. Hayashi Y, Sunthornthepvrakul T, Refetoff S 1994 Muta-tions of CpG dinucleotides located in the triiodothyronine(T3)-binding domain of the thyroid hormone receptor (TR)b gene that appears to be devoid of natural mutationsmay not be detected because they are unlikely to pro-duce the clinical phenotype of resistance to thyroid hor-mone. J Clin Invest 94:607–615
29. Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP,Haefner P, Dawson MI, Pfahl M 1992 Retinoids selectivefor retinoid X receptor response pathways. Science258:1944–1946
30. Zhang X, Lehmann J, Hoffmann B, Dawson MI, CameronJ, Graupner G, Hermann, Tran P, Pfahl M 1992 Ho-modimer formation of retinoid X receptor induced by9-cis retinoic acid. Nature 358:587–591
31. Hollenberg AN, Monden T, Flynn TR, Boers ME, CohenO, Wondisford FE 1995 The human thyrotropin-releasinghormone gene is regulated by thyroid hormone throughtwo distinct classes of negative thyroid hormone re-sponse elements. Mol Endocrinol 9:540–550
32. Hsu JH, Zavacki AM, Harney JW, Brent GA 1995 Retin-oid-X receptor (RXR) differentially augments thyroid hor-mone response in cell lines as a function of the responseelement and endogenous RXR content. Endocrinology136:421–430
33. Banahmad A, Tsai SY, O’Malley BW, Tsai MJ 1992 Kin-dred S thyroid hormone receptor is an active and con-stitutive silencer and a repressor for thyroid hormone andretinoic acid responses. Proc Natl Acad Sci USA89:10633–10637
34. Nagaya T, Jameson JL 1993 Thyroid hormone receptordimerization is required for dominant negative inhibitionby mutations that cause thyroid hormone resistance.J Biol Chem 268:15766–15771
35. Hao E, Menke JB, Smith AM, Jones C, Geffner ME,Hershman JM, Wuerth JP, Samuels HH, Ways DK, UsalaSJ 1994 Divergent dimerization properties of mutant b1thyroid hormone receptors are associated wit differentdominant negative activities. Mol Endocrinol 8:841–851
36. Hayashi Y, Weiss RE, Sarne DH, Yen PM, Sunthornthep-varakul T, Marcocci C, Chin WW, Refetoff S 1995 Doclinical manifestations of resistance to thyroid hormonecorrelate with the functional alteration of the correspond-ing mutant thyroid hormone-b receptors? J Clin Endo-crinol Metab 80:3246–3256
37. Weinberger C, Thompson CC, Ong ES, Lebo R, GruolDJ, Evans RM 1986 The c-erb-A gene encodes a thyroidhormone receptor. Nature 324:641–646
38. Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM 1990Nuclear receptor that identifies a novel retinoic acid re-sponse pathway. Nature 345:224–229
39. Baniahmad A, Steiner C, Kohne AC, Renkawitz R 1990Modular structure of a chicken lysozyme silencer: in-volvement of an unusual thyroid hormone receptor bind-ing site. Cell 61:505–514
MOL ENDO · 1997 Vol 11 No. 126
Related Documents











