Investigation of the binding of spermine and derivative polyamines on the 1U6M active site in the presence of Acetyl coenzyme-A Lindsay Mitchell [email protected] Pace University – NYC Department of Chemistry and Physical Sciences Advisor: Dr. Demosthenes Athanasopoulos

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Investigation of the binding of spermine and derivative polyamines
on the 1U6M active site in the presence of Acetyl coenzyme-A
Lindsay Mitchell [email protected]
Pace University – NYC Department of Chemistry and Physical Sciences
Advisor: Dr. Demosthenes Athanasopoulos

2
TABLE OF CONTENTS
LIST OF FIGURES AND TABLES……………………………………………………3
ABSTRACT……………………………………………………………………………...4
INTRODUCTION……………………………………………………………………….5
MATERIALS & METHODS………………………………………………………….13
RESULTS………….……………………………………………………………………16
DISCUSSION…………………………………………………………………………...26
REFERENCES………………………………………………………………………....28

LIST OF FIGURES AND TABLES
Figure 1. PyRx User Interface……………………………............………………..…...7
Figure 2. Polyamine biosynthetic pathway in C. parvum…..…….…….…………….10
Figure 3. Polyamine Structures………………………………….....………………….11
Figure 4. Docking Quadrants………………………………………………………….14
Table 1. Polyamine Binding Energies…………………….…………………………...16
Table 2. GNAT Binding Energies……………………………………………………...17
Table 3. Binding Positions Relative to Spermine……………………………………..18
Table 4. Binding Site Locations Compared With and Without Coenzyme………...20
Figure 4. Spermine (Quadrant A) without Coenzyme……………………………….22
Figure 5. Spermine (Quadrant A) with Coenzyme…………………………………...22
Figure 6. Spermine (Quadrant B) without Coenzyme………………………………..23
Figure 7. Spermine (Quadrant B) with Coenzyme…………………………………...23
Figure 8. Spermine (Quadrant C) without Coenzyme……………………………….24
Figure 9. Spermine (Quadrant C) with Coenzyme…………………………………..24
Figure 10. Diaminobutane (Quadrant C) without Coenzyme……………………….25
Figure 11. Diaminobutane (Quadrant C) with Coenzyme………………………..…25

4
ABSTRACT Cryptosporidiosis is a disease caused by Cryptosporidium parvum that is not curable for
immune-compromised individuals. Spermidine/spermine N1-acetyltransferase (SSAT) is
an enzyme in C. parvum that retro converts spermine to spermidine and spermidine to
putrescine. Since a treatment for the disease does not exist, studies are ongoing to
establish a difference between SSAT in C. parvum and SSAT in the human host cell to
determine whether CpSSAT could be a good drug target. This study investigates binding
sites of the 1U6M active site of the CpSSAT enzyme using various computer programs
including PythonPrescription, AutoDockVina, and AutoDockTools. These programs
analyze the binding affinity for spermine and derivative polyamines to specific binding
sites within the 1U6M active site to investigate how the different ligands will bind,
especially in the presence of acetyl-Coenzyme A (CoA). It was found that the presence
of acetyl-CoA did not affect the binding energies of the polyamines. A direct trend
between polyamine size and binding energies was also found. As polyamine size
increased (closer to size or spermine) the binding energies decreased. Polyamines with
similar sizes to spermine were found to have similar binding energies as well.
Additionally, it was found that the enzyme was more specific for spermine and
spermidine than lysine, Dapsone, and para-aminobenzoic acid indicating that the enzyme
belongs to the SSAT protein family, not the general N-acetyltransferase (GNAT) group.

5
INTRODUCTION
Molecular Docking
Virtual docking, a computational method for determining possible binding sites of
ligands on enzymes, is employed in this experiment to determine where spermine and it’s
derivative polyamines will bind to the spermidine/spermine N1-acetyltransferase (SSAT)
enzyme in the presence of acetyl Co-enzyme A (acetyl-CoA). There are many different
virtual docking programs that exist but these computational packages all utilize two key
functions, search algorithms and a scoring function, in order to determine the best
possible docking positions for a ligand on the enzymatic protein (Onodera 2007).
The docking methodology consists of scanning of the surface of the protein by the
potentially bound ligand. In the beginning, we define a grid of points on the surface of
the protein. This step requires a balanced approach: the resolution of the grid points must
be enough for a thorough scanning of the surface of the protein, but not excessive since
this would require long computing time. During the docking calculation, the ligand
molecule is placed sequentially in each one of these points and the interactions between
the ligand and the protein at this particular relative position is allowed to reach a
minimum of the potential energy. At the end of the calculation, the program will select
these grid points and conformations of the ligand that correspond to the lowest potential
energy and therefore constitute probably binding sites.

6
The search algorithm in the AutoDock program, one of the programs used in this
experiment, is comprised of a combination of a genetic algorithm and the Lamarkian
genetic algorithm, which is a local search algorithm (Onodera 2007). In the case of
molecular modeling, the algorithm searches all possible conformations of the ligand and
scans it against the enzyme (Trott 2009). The user designates a search space, which is the
space on the enzymatic protein that the computer will test within, and all the possible
conformations are scanned against all possible locations within that search space.
Smaller search spaces are better because it is easier for the algorithm to explore it (Trott
2009).
The next part of the virtual screening process is assigning a scoring function, which is the
evaluation of the local minima of the ligand-protein interaction. After scanning, the
results are given a fitness value and the results with the highest values are reported.
These fitness values correlate to the binding energy, in kcal/mol, required to bind a
specific conformation of a ligand to the enzyme in any given location (Onodera 2007).
AutoDock’s scoring functions produces two results, which are the final docking energy
(FDE) and the estimated final energy of binding (EFEB). The FDE is the first result,
while the EFEB includes other factors such as van der Waals interactions and hydrogen
binding (Onodera 2007).
Python Prescription, PyRx, is a graphical interface for the preparation of the input data
and the performance of the calculation in an integrated user friendly environment. It uses
open source software including AutoDock4, AutoDock Vina, and AutoDockTools.
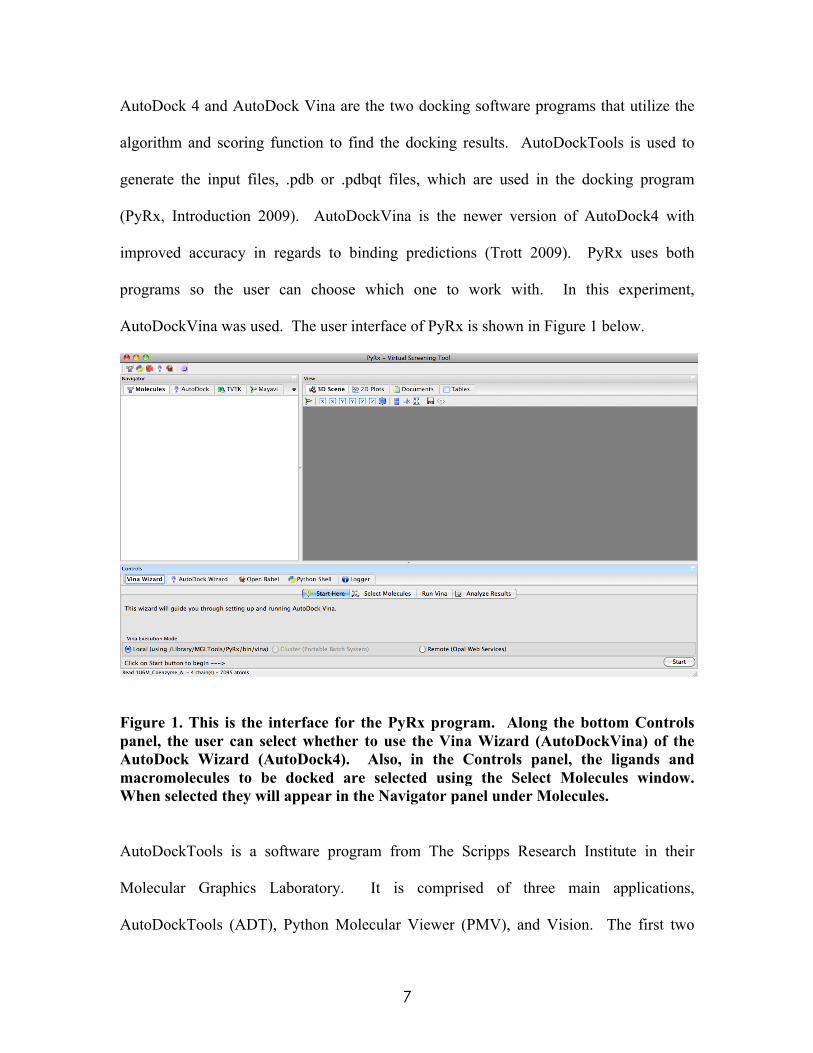
7
AutoDock 4 and AutoDock Vina are the two docking software programs that utilize the
algorithm and scoring function to find the docking results. AutoDockTools is used to
generate the input files, .pdb or .pdbqt files, which are used in the docking program
(PyRx, Introduction 2009). AutoDockVina is the newer version of AutoDock4 with
improved accuracy in regards to binding predictions (Trott 2009). PyRx uses both
programs so the user can choose which one to work with. In this experiment,
AutoDockVina was used. The user interface of PyRx is shown in Figure 1 below.
Figure 1. This is the interface for the PyRx program. Along the bottom Controls panel, the user can select whether to use the Vina Wizard (AutoDockVina) of the AutoDock Wizard (AutoDock4). Also, in the Controls panel, the ligands and macromolecules to be docked are selected using the Select Molecules window. When selected they will appear in the Navigator panel under Molecules.
AutoDockTools is a software program from The Scripps Research Institute in their
Molecular Graphics Laboratory. It is comprised of three main applications,
AutoDockTools (ADT), Python Molecular Viewer (PMV), and Vision. The first two

8
were used in this experiment. ADT is used for making the molecules run in AutoDock.
PMV is a more advanced molecular viewer that can show molecules displayed in
multiple ways, such ball and stick, ribbon, and molecular surface configurations (MGL
Tools 2010).
Enzyme Background
An enzyme is a protein that speeds up the rate of a reaction by decreasing the activation
energy (Dolphin 2008). The amino acid groups that make up the protein, as well as the
secondary and tertiary structures, help determine the shape of the enzyme. It has an
active site, where the substrate binds, which is specific to certain ligands present on the
substrate (Dolphin 2008). When the substrate enters the active site of the enzyme it binds
with the enzyme and sometimes the binding is facilitated by the presence of a coenzyme,
which are usually metallic ions that help the substrate bind to the enzyme. After the
ligand is bound to the active site, the enzyme changes its shape so that the substrate fits
better in the enzyme. When bound to the active site of the enzyme an enzyme-substrate
complex forms weak, non-covalent bonds. The covalent bonds in the substrate are
stressed, breaking the bonds in the substrate and converting it to the products (Dolphin
2008).
Enzymatic activity is dependent on multiple factors including temperature, pH,
concentration of enzyme or substrate, and presence of inhibitors. Competitive inhibitors
are compounds that have structures similar to the ligand, so the inhibitor can bind to the
active site. When a competitive inhibitor binds to an active site, the substrate ligand

9
cannot bind to that same active site, resulting in reversible inhibition (Campbell and
Reece 2008). Irreversible inhibitors covalently bind to the active site or they damage a
functional group so that the intended ligand can no longer bind. Reversible inhibitors,
however, are non-covalently bonded and do not change the active site so they can be
removed (Nelson 2008).
Cryptosporidium parvum Background
Cryptosporidium parvum is a parasite that causes cryptosporidiosis, a diarrheal disease
with symptoms such as dehydration, nausea/vomiting, stomach pains, and weight loss
that affects approximately 748,000 people per year in the United States alone (CDC,
Disease 2010 & CDC, Epidemiology & Risk Factors 2011). The different species of C.
parvum, which can infect both humans and animals, are spread many different ways, the
most common being through water (CDC, Parasite-Cryptosporidium). While treatment is
usually not necessary for people with healthy immune systems, infection can last longer
and be more severe for people with weaker immune systems. For example, for
individuals with AIDS, cryptosporidiosis is not curable. Even if the symptoms disappear,
they could return with the weakening of the immune system over time (CDC, Treatment
2012).
Spermidine/spermine N1-acetyltransferase (SSAT) is an important enzyme for the
metabolism of polyamines by C. parvum (Yarlett 2007). It was determined that this
enzyme, along with polyamine oxidase, converts spermine to lower polyamines. SSAT in
C. parvum (CpSSAT) has been compared to SSAT in the host cell (HsSSAT) to

10
determine whether SSAT would be a good target for drug design, and the two SSAT’s
were in fact found to have different properties making it a good target (Yarlett 2007).
Biochemical Mechanism of Spermine Interaction with SSAT
As was mentioned before, CpSSAT metabolizes polyamines in C. parvum. More
specifically, CpSSAT retro-converts spermine to spermidine and spermidine to
putrescence (diaminobutane) in the presence of acetyl-CoA (Yarlett 2007). This pathway
is shown below in Figure 2. While some information about pathway is known, it is not
known if the complex is ternary or substituted. It is also unknown whether the reaction is
ordered or random. Using the 1U6M active site of the CpSSAT, docking can be
performed on the enzyme to help determine whether or not it is ternary or substituted as
well as random or ordered.
Figure 2. Polyamine biosynthetic pathway in C. parvum. SSAT retro-converts spermine to spermidine then to putrescine.
The 1U6M active site is chosen for two main reasons. First, since it is represents the
active site, it contains the substrate binding domain of CpSSAT. Second, the entire
SSAT is so large that the computer cannot determine the fold of the entire molecule.
With this particular part however, the computer can use HsSSAT as a guide for folding

11
because the two SSAT’s share a high degree of amino acid similarity in the active site.
The HsSSAT contains five arginine residues, while the CpSSAT contains eight arginine
residues (five of which are in the same location as the human enzyme).
Investigation of Activity of Other Polyamines
Figure 3. The structures of the three main polyamines used in this experiment. From top to bottom: spermine, spermidine and 1,4-diaminobutane. These are the polyamines that are used in the SSAT pathway shown in Figure 2. Spermine and Spermidine (shown above in Figure 3) are both cationic molecules
involved in cellular metabolism. Spermine is the preferred polyamine in C. parvum
SSAT, while spermidine is preferred in the human enzyme meaning that CpSSAT
exhibited higher enzyme activity for spermine, while HsSSAT exhibited higher enzyme
activity for spermidine (Yarlett 2007). Since it is known that spermine is acetylated to N-
acetyl spermine, which can be further metabolized to spermidine, N-acetylspermidine or
putrescine, it is of interest to note whether or not the other polyamines of different sizes
can also bind to the 1U6M active site. The additional polyamines scanned are 1,3-
diaminopropane, 1,5-diaminopentane, 1,6-diaminohexane, 1,7-diaminoheptane, 1,8-
diaminooctane, 1,9-diaminononane, 1,10-diaminodecane, and 1,11-diaminendecane. It

12
was already found that a molecule larger than spermine was able to dock because it made
boomerang curve allowing the amine groups on the end of the molecule to interact with
the active site. By using a series of larger polyamines that only contain amino groups on
the end, it is possible to determine whether the size affects binding with the enzyme, and
if so, at what size is there a change in ability to bind.
Activity of Non-Polyamine Ligands
Another use for the molecular docking is to determine whether or not the enzyme in
question is actually CpSSAT or a Gcn5-related N-acetyltransferases (GNAT). GNAT’s
are a group of enzymes that function similarly to SSAT in that they also transfer the
acetyl group to the ligand (Vetting 2005). There are three molecules, Histone II A,
Dapsone, and p-aminobenzoic acid that typically bind with GNAT’s. Lysine will be
docked in place of Histone II A because it is the active residue of Histone II A that binds
to the enzyme active site. If these ligands selectively bind to the 1U6M active site, it
infers’ the enzyme belongs to the GNAT class. If these ligands do not selectively bind
however, it would support previous enzymatic data that the enzyme is a true SSAT.
Our Computational Approach
Our calculations aim to investigate, at the molecular level, the interactions of all the
above mentioned ligands with the SSAT. In particular, to identify the possible binding
sites on the protein surface, the binding affinities for each ligand and each binding
position. Furthermore, we address the effect of the pre-bonded coenzyme in the
ligand/protein interactions.

13
MATERIALS & METHODS
1. PyRx/Vina Computational Package
Polyamines
The Vina Computational Package is used to run a virtual screening of the possible
binding sites of spermine, spermidine, diaminodecane, diaminoendecane,
diaminopentane, diaminoheptane, diaminohexane, diaminononane, diaminooctane,
diaminopentane, and diaminopropane, and acetyl-CoA on the enzyme. The enzyme is
separated into four quadrants, A, B, C, and D (shown on the next page in Figure 4), and
the 9 binding sites with the highest binding energies are found in each part totaling 32
binding sites for each polyamine.
First, the enzyme was screened in the presence of spermine in quadrant A and the 9
binding sites were obtained. Then it was screened in quadrant B, followed by quadrant C
and D. This same process was repeated for spermidine, the rest of the polyamines, and
acetyl-CoA. Next, the polyamines were all screened again in quadrant A in the presence
of both the polyamine and acetyl-CoA. This is used to determine whether or not there is
a difference between the binding sites and energies with or without the presence of
acetyl-CoA.
GNAT Experiment
The same procedures will also be followed using lysine, Dapsone, and para-
aminobenzoic acid.

14
Figure 4. This figure shows the four docking quadrants in respect to the enzyme as well as the coordinates of each quadrant. Quadrant A is shown in the top left, B in the top right, C in the bottom left and D in the bottom right.

15
2. AutoDockTools
The runs from the Vina computational program are then analyzed using
AutoDockTools1.5.4 by AutoDockTools. This program visually shows the binding
energies and locations of the polyamines in regard to the enzyme. The highest binding
energies are recorded for each polyamine/ligand in quadrants A, B, C, and D without the
coenzyme. Then they are recorded for quadrant A in the presence of the coenzyme. In
the case that all nine locations are similar, only the lowest binding energies are recorded.
If the locations are scattered within the quadrant, the lowest binding energy at each
location is recorded.

16
RESULTS
Table 1. Polyamine Binding Energies
Polyamine Binding Energy w/o
Coenzyme Binding Energy w/
Coenzyme Spermine A -4.6 -4.5 Spermine B -4.4, -4.2 -4.5, -4.4, -4.2 Spermine C -4.4, -4.1, -4.1 -4.3, -4.1 Spermine D -4.3, -4.1, -4.5, -4.2 Spermidine A -3.9, -3.9, -3.7 -4.0, -4.0, -3.8 Spermidine B -4.0, -3.5 -3.9, -3.8, -3.8, -3.7 Spermidine C -3.9, -3.8 -3.7, -3.7 Spermidine D -4.0, -3.6, -3.5 -3.8, -3.7 Diaminopropane A -3.1, -3.1, -2.9 -3.2, -3.1, -3.0, -2.9, -2.9 Diaminopropane B -3.1, -3.0, -2.8, -2.8 -3.1, -3.1, -2.9, -2.9, -2.8 Diaminopropane C -3.0, -2.8, -2.7 -3.1, -3.0, -2.8, -2.7 Diaminopropane D -3.1, -2.9, -2.8 -3.1, -3.0, -2.8, Diaminobutane A -3.4, -3.3, -3.1 -3.4, -3.4, -3.3 Diaminobutane B -3.3, -3.3, -3.2 -3.4, -3.4, -3.4 Diaminobutane C -3.3, -3.2, -3.1, -3.3, -3.1, -2.9, -2.9 Diaminobutane D -3.4, -3.2, -3.2 -3.4, -3.4, -3.2 Diaminopentane A -3.6, -3.5, -3.4, -3.4 -3.6, -3.5, -3.4, -3.3 Diaminopentane B -3.5, -3.3, -3.2 -3.5, -3.5, -3.5, -3.4 Diaminopentane C -3.5, -3.5, -3.3, -3.3 -3.6, -3.6, -3.3 Diaminopentane D -3.6, -3.5, -3.3, -3.3 -3.6, -3.5, -3.3 Diaminohexane A -3.8, -3.7 -3.7, -3.6 Diaminohexane B -3.7, -3.5, -3.3 -3.8, -3.5, -3.5 Diaminohexane C -3.7, -3.6, -3.6, -3.6 -3.7, -3.7, -3.7 Diaminohexane D -3.7, -3.7, -3.3 -3.6, -3.6, -3.5, -3.4, -3.4 Diaminoheptane A -4.0, -3.8 -3.9, -3.7, -3.7 Diaminoheptane B -4.0, -3.6, -3.6 -3.9, -3.7, -3.7, -3.7 Diaminoheptane C -4.0, -3.8, -3.8, -3.6 -3.9, -3.7, -3.5 Diaminoheptane D -3.9, -3.8, -3.6 -3.7, -3.7, -3.6, -3.5 Diaminooctane A -4.3, -4.2 -4.3 Diaminooctane B -3.9, -3.7, -3.7 -4.0, -3.9, -3.7 Diaminooctane C -4.4, -4.1, -3.9, -3.9 -4.0, -3.7 Diaminooctane D -3.7, -3.6 -3.9, -3.9, Diaminononane A -4.6 -4.6

17
Diaminononane B -4.0, -3.8 -4.1, -4.0, -3.8, -3.8 Diaminononane C -4.0, -3.9, -3.8 -4.2, -4.0, -3.8 Diaminononane D -4.2, -3.7 -3.9, -3.8, -3.7 Diaminodecane A -4.9, -4.2 -4.7 Diaminodecane B -4.2, -4.1 -4.5, -4.3 Diaminodecane C -4.2, -4.2, -4.0, -4.0 -4.2, -3.9, Diaminodecane D -4.2, -4.2 -4.3, -3.7, Diaminoendecane A -5.0 -4.7 Diaminoendecane B -4.7, -4.6 -4.3, -3.9, -3.9, -3.8 Diaminoendecane -4.6, -4.5, -4.5 -4.2, -4.2, -4.0, -4.0 Diaminoendecane D -4.2, -3.9 -4.1, -4.0
This table shows the binding energies obtained without the presence of the coenzyme (first column) as well as in the presence of the coenzyme (column 2). A, B, C, and D refers to the quadrant of the macromolecule in which it was tested. Multiple binding energies per polyamine correlate to multiple possible binding sites. The green highlighting indicates that the first binding posiiton of the polyamine was the same with or without the coenzyme.

18
Table 2. GNAT Experiment binding energies
Polyamine Binding Energy w/o
Coenzyme Binding Energy w/
Coenzyme Spermine A -4.6 -4.5 Spermine B -4.4, -4.2 -4.5, -4.4, -4.2 Spermine C -4.4, -4.1, -4.1 -4.3, -4.1 Spermine D -4.3, -4.1 -4.5, -4.2 Spermidine A -3.9, -3.9, -3.7 -4.0, -4.0, -3.8 Spermidine B -4.0, -3.5 -3.9, -3.8, -3.8, -3.7 Spermidine C -3.9, -3.8 -3.7, -3.7 Spermidine D -4.0, -3.6, -3.5 -3.8, -3.7 Histone A -4.7, -4.3, -4.0 -4.8, -4.1, -4.0, -4.0 Histone B -4.3, -4.2 -4.8, -4.3, -4.3, -4.3, -4.0, -4.0 Histone C -4.3, -4.0, -3.8, -3.8 -4.5, -4.5, -4.5 Histone D -4.7, -4.1, -4.3, -4.1, -4.0, -4.0 Dapsone A -7.2 -7.3, -6.7 Dapsone B -6.5, -5.7, -5.7 -6.5, -6.4, -6.1, -6.1 Dapsone C -6.9, -6.0 -6.5, -6.0, -5.9, -5.9 Dapsone D -7.0, -5.8, -5.6, -5.6 -7.0, -5.7, -5.6, -5.6 p-aminobenzoic acid A -5.5, -5.3, -5.1, -4.9 -5.5, -5.3, -4.8 p-aminobenzoic acid B -5.3, -5.1, -5.0, -4.7, -4.5 -5.4, -5.3, -5.3, -5.0, -4.9 p-aminobenzoic acid C -5.1, -5.0, -4.8, -4.6 -5.1, -5.0, -4.9, -4.6 p-aminobenzoic acid D -5.4, -5.0, -4.9 -5.1, -5.0
These are the binging energies obtained with and without the coenzyme for the following ligands: Histone IIa, Dapsone, and p-aminobenzoic acid. Multiple binding energies per polyamine correlate to multiple possible binding sites. The ligands highlighted in green had a similar first binding position with and without the coenzyme present.

19
Table 3. Binding Positions Relative to Spermine QUAD A QUAD B
POLYAMINES w/o
coenzyme w/
coenzyme POLYAMINES w/o
coenzyme w/
coenzyme Spermidine Spermidine Diaminopropane Diaminopropane Diaminobutane Diaminobutane Diaminopentane Diaminopentane Diaminohexane Diaminohexane Diaminoheptane Diaminoheptane Diaminooctane Diaminooctane Diaminononane Diaminononane Diaminodecane Diaminodecane Diaminoendecane Diaminoendecane LIGANDS LIGANDS Histone Histone Dapsone Dapsone p-aminobenzoic acid
p-aminobenzoic acid
QUAD C QUAD D
POLYAMINES w/o
coenzyme w/
coenzyme POLYAMINES w/o
coenzyme w/
coenzyme Spermidine Spermidine Diaminopropane Diaminopropane Diaminobutane Diaminobutane Diaminopentane Diaminopentane Diaminohexane Diaminohexane Diaminoheptane Diaminoheptane Diaminooctane Diaminooctane Diaminononane Diaminononane Diaminodecane Diaminodecane Diaminoendecane Diaminoendecane LIGANDS LIGANDS Histone Histone Dapsone Dapsone p-aminobenzoic acid
p-aminobenzoic acid
All outputs 5 or more outputs 4 outputs less than 4 This table shows the binding positions of the polyamines/ligands relative to spermine. Light blue indicates that all ligand output positions were the same as

20
spermine positions. Purple indicated 5 or more outputs were the same, purple indicates 4, and orange indicated less than 4 outputs were the same as spermine.
Table 4. Binding Site Locations Compared With and Without Coenzyme Ligand Site Locations Spermine A same site Spermine B introduces 1 new binding site Spermine C one binding site missing Spermine D same sites Spermidine A same sites Spermidine B introduces 2 new binding sites Spermidine C 2nd binding site moved Spermidine D one site missing Diaminopropane A introduces 2 new binding sites in relative same area Diaminopropane B one site missing; one introduced Diaminopropane C introduces 1 new binding site Diaminopropane D same sites Diaminobutane A same sites Diaminobutane B roughly same sites Diaminobutane C 3rd binding site moved, introduces 4th site Diaminobutane D 1 binding site moved Diaminopentane A same sites Diaminopentane B 1 binding site moved; 1 binding site splits into two Diaminopentane C one site missing; one introduced Diaminopentane D introduces 1 new binding site Diaminohexane A same sites Diaminohexane B one site moved Diaminohexane C one site missing Diaminohexane D introduces 2 new binding sites Diaminoheptane A introduces 1 new binding site Diaminoheptane B 1 binding site moved; 1 introduced Diaminoheptane C 2 binding sites condense into 1 Diaminoheptane D introduces 1 new binding site Diaminooctane A one site missing Diaminooctane B 2 sites missing; 2 introduced Diaminooctane C 2 binding sites missing Diaminooctane D same sites Diaminononane A same site Diaminononane B introduces 2 new binding sites

21
Diaminononane C one site moved slightly, one site introduced Diaminononane D binding sites become 1, introduced 2 new sites Diaminodecane A 2nd binding site disappears Diaminodecane B one binding site moves Diaminodecane C 2 binding sites disappear Diaminodecane D same sites Diaminoendecane A same site Diaminoendecane B 2 sites introduced Diaminoendecane C introduces 1 new binding site Diaminoendecane D same sites Histone A one site moved, introduces new site Histone B introduces 4 new sites Histone C 1 binding site disappears Histone D introduces 2 new sites Dapsone A introduces 1 new site Dapsone B one site remains the same Dapsone C one site remains the same Dapsone D one site moves p-aminobenzoic acid A one site moves, 1 introduced p-aminobenzoic acid B 3 binding sites are the same p-aminobenzoic acid C same sites p-aminobenzoic acid D one site missing
This table indicates how the presence of the coenzyme affected binding positions.

22
Figure 4. Spermine (Quadrant A) without Coenzyme
Figure 5. Spermine (Quadrant A) with Coenzyme
These figures show that the same binding site exist with or without the coenzyme.

23
Figure 6. Spermine (Quadrant B) without Coenzyme
Figure 7. Spermine (Quadrant B) with Coenzyme
These figures show the addition of a new binding site in the presence of the coenzyme.

24
Figure 8. Spermine (Quadrant C) without Coenzyme
Figure 9. Spermine (Quadrant C) with Coenzyme
These two figures indicate that the presence of the coenzyme takes away a binding position.

25
Figure 10. Diaminobutane (Quadrant C) without Coenzyme
Figure 11. Diaminobutane (Quadrant C) with Coenzyme
These figures show the removal of one site and addition of two more sites in the presence of the coenzyme.

26
DISCUSSION
Polyamine Binding Energies
Overall, it was found that the presence of the acetyl-CoA did not significantly affect the
binding energies of the polyamines. Additionally, all except 15 polyamines had the same
first binding position regardless of the presence of acetyl-CoA. In the cases where the
polyamine did not bind in the same position first, the second or third binding positions
were similar. Compared to spermine, the positions of the ligands were relatively similar.
In only 13 out of the 104 cases, did less than half of the outputs not match up with the
spermine outputs. This indicates that the reaction is most likely a ternary reaction since
the introduction of a coenzyme does not significantly affect the binding energy or
positions.
A direct trend between polyamine size and binding energies was also found. As the size
of the polyamine increases, the binding energies become more negative and therefore
stronger. The binding energies of the polyamines are consistent with that of spermine.
Spermine has a 10-Carbon backbone (with 2 amine groups in the carbon chain and 2
more amine groups on the ends) and its energies are consistent with 1,9-diaminononane
and 1,10-diaminodecane, which are molecules with 9-carbon and 10-carbon backbones
respectively.
GNAT Experiment
Three ligands, lysine, Dapsone, and para-aminobenzoic acid were docked in order to
determine whether or not the enzyme being worked with is in fact SSAT or if it is a

27
GNAT, an enzyme that functions similarly to SSAT. These three ligands were tested
because they are known to bind well with GNAT’s. The binding energies for the three
ligands were more negative than spermine and spermidine, which could possibly indicate
that they bind stronger. However, all the binding energies are so similar, it is not
possibly to say that one definitely binds stronger than another. It is possible to determine,
though, that spermine and spermidine are more specific for this enzyme than the lysine,
Dapsone, and p-aminobenzoic acid. This would indicate that the enzyme is in fact SSAT,
and not a GNAT.

28
REFERENCES
1. Onodera K, Satou K, Hirota H. Evaluations of Molecular Docking Programs for
Virtual Screening. J Chem Inf Model 2007;47:1609-18
2. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking
with a new scoring function, efficient optimization and multithreading. Journal of
Computational Chemistry 2010;31: 455-461
3. Introduction. Python Prescription Virtual Screening Tool. PyRx, 22 September 2011.
Web
4. Dallakyan Sargis. MGL Tools Website-Welcome. MGL Tools. The Scripps Research
Institute, 19 April 2010. Web.
5. Dolphin, W.D. Biological Investigations: Form, Function, Diversity and Process.
2008; 8.
6. Campbell, N.A., Reece, J.B. Biology. 2008; 8
7. Nelson DL, Cox MM. Principles of Biochemistry. 2008; 5.
8. Disease. Centers for Disease Control and Prevention. CDC, 2 November 2010. Web.
9. Epidemiology & Risk Factors. Centers for Disease Control and Prevention. CDC, 9
March 2011. Web.
10. Parasite-Cryptosporidium. Centers for Disease Control and Prevention. CDC, 16
January 2013. Web.
11. Treatments. Centers for Disease Control and Prevention. CDC, 1 March 2012. Web.
12. Yarlett N, Wu G, Waters WR, Harp JA, Wannemuehler MJ, Morada M,
Athanasopoulos D, Martinez MP, Upton SJ, Marton LJ, Frydman BJ.

29
Cryptosporidium parvum spermidine/spermine N1-acetyltransferase exhibits different
characteristics from the host enzyme. Mol Biochem Parisitol. 2007;152:170-180
13. Vetting MW. Structure and functions of the GNAT superfamily of acetyltransferases.
Archives of Biochemistry and Biophysics. 2005;433: 212–226
Related Documents








![DOI: 10.1002/minf.201300143 FindSite: Enhanced ......pare eFindSite to AutoDock Vina,[5] which is one of the most widely used tools for structure-based virtual screen-ing. We show](https://static.cupdf.com/doc/110x72/5e9dec2cbc228c73c608afb6/doi-101002minf201300143-findsite-enhanced-pare-efindsite-to-autodock.jpg)


