Introduction to Short Read Sequencing Analysis Jim Noonan GENE 760

Introduction to Short Read Sequencing Analysis Jim Noonan GENE 760.
Dec 18, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Introduction to Short Read Sequencing Analysis
Jim NoonanGENE 760

Sequence read lengths remain limiting
• For most applications reads are aligned to a reference genome• Short reads contain inherently limited information• De novo assembly of short reads is difficult
Chr1: 249 Mb
249 Mb sequencing read
Current platforms:• A moderate number (~500,000) of long reads (~10 kb)• A very large number (>200 M) of short reads (100 bp)

Determining the identity and location of short sequence reads in the genome/exome/transcriptome
@HWI-ST974:58:C059FACXX:2:1201:10589:110434 1:N:0:TGACCATGCACACTGAAGGACCTGGAATATGGCGAGAAAACTGAAAATCATGGAAAATGAGAAATACACACTTTAGGACGTG
Need a computationally efficient method to perform accurate alignments of millions of reads
Aligning short reads to much larger reference
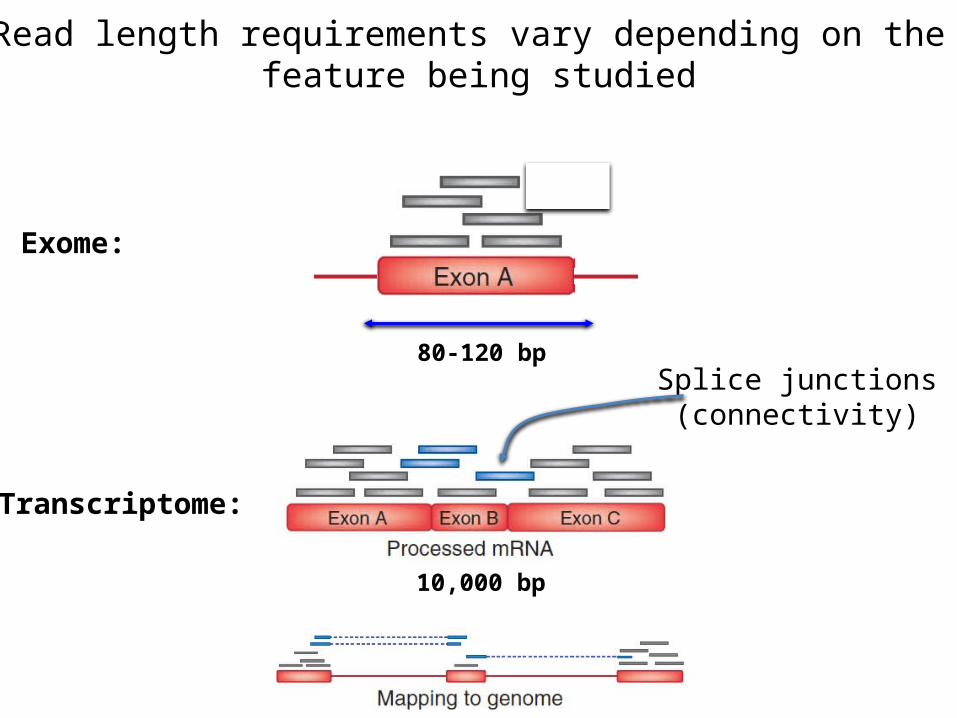
Read length requirements vary depending on the feature being studied
Exome:
80-120 bp
Transcriptome:
10,000 bp
Splice junctions(connectivity)

Determining the identity and location of short sequence reads in the genome/exome/transcriptome
@HWI-ST974:58:C059FACXX:2:1201:10589:110434 1:N:0:TGACCATGCACACTGAAGGACCTGGAATATGGCGAGAAAACTGAAAATCATGGAAAATGAGAAATACACACTTTAGGACGTG@HWI-ST974:58:C059FACXX:2:1201:10589:110434 1:N:0:TGACCATGCACACTGAAGGACCTGGAATATGGCGAGAAAACTGAAAATCATGGAAAATGAGAAATACACACTTTAGGACGTG
Exome or Genome
TranscriptomeConsiderations•Alignment scoring•Source of the reads•Sequencing format (PE or SE)•Read length•Error rates
Aligning short reads to much larger reference

Topics
•Mapability
•Error rates and quality scores for short read sequencing
•Common algorithms for short read sequence alignment
•Scoring short read sequence aligments
•Uniform data output formats
•Scoring alignments

Scoring alignments
TAGATTACACAGATTAC|||||||||||||||||TAGATTACACAGATTAC
TAGATTACTCAGA-TAC|||||||| |||| |||TAGATTACACAGATTAC
Adapted from Mark Gerstein
Correct:
Wrong:
C|C
Match (+1)
Mismatch (-1, -2, etc.)C
T
Gap penalty:
P = a +bNa = cost of opening a gapb = cost of extending gap by 1N = length of gap
A-TAC|||||ATTAC
A--AC|||||ATTAC
Many short read alignment algorithmsallow a fixed number of mismatches

Scoring alignments
TAGATTACTCAGATTAC|||||||| ||||||||TAGATTACACAGATTAC
TAGATTACTCAGA-TAC|||||||| |||| |||TAGATTACACAGATTAC
Adapted from Mark Gerstein
Correct (polymorphism):
Wrong:
C|C
Match (+1)
Mismatch (-1, -2, etc.)C
T
Gap penalty:
P = a +bNa = cost of opening a gapb = cost of extending gap by 1N = length of gap
A-TAC|||||ATTAC
A--AC|||||ATTAC
Many short read alignment algorithmsallow a fixed number of mismatches

Quality scores
A quality score (or Q-score) expresses the probability that a basecall is incorrect. Given a basecall, A:
• The estimated probability that A is not correct is P(~A);
• The quality score for A is Q (A) = -10 log10 (P(~A))
A quality score of 10 means a probability of 0.1 that A is the wrong basecall.
Quality scores are logarithmic:
P(~A) is platform-specific; Q-scores can be compared across platforms.
Q-score Error probability
10 0.1
20 0.01
40 0.0001

Sequencingby synthesiswith reversibledye terminators
1 cycle
Scan flow cell
Add base
Reverse terminationAdd next base, etc.
Error rates in lllumina sequencing reads
Individual synthesis reactions go out of phase

Error rates in lllumina sequencing reads
• Error rates are mismatch rates relative to reference genome
• Reads may be trimmed to improve alignment quality
• Error rates increase with increasing cycle number
• Contingent on reference genome quality

Illumina quality score encoding in FASTQ format(CASAVA v1.8)
>90% Q30 bases in high quality run>80% mappable reads

Sources of error in single-molecule sequencing
Illumina:
PacBio:
TAGATTACACAGATTAC|||||||||||||||||TAGATTACACAGATTAC
Consensus signal
TAGATTA-ACAG-TT-C||||||| |||| || |TAGATTACACAGATTAC
One molecule, one read
Sequence templates multiple times

Mapability
•The genome contains non-unique sequences (repeats, segmental duplications)•Short reads derived from repetitive regions are difficult to map
Chr3 Chr7repeat repeat
Longer reads:
Paired reads:

Mapability scores at UCSC
•The genome contains non-unique sequences (repeats, segmental duplications)•Short reads derived from repetitive regions are difficult to map
36mers, 2 mismatches
75mers, 2 mismatches
100mers, 2 mismatches
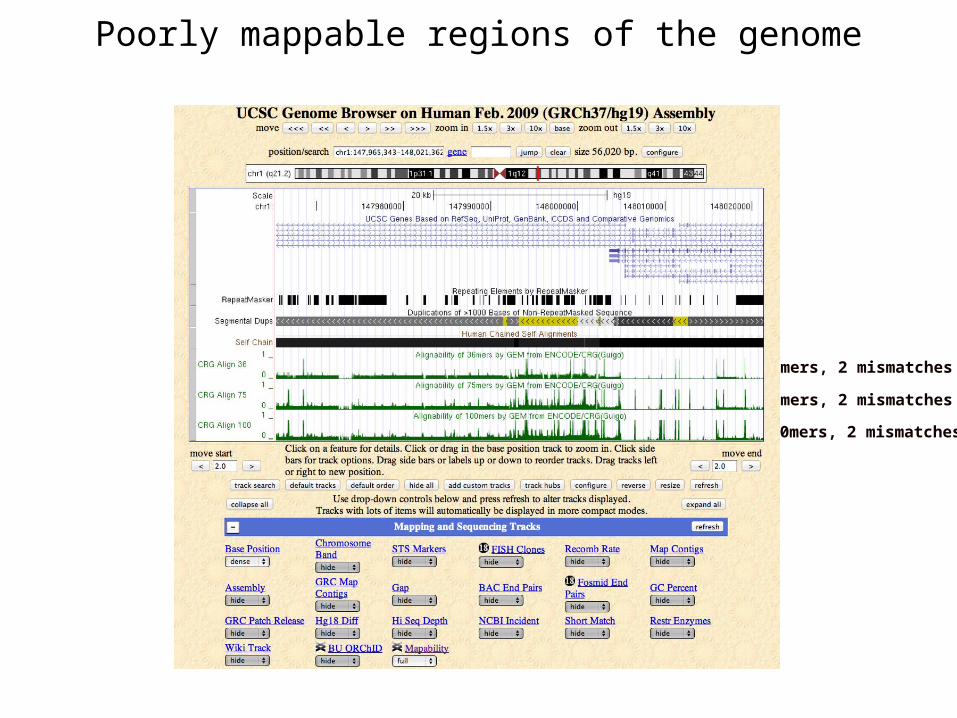
Poorly mappable regions of the genome
36mers, 2 mismatches
75mers, 2 mismatches
100mers, 2 mismatches

Program WebsiteELAND (v2) N/A – integrated into Illumina pipelineBowtie http://bowtie-bio.sourceforge.net/BWA http://bio-bwa.sourceforge.net/Maq http://maq.sourceforge.net/
Common algorithms for mapping short reads to a reference genome
Considerations•Alignment scoring method•Speed•Quality aware•Seeding•Gapped alignment
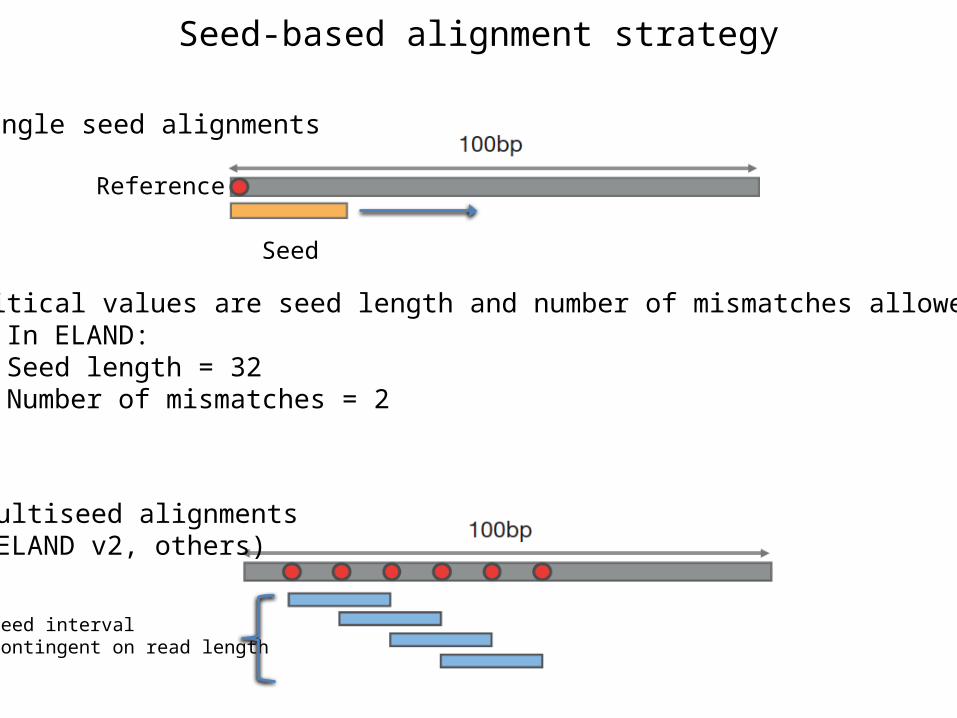
Seed-based alignment strategy
Reference
Seed
Critical values are seed length and number of mismatches allowedIn ELAND:Seed length = 32Number of mismatches = 2
Single seed alignments
Multiseed alignments(ELAND v2, others)
Seed intervalcontingent on read length

Implementation in ELAND v2
A read must have at least one seed with no more than 2 mismatches and no gaps
Gapped alignment: extend each alignment to full length of read, allowing gaps up to 10 bp
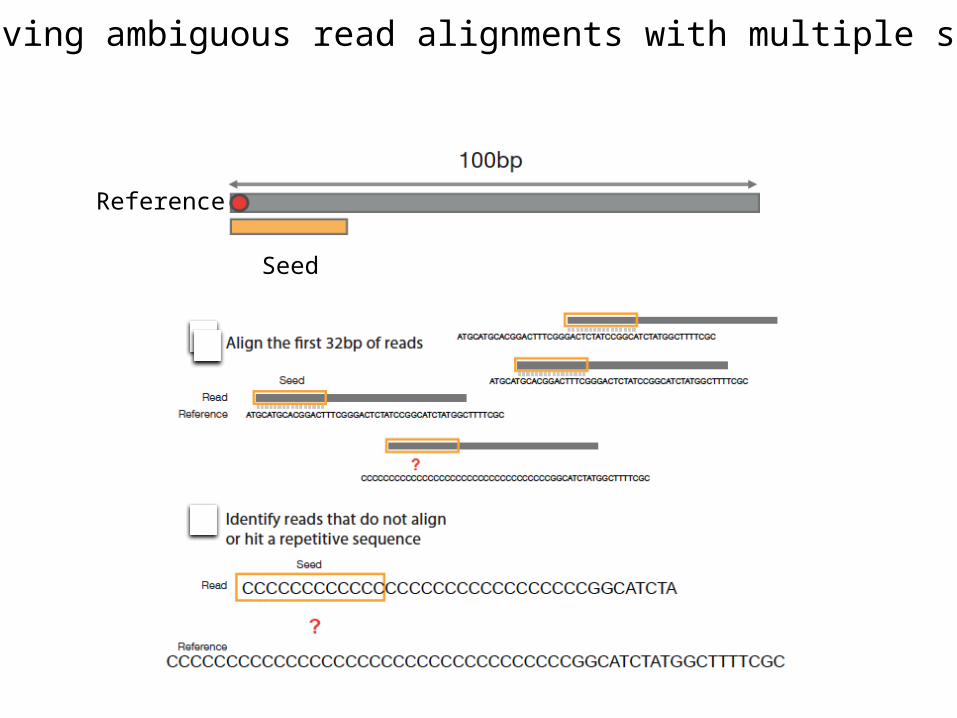
Resolving ambiguous read alignments with multiple seeds
Reference
Seed
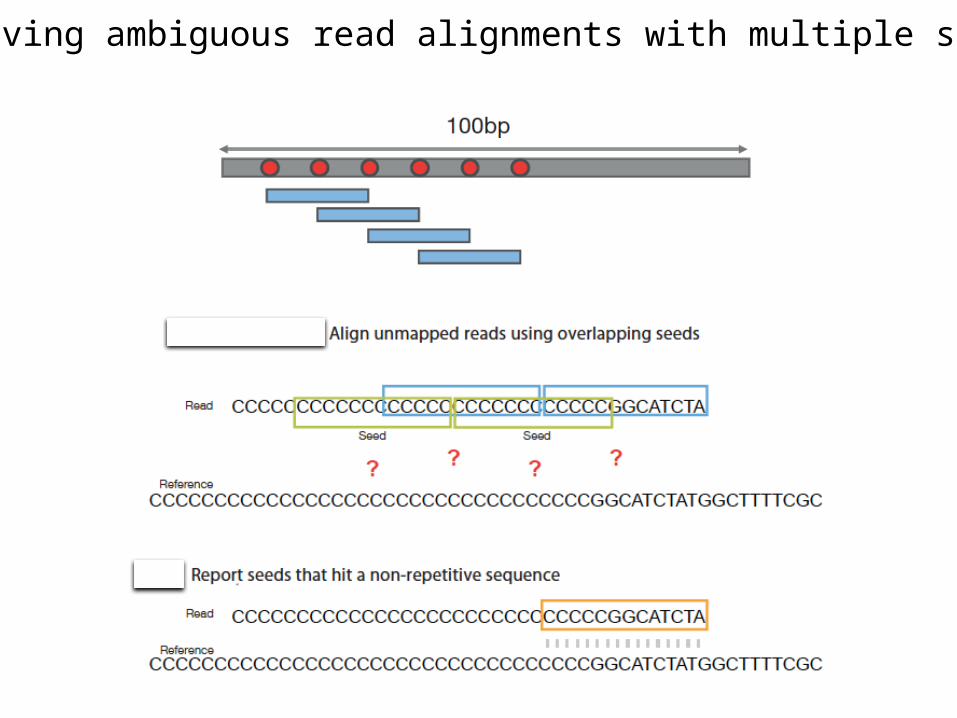
Resolving ambiguous read alignments with multiple seeds

Utility of gapped alignments
RNA-seq Insertions and deletion variants in exome and whole genome sequencing

Mapping paired end reads
Read 1 Read 2
Insert size
Insert size within specified range


ELAND alignment scoring
Base quality values and mismatch positions in a candidate alignment are used to assign a p value
P values reflect probability that candidate position in genome would give rise to the observed read if its bases were sequenced at error ratescorresponding to the read’s quality values
Alignment score for a read is computed from p values of all candidatealignments
If there are two candidates for a read with p values 0.9 and 0.3:
• 0.9/(0.9+0.3) = 0.75, chance highest scoring alignment is correct
• 1- 0.75, chance highest scoring alignment is wrong
• Alignment score = -10 log(0.25) = 6.
BaseSpace
https://basespace.illumina.com/
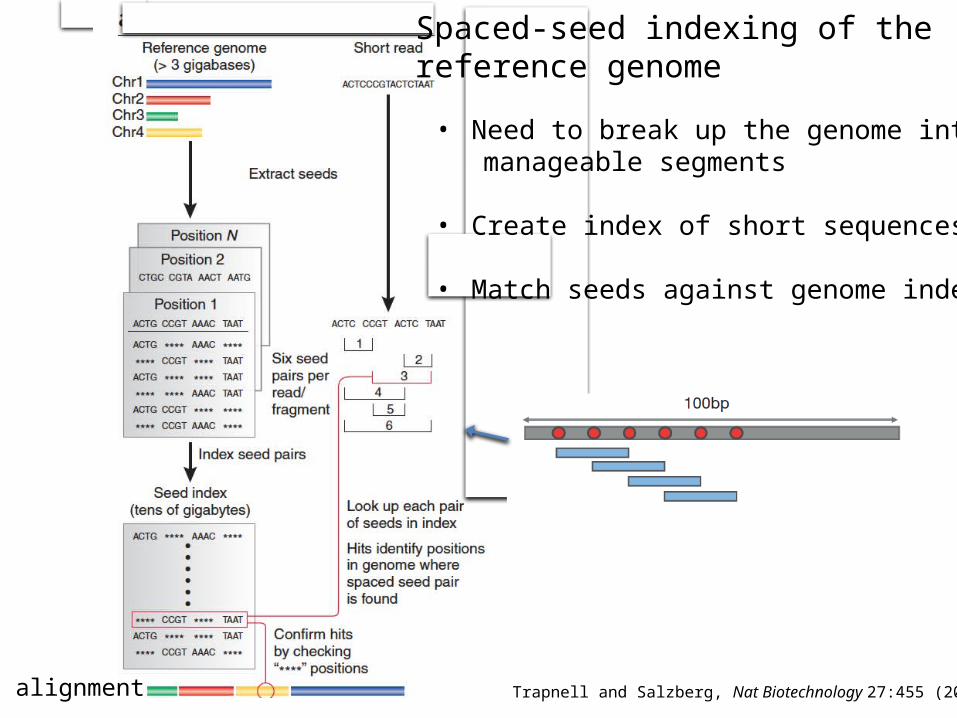
alignment
Spaced-seed indexing of the reference genome
Trapnell and Salzberg, Nat Biotechnology 27:455 (2009)
• Need to break up the genome intomanageable segments
• Create index of short sequences
• Match seeds against genome index

Reference genome indexing usingBurrows-Wheeler transform
alignment Trapnell and Salzberg, Nat Biotechnology 27:455 (2009)
• Reversible encoding scheme• Simplifies genome sequence• Results in “indexed” genome• Very rapid alignments

Bowtie 2
Pre-built Indexed genomes
Bowtie 1 and Bowtie 2indexes are not compatible

Alignments in Bowtie 2
@HWI-ST974:58:C059FACXX:2:1201:10589:110434 1:N:0:TGACCATGCACACTGAAGGACCTGGAATATGGCGAGAAAACTGAAAATCATGGAAAATGAGAAATACACACTTTAGGACGTG
Multiseed alignment (ungapped) Seed length: 16 nt, every 10 nt# mismatches: 0
Mismatch = -6
TGCACACTGAAGGACCTGGAATATGGCGAGAAAACTGAAAATCATGGAAAATGAGAAATACACACTTTAGGACGTGTGCACACTGAAGGTCCTGGAATATGGCGAGAAAACTGAAAATCATGGAAA--GAGAAATACACACTTTAGGACGTG
RefRead
Gap = -11-5 to open
-3 to extend by 1 bp
Seeds are extended (gaps allowed) to generate alignment Match = 2

http://bowtie-bio.sourceforge.net/manual.shtmlhttp://bowtie-bio.sourceforge.net/bowtie2/manual.shtml
Mapping in highly repetitive regions
ELAND is conservative• Non-unique alignments are flagged; only one is reported in export.txt• Post-alignment CASAVA analyses ignore these
Bowtie will report non-unique alignments• User-specified options determine how these are reported

Sequence Alignment/Map (SAM) format
Standard format for reporting short read alignment data• BAM is compressed version
Header
Alignment info
http://samtools.sourceforge.net/

Summary
•Read the material posted for this lecture on the class wiki
•Next week: first Regulomics lecture
Related Documents











