1 | Page Romeo Teodor CRISTINA Professor PhD. DVM, Head of Pharmacology & Pharmacy Depts. to Faculty of Veterinary Medicine Timisoara Introduction in Veterinary Pharmacology Electronic Course Support for Year III – English class students Speciality - Veterinary Medicine Speciality: Veterinary Pharmacology

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
1 | P a g e
Romeo Teodor CRISTINA Professor PhD. DVM, Head of Pharmacology & Pharmacy Depts. to Faculty of Veterinary Medicine Timisoara
Introduction in Veterinary Pharmacology
Electronic Course Support for Year III – English class students
Speciality - Veterinary Medicine
Speciality: Veterinary Pharmacology

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
2 | P a g e
Part I – Veterinary Pharmacology
Scientific Referee
Prof. Univ. Dr.Hc. Alexandra Trif, F.M.V. Timisoara
©2014 All rights reserved
Piracy is theft and criminal law covered!
This work is protected by Copyright
No part of this material may be reproduced in any form, by any mechanical or electronic mean, or stored in a database without prior consent of the author: Prof. Romeo T. Cristina
Editure Waldpress Timisoara is NURC accredited
Computerised Editing: R.T. Cristina Layout: R.T. Cristina Mastering: Youlian©
Descrierea CIP a Bibliotecii Na ionale a României
CRISTINA, Romeo Teodor
/Romeo – Teodor Cristina
Editura Solness Timi oara, 2008
Suport electornic
Introduction in Veterinary Pharmacology,
Partea a I-a. Farmacologia generala
ISBN (13)xxxxxxxxxxx

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
3 | P a g e
Part I General Pharmacology
1. Introduction to Veterinary Pharmacology 1.1. Pharmacology, evolution and subdivisions 1.1.1. Biopharmacy (biopharmaceutics) 1.2. The concept of medicinal product 1.2.1. Relationship: food - drug - toxic 1.2.1. Drugs denomination and classification 1.3. Pharmacopoeia 1.4. Classification of the medicinal active substances 1.5. The veterinarian & the drugs 1.5.1. Drugs Conditioning 1.6. Pharmaco – clinical studies in veterinary medicine 1.6.1. Biomedical research 2. Administration & Drug absorption 2.1. The formulation for administration (dosage) 2.1.1. The correlation between diffusion into the tissues and the effect installation 2.3. Local or topical treatment 2.3.1. The oral way (Per os, p.o. or P.O.) 2.3.1.1. Per lingual or sublingual way 2.3.2. The ruminal space 2.3.3. Gastric mucosa in monogastrics 2.4. Intestinal mucosa 2.4.1. The intestinal mucosa and absorption 2.5. The large intestine absorption 2.6. Administration on the external ways 2.6.1. Inhalation way 2.6.2. Intratracheal injections 2.6.3. Absorption through the apparent mucosa 2.6.4. Absorption through the skin 2.7. The parenteral ways 2.7.1. Intradermal way (i.d.) 2.7.2. Subcutaneous way (s.c.) 2.7.3. Intramuscular way (i.m.) 2.7.4. The intravenous way 2.7.6. The intraperitoneal way (i.p.) 2.7.7. Intrathoracic and intracardiac injections 2.7.8. Intrathecal injections (subarahcnoidal) 2.7.9. Epidural injections 2.7.10. Intraarticular injections 2.7.11. Rectal, vaginal and intramamar injections 3. Drug blood transport & Drugs distribution 3.1. Factors that influence drug transport

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
4 | P a g e
4. Diffusion in the body's hydric regions 4.1. The role of cell membranes 4.1.1. The diffusion mechanisms 4.2. Relation pH, pKa and drug diffusion 4.3. Diffusion through barriers 4.3.1. Haemato-encephalic (Blood-brain) barrier 4.3.2. Hemato-oftalmic barrier 4.3.3. Placentary barrier 4.3.4. Coetaneous barrier 4.4. Drugs’ Redistribution 4.4.1. Consequences of uneven distribution 5. Drug-receptor binding 5.1. Preliminary aspects of drug-receptor interaction 5.1.1. Activity and receptor’s characterization 5.1.2. Receptors’ mode of action 5.1.3. The nature of receptors 5.1.4. Isolation and receptor’s identification 5.1.5. The definition of agonists and antagonists 6. Coupling response quantification 6.1. Clark's theory (of occupation) and its variant 6.2. Ariens theory 6.2.1. Stephenson’s theory 6.3. Paton’s theory 6.4. Activation theory and other recent postulates 6.5. Enzymology theories 7. Drug metabolism 7.1. Factors that influence drug metabolism 7.1.1. Physiological (pharmacokinetic) factors 7.1.2. Urinary pH 7.1.3. Coupling with plasma proteins 7.1.4. Enzymatic induction 7.1.5. Enzymatic inhibition 7.2. Animal related factors 7.2.1. Species 7.2.2. Individuality / breed 7.2.3. Age 7.2.4. Gender 7.2.5. Gestation 7.2.6. Feeding 7.2.7. Health status 7.2.8. Genetic factors 7.3. Exogenous Factors 7.3.1. The circadian rhythm 7.3.2. Exogenous compounds 7.3.3. Stress factors

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
5 | P a g e
8. Stages of metabolism 8.1. Drug biotransformation 8.1.1. Microsomal biotransformation 8.1.1.1. Microsomal oxidation 8.1.1.2. Microsomal reduction 8.1.2. Non microsomal biotransformations 8.1.2.1. Non microsomal oxidation 8.1.3. Biotransformation by the action of digestive microflora 8.2. Conjugation of drugs 8.2.1. Acetylation. 8.2.2. Methylation 8.2.3. Sulphono-conjugation 8.2.4. Glucuronide conjugation. 8.2.5. Peptide conjugation. 8.2.6. Mercaptation. 10. Elements of theoretical pharmacokinetics 10.1. Pharmacokinetics modelling 10.1.1. Kinetics redundancy 10.1.1.1. The monocompartmental open model 10.1.1.2. The bicompartmental model 10.1.1.3. The tricompartmental model 10.2. Bateman’s function 10.2.1. The absorption and elimination constants (invasion and evasion) 10.2.2. The minimum blood level 10.2.3. The discontinuation of a drug administration 10.2.4. Enzyme induction and blood level 10.3. The parameters of pharmacokinetic quantification 11. Main pharmacodynamic factors that influence the drugs effect - dose theory 11.1. Factors establishing a dose 11.1.1. Genetic factors 11.1.2. Susceptibility 11.1.3. Species 11.1.4. Anatomy of the digestive system 11.1.5. Age 11.1.6. Gender 11.1.7. Time administration and pathology 11.2. Tolerance and intolerance 11.2.1. Therapeutic indications 11.2.2. Concomitant drug therapy 11.2.3. Amplified response 11.2.4. Diminished response 11.2.5. Incompatibilities 11.2.6. Amplified toxicity 11.2.7. Reduced toxicity 11.3. Factors determining the frequency of administration 11.3.1. Concentration stability 11.4. Establishing rates of drug dosing

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
6 | P a g e
11.5. Establishing the frequency of administration 11.5.1. Establishing intravenous infusion rate 11.5.3. Plateau effect 11.6. The effect of repeated administrations 11.7. Stereo specificity of drug action 11.7.1. Different spatial structure 11.8. Zero-order kinetics influence 12. Other pharmacodynamic elements that can influence the drugs’ effect 12.1. Drug residues 12.2. The risk - benefit ratio 12.2.1. Dose-effect relationship 12.2.2. The potency of a drug: 12.2.3. Latency and intensity 12.2.4. The duration of action of a pharmacon 12.2.5. The duration of drug effect 12.2.6. The plasma concentration 12.2.7. First-pass effect 12.2.8. Veterinary pharmacovigilance 13. The animal body's response to medication – Main pharmaceutical aspects 13.1. Practical pharmacokinetic issues of drug administration and absorption 13.1.1. Bioavailability of a.u.v. drugs 13.1.2. Polymorphism: 13.1.3. Particle size 13.2. Bioequivalence of a.u.v. drugs 14. Practical elements of veterinary therapeutics 14.1. Drug formulation kinds 14.1.1. Drug combinations 14.1.2. Drug interactions 14.3. Pharmacokinetic interactions 14.3.1. Interactions on absorption phase 14.3.2. Interactions on the distribution phase 14.3.3. Interactions on metabolization phase 14.3.4. Interactions on urinary excretion phase 14.4. Interactions of pharmacodynamic order 14.5. Synergistic combinations 14.5.1. Direct synergism 14.5.2. Drug potentiation 14.6. Attenuation associations 14.7. Indifferent associations 14.8. Antagonistic associations 14.8.1. Biological antagonism 14.10. Pharmacodynamic ambivalence 14.11. Undesirable reactions to medications 14.11.1 Adverse reactions

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
7 | P a g e
1. Introduction to Veterinary Pharmacology

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
8 | P a g e
Definitions
Pharmacology (pharmakon = drug; logos = knowledge) is the science concerned with the study
of drugs, including their origin, physic and chemical properties, composition, uses, modes of action
and their effects on living organisms. Pharmacology can also be defined as the study of the interaction
between pharmacons and biological systems.
Pharmacons (or drugs) are chemical agents that affect the function of biological systems. The
Veterinarian is interested in the rational and optimal use of the drugs for the prevention, diagnosis and
treatment of disease. This branch of pharmacology is called pharmaco-therapeutics. In the middle ages
the discipline was called De materia medica and it included elements of:
pharmacology,
therapeutics and
pharmacy
It displayed advanced principles of therapeutics, and was divided into two categories:
rational (when the nature of disease and mode of action of the substance was known).
empirical (if the above knowledge was nonexistent or incomplete), which became an
experimental field for clinicians.
Two systems of medical practice have established themselves over the centuries, which are also
generally accepted to this day. These are:
allopathy, and
homeopathy
Allopathy (allos = other; pathos = disease). Principle of allopathy it was introduced in 400 BC,
by the famous Greek physician Hippocrates of Kos, called the “Father of medicine”. Represents a
treatment system based on the principle “Contraria contrariis curantur” (opposites are cured by
opposites), which advocates the use of drugs that produce effects, opposite to the symptoms.
Principles of homeopathy (homoios = similar; pathos = disease) were enunciated at the end of
XVIII century by the Saxon doctor Samuel Hahnemann (1755-1843) (who was a librarian at the
Bruckenthal Palace in Sibiu). Homeopathy is based on the principle: “Similia similibus curantur”
(likes are cured by likes), which advocates the use of drugs that produce effects similar to the
symptoms. It is the exact opposite to allopathy and is based on three fundamental principles:
similarity
infinitesimal dose (high dilutions);
treatment individualization

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
9 | P a g e
1.1. Pharmacology, evolution and subdivisions
Pharmacognosy (pharmakon = drug; gnosis = knowledge). Is the branch of pharmacology
concerned with medicinal substances obtained from plants or other natural sources, their main
characteristics or origin of the medicinal substances, which can be: vegetal, animal or mineral
Pharmaceutical chemistry deals with the composition and preparation of medicinal active
substances (drugs) and studies their physico-chemical properties.
Pharmacodynamics (dynamis = power). The branch of pharmacology concerned with the effects
of drugs and the mechanism of their action.
Experimental pharmacodynamics. Is the study of drugs effect on laboratory animals, or on the
organs and isolated systems, which serves a research objective.
Clinical pharmacodynamics. It follows the drugs effect during the treatment period in animals
or humans.
Pharmacokinetics (kinetikos = motion, movement). Important branch of pharmacology
concerned with the drugs circulation within the body, and the determination of the fate of all
substances administered externally to a living organism, in order to describe how the body affects a
specific drug after administration.
Pharmacometrics It analyze interactions between drugs and patients and study methods of
measuring the intensity of drug effects.
Pharmacotherapy (therapoeia = care) or clinical pharmacology, studies the clinical application
of drugs in different diseases, insisting on the mechanism of action, therapeutic efficacy, adverse
reactions and toxic potential. Therapy is a wider notion that includes other non-pharmacological
methods of treatment intended to relieve or heal a disorder (physical agents, diet… etc).
Prescribing Advise and authorize the use of a medicine or treatment, especially in writing. It has
two subdivisions:
Pharmacography (graphein = to write), studies the prescription of medicines in the form of a
recipe.
Pharmaceutical technique (or galenic technique) Studies the drug formulation and preparation
methods.
Pharmacotoxicology. It deals with the study of acute or chronic intoxications and the adverse
reactions produced by the drugs.
Molecular pharmacology. Is a branch of pharmacology which is concerned with the study of
pharmacology on a molecular basis.
Pharmacogenetics. It is a branch of pharmacology, concerned with the effect of genetic factors
on reactions to drugs. Extensive research in this area has led to the emergence of new sub-branches of
the pharmacology domain like: immunopharmacology, chronopharmacology, neuropharmacology,
citopharmacology, biochemo-morphology etc.

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
10 | P a g e
1.1.1. Biopharmacy (biopharmaceutics)
Deals with the study of:
physico-chemical properties of the biologic active substances,
them conditioning form and administration,
them pharmacokinetic parameters
the obtained bio-pharmacologic effects.
Bioavailability is a basic notion of biopharmacy, which refers to the proportion of a drug or other
substance which enters the circulation when introduced into the body and so is able to have an active
effect.
1.2. The concept of medicinal product
1.2.1. Relationship: food - drug - toxic
By food we understand generaly: “any nutritious substance of vegetal, animal or mineral origins,
which enters the body’s metabolism, in order to maintain life and growth”.
The Drug as defined by WHO (World Health Organization), means: “any product used in
diagnostics, treatment, attenuation or prevention of diseases and abnormal physical states, or their
symptoms, in humans or animals”.
A medicine or other substance which has a physiological effect when ingested or otherwise
introduced into the body in order to:
a) diagnose, cure, mitigate, treat or prevent diseases.
b) recognize and affect the structure or function of organic structures.
So, the pharmacon is any biologically active substance or product used or proposed for use, in
order to influence or investigate physiological systems or pathological states, in the patient's benefit.
An “ideal” drug, will present:
an accurate activity,
a known mechanism of action,
a constant effectiveness,
the absence of adverse effects
economic accessibility.
Drugs (medicines) can be obtained from the following sources: vegetal, animal, mineral and
synthetics By toxic we understand: “any substance which introduced into the body produces general
disorders known as intoxication”. All drugs which are absorbed in the body can become toxic, when
significantly exceeding the therapeutic dosages.
Long before the appearance of modern pharmacology, Paracelsus (1493-1541) showed that all

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
11 | P a g e
substances are “poisons” and everything depends on the dose affirming that: “Dosis sola facit
venenum” (The dose alone makes the poison).
1.2.1. Drugs denomination and classification
Questions that pharmacologists are preoccupied with:
what pharmaceutical preparation should be used?
what is the optimal dose?
which is the optimal frequency of drug administration?
The answers to these questions depend on the pharmacist and the manufacturer's ability to prepare
compounds from raw materials and to calculate the correct dosages, so that further recommendations
can be made. This ideal has been achieved by standardizing drugs and remedies. The essential
elements of such a system are:
definition of tests in order to establish identity, purity and strength of a medicinal source, of a
substance or a preparation.
Recommendations on dosage, administration frequency and indications for each drug.
However there it is still a high degree of confusion over drug nomenclature, because each
chemical may be known under a variety of different names worldwide.
The “blame” lies on the drug manufacturers who, in order to protect and standardize their
products, consider convenient to use brand names or trademarks to name their products. Brand names
are, most of the time, registered as a trademark.
Thus, chemical compounds with different formulations, can be produced in a number of unrelated
names by several manufacturers. An even greater confusion is created by the fact that the same drug
can be used as a component in a number of compounds that contain multiple active ingredients. In an
attempt to clarify this situation, the drafting committees of pharmacopoeia give each compound an
accepted name (known as official or generic name).
Most of the time the approved name, is an abbreviation that derives from the chemical name of
the substance, because most of the time, chemical names are long and difficult to memorize.
There are several "versions" of the names used by the chemists.
Therefore a compound can have multiple chemical names (different, but correct) (therefore, the
best solution is the one accepted and internationally approved name).
Due to these considerations we see a multitude of drug names (received after various criteria). For
example, medications extracted from vegetal drugs have names close to the plants or yeasts from
which they are extracted:
atropine (Atropa belladonna),
strychnine (Strychnos nux vomica),
caffeine (Coffea arabica),

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
12 | P a g e
digitalin (Digitalis purpurea),
penicillin (Penicillium notatum,…),
streptomycin (Streptomyces griseus)…. etc.
The chemical name. It is referring to the chemical makeup of a drug rather than to the advertised
brand name under which the drug is sold. (ex: phenyl-ethyl barbituric acid is the chemical name of
barbiturate derivative, known in over 120 commercial names).
Officinal name. Is the name provided by the pharmacopoeia and is expressed in Latin (ex:
Coffeinum et natrii benzoas for caffeine sodium benzoate). The officinale name it is used mostly by
researchers and by those working in the preclinical stage.
To put order into medicine nomenclature, the W.H.O. through its specialized committees, has
agreed on an easier to remember, Common International Name (or DCI) for each substance, based on
the chemical structure or on other criteria (ex: aminophenazonum for piramidone, methenaminum for
urotropine, pethidinum for mialgin, etc.)
1.3. Pharmacopoeia
Pharmacopoeia is the basic book for the preparation of medicinal forms, whose name derives
from the Greek words: pharmacon = remedy and poise = to make.
Pharmacopoeia can be considered an official publication containing a list of drugs, their formulas,
methods for making medicinal conditionings, and other related information.
The first reference dates to 2100BC in Sumer (Pharmacopoeia from Nippur, written on burned
clay). In Japan the first pharmacopoeia appeared around 900 AD, describing 1025 products, from
Chinese sources.
The first Arab pharmacopoeia includes over 200 medicinal plants, many still in use today.
The first European Pharmacopoeia appeared in the late XVII and early XIX centuries. In 1865
the first International Congress of Pharmacology took place in Paris, France where the need of a
unitary Pharmacopoeia was determined for the first time.
The first Romanian Pharmacopoeia appeared in 1862, during the ruler Alexandru I. Cuza,
under the care of Constantinos C. Hepites (a Greek origin pharmacist who opened his first pharmacy
in Iasi), being one of the first works of its kind in Eastern Europe.
The ancient pharmacopoeias were abundant in preparations of natural origin, mainly vegetal.
The development of biological simulation systems on which the expressions of potency were based,
was a major contribution from the pharmacologists.
The first edition of the Veterinary Pharmacopoeia appeared in 1977, and was published in Great
Britain.
In the USA the equivalent of the European pharmacopoeia is the United States Pharmacopoiea
National Formulary (USP).

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
13 | P a g e
The biological simulation will continue to be a standard methodology in the analysis of
qualitative and quantitative pharmacology for years to come. Quantitative biological simulation
expresses: “the potency of a batch of medicinal products in relation to the ability to produce selective
biological responses, related to a standard preparation of the same product”
The first synthetic organic drugs introduced in medicine were volatile anesthetics, followed by
phenolic antiseptics. Another big step was the molecular modification of natural products (ex: 6-
aminopenicillanic acid, product of fermentation, which was the starting point for semi synthetic
penicillins).
1.4. Classification of the medicinal active substances
Medicinal substances are categorized by origin in:
vegetal,
animal,
mineral or
synthetic.
At the present, most drugs are either synthetic or of vegetal origin.
From a toxicity point of view, drugs are divided into three major groups:
Venena (highly poisonous). Includes the toxic substances, with a very strict regime of
keeping, release, use, and which are usually to be kept locked away in special storage places.
In these medications the toxic dose is very close to the maximal therapeutic dosage and is
usually expressed in milligrams or fractions of. Drugs in this group require special recipes to
be released.
Separanda (to be kept separately). Includes highly active substances whose manipulation and
use are highly dangerous. They do not have the same degree of toxicity as the venena group,
but their administration requires strict supervision. And they also must be kept locked in
separate cabinets.
Anodina (anodyne, which means “painless” or in this case “harmless”). Includes non-toxic or
substances of reduced toxicity, generally without risk in actual use.
By the prescribing and manufacturing way, medicinal forms can be classified as:
Magisterial. In pharmacy, after a doctor's prescription, composition can be different in each
case.
Officinal. In pharmacy, the prescriptions from the Pharmacopoeia have a fixed composition.
These are prescribed by enouncing the exact name, without explanation of the formula
Standardized (pharmaceutical specialty, industrial medicines). They are industrially made, in
drug factories and have a fixed composition and preparation.
By drug formulation, we understand: „the finite form of presentation of a drug for

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
14 | P a g e
administration”. From this point of view drugs are:
Solid: powders, granules, tablets, pills, bolts, capsules, etc.
Soft: ointments, pastes, plasters, electuaries.
Liquid: - of extraction (macerates, infusions, decoctions, tinctures) or
- of preparation (molecular solutions, colloids, mixtures, emulsions).
The biologic drug is the product containing biological substances that are used for: diagnostic,
prophylactic and/or - curative purposes. This category includes:
serums,
vaccines and
immunostimulating products.
1.5. The veterinarian & the drugs
The clinician characterizes a drug based on its effect (ex: bacteriostatic, diuretic, stimulant etc.),
based on the symptoms from the indications for use (ex: analgesic, antacid, antispasmodic etc.).
The chemist is more “interested” in the chemical structure than in the activity of the pharmacon.
Activity of the drug often forms the basis for different criteria of classification. For example, to
describe a drug as being a surfactant, diuretic osmotic, emollient etc. reference is made on their
physical terms. Description of a drug as being for example: parasympathomimetics, adrenergic,
neuromuscular blocking agent require a functional physiological terminology.
Another but now, old classification was based on the source and preparation of the product (for
example, identification by naming the plant sources and vegetal structure ex.: Gentiana root
(Gentianae), Juniper berries (Juniperis), chrysanthemum flowers (Pyrethrum).
Drugs developing with similar activities, led to obtaining of the type-compounds, against which
new compounds are compared. This practice led to the expression of terms such as: histamines,
atropines, chlorpromazine etc.
The discovery that drugs act by binding to the active macromolecular sites and the development
of radio labeled ligands for these sites, made possible the use of ligands for revealing unidentified
binding sites, or the use of one binding site for the identification of endogenous ligands (ex:
enkephalins, opioid receptors, etc.).
1.5.1. Drugs Conditioning
Knowledge of the molecular structure of drugs allows observations regarding the:
conformation of the sites at which level they link and act,
offers a basis in order to suggest hypothetical structures of drugs with “high potency”,
selective activity and specific antagonism.

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
15 | P a g e
In order to facilitate discovery and to lower the cost of new remedies, computerized techniques
are employed in an attempt to specify the best possible parameters, before the synthesis of a molecule.
By doing this, it's possible to recognize the portions of the drug molecule responsible for directing
the therapeutic action, and synthesize only the substances that correspond in this regard, excluding
those that prevent the drug association with its molecular target.
When the coupling is engaged in only a small portion of the molecule, specificity will allow the
identification of a group of structurally different compounds, with common biological activity. This
creates the potential for discovering structurally simpler analogs, easier to synthesize (ex: pethidin
instead of morphine).
The opposite phenomenon is increasing the size (volume) of the molecule and therefore, convert a
product into an antagonist (ex: beta blocker drugs), or make the part of a structure that is uncovered
for unwanted enzymatic attack, inaccessible, for example, the modification of the natural product (ex.
penicillin in semi synthetic penicillin that is resistant to penicillinase attack).
These indicators are used in some methods based on analytical regression, defining correlations
between them and the activity of the drug (ex: Hansch analysis and Hammet correlations).
These have established QSAR (Quantitative Structure Activity Relations)
The presence of a drug in the body and the resulting chemical responses, are of a great
importance to clinical utility. Knowledge gained in this field enables administration of drugs which are
biologically inactive until their activation in the body and sometimes at the site of action.
Such drugs are referred to as pro-drugs. It is obvious that rational therapy can only exist when
establishing a certain diagnostic procedure. However, the effective use of drugs involves a little more
than selecting the best drug and the use of a potent formulation.
After the diagnosis, before administrating any drug, the clinician has to consider: benefits /
disadvantages; stopping the treatment with a drug and/or switching to another; correct dosage which
will lead to the desired effect
Pharmaceutical Science is evolving rapidly and constantly. Currently the drug market faces an
excess of “copies” of “renowned” preparations (cases where patent copyright infringement is
involved). Unfortunately, the sales of a product depend directly on advertising and not on its qualities.
This is where “aggressive advertising” interferes.
1.6. Pharmaco – clinical studies in veterinary medicine
1.6.1. Biomedical research
In human and / or animal subjects the recognized scientific rules must be followed.
The research has to be based on laboratory studies on a sufficient number of animals and
complete knowledge of the literature.

Introduction in Veterinary Pharmacology Chapter 1 Romeo – Teodor CRISTINA
16 | P a g e
Planning and conducting the experiment
A research protocol made by a specialized committee must be established, (this committee must
be neutral to the experiment and is empowered to supervise, make comments and give advice
regarding the experiment).
Any project must be preceded, by establishing with certainty the risks involved, in relation to the
benefits they bring to the subjects.
Veterinarians should be cautious when performing experiments whose risks cannot be assessed.
Any experiments, in which potential risks outweigh the potential positive results, should be
abandoned. Veterinarians have the duty to publish the experiment results, unaltered.
Clinical research
Concerning treatments, the practitioner must feel free to use new diagnostic and therapeutical
methods, when in his opinion they increase the chance of survival or cure, or reduce suffering.
Possible advantages, risks or side effects of new methods must be reported to the benefits of the
best known methods. For every medical experiment, each patient or control group must be provided
with the best methods of diagnosis and therapy. A veterinarian can perform clinical trials in order to
obtain new scientific information, only when these experiments are in accordance with the medical act
itself.
Placebo therapy in veterinary medicine
The therapeutic effect of the placebo drug is dependent on how is administered by the physician:
optimistic or pessimistic.
Administration of placebo drugs (apparent drugs) to humans, may lead to improvement or
healing, but in veterinary medicine, especially for pets where the level of owner - animal affection is
raised, the therapeutic effect may be sometimes dependent on the owner’s state of mind. In these
situations, one can speak of "placebo therapy“(closely dependent on the owner's affection and
personality which can be influenced by the suggestion of the veterinarian). Placebo substances in
veterinary medicine may be used only in two circumstances:
when a real pharmacotherapy is not necessary;
the veterinarian is aware that he/she can perform psychotherapy on the owner whit the help of
the drugs administered to the pet.
However : in contrast to placebo therapy in human medicine, which could be monitored in
veterinary medicine it was not possible to monitor the occurrence of conditioned reflexes or some side
effects in animals.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
17 | P a g e
2. Administration & Drug absorption

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
18 | P a g e
The Invasion and Evasion phases scheme (Cristina RT, 2000)
2.1. The formulation for administration (dosage)
Is the pharmaceutical preparation in which the active ingredient can be found and which is
administered in the body as administering formulation. In order to obtain therapeutic effects, the drug
should come in contact with the body, specifically with the sensitive cells (responsible for the effect)
of the body. This contact can be accomplished using a variety of ways of administration.
The ways of drug administration are chosen depending on the:
substance’s physical-chemical properties
place of action
animal’s condition and
the speed and intensity with which drugs are expected to act
The period that elapses from: the moment the substance is administered until the substance starts
acting = the latent period
The latent period is based on the way of administration and depends on:
absorption speed,
transport time in the organic liquid mediums,
diffusion duration in tissues,
time needed to produce biological changes that will trigger the therapeutic effect.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
19 | P a g e
The correlation between diffusion into the tissues and the effect installation (Cristina RT, 2000)
2.1.1. The correlation between diffusion into the tissues and the effect installation
The way of drug administration has a particular importance for the success of a treatment and
should be chosen wisely. If an urgent pharmacodynamic action is needed, the I.V. way is preferred.
But at the same time, it is to be considered, that in this way drugs can come very quickly in
contact with the tissues. So, the action can become brutal and potentially dangerous. Some drugs can
be administered only in one way, (for example: Suzotril, a sulphonamide can be administered only
I.V., Acaprin, a chemiotherapic only S.C.).
Sometimes the drug effect varies depending on the way of administration. For example,
magnesium sulphate, administered in an: enteric way = generates a purgative effect, while in a
parenteral (depending on dose) = the CNS depressing effect.
2.3. Local or topical treatment
This administration way is represented by:
application of powders and ointments on the skin,
instillation of drops in the eyes and ears,
injection through mammelons with solutions and / or soft formulations,
introduction of pessaries in the lumen of uterus.
Topical administration puts the remedy in direct contact with the site of action in the highest
possible concentration, reducing the risk of damage to the other organs. In many cases the absorption
of the drug at the administration site, is not desired.
On the contrary, when a generalized or systemic response, is followed, or when the target organ
is far from the administration site, the drug absorption is essential.
Systemic effect can be achieved by oral or parenteral administration of the medicinal preparations.
As such, the method of the drug preparation will determine the route of administration.
For example, percutaneous absorption is sufficient to secure the systemic effect of the pour-on
(ex: Ivomec pour-on) ectoparasiticides.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
20 | P a g e
Drug formulations are prepared by taking into account, biopharmaceutical and pharmacokinetic
considerations. The selection of the remedy of choice is made by the clinician, depending on the
intensity and duration of the desired effect. Each administration route has its own advantages and
disadvantages. The nature and number of different membrane barriers that the drug must cross,
largely influences the absorption rate.
Doses can vary depending on the administration route. Sometimes these variations are very high:
for example strophantines dose in rabbits / kg.bw. is: 0.0003 g, for the i.v. way, 0.001 g, for the s.c.
way and 0.040 g, for the oral way. This ratio of 1:3:133 between these ways of administration is
suggestive!
The ways of administration are classified into:
internal (oral and rectal) and
external (all other pathways).
There are: natural and artificial ways of administration
The natural ways consist of drug administration’s to the surfaces of the body that physiologically
come in contact with the exterior environment.
These are skin and mucosa (divided in):
apparent (conjunctive, nasal, oral, vaginal);
unapparent (bronchial, tracheal, esophageal, gastric, intestinal).
The mucous ways are the following:
digestive,
respiratory,
genito-urinary,
galactophore and
conjunctive.
Artificial ways are known as: parenteral (para = beyond; enteron = intestine) and these ways are
artificially created for drug introduction into the body. They involve forming of the continuity
solutions in which active substances will be introduced into the dermis, subcutaneously, in muscles,
veins, arteries, serous cavities and other different organs: i.d. or I.D., s.c., or S.C., i.m. or I.M., i.v. or
I.V., • i.a. I.A., intraosseous, intraarticular, intrasynovial etc.
Artificial ways started in XIX century, being used once with the invention of the syringe by the
Czech Pravaz (in 1835). This way was meant to put the active substance in direct contact with the
tissues inside the body, avoiding the external barriers.
Absorption is the process in which the active substance: emerges out from its formulation and
passes from the administration site into the bloodstream. Absorption has a more important role when
the pharmacons are not injected directly into the bloodstream and relies on the physical processes of
diffusion and distribution, which are influenced by active biological processes (ex. transport against

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
21 | P a g e
the concentration gradient of the potassium, selective transport of the carbohydrates etc.).
The absorption rate depends on:
way of administration,
preparation form and
The drug’s physicochemical properties.
The absorption of the drug is considered complete when it reaches the site of action or the
bloodstream. In this aim the main factors that favor the absorption:
molecule size,
low polarity,
high liposolubility,
rich blood irrigation and
good permeability at administration site.
2.3.1. The oral way (Per os, p.o. or P.O.)
This administration way is more often used in human medicine, but it is also common in
veterinary medicine, where in most cases, a forced administration must be performed. Oral way is
useful for tasteless drugs or with a taste that can be easily masked, especially for mass administration
(in forages or in water). Orally can be administered the:
biostimulators,
anthelmintic and coccidiostatic substances,
• antiinfectious ones,
vitamins,
minerals, etc
Oral way can have also disadvantages. In the digestive tract, drugs can suffer modifications like
for example: penicillin G, adrenaline, most hormones; inactivations determined by the gastric acid),
digestive mucosal modifications like gastroenteritis lead to absorption rate modification, introducing
the phenomenon of malabsorption
The main mechanisms of absorbtion (Cristina RT, 2000)
Main mechanism Organ involved
Passive diffusion Mouth (M), Stomach (S), Small intestine (Si), Large intestine (Li), Rectum (R)
Absorption by connection M, S, Si, Li, R
Active transport S, Si, Li
Passive transport Si
Tonic Si
Pinocitosis Si, Li, R

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
22 | P a g e
Remedies for oral administration in veterinary medicine include: solutions, suspensions,
mixtures, pills, capsules, tablets, powders, granules, boluses and premixes.
Oral mucosa though it is not a mucosa with an absorptive profile, it allows the absorption of
hydrosoluble substances
Watering System VAL adaptable to Medicator type system for drug administration
Drug administration into milk in calves and administration with dosing piston and simple drencher in sheep
Between the portions of oral mucosa, sublingual mucosa, thin and richly vascularized, absorbs
the best.
2.3.1.1. Per lingual or sublingual way
This method is used exclusively in human medicine for a relatively low number of substances (for
example: nitroglycerin, trinitrin, isoprenaline, sexual hormones etc).
Drugs that are absorbed in the oral cavity escape to the gastric acid. In the veterinary medicine the
oral cavity it is used in order to obtain a local effect in the case of oral cavity diseases or pharynx.
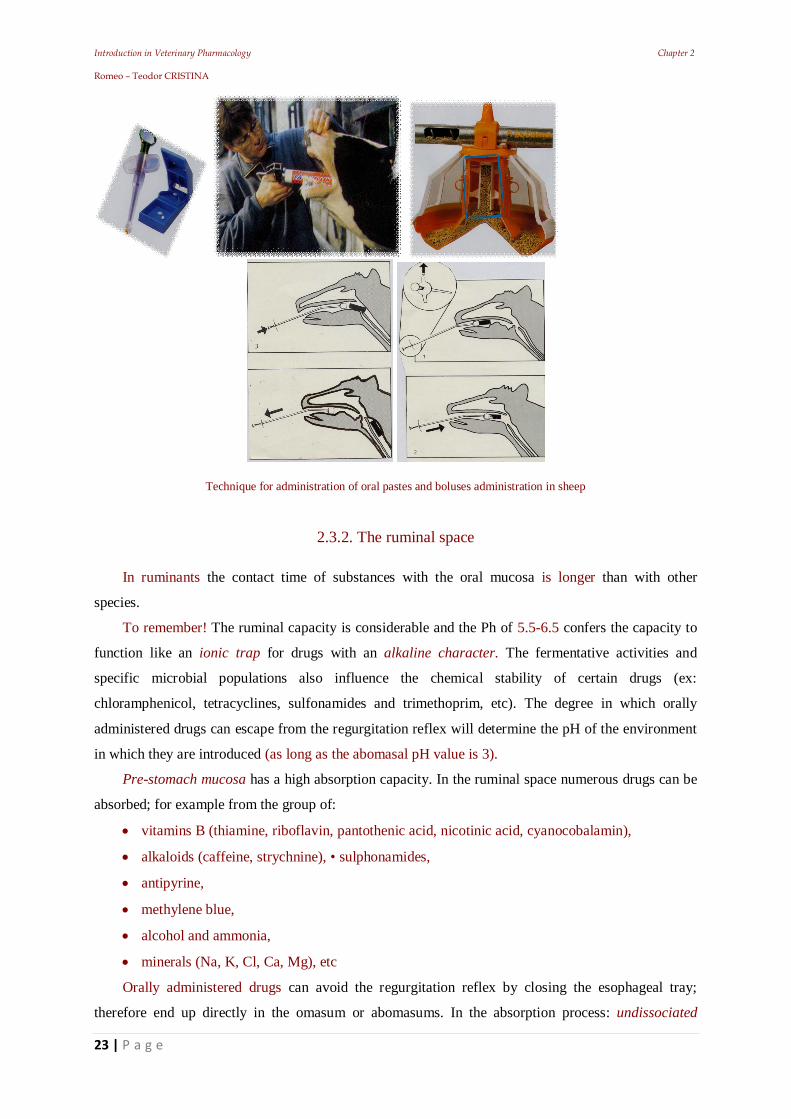
Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
23 | P a g e
Technique for administration of oral pastes and boluses administration in sheep
2.3.2. The ruminal space
In ruminants the contact time of substances with the oral mucosa is longer than with other
species.
To remember! The ruminal capacity is considerable and the Ph of 5.5-6.5 confers the capacity to
function like an ionic trap for drugs with an alkaline character. The fermentative activities and
specific microbial populations also influence the chemical stability of certain drugs (ex:
chloramphenicol, tetracyclines, sulfonamides and trimethoprim, etc). The degree in which orally
administered drugs can escape from the regurgitation reflex will determine the pH of the environment
in which they are introduced (as long as the abomasal pH value is 3).
Pre-stomach mucosa has a high absorption capacity. In the ruminal space numerous drugs can be
absorbed; for example from the group of:
vitamins B (thiamine, riboflavin, pantothenic acid, nicotinic acid, cyanocobalamin),
alkaloids (caffeine, strychnine), • sulphonamides,
antipyrine,
methylene blue,
alcohol and ammonia,
minerals (Na, K, Cl, Ca, Mg), etc
Orally administered drugs can avoid the regurgitation reflex by closing the esophageal tray;
therefore end up directly in the omasum or abomasums. In the absorption process: undissociated

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
24 | P a g e
component is the one that penetrates freely, according to the concentration gradient.
Dissociated components will be submitted to the restrictions through electric charges and
therefore, they will not be absorbed
2.3.3. Gastric mucosa in monogastrics
The stomach condition can determine a delayed absorption from the result of feeding, For
example: the pylorus can be closed a time period after feeding, thereby the drugs selectively absorbed
in the small intestine, would be delayed from their action. Knowing the dissociation constant of the
drug (pKa) and pH for the digestive tract compartment, we can calculate the absorption percentage
using the Henderson - Hasselbach equation:
weak acid: pKa = pH + log (Cn/Ci)
weak base: pKb = pH + log (Ci/Cn)
Where: Cn = deionized concentration
Ci = ionized concentration
For example, sulphadimerazine, having pKa = 7.4 will be present in the rumen (pH = 5.4)
undissociated, almost entirely, which will allows a good absorption.
So, in order to be absorbed, a drug needs to be soluble in the fat drops, as well as in the aqueous
phase of the intestinal content. The insoluble compounds will not be absorbed (ex: barium sulfate).
It is to remember that the gastric mucosa is considered mainly an excretion mucosa and not an
absorption one! For this reason, absorption on this level will be, in general, slow and reduced.
Although, some substances can be absorbed here (ex: aspirin, alcohol, caffeine, strychnine, PP
vitamin).
The plenitude of the stomach can influence the absorption processes! Inside a full stomach, drugs
will combine with some organic substances.
The absorption will be the best when the stomach is empty. The active substances covered with
layers of keratin, gluten, salol, or formalingelatin, do not dissolve in the stomach and because of this,
these substances are prepared in gastro-resistant tablets and/or pills.
The gastric absorption duration depends on a series of factors:
drug type (liposoluble, hydrosoluble),
particles size,
ionization constant,
pH of the gastric content,
physiological conditions (vascularization, secretion, tonus, motility) and
state of stomach plenitude
Liposoluble substances can be absorbed much easier than the hydrosoluble substances (in the

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
25 | P a g e
ionized formulas they are not absorbed at all).
Dissociation coefficient of the drug (pKa) and the pH of the gastric content are the most important
factors of absorption. At a strong acid Ph of the gastric juice the most absorbed are the weak acids
while the basic ones do not absorb.
Therefore, in the stomach: salicylic acid, aspirin and barbiturates will be best absorbed, which at
this pH will not dissociate, or only in a much reduced percentage.
For example: if we consider the theoretic distribution of a weak acid drug (having the pKa = 4
value), it can be found that in the stomach (at pH = 1), 99,9% will be found undissociated and it
absorbs and only 0,1% will be ionized, while in the plasma the exact opposite will happen.
Drug absorption can also be hastened or delayed through other ways.
Therefore, concomitant administration of isotonic solutions at the body temperature, hastens
absorption through “solvent drag”. For example: alcohol, saponins, bile salts, are producing the
hyperemia of the gastric mucosa and raising the absorption.
Because the gastric pH is usually placed between 1 and 3, and the intestinal pH exceeds the value
of 5, it is expected that the absorption rate of the same drug, will vary a lot in both instances.
The difference will depend on the drug's pKa.
The limits of classification for acids and bases
pKa - pH % Undissociated
Weak acid Weak base
-3 0,10 99,90
-2 0,99 99,01
-1 9,09 90,91
-0,7 16,60 83,40
-0,5 24,00 76,00
-0,2 38,70 61,3
0 50,00 50,00
+0,2 61,30 38,70
+0,5 76,00 24,00
+0,7 83,40 16,60
+1 90,91 9,09
+2 99,01 0,99
+3 99,90 +0,10
Esophageal mucosa it does not matter for absorption! In special cases (e.g. esophageal
diverticulum in birds or esophageal obstructions in mammals), absorption can occur due to prolonged
stagnation of drugs in this segment.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
26 | P a g e
2.4. Intestinal mucosa
Is responsible with the drug absorption capacity! In case of oral drug administration, the rich
vascularization and the large absorption surface of the small intestine makes it the most important
absorption place.
The intestine behaves like a lipoid membrane with pores and transport systems. Intestinal
absorption can occur in the entire length of the intestine, regardless of the histologic differences or pH
between the different segments of the intestine.
2.4.1. The intestinal mucosa and absorption
The large surface, the presence of numerous villi and the rich vascularization (presence of a
massive lymphatic and blood vessel network) ensures a high absorption capacity.
Absorption mechanisms through intestinal mucosa are grouped into two categories:
unsaturable passage (as passive transport);
saturable passage (as active transport).
The majority of drugs are absorbed through passive diffusion in the gradient sense of
concentration (based on Fick’s law).
The correlation between pH of the intestinal medium and the drug pKa is important in absorption,
due to the Henderson-Hasselbach equation.
In the intestine especially the weak bases (with pKa lower than 8) are absorbed, and in some
extent, organic acids with pKa lower than 3.
So, the absorption through the intestinal mucosa is selective!
Thereby, from the inorganic substances, monovalent ions are much easier absorbed, while the
bivalent ions will be absorbed with much more difficulty. The organic substances are better absorbed
in a liposoluble undissociated form than as a dissociated form.
When the intestinal mucosa is damaged the absorption will be unselective!
In the case of hemorrhagic gastroenteritis for example, the substances that normally are not
absorbed, or only in a low percentage (acting locally), can pass into the bloodstream causing poisoning
(ex: nitrofurane, furazolidone, anthelmintics etc.).
The factors that influence the bloodstream and the intestinal mobility can rush or delay the
absorption. The substances that produce intestinal vasoconstriction decrease the absorption, while
vasodilatation correlates with a faster absorption.
The intestinal absorption also influences the drugs mode of action.
Thereby, the orally administered streptomycin in the digestive tract will act locally, with an
absorption rate of only 5% (in dogs, up to 10%) and that’s why it cannot be used in generalized
infections.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
27 | P a g e
The substances that are absorbed in the stomach and intestine get into the portal circulation where
they will meet the hepatic barrier.
Here:
a part of the drug will be metabolized and then eliminated and
a part enters into the bloodstream, another being eliminated through bile, getting back into the
intestine, forming the gastro-entero-hepatic circuit.
For example tetracycline enters into the enterohepatic circuit and can accumulate into the body by
overdosing.
2.5. The large intestine absorption
Through the mucosa of the large intestine, substances with low molecular weight and residues of
drugs that have not been absorbed in the small intestine can be absorbed.
The rectal mucosa is used for absorption, being considered an internal administration way.
The substances that are administered in a rectal way (enemas and suppositories) are absorbed and
enter into the posterior hemorrhoidal veins, arriving into the vena cava, traversing the hepatic barrier.
Because of that, the consequence will be a faster diffusion into the body and a metabolisation delay.
In the veterinary medicine, rectal administrations are used as enemas or suppositories, usually in
pets. For example, chloralhydrate is administered generally as narcotic enemas in horses, or also as an
antidote in strychnine poisoning in dogs.
Research showed that, after rectal administration the blood concentration is not predictable and
most of the times it is much less than required.
When a substance is decomposed quickly in the liver, a significant difference can appear between
the determined effects in the case this substance was administrated sublingually or enteric. As a
conclusion, the absorption at the intestinal level is dependent to the following main factors:
a). Molecules physicochemical properties:
the size,
solubility,
dissociation degree of acids or bases,
the characteristics for a specific physiological transport mechanism etc.
b). The form and availability of Galenic preparation (solution, powders, tablets, pills) and features
like:
particle size,
the rate of decomposition
drug consistency (mass of incorporation).

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
28 | P a g e
2.6. Administration on the external ways
2.6.1. Inhalation way
It is an important way of administration for some specific drugs, especially for the ones from the
sphere of anesthesiology. Using this way active substances under a gaseous form, liquid, or even very
fine solid particles, are administered to animals.
The absorption can be produced at a respiratory level, or in the pulmonary alveoli.
The respiratory mucosa has the advantage of a large area of absorption, with rich vascularization
and in direct contact with the alveolar epithelium of the capillaries. This way, gaseous substances like:
oxygen, carbon dioxide or the mixture of CO2 (5%) and O2 (95%), known as carbogene, are
administered
The carbon dioxide is the physiological stimulator of the respiratory center! Inhaled in the
concentration of 5% of the atmospheric air, this will amplify the respiratory movements. Volatile
drugs are administered largely by the respiratory route. Currently, in narcosis, a series of substances
are used, like: chloroform, ether, ethyl chloride, halothane etc.
Numerous volatile oils (ex. eucalyptol gomenole, are applied locally under the form of drops in
the nasal mucosa or they are administered under the inhalation or fumigation formulas.
Inhalations are formulations in which volatile substances are activated by water vapors and then
inhaled by the respiratory system.
Fumigations suggest the burning of antiseptic substances and the inhalation of the produced
smoke.
Aerosols are small liquid or solid particles suspended in air, administered by the respiratory way.
Profundity of penetration of aerosols inside the respiratory system depends on the particle size.
Thereby, particles:
over 30 micrometers remain into the nasal cavity, pharynx and larynx;
between 20-30micrometers remain into the trachea;
between 10-20 micrometers into the bronchi;
between 3-5 micrometers into the bronchioles,
under 3 micrometers enters into the pulmonary alveoli.
So, the optimal penetration size for the pulmonary alveoli is of: 1-3 micrometers. Bigger particles
cannot enter, and the ones under 1mcm will be eliminated through exhalation.
2.6.2. Intratracheal injections
Are considered to be used the respiratory route of administration. The substance is deposited on
the respiratory mucosa and after placing the animal in lateral decubitus on an inclined plane, it is
allowed to escape through the gradient in the lung.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
29 | P a g e
This is the way the Lügol solution can be administered in sheep dictiocaulosis, rarely in calves,
(iodine 1.0; iodine 1.5; distilled water ad 1500.0), the first administration it is made in one lung and
the second is made after 24 hours in the opposite lung.
Intratracheal administration: puncture and the catheter introduction; the catheter route into the trachea.
Inhalation systems Oral spray with two or three phases - for big animals (Nebul 101) - for average animals (Nebul 81)
2.6.3. Absorption through the apparent mucosa
Drugs that are administered on the apparent mucosa will have a differential absorption.
Conjunctive mucosa it is easily permeable for drugs. It is used for local applications, especially
antiseptics, chemotherapeutics, antibiotics, anesthetics, meiotics and mydriatics. The administrations
are made under the form of eye washes. The solutions should be neutral and isotonic.
Nasal mucosa it is used for local applications or for inhalations of the volatile oils through the
airways. Generally, the nasal mucosa absorbs drugs well and for this reason, it can be used with
efficacy in small animals for general treatments.
Vaginal mucosa it is a less permeable layer to drugs, but can be traversed by liposoluble
substances.
Uterine mucosa especially in the puerperium, it absorbs well chemotherapeutics, antibiotics or
other substances that need to be locally applied.
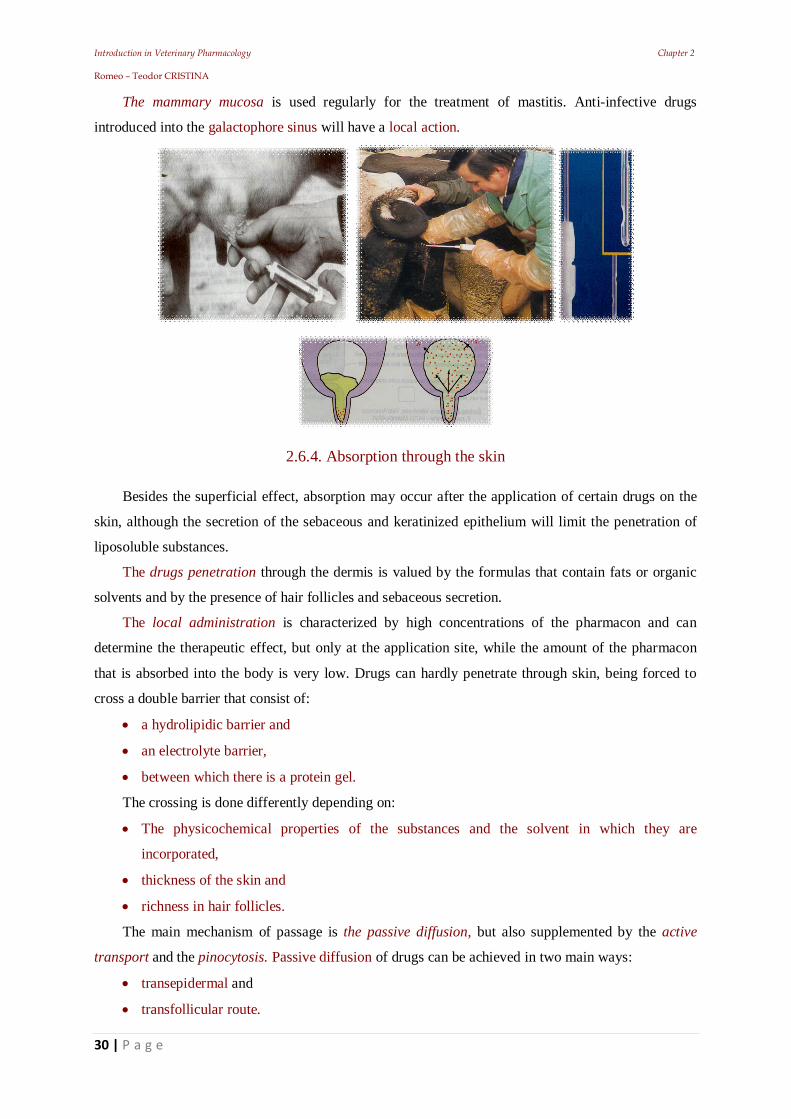
Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
30 | P a g e
The mammary mucosa is used regularly for the treatment of mastitis. Anti-infective drugs
introduced into the galactophore sinus will have a local action.
2.6.4. Absorption through the skin
Besides the superficial effect, absorption may occur after the application of certain drugs on the
skin, although the secretion of the sebaceous and keratinized epithelium will limit the penetration of
liposoluble substances.
The drugs penetration through the dermis is valued by the formulas that contain fats or organic
solvents and by the presence of hair follicles and sebaceous secretion.
The local administration is characterized by high concentrations of the pharmacon and can
determine the therapeutic effect, but only at the application site, while the amount of the pharmacon
that is absorbed into the body is very low. Drugs can hardly penetrate through skin, being forced to
cross a double barrier that consist of:
a hydrolipidic barrier and
an electrolyte barrier,
between which there is a protein gel.
The crossing is done differently depending on:
The physicochemical properties of the substances and the solvent in which they are
incorporated,
thickness of the skin and
richness in hair follicles.
The main mechanism of passage is the passive diffusion, but also supplemented by the active
transport and the pinocytosis. Passive diffusion of drugs can be achieved in two main ways:
transepidermal and
transfollicular route.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
31 | P a g e
The classification of excipients according to the factor of acanthosis (After: Cristina RT, 1996)
Non acanthogene
Medium acanthogene
Strongly acanthogene
silicone oil, cetaceea, sesame oil, methylcellulose, stearyl alcohol, cetylalcohol, paraffin, glycerin, propylene glycol, stearin, lanolin hydrate (50%), wax, PEG 400, 1500, 4000
vaseline, animal fats eucerin; anhydrous and hydrated, yellow Vaseline, axungia, olive oil, paraffin oil, sorbitol (70%) Undecilenat acid, cocoa butter 70%
Classification of the pharmaceutical forms by: penetration degree, action of vehicle and stage of disease
(After: Cristina RT, 1996)
Pharmaceutical form and mode of application
The degree of action in depth
The current direction (flux)
The action of the vehicle
Stage of disease
Powders Open compressor
Wet bandages Solutions
Emulsion U / AA Suspension-Emulsion U/A (emollient paste) Hydrophilic ointment
(Emulsion U / A) Hydrogels
Pastes Emulsifying ointments
A / U Lipogels
Occlusive dressing
From the inside to outside
the outside to inside
Refreshing Decongestants Superficial Antiinflammatory Antiinflamatory
congested Penetration increases the inflammation
Acute
Subacute
Chronic
Transepidermic (transcellular) way is important because of its great surface.
It involves the:
crossing of the lipid film on the surface and
penetration through, or between of the stratum corneum of the epidermis.
Unionized substances with a balanced partition coefficient (around value 1), with small
molecules, cross the transepidermal layer more easily.
Transfollicular route (intercellular) is accomplished through epithelium of the:
hair follicle,
sebaceous glands and
sweat gland ducts.
Penetration by this route is considered easy but the absorption area is much smaller compared to
the trans-epidermal way. The crossing is often made by passive diffusion.
Rubbing or massaging the skin will amplify the percutaneous absorption by removing the stratum
corneum and by activating the local circulation.
Ointments using excipients with high penetrating power will be highly absorbed acting into the
depth. For example: dimethylsulfoxide (DMSO), dimethylformamide (DMFA) and dimetillactamide

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
32 | P a g e
(DMLA) are helping the penetration by the emollient effect and increasing the stratum corneum
hydration with destruction and dissolution of the lipoproteins. These substances facilitate the
absorption of some drugs (chemotherapeutics, antibiotics), with whom they are associated.
Scheme for the proper use in the external treatment of preparations composed of two-or three-phase systems
(After: Cristina, R.T. 1996)
2.7. The parenteral ways
Parenteral drug products are reabsorbed non selectively being stored directly into the tissues or
into the bloodstream. If by oral administrations, inappropriate systemic concentrations are reached
(probably due to incomplete absorption or to the degradation into the intestine), parenteral
administration will be required
The preparations intended for injection should be:
non-pyrogenic,
sterile,
adjusted to the osmolarity and
to the body’s pH.
The correlation between pH values and solutions reaction
The pH value The solutions reaction
under 2 Strongly acid
2 – 4 Acid
4 – 6,5 Weakly acid
6,5 - 7,5 Neutral
7,5 – 10 Slightly alkaline
10 – 12 Alkaline
over 12 Strongly alkaline
The pH value of a solution gives an indication of acidity or alkalinity
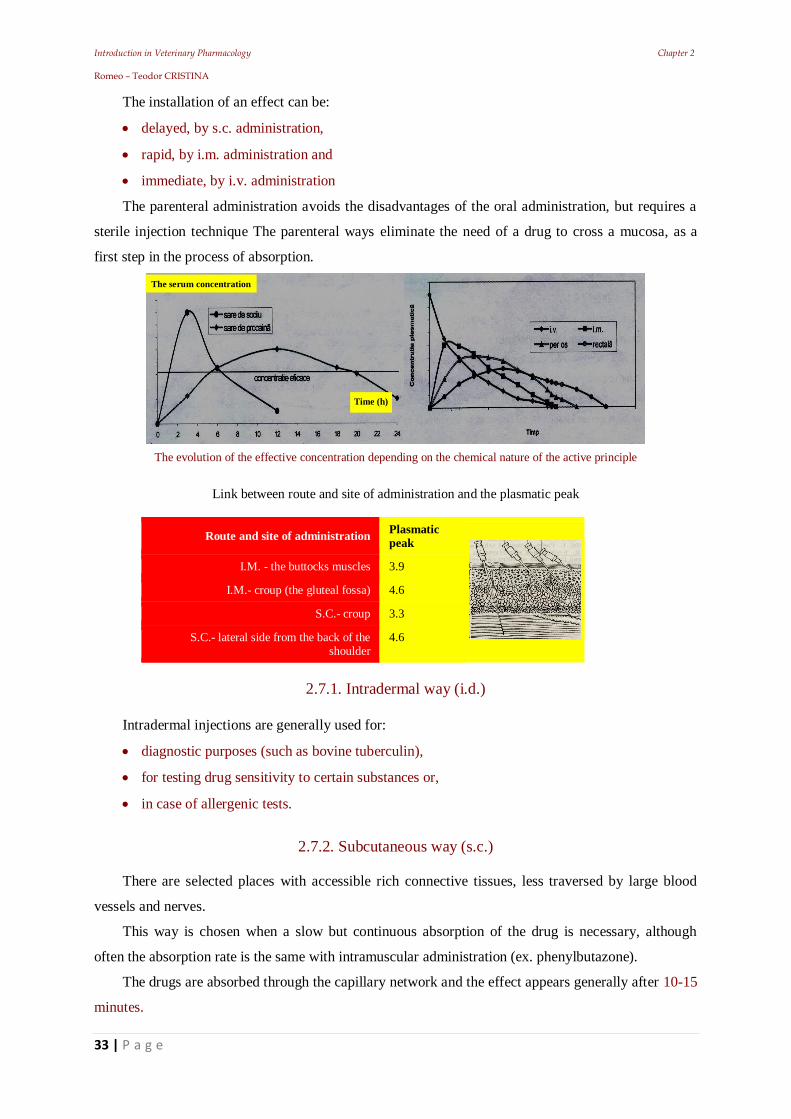
Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
33 | P a g e
The installation of an effect can be:
delayed, by s.c. administration,
rapid, by i.m. administration and
immediate, by i.v. administration
The parenteral administration avoids the disadvantages of the oral administration, but requires a
sterile injection technique The parenteral ways eliminate the need of a drug to cross a mucosa, as a
first step in the process of absorption.
The evolution of the effective concentration depending on the chemical nature of the active principle
Link between route and site of administration and the plasmatic peak
Route and site of administration Plasmatic peak
I.M. - the buttocks muscles 3.9
I.M.- croup (the gluteal fossa) 4.6
S.C.- croup 3.3
S.C.- lateral side from the back of the shoulder
4.6
2.7.1. Intradermal way (i.d.)
Intradermal injections are generally used for:
diagnostic purposes (such as bovine tuberculin),
for testing drug sensitivity to certain substances or,
in case of allergenic tests.
2.7.2. Subcutaneous way (s.c.)
There are selected places with accessible rich connective tissues, less traversed by large blood
vessels and nerves.
This way is chosen when a slow but continuous absorption of the drug is necessary, although
often the absorption rate is the same with intramuscular administration (ex. phenylbutazone).
The drugs are absorbed through the capillary network and the effect appears generally after 10-15
minutes.
The serum concentration
Time (h)

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
34 | P a g e
Resorption is amplified by hyaluronidase that can be added to the injection solution.
This will depolarize the hyaluronic acid, found in the intercellular substance. Resorption rate can
be increased by heat and by massaging the injection site. These measures can be applied also when
administering large volumes of saline.
Nr. The length Indications The code
1 0,90 x 40 i.m., i.v., venesection yellow
2 0,80 x 40 i.m., i.v., venesection green
12 0,70 x 30 i.m., i.v. black
14 0,60 x 30 i.m., i.v. in small animals blue
16 0,60 x 25 i.m., i.v. in small animals transparent
17 0,55 x 25 i.v., s.c. in small animals and birds violet
18 0,45 x 23 i.v., s.c. in small animals and birds brown
20 0,40 x 19 i.m. in small animals and birds white
Regarding the absorption mechanism, this is different for oils and aqueous solutions. The oily
solutions reach the lymphatic vessels by:
penetrating the endothelial cells, or
passing on (firstly the substance, after the oil).
Isotonic substances are absorbed faster than the isotonic solutions, and they are more easily
absorbed than the hypertonic solutions. The drugs are typically soluble in saline or distilled water,
rarely into the polyvinylpyrrolidone (PVP). Subcutaneously, organic and tissue implants may also be
administered by the form of hormonal micro tablets with a slow absorption rate.
2.7.3. Intramuscular way (i.m.)
The veterinarian chooses the intramuscular route when:
he administers irritating substances;
when absorption rate of drug administered subcutaneously is unsatisfactory
for the administration of deposit type preparations (ex: iron-dextrane in piglets with iron
deficiency anemia);
when the injection substance is not a solution but it is a suspension.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
35 | P a g e
The diffusion of solutions occurs over a wide area and the osmotic balancing in the case of
slightly hypertonic solutions is fast. The fact that the sensory innervations are reduced makes the local
tolerance to be higher.
The solutions with a high acid or basic pH, those highly hypertonic and the caustic ones cannot be
administered, because they can produce: indurations, phlegmons, abscess or necrosis. Besides, in
animals, unlike humans, the intramuscular way is much more painful.
The intramuscular way can be used for the administration of medical substances into aqueous
solutions, oily solutions and fine suspensions. It is the best way of administering oily solutions and
deposit medication (ex: procaine penicillin, benzathine penicillin, hormones, etc.). The injections are
made profoundly intramuscular, this way being less painful and avoiding the risk of the substances
entering the blood vessels, which always leads to serious consequences.
The intramuscular administration can be made to every species, into:
the gluteus muscle or
the superiors thigh muscles;
The administration can be made also into the superior cervical muscles in pigs, cows and horses.
The volume of liquid injected in a single place should not exceed 20-40 ml in large animals and
proportionally smaller quantities to other animals.
2.7.4. The intravenous way
It is the fastest way to introduce drugs into the general circulation, because it eliminates the need
for the active substance to cross the endothelial barrier, therefore the total amount administered is
immediately available. The intravenous way is used for:
plasma or blood transfusion
when a rapid effect is needed
when a drug is too irritating to be administered in another way
for an accurate control of the dose
for a longer-term administration, using an intravenous cannula for drugs with a transient action The specific conditions that a solution needs to satisfy in order to be administered using the i.v
way, except the usual ones (sterile, non-pyrogenic) are:
should not be hemolytic, coagulant or precipitant, should not be toxic for the myocardium,
should not harm the vascular endothelium,
should not cause embolia and
to be close to the body temperature.

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
36 | P a g e
In veterinary medicine, as an exception, the i.v. injection of the oiled camphor is allowed, in colic
therapy in horses, but in low-doses (3-5 ml) administered slowly.
The i.v. way allows the administration of the substances that are not tolerated by the tissues:
irritant; hypertonic, or alkaline solutions.
Macromolecular substances can be introduced intravenously:
gelatins (Marisang) or
dextrans (Vetoplasm)
colloidal plasma substitutes etc.
The injection is usually made
in the jugular vein in: horses, cow, sheep and goats.
in auricular veins: in pigs.
in the cephalic vein and the recurrent tarsal veins: in cats and dogs.
The water renewal rate represents a "turn over", and in mammals the complete water renewal is
made in 20 days. In 24 hours the “turn over” varies depending on the species:
143ml / kgbw in cows
150ml / kgbw in sheep,
73ml / kgbw in goats,
75ml / kgbw in donkey.
Depending on the intended therapeutic purpose, the infusions can be:
with electrolytes
for the acid-base equilibrium;
with energetic and reconstructing substances;
substitute solution for colloidal plasma
as drugs dilutor.
Remember that:

Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
37 | P a g e
2.7.5. Intraarterial way (i.a.)
It is rarely used in the veterinary field.
The major disadvantage being that use of this can achieve high drug concentrations in some
peripheral areas.
2.7.6. The intraperitoneal way (i.p.)
Is commonly used, especially in dogs, cats, pigs and large animal younglings, but may be useful
in other animals as well.
Due to great surface and the high absorption rate of the peritoneum, his route is advantageous
for the administration of large volumes of liquids.
The injections will be done into the lumbar fossa (needs to be made carefully in order to avoid
injecting the solutions into the abdominal organs).
2.7.7. Intrathoracic and intracardiac injections
This ways are used occasionally in small animal euthanasia
2.7.8. Intrathecal injections (subarahcnoidal)
These are involving the penetration of the CNS lining, the special technique of administration
being learned at anesthesiology.
2.7.9. Epidural injections
This technique is used more often in cattle, in case of birth, when the abolition of the uterine
contractions is desired.
The local anesthetic is introduced into the space between the first two coccidian vertebrae
respecting the technique learned at anesthesiology.
2.7.10. Intraarticular injections
This technique is used generally when administering anti-inflammatory drugs and antibiotics into
the intraarticular space (especially in horses).
2.7.11. Rectal, vaginal and intramamar injections
These administrations are used only when the therapy is needed in this region
Introduction in Veterinary Pharmacology Chapter 2 Romeo – Teodor CRISTINA
38 | P a g e
The differences between injections and infusions
(Synthesis: Cristina RT, 1999)
Injectable preparation Infusion preparation
Containing drug substances with a pharmacodynamic activity
Rarely serves as a mode of a drug administration
May have as carrier besides water: oil and various organic solvents. The exclusive carrier will be the water.
The active substances may be dispersed in the form of suspensions.
The active substances are dispersed molecular, colloidal, and rarely emulsions.
Administrations are made in small or medium units (usually 1 20ml).
They are prepared and administered in large amounts (usually up to 100 ml).
They can be administered using the i.m., s.c., i.v., i.d., i.p. way.
The administration is made strictly on the iv way.
The duration of the administration is short (seconds, minutes), so it is more comfortable
in animals.
The duration of the administration is large (tens of minutes, even hours), are difficult to animals.
The isotonic and the isohydric are not always required.
The isotonic is required, the pH of 7.4 and the ionic composition, needs to be as close as possible to the body fluids.
The preparation is made into ampoules, rarely into vials with a low volume.
The preparation is made using vials or packaging with 200-1000 ml without preservatives. For peritoneal dialysis the packaging can be barrels with a capacity of 10-20 liters.
Theoretic the condition of the pyrogenic (especially for the small amounts of the
injected solutions ) is less important.
Preparation conditions should provide solutions perfect sterile, without pyrogenic substances.

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
39 | P a g e
3. Drug blood transport & Drugs distribution

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
40 | P a g e
Introduction
Drug substances and most exogenous or endogenous compounds (e.g. hormones, bilirubin, etc.),
bind in the body to:
plasmatic or
tissular proteins.
They will result in large complexes that cannot cross the biological membranes.
The biologic membranes are functional units, of 5 to 8 nm. thick, composed mainly by lipoproteic
and phospholipidic complexes. They have a perpendicular orientation on the membranal surface thus
forming a hidrofobic chain.
Proteins are incorporated into the membranes as globular molecule groups, providing the contact
of the average extra- and intra- cellular environment. Individual lipidic molecules have the ability to
move laterally, ensuring the membrane’s specific flexibility & fluidity.
In the middle of aqueous channels can be finded the globular molecules, which can open and
close, depending on the electric resistance, allowing the exchange of substances. In the blood, drugs
can be found under two forms: free and coupled.
The coupled form is reversible, fixed on the plasmatic proteins (or to the sanguine elements).
Generally, the drugs have thre3 main caracteristics:
one part of the active substance is linked and one part is free;
the link is reversible;
only unlinked substances can pass biologic membranes
Drugs bind to proteins by interacting with the: ionisant, polar or non-polar groups, generating the
following bonds:
a). covalent bonds (electrons are shared between two atoms; this kind are sparse and much more
common for the toxic drugs)
b). ionic bonds (energy = cca. 5 Kcal / mol) (accomplished between oppositely charged electric
ions. Such a bond is proportional with the task size and square of the distance between the centers of
particles)
c). hydrogen bonds (energy = cca. 0.5 Kcal/mol) (which are achieved when two atoms come very
close. These are weak links with low energy, forming less stable complexes).
3.1. Factors that influence drug transport
Chemical structure: It is very important for the drug coupling and transport because it is
influencing the affinity of the organic molecules for proteins. For example: phenylbutazone,
oxphenbutazone, dicoumarinic derivatives, long-active sulphonamides, some penicillins, salicilates,

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
41 | P a g e
etc. are binding heavily on the plasmatic proteins. Changes in the chemical structure of drugs can
cause large differences in terms of coupling to plasma proteins.
Bounding of some drugs to plasmatic proteins (After: Dobrescu, 1977)
Species % of drug bound to blood proteins
Penicillin G Cloxacillin Sulphadiazin Sulphafurasol
Humans 49 7 67 16
Horse 59 30 - -
Rabbit 65 22 45 18
Rat - - 55 16
Mouse - - 93 69
Drug binding for the transport is accomplished with a preference on the proteins, because they are
the only peptide chains with a large contact surface compared to other blood proteins.
Theoretically, each molecule can carry approx. 100 positive or negative charges.
Drugs bind to groups consisting of amino acid residues of albumin, surface oriented:
R –COO-,
R –O-,
R –S-,
R –NH3+
In solution they interact with polar molecules of the drug. Ions have a different affinity depending
on the nature of the group to which they refer, for example:
Mn (for the sulhidril groups),
Zn, Cd (for the imidazole groups).
The anion affinity order seems to be: bicarbonates < acetates < chlorures < citrates < nitrates
The amount of the drug coupled protein is determined by:
the concentration of the drug;
drug affinity and
capacity up to saturation of these coupling sites.
Serum albumin provides:
a) some coupling places for the basic drugs;
b) for the binding of acid drugs , there are no more than two primary (usually only one) coupling
sites.
Globulins compared to the albumins, they have a relatively small importance for the drug
coupling. Very few drugs have an affinity for them. for example it is well known that thyroxine and
cortisol have a high affinity for the a-globulins, but with relatively low coupling capacity.
When the coupling capacity is saturated, the exceeding drug is fixed to the albumins.
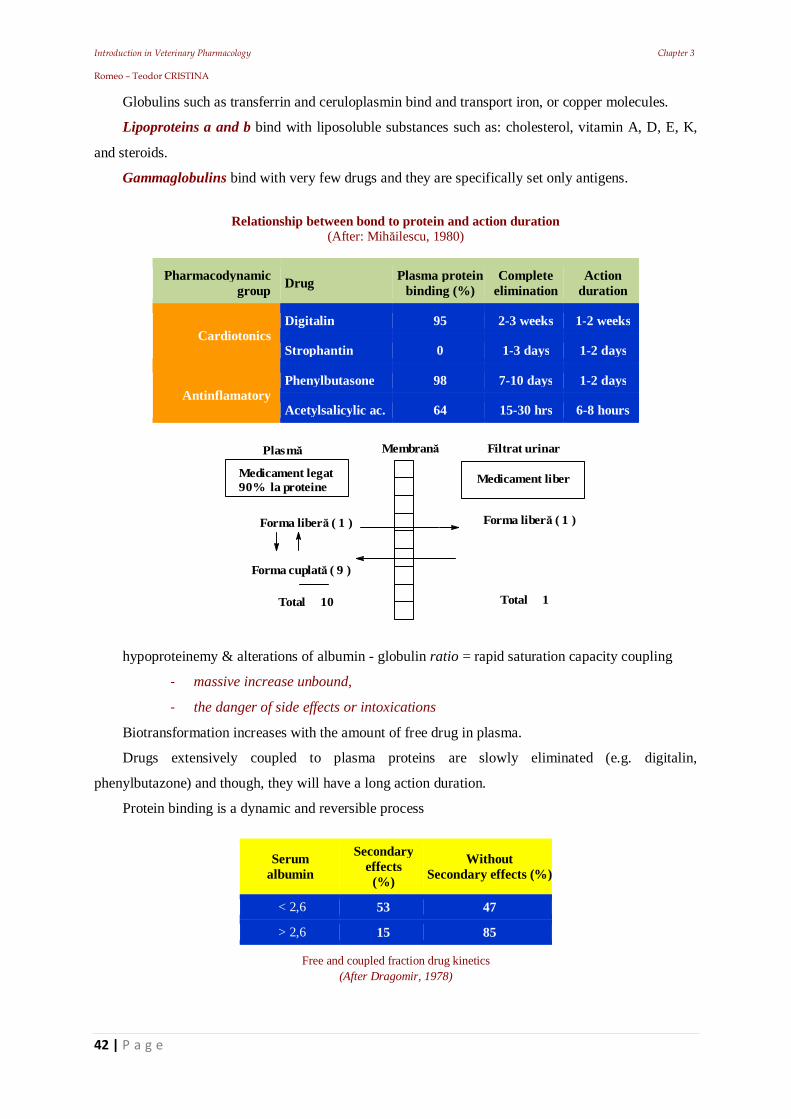
Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
42 | P a g e
Globulins such as transferrin and ceruloplasmin bind and transport iron, or copper molecules.
Lipoproteins a and b bind with liposoluble substances such as: cholesterol, vitamin A, D, E, K,
and steroids.
Gammaglobulins bind with very few drugs and they are specifically set only antigens.
Relationship between bond to protein and action duration
(After: Mih ilescu, 1980)
Pharmacodynamic group Drug Plasma protein
binding (%) Complete
elimination Action
duration
Cardiotonics Digitalin 95 2-3 weeks 1-2 weeks
Strophantin 0 1-3 days 1-2 days
Antinflamatory Phenylbutasone 98 7-10 days 1-2 days
Acetylsalicylic ac. 64 15-30 hrs 6-8 hours
Medicament legat 90% la proteine
Medicament liber
Plasm Membran Filtrat urinar
Forma liber ( 1 )
Forma cuplat ( 9 )
Total 10
Forma liber ( 1 )
Total 1
hypoproteinemy & alterations of albumin - globulin ratio = rapid saturation capacity coupling
- massive increase unbound,
- the danger of side effects or intoxications
Biotransformation increases with the amount of free drug in plasma.
Drugs extensively coupled to plasma proteins are slowly eliminated (e.g. digitalin,
phenylbutazone) and though, they will have a long action duration.
Protein binding is a dynamic and reversible process
Serum albumin
Secondary effects
(%)
Without Secondary effects (%)
< 2,6 53 47
> 2,6 15 85
Free and coupled fraction drug kinetics (After Dragomir, 1978)

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
43 | P a g e
Saturation of plasma protein binding capacity and increased free fraction, leads to a quicker
metabolism and elimination of the drug, resulting equilibrium between the two factions.
The states of hyperproteinemia and alterations of the: albumin - globulin ratio have as a result:
a faster saturation coupling capacity,
a massive increase of the unbound fraction,
danger of the side effects or of poisoning.
For example, in newborn animals, plasma proteins are reduced. For this reason, the unbound
fraction of the drugs is higher than in the adults, a fact which explains the sensitivity of newborns and
the risk of poisoning.
In pregnant females, a large part of the plasma protein's ability to couple endogenous compounds
is occupied, a fact that will increase the drug unbound fraction in the blood.
Among substances there is a competition for the coupling sites. Some acidic drugs compete for
the same binding sites on plasma proteins. Sometimes movement may be therapeutic advantageous,
sometimes in contrast, toxicities occur.
Corticosteroids present in plasma are circulating coupled to a specific globulin, named
transcortin. Anti-inflammatory substances (such as, phenylbutazone or salicylic acid derivatives) are
able to move the corticosteroids, accomplishing the therapeutic effect.
Stages of drug blood diffusion Blood represents a central compartment responsible for the distribution of drugs, while
representing a small proportion compared to the other two great diffusion compartments (intra- and
extra-cellular) of the body. Circulatory, the absorbed drug is able to access all body compartments in
different concentrations.
Diffusion phases are beginning with the vascular wall crossing and ends with the drug penetration
to the site of action, a phase, also known as the drug distribution phase.
In addition to these three compartments, there are also a number of special sections whom
accessibility is regulated by key barriers as:
• CNS blood-brain barrier,
• fetal placental-aqueous humor and
• Inner’s ear endolymph.
Histo-morphologic features The morphological boundary between blood plasma and the extracellular compartment is
represented by the vascular endothelium.
There are three main endothelial types:
1) high active transport by pinocytosis

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
44 | P a g e
This form of endothelium is present in almost all organs and allows for the rapid transfer of
substances in both directions;
2) Fenestrated epithelia
Endocrine organs and intestinal capillaries constitute this type of endothelium.
This allows the exchange of substances very quickly. Here, the renal glomerule capillary
endothelium can be also included.
3) Endothelia that have no transport activity by pinocytosis and present so called Zonulae
occludentes (or tight junctions), continuous type connections between cells, preventing the
intercellular exchange of substances.
The blood-brain barrier basis is located in the CNS; it is also met in the case of peripheral
nerves. From a kinetic standpoint, the plasma compartment and the extracellular compartment are
considered as a unit.
The fact that membranes are composed of a double lipid layer is of particular importance to the
phenomenon of distribution, since membranes are impermeable to water-soluble substances.
Only few substances in the body are distributed in proportion to the percentage that represents
each compartment. Most pharmacons and toxins have a complicated behavior, as additional
phenomena can be induced depending on the nature of the molecule. Physico-chimic factors involved in drug distribution Pharmacon solubility is a significant feature for drug distribution, absorption and elimination.
Substances can be divided into three groups: a) Strictly water-soluble compounds)
hardly absorbed after the p.o. administration
after i.v. administration they are distributed only in the extracellular compartment, being easily
eliminated by the kidney. In this group, there are few substances (e.g. the osmotic diuretics). b) strictly fat-soluble compounds
are placed in body fat, where the partition coefficient, water / octenol is in function, especially in
the neutral fat of fat cells (e.g. chlorinated hydrocarbons). c) amphiphylic compounds
A molecule is considered amphiphylic, when it presents:
a hydrophilic part and
a hydrophobic part, positioned close to one another.
In the case of larger distances between these components, they will enter the surfactants category.

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
45 | P a g e
C l
C H 3
C H 3 N + H C H 2 C H 2 C H 2
N
S
Amphiphilic character of chlorpromazine
(After: Kuschinsky, 1989)
Amphiphylic substances accumulate properly in the interphase (i.e. where the aqueous phase
meets the lipid phase).
This is the case for all cellular membranes: either plasmalemma or intracellular membranes (e.g.
mitochondria, nucleus, ER, lysosomes). This accumulation has already been demonstrated for
membranes in the case of numerous drugs and is of practical importance (i.e. the ratio of the cell and
plasma concentration can reach values of 150 or higher).
Therefore amphiphylic drugs are found only at a very small extent in the neutral lipids of the fat
cells, because they are not lypophilic. Since most of drugs are weak acids or bases they are found as
unionized forms (in case of a biological pH). The size of the dissociation constant is, therefore,
important for the distribution phenomenon.
Another phenomenon that depends on the hydrophobic drug molecule and plays an important role
in drug distribution (and in drug interactions) is the coupling to the plasma proteins and to the
extracellular fluids, based on the hydrophobic interactions. Since the drug came into use, there are
many factors that tend to decrease its active concentration.
These phenomena are mainly determined by:
storing drugs in the body;
binding to the proteins;
dilution in the body fluids.
A drug is able to leave the vascular space by:
diffusion through the lipoid membranes,
the large size pores (4nm) or
the capillary wall fenestrations.
These "openings" allow passage to albumins, so that all, even the biggest drug molecules (e.g.
dextrin 70,000 Da.) can quickly get out of the vascular bed. Balance will occur:
rapidly in: heart, liver, kidney and brain
slowly in: skin, bones and fat stores.
Even after the passing of sufficient time to achieve equilibrium, there are differences in drug
concentrations in different parts of the body.

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
46 | P a g e
For this reason all molecules, even the greatest are able to distribute the ECL. The speed of drug
plasma balancing, achieved concentrations and ECL depends on: the degree of vascular tissue
infusion.
Unionized lipid-soluble fraction is shown as being in balance between the different
compartments. Although there is a balance between the concentrations of substances uncoupled from
each compartment, the total drug concentration may differ significantly between the compartments.
There may be also significant differences in pH between compartments which will cause different
ratios between the unionized and ionized fraction.
For example, significant pH difference between compartments is important: e.g. stomach pH =
2/ECL (pH = 7).
A weak acid with pKa = 4, it will be almost exclusively in a non-ionized state in the stomach,
while the CEL will be mainly in the ionized state. Generally the acidic drugs tend to accumulate in the
phases where the pH is high, and the alkaline drugs tend to be concentrated in areas with low pH.
Available binding site distribution in compartments exercise also affects on the total amount of
the drug present in each compartment when there is balance between them.
Because of the coupling variations in concentration between the two compartments, even if the
pH has the same value and thus the concentration of unionized drug, it is the same in both
compartments.
FenomenLoc
Administrare Absorb ie Distribu ie Ac iuneIntestin Sânge LEC int
Frac iune cuplat
Frac iune liber
Formulare Cuplatplasmatic
Cuplattisular
Mucoas Endoteliu Perete celular
Neionizat Neionizat Neionizat Neionizat
Barier
Ioni Ioni Ioni Ioni
Loc deac iune
Equilibrium diagram of a drug that is found in a compartment disposed in and between the different fluid body
compartments. In this example, drug was orally administered, and its growth is monitored until it reaches the site of action (Brander, 1991).
Another factor that can cause an uneven distribution of the drug between compartments is the
presence of an active transport mechanism suitable to the membrane that separates them (e.g. that is
allowing the thyroid gland to avidly accumulate iodine)
Coupling influence of drugs on the proteins
A variable proportion of an absorbed drug can be reversibly coupled to the plasma proteins.
Active drug concentration will stay in the uncoupled fraction, since it will be able to leave the
plasmatic space and reach the action site.
Between the coupled and free fraction, equilibrium is forming

Introduction in Veterinary Pharmacology Chapter 3 Romeo – Teodor CRISTINA
47 | P a g e
When the free substance leaves the circulation, the coupled fraction will be released, in order to
restore balance.
Protein couplings reduce the loss of substance rate in plasma, to the extent that it lowers the free
fraction plasma concentration.
This will decrease the concentration gradient on which the drug diffusion occurs. It will reduce
the loss rate of the drug through the kidneys (because only the free fraction is filtered).
When a drug is actively excreted, coupling to protein does not confer protection (e.g. penicillin is
excreted almost entirely in the first-pass renal).
The practical consequence of coupling to plasma proteins is that the toxicity and efficacy of the
drugs that are coupling, are greatly intensified in a substantial portion of the proteins in the case of
hypo-proteinemia.
The unbound fraction concentration of a drug coupled in a large proportion may be increased
when administering a higher affinity drug for the same coupling sites.
Drug coupling in blood, most commonly, but not exclusively, occurs with the serum albumins,
but can be held also at figurate elements or to: -1 acid glycoproteins.
Albumin is able to achieve the following couplings:
high affinity - low capacity or
high capacity - low affinity.
Concentration estimation of unbound and total concentration is feasible in experiments where the
total drug concentration is gradually increased.
Studies of this type provide information on the number of coupling sites on an albumin molecule
and about the value of the coupling constant affinity, being important for example when searching for
a suitable dose for an antimicrobial drug.

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
48 | P a g e
4. Diffusion in the body's hydric regions

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
49 | P a g e
Introduction
In adult animals, body water can be found in percentages of 70-75% (depending on the age and
species) of the body weight, being included in fluid or distribution regions, separated by tissue barriers
with a variable component. In each of these compartments, a drug reaches steady state surprisingly
quickly.
In terms of drugs distribution, the body is divided into three major areas:
blood plasma (intravascular), aprox. 4-5% of bw.;
extracellular (intercellular), approx. 15-20% of bw., which bathes the cells (ECL)
intracellular aprox. 50% of body weight (ICL).
Also known, is the:
the luminal intestinal space, aprox. 25-30% of bw.
The drug distribution volume is the portion of the total body water in which a drug can be
successfully diffuse.
Solubility and diffusion in the aqueous phase are medicinal properties that give to the drug the
ability to come into contact with the first membrane.
The degree to which a specific dose of a medicine will be diluted, depends on the number of
compartments it can penetrate in the body. Since the elimination mechanisms cause a lowering of the
plasma level, drugs tend to revert back from the distribution volume in the plasma.
Transcellular fluids are separated by the interstitial fluid that surrounds the epithelium cells.
Transcellular fluids are considered the:
liquids from the intestinal lumen,
urinary tract
CNS
glands
joints and body cavities.
When drugs diffuse in these fluids, they must overcome all these spaces.
The capillary wall is a membrane which has a different permeability for different drugs.
Their penetration will depend on the:
liposolubility,
physiological state and
molecular size.

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
50 | P a g e
The more liposoluble the drugs are, the easier they will penetrate the capillary walls. Substances
coupled with the plasmatic proteins cannot diffuse transcapillary, until after they get back into free
form. Passing through the capillary wall is influenced by the capillary permeability changes, under the
influence of some drugs or tissular metabolites.
Drugs that can cross cell membranes are distributed into the intracellular space, or in the
constitution water (representing about 50% of the body weight).
All drugs with a low molecular weight (incl. acids) will be filtered at a glomerular level,
according to their plasma concentration. In the frame of this mechanism, an active process which lacks
specificity towards its substrate and a high capacity transport are involved.
Acidic active substances will be transported again by this mechanism, which can lead to
distribution and renal elimination will not be adjusted only by the physico-chemical parameters, but
will be determined, also by the active transport processes.
The kinetic behavior of drugs is not only influenced by modifications to the acid transport
mechanism, but also the kinetic behavior of the body's own substances versus some medicines.
A good example is the uric acid: which is filtered at the glomerule, and then, quantitatively
reabsorbed. Any reduction in acid secretion capacity due to the involvement of this mechanism in the
elimination of other drugs will influence the rate of uric acid secretion.
Proximal portion of nephron representation: acids active resorption mechanisms, absorption & secretion in drug distribution (After: Kuschinsky i L llmann, 1989)
4.1. The role of cell membranes
These components are important from a functional standpoint (membranes of organelles,
citoplasmatic ones and plasmatic) represent about 80% of the cell’s dry matter.
Plasmatic membrane works as an interface between the cell and ECL (extracellular fluid) and
possesses qualities and properties that allow the transfer, from and to the cell. The phospholipids
fluidity in double layer explains the surface mobility of the cell components (e.g. of the receptors).

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
51 | P a g e
This vision of cell membranes is known as the fluid mosaic model and it is fully compatible with
the known behavior of the membrane medicines.
Biological membranes behave as punctured lipidic pores, allowing the drugs and physiological
substances to penetrate though passive or carrier (intermediate) processes.
4.1.1. The diffusion mechanisms
The simplest case is that of a small water-soluble molecule who has a concentration controlled
rate across the membrane. Since water-soluble molecules larger than the urea penetrate more slowly,
the presence of membrane pores or channels of small diameter was supposed (aprox. 0.4 nm).
Because of the water passing through and its dependency on the differences in hydrostatic and
osmotic pressure, this process has been called filtration.
Liposoluble drugs must cross from the aqueous ECL into the lipidic membranes and then into the
aqueous phase after this barrier.
The drug is partitioned between the aqueous and lipid phases and the fat penetration rate will
depend on concentration difference and the contact surface with the barrier.
Concerning the penetration of medicinal substances through the membranes, several mechanisms
are involved: Some of them are carried out passively without energy sources, while others are active
mechanisms requiring energy sources.
Simple diffusion. The aqueous substances pass through the aqueous pores of the cell
membranes. The penetration is achieved by random movements with no interaction with other
molecules.
The solvent involving ("solvent drag"). The aqueous substances penetrate the aqueous pores
of a membrane as a result of increased water circulation.
Diffusion limited by electrical charges. The polarity of membranes cause the ionized forms of
the drugs, to meet barrier electric charges. Nonetheless, small anions (Cl-) can pass through the
positively charged aqueous channels excluding the cations.
Lipidic barrier limited diffusion. Penetrating molecules can enter into the cell, if it has an
appropriate solubility, which would allow the dissolution of the membrane site first, and afterwards in
the aqueous phase.
Facilitated diffusion. Is a selective, saturable, transport system subjected to competition
between substrates. The transported molecule is combined reversiverly with a carrier. Mechanisms
listed do not require energy and do not usually lead to the concentration against an electrochemical
gradient. Mechanisms that require energy are carried out against the concentration gradient.
Exchange diffusion. In this mechanism, a specific carrier is present that can cross the
membrane, but only under complex form.
Active transport by carrier. Is the most common mechanism, although energy consuming.

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
52 | P a g e
The penetrating molecule combines with a carrier that is subjected to some chemical changes in
the membrane. Trough a reaction that requires energy (ATP), the carrier is modified on one side of the
membrane, to have a greater affinity for the molecule. On this basis it links the substance and
transports it trough the membrane, then with another chemical reaction it loses the affinity and releases
the substance, to finally return either empty or in combination with other substances, repeating the
cycle. Numerous active substance diffuse through this mechanism.
Pinocytosis. It is a mechanism in which the cell membrane develops invaginations with the
incorporation of the substance, followed by the integration as intracellular vesicles.
External substances are taken under this form and then released into the cell, after the dissolution
of the vesicle.
Active transport. It is occurring when, in addition to the functions of: selectivity, satiability and
competition, the system is also dependent on energy (as such, it is rapidly inactivated by metabolic
inhibitors) and so, it is capable to transport the substrate against the concentration and the
electrochemical gradients.
4.2. Relation pH, pKa and drug diffusion
Only a few drugs are exclusively, hydrosoluble or liposoluble. On the other hand, many drugs
are able to solubilize both in water and fat (or other lypophilic solvents).
4.2.1. Molecular and biochemical aspects
ions, if they have sufficiently small molecular sizes, can cross the membranes via the hydric
channels,
unionized liposoluble fractions can diffuse through the lipidic portions of the membranes.
the drug ionization degree is dependent on the pH of the aqueous phase in which they are
found in the solution.
The consequence of the partition effect on the pH - pKa difference - on the balance of ionic
diffusion, is called ionic capture.
Only non-ionized molecules that are able to diffuse through the lipidic membranes have a
tendency to equalize the concentrations on both sides of the membrane.
The presence of a pH difference between the two sides of the membrane, allows a drug with
suitable pKa, to develop different ionization ratios for the two liquid phases.
So, although the ionized fraction concentration levels are almost equal, the total concentration
of dissociated and undissociated forms can be very different from one side of the membrane to the
other.
Electivity for certain tissues may lead to a substance concentration with a uniform distribution.
Most of the drugs are distributed unevenly, being able to accumulate selectively in some tissues.

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
53 | P a g e
pKa values of acidic or basic drugs (After: Brander, 1991)
Acid drugs pKa Alcaline drugs pKb
Ampicillin 2.5 Teophilin 0.7
Aspirin 3.5 Strichnine 2.3
Phenilbutasone 4.5 Methilene blue 3.8
Sulphacetamide 5.4 Chinidin 4.4
Sulphadiazine 6.5 Piperazine 5.7
Sulphadimidine 7.4 Trimethoprim 6.4
Penthobarbital 8.1 Ampicillin 7.2
Teophillin 8.8 Strichnine 8.0
Adrenalin 10.2 Adrenalin 8.7
Ascorbic acid 11.5 Atropin 9.7
4.3. Diffusion through barriers
In veterinary medicine, three main drug substance body barriers are recognized, namely:
• blood-brain (hematoencefalic),
• blood-ocular and the
• placental barrier.
4.3.1. Haemato-encephalic (Blood-brain) barrier
The blood vessels that are crossing the brain and bone marrow are lined with a specialized
endothelium with cells linked together by impermeable formations named zonulae occludentes, with
no pinocytosis activity.
This barrier is placed between the plasma of the encephalon and extracellular space.
Anatomically, the cerebrospinal fluid barrier (CSF) it is placed to the level of the choroid
plexus. Drugs that are not soluble or those that are highly ionized penetrate slowly into the forebrain,
while fat-soluble agents (e.g. volatile anesthetics) penetrate this space rapidly.
The barrier exists due to the fact that the encephalon’s capillaries are free of pores, which in other
parts of the body facilitate the drug out of the plasmatic area.
Endothelial cells are accompanied by tight junctions of brain substance and not by the usual gap
(button) type junctions. In addition, the capillaries of the encephalon are very closely wrapped by the
glial cells. In the absence of channels, the diffusion in the brain’s ECL is only easy for fat-soluble
drugs. The blood-brain barrier of the newborn is inefficient compared to an adult one.
The blood-brain barrier efficiency reduction is considered a chemical toxicity mechanism, which
is still under investigation. The CNS is separated from the interior fluid space by the ependymal and

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
54 | P a g e
from the outside, by the glial cells. Both structures present intercellular spaces, which allow
communication between the extracellular fluid and CSF. A particular interest in terms of physiology
and pharmacology is given to those small areas of the brain that are not located "after" the blood -
brain barrier but belong to the plasmatic network. They are called: circumventricular organs.
Of these, the most important are:
• Area postrema and
• Eminentia mediana
The limit between CSF and plasmatic network is represented by the surface coating.
The area postrema can be regarded as an assembly of chemoreceptors.
Through these "sensors" the CNS can directly receive information through the network of blood,
which is important, among other things, for the function of the respiratory center.
In the area postrema are positioned the vomiting chemoreceptors, and their excitement can cause
the act.
In the eminentia mediana, the neuro-secretor axons are placed, which release prior regulator
hormones of the pituitary function. These hormones are taken up by the fenestrated endothelium
capillaries. Many substances, (e.g., chemotherapeutics and antibiotics have difficulties in their CNS
penetrating (e.g. tetracycline, penicillin, even streptomycin).
When crossing the Central Nervous System, drugs meet two main barriers: blood-brain barrier
and blood-cerebrospinal fluid (CSF). Blood-brain barrier through which the drug passes into the
encephalon’s extracellular fluid is constituted by the capillaries surrounding walls and glial cell layers.
Blood - CSF barrier is composed mainly of the choroid plexus epithelium. Studies have shown
that the two barriers often act as lipidic membranes. The intravenous drugs pass into the brain or CSF
at rates proportional to their partition coefficient and its dissociation constant at a pH of 7.4.
Among the two barriers; blood - brain and blood - CSF can pass a series of drugs, like:
chloroform, ether, halothane, chloralhydrate, barbiturates etc.
4.3.2. Hemato-oftalmic barrier
The passing of drugs, through the plasma in the aqueous chamber of the eye is performed by the
ciliary body epithelium. Substances cross with difficulty, because of the eye’s much lower vasculature,
compared to other tissues.
4.3.3. Placentary barrier
The placenta is placed between maternal blood and fetal circulation.
This barrier comes from the syncytial trophoblast formed by the merger of several cells. In this
situation, the intercellular spaces are missing, but transcellular exchanges are present.
The placental barrier’s permeability is higher than that of the blood-brain barrier.

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
55 | P a g e
All pharmacons that are having central effects, namely, those who cross the blood-brain barrier,
enter relatively easy in the fetal circulation. Drug effects will last longer in the newborn animals
compared with adults because the specific removal mechanisms are not yet defined.
Liposoluble drugs diffuse through the placenta easily and, therefore, most anesthetics may cause
respiratory depression in the newborn. The original concept that the placenta is an important barrier to
protect the fetus from the action of medicinal substances proved to be illusory.
4.3.4. Coetaneous barrier
It generally prevents substances from entering in the body, which limits their effect substantially.
The exceptions are the: liposoluble and volatile drugs (e.g. iodine, guaiacolum, eucalyptol, etc.), which
can have a deep, diadermic penetration.
Most drugs, to exert their pharmacodynamic effect, must penetrate the body humors from which
they are directed towards farmacoceptors. Insoluble compounds are considered as inert, from a
pharmacological standpoint.
Theoretical distribution of drugs in tissues and organs (After Cristina, 2000)

Introduction in Veterinary Pharmacology Chapter 4 Romeo – Teodor CRISTINA
56 | P a g e
4.4. Drugs’ Redistribution
This phenomenon is illustrated for example, by the pharmacokinetics of thiopental. When this
lypophilic drug is administered I.V., it will rapidly diffuse in the CNS (because it is a well-
vascularized and rich lipidic tissue), so the general anesthesia is rapidly induced. The initial
equilibrium between blood and brain will change, because the drug is more slowly equilibrating in the
other tissues. Because of this, the drug will diffuse back into the blood from the CNS, to recreate a
new blood-brain balance.
4.4.1. Consequences of uneven distribution
These mechanisms contribute to variations in drug concentration between the different body areas
at the moment of equilibrium. Drug concentration in tissues, at known established time intervals after
the last administration (the so-called "residue studies"), is essential to establish the withdrawal period
i.e. the time that must elapse after the last administration to slaughter for human consumption. If the
ability to attach or to seize the drug in other places than the action site (on the so-called loss sites, drug
acceptor sites or silent receptors) is significant, high initial doses may be necessary. It is possible for a
pharmacon’s high local concentration to produce changes (e.g. nitrofurantoin causes yellowing of the
teeth), undesirable side effects (e.g. cloroquins, causes retinal dystrophies), or even accidental large
values (e.g. arsenic and heavy metals, etc.).
Conclusions
Regardless of the route of administration, a medicinal product must:
be absorbed and leave the administration site,
enter into the circulatory stream and then,
diffuse into the body.
A drug can be: fat soluble (or liposoluble); water soluble (or hydrosoluble) and amphiphylic
(even phases).
The rate of absorption will depend primarily on the:
• pH of the absorption surface,
• pKa of the drug,
• oil-water partition coefficient,
• degree of blood irrigation of the absorption area and of the
• absorption area surface.
The concentration that a drug can reach into the diffused compartments depends on the:
- pH difference between the two spaces separated by the traversed membrane
- various coupling capacities on both sides of the membrane
- The existence of an adequate transport system, or
- The existence of specific membrane barriers

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
57 | P a g e
5. Drug-receptor binding
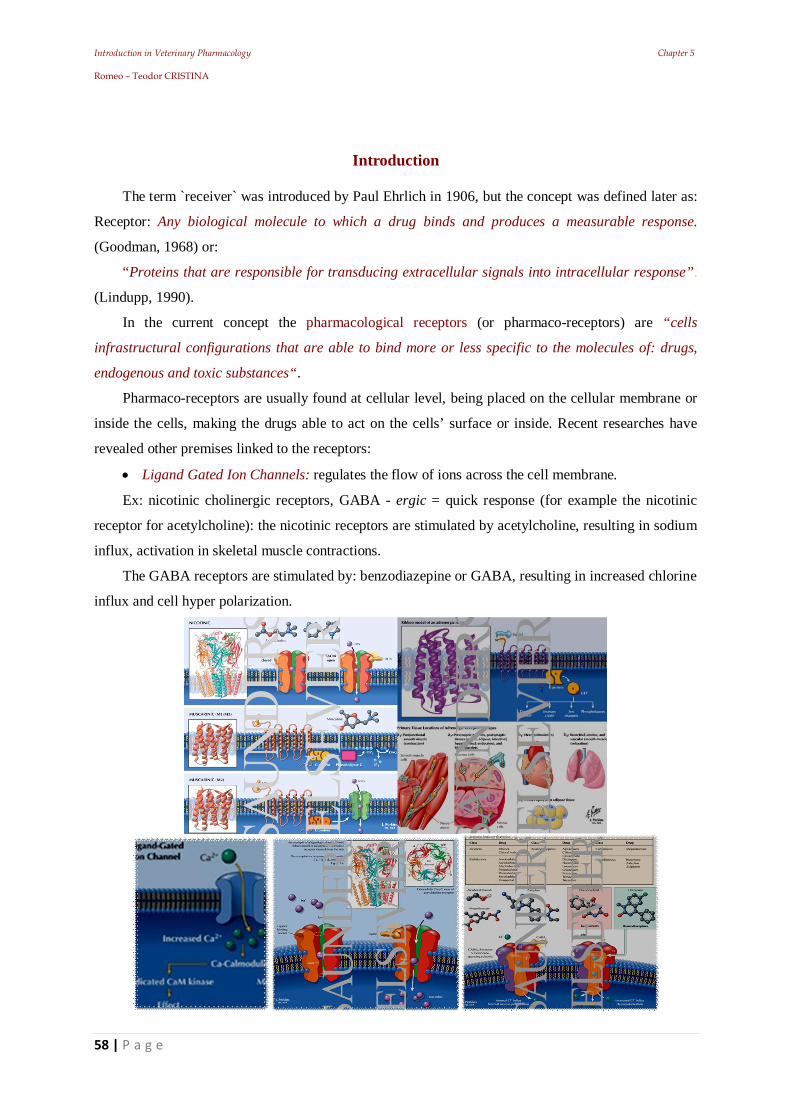
Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
58 | P a g e
Introduction
The term `receiver` was introduced by Paul Ehrlich in 1906, but the concept was defined later as:
Receptor: Any biological molecule to which a drug binds and produces a measurable response.
(Goodman, 1968) or:
“Proteins that are responsible for transducing extracellular signals into intracellular response”.
(Lindupp, 1990).
In the current concept the pharmacological receptors (or pharmaco-receptors) are “cells
infrastructural configurations that are able to bind more or less specific to the molecules of: drugs,
endogenous and toxic substances“.
Pharmaco-receptors are usually found at cellular level, being placed on the cellular membrane or
inside the cells, making the drugs able to act on the cells’ surface or inside. Recent researches have
revealed other premises linked to the receptors:
Ligand Gated Ion Channels: regulates the flow of ions across the cell membrane.
Ex: nicotinic cholinergic receptors, GABA - ergic = quick response (for example the nicotinic
receptor for acetylcholine): the nicotinic receptors are stimulated by acetylcholine, resulting in sodium
influx, activation in skeletal muscle contractions.
The GABA receptors are stimulated by: benzodiazepine or GABA, resulting in increased chlorine
influx and cell hyper polarization.

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
59 | P a g e
G-Protein Coupled Receptors: the peptides are linked to G Protein by three subunits: alpha
(linked with GTP), beta and gamma. The linkage of the appropriate extracellular ligand = G protein
activation. GTP replaces GDP on the alpha subunit.
During the dissociation of the G Protein the subunits interact with secondary messengers,
resulting in a response in seconds to minutes (example, the alpha and beta adrenoceptors).
Enzyme-Linked Receptors are: a specific cytosolic enzymatic activity as an integral
component of the structure / function. Binding an extracellular ligand activates or inhibits the activity
of cytosolic enzymes (ex: insulin receptors or Tyrosine Kinase Receptors).
Duration of response in this case: minutes to hours.
Intracellular Receptors: are completely intracellular, specific ligands, which to act, must
diffuse into the cell in order to interact with it.
For example the steroid receptors; in these receptors, ligand must have a good liposolubility in
order to be able to cross the cell membrane.
In the case of steroid receptor, the activated receptor-ligand complex will migrate to the nucleus,
where it binds to a specific DNA sequence resulting regulation of gene expression.
Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
60 | P a g e
The response duration is long: hours or days. Nuclear receptors have in them structure: one or
more active centers and the active groups of the pharmacon will be fixed on these active centers.
5.1. Preliminary aspects of drug-receptor interaction
The Receptor theory starts from the following principle: a substance will become active at a
cellular level when a specific “molecular reaction partner” will be present.
This reaction partner (Receiver-R) must have specific qualities, so that a substance (or
substances group) can form a chemical bond with it (whose type does not play, usually, any
pharmacodynamic role). As a result, changes in physicochemical properties of biological response
from the action place will constitute an "excitation" that will trigger "the effect".
In general, in case of fixation, the following types of connections are formed:

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
61 | P a g e
hydrogen bonds,
non-polar (Van der Waals),
ionic and
covalent.
The binding process has two stages; the first one, pharmacokinetic: the accumulated drug binds to
the receptor in order to form drug-receptor complexes. The reaction is reversible and depends on the
affinity between the substance and pharmaco-receptors.
The second stage is pharmacodynamic: The drugs’ effect is the result of substance-receptor
interaction, due to the substrate’s biophysical, biochemical and physiological modifications, on which
the receptors are fixed.
Of greater importance for determining the number of receptors and their properties is the
bindingrate of agonists and antagonists. This consists in: measurement of the binding specific capacity
using radio-labeled material (of radioactive isotopes H3, C14).
5.1.1. Activity and receptor’s characterization
The receptors structure-function relationship is based on the receptors representation (because
when the receptor assigned specific chemical physicochemical and physical properties, it is self
explanatory that the agonist has an additional structure). Possible situations:
1. an enantiomer can occupy an entire binding location.
2. an enantiomer can occupy only a part of a binding location.
1 2

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
62 | P a g e
About the biological effect of a chemical coupling certain assumptions can be made, but with
reservations. However, one type of principle can be applied: modified structure = an effect that is
applied to obtain analogous preparations in pharmacology.
If a substance is found to be effective, then the active parts of the molecule should not be
modified. For example, phenothiazine (neuroleptic substance) insignificant changes in the cycle and
the carbon chain in position 10 of the phenothiazine molecule will give either: benzodiazepine, saline
diuretics etc.
5.1.2. Receptors’ mode of action
The receptor concept was sustained in analogy with enzymes, comparing receptors with the
active centers of enzymes. In this way, the classic model "key-lock" was applied in drug action
explanation:
• the “key" is the drug, and
• the "lock" is the receiver.
Changing the form the “key”, will clearly affect its ability to "come and open the lock."
Similarly, an antagonist is a drug able to enter into the lock, but unable to trigger the response (in our
case to open the lock). It will remain there to prevent the entry of the "appropriate" key.
Possible situation: drugs’ specificity for the receptor (It happens often that a drug’s name is used
to identify the population of receptors of cells in which they react). A problem of particular interest
was to explain the drug actions immediate reversibility (e.g. by successive washes in isolated tissue
preparations), when the drug action is dependent on a chemical bond between the drug and target.
The current hypothesis is that: drug molecules that are disorderly moving in biophases will
couple with their receptors only when they are close enough to them.
For example, covalent and coordinative links are formed by certain drugs, as a feature of their
own specific action (ex: organophosphate anticholinesterases).

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
63 | P a g e
Energetic links of this type are stable and many times characterize the drugs that have higher
action duration. Whereas the drug approaching to the receptor is favorized by the electrostatic forces,
that function on long distances (e.g. ionic coupling potential), short distance forces of weaker bonds
are numerous, and thereby, more important for the association recovering when there is a high degree
of comlementarity.
Coupling of drug to receptor is classified as: with high affinity / low capacity.
Demonstration of the coupling process is not sufficient to highlight the place where the drug acts.
The sites where drugs are bonded, but where no type of associated effect is produced, are most of the
time, with: high affinity / low capacity (e.g. the serum albumins).
They are also known as: drug “acceptors” or “mute receptor”.
A drug molecule can be switched from the receptor when its kinetic energy is increased by
thermal collisions, at a level that exceeds the coupling power.
Neuromuscular junctions are among the few detectable histological entities that contain numerous
receptors placed on the cell surface. Changeux et al. showed that nicotinic receptor for acetylcholine
(250 kDa glycoprotein), contains five functional subunits, where two are identical.
The transport mechanism (e.g. pores, channels or ionophores) through which’s opening and
closing pulse allows the sodium ions passage through the membrane is the one who initiates the
response.
The authors suggested that the affinity of coupling sites is not constant; in the presence of the
agonist, the conformation of the binding sites is changing, meaning that the affinity for the agonist will
grow.
During the active phase, the ionic gate will open transitory and the physiological /
pharmacological response will be able to install. If the agonist is still present, the coupling affinity will
reach a high level, while the ionic gates will be closed.
The reduction or absence of a response, that follows it, is called desensitization. Both activation
and desensitization are prevented in the presence of a competitive antagonist, for which the site found
in state of repose has a higher affinity than the activated or desensitized site.
A big part of the available information regarding the trans-membranal ion channel activity and
the ways in which drugs or other ligands can modulate their activity, were recently discovered (an
ultrafine micropipette is applied on the plasma membrane area that contains nicotinic receptors (for
example the terminal motor neurons).
Using this technique it was possible to define the concentration of acetylcholine required:
to produce the ion channels opening,
to establish the opening duration,
to measure the size of the flow of sodium ions that enters the cell;
to measure the drugs’ effect.

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
64 | P a g e
In the attempt to propose some models that are able to explain the action of drugs, the mobility
term was introduced, with reference to the individual receptors from the membrane’s surface. It has
been suggested that the coupling phenomenon may induce a change in the membranal receptor’s
conformation (or of a group of associated receptors).
The expected result should be the opening of a pore in the membrane, allowing the ionic flow
realization. Further, membrane depolarization will be produced.
The ability to move laterally is totally compatible with the fluid mosaic model of the structure of
plasma membrane.
Drugs that can cause opening or blocking the membrane channels for potassium are, also, of
interest for regulating muscular tonus of the blood vessels.
Some drugs can affect the muscular tonus, although each class works on the other group of
receptors. Though an antagonist can inhibit the action of a group of drugs by blocking its receptors,
the tissue may still respond in a characteristic manner if a drug that will activate another type of
receptor is administrated.
In the case of nicotinic choline receptors, a demonstration often cited, is about alpha-
bungarotoxin, a toxin extracted from snake venom that will connect to the receptor with high affinity
and specificity.
5.1.3. The nature of receptors
The accessibility of studying enzymes, as the facility to estimate the concentration of substrate
and product has allowed enzymology, as a new science, to advance and provide valuable concepts in
the study of receptors.
The classic receptor from the cell surface is, normally, an included lipoprotein or a lipoprotein

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
65 | P a g e
that penetrates the plasma membrane of the cell, such as the active site of an enzyme is known to be a
small portion located within a folded protein.
The substrate will bind with the active site that will catalyze a change in the substrate structure. In
this aim, the key - lock analogy requires a "rigidity" of the reactants and, therefore, it is not entirely
compatible, since the vast majority of drug molecules are "flexible" as structures.
Some enzymes suffer changes of the chemical conformation, and the induction of a change in the
conformation of the receptor can be necessary for the drug action.
Clearly, the "rigidity" implies (requires) the drug’s complete specificity for the receptor. Such
specificity is not absolutely necessary; the enzymes can be inhibited irreversibly, just like some
receptors, by molecules that are covalently bound to their active site. A false substrate can be coupled
with an enzyme, which later will be separated at a much lower rate than the right substrate.
Likewise, an antagonist who binds to the receptor, does not cause any response and remains
coupled to the receiver a relatively long time.
Various enzymes that can catalyze the same reaction were called isoenzymes. The differences in
the apparent sensitivity of the receptor at a number of activator drugs had led to the concepts of:
isoreceptors (e.g. alpha & beta receptors) and
allosteric receptors which describes a change in shape induced by an enzyme, after coupling
to a site other than the active one, with a different substance than the normal substrate.
The combination of the so-called allosteric site may result in allosteric activation / or inhibition of
the enzyme by changing the access to the active site.
This mechanism was (also) used to explain the ability of a drug to alter the action of another drug
at the receptor site.
Therefore, it was not unexpected that the cases that demonstrates that drugs are able to inhibit "in
vitro“ Although it does not automatically follow that this mechanism is relevant for the same drug "in
vivo".
Demonstration of the process depends on establishing a relationship between:
dose,
local concentration and
the degree of inhibition of the response.
The inhibition of enzymes can be accomplished through several mechanisms.
The active site can be irreversibly blocked by an antagonist who binds covalently (e.g. heavy
metals)
Reversible inhibition can be achieved by using agents that are structurally related to the
physiologic substrate, but dissociate slowly from the enzyme (e.g. physostigmine and cholinesterase).
Such agents can act (also) as "false substrates” that can be "processed" by enzymes in a fake
product. Drugs can inhibit enzymes by interfering their synthesis or by removing essential cofactors.
An example is the case of benzimidazoles, where they interfere with enzymatic systems (fumarate

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
66 | P a g e
reductase and succinate decarboxylase), the ATP synthesis sites, being essential for the energy
metabolism of helminths.
Main Ligands/ Receptors
One way to characterize the receptor consists in isolating and studying them.
Initially researchers tried to isolate the nicotinic acetylcholine receptors in a tissue, in which they
are in a high density namely the fish’s electric organ.
The "in vitro" binding of an agonist cannot be made in the same manner as for the "in vivo" and,
therefore, an accurate assessment of the functional capacity of receptors is not yet possible.
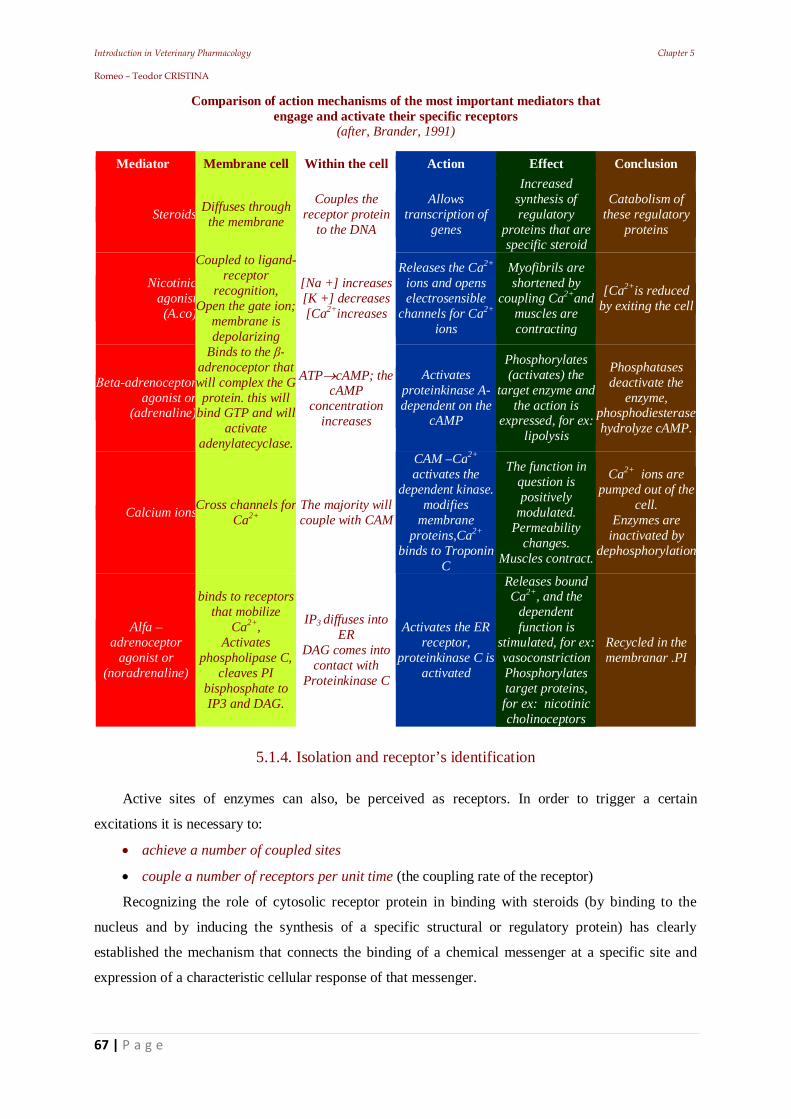
Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
67 | P a g e
Comparison of action mechanisms of the most important mediators that engage and activate their specific receptors
(after, Brander, 1991)
Mediator Membrane cell Within the cell Action Effect Conclusion
Steroids Diffuses through the membrane
Couples the receptor protein
to the DNA
Allows transcription of
genes
Increased synthesis of regulatory
proteins that are specific steroid
Catabolism of these regulatory
proteins
Nicotinicagonist (A.co)
Coupled to ligand-receptor
recognition, Open the gate ion;
membrane is depolarizing
[Na +] increases [K +] decreases [Ca2+increases
Releases the Ca2+
ions and opens electrosensible
channels for Ca2+
ions
Myofibrils are shortened by
coupling Ca2+and muscles are contracting
[Ca2+is reduced by exiting the cell
eta-adrenoceptor agonist or
(adrenaline)
Binds to the - adrenoceptor that will complex the G protein. this will
bind GTP and will activate
adenylatecyclase.
ATP cAMP; the cAMP
concentration increases
Activates proteinkinase A-dependent on the
cAMP
Phosphorylates (activates) the
target enzyme and the action is
expressed, for ex: lipolysis
Phosphatases deactivate the
enzyme, phosphodiesterase hydrolyze cAMP.
Calcium ionsCross channels for Ca2+
The majority will couple with CAM
CAM –Ca2+ activates the
dependent kinase. modifies
membrane proteins,Ca2+
binds to Troponin C
The function in question is positively
modulated. Permeability
changes. Muscles contract.
Ca2+ ions are pumped out of the
cell. Enzymes are
inactivated by dephosphorylation
Alfa – adrenoceptor
agonist or (noradrenaline)
binds to receptors that mobilize
Ca2+, Activates
phospholipase C, cleaves PI
bisphosphate to IP3 and DAG.
IP3 diffuses into ER
DAG comes into contact with
Proteinkinase C
Activates the ER receptor,
proteinkinase C is activated
Releases bound Ca2+, and the
dependent function is
stimulated, for ex: vasoconstriction Phosphorylates target proteins, for ex: nicotinic cholinoceptors
Recycled in the membranar .PI
5.1.4. Isolation and receptor’s identification
Active sites of enzymes can also, be perceived as receptors. In order to trigger a certain
excitations it is necessary to:
achieve a number of coupled sites
couple a number of receptors per unit time (the coupling rate of the receptor)
Recognizing the role of cytosolic receptor protein in binding with steroids (by binding to the
nucleus and by inducing the synthesis of a specific structural or regulatory protein) has clearly
established the mechanism that connects the binding of a chemical messenger at a specific site and
expression of a characteristic cellular response of that messenger.

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
68 | P a g e
5.1.5. The definition of agonists and antagonists
Substances that stimulate receptors are called agonists. For example, morphine is fixed on the
opioid receptors (OR), with analgesic effects and respiratory center depression.
For example, nalorphine, its antagonist, will keep the analgesic action but a stimulating effect of
the respiratory center will produce (so it is used as antidote in the morphine poisoning).
The agonist, is a drug that can be coupled with a receptor and cause a positive response in the
tissue where receptors are located.
The maximal response is considered the response with an intensity that can’t be overcome by
subsequent agonist administration (by increasing the agonist concentration).
Body potent agonists (such as acetylcholine, norepinephrine and histamine) are identified by a
high coupling / decoupling speed. In this context, the image about the receptors utility, regarding the
substances competitive antagonism is clarified. Agonists are substances that are bound to receptors
and that will induce modifications of cell properties (high affinity and “intrinsic” activity)
Competitive antagonism binds reversibly to the same receptor and will not induce any change
(high affinity and absent “intrinsic” activity), but will block some receptors (e.g. decreases the
concentration of active receptors) so that the agonist will lose efficiency (e.g. acetylcholine- atropine,
acetylcholine- D-tubocurarine,
Agonists
Whether a cell will respond to the administration of a chemical messenger type will depend on
the presence (or absence) of the appropriate receptors:
exogenous or
endogenous.
The nature of any cell response to its receptor activation depends on the cell. It produces the
response that represents its usual function (e.g. muscle cells are contracting if membrane
depolarization at the neuromuscular junction exceeds a critical level).
Antagonists Antagonist is considered a drug that, when is administered before or concurrently with the
agonist’s administration will diminish or abolish the agonist response.

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
69 | P a g e
It is said that antagonism is:
permanent,
irreversible or
non-competitive, if the intensity will remain unaffected in the presence of
increasing concentrations of agonist.
The antagonism of a drug toward generating capacity of response, at another medication, is a
negative response that can be played using the dose - effect curves.
By definition, the antagonism is non-competitive when, in the presence of an antagonist, the
agonist is no longer able to produce the maximum effect, regardless
of the increase of its concentration.
In this situation, dose-response curves of agonist in the presence of antagonist’s gradually
increasing concentrations will become progressively less inclined, and the maximum possible effect
will decrease, as the
concentration of the antagonist increases. If the increasing agonist concentration reduces or
exceeds antagonism, the antagonism is called:
temporary,
reversible or
competitive.
Competitive or non-competitive reduction of the response to an agonist are types of
pharmacological antagonism. When a drug reduces the effect of another drug by inducing a contrary
response, by activation of other receptors, we talk about a physiologic antagonism.
For example, antihistaminic drugs pharmacologically can block the action of histamine, but
effects of histamine can be obtained also with adrenaline.
In the treatment of poisoning, the continuous drug absorption through the gastro-intestinal tract,
sometimes, may be prevented by transforming the toxic substance into a insoluble form.
The secondary messengers
There where many progresses accomplished during the last years, concerning the identification
of the binding path for certain chemical messengers and specific receptor proteins from the membrane,

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
70 | P a g e
the way this path can intensify or diminish the cellular function.
In most cases this phenomenon consists of a modified rate of entrance or synthesis of so-called
secondary messengers. In this case, in contrast with the complex steroid hormone – cytosolic receptor
protein, the drug receptor complex will no longer be the final intracellular effector.
The integration of the drug - receptor couple and the activation of the cells functional apparatus
will be made by an intracellular secondary messenger. Several chemical mediators, including:
• neurotransmitters,
• endocrine hormones and
• tissue hormones,
After the complexation with the membrane`s receptors, they can determine the activation /
inhibition of adenylate cyclase enzyme at the membrane level.
The concentration of cyclic AMP from the cell will increase, and consequently, the mediator
will react as an intracellular messenger, and with the help of protein-kinase A, will adjust those
enzymes that mediate cells’ characteristic response.
This is the process from where the secondary messenger term derives.
Essentially, drug-receptor complex has an increased affinity for adenylate cyclase, forming a
large complex. In fact, a protein realizes the bond between the complex: drug + receptor + adenylate
cyclase and binds nucleotide guanosine triphosphate (GTP).
Adenylate cyclase’s maximum activity requires the presence of the combination: drug-receptor
+ coupled protein + GTP + enzyme.
Cyclic GMP is another intracellular messenger synthesized by the guanylate cyclase enzyme. A
practical consequence of this is that the levels of cyclic nucleotide concentrations can be monitored.
Muscarinic choline receptors can be enzymatic mediated. They are able, after coupling a
muscarinic agonist, to bind at the cell’s membranes interior with the protein that binds guanosine 5'-
phosphate (G protein). It has been demonstrated that this complex is able to:
• inhibit adenylate cyclase,
• activate guanylate cyclase, \
• increase the potassium ions conductance (in the heart)
• reduce the conductance of potassium ions (in CNS)
• ease triphosphate-inositol-phospholipase activity.
So: the action will be dependent to the site, in each of these cases, because the receiver will be
coupled to a different G protein.
Calcium ions
They are considered also intracellular secondary messengers with a great importance. Free
concentrations may be increased not only by membrane depolarized agents acting on receptors
coupled to the membrane pores (e.g. acetylcholine) but also through several other mechanisms (e.g.

Introduction in Veterinary Pharmacology Chapter 5 Romeo – Teodor CRISTINA
71 | P a g e
medications, hormones or neuro-transmitters) that act on mobilization receptors of the calcium ions.
For example, calcium ions are involved in the arahidonic acid release from the phospholipidic
membrane by activated phospholipase, initiating the synthesis of prostaglandins and leukotrienes
(which are extracellular messengers).
While previous researches were focused on the calcium entry through the electro-sensitive
channels in the electrically excitable cells, as a mechanism to increase the calcium ion concentration in
the cytosol, the current studies, performed on endoplasmic reticulum (ER) from muscle fibers, have
revealed that the release of calcium coupled inside the cell, can act as an alternative mechanism that
leads to the same result (increased intracellular concentration).
Later, it was revealed the involvement of membrane phospholipids in the cellular turnover of
calcium ions. In this case, phospholipase C (activated enzyme by the receptors located in membrane)
cleaves the phosphatidyl inositol (PI) in:
• diacylglycerol (DAG) and
• inositol-1-phosphate, stage in which the intracellular calcium ions will be released.
DAG is a secondary messenger that activates protein kinase C before it is resynthesized in the
PI.
Recently, it has been demonstrated that: phospholipase C hydrolyzes a second phospholipidic
membrane, phosphatidyl inositol-bisphosphate (PIP2), releasing: DAG and inositol triphosphate (IP3),
the latter is a calcium bound powerful "liberator" to the endoplasmic reticulum.
Like DAG, IP3 is recycled to the membranal PI.
Conclusion
The molecular structure is the first determinant of drug activity of a chemical substance.
The response type produced depends on the location’s normal functioning of which the
molecular properties of the drug allow it to:
• persist,
• accumulate and
• bind.

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
72 | P a g e
6. Coupling response quantification

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
73 | P a g e
Introduction
Currently there are several hypotheses regarding the relationship between drug and
receptor, from which three theories have been established:
The “Occupancy” theory (Clark’s), where the intensity of a drug's effect depends on the
number of occupied receptors (this theory has many variations)
The “Speed” theory (Paton’s), where the intensity of the effect is the result of speed
combined with the receiver.
The “Activation” theory, claims that the pharmacodynamic effect is produced by
converting receptors from an "inactive" into an "active" form.
Today it is considered, that the allosteric activation of the receptors, is a necessary
process to obtain the drug effect.
Knowing the type of responses produced by a drug they can also be completed by
measuring the:
response size and respectively the
drug quantity, in order to make it possible to study the dose-effect relationship.
The procedure known as “quantitative bioassay” has allowed for several active
substances to be used with a reasonable accuracy from a dosing point of view.
The bioassay can also be applied for the identification of a drug whose effect is
characteristic or, for which an identified antagonist is known.
6.1. Clark's theory (of occupation) and its variant
Clark noted the similarity between Langmuir’s adsorption isotherm form and many dose-
response curves. He applied to drug-receiver interactions, principles and kinetics used to
reproduce a hypothetical model for the reactions occurring at a surface level.
Clark was pleased with this model because, in many cases, the form of dose - effect
curves experimentally obtained matched the obtained form of a relationship that involved:
interaction type: molecule - molecule;
tiny proportion from administered drugs that bind to receptors;
the proportion from the maximum effect that can be achieved: depending on the
administered drug concentration (its affinity is constant).

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
74 | P a g e
Explanation:
The adsorption isotherm has derived as a result of studying the adsorption rate of a gas at
a constant temperature by the material prepared (considered impermeable surface).
The curve shows how the adsorbed gas concentration increases, once the applied
concentration increases:
first, very quickly, then,
much more slowly, as the adsorbent is approaching saturation level.
In the vicinity of a certain threshold it will no longer reach a higher adsorbed
concentration, regardless of how much the applied concentration increases.
Drug binding on the cell surface was assumed to be based on the principle:
“A drug molecule per receptor”, similarly with the case of a molecular reaction between
a gas and a solid surface.
Adsorbent surface saturation has been equated with: occupying all available receptors,
corresponding to the point of dose-response curve, in which the effect is maximal.
From this it was deduced that: a lower proportion of receptor occupancy could produce a
weaker effect. Assuming that each occupied receiver produces a constant unit of response, the
effect would be directly proportional to the receptor occupancy, because the occupying rate
and stimulus are cumulating.
Another assumption was that: adsorption on the surface did not produce a significant
reduction in the concentration of a gas or free drug.
When various gases are present, in the same molecular concentrations at the adsorbent
surfaces (that have the same area and are from the same structure) under standard conditions
of temperature and pressure, it is obvious that the extent to which different concentrations of
adsorbed gas differ at the time of equilibrium, depends on its individual coefficients of
adsorption.
If it is considered that drug - receptor interaction is accomplished in a closed system ("in
vitro") it can be assumed that the process follows the principles of the mass action law: when
the effect level is stable in the presence of an administered drug, there exist balances at which
the association and dissociation rates of complex are equal.
A difficulty in this theory was found for “dualists” also called partial agonists.
A partial agonist is unable to produce the maximum effect compared to a full agonist,
when reacting with the same population of receivers (it does not produce the same level of
response).

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
75 | P a g e
6.2. Ariens theory
Clark's theory assumed that: each coupling of any drug with any receiver from the same
population will have the same efficiency in producing response units.
Partial agonists have shown that it is not like that!
In order to introduce this observation in the theory, a second property of drugs, was
invoked: one that is quantitative non-dependent on affinity!
While the affinity remains the property which determines the ability of a drug to bind to
receptors, Ariens proposed another property: the intrinsic activity.
The concept of intrinsic activity allows the understanding of another set of observations
that do not correspond to theory of occupancy.
Like, when agonists are administered in mixtures and the result (answer) is sometimes
weaker than the one that is expected (due to an addition of effects of receptor occupancy).
In the presence of a partial agonist, for example, the concentration of a potent agonist
(which under normal circumstances is sufficient to cause a maximum response) turns out to
be, not as effective.
This is due to the occupancy of a receptor fraction by a partial agonist and that it will no
longer be able to produce only sub-maximal stimuli.
6.2.1. Stephenson’s theory
Another alternative extension of Clark's theory was the one of Stephenson who proposed
a modified theory that incorporates the following observation: for some drugs, the production
of a maximum effect can be achieved by receptor occupancy in a proportion of less than
100%. He invoked the concept of efficiency, whose value expresses the relative ability of the
receptor occupied by the drug in question to “donate” a biological stimulation unit (S) to the
cell.
A drug with high efficiency can produce a maximum response after occupying only a
small proportion of the receptor population leaving a number of spare receptors.
On the contrary, a drug with low efficiency has to deal with a higher proportion of
receptor, in order to produce a maximal response.
A partial agonist does not manage to induce a maximal response even when all receptors
are occupied, because the value of its efficacy (e) is too low for as the critical value of S to be
reached. And in this case, for an antagonist the value of e is zero.

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
76 | P a g e
6.3. Paton’s theory
The inclusion of “intrinsic” and “effectiveness activity” in the occupancy theory of drug -
receptor interactions has successively reduced the importance of the idea of a stable
occupation of receptors, as a major determinant of response.
Rate theory for relations: drug - receptor - response (introduced by WDM Paton) led to a
point where, the occupancy alone was considered unimportant for agonist activity. Paton
proposed the idea that the formation of a complex drug - receptor generates a stimulation unit
of the production response towards the cell. So:
after drug administration,
all tissular receptors are available for coupling,
the tissue will receive the maximum stimulus and
will generate a maximal response.
For the answer to be maintained:
the complex must be decoupled and recovered
when the complex is quickly disentangled, couples will be much more quickly
recovered.
So, for an agonist, the rate of decoupling complexes (governed by the dissociation
constant) is the one that determines potency, because it dictates the rate at which new
complexes can be achieved.
According to this hypothesis, the antagonist binds fast, but the dissociation will happen
slowly. Embracing this idea, Paton expressed some observations inconsistent with the theory
of occupancy (Clark).
For example, the fact that:
1). the same receptor set can be stimulated at first and then blocked.
2). many active substances produce a maximum effect only after the first administration
(phenomenon called tachyphylaxis).
The first situation is considered “partial agonism”:
When the drug stimulates at the moment of coupling, but because it dissociates
relatively slowly, it will persist on receptors and thereby will succeed in exerting an
antagonistic activity.
The second situation occurs, after repeated exposures to a drug, when a part of the
previously administered dose has remained coupled to receptors (reducing the number of
receptors available for coupling).

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
77 | P a g e
For practical reasons, the experiments that are meant to test the rate theory are difficult to
perform. Instantaneous registration of the response is required to confirm that the installation
of the maximal effect was achieved only under experimental conditions.
The instantaneous termination of the response (consecutive removal of the drug from the
tissue) requires instant removal, but the diffusion based on the concentration gradient is a
lengthy process.
6.4. Activation theory and other recent postulates
The continuing response requires the release and regeneration of receptors.
Recent comments have pushed this dynamic vision further, even questioning the very
existence of receptors. As populations fixed, in terms of numbers, location or affinity, more
complex patterns of interaction and modern drug - receiver are being proposed.
For all the theory variants of the receptor, the postulate theory is common, which states:
an agonist drug is combined with a site on the receptor, and the receptor is activated,
thereby achieving a response from the cell.
when the drug disappears, the receptor returns to inactive status (meaning, it
regenerates).
This is essential for the subsequent cycles of response. Upward or downward adjustment
of the number of receptors is another mechanism by which some drugs can act.
6.5. Enzymology theories
Expanding the concept of allosteric sites for drug-receptor interactions is another modern
thesis suggested by researchers.
In enzymology, allosteric sites are recognized as adjacent locations to active sites of
enzymes, to which antagonists can bind, covering or distorting the active site so that it can no
longer complex with the substrate.
In pharmacology, the involvement of allosteric binding sites for antagonists was
postulated for situations where the antagonism between two drugs moves from competitive to
non - competitive as the antagonist concentration increases (e.g. acetylcholine and atropine
acts on smooth muscle). Although the study of receptors has become a real science
(Receptology), many questions about the receptors, still remain unanswered.
For example: why is it necessary that the cells possess specific structures that allow them
to react to foreign substances?

Introduction in Veterinary Pharmacology Chapter 6 Romeo – Teodor CRISTINA
78 | P a g e
The existence of receptors has already been demonstrated, and in time, all the attention
has been focused on the temporal, qualitative and quantitative aspects of drug action, at
cellular and molecular level, with advantageous consequences for pharmaceutical and
pharmacological research
For example:
some pathological conditions in humans or animals have been demonstrated to be
caused by receptor depletion (e.g. Myasthenia gravis)
the efficiency loss of some drugs due to consecutive prolonged administration was
explained by desensitization and/or depletion of receptors (e.g. - adrenoceptors,
bronchodilators).
Conclusion The term receptor identifies a location where a drug can bind and induce an alteration
that is expressed under the form of an observed drug effect.
Different types of receptors have been identified:
cytosolic,
coupled to pores
or coupled to enzymes and
different mechanisms of production of effects on cell:
membrane depolarization,
selective permeability changes,
increasing or decreasing level of various cell regulators
(enzymes, ions, degradation of the phospholipids).
This makes it possible to understand why:
some chemical messengers are acting extremely fast,
(e.g. neurotransmitters through receptors coupled to pore over 1-2 ms) others,
in a longer time, but a longer period (e.g. minutes for peptide hormones and receptor
mediated enzymes), while
others produce an effect only after several hours but have an extended duration of
action (e.g. steroid hormones and functional proteins synthesized de novo by target cells).

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
79 | P a g e
7. Drug metabolism

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
80 | P a g e
Introduction
While absorption is of prime importance in the growth and maintenance of a constant
concentration of a pharmacon in the plasma, thus determining the intensity and duration of action of
the drug, other processes operate to reduce this concentration.
The influence of distribution, in the dilution of the drug is reinforced by the removal of the free
active drug trough the process of elimination.
This includes the:
metabolic inactivation of the pharmacon and
excretion of both the intact molecules of the pharmacon as well as the modified one’s.
These changes often reduce or even block the activity of the pharmacon.
Most drugs are metabolized in the liver and excreted by the kidneys.
The microsomal enzyme system in the liver has a role in metabolizing fat soluble drugs.
Drug metabolism may also occur in blood plasma and in the intestinal lumen where hydrolytic
and reduction reactions occur.
After the metabolism phase, the appearance of metabolites with favorable physico-chemical
properties for excretion is observed. Ionized (polarized) drugs and their metabolites are excreted by
the kidneys.
This includes:
metabolic inactivation of the pharmacon and
excretion, of both the intact molecules of the pharmacon as well as the modified one’s.
These changes often reduce or even block the activity of the pharmacon

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
81 | P a g e
7.1. Factors that influence drug metabolism
The factors that influence drug metabolism and elimination are pharmacokinetic and
pharmacodynamic whose interweaving causes the direct activity of the pharmacon after the invasive
phase.
7.1.1. Physiological (pharmacokinetic) factors
Renal blood flow effective hemodynamic is essential for renal function, this function influences
the rate of drug excretion the most, trough the fact that glomerular ultra filtration is dependent on the
pressure of filtration. In the healthy animal, the kidney receives about 25% of the cardiac output,
converts about 1/5 of this in glomerular ultra filtrate, then reabsorbs about 99% of the filtrate.
For a drug that is eliminated massively by excretion, the rate of blood flow through the kidneys is
an important determinant of its existence in the body, (ex. digoxin and gentamicin).
Solubility in the ultra filtrate. Drugs with hydrophilic character are most commonly excreted in
urine, in their unaltered state, while fat soluble pharmacons may be subject to metabolism (to form
water-soluble compounds before being excreted).
Occasionally (e.g., quinolones and old acetyl sulphonamides) a metabolite is less soluble than the
parenteral pharmacon in the concentrate acid ultra filtrate from the proximal convoluted tubule. In
this case, there is the risk of precipitation of the drug in the convoluted tubules preventing renal
function. This situation may be avoided by urine alkalinization, by unrestricted administration of water and
by using sulphonamidic mixtures. Corresponding with the multitude of chemical compounds that are administered to the body as
pharmacons (or toxics), there are numerous possibilities for biotransformation, which leads to the
formation of active or inactive metabolites. If a change occurs in a toxic, decreasing the intensity of
the effect, one speaks of detoxification.
If a substance is changed in the body, and turned into a toxic, then this process is called
intoxication (e.g. conversion of methanol to formaldehyde of insecticide diethyl-p-nitro phenyl-
thiophosphate in diethyl-p-nitro phenyl-phosphate etc).
The same thing happens in the case of drugs, which are primarily inactive, they become
pharmacologically active only after their metabolic conversion or cyclization (eg: chlordiazepoxide, an
antidepressant), opiates, levodopa, enalapril, pro-benzimidazoles etc.).
The coupling with activated glucuronic acid is of great importance.
Hydroxyl groups of alcohols and phenols, carboxyl, amino and amide groups are conjugated with
glucuronic acid = increases hydro solubility.
In drug metabolism the reactions that occur are:
hydroxylation,

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
82 | P a g e
demethylation,
oxidation and finally, de
glucuronidation (in the phase of conjugation).
This last step will increase the hydro solubility and will ease the removal.
We know some principal pathways of decomposition:
scission and
burning up to CO2 and water (ex: ethanol);
Partial decomposition by:
decarboxylation,
deamination (ex: catecholamines, -methyldopa, histamine, serotonin)
N-demethylation (chlorpromazine, morphine, pethidine).
Oxidation (ex: chlorpromazine)
Reduction (ex: nitrazepam);
Hydrolysis (spontaneous or fermentative) (e.g. succinyl-choline, ester-type local
anaesthetics);
Coupling to acids (ex: acetylation of sulphonamides, coupling with glucuronic acid).
Metabolic pathways of chlorpromazine (after Kuschinsky, 1989)
Representation of coupling and scission of lipophilic drugs in the liver cell (after Kuschinsky, 1989)

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
83 | P a g e
7.1.2. Urinary pH
Renal excretion of weak acidic or basic drugs is closely related to urinary pH. So, weak acids are
eliminated better when the urine is alkaline, while the weak bases in acidic urine.
When the elimination is reduced (due to unfavorable pH), it will activate the metabolic processes
(to make substances more soluble), thereby increasing the rate of conjugated compounds.
7.1.3. Coupling with plasma proteins
Medicinal substances coupled to plasma proteins cannot be metabolized until they are severed
from their links and transformed in free fraction. As a result, their half-life is even longer as the
medicine is of a higher percentage of couplings.
7.1.4. Enzymatic induction
Enzymatic induction means the stimulation of the activity of liver enzymes; under the action of
xenobiotics (non-biological) this includes drugs and pesticides, etc.
These inductors speed up the metabolism, by increasing the rate of synthesis of the enzymes.
So far, we know more than 200 substances that are considered enzyme inducers, having very
different chemical structures. A correlation cannot be established between the chemical structure and
the inductive effect.
The most studied enzyme inducer, phenobarbital, is considered to be the prototype of this action,
given that it boosts the metabolic activity for numerous medicinal substances. Through enzyme self-
inductance, some drugs after repeated administration can stimulate their own metabolism.
Most of the enzymes responsible for the biotransformation are in the liver, specifically in the
endoplasmic reticulum (ER), in the microsomes.
These enzymes can be multiplied by a number of different chemicals of pharmacons, even when a
pharmacon interacts only with one enzyme of the ER.
The consequence of this enzyme induction = faster and easier decomposition of that pharmacon.
The best known enzyme inducers are: barbiturates, psycho-pharmacons, rifampicine,
chlorphenotane, HCH, tolbutamide, carcinogenic substances etc. In the case of these substances will
cause a different enzyme induction comparable in quality to the induction caused by barbiturates.
So we can speak of two types of induction:
a “phenobarbital type” and
a “methylcholanthrene type”.
An important enzyme system, which is enabled by the phenobarbital type of mechanism of
induction, is the plurifunctional oxidaze which is responsible for the oxidation of organic links.
Terminal oxidaze of the system is cytochrome P-450.

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
84 | P a g e
7.1.5. Enzymatic inhibition
There are some substances that inhibit the activity of hepatic microsomal enzymes, for example:
piperonylbuthoxid, piperonyl-sulfoxide, sesamex, cloramphenicol, ketoconazole, cimetidine etc.
For example long-term administration of chlortione will lead to a marked inhibition of
microsomal rat liver enzymes. In addition to the possibility of decomposition which is general and
non-specific, there are a number of specific mechanisms for some pharmacons, within which a series
of the body’s own substances are involved.
So, for example,
acetylcholine, is hydrolyzed by acetyl cholinesterase,
norepinephrine, under the action of O-methyltransferase is methylated, both
substances being inactivated.
The lung has the ability to inactivate the body's own substance (serotonin,
noradrenaline) and the results from their synthesis (ex: angiotensin II, prostaglandins
E and F).
A series of amphiphylic pharmacons can accumulate in the lung (ex: neuroleptics,
thimoleptic) and thus temporarily or permanently disappear from circulation (by
presystemic elimination).
7.2. Animal related factors
7.2.1. Species
Comparative studies on animal species revealed a wide variety of metabolic pathways.
The differences are mainly related to the development on the phylogenetic scale but are evident
within the same group of species: in mammals there are large variations in the speed of metabolism
and biotransformation or conjugation pathways.
Examples:
Rabbits have significant amounts of tropinesterase, which explains the great resistance
of this species to atropine and atropinics.
Cats have a low activity of hepatic glucuronyl transferase, resulting in a deficiency in
the formation of the glucuronide conjugates.
In dogs and foxes, acetylation of sulfonamides is done at N1 (amidic nitrogen) and not
at N4 (amino nitrogen), as occurs in other species.
The most important feature of the specie differences in drug metabolism is represented
by the quantitative aspects.

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
85 | P a g e
Thereby, in animals we can observe variations not only within the nature of the enzyme systems
which they have, but especially in the quantitative distribution of their activity.
As a consequence, there are variations in the metabolic pathways both in the biotransformation
processes, as well as in the conjugation ones.
For example, specific differences in sheep and goats in the metabolism of benzimidazole
nematocides. In the most of the times unawareness of these aspects can lead to the development of
drug resistance. A lack of the enzyme system can be regarded as a particular sensitivity of the cat to
the phenolic products.
For example amphetamine in rabbits, suffers oxidative deamination processes, while, many other
species have the hydroxylation process.
Sulphadimethoxin (a retarded sulfonamide) metabolized by acetylation in the proportion of: 80%
in cattle, 20% in goats, 80% in rabbits and 10% in humans.
7.2.2. Individuality / breed
In veterinary practice, the type of nervous activity of an animal should be taken into account,
because it can influence the rate of drug metabolism.
Examples:
the use of strychnine in therapeutic doses in animals may be followed by poisoning;
apomorphine in some breeds of pigs induces vomiting, while in others it doesn't.
7.2.3. Age
Newborns, especially premature ones, are put in danger by the administration of drugs, because
the hepatic enzymes are still in small quantities, or have not yet been synthesized, and the renal
elimination capacity is limited.
For example: oxidative enzymes from hepatic microsomes are missing in fetuses.
They are formed from the first day of life and they reach the adult limits after a month in rats and
after 3 month in children.
Similarly the synthesis ability of conjugates is reduced, or it may even be missing in the case of:
glucuronic acid, glycine and glutathione.
In case of old age elimination of pharmacons is prevented by: reduced renal function and by
decreased speed of hepatic metabolic processes = proper evaluation of required dose.
While the process of conjugation takes place in the future without any impediment, the
dealkylation and hydrolysis processes will be slowed down.
The elimination rate of drugs is dependent on the secretory and metabolic function of the: liver,
kidney and lung. Any change in their function leads to high blood levels, with lower tendency to
decrease. This results in a more prolonged effect, potential toxicity and tendency to accumulate.

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
86 | P a g e
Acute toxicity of drugs in neonate and adult rats (Yeary, Benish and Finkelstein, cit. Gherdan)
Drug Oral LD50 (mg / kg)
Neonate (1-3 days) Adult
d-Amphetamine 80 140
Aspirin 560 1500
Paracetamol 420 2400
Meprobamate 350 1500
Phenobarbital 120 320
Dicumarol 70 700
The hepatic metabolism increases progressively from birth to adulthood, then decreases
gradually. In aged animals, oral drug absorption and distribution is slow: gastric pH increases and the
intestinal transit, GI motility and the area of absorption are reduced.
Metabolism and drug elimination are diminished due to reduced renal and hepatic clearance.
There are some exceptions:
In dogs the oxidation function peaks at 8 weeks after birth and disappears after weaning.
In ruminants, metabolic changes occur when they change from pre-ruminants to ruminants,
due to changes in feeding. Eg., ceftiofur will be metabolized into dis-fluoril-ceftiofur, a metabolite that
is much higher in ruminants than in pre-ruminants.
7.2.4. Gender
Females metabolize drugs more slowly and are more susceptible to poisoning.
Studies in rats show that males have higher metabolic capacity for: alkaloids, pyramidon,
morphine, hexobarbital and pentobarbital.
For example, female rats are more sensitive to strychnine sulphate than males: 82% of females
die from a 2mg/kgc dose strychnine sulphate, compared with only a 30% of males.
7.2.5. Gestation
Administration of drugs during gestation is contraindicated, because they will cross the
placental barrier and reach the fetal circulation. In pregnant female rats and rabbits, glucuronidation
(major route of metabolism) is reduced to 50%.
Cause: high levels of progesterone and pregnanediol (considered glucuronyl-transferase
inhibitors). Similar findings were made also over sulphonated conjugates (a reduction in oxidative
biotransformation has been observed for phenacetin and aminophenazone, in gestating females).

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
87 | P a g e
7.2.6. Feeding
Undernourishment, reducing protein intake, deficiency states (lack of minerals and vitamins)
decrease the ability of metabolism.
Microsomal enzymes are most commonly affected by dietary factors.
By reducing the amount of drug substance after oral administration results in decreasing the
efficacy, due to gastro-intestinal pH change, formation of chelates, etc. For example, in the case of
oral administration of: penicillin, diazepam, codeine, increased gastric acidity diminishing the
absorption of these drugs. Feeding with lipids = stimulating bile secretion and increasing
bioavailability of liposolubles, e.g.: griseofulvin, albendazole, mebendazole, etc.
7.2.7. Health status
The normal functioning of the organs involved in metabolism, especially the liver, is an
essential condition for carrying out this process. In animals with hepatic diseases, blood flow reduces
= slow metabolism. In this case it is not recommended to administer: lincosamides, -lactams,
macrolids and chloramphenicol. Chloramphenicol for e.g., metabolizes difficult especially in cirrhosis
= hematopoietic accidents, result of reduced capacity for glucuronide conjugation.
Renal disease decreases the renal clearance for creatinin, frequently creating drug accumulations
that can cause adverse reactions and intoxications.
In the case of uremia, binding to plasma proteins and hepatic metabolism decrease significantly.
Metabolism of chloramphenicol

Introduction in Veterinary Pharmacology Chapter 7 Romeo – Teodor CRISTINA
88 | P a g e
7.2.8. Genetic factors
Important differences in the metabolic capacity, conditioned by the specific enzymatic equipment
for each individual: sensitizing genetic factors.It is known that (due to enzyme polymorphism) there
are individuals who genetically metabolize drugs more easily compared to others, who because of this,
are more susceptible to drug poisoning. The presence of sensitizing genetic factors has been
demonstrated especially for improved breeds (ex: Arabian thoroughbred horses, Merinos sheep,
Landrace pigs, Supercuni rabbits, Cocker dogs etc.).
7.3. Exogenous Factors
7.3.1. The circadian rhythm
Chronopharmacology revealed differences in drug metabolism related to circadian rhythms in
humans. It has been found that the most active metabolism is reported around 2 AM, and the lowest at
about 2 PM. These metabolic phases have a maximum and a minimum specific value in animals too.
In the case of sleep, the effect of the drug is closely related to the type of nervous activity of the
animal. This has been demonstrated by administering to animals of the same species, same gender,
weight and age of CNS, depressants (hypnotic or narcotic), it was found that the intensity and duration
of the effect was different. In most animals, narcotic sleep duration was average, but there were also
identified limit situations (too long or too short sleep).
7.3.2. Exogenous compounds
Chemical substances from the environment, (ex. insecticides, dyes, feed additives, auto oxidant
substances etc.), ingested by animals through food and water, or entering the body in other ways, have
a definite influence on the processes of metabolism. Many of them have the effect of enzyme
induction, especially after repeated contacts, when they produce a higher rate of metabolism (2 to 10
times).
7.3.3. Stress factors
Adverse conditions: cold, humidity, agglomeration, noise, increase the metabolic activity of
microsomal enzymes, by stimulating the pituitary - adrenal reflex arch. Stress increases the adrenal
ascorbic acid, observed in treatments with phenobarbital (enzyme inducer).
Small amounts of radiation (ionizing, in particular) can act as stressors increasing the activity of
microsomal enzyme systems. Radiation reduces drug metabolization by their effect on NADPH
formation (nicotinamide adenine dinucleotide phosphate oxidaze) and glucuronide conjugation.

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
89 | P a g e
8. Stages of metabolism

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
90 | P a g e
Introduction
Drugs entering the organism undergo transformation processes which lead to their elimination.
Metabolizing takes place, generally, after the pharmacodynamic action of the drug, but in some cases
the active substance is produced from the metabolic transformation. In such cases we say that the
substance was “activated”.
For example, acetanilide and phenacetin, two structurally close antipyretic analgesic substances,
are metabolized in acetaminophen (which is actually the active ingredient that produces the
pharmacodynamic effect.
The general rule is to "deactivate" or "detoxify" by which drugs are inactivated, especially in the
second phase of metabolism. Some substances can be removed from the body without undergoing
transformation processes.
Such as: some inorganic substances, bromides, volatile narcotics, phenolphthalein, etc.
Others are metabolized in a smaller proportion, and can be recovered from urine as their active
(ex: penicillin, streptomycin).
The purpose of the process of metabolism, is to increase the polarity and aqueous solubility of the
substances, to increase renal elimination and to decrease tubular absorption. It is known that both the
kidney, and the bile, excrete the polar compounds.
Oxidation of CH3 in COOH (biotransformation) makes renal or biliary excretion of the drug,
easier. Similar effects are observed by binding to the molecule of the radical sulfate (conjugation).
Some drugs, that were initially considered not metabolized, were later found to be subject in a
lower rate to the transformation process.
For example veronal is eliminated in 95% of its unchanged form in urine, but 5% will undergo
biotransformation, of which 3% by hydroxylation and 2% by oxidative dealkylation.
Biotransformation of barbital

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
91 | P a g e
Metabolization is achieved by two main processes:
biotransformation and
conjugation.
Biotransformation: a process by which drugs are processed by oxidation, reduction or hydrolysis.
During these processes the molecule will either maintain its size, or simplify it.
Conjugation: a synthesis process in which the substance is amplified by binding a compound or a
radical to the molecule.
Current drugs are generally more complex synthetic substances that are metabolized in several
ways, resulting in a large number of metabolites. For example, chlorpromazine, suffers many
transformation processes, finally resulting in a total of more than 20 metabolites.
8.1. Drug biotransformation
Biotransformation processes = activation / inactivation of the drug that renders non-polar
compounds polar, which will be removed as such or be subjected to processes of conjugation.
In general, biotransformation precede conjugation reactions and sometimes medicinal substances
undergo several successive biotransformation processes followed or not by conjugation.
Drug metabolism can begin:
after administration, before their resorption (in the digestive tract) or,
in the internal medium of the body, immediately after resorption (in the blood) or,
in the metabolization organs, the liver is the most important
The metabolic processes from the digestive tract take place under the action of this organ's own
enzymes, but numerous transformations are produced by enzymes from the digestive microflora and
micro fauna encountered in all species, especially in ruminants.
The presence of numerous esterases, in the blood, leads to biotransformation by hydrolysis.
Thereby, intravenously administered procaine to horses with colic, is rapidly decomposed in: para-
amino benzoic acid and diethyl-amino ethanol, (considered responsible for the calming, antispasmodic
effect).
Most biotransformation processes take place in the main metabolic organ, the liver, but they may
also occur in different proportions in the:
kidney,
spleen,
lung,
intestinal mucosa,
blood and
skin.

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
92 | P a g e
They occur under the action of metabolizing enzymes of xenobiotic substances (xenos = foreign;
bios = life), but, as we have seen, also under the action of enzymes which metabolize nutrients.
Microsomal oxidation reactions require cytochrome P-450 reduced nicotinamide adenine
dinucleotide phosphate (NADPH2) and O2, being mediated by a coupled redox system consisting of:
NADPH2,
a flavoprotein,
nonhemic Fe-protein and
a hemoprotein known as cytochrome P-450. NADPH2 acting as a hydrogen donor for the
reduction of cytochrome P-450.
It oxidizes rapidly under the action of molecular oxygen (O2) and then gives O2 to the
metabolized substance (after they previously coupled with the drug).
Microsomal drug oxidation general scheme
The coupled redox system involved in the microsomal oxidation of drugs
The processes of biotransformation are classified in processes of:
oxidation,
reduction and
hydroxylation,
these may be catalyzed by microsomal enzymes, nonmicrosomal enzymes (mitochondria, cytoplasm,
plasma blood) or enzymes from the intestinal flora.

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
93 | P a g e
Classification of drug biotransformation (Parke, cit. Gherdan)
Type of reaction
Microsomal enzymes of metabolisation
Nonmicrosomal enzymes of metabolisation
Enzymes from intestinal flora
Oxidation
Aromatic hydroxylation Acyclic hydroxylation Alicyclic hydroxylation Epoxidation N-oxidation S-oxidation Desulphurisation Dealkylation Deamination
Oxidation of alcohols (cytoplasm) Oxidation of aldehydes (cytoplasm) Acyclic aromatization (mitochondria) Deamination (mitochondria & blood plasma)
-
Reduction Nitro reduction Azo reduction Reductive dehalogenation
Reduction of sulfur-oxides and N-oxides (cytoplasm) Reduction of disulfide
Reduction of N-oxizides Azo reduction Dehydroxylation
Hydrolysis Ester hydrolysis
Hydrolysis of esters and amides (blood plasma) Hydrolytic dissolution of cyclic compounds Hydrolytic dehalogenation
Hydrolysis of esters Hydrolytic dissolution of heterocyclic compounds
8.1.1. Microsomal biotransformation
8.1.1.1. Microsomal oxidation
The microsomal oxidation process takes place under the action of some mixed-function oxidase
of the smooth endoplasmic reticulum. In “teaching terms” we are talking about : ten types of
microsomal oxidation.
Aromatic hydroxylation is introducing a hydroxyl radical in an aromatic ring. Hydroxylation of
aromatic rings to give phenols is preceded by two mechanisms:
first, gives monophenols = hydroxylation via free radical transfer mechanism or passing
through the phase of epoxide followed by intra-molecular rearrangement;
second = epoxide formation which reacts with water and gives: dihydrodiols, phenols and
catechols (e.g. case of naphthalene, amphetamine, phenacetin, ox. salicylic acid, chlorpromazine, etc.).
Aromatic hydroxilation
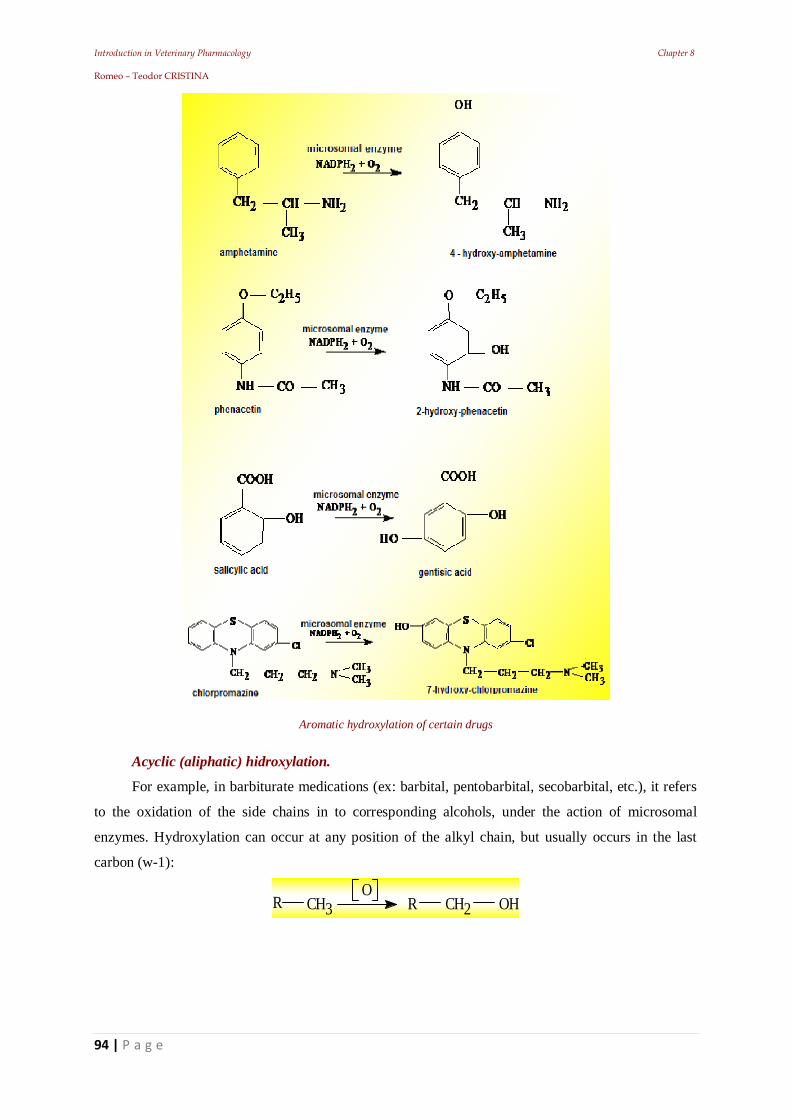
Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
94 | P a g e
Aromatic hydroxylation of certain drugs
Acyclic (aliphatic) hidroxylation.
For example, in barbiturate medications (ex: barbital, pentobarbital, secobarbital, etc.), it refers
to the oxidation of the side chains in to corresponding alcohols, under the action of microsomal
enzymes. Hydroxylation can occur at any position of the alkyl chain, but usually occurs in the last
carbon (w-1):
R CH3O
CH2 OHR
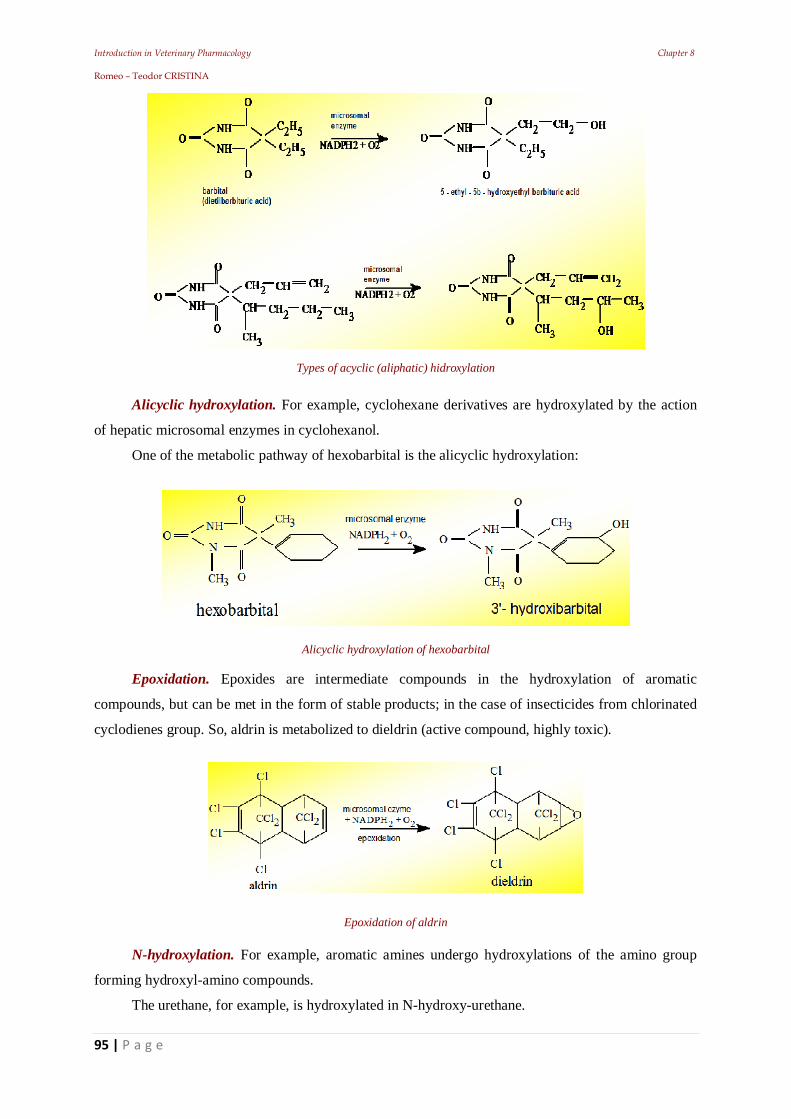
Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
95 | P a g e
Types of acyclic (aliphatic) hidroxylation
Alicyclic hydroxylation. For example, cyclohexane derivatives are hydroxylated by the action
of hepatic microsomal enzymes in cyclohexanol.
One of the metabolic pathway of hexobarbital is the alicyclic hydroxylation:
Alicyclic hydroxylation of hexobarbital Epoxidation. Epoxides are intermediate compounds in the hydroxylation of aromatic
compounds, but can be met in the form of stable products; in the case of insecticides from chlorinated
cyclodienes group. So, aldrin is metabolized to dieldrin (active compound, highly toxic).
Epoxidation of aldrin
N-hydroxylation. For example, aromatic amines undergo hydroxylations of the amino group
forming hydroxyl-amino compounds.
The urethane, for example, is hydroxylated in N-hydroxy-urethane.
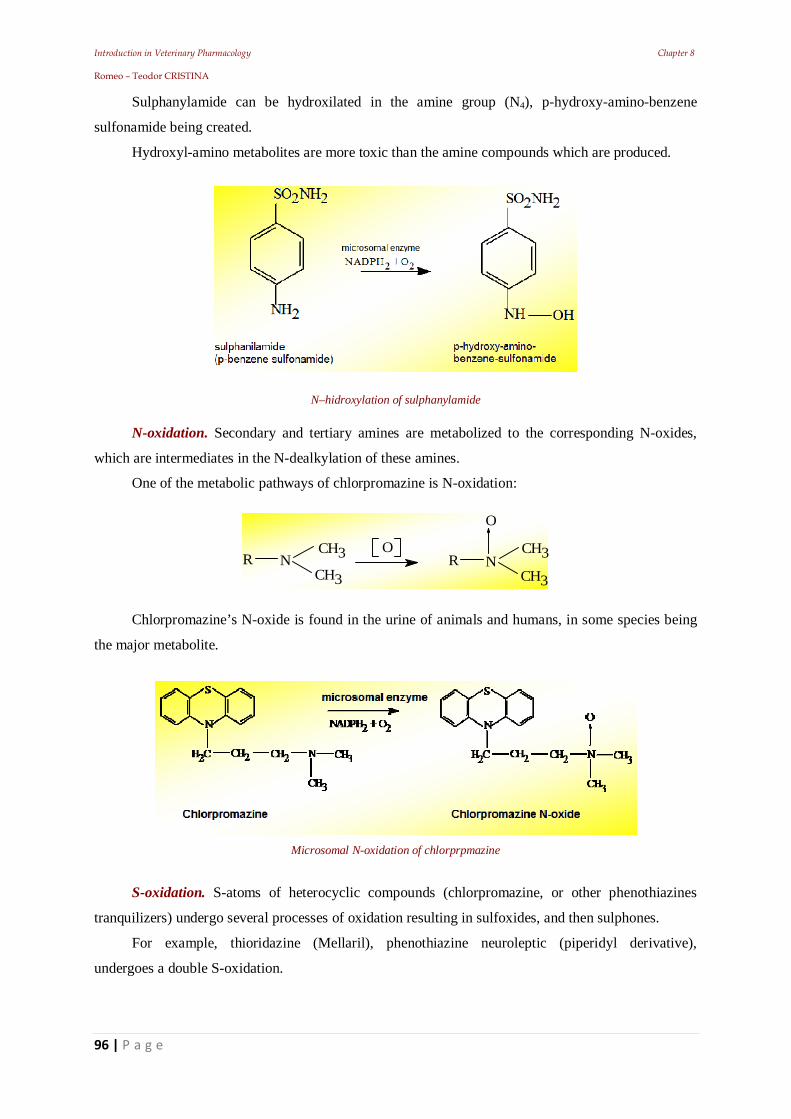
Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
96 | P a g e
Sulphanylamide can be hydroxilated in the amine group (N4), p-hydroxy-amino-benzene
sulfonamide being created.
Hydroxyl-amino metabolites are more toxic than the amine compounds which are produced.
N–hidroxylation of sulphanylamide
N-oxidation. Secondary and tertiary amines are metabolized to the corresponding N-oxides,
which are intermediates in the N-dealkylation of these amines.
One of the metabolic pathways of chlorpromazine is N-oxidation:
RCH3
ON
CH3 RCH3
NCH3
O
Chlorpromazine’s N-oxide is found in the urine of animals and humans, in some species being
the major metabolite.
Microsomal N-oxidation of chlorprpmazine
S-oxidation. S-atoms of heterocyclic compounds (chlorpromazine, or other phenothiazines
tranquilizers) undergo several processes of oxidation resulting in sulfoxides, and then sulphones.
For example, thioridazine (Mellaril), phenothiazine neuroleptic (piperidyl derivative),
undergoes a double S-oxidation.
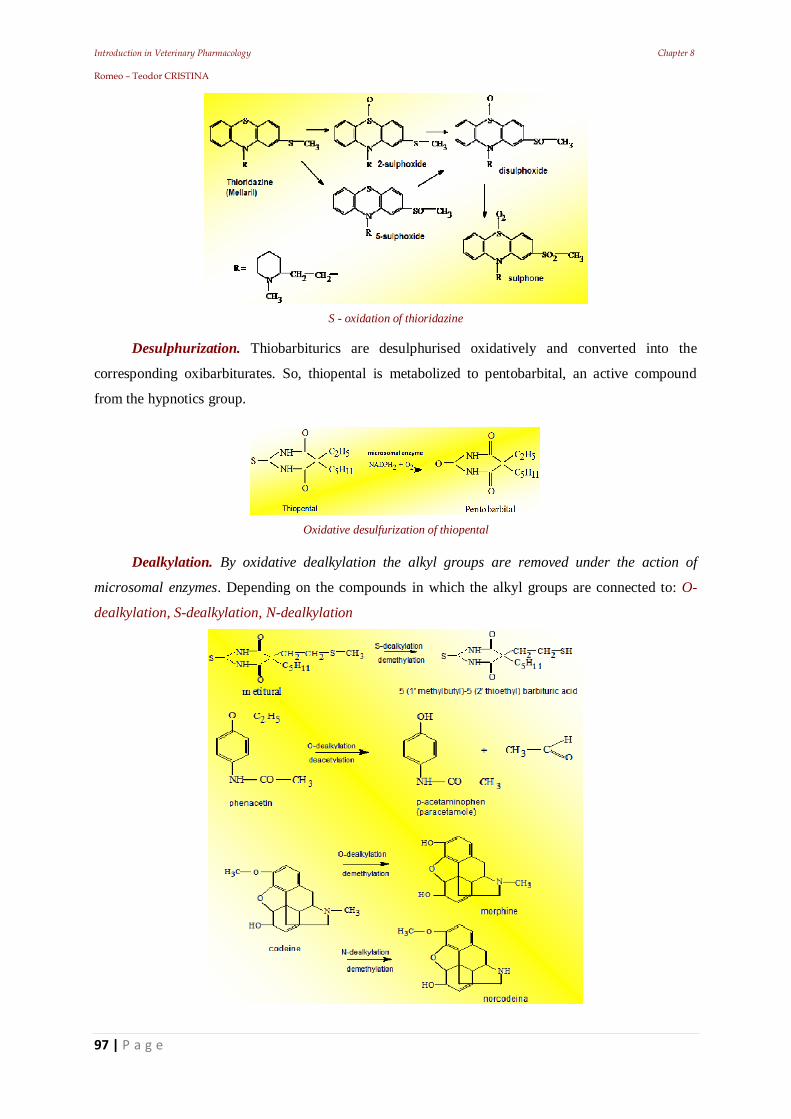
Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
97 | P a g e
S - oxidation of thioridazine
Desulphurization. Thiobarbiturics are desulphurised oxidatively and converted into the
corresponding oxibarbiturates. So, thiopental is metabolized to pentobarbital, an active compound
from the hypnotics group.
Oxidative desulfurization of thiopental
Dealkylation. By oxidative dealkylation the alkyl groups are removed under the action of
microsomal enzymes. Depending on the compounds in which the alkyl groups are connected to: O-
dealkylation, S-dealkylation, N-dealkylation

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
98 | P a g e
Oxidative dealkylation of drugs Oxidative deamination. Aside from mono-amino-oxidaze (MAO), a mitochondrial enzyme, in
the liver is known as a microsomal enzyme too, which de-aminates amphetamine.
Oxidative deamination of amphetamine
8.1.1.2. Microsomal reduction
Microsomal reduction reactions occurs under the action of reductases in the smooth
endoplasmic reticulum of liver cells. There are three processes, that catalyzes the reduction of azo and
nitro groups, as well as the dehalogenation processes.
Azo-reduction. Under the action of microsomal azo-reductase azoic bonds are undone, after
passing through an intermediate hydrazo compound: e.g. prontosil and old sulphonamide is
metabolized by azo-reduction in a sulphanilamide (active compound) and a triaminobenzene.
After the discovery of this pathway prontosil was abandoned, and nowadays only
sulphanilamide derivatives are used.
R NH NH2N N R R HN R R R+ NH2
Azo-reduction of prontosil
Nitro-reduction. Aromatic nitro compounds (ex. nitrobenzene, chloramphenicol) are reduced by
the action of microsomal nitro-reductase.

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
99 | P a g e
Microsomal nitro-reduction of drugs
Dehalogenation. Halogenated compounds are metabolized by microsomal enzymes. Ions of: Cl,
Br and I are undone, and replaced with H. Ions of Fluorine remain stable! Halothane, a volatile
narcotic, is metabolized by reductive dehalogenation:
Schematic reductive dehalogenation of halothane
Microsomal hydrolysis. Esters and amides are metabolized by hydrolysis, catalyzed by
esterases and amidases in the blood and liver.
Pethidine (Mialgin, Dolantin), widely used analgesic substance, is metabolized by microsomal
esterases.
8.1.2. Non microsomal biotransformations
8.1.2.1. Non microsomal oxidation
Oxidation of alcohols. Primary alcohols are metabolized to aldehydes. The process is catalyzed
by alcohol dehydrogenase, found in the cytoplasm of liver cells, kidney and lung and that uses NAD
or NADP as coenzyme. This reaction is reversible:
Oxidation of aldehydes. The aldehydes are oxidized in the corresponding carboxylic acid,
through this enzymes: aldehyde oxidase, xanthine oxidase and NAD-specific aldehyde dehydrogenase.
Oxidative deamination. Non microsomal tissue or plasma enzymes (mono and di-amino
oxidases) deaminates primary, secondary and tertiary amines.
Histamine is deaminated by a non micrsomal diamine oxidase and is converted to aldehyde, and
then under the action of aldehyde oxidase in imidazole acetic acid:

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
100 | P a g e
Non microsomal oxidative deamination of histamine
Oxidative cleavage of arsenobenzenes. Arsenobenzenes are split in the organism by the action
of an oxidase, with an unspecified location, in the corresponding arsenoxides, and then into arsenic
acids :
8.1.3. Biotransformation by the action of digestive microflora
Microflora of the digestive tract is able to mediate the metabolic transformations by hydrolysis
and reduction reactions.
For example, phthalylsulphathiazole is transformed by GI microflora in sulfathiazole.
Antibiotics and chemotherapies may affect the eubiosis of the environment and destroy GI
microflora, affecting the metabolism of other drugs.
Here we can mention:
reduction of azo-compounds,
hydrolysis of esters and glycosides,
splitting of cyclic and heterocyclic compounds.
8.2. Conjugation of drugs
Conjugation or synthesis reactions represents phase II of metabolization, because many
substances have undergone a process of biotransformation (phase I), so that subsequently undergo
conjugation reactions.
During the biotransformation, drugs can be subjected to various reactions:
oxidation,
reduction sau
hydrolysis, resulting in the introduction or dissolution of functional groups which increase
the polarity of the molecule and serve as centers for the second phase of the metabolic response,
conjugation.
By synthetic reactions of conjugation drugs (in their original form) or their metabolites are
combined with endogenous compounds, as: glucuronic acid, glycine, glutathione, sulfate, methyl,
acetyl groups etc.

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
101 | P a g e
Conjugation pathways of drug (synthesis Cristina, R.T. 2006)
Type of reaction
Conjugated group or compound
Functional group to which links
Acetylation Acetyl radical Amino, sulfonamide, hydrazino
Methylation Methyl radical Hydroxyl, amino, thiol
Sulphono-conjugation Sulfate radical Hydroxyl, amino
Glucuronide conjugation Glucuronic acid Hydroxyl, carboxyl, amino, thiol
Peptide conjugation Glycine, glutamine and other amino acids. carboxyl
Mercaptation Cysteine or glutathione Epoxy, halogen, nitro, sulfonamide
8.2.1. Acetylation
Metabolic pathway of compounds with -NH2 and –OH groups.
The most important acetylation is found in primary amines, in: aniline derivatives, sulfonamides,
aminophenazone ,etc. Acetylation involves the enzymatic transfer of the acetyl group from acetyl-CoA
with the help of acetyl-transferase.
Sulphonamides, especially classic ones, are metabolized by acetylation at N4 (amino nitrogen).
Dogs, for example are unable to acetylate aromatic amines, producing instead a reverse reaction of
“deacylation” of acetylated aliphatic aminoacids.
Chikens manage “deacetylation” of acetylated aryl amines.
Acetylation of sulfonamides
8.2.2. Methylation
Is a conjugation reaction which transfers methyl groups from coenzyme S-adenosyl-methionine,
under the action of methyltransferases.
Numerous endogen compounds and some drugs (as phenols, amines, thiols etc) are metabolized
this way. Histamine and adrenaline can be metabolized through methylation process.
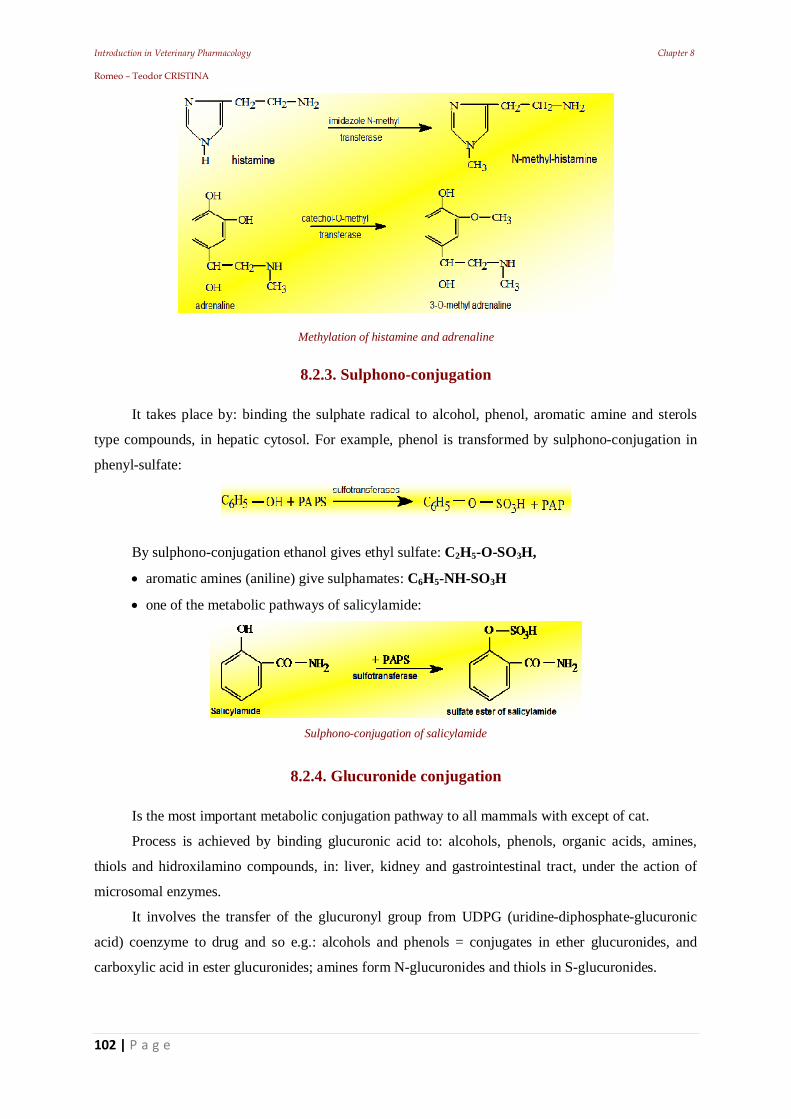
Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
102 | P a g e
Methylation of histamine and adrenaline
8.2.3. Sulphono-conjugation
It takes place by: binding the sulphate radical to alcohol, phenol, aromatic amine and sterols
type compounds, in hepatic cytosol. For example, phenol is transformed by sulphono-conjugation in
phenyl-sulfate:
By sulphono-conjugation ethanol gives ethyl sulfate: C2H5-O-SO3H,
aromatic amines (aniline) give sulphamates: C6H5-NH-SO3H
one of the metabolic pathways of salicylamide:
Sulphono-conjugation of salicylamide
8.2.4. Glucuronide conjugation
Is the most important metabolic conjugation pathway to all mammals with except of cat.
Process is achieved by binding glucuronic acid to: alcohols, phenols, organic acids, amines,
thiols and hidroxilamino compounds, in: liver, kidney and gastrointestinal tract, under the action of
microsomal enzymes.
It involves the transfer of the glucuronyl group from UDPG (uridine-diphosphate-glucuronic
acid) coenzyme to drug and so e.g.: alcohols and phenols = conjugates in ether glucuronides, and
carboxylic acid in ester glucuronides; amines form N-glucuronides and thiols in S-glucuronides.
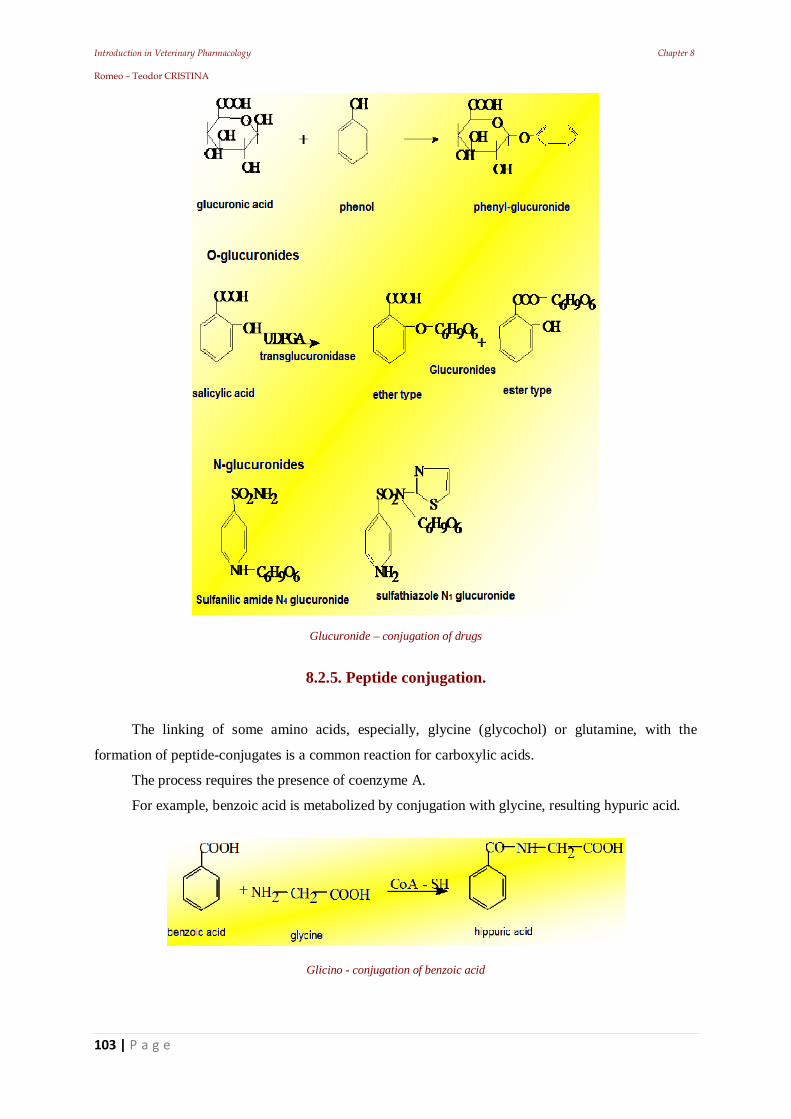
Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
103 | P a g e
Glucuronide – conjugation of drugs
8.2.5. Peptide conjugation.
The linking of some amino acids, especially, glycine (glycochol) or glutamine, with the
formation of peptide-conjugates is a common reaction for carboxylic acids.
The process requires the presence of coenzyme A.
For example, benzoic acid is metabolized by conjugation with glycine, resulting hypuric acid.
Glicino - conjugation of benzoic acid

Introduction in Veterinary Pharmacology Chapter 8 Romeo – Teodor CRISTINA
104 | P a g e
8.2.6. Mercaptation
Conjugating drugs with cistern or glutathione gives rise to mercapturic acids.
This reaction takes place within the kidney. Among the best-known drugs that are metabolized
by mercaptation are: arecoline, nitrofurane, some sulfonamides.
Phenacetin (substance which is metabolized in several ways) suffers among other things, and a
process of mercaptare through glutation conjugation.
Pathways of metabolism for phenacetin

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
105 | P a g e
C.9. Drug excretion

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
106 | P a g e
Introduction
By drug elimination (excretion) we mean all processes that lead to drug inactivation and their
removal through:
kidney,
bile digestive tract,
respiratory tract,
skin and mucous membranes,
mammary gland and placenta,
as well as the chemical transformations of the molecule.
The measuring unit for the rate of elimination is: the biologic half-life (elimination half-life).
This represents the elapsed period until the amount of the substance in the body drops to half of
the initial amount and is a temporal measure that underlies the mathematical transposition of the
elimination process.
Plasma half-life time t1/2 represents the velocity constant of an exponential process.
To express the removal rate via the kidneys, veterinary medicine has adopted the term: renal
clearance. This is expressed in volume of plasma (ml) occupied by the drug substance excreted over
an established period of time (1min.) through the kidneys. Clearance formula will be:
Cu Cl = VuE × ------, Cp
Where:
Cl = clearance expressed in ml;
VuE = excreted urine volume / minute (ml);
Cu = concentration (mg %) of the drug substance in urine;
Cp = concentration (mg %) of drug substance in plasma.
Of course, the expression of clearance can be used by extrapolation, for the: liver, lung etc. The
half-life of a drug in the body can be influenced by:
serric proteins capable of binding drugs in the form of complexes,
the possibility of tissue storage or
the metabolism rate.
Kidney and bile (through the liver) are considered the main organs for drug excretion, and saliva,
sweat, mammary gland and the lung are secondary pathways of excretion.

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
107 | P a g e
Pharmacons may be removed by urine and faeces, where the highest amount of the administered
substance or by-products resulted after metabolism, can be found.
Liposoluble substances are difficult to be expelled by the kidney (due to the tubular passage
which leads to permanent reabsorbing). If there is a strong binding to serum albumin, glomerular
filtration rate remains at a low level. Most of the amount of the substance that was filtered suffers a
diffusion process in reverse at a tubular level, due to the hydrophobic characteristics of molecules.
Renal elimination rate is increased when renal function is impaired.
Substances that are easily removed through the urinary system can cause elevated blood levels in
the case of renal insufficiency. Substances that have decreased renal clearance are indicated in such
conditions.
In the faeces will appear substances that are eliminated via biliary route or are secreted by the
intestinal mucosa. Elimination by sweat, saliva and milk are less important quantitatively, respiration
being considered a major route of elimination for narcotics.
Some substances can concentrate at the elimination site and reach local toxic concentrations, e.g.
renal disorders caused by the compounds based on: mercury, phenols and by aminoglycosidic
antibiotics. The route of elimination changes, depending on the properties of the drugs:
those insoluble P.O. will be eliminated through the digestive tract,
those soluble are excreted by the kidneys,
volatile or gaseous substances by the lungs.
Most of active substances that reach the general circulation are eliminated after previously
undergoing the processes of metabolism. The form under which drugs will be eliminated depends on
what transformations they will go through in the body.
For example: penicillin, most part (80%) is eliminated on renal way as: 20% through glomerular
filtration and 80% by tubular excretion, almost fully recovering its active form. Streptomycin and
oxytetracycline can be, also, largely recovered in the urine in their active form.
In other cases, substances that are excreted unchanged produce kidney damage (e.g. cantharides).
The same thing appears in the action of some metabolites. Acetyl sulphonamide precipitates in the
acid medium of urine under the form of sharp micro crystals which will damage the renal tubules or
they will agglomerate in their lumen.
A substance administered by the respiratory route (volatile or gaseous) is rapidly absorbed and
eliminated. An orally administered substance, which passes into the general circulation and binds
massively to plasma proteins (e.g. retard sulphonamides) or to tissular ones (e.g. digitoxin) will be
eliminated slowly, being maintained in the body for several days.
The drug elimination rate depends on:
the route of administration,
physicochemical properties,

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
108 | P a g e
fixation to plasma proteins or tissue,
transformations suffered in body,
elimination pathway.
When a medicine (with renal elimination) is not metabolized, its half life may reach 20-30 days.
A drug that has good distribution, in all body compartments and is secreted by the renal tubules will
have (generally) a half-life of 60 minutes.
In drug elimination various situations can emerge, like:
the removal of all the administered substances is complete (when we speak of a real
elimination), or when,
due to various causes, elimination is incomplete and that is usually apparent.
9.1. The renal elimination
The kidneys are the most important route of elimination; most drugs are partially or totally
eliminated here. The physiological kidney purge takes place in three ways:
a) glomerular filtration;
b) tubular excretion;
c) glomerular filtration and tubular excretion
During circulation, plasma will be filtered by the renal glomerule. The glomerular membrane, (as
the capillary wall) will allow the passage of small molecule solutions and substances.
The filtrate passes in the renal tubules (lined with interlinked epithelial cells, forming a
continuous membrane with lipoid features). During its passage through the tubes and collecting
tubules, 99% of the filtered water is reabsorbed and the urine will be concentrated. At the same time,
non ionized liposoluble substances may be reabsorbed, creating equilibrium between plasma and renal
tubules.
Organic molecules pass through the tubular membrane, respecting the same conditions that apply
to other membranes of the body (according to the concentration gradient). They depend on the:
physicochemical properties of the substances (molecule size, partition coefficient,
pKa) and
pH of the urinifere tubes.
only non-ionized drugs may be reabsorbed in the tubular epithelium (ionized form of
the majority of weak acids and bases is liposoluble).
as such, the amount of drug excreted will be consistent with the pH of the content of
the tubules (Henderson- Hasselbach).

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
109 | P a g e
the organic molecules are weak electrolytes and at the urinary pH are partially ionized
and partially unionized.
In general, the excretion of drugs with a pKa of: 3.0 – 7.5 for weak acids and 7.5 – 10.5 for weak
bases; is pH dependent and deeply affected by the urinary pH.
Alkaline drug excretion is:
increased: by acidification of urine and
decreased: through its alkalinization (and vice versa).
Thereby,
Procaine (a weak base) with pKa = 8.95 is eliminated 10 times more in acid urine.
Amphetamine is eliminated less (5%) when the urine pH = 8 (at this pH is almost entirely
unionized and is reabsorbed) and over 50% in urine with a pH = 5
Aspirin (weak acid) with pKa = 3 is eliminated 80 times more in alkaline urine than in acid
urine.
Changing the urinary pH by acidification or alkalinization of urine, will lead to the alteration
of the elimination process.
Tubular excretion is performed by specialized transport in the epithelium of the proximal tubule
and is the most important route of elimination. There are two specialized transport mechanisms:
a. one which transports ionized forms of acid drugs (ex. salicylic acid, penicillin, probenecide,
sulpha acetylated, glucuronids and ester sulphates, etc.) and
b. one which transports ionized forms of basic drugs (for histamine, thiamine, hexamethonium
and other quaternary ammonium derivatives etc).
These two mechanisms have the characteristics of the active transport:
against the concentration gradient (dependent on the energy supply).
there is a competition between weak acids and weak bases.
Renal elimination can be accelerated by:
increasing diuresis,
urinary pH change
preventing tubular reabsorption.
Drugs metabolized by the body are more easily removed, because they are transformed in water-
soluble compounds. The function status of the kidneys influences the rate of elimination.
In case of kidney failure, the removal process takes much longer.
Thus, streptomycin is eliminated normally 60-80% during the first 24 h, by glomerular filtration.
In the case of renal failure the elimination decreases to a rate of 2% in 24 h.
Fullness of the digestive tract may influence the half life of a drug. A water-soluble drug
administered prior to feeding can be eliminated renal after 10 min.

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
110 | P a g e
The maximum removal will be at 60 min., it then decreases gradually (due to glomerular filtration
and partial reabsorption).
9.2. Elimination through the digestive tract
Many insoluble or poorly soluble drugs administrated orally, have a local action in the digestive
tract, where they achieve significant concentrations and are eliminated in the faeces.
Through the digestive tract, a number of substances are eliminated, that diffuse passively more
often than not, from plasma through mucous membranes or glands of the digestive tract.
In the saliva, a number of drugs can be eliminated. Such as bromine, iodine, Hg, Bi and Pb salts.
Through the gastric mucosa and gastric juice, a number of drugs such as: alkaloids and halogenated
derivatives are also eliminated.
The factors that increase elimination through the gastric mucosa are:
those who activate the local circulation and
those who increase the gastric secretion.
The mucosa of the fore stomach, glandular stomach and intestine, behave as semi permeable
membranes for drugs in blood plasma. They are crossed in both directions by the free fraction,
depending on the concentration gradient. The amount of drug that, after oral administration, can be
found in the faeces is composed of:
unabsorbed drug and
the product recovered in the intestine.
Drugs access the digestive tract through:
the intestinal wall,
its secretions
bile, by diffusion or by active transport mechanisms.
9.2.1. Biliary excretion
Low molecular weight drugs (under 150kD) are excreted by the kidneys.
Biliary excretion of large molecules, insoluble in lipids, indicate that the membrane of the bile
and the hepatic sinusoidal blood, is highly porous and allows the penetration of molecules and ions
weighing less than plasma proteins. The vast majority of the drug excreted in bile, is found in
conjugated form. Glucuronide conjugates are excreted in bile.
9.2.2. Pancreatic juice
It may lead, in the case of oral administration by passing inside the the duodenum, tinctures or
chemotherapeutics.

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
111 | P a g e
9.2.3. Hepatic cell influence
Is permeable for liposoluble drugs and has limited permeability for polar drugs, e.g. antibiotics
that achieve high proportions in biliary secretion: tetracycline, chloramphenicol, rifampicine etc.
Drugs eliminated in the bile are reabsorbed in the intestine and once again reach the liver, where
part of them:
enters the general circulation,
is metabolized,
is once again removed through the bile into the intestine.
9.2.4. The entero-hepatic circuit influence
It delays the elimination of drugs (e.g. tetracycline, chloramphenicol, ampicillin and other
substances forming this circuit, maintaining therapeutically useful levels in the body).
Derivatives of: salicylic acid, tetrachloride, halogenated, some antibiotics (tetracyclines), dyes,
contrast agents etc, are excreted in bile.
The fact that most substances excreted in bile are reabsorbed and once again reach the liver will
establish a double absorption circuit and elimination, known as gastro-entero-hepatic circulation.
Ions of: calcium, phosphorus, iron, salts of heavy metals are eliminated in the colon.
Insoluble drugs administered orally, which are not absorbed through the digestive tract (e.g. the
medicinal coal, paraffin oil, bentonites, kaolin, bismuth salts, magnesium sulphate, neomycin,
streptomycin, digestive sulphonamides etc.) will be eliminated in faeces.
In the veterinary medicine there is the special case of the anthraquinone purgatives, which are
absorbed by the small intestine, but will be removed through the large intestine, through this
segment's own circulation.
9.3. Elimination through the respiratory route
The alveolar surface, together with high pulmonary vasculature allows a rapid equilibration of the
volatile agent from the blood and from the alveolar air.
Mostly gaseous substances are eliminated through the alveolar epithelium like:
ammonia,
hydrogen sulphide,
hydrocyanic acid,
carbon dioxide,
narcotic gases (e.g. nitrous oxide, cyclopropane, ethylene),

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
112 | P a g e
volatile anaesthetics (e.g. chloroform, ether, halothane, ethyl chloride),
volatile oils (e.g. oleum eucalypti, menthae, carvi, thymi, pini),
alcohol,
guaiacolum,
camphor and others.
Some substances that are partially eliminated by the respiratory route are used in the treatment of
pulmonary parasites.
Drugs administered orally or parenteral can undergo metabolic processes in the body which
transform them into volatile substances which are eliminated via the respiratory route. Some drugs can
be partially oxidized in carbon dioxide and they are excreted by the respiratory route. In the case
where the carbon is radio labelled, excretion can be measured this way.
9.4. Elimination through the skin
The dermal route may represent a significant excretion organ, which completes renal elimination.
Sweat glands (in species where they are present) and partly sebaceous glands represent the major
routes of cutaneous elimination. In general, cutaneous elimination is based on: sweat hyper secretion
and less on: sebaceous secretion (only important in sheep) having a favourable effect of the cutis and
skin annexes (ex: antimycotic, sulphurs, arsenics etc.), also unfavourable (ex: elimination dermatitis,
produced in general by halogenated compounds).
Arsenics and sulphurs are eliminated through hair, skin appendages and stratum corneum.
9.5. Elimination through the mammary gland
Milk is a current food for humans. Drug elimination in milk has a particular significance.
The following substances are all eliminated through milk: chloroform, phenazone, lead, mercury
and other heavy metals, caffeine, barbiturates, colistin, bromides and halogenated, phenylbutazone,
cortisone, ether, camphor, substances that give milk an odour. Excretion of radioactive metals can be a
risk, as a consequence of nuclear accidents, which may contaminate pastures.
9.5.1. Cow milk
Is usually weak acid (pH: 6.5-6.9) compared to the plasma (pH 7.2-7.4) and therefore tends to
concentrate alkaline liposoluble drugs. Most of drugs are able to pass from plasma into milk =
problems of toxicity in children.

Introduction in Veterinary Pharmacology Chapter 9 Romeo – Teodor CRISTINA
113 | P a g e
9.6. Elimination through the egg
In birds some drugs diffuse:
in the ovary and
oviduct being incorporated into eggs. This phenomenon is reported for sulphonamides.
Conclusions
The excretion of a drug will be fast when it or its metabolite in the blood is in ionized form,
highly polarized, because this is poorly reabsorbed from tubular ultra filtrate.
The excretion of weak acids or weak bases is influenced by the pH and the concentration
differences at the level of the walls of renal convoluted tubules
The excretion of drugs is much accelerated by active transport systems.
Maintaining a good blood supply to the healthy kidney and, if not actively excreted, the extent to
which the drug is coupled to plasma proteins.
The end of drug action is achieved by metabolic inactivation and storage in the body, away from
the site of action and by simple excretion. This process starts as soon as the absorbed drug will enter
the circulation.

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
114 | P a g e
10. Elements of theoretical pharmacokinetics

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
115 | P a g e
10.1. Pharmacokinetics modelling
Pharmacokinetics provide information about the “fate” of substances administered externally to
a living organism and explores the interactions of absorption, distribution, metabolism and excretion
by analyzing the relationships between: plasma concentration and time elapsed after administration.
Studies so far show that it is possible to know:
the plasma concentration at which the effect becomes apparent;
correlation between: intensity of the effect and plasma concentration.
correlation between: the effect duration and period when plasma exceeds a certain value,
and the effect disappears when plasma concentration drops under this value.
This situation can be applied to most drugs that:
act instantaneously,
do not require metabolizing into an active form
have a reversible coupling with receptors and
do not have an irreversible effect.
Hypothetical representation of a plasma concentration-time curve
(Brander, 1991)
10.1.1. Kinetics redundancy
Absorption is the unique factor that determines the initial growth of drug plasma concentration.
Distribution, metabolization and excretion will remove the free pharmacon from plasma resulting in a
decrease of its concentration.
Instead of identifying and measuring the individual contribution of these three processes
mentioned above, an act of kinetic simplification would be to unite them, in a process called
redundancy. This it defines the kinetic disposition of a drug by calculating the equation that fits the
plasma concentration-duration curve.
Generating curves (while searching the best equation to fit the gross data) is known as:
mathematical modeling. Drug concentration is essential for its efficiency in the place where the action

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
116 | P a g e
between its molecules and biological partner of reaction unfolds. Depending on the action mechanism,
this reaction can occur:
intracellularly,
extracellularly,
in the blood,
in the CSF,
in the urine etc.
The obtained values provide information about:
the elimination rate and
the apparent volume dissemination
10.1.1.1. The monocompartmental open model
The simplest model is being viewed as a simple fluid space where the pharmacon is administered
and where it will diffuse until it reaches a state of equilibrium.
The term „open” describes the continuing loss of drug from the compartment, and represents the
organism’s „opening”, meaning that the consequences associated with drug loss during metabolization
and excretion, will determine the uncoupling of the receptor molecules and therefore, put an end to the
drug’s action. The drug concentration in the biophase stops increasing when: the addition rate through
absorption of the pharmacon in the biophase is exceeded by the removal rate through the elimination
process.
Therefore:
the intensity of the response depends on the quantity of the pharmacon present on
receptors.
the duration of the response depends on the elimination kinetics of the uncoupled
fraction.
After studying the drug’s plasma concentration, it can be demonstrated that in some cases, the
rate of drug loss from plasma after reaching the dissemination equilibrium is constant in terms of mass
unit / time unit.
In these conditions, it can be said that: “the plasma concentration per unit of time, is represented
by a straight line”.
More often than not, the decrease of plasma concentration follows the first order kinetics, where
“a constant fraction of the drug is eliminated per unit of time”.
Such a relationship arithmetically represented would offer an exponential curve that could turn
into a straight line if the representation would be semilogarithmic.

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
117 | P a g e
The monoexponential process can be defined by its constant rate that expresses the fraction’s
modification per time unit or by the half-life (t1/2), that means the time it takes for the blood plasma
concentration of a substance to halve its steady state. Both values are independent from plasma
concentration, meaning that they are constant no matter the concentration.
Continuous i.v. Injection: the organism behaves like an open monocompartmental system (Kuschinsky, 1989)
10.1.1.2. The bicompartmental model
This model describes the behavior of a pharmacon that disseminates extracellularly after entering
the blood and concomitantly, is eliminated through renal excretion. The size of the two areas represent
in adults: first area, approx. 4% (PS) and second area, 16% (ECS) of total body weight, therefore, a ¼
ratio exists between the two areas:
the plasmatic area (PS) and
the extracellular area (ECS),
whose separation barrier is easily crossed by the pharmacon.
The pharmacon reaches:
1. the central compartment, and from here,
2. the second compartment (SEC).
There are two known outcomes:
a). the drug passes into the ECS and back, that is very fast regarding the elimination process (k12
= k21 > k3) and
b). the elimination speed is in the same domain with the diffusion speed from the central space
into the peripheral space. (k12 = k21 » k3).
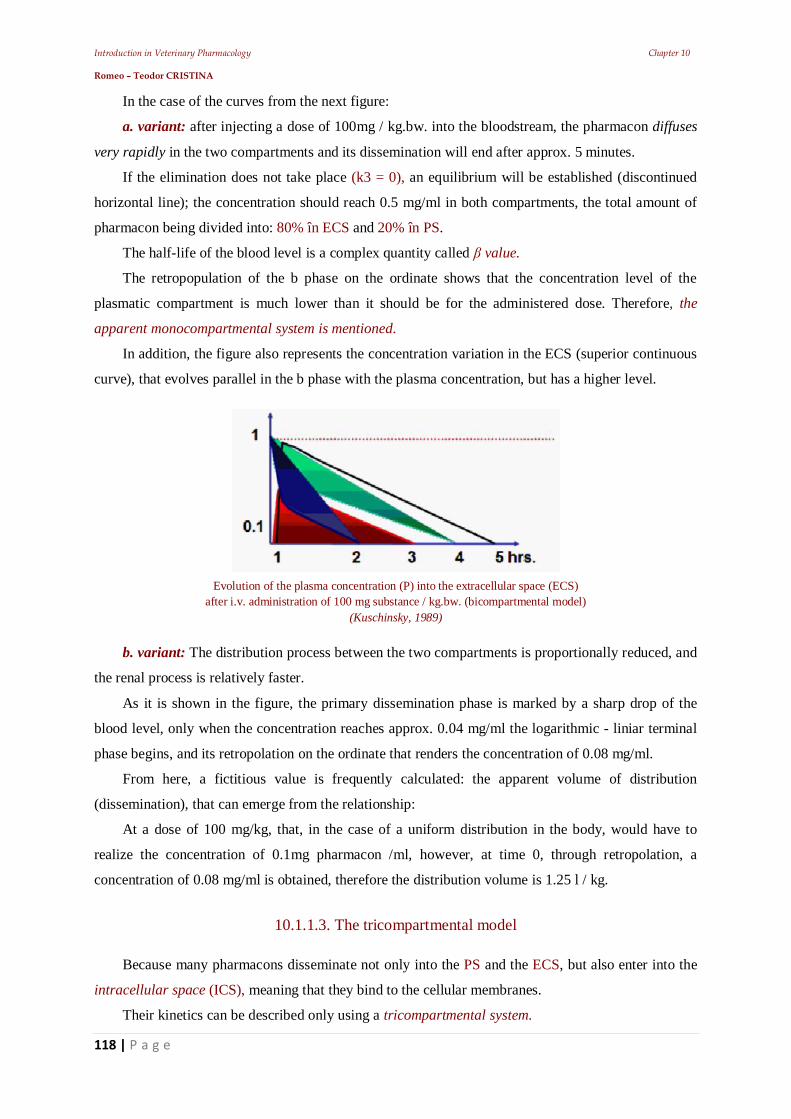
Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
118 | P a g e
In the case of the curves from the next figure:
a. variant: after injecting a dose of 100mg / kg.bw. into the bloodstream, the pharmacon diffuses
very rapidly in the two compartments and its dissemination will end after approx. 5 minutes.
If the elimination does not take place (k3 = 0), an equilibrium will be established (discontinued
horizontal line); the concentration should reach 0.5 mg/ml in both compartments, the total amount of
pharmacon being divided into: 80% în ECS and 20% în PS.
The half-life of the blood level is a complex quantity called value.
The retropopulation of the b phase on the ordinate shows that the concentration level of the
plasmatic compartment is much lower than it should be for the administered dose. Therefore, the
apparent monocompartmental system is mentioned.
In addition, the figure also represents the concentration variation in the ECS (superior continuous
curve), that evolves parallel in the b phase with the plasma concentration, but has a higher level.
Evolution of the plasma concentration (P) into the extracellular space (ECS) after i.v. administration of 100 mg substance / kg.bw. (bicompartmental model)
(Kuschinsky, 1989)
b. variant: The distribution process between the two compartments is proportionally reduced, and
the renal process is relatively faster.
As it is shown in the figure, the primary dissemination phase is marked by a sharp drop of the
blood level, only when the concentration reaches approx. 0.04 mg/ml the logarithmic - liniar terminal
phase begins, and its retropolation on the ordinate that renders the concentration of 0.08 mg/ml.
From here, a fictitious value is frequently calculated: the apparent volume of distribution
(dissemination), that can emerge from the relationship:
At a dose of 100 mg/kg, that, in the case of a uniform distribution in the body, would have to
realize the concentration of 0.1mg pharmacon /ml, however, at time 0, through retropolation, a
concentration of 0.08 mg/ml is obtained, therefore the distribution volume is 1.25 l / kg.
10.1.1.3. The tricompartmental model
Because many pharmacons disseminate not only into the PS and the ECS, but also enter into the
intracellular space (ICS), meaning that they bind to the cellular membranes.
Their kinetics can be described only using a tricompartmental system.

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
119 | P a g e
Usually the process of analysis of the distribution of a drug into the body is based, in principle, on
the establishment of plasma concentrations and assigning these values to the other compartments.
If a pharmacon accumulates or binds to a specific location in the body, the apparent volume of
distribution values will exceed the unit.
A very important biological principle, is to consider the body compartments as given sizes from
the beginning, because they are known and can be established independently.
The pharmacon concentration in these spaces should be considered as a variable. This way we
can calculate the amount of pharmacon from each compartment.
For example: the administered dose is shown in a proportion of 100%, in addition to the amount
of plasma (4% of body weight) and ECS (16% of body weight) is rendered also the quantity of
substance excreted by the kidneys.
Immediately after the injection, the pharmacon leaves quickly the PS, after approx. 10 minutes
about 40% of the substance reaches the ECS, 20% is eliminated by the kidneys and after approx. 20
minutes, the ECS contains 50% of the dose and 30% has been eliminated. After 40 minutes the
distribution phase is finished.
In the PS there is only 10% of the dose, but in the ECS, 50% of the dose can be found. During the
terminal phase, the quantity of substance in the ECS is 6 times higher than the one from PS.
When taking into account the sizes of the compartments, the concentration in the ECS, from a
biological point of view, is 1.5 times higher.
In this case, such an example is imagined following: The quantitative influence of the ECS (of
high capacity) is obvious, because it represents 50% of the body weight, compared with 16% of ECS
and 4% of PS.
In the case of a proportional distribution of a pharmacon, it is expected to be found approx. 5% of
the administered dose in the PS (if the elimination does not occur).
The percentage of the temporal distribution of a drug after iv administration into the plasma (continuous line) ECS (dotted
line) and urine (dashed line). The amount injected was immediately available in a percentage of 100%. On the ordinate there are represented the logarithmic units, and on the abscissa the hours
(Kuschinsky).
it is apparent that :
within one hour after the administration, the quantity of pharmacon from the PS has decreased
to 3%;

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
120 | P a g e
in the ECS, during the period of time between the 5th minute and the 10th minute, a maximum
value is attained (approx. 35% of dose);
the quantity of substances from the ECS increases relatively fast during the distribution period
and reaches a maximum value, after approx. 30 minutes.
At that moment, 50% of the administered dose is in ECS. The dynamics of the concentration, resulting in SP and in the second compartment after repeated
administration. Meanwhile the blood picture does not render, almost at all, the accumulation
phenomenon, in the tissular compartment the concentration increases abruptly.
This kinetic compartment of a pharmacon (with increasing quantity in the neighboring
compartment, in conditions of fast blood elimination) is important for the practical therapy, because
the compartment where the therapeutic effect is taking place is almost always a compartment that is
adjoined to blood.
a. The quantity determination of substances in blood, in these conditions, does not
provide information about the pharmacon concentration and about the temporal
modifications from the place of action.
b. The half-life of PS does not reflect changes of the concentration in the action site.
The percentage of temporal distribution after iv administration of a drug, in plasma (thick solid line), ECS (dotted line), ICS
(thin solid line), urine (dashed line) (tricompartmental model) (Kuschinsky, 1989).
This assumption is demonstrated by an example:
Thiopental, is lipid-soluble and accumulates in fat tissue:
Three hours after administration it is still found (70%) in the fat tissue, although the blood level
has dropped below the level that obtains the effect, and the narcotic effect disappeared.
The concentration in the cerebral compartment is closer to the evolution of blood concentration
than the concentration of fatty tissue. The concentration from the fatty tissue remains increased for a
long time, compared with the blood concentration. Therefore, in the case of a re-administration, this
fact is easy to observe, because Thiopental reaches a presaturated deposit tissue.
The result of this fact is a persistent and a higher blood level, and the risk of poisoning when re-
administering a higher dose. Due to their physico-chemical characteristics, drugs are hydrophobic, and
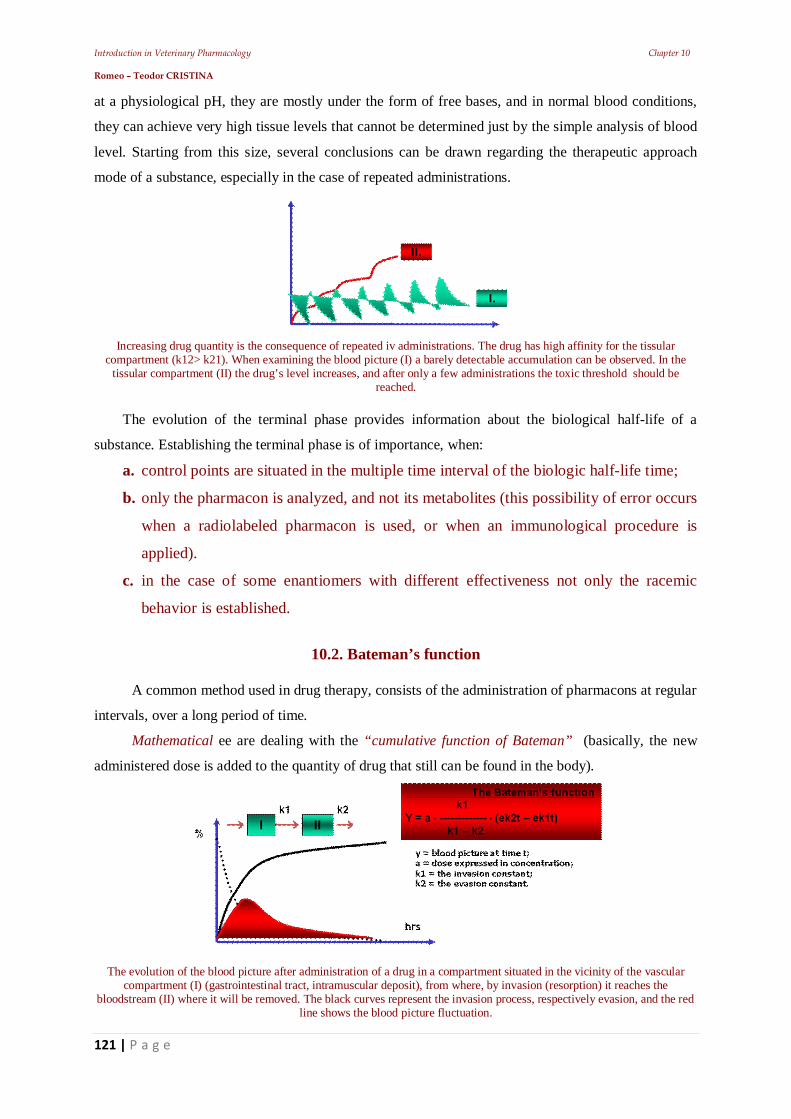
Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
121 | P a g e
at a physiological pH, they are mostly under the form of free bases, and in normal blood conditions,
they can achieve very high tissue levels that cannot be determined just by the simple analysis of blood
level. Starting from this size, several conclusions can be drawn regarding the therapeutic approach
mode of a substance, especially in the case of repeated administrations.
Increasing drug quantity is the consequence of repeated iv administrations. The drug has high affinity for the tissular compartment (k12> k21). When examining the blood picture (I) a barely detectable accumulation can be observed. In the
tissular compartment (II) the drug’s level increases, and after only a few administrations the toxic threshold should be reached.
The evolution of the terminal phase provides information about the biological half-life of a
substance. Establishing the terminal phase is of importance, when:
a. control points are situated in the multiple time interval of the biologic half-life time;
b. only the pharmacon is analyzed, and not its metabolites (this possibility of error occurs
when a radiolabeled pharmacon is used, or when an immunological procedure is
applied).
c. in the case of some enantiomers with different effectiveness not only the racemic
behavior is established.
10.2. Bateman’s function
A common method used in drug therapy, consists of the administration of pharmacons at regular
intervals, over a long period of time.
Mathematical ee are dealing with the “cumulative function of Bateman” (basically, the new
administered dose is added to the quantity of drug that still can be found in the body).
The evolution of the blood picture after administration of a drug in a compartment situated in the vicinity of the vascular compartment (I) (gastrointestinal tract, intramuscular deposit), from where, by invasion (resorption) it reaches the
bloodstream (II) where it will be removed. The black curves represent the invasion process, respectively evasion, and the red line shows the blood picture fluctuation.
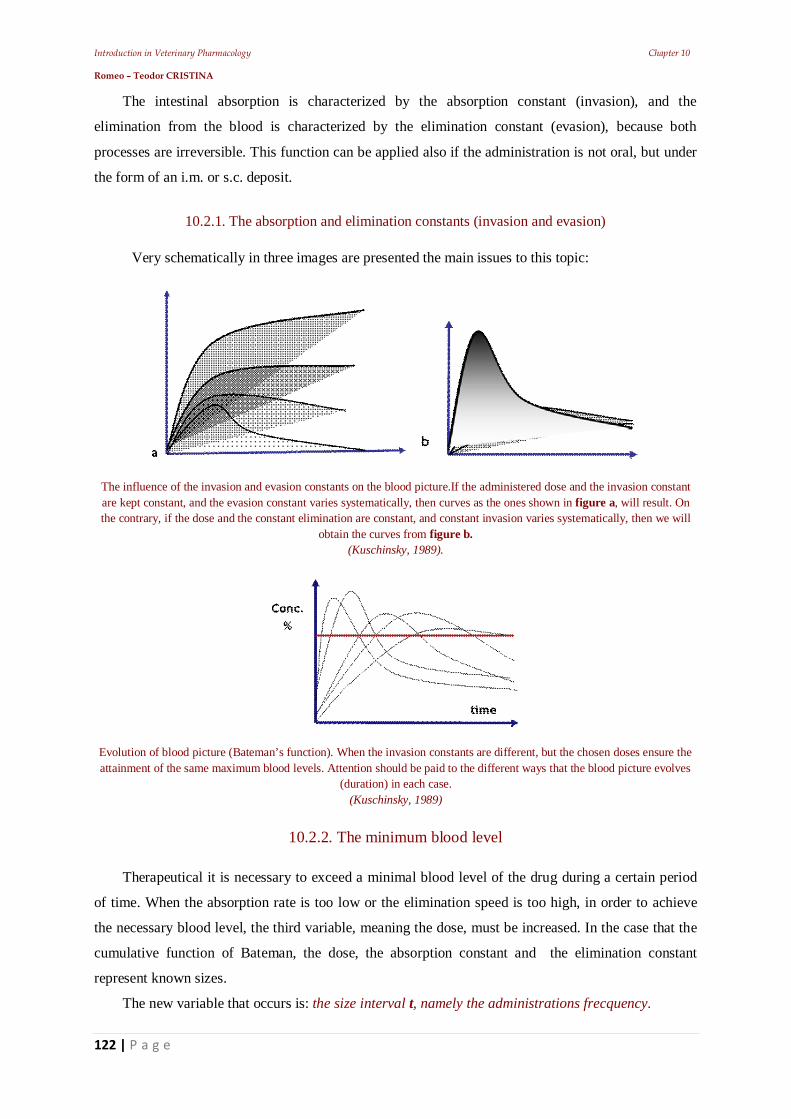
Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
122 | P a g e
The intestinal absorption is characterized by the absorption constant (invasion), and the
elimination from the blood is characterized by the elimination constant (evasion), because both
processes are irreversible. This function can be applied also if the administration is not oral, but under
the form of an i.m. or s.c. deposit.
10.2.1. The absorption and elimination constants (invasion and evasion)
Very schematically in three images are presented the main issues to this topic:
The influence of the invasion and evasion constants on the blood picture.If the administered dose and the invasion constant are kept constant, and the evasion constant varies systematically, then curves as the ones shown in figure a, will result. On the contrary, if the dose and the constant elimination are constant, and constant invasion varies systematically, then we will
obtain the curves from figure b. (Kuschinsky, 1989).
Evolution of blood picture (Bateman’s function). When the invasion constants are different, but the chosen doses ensure the attainment of the same maximum blood levels. Attention should be paid to the different ways that the blood picture evolves
(duration) in each case. (Kuschinsky, 1989)
10.2.2. The minimum blood level
Therapeutical it is necessary to exceed a minimal blood level of the drug during a certain period
of time. When the absorption rate is too low or the elimination speed is too high, in order to achieve
the necessary blood level, the third variable, meaning the dose, must be increased. In the case that the
cumulative function of Bateman, the dose, the absorption constant and the elimination constant
represent known sizes.
The new variable that occurs is: the size interval t, namely the administrations frecquency.
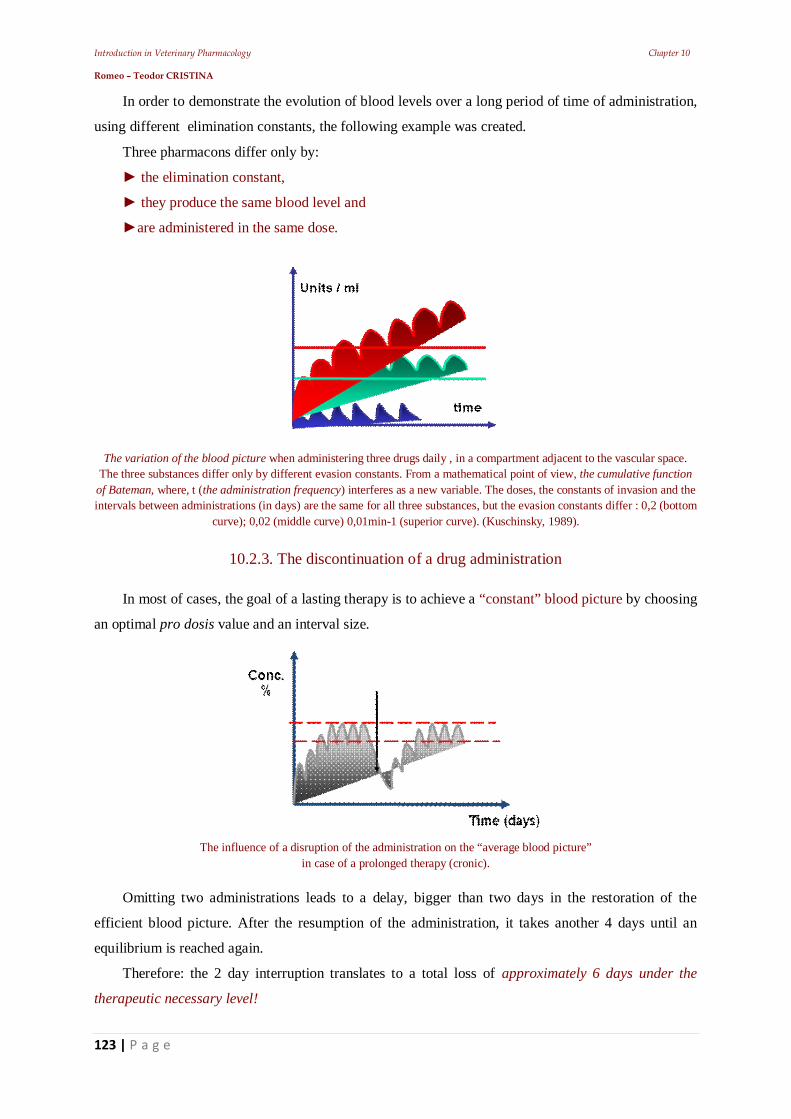
Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
123 | P a g e
In order to demonstrate the evolution of blood levels over a long period of time of administration,
using different elimination constants, the following example was created.
Three pharmacons differ only by:
the elimination constant,
they produce the same blood level and
are administered in the same dose.
The variation of the blood picture when administering three drugs daily , in a compartment adjacent to the vascular space. The three substances differ only by different evasion constants. From a mathematical point of view, the cumulative function
of Bateman, where, t (the administration frequency) interferes as a new variable. The doses, the constants of invasion and the intervals between administrations (in days) are the same for all three substances, but the evasion constants differ : 0,2 (bottom
curve); 0,02 (middle curve) 0,01min-1 (superior curve). (Kuschinsky, 1989).
10.2.3. The discontinuation of a drug administration
In most of cases, the goal of a lasting therapy is to achieve a “constant” blood picture by choosing
an optimal pro dosis value and an interval size.
The influence of a disruption of the administration on the “average blood picture”
in case of a prolonged therapy (cronic).
Omitting two administrations leads to a delay, bigger than two days in the restoration of the
efficient blood picture. After the resumption of the administration, it takes another 4 days until an
equilibrium is reached again.
Therefore: the 2 day interruption translates to a total loss of approximately 6 days under the
therapeutic necessary level!

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
124 | P a g e
10.2.4. Enzyme induction and blood level
For example: in one case an optimal blood level is obtained (but from the 12th day of treatment,
the patient is given a second drug, which causes an enzyme induction in the liver.
Corresponding to this administration, the elimination rate of the first pharmacon will expand.
The influence of the increased rate of evasion over the medium blood picture in case of chronic therapy. The decrease of blood levels is due to the enzyme induction caused by another drug. The increased elimination rate causes the decrease of the
blood level and the establishment of a new equilibrium level, but situated under the therapeutic one. (Kuschinsky, 1989).
10.3. The parameters of pharmacokinetic quantification
The pharmacokinetic evaluation of the quantitative consequences determined by the absorption
process and the elimination of drugs, are obtained by viewing the body as a “machine that works
mechanically”. This machine is seen as performing two actions after the administered drug dose:
- first dilute the drug and
- then remove it.
The drug concentration at any moment, is the measure of the diluted fraction remaining at that
time from the administered drug.
The rate at which the concentration decreases over time is the measure of the “machine's”
capacity to eliminate the drug.
Furthermore, they are independent of the size of the dose until one of the involved mechanisms
becomes saturated (e.g.: coupling capacity and the degradation paths).
After the iv administration, the dilution includes:
the pharmacon mixes with the blood,
the pharmacon exits the vascular space into the distribution volume and
the loss of free drug to the receptors by coupling, lipid solubilization and ionic
capture.
Determination of the dilution capacity and the speed constant elimination can be determined
experimentally by administering a single dose iv.
The correlation between the administration of a drug and the pharmacological or toxicological
final effect is determined by many factors.

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
125 | P a g e
The transformation into biological effects is closely related with the coupling of the pharmacon
on specific or nonspecific sites (transformation kinetics).
Transforming receptor occupancy in effect is probably directly proportional only in exceptional
cases, in the rest of the cases it submits to complicated functions.
It results that there are different growth rates of the dose-effect curves that represent the effect
dependence of concentration.
The transformation can take place quickly and directly (e.g.: increasing the ionic permeability of
the plate terminal membrane after binding acetylcholine to acetylcholine receptors), but it requires a
sequence of processes (or may even be a slow process).
Examples in this regard are:
the effects of hormones with a steroidic structure on the synthesis of proteins or
the inhibition of blood clotting factors by coumarin.
In these cases, the transformation takes place at a much slower rate compared to the two previous
kinetic processes.
Factors and processes involved in the onset of the activity of a drug (Synthesis, Cristina)
Pharmacokinetics
Dose; administration method; galenic disponibility; invasion into the venous system; presystemic elimination (liver, lung) the great arterial circulation volume, distribution, elimination (metabolization and excretion) concentration into biophase.
Receptor kinetics Biophase: concentration, the receptors affinity, binding site;
The transformation kinetics coupling transformation of drugs in pharmacologic or toxic effect.
Generally, the drug gets distributed in the body and reaches the target organs, through the blood
pathway. There are two main types of administration: oral and parenteral.
After oral administration, in general, the pharmacon gets reabsorbed by the gastrointestinal
mucosa.
Blood drainage it is done through the portal vein that develops a new capillary territory in the
liver, leading to the decrease of the flow rate in this zone, implying a prolonged contact of the liver
cells with the blood.
Therefore, an intensive exchange of substances can be possible.
Some part of the quantity of the reabsorbed substance can thus be captured = lost at the first
hepatic passage or “first pass efect”.
The fact that a part of the pharmacon’s quantity that is reabsorbed at an intestinal level is retained
in the lung and liver, before reaching the big circulation, can be called: presystemic elimination.
From here, the blood passes the right heart and then into the lung, where due to capilarization an
intensive contact with the tissue cells takes place.

Introduction in Veterinary Pharmacology Chapter 10 Romeo – Teodor CRISTINA
126 | P a g e
Here, a part of the quantity of the substance absorbed from the gastrointestinal system can remain,
because the lung has a high coupling capacity for amphiphilic and lipophilic substances.
When administering intravenous injections, the pharmacon goes straight into the blood, but must
pass the lung barrier before reaching the big circulation.
When rapidly administering a drug, by i.v. injection, the lung can act as a buffer, in order to
protect the following organs from excessive concentrations, such as the myocardium, which is directly
irrigated by the coronary system.
Representation of the major kinetic processes that can influence the speed of installation of the pharmacologic effect of a drug product.
Conclusion
Pharmacokinetics is a pharmacology sub domain that studies the temporal changes in the
concentration of the pharmacon in different compartments of the body.
Because the power of the effect has a parallel dynamic with the dynamic of the concentration,
knowing the concentration of a pharmacon at the action site is particularly important.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
127 | P a g e
11. Main pharmacodynamic factors that influence the drug effect - Dose theory

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
128 | P a g e
Introduction Dose is:
the amount of drug used in one administration.
one of the decisive factors of the drug effect.
depending on the administered amount, drugs may have different actions.
By dose: we understand the quantity of drug which produces a certain pharmacodynamic effect.
From the point of view of the intensity of effects, three main types of dose are distinguished:
Effective dose (ED) (sin. therapeutic dose) which produces a useful, efficient
pharmacodynamic effect;
Toxic dose (TD) that determines the appearance of toxic phenomena;
Lethal dose (LD) which produces the animal's death.
We also know about the threshold dose (sin. subliminal dose) the amount of drug that does not
produce visible effects (or possibly at cellular level). Therapeutic range
The therapeutic index of a drug is the measure of security of the drug.
The term of: safe area that a drug ensures in its use actually means the therapeutic range.
Quantitative measures for the therapeutic range are represented by the ratio of different points on
the lethality and dose - effect curve.
The therapeutic index is defined as: LD50 (median lethal dose) T.I. = ----------------------------------------; ED50 (median effective dose) The higher is the value of this ratio, respectively the more distant are the curves from each other,
the therapeutic range is higher. This measure has a drawback because it only renders the existing
relations when the curves are parallel. If the curves are not exactly in the same inclination, the I.T.
index defined above is not an accurate measure of the therapeutic range.
11.1. Factors establishing a dose
A satisfactory therapeutical answer can be expected only: in case the drug reaches the place
where it will act, or at an adequate concentration. In this context, the individuality of the animal can
influence the effect of a treatment.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
129 | P a g e
Examples:
The use of strychnine, in nervous individuals can induce poisoning,
Apomorphine may induce vomiting, only in some breeds of pigs (Landrace, Duroc)
but in other rustic breeds no!
Dose variation depending on the route of administration (after W. Cooke, 1994)
Route of administration Standard Increased dose (%)
Decreased dose (%)
Oral (p.o.) 1 - -
Rectal (p.r.) - 150-200 -
Subcutaneous (s.c.) - - 75-50
Intramuscular (i.m.) - - 75-50
Intravenous (i.v.) - - 50
Intraperitoneal (i.p.) - - 50
Intratracheal (i.t.) - - 50
11.1.1. Genetic factors
Some breeds may be sensitive to the action of drugs.
This can be explained by the absence of some specific enzymes (ex: deficiency in glucose-6-
phosphate dehydrogenase in some breeds is associated with toxicity).
Such anomalies have led to the emergence of a new branch, Pharmacogenetics.
When response to a drug is abnormal qualitatively or quantitatively, idiosyncrasy intervenes.
Sometimes idiosyncrasy can be explained genetically. In the case of improved breeds, the effect
of the drug may be altered due to sensitizing genetic factors
Examples:
Arabian thoroughbred horses,
Supercuni rabbits,
Cocker`s etc.
11.1.2. Susceptibility
Is the term used to describe an abnormal quantitative response and is demonstrated by the so-
called hyperactive (a patient particularly sensitive to the action of a drug).
Such variations are frequently dependent on the atypical elimination rates.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
130 | P a g e
11.1.3. Species
Among the species of animals, there are some examples of extreme resistance or sensitivity to
drugs. Species influence the effect, the cause being mainly: genetic or morphopathological factors.
There are species that react differently to the same drug.
Examples:
dogs react to morphine through hypnosis or vomiting, whilst
cats and large ruminants will react to the same drug through over excitement /
hyperactivity
in cows alcohol is well supported as a narcotic, whilst horses are sensitive,
chloralhydrate, is very effective in horses, but it is hardly supported by cows.
apomorphine in dogs, produces vomiting constantly, whilst in pigs its action is
inconsistent.
vomitive drugs in omnivores and carnivores can become ruminatories in ruminants.
Sensitivity depending on the species:
pigs and poultry to salt,
large ruminants to mercury
cats to phenolic drugs.
doses in ruminants, increased by 20 - 40% compared to equines, (drugs stagnate and
even suffer decomposition in the fore stomach.
Equines and some dog breeds are sensitive to injectable Ivomec, due to the permeability of the
meningeal blood brain barrier common in some individuals.
In the case of using drugs that are common for human and veterinary use, the doses for animals
are:
Correspondence animal-human dose (after Suciu, 1990)
Species Increasing the dose from humans
Cow x 24
Horse x 16
Sheep x 3
Goat x 3
Pig x 3
Dog Equal to that of humans
Cat ½ of the human dose

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
131 | P a g e
Examples:
if we would take a standard adult man (aprox. 70 kg) then the required dose is
equivalent with a dose for a 10 kg dog.
even if small ruminants (circa 40 kg) are four times as heavy as the dog above, they
will only need a dose twice as high.
a pig (approx. 100 kg) will receive a dose, not ten times higher but only four times
higher compared to a 10 kg dog.
a horse (approx. 400 kg) will require doses, only ten times higher than the dog from
example,
large ruminants (100-400 kg) will be treated with doses 10-15 times higher.
So, the more the species have smaller sizes can handle higher doses reported per kg body weight
corp. For example, a 2 kg cat will not receive, 20% from the dog's (10 kg) dose, but much more, 50%.
The same is true for birds (2 kg) who will receive 40-50% from the dog dose.
11.1.4. Anatomy of the digestive system
In ruminants, food passage rate is slow, and the intestinal content is large, in comparison to the
rate of absorption.
Therefore, there is much time available for absorption, while the large volume of intestinal
contents dilutes the orally administered drug, thus slowing the rate of absorption.
One problem is related to the compartment into which the orally administered drug enters,
influenced by the work of the esophageal tray.
Drugs with weakly alkaline character tend to accumulate in weak acid ruminal juice, which has a
very large volume in ruminant species.
11.1.5. Age
Very young and very old animals generally require the administration of reduced doses due to the
possibility of organ dysfunctions. In old animals dysfunctions are mostly degenerative, at the hepatic
and renal level.
In young animals excretory and metabolic functions are not yet developed (ex: chloramphenicol
is toxic for piglets due to the absence of suitable enzymatic equipment).
Youngsters, infants, will receive reduced doses with 30-40% (small animals) or even 50-70%
(youngsters up to 1 year old in large animals).
There are situations when, in compared to adults, youngsters are more resistant to therapeutic
doses (ex: barbiturates in piglets). They will receive doses reduced by 20-40% because the activity of
some enzymatic systems may be reduced or even abolished.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
132 | P a g e
Doses by age categories (after Balaci)
Species Category Expected dose
Equines
3 - 15 years 1 dose
15 - 20 years ¾ dose
20 – 25 years ½ dose
Foals 2 years ½ dose
Foals 1 year 1/12 dose
Foals2-6 months 1/24 dose
Cattle
3-8 years 1 dose
10–15 years ¾ dose
15-20 years ½ dose
Calves 4 – 8 months 1/8 dose
Calves1-4 months 1/16 dose
Sheeps and goats
Over 2 years 1 dose
1-2 years ½ dose
Lambs & kids 6-12 months ¼ dose
Swines
Over 1,5 years 1 dose
8 – 18 ½ dose
Youngsters 4-9 months ¼ dose
11.1.6. Gender
Gestation involves contraindications (ex: purgatives or corticosteroids, which can induce
abortion). Teratogenic effects are investigated and taken into account in the evaluation of each new
drug (for veterinary use too). The elimination of drugs by drinking milk is another example for
toxicity risk related to animal gender.
11.1.7. Time administration and pathology
A drug administered orally, is more rapidly and completely absorbed if the anterior digestive
segment is empty, but often, it is irritating to the tissue.
The recognition of the existence of the circadian rhythm within physiological functions has
already found application in drug administration.
Generally, sick animals have a diminished drug detoxification capacity. An increased or
decreased rate of intestinal passage will change:
absorption period, and therefore,
the proportion of the absorbed dose. Also:
hypoalbuminemia decreases the coupling rate.
heart failure will be accompanied by liver and kidney failure.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
133 | P a g e
enteritis reduces intestinal transit time and therefore may reduce the absorption of drugs.
peripheral circulation is inadequate in states of shock of any origin, preventing absorption of
s.c. injections.
11.2. Tolerance and intolerance
Tolerance to a drug disappears with the discontinuation of the treatment (ex: dogs may exhibit
tolerance to the narcotic effect of barbiturates).
Resistance to drugs can occur for many reasons:
when a drug is a specific antigen, and antibodies may be produced for it, inactivating
it;
(metabolic) resistance of Trichostrongylus population to therapeutic doses of benzimidazoles.
11.2.1. Therapeutic indications This assessment is purely therapeutic and includes :
dosage adjustment based on the nature of the disease and
depending on the causative agent (ex: therapy of acute fasciolosis requiring higher doses of the
same drug as in the chronic form).
often the use of high doses is similar to increasing the risk of toxicity, to the benefit of
receiving increased effects.
certain antibiotics are so toxic that their systemic administration is only done case of
emergency (ex: polymyxin).
11.2.2. Concomitant drug therapy
Concomitant use of several remedies requires the introduction of several variables in calculating
doses, because of the potential interactions between administered components and patient.
Use of ”shot-gun” type products or polypharmacy (active substances associated without a certain
diagnosis) is a simple substitute to a certain, professional diagnosis, often with undesirable
implications.
11.2.3. Amplified response
To reduce the incidence of toxicity, one or more drugs can be administered simultaneously.
The final answer can be quantitative = with the amount of expected responses in the case of
independent administration = medication summation
If the answer is higher than what can be explained by simple summation, We are dealing with
the effect of potentiation or synergism.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
134 | P a g e
11.2.4. Diminished response
In multidrug therapy it happens that the observed response is less than the sum of components
responses = antagonism between the drugs used.
sometimes the antagonism can be explained by the fact that a drug interferes or performs an
action, opposite to the other.
the antagonism is often dependent on a mechanism that involves pharmacological or
physiological incompatibility.
11.2.5. Incompatibilities
The associated components may be incompatible:
physically or
chemically, when reacting with each other
Often, the need for administration tempts the clinician to combine remedies. In the case that, the
compatibility of the remedies is unknown, concomitant use is contraindicated.
Pharmacodynamic incompatibility is the use of adrenaline as a cardiac stimulant in the
anesthetized animals with a drug that sensitizes the heart to adrenaline action (e.g. cyclopropane).
11.2.6. Amplified toxicity
The toxicity of a drug can increase several times, depending on the situation.
Two drugs whose degradation pathways are the same, can enter in competition if, the metabolic
pathway has limited capacity. If one of them has a narrow therapeutic range, toxicity is facilitated.
Competition for coupling sites is another mechanism which may increase the risk of toxicity of
drugs which engage massively to proteins.
Drugs whose plasma half-life is much shorter than the biological half-life, so-called: “hit-and-
run” (achieve plasma levels rapidly, but are eliminated as quickly) causes increased responses to other
drugs.
11.2.7. Reduced toxicity
A common example of low toxicity can be the premedication with tranquilizers before the
induction of anesthesia.
This simplifies the process of induction and reduces the dose of barbiturate required; therefore it
is useful in reducing the risk of anesthesia.
The antidote in poisonings exploits both pharmacokinetic and pharmacodynamic interactions
(competitive antagonism) in the benefit of the patient.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
135 | P a g e
11.3. Factors determining the frequency of administration In the treatment of diseases, the initial goal is to achieve an adequate response. This fact depends
on the suitable concentration of a drug in the biophase. An appropriate therapeutic effect often asks the
drug to act over a longer period of time.
The shorter the half-life is, the quicker the product will be removed from the body and the shorter
the interval will be between administrations (when necessary to maintain a constant level of effect).
Because, the belief that there is one standard interval between administrations is incorrect, the
size of repeatedly administered doses will vary according to the benefits.
For example, an initial attack dose, followed by a daily maintenance dose is a procedure often
used with sulfonamides therapy
The probable plasma level originally obtained by administering an attack dose, which reaches desired plasma concentration, and then, by administration of lower maintenance doses;
When the level obtaind by administering a dose, does not return to te initial value before the next dose, concentration may increase successively with each dose, this phenomenon can produce a cumulative toxicity.
Coupling to plasma proteins will suppress inactivation and excretion rates,
Extensivity or the power of coupling can vary considerably for the same drug in several species
or within a family of drugs, when it is tested on different individuals. Cumulative toxicity is
characteristic of compounds with half-lives exceeding the interval between administrations and when
dose size allows the cumulating phenomenon to progress beyond the therapeutic level, (thus falling
within the toxic concentration).

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
136 | P a g e
11.3.1. Concentration stability
Besides the mentioned factors, the frequency of administration can exert considerable influence,
not only on the duration of action of drugs but also on the quality of drug action.
Generally, greater concentration stability is achieved when a pro die dose is administered on
several occasions over a period of 24h.
11.4. Establishing rates of drug dosing
The rate of: absorption, distribution and elimination can be experimentally quantified through the
apparent volume of distribution, where the level of a drug in the body, can be estimated in any time
when plasma concentration is known.
Using these pharmacokinetic parameters and on the basis of the calculations it is possible to issue
rational recommendations about the size and frequency of the dose.
If a drug acts quickly, achieving immediately observable effects in an animal, dose determination
is possible only through the continued use of the drug until the desired level of response is reached.
Dose titration according to the response is easy, for instance, when administering i.v. anesthetics.
The only requirement is to know the exact intensity of the desired effect, before starting the
administration. In the case of a drug whose effect is manifested slowly or cannot be measured
clinically, the approach will be different.
For some groups, the concentrations may be set based on "in vitro" studies (ex: identification of
concentration at which antimicrobial agents inhibit the growth of bacterial cultures).
This, multiplied by an appropriate safety factor (generally = 5) = necessary concentration in the
body fluids. For the other groups of drugs, the study is based on the measurement of plasma
concentrations, when it is assumed that the response has reached the desired level. In each case, the
dose calculation which implies reaching such concentrations is made using this relation:
D = Cpd × Vd
where: D = dose (mg) Cpd = desired plasma concentration (mg l-1), Vd = apparent volume of distribution (l.).
When the drug is not administered i.v., it may be necessary to apply a correction factor that takes
into account the incomplete bioavailability of the dose.
11.5. Establishing the frequency of administration
The single dose has duration of action determined by:
- the size of the dose,
- the elimination rate constant and by
- the apparent volume of distribution.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
137 | P a g e
If the minimum plasma concentration required to gain a therapeutic effect is known, it will be
possible to calculate: the time required to decrease the initial concentration to this level.
The elimination of half of the amount of the drug in the body by the end of a half-life, will lead to a drastic exponential decrease, of the percentage of the dose which exist in the body
(after Brander, 1991)
11.5.1. Establishing intravenous infusion rate
When a therapeutic effect of constant intensity is necessary, this requirement can be satisfied by
iv infusion at a suitable dosage rate.
The rate at which the drug is lost from the body may be most useful, expressed as total clearance,
when the desired plasma concentration is known or can be found out (Cpd):
R = Cpd x Vd x ß
Where: R = loss rate of the drug(mg h-1).
So, to maintain the level of an already achieved concentration of a drug in the body, constant, it is
only necessary to perfuse the drug with an hourly rate equal to the rate of elimination.
11.5.3. Plateau effect Achieving a stable plateau concentration is possible without the administration of an attack dose.
The disadvantage is that, the time required for the therapy may be incompatible with the
desideratum of a favorable therapeutic outcome.
The amount of drug excreted per time unit increases progressively as long as a continuous
infusion of the drug will cause progressive increase in plasma concentration.
The time to reach the plateau concentration and that required for complete removal, is
approximately equal to = 6 x t½.
The progressive achieving of a plateau concentration is illustrated in the figure, where: the total
amount of the drug in the body represented on each half-life period is equal to the amount infused over
the duration of the half-life plus the residue of infusion until then.
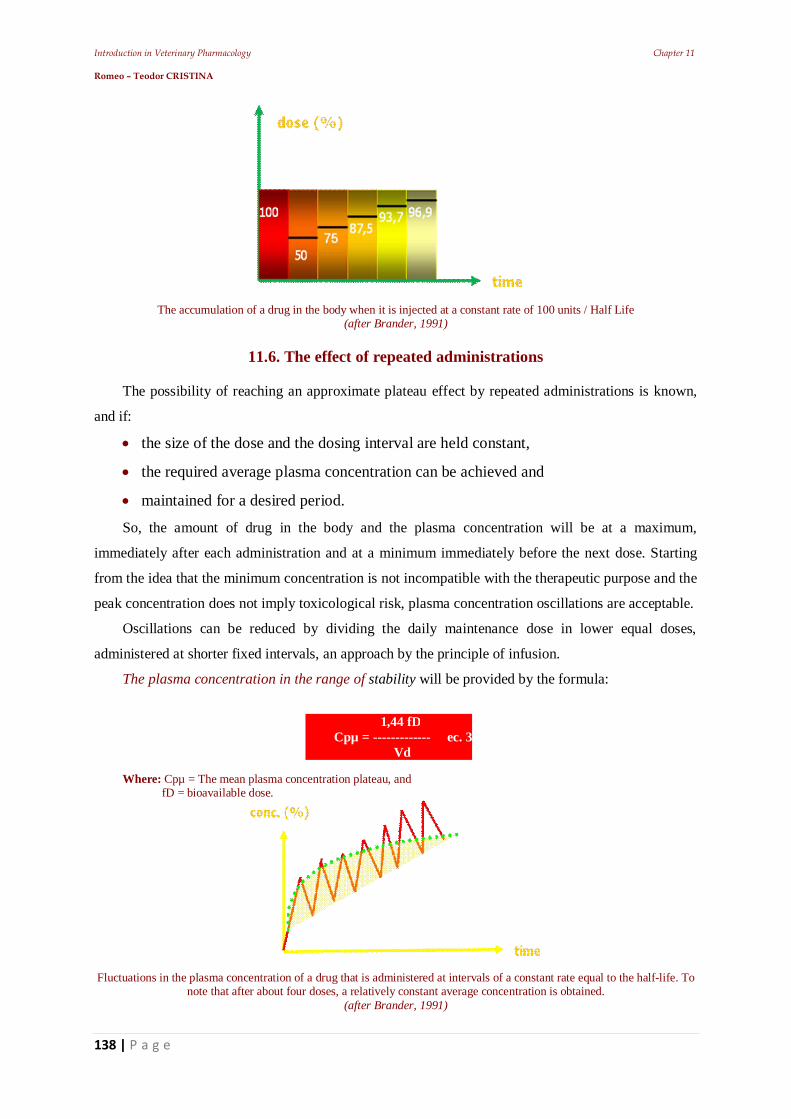
Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
138 | P a g e
The accumulation of a drug in the body when it is injected at a constant rate of 100 units / Half Life (after Brander, 1991)
11.6. The effect of repeated administrations
The possibility of reaching an approximate plateau effect by repeated administrations is known,
and if:
the size of the dose and the dosing interval are held constant,
the required average plasma concentration can be achieved and
maintained for a desired period.
So, the amount of drug in the body and the plasma concentration will be at a maximum,
immediately after each administration and at a minimum immediately before the next dose. Starting
from the idea that the minimum concentration is not incompatible with the therapeutic purpose and the
peak concentration does not imply toxicological risk, plasma concentration oscillations are acceptable.
Oscillations can be reduced by dividing the daily maintenance dose in lower equal doses,
administered at shorter fixed intervals, an approach by the principle of infusion.
The plasma concentration in the range of stability will be provided by the formula:
Where: Cpµ = The mean plasma concentration plateau, and fD = bioavailable dose.
Fluctuations in the plasma concentration of a drug that is administered at intervals of a constant rate equal to the half-life. To note that after about four doses, a relatively constant average concentration is obtained.
(after Brander, 1991)
1,44 fD Cpµ = ------------- ec. 3 Vd
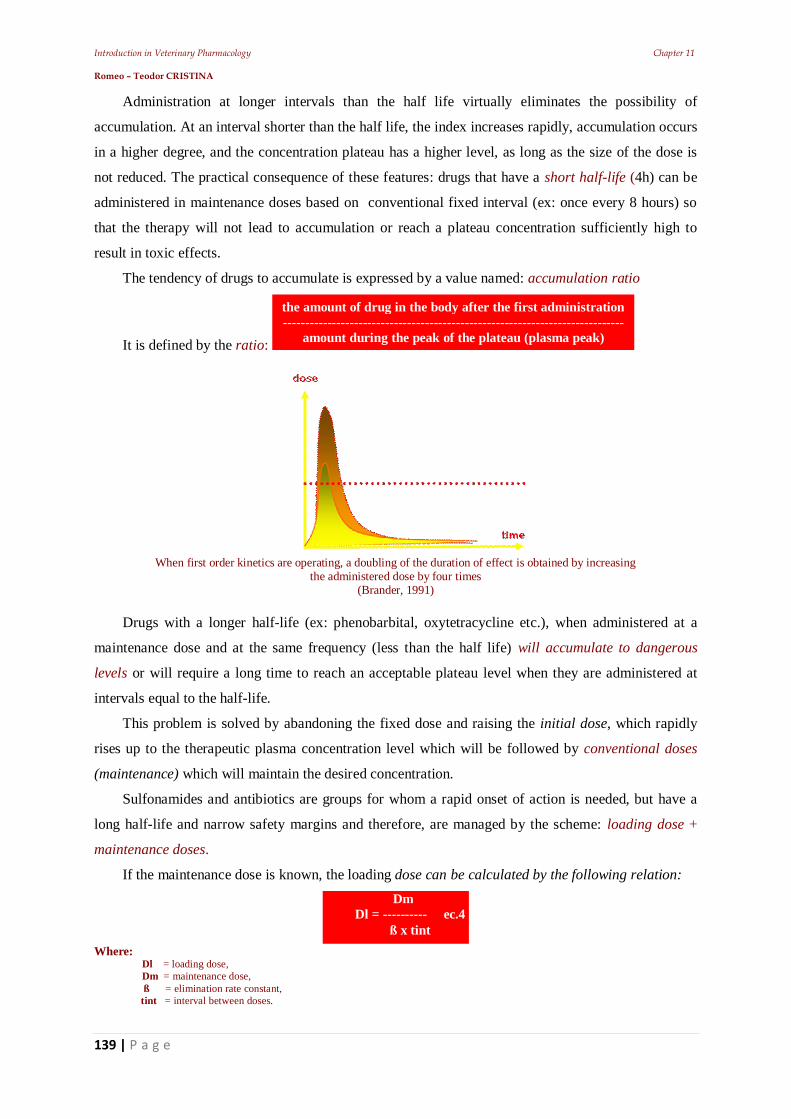
Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
139 | P a g e
Administration at longer intervals than the half life virtually eliminates the possibility of
accumulation. At an interval shorter than the half life, the index increases rapidly, accumulation occurs
in a higher degree, and the concentration plateau has a higher level, as long as the size of the dose is
not reduced. The practical consequence of these features: drugs that have a short half-life (4h) can be
administered in maintenance doses based on conventional fixed interval (ex: once every 8 hours) so
that the therapy will not lead to accumulation or reach a plateau concentration sufficiently high to
result in toxic effects.
The tendency of drugs to accumulate is expressed by a value named: accumulation ratio
It is defined by the ratio:
When first order kinetics are operating, a doubling of the duration of effect is obtained by increasing
the administered dose by four times (Brander, 1991)
Drugs with a longer half-life (ex: phenobarbital, oxytetracycline etc.), when administered at a
maintenance dose and at the same frequency (less than the half life) will accumulate to dangerous
levels or will require a long time to reach an acceptable plateau level when they are administered at
intervals equal to the half-life.
This problem is solved by abandoning the fixed dose and raising the initial dose, which rapidly
rises up to the therapeutic plasma concentration level which will be followed by conventional doses
(maintenance) which will maintain the desired concentration.
Sulfonamides and antibiotics are groups for whom a rapid onset of action is needed, but have a
long half-life and narrow safety margins and therefore, are managed by the scheme: loading dose +
maintenance doses.
If the maintenance dose is known, the loading dose can be calculated by the following relation:
Where:
Dl = loading dose, Dm = maintenance dose, ß = elimination rate constant, tint = interval between doses.
Dm Dl = ---------- ec.4
ß x tint
the amount of drug in the body after the first administration -----------------------------------------------------------------------------
amount during the peak of the plateau (plasma peak)

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
140 | P a g e
11.7. Stereo specificity of drug action The alleged action of a particular drug is based on the preferential binding of a substance to a
specific molecular reaction partner, namely to a receptor.
The special affinity of a pharmacon to its “own” receptor implies that it has a configuration that
fits very well and that there is some degree of complementation between the two partners.
A form of stereoisomerism is enantiomery. Is the isomerism in which the spatial structures of two
substances (enantiomers) are symmetrical to a plane = “mirror image” and their images are not
"congruent".
Enantiomery is based on the fact that in a molecule there is a carbon atom bearing four different
substituents
Stereoselectivity receptor occupancy. Only one of the two enantiomers (left) has features complementary to the site of receptor coupling.
(after Kuschinsky) Distances between a given atom and neighboring atoms are identical in enantiomers.
Enantiomers are comparable to one another in almost all chemical and physical properties.
They differ however, in their optical activity, because they rotate the polarized plans of a beam of
polarized light in different directions. The beam of polarized light will be rotated to:
right by the (+, dextrorotatory) form and
left by the (-, levorotatory) form.
Independent from the direction of rotation of polarized light, the characterization of both
enantiomers is possible by means of two classification systems.
The classification is done by dividing the substances in:
D-(dextrorotatory) and
L-(levorotatory) glycerin aldehyde, thus obtaining Series D and Series L.
Taking into account the location of substituents on the asymmetric carbon atom and their number,
it is possible to classify using the R-S system.
In the chemical synthesis of a substance with an asymmetric carbon atom, often the result is a
mixture (racemate), in which the enantiomers have the ratio of 1:1 and they do not produce the
rotation of plane polarized light.

Introduction in Veterinary Pharmacology Chapter 11 Romeo – Teodor CRISTINA
141 | P a g e
In nature, controlled enzymatic synthesis takes place stereo selectively, such that only one of the
enantiomers is synthesized: (-), D,R-adrenalin, (-),L,S- hyoscyamine).
When the asymmetric center of a molecule`s pharmacon is found in the area of its coupling with
the receptor, and three of the groups linked to the asymmetric carbon participate in the binding, then
only one of the enantiomers will present optimal comlementarity with the receptor.
11.7.1. Different spatial structure
Can influence the comlementarity of enzymes involved in the metabolism of the drug, so that the
metabolic transformation of the enantiomers will be carried by different routes, stereo selectively.
Example:
S (-), enantiomer of warfarin (oral anticoagulant) is the more active which will be decomposed in
the liver, at the level of the coumarinic cycle.
During this time the (+), R enantiomer will be changed especially at the level of the carbon atoms
chain. Therefore, the elimination of the form S occurs faster.
11.8. Zero-order kinetics influence
As presented before, describing the kinetics of absorption and elimination and their consequences
on the size of the dose and their frequency, it started from the idea that these processes apply first
order kinetics. One of its characteristics is that the half life for the influenced process (usually
elimination) is dose-dependent.
When zero-order kinetics is applied, a constant amount is circulated, rather than a constant
fraction per time unit.
It is also assumed that when the rate constant for a given drug in a particular individual stabilizes,
it will remain unchanged. Such a change during treatment would inevitably produce a significant
deviation of the initial plasma concentration, with unwanted consequences (ex. inadequate absorption,
excessive drug action or toxicity).
It is possible for a drug whose elimination is dependent on the transport processes through
carriers, to be maneuvered through first order kinetics until the carrier is saturated. In that moment the
kinetics become order zero.

Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
142 | P a g e
12. Other pharmacodynamic elements that can influence the drugs’ effect

Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
143 | P a g e
12.1. Drug residues
The existence of drug residues in milk or edible tissues of animals is a concern of public health
interest. To reduce the risk involved by residues in food of animal origin, the legislation requires for a
period called: the waiting period, period of prohibition or withdrawal.
Initially problems caused by persistence organochlorurates (O.C.) in the body fat stores and
undesirable effects on the farm animals initiated the domain. O.C. has a high partition coefficient, thus
large amounts are entering the body fat where it remains stable and can be released in time.
The persistence of chemical substances and the realization that the milk that contains antibiotics
(ex: betalactams) induces sensitization phenomena in humans, and focuses the attention on the toxic
potential residues. Pesticide residues in dairy products, antibiotics, growth promoters, hormones in
meat are detected frequently.
These are a safety concern for consumers regularly exposed to chemicals led to define a unit
called: ADI = acceptable daily intake
The value of this quantity for a human represents the daily intake of a substance through food
which, per unlimited period, cannot produce undesirable effects.
This depends on the known toxicity of the substance and is calculated by the relationship:
Where:
ADI = ineffective quantity (mg kg-1); NEL = “no-effect level” deducted from p.o. administration of the substance to rodents, per long-term; Factor 70 = derived from the average body weight of human (adult humans is considered to be 70 kg); 100 = arbitrary factor of safety (can increase up to 2000 in the case of carcinogens).
Residues may occur, not only because of the physicochemical properties of the substances
themselves but also because of the effectiveness of pharmaceutical and bio-engineering devices that
try to increase the half-life of the drug in question (bowls whit sequential or continuous removal,
implants etc).
12.2. The risk - benefit ratio
Before starting a treatment, the physician should choose the drug that will produce favorable
changes in the health status. Also, he must be sure that he knows how to properly use the drug to
obtain not only the type but also the level of response he wants to get.
He must know the disadvantages that might be involved in the therapy with the product in
question or the discontinuation of treatment. If the doctor has information, he can decide if the benefits
of using a drug, outweigh the disadvantages of administering or with-holding the drug.
NEL × 70 ADI = ----------------
100

Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
144 | P a g e
This is the risk – benefit ratio.
If the veterinarian decides in favor of therapy, he can also make a decision regarding the cost-
benefit ratio, in case of farm animals. Obviously, this decision involves an adequate knowledge of the
available information and the ability to consider these and other factors.
The term: hazard is used to describe the nature of any possible disadvantage produced by drug
use (ex: hypersensitivity to penicillin)
The term: risk is used to describe the likelihood of the hazard to occur in that case
Examples:
occasionally, phenylbutazone in horses causes death
diethylstilbestrol (DES) hormone, used as a growth promoter, has been reported to
induce cancer in humans and animals (as same as clenbuterol).
anticancerous substances can cause cancer and, in any case have numerous side
effects.
oxytetracycline produced fatal colitis in horses.
Estimation of the risk-benefit situation requires numerous data and, In European countries, the
recording of all adverse effects that may occur in animals began in 2000 or: Veterinary
pharmacovigilance
12.2.1. Dose-effect relationship
The investigation of dose – dependent response variation was one of the starting points of
pharmacotherapeutics. This fundamental relationship has made the development of quantitative
biodetermination possible and stimulated the proposals for the description of the quantitative
consequences of couplings between drug and receptor.
Two types of responses are known:
in the first case, the answer is an event whose frequency of occurrence in a population is dose-
dependent;
secondly, the intensity of response in an individual is dose-dependent.
The dependence of a drug effect, on the dosage or on its concentration, is a characteristic function
for each substance.
In drug testing, a positive result is, conventionally, the occurrence of a response of a
predetermined Intensity, while an increased dose will determine that a higher percentage of the treated
animals respond with the same intensity
This function is represented graphically by a dose - effect curve from which the following three
values can be extracted:
affinity,
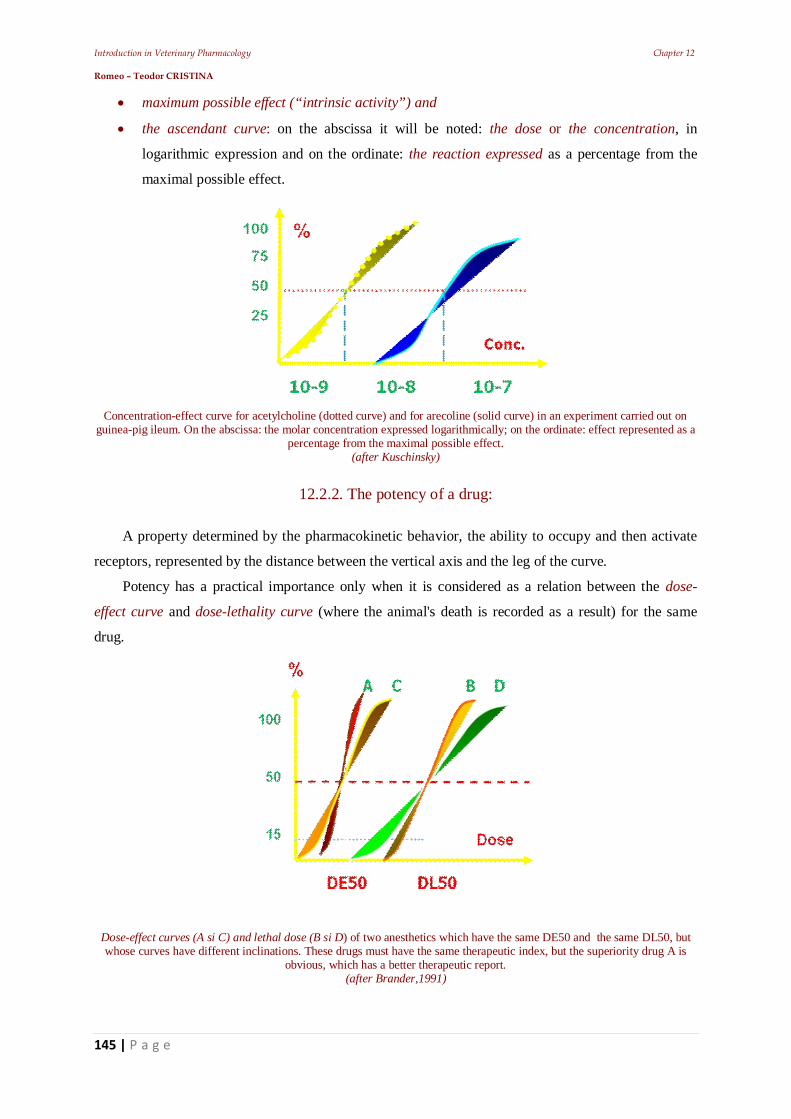
Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
145 | P a g e
maximum possible effect (“intrinsic activity”) and
the ascendant curve: on the abscissa it will be noted: the dose or the concentration, in
logarithmic expression and on the ordinate: the reaction expressed as a percentage from the
maximal possible effect.
Concentration-effect curve for acetylcholine (dotted curve) and for arecoline (solid curve) in an experiment carried out on
guinea-pig ileum. On the abscissa: the molar concentration expressed logarithmically; on the ordinate: effect represented as a percentage from the maximal possible effect.
(after Kuschinsky)
12.2.2. The potency of a drug: A property determined by the pharmacokinetic behavior, the ability to occupy and then activate
receptors, represented by the distance between the vertical axis and the leg of the curve.
Potency has a practical importance only when it is considered as a relation between the dose-
effect curve and dose-lethality curve (where the animal's death is recorded as a result) for the same
drug.
Dose-effect curves (A si C) and lethal dose (B si D) of two anesthetics which have the same DE50 and the same DL50, but whose curves have different inclinations. These drugs must have the same therapeutic index, but the superiority drug A is
obvious, which has a better therapeutic report. (after Brander,1991)
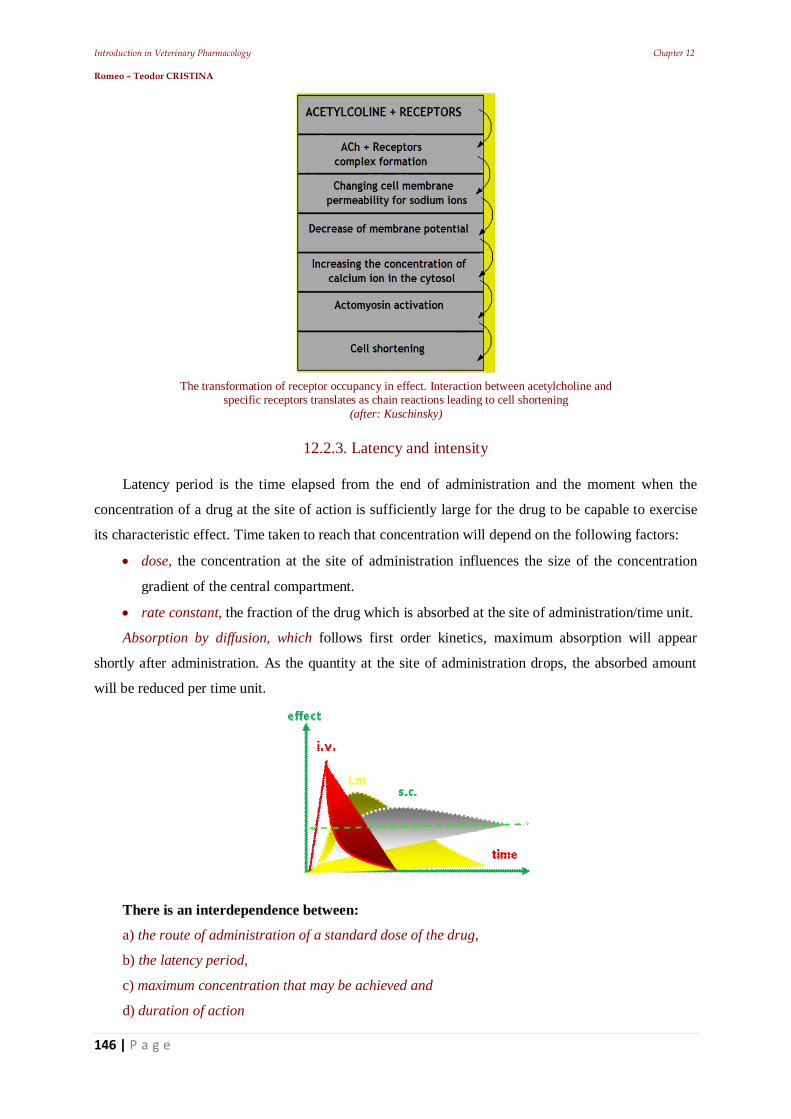
Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
146 | P a g e
The transformation of receptor occupancy in effect. Interaction between acetylcholine and specific receptors translates as chain reactions leading to cell shortening
(after: Kuschinsky)
12.2.3. Latency and intensity
Latency period is the time elapsed from the end of administration and the moment when the
concentration of a drug at the site of action is sufficiently large for the drug to be capable to exercise
its characteristic effect. Time taken to reach that concentration will depend on the following factors:
dose, the concentration at the site of administration influences the size of the concentration
gradient of the central compartment.
rate constant, the fraction of the drug which is absorbed at the site of administration/time unit.
Absorption by diffusion, which follows first order kinetics, maximum absorption will appear
shortly after administration. As the quantity at the site of administration drops, the absorbed amount
will be reduced per time unit.
There is an interdependence between:
a) the route of administration of a standard dose of the drug,
b) the latency period,
c) maximum concentration that may be achieved and
d) duration of action

Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
147 | P a g e
12.2.4. The duration of action of a pharmacon
After a unique iv administration, the entire dose will be present in the central compartment and,
as so, will be exposed to first order elimination process.
As the concentration in such a process is higher, the proportion removed per time unit, is greater
(concentration decreases exponentially and fast).
Longer duration for other injectable routes and lower maximum concentrations reflect the full
absorption period required. For both the s.c. as well as the i.m. path, the plasma peak coincides with
the period the rate of introduction of the drug into the central compartment coincides with the
elimination rate through processes of elimination.
Applying the elimination rate constant to the quantity of drug in the body, the eliminated fraction
per time unit can be calculated.
12.2.5. The duration of drug effect
It depends on the time required to decrease the plasma concentrations below the minimum active
value. This in turn, is determined by the distribution volume of the drug (the greater the volume, the
higher the time for elimination) and the relative contributions of the various mechanisms of the
removal process.
The expression of inconstancy, in relation to metabolism and excretion can be observed in the
variation of the plasma half-life, in a particular species and a particular drug.
Examples:
oxytetracycline has a 6h half-life (t1/2) in dogs and about 10h in horses, while
t 1/2 of chloramphenicol is 6 h in dogs, and only 1h in horses
This highlights the lack of safety when extrapolating doses from .one species to another based on
body weight.
Influence on the duration of drug action, mechanism of distribution & excretion
(after Brander, 1991)
Distribution volume Excretion mechanism Half-life
Plasma Glomerular + active tubular 3 minutes
SEC Glomerular + active tubular 15 minutes
Total water
Glomerular + active tubular 50 minutes
Glomerular 4 – 5 hours
Glomerular but with 99% resorption 25 days
SEC (High coupling affinity) Glomerular + active tubular 50 days

Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
148 | P a g e
12.2.6. The plasma concentration
This is frequently measured as an indicator of the useful length of life of a drug in the body and
one must not forget, that the forces that strive to alter this level, operate continuously.
These forces can be divided into:
those which tend to increase concentration (absorption, biotransformations for activation and
release of the coupled drug) respectively,
those that tend to decrease concentration (biotransformations for inactivation, storage in
tissues and excretion of drugs.
12.2.7. First-pass effect
This effect contributes almost in all cases, to the appearance of differences in the bioavailability
of an administered drug. Intestinal absorption (except the sublingual or rectal ones), before reaching
the systemic circulation, leads the drug through the portal circulation.
Therefore, it will be exposed to extraction from circulation and inactivation. For some drugs (e.g.
griseofulvin) even a single hepatic pass, can lead to extensive loss of substance.
Gastric acidity
destroys penicillin G,
proteolythyc enzymes attack the polypeptide drugs (e.g. insulin).
tetracyclines form chelates with the calcium in milk, and
penicillinase secreted by E. coli, reduces availability of penicillin quantity (e.g.
benzylpenicillin).
Intestinal mucosa
may inactivate some orally administered drugs in a substantial degree through :
hydrolysis (ex: glycerol trinitrate),
decarboxylation (ex: levodopamine), or
formation of sulfates (ex: isoprenaline).
12.2.8. Veterinary pharmacovigilance
Alerts about adverse reactions to drugs as a result of the treatment in animals urged the doctors to
classify the main causes of these shortcomings.
Trying a definition, veterinary pharmacovigilance is a operation of registration, monitoring and
systematic evaluation of side effects occurring as a result of inappropriate medication with some
veterinary drugs.

Introduction in Veterinary Pharmacology Chapter 12 Romeo – Teodor CRISTINA
149 | P a g e
Main reasons of unwanted installation are:
poor (clinical phase) experimentation for drugs advertised "aggressively" by drug firms
producing.
excessive prescribing of only a few groups of drugs (polypragmasia) to the same herd (e.g.
sulfonamides, chemotherapeutic agents, antibiotics, antiparasitic).
increased “aggressiveness” of the new syntheses;
biological peculiarities of each case;
interactions between drugs resulted from combinations.
Veterinary Pharmacovigilance has two main objectives:
the quickest possible detection of unexpected reactions (in animals);
determining the frequency and incidence of all already known or recently found adverse
reactions, that are produced by veterinary drugs.

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
150 | P a g e
13. The animal body's response to medication – Main pharmaceutical aspects

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
151 | P a g e
Introduction
In the study of drugs, a lot of tests are performed in vitro.
One of the most important is the dissolution rate, because it determines the rate of absorption.
These are corroborated using in vivo experiments.
Tracking a drug concentration in the blood or urine one can determine:
dissolution constant;
absorption constant;
biological half-life;
elimination constant;
other parameters (using equations or graphical methods). Generally, biological availability increases in direct proportion to the rate of dissolution.
Each type of pharmaceutical formulation has its own behavior (ex. Oral formulations raise their
biological availability from dragees aqueous solutions).
In the hygroscopicity testing: bioequivalence and the clinical results of the treatment, are taken
into consideration.
In what concerning the doses used:
Median effective dose (ED50) represents the dose inducing positive effect for 50% of the
individuals of a population.
Ideal dose (ID99) represents the dose inducing a positive effect for 99% of the individuals of
a population.
Lethal dose (LD50) represents the dose inducing lethal effect for 50% of the individuals of a
population.
The unjustified increase of the doses cause the increase of side effects, toxicity and even
mortality, determining at the same time the decrease of the safety margins between ED and LD50.
The higher the difference between ED and LD, the safer the product will be the ratio between the
dose that produces side effects and the dose that has a therapeutic effect, is called therapeutic index
(TI) or safety limit and indicates the selectivity of a drug.
A medication can have multiple therapeutical indexes if it has more side effects or more
therapeutic effects. The correct dosage is found by determining the safety limit and the relationship
between the maximum therapeutic dose and the minimum dose that induces toxicity.
Dose that produces side effects T. I. = -------------------------------------------- Dose that has therapeutic effect
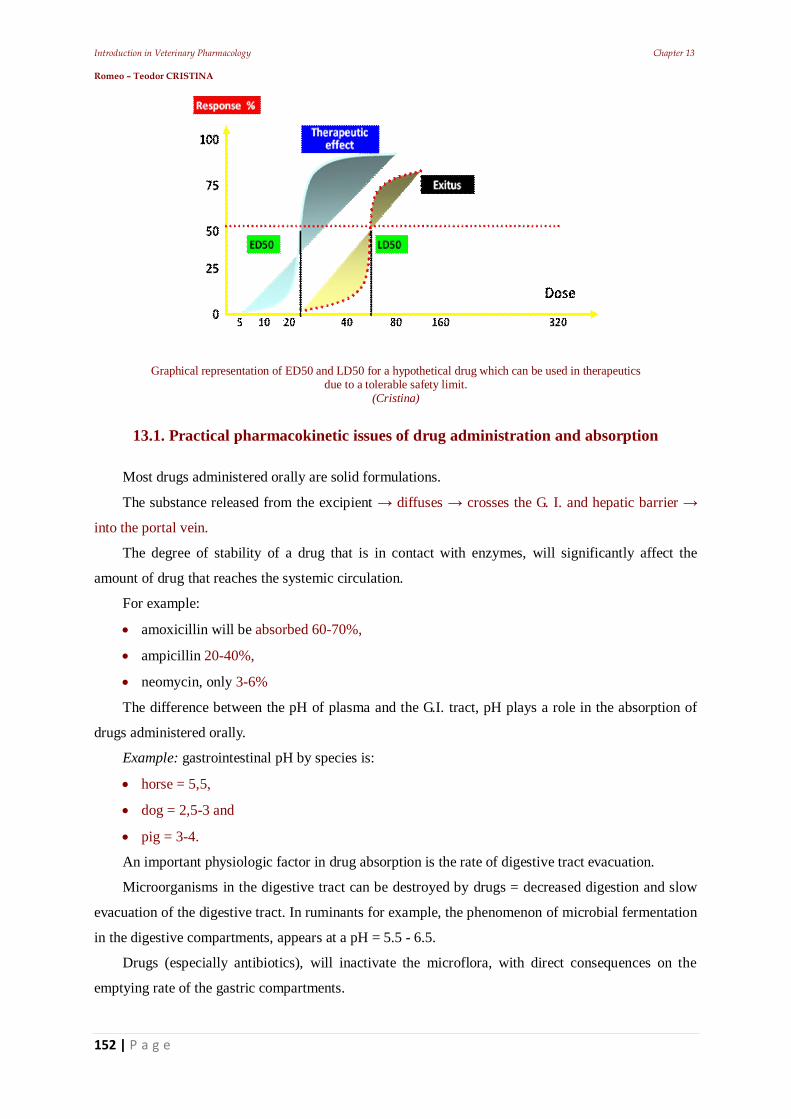
Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
152 | P a g e
Graphical representation of ED50 and LD50 for a hypothetical drug which can be used in therapeutics due to a tolerable safety limit.
(Cristina)
13.1. Practical pharmacokinetic issues of drug administration and absorption
Most drugs administered orally are solid formulations.
The substance released from the excipient diffuses crosses the G. I. and hepatic barrier
into the portal vein.
The degree of stability of a drug that is in contact with enzymes, will significantly affect the
amount of drug that reaches the systemic circulation.
For example:
amoxicillin will be absorbed 60-70%,
ampicillin 20-40%,
neomycin, only 3-6%
The difference between the pH of plasma and the G.I. tract, pH plays a role in the absorption of
drugs administered orally.
Example: gastrointestinal pH by species is:
horse = 5,5,
dog = 2,5-3 and
pig = 3-4.
An important physiologic factor in drug absorption is the rate of digestive tract evacuation.
Microorganisms in the digestive tract can be destroyed by drugs = decreased digestion and slow
evacuation of the digestive tract. In ruminants for example, the phenomenon of microbial fermentation
in the digestive compartments, appears at a pH = 5.5 - 6.5.
Drugs (especially antibiotics), will inactivate the microflora, with direct consequences on the
emptying rate of the gastric compartments.

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
153 | P a g e
Quantifying the absorption is based on the plasma concentration (Pc) of the drug, and the
graphical representation is shown in the figure below.
A. The rate of absorption - is the time necessary for one half (T1/2) of the administered dose to be absorbed and found in
the systemic circulation.
B. The level of absorption (bioavailability) - represents the extent in which a drug administered in a particular dose enters
the systemic circulation unchanged (as active form).
For the same drug bioavailability varies from individual to individual or from one species to
another. For exemple, in dogs oeally administeres drugs have different BD:
digoxin cpr. - 1mg/animal = 80% systemic BD
propanolol cpr. 80mg/animal = 2-7% systemic BD
lidocaine solution 10mg/kgbw = 15% systemic BD
13.1.1. Bioavailability of a.u.v. drugs
The amount of influence that a formulation has on the action of active substances is of
fundamental importance.
In the last period, an increasing importance is given to the notion of: biological bioavailability.
Biological bioavailability plays a part in knowing the changes those drugs are submitted to in the
body, and is expressed as a percentage following the relation:
where: S1=blood levels obtained for the test preparation S2=the blood concentration of a reference preparation
S1 % availability = ----- S2
AUC oral B.D. = --------------- AUC IV

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
154 | P a g e
A pharmaceutical formulation should have a specific, predictable speed of action depending on
the variability of individual response
13.1.2. Polymorphism:
The property of a substance to crystallize in different ways. Polymorphs have the same chemical
properties, but different physical properties.
Crystalline polymorphism
Is the ability of a substance to crystallize in two, or more crystalline forms with a different spatial
arrangement. Amorphous substances usually have small particles and can be found either in
anhydrous form, or with crystalline water molecules.
For example:
The anhydrous substances of caffeine and theophylline have a higher dissolution rate than the
crystalline forms. Therefore: the degree of solubility, toughness, configuration, dimensions, optical
and electrical properties etc. are different from one polymorph to another, altering the biological
availability.
Hydrocortisone acetate may exist under five crystalline forms, each with its own biological
availability;
Chloramphenicol, under its forms palmitate and stearate, exists in three polymorphic forms: two
crystalline and one amorphous, the more active being the second crystalline form, otherwise known as
the most common of the chloramphenicol esters (Leucu a, 1975).
Other active substances (ex. acetyl salicylic acid, barbiturates, corticoids, sulfathiazole, steroid
derivatives etc.) may be encountered under two or more crystalline forms, each with their own
different speed of dissolution, therefore different absorbtion.
Designation of polymorphs
Is made with roman numbers (the digit I corresponding to the most stable form) or in capital
letters (in the order in which they were discovered).
Thermodynamically unstable forms are called metastable.
Metastable polymorphs are preferred, because the use of forms with high energy and solubility,
constitutes an advantage.
For example, novobiocin (amorphous, acid form) cannot be processed in suspension, because in
less than six months at room temperature, it turns into crystallized form.
13.1.3. Particle size
For some substances with low solubility, particle size determines gastrointestinal absorption,
therefore, therapeutic efficacy is directly proportional to the logarithm of the particle surface area
(Leucu a, 1975).

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
155 | P a g e
For example:
Particles of griseofulvin, sulfur, cortisone (prednisolone, medroxyprogesterone), sulfadiazine,
barbiturates (finely pulverized). Sometimes the use of particles with different sizes is required, in order
to achieve optimal activity (reduced side effects, eliminate intolerance, maximum effect).
Particle size directly influences the activity of drugs that are administered parenteral as
suspensions (ex. penicillins, testosterone esters).
Particle sizes in these cases are between 1 and 20mm and generally are micronized powders
composed of very small particles (mono-particles).
It has been found that the tolerability of a drug is closely related to the particles size.
For example:
Aspirin administered in monoparticles causes less gastric bleeding than in the form of large
particles.
Nitrofurantoin in the form of small crystals easily causes side effects (vertigo, nausea), compared
to when used in the form of larger particles (more difficult to absorb).
That is why, in this case, the optimum particle size (to avoid having a too rapid dissolution) is 50-
100 mm.
In order for some digestive antiseptics and anthelmintics to have a complete action, they should
not have a rapid dissolution.
That is why the most effective particles have a size of 100 mm (Leucu a, 1975).
Determining the particle size
Both the aspects of the pharmaceutical technique and the pharmacological implications must be
taken into account.
It is important in the case of some pharmaceutical forms (ex. powders, tablets, dragees,
suspensions, suppositories, ointments) because it helps to establish measurement methods and
permissible limits for particles for each drug substance.
13.2. Bioequivalence of a.u.v. drugs
Two drugs are bioequivalent when they produce the same therapeutic effect and have the same
rate and extent of absorption.
To determine the bioequivalence of drugs a drug is compared with another, or with a set of
predefined quality requirements, including:
chemical bioequivalence
The amount of active substance in two or more preparations;
clinical bioequivalence

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
156 | P a g e
Two preparations containing the same active substance give identical results, measured through a
pharmacological response or appraised by controlling a symptom or a disease
therapeutic bioequivalence
structurally different drugs, give the same clinical outcome;
Bioequivalence
Two preparations containing the same substance in equal amounts, allow the active substance to
enter the general circulation at the same relative speed, achieving the same blood concentration
The following, are of biopharmaceutical interest:
selection of chemical status (acid, base, salt, ether, ester etc.);
choice of physical state (amorphous, crystalline, polymorph, hydrated, the particle size);
choice of pharmaceutical form (liquid, soft, solid);
choice of adjuvants (solvents, diluents, thickening agents, disintegrants, antioxidants,
antiseptics, etc.);
choice of a suitable pharmaceutical technique.
The purity of the active substance
The tolerated impurities will vary depending on the: substance type, origin and destination.
Therefore: organic substances of synthetic origin are considered satisfactory and compatible with
therapeutic use, if impurities do not exceed 0.5-1%, because the variability of responses in the body is
sufficiently broad to compensate for small percentages of impurities.
The condition is valid when the impurities are biologically inert.
Some weak bases (alkaloids) or some acids (barbiturates), will be converted into salts, through
the so-called salification with inert ions, in order to turn into products with a higher solubility.
So, a specific form must be selected carefully depending on the induced effect.
Therefore:
phenobarbital administered orally, will be absorbed more slowly than its sodium salt (observed
in almost all barbiturates);
polymyxin sulfate is more toxic than the metasulfonate, because of the gastric acidity that is
produced (fine precipitate appears, well dissolved which allows for a faster absorption)
(Leucu a, 1995).
In many cases, significant differences were found between the effects of the salified form,
compared to the non-salified form.
The pharmaceutical form
The use of substances as such is extremely rare.
A drug compound is also formed from supporting materials, in addition to the active substance
(solvents, emulsifiers, antioxidants, correctors, preservatives, dyes, thickeners, etc)

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
157 | P a g e
By definition, they should be inactive, biologically inert and should not produce any
physicochemical changes, harmful to the active substances.
This is only theoretical, supporting substances can have a role in modifying drug absorption
(either increasing or decreasing the speed of absorption), additionally claiming knowledge of the
pharmacological effects of adjuvant.
For example:
excipients of dermatological preparations, can influence the degree of i.d. penetration of the
active substance
surfactants (complexed of hydrophilic solvents, PEG), used to realize a suitable
pharmaceutical form from the solubilized systems of drugs (poorly soluble before) and thus
favoring absorption.
These substances (often used as solubilizers and emulsifiers), generally act in three ways:
1. moistening in low concentrations, surfactants lower the surface tension. The effect occurs
within the biophysical system (solid / liquid) consisting of the drug found in the GI fluids and
supports the dissolution of poorly soluble substances. Also certain surfactants improve
absorption of drugs administered as: eye drops, erins, suppositories or ointments.
2. complexation. In high concentrations, surfactants form complexes between drugs with
lipophilic character. This phenomenon has been reported in the case of associating antiseptic
surfactants.
3. influence on physiological processes
Certain surfactants can affect physiological processes, of which the most important are:
changing cell permeability,
inhibition of gastric activity or
Delaying stomach evacuation.
Work technique
It influences the biological availability of a product. It has been found that different batches of the
same drug different characteristics, this happens due to some insignificant and usually hard to spot
changes of the working conditions.
For example: tablets and dragees because of these "small" changes not disintegrate in time,
missing the drug release.
When it comes to instability one can speak of a chemical degradation of the active substance,
accompanied by reduced activity by a change of the drug, that hinder the release of the active principle
or, cause destructive change in the characteristics of the drug.
A task of the formulation and of the work technique is the realization of preparations which
maintain their quality for a long time.

Introduction in Veterinary Pharmacology Chapter 13 Romeo – Teodor CRISTINA
158 | P a g e
This is not an easy to solve problem, because the studied factors (temperature, light, humidity, air,
microorganisms) cause unpredictable interactions.
Because of bioavailability (BA), manufacturers will select the most appropriate form of
administration, not only from a medical point of view (route of administration and dosage), but also
taking biological activity into account according to the individual parameters: age, sex, disease, route
of administration etc.
There are differences between monogastrics and polygastric animals.
For example: salicylates administered orally in gelatin capsules to different species in doses of:
18.5; 50 and respectively 133mg / kg.bw, in: dogs, pigs, ponies and goats indicated that BA is:
very high in: dogs and pigs, while is
very low in: goats and especially in ponies.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
159 | P a g e
14. Practical elements of veterinary therapeutics

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
160 | P a g e
Introduction
The choice of a rational therapy is not possible without an accurate diagnosis. In this context, a
rational therapy must take into account:
drug formulation,
dosage scheme,
the safety limit (IT),
counter-indications,
side effects,
drug interactions, but also
the associations of good nature.
14.1. Drug formulation kinds
protected label: belonging to pharmaceutical companies which hold the license of
manufacturing and can not be copied by other companies;
generic name: drugs with an expired patent that can also be produced by other companies;
used off-label (Extra label): following a doctor's prescription, a drug can be used outside the
manufacturer's indications;
for human use, which can be used a.u.v., on the responsibility of the veterinarian and only for
species whose products and by-products are not for human consumption;
unregistered, used on the risk of the clinician and owner (drugs in clinical testing).
prohibited, which are prohibited to be used in certain species or for all.
In veterinary practice, a situation where two or more drug substances must be administered to a
single animal, either separately or associated in the same pharmaceutical compound is fairly common.
The higher is the number of concomitantly administered substances, the higher is the risk of
incompatibility. In the case of interactions and incompatibilities, the effect they have on the animal
may be manifested through:
mutual annihilation (total or partial) or decrease of the effect of the associated substances and
treatment failure;
negative effects on the animal manifested by deterioration or worsening of the general
condition, and sometimes death of the animal by the appearance of toxic products.
Interactions and incompatibilities may occur before the drugs enter into the organism and they are
due to physicochemical phenomena, such as:
precipitation,

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
161 | P a g e
hydrolysis,
liquefaction,
effervescence,
oxidation etc.
These interactions are common with i.m. injectable solutions, or in the case of intravenous
infusion preparations. In this context, we must recall some essential terms: affinity, efficacy, potency.
14.1.1. Drug combinations
Drug combinations influence the pharmacodynamic effects through interactions.
The drugs may be associated in a single preparation or administered separately.
Through associations we seek to:
enhance the action of a drug,
extend the action of a drug,
reduce / cancel a joint action
The pharmacodynamic interactions can be:
Synergistic, when the drugs act in the same way (ex. lincomycin + spectinomycin
mycoplasma).
Antagonistic, when the drugs act in an opposite way (ex. effects of histamine / antihistamine).
Synergistic interactions can be classified into two categories:
Addition: the effects of two or more drugs are summed (ex. anti-inflammatory analgesics +
antipyretic).
Potentiation: the global effect is is greater than the sum of partial effects. Example:
lincomycin Gram positive and Mycoplasma; spectinomycin Gram-negative, and
Mycoplasma; lincomycin + spectinomycin Gram positive, Gram negative, and
Mycoplasma
Antagonistic interactions can be:
Partial when the the global effect is smaller than the partial effects. Example: non-steroidal
anti-inflammatory + diuretics; heparin + aspirin etc.
Total, when the the global effect is null. Example: atropine + pilocarpine.
14.1.2. Drug interactions
Drug interactions are changes in the nature and intensity of the therapeutic response to a drug
compared to another drug administered at a separate time or simultaneously, in an animal.
The result of the interaction = either increase or decrease of active substance. or a specific
metabolite, at the site of the biologic activity, called: pharmacokinetic interactions or those installed by
other mechanisms = pharmacodynamic interactions.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
162 | P a g e
“Physiological type” incompatibilities are the result of proper or improper association of active
substances used therapeutically (or diverted from the desired effect).
Generally, this type of incompatibilities is the result of the veterinarian that causes them,
inadvertently or by ignorance. (from here the word iatropathy: iathros = therapist, pathos = distress).
Pharmacodynamic incompatibilities are connected to the pharmacodynamic action produced by
the drug. They are divided into those that occur in the:
• outside of the body (therapeutic technique)
• inside the body :
- changes in pharmacokinetic parameters determine the presence of another medicinal product;
- alterations of pharmacodynamics, are due to the presence of another medicinal product.
14.3. Pharmacokinetic interactions
The pharmacokinetics of a drug can be modified at all levels: absorption, distribution,
metabolism, excretion, with implications for the therapeutic efficacy (influencing the availability of
the substance at the site of action).
Associated drugs may alter the pharmacokinetic behavior of each other in the processes of
absorption, distribution, metabolism and excretion.
Changes can lead to:
- decrease of therapeutic efficacy or
- occurrence of side effects, because the changes affect substances’ availability at the action site
Pharmacokinetic interactions which affect the availability at the site of action, are generally
driven by a decrease in the rate of absorption or by a decrease in the rate of metabolism or excretion
Increased bioavailability in the biophase may be determined by pharmacokinetic interactions like:
- protein coupling rate change,
- blocking the metabolism or
- blocking the renal excretion.
14.3.1. Interactions on absorption phase
The mechanisms by which these processes become viable are:
• the drug movement from the plasma protein binding sites;
• enzymatic induction / inhibition (change of biotransformation).
Resulting = modification of half-life(t1/2).
Gastrointestinal absorption of a drug may be accelerated by the intervention of the second drug

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
163 | P a g e
Examples: phenacetin, paracetamol, vitamin B12 will be absorbed more rapidly in the presence of
sorbitol, epinephrine administered i.m. in combination with a local anesthetic will enhance its
absorption by a local vasoconstrictor effect.
Influence of the digestive tract on absorption
In the intestine, metal and molecules salts (ex: Ca+2, Mg+2, Al+3, Fe+3) produce, in the
presence of other drugs, poorly soluble combinations thereby, poorly absorptive (ex: powder
tetracycline administered in milk or feeding the young simultaneously with milk; this drawback can be
removed by distancing administrations of milk to 2-3 h after drug administration).
Tetracycline administration simultaneously with preparations based on aluminum hydroxide (ex.
antacids, digestive bandages) determines the net decrease of the plasma level of tetracyclines (even
90%), due to the formation of chelates.
Administration of anions with increased affinity for acid molecules may influence the absorption,
especially of: fat, thyroid hormones, cardiac glycosides, iron salts, warfarin, phenylbutazone, vitamins
A, D, E, K, etc.).
Anticholinergics and opiates delay gastric emptying significantly, slowing the absorption of other
p.o. drugs. The alteration of the peristalsis, may delay the dissolution of some tablets.
Lingering in the intestine, can cause incorrect absorption of some drugs.
Examples:
Metoclopramide (stimulator of gastric emptying), increases the absorption of paracetamol but
decreases the gastric concentration of digoxin.
Neostigmine (anticholinesterase drug) alters the rate of absorption of many drugs it is associated
with, due to the stimulation of the intestinal peristalsis.
Barbiturates (e.g. phenobarbital) decrease the antifungal effect of griseofulvin.
Saline purgatives decrease absorption by diluting intestinal contents and accelerating the transit.
The intestinal flora modifies the activity of several active substances, due to the biochemical
changes that take place.
Antibiotics may act directly on the intestinal flora and thus indirectly, modify the plasma
concentrations of other drugs.
Examples:
Increasing the effect of anticoagulants: by destroying the flora producing vitamin K, in
concomitant administrations of chloramphenicol, neomycin, oral sulfonamides and penicillin
derivatives.
Concomitant administration of broad-spectrum antibiotics can cause enteritis in some cases (and
therefore, due to reduced absorption, decrease the effect of anticoagulants).

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
164 | P a g e
Enzymes can also be inhibited by drugs: folic acid absorption occurs in the presence of glutamate
(originating from food) resulting from hydrolysis.
Diphenylhydantoin or nitrofurantoine may prevent this hydrolysis, resulting in the decrease of
folic acid absorption rates.
Associations with adsorbents (ex: activated charcoal, kaolin, bentonites, powdered liquoric etc.)
or mucilages can determine physicochemical interactions, which will delay the absorption due to the
retention of active principles on their molecules and their gradual disposal
Examples:
kaolin reduces the absorption of lincomycin;
cationic antacids decrease absorption of tetracyclines,
tensioactives increase the poorly soluble drug toxicity, by solubilization.
In the case of parenteral administration, therapeutically useful interactions are possible.
Subcutaneous medications will be absorbed more quickly in association with vasodilator drugs
or by the addition of substances that increase the permeability of connective tissue (ex: hyaluronidase).
The delay of absorption rate in drugs that are administered s.c. or i.m. is achieved by combining
them with heavy absorbable macromolecular substances (ex: PVP) or with vasoconstrictors (ex:
adrenaline, procaine).
Some drug interactions may decrease the permeability of connective tissue.
Example: estrogens can increase the content of hyaluronic acid in the fundamental substance and
thus decrease the rate of absorption of associated drugs.
Influence of pH
Digestive and intestinal juices may influence absorption in a unpredictable manner through pH.
There are several mechanisms by which the absorption is affected:
changes in the pH of gastric juice and intestinal = alkalizing substances, decrease the
absorption of weak acid drugs.
formation of complexes: tetracyclines form reversible or irreversible chelate complexes, with
metal ions (especially: calcium, aluminum, iron; furosemide forms a non-resorbable complex
with the magnesium hydroxide.
Changing gastrointestinal motility
There are drugs that accelerate G.I. transit decreasing absorption (metoclopramide, sodium
bicarbonate) or delaying intestinal transit (atropine, morphine analgesics, aluminum hydroxide and
magnesium).
Competition for the same transport system: substances which are transported by the same
system of membrane active transport, tend to compete for the binding sites on the transporting plasma

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
165 | P a g e
proteins.Through the saturation of these sites, a mutual reduction of absorption of the attached
substances may occur (ex. phenytoin prevents absorption of folic acid).
Intestinal metabolization influencing. The intestinal enzymes can metabolize drugs, as is the case
for chlorpromazine, which is metabolized 50% at this level.
Impairment of intestinal blood flow: there are substances with high fat solubility that quickly
cross the intestinal barrier. In this case, the blood flow at the intestinal level limits the factor of
absorption. When combined with a product that decreases or increases intestinal blood flow,
absorption may decrease or increase.
Toxic effects on the digestive tract:the drugs or the terms of their administration may lead to toxic
phenomena or malabsorption. Ex. Neomycin p.o. for long term = atrophies the intestinal villi =
malabsorption.
14.3.2. Interactions on the distribution phase
Therapeutic plasma concentration will depend on:
the size of the dose
interval between administrations
dosage form
administration route
systemic distribution
rate & degree of absorption
degree of plasma protein binding
rate of elimination If a drug is highly bound to plasma proteins, then its displacement by an associated drug
substance, capable to displace it competitively, will lead to a large increase of the free concentration of
the first drug substance. The result: effect potentiation, because of the fact that, unbound free fraction
is the one that has pharmacokinetic action (because it accesses the pharmaco-receptors).
Phenylbutazone and sulphaphenasole moves tolbutamide from the plasma proteins, inhibiting its
metabolism (leading to hypoglycemia).
Valproic acid lowers the plasma level of phenytoin by moving it from the protein and speeding
up the metabolism of the free fraction.
For drugs to be involved in interactions that displace substances from proteins (in such a way that
the movement is clinically significant) they must possess, in addition to:
a high protein binding,
a reduced apparent volume of distribution (Vd) and
a low therapeutic index.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
166 | P a g e
Distribution ratio in different organs and tissues (Synthesis, Cristina)
Organ / tissue Horse Dog Goat Cow Human
Blood 8,60 - 4,70 7,80 -
Brain 0,21 0,51 0,29 0,06 2,00
Heart 0,66 0,82 0,48 0,37 0,47
Lung 0,89 0,89 0,88 0,71 1,40
Liver 1,30 2,32 0,95 1,22 2,60
Spleen 1,11 0,26 0,25 0,16 0,26
Kidneys 0,36 0,61 0,35 0,24 0,44
Intestine 5,80 3,90 6,40 3,80 1,70
Intestinal content 12,30 0,72 13,90 18,40 1,40
Skin 7,45 9,30 9,20 8,30 3,70
Muscle 40,10 54,50 45,50 38,50 40,00
Bones 14,60 8,70 6,30 12,70 14,00
Tendons 1,71 - - - 2,00
Adipose tissue 5,10 - - 18,09 18,10
Average weight 380,0 16,0 39,0 620,0 70,0
The affinity for plasma proteins can be modified by binding another drug.
Aspirin, besides the fact that it is reversibly bound to serum albumin, it acetylates it as well.
Acetylated albumin will present a high affinity for phenylbutazone (and a somewhat lower
affinity). In this way it is possible that the effect of such an interaction manifests long after the
substance that produced it (in this case aspirin) is eliminated. Reversible fixation is the condition for
drug action, irreversible binding causing particularly toxic action.
Changes in blood flow. At hepatic levels, affect the bioavailability of some medicinal substances
metabolised by liver (noradrenaline administered i.v. decreases hepatic lidocaine’s clearance).
Displacement from plasma proteins. Competition becomes critical when the binding sites on the
protein macromolecules become almost saturated.
Examples:
• phenylbutazone oral anticoagulants;
• Non-steroidal anti-inflammatory drugs antidiabetic sulfonamides;
• antibacterial sulfonamides oral anticoagulants.
Binding to plasma proteins plays a role in limiting the distribution and influencing drug
elimination from the body. The link is reversible, constituting a reservoir of active substance (when
releasing the specific receptors).
Examples: ceftiofur is metabolized in desfluorilceftiofur having a high binding affinity to plasma
proteins causing the increase of T1/2 from 1 hour to 10 hours.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
167 | P a g e
• hypoalbuminemia (nephrotic syndrome) = increasing the amount of free plasmatic drug (more
than 80% for phenilbutazone, furosemide, ceftiofur).
14.3.3. Interactions on metabolization phase
Interactions by enzyme induction
Enzyme induction causes:
increased clearance and
decreases plasma concentrations of drugs.
The consequence of enzymatic induction is manifested by the reduction or cancellation of all
therapeutic effects.
Interactions by enzymatic inhibition
Some drugs can inhibit the metabolizing enzymes: chloramphenicol, phenylbutazone, estrogens,
imidazoles, some sulfonamides and therefore reducing hepatic clearance and increasing the
concentration of substances that would have been metabolized under the action of those enzymes.
These drugs can also influence the metabolism of endogenous substances (ex: bilirubin, steroid
hormones etc.).
Drugs that stimulates biotransformation of other drugs or substances (after Safta, 1984)
Inductor Medicament(e) sau substan a(e) a(ale) c ror metabolism este
stimulatFenobarbital ialte barbiturice
Deriva ii ac. salicilic, fenazona, aminofenazona, fenilbutazona,barbiturice, fenitoina, clorpromazina, dismetilimipramina,anticoagulante de sintez (indirecte), digitoxina, digoxina,testosteron, androsteron, estradiol, progesteron, anticoncep ionaleorale, hidrocortizon, dexametazona, tiroxina, chinina,cloramfenicol, doxicilina, griseofulvina, ciclofosfamida, bilirubina
Fenitoina Fenazona, corticosteroizi, hormoni sexuali, tiroxina,anticoagulante indirecte, digitoxina, doxicilina, vitamina D
Fenilbutazona Corticoizi, hormoni sexuali, aminofenazona, digitoxina, digoxinaFenazona Anticoagulante indirecte, hormoni steroiziFenotiazinele Anticoagulante indirecte, benziprenHaloperidol Anticoagulante indirecte, benziprenMeprobamat Meprobamat, anticoagulante indirecteDiazepam DiazepamClorciclizina Hormoni steroiziPrometazina FenilbutazonaPrednison Fenilbutazona, ciclofosfamidaTolbutamida FenilbutazonaSpironolactona Fenazona, anticoagulante indirecte, hexobarbital, cortisolRifampicina Rifampicina, fenazona, tolbutamida, hexobarbital, metadona,
digitoxina, anticoagulante de sintez , hormoni steroiziGriseofulvina Anticoagulante de sintezDDT Corticosteroizi, hormoni sexuali, tiroxina
Enzymatic inductors also can stimulate microsomal oxygenase systems:
NADPH-cytochrome P-450 reductase,

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
168 | P a g e
cytochrome P-450,
UDP-glucuronyltransferase and
glucose-6-phosphate dehydrogenase.
Nonspecific inducers can stimulate their own metabolism or the metabolism of other chemical
compounds. From the category of nonspecific inducers it is worth mentioning: phenobarbital,
glutethimide, meprobamate, hexobarbital, pentobarbital, carbamazepine, phenytoin, ethanol,
chlorpromazine, tricyclic antidepressants, phenylbutazone, aminophenazone chlorcyclizine,
tolbutamide, probenecid, halothane, rifampin, DDT etc.
Inhibition of drug metabolism
This will lead to extension and exaggeration of the pharmacodynamic effect and/or to the
occurrence of adverse effects. The inhibition of enzymes involved in the biotransformation may affect
the inhibition of the activity or their synthesis.
The interactions affect hepatic microsomal enzymes, with the possibility of interaction with other
enzyme systems. Blocking enzymatic synthesis = less common as induction. Drug interactions often
determine inhibition by competition, compared to the same enzyme system.
Interaction through the inhibition of biotransformation of drugs and clinical consequences in animals (after Safta, 1984)
Medicament cumetabolismul
inhibat
Inhibitor Consecin e
Fenitoina Anticoagulante indirecte, PAS,Cicloserin , Fenilbutazona,Clorpromazina, Diazepam,Halotan, Carbamazepin, Estrogeni,Sulfafenazona
Tulbur ri neurologiceHiperplazie gingival
Bishidroxicumarina Cloramfenicol, Feniramidol,Fenilbutazona, Oxifenilbutazona,Clorpropamida, Chinidina, Etanol,Steroizi anabolizan i
Accidente hemoragice
Tolbutamida Cloramfenicol, FenilbutazonaProbenecid, Salicila iParacetamol, SulfafenazolAnticoagulante indirecte
Hipoglicemie
Clorpropamida Cloramfenicol, Dicumarol HipoglicemieHexobarbital Ac. aminosalicilic, Testosteron,
Progesteron, HidrocortizonPrelungirea ac iunii
Fenobarbital Fenitoina Diverse reac ii adversePromazina Dietilstibestrol
ProgesteronPrelungirea efectului
Perfenazina Nortriptilina Prelungirea efectuluiFenazona Fenilbitazona
Ac. nalidixicPrelungirea timpuluide înjum ire
FenilbutazonaOxifenilbutazona
Steroizi anabolizan i Prelungirea efectuluiReac ii adverse
Etilmorfina Estradiol Prelungirea efectuluiCiclofosfamida Cloramfenicol
PrednisonDiminuarea efectuluiReac ii adverse

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
169 | P a g e
14.3.4. Interactions on urinary excretion phase
The excretion of the drug is linked to the: glomerular filtration, tubular reabsorption and tubular
secretion. Many pharmacons are eliminated through several mechanisms.
Free drugs, unfixed on plasma protein: are eliminated through glomerular filtration. Ultra
filtration can be followed by tubular resorption.
Unionized, liposoluble forms of drugs can be reabsorbed by the membrane of renal tubules. An
important factor in directing resorption is the pH of the tubular urine:
• in acid urine, weak acid drugs are under a unionized liposoluble (HX) form, which gets
diffused easily from the renal tubule towards the plasma, having a low-clearance;
• in alkaline urine, they are under an ionized, non-diffusible form (X- + H+), which is excreted
in urine. In the case of weak alkaline drugs, the situation is reversed.
Urine pH may vary, with values of 4.5-8.0 its changes considerably influences the tubular
resorption and, consecutively, the renal excretion of drugs. In the case of drugs that are weak acids
(pKa 3-7) (ex: anticoagulants, nalidixic acid, barbiturates, indomethacin, phenylbutazone, salicylates,
streptomycin, sulfonamides, penicillin etc.) excretion will decrease if the urine is acidic and will
increase if the urine becomes alkaline.
Drugs that are weak bases (pKa 7,5-10,5) (ex: caffeine, antihistamines, antipyrin, nicotine,
pethidin, procaine, theophylline, etc.) show an increased urinary excretion in the case of acid urine. In
alkaline urine, their elimination decreases (Safta, 1984). From the drugs which may lead to change in
the urinary pH, it is worth mentioning: sodium bicarbonate, which alkalinizes urine, as well as
ascorbic acid and ammonium chloride, which produces the acidification of urine. Urine alkalinization
is necessary when administering sulfonamides that acetylated (ex: sulphathiazole, sulphadiazine etc.)
to avoid intra-tubular crystallization. In urine with alkaline pH, the solubility of sulphonamides in
increased
Drug interactions at the level of the active transport systems in the renal tubules (after Safta, 1984)
Medicament Interac ioneaz cu:Sulfamide Penicilina G, TolbutamidaProbenecid Penicilina G, Ampicilina
Oxacilina, CarbenicilinaCefalotina, IndometacinaClorotiazina
Salicila i Probenecid, FenilbutazonaSulfinpirazona, PenicilinaMetotrexat
Fenilbutazona Penicilina G, TolbutamidaAcetohexamida
Oxifenilbutazona Penicilina GAminofenazona PenicilinaDeriva i cumarinici ClorpropamidaSulfinpirazona Salicila i

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
170 | P a g e
Influence of pH on absorbtion, distribution and elimination of certain drugs (after Safta, 1984)
Procesul Medicamente
acizi slabiMedicamentebaze slabe
Absorb ia gastric relativ rapid relativ lentAbsorb ia în intestinul sub ire relativ lent relativ rapidRaportul concentra ie plasmatic /concentra ie intracelular
Ridicat sc zut
Clearance renal în:- urina acid- urina alcalin
- redus- ridicat
- ridicat- redus
14.4. Interactions of pharmacodynamic order
A pharmacodynamic effect (sin. pharmacological action) is the amount of the body’s responses
reflected functionally, after the administration of drugs.
Body – drug interaction = the amplification or reduction of some specific functions of the
organism with a (generally) reversible character, that does not result in the creation of new
physiological functions. The occurrence of a “visible” effect = linked to the existence of a minimum
dose of the drug.
Main effect
Is the most visible response occurs after the administration of a drug (ex: narcosis after
administration of a narcotic, healing a bacteriosis after a treatment with an antibiotic etc.).
Secondary effect
Is a less intense response or responses that can accompany a main effect (ex: the sedative effect,
even hypnotic of antihistamines). Generally the secondary effects are unwanted in the practice of
veterinary therapy, being usually harmful (adverse reactions)
Another classification refers to the functional changes which a drug produces in the body:
Stimulating effect
when a drug increases the functional status of an organ, apparatus or system :
directly, through an excitatory stimuli directed towards them
indirectly, by blocking or reducing an inhibitory function
Example:
adrenaline stimulates the beta-adrenergic cardiac receptors = tachycardia, change that can be
achieved also by inhibiting M-cholinergic cardiac receptors by atropine).
digitalin can stimulate the myocardium (stimulating activity) but at the same time, it may
decrease the conductivity of Hiss bundles (therefore having an inhibitory activity).
Depressing effect
Produced by drugs which have the ability to reduce the operational state of an organ, apparatus or
system by actual inhibition or by excitation of an inhibitory function:

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
171 | P a g e
Example: the paralysis of adrenergic catecholamine endings (tachycardia) is achieved directly
through the administration of guanethidine. The physiological result = bradycardia. In the same time
acetylcholine can achieve the same effect indirectly through the excitation of M-cholinergic cardiac
receptors (bradycardic role).
Some drugs can cause depressant effects which can abolish (paralyze) the targeted function.
This medication is called on, when the exaggeration of some functions is found during some
diseases (ex: CNS depressant substances, in the case of an abnormal excitation, antispasmodic
substances, anti-diarrheal, astringents etc.).
Related to this classification, physiological changes can be expressed through:
Direct effect
The active substance acts on the target directly (ex: bulbar center excitation by CO2 or cortical
nerve center excitation by caffeine);
Elective effect
When drugs act in a particular manner, selectively, only on an organ or a function (ex: digitalis
acts selectively on the heart and its function). Actually, a few drugs possess this feature, they influence
other organs or systems of the body too (ex: same digitalis = effects on CNS and kidney too),
therefore, one cannot speak of an actual selective effect. Also the expression: preponderant or
dominant effect is more accurate.
Indirect effect
Is characterized by the induction of the same changes (as in the case of direct action), but in a
different manner. Caffeine, can cause vasoconstriction by direct action, but may also cause “indirect
vasoconstriction” by stimulating the bulbar vasomotor center. So caffeine can reduce diuresis both
through direct action (on the kidneys), and indirectly (acting on the cardiovascular system).
A classification of the effects may relate the location:
Local effect
Is identifiable at the administration site (considered that is not reaching the vascular bed). The
best known "producer" of local effects = topical medications.
This classification is purely theoretical, because topical medications can achieve more than local
effects, interacting with adjacent tissues.
General effect
It is produced when the drug enters into the general circulation and then, because of the
distribution will determine effects in all tissues and organs.
Examples:
strychnine, triggers general effects, through excitation of CNS,
adrenaline, through its hypertensive action,
papaverine through its antispasmodic action.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
172 | P a g e
Drugs with a generic action can act locally: ex. local irritant activity of chloralhydrate, (a
narcotic).
Depending on specific activity, antibacterial, antipathogenic or symptomatic, the effects can be:
Symptomatic effect
It is produced consecutively after the intervention of a drug on the symptoms of a disease. The
result = increases the body’s resistance, even if the cause is not addressed.
Example:
caffeine prevents collapse increases low blood pressure,
opiates suppress acute pain which can install shock.
also symptomatic (indirect) effects immunostimulants (e.g. tissular extracts, polidin,
iodisept, vaccines etc.)
It is known that one drug can relate with multiple types of receptors, and stimulation = different
effects, sometimes opposing (ex: adrenaline produces vasodilatation in small doses by binding to beta
receptors and vasoconstriction in therapeutic doses by binding to alpha receptors).
Etiotrope effect
It refers to the action of drugs on the pathogens (ex: chemotherapeutics, antibiotics, antiparasitic).
In veterinary medicine etiotrope therapy is considered to be the most rational.
Ant pathogenic effect
Is the result of a the action of a drug on the installed pathogenic mechanisms of a disease.
Therefore, it relates to the other causes of a disease, different from the etiologic agents (ex:
hypovitaminosis, hipomineralosis, histaminemia etc.) that are controlled through specific medication
(vitamins, oligominerals, calcium, antihistamines etc.).
14.5. Synergistic combinations
Are the associations of “good nature” = drug synergism.
Advantages:
detectable therapeutic effect at low doses
reduce the incidence of adverse effects
obtaining efficient conditionings
Disadvantages:
potentiating effect with CNS depressant activity
Drug synergism (sin = together, ergon = action) is of direct practical importance because,
through “thought associations”. The same therapeutic effect can be obtained, but with much smaller
doses, thus reducing the risk of adverse reactions. The synergism can be direct and it is also called

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
173 | P a g e
summation (or addition). It determines the final effects (E) which can be at most, equal to the algebraic
sum of the partial effects of the two associated drugs, A and B (E < A+B).
14.5.1. Direct synergism
Is a characteristic of active substances, related as mode of action who exercise their mechanism
on the same “targets”. For this synergism (summation or addition) associated drugs do not affect each
other, the rate of fixation on the receptors being equal.
Expanding the spectrum of activity association:
antibiotics: penicillin + streptomycin, ampicillin + cloxacillin, gentamicin + vancomycin or
antiparasitics: rafoxanide + thiabendazole, oxiclozamide + tetramisole, albendazole +
levamisole etc.
In the case they do not have the same mechanism of action and have different morphological
“targets”, one speaks of indirect synergism (e.g. pilocarpine and saline purgatives).
14.5.2. Drug potentiation
Is also a drug synergism but which follows with a final intensified effect, superior of the sum of
partial effects caused by the associated drugs A i B (E > A+B).
Potentiation is a super addition of the effects of some drugs from different therapeutic classes that
may have similar effects (ex: association of sulphonamides + potentializators leads to increase x 5-10
times of the antibacterial activity,
Trifadoxin = Sulphadoxine + Trimethoprim in a ratio of has an activity x 8-10 times stronger
than each component.
Sulfaveridin = Sulfaquinoxaline + Etoxidiaveridine = amplified anticoccidian effect;
Magnesium sulfate potentiates the hypnotic action of chloral hydrate,
Penicillins are potentiated by some sulfonamides etc.).
Potentiation may also follow the intensification of the effects of a component of the association
through another component of the association (that doesn't have the same effect)
Examples:
Neuroleptics potentiate narcotics:
Droperidol enhances fentanyl analgesia (in NLA).
Through potentiation, the usual doses of a drug may be reduced, the effect of this combination is
the same or broader
Example:
Therapeutic association between atropine and papaverine produce the same effect as the
individual therapeutic doses, even at a 50% of the avarege therapeutic dose for atropine and 33% of
the avarege therapeutic dose for papaverine).

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
174 | P a g e
Potentiating associations are the most useful in veterinary medicine, clinical synergism is often
used in the cases of veterinary therapy.
In conclusion, potentiating associations can be the result of these mechanisms :
inhibiting the inactivation of some drugs
example: potentiate the activity of acetylcholine and choline esters by anticholinesterases;
Antagonize the biosynthesis of a key component of the metabolism
example: sulfonamides and potentializators inhibit the microbial biosynthesis of
tetrahydrofolic acid;
sensitization of substrates subsequent action of some drugs
example: chlorpromazine causes changes in the CNS that will sensitize the neurons to the
activity of some CNS inhibitors.
14.6. Attenuation associations
They are not very frequent in V.M. being only used in the case of drugs with “too drastic”
actions. Example: Irritant and purgative activity of croton oil diminished by sunflower oil (or castor
oil), known to have a more "gentle" activity.
14.7. Indifferent associations
It is made between drugs that do not influence each other. These associations are frequent in the
case of the magisterial or standardized preparations.
14.8. Antagonistic associations
Antagonism is represented by the contrary, opposite activity, of two or more drugs, which cancels
partially or totally the pharmacodynamic action. The situation requires the presence of an agonist
(influencing the pharmacodynamic effect) and an antagonist (which influences the effect of the
agonist, for the purpose of reducing or annihilating). Antagonistic interactions may occur in coupling
on the cellular receptors or in different enzymatic processes
Example:
Sulfonamides are competitively antagonized by PABA or by substances with a similar
structure which derive from this compound: procaine, anestesine;
Concomitant administration of a bacteriostatic and a bactericide is = drug antagonism.
Clinical drug antagonism can be classified as:
Direct
Two or more substances having an opposite action, but on the same morphological objective

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
175 | P a g e
Example:
The circular muscles of the iris can be paralyzed by atropine, causing mydriasis, while ezerine
acts on the same muscles resulting in its stimulation, causing miosis.
Indirect
Substances with an opposite activity acting on different morphological objectives
Example:
Pilocarpine contracts the pupil by stimulating the circular muscle of the iris, while adrenaline
dilates the pupil acting on the radial muscle of the iris.
Depending on the intensity of the pharmacodynamic action the known antagonisms are:
Unilateral
When one of the antagonist active substances has a more intense pharmacodynamic activity. It is
the most common situation in veterinary therapeutics (depressor substances usually have a stronger
activity than the stimulants have);
Bilateral
When the activities of antagonistic substances have the same intensity. These two types of
antagonism are also called:
physiologic or
pharmacodynamic.
Antagonism, is also considered the :
physical and
chemical
Forms that occur from a direct physical or chemical agonist-antagonist type of reaction.
Example:
EDTA, the ability to fix heavy metals,
Acid + alkali in states of acidosis of the digestive tract,
Albumins in contact with salts of heavy metals,
Tannins in presence of alkaloids etc.
14.8.1. Biological antagonism
It can be subdivided in:
1. Competitive antagonism
2. Non-competitive antagonism
3. Functional antagonism
4. Physiological antagonism

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
176 | P a g e
Competitive antagonism
It occurs when the agonist and antagonist act on the same effector receptor.
Competitive antagonism can be:
specific,
reversible and
mutual.
Non-competitive antagonism
The antagonist will not be attached on the same receptor like the agonist or will act on different
areas of the receptor.
Example:
Excessive administration of corticosteroids leads to a major depression of adrenocortical
hypothalamic - hipophysal depression = corticotrophin depletion.
Essential in assessing the receptor - drug relationship:
affinity and
intrinsic activity (the "attraction" between receptor and drug as well as the ability of the
stimulated receptor to determine the pharmacodynamic effects).
Non-competitive antagonism may lead to:
inhibition of stimulants.
drugs with a powerful nonspecific, blocking effect, inhibit the action of stimulant pharmacons,
for certain effects
Example:
papaverine antagonizes serotonin,
histamine or acetylcholine contracted smooth muscle relaxation (myotropic mechanism)
not at the level of specific receptors;
the convulsing effect of strychnine can be counteract by blocking motor nerves with local
anesthetics or by blocking neuromuscular junctions with currarisants).
Allosteric inhibition
Are changes taking place around the receptor (after which, its spatial conformation will change).
In this situation, the agonist will no longer act, or will no longer be able to produce only a partial
interaction with the receptors. This inhibition takes place at an enzymatic level

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
177 | P a g e
Covalent attachment
Is a type of non-competitive antagonism, attachment by strong (covalent) bonds to the
pharmacological receptors. These processes are irreversible (ex: anticholinesterases).
Functional antagonism
In this case, the agonist and the antagonist act on different receptors of the same organ.
It is about the activity as an agonist, on different receptors, in the opposite sense
Example:
interaction between bacteriostatic - bactericide groups: the first group prevents bacterial
multiplication and the second one interferes and prevents the bacterial growth phase,
histamine contracts the bronchial smooth muscles, while isoprenaline will relax (ß-adrenergic
mechanism) the same muscles, acting on the same type of receptor (histaminergic).
Physiological antagonism
It is different from the functional type, because the agonists and antagonists are acting on
different tissues.
Example:
Increased cardiac output can be counteracted by hypotensive substances, reducing peripheral
resistance.
To remove the toxic side effects of drugs (by overdose), the identification of the type of
antagonism is very important, the antagonism may evolve and cause various types of incompatibilities
(pharmacodynamic, of administration etc.).
14.10. Pharmacodynamic ambivalence
Two substances can be both synergistic and antagonistic:
Papaverine is:
synergistic with morphine when it comes to analgesia,
antagonistic on the digestive sphincters that morphine contracts, and papaverine relaxes;
Narcotine and morphine are:
synergistic as an activity on pain,
antagonistic regarding the respiratory center, narcotine stimulates, and morphine depresses it.
The body as a "whole" receives "information" from the outside and inside, the administration of
drugs being a “stimulus” adjusted by feedback. When drugs "depress" physiological systems, the
dependent morphological substrates will adapt, in the first phase by enhancing the functionality. In the

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
178 | P a g e
case of the suppression of administration of the respective drug, the compensation mechanisms remain
present.
14.11. Undesirable reactions to medications
Occurs due to failure to comply with the general principles of therapeutics:
inappropriate therapies;
not analyzing the risk-benefit relationship;
not monitoring therapeutic response;
not analyzing the impact + effect of the disease/ pharmacodynamics and pharmacokinetics of
drugs.
Undesirable effects may be:
predictable or
unpredictable.
Occur mainly in: young, old, obese, emaciated, pregnant animals, or with hepatic disease /
nephropathy.
Classification of adverse reactions:
Low (limited) efficacy;
Secondary effects
Extension of the pharmacodynamic properties,
Allergic reactions
They cannot be anticipated and are not related to dose size and they are manifested by:
skin reactions,
hemolytic anemia,
anaphylactic reactions etc.
14.11.1 Adverse reactions
Undesirable or even dangerous effects, triggered by an inappropriate dosage, often are
accompanied by immune reaction. The frequency of AR is reduced compared to the one encountered
in human medicine. In animals, it is estimated that the frequency of adverse reactions is: 5 to 8%.
The most common adverse reactions are:
toxic;
idiosyncratic;
allergic;
mutagenic-teratogenic;
carcinogenic.
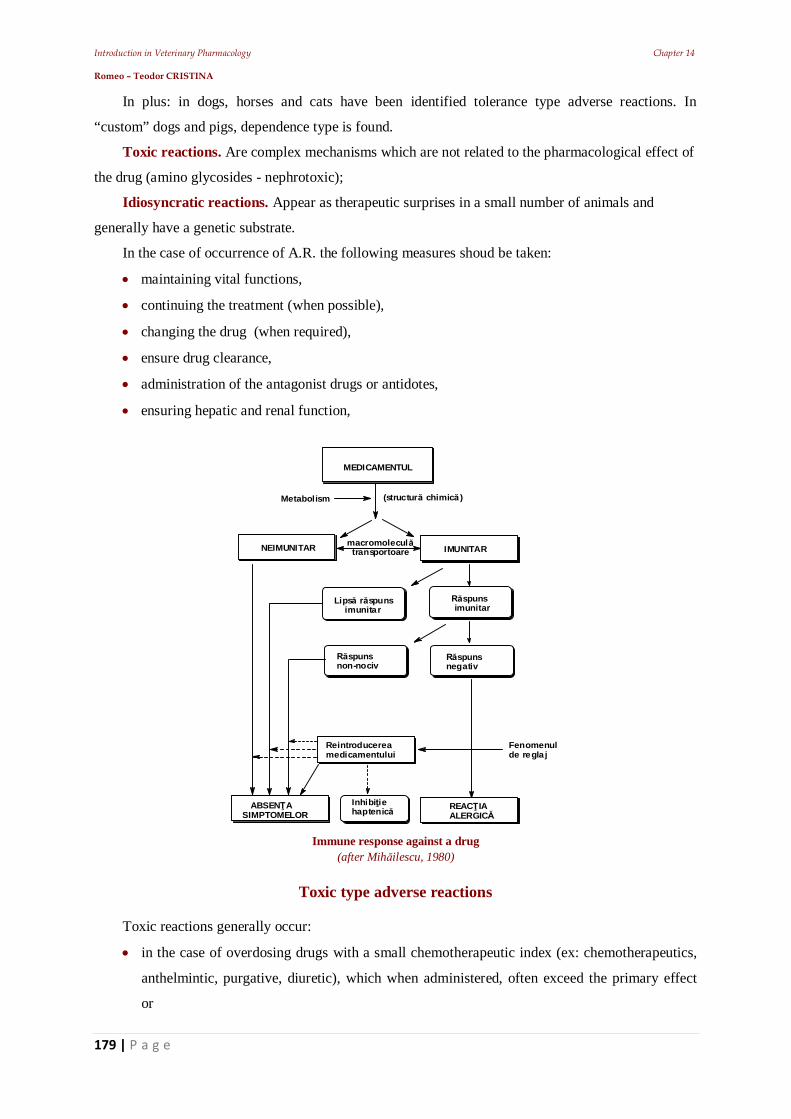
Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
179 | P a g e
In plus: in dogs, horses and cats have been identified tolerance type adverse reactions. In
“custom” dogs and pigs, dependence type is found.
Toxic reactions. Are complex mechanisms which are not related to the pharmacological effect of
the drug (amino glycosides - nephrotoxic);
Idiosyncratic reactions. Appear as therapeutic surprises in a small number of animals and
generally have a genetic substrate.
In the case of occurrence of A.R. the following measures shoud be taken:
maintaining vital functions,
continuing the treatment (when possible),
changing the drug (when required),
ensure drug clearance,
administration of the antagonist drugs or antidotes,
ensuring hepatic and renal function,
MEDICAMENTUL
(structur chimic )Metabolism
NEIMUNITAR IMUNITARmacromolecul transportoare
Lips r spuns imunitar
spuns imunitar
spunsnegativ
spunsnon-nociv
ABSEN ASIMPTOMELOR
Reintroducereamedicamentului
Fenomenulde regla j
REAC IAALERGIC
Inhibi iehaptenic
Immune response against a drug (after Mih ilescu, 1980)
Toxic type adverse reactions
Toxic reactions generally occur:
in the case of overdosing drugs with a small chemotherapeutic index (ex: chemotherapeutics,
anthelmintic, purgative, diuretic), which when administered, often exceed the primary effect
or

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
180 | P a g e
may also be determined by phenomena that are not derived from the primary effect (ex:
auditory and vestibular organ damage by the aminoglycosidic antibiotics) products that once
appeared on the market.
Example:
Florosil in pigs is not allowed to be taken in water or moist feed, due to the increased toxicity
of fluorosil in water (1:25); santonin in pigs’ parasites etc.).
Neguvon in p.o. Administration in nematodosis in horses
An example of a toxic reaction = Herxheimer reaction = effect of endotoxins released by
microorganisms, which die by the action of antibiotics. Generally bacterial metabolites are
considered to be primary toxins, but these also lead to allergic sensitization. The Herxheimer
reaction cannot be produced in the absence of bacterial infection and should not lead to the
abandonment of the treatment with antibiotics.
If the likelihood of such reactions is anticipated, therapy should be started (contrary to usage)
with low doses.
Neuro-toxic effects
Antibiotics can often be neurotoxic.
Example:
beta-lactams and polymyxins = hyperexcitation and convulsions.
Neurotoxic effects are also at these levels:
acustico-vestibular, as a result of damaging the eighth pair of cranial nervs, the vestibulo-
cochlear, produced by streptomycin, kanamycin and neomycin, or
vestibular, by: gentamicin, streptomycin and kanamycin.
polymyxins and chloramphenicols, affects the optic nerve and thus decreasing visual acuity.
arsanilic acid = neurotoxic effects on the optic nerve.
high doses of barbiturates (barbital, ciclobarbital, phenobarbital, pentobarbital, Inactin,
Medinal etc.) = strong depressing activity on the CNS
Antibiotics can often be neurotoxic.
Example:
betalactams and polimyxyns = hyperexcitation and convulsions.
Neurotoxic effects are also found at these levels:
acustico-vestibular, as a result of damaging the eighth pair of cranial nerves, the vestibule-
cochlear, produced by streptomycin, kanamycin and neomycin, or
vestibular, by: gentamicin, streptomycin and kanamycin.
polymyxin and chloramphenicol, affect the optic nerve and thus decreasing visual acuity.
arsanilic acid = neurotoxic effects on the optic nerve.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
181 | P a g e
high doses of barbiturates (barbital, ciclobarbital, phenobarbital, pentobarbital, Inactin,
Medinal etc.) = strong depressing activity on the CNS.
Butyrophenones (haloperidol, droperidol, azaperone) generates:
more intense action that phenothiazines regarding: hyperthermia and extrapyramidal syndrome
and
enhance barbiturates, narcotics and benzodiazepines.
Diphenylmethane tranquilizers (hydroxyzine, benactyzine):
depress the respiratory centers in the CNS.
m-colinolytic action (cardiovascular and digestive):
enhance the activity of barbiturates and opiates.
The effects of hydroxyzine are amplified by phenothiazines.
Carbamate tranquilizers (ex. meprobamate):
increase muscle relaxant activity and depress the CNS.
depress the respiratory centers and cause the paralysis of respiratory muscles,
hypnotics are enhanced.
Benzodiazepines (Diazepam, Chlordiazepoxide):
are CNS depressants, and affect vision.
enhance the action of CNS depressants, serious evolutions
Amphetamines are powerful CNS stimulants prolonged use of high doses = they cause
exhaustion after the end of the initial stimulation phase depression of vital centers, due to lower
functioning of the nerve cells. Also:
enhance the activity of tricyclic antidepressants.
induce pronounced adrenomimetic hypoactivity (x 100 times lower, compared to adrenaline).
Purinic cortical stimulants (ex. caffeine, theobromine, theophylline):
high doses affect CNS = excitation
Medullar and bulbar stimulants (ex. strychnine):
excite medullar reflectivity lowering the excitability threshold = exaggerated responses
(ex: seizures).
block intercalary neurons in the control of responses.
at the level of the bulb depression of vital centers,
at the peripheral nerves currarisants type of action (decrease of cronaxia).
Haemato-toxicity
Antibiotics:
in high doses = medullo-toxic mechanism
in low doses= hematotoxics

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
182 | P a g e
Example: Chloramphenicol in high doses gives:
depression of the spinal cord
inhibition of hematopoiesis.
Hematotoxic disorders are recognized by:
anemia (even aplastic) to: chloramphenicol [chloramphenicol is already banned to veterinary
use in farm animals in RO], streptomycin,
leukopenia to chloramphenicol and novobiocin,
granulocytopenia and agranulocytosis to chloramphenicol and ristocetin
thrombocytopenia to rifampicin and novobiocin).
haemolysis may be caused by novobiocin and rifampicin.
Sulphonamides generates haemolyzant and methemoglobinizant, hematologic disorders =
medullar depression. Frequent haematological disorders are:
leukopenia,
granulocytopenia
thrombocytopenia, manifested clinically by severe anemia.
Nitrofurans and furazolidone are:
hematotoxics (thrombocytopenia and agranulocytosis).
generating haemorrhagic diathesis consecutive to using furazolidone in aviculture.
Benzodiazepines (eg. Diazepam, Chlordiazepoxide) can give:
eosinophilia,
anemia and
thrombocytopenia.
High doses of salicylic acid in dogs generating:
anemia by suppressing erythropoiesis,
hypotrombinemia and disorders of blood coagulation.
Following the destruction of the gastric mucosa = ulceration and hemorrhages.
Indirectly, histamine release = increase of acidity + local vasodilatation.
Aspirin = coagulability disorders in newborns.
Phenacetin (paraaminophenolic derivative) = methemoglobinemia (aniline metabolites) S-
hemoglobin = hemolytic anemia.
In enzyme deficient subjects (glucose-6-phosphate dehydrogenase):
methemoglobinemia, massive hemolysis, even exitus.
Paracetamol = phenacetin, but the methemoglobinisant action is 2-3 times lower.
Phenylbutazone în excess = haemorrhages, reactivation of stomach ulcers, haematological
disorders.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
183 | P a g e
Dermatotoxic effects
Sulphonamides after an extensive use (1-2 weeks):
intense pruritus,
erythema,
exfoliative dermatitis,
angioneurotic edema (especially petechiael and purpuric) which,
can give serious anaphylactic episodes.
Meprobamate can cause, in increased doses: allergic manifestations.
Morphine causes eruptions, urticaria, pruritus.
Aspirin causes: hypersensitivity and skin reactions, edema etc.
Aminophenazone and phenylbutazone produce, in increased doses: cutaneous allergic
manifestations.
Hepatotoxicity
Antibiotics act hepatotoxic through the mechanisms of:
cytolysis (e.g. oxytetracycline, chlortetracycline)
steatosis (eg tetracyclines) or
cholestatic (streptomycin, rifampin, tetracycline).
Arsanilic acid, a chemotherapeutic used in many enteropathy in pigs and poultry produces
massive hepatotoxic effects even at slightly increased therapeutic doses (300-400 mg/kg fodder).
Chemotherapeutics in increased doses, cause:
enteritis, paresis and even paralysis in pigs.
Tranquilizing carbamates (ex. Meprobamate):
may affect morpho-functionally in the hepatic tissue.
Diazepam (benzodiazepines) produces hepatic disorders, initially weaker but which increase in
the case of repeated treatments with high doses.
Salicylic acid is dangerous to administrate to: cats, a species in which it causes toxic hepatitis and
gastric lesions (eg: doses of 30 mg / kgbw, several days = in over 50% of individuals, toxic hepatitis).
Phenacetine and paracetamol, in increased doses and used for an extensive time period:
- produces hepatic necrosis (after a serious evolution).
Aminophenazone and phenylbutazone can become: hepatotoxic and cause biliary stasis,
cholestatic icter.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
184 | P a g e
Toxic respiratory effects
Hypnotics, in increased doses, may induce: hypoventilation progressing to apnea. Changes in
respiratory physiological parameters can trigger: bronchitis and bronchopneumonia. In newborns
respiratory disorders are always serious.
Phenothiazine can: depress the respiratory nervous centrals and induce trachea bronchial
paralysis. Narcotic analgesics: depress the respiratory centers from the physiological stimulant (CO2)
and produce bronchial muscle spasm, often followed by exitus.
Nephrotoxicity
Approximately 40% of antibiotics (e.g. oligosaccharides and polypeptides) can generate
nephrotoxicity. They achieve a renal concentration, 10-50 times higher than the usual blood
concentration, and thus affect glomerular ultra filtration and tubular resorption. More frequent clinical
manifestations are:
albuminuria and
cylindruria.
The highest nephro-toxicity from antibiotics is caused by: kanamycin, neomycin, bacitracin,
gentamicin and polymyxin. Renal disorders caused by sulphonamides are recognized by severe colic
consecutive nephron damage (tubular lesions)
Paraclinical, the nephrotoxic effects produced by sulphonamides can be identified by: hematuria,
crystalluria and albuminuria.
Barbiturate derivatives induce: renal failure, resulting in dehydration and shock, death through
respiratory arrest (in 1-3 days after administrations). When evolutions are more longer = pulmonary
complications. In subjects with renal disorders (manifested hormonally) = urinary retention.
Morphine and opium: decrease in diuresis by stimulating the release of antidiuretic hormone.
Salicylic acid derivatives cause: fluid and electrolyte imbalance + metabolic acidosis (and the
appearance of ketonic bodies by disrupting the cycle of carboxylic acids) and production of functional
renal failure.
Phenacetin generates: nephrotoxic effects in long treatments: interstitial nephritis, papillary
necrosis, nephritis (irritation produced by metabolites in the glomerule).
Abacterian nephritis by associating bacterial factor can give bacterial nephritis.
Cardio-circulatory toxicity
Barbiturates can induce: tachycardia, hypotension even (in serious forms) tension failure and
shock (consecutive to hypoxia).
In general anesthesia, besides hypotension and thrombophlebitis will generate extension of
hypnotic action.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
185 | P a g e
m-colinolytics and alfa-adrenolytics in excess tachycardia and finally, hypotension.
Also acute respiratory failure can occur (consecutive to hypoxia due to the inhibition of
mitochondrial oxido-reductions.
Diphenylmethane tranquilizers in association with dicoumarinic anticoagulants = bleeding,
hypotension and tachycardia.
Carbamate tranquilizers can become cardio-vascular toxics: through arterial hypotension.
Antidotism
The sum of the measures used to annihilate toxins which enter the body, as well as their effects.
Substances used in combating the toxic effects = antidotes.
These can be: a single substance or a mixture, their action is based on the incompatibilities that
are produced in the reaction with the toxic substances. Depending on their specificity to the toxics,
antidotes can be:
general – with a wide range of action, used when the substance that caused this intoxication is
not known precisely;
specific - well established for each toxic substance and which are used whenever the nature of
the poisoning is known.
Idiosyncratic adverse reactions
Idiosyncrasy (idios = own; sincrazis = interference, mixture, blend,) refers to congenital
intolerance, characterized by qualitatively changed responses following the administration of a
particular drug.
Variations of intolerance are the expression of biological dissipation, idiosyncrasy was
mistakenly compared with drug allergy but, in the case of idiosyncrasy, one does not have to deal with
antigen-antibody type of allergic reactions but with ones with some degenerative somatic properties:
In this way, a reaction to a drug can be obtained, even from its first administration and not after a
sensitization (allergy). The symptoms of idiosyncratic intoxication are different from the allergy type
ones. This incompatibility is due to some genetic particularities, most often due to enzyme
deficiencies. These may cause the metabolic degradation of the drugs.
Examples:
Nitrofurantoin engages haemolytic anemia (glucose-6-phosphate deficiency in red blood
cells).
Succinylcholine can give apnea, animals with "atypical" plasma cholinesterase.
The best known example for the production of idiosyncratic effects is in: youngsters (premature,
newborns) toxic effects, even fatal, due to ontogeny absence of some enzymes when administering
sulfonamides (hepatocellular icter) and chloramphenicol (“Grau syndrome” in animals and humans
lethal cardio-circulatory collapse).

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
186 | P a g e
Drug allergy
Active substances, even if they are not allergens, may cause allergic reactions.
The reactions will be mediated by humoral antibodies (Ig) ce which will lead to antigen –
antibody reactions. Actually, it is an altered response to the drug as a result of prior exposure and
involves immunological mechanisms.
Allergic reactions are caused by drugs with a protein structure and also by compounds that can
couple proteins and which can trigger antigen-antibody type reactions.
Example:
penicillamic acid can couple with peptides,
sulphonamides, dextrin may trigger allergic reactions due to the formation of complexes with
proteins.
Drugs act like a haptene that couples with the body's own albumin and acts like an antigen. The
process can occur at the surface of:
erythrocytes,
platelet and
granulocytes, in this case, the haptene (by coupling) transforms a component of the membrane
into an antigen.
This, in turn, stimulates the production of antibodies that persist for a long time. This means that
the body retains the ability to instantly synthesize antibodies, in the case of a new contact with the
haptene. If the administration of the haptene is continued (re-exposure), it will connect back to the cell
surface and form the antigen that will react with the antibody (This combination at cell surface cell
lysis). Instead of a primary haptene, modified active substances or their metabolites can also enter in
the reaction, responsible for: allergy to medication group (e.g. sulfonamides, betalactams,
benzothiadiazine etc.).
In VM drug allergy is caused after a topical application, after p.o. Administration, rarely post-
injection (but much more serious).
Reactions = histamine and serotonin release are known to appear only in a part of individuals.
The bodies respond in form of severe reactions (even in low doses). The recorded reactions are
different from the possible usual effect.
There is an initial period, preceding a violent reaction. Paraclinic analyze may reveal: circulating
antibodies in previously sensitized animals. Patch-tests can demonstrate positive reactions to the tested
drugs (ex: scarification test for penicillin highlights anaphylactic hypersensitivity leisure species).

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
187 | P a g e
M M
M
A A AC C
P
P P
Medicamentul Protein -Medicament
I. FORMAREACOMPLEXULUI ANTIGENIC
II. FAZA SENSIBILIZ RII
III. FAZA ALERGIC
Complexantigenic
+
Anticorp Celul
A AC M A AC
Simplified mechanism of drug allergy
(after Drago 1978, modif. Cristina) Symptoms associated with allergic reaction may occure:
immediately or
after 7-14 days (serum diseases with hyperthermia, arthritis, glomerulonephritis etc., as a
result of a disparity between the amounts of antigen and antibody).
Clinic and paraclinic drug allergy can be identified by:
skin rash
itching and hives,
asthmatic phenomena,
haematological changes,
edema (angioneurotic)
febrile reaction,
anaphylactic shock,
dermatitis (delayed allergic reactions),
is the majority of allergic sensitization reactions to antibiotics = a consequence of impurities in the
manufacture of antibiotics and the specific toxicity of antibiotics and of the compounds in
decomposition. Reactions to antibiotics occur quickly than 10-30min. After administration
anaphylactic shock occur being treated with:
antihistamines,
cortisone,
oxygen and
noradrenaline (i.v.).
Certain pharmacons can cause serum disease (ex. alfa-metil-DOPA) (reactions, the immediate
and the delayed one, similar to those caused by allergens: albumin, pollen etc.).
In the subcategory of serum disease:
skin reactions consequence of photosensitization or
of hematopoietic system,
fever or allergic arthritis.

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
188 | P a g e
Medications frequently responsible for allergic reactions in veterinary medicine (Synthesis Cristina)
Medicamentul Manifestarea clinic /
paraclinicMecanismul
Aspirina Urticarie generalizat , edemangioneurotic
Prin anticorpi IgE
Penicilinele oc anafilactic, anemiehemolitic
Prin anticorpi IgE,IgE/IgG
Digitoxina, Novobiocina,Chinina
Trombocitopenie IgE / IgG
Aminofenazona,Ampicilina, sulfamidele
Agranulocitoz , exantemmorbiliform
Tip celular
ruri de argint sau aur Eritrodermie, febrilitate Tip celular
Drug photo allergies Photo allergy: all photosensitivity reactions appearing in a conflict of photo-antigen or photo-
allergen – photo-antibody. After Longhin, formation of photo-allergens is influenced by:
substances that have different chemical composition and have: animal, vegetable, mineral and
pharmaceutical origins.
the mechanism of skin photosensitization is: photodynamic and photo allergic.
Photo reactive substances are complex substances that increase skin reactivity to UV or visible
radiations (between 2900–7900 Å). Generally, molecules that are capable to induce photosensitivity
are able to absorb energy from:
photons (high)
UV-radiation and
visible radiations (due to selective absorption of radiation, many of them being colored).
The majority of substances have a structure of three benzene rings arranged linearly (those
ordered in angles have a reduced activity) and wavelength of 310 to 430 nm.
Many are fluorescent and can easily form free radicals. Some are: contact allergens (cause contact
dermatitis), others are carcinogenic. Some (the photodynamic ones) can kill fungal cultures (ex:
Candida albicans) by phototoxic processes.
Photo allergic reactions have the following main features:
are individual,
occur in animals with lighter hair and skin;
reactions do not occur at the first irradiation, but after successive exposures;
the incubation period lasts a few days;
eruptions occur away from the irradiated, as outbreaks of eczema or urticaria (without
residual pigmentation).

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
189 | P a g e
The main substances that are recognized as photoallergens (Synthesis Cristina)
ExterneBitionolul, eozina, gudroanele, hexaclorfenul, lavanda, plantele din familiaUmbelifere i Rutacee care con in furocumarine (angelica, bergamotul,
unile, coada oricelului, fr sinelul, morcovul, mu tarul, p stârnacul,rarul, mohorul, p dia, p trunjelul, pintenul coco ului, portocalul,
rapi a, sun toarea, teiul, troscotul, elina, volbura, etc), rivanolul,tripaflavina, sulfamidele, uleiurile volatile, vanilia.
Subs
tan
e exo
gene
Interne
Acridina, albastrul de metilen, antihistaminicele de sintez (fenergan,romergan), antipirina, argintul, atebrina, aurul, barbituricele, chinina,chinidina, fenocumarinele, fenotiazinicele, griseofulvina, hematoporfirina,neoxazolul, PAS – sodic, sulfacetamida, sulfadiazina, sulfamerazina,sulfametinul, sulfanilamida, sulfapiridina, tetraciclinele,
Substan eendogene
Rezult din metabolismul viciat (dintre care cele mai numeroase suntporfirinele i deriva ii indolici)
Mutagenic & Teratogenic reactions
Some a.u.v. Drugs can cause permanent mutations. These can interfere with:
DNA replication or
chromosomal configurations (teratogenic)
Example: alkylation agents can engage mutagenic effects due to: alteration of pair nitrogenous
bases or cracking the chromosomes. Many known drugs, that are administered to gestating animals:
pyrimidone, phenytoin, CBZ, PBZ, ABZ in ruminants may cause fetal malformations especially in the
first part of gestation. These malformations are translated through:
fetal growth retardation;
absence of the soft palate,
hydrocephalus
minor malformations or serious and even embryonic death;
extremity malformations (shortening of the bones)
skeletal abnormalities.
teratogenic reactions in animals have been described for:
CNS. inhibitors,
immunosuppressant's (antifolates)
antivitamins (K),
phenothiazines,
diazepam and chlordiazepoxides,
morphine
heroin
meperidine,
methadone
glucocorticoids,

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
190 | P a g e
antibiotics (streptomycin, tetracycline) etc..
sulfonamides
Carcinogenic reactions
Some substances can promote the proliferation of cancerous process. They can act:
either at the site of injection
either in the digestive tract, in the case p.o. administration,
either systemically.
Cancer in animals can cause:
alkylating agents,
organochlorinated products,
urethane, etc..
phenacetin (ureters and bladder cancer)
phenylbutazone causes leukemia.
Tolerance (habituation) type adverse reactions
Tolerance (habituation) is a reduced sensibility to some drugs requires teh increase of the dose in
order to obtain the same therapeutic effect, as to another individual who received the usual dose.
The change that occurs is of pharmacokinetic nature. The incriminated drug will not reach the
receptors or the targeted tissues in active concentrations, identifiable by a clinical effect.
Tolerance may be divided in:
congenital,
connected to the species (ex: rabbit’s insensitivity to atropine (Atropa belladonna) an ability
owed to the capacity of atropine-specific esterase to metabolize the alkaloid);
acquired (actual habit), as the result of repeated administrations which will lead, in time, to
minor pharmacodynamic responses.
Generally, in the case of the second type of drug tolerance, the decreased effect is due to:
decrease in receptor responsiveness,
or to the interference of some enzymatic systems.
In animals, this type of adverse reaction can be identified for:
sympathomymethic amines (e.g., ephedrine),
vasodilators cholinergic
hypertensive etc.
In this situations tachyphylaxis is produced (tachis = fast, phylaxys = protection) = rapid
tolerance. This type o tolerance is the result of rapid development of responsiveness by diminishing

Introduction in Veterinary Pharmacology Chapter 14 Romeo – Teodor CRISTINA
191 | P a g e
the effects within minute - or after repeated administrations. The mechanism of tachyphylaxis in
veterinary medicine is still under study, it is fully known only in experimental cases.
Another type of gained tolerance is: Mitridatism it has been identified in individuals which were
treated for a long period of time with atropine, arsenic derivatives etc.
The change of route of administration leads to the loss of this capacity. In human medicine, cross-
tolerance is considered a fact (ex: ethylic persons can became less sensitive to narcotics). Probably this
phenomenon exists in veterinary medicine too, but not many incidents have been yet recorded.
Pharmacodependence
According to WHO Pharmacodependence is: ”a complex medical condition psychological,
sometimes physical, resulting from the interaction between the living organism and a drug substance,
characterized by behavioral changes and other reactions that require continuous or periodic
administration of the substance, for the purpose of finding the psychological effects and sometimes to
avoid the morbid condition resulting from deprivation”.
This condition can often be accompanied by a state of tolerance; same individual may become
dependent on several drugs. Repeated administration, with the tendency to overcome the usual doses
may cause addiction or eufomania.
These types of adverse reactions do not exist in veterinary medicine, only in animals specially
trained to identify drugs (dogs and pigs) or more rarely in horses doped for competition. It seems that
tolerance and dependence (opiates, morphine) is caused by retrograde inhibition of the synthesis or
release of endorphins.
Tolerance = endorphin deficiency (that will leave a growing number of free receptors that will
attach opiates). When administration is suddenly suppressed = withdrawal syndrome.
In connection with this fact a hypotheses was issued, some individuals are predisposed to the
opiate habit, precisely due to a genetic deficiency in endorphin.
Also considered as a side effect is pharmaco-thesauriosmosis it consists in the accumulation of
drugs in tissues for extended periods of time (months - years), which can cause serious effects and
injuries: hemorrhage, sclerosis, tumors). Generally, in animals the locations are:
adipose tissue (e.g. organochlorinated insecticides)
skin and appendage (e.g arsenic, sulfur, etc..)
kidney (e.g. salts, especially of calcium),
the reticuloendothelial system in the liver and spleen (eg salts of heavy metals, salts of gold,
iron, PVP etc.) and less frequently.
central nervous system (e.g., phenytoin).
To alkaloids, habit is installed by enhancing the possibilities of organisms to inactivate (usual
through oxidation) the substance

Introduction in Veterinary Pharmacology References Romeo – Teodor CRISTINA
192 | P a g e
Bibliography
Pop P., R.T. Cristina
Dermatologie medical veterinar
Mirton, Timi oara 973-578-025-9
Ed. 1 1995, Ed. 2 1996
267 pp.
R.T. Cristina
Bazele farmacologiei veterinare
Brumar Timi oara 973-9295-78-9
2000 254 pp.
R.T. Cristina
Receptur , Calcul i Interac iuni în Medicina Veterinar
Sedona Timi oara 973-9345-42-5
2001 304 pp.
R.T. Cristina Ghid de dozaj i teste de Farmacologie veterinar Ed. 2 ad ugit
Brumar Timi oara 973-8057-46-9 973-602-020-7
Ed.1 2001
Ed. 2 ad. 2004
224 pp. 248 pp.

Introduction in Veterinary Pharmacology References Romeo – Teodor CRISTINA
193 | P a g e
R.T. Cristina, P.A. Dar u, E. Dumitrescu
Practicum de farmacologie i terapie veterinar
Vasile Goldi Univ. Press, Arad
973-664-071-X 2005
205 pp.
R.T. Cristina, P.A. Dar u
Biotehnologii farmaceutice i Industrializarea medicamentului de uz veterinar
Vasile Goldi Univ. Press, Arad
973-664-072-8 200
415 pp.
R.T. Cristina.
Introducere în farmacologia i terapia veterinara
Ed. Solness, Timi oara, 2006,
(10)973-729-064-X, (
13)978-973-729-064-9.
2006. 750 pp.
R.T. Cristina, V. Teusdea Ghid de farmacie si terapeutica veterinara
Ed. Brumar Timisoara

Introduction in Veterinary Pharmacology References Romeo – Teodor CRISTINA
194 | P a g e
978-973-602-345-5
2008 427 pp.
R.T. Cristina, V. Teusdea, Maria Andre escu, I. Hu u,
GHID LEGISLATIV EUROPEAN
Farmacovigilen i Legisla ia medicamentului veterinar
Ed. Mirton Timi oara, Colec ia Ceres
ISBN 978-973-52-0579-9.
2009 147 pp.
Electronic sources
R.T. Cristina
Biotehnologii farmaceutice i Industrializarea medicamentului de uz veterinar
Suport de curs Impact Media Timi oara ISBN 978-973-7996-24-4
Suport de curs Partea I . Farmacologie general - ISBN(13)978-973-729-064-9, 270 pag..
Related Documents











