Intracellular Levels of the LIS1 Protein Correlate with Clinical and Neuroradiological Findings in Patients with Classical Lissencephaly Antonella Fogli, PhD,*² Renzo Guerrini, MD,² Francesca Moro, PhD,*‡ Emilio Fernandez-Alvarez, MD,§ Marie Odile Livet, MD, i Alessandra Renieri, MD,¶ Maddalena Cioni, MD,# Daniela T. Pilz, MD,** Pierangelo Veggiotti, MD,²² Elena Rossi, PhD,‡ Andrea Ballabio, MD,* and Romeo Carrozzo, MD*‡ We report on the genotype–phenotype correlation in 7 patients with classical lissencephaly carrying a heterozygous subtle mutation in the LIS1 gene. Six patients showed a mutation predicted to encode for a truncated protein, and one mutation altered a splicing site, resulting in skipping of exon 4. Western blot analysis performed on the lymphoblastoid cell line of 2 patients bearing truncating mutations indicated that the mutated allele did not produce a detectable amount of the LIS1 protein; whereas the analysis performed on the fibroblasts from the patient with a splice-site mutation was suggestive of partial protein synthesis from the mutated allele. Although clinical and magnetic resonance imaging find- ings of patients with truncating mutations did not differ from those observed in patients with a heterozygous deletion, the patient bearing the exon-skipping mutation had less severe clinical and brain involvement. Our data suggest that truncating mutations in the LIS1 gene are relatively common among patients with classical lissencephaly not bearing a heterozygous deletion at 17p13.3, and strengthen the relevance of correct intracellular dosage of the LIS1 protein in the neuronal migration process. Fogli A, Guerrini R, Moro F, Fernandez-Alvarez E, Livet MO, Renieri A, Cioni M, Pilz DT, Veggiotti P, Rossi E, Ballabio A, Carrozzo R. Intracellular levels of the LIS1 protein correlate with clinical and neuroradiological findings in patients with classical lissencephaly. Ann Neurol 1999;45:154 –161 Classical lissencephaly or generalized agyria–pachygyria is a severe human brain malformation manifested by a smooth cerebral surface, resulting from an arrest in neuronal migration at 8 to 14 weeks of gestation. 1,2 The neocortex is abnormally thick and poorly orga- nized, with four to six primitive layers, and diffuse neuronal heterotopia. 3,4 Classical lissencephaly is either found in patients with Miller-Dieker syndrome in association with a characteristic facial dysmorphism, or occurs without peculiar facial features in patients with isolated lissen- cephaly sequence (ILS). 2 Almost all of the patients with Miller-Dieker syndrome have either cytogenetically visible or submicroscopic deletions involving chromo- some 17p13.3, whereas about 40% of patients with ILS show submicroscopic deletions occurring at the same locus (Pilz DT and Ledbetter DH, personal communication). In 1993, a gene mapping to the critical region for lissencephaly and indicated as LIS1 was isolated. This gene was initially considered to be involved in classical lissencephaly, based on the observation of nonoverlap- ping deletions detected by the LIS1 cDNA in 2 inde- pendent patients. 5 Subsequently, the finding of an ap- parently balanced translocation disrupting the LIS1 gene, and of three heterozygous subtle mutations (one missense, one frameshift, and one exon-skipping) in patients not bearing a deletion at 17p13.3, confirmed LIS1 as the “chromosome 17” lissencephaly gene. 6,7 The LIS1 gene is ubiquitously expressed 5 and it en- codes for the 45-kd subunit of the intracytoplasmic From the *Telethon Institute of Genetics and Medicine (TIGEM), San Raffaele Biomedical Science Park and ‡Cytogenetic Laboratory, San Raffaele Hospital, Milan; ²Institute of Child Neurology and Psychiatry, University of Pisa, and Institute for Medical Research Stella Maris Foundation, Pisa; ¶Department of Molecular Biology and #Institute of Pediatrics, University of Siena, Siena; and ²²Insti- tute of Child Neurology and Psychiatry, Institute for Medical Re- search Casimiro Mondino Foundation, Pavia, Italy; §Pediatric Neu- rology, Sant Joan de De ´u Hospital, Barcelona, Spain; i Pediatrics and Pediatric Neurology, Regional Hospital and University of Mar- seille, Marseille, France; and **Department of Human Genetics, University of Chicago, Chicago, IL. Received Aug 25, 1998, and in revised form Oct 21. Accepted for publication Oct 23, 1998. Address correspondence to Dr Carrozzo, Servizio di Genetica Med- ica, Ospedale San Raffaele, Via Olgettina 60, 20132 Milan, Italy. 154 Copyright © 1999 by the American Neurological Association

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Intracellular Levels of the LIS1 ProteinCorrelate with Clinical and
Neuroradiological Findings in Patientswith Classical Lissencephaly
Antonella Fogli, PhD,*† Renzo Guerrini, MD,† Francesca Moro, PhD,*‡ Emilio Fernandez-Alvarez, MD,§Marie Odile Livet, MD,i Alessandra Renieri, MD,¶ Maddalena Cioni, MD,# Daniela T. Pilz, MD,**Pierangelo Veggiotti, MD,†† Elena Rossi, PhD,‡ Andrea Ballabio, MD,* and Romeo Carrozzo, MD*‡
We report on the genotype–phenotype correlation in 7 patients with classical lissencephaly carrying a heterozygous subtlemutation in the LIS1 gene. Six patients showed a mutation predicted to encode for a truncated protein, and onemutation altered a splicing site, resulting in skipping of exon 4. Western blot analysis performed on the lymphoblastoidcell line of 2 patients bearing truncating mutations indicated that the mutated allele did not produce a detectable amountof the LIS1 protein; whereas the analysis performed on the fibroblasts from the patient with a splice-site mutation wassuggestive of partial protein synthesis from the mutated allele. Although clinical and magnetic resonance imaging find-ings of patients with truncating mutations did not differ from those observed in patients with a heterozygous deletion,the patient bearing the exon-skipping mutation had less severe clinical and brain involvement. Our data suggest thattruncating mutations in the LIS1 gene are relatively common among patients with classical lissencephaly not bearing aheterozygous deletion at 17p13.3, and strengthen the relevance of correct intracellular dosage of the LIS1 protein in theneuronal migration process.
Fogli A, Guerrini R, Moro F, Fernandez-Alvarez E, Livet MO, Renieri A, Cioni M, Pilz DT, Veggiotti P, Rossi E,Ballabio A, Carrozzo R. Intracellular levels of the LIS1 protein correlate with clinical and
neuroradiological findings in patients with classical lissencephaly. Ann Neurol 1999;45:154–161
Classical lissencephaly or generalized agyria–pachygyriais a severe human brain malformation manifested by asmooth cerebral surface, resulting from an arrest inneuronal migration at 8 to 14 weeks of gestation.1,2
The neocortex is abnormally thick and poorly orga-nized, with four to six primitive layers, and diffuseneuronal heterotopia.3,4
Classical lissencephaly is either found in patientswith Miller-Dieker syndrome in association with acharacteristic facial dysmorphism, or occurs withoutpeculiar facial features in patients with isolated lissen-cephaly sequence (ILS).2 Almost all of the patientswith Miller-Dieker syndrome have either cytogeneticallyvisible or submicroscopic deletions involving chromo-some 17p13.3, whereas about 40% of patients withILS show submicroscopic deletions occurring at the
same locus (Pilz DT and Ledbetter DH, personalcommunication).
In 1993, a gene mapping to the critical region forlissencephaly and indicated as LIS1 was isolated. Thisgene was initially considered to be involved in classicallissencephaly, based on the observation of nonoverlap-ping deletions detected by the LIS1 cDNA in 2 inde-pendent patients.5 Subsequently, the finding of an ap-parently balanced translocation disrupting the LIS1gene, and of three heterozygous subtle mutations (onemissense, one frameshift, and one exon-skipping) inpatients not bearing a deletion at 17p13.3, confirmedLIS1 as the “chromosome 17” lissencephaly gene.6,7
The LIS1 gene is ubiquitously expressed5 and it en-codes for the 45-kd subunit of the intracytoplasmic
From the *Telethon Institute of Genetics and Medicine (TIGEM),San Raffaele Biomedical Science Park and ‡Cytogenetic Laboratory,San Raffaele Hospital, Milan; †Institute of Child Neurology andPsychiatry, University of Pisa, and Institute for Medical ResearchStella Maris Foundation, Pisa; ¶Department of Molecular Biologyand #Institute of Pediatrics, University of Siena, Siena; and ††Insti-tute of Child Neurology and Psychiatry, Institute for Medical Re-search Casimiro Mondino Foundation, Pavia, Italy; §Pediatric Neu-rology, Sant Joan de Deu Hospital, Barcelona, Spain; iPediatrics
and Pediatric Neurology, Regional Hospital and University of Mar-seille, Marseille, France; and **Department of Human Genetics,University of Chicago, Chicago, IL.
Received Aug 25, 1998, and in revised form Oct 21. Accepted forpublication Oct 23, 1998.
Address correspondence to Dr Carrozzo, Servizio di Genetica Med-ica, Ospedale San Raffaele, Via Olgettina 60, 20132 Milan, Italy.
154 Copyright © 1999 by the American Neurological Association

platelet-activating factor (PAF) acetylhydrolase isoformIb (approved gene symbol PAFAH1B1).8
A second gene playing a crucial role in neuronal mi-gration, and indicated as “doublecortin” (approvedgene symbol DCX), mapping to chromosome Xq22.3–q23, has recently been demonstrated to harbor muta-tions in 6 boys with classic lissencephaly, in their moth-ers having subcortical band heterotopia, and in mostwomen with sporadic subcortical band heterotopia.9–11
In the present study, we searched for mutations in21 patients with classical lissencephaly not showing adeletion at the LIS1 locus, identifying six novel muta-tions in 7 unrelated patients. Five mutations were pre-dicted to encode for a truncated protein, and one re-sulted in the skipping of the entire exon 4. We alsostudied the expression of mutations in the cell line of 3patients by western blot analysis, and found that theintracellular levels of the LIS1 protein were correlatedwith the severity of neuronal migration abnormality onmagnetic resonance imaging (MRI) study of the brain.
Patients and MethodsPatientsBetween 1995 and June 1998, 24 patients with classical lis-sencephaly were referred to the Center for Medical Geneticsat the San Raffaele Hospital for genetic counseling and cy-togenetic analysis. Classical lissencephaly had been diagnosedin all patients by using standard clinical and MRI criteria.3,4
We performed cytogenetic analysis in all patients and molec-ular studies in 21 patients in whom no cytogenetic deletioncould be demonstrated. For the 7 patients in whom we coulddemonstrate a mutation of the LIS1 gene, we correlated theseverity of the clinical picture and of lissencephaly with thatof the mutations observed.
Cytogenetic and Molecular AnalysisFluorescence in situ hybridization studies were undertakenby using cosmid c120A7 as probe on metaphase chromo-somes.6 Twenty-one patients who did not show a submicro-scopic rearrangement at 17p13.3 were selected for molecularanalysis.
Blood samples were collected for DNA and lymphoblas-toid cell lines after a consent form was signed by the parents.For Patient ILS-I07, skin fibroblasts were obtained during aminor surgery procedure. Genomic DNA was prepared fromperipheral blood leukocytes and RNA was obtained fromlymphoblastoid cell lines or fibroblasts according to standardtechniques.12
Southern blot analysis was performed according to stan-dard procedures,12 digesting 7 mg of genomic DNA from thepatients and from normal individuals with both EcoRI andPstI. Hybridization probes were polymerase chain reaction(PCR)-amplified from LIS1 cDNA and reactions were per-formed as previously described.7 Genomic DNA from Pa-tient ILS-F01 was also digested with HindIII, BamHI andEcoRV.
Single-strand conformation polymorphism (SSCP) analy-sis was performed on exons 2 through 11 of the LIS1 gene,as described.7
In case of an altered electrophoretic bandshift, genomicamplification and SSCP analysis were repeated on a secondDNA sample, to rule out Taq polymerase artifacts. Productsthat showed a gel-mobility shift were excised directly fromthe SSCP gel, reamplified, and sequenced. PCR productsfrom genomic DNA, containing both the normal and mu-tant alleles, were also directly sequenced. PCR sequence anal-ysis was performed on an ABI 377 automated DNA se-quencer (Perkin-Elmer ABI, Milan, Italy).
For Patient ILS-07, 1 mg of total RNA from fibroblastswas reverse transcribed using the GeneAmp RNA PCR kit(Perkin-Elmer Cetus, Norwalk, CT). The reverse-transcribedproduct was then PCR-amplified, using 0.5 mM concentra-tions of primers LIS1(71)269F (59-TATCTTCGTTCAAA-TGGCTATGAAG-39) and LIS1(71)505R (59-GGAATCCA-TTCTTTTGGGT-39) in a 100-ml volume. Amplificationswere performed as described.7 PCR products were separatedin a 6% polyacrylamide gel and the 162-bp mutant fragmentwas excised from the gel, reamplified, and directly sequenced.
Antibody Preparation and Western Blot AnalysisThe peptide MVLSQRQRDELNRC (N1), corresponding tothe amino acid residues 1 to 13, deduced from the LIS1cDNA, was used to immunize white rabbits (Primm, Milan,Italy).13 The polyclonal antiserum was purified by immuno-affinity chromatography, using the peptide antigen accordingto standard procedures.14
To evaluate the amount of cross-reacting material in thepatients’ cell lines, Epstein-Barr virus–transformed lympho-blastoid cell lines were established from Patients ILS-I05,ILS-S03, and ILS-I01 (an ILS patient with a heterozygousdeletion of the LIS1 gene). Cells culture and cell extractspreparation were performed as previously described.15 Fibro-blasts from Patient ILS-I07 were grown in minimum essen-tial medium Eagle Alpha (GibcoBRL) supplemented with10% fetal calf serum. The coding region of the LIS1 cDNAwas introduced into a modified pMT21 vector containingthe c-myc epitope (a kind gift from Dr Marc Tessier-Lavigne), to yield expression of a c-myc–tagged LIS1 protein.Transient transfections of pMT21-LISmyc into Cos7 cellswere performed using LipofectAMINE (GibcoBRL) follow-ing the manufacturer’s instructions. Cells were harvested 48hours after transfection and lysated for sodium dodecyl sul-fate (SDS)–polyacrylamide gel electrophoresis analysis.
For cell extracts, 40 mg of soluble cellular proteins fromlymphoblastoid cell lines of Patients ILS-I05, ILS-S03, andILS-I01, and of a normal control, and 30 mg of cellular ex-tracts from mock-transfected and pMT21-LISmyc–transfectedCos7 cells were electrophoresed through 10% SDS–poly-acrylamide electrophoresis gels and electroblotted onto nitro-cellulose membrane (Bio-Rad).16 The protein content wasdetermined by using crystalline bovine serum albumin as thestandard.17 For Patient ILS-I07, the electrophoretic resolu-tion of proteins was increased by using an 8 to 15% gradientpolyacrylamide gel.18
Western blot analysis was performed as described16 andvisualization of antibody binding was performed by en-hanced chemiluminescence (ECL, Amersham), according tothe manufacturer’s recommendations. X-ray films of westernblots were scanned and densitometry was performed by using
Fogli et al: Mutations in Classical Lissencephaly 155
a computing densitometer (Molecular Dynamics). The den-sity of each band was measured as pixels per unit area andthe background was subtracted.
Magnetic Resonance Imaging andClinical EvaluationPatients with a mutation of the LIS1 gene underwent brainMRI, using 0.5- or 1.5-T apparatus. Non–contrast-enhanced spin-echo, inversion recovery, and gradient-echosequences were performed in the axial, sagittal, and coronalplanes. All patients were studied with standard 5-mm slicethickness. Severity of lissencephaly was labeled, as follows,according to the lissencephaly grading system proposed byDobyns and Truwit4: grade 1 5 agyria only; grade 2 5widespread agyria with limited areas of pachygyria primarilyinvolving the frontal and temporal poles; grade 3 5 fronto-temporal pachygyria and posterior agyria–pachygyria; grade4 5 diffuse pachygyria with no areas of agyria. All patientshad undergone dysmorphological and repeated neurologicalexaminations.
ResultsMutation AnalysisAs a first step, the DNA from each patient was exam-ined by Southern blot analysis in the search for grossrearrangements of the LIS1 gene. An altered hybridiza-tion band for Patient ILS-F01 could be detected byfive different restriction enzymes, using a probe encom-passing the 39 end of the LIS1 gene (data not shown).Neither of the parents showed an altered hybridizationpattern. No altered band could be detected on South-ern blot analysis for the other patients.
Screening by SSCP was subsequently performed inthe search for mutations in the LIS1 gene by using 10
pairs of PCR primers designed to amplify each codingexon separately.
The list of mutations identified in this study is re-ported in the Table. The insertion of a single A at nu-cleotide 154 in exon 4, found in Patient ILS-E03, leadsto a frameshift and a premature stop codon 23 bpdownstream. The 10-bp deletion with the insertion ofAA at nucleotide 520 in exon 6, found in Patients ILS-I02 and ILS-I05, causes a frameshift with a stop codon16 bp downstream. In Patient ILS-S03, the 46-bp in-sertion at nucleotide 558 in exon 6 is the result of aperfect tandem duplication from position 513 to 558and introduces a frameshift and a stop codon 10 bpdownstream. The mutation in Patient ILS-I12, a C 3T transition at nucleotide 430 in exon 6, converts anarginine at position 144 into a stop codon.
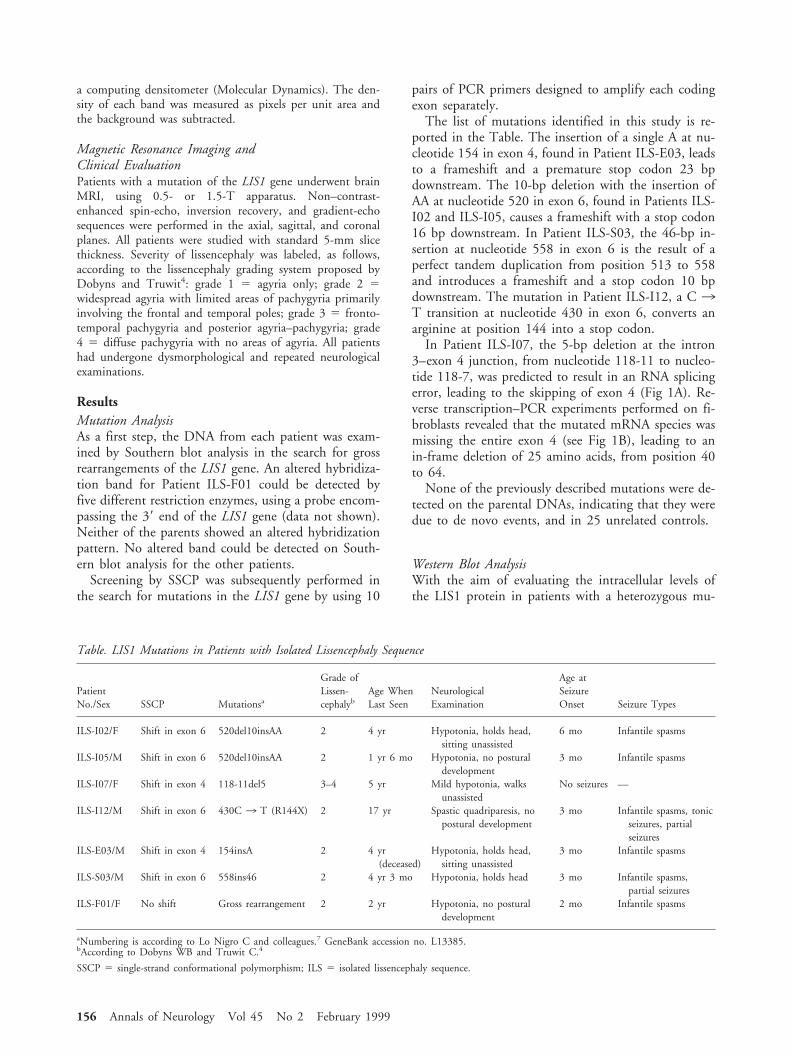
In Patient ILS-I07, the 5-bp deletion at the intron3–exon 4 junction, from nucleotide 118-11 to nucleo-tide 118-7, was predicted to result in an RNA splicingerror, leading to the skipping of exon 4 (Fig 1A). Re-verse transcription–PCR experiments performed on fi-broblasts revealed that the mutated mRNA species wasmissing the entire exon 4 (see Fig 1B), leading to anin-frame deletion of 25 amino acids, from position 40to 64.
None of the previously described mutations were de-tected on the parental DNAs, indicating that they weredue to de novo events, and in 25 unrelated controls.
Western Blot AnalysisWith the aim of evaluating the intracellular levels ofthe LIS1 protein in patients with a heterozygous mu-
Table. LIS1 Mutations in Patients with Isolated Lissencephaly Sequence
PatientNo./Sex SSCP Mutationsa
Grade ofLissen-cephalyb
Age WhenLast Seen
NeurologicalExamination
Age atSeizureOnset Seizure Types
ILS-I02/F Shift in exon 6 520del10insAA 2 4 yr Hypotonia, holds head,sitting unassisted
6 mo Infantile spasms
ILS-I05/M Shift in exon 6 520del10insAA 2 1 yr 6 mo Hypotonia, no posturaldevelopment
3 mo Infantile spasms
ILS-I07/F Shift in exon 4 118-11del5 3–4 5 yr Mild hypotonia, walksunassisted
No seizures —
ILS-I12/M Shift in exon 6 430C 3 T (R144X) 2 17 yr Spastic quadriparesis, nopostural development
3 mo Infantile spasms, tonicseizures, partialseizures
ILS-E03/M Shift in exon 4 154insA 2 4 yr(deceased)
Hypotonia, holds head,sitting unassisted
3 mo Infantile spasms
ILS-S03/M Shift in exon 6 558ins46 2 4 yr 3 mo Hypotonia, holds head 3 mo Infantile spasms,partial seizures
ILS-F01/F No shift Gross rearrangement 2 2 yr Hypotonia, no posturaldevelopment
2 mo Infantile spasms
aNumbering is according to Lo Nigro C and colleagues.7 GeneBank accession no. L13385.bAccording to Dobyns WB and Truwit C.4
SSCP 5 single-strand conformational polymorphism; ILS 5 isolated lissencephaly sequence.
156 Annals of Neurology Vol 45 No 2 February 1999
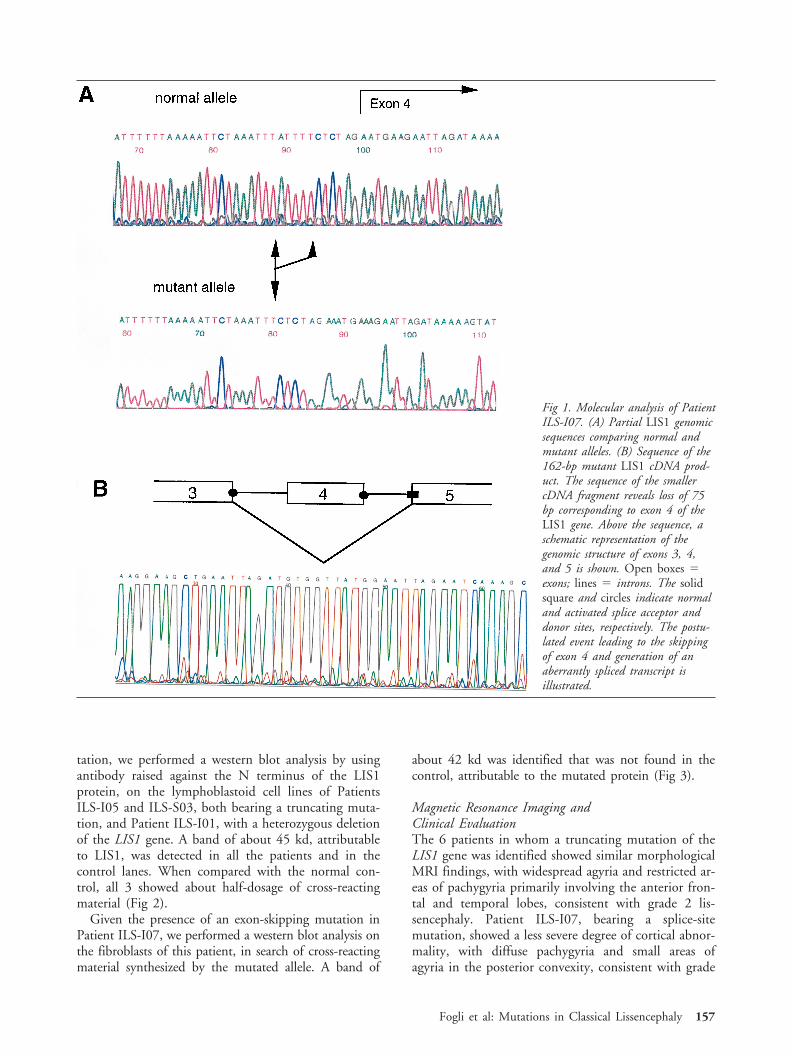
tation, we performed a western blot analysis by usingantibody raised against the N terminus of the LIS1protein, on the lymphoblastoid cell lines of PatientsILS-I05 and ILS-S03, both bearing a truncating muta-tion, and Patient ILS-I01, with a heterozygous deletionof the LIS1 gene. A band of about 45 kd, attributableto LIS1, was detected in all the patients and in thecontrol lanes. When compared with the normal con-trol, all 3 showed about half-dosage of cross-reactingmaterial (Fig 2).
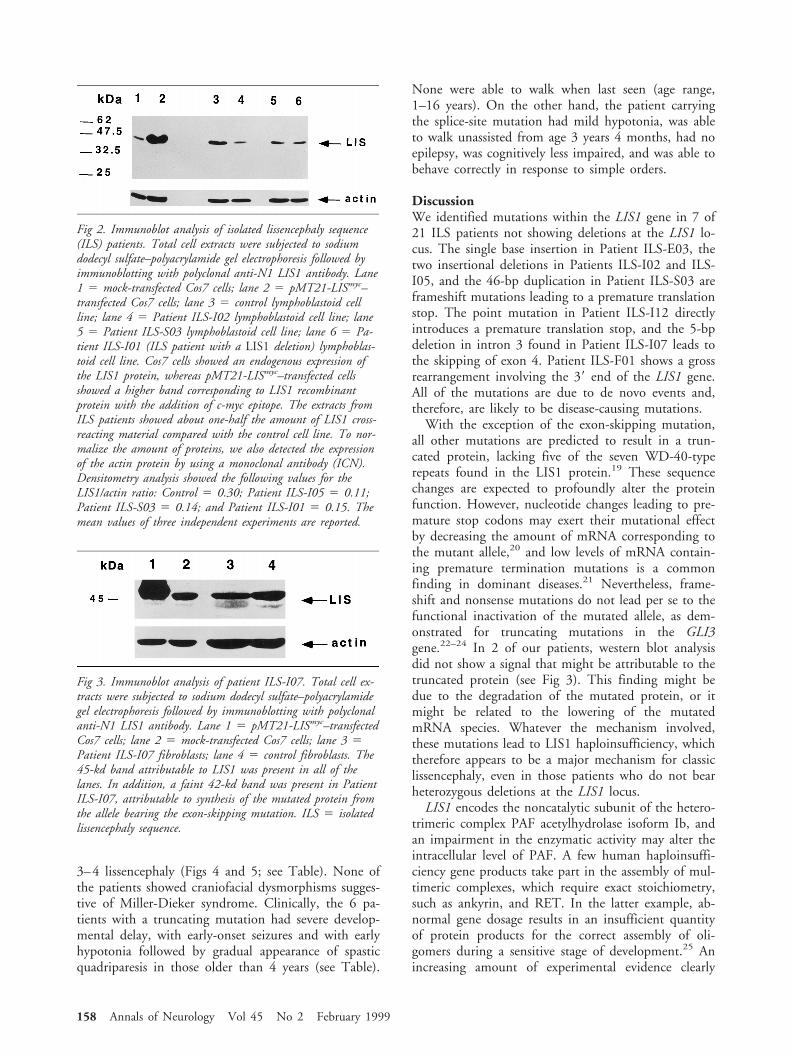
Given the presence of an exon-skipping mutation inPatient ILS-I07, we performed a western blot analysis onthe fibroblasts of this patient, in search of cross-reactingmaterial synthesized by the mutated allele. A band of
about 42 kd was identified that was not found in thecontrol, attributable to the mutated protein (Fig 3).
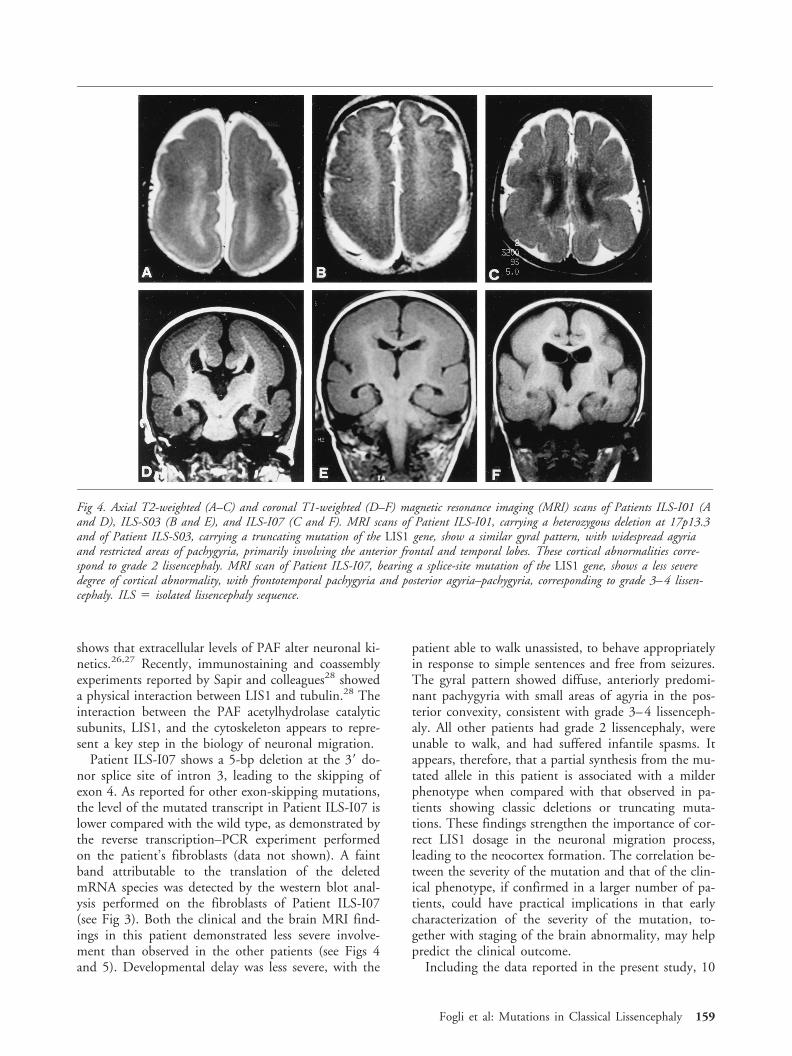
Magnetic Resonance Imaging andClinical EvaluationThe 6 patients in whom a truncating mutation of theLIS1 gene was identified showed similar morphologicalMRI findings, with widespread agyria and restricted ar-eas of pachygyria primarily involving the anterior fron-tal and temporal lobes, consistent with grade 2 lis-sencephaly. Patient ILS-I07, bearing a splice-sitemutation, showed a less severe degree of cortical abnor-mality, with diffuse pachygyria and small areas ofagyria in the posterior convexity, consistent with grade
Fig 1. Molecular analysis of PatientILS-I07. (A) Partial LIS1 genomicsequences comparing normal andmutant alleles. (B) Sequence of the162-bp mutant LIS1 cDNA prod-uct. The sequence of the smallercDNA fragment reveals loss of 75bp corresponding to exon 4 of theLIS1 gene. Above the sequence, aschematic representation of thegenomic structure of exons 3, 4,and 5 is shown. Open boxes 5exons; lines 5 introns. The solidsquare and circles indicate normaland activated splice acceptor anddonor sites, respectively. The postu-lated event leading to the skippingof exon 4 and generation of anaberrantly spliced transcript isillustrated.
Fogli et al: Mutations in Classical Lissencephaly 157
3–4 lissencephaly (Figs 4 and 5; see Table). None ofthe patients showed craniofacial dysmorphisms sugges-tive of Miller-Dieker syndrome. Clinically, the 6 pa-tients with a truncating mutation had severe develop-mental delay, with early-onset seizures and with earlyhypotonia followed by gradual appearance of spasticquadriparesis in those older than 4 years (see Table).
None were able to walk when last seen (age range,1–16 years). On the other hand, the patient carryingthe splice-site mutation had mild hypotonia, was ableto walk unassisted from age 3 years 4 months, had noepilepsy, was cognitively less impaired, and was able tobehave correctly in response to simple orders.
DiscussionWe identified mutations within the LIS1 gene in 7 of21 ILS patients not showing deletions at the LIS1 lo-cus. The single base insertion in Patient ILS-E03, thetwo insertional deletions in Patients ILS-I02 and ILS-I05, and the 46-bp duplication in Patient ILS-S03 areframeshift mutations leading to a premature translationstop. The point mutation in Patient ILS-I12 directlyintroduces a premature translation stop, and the 5-bpdeletion in intron 3 found in Patient ILS-I07 leads tothe skipping of exon 4. Patient ILS-F01 shows a grossrearrangement involving the 39 end of the LIS1 gene.All of the mutations are due to de novo events and,therefore, are likely to be disease-causing mutations.
With the exception of the exon-skipping mutation,all other mutations are predicted to result in a trun-cated protein, lacking five of the seven WD-40-typerepeats found in the LIS1 protein.19 These sequencechanges are expected to profoundly alter the proteinfunction. However, nucleotide changes leading to pre-mature stop codons may exert their mutational effectby decreasing the amount of mRNA corresponding tothe mutant allele,20 and low levels of mRNA contain-ing premature termination mutations is a commonfinding in dominant diseases.21 Nevertheless, frame-shift and nonsense mutations do not lead per se to thefunctional inactivation of the mutated allele, as dem-onstrated for truncating mutations in the GLI3gene.22–24 In 2 of our patients, western blot analysisdid not show a signal that might be attributable to thetruncated protein (see Fig 3). This finding might bedue to the degradation of the mutated protein, or itmight be related to the lowering of the mutatedmRNA species. Whatever the mechanism involved,these mutations lead to LIS1 haploinsufficiency, whichtherefore appears to be a major mechanism for classiclissencephaly, even in those patients who do not bearheterozygous deletions at the LIS1 locus.
LIS1 encodes the noncatalytic subunit of the hetero-trimeric complex PAF acetylhydrolase isoform Ib, andan impairment in the enzymatic activity may alter theintracellular level of PAF. A few human haploinsuffi-ciency gene products take part in the assembly of mul-timeric complexes, which require exact stoichiometry,such as ankyrin, and RET. In the latter example, ab-normal gene dosage results in an insufficient quantityof protein products for the correct assembly of oli-gomers during a sensitive stage of development.25 Anincreasing amount of experimental evidence clearly
Fig 2. Immunoblot analysis of isolated lissencephaly sequence(ILS) patients. Total cell extracts were subjected to sodiumdodecyl sulfate–polyacrylamide gel electrophoresis followed byimmunoblotting with polyclonal anti-N1 LIS1 antibody. Lane1 5 mock-transfected Cos7 cells; lane 2 5 pMT21-LISmyc–transfected Cos7 cells; lane 3 5 control lymphoblastoid cellline; lane 4 5 Patient ILS-I02 lymphoblastoid cell line; lane5 5 Patient ILS-S03 lymphoblastoid cell line; lane 6 5 Pa-tient ILS-I01 (ILS patient with a LIS1 deletion) lymphoblas-toid cell line. Cos7 cells showed an endogenous expression ofthe LIS1 protein, whereas pMT21-LISmyc–transfected cellsshowed a higher band corresponding to LIS1 recombinantprotein with the addition of c-myc epitope. The extracts fromILS patients showed about one-half the amount of LIS1 cross-reacting material compared with the control cell line. To nor-malize the amount of proteins, we also detected the expressionof the actin protein by using a monoclonal antibody (ICN).Densitometry analysis showed the following values for theLIS1/actin ratio: Control 5 0.30; Patient ILS-I05 5 0.11;Patient ILS-S03 5 0.14; and Patient ILS-I01 5 0.15. Themean values of three independent experiments are reported.
Fig 3. Immunoblot analysis of patient ILS-I07. Total cell ex-tracts were subjected to sodium dodecyl sulfate–polyacrylamidegel electrophoresis followed by immunoblotting with polyclonalanti-N1 LIS1 antibody. Lane 1 5 pMT21-LISmyc–transfectedCos7 cells; lane 2 5 mock-transfected Cos7 cells; lane 3 5Patient ILS-I07 fibroblasts; lane 4 5 control fibroblasts. The45-kd band attributable to LIS1 was present in all of thelanes. In addition, a faint 42-kd band was present in PatientILS-I07, attributable to synthesis of the mutated protein fromthe allele bearing the exon-skipping mutation. ILS 5 isolatedlissencephaly sequence.
158 Annals of Neurology Vol 45 No 2 February 1999
shows that extracellular levels of PAF alter neuronal ki-netics.26,27 Recently, immunostaining and coassemblyexperiments reported by Sapir and colleagues28 showeda physical interaction between LIS1 and tubulin.28 Theinteraction between the PAF acetylhydrolase catalyticsubunits, LIS1, and the cytoskeleton appears to repre-sent a key step in the biology of neuronal migration.
Patient ILS-I07 shows a 5-bp deletion at the 39 do-nor splice site of intron 3, leading to the skipping ofexon 4. As reported for other exon-skipping mutations,the level of the mutated transcript in Patient ILS-I07 islower compared with the wild type, as demonstrated bythe reverse transcription–PCR experiment performedon the patient’s fibroblasts (data not shown). A faintband attributable to the translation of the deletedmRNA species was detected by the western blot anal-ysis performed on the fibroblasts of Patient ILS-I07(see Fig 3). Both the clinical and the brain MRI find-ings in this patient demonstrated less severe involve-ment than observed in the other patients (see Figs 4and 5). Developmental delay was less severe, with the
patient able to walk unassisted, to behave appropriatelyin response to simple sentences and free from seizures.The gyral pattern showed diffuse, anteriorly predomi-nant pachygyria with small areas of agyria in the pos-terior convexity, consistent with grade 3–4 lissenceph-aly. All other patients had grade 2 lissencephaly, wereunable to walk, and had suffered infantile spasms. Itappears, therefore, that a partial synthesis from the mu-tated allele in this patient is associated with a milderphenotype when compared with that observed in pa-tients showing classic deletions or truncating muta-tions. These findings strengthen the importance of cor-rect LIS1 dosage in the neuronal migration process,leading to the neocortex formation. The correlation be-tween the severity of the mutation and that of the clin-ical phenotype, if confirmed in a larger number of pa-tients, could have practical implications in that earlycharacterization of the severity of the mutation, to-gether with staging of the brain abnormality, may helppredict the clinical outcome.
Including the data reported in the present study, 10
Fig 4. Axial T2-weighted (A–C) and coronal T1-weighted (D–F) magnetic resonance imaging (MRI) scans of Patients ILS-I01 (Aand D), ILS-S03 (B and E), and ILS-I07 (C and F). MRI scans of Patient ILS-I01, carrying a heterozygous deletion at 17p13.3and of Patient ILS-S03, carrying a truncating mutation of the LIS1 gene, show a similar gyral pattern, with widespread agyriaand restricted areas of pachygyria, primarily involving the anterior frontal and temporal lobes. These cortical abnormalities corre-spond to grade 2 lissencephaly. MRI scan of Patient ILS-I07, bearing a splice-site mutation of the LIS1 gene, shows a less severedegree of cortical abnormality, with frontotemporal pachygyria and posterior agyria–pachygyria, corresponding to grade 3–4 lissen-cephaly. ILS 5 isolated lissencephaly sequence.
Fogli et al: Mutations in Classical Lissencephaly 159

subtle mutations have been identified in the LIS1 genein patients with ILS and, among these, only one re-sulted in a missense mutation.7 A bias of ascertainmentof mutations in the LIS1 gene might be attributable tothe peculiar phenotype associated to haploinsufficiency.It is plausible that missense mutations in the LIS1 genemight cause other neuronal migration disorders differ-ent from classic lissencephaly, and for which a geneticbasis has been hypothesized.29
Fourteen of our patients did not show a mutation inthe LIS1 gene. Some of these patients may bear muta-tions in the LIS1 exons that have not been detectedbecause of the lack of full sensitivity of our screeningtechnique. Alterations in the intronic sequences thatwere not screened, or in the 59 upstream sequences aswell as 39 downstream sequences involved in the regu-lation of transcription, would not be detected by thepresent methodology. Some of these patients, however,might bear mutations involving the doublecortin gene,recently associated with classical lissencephaly and sub-cortical band heterotopia.9–11 In contrast, they couldbear a mutation in a still unidentified “lissencephaly”gene. The genes encoding the two catalytic subunits ofPAF acetylhydrolase,30,31 and the human gene encod-ing the homologue of NudC, a protein required fornuclear movement in Aspergillus nidulans, recentlydemonstrated to interact with LIS1,32 are potentialcandidates for being involved in neuronal migrationdisorders.
In conclusion, our data indicate that haploinsuffi-ciency is a relatively common mechanism for classicallissencephaly, even in those patients who do not bearheterozygous deletions at the LIS1 locus. The findingof a milder phenotype in the patient showing partialsynthesis of the mutated LIS1 protein in fibroblastspoints to the relevance of the correct LIS1 dosage inthe neuronal migration process.
The contribution of the Italian Telethon Foundation (grant E.512)is gratefully acknowledged.
We thank Drs C. Lo Nigro and R. Tonlorenzi for participating inthe early stages of this project, and Dr M. Zollo for sequencing
analysis. We also thank M. Smith for helping in the preparation ofthis manuscript.
References1. Barkovich AJ, Koch T, Carrol C. The spectrum of
lissencephaly: report of ten patients analyzed by magnetic reso-nance imaging. Ann Neurol 1991;30:139–146
2. Dobyns WB, Elias ER, Newlin AC, et al. Causal heterogeneityin isolated lissencephaly. Neurology 1992;42:1375–1388
3. Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissen-cephaly. A human brain malformation associated with deletionof the LIS1 gene located at chromosome 17p13. JAMA 1993;270:2838–2842
4. Dobyns WB, Truwit C. Lissencephaly and other malformationsof cortical development: 1995 update. Neuropediatrics 1995;26:132–147
5. Reiner O, Carrozzo R, Shen Y, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein b-subunit-likerepeats. Nature 1993;364:717–721
6. Chong SS, Pack SD, Roschke AV, et al. A revision of the lis-sencephaly and Miller-Dieker critical regions in chromosome17p13.3. Hum Mol Genet 1997;6:147–155
7. Lo Nigro C, Chong SS, Smith ACM, et al. Point mutationsand an intragenic deletion in LIS1, the lissencephaly causativegene in isolated lissencephaly sequence and Miller-Dieker syn-drome. Hum Mol Genet 1997;6:157–164
8. Hattori M, Adachi H, Tsujimoto M, et al. Miller-Dieker lis-sencephaly gene encodes a subunit of brain platelet-activatingfactor. Nature 1994;370:216–218
9. des Portes V, Pinard JM, Billuart P, et al. A novel CNS generequired for neuronal migration and involved in X-linked sub-cortical laminar heterotopia and lissencephaly syndrome. Cell1998;92:51–61
10. Gleeson JG, Allen KM, Fox JW, et al. doublecortin, a brain-specific gene mutated in human X-linked lissencephaly anddouble cortex syndrome, encodes a putative signaling protein.Cell 1998;92:63–72
11. des Portes V, Francis F, Pinard JM, et al. doublecortin is themajor gene causing X-linked subcortical laminar heterotopia(SCLH). Hum Mol Genet 1998;7:1063–1070
12. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a lab-oratory manual, 2nd ed. Cold Spring Harbor, NY: Cold SpringHarbor Laboratory Press, 1989
13. Mizuguchi M, Takashima S, Kakita A, et al. Lissencephaly geneproduct. Localization in the central nervous system and loss ofimmunoreactivity in Miller-Dieker syndrome. Am J Pathol1995;147:1142–1151
14. Harlow E, Lane D. Antibodies: a laboratory manual. ColdSpring Harbor, NY: Cold Spring Harbor Laboratory, 1988
15. Daniele A, Parenti G, d’Addio M, et al. Biochemical character-
Fig 5. Sagittal T1-weighted magnetic resonance imaging(MRI) scans (passing through the brain midline) of Pa-tients ILS-S03 (A) and ILS-I07 (B). Analysis of the gyralpattern in the mesial hemispheric surface confirms themore severe degree of cortical abnormality in Patient ILS-S03 in whom no sulci are recognizable in the posteriortwo-thirds of the mesial brain surface, with the exceptionof the parieto-occipital sulcus and the calcarine fissure.ILS 5 isolated lissencephaly sequence.
160 Annals of Neurology Vol 45 No 2 February 1999

ization of arylsulfatase E and functional analysis of mutationsfound in patients with X-linked chondrodysplasia punctata.Am J Hum Genet 1998;62:562–572
16. Laemmli UK. Cleavage of structural proteins during the assem-bly of the head of bacteriophage T4. Nature 1970;227:680–685
17. Bradford M. A rapid and sensitive method for the quantitationof microgram quantities of protein utilizing the principle ofprotein-dye binding. Anal Biochem 1976;72:248–254
18. Matsudaira P, Burgess D. SDS microslab linear gradient poly-acrylamide gel electrophoresis. Anal Biochem 1978;87:386–396
19. Neer E, Schmidt C, Nambudripad R, Smith T. The ancientregulatory-protein family of WD-repeat proteins. Nature 1994;371:297–300
20. Maquat L. When cells stop making sense: effects of nonsensecodons on RNA metabolism in vertebrate cells. RNA 1995;1:453–465
21. McIntosh I, Hamosh A, Dietz H. Nonsense mutations and di-minished mRNA levels. Nat Genet 1993;4:219 (Letter)
22. Wild A, Kalff SM, Vortkamp A, Bornholdt D, Konig R,Grzeschik K. Point mutations in human GLI3 cause Greig syn-drome. Hum Mol Genet 1997;6:1979–1984
23. Kang S, Graham JJ, Olney A, Biesecker L. GLI3 frameshift mu-tations cause autosomal dominant Pallister-Hall syndrome. NatGenet 1997;15:266–268
24. Radhakrishna U, Wild A, Grzeschik K, Antonarakis S. Muta-tion in GLI3 in postaxial polydactyly type A. Nat Genet 1997;17:269–271
25. Fisher E, Scambler P. Human haploinsufficiency: one for sor-row, two for joy. Nat Genet 1994;7:5–7
26. Adachi H, Aoki J, Manya H, et al. PAF analogues capable ofinhibiting PAF acetylhydrolase activity suppress migration ofisolated rat cerebellar granule cells. Neurosci Lett 1997;235:133–136
27. Bix G, Clark G. Platelet-activating factor receptor stimulationdisrupts neuronal migration In vitro. J Neurosci 1998;18:307–318
28. Sapir T, Elbaum M, Reiner O. Reduction of microtubule ca-tastrophe events by LIS1, platelet-activating factor acetylhydro-lase subunit. EMBO J 1997;16:6977–6984
29. Barkovich AJ, Kuzniecky RI, Dobyns WB, et al. A classificationscheme for malformations of cortical development. Neuropedi-atrics 1996;27:59–63
30. Adachi H, Tsujimoto M, Hattori M, et al. cDNA cloningof human cytosolic platelet-activating factor acetylhydrolasegamma-subunit and its mRNA expression in human tissues.Biochem Biophys Res Commun 1995;214:180–187
31. Adachi H, Tsujimoto M, Hattori M, et al. Differential tissuedistribution of the beta- and gamma-subunits of human cyto-solic platelet-activating factor acetylhydrolase (isoform I). Bio-chem Biophys Res Commun 1997;233:10–13
32. Morris SM, Albrecht U, Reiner O, et al. The lissencephaly geneproduct Lis1, a protein involved in neuronal migration, inter-acts with a nuclear movement protein, NudC. Curr Biol 1998;8:603–606
Fogli et al: Mutations in Classical Lissencephaly 161
Related Documents


![48, XXXY/49, XXXXY mosaic: new neuroradiological features ... · speech delay, multiple skeletal anomalies, and cardiac defects [1, 3]. SCA patients have lower intellectual quotient](https://static.cupdf.com/doc/110x72/5f97ac9958d69c3e907f9c48/48-xxxy49-xxxxy-mosaic-new-neuroradiological-features-speech-delay-multiple.jpg)








