1 International Human Microbiome Standards Grant Agreement: HEALTH-F4-2010-261376 DELIVERABLE REPORT Work package WP3 – Improved standards for sequencing Work package leader Partner 5 – CEA Genoscope Deliverable D3.2 – Improved standards for sequencing Delivery date* 01/08/2013 Dissemination level** PU (Public) * Please refer to IHMS Calendar on IHMS intranet * *Please highlight the dissemination level appropriate for the deliverable. You can find the corresponding information in the IHMS Calendar Summary report From January 2012 until now, we have received some other 22 DNA extractions from faecal samples from INRA partner and 217 DNA extractions from the other partners. All the samples have been treated according to our validated pipeline which includes: i) sample quality control at arrival; 2) Illumina sequencing library preparation from samples which passed the QC, by applying our standardized protocol; iii) 100 bp lenght paired end sequencing of each library; iv) sequence quality control and validation; v) data delivery to partner 7. In order to help in the establishment of standards for faecal sample extraction protocol, a particular attention has been paid to the check of quality of the DNA samples. In this report we will describe the analysis applied to sample QC and the exclusion criteria used. All the INRA samples passed the QC and were sequenced. Of the other 217 samples, 192 passed the QC and were sequenced. All sequencing data have been transferred to partner 7 and analyses are under progress.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
International Human Microbiome
Standards
Grant Agreement: HEALTH-F4-2010-261376
DELIVERABLE REPORT
Work package WP3 – Improved standards for sequencing
Work package leader Partner 5 – CEA Genoscope
Deliverable D3.2 – Improved standards for sequencing
Delivery date* 01/08/2013
Dissemination level** PU (Public)
* Please refer to IHMS Calendar on IHMS intranet
* *Please highlight the dissemination level appropriate for the deliverable. You can find the
corresponding information in the IHMS Calendar
Summary report
From January 2012 until now, we have received some other 22 DNA extractions from faecal
samples from INRA partner and 217 DNA extractions from the other partners. All the samples
have been treated according to our validated pipeline which includes: i) sample quality control at
arrival; 2) Illumina sequencing library preparation from samples which passed the QC, by
applying our standardized protocol; iii) 100 bp lenght paired end sequencing of each library; iv)
sequence quality control and validation; v) data delivery to partner 7.
In order to help in the establishment of standards for faecal sample extraction protocol, a
particular attention has been paid to the check of quality of the DNA samples. In this report we will
describe the analysis applied to sample QC and the exclusion criteria used. All the INRA samples
passed the QC and were sequenced. Of the other 217 samples, 192 passed the QC and were
sequenced. All sequencing data have been transferred to partner 7 and analyses are under
progress.

2
sd3.2.1 – Improved inventory of standards for genomic sequencing
sd3.2.2 – Improved standards and recommendation for metagenomic long contiguous reference
sequence
In the period January 2012 – January 2013, the INRA partner sent to Genoscope 239 DNA
extractions. The INRA partner extracted 22 of them by using the same protocol applied for
extraction of the 20 samples previously processed. The other 217 samples were extracted by the
other IHMS partner starting from the same two faecal samples aliquots (A and B) by using their
own extraction protocols.
Upon arrival at Genoscope, all the samples were recorded in our LIMS system for internal follow
up at any stage of the processing. They were stored at –20°C until processing according to our
well established and standardized pipeline described below.
defines stopping points : the
experiment must fill some well defined
criteria, otherwise it is stopped

3
i) Sample quality control
Our standardized protocol for genomic DNA quality control was initially applied on all the samples.
A SOP for genomic DNA QC is described in appendix 1. We recommend to the laboratory where
extractions are performed to use this protocol in order to evaluate DNA quality.
Briefly, the protocol includes two steps:
- Quantity evaluation: quantification by two independent measures by Qbit BR Assay kit is
performed. A mean concentration is calculated. For library preparation protocol established for
IHMS project, 250 ng input DNA are required. In our standard procedure, if total DNA quantity is
less than 500 ng (2 fold the minimal quantity), the sample is not valid and the QC ends at this
stage. In the context of this project, we have decided to check the quality also of samples with an
insufficient quantity (<250 ng) to perform the library.
- Quality evaluation: samples are loaded on a 0,4 % agarose gel and migration is performed at
100V during one hour. A photo is taken and quality of DNA is visually checked. If RNA
contamination is present, an RNAse treatment is applied to the sample, after which the sample
repeats the QC from the beginning. DNA integrity is visually checked. For standard paired end
library preparation, DNA passes the QC if the majority of the DNA is located on a tight band at high
molecular weight. Anyway, we wanted to check the IHMS samples quality much more carefully in
order to produce the most of information about DNA quality. This should be helpful in order to
evaluate the different extraction protocols used by the IHMS partner and to establish a
standardized protocol to produce good quality DNA. For this aim, we took advantage of the
availability in our laboratory of a gel image analysis system (GeneTools, Syngene) which is able to
calculate the % of DNA present at different size ranges selected by the user. Based on the size of
the DNA ladder bands as reference, we have chosen four size ranges: > 9 kb, between 9 and 5
kb, between 5 and 1,8kb and < 1,8kb. We have manually delimited these size regions on each gel
image and the analysis system has calculated the % of DNA for each region. We have combined
the results of the software analysis with our visual interpretation of the images and finally we have
established four DNA quality categories:
Qualitative classification colour code
Group 1
Very good quality DNA.
Optimal for sequencing Group 2
Majority of high molecular weight DNA.
Good for sequencing
Group 3
Presence of degraded DNA mostly > 1.8kb.
Acceptable for standard PE sequencing (not for MatePair)
Group 4
Presence of degraded DNA with most fragments < 1.8kb.
Not suitable for standard sequencing.
Group 5
Totally degraded DNA.
Not acceptable for sequencing
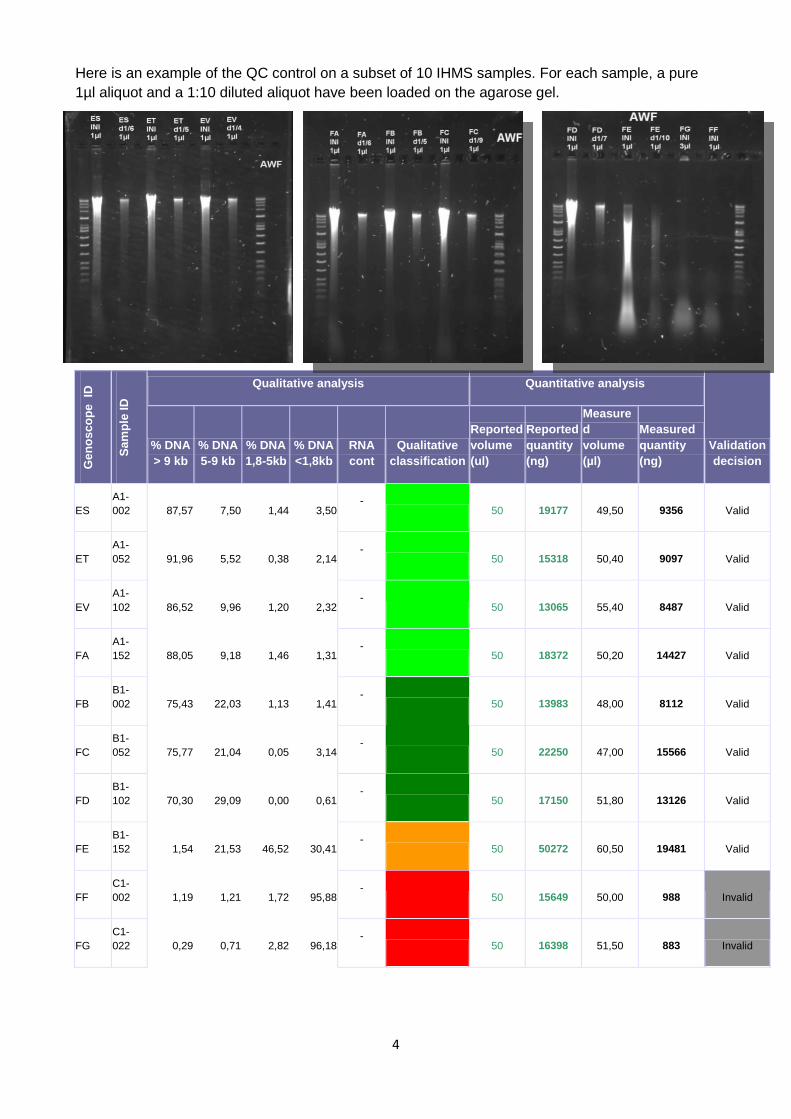
4
Here is an example of the QC control on a subset of 10 IHMS samples. For each sample, a pure
1µl aliquot and a 1:10 diluted aliquot have been loaded on the agarose gel.
Ge
no
sc
op
e
ID
Sa
mp
le ID
Qualitative analysis Quantitative analysis
Validation
decision
% DNA
> 9 kb
% DNA
5-9 kb
% DNA
1,8-5kb
% DNA
<1,8kb
RNA
cont
Qualitative
classification
Reported
volume
(ul)
Reported
quantity
(ng)
Measure
d
volume
(µl)
Measured
quantity
(ng)
ES
A1-
002 87,57 7,50 1,44 3,50 -
50 19177 49,50 9356 Valid
ET
A1-
052 91,96 5,52 0,38 2,14 -
50 15318 50,40 9097 Valid
EV
A1-
102 86,52 9,96 1,20 2,32 -
50 13065 55,40 8487 Valid
FA
A1-
152 88,05 9,18 1,46 1,31 -
50 18372 50,20 14427 Valid
FB
B1-
002 75,43 22,03 1,13 1,41 -
50 13983 48,00 8112 Valid
FC
B1-
052 75,77 21,04 0,05 3,14 -
50 22250 47,00 15566 Valid
FD
B1-
102 70,30 29,09 0,00 0,61 -
50 17150 51,80 13126 Valid
FE
B1-
152 1,54 21,53 46,52 30,41 -
50 50272 60,50 19481 Valid
FF
C1-
002 1,19 1,21 1,72 95,88 -
50 15649 50,00 988 Invalid
FG
C1-
022 0,29 0,71 2,82 96,18 -
50 16398 51,50 883 Invalid
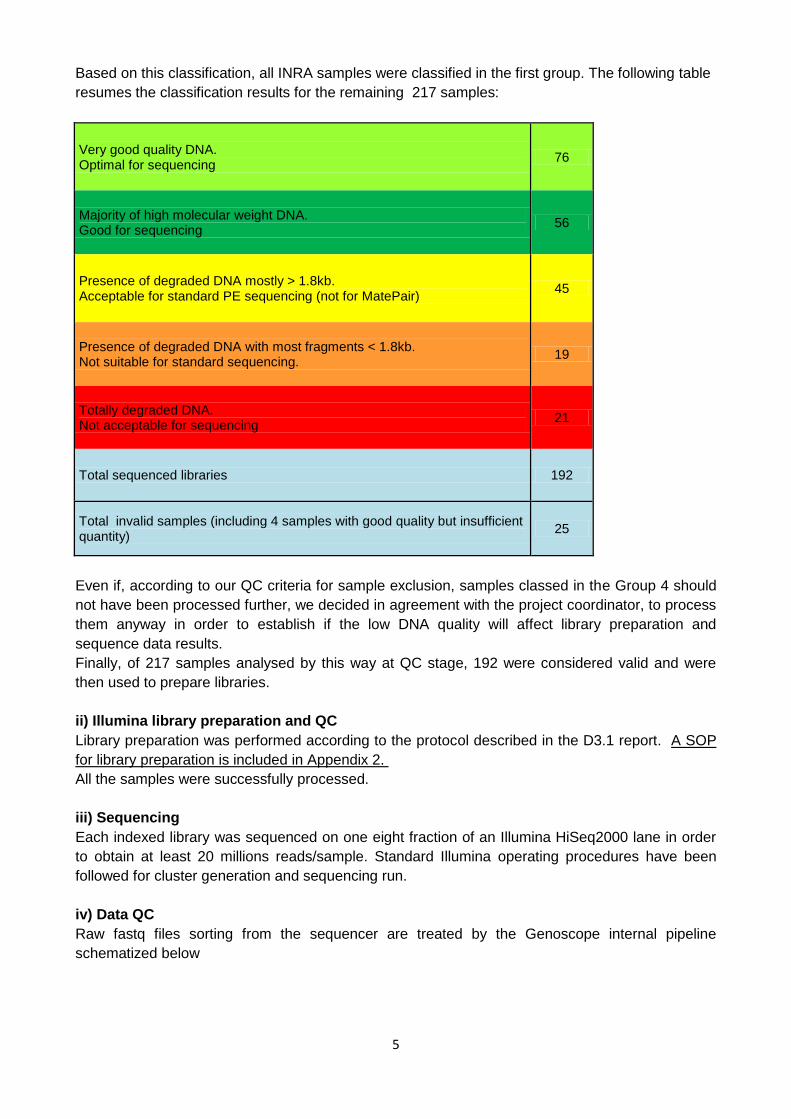
5
Based on this classification, all INRA samples were classified in the first group. The following table
resumes the classification results for the remaining 217 samples:
Very good quality DNA. Optimal for sequencing
76
Majority of high molecular weight DNA. Good for sequencing
56
Presence of degraded DNA mostly > 1.8kb. Acceptable for standard PE sequencing (not for MatePair)
45
Presence of degraded DNA with most fragments < 1.8kb. Not suitable for standard sequencing.
19
Totally degraded DNA. Not acceptable for sequencing
21
Total sequenced libraries 192
Total invalid samples (including 4 samples with good quality but insufficient quantity)
25
Even if, according to our QC criteria for sample exclusion, samples classed in the Group 4 should
not have been processed further, we decided in agreement with the project coordinator, to process
them anyway in order to establish if the low DNA quality will affect library preparation and
sequence data results.
Finally, of 217 samples analysed by this way at QC stage, 192 were considered valid and were
then used to prepare libraries.
ii) Illumina library preparation and QC
Library preparation was performed according to the protocol described in the D3.1 report. A SOP
for library preparation is included in Appendix 2.
All the samples were successfully processed.
iii) Sequencing
Each indexed library was sequenced on one eight fraction of an Illumina HiSeq2000 lane in order
to obtain at least 20 millions reads/sample. Standard Illumina operating procedures have been
followed for cluster generation and sequencing run.
iv) Data QC
Raw fastq files sorting from the sequencer are treated by the Genoscope internal pipeline
schematized below
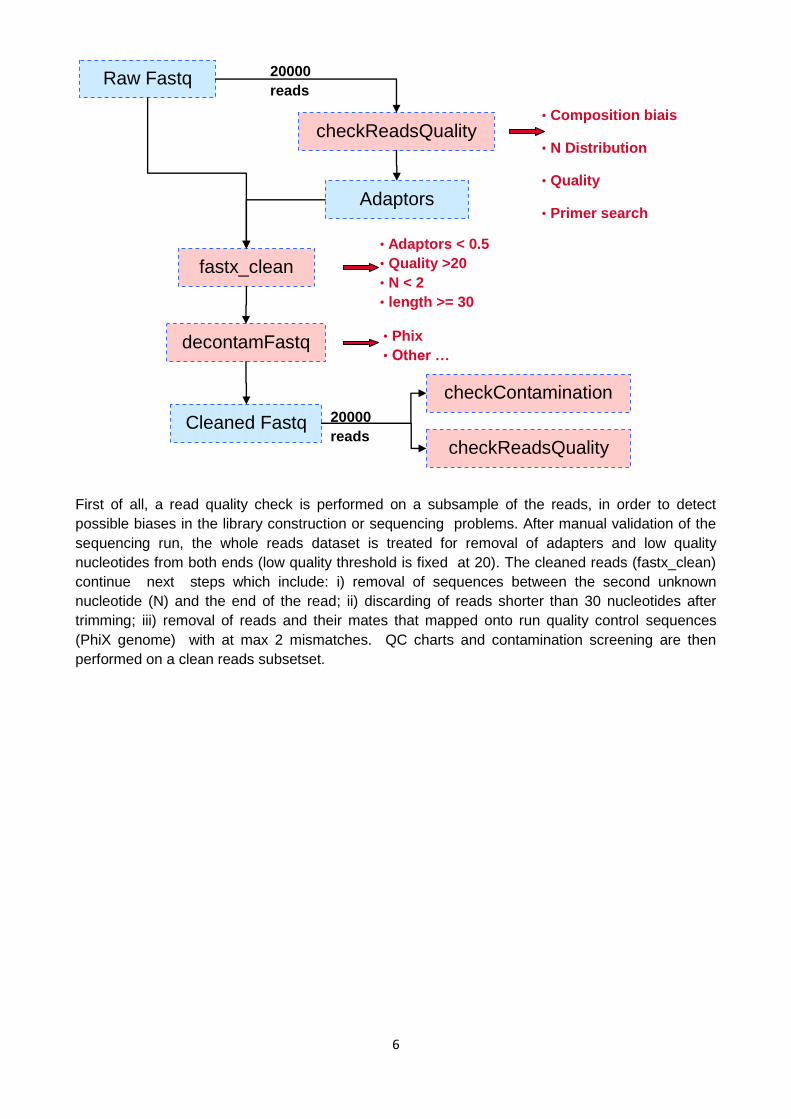
6
First of all, a read quality check is performed on a subsample of the reads, in order to detect
possible biases in the library construction or sequencing problems. After manual validation of the
sequencing run, the whole reads dataset is treated for removal of adapters and low quality
nucleotides from both ends (low quality threshold is fixed at 20). The cleaned reads (fastx_clean)
continue next steps which include: i) removal of sequences between the second unknown
nucleotide (N) and the end of the read; ii) discarding of reads shorter than 30 nucleotides after
trimming; iii) removal of reads and their mates that mapped onto run quality control sequences
(PhiX genome) with at max 2 mismatches. QC charts and contamination screening are then
performed on a clean reads subsetset.
Raw Fastq
checkReadsQuality
20000
reads
Adaptors
fastx_clean
Cleaned Fastq
decontamFastq
checkContamination
checkReadsQuality
• Composition biais
• N Distribution
• Quality
• Primer search
• Adaptors < 0.5
• Quality >20
• N < 2
• length >= 30
• Phix
• Other …
20000
reads

7
APPENDIX 1 : Genomic DNA QC using standard electrophoresis
Summary
This protocol describes how to evaluate the quality and quantity of genomic DNA samples using
run a standard agarose gel as well as Qubit™ fluorometer
Reagents and consumables
Reagent / consommable Supplier
Seakem Agarose Biorad
50x TBE buffer Biorad
SYBR® Safe DNA gel stain (10,000X concentrate in DMSO) Invitrogen
5x loading dye General lab supplier
RNAse A 100 mg/ml Qiagen
0.1x TE buffer General lab supplier
Resuspension buffer (10mM TrisHCl, pH 7,5) General lab supplier
Agilent DNA HS kit Agilent
Quant-iTTM
dsDNA BR assay kit Life Technologies
DNA molecular weight marker II (0,1 – 23 kb) Roche
Equipment
Equipment Supplier
Mini horizontal device 15-wells combs Biorad
Mini horizontal Gel electrophoresis device with 7x10
cm tray Biorad
Gel imager system Different lab suppliers
Qubit™ fluorometer 1.0 or 2.0 Life Technologies

8
Procedure
Upon arrival, store the sample at –20 °C until use.
STEP 1: gDNA quantification using Qbit™ fluorometer
Use the Quant-iTTM
dsDNA BR assay kit following the manufacturer instructions for use of the
kit and the Qbit fluorometer. Perform two independent measurements using 1 µl of the DNA
sample for each measure. Calculate the mean concentration in ng/µl.
STEP2: gDNA integrity check by agarose gel electrophoresis
All reagents and stock solution should be prepared prior to the start of the procedure.
Gel & Sample Preparation
a) Cast a ~40ml 0,6% Seakem agarose gel with 1X TBE and 10 µl SYBR® Safe DNA gel stain
(10,000X concentrate in DMSO). Use a narrow well comb.
b) For each sample to be tested prepare two clean labeled tubes
Tube 1: transfer 1µl DNA and complete with 5 µl H2O and 2µl 5x loading dye
Tube 2: prepare a 1:10 dilution of the initial sample in TE buffer and use 1 µl of the dilution.
Complete with 5 µl H2O + 2µl 5x loading dye
Gel Electrophoresis a) Load the gel by leaving an empty well between two samples. Load 100-150 ng of the DNA
molecular weight marker II in the two wells located on the left and right edgex of the gel
b) Run gel for 30 min at ~100V in 1X TBE buffer.
c) Remove gel from gel box and image.
This first image capture allows to better evaluate the presence of RNA contamination
d) Return gel to gel box and run again for 30 min at 100V
e) Remove gel from gel box and image
DNA QC Gel Analysis
Evaluate genomic DNA integrity and RNA contamination
a) RNA contamination
If RNA is massively present in the sample (visible as a cloud at < 1 kb and /or two bands at at
~ 5kb and 1,8 kb corresponding to rRNA), treat the initial sample with RNAse A: use 1 µl
RNAse A for each 100 µl sample, incubate 90 min at 37 °C and reload 1µl of the treated
sample on the gel. If RNA has disappeared, perform a new quantification by Qbit assay as
previously described. If RNA is still present, retreat sample with RNAse A.
b) DNA integrity
The majority of DNA shoud appear as a tight band > 23 kb. If a smear is present, this means
that DNA is partially degraded. If no tight high molecular weight band is visible and DNA is
present only in the smear, the degradation is massive and DNA is not suitable for sequencing.
If a quantification software system is available, refer to the software instructions analyze DNA
quality on gels.
If DNA has to be used for large long mate-pair library construction, the size of DNA needs to
be in the high molecular weight. In this case, DNA band should be above the 23kb band. It is
highly recommended to check the integrity of DNA by pulsed field electrophoresis to properly
determine the molecular weight.

9
APPENDIX 2: Library Preparation Recommendations for Illumina sequencing
of metagenomic samples
Summary
The purpose of this procedure is to generate a 180-480 bp insert size DNA library that will be
used for sequencing on the Illumina HiSeq2000 on 100 bp paired end lengths. Starting material is
500 ng genomic DNA extracted from fecal samples. Genomic DNA is broken into smaller
fragments via Covaris instrument and barcoded adapters are added so that the DNA can be
hybridized to a FlowCell before being put on the HiSeq instrument. During library preparation,
end repair, A tailing, adaptors ligation and size selection are perfomed by a semi automatized
instrument, the SPRI TE instrument supplied by Beckmann Coulter
Reagents and consumables
Reagent / consommable Supplier
6-mm × 16-mm AFA microtubes and snap caps Covaris
LoBind tubes, 1.5 mL Eppendorf
Agencourt AMPure XP beads Beckman Coulter
SPRI Works Fragment Library System I Beckmann Coulter
Platinum Pfx Taq Polymerase kit Life Technologies
0.1x TE buffer
Resuspension buffer (10mM TrisHCl, pH 7,5) General lab supplier
Agilent DNA HS kit Agilent
Quant-iT dsDNA HS assay kit Life Technologies
Illumina adapters Bioo Scientific
Illumina Library quantification kit KAPA Biosystems
Equipment
Equipment Supplier
Covaris AFA™ Ultrasonicator Covaris
SPRI-TE Instrument Beckmann Coulter
2100 Bioanalyzer Agilent
Thermal cycler General lab supplier
Qbit fluorometer or equivalent Life Technologies
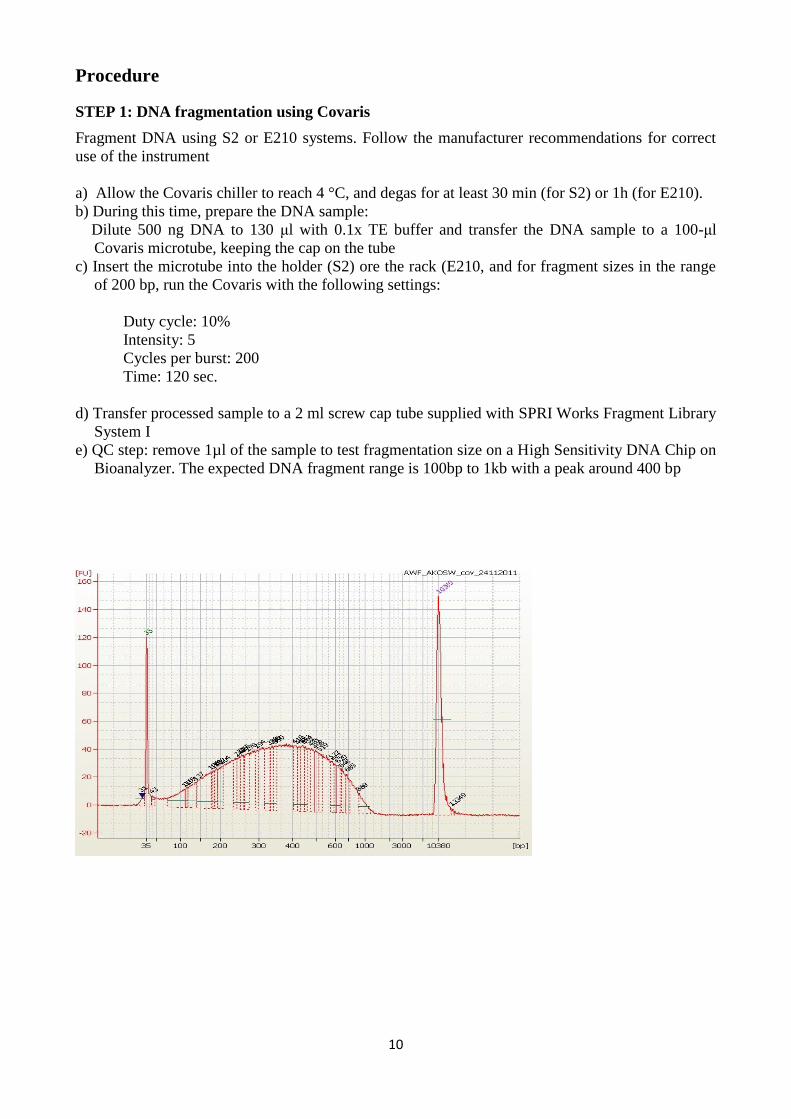
10
Procedure
STEP 1: DNA fragmentation using Covaris
Fragment DNA using S2 or E210 systems. Follow the manufacturer recommendations for correct
use of the instrument
a) Allow the Covaris chiller to reach 4 °C, and degas for at least 30 min (for S2) or 1h (for E210).
b) During this time, prepare the DNA sample:
Dilute 500 ng DNA to 130 μl with 0.1x TE buffer and transfer the DNA sample to a 100-μl
Covaris microtube, keeping the cap on the tube
c) Insert the microtube into the holder (S2) ore the rack (E210, and for fragment sizes in the range
of 200 bp, run the Covaris with the following settings:
Duty cycle: 10%
Intensity: 5
Cycles per burst: 200
Time: 120 sec.
d) Transfer processed sample to a 2 ml screw cap tube supplied with SPRI Works Fragment Library
System I
e) QC step: remove 1µl of the sample to test fragmentation size on a High Sensitivity DNA Chip on
Bioanalyzer. The expected DNA fragment range is 100bp to 1kb with a peak around 400 bp

11
STEP 2: SPRI TE run
This step includes end repair, A tailing, adaptors ligation and size selection performed using the
preloaded SPRIWorks reagents cartridge and SPRI TE instrument
a) Remove SPRIworks Fragment Library I cartridges from-20°C storage and allow the cartridges to
thaw during shearing. Remove one cartridge for each library to be
constructed. Thaw cartridges at room temperature for approximately one hour, or until all
contents are completely thawed.
b) Set up the SPRI-TE instrument and prepare the reagent rack, following the manufacturer
instructions included with the SPRI Works Library System I.
Use barcoded Illumina compatible adapters (they can be home made or purchased from various
suppliers as Bioo Scientific)
**Note: depending on the initial adapter concentration, dilute the adapters with resuspension
buffer to adjust for 500 ng input DNA. Excess adapters can interfere with sequencing. The adapters
may have to be titrated relative to starting material.
If using 15 µM adapters, you have to dilute 1:10 for about 500 ng input DNA
c) Start the run by selecting the option 300-600 bp size selection
d) At the end of the run, retrieve the tube containing the library and clean up the reaction using
AMPure XP beads.
This step allows to additionally remove fragments <300 bp and remaining adapters dimers that can
interfere during PCR enrichment. Before performing clean up, rewiev AMPure XP handling
recommendations of the manufacturer (Beckmann Coulter).
Measure the library volume and adjust to 50µl with Resuspension buffer. Add 32,5µL (0,65
volumes) AMPure XP beads, mix by short vortexing. Incubate for 5 minutes, then bind the beads
and remove the supernatant. Add 500 µL 70% ethanol (made fresh each time), incubate 30
seconds and remove. Repeat wash once. Let the pellet dry completely (5-10 minutes), then elute
in 40 µL Resuspension buffer
e) Remove 1 µl form the sample and perform a Qbit quantification.
STEP 3: PCR enrichment
Perform enrichment of the library using Platinum Pfx Taq Polymerase (Life Technologies) and P5
and P7 primers. Other protocols suitable for use with the Illumina HiSeq2000 may also be used.
Primer P5
5' AATGATACGGCGACCACCGAG
Primer P7
5’CAAGCAGAAGACGGCATACGAG
This protocol is based on 10 ng ligated DNA input as matrix for 12 cycles enrichment
12
a) Combine and mix the following components in two sterile 0,2 ml tubes
Ligated DNA (10 ng) x µl
Pfx amplification buffer 10x Reaction Buffer 5 µl
P5 primer 50 µM 1 µl
P7 primer 50 µM 1 µl
MgSO4 50mM 2 µl
dNTP 10mM 2 µl
Pfx Platinum Taq polymerase 0.8 µl
H2O 18.2-x µl
Total volume 50 µl
b) Amplify using the following PCR cycling conditions:
30 sec at 98 °C
[10 sec at 98 °C, 30 sec at 60 °C, 30 sec at 72 °C] 12 cycles total
5 min at 72 °C
Hold at 4 °C
c) Clean up the reaction using AMPure XP beads (Agencourt).
Add 40 µL (0,8 volumes) AMPure XP beads, mix by short vortexing. Incubate for 5 minutes,
then bind the beads and remove the supernatant. Add 500 µL 70% Ethanol (made fresh each
time), incubate 30 seconds and remove. Repeat once. Let the pellet dry completely (5-10
minutes), then elute in 30 µL Resuspension buffer.
STEP 4: Quantitative and qualitative assessment of the library
The sample must be accurately quantified in order to optimize yield. This step is absolutely crucial
to the success of any experiment.
a) Measure the concentration using the Qubit using the HS kit.
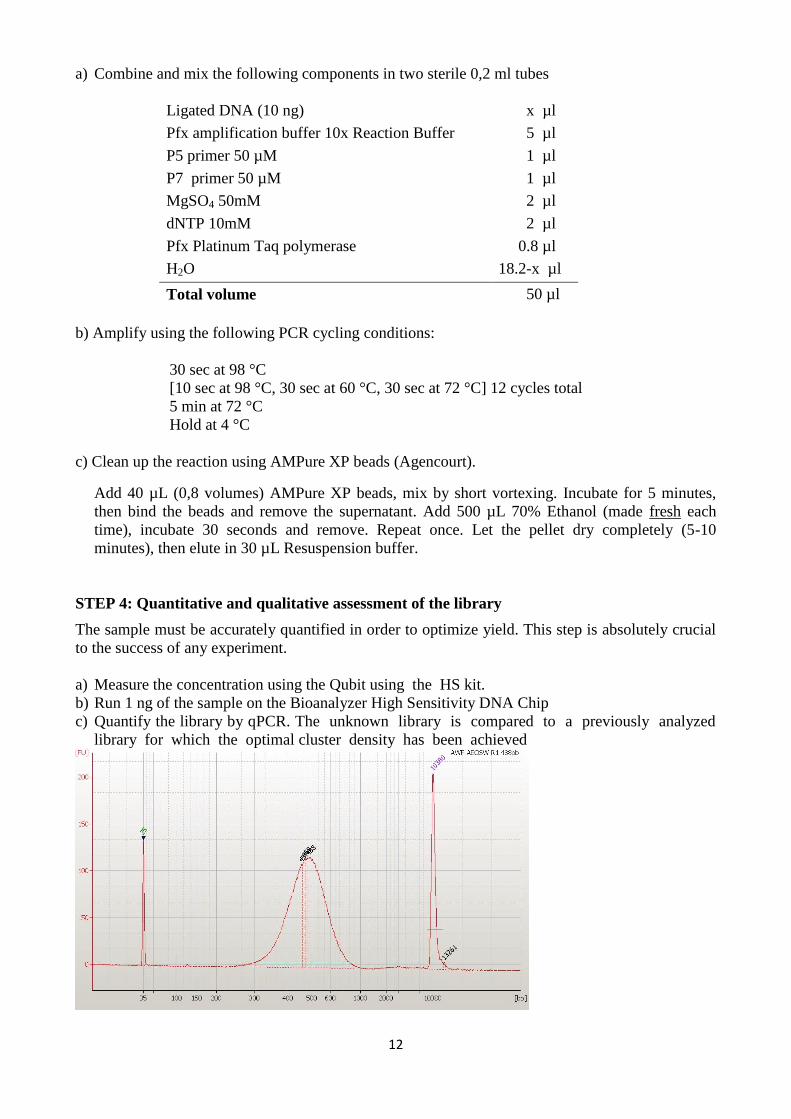
b) Run 1 ng of the sample on the Bioanalyzer High Sensitivity DNA Chip
c) Quantify the library by qPCR. The unknown library is compared to a previously analyzed
library for which the optimal cluster density has been achieved
Related Documents











