ORIGINAL PAPER Interdomain communication in the endonuclease/motor subunit of type I restriction-modification enzyme EcoR124I Dhiraj Sinha & Katsiaryna Shamayeva & Vyas Ramasubramani & David Řeha & Vitali Bialevich & Morteza Khabiri & Alena Guzanová & Niv Milbar & Marie Weiserová & Eva Csefalvay & Jannette Carey & Rüdiger Ettrich Received: 19 December 2013 /Accepted: 3 June 2014 # Springer-Verlag Berlin Heidelberg 2014 Abstract Restriction-modification systems protect bacteria from foreign DNA. Type I restriction-modification enzymes are multifunctional heteromeric complexes with DNA- cleavage and ATP-dependent DNA translocation activities located on endonuclease/motor subunit HsdR. The recent structure of the first intact motor subunit of the type I restric- tion enzyme from plasmid EcoR124I suggested a mechanism by which stalled translocation triggers DNA cleavage via a lysine residue on the endonuclease domain that contacts ATP bound between the two helicase domains. In the present work, molecular dynamics simulations are used to explore this pro- posal. Molecular dynamics simulations suggest that the Lys– ATP contact alternates with a contact with a nearby loop housing the conserved QxxxY motif that had been implicated in DNA cleavage. This model is tested here using in vivo and in vitro experiments. The results indicate how local interac- tions are transduced to domain motions within the endonuclease/motor subunit. Keywords DNA restriction enzymes . Molecular modeling . QM/MM calculations . Principal components analysis . E. coli . Multisubunit enzyme complex . Correlated loop motions Introduction Restriction-modification systems protect bacteria from foreign DNA. Type II restriction enzymes, which are relatively sim- ple, are widely used in molecular biology and are well under- stood. Type I enzymes, which were discovered first, are not used commercially because of their complicated subunit struc- ture and cleavage behavior. They are encoded by the host specificity of DNA (hsd) genes; for a review see [1]. Two copies of the HsdM methylation subunit and one copy of specificity subunit HsdS bind at a specific asymmetric DNA site and form a methyltransferase complex that modifies the host DNA, protecting it from cleavage. When invading DNA is detected by the methyltransferase based on its methylation status, two HsdR endonuclease/motor subunits are recruited to the complex, which then translocates up to thousands of duplex base pairs through the stationary enzyme without unwinding the DNA, consuming ∼1 ATP per base pair. If translocation is blocked, the endonuclease activity of HsdR is triggered, and DNA is cleaved nonspecifically at sites distant from the original binding site. These un- usual activities have prompted many years of genetic and biochemical study aimed at understanding the molecular Dhiraj Sinha and Katsiaryna Shamayeva contributed equally. This paper belongs to Topical Collection MIB 2013 (Modeling Interactions in Biomolecules VI) Electronic supplementary material The online version of this article (doi:10.1007/s00894-014-2334-1) contains supplementary material, which is available to authorized users. D. Sinha : K. Shamayeva : D. Řeha : V. Bialevich : M. Khabiri : E. Csefalvay : J. Carey : R. Ettrich Institute of Nanobiology and Structural Biology, Global Change Research Center, Academy of Sciences of the Czech Republic, Zamek 136, 373 33 Nove Hrady, Czech Republic D. Sinha : K. Shamayeva : D. Řeha : V. Bialevich : R. Ettrich (*) Faculty of Sciences, University of South Bohemia in Ceske Budejovice, Zamek 136, 373 33 Nove Hrady, Czech Republic e-mail: [email protected] V. Ramasubramani : N. Milbar : J. Carey Chemistry Department, Princeton University, Princeton, NJ 08544-1009, USA A. Guzanová : M. Weiserová Laboratory of Molecular Genetics of Bacteria, Institute of Microbiology, Academy of Sciences of the Czech Republic, Vídeňská 1083, 142 20 Praha 4, Czech Republic J Mol Model (2014) 20:2334 DOI 10.1007/s00894-014-2334-1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL PAPER
Interdomain communication in the endonuclease/motor subunitof type I restriction-modification enzyme EcoR124I
Dhiraj Sinha & Katsiaryna Shamayeva & Vyas Ramasubramani & David Řeha &
Vitali Bialevich & Morteza Khabiri & Alena Guzanová & Niv Milbar &
Marie Weiserová & Eva Csefalvay & Jannette Carey & Rüdiger Ettrich
Received: 19 December 2013 /Accepted: 3 June 2014# Springer-Verlag Berlin Heidelberg 2014
Abstract Restriction-modification systems protect bacteriafrom foreign DNA. Type I restriction-modification enzymesare multifunctional heteromeric complexes with DNA-cleavage and ATP-dependent DNA translocation activitieslocated on endonuclease/motor subunit HsdR. The recentstructure of the first intact motor subunit of the type I restric-tion enzyme from plasmid EcoR124I suggested a mechanismby which stalled translocation triggers DNA cleavage via alysine residue on the endonuclease domain that contacts ATPbound between the two helicase domains. In the present work,molecular dynamics simulations are used to explore this pro-posal. Molecular dynamics simulations suggest that the Lys–ATP contact alternates with a contact with a nearby loop
housing the conserved QxxxY motif that had been implicatedin DNA cleavage. This model is tested here using in vivo andin vitro experiments. The results indicate how local interac-tions are transduced to domain motions within theendonuclease/motor subunit.
Keywords DNA restriction enzymes .Molecular modeling .
QM/MM calculations . Principal components analysis .
E. coli . Multisubunit enzyme complex .
Correlated loop motions
Introduction
Restriction-modification systems protect bacteria from foreignDNA. Type II restriction enzymes, which are relatively sim-ple, are widely used in molecular biology and are well under-stood. Type I enzymes, which were discovered first, are notused commercially because of their complicated subunit struc-ture and cleavage behavior. They are encoded by the hostspecificity of DNA (hsd) genes; for a review see [1]. Twocopies of the HsdM methylation subunit and one copy ofspecificity subunit HsdS bind at a specific asymmetric DNAsite and form a methyltransferase complex that modifies thehost DNA, protecting it from cleavage. When invading DNAis detected by the methyltransferase based on its methylationstatus, twoHsdR endonuclease/motor subunits are recruited tothe complex, which then translocates up to thousands ofduplex base pairs through the stationary enzyme withoutunwinding the DNA, consuming ∼1 ATP per base pair. Iftranslocation is blocked, the endonuclease activity ofHsdR is triggered, and DNA is cleaved nonspecificallyat sites distant from the original binding site. These un-usual activities have prompted many years of genetic andbiochemical study aimed at understanding the molecular
Dhiraj Sinha and Katsiaryna Shamayeva contributed equally.
This paper belongs to Topical Collection MIB 2013 (ModelingInteractions in Biomolecules VI)
Electronic supplementary material The online version of this article(doi:10.1007/s00894-014-2334-1) contains supplementary material,which is available to authorized users.
D. Sinha :K. Shamayeva :D. Řeha :V. Bialevich :M. Khabiri :E. Csefalvay : J. Carey :R. EttrichInstitute of Nanobiology and Structural Biology, Global ChangeResearch Center, Academy of Sciences of the Czech Republic,Zamek 136, 373 33 Nove Hrady, Czech Republic
D. Sinha :K. Shamayeva :D. Řeha :V. Bialevich : R. Ettrich (*)Faculty of Sciences, University of South Bohemia in CeskeBudejovice, Zamek 136, 373 33 Nove Hrady, Czech Republice-mail: [email protected]
V. Ramasubramani :N. Milbar : J. CareyChemistry Department, Princeton University, Princeton,NJ 08544-1009, USA
A. Guzanová :M. WeiserováLaboratory of Molecular Genetics of Bacteria, Institute ofMicrobiology, Academy of Sciences of the Czech Republic,Vídeňská 1083, 142 20 Praha 4, Czech Republic
J Mol Model (2014) 20:2334DOI 10.1007/s00894-014-2334-1

mechanisms and physiological role of type I enzymes, butmany questions remain unanswered.
The X-ray crystal structure of HsdR with bound ATP fromthe type I enzyme system from plasmid EcoR124I (Fig. 1),solved in 2009, shed some light on these questions and raisednew ones [2]. Sequence similarities among type I enzymes arevery weak, and thus only limited assignments of functioncould be made for EcoR124I HsdR prior to structure determi-nation. Two so-called DEAD-box helicase motifs with variantsequences [3] could be identified in the primary structure, aswell as three endonuclease active-site residues [4] and aQxxxY motif common to the RecB-like nucleases [5]. Thestructure revealed that EcoR124I HsdR is organized into fourdistinct globular domains in a square-planar arrangement: anN-terminal endonuclease domain, two RecA-like helicasedomains that together carry out the translocase function, anda C-terminal helical domain that is presumed to contact meth-yltransferase. An additional ∼150 C-terminal residues—con-firmed to be present in the crystallized protein but unresolvedin the structure—are presumed to be part of the helicaldomain.
The endonuclease domain contains a typicalαβα core foldshared by many type I and II enzymes [6], with the catalyticresidues identified in the sequence forming a typical endonu-clease active site, as expected. The QxxxY motif, which wasassigned an auxiliary role during cleavage [5], is located nearthe endonuclease active site, although part of it resides on achain segment that was unresolved in the crystal structure. Thevariant DEAD-box motifs are found in typical locations in theRecA-like helicase domains that form the translocase, withone ATP bound between them. Unexpectedly, this ATP also
contacts the endonuclease domain via Lys220, a residue lying∼20 Å from the endonuclease active site on a short helicalsegment that is not part of the core fold. Such a contact had notbeen detected in other structures of helicases or translocases,where additional domains or subunits are absent either fromthe enzymes themselves or from the solved structures.
The Lys–ATP contact suggests a potential means for cou-pling helicase and endonuclease activities during enzymeactivation. However, the structure itself represents only asingle snapshot of the conformers the protein must exploreduring the catalytic cycle. Molecular dynamics simulations(MD) can help to fill out the picture by exploring how theprotein responds as it samples conformational space. In thepresent work, MD suggests a role for the Lys–ATP contact,which is then tested with in vivo and in vitro experiments. Theresults indicate how local interactions are transduced to do-main motions within the endonuclease/motor subunit.
Materials and methods
Molecular dynamics The crystal structure of the motor sub-unit HsdR from the restriction-modification system EcoR124I(PDB entry 2 W00) and a recent crystal structure of HsdRmutant Lys220Ala (PDB entry 4BEC; manuscript in prepara-tion) were used to prepare a wild-type (WT) protein structurefor the simulations by modeling four short segments missingfrom the published crystal structure. In a first step, threesegments (residues 142–147, 585–590, and 859–869) werebuilt using standard loop modeling in YASARA [7, 8] andadded to the modeled WT structure. The missing segment
Fig. 1 HsdR. Structure of themotor subunit. The four domainsare color-coded: yellowendonuclease, cyan helicase 1,magenta helicase 2 (whichtogether form the translocase);green helical. The structurecomprises residues 13–892 of thepolypeptide sequence of nativeHsdR. ATP is shown as a skeletalmodel with cyan carbons.Selected side chains relevant forthis work are also indicated asskeletal models: Lys220, whichcontacts ATP, as well as the threeconserved catalytic site residues(Asp151, Lys165, and Glu167)are shown in multiple colors withcyan carbons; and Arg182,together with the modeled 180sloop comprising residues 182–189 that contain part of theQxxxY motif, is shown in red
2334, Page 2 of 13 J Mol Model (2014) 20:2334

from residue 182 to residue 189 is resolved in the Lys220Alamutant crystal structure and thus was built into the modeledWT structure by adding the coordinates from the mutantcrystal structure, followed by steepest-descent energy minimi-zation with a maximum step size of 0.05 Å. GROMACSversion 4.55 [9–11] was used to prepare the system andperform MD simulations using the AMBER99SB force field[12]. ATP was parameterized by applying the standard RESPprocedure using Antechamber [13], with charges for freeMgATP derived from HF/6-31G* calculation in Gaussian03[14]. The resulting parameter file in Gromacs format is at-tached as “Electronic supplementary material” (ESM). Histi-dine was assumed to be charged, with the Nδ and Nε atomsprotonated; arginine and lysine residues were assumed to beprotonated. The Arg182Ala mutant was generated in silicofrom the above WT structure by replacing the Arg side chainwith a methyl group.
Both the WT and Arg182Ala structures were solvated byexplicit TIP3P water [15], and the systems were brought toneutrality by adding sodium counterions. The particle-meshEwald method [16] was applied to calculate long-range elec-trostatic interactions with a cutoff distance of 10 Å, and aLennard–Jones 6–12 potential was used to evaluate van derWaals interactions within 10-Å cutoff distance. The LINCSalgorithm of fourth-order expansion was used to constrainbond lengths [17]. After solvation and neutralization steps,each system was optimized for 10,000 steps using thesteepest-descent method to remove steric clashes betweenatoms, and then equilibrated for 1 ns with position restraintsof 1,000 kJ/mol on all heavy atoms. A constant temperature of300 K was maintained using the V-rescale algorithm [18],with a coupling time of 0.1 ps and separate baths for the soluteand the solvent. The pressure was kept constant at 1 bar usingthe Parrinello–Rahman pressure coupling scheme [19] with atime constant of 2 ps. Initial velocities were generated ran-domly using a Maxwell–Boltzmann distribution correspond-ing to 300 K. Neighbor lists were updated every 10 fs using agroup cutoff scheme. The production runs were performed for100 ns without restraints at 300 K in the isothermal–isobaric(NPT) ensemble.
DynDom [20, 21] was used to carry out a protein domainmotion analysis, which permits the identification of movingdomains, defines the screw axis, and measures the degree ofrotation between two conformers. Principal-components anal-ysis [22] was used to identify the global motions of thesystems, taking into account only Cα atoms.
QM/MM calculations QM/MM calculations were performedby the program QSite from the Schrödinger package [23].ATP was selected as the QM region and the rest of the systemas the MM region. Solvation was treated implicitly using thePoisson–Boltzmann approach implemented in QSite. The QMenergies as well as the electrostatic contribution to QM/MM
coupling energies were calculated using Gaussian03 by thedensity functional theory method with a B3LYP functionalincluding additive dispersion (treated by DFTD3 [24]), usingthe 6–31G* basis set. The basis set superposition error wastreated by the counterpoise correction method [25]. Contribu-tions to the QM/MM coupling energy from van der Waalsinteractions were calculated by the MM program Impact [26]using the OPLS2005 force field [27]. To calculate the contri-bution of each residue in the ATP binding pocket, eachcontacting residue was replaced in turn by Gly. All molecularstructures were inspected using a molecular visualization pro-gram, either VMD [28] or YASARA [7].
Site-directed mutagenesis, protein expression andpurification Plasmid pTrcR124 [29] carrying the hsdR genewas used for site-directed mutagenesis. Oligonucleotideprimers were used in the polymerase chain reaction (forwardprimer sequence is given, reverse primer is its complement;mutated codon underlined, changed nucleotides in bold: 5′-CCAGATACATGCTTACAGTAAAGAGAG-3′). The poly-merase chain reaction was performed in 200-μl PCR tubes inan Eppendorf (Hamburg, Germany) mastercycler. The 20-μlreaction mixture contained 1.5 U Expand Long Range poly-merase (Roche, Basel, Switzerland); 1X Expand Long RangeBuffer with MgCl2; 100 ng of plasmid DNA pTrcR124 124;0.3 μM each of the forward and reverse primers; 200 μMdNTPs; and 3 % or 6 % DMSO. PCR cycles were: 2 min at96 °C; 30 × (30 s at 96 °C, 90 s at 55 °C, 8min at 68 °C); 7 minat 68 °C. DpnI (20 U) was used to degrade methylatedparental plasmid. The reaction mixture was transformed intoE. coli DH5α competent cells grown on LB-agar plates con-taining 100 μg/ml of ampicillin. Plasmid DNA was isolatedusing a Zyppy plasmid miniprep kit (Zymo Research, Irvine,CA, USA) and sequenced.
E. coliBL21(DE3) Gold competent cells were transformedwith plasmid DNA containing the desired mutation. Over-night cultures were diluted 1:100 into 0.5 l of fresh LBmedium in a 3-l flask supplemented to a final concentrationof 100 μg ml−1 with ampicillin and incubated at 180 rpm at37 °C until OD600 was about 0.5–0.6. IPTG was added to afinal concentration of 1 mM and the culture was incubated foran additional 4 h at 37 °C. Harvested cells were washed insodium chloride–Tris-EDTA buffer, pH 8.0. Both the WTandArg182Ala mutant HsdR subunits were purified as describedpreviously [29]. EcoR124I methyltransferase was expressedfrom pAC15M [30] and pJS491 [31] plasmids encodingHsdM and HsdS subunits, respectively, and purified as de-scribed previously [32]. Purified methyltransferase and HsdRsubunits were mixed together in a 1:6 ratio to reconstitute theEcoR124I enzyme complex in vitro.
In vivo restriction activity assays Positive and negative com-plementation analyses were used to determine the restriction
J Mol Model (2014) 20:2334 Page 3 of 13, 2334

phenotype of EcoR124I bearing theWTor Arg182Ala mutantHsdR subunit in a plate assay measuring the ability of virulentλ phage [33] to lysate E. coli. For negative complementation,the pTrcR124 plasmid expressing WT or Arg182Ala mutantHsdR was transformed into E. coli JM109(DE3)[pKF650], arestricting (r+m+) host containing all three hsd genes ofEcoR124II. For positive complementation, pTrcR124 wastransformed into E. coli JM109(DE3)[pACMS] expressingonly the EcoR124II methyltransferase (r−m+) [31]. Soft agarat 45 °C was mixed with 0.5 ml of each fresh overnightculture, gently mixed, and immediately poured onto agarplates with appropriate antibiotics. Solidified soft agar wasspotted with 30 μl each of tenfold serial dilutions of λvir.0phage lysate at 102 to 106 plaque-forming units/ml. The spotswere dried at room temperature and the plates incubatedovernight at 37 °C [34]. The efficiency of plating was deter-mined as the number of plaques on the tested strains comparedto the number of plaques on the non-restricting control strainE. coli JM109(DE3) [35]. Values in the range 0.0001–0.01correspond to the restriction-competent (r+) phenotype, thosein the range 0.01–0.1 to the mixed-competence (r±) pheno-type, and those in the range 0.1–1 to the restriction-deficient(r−) phenotype.
In vitro cleavage activity assays Cleavage activity wasassayed in vitro using supercoiled plasmid DNA substratepRK [32] carrying a single recognition site for EcoR124I, orlinear plasmid DNA substrate pDRM-2R [29] carrying tworecognition sites for EcoR124I. Supercoiled plasmid DNApDRM-2R was linearized with ScaI (Fermentas) restrictionenzyme and the digested product was gel-purified with DNAClean and Concentrator from Zymo Research. Following[29], reaction mixtures contained restriction buffer (50 mMTris–HCl pH 8.0, 1 mM DTT, 10 mMMgCl2, 50 mM NaCl)with either 15 nM supercoiled pRK plasmid, 15 nMmethytransferase, and 90 nM HsdR, or 5 nM linear pDRM-2R plasmid, 50 nM methytransferase, and 300 nM HsdR.After 1 min of pre-incubation at 37 °C, reactions were startedby adding ATP and SAM to final concentrations of 4 mM and0.2 mM, respectively. Reactions were stopped by adding 0.25vol of stop solution (3 % SDS, 0.15 M EDTA, 10 % glycerol,0.1 % bromophenol blue) and heating at 65 °C for 5 min.Samples were loaded into 1 % agarose gels in Tris-acetate-EDTA buffer and run at 5 V/cm for 2 h for supercoiledsubstrates or 1.25 V/cm for 5 h for linear substrates. Gels werestained with 2 μg/ml ethidium bromide, destained in water,and photographed under UV illumination. The relativeamounts of DNA were evaluated by densitometry usingImageJ software [36].
In vitro ATPase activity assays Assays of DNA-dependentATPase activity were carried out in 20 μl of reaction mixturecontaining restriction buffer, 90 nM supercoiled pRK plasmid,
15 nM methyltransferase, and 90 nM HsdR subunit. After1 min of pre-incubation at 37 °C, the time course was initiatedby adding ATP to a final concentration of 4 mM. Aliquots of20 μl withdrawn at the indicated time points were stopped byadding 20 μl of 0.1 M EDTA (pH 8.0). The concentration ofinorganic phosphate released was measured by spectrophoto-metric quantification of a phosphomolybdate–malachite greencomplex [37]. The 40-μl samples were mixed in ELISAmicroplate wells with 150 μl of reagent (5.72 % ammoniummolybdate in 6 M HCl, 0.0812 % aqueous malachite green,and H2Omixed in a 1:2:3 ratio immediately before use). After5 min, the plates were scanned at 620 nm in a microplateELISA reader. The quantity of inorganic phosphate was de-termined with a calibration curve derived from solutions ofknown KH2PO4 concentration.
Results
QM/MM calculations Quantum mechanical/molecular me-chanical calculations were used to assess the energy contribu-tions of the contacting side chains to the binding of ATP in thecrystal conformation of EcoR124I HsdR. Solvation was treat-ed implicitly using the Poisson–Boltzmann approach, andATP and Mg ion were taken as the QM region at the DFTB3LYP/6-31G level of theory with DFTD3 dispersion correc-tion [24]. The protein was taken as the MM region in theOPLS force field [27]. The contribution of each side chain wascalculated as the difference between the summed electrostatic,van der Waals, and solvent contributions when the side chainis present and when it is replaced by a hydrogen atom. Thetotal binding energy of ATP is −118.5 kcal/mol (Table 1),
Table 1 Energy contributions (kcal/mol) to the ATP binding of aminoacid residues in the binding pocket
ΔEelec(QM/MM) ΔEvdw(MM) ΔEsolv(MM) ΔEbinding
WT −480.8 −51.9 414.2 −118.5Lys220a −91.8 −1.3 76.9 −16.2Gln276a −17.8 −1.4 −4.7 −23.9Lys313b −160.7 −0.5 70.0 −91.2Thr314b −33.0 1.0 −19.8 −51.8Asp408b 52.9 −0.5 −44.7 −7.7Glu409b 83.1 −0.5 −117.8 −35.2Asp664b 65.7 −0.19 −57.4 8.1
Arg688b −129.5 0.8 100.5 −28.2Arg691c −149.2 −3.1 112.4 −39.9
a Residue in contact with the adenine ringb Residue in contact with phosphate/Mgc Residue in contact with the ribose
2334, Page 4 of 13 J Mol Model (2014) 20:2334

reflecting interactions from nine residues. Each of these resi-dues was replaced in turn by a hydrogen atom to determine itsindividual contribution. The majority of the ATP bindingenergy is contributed by contacts to Mg ion and phosphatefrom six residues (Arg691, Asp408, Glu409, Lys313, Arg688,and Asp664), which each contribute total energies ranging upto −91 kcal/mol. One residue, Arg691, also contacts the ribosering with an energy contribution ofapproximately−40 kcal/mol. Two residues (Gln276 and Lys220) contact the adeninebase, with contributions of −23.9 kcal/mol and −16.2 kcal/mol, respectively. Thus, the energy contribution of Lys220 toATP binding is of a magnitude comparable to that of the onlyother contact to the adenine ring, indicating that the Lys220–ATP contact is likely to be functionally relevant rather than afortuitous contact or an artifact of crystallization.
Tyr183 Residue Tyr183 in the QxxxY motif of EcoR124I isconserved among HsdR homologs [38]. Biochemical charac-terization of a Tyr183Ala mutant of EcoR124I HsdR [5]revealed that the rate of nicking of the first DNA strand isdecreased more than twofold due to a >3.5-fold increase in thelag time before the first successful nicking event, leading toseverely impaired cleavage of the second strand. DNA-dependent ATPase activity of the Tyr183Ala mutant is similarto that of the wild-type enzyme, implying that translocationactivity is unaffected. MD was used in the present work toexplore the role of Tyr183 in enzyme function.
In the published X-ray crystal structure of EcoR124I HsdRwith bound ATP (Fig. 1) [PDB entry 2 W00; 2], the QxxxYmotif at residues 179–183 is only partially resolved, withresidues Arg182 and Tyr183 of the motif in unresolved seg-ment 182–189. A recent crystal structure of HsdR mutantLys220Ala (PDB entry 4BEC; manuscript in preparation)resolves this region completely, revealing that the loop liesabove the endonuclease active site with the motif pointingtoward the 220s loop bearing Lys220, which contacts ATP.The crystal structure of Lys220Ala HsdR was thus used as atemplate to model the unresolved 180s loop. Three other shortloop regions that were unresolved in the published crystalstructure (residues 142–147, 585–590, and 859–869) wereadded by standard loop modeling in YASARA, as describedin “Methods.” The resulting modeled structure (Figs. 1 and 2a)is referred to here as “WT HsdR” to distinguish it from themutant Arg182Ala discussed later, although the WT HsdRdiscussed here is distinct from the original crystal structure ofthe wild-type protein in that its missing short loop segments aremodeled. Themodeled structure is complete from residue 13 toresidue 892, and was used to prepare systems for MD simula-tion using standard methods.
Typical simulations of WT HsdR, run for 100 ns, achievedequilibrium after ∼50 ns as judged from the Cα root-mean-square deviation (RMSD) (see Fig. 1 in the ESM). RMSDvalues relative to the starting structure stabilize at ∼3 to
∼3.5 Å, a reasonable value considering the very large sizeand multiple domains of HsdR. Root-mean-square fluctuation(RMSF) analysis (Fig. 2 in the ESM) shows that, except forthe helical C-terminal domain, RMSF values are ∼0.5 Å with-in secondary structure elements, and the highest values ofmean fluctuation for Cα atoms are ∼2 Å in segments locatedbetween secondary structure elements. The correspondencebetween RMSF values and secondary structure is shown inFig. 3 of the ESM for the endonuclease domain, the mainfocus of this work. Starting around residue 725, RMSF valuesbecome much larger, ranging from 1 to >4 Å (Fig. 2 of theESM). These values presumably reflect the fact that part of thehelical domain is disordered in the crystal structure. Thus,useful information about the helical domain is not availablefrom these simulations.
In the initial modeled structure of WT HsdR used here, the180s loop borrowed from the Lys220Ala mutant structurepresents Tyr183 of the QxxxYmotif facing the solvent shortlybeyond the C-terminus of helical segment 6. Secondary struc-ture assignment in the PDB by the standard algorithm DSSP(Define Secondary Structure of Proteins) places residues 172–180 in helix 6. Residues 181–183 are still within the allowedhelical region of (φ,ψ) space, and form a helix-like structure.Residue 184 is in the generously allowed beta region of (φ,ψ)space, and begins an irregularly structured loop segment.Thus, Tyr183 is the last residue in the helix-like conformation,marking the border between a helix-like prolongation of helix6 and the subsequent loop. The outward orientation of Tyr183is maintained throughout the simulations; at no time is thehydroxyl of this residue observed to approach closer than 8 Åto nearby elements of the endonuclease domain that couldexplain the pronounced effects of the Tyr183Ala mutationin vivo and in vitro. However, during inspection of the behav-ior of Tyr183 in these trajectories, it was observed that thebehavior of the adjacent residue, Arg182, is strongly coupledto the behavior of the loop bearing Lys220. This observationled to the detailed analysis of Arg182 in these simulations thatis presented below; to wet experiments analyzing the behaviorof an Arg182Ala mutant HsdR in vivo and in vitro; and tosimulations with an Arg182Ala virtual mutant.
Lys220 The initial hydrogen-bonding interaction between theLys220 ε-amino group (Nζ) and adenine ring atom N3(AdeN3) observed in the crystal structure is rapidly lost andnot regained in all production runs of the WT HsdR structureanalyzed in this work. Other ATP interactions with compara-ble energies are maintained; in fact, no other ATP contact islost during these simulations. QM/MM calculations indicatethat this interaction (interatomic distance Lys220Nζ to AdeN3is 3.09 Å in the published crystal structure, contributingapprox.−16 kcal/mol) is of similar magnitude to, e.g.,Gln276 (approx.−24 kcal/mol). The mean±s.d. distance be-tween Lys220Nζ and AdeN3 during the equilibrated last 50 ns
J Mol Model (2014) 20:2334 Page 5 of 13, 2334

of the WT simulation is ∼7±∼1 Å (Fig. 2b). Instead ofinteracting with AdeN3, the Lys220 side chain turns towardthe adjacent Asp219 and forms a hydrogen bond with itsside-chain carboxylate Oδ1 atom that persists for theentire simulation. The motion of the 220s loop is in thedirection of the 180s loop, and brings the backbonecarbonyl Ο atom of Asn221 within hydrogen-bondingdistance of the Arg182 guanidino Nε atom, from aninitial distance of 3.24 Å to a mean equilibrated distanceof ∼2.9±0.3 Å. A hydrogen bond is formed betweenthese two atoms that persists for the entire simulation.The distance between Lys220Cα and Arg182Cα duringthe equilibrated part of WT simulations is ∼11±∼0.5 Å,with the respective side chains coming no closer to eachother than ∼9.5 Å.
Loss of the Lys220–ATP contact involves not onlymovement of the Lys220 side chain and the 220s loopbut also rotational movement of the entire endonucleasedomain relative to the other three domains (Fig. 3a, c).Principal-components analysis shows that the domainrotates by ∼18° about a screw axis defined by the vector(0.75, 0.5, −0.43) through the center of mass of the
endonuclease domain in a coordinate system in whichthe domain is positioned as in Fig. 3a, with the z-axisorthogonal to the plane of the page. This screw axisapplies only to the endonuclease domain and not to theother three domains, which show no rotational motion andstay in place during the simulations, as is evident inFig. 3a and b. This rotation is in the same directionthough it is much smaller in magnitude than the rotationof the endonuclease domain observed in the X-ray crys-tal structure of a fragment of a putative HsdR proteinfrom Vibrio vulnificus (PDB ID 3H1T; [39]). Relative tothe crystal structure of EcoR124I HsdR, the Vibrioendonuclease domain is rotated by ∼180°; the screwaxis would be described by the same vector, but withthe axis shifted toward the center of mass of the endo-nuclease domain, i.e., downward toward the lower rightin Fig. 3a. Although the Vibrio HsdR polypeptide chainwas incomplete, the structure resolved both helicasedomains and the entire endonuclease domain, and thussuggests that a range of motion is possible for the latterdomain that is much greater than that observed in thepresent simulations.
Fig. 2a–c Local interactions in the ATP-binding site. The center of theplanar array of the four HsdR domains is shown in a zoomed-in view withMg-ATP in the top center. This viewpoint was chosen to visualize as manyof the relevant interactions as possible, and is the same in all three panels.The polypeptide chain is shown as a gray tube, and residues involved inhydrogen-bonding interactions discussed in the text are shown as sticks.Because some hydrogen bonds (black dashed lines) are obscured regard-less of viewpoint, residues of interest are identified by unique solid colorswhen they are not involved in hydrogen bonding, or atomic colors withcyan carbons when they are involved in hydrogen bonds, except for their
Cα atoms, which retain the unique color in order to permit the identifica-tion of each residue. Gold Mg-ATP, orange Asp219, green Lys220,magenta Asn221, purple Arg182, gray Tyr183. a. The initial modelstructure used for simulations of WT HsdR, prepared from the X-raycrystal structure of HsdR by adding four short loop structures as describedin the text. The hydrogen bond indicated between the Lys220 Nζ andadenine N3 atoms is inferred from the distance and angle in the crystalstructure. b. A typical snapshot from the equilibrated part of the simulationof WT HsdR. AdeN3 is marked with an asterisk. c. A typical snapshotfrom the equilibrated part of the simulation of restrained WT HsdR
2334, Page 6 of 13 J Mol Model (2014) 20:2334

Arg182 The conformation of the 180s loop borrowed fromthe Lys220Ala mutant structure, when modeled into the WTstructure, places the 180s loop in proximity of the 220s loop,where Lys220 is engaged with ATP. The loss of the Lys220–ATP contact during simulations starting from this structurecould reflect that this conformation of the two loops might beincompatible. The persistence of the Arg182–Asn221 interac-tion during the entire WT simulation upon the loss of theLys220–AdeN3 contact further suggests that these two inter-actions may reflect alternative states of the system. Thus, toenable observation of the behavior of the 180s and 220s loopswhile ATP is engaged by the endonuclease domain (the stateof the enzyme detected crystallographically), additional sim-ulations were prepared and run in which the Lys220–AdeN3interaction was maintained artificially via a restraining force.Due to technical difficulty in applying an intermolecular dis-tance restraint in Gromacs, the restraint was accomplished byidentifying the backbone nitrogen atom of Arg273 as a locuswhere an applied distance restraint to Lys220Nζ would keepthe Lys220 ε-amino group within hydrogen-bonding dis-tance of AdeN3. The force constant of the distancerestraint was incremented until the Lys220Nζ–AdeN3interaction was maintained at a distance of ∼3.5±∼0.5 Å
during the entire simulations, which are hereafter referred toas restrained WT simulations. The final force constant usedwas 5,000 (kJ/mol nm2). A representative snapshot from therestrained WT simulation is shown in Fig. 2c. The position ofthe endonuclease domain corresponds to the crystal structureand no global rotational movement is observed, i.e., rotationabout the screw axis in Fig. 3b and d is 0°. In these simula-tions, the distance from Arg182Cα to Lys220Cα is ∼15±0.5 Å, with the respective side chains coming no closer toeach other than ∼13.5 Å.
Figure 4 compares relevant distances that illustrate themovements of the 180s and 220s loops in the final equilibratedstructures of the WT (blue lines) and restrainedWT (red lines)simulations. Panel a shows the mean±s.d. distance betweenLys220Nζ and AdeN3 in the two simulations, and panel bshows the mean±s.d. distance between Asn221Cα andArg182Cα. When the Lys220–ATP contact is engaged, as inthe restrainedWTsimulation (red line in panel a), the 180s and220s loops are far apart (red line in panel b), with a well-defined and narrowly distributed distance between Asn221Cαand Arg182Cα (∼10.8±∼0.4 Å). This distance is too large topermit atomic contacts between the loops, and indeed nohydrogen bonds or salt bridges between them are detected in
Fig. 3a–d Endonuclease domain rotation. The planar array of four HsdRdomains is shown in tube representation. The coordinate system used todefine the rotation is indicated at the center of the panels: red x-axis, greeny-axis, blue z-axis. The vector describing the rotation of the endonucleasedomain is shaded in black to gray from near to far, and the direction ofrotation is indicated by the circle arrows at the origin of the vector. For thesake of orientation in Figs. 1 and 2, the center of the vector shown by thegray sphere is approximately the position of the bound Mg ion. a. Theaverage conformation in the equilibrated part of the restrained simulationof WT HsdR is shown in gray. The conformation in the equilibrated part
of the WT HsdR simulation is overlaid and, except for the endonucleasedomain (red), it is not shown because it is identical. b. Extreme positionsalong the first eigenvector calculated by principal-components analysisfor the second half of the simulation of Arg182Ala HsdR. The extremeshown in gray corresponds to the conformer at 56 ns. When it is overlaidon the other extreme corresponding to the conformer at 39 ns (notshown), it is identical, except in the endonuclease domain (red). c As inpanel a with a view along the screw axis from the near end. dAs in panelb with a view along the screw axis from the near end
J Mol Model (2014) 20:2334 Page 7 of 13, 2334

the restrained WT simulation. On the other hand, when theLys220–ATP contact is lost and Lys220 forms a persistentinteraction with Asp219, as in the WTsimulation (blue line inpanel a), the 180s and 220s loops are in close contact with anarrowly distributed distance from Asn221Cα to Arg182Cαof ∼7.9±∼0.5 Å. The finding that loss of the Lys220–ATPcontact is coupled to domain movement, together with thefinding that the behavior of the 180s loop is coordinated viaArg182 with the behavior of Lys220, suggests a role forArg182 in communicating the ATP-ligation state through theendonuclease/motor subunit.
Ala182 To test this hypothesis, a mutation of Arg182 to Alawas introduced both in silico and by in vitro mutagenesis. Theinitial conformation of the virtual mutant is identical to theinitial WT conformation except for the missing Arg182 sidechain, which was replaced by in silico mutation with a methylgroup. Simulations using the Arg182Ala virtual mutant struc-ture were prepared and run for at least 75 ns, with equilibrationduring the final ∼20 ns as judged from the RMSD (Fig. 1c inthe ESM). RMSF values (Fig. 2c in the ESM) are similar tothose reported above for WT and restrained WT simulations.
The Lys220Nζ–AdeN3 and Ala182Cα–Asn221Cα dis-tances in the Arg182Ala simulation are superimposed ontothe WT and restrained WT simulation distances in Fig. 4(black traces). As in WT, the contact of Lys220 with ATP islost early in the Arg182Ala simulation (panel a). This resultindicates that the 220s loopmoves away fromATP even whenthere is no long charged side chain at residue 182. Themovement is toward the 180s loop initially, and for the first∼30 ns the simulation approximately follows the course of theWT simulation. However, after ∼30 ns the distance between
Lys220Nζ and AdeN3 increases further, reaching a maximumof ∼17.4 Å at ∼40 ns. The fact that the Lys–ATP distancecontinues to increase beyond the distance observed in the WTsimulation suggests that the 220s loop enjoys more degrees offreedom than in the WT structure, perhaps because of theabsence of the Arg182 side chain that in the WT simulationinteracts persistently with Asn221.
This picture of the role of Arg182 is reinforced by analysisof the Ala182Cα–Asn221Cα distance (Fig. 4b), which showsthat the two loops explore a range of distances but with nocorrelation in time between their movements. Thus, in theArg182Ala mutant, the interloop distance is insensitive tothe contact between Lys220 and ATP. As in the WT simula-tion, the Arg182Ala simulation also develops a pronouncedrotation of the endonuclease domain (maximum indicated byarrow at 39 ns) about the same screw axis as in the WTsimulation, but the rotation is only ∼11° (Fig. 3b and d). Therotation is not found in the equilibrated part of the mutantsimulation, where the rotation is zero, but occurs only in thefirst 40 ns, when the system is not yet equilibrated. Themaximum rotation of the endonuclease domain coincides withthe maximum in the distance between Lys220Nζ andAdeN3 at the ∼40-ns time point. Thus, the degree of rotationis also insensitive to the contact between Lys220 and ATP.
In the second half of the Arg182Ala simulation, Lys220slowly returns to a position close to the initial one, and after∼55 ns it approaches ATP again. During this time period,rotation occurs in the opposite direction, toward the non-rotated position of the endonuclease domain observed in thecrystal (arrow at 56 ns). During the short (20 ns) equilibratedpart of the simulation that could be observed, the Lys220Nζ–AdeN3 hydrogen bond is not fully re-established and
Fig. 4a–b Correlated and uncorrelated loop motions. Interatomic dis-tances are plotted as a function of simulation time during the Arg182Alamutant simulation (black traces). Horizontal heavy lines are the meandistances over the equilibrated part of the WT simulation (blue lines) orthe restrained WT simulation (red lines). Lighter horizontal lines mark
one standard deviation from the mean distance. Conformers correspond-ing to the extremes along the first eigenvector in the principal-compo-nents analysis are indicated by arrows: 39 ns rotated conformer, 56 nsunrotated conformer. a Lys220Nζ–AdeN3. b Arg182Cα–Asn221Cα
2334, Page 8 of 13 J Mol Model (2014) 20:2334

hydrogen-bond distances are seen in only a few snapshots.These results indicate that the rotated conformation with themaximum Lys220–ATP distance does not persist in theArg182Ala mutant simulation as it does in theWTsimulation.Thus, the mutant is apparently not trapped in the alternativerotated conformation that traps theWTstructure. These resultsfor the Arg182Ala virtual mutant indicate that Arg182 plays astrong role in the switch-like behavior observed in WT simu-lations, and that this role involves communicating the ATP-ligation state through the endonuclease/motor subunit by con-trolling a switch between mutually exclusive alternativestructures.
The rotational transition in the first half of the Arg182Alasimulation, and its reversal as the system equilibrates, raise thequestion of whether the presence of the Lys220–ATP contactis sufficient to prevent the endonuclease domain from rotating,or if the contact between the 180s and 220s loops plays a role.The first 40 ns of the WT simulation suggest that loss of theLys220–ATP contact initiates domain rotation. Mutation ofArg182 does not alter this behavior, and after 40 ns theendonuclease domain is rotated in both simulations. However,in the mutant, the rotated state does not equilibrate as in WT,but after 50 ns the domain rotates in the opposite direction inconcert with the movement of Lys220 toward ATP. This resultsuggests that a factor stabilizing the rotated conformation ispresent in WT HsdR but absent in the mutant, and that thisfactor involves Arg182. Comparison of panels a and b showsthat in the Arg182Ala virtual mutant the 180s to 220sinterloop distance changes independently of the Lys220–ATP distance and the endonuclease domain rotation, whereasin the WT simulation the interloop distance is dependent onthe Lys220–ATP distance. The presence of the Lys220–ATPcontact therefore appears to be insufficient by itself to preventendonuclease domain rotation unless a restraining force isapplied to maintain it or the contact made by Arg182 withthe 220s loop in the rotated conformation is absent.
Endonuclease activity in vivo The Arg182Ala mutant wascreated by site-directed mutagenesis. Its restriction phenotypewas determined in vivo by testing the ability of cells express-ing mutated or unmutated HsdR to restrict the growth ofunmodified bacteriophage λ. Positive and negative comple-mentation tests [40] were used to distinguish between defectsin DNA cleavage and defects in the interaction of mutantHsdR with HsdS-HsdM2 methyltransferase. These comple-mentation tests used plasmid pTrcR124 [29] expressing theunmutated or mutated hsdR gene transformed into restriction-proficient JM109(DE3)/pKF650 r+m+ or restriction-deficientJM109(DE3)/pACMS r−m+ cells, and the efficiency of platingof λ bacteriophage on these strains was determined using aplate assay, as described in “Methods.” Positive complemen-tation uses an r− host to test if a mutant HsdR subunit isdefective in DNA cleavage. Negative complementation uses
an r+ host to test if a mutant HsdR subunit that is defective inDNA cleavage is competent for assembly with methyltrans-ferase, allowing it to compete with unmutated HsdR subunits,reducing their restriction activity. This test showing a so-called trans-dominant effect can thus confirm a restriction-deficient mutant phenotype in an r− host based on the reduc-tion in restriction activity of the unmutated subunits producedin the r+ host.
The results of the positive complementation test (Table 2,r− host) reveal that the Arg182Ala mutant fails to complementrestriction in the r− host (value 0.2, within the range 0.1–1 forthe restriction-deficient phenotype) and is thereforerestriction-deficient. Unmutated HsdR (referred to in the tableas WT, but in this case indicating the natural, full-lengthprotein) complements restriction in the r− host, as expected(value 0.002, within the range 0.0001–0.01 for the restriction-proficient phenotype). The negative complementation test(Table 2, r+host) confirms this result, with 500-fold reducedrestriction by the mutant compared to unmutated HsdR in ther+host, consistent with a failure of restriction by the mutant inthe r− host. The reduced restriction activity of the Arg182Alamutant in the r+ host also confirms that the mutant subunit isfully competent for assembly with methyltransferase to formthe endonuclease complex. The in vivo tests thus indicate thatthe Arg182Ala mutation of HsdR exerts an effect on the DNAcleavage activity of the enzyme similar to that observed withTyr183Ala [5].
Endonuclease activity in vitro The Arg182Ala mutant wasoverexpressed and purified under the conditions used fornatural, full-length HsdR. The endonuclease activity ofunmutated and Arg182Ala mutant HsdRs was evaluatedin vitro using circular plasmid DNA with one EcoR124Irecognition site or linearized DNAwith two recognition sites.Following Janscak et al. [29], excess unmutated or Arg182AlaHsdRwas mixed with separately purified methyltransferase toreconstitute the EcoR124I enzyme complex in vitro. As
Table 2 In vivo restriction phenotype
Restrictiona
HsdR r− hostb r+ hostc
WT 0.002 0.0001
R182A 0.2 0.05
a Restriction activity is reported as the efficiency of plating of λvir.0 onthe indicated strains relative to the efficiency of plating of λvir.0 on thenonrestricting indicator strain E. coli JM109(DE3), as described in“Methods”b Positive complementation of r− m+ host E. coli JM109(DE3)[pACMS]by plasmid pTrc124 expressing WT or Arg182Ala mutant HsdRcNegative complementation of r+ m+ host E. coli JM109(DE3)[pKF650]by plasmid pTrc124 expressing WT or Arg182Ala mutant HsdR
J Mol Model (2014) 20:2334 Page 9 of 13, 2334
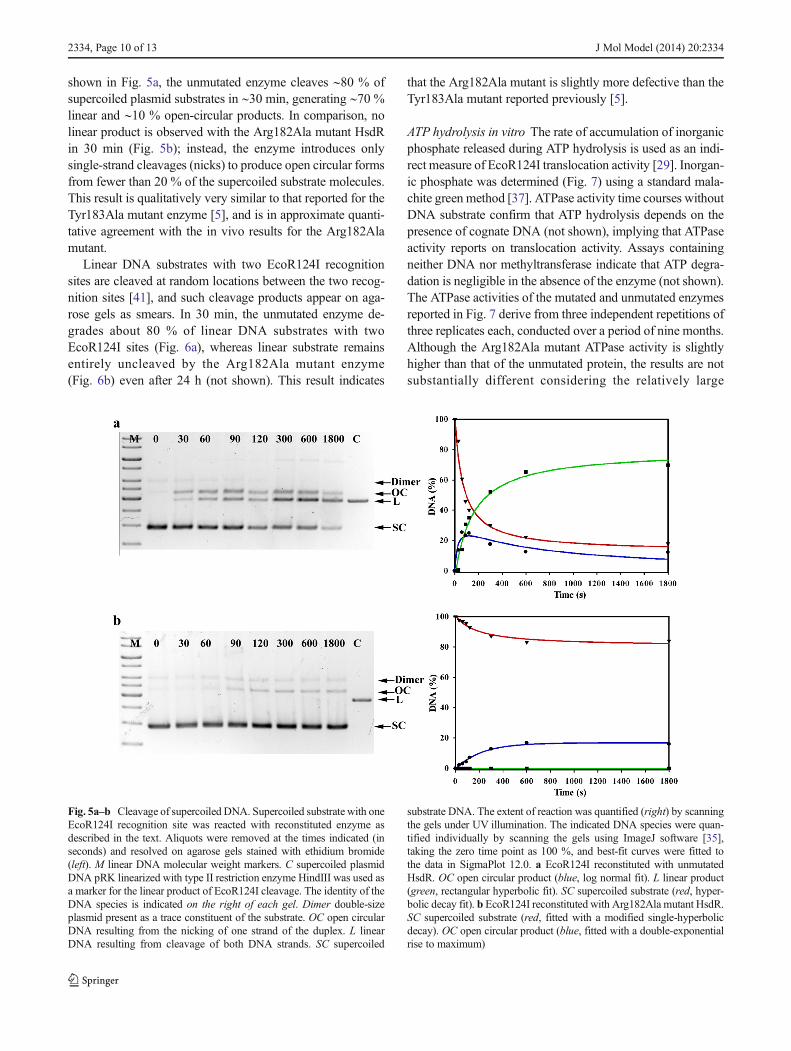
shown in Fig. 5a, the unmutated enzyme cleaves ∼80 % ofsupercoiled plasmid substrates in ∼30 min, generating ∼70 %linear and ∼10 % open-circular products. In comparison, nolinear product is observed with the Arg182Ala mutant HsdRin 30 min (Fig. 5b); instead, the enzyme introduces onlysingle-strand cleavages (nicks) to produce open circular formsfrom fewer than 20 % of the supercoiled substrate molecules.This result is qualitatively very similar to that reported for theTyr183Ala mutant enzyme [5], and is in approximate quanti-tative agreement with the in vivo results for the Arg182Alamutant.
Linear DNA substrates with two EcoR124I recognitionsites are cleaved at random locations between the two recog-nition sites [41], and such cleavage products appear on aga-rose gels as smears. In 30 min, the unmutated enzyme de-grades about 80 % of linear DNA substrates with twoEcoR124I sites (Fig. 6a), whereas linear substrate remainsentirely uncleaved by the Arg182Ala mutant enzyme(Fig. 6b) even after 24 h (not shown). This result indicates
that the Arg182Ala mutant is slightly more defective than theTyr183Ala mutant reported previously [5].
ATP hydrolysis in vitro The rate of accumulation of inorganicphosphate released during ATP hydrolysis is used as an indi-rect measure of EcoR124I translocation activity [29]. Inorgan-ic phosphate was determined (Fig. 7) using a standard mala-chite green method [37]. ATPase activity time courses withoutDNA substrate confirm that ATP hydrolysis depends on thepresence of cognate DNA (not shown), implying that ATPaseactivity reports on translocation activity. Assays containingneither DNA nor methyltransferase indicate that ATP degra-dation is negligible in the absence of the enzyme (not shown).The ATPase activities of the mutated and unmutated enzymesreported in Fig. 7 derive from three independent repetitions ofthree replicates each, conducted over a period of nine months.Although the Arg182Ala mutant ATPase activity is slightlyhigher than that of the unmutated protein, the results are notsubstantially different considering the relatively large
Fig. 5a–b Cleavage of supercoiled DNA. Supercoiled substrate with oneEcoR124I recognition site was reacted with reconstituted enzyme asdescribed in the text. Aliquots were removed at the times indicated (inseconds) and resolved on agarose gels stained with ethidium bromide(left). M linear DNA molecular weight markers. C supercoiled plasmidDNA pRK linearized with type II restriction enzyme HindIII was used asa marker for the linear product of EcoR124I cleavage. The identity of theDNA species is indicated on the right of each gel. Dimer double-sizeplasmid present as a trace constituent of the substrate. OC open circularDNA resulting from the nicking of one strand of the duplex. L linearDNA resulting from cleavage of both DNA strands. SC supercoiled
substrate DNA. The extent of reaction was quantified (right) by scanningthe gels under UV illumination. The indicated DNA species were quan-tified individually by scanning the gels using ImageJ software [35],taking the zero time point as 100 %, and best-fit curves were fitted tothe data in SigmaPlot 12.0. a EcoR124I reconstituted with unmutatedHsdR. OC open circular product (blue, log normal fit). L linear product(green, rectangular hyperbolic fit). SC supercoiled substrate (red, hyper-bolic decay fit). b EcoR124I reconstituted with Arg182Ala mutant HsdR.SC supercoiled substrate (red, fitted with a modified single-hyperbolicdecay). OC open circular product (blue, fitted with a double-exponentialrise to maximum)
2334, Page 10 of 13 J Mol Model (2014) 20:2334

variability in this manual method, much of which is attribut-able to unavoidable time variance in initiating the reactions.The ATPase results thus imply that the translocation properties
of the Arg182Ala mutant enzyme are similar to those of theunmutated enzyme, and to those reported for the Tyr183Alamutant enzyme [5].
Conclusions
This work has elucidated a role for the 180s and 220s loops inswitching between two conformations that represent alterna-tive states of ATP engagement by the HsdR subunit. Thealternate interactions of the 180s and 220s loops are connectedto rotation of the endonuclease domain, which may conveyinformation about ATP-ligation status to the enzyme complex.The results suggest that Arg182, though less strictly conservedthan Tyr183, plays a key role in the switch by its ability toengage in persistent distant interactions involving its longcharged side chain. The QxxxY sequences of other HsdRendonuclease/motor subunits typically present at least oneLys or Arg residue that may fulfill the role suggested herefor Arg182. In contrast, the solvent-exposed location ofTyr183 close to the catalytic site of HsdR in the presentsimulations, even in the absence of DNA, supports the view
Fig. 6a–c Cleavage of linear DNA. Linear substrate with two EcoR124Irecognition sites was reacted with reconstituted enzyme and analyzed asdescribed in Fig. 5. The parenthesis on the right side of panel amarks theregion of the smeared products that result from cleavage at randomlocations between the two recognition sites. a. EcoR124I reconstitutedwith unmutated HsdR. b. EcoR124I reconstituted with Arg182Ala mu-tant HsdR. c. Quantitation of the linear substrate DNA for unmutated
enzyme (black points, black line) and for Arg182Ala mutant enzyme(black points, red line). The decrease in the amount of DNA substrate wasquantified by scanning the gels using ImageJ software [35], taking thezero time point as 100 %. A single-exponential curve (black line) wasfitted to the data for the unmutated enzyme in SigmaPlot 12.0, taking thezero time point as 100 %
Fig. 7 ATPase activity. The concentration of inorganic phosphate re-leased in the DNA-dependent reaction is plotted as a function of time forenzyme reconstituted with unmutated (blue) or Arg182Ala mutant (red)HsdR. The error bars represent one standard deviation calculated fromthree independent replicates, each with triplicate determinations
J Mol Model (2014) 20:2334 Page 11 of 13, 2334

[5] that the common role of this residue in the RecB-familyhelicases and HsdRs may be its engagement with DNA. Thus,the present results suggest that in HsdRs the QxxxY motifgained an additional function in communicating a signalthrough the protein about its ATP-ligation status. The furtherpropagation of that switch through the multisubunit enzymecomplex remains a difficult challenge for future study.
Acknowledgments We gratefully acknowledge support from theCzech Science Foundation (P207/12/2323 to RE and MW), the institu-tional research project RVO 61388971, and joint Czech–US NationalScience Foundation international research cooperation (DBI10-04830).
References
1. Murray NE (2002) Immigration control of DNA in bacteria: selfversus non-self. Microbiology 148:3–20
2. Lapkouski M, Panjikar S, Janscak P, Smatanova IK, Carey J, EttrichR, Csefalvay E (2009) Structure of the motor subunit of type Irestriction-modification complex EcoR124I. Nat Struct Mol Biol16:94–105
3. Gorbalenya AE, Koonin EV (1991) Endonuclease (R) subunits oftype-I and type-III restriction–modification enzymes contain ahelicase-like domain. FEBS Lett 291:277–281
4. Obarska-Kosinska A, Taylor JE, Callow P, Orlowski J, Bujnicki JM,Kneale GG (2008) HsdR subunit of the type I restriction-modification enzyme EcoR124I: biophysical characterisation andstructural modelling. J Mol Biol 376(2):438–452
5. Sisakova E, Stanley LK, Weiserova M, Szczelkun MD (2008) ARecB-family nuclease motif in type I restriction endonucleaseEcoR124I. Nucleic Acids Res 36:3939–3949
6. Niv MY, Ripoll DR, Vila JA, Liwo A, Vanamee ES, Aggarwal AK,Weinstein H, Scheraga HA (2007) Topology of type II REasesrevisited; structural classes and the common conserved core.Nucleic Acids Res 35:2227–2237
7. Krieger E, KoraimannG, VriendG (2002) Increasing the precision ofcomparative models with YASARA NOVA; a self-parameterizingforce field. Proteins 47:393–402
8. Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM (2006)MUSTANG: a multiple structural alignment algorithm. Proteins 64:559–574
9. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE,Berendsen HJC (2005) GROMACS: fast, flexible, and free. JComput Chem 26:1701–1718
10. Berendsen HJC, van der Spoel D, van Drunen R (1995) GROMACS:a message-passing parallel molecular dynamics implementation.Comp Phys Comm 91:43–56
11. Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, ShirtsMR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E(2013) GROMACS 4.5: a high-throughput and highly parallel opensource molecular simulation toolkit. Bioinformatics 29(7):845–854
12. Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C(2006) Comparison of multiple AMBER force fields and develop-ment of improved protein backbone parameters. Proteins 65:712–725
13. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004)Development and testing of a general AMBER force field. JComput Chem 25:1157–1174
14. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA,Cheeseman JR, Pople JA et al. (2004) GAUSSIAN 03 (revisionC.02). Gaussian, Inc., Wallingford
15. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML(1983) Comparison of simple potential functions for simulatingliquid water. J Chem Phys 79:926
16. Darden T, York D, Pedersen L, Ewald P (1993) An N·log(N)method for Ewald sums in large systems. J Chem Phys 98(12):10089–10092
17. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: alinear constraint solver for molecular simulations. J Comp Chem18(12):1463–1472
18. Bussi G, Donadio D, Parrinello M (2007) Canonical samplingthrough velocity rescaling. J Chem Phys 126:014101
19. Parrinello M, Rahman A (1981) Polymorphic transitions in singlecrystals: a new molecular dynamics method. J Appl Phys 52:7182
20. Hayward S, Kitao A, Berendsen HJC (1997) Model-free methods ofanalyzing domain motions in proteins from simulation: a comparisonof normal mode analysis and molecular dynamics simulation oflysozyme. Proteins 27:425–437
21. Hayward S, Berendsen HJC (1998) Systematic analysis of domainmotions in proteins from conformational change; new results oncitrate synthase and T4 lysozyme. Proteins 30:144–154
22. Amadei A, Linnssen AB, Berendsen HJ (1993) Essential dynamicsof proteins. Proteins 17:412–25
23. Schrödinger LLC (2011) QSite 5.7. Schrödinger LLC, New York24. Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and
accurate ab initio parametrization of density functional dispersioncorrection (DFT-D) for the 94 elements H–Pu. J Chem Phys 132:154104–154123
25. Boys SF, Bernardi F (1970) Calculation of small molecular interac-tions by differences of separate total energies—some procedures withreduced errors. Molec Phys 19:553–556
26. Schrödinger LLC (2011) Impact 5.7. Schrödinger LLC, New York27. Jorgensen WL, Tirado-Rives J (2005) Potential energy functions for
atomic-level simulations of water and organic and biomolecularsystems. Proc Natl Acad Sci USA 102:6665–6670
28. Humphrey W, Dalke A, Schulten K (1996) VMD—visual moleculardynamics. J Mol Graph 14:33–38
29. Janscak P, Abadjieva A, Firman K (1996) The type I restrictionendonuclease R.EcoR124I: over-production and biochemical prop-erties. J Mol Biol 257(5):977–991
30. Holubova I, Vejsadová Š, Firman K, Weiserova M (2004) Cellularlocalization of type I restriction-modification enzymes is familydependent. Biochem Biophys Res Commun 319:375–380
31. Patel J, Taylor I, Dutta CF, Kneale G, Firman K (1992) High-levelexpression of the cloned genes encoding the subunits of and intactDNA methylase, M.EcoR124. Gene 112:21–27
32. Taylor I, Patel J, Firman K, Kneale GG (1992) Purification andbiochemical characterization of the EcoR124 type I modificationmethylase. Nucleic Acids Res 20:179–186
33. Jacob F, Wollman EL (1954) Etude génétique d’un bactériophagetempéré d’Escherichia coli. III. Effet du rayonnement ultraviolet surla recombinaison génétique. Ann Inst Pasteur 87:653–673
34. Colson C, Glover SW, Symons N, Stanley KA (1965) The location ofthe genes for host-controlled modification and restriction inEscherichia coli K-12. Genetics 52:1043–1050
35. Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phagecloning vectors and host strains: nucleotide sequences of theM13mp18 and pUC19 vectors. Gene 33:103–119
36. Abramoff MD, Magalhaes PJ, Ram SJ (2004) Image processing withImageJ. Biophoton Int 11(7):36–42
37. Chan KM, Delfert D, Junger KD (1986) A direct calorimetric assayfor Ca2+ stimulated ATPase activity. Anal Biochem 157:375–380
38. Davies GP, Martin I, Sturrock SS, Cronshaw A, Murray NE, DrydenDTF (1999) On the structure and operation of type I DNA restrictionenzymes. J Mol Biol 290:565–579
39. Uyen NT, Park S, Choi J, Lee HJ, Nishi K, Kim JS (2009)The fragment structure of a putative HsdR subunit of a type I
2334, Page 12 of 13 J Mol Model (2014) 20:2334

restriction enzyme from Vibrio vulnificus YJ016: implicationsfor DNA restriction and translocation activity. Nucleic AcidsRes 37:6960–6969
40. Sisakova E, Weiserova M, Dekker C, Seidel R, SzczelkunMD (2008) The interrelationship of helicase and nuclease
domains during DNA translocation by the molecular motorEcoR124I. J Mol Biol 384:1273–1286
41. Studier FW, Bandyopadhyay PK (1988) Model for how type Irestriction enzymes select cleavage sites in DNA. Proc Natl AcadSci USA 85:4677–4681
J Mol Model (2014) 20:2334 Page 13 of 13, 2334
Related Documents