In Out side inform + connect + improve 3101 Gaylord Parkway Frisco, Texas 75034 www.asdhealthcare.com Bad Bites Ahead gives you the knowledge you need to stay safe this snake bite season. Which B Strain? Find out what the experts advise for the 2011/2012 flu season. Save One Life brings hope to hemophilia patients in developing countries. Accountable Care Organizations? Discover opportunities and challenges for you. Also in this issue, see what’s In the Works, In the News and what’s happening in healthcare that affects your world – inside and out. This quarter, InsideOut magazine takes you into the great outdoors to enjoy nature – safely. Plus, you’ll get these stories to help you stay informed and connected to your healthcare communities. LOOK WHAT’S NEW in InsideOut! Hi Bernadette Rospigliosi, We hope you find the new issue of InsideOut and this reminder helpful in your healthcare community. — Compliments of your ASD Healthcare Sales Team. Yes, it’s time to plan this year’s flu programs, and ASD Healthcare can help. We offer advantages that reduce your purchasing risks. Now you can protect your community while protecting your facility’s bottom line with: FLU ALERT: PREBOOKED IS PREPARED! For more information, see Page 25 in this issue of InsideOut magazine. Call ASD Healthcare at 866.281.4FLU (4358) to prebook your vaccines today! Price protection. Best return policy. Most favorable delivery schedules. In Out side inform + connect + improve Inhibitor Insights: When to suspect an inhibitor page 18 Accountable Care Organizations Opportunities and Challenges page 36 Warning: BAD BITES AHEAD! an ASD Healthcare publication spring 2011 / 2012 Expert Flu Advice page 12 Social Media Does it work for pharma? page 17 48 page

InsideOut Spring Edition
Mar 08, 2016
ASD Healthcare's e-edition to quarterly publication
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

In Outsideinform+connect+improve
3101 Gaylord ParkwayFrisco, Texas 75034
www.asdhealthcare.com
Bad Bites Ahead gives you the knowledge you need to stay safe this snake bite season.
Which B Strain? Find out what the
experts advise for the 2011/2012 flu season.
Save One Life brings hope to
hemophilia patients in developing countries.
Accountable Care Organizations?
Discover opportunities and challenges for you.
Also in this issue, see what’s In the Works, In the News and what’s happening in healthcare that affects your world – inside and out.
This quarter, InsideOut magazine takes you into the great outdoors
to enjoy nature – safely. Plus, you’ll get these stories to help you stay
informed and connected to your healthcare communities.
Look what’s
NEWin InsideOut!
Hi Bernadette Rospigliosi,We hope you find the new issue of InsideOut and this reminder helpful in your healthcare community.— Compliments of your ASD Healthcare Sales Team.
Yes, it’s time to plan this year’s flu programs, and ASD Healthcare can help.
We offer advantages that reduce your purchasing risks. Now you can protect
your community while protecting your facility’s bottom line with:
FLU ALERT: PreBOOked IS PrePAred!
For more information, see Page 25 in this issue of InsideOut magazine.
Call ASd Healthcare at 866.281.4FLU (4358) to prebook your vaccines today!
Price protection. Best return policy. Most favorable delivery schedules.
In Outsideinform+connect+improve
Inhibitor Insights: When to suspect an inhibitor page 18
Accountable Care Organizations Opportunities and Challenges page 36
Warning: Bad Bites AheAd!
an ASD Healthcare publication s p r i n g
2011 / 2012Expert Flu Advice page 12
Social Media Does it work for pharma? page 17
48page

XYNTHADILUENT
Indication for XYNTHAXyntha® Antihemophilic Factor (Recombinant), Plasma/Albumin-Free is indicated for the control and prevention of bleeding episodes in patients with hemophilia A (congenital factor VIII defi ciency or classic hemophilia) and for surgical prophylaxis in patients with hemophilia A.
XYNTHA does not contain von Willebrand factor and, therefore, is not indicated in von Willebrand’s disease.
Important Safety Information for XYNTHA• Anaphylaxis and severe hypersensitivity reactions are
possible. Should such reactions occur, treatment with the product should be discontinued, and appropriate treatment should be administered.
For illustration of size only. Please see full Prescribing Information for reconstitution instructions.
• Patients using coagulation factor VIII products should be monitored for inhibitors, which have been detected in patients receiving factor VIII-containing products, including XYNTHA.
• The most common adverse reaction in study 1 (safety and effi cacy study) is headache (24% of subjects) and in study 2 (surgery study) is fever (41% of subjects). The most common adverse reactions (≥5% of subjects) in clinical studies were headache, fever, nausea, diarrhea, vomiting, and weakness.
• Patients may develop hypersensitivity to hamster protein, which is present in trace amounts in XYNTHA.
• XYNTHA is an injectable medicine administered by intravenous (IV) infusion.
Please see brief summary of Prescribing Information.
Available Now in 3000 IU.
Additional dosing options in 2011.Visit Pfi zerHemophilia.com to learn more.
The only onethat’s all-in-oneNext-generation purifi cation now in an innovative reconstitution-ready device.1,2 Zero transfer step. More convenience. For the fi rst time ever, factor VIII and diluent come preloaded in a single device for your patients.3
New for your hemophilia A patients
Manufactured by Wyeth Pharmaceuticals Inc. Marketed by Pfizer Inc.
271246-01 Copyright © 2010 Pfizer Inc. All rights reserved. December 2010
References: 1. Xyntha™ Antihemophilic Factor (Recombinant), Plasma/Albumin-Free Prescribing Information, Wyeth Pharmaceuticals Inc. April 2008 2. Kelley B, Jankowski M, Booth J. An improved manufacturing process for Xyntha/ReFacto AF. Haemophilia. 2009. doi:10.1111/j.1365-2516.2009.02160.x. 3. Xyntha™ Antihemophilic Factor (Recombinant), Plasma/Albumin-Free Prescribing Information, Wyeth Pharmaceuticals Inc. August 2010.
271246-01.indd 1-2 3/3/11 11:44 AM

in the news
Spring 2011 54 i n s i d e o u t
35
in t
he
spot
ligh
t
Article and Advertising Submissions
Article submissions and suggestions, as well as advertising inquiries may be sent to:
ASD Healthcare attn: InsideOut Marketing 3101 Gaylord Parkway Frisco, Texas 75034
or by email: [email protected]
ASD Healthcare is committed to providing our customers with timely, relevant information. In the coming months, InsideOut will include articles and sections that are important to you and your business. As we move forward, we are reaching out to you, our valued customers, for ideas and input on topics you would like to see covered.
Please send your thoughts, ideas and suggestions for making this a dynamic and interesting publication to ASD Healthcare’s Marketing Coordinator — Sarah Millecker ([email protected]).
Editorial StaffMarketing Manager
Teri BurgessCopywriter
Christina McFarlandContributing writers
Dale DirksGavin Lindberg
advertising salesBernadette Rospigliosi
advertising CoordinatorJuli Phillips
graphiC designerWes Geiger
Information presented in this publication is not intended as a substitute for the personalized advice given by a healthcare provider. Although ASD Healthcare strives to present only current and accurate information, readers should not consider it as professional advice or endorsement of any position. Although great care has been taken in compiling and checking the information given in this publication to ensure accuracy, the authors, ASD Healthcare, and its employees or agents shall not be responsible or in any way liable for the continued currency of the information or for any errors, omissions, or inaccuracies in this catalog, whether arising from negligence or otherwise or for any consequence arising therefrom.
12 44
in t
he
new
s
81
in t
he
wor
ks
in t
he
com
mu
nit
y
12“Keep Current
Flu Strains in Vaccine”
18Inhibitor Insights:
When to Suspect an Inhibitor
17Radiologist reviews iPad imaging apps
22Considering Long-Term
Health in Your 20s
s p r i n g
Brief Summary
See package insert for full Prescribing Information. For further product information and current package insert, please visit XYNTHA.com or call Wyeth Pharmaceuticals toll-free at 1-800-934-5556.
INDICATIONS AND USAGE
Control and Prevention of Bleeding Episodes in Hemophilia A
XYNTHA Antihemophilic Factor (Recombinant), Plasma/Albumin-Free is indicated for the control and prevention of bleeding episodes in patients with hemophilia A (congenital factor VIII deficiency or classic hemophilia). XYNTHA does not contain von Willebrand factor, and therefore is not indicated in patients with von Willebrand’s disease.
Surgical Prophylaxis in Patients with Hemophilia A
XYNTHA Antihemophilic Factor (Recombinant), Plasma/Albumin-Free is indicated for surgical prophylaxis in patients with hemophilia A.
DOSAGE FORMS AND STRENGTHS
XYNTHA is supplied as a white to off-white freeze-dried powder in the following dosages: 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU.
CONTRAINDICATIONS—None.
WARNINGS AND PRECAUTIONS
Anaphylaxis and Severe Hypersensitivity Reactions—Allergic type hypersensitivity reactions are possible. Patients should be informed of the early signs or symptoms of hypersensitivity reactions [including hives (rash with itching), generalized urticaria, tightness of the chest, wheezing, and hypotension] and anaphylaxis. Patients should be advised to discontinue use of the product and contact their physicians if these symptoms occur.
Neutralizing Antibodies—The occurrence of neutralizing antibodies (inhibitors) is well known in the treatment of patients with hemophilia A. Inhibitors have been detected in patients receiving factor VIII-containing products. Inhibitors are common in previously untreated patients and have been observed in previously treated patients on factor VIII products. Patients using coagulation factor VIII products, including XYNTHA, should be monitored for the development of factor VIII inhibitors. If expected factor VIII activity plasma levels are not attained, or if bleeding is not controlled with an appropriate dose, an assay should be performed to determine if a factor VIII inhibitor is present. If detected, inhibitors should be titered in Bethesda Units (BU).
Formation of Antibodies to Hamster Protein—XYNTHA contains trace amounts of hamster proteins. Patients treated with this product could develop hypersensitivity to these non-human mammalian proteins.
Monitoring: Laboratory Tests—Monitor plasma factor VIII activity levels by the one-stage clotting assay to confirm that adequate factor VIII levels have been achieved and are maintained, when clinically indicated [see Dosage and Administration in full Prescribing Information].
It is recommended that individual factor VIII values for recovery and, if clinically indicated, other pharmacokinetic characteristics be used to guide dosing and administration.
Monitor for development of factor VIII inhibitors. Perform assay to determine if factor VIII inhibitor is present when expected factor VIII activity plasma levels are not attained, or when bleeding is not controlled with the expected dose of XYNTHA. Use Bethesda Units (BU) to titer inhibitors.
ADVERSE REACTIONS
Clinical Trials Experience—Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Study 1 is a pivotal phase 3 (safety and efficacy) study in which previously treated patients (PTPs) with hemophilia A received XYNTHA for routine prophylaxis and on-demand treatment, 94 subjects received at least one dose of XYNTHA, resulting in a total of 6,775 infusions [see Clinical Studies in full Prescribing Information]. In Study 1, the most frequently reported treatment-emergent adverse reaction was headache (24% of subjects). Other adverse reactions reported in ≥ 5% of subjects were: nausea (6%), diarrhea (5%), asthenia (5%) and pyrexia (5%). No subject developed anti-CHO or anti-TN8.2 antibodies.
Study 2 (surgery) is an open-label, single-arm study of at least 25 evaluable PTPs with severe or moderately severe hemophilia A (factor VIII activity in plasma [FVIII:C] ≤ 2%) who required elective major surgery and were planned to receive XYNTHA replacement therapy for at least 6 days post-surgery. Twenty-two subjects received at least one dose of XYNTHA, resulting in 766 infusions [see Clinical Studies in full Prescribing Information]. In Study 2, the most frequently reported treatment-emergent adverse reaction was pyrexia (41% of subjects). Other adverse reactions reported in ≥ 5% of subjects were: headache (9%), nausea (9%), diarrhea (5%), vomiting (5%) and asthenia (5%). The adverse reactions reported in either study were considered mild or moderate in severity.
Immunogenicity
In Study 1, the incidence of FVIII inhibitors to XYNTHA was the primary safety endpoint. Two subjects with inhibitors were observed in 89 subjects (2.2%) who completed ≥ 50 exposure days. These results were consistent with the pre-specified endpoint that no more than 2 inhibitors may be observed in at least 81 subjects.
In a Bayesian statistical analysis, results from this study were used to update PTP results from a prior supporting study using XYNTHA manufactured at the initial facility, where one de novo and two recurrent inhibitors were observed in 110 subjects, and the experience with predecessor product (1 inhibitor in 113 subjects). This Bayesian analysis indicates that the population (true) inhibitor rate for XYNTHA, the estimate of the 95% upper limit of the true inhibitor rate, was 4.17%.
DRUG INTERACTIONS—None known.
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Category C - Animal reproduction studies have not been conducted with XYNTHA Antihemophilic Factor (Recombinant), Plasma/Albumin-Free. It is also not known whether XYNTHA can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. XYNTHA should be given to a pregnant woman only if clinically indicated.
Labor and Delivery—There is no information available on the effect of factor VIII replacement therapy on labor and delivery. XYNTHA should be used only if clinically indicated.
Nursing Mothers—It is not known whether this drug is excreted into human milk. Because many drugs are excreted into human milk, caution should be exercised if XYNTHA is administered to nursing mothers. XYNTHA should be given to nursing mothers only if clinically indicated.
Pediatric Use—Pharmacokinetics of XYNTHA was studied in 7 previously treated patients 12-16 years of age. Pharmacokinetic parameters in these patients were similar to those obtained for adults after a dose of 50 IU/kg. For these 7 patients, the mean (± SD) Cmax and AUC∞ were 1.09 ± 0.21 IU/mL and 11.5 ± 5.2 IU·h/mL, respectively. The mean clearance and plasma half-life values were 5.23 ± 2.36 mL/h/kg and 8.03 ± 2.44 hours (range 3.52 – 10.6 hours), respectively. The mean K-value and in vivo recoveries were 2.18 ± 0.41 IU/dL per IU/kg and 112 ± 23%, respectively.
Geriatric Use—Clinical studies of XYNTHA did not include subjects aged 65 and over. In general, dose selection for an elderly patient should be individualized.
STORAGE AND HANDLING
Product as Packaged for Sale - Store XYNTHA under refrigeration at a temperature of 2° to 8°C (36° to 46°F) for up to 36 months from the date of manufacture until the expiration date stated on the label. XYNTHA may also be stored at room temperature not to exceed 25°C (77°F) for up to 3 months. The starting date at room temperature storage should be clearly recorded in the space provided on the outer carton. At the end of the 3-month period, the product must not be put back into the refrigerator, but must be used immediately or discarded. Do not use XYNTHA after the expiration date stated on the label or after 3 months when stored at room temperature, whichever is earlier. Do not freeze, to prevent damage to the XYNTHA prefilled syringe. During storage, avoid prolonged exposure of XYNTHA to light.
Product After Reconstitution - Administer XYNTHA within 3 hours after reconstitution or after removal of the grey rubber tip cap from the XYNTHA prefilled syringe. The reconstituted solution may be stored at room temperature prior to administration.
This brief summary is based on the Xyntha® [Antihemophilic Factor (Recombinant), Plasma/Albumin-Free] Prescribing Information W10528C004, revised 04/08, and W10547C002, revised 08/10.
Manufactured by Wyeth Pharmaceuticals Inc. Marketed by Pfizer Inc.
270327-01 Copyright © 2010 Pfizer Inc. All rights reserved. September 2010
271246-01.indd 3 3/3/11 11:44 AM
Drug trial delivers positive results for hemophilia
patients in poor countries
Payer Trends: specialty infusible Management
20
28
WARNING! SNAke bIte season!
page
52
In Outsideinform+connect+improve

6 i n s i d e o u t Spring 2011 7
Say “hello” to the great outdoors. With summer on its way and the kids out of school, it’s the perfect time to step outside, go on an adventure and enjoy nature.
It’s also a time to remember that the season does come with a mixture of the good (outdoor adventures), the bad (this year, it’s certain to be gas prices) and in some cases the deadly – specifi-cally snake bites. To help you “be prepared,” this issue of InsideOut gives you information you need to know in Bad Bites Ahead: Staying Safe this Snake Bite Season (Page 52).
Most of us don’t spend our time thinking about snake bites, but when we venture out into areas where they live, it does happen. In fact, there are about 8,000 snake bites in the U.S. each year, some resulting in death. And those most vulnerable to lethal bites are children.
That number could rise this year due to the wide spread flooding in the North Central United States and along the Mississippi River through the South. Flood waters have flushed snakes and other animals out of their natural habitats and into areas where we don’t expect to encounter them. Bad Bites Ahead will show you how to play it safe and what to do in an emergency. Plus, you’ll find updates on anti-venom vaccines that save lives.
ASD Healthcare is proud to support Save One Life in this issue by featuring it and its upcoming fundraiser, Climb for a Cause. This August, I will have the privilege of joining Save One Life in a true outdoor adventure. I’ll be joining a group, including Save One Life Founder Laurie Kelley, and scaling the highest mountain in Africa – 19,340 foot Mt. Kilimanjaro. It’s an adventure with a purpose – to raise funds and put a spotlight on hemophilia care in Africa.
I’d like to encourage readers and professionals who serve the hemophilia community to reach the top with us by supporting your favorite climber in this event. (Did I mention that I will be one of the climbers?) The article on page 44 will give you more details and how you can support a climber in this life-saving mission.
May 13 proved to be a lucky day for the three winners of our MP3-Player drawing (see You’re a Winner on Page 49). Their names were drawn from the hundreds of content survey cards sent in by readers like you. It was my pleasure to call the winners, tell them the good news and thank them for their comments. I’d also like to say, “thank you” to all of you for sending in your survey cards and sharing your thoughts. Your input is vital in helping us make InsideOut a magazine that works for your profession and your life. As ASD Healthcare looks for new ways to customize its content for you, we welcome your ongoing ideas, suggestions and in-sights about the news you want to read.
This vision for InsideOut is an extension of our True Blue philosophy at ASD Healthcare. It reflects our commitment to building superior customer relationships and delivering the superior support you turn to in business and in your communities.
We hope you’ll share this InsideOut with your patients, family and friends for a season of outdoor adventures that are both safe and rewarding.
Sincerely,
Neil Herson, Presidenton behalf of ASD Healthcare
inside out: (adv) to a thorough degree <knows the subject inside out>
With Alphanate® you have a choice!Packaged with Mix2Vial® Filter Transfer Set
VWF:RCo and FVIII potency on vial labels and folding cartons
Available in the following potencies and color coded assay ranges
5 mL
5 mL
10 mL
10 mL
Diluent SizePotency
250 IU FVIII Range
500 IU FVIII Range
1000 IU FVIII Range
1500 IU FVIII Range
A8L1
0-23
-US-
10
For further information call: Grifols USA, LLC Professional Service: 888 GRIFOLS (888 474 3657)Customer Service: 888 325 8579 Fax: 323 441 7968 www.grifols.com
Grifols Biologicals Inc.5555 Valley Boulevard, Los Angeles, California 90032, USA

Introducing
(1) Data on file, Instituto Grifols, S. A.(2) Berger M. et al. Efficacy, Pharmacokinetics, Safety and Tolerability of Flebogamma® 10% DIF, a high purity human intravenous immunoglobulin in primary immunodeficiency. J Clin Immunol 2010; 30 (2): 321-9. (3) Diez JM, et al. Capacity of the manufacturing process of Flebogamma® DIF, a new human high purity intravenous immunoglobulin, to remove a TSE model-agent. Biologicals (2010), doi:10.1016/j.biologicals.2010.08.003.
Shaping the future
Please see reverse for Important Safety Information and Black Box Warning.
FDA0
4-1-
US-1
0
See the difference today
For your convenience• Liquid
• Room temperature storage 2-25° C (36-77° F)
for the entire 2-year shelf life
• Three presentations: 5, 10 and 20 gram vials
Enhancing our commitment to you• Every vial is laser etched with its own unique
identifier number*, which helps to deter
tampering and counterfeiting
• PediGri® On Line, unique to Grifols, offers full
traceability from donation to the final product at
www.pedigri.grifols.com
Flebogamma® 10% DIF is contraindicated in patients who have had a history of anaphylactic or severe systemic reactions to the administration of human immune globulin and in IgA deficient patients with antibodies to IgA and a history of hypersensitivity. In case of hypersensitivity, discontinue Flebogamma® 10% DIF infusion immediately and institute appropriate treatment. Medications such as epinephrine should be available for immediate treatment of acute hypersensitivity reactions.
In patients at risk for developing acute renal failure, monitor renal function, including blood urea nitrogen, serum creatinine, and urine output.
Hyperproteinemia, increased serum viscosity, and hyponatremia may occur in patients receiving Flebogamma® 10% DIF therapy.
Thrombotic events may occur during or following treatment with Flebogamma® 10% DIF. Monitor patients at risk for thrombotic events, including those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, coagulation disorders, prolonged periods of immobilization, and known or suspected hyperviscosity.
Aseptic meningitis syndrome (AMS) may occur infrequently with Flebogamma® 10% DIF treatment. AMS may occur more frequently following high doses and/or rapid infusion of IGIV.
Flebogamma® 10% DIF may contain blood group antibodies that can act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin reaction and hemolysis.
Non-cardiogenic pulmonary edema [Transfusion-Related Acute Lung Injury (TRALI)] may occur in patients following Flebogamma® 10% DIF
treatment. If TRALI is suspected, perform appropriate tests for the presence of antineutrophil antibodies and anti-HLA antibodies in both the product and patient serum.
All patients, but especially individuals receiving Flebogamma® 10% DIF for the first time or being restarted on the product after a treatment hiatus of more than 8 weeks, may be at a higher risk for the development of fever, chills, nausea, and vomiting. Careful monitoring of recipients and adherence to recommendations regarding dosage and administration may reduce the risk of these types of events.
Because Flebogamma® 10% DIF is made from human plasma, it may carry a risk of transmitting infectious agents, e.g. viruses, and theoretically, the Creutzfeldt-Jakob (CJD) agent. No cases of transmission of viral diseases or CJD have ever been identified for Flebogamma® 10% DIF.
The most common adverse reactions (reported in ≥ 5% of clinical trial subjects) occurring during or within 72 hours of the end of an infusion were headache, chills, fever, shaking, fatigue, malaise, anxiety, back pain, muscle cramps, abdominal cramps, blood pressure changes, chest tightness, palpitations, tachycardia, nausea, vomiting, cutaneous reactions, wheezing, rash, arthralgia, and edema. The most serious adverse reactions observed with Flebogamma® 10% DIF were back pain, chest discomfort, and headache (2 patients); and chest pain, maculopathy, rigors, tachycardia, bacterial pneumonia, and vasovagal syncope (1 patient).
Please refer to enclosed Flebogamma® 10% DIF full prescribing information for full prescribing details, including comprehensive adverse event profile and black box warning.
Important Safety InformationFlebogamma® 10% DIF is a human immune globulin intravenous (IGIV) that is indicated for the treatment of primary immune deficiency (PI), including the humoral immune defect in common variable immunodeficiency, x-linked agammaglobulinemia, severe combined immunodeficiency, and Wiskott - Aldrich syndrome.
• Use of immune globulin intravenous (IGIV) products, particularly those containing sucrose, has been reported to be associated with renal dysfunction, acute renal failure, osmotic nephropathy, and death (1). Patients at risk of acute renal failure include those with any degree of pre-existing renal insufficiency, diabetes mellitus, advanced age (above 65 years of age), volume depletion, sepsis, paraproteinemia, or those receiving known nephrotoxic drugs (see Warnings and Precautions [5.2]). Flebogamma® 10% DIF does not contain sucrose. • For patients at risk of renal dysfunction or failure, administer Flebogamma® 10% DIF at the minimum infusion rate practicable (see Dosage and Administration [2.3], Warnings and Precautions [5.2]).
* Laser etched identifier number may at times be covered by the label.
WARNING: ACUTE RENAL DYSFUNCTION AND ACUTE RENAL FAILURE
Flebogamma 10%DIFImmune Globulin Intravenous(Human)
®
Highly purified IGIV• Trace amounts of IgA: <0.006 mg/mL 1
(specification value: <0.1 mg/mL)
• Very low sodium content
• Sorbitol stabilized
Demonstrated benefits in replacement therapy • In the pre-approval clinical trial: 2
- 0.025 serious bacterial infections/patient/year
- Well tolerated: Does not put patients at increased risk
for any adverse events other than those that could be
reasonably expected in primary immune deficiency
patients who are receiving an infusion of intravenous
immune globulin
Broad pathogen safety margin• Seven validated pathogen elimination steps including:
- 20 nm nanofiltration
- Dual specific inactivation:
pasteurization and solvent detergent
• Highly effective process:
- 15.0 log reduction of PPV (PVB19 model)
- ≥ 13.3 log reduction of EMCV (HAV model)
- ≥ 6.2 log reduction through 4% PEG precipitation
and ≥ 5.5 log reduction through 20 nm nanofiltration
of an experimental agent considered a model for the
vCJD and CJD agents 3
Introducing
(1) Data on file, Instituto Grifols, S. A.(2) Berger M. et al. Efficacy, Pharmacokinetics, Safety and Tolerability of Flebogamma® 10% DIF, a high purity human intravenous immunoglobulin in primary immunodeficiency. J Clin Immunol 2010; 30 (2): 321-9. (3) Diez JM, et al. Capacity of the manufacturing process of Flebogamma® DIF, a new human high purity intravenous immunoglobulin, to remove a TSE model-agent. Biologicals (2010), doi:10.1016/j.biologicals.2010.08.003.
Shaping the future
Please see reverse for Important Safety Information and Black Box Warning.
FDA0
4-1-
US-1
0
See the difference today
For your convenience• Liquid
• Room temperature storage 2-25° C (36-77° F)
for the entire 2-year shelf life
• Three presentations: 5, 10 and 20 gram vials
Enhancing our commitment to you• Every vial is laser etched with its own unique
identifier number*, which helps to deter
tampering and counterfeiting
• PediGri® On Line, unique to Grifols, offers full
traceability from donation to the final product at
www.pedigri.grifols.com
Flebogamma® 10% DIF is contraindicated in patients who have had a history of anaphylactic or severe systemic reactions to the administration of human immune globulin and in IgA deficient patients with antibodies to IgA and a history of hypersensitivity. In case of hypersensitivity, discontinue Flebogamma® 10% DIF infusion immediately and institute appropriate treatment. Medications such as epinephrine should be available for immediate treatment of acute hypersensitivity reactions.
In patients at risk for developing acute renal failure, monitor renal function, including blood urea nitrogen, serum creatinine, and urine output.
Hyperproteinemia, increased serum viscosity, and hyponatremia may occur in patients receiving Flebogamma® 10% DIF therapy.
Thrombotic events may occur during or following treatment with Flebogamma® 10% DIF. Monitor patients at risk for thrombotic events, including those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, coagulation disorders, prolonged periods of immobilization, and known or suspected hyperviscosity.
Aseptic meningitis syndrome (AMS) may occur infrequently with Flebogamma® 10% DIF treatment. AMS may occur more frequently following high doses and/or rapid infusion of IGIV.
Flebogamma® 10% DIF may contain blood group antibodies that can act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin reaction and hemolysis.
Non-cardiogenic pulmonary edema [Transfusion-Related Acute Lung Injury (TRALI)] may occur in patients following Flebogamma® 10% DIF
treatment. If TRALI is suspected, perform appropriate tests for the presence of antineutrophil antibodies and anti-HLA antibodies in both the product and patient serum.
All patients, but especially individuals receiving Flebogamma® 10% DIF for the first time or being restarted on the product after a treatment hiatus of more than 8 weeks, may be at a higher risk for the development of fever, chills, nausea, and vomiting. Careful monitoring of recipients and adherence to recommendations regarding dosage and administration may reduce the risk of these types of events.
Because Flebogamma® 10% DIF is made from human plasma, it may carry a risk of transmitting infectious agents, e.g. viruses, and theoretically, the Creutzfeldt-Jakob (CJD) agent. No cases of transmission of viral diseases or CJD have ever been identified for Flebogamma® 10% DIF.
The most common adverse reactions (reported in ≥ 5% of clinical trial subjects) occurring during or within 72 hours of the end of an infusion were headache, chills, fever, shaking, fatigue, malaise, anxiety, back pain, muscle cramps, abdominal cramps, blood pressure changes, chest tightness, palpitations, tachycardia, nausea, vomiting, cutaneous reactions, wheezing, rash, arthralgia, and edema. The most serious adverse reactions observed with Flebogamma® 10% DIF were back pain, chest discomfort, and headache (2 patients); and chest pain, maculopathy, rigors, tachycardia, bacterial pneumonia, and vasovagal syncope (1 patient).
Please refer to enclosed Flebogamma® 10% DIF full prescribing information for full prescribing details, including comprehensive adverse event profile and black box warning.
Important Safety InformationFlebogamma® 10% DIF is a human immune globulin intravenous (IGIV) that is indicated for the treatment of primary immune deficiency (PI), including the humoral immune defect in common variable immunodeficiency, x-linked agammaglobulinemia, severe combined immunodeficiency, and Wiskott - Aldrich syndrome.
• Use of immune globulin intravenous (IGIV) products, particularly those containing sucrose, has been reported to be associated with renal dysfunction, acute renal failure, osmotic nephropathy, and death (1). Patients at risk of acute renal failure include those with any degree of pre-existing renal insufficiency, diabetes mellitus, advanced age (above 65 years of age), volume depletion, sepsis, paraproteinemia, or those receiving known nephrotoxic drugs (see Warnings and Precautions [5.2]). Flebogamma® 10% DIF does not contain sucrose. • For patients at risk of renal dysfunction or failure, administer Flebogamma® 10% DIF at the minimum infusion rate practicable (see Dosage and Administration [2.3], Warnings and Precautions [5.2]).
* Laser etched identifier number may at times be covered by the label.
WARNING: ACUTE RENAL DYSFUNCTION AND ACUTE RENAL FAILURE
Flebogamma 10%DIFImmune Globulin Intravenous(Human)
®
Highly purified IGIV• Trace amounts of IgA: <0.006 mg/mL 1
(specification value: <0.1 mg/mL)
• Very low sodium content
• Sorbitol stabilized
Demonstrated benefits in replacement therapy • In the pre-approval clinical trial: 2
- 0.025 serious bacterial infections/patient/year
- Well tolerated: Does not put patients at increased risk
for any adverse events other than those that could be
reasonably expected in primary immune deficiency
patients who are receiving an infusion of intravenous
immune globulin
Broad pathogen safety margin• Seven validated pathogen elimination steps including:
- 20 nm nanofiltration
- Dual specific inactivation:
pasteurization and solvent detergent
• Highly effective process:
- 15.0 log reduction of PPV (PVB19 model)
- ≥ 13.3 log reduction of EMCV (HAV model)
- ≥ 6.2 log reduction through 4% PEG precipitation
and ≥ 5.5 log reduction through 20 nm nanofiltration
of an experimental agent considered a model for the
vCJD and CJD agents 3

Immune Globulin Intravenous (Human)Flebogamma® 10% DIFFor intravenous use onlyRX onlyBRIEF SUMMARYCONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
INDICATIONS AND USAGEFlebogamma® 10% DIF is a human immune globulin intravenous (IGIV) that is indicated for the treatment of primary immune deficiency (PI), including the humoral immune defect in common variable immunodeficiency, x-linked agammaglobulinemia, severe combined immunodeficiency, and Wiskott - Aldrich syndrome.
DOSAGE AND ADMINISTRATIONThe recommended dose of Flebogamma® 10% DIF for patients with PI is 300 to 600 mg/kg body weight (3.0 to 6.0 mL/kg), administered every 3 to 4 weeks.
The infusion of Flebogamma® 10% DIF should be initiated at a rate of 0.01 mL/kg body weight/minute (1.0 mg/kg/minute). If there are no adverse drug reactions, the infusion rate for subsequent infusions can be slowly increased to the maximum rate of 0.08 mL/kg/minute (8 mg/kg/minute).
Ensure that patients with pre-existing renal insufficiency are not volume depleted. For patients judged to be at risk for renal dysfunction or thrombotic events, administer Flebogamma® 10% DIF at the minimum infusion rate practicable, and consider discontinuation of administration if renal function deteriorates.
CONTRAINDICATIONSFlebogamma® 10% DIF is contraindicated in patients who have had a history of anaphylactic or severe systemic reactions to the administration of human immune globulin and in IgA deficient patients with antibodies to IgA and a history of hypersensitivity.
WARNINGS AND PRECAUTIONS
• Weigh the potential risks and benefits of Flebogamma® 10% DIF against those of alternative therapies in all patients for whom Flebogamma® 10% DIF is being considered.• Before prescribing Flebogamma® 10% DIF, the physician should discuss risks and benefits of its use with patients.
HypersensitivitySevere hypersensitivity reactions may occur. In case of hypersensitivity, discontinue Flebogamma® 10% DIF infusion immediately and institute appropriate treatment. Medications such as epinephrine should be available for immediate treatment of acute hypersensitivity reactions.
Renal Dysfunction/FailurePeriodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure. Assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of Flebogamma® 10% DIF and at appropriate intervals thereafter. If renal function deteriorates, consider discontinue use of Flebogamma® 10% DIF.
In patients who are at risk of developing renal dysfunction because of pre-existing renal insufficiency or predisposition to acute renal failure, administer Flebogamma® 10% DIF at the minimum rate of infusion practicable.
HyperproteinemiaHyperproteinemia, increased serum viscosity, and hyponatremia may occur in patients receiving Flebogamma® 10% DIFtherapy. It is clinically critical to distinguish true hyponatremia from a pseudo-hyponatremia that is temporally or causally related to hyperproteinemia with concomitant decreased calculated serum osmolarity or elevated osmolar gap, because treatment aimed at decreasing serum free water in patients with pseudohyponatremia may lead to volume depletion, a further increase in serum viscosity and a higher risk of thrombotic events.
Thrombotic events may occur during or following treatment with Flebogamma® 10% DIF. Monitor patients at risk for thrombotic events, including those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, coagulation disorders, prolonged periods of immobilization, and known or suspected hyperviscosity.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients judged to be at risk of developing thrombotic events, administer Flebogamma® 10% DIF at the minimum rate of infusion practicable (see Dosage and Administration [2.3]).
Aseptic Meningitis Syndrome (AMS)AMS may occur infrequently with Flebogamma® 10% DIF treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae (3-4).
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting (see Patient Counseling Information [17]). Cerebrospinal fluid (CSF) studies frequently reveal pleocytosis up to several thousand cells per cubic millimeter, predominantly from the granulocytic series and elevated protein levels up to several hundred mg/dL, but negative culture results. Conduct a thorough neurological examination to patients exhibiting such signs and symptoms, including CSF studies, to rule out other causes of meningitis.
AMS may occur more frequently following high doses (2 g/kg) and/or rapid infusion of IGIV.
HemolysisFlebogamma® 10% DIF may contain blood group antibodies that can act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin reaction and hemolysis (5-6). Delayed hemolytic anemia may develop subsequent to Flebogamma® 10% DIF therapy due to enhanced RBC sequestration (7), and acute hemolysis, consistent with intravascular hemolysis, has been reported.
Monitor patients for clinical signs and symptoms of hemolysis. If signs and/or symptoms of hemolysis are present after Flebogamma® 10% DIF infusion, perform appropriate confirmatory laboratory testing (see Patient Counseling Information [17]). Transfusion-Related Acute Lung Injury (TRALI) Non-cardiogenic pulmonary edema may occur in patients following Flebogamma® 10% DIF treatment (11). TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Symptoms typically appear within 1 to 6 hours following treatment.
Monitor patients for pulmonary adverse reactions (see Patient Counseling Information [17]). If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies and anti-HLA antibodies in both the product and patient serum. TRALI may be managed using oxygen therapy with adequate ventilatory support. Infusion ReactionsAll patients, but especially individuals receiving Flebogamma® 10% DIF for the first time or being restarted on the product after a treatment hiatus of more than 8 weeks, may be at a higher risk for the development of fever, chills, nausea, and vomiting. Careful monitoring of recipients and adherence to recommendations regarding dosage and administration may reduce the risk of these types of events (see Dosage and Administration [2.3]).
Transmissible Infectious AgentsBecause Flebogamma® 10% DIF is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, and theoretically, the Creutzfeldt-Jakob (CJD) agent. No cases of transmission of viral diseases or CJD have ever been identified for Flebogamma 10% DIF. All infections suspected by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Grifols Biologicals at 1-888-474-3657. Before prescribing or administering Flebogamma® 10% DIF, the physician should discuss the risks and benefits of its use with the patient (see Patient Counseling Information [17]).
Monitoring: Laboratory Tests• Periodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure. Assess renal function, including measurement of BUN and serum creatinine, before the initial infusion of Flebogamma® 10% DIF and at appropriate intervals thereafter.• Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies, because of the potentially increased risk of thrombosis.• If signs and/or symptoms of hemolysis are present after an infusion of Flebogamma® 10% DIF, perform appropriate laboratory testing for confirmation.• If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies and anti-HLA antibodies in both the product and patient’s serum. Interference with Laboratory TestsAfter infusion of IgG, the transitory rise of the various passively transferred antibodies in the patient’s blood may yield positive serological testing results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g., A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs’) test.
Adverse ReactionsThe most common adverse reactions (reported in ≥ 5% of clinical trial subjects) occurring during or within 72 hours of the end of an infusion were headache, chills, fever, shaking, fatigue, malaise, anxiety, back pain, muscle cramps, abdominal cramps, blood pressure changes, chest tightness, palpitations, tachycardia, nausea, vomiting, cutaneous reactions, wheezing, rash, arthralgia, and edema. The most serious adverse reactions observed with Flebogamma® 10% DIF were back pain, chest discomfort, and headache (2 patients); and chest pain, maculopathy, rigors, tachycardia, bacterial pneumonia, and vasovagal syncope (1 patient).
Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In a multicenter, open-label, non-randomized, historically controlled clinical study, 46 individuals with primary humoral immunodeficiency received infusion doses of Flebogamma 10% DIF at 300 to 600 mg/kg body weight every 3 weeks (mean dose 469 mg/kg) or 4 weeks (mean dose 457 mg/kg) for up to 12 months (see Clinical Studies [14.1]). Routine pre-medication was not allowed. Of the 601 infusions administered, 130 infusions (22%) in 21 (47%) subjects were given pre-medications (antipyretic, antihistamine, or antiemetic agent) because of experience with consecutive infusion-related adverse reactions.
One subject experienced four serious adverse events (AEs, bacterial pneumonia, subcutaneous abscess and two episodes of cellulitis) and withdrew from the study. Two other subjects who participated in the study discontinued prematurely due to AEs (back pain/chest pain/headache; and chills/tachycardia). Three subjects experienced four serious non-related AEs (drug abuse/depression; hernia; and sinusitis).
Forty-five (98%) subjects experienced at least 1 AE irrespective of the relationship with the product, and these subjects reported a total of 723 AEs. Thirty-eight subjects (83%) had an adverse reaction at some time during the study that was considered product-related. Of the 21 subjects receiving pre-medications, 12 (57%) subjects reported adverse reactions during or within 72 hours after the infusion in 48 of the 130 pre-medicated infusions (37%).
Table 2. Treatment-related Adverse Events Occurring in ≥ 5% of Subjects with PI during a Flebogamma® 10% DIF Infusion or within 72 Hours after the End of an infusion
• Use of immune globulin intravenous (IGIV) products, particularly those containing sucrose, has been reported to be associated with renal dysfunction, acute renal failure, osmotic nephropathy, and death (1). Patients at risk of acute renal failure include those with any degree of pre-existing renal insufficiency, diabetes mellitus, advanced age (above 65 years of age), volume depletion, sepsis, paraproteinemia, or those receiving known nephrotoxic drugs (see Warnings and Precautions [5.2]). Flebogamma® 10% DIF does not contain sucrose. • For patients at risk of renal dysfunction or failure, administer Flebogamma® 10% DIF at the tminimum infusion rate practicable (see Dosage and Administration [2.3], Warnings and Precautions [5.2]).
WARNING: ACUTE RENAL DYSFUNCTION AND ACUTE RENAL FAILURE
The total number of adverse events occurring during or within 72 hours after the end of an infusion, irrespective of causality, was 359, excluding non-serious infections.
Table 3 lists the AEs that occurred in greater than 5% of subjects during a Flebogamma® 10% DIF infusion or within 72 hours after the end of an infusion, irrespective of causality.
Table 3. Adverse Events Occurring in ≥ 5% of Subjects with PI during a Flebogamma® 10% DIF Infusion or within 72 Hours after the End of an infusion, Irrespective of Causality
Adverse Event Subjects (%) [N=46]
Infusions (%) [N=601]
Headache 28 (61%) 71 (12%)
Pyrexia 17 (37%) 27 (5%)
Rigors 17 (37%) 37 (6%)
Back pain 13 (28%) 29 (5%)
Cough or Productive cough 12 (26%) 5 (1%)
Nausea 12 (26%) 8 (1%)
Hypotension 10 (22%) 13 (2%)
Tachycardia 10 (22%) 19 (3%)
Myalgia 9 (20%) 17 (3%)
Diarrhea 8 (17%) 2 (0.3%)
Infusion site reaction 8 (17%) 8 (1%)
Pharyngolaryngeal pain 7 (15%) 3 (1%)
Nasal congestion 7 (15%) 2 (0.3%)
Postnasal drip 7 (15%) 4 (1%)
Arthralgia 6 (13%) 2 (0.3%)
Conjunctivitis 6 (13%) 2 (0.3%)
Pain 6 (13%) 10 (2%)
Vomiting 6 (13%) 0 (0%)
Dizziness 5 (11%) 3 (1%)
Fatigue 5 (11%) 1 (0.2%)
Urinary tract infection 5 (11%) 4 (1%)
Chest pain 5 (11%) 4 (1%)
Ear pain 5 (11%) 1 (0.2%)
Pain in extremity 5 (11%) 2 (0.3%)
Dyspnea 5 (11%) 0 (0%)
Rhinorrhea 4 (9%) 1 (0.2%)
Wheezing 4 (9%) 4 (1%)
Body temperature increased 4 (9%) 6 (1%)
Neck pain 4 (9%) 2 (0.3%)
Sinus pain 4 (9%) 1 (0.2%)
Chest discomfort 4 (9%) 4 (1%)
Crackles lung 4 (9%) 2 (0.3%)
Abdominal pain 3 (7%) 2 (0.3%)
Dyspepsia 3 (7%) 1 (0.2%)
Toothache 3 (7%) 0 (0%)
Gastroesophageal reflux disease 3 (7%) 0 (0%)
Lymphadenopathy 3 (7%) 3 (1%)
Respiratory tract congestion 3 (7%) 0 (0%)
Fall 3 (7%) 1 (0.2%)
Hypertension 3 (7%) 4 (1%)
Adverse Event Subjects (%) [N=46]
Infusions (%) [N=601]
Headache 24 (52%) 67 (11%)
Rigors 17 (37%) 37 (6%)
Pyrexia 15 (33%) 27 (5%)
Tachycardia 10 (22%) 18 (3%)
Hypotension 9 (20%) 11 (2%)
In this study, the upper bound of the 1-sided 95% confidence interval for the proportion of Flebogamma® 10% DIF infusions associated with one or more AEs was 37.8% (total infusions: 208; actual proportions: 34.6%). The average percent of infusions with AEs during or within 72 hours after the end of an infusion for each individual subject was 36.7% and the upper bound of the 1-sided 95% confidence interval was 43.9%.AE reporting was based upon a clinical protocol precluding pre-medication against AEs. Pre-medication could be utilized only after the first 2 infusions only in those patients that exhibited adverse events.
Forty-three of the 46 subjects enrolled in this study had a negative Coombs test at baseline. Of these 43 subjects, 10 (23.3%) developed a positive Coombs test at some time during the study. However, no subjects showed evidence of hemolytic anemia.
Post-marketing ExperienceBecause adverse reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure. The following adverse reactions have been identified during post approval use of intravenous immune globulins, including Flebogamma 5% (see References [15]). Infusion reactions Hypersensitivity (e.g., anaphylaxis), headache, diarrhea, tachycardia, fever, fatigue, dizziness, malaise, chills, flushing, urticaria or other skin reactions, wheezing or other chest discomfort, nausea, vomiting, rigors, back pain, myalgia, arthralgia, and changes in blood pressureRenal Acute renal dysfunction/failure, osmotic nephropathyRespiratory Apnea, Acute Respiratory Distress Syndrome (ARDS), Transfusion-Related Acute Lung Injury (TRALI), cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasmCardiovascular Cardiac arrest, thromboembolism, vascular collapse, hypotensionNeurological Coma, loss of consciousness, seizures, tremor, aseptic meningitis syndromeIntegumentary Stevens-Johnson Syndrome, epidermolysis, erythema multiformae, dermatitis (e.g., bullous dermatitis) Hematologic Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs) testMusculoskeletal Back painGastrointestinal Hepatic dysfunction, abdominal painGeneral/Body as a Whole Pyrexia, rigors
DRUG INTERACTIONSPassive transfer of antibodies may transiently impair the immune response to live attenuated virus vaccines such as measles, mumps, and rubella. Inform the immunizing physician of recent therapy with Flebogama® 10% DIF so that appropriate measures may be taken (see Patient Counseling Information [17]). USE IN SPECIFIC POPULATIONS PregnancyPregnancy Category C. Animal reproduction studies have not been performed with Flebogamma® 10% DIF. It is also not known whether Flebogamma® 10% DIF can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Flebogamma® 10% DIF should be given to a pregnant woman only if clearly needed. Immunoglobulins cross the placenta from maternal circulation increasingly after 30 weeks of gestation. Nursing Mothers Use of Flebogamma® 10% DIF has not been evaluated in nursing mothers. Pediatric UseThree (3) pediatric patients with primary humoral immunodeficiency (two between the ages of 6 and 10, and one 16 year old) were included in the clinical evaluation of Flebogamma® 10% DIF. This number of subjects is too small to establish safety and efficacy in the pediatric population (see Clinical Studies [14]).
Geriatric UseUse caution when administering Flebogamma® 10% DIF to patients over 65 years of age who are judged to be at increased risk for developing certain adverse reactions such as thromboembolic events and acute renal failure (see Boxed Warning, Warnings and Precautions [5.2]). Do not exceed the recommended dose, and infuse Flebogamma® 10% DIF at the minimum infusion rate practicable.
One (1) patient with primary humoral immunodeficiency at or over the age of 65 was included within the clinical evaluation of Flebogamma® 10% DIF. This number of geriatric patients was too small for separate evaluation from the younger patients for safety or efficacy (see Clinical Studies [14]).
HOW SUPPLIED/STORAGE AND HANDLINGFlebogamma® 10% DIF is supplied in single-use, individually laser etched vials containing the labeled amount of functionally active IgG.
The following presentations of Flebogamma® 10% DIF are available:
NDC Number Fill Size Grams Protein
61953-0005-1 50 mL 5g
61953-0005-2 100 mL 10g
61953-0005-3 200 mL 20g
Each vial has an integral suspension band and a label with two peel-off strips showing the product name and lot number.
DO NOT FREEZE.When stored at room temperature (up to 25 ºC [77 ºF]), Flebogamma® 10% DIF is stable for up to 24 months, as indicated by the expiration date printed on the outer carton and container label.
Keep Flebogamma® 10% DIF in its original carton to protect it from light.
Manufactured by INSTITUTO GRIFOLS, S.A.Barcelona - SpainU.S. License No. 1181Distributed by GRIFOLS BIOLOGICALS Inc.Los Angeles - CA 90032Phone: 888-GRIFOLS (888-474-3657)
Adverse Event Subjects (%) [N=46]
Infusions (%) [N=601]
Back pain 8 (17%) 27 (5%)
Myalgia 8 (17%) 17 (3%)
Body temperature increased 4 (9%) 6 (1%)
Nausea 4 (9%) 6 (1%)
Pain 4 (9%) 8 (1%)
Chest discomfort 3 (7%) 4 (1%)
Chest pain 3 (7%) 5 (1%)
Infusion site reaction 3 (7%) 4 (1%)
Pain in extremity 3 (7%) 3 (0.5%)
Immune Globulin Intravenous (Human)Flebogamma® 10% DIFFor intravenous use onlyRX onlyBRIEF SUMMARYCONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
INDICATIONS AND USAGEFlebogamma® 10% DIF is a human immune globulin intravenous (IGIV) that is indicated for the treatment of primary immune deficiency (PI), including the humoral immune defect in common variable immunodeficiency, x-linked agammaglobulinemia, severe combined immunodeficiency, and Wiskott - Aldrich syndrome.
DOSAGE AND ADMINISTRATIONThe recommended dose of Flebogamma® 10% DIF for patients with PI is 300 to 600 mg/kg body weight (3.0 to 6.0 mL/kg), administered every 3 to 4 weeks.
The infusion of Flebogamma® 10% DIF should be initiated at a rate of 0.01 mL/kg body weight/minute (1.0 mg/kg/minute). If there are no adverse drug reactions, the infusion rate for subsequent infusions can be slowly increased to the maximum rate of 0.08 mL/kg/minute (8 mg/kg/minute).
Ensure that patients with pre-existing renal insufficiency are not volume depleted. For patients judged to be at risk for renal dysfunction or thrombotic events, administer Flebogamma® 10% DIF at the minimum infusion rate practicable, and consider discontinuation of administration if renal function deteriorates.
CONTRAINDICATIONSFlebogamma® 10% DIF is contraindicated in patients who have had a history of anaphylactic or severe systemic reactions to the administration of human immune globulin and in IgA deficient patients with antibodies to IgA and a history of hypersensitivity.
WARNINGS AND PRECAUTIONS
• Weigh the potential risks and benefits of Flebogamma® 10% DIF against those of alternative therapies in all patients for whom Flebogamma® 10% DIF is being considered.• Before prescribing Flebogamma® 10% DIF, the physician should discuss risks and benefits of its use with patients.
HypersensitivitySevere hypersensitivity reactions may occur. In case of hypersensitivity, discontinue Flebogamma® 10% DIF infusion immediately and institute appropriate treatment. Medications such as epinephrine should be available for immediate treatment of acute hypersensitivity reactions.
Renal Dysfunction/FailurePeriodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure. Assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of Flebogamma® 10% DIF and at appropriate intervals thereafter. If renal function deteriorates, consider discontinue use of Flebogamma® 10% DIF.
In patients who are at risk of developing renal dysfunction because of pre-existing renal insufficiency or predisposition to acute renal failure, administer Flebogamma® 10% DIF at the minimum rate of infusion practicable.
HyperproteinemiaHyperproteinemia, increased serum viscosity, and hyponatremia may occur in patients receiving Flebogamma® 10% DIFtherapy. It is clinically critical to distinguish true hyponatremia from a pseudo-hyponatremia that is temporally or causally related to hyperproteinemia with concomitant decreased calculated serum osmolarity or elevated osmolar gap, because treatment aimed at decreasing serum free water in patients with pseudohyponatremia may lead to volume depletion, a further increase in serum viscosity and a higher risk of thrombotic events.
Thrombotic events may occur during or following treatment with Flebogamma® 10% DIF. Monitor patients at risk for thrombotic events, including those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, coagulation disorders, prolonged periods of immobilization, and known or suspected hyperviscosity.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients judged to be at risk of developing thrombotic events, administer Flebogamma® 10% DIF at the minimum rate of infusion practicable (see Dosage and Administration [2.3]).
Aseptic Meningitis Syndrome (AMS)AMS may occur infrequently with Flebogamma® 10% DIF treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae (3-4).
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting (see Patient Counseling Information [17]). Cerebrospinal fluid (CSF) studies frequently reveal pleocytosis up to several thousand cells per cubic millimeter, predominantly from the granulocytic series and elevated protein levels up to several hundred mg/dL, but negative culture results. Conduct a thorough neurological examination to patients exhibiting such signs and symptoms, including CSF studies, to rule out other causes of meningitis.
AMS may occur more frequently following high doses (2 g/kg) and/or rapid infusion of IGIV.
HemolysisFlebogamma® 10% DIF may contain blood group antibodies that can act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin reaction and hemolysis (5-6). Delayed hemolytic anemia may develop subsequent to Flebogamma® 10% DIF therapy due to enhanced RBC sequestration (7), and acute hemolysis, consistent with intravascular hemolysis, has been reported.
Monitor patients for clinical signs and symptoms of hemolysis. If signs and/or symptoms of hemolysis are present after Flebogamma® 10% DIF infusion, perform appropriate confirmatory laboratory testing (see Patient Counseling Information [17]). Transfusion-Related Acute Lung Injury (TRALI) Non-cardiogenic pulmonary edema may occur in patients following Flebogamma® 10% DIF treatment (11). TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Symptoms typically appear within 1 to 6 hours following treatment.
Monitor patients for pulmonary adverse reactions (see Patient Counseling Information [17]). If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies and anti-HLA antibodies in both the product and patient serum. TRALI may be managed using oxygen therapy with adequate ventilatory support. Infusion ReactionsAll patients, but especially individuals receiving Flebogamma® 10% DIF for the first time or being restarted on the product after a treatment hiatus of more than 8 weeks, may be at a higher risk for the development of fever, chills, nausea, and vomiting. Careful monitoring of recipients and adherence to recommendations regarding dosage and administration may reduce the risk of these types of events (see Dosage and Administration [2.3]).
Transmissible Infectious AgentsBecause Flebogamma® 10% DIF is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, and theoretically, the Creutzfeldt-Jakob (CJD) agent. No cases of transmission of viral diseases or CJD have ever been identified for Flebogamma 10% DIF. All infections suspected by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Grifols Biologicals at 1-888-474-3657. Before prescribing or administering Flebogamma® 10% DIF, the physician should discuss the risks and benefits of its use with the patient (see Patient Counseling Information [17]).
Monitoring: Laboratory Tests• Periodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk of developing acute renal failure. Assess renal function, including measurement of BUN and serum creatinine, before the initial infusion of Flebogamma® 10% DIF and at appropriate intervals thereafter.• Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies, because of the potentially increased risk of thrombosis.• If signs and/or symptoms of hemolysis are present after an infusion of Flebogamma® 10% DIF, perform appropriate laboratory testing for confirmation.• If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies and anti-HLA antibodies in both the product and patient’s serum. Interference with Laboratory TestsAfter infusion of IgG, the transitory rise of the various passively transferred antibodies in the patient’s blood may yield positive serological testing results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g., A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs’) test.
Adverse ReactionsThe most common adverse reactions (reported in ≥ 5% of clinical trial subjects) occurring during or within 72 hours of the end of an infusion were headache, chills, fever, shaking, fatigue, malaise, anxiety, back pain, muscle cramps, abdominal cramps, blood pressure changes, chest tightness, palpitations, tachycardia, nausea, vomiting, cutaneous reactions, wheezing, rash, arthralgia, and edema. The most serious adverse reactions observed with Flebogamma® 10% DIF were back pain, chest discomfort, and headache (2 patients); and chest pain, maculopathy, rigors, tachycardia, bacterial pneumonia, and vasovagal syncope (1 patient).
Clinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In a multicenter, open-label, non-randomized, historically controlled clinical study, 46 individuals with primary humoral immunodeficiency received infusion doses of Flebogamma 10% DIF at 300 to 600 mg/kg body weight every 3 weeks (mean dose 469 mg/kg) or 4 weeks (mean dose 457 mg/kg) for up to 12 months (see Clinical Studies [14.1]). Routine pre-medication was not allowed. Of the 601 infusions administered, 130 infusions (22%) in 21 (47%) subjects were given pre-medications (antipyretic, antihistamine, or antiemetic agent) because of experience with consecutive infusion-related adverse reactions.
One subject experienced four serious adverse events (AEs, bacterial pneumonia, subcutaneous abscess and two episodes of cellulitis) and withdrew from the study. Two other subjects who participated in the study discontinued prematurely due to AEs (back pain/chest pain/headache; and chills/tachycardia). Three subjects experienced four serious non-related AEs (drug abuse/depression; hernia; and sinusitis).
Forty-five (98%) subjects experienced at least 1 AE irrespective of the relationship with the product, and these subjects reported a total of 723 AEs. Thirty-eight subjects (83%) had an adverse reaction at some time during the study that was considered product-related. Of the 21 subjects receiving pre-medications, 12 (57%) subjects reported adverse reactions during or within 72 hours after the infusion in 48 of the 130 pre-medicated infusions (37%).
Table 2. Treatment-related Adverse Events Occurring in ≥ 5% of Subjects with PI during a Flebogamma® 10% DIF Infusion or within 72 Hours after the End of an infusion
• Use of immune globulin intravenous (IGIV) products, particularly those containing sucrose, has been reported to be associated with renal dysfunction, acute renal failure, osmotic nephropathy, and death (1). Patients at risk of acute renal failure include those with any degree of pre-existing renal insufficiency, diabetes mellitus, advanced age (above 65 years of age), volume depletion, sepsis, paraproteinemia, or those receiving known nephrotoxic drugs (see Warnings and Precautions [5.2]). Flebogamma® 10% DIF does not contain sucrose. • For patients at risk of renal dysfunction or failure, administer Flebogamma® 10% DIF at the tminimum infusion rate practicable (see Dosage and Administration [2.3], Warnings and Precautions [5.2]).
WARNING: ACUTE RENAL DYSFUNCTION AND ACUTE RENAL FAILURE
The total number of adverse events occurring during or within 72 hours after the end of an infusion, irrespective of causality, was 359, excluding non-serious infections.
Table 3 lists the AEs that occurred in greater than 5% of subjects during a Flebogamma® 10% DIF infusion or within 72 hours after the end of an infusion, irrespective of causality.
Table 3. Adverse Events Occurring in ≥ 5% of Subjects with PI during a Flebogamma® 10% DIF Infusion or within 72 Hours after the End of an infusion, Irrespective of Causality
Adverse Event Subjects (%) [N=46]
Infusions (%) [N=601]
Headache 28 (61%) 71 (12%)
Pyrexia 17 (37%) 27 (5%)
Rigors 17 (37%) 37 (6%)
Back pain 13 (28%) 29 (5%)
Cough or Productive cough 12 (26%) 5 (1%)
Nausea 12 (26%) 8 (1%)
Hypotension 10 (22%) 13 (2%)
Tachycardia 10 (22%) 19 (3%)
Myalgia 9 (20%) 17 (3%)
Diarrhea 8 (17%) 2 (0.3%)
Infusion site reaction 8 (17%) 8 (1%)
Pharyngolaryngeal pain 7 (15%) 3 (1%)
Nasal congestion 7 (15%) 2 (0.3%)
Postnasal drip 7 (15%) 4 (1%)
Arthralgia 6 (13%) 2 (0.3%)
Conjunctivitis 6 (13%) 2 (0.3%)
Pain 6 (13%) 10 (2%)
Vomiting 6 (13%) 0 (0%)
Dizziness 5 (11%) 3 (1%)
Fatigue 5 (11%) 1 (0.2%)
Urinary tract infection 5 (11%) 4 (1%)
Chest pain 5 (11%) 4 (1%)
Ear pain 5 (11%) 1 (0.2%)
Pain in extremity 5 (11%) 2 (0.3%)
Dyspnea 5 (11%) 0 (0%)
Rhinorrhea 4 (9%) 1 (0.2%)
Wheezing 4 (9%) 4 (1%)
Body temperature increased 4 (9%) 6 (1%)
Neck pain 4 (9%) 2 (0.3%)
Sinus pain 4 (9%) 1 (0.2%)
Chest discomfort 4 (9%) 4 (1%)
Crackles lung 4 (9%) 2 (0.3%)
Abdominal pain 3 (7%) 2 (0.3%)
Dyspepsia 3 (7%) 1 (0.2%)
Toothache 3 (7%) 0 (0%)
Gastroesophageal reflux disease 3 (7%) 0 (0%)
Lymphadenopathy 3 (7%) 3 (1%)
Respiratory tract congestion 3 (7%) 0 (0%)
Fall 3 (7%) 1 (0.2%)
Hypertension 3 (7%) 4 (1%)
Adverse Event Subjects (%) [N=46]
Infusions (%) [N=601]
Headache 24 (52%) 67 (11%)
Rigors 17 (37%) 37 (6%)
Pyrexia 15 (33%) 27 (5%)
Tachycardia 10 (22%) 18 (3%)
Hypotension 9 (20%) 11 (2%)
In this study, the upper bound of the 1-sided 95% confidence interval for the proportion of Flebogamma® 10% DIF infusions associated with one or more AEs was 37.8% (total infusions: 208; actual proportions: 34.6%). The average percent of infusions with AEs during or within 72 hours after the end of an infusion for each individual subject was 36.7% and the upper bound of the 1-sided 95% confidence interval was 43.9%.AE reporting was based upon a clinical protocol precluding pre-medication against AEs. Pre-medication could be utilized only after the first 2 infusions only in those patients that exhibited adverse events.
Forty-three of the 46 subjects enrolled in this study had a negative Coombs test at baseline. Of these 43 subjects, 10 (23.3%) developed a positive Coombs test at some time during the study. However, no subjects showed evidence of hemolytic anemia.
Post-marketing ExperienceBecause adverse reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure. The following adverse reactions have been identified during post approval use of intravenous immune globulins, including Flebogamma 5% (see References [15]). Infusion reactions Hypersensitivity (e.g., anaphylaxis), headache, diarrhea, tachycardia, fever, fatigue, dizziness, malaise, chills, flushing, urticaria or other skin reactions, wheezing or other chest discomfort, nausea, vomiting, rigors, back pain, myalgia, arthralgia, and changes in blood pressureRenal Acute renal dysfunction/failure, osmotic nephropathyRespiratory Apnea, Acute Respiratory Distress Syndrome (ARDS), Transfusion-Related Acute Lung Injury (TRALI), cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasmCardiovascular Cardiac arrest, thromboembolism, vascular collapse, hypotensionNeurological Coma, loss of consciousness, seizures, tremor, aseptic meningitis syndromeIntegumentary Stevens-Johnson Syndrome, epidermolysis, erythema multiformae, dermatitis (e.g., bullous dermatitis) Hematologic Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs) testMusculoskeletal Back painGastrointestinal Hepatic dysfunction, abdominal painGeneral/Body as a Whole Pyrexia, rigors
DRUG INTERACTIONSPassive transfer of antibodies may transiently impair the immune response to live attenuated virus vaccines such as measles, mumps, and rubella. Inform the immunizing physician of recent therapy with Flebogama® 10% DIF so that appropriate measures may be taken (see Patient Counseling Information [17]). USE IN SPECIFIC POPULATIONS PregnancyPregnancy Category C. Animal reproduction studies have not been performed with Flebogamma® 10% DIF. It is also not known whether Flebogamma® 10% DIF can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Flebogamma® 10% DIF should be given to a pregnant woman only if clearly needed. Immunoglobulins cross the placenta from maternal circulation increasingly after 30 weeks of gestation. Nursing Mothers Use of Flebogamma® 10% DIF has not been evaluated in nursing mothers. Pediatric UseThree (3) pediatric patients with primary humoral immunodeficiency (two between the ages of 6 and 10, and one 16 year old) were included in the clinical evaluation of Flebogamma® 10% DIF. This number of subjects is too small to establish safety and efficacy in the pediatric population (see Clinical Studies [14]).
Geriatric UseUse caution when administering Flebogamma® 10% DIF to patients over 65 years of age who are judged to be at increased risk for developing certain adverse reactions such as thromboembolic events and acute renal failure (see Boxed Warning, Warnings and Precautions [5.2]). Do not exceed the recommended dose, and infuse Flebogamma® 10% DIF at the minimum infusion rate practicable.
One (1) patient with primary humoral immunodeficiency at or over the age of 65 was included within the clinical evaluation of Flebogamma® 10% DIF. This number of geriatric patients was too small for separate evaluation from the younger patients for safety or efficacy (see Clinical Studies [14]).
HOW SUPPLIED/STORAGE AND HANDLINGFlebogamma® 10% DIF is supplied in single-use, individually laser etched vials containing the labeled amount of functionally active IgG.
The following presentations of Flebogamma® 10% DIF are available:
NDC Number Fill Size Grams Protein
61953-0005-1 50 mL 5g
61953-0005-2 100 mL 10g
61953-0005-3 200 mL 20g
Each vial has an integral suspension band and a label with two peel-off strips showing the product name and lot number.
DO NOT FREEZE.When stored at room temperature (up to 25 ºC [77 ºF]), Flebogamma® 10% DIF is stable for up to 24 months, as indicated by the expiration date printed on the outer carton and container label.
Keep Flebogamma® 10% DIF in its original carton to protect it from light.
Manufactured by INSTITUTO GRIFOLS, S.A.Barcelona - SpainU.S. License No. 1181Distributed by GRIFOLS BIOLOGICALS Inc.Los Angeles - CA 90032Phone: 888-GRIFOLS (888-474-3657)
Adverse Event Subjects (%) [N=46]
Infusions (%) [N=601]
Back pain 8 (17%) 27 (5%)
Myalgia 8 (17%) 17 (3%)
Body temperature increased 4 (9%) 6 (1%)
Nausea 4 (9%) 6 (1%)
Pain 4 (9%) 8 (1%)
Chest discomfort 3 (7%) 4 (1%)
Chest pain 3 (7%) 5 (1%)
Infusion site reaction 3 (7%) 4 (1%)
Pain in extremity 3 (7%) 3 (0.5%)

12 i n s i d e o u t Spring 2011 13
During the meeting of the Federal Drug Administration’s Vaccines and Related Biological Products
Advisory Committee, held in Bethesda, Md., the 16 advisers voted unanimously to retain the current influenza type A H3N2 and influenza B strains in next season’s (2011-2012) vaccine formulation.
The specific recommendations are for an A/California/7/2009-like H1N1 virus, an A/Perth/16/2009-like H3N2 virus, and a /Brisbane/60/2008-like virus. This is the same Northern Hemisphere recommenda-tion made days earlier by the World Health Organization.
Influenza ActivityAt the time of the late February meeting, 44 states had widespread influenza activity, and 35 pediatric deaths from influenza had been reported to the Centers for Disease Control and Prevention (CDC).
Nancy Cox, director of CDC’s Influenza Division, said the pandemic-causing H1N1 influenza strain that emerged in 2009 has essentially supplanted the seasonal H1N1 viruses that caused illness before the pandemic. From Aug. 29, 2010, through Jan. 29, 2011, Cox said, global surveillance laboratories identified a total of six speci-mens of former seasonal H1N1 viruses.
Cox said these viruses came from five countries, including the United States, and “were resistant to oseltamivir but sen-sitive to zanamivir.”
Which B Strain? As is often the case, making a decision about which influenza B strain to recom-mend for the upcoming flu season caused problems for the expert panel.
“We are always struggling with this B issue,” said Pamela McInnes of the National Institutes of Health.
Two distinct types of influenza B strains, known as Victoria-lineage and Yamagata-lineage viruses, typically circu-late each year.
FDA’s advisers must predict, on the basis of recent surveillance data, which lineage is likely to circulate in the United States in the upcoming flu season and recom-mend a corresponding vaccine strain.
In most of the world, Victoria-lineage strains similar to those in this season’s U.S. vaccines have been the main strains circulating. But in China, strains of the Yamagata-lineage have been responsible for most influenza B infections.
A big unknown factor is whether Yamagata-lineage viruses will overtake Victoria-lineage viruses in the Southern Hemisphere this summer and then surge into the Northern Hemisphere in the fall. By midsummer, U.S. vaccine production is well underway, and introducing a new strain into the vaccine would be problem-atic for manufacturers.
“It’s very difficult to predict what’s going to happen with the influenza B virus,” Cox said. “We simply have to make the best judgment we can based on the in-formation that we have at the time.”
Questions about effectiveness Worldwide surveillance data from the Department of Defense (DOD) suggest that this season’s vaccine is not performing ro-bustly, said Navy Captain Kevin Russell, di-rector of DOD’s Global Emerging Infectious Surveillance and Response System.
“We’re seeing a dramatic increase in H1N1 among our service members, par-ticularly among our recruits,” Russell said. He said live attenuated influenza virus (LAIV) appears to be less effective among the military than trivalent inactivated vac-cine formulations.
Russell said one study of new re-cruits, all of whom were vaccinated with LAIV, indicated that it was 81% effec-tive overall but just 41% effective against H1N1. A small study of active-duty per-sonnel that excluded recruits suggested a 59% overall effectiveness for trivalent inactivated influenza vaccines, compared with 30% for LAIV.
Russell said the studies used different methods but all suggest the same thing: “There appears to be poor protection against H1N1, particularly with LAIV.”
source: “advisers say keep Current Flu strains in vaccine,” american Journal of health-system pharmacy, 1 apr. 2011, http://www.ajhp.org
Advisers Say:
“ Keep Current Flu Strains in Vaccine”
in the news
Bayer HealthCare Pharmaceuticals has re-ceived U.S. Food and Drug Administration’s approval for its Gadavist (gadobutrol) injection for the detection and visualization of areas with disrupted blood brain barrier and/or abnormal vascularity of the central nervous system in diagnostic magnetic resonance imaging in adults and children (2 years of age and older).
According to Bayer HealthCare, Gadavist demonstrated improvement of visualization endpoints in paired Gadavist images com-pared to pre-contrast images resulted in improved assessment of normal and ab-normal CNS anatomy in two Phase III trials.
John Rotondo, Bayer HealthCare Commercial Operations, said the approval of Gadavist enriches their portfolio of MRI contrast media and provides a new option for U.S. healthcare providers in contrast-enhanced imaging of the CNS.
source: © 1994-2011 by Medscape. all rights reserved. (http://www.medscape.com/public/copyright)
In February the FDA announced the licensing of Corifact for the prevention of bleeding in people with congenital factor XIII deficiency.
The CSL Behring product is made from purified plasma from healthy donors, according to the FDA. New U.S. labeling for Corifact is not yet available.
According to FDA, congenital factor XIII deficiency is a rare disease that may cause soft-tissue bruising, mucosal bleed-ing and fatal intracranial bleeding. About 1
of every 3–5 million people are estimated to have the congenital deficiency.
Corifact was approved under FDA’s ac-celerated approval process. According to the agency, the approval was granted on the basis of a study involving 14 adults and chil-dren with congenital factor XIII deficiency.
According to the FDA, the most common adverse events associated with Corifact use in clinical studies were hyper-sensitivity reactions, chills, fever, arthralgia, headache, elevated thrombin–antithrombin levels and elevated liver enzymes.
Bayer HealthCare Gadavist injection gets FDA approval
Corifact Licensed for Factor XIII Deficiency
“ As is often the case,
making a decision about
which influenza B strain
to recommend for the
upcoming flu season
caused problems.”
CSL Behring, a global biotherapeutics company specializing in plasma-derived and recombinant therapies and a sub-sidiary of CSL Limited (ASX:CSL), is a recipient of the National Organization for Rare Disorders (NORD) 2011 Corporate Award. The award was presented at the NORD Partners in Progress Celebration 2011 for “new treatments brought to market for patients with rare diseases.”
CSL Behring’s new treatment, factor XIII concentrate (human), is ap-proved for the routine prophylactic treat-ment of congenital factor XIII deficiency, an extremely rare and potentially life-threatening bleeding disorder. Factor
XIII concentrate (human) is the first and only dual sub-unit FXIII concentrate. It contains both A and B sub-units to treat FXIII patients regardless of sub-unit de-ficiency. Congenital factor XIII deficiency is estimated to affect one person in two million, with an incidence in the U.S. of approximately 150 people.
“CSL Behring is honored to receive this NORD Corporate Award,” said Paul Perreault, CSL Behring Executive Vice President for Worldwide Commercial Operations and incoming president. “People with rare diseases often face a host of challenges in being accu-rately diagnosed and in gaining ongo-ing access to appropriate medical care. CSL Behring focuses on these areas and partners with groups such as NORD to improve patients’ lives. We commend NORD for their outstanding achievements
and dedication to supporting people with rare diseases.”
This award is the most recent in a series of recognitions for CSL Behring’s innovative therapies used to treat rare and serious diseases. NORD previously honored CSL Behring for developing fi-brinogen concentrate (human) for treat-ing acute bleeding episodes in patients in the U.S. with congenital fibrinogen de-ficiency (CFD).
NORD is a leading patient advoca-cy organization whose mission is to ad-vance the causes of people with rare dis-eases. They provide support for orphan product research – products used to treat serious conditions that affect fewer than 200,000 people – and they develop and advocate on public policy issues before Congress and health agencies. source: www.cslbehring.com
NORD Presents
CSL Behring with 2011 Corporate Award

14 i n s i d e o u t Spring 2011 15
Switch to Privigen
Important Safety Information
Privigen is indicated for the treatment of patients with primary immunodeficiency (PI) associated with defects in humoral immunity, including but not limited to common variable immunodeficiency (CVID), X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
WARNING: Renal dysfunction, acute renal failure, osmotic nephrosis, and death may be associated with the administration of Immune Globulin Intravenous (Human) (IVIg) products in predisposed patients. Administer IVIg products at the minimum infusion rate possible. Renal dysfunction and acute renal failure occur more commonly in patients receiving IVIg products containing sucrose. Privigen does not contain sucrose. See full Prescribing Information for complete Boxed Warning.
Privigen is contraindicated in patients who have had an anaphylactic or severe systemic reaction to the administration of human immune globulin, in patients with hyperprolinemia, and in IgA-deficient patients with antibodies to IgA and a history of hypersensitivity.
In patients at risk for developing renal failure, monitor urine output and renal function, including blood urea nitrogen and serum creatinine. Thrombotic events have been reported with Privigen and other IVIg treatments. Monitor patients with risk factors for thrombotic events, including a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, hypercoagulable disorders, prolonged periods of immobilization, and/or known or suspected hyperviscosity.Aseptic meningitis syndrome (AMS) may occur infrequently with Privigen and other IVIg treatments; AMS may occur more frequently with high doses and/or rapid infusion of IVIg. Hemolysis, hemolytic anemia, and pulmonary adverse events have also been reported. There have been reports of noncardiogenic pulmonary edema in patients administered IVIg. If transfusion-related acute lung injury is suspected, test product and patient for antineutrophil antibodies.
Privigen is derived from human plasma. The risk of transmission of infectious agents, including viruses and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent, cannot be completely eliminated.
In clinical studies, the most common adverse reactions with Privigen were headache, pain, nausea, pyrexia/hyperthermia, fatigue, and chills.
Please see brief summary of full Prescribing Information on following pages.
Privigen is manufactured by CSL Behring AG and distributed by CSL Behring LLC. Privigen is a registered trademark of CSL Behring AG.
©2010 CSL Behring LLC 1020 First Avenue, PO Box 61501, King of Prussia, PA 19406-0901 USA www.CSLBehring-us.com www.Privigen.com 09-PVG-051 4/2010
Choose the IVIg therapy that is:
Simple. Ready-to-use 10% liquid IVIg 36-month room temperature storage
Sophisticated. First and only IVIg stabilized with proline
Sucrose-free
IgA 25 mcg/mL
Safe. In clinical trials, 97% of related adverse events were non-serious; 95% of 1038 infusions were administered without premedication. The most common adverse reactions were headache, pain, nausea, pyrexia/hyperthermia, fatigue, and chills
3-step virus inactivation/removal process, including nanofiltration to 20 nanometers, reduces the risk of pathogen transmission
Guarantee your IVIg supply
For more information, call 1-888-310-2525or visit www.Privigen.com
Guarantee your IVIg supply for up to 5 years
Minimize your hospital’s supply risk
Ensure your patients’ needs are met

in the news
Spring 2011 1716 i n s i d e o u t
Expect “a lot of the Apple® iPad® in our departments –
and, more importantly, outside the radi-ology suite,” says Sam Friedman, CTO and Director for Nuclear Medicine at Pitts Radiology in South Carolina. Friedman also is a radiology blogger at Doctor Dalai’s.
He doesn’t expect that radiolo-gists will replace their PC-based pic-ture archiving and communications sys-tems overnight, but rather at the pace of software and hardware developments. He reviews current radiology viewing apps for the iPhone in advance. They in-clude offerings from Merge Healthcare, OsiriX, iCRco Inc, MIM Software and Calgary Scientific.
The current offerings are “bare-bones programs,” he writes, with much code devoted to file management. Most func-tion as extensions of their desktop viewer counterparts. He does find that the current crop functions well in magnified mode.
The viewers have disclaimers against their use for diagnosis – something that Friedman says he won’t dispute. And he notes that computed radiography exams “don’t look as good on the iPad as the cross-sectional modalities.” But the iPad screen should be adequate for diagnosis, he reasons. “How many of us use stan-dard Dell consumer-grade monitors here and there instead of ‘proper’ medical-grade equipment?”
source: “radiologist reviews ipad imaging apps,” by george Miller, www.fiercebiotechit.com
Radiologist reviews iPad® imaging apps
LIfe ScIeNce compANIeS Not RuShING to SocIAL NetWoRkING
The FDA hasn’t yet weighed in on how pharma should be using social media without running afoul of the agency’s rules on drug marketing.
And that, says a report from Deloitte, is one of the chief reasons more life sciences companies haven’t embraced social networking more aggressively.
The results are based on interviews with life sciences company executives and an online survey of 208 life sciences marketing and risk management professionals.
Even after the FDA issues guidance, 53% of respondents expect there to be confusion about how life sciences companies can use things like Facebook and Twitter. Some 28% say their companies are waiting to see from others’ experiences what the return on investment in social media efforts will be. Of those respondents who are already engaged in social media, more than half say they’re using them to disseminate information, 42% to proactively seek information and 23% to react or respond to infor-mation posted on an online social network.
There are “significant” risks for companies using social media. Deloitte says data collection is least risky since most social network users have already agreed to swap personal data for the free use of the network. It’s the information dissemination that has the big risks, because of the FDA’s marketing rules. The agency “worries that conversation could result in off-label promotion, unfair or unbalanced portrayals of therapy risks and benefits, and failure to report adverse events,” the report says. source: healthblog at blogs.wsj.com/health
41%Company currently uses online
social networks to seek or disseminate information
21%plans to
38%no plans to do so

in the news
18 i n s i d e o u t Spring 2011 19
Unresolved bleedsYou may suspect an inhibitor the hard way: when factor no longer works well to stop bleeds. If you’re new to hemophilia, this may be difficult to judge – how long should it take for a bleed to stop after an infusion? It may not immediately dawn on you that the infused factor isn’t work-ing properly, especially if your child is already well into a muscle or joint bleed. It’s common for parents to think that they simply need to give their child more frequent infusions, or give a higher dose per infusion. If you think your child’s bleed is not resolving normally, or wonder whether you should dose higher or more frequently, please call your hemophilia treatment center (HTC).
Increased bruisingBruising in young children with severe hemophilia is common. But if your child is on prophylaxis and you notice increased bruising, this may be a sign of an inhibitor.
Routine clinic visitA blood test at your child’s HTC comprehen-sive clinic visit can identify an inhibitor. Low-level inhibitors are often diagnosed in this way. It’s wise to have a child with hemophilia tested for inhibitors routinely. Learning that he has an inhibitor prior to surgery or a major bleed allows parents to have a plan in place and the correct treatment on hand.
Breakthrough bleeds while on prophylaxisProphylaxis is the scheduled infusion of factor to help prevent bleeding. Many children with hemophilia on prophylaxis receive factor two to three times a week, enough to allow circulating factor to pre-vent most spontaneous bleeds and abnor-mal bruising. When a child on prophylaxis starts bruising or bleeding more often than usual, an inhibitor may be inactivating some of the factor, lowering his factor level and increasing his risk of bleeding.
Bleeding after surgeryAny kind of surgery on a child with hemophilia requires careful planning and monitoring of factor levels, and any child who continues to bleed following surgery,
even with adequate factor, should be im-mediately tested for an inhibitor. Ideally, all children with hemophilia should be tested for an inhibitor before any surgery. If you see any kind of bleeding following surgery, call your HTC immediately.
Reaction following infusionAn allergic reaction is a response by the immune system to environmental contam-
inants such as pollen, animal dander, or food. It can also happen after an infusion of factor. Symptoms may include sneezing; itching; hives; rapid swelling of the skin, neck or face; wheezing; faintness; fast heart rate; low blood pressure. Allergic reactions are especially worrisome with hemophilia B. An allergic reaction after a factor IX infusion is sometimes the first sign that an inhibitor to factor IX has de-veloped. A whopping 45% of people with hemophilia B and inhibitors also develop allergic reactions at about the same time that they develop inhibitors.
Don’t downplay allergic reactions. They may start out mild but then increase in severity after repeated exposure to products containing factor IX, often to a serious, life-threatening allergic reac-tion called anaphylaxis. If your child has severe hemophilia B, his first 20 infusions of factor IX concentrate should be done in a hospital or clinic with expertise in treating severe allergic reactions.
Following a major bleed or emergencyWhenever your child receives large amounts of factor – in response to a major bleed or during and after surgery – he may be at higher risk of developing an inhibitor. Experts aren’t sure if large amounts of factor stimulate inhibitor development, or if the body is more sus-ceptible to inhibitor development during a medical crisis because the immune
system is on high alert. But whatever the reasons, be aware that the risk of developing an inhibitor is slightly higher during an illness or surgery.
Later in lifeIf a person with hemophilia develops an inhibitor, it’s usually while he’s a child, almost always before exposure day 100. And he probably has severe hemophilia. But in rare cases, an inhibitor develops in a teen or an adult – usually in people with mild or moderate hemophilia, and usually after intensive exposure to factor during and after surgery or traumatic injury.
Inhibitors are scary to contemplate. Even when your child passes exposure day 100, don’t be lulled into thinking that he may never develop one. You can always request an inhibitor test from your HTC. Never try to diagnose on your own, or change your child’s dosing regimen on your own. source: “inhibitor insights: when to suspect an inhibitor,” the parent empowerment newsletter, February 2011. www.kelleycom.com © la kelley Communications, inc. 2011
Inhibitor Insights:
Let’s face it: an inhibitor is a major
complication of hemophilia. It develops
when the body’s immune system does
not recognize infused factor as a normal
part of blood. Instead, the body thinks
that factor is a foreign invader, like a
virus or germ, and it develops antibodies
to attack the factor and make it harm-
less – and useless. So despite an infusion
of factor, your child continues to bleed.
Prolonged bleeding, even after a
factor infusion, is the most common sign
that your child may have an inhibitor.
But other symptoms may also tip you off.

in the news
20 i n s i d e o u t Spring 2011 21
The Pain of PovertyI’ve treated patients with hemophilia for ten years, and I’m frustrated that I can infuse factor only during an active bleed, due to the exorbitant cost. We can do nothing between bleeding episodes except counseling or physical therapy for patients. Most patients do not receive factor because they are too poor to travel to the hemo-philia treatment center (HTC). We have few HTCs, so most patients must travel long distances. When patients can’t come to the HTC, due to either ignorance or poverty, they may consult a local health professional who is unaware of their special needs. The result is usually incorrect or inadequate treatment, which may cause permanent damage – sometimes death. As a result, we often see patients with severely damaged joints when they arrive at our HTC for the first time.
This is the situation at almost all HTCs in India. Hemophilia patients here lead lives filled with pain. If patients can’t afford a single visit to the HTC, how can we expect them to take regular physical therapy?
Replacement Factor The best treatment is factor, but this is impossible to prescribe for most patients here. Yet, India is home to many alterna-tive therapies: Ayurveda, Unani, Siddha, different forms of traditional comple-mentary and alternative medicine, and homeopathy. One government sector (AYUSH) even encourages research into these branches of medicine. Watching my patients suffer, I seriously considered alternative medicines for hemophilia.
Following one of my presentations to create awareness about hemophilia at Homeopathy Medical College in Nashik, Dr. Kundu, head of homeopathy medicine,
offered immediately to start a drug trial with me for hemophilia patients. It would be a double-blind placebo control trial with some patients receiving homeo-pathic medicine and others a placebo. In December 2007, we began the trial. Our main concern was relieving severe pain after joint and muscle bleeds. We applied these remedies:
❉ Lachesis 30 for pain and bleeding❉ Silicea 30 (silica) for patients requiring
repeated factor❉ Bryonia 30 + calcaria carb 30 (calcium
carbonate) for excruciating pain❉ Hamamelis virginiana (witch hazel) for
local application and gum bleeds
The selection and combination of each drug depended on each patient’s history and personality type. We have found that generalized use for any person with hemo-philia is not effective.
The evaluation and follow-up of patients was based on the Wong-Baker FACES Pain Rating Scale, the Child Adaptive-Maladaptive Behavior Scale and the Joint Mobility Assessment SF-32 scale.
Results with Unexpected BenefitsWe observed significant pain relief in the majority of cases, along with other ben-efits. In the past ten years, I have often observed aggression and depression in children with hemophilia. But following our homeopathic drug trial, the changes were amazing. Alleviation of pain was accomplished and accompanied by:
❉ Reduced duration of bleeding❉ Reduced frequency of bleeds❉ Reduced school or work absenteeism❉ Improved range of motion in the
affected joints
The most significant change: children were more friendly and cheerful, less irritable and depressed during painful bleeding episodes. Families reported a positive improvement in attitude toward living with hemophilia.
Sharing SuccessEncouraged by these promising results, we have extended this trial to three more chapters in western India. I’d like to expand the database and include more patients and chapters. If similar results are obtained in more patients, we can continue the trial throughout India and perhaps in other developing countries where factor is unaffordable.
As an allopathic doctor, I believe strongly in holistic health. I believe the majority of patients will benefit from appropriate homeopathic medicine pre-scribed by homeopathic physicians. My faith is based on the results of my personal follow-up of patients for the past two years. Though this is a short period, I believe that the results of our trial invite a larger, multi-centric trial on a bigger scale.
A Vision of HopeSupport in any form for this unique project will help to open some unexpected and promising new doors in hemophilia treat-ment, especially in developing countries. We someday want to see, Hemophilia without disability; children free of pain; the vision of the Hemophilia Federation (India).
Dr. Ranjana Kulkarni is a pathologist and President of Hemophilia Society Nashik, India.
source: “as i see it,” dr. ranjana kulkarni, the parent empowerment newsletter, February 2011, www.kelleycom.com
THE HEMOPHILIA SOCIETY, NASHIK (INDIA) CHAPTER, AFFILIATED WITH THE HEMOPHILIA
FEDERATION IS LOCATED ABOUT 150 MILES NORTHEAST OF MUMBAI (BOMBAY). WE
SUPPORT 150 PATIENTS WITH HEMOPHILIA AND THEIR FAMILIES. IN THIS PART OF INDIA,
BECAUSE OF POOR PUBLIC AWARENESS AND LACK OF MEDICAL TRAINING, WE HAVE
FEW HEALTH PROFESSIONALS WITH EVERYDAY EXPERIENCE TREATING HEMOPHILIA.
Out Of IndIa Drug trial delivers
positive results for
hemophilia patients
in poor countries.
“I’ve treated patients with hemophilia for ten years, and I’m frustrated that I can infuse factor only during an active bleed, due to the exorbitant cost. We can do nothing between bleeding episodes except
counseling or physical therapy for patients.”

in the news
22 i n s i d e o u t Spring 2011 23
Transitions:
Independent at Last! Perhaps more than any other time in our lives, the 20s are a decade of major transformation. Many young adults move out of their parents’ homes. Others grad-uate from college. Even those who’ve diligently managed their hemophilia may be thrown for a loop by all the changes in their lives.
Your hemophilia treatment center (HTC) may no longer be in the same town or even the same time zone. And finding an HTC is just one of the myriad
issues you’ll need to address. You’ll also have to ask yourself some of the basics. “Where will I buy groceries? How long will my commute be? When will I find time to renew my driver’s license?”
Weighty IssuesEntering the 9-to-5 world for the first time is a big deal that can have a major impact on your health—and not just for those in Deadliest-Catch-type occupations. For many, this is their first experience sitting behind a desk for several hours a day. A
tangible and common consequence is weight gain. Not only is extra weight bad for your joints, but obesity is a contributing factor to several leading causes of death, including heart disease, stroke and certain types of cancer.
The effects of “a few extra pounds” may not be immediately apparent, but that’s the point. In your 20s, you need to establish good habits to protect your body for the long run. This means adjust-ing what you’re eating while maintaining some form of routine exercise.
Ian Muir, a 25-year-old with he-mophilia, took a new job recently and is slowly figuring out how to get all the pieces of his life to mesh. In college, Ian competed in triathlons, training 25 hours a week. “In school, you had the motiva-tion of working out with your teammates,” says Ian. “And you had a relatively flexible
Considering Long-Term Health in Your 20s
Shaping Your FutureThroughout your 20s, medical concerns may surface that have nothing to do with hemophilia, and everything to do with the natural aging process. You’ll begin to shed the cloak of invincibility you donned as a teen, and realize that you need to act with an eye to your future. So manage your total health as attentively as you manage your hemophilia:
Eat a healthy, balanced diet. Maintain your optimal body weight. Make exercise a priority. If you drink or smoke, reduce your
alcohol consumption – and quit smoking! Schedule all the exams you’ve neglected
for so long. If you don’t have a local dentist, eye
doctor, or physician, contact your HTC or insurance company. They can help you find one.
schedule.” Now he’s struggling to find the hours to train for just a fraction of that time. And like many of his peers, Ian has let his diet suffer: “I know I need to get back to eating food that’s good for me, and not just what’s convenient.”
Living healthy in your 20s doesn’t necessarily require big time commitments. In some cases, you just have to make better choices. When you can, take the stairs instead of the elevator. Cut back on the amount of alcohol and caffeine you consume. And if you’re among the 1 in 5 Americans who smoke, quit now.
Get a Health NetWhile you lived at home with your family, you probably had a stable network of medical resources. But once you’re on your own, you may need to rebuild that network by establishing a relationship
with your new HTC, plus maintaining all facets of your health.
Dental health is one of the most neglected aspects of overall well-being. By the time you’re in your 20s, Mom or Dad probably don’t schedule your dental appointments, which means that nobody does… until a minor toothache becomes something worse.
As Ian assembles the medical re-sources he needs near his new home, he sees the value in finding the right Primary Care Physician. “You want someone you can talk to about health concerns that aren’t hemophilia related,” he says. “Someone who knows you and your medical history and if necessary, who can point you to the right specialists.”
source: “transitions: Considering long-term health in your 20s,” the parent empowerment newsletter, February 2011, www.kelleycom.com
By the time teens with hemophilia transition into their 20s, ideally they’ve built a firm foundation on which to manage their disorder. Among the many skills needed, young adults should be able to self-infuse and negotiate health insurance. But when you’re healthy and your hemophilia is under control, it’s easy to overlook general health maintenance beyond hemophilia.
It’s uncommon for 20-somethings to consider the long-term impact of their lifestyle choices. But young adulthood is the time to develop the habits that will help maintain a healthy body in the decades ahead.

Name: Joseph Miller Age: 62 years
if you spot it, you can stop it
Novo Nordisk Inc., 100 College Road West, Princeton, New Jersey 08540 U.S.A.
© 2011 Novo Nordisk Printed in the U.S.A. 0211-00001711-1 March 2011
References: 1. Huth-Kühne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009;94(4):566-575. 2. Collins PW, Hirsch S, Baglin TP, et al; for UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109(5):1870-1877. 3. Collins PW, Percy CL. Advances in the understanding of acquired haemophilia A: implications for clinical practice. Br J Haematol. 2010;148(2):183-194. 4. Bitting RL, Bent S, Li Y, Kohlwes J. The prognosis and treatment of acquired hemophilia: a systematic review and meta-analysis. Blood Coagul Fibrinolysis. 2009;20(7):517-523.
Symptoms1,2: • Arrives at the ER with spontaneous,
severe gastrointestinal bleeding
• No prior history of bleeding
Labs1,3: • Prothrombin time (PT) and activated
partial thromboplastin time (aPTT) tests and additional testing ordered by the attending physician
Treatments1: • Did not respond to treatments, including
platelets and fresh frozen plasma
Diagnosis:
Joe has acquired hemophilia (acquired inhibitors), which can be very diffi cult to diagnose and is fatal in more than 20% of all cases.4
You can help patients like Joe by being aware of the red fl ags of acquired hemophilia and bringing them up to the physician.
Find out more about acquired hemophilia and treatment at CoagsUncomplicated.com/Joe.
Model is used for illustrative purposes only.
When you see an unusual order of factor VIII (FVIII), ask some simple questions:
• What is the reason for your recent unusual order of FVIII?
• Do you have a patient with congenital hemophilia?
• Is bleeding under control?
• What diagnostic tests, such as an aPTT or a mixing study, have been performed?
• Was the aPTT prolonged?
• Have you consulted a hematologist?
• Have you considered acquired hemophilia?
NOSV0J0807_AH_DM_Full_Page_Ad_r10.indd 1 3/1/11 1:15 PM
It’s just the combination to help take the sting out of getting the flu vaccine. Order some of these colorful, complimentary “Stop the Flu” stickers for your next vaccine program. Just call ASD Healthcare at 866.281.4FLU or order online at www.asdhealthcare.com and ask for Item #10953 (30 stickers per sheet).
Protect yourself this flu season.
Let’s face it. Flu season is risky – especially for pharmacy professionals, like you. That’s why asD Healthcare is helping you protect your community while protecting your facility’s bottom line. order from the largest range of vaccine options, plus get the
asD Healthcare advantage on price, returns and deliveries. It’s going the extra mile to keep your vaccine programs on-time and on-budget, so everyone gets the protection they need – including you. That’s an example of our True Blue customer support.
Protection’s just a call away. Start reducing your flu season risks by calling ASD Healthcare at 866.281.4FLU (4358) today and visit www.asdhealthcare.com.
Reduce your risks with the asD Healthcare advantage:Price protection Best return policyMost favorable delivery schedules
Fun and Free!

in the news
26 i n s i d e o u t Spring 2011 27
March 23 marked the one-year anniversary of the enactment of the Affordable Care Act. Notwithstanding the repeal vote in the House and pending action in the fed-eral courts to overturn the law, Health and Human Services is moving forward with full implementation of the statute. The fol-lowing provisions went into effect at the beginning of this year:
a 10% Medicare bonus payment for primary care services. a 10% Medicare bonus payment to general surgeons practicing in health professions shortage areas.a requirement that health insurers provide re-bates to consumers if the share of premium dol-lars spent on clinical and quality services is less than 85% for plans in the large group market, and 80% for plans in the individual and small group markets.a requirement that pharmaceutical companies provide a 50% discount on brand-name prescrip-tions filled in the Medicare part d coverage gap.an elimination of beneficiary cost sharing for Medicare-covered preventive screenings and services.authorization of $50 million in demonstration grants for states to develop and implement alter-natives to medical malpractice litigation.an increase in the number of graduate Medical education training positions through a redistribu-tion of currently unused slots.
Congressmen Ed Whitfield (R-KY) and Gene Green (D-TX) introduced legislation in March to address a chronic shortfall in the Medicare drug reimbursement system for physicians. The bill (H.R. 905) would exclude customary prompt-pay discounts extended to wholesalers from the manufacturer’s Average Sales Price. Similar legis-lation was introduced in the last Congress and attracted 74 bipartisan co-sponsors in the House.
ASP is the price Medicare uses to set reimbursement rates for Part B drugs administered in the physician’s office. Inclusion of prompt-pay discounts in the ASP methodology effectively reduces Part B drug reimbursement to a level that hinders the ability of many providers to meet the therapeutic needs of their patients. Inclusion of the discounts also threatens the secure and highly efficient U.S. phar-maceutical distribution model resulting in increased costs and inefficiencies. It is esti-mated that distributors save consumers $34 billion a year by distributing products to local doctors and clinics.
Medicare Part B “PROMPT-PAY” LEGISLATION INTRODUCED
The 112th Congress convened in early January with Republicans holding a historic 49 seat majority in the House of Representatives. Speaker of the House John Boehner (R-OH) and his GOP colleagues wasted little time fol-lowing through on their campaign pledge to repeal the health care reform bill and cut federal spending. On Jan. 19, the House voted 245-189 to repeal the Affordable Care Act (ACA). On Feb. 19, the House passed an appropriations package for the remainder of fiscal year 2011 that cuts $60 billion in discretionary spending. Both bills were quickly rejected by the Senate as the Democratic majority in that chamber banded together in opposition to the Tea Party inspired proposals.
Medicare Physician Payment In it’s annual report to Congress, the Medicare Payment Advisory Commission (MedPAC) recommended that lawmakers increase reimbursement to Medicare physicians by 1% in FY12. Under the “Sustainable Growth Rate” formula used to calculate the annual physician fee schedule, payments to Medicare provid-ers are slated for a 25% reduction unless Congress acts to avert the cuts.
FDA Workshop on IVIG On May 17 and 18, the Food and Drug Administration will sponsor a public work-shop in Rockville, Md. on risk mitigation strategies for pro-coagulant activity
that may be present in some intravenous immune globulin (IVIG) products. The purposes of the public workshop are:
1) Identify the most likely causes
of IVIG-associated thrombotic events,
2) determine which pro-coagulant
proteins may be involved, and; 3) identify tests to assess the activity
of the proteins in IVIG products.
The workshop will feature presentations by national and international experts from government, academia and industry.
Erythropoiesis Stimulating Agents (ESAs)After a nine month review, the Centers for Medicare and Medicaid Services announced in March their decision to forgo a National Coverage Determination for ESAs used to treat anemia in chronic kidney disease patients. The agency said that published studies on the use of ESAs are insufficient to identify the risk/benefit of the therapy for the Medicare popula-tion. The FDA is expected to announce a label change for the use of ESAs in dialysis and pre-dialysis patients this spring which may trigger another review of Medicare reimbursement policy. Until then, local Medicare coverage policies for ESAs remain in effect.
Ove
rvie
w Washington updates provided by
Dale Dirks & Gavin LindbergWashington Representatives for ASD Healthcare
Regulatory Update
Although Congress has yet to finalize the FY11 appropriations process, President Obama sent his FY12 budget request to Capitol Hill on Feb. 14. The proposal calls for $3.7 trillion in federal spending. The White House projects that the plan would reduce the federal deficit by $1.1 trillion over 10 years.
$400 billion of that savings would result from a five-year freeze in “non-defense dis-cretionary” spending. The Administration did not call for any major reforms to entitle-ment programs such as Medicare and Social Security.
The President’s budget is non-binding and serves primarily as a blueprint for Congress to consider as it crafts its own budget. Congressional Republicans, and several prominent Democrats, have deemed the President’s proposal insufficient to combat the nation’s budget challenges. Despite the inherent political risks, a growing number of lawmakers on both sides of the aisle want to address the disproportionate burden that entitlement spending has on the ballooning national debt. Sixty-four senators from both parties sent a letter to President Obama in March urging him to support a comprehensive solution to the budget crisis through a combination of entitlement reforms, discretionary spending reductions, and tax policy changes.
Supreme court hears 340B drug discount caseOn Jan. 19, the Supreme Court heard oral arguments in Astra USA, Inc. v. Santa Clara County, a case involving whether safety net hospitals and clinics have a pri-vate right to sue drug manufacturers to enforce requirements of the federal 340B drug discount program. Under the 340B program, pharmaceutical companies are required to provide outpatient drugs at a discounted price to more than 15,000 “covered entities.”
The U.S. Department of Health and Human Services enters into agreements with manufacturers to establish maximum prices for drugs sold to 340B providers. Santa Clara County, California brought suit on behalf of several 340B covered entities that it operates. Counsel for the drug companies and the U.S. Solicitor General (representing the federal government) argued that 340B entities do not have a private right to sue for breach of 340B agreements. A ruling will be handed down by the Supreme Court soon.
HEALTH REFORM
IMPLEMENTATION
PResIDenT oBaMa’s fY12 buDGet pRopoSAL

in the news
28 i n s i d e o u t Spring 2011 29
As payers are faced with significant challenges related to coverage, cost, clinical management, and reimburse-ment, a variety of strategies has been utilized to address these challenges. These strategies will continue to evolve as payers analyze key areas. Currently, payers and employers are engaging more intense management techniques in specialty drug areas, like:
> Specialty pharmacy contracts and agreements
> Cost and reimbursement issues > Medical policy coverage challenges/limitations > Clinical outcomes > Compliance and adherence –
Medication Possession Ratio (MPR) > Site of care of administration > Office Based Injection programs (OBI) > Benefit design > Provider/employer partnerships > Technologic enhancements to claims
visibility for infusible medications
Specialty pharmacies are sensing the pressures from payers through narrow-ing of the specialty pharmacy networks and other restrictive utilization tech-niques, such as:
> Correlations between payer policy and specialty pharmacy – building commonalities across plan designs
> Preferred Specialty Pharmacy Networks > PHS centers taking share from specialty
pharmacies > Payer waste management – dose optimization
programs and proactive refill compliance assessments
> Payer adherence programs > Payer precertification, predetermination,
revaluation policies > Medication Therapy Management or
Pharmacy Therapy Management programs that focus on dosing, duration, therapy interval, diagnosis and drug
> Movement to ASP reimbursement methodology > Preferred product selection
Manufacturers need to consider how to navigate the complexities and degree of their relationships with specialty pharmacies and payers as they are rec-ognizing ways to manage these areas by gathering data on specialty costs. Look for more tiers, preferred product tiers within specialty tiers, and even other specialty disease-related programs.
About the author: Mike baldzicki is a senior director at Xcenda within the Customer relations team. with more than 14 years of experience in the pharmaceutical and biotech industry, Mr. baldzicki offers manufacturers strategic insight into payer marketing, contracting, reimbursement, and the application of health outcomes and economic evidence to prove product value.
In today’s healthcare environment, payers have a greater interest in determining the total cost of care for various disease states and in establishing guidelines to ensure the appropriate use of specialty infusible product. To accomplish this, payers use cost and utilization management strategies that focus on the use of
benefit design to alter prescribing patterns, ensure affordability, utilize the most ap-propriate drug and prevent waste. Payers expect prescribers to follow evidence-based clinical guidelines to ensure appropriate use of specialty drugs and minimize the risk to patient safety. The list of high-cost infusible medications based on disease states that payers recognize includes:
Strategic ImperativesXcenda, a member of the AmerisourceBergen Consulting Services family of companies, is the premier provider of services that develop, com-municate and deliver the value of healthcare technologies and pharmaceuticals. Xcenda recommends the following strategic imperatives for Specialty Pharmacy Market Access Success:
> Analyze payer trends in specialty phar-macy management
> Contracting - develop integrated strategy models
> Assess specialty pharmacy network segmentationPayer Trends:
SPecIALTy InFUSIBLe MAnAgeMenT
> Oncology > Human Growth Hormone > Multiple Sclerosis > Hemophilia > Rheumatoid Arthritis > Hepatitis > HIV/AIDS > Kidney Disease > Ophthalmology > Other selected major conditions
targeted by specialty pharmacies
• Timely payer and provider market research data• Strategic recommendations • Resources to help your team take action
Are you responsible for the design and/or implementation of a value-based commercial plan? Want to be the first to know how a group of payers who collectively represent nearly 200 million covered lives responds to industry changes and trends?
Join other senior decision makers at pharmaceutical, biotech, and device firms who already receive “Now You Know,” a complimentary publication from our managed markets agency.
Read and subscribe to “Now You Know” at www.xcenda.com.
Areas of expertise:Managed Markets
Market ResearchOutcomes Research
Reimbursement Strategy & Field SupportEstablished Brands & Pre-commercialization Phase Products
now you
KNOWA new monthly publication that delivers actionable insights from the stakeholders who mean the most to your business.
Xcenda_now You Know_5.5x8.5Ad.indd 1 2/24/11 9:22 AM

30 i n s i d e o u t Spring 2011 31
Novartis Vaccines and Diagnostics, Inc. Cambridge, MA 02139
© 2011 Novartis Vaccines Printed in USA February 2011 NVDFLU286-1
Important Safety InformationSerious allergic reactions, including anaphylactic shock, have been observed in people receiving FLUVIRIN Influenza Virus Vaccine. FLUVIRIN vaccine should not be administered to individuals with a history of systemic hypersensitivity reaction to eggs or egg proteins or other components of FLUVIRIN vaccine, including thimerosal, or to anyone who has had a life-threatening reaction to previous influenza vaccination.
Pre-filled syringes of 2010/2011 FLUVIRIN influenza vaccine are tipped with caps which may contain natural rubber latex in trace amounts. Do not administer pre-filled syringe doses of FLUVIRIN vaccine to any patients with a demonstrated history of hypersensitivity to latex. Multi-dose vial presentations of FLUVIRIN are latex-free.
In clinical trials, the most common adverse events in adults were headache, fatigue, injection site reactions (pain, mass, redness, and induration), and malaise. These adverse events were generally mild/moderate and transient. Vaccination with FLUVIRIN vaccine may not protect all individuals who are susceptible to influenza.
Immunocompromised persons, including individuals receiving immunosuppressive therapy, may have a reduced immune response to FLUVIRIN vaccine. If Guillain-Barré syndrome has occurred within 6 weeks of receipt of prior influenza vaccine, the decision to use FLUVIRIN vaccine should be based on careful consideration of the potential benefits and risks. All people, including those who are pregnant, nursing, and/or taking other medications, should consult their healthcare providers before receiving FLUVIRIN vaccine.
Please see a Brief Summary of the FLUVIRIN Prescribing Information on the following pages.
References: 1. Avert. United States HIV & AIDS Statistics Summary. Avert Web site. http://www.avert.org/usa-statistics.htm. Accessed October 27, 2010. 2. American Cancer Society. Breast Cancer Overview: How Many Women Get Breast Cancer? American Cancer Society Web site. http://www.cancer.org/Cancer/BreastCancer/OverviewGuide/breast-cancer-overview-key-statistics. Accessed November 1, 2010. 3. Centers for Disease Control and Prevention. Questions & Answers: Seasonal Influenza. CDC Web site. http://www.cdc.gov/flu/about/qa/disease.htm. Accessed October 26, 2010. 4. Centers for Disease Control and Prevention (MMWR). Prevention and Control of Influenza with Vaccines. Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. CDC Web site. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr59e0729a1.htm. Accessed November 17, 2010.
FVN6566-B_JournalAd_FFF_DR3_ASD-Rework.indd 2 6/7/11 4:02 PM
Influenza Virus VaccineFluvirin®
Influenza can kill almost as many people a year as AIDS or breast cancer.1,2
Order FLUVIRIN® now and help protect your patients for the 2011-2012 flu season.
In 2010, more than 17,000 people are expected to die from AIDS1 and nearly 40,000 women from breast cancer.2 Though influenza may not seem like a serious disease, in any given flu season it may cause 3,000 to 49,000 flu-associated deaths.3
The ACIP recommendation for annual influenza vaccination now includes all persons aged 6 months and older.4 FLUVIRIN is indicated for persons 4 years of age and older.
Novartis Vaccines is committed to providing seasonal flu vaccine doses on time. In fact, in 2010 Novartis Vaccines completed the shipping of ~40 million seasonal flu vaccine doses ahead of schedule, allowing for early and convenient administration.
Make sure you have your supply of vaccine ready for the next flu season. Contact your ASD Healthcare representative today at 866.281.4FLU or visit www.asdhealthcare.com
IndicationFLUVIRIN is an inactivated influenza virus vaccine indicated for active immunization of persons 4 years of age and older against influenza disease caused by influenza virus subtypes A and type B contained in the vaccine.
FLUVIRIN vaccine is not indicated for children less than 4 years of age because there is evidence of diminished immune response in this age group.
Please see reverse for Important Safety Information.
FVN6566-B_JournalAd_FFF_DR3_ASD-Rework.indd 1 6/7/11 4:02 PM

32 i n s i d e o u t Spring 2011 33
Fonts: NoneImages: 73526-10 Fluvirin Brief Reflow.pdf (CW_150:Users:mlove:Desktop:FVN...73526-10 Fluvirin Brief Reflow.pdf)
FVN6566-B_JournalAd_FFF_DR3.indd Melissa Love
app
rova
ls
2-22-2011 12:43 PM _______________
______________
_______________
_______________
_______________
_______________
_______________
Print Scale: NoneInk Density: 300%
Bleed: 8.625" x 10.75"Trim: 8.375" x 10.5"Safety: 7.125" x 9.125"
Folded Size: NoneGutter: NoneScale: 1" = 1"
Colors: Black GS
GSM
ED
CW
AD
AE
PD
Client: NOVARTIS MS
Job Description: JournalAd FFF
Job #: FVN6566B
Stage: DISK RELEASE
Round: 3
Three clinical studies were carried out between 1995 and 2004 in a total of520 pediatric subjects (age range 6-47 months). Of these, 285 healthy sub-jects plus 41 ‘at risk’ subjects received FLUVIRIN®. No serious adverseevents were reported.FLUVIRIN® should only be used for the immunization of persons aged 4 yearsand over.6.3 Postmarketing ExperienceThe following additional adverse reactions have been reported during post-approval use of FLUVIRIN®. Because these reactions are reported voluntarilyfrom a population of uncertain size, it is not always possible to reliably esti-mate their frequency or establish a causal relationship to vaccine exposure.Adverse events described here are included because: a) they represent reactions which are known to occur following immunizations generally orinfluenza immunizations specifically; b) they are potentially serious; or c) the frequency of reporting.• Body as a whole: Local injection site reactions (including pain, pain limiting
limb movement, redness, swelling, warmth, ecchymosis, induration), hotflashes/flushes; chills; fever; malaise; shivering; fatigue; asthenia; facialedema.
• Immune system disorders: Hypersensitivity reactions (including throatand/or mouth edema). In rare cases, hypersensitivity reactions have lead toanaphylactic shock and death.
• Cardiovascular disorders: Vasculitis (in rare cases with transient renalinvolvement), syncope shortly after vaccination.
• Digestive disorders: Diarrhea; nausea; vomiting; abdominal pain.• Blood and lymphatic disorders: Local lymphadenopathy; transient
thrombocytopenia.• Metabolic and nutritional disorders: Loss of appetite.• Musculoskeletal: Arthralgia; myalgia; myasthenia.• Nervous system disorders: Headache; dizziness; neuralgia; paraesthesia;
confusion; febrile convulsions; Guillain-Barré Syndrome; myelitis (includingencephalomyelitis and transverse myelitis); neuropathy (including neuritis);paralysis (including Bell’s Palsy).
• Respiratory disorders: Dyspnea; chest pain; cough; pharyngitis; rhinitis.• Skin and appendages: Stevens-Johnson syndrome; sweating; pruritus;
urticaria; rash (including non-specific, maculopapular, and vesiculobullous).6.4 Other Adverse Reactions Associated with Influenza VaccinationAnaphylaxis has been reported after administration of FLUVIRIN®. AlthoughFLUVIRIN® contains only a limited quantity of egg protein, this protein caninduce immediate hypersensitivity reactions among persons who have severeegg allergy. Allergic reactions include hives, angioedema, allergic asthma,and systemic anaphylaxis [see CONTRAINDICATIONS (4)].The 1976 swine influenza vaccine was associated with an increased frequencyof Guillain-Barré syndrome (GBS). Evidence for a causal relation of GBS withsubsequent vaccines prepared from other influenza viruses is unclear. Ifinfluenza vaccine does pose a risk, it is probably slightly more than 1 additionalcase/1 million persons vaccinated.Neurological disorders temporally associated with influenza vaccination suchas encephalopathy, optic neuritis/neuropathy, partial facial paralysis, andbrachial plexus neuropathy have been reported.Microscopic polyangiitis (vasculitis) has been reported temporally associatedwith influenza vaccination.
7 DRUG INTERACTIONS7.1 Concomitant Administration with Other VaccinesThere are no data to assess the concomitant administration of FLUVIRIN®
with other vaccines. If FLUVIRIN® is to be given at the same time as anotherinjectable vaccine(s), the vaccines should always be administered at differentinjection sites.FLUVIRIN® should not be mixed with any other vaccine in the same syringeor vial.7.2 Concurrent Use with Immunosuppressive TherapiesImmunosuppressive therapies, including irradiation, antimetabolites, alkylating agents, cytotoxic drugs, and corticosteroids (used in greater than physiologic doses), may reduce the immune response to FLUVIRIN®.
8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category C: Animal reproduction studies have not been conductedwith FLUVIRIN®. It is also not known whether FLUVIRIN® can cause fetalharm when administered to a pregnant woman or can affect reproductioncapacity. FLUVIRIN® should be given to a pregnant woman only if clearlyneeded.8.3 Nursing MothersIt is not known whether FLUVIRIN® is excreted in human milk. Becausemany drugs are excreted in human milk, caution should be exercised whenFLUVIRIN® is administered to a nursing woman.8.4 Pediatric UseThe safety and immunogenicity of FLUVIRIN® have not been established inchildren under 4 years of age.The safety and immunogenicity of FLUVIRIN® have been established in theage group 4 years to 16 years. The use of FLUVIRIN® in these age groups is supported by evidence from adequate and well controlled studies ofFLUVIRIN® in adults that demonstrate the immunogenicity of FLUVIRIN®
[see ADVERSE REACTIONS (6) and CLINICAL STUDIES (14) in the full pre-scribing information].8.5 Geriatric UseSince 1997, of the total number of geriatric subjects (n=397) in clinical stud-ies of FLUVIRIN®, 29% were 65 years and over, while 2.1% were 75 yearsand over.Antibody responses were lower in the geriatric population than in youngersubjects. Adverse events occurred less frequently in geriatric subjects (≥65 years) than in younger adults. Other reported clinical experience hasnot identified differences in responses between the elderly and youngerpatients. [See ADVERSE REACTIONS (6) and CLINICAL STUDIES (14) in thefull prescribing information.]FLUVIRIN® is a registered trademark of Novartis Vaccines and DiagnosticsLimited.Manufactured by: Novartis Vaccines and Diagnostics Limited, Speke, Liverpool, UKAn affiliate of: Novartis Vaccines and Diagnostics, Inc., 350 Massachusetts Avenue, Cambridge, MA 02139 USA1-800-244-7668
S:7.125”S:9.125”
T:8.375”T:10.5”
B:8.625”B
:10.75”
Fonts: NoneImages: 73526-10 Fluvirin Brief Reflow.pdf (CW_150:Users:mlove:Desktop:FVN...73526-10 Fluvirin Brief Reflow.pdf)
FVN6566-B_JournalAd_FFF_DR3.indd Melissa Love
app
rova
ls
2-22-2011 12:43 PM _______________
______________
_______________
_______________
_______________
_______________
_______________
Print Scale: NoneInk Density: 300%
Bleed: 8.625" x 10.75"Trim: 8.375" x 10.5"Safety: 7.125" x 9.125"
Folded Size: NoneGutter: NoneScale: 1" = 1"
Colors: Black GS
GSM
ED
CW
AD
AE
PD
Client: NOVARTIS MS
Job Description: JournalAd FFF
Job #: FVN6566B
Stage: DISK RELEASE
Round: 3
FLUVIRIN® (Influenza Virus Vaccine) Suspension for Intramuscular Injection2010-2011 FormulaInitial U.S. Approval: 1988
BRIEF SUMMARY: Please see package insert for full prescribing information. 1 INDICATIONS AND USAGE
FLUVIRIN® is an inactivated influenza virus vaccine indicated for immunizationof persons 4 years of age and older against influenza virus disease caused byinfluenza virus subtypes A and type B contained in the vaccine [see DOSAGEFORMS AND STRENGTHS (3) in the full prescribing information].FLUVIRIN® is not indicated for children less than 4 years of age becausethere is evidence of diminished immune response in this age group.
4 CONTRAINDICATIONS4.1 HypersensitivityFLUVIRIN® should not be administered to anyone with known systemichypersensitivity reactions to egg proteins (eggs or egg products), or to anycomponent of FLUVIRIN®, or who has had a life-threatening reaction to pre-vious influenza vaccinations.
5 WARNINGS AND PRECAUTIONS5.1 Guillain-Barré SyndromeIf Guillain-Barré syndrome has occurred within 6 weeks of receipt of priorinfluenza vaccine, the decision to give FLUVIRIN® should be based on carefulconsideration of the potential benefits and risks.5.2 Altered ImmunocompetenceIf FLUVIRIN® is administered to immunocompromised persons, includingindividuals receiving immunosuppressive therapy, the expected immuneresponse may not be obtained.5.3 Preventing and Managing Allergic ReactionsPrior to administration of any dose of FLUVIRIN®, the healthcare providershould review the patient’s prior immunization history for possible adverseevents, to determine the existence of any contraindication to immunizationwith FLUVIRIN® and to allow an assessment of benefits and risks. Appropri-ate medical treatment and supervision must be available to manage possibleanaphylactic reactions following administration of the vaccine.The tip caps of the FLUVIRIN® prefilled syringes may contain natural rubberlatex which may cause allergic reactions in latex sensitive individuals.5.4 Limitations of Vaccine EffectivenessVaccination with FLUVIRIN® may not protect all individuals.
6 ADVERSE REACTIONS6.1 Overall Adverse Reaction ProfileSerious allergic reactions, including anaphylactic shock, have been observedin individuals receiving FLUVIRIN® during postmarketing surveillance.6.2 Clinical Trial ExperienceAdverse event information from clinical trials provides a basis for identifyingadverse events that appear to be related to vaccine use and for approximat-ing the rates of these events. However, because clinical trials are conducted under widely varying condi-tions, the adverse reaction rates observed in the clinical trials of a vaccinecannot be directly compared to rates in the clinical trials of another vaccine,and may not reflect rates observed in clinical practice.Adult and Geriatric SubjectsSafety data were collected in a total of 2768 adult and geriatric subjects (18 years of age and older) who have received FLUVIRIN® in 29 clinical stud-ies since 1982.In 9 clinical studies since 1997, among 1261 recipients of FLUVIRIN®, 745 (59%) were women; 1211 (96%) were White, 23 (2%) Asian, 15 (1%)Black and 12 (1%) other; 370 (29%) of subjects were elderly (≥65 years ofage). All studies have been conducted in the UK, apart from a study run inthe US in 2005-2006 where FLUVIRIN® was used as a comparator for anunlicensed vaccine.After vaccination, the subjects were observed for 30 minutes for hypersensi-tivity or other immediate reactions. Subjects were instructed to complete adiary card for three days following immunization (i.e. Day 1 to 4) to collectlocal and systemic reactions (see Tables 1 and 2). All local and systemicadverse events were considered to be at least possibly related to the vaccine.Local and systemic reactions mostly began between day 1 and day 2. Theoverall adverse events reported in clinical trials since 1998 in at least 5% ofthe subjects are summarized in Table 3.
TABLE 1Solicited Adverse Events in the First 72-96 Hours After
Administration of FLUVIRIN® in Adult (18-64 years of age) and Geriatric (≥65 years of age) Subjects
1998-1999*§ 1999-2000*§ 2000-2001*§
18-64 yrs ≥65 yrs 18-64 yrs ≥65 yrs 18-64 yrs ≥65 yrsN=66 N=44 N=76 N=34 N=75 N=35
Local Adverse EventsPain 16 (24%) 4 (9%) 16 (21%) - 9 (12%) -Mass 7 (11%) 1 (2%) 4 (5%) - 8 (11%) 1 (3%)Inflammation 5 (8%) 2 (5%) 6 (8%) - 7 (9%) 1 (3%)Ecchymosis 4 (6%) 1 (2%) 3 (4%) 1 (3%) 4 (5%) -Edema 2 (3%) 1 (2%) 1 (1%) 2 (6%) 3 (4%) 1 (3%)Reaction 2 (3%) - 2 (3%) - 4 (5%) 1 (3%)Hemorrhage - - 1 (1%) - - -Systemic Adverse EventsHeadache 7 (11%) 1 (2%) 17 (22%) 3 (9%) 4 (5%) -Fatigue 3 (5%) 2 (5%) 4 (5%) 1 (3%) 3 (4%) -Malaise 2 (3%) 1 (2%) 2 (3%) 1 (3%) 1 (1%) -Myalgia 1 (2%) - 2 (3%) - - -Fever 1 (2%) - 1 (1%) - - -Arthralgia - 1 (2%) - 1 (3%) - -Sweating - - 3 (4%) - 1 (1%) 1 (3%)
2001-2002*^ 2002-2003*^ 2004-2005*^
18-64 yrs ≥65 yrs 18-64 yrs ≥65 yrs 18-64 yrs ≥65 yrsN=75 N=35 N=107 N=88 N=74 N=61
Local Adverse EventsPain 12 (16%) 1 (3%) 14 (13%) 7 (8%) 15 (20%) 9 (15%)Mass 4 (5%) 1 (3%) - - - -Ecchymosis 2 (3%) - 3 (3%) 3 (3%) 2 (3%) 1 (2%)Edema 2 (3%) 1 (3%) 6 (6%) 2 (2%) - -Erythema 5 (7%) - 11 (10%) 5 (6%) 16 (22%) 5 (8%)Swelling - - - - 11 (15%) 4 (7%)Reaction - - 2 (2%) - - -Induration - - 14 (13%) 3 (3%) 11 (15%) 1 (2%)Pruritus - - 1 (1%) - - -Systemic Adverse EventsHeadache 8 (11%) 1 (3%) 12 (11%) 9 (10%) 14 (19%) 3 (5%)Fatigue 1 (1%) 1 (3%) - - 5 (7%) 2 (3%)Malaise 3 (4%) - 3 (3%) 4 (5%) 1 (1%) 1 (2%)Myalgia 3 (4%) - 5 (5%) 3 (3%) 8 (11%) 1 (2%)Fever - - - 1 (1%) - -Arthralgia - - 2 (2%) - 1 (1%) -Sweating 3 (4%) 1 (3%) - 2 (2%) - -Shivering - - - 1 (1%) - -Results reported to the nearest whole percent; Fever defined as >38°C- not reported* Solicited adverse events in the first 72 hours after administration of FLUVIRIN®
§ Solicited adverse events reported by COSTART preferred term^ Solicited adverse events reported by MedDRA preferred term
TABLE 2Solicited Adverse Events in the First 72 Hours After Administration of
FLUVIRIN® in Adult Subjects (18-49 years of age)2005-2006 US Trial
FLUVIRIN®
N=304Local Adverse EventsPain 168 (55%)Erythema 48 (16%)Ecchymosis 22 (7%)Induration 19 (6%)Swelling 16 (5%)
(continued)
S:7.125”
S:9.125”
T:8.375”
T:10.5”
B:8.625”
B:10.75”

in the spotlight
Spring 2011 3534 i n s i d e o u t
Fonts: NoneImages: 73526-10 Fluvirin Brief Reflow.pdf (CW_150:Users:mlove:Desktop:FVN...73526-10 Fluvirin Brief Reflow.pdf)
FVN6566-B_JournalAd_FFF_DR3.indd Melissa Love
app
rova
ls
2-22-2011 12:43 PM _______________
______________
_______________
_______________
_______________
_______________
_______________
Print Scale: NoneInk Density: 300%
Bleed: 8.625" x 10.75"Trim: 8.375" x 10.5"Safety: 7.125" x 9.125"
Folded Size: NoneGutter: NoneScale: 1" = 1"
Colors: Black GS
GSM
ED
CW
AD
AE
PD
Client: NOVARTIS MS
Job Description: JournalAd FFF
Job #: FVN6566B
Stage: DISK RELEASE
Round: 3
Three clinical studies were carried out between 1995 and 2004 in a total of520 pediatric subjects (age range 6-47 months). Of these, 285 healthy sub-jects plus 41 ‘at risk’ subjects received FLUVIRIN®. No serious adverseevents were reported.FLUVIRIN® should only be used for the immunization of persons aged 4 yearsand over.6.3 Postmarketing ExperienceThe following additional adverse reactions have been reported during post-approval use of FLUVIRIN®. Because these reactions are reported voluntarilyfrom a population of uncertain size, it is not always possible to reliably esti-mate their frequency or establish a causal relationship to vaccine exposure.Adverse events described here are included because: a) they represent reactions which are known to occur following immunizations generally orinfluenza immunizations specifically; b) they are potentially serious; or c) the frequency of reporting.• Body as a whole: Local injection site reactions (including pain, pain limiting
limb movement, redness, swelling, warmth, ecchymosis, induration), hotflashes/flushes; chills; fever; malaise; shivering; fatigue; asthenia; facialedema.
• Immune system disorders: Hypersensitivity reactions (including throatand/or mouth edema). In rare cases, hypersensitivity reactions have lead toanaphylactic shock and death.
• Cardiovascular disorders: Vasculitis (in rare cases with transient renalinvolvement), syncope shortly after vaccination.
• Digestive disorders: Diarrhea; nausea; vomiting; abdominal pain.• Blood and lymphatic disorders: Local lymphadenopathy; transient
thrombocytopenia.• Metabolic and nutritional disorders: Loss of appetite.• Musculoskeletal: Arthralgia; myalgia; myasthenia.• Nervous system disorders: Headache; dizziness; neuralgia; paraesthesia;
confusion; febrile convulsions; Guillain-Barré Syndrome; myelitis (includingencephalomyelitis and transverse myelitis); neuropathy (including neuritis);paralysis (including Bell’s Palsy).
• Respiratory disorders: Dyspnea; chest pain; cough; pharyngitis; rhinitis.• Skin and appendages: Stevens-Johnson syndrome; sweating; pruritus;
urticaria; rash (including non-specific, maculopapular, and vesiculobullous).6.4 Other Adverse Reactions Associated with Influenza VaccinationAnaphylaxis has been reported after administration of FLUVIRIN®. AlthoughFLUVIRIN® contains only a limited quantity of egg protein, this protein caninduce immediate hypersensitivity reactions among persons who have severeegg allergy. Allergic reactions include hives, angioedema, allergic asthma,and systemic anaphylaxis [see CONTRAINDICATIONS (4)].The 1976 swine influenza vaccine was associated with an increased frequencyof Guillain-Barré syndrome (GBS). Evidence for a causal relation of GBS withsubsequent vaccines prepared from other influenza viruses is unclear. Ifinfluenza vaccine does pose a risk, it is probably slightly more than 1 additionalcase/1 million persons vaccinated.Neurological disorders temporally associated with influenza vaccination suchas encephalopathy, optic neuritis/neuropathy, partial facial paralysis, andbrachial plexus neuropathy have been reported.Microscopic polyangiitis (vasculitis) has been reported temporally associatedwith influenza vaccination.
7 DRUG INTERACTIONS7.1 Concomitant Administration with Other VaccinesThere are no data to assess the concomitant administration of FLUVIRIN®
with other vaccines. If FLUVIRIN® is to be given at the same time as anotherinjectable vaccine(s), the vaccines should always be administered at differentinjection sites.FLUVIRIN® should not be mixed with any other vaccine in the same syringeor vial.7.2 Concurrent Use with Immunosuppressive TherapiesImmunosuppressive therapies, including irradiation, antimetabolites, alkylating agents, cytotoxic drugs, and corticosteroids (used in greater than physiologic doses), may reduce the immune response to FLUVIRIN®.
8 USE IN SPECIFIC POPULATIONS8.1 PregnancyPregnancy Category C: Animal reproduction studies have not been conductedwith FLUVIRIN®. It is also not known whether FLUVIRIN® can cause fetalharm when administered to a pregnant woman or can affect reproductioncapacity. FLUVIRIN® should be given to a pregnant woman only if clearlyneeded.8.3 Nursing MothersIt is not known whether FLUVIRIN® is excreted in human milk. Becausemany drugs are excreted in human milk, caution should be exercised whenFLUVIRIN® is administered to a nursing woman.8.4 Pediatric UseThe safety and immunogenicity of FLUVIRIN® have not been established inchildren under 4 years of age.The safety and immunogenicity of FLUVIRIN® have been established in theage group 4 years to 16 years. The use of FLUVIRIN® in these age groups is supported by evidence from adequate and well controlled studies ofFLUVIRIN® in adults that demonstrate the immunogenicity of FLUVIRIN®
[see ADVERSE REACTIONS (6) and CLINICAL STUDIES (14) in the full pre-scribing information].8.5 Geriatric UseSince 1997, of the total number of geriatric subjects (n=397) in clinical stud-ies of FLUVIRIN®, 29% were 65 years and over, while 2.1% were 75 yearsand over.Antibody responses were lower in the geriatric population than in youngersubjects. Adverse events occurred less frequently in geriatric subjects (≥65 years) than in younger adults. Other reported clinical experience hasnot identified differences in responses between the elderly and youngerpatients. [See ADVERSE REACTIONS (6) and CLINICAL STUDIES (14) in thefull prescribing information.]FLUVIRIN® is a registered trademark of Novartis Vaccines and DiagnosticsLimited.Manufactured by: Novartis Vaccines and Diagnostics Limited, Speke, Liverpool, UKAn affiliate of: Novartis Vaccines and Diagnostics, Inc., 350 Massachusetts Avenue, Cambridge, MA 02139 USA1-800-244-7668
S:7.125”
S:9.125”
T:8.375”
T:10.5”
B:8.625”
B:10.75”
And the winners are …
The envelope Please: Chief Operating Officer, Chris Myers, at ASD Healthcare, selected the winners of the MP3 Players by drawing three names from the survey cards sent in by InsideOut readers.
Making the Personal connection:Sharing the good news with the winners of the MP3 Players, Neil Herson, President of ASD Healthcare, called to personally thank them for their comments and their business.
Penny StephanMercy Hospital
Grayling, Michigan
Top Topics: Articles on social media and nutrition top Penny’s InsideOut
editorial list.
Richard WestNorthside Home Infusion
Zanesville, Ohio
Richard’s Picks: He’d like to see disease specific articles and articles
on nutrition in upcoming issues.
Stacey HarsyManteno Dialysis Center
Manteno, Illinois
Most Helpful Source: Stacy finds ASD Healthcare’s customer service
the most helpful to her.
In the premier issue of InsideOut magazine, we made a winning proposition: “Tell us what articles you’d like to see in the magazine, and we’ll give MP3 Players to three lucky winners.” Well the results are in! And even though only three names were drawn – everyone is a winner.
The information each of you shared in the survey will help us create a magazine that’s an even better resource for you. That’s a win/win for everyone. The editorial staff at InsideOut thanks you for your comments and welcomes your ongoing ideas, suggestions and insights about the news that matters to you. Your input will help ASD Healthcare create a magazine that’s truly in touch with your professional and lifestyle interests.
Each quarter InsideOut will continue to include news-worthy articles that make a difference to your practice and to the patients you serve. Our goal is to help you serve them better by keeping you informed and connected to your world – inside and out.
You’re a winner!Announcing the winners of the InsideOut contest survey

in the news
36 i n s i d e o u t Spring 2011 37
Healthcare reform’s needle-in-the-haystack has quickly become its
elephant-in-the-room. In the entire 1,000-page reform bill, one of the
most thoroughly discussed and highly debated provisions spans only seven pages. Welcome to
Accountable Care Organizations (ACOs). The loosely written guide-
lines serve as a vehicle for the Medicare Shared Savings Program
and yield both headaches and valuable opportunities for stake-
holders across each sector of healthcare.
For providers, ACOs may re-quire business model overhaul and
delivery of care realignment, but they also may generate significant cost savings. For pharmaceutical
and device manufacturers, the next steps are entirely contingent upon
provider reaction toward ACOs.
What is an ACO?An ACO is a network of providers who are responsible for the quality, cost, and overall care of its assigned Medicare beneficiaries. Structurally, ACOs may consist of physi-cians, group practices, networks of prac-tices, hospitals, and/or joint ventures be-tween hospitals and physicians. Each ACO must encompass at least 5,000 Medicare beneficiaries and possess the formal legal structure to distribute shared savings. ACOs will be measured by predetermined quality-of-care benchmarks and will receive a percentage of the ACO’s shared savings for performing below the benchmark (ie, reducing unnecessary spending by align-ing delivery of care and coordinating with network providers).1
Valuable opportunities and key chal-lenges are prominent throughout the great ACO debate. ACO implementation in 2012 will generate both direct and underlying ripple effects for which providers and manufacturers must be prepared.
Opportunities for ProvidersOrganizational flexibility ✻ The health care reform bill purposely sets forth vague ACO language as to not stifle innovation for pilot programs. Therefore, providers have the freedom to outfit their organi-zation with the best fit business model. Currently, providers who establish the ACO are responsible for setting key per-formance metrics and alignment incen-
tives. Additionally, providers can choose from a variety of payment structures
that offer flexibility in the amount of risk the ACO chooses to assume.2
Ongoing patient relationship ✻ Specialists are likely to experience an ongoing relationship with patients once ACOs are implemented due to their alignment and network of care. Each ACO will need to determine prospectively whether these specialists are members and whether they will participate in its shared savings. Specialists may even prefer participating in bundled payment arrangements over participating in shared savings arrangements.3
elevated physician engagement ✻ Within ACOs, clinical care delivery requires not only strong physician relationships but also physician leadership presence in decisions affecting strategy. In large part, success in obtaining strategic input arises from internal inclusivity.4
Challenges for ProvidersThe payer market ✻ There are hundreds of different health insurers and healthcare payment programs in the United States, each with its own reimbursement policies, payment methodologies, and rates. There is no assurance that any particular payer will adopt the same method as any other payer for paying or incentivizing an ACO.5
Patient attribution ✻ No provision as-signs patients to a unique ACO. Patients who choose their provider must be attrib-
uted to a provider organization via their healthcare claims. A provider can be held accountable for the cost and outcomes of a patient who receives the majority of his or her care from other providers that may or may not belong to the same ACO.6
Shared savings allocation ✻ ACOs may require a transition step from the fee-for- service payment systems toward full risk-based payments. This transition may be necessary because ACOs provide the po-tential for upside reward (eg, shared cost savings and pay-for-performance bonuses) without the downside risk of exceeding a preset cost or utilization budget.5
Opportunities for Manufacturers 7electronic health records (EHR) ✻ As a prerequisite for ACOs, EHR will make the real-world impact of drugs on provid-er quality of care more readily available. Manufacturers that offer products with the ability to demonstrate improvements in clinical outcomes will find a much more receptive institutional audience.
Formulary influence ✻ Manufacturers armed with compelling data will be able
to influence the inclusion of their prod-ucts on formularies and in treatment guidelines, especially for value-priced preventive medicines, vaccines, and com-panion diagnostics.
collaborative opportunities ✻ If ACOs feel a product will assist their quality of care and cost-savings goals, manufactur-er representatives may be able to enlist. Representatives may be encouraged to help educate physicians and other healthcare providers about the appropri-ate use of their product within treatment guidelines.
Challenges for Manufacturers 7Access to primary care physicians ✻ Sales representatives will more than likely experience reduced access to pri-mary care physicians as they join larger, busier practices. If representatives can demonstrate value by favorably impacting quality of care or cost reductions, physi-cian access will prove less challenging.
governance structure ✻ Selective product recommendations and admin-istrative policies developed by the ACO
are likely to govern manufacturer access to physicians. Healthcare provider teams with therapeutic and cost-benefit exper-tise will determine formulary and policy guidelines, which may create a communi-cation barrier for representatives who are not armed with matching expertise.
Financial vested interest ✻ Due to the ACO’s shared savings arrangement, pro-viders will have a financial vested interest in prescribing cost-effective treatments, complying with formularies, and following recommended treatment guidelines es-tablished by the ACO. This vested interest will increase the difficulty for manufactur-ers to directly influence the prescribing of products not supported by ACOs.
As providers and manufacturers embark upon the journey of ACO imple-mentation, additional opportunities and challenges will arise. With a proactive approach, careful due diligence, and em-phasis on improving patient quality of care, each stakeholder across the healthcare spec-trum will find opportunities to succeed.
About the author: harrison burns is an analyst in reimbursement strategy and trends at Xcenda.
references: accountable Care organizations: seizing the aCo opportunity1. Medicare accountable Care organizations preliminary Questions and answers. CMs website. https://www.cms.gov/officeoflegislation/downloads/accountableCareorganization.pdf. accessed March 10, 2011.2. accountable Care organizations: a new Model for sustainable innovation. deloitte website. http://www.deloitte.com/assets/dcom-unitedstates/local%20assets/documents/us_Chs_accountableCareorganizations_041910.pdf. accessed
March 9, 2011.3. aCo learning network toolkit. brookings website. https://xteam.brookings.edu/bdacoln/documents/aCo%20toolkit%20January%202011.pdf. accessed March 7, 2011.4. Contemplating the aCo opportunity—an hFM Compendium. hFMa website. http://www.hfma.org/workarea/linkit.aspx?linkidentifier=id&itemid=24081. accessed March 8, 2011.5. transforming health Care through accountable Care organizations. Foley & lardner website. http://www.foley.com/files/tbl_s84highlights/Fileupload562/381/aCo_white_paper.pdf. accessed March 10, 2011.6. McCracken J. the Fatal Flaw of aCos. ut dallas website. http://amme.utdallas.edu/2011/02/the-fatal-flaw-of-acos/. accessed March 9, 2011.7. wokasch M. how accountable Care organizations (aCos) will affect pharmaceutical sales representatives. pharma reform website. .
Accountable Care Organizations:Seizing the Opportunity HOSPITAL
PHYSICIAN
GROUPPRACTICE
LOWERCOSTS
?
??MEDICARE BENEFICIARIES
HOSPITAL
PHYSICIAN
GROUPPRACTICE
LOWERCOSTS
?
??MEDICARE BENEFICIARIES

38 i n s i d e o u t Spring 2011 39
Important Safety Information
Hizentra and Vivaglobin are indicated as replacement therapy for patients with primary humoral immunodeficiency (PI). This includes but is not limited to the humoral immune defect in congenital agammaglobulinemia, common variable immunodeficiency, X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
Hizentra and Vivaglobin are contraindicated in patients with a history of anaphylactic or severe systemic reaction to human immune globulin preparations or their components. Because it contains the stabilizer L-proline, Hizentra is also contraindicated in patients with hyperprolinemia. Both treatments are contraindicated in patients with immunoglobulin A deficiency who have antibodies against IgA and a history of hypersensitivity.
Hizentra and Vivaglobin should be administered subcutaneously only. Do not administer intravenously.
All IgA-deficient patients with anti-IgA antibodies are at greater risk of developing potentially severe hypersensitivity and anaphylactic reactions. If hypersensitivity occurs or anaphylactic reactions are suspected, discontinue administration of these treatments immediately and treat as medically appropriate.
Hizentra and Vivaglobin are derived from human plasma. The risk of transmission of infectious agents, including viruses and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent, cannot be completely eliminated.
The most common drug-related adverse reactions with Hizentra (observed in 5% or more of subjects in the clinical trial) were local reactions (ie, swelling, redness, heat, pain, and itching at the injection site), headache, vomiting, pain, and fatigue.
The most common drug-related adverse reactions with Vivaglobin (observed in 5% or more of subjects in the clinical trial) were local injection-site reactions (eg, swelling, redness, and itching), headache, nausea, rash, asthenia, and gastrointestinal disorders.
Aseptic meningitis syndrome (AMS) has been reported to occur infrequently with IVIg treatment and treatment with Vivaglobin, and might also occur with Hizentra. AMS is usually evidenced within 2 days of administration. Also monitor patients for other reactions reported to occur with IVIg treatment that might also occur with Vivaglobin or Hizentra, including renal dysfunction/failure, thrombotic events, hemolysis, and transfusion-related acute lung injury (TRALI).
Ig administration can transiently impair the efficacy of live attenuated virus vaccines, such as measles, mumps and rubella. It can also lead to misinterpretation of serologic testing.
Please see brief summary of full Prescribing Information for Hizentra and Vivaglobin on following pages.
Hizentra is manufactured by CSL Behring AG and distributed by CSL Behring LLC. Hizentra is a registered trademark of CSL Behring AG.
Vivaglobin is manufactured by CSL Behring GmbH and distributed by CSL Behring LLC. Vivaglobin is a registered trademark of CSL Behring GmbH.
©2010 CSL Behring LLC 1020 First Avenue, PO Box 61501, King of Prussia, PA 19406-0901 USA www.CSLBehring-us.com www.Hizentra.com 11-HIZ-132 10/2010
When treating patients with PIDD…
Make the leap to Hizentra The first and only FDA-approved 20% SCIg therapy
Important Safety Information
Hizentra and Vivaglobin are indicated as replacement therapy for patients with primary humoral immunodeficiency (PI). This includes but is not limited to the humoral immune defect in congenital agammaglobulinemia, common variable immunodeficiency, X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
Hizentra and Vivaglobin are contraindicated in patients with a history of anaphylactic or severe systemic reaction to human immune globulin preparations or their components. Because it contains the stabilizer
L-proline, Hizentra is also contraindicated in patients with hyperprolinemia. Both treatments are contraindicated in patients with immunoglobulin A deficiency who have antibodies against IgA and a history of hypersensitivity.
Hizentra and Vivaglobin should be administered subcutaneously only. Do not administer intravenously.
Please see Important Safety Information on reverse and brief summary of full Prescribing Information for Hizentra and Vivaglobin on following pages.
Get helpful materials for you and your patients at www.HizentraResources.com
* Initial infusion rate not to exceed 15 mL/hr/site. Total infusion rate must not exceed 50 mL/hr for all sites combined (maximum of 4 sites).
20%concentration delivers more Ig in less volume
Up to
77°F (25°C)room temperature storage for up to 24 months
Convenient storage
Up to
25 mL/hr/site*and a volume of 25 mL/site,
as tolerated
25mL / hr
25mL / hr
25 mL / hr
Fast infusion rate
Similar patient freedom to Vivaglobin®, Immune Globulin Subcutaneous (Human), with the added convenience of room temperature storage
High concentration

40 i n s i d e o u t Spring 2011 41
The following adverse reactions have been identified and reported during the postmarketing use of IGIV products11:
Infusion reactions: Hypersensitivity (e.g., anaphylaxis), headache, diarrhea, tachycardia, fever, fatigue, dizziness, malaise, chills, flushing, urticaria or other skin reactions, wheezing or other chest discomfort, nausea, vomiting, rigors, back pain, myalgia, arthralgia, and changes in blood pressure
Renal: Acute renal dysfunction/failure, osmotic nephropathyRespiratory: Apnea, Acute Respiratory Distress Syndrome (ARDS), TRALI,
cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasmCardiovascular: Cardiac arrest, thromboembolism, vascular collapse,
hypotensionNeurological: Coma, loss of consciousness, seizures, tremor, aseptic
meningitis syndromeIntegumentary: Stevens-Johnson syndrome, epidermolysis, erythema
multiforme, dermatitis (e.g., bullous dermatitis)Hematologic: Pancytopenia, leukopenia, hemolysis, positive direct
antiglobulin (Coombs’) testGastrointestinal: Hepatic dysfunction, abdominal painGeneral/Body as a Whole: Pyrexia, rigors
To report SUSPECTED ADVERSE REACTIONS, contact CSL Behring Pharmacovigilance at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
7 DRUG INTERACTIONS
7.1 Live Virus VaccinesThe passive transfer of antibodies with immunoglobulin administration may interfere with the response to live virus vaccines such as measles, mumps, rubella, and varicella (see Patient Counseling Information [17]).
7.2 Serological TestingVarious passively transferred antibodies in immunoglobulin preparations may lead to misinterpretation of the results of serological testing.
8 USE IN SPECIFIC POPULATIONS
8.1 PregnancyPregnancy Category C. Animal reproduction studies have not been conducted with Hizentra. It is not known whether Hizentra can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Hizentra should be given to pregnant women only if clearly needed.
8.3 Nursing MothersHizentra has not been evaluated in nursing mothers.
8.4 Pediatric UseHizentra was evaluated in 10 pediatric subjects (3 children and 7 adolescents) with PI. No pediatric-specific dose requirements were necessary to achieve the desired serum IgG levels.
Hizentra was not evaluated in neonates or infants.
8.5 Geriatric UseOf the 49 subjects evaluated in the clinical study of Hizentra, 6 subjects were 65 years of age or older. No overall differences in safety or efficacy were observed between these subjects and younger subjects.
15 REFERENCES1. Cayco AV, Perazella MA, Hayslett JP. Renal insufficiency after intravenous immune
globulin therapy: a report of two cases and an analysis of the literature. J Am Soc Nephrol 1997;8:1788-1793.
2. Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology 1994;44:223-226.
3. Woodruff RK, Grigg AP, Firkin FC, Smith IL. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet 1986;2:217-218.
4. Wolberg AS, Kon RH, Monroe DM, Hoffman M. Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol 2000;65:30-34.
5. Gabor EP, Meningitis and skin reaction after intravenous immune globulin therapy. Ann Intern Med 1997;127:1130.
6. Copelan EA, Strohm PL, Kennedy MS, Tutschka PJ. Hemolysis following intravenous immune globulin therapy. Transfusion 1986;26:410-412.
7. Thomas MJ, Misbah SA, Chapel HM, Jones M, Elrington G, Newsom-Davis J. Hemolysis after high-dose intravenous Ig. Blood 1993;15:3789.
8. Wilson JR, Bhoopalam N, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle Nerve 1997;20:1142-1145.
9. Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H. In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmun 1999;13:129-135.
10. Rizk A, Gorson KC, Kenney L, Weinstein R. Transfusion-related acute lung injury after the infusion of IVIG. Transfusion 2001;41:264-268.
11. Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Trans Med Rev 2003;17:241-251.
Manufactured by: Distributed by:CSL Behring AG CSL Behring LLCBern, Switzerland Kankakee, IL 60901 USAUS License No. 1766 Based on March 2010 version
AE ( 4 Subjects)
All AEs*AEs* Occurring During or Within 72 Hours of
Infusion
Number (%) of
Subjects(n=49)
Number (Rate†)of AEs
(n=2264 Infusions)
Number (%)
of Subjects(n=49)
Number (Rate†)of AEs
(n=2264 Infusions)
Other AEs:HeadacheCoughDiarrheaFatigueBack painNauseaAbdominal pain, upperRashPain in extremityMigrainePainEpistaxisPharyngolaryngeal painArthralgia
13 (26.5)8 (16.3)7 (14.3)6 (12.2)5 (10.2)5 (10.2)5 (10.2)5 (10.2)4 (8.2)4 (8.2)4 (8.2)4 (8.2)4 (8.2)4 (8.2)
40 (0.018)9 (0.004)8 (0.004)6 (0.003)11 (0.005)5 (0.002)5 (0.002)7 (0.003)7 (0.003)5 (0.002)5 (0.002)6 (0.003)6 (0.003)5 (0.002)
12 (24.5)5 (10.2)5 (10.2)4 (8.2)4 (8.2)4 (8.2)3 (6.1)2 (4.1)4 (8.2)3 (6.1)3 (6.1)2 (4.1)2 (4.1)2 (4.1)
32 (0.014)6 (0.003)6 (0.003)4 (0.002)5 (0.002)4 (0.002)3 (0.001)3 (0.001)6 (0.003)4 (0.002)4 (0.002)3 (0.001)
2 (<0.001)3 (0.001)
* Excluding infections.† Rate of AEs per infusion.‡ Includes injection-site reactions as well as bruising, scabbing, pain, irritation, cysts, eczema, and nodules at the injection site.
The ratio of infusions with temporally associated AEs, including local reactions, to all infusions was 1338 to 2264 (59.1%; upper 95% confidence limit of 62.4%). Excluding local reactions, the corresponding ratio was 173 to 2264 (7.6%; upper 95% confidence limit of 8.9%).
Table 3 summarizes the most frequent ARs (i.e., those AEs considered by the investigators to be “at least possibly related” to Hizentra administration) experienced by at least 2 subjects.
Table 3: Incidence of Subjects With Adverse Reactions (Experienced by 2 or More Subjects) to Hizentra and Rate per Infusion (ITT Population)
Adverse Reaction ( 2 Subjects)
Number (%) of Subjects(n=49)
Number (Rate*) of Adverse Reactions (n=2264 Infusions)
Local reactions† 49 (100) 1338 (0.591)Other ARs:
HeadacheVomitingPainFatigueContusionBack painMigraineDiarrheaAbdominal pain, upperNauseaRashArthralgia
12 (24.5)3 (6.1)3 (6.1)3 (6.1)2 (4.1)2 (4.1)2 (4.1)2 (4.1)2 (4.1)2 (4.1)2 (4.1)2 (4.1)
36 (0.016)3 (0.001)4 (0.002)3 (0.001)3 (0.001)3 (0.001)3 (0.001)
2 (<0.001)2 (<0.001)2 (<0.001)2 (<0.001)2 (<0.001)
* Rate of ARs per infusion.† Includes injection-site reactions as well as bruising, scabbing, pain, irritation, cysts, eczema, and nodules at the injection site.
Table 4 summarizes injection-site reactions based on investigator assessments 15 to 45 minutes after the end of the 683 infusions administered during regularly scheduled visits (every 4 weeks).
Table 4: Investigator Assessments* of Injection-Site Reactions by Infusion
Injection-Site Reaction Number† (Rate‡) of Reactions(n=683 Infusions§)
Edema/indurationErythemaLocal heatLocal painItching
467 (0.68)346 (0.50)108 (0.16)88 (0.13)64 (0.09)
* 15 to 45 minutes after the end of infusions administered at regularly scheduled visits (every 4 weeks).† For multiple injection sites, every site was judged, but only the site with the strongest reaction was recorded.‡ Rate of injection-site reactions per infusion.§ Number of infusions administered during regularly scheduled visits.
Most local reactions were either mild (93.4%) or moderate (6.3%) in intensity.
6.2 Postmarketing ExperienceBecause postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
Table 2: (Continued)of IGIV treatment has resulted in remission of AMS within several days without sequelae.
HemolysisHizentra can contain blood group antibodies that may act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin (Coombs’) test result and hemolysis.6-8 Delayed hemolytic anemia can develop subsequent to immune globulin therapy due to enhanced RBC sequestration, and acute hemolysis, consistent with intravascular hemolysis, has been reported.9
Monitor recipients of Hizentra for clinical signs and symptoms of hemolysis. If these are present after a Hizentra infusion, perform appropriate confirmatory laboratory testing. If transfusion is indicated for patients who develop hemolysis with clinically compromising anemia after receiving Hizentra, perform adequate cross-matching to avoid exacerbating on-going hemolysis.
Transfusion-Related Acute Lung Injury (TRALI)Noncardiogenic pulmonary edema may occur in patients administered human immune globulin products.10 TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Typically, it occurs within 1 to 6 hours following transfusion. Patients with TRALI may be managed using oxygen therapy with adequate ventilatory support.
Monitor Hizentra recipients for pulmonary adverse reactions. If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies in both the product and patient’s serum.
5.3 Transmissible Infectious AgentsBecause Hizentra is made from human plasma, it may carry a risk of transmitting infectious agents (e.g., viruses, and theoretically, the Creutzfeldt-Jakob disease [CJD] agent). The risk of infectious agent transmission has been reduced by screening plasma donors for prior exposure to certain viruses, testing for the presence of certain current virus infections, and including virus inactivation/removal steps in the manufacturing process for Hizentra.
Report all infections thought to be possibly transmitted by Hizentra to CSL Behring Pharmacovigilance at 1-866-915-6958.
5.4 Laboratory TestsVarious passively transferred antibodies in immunoglobulin preparations may lead to misinterpretation of the results of serological testing.
6 ADVERSE REACTIONSThe most common adverse reactions (ARs), observed in 5% of study subjects receiving Hizentra, were local reactions (i.e., swelling, redness, heat, pain, and itching at the injection site), headache, vomiting, pain, and fatigue.
6.1 Clinical Trials ExperienceBecause clinical studies are conducted under widely varying conditions, AR rates observed in clinical studies of a product cannot be directly compared to rates in the clinical studies of another product and may not reflect the rates observed in clinical practice.
The safety of Hizentra was evaluated in a clinical study for 15 months in subjects with PI who had been treated previously with IGIV every 3 or 4 weeks. The safety analyses included 49 subjects in the intention-to-treat (ITT) population. The ITT population consisted of all subjects who received at least one dose of Hizentra (see Clinical Studies [14]).
Subjects were treated with Hizentra at weekly doses ranging from 66 to 331 mg/kg body weight during the wash-in/wash-out period and from 72 to 379 mg/kg during the efficacy period. The 49 subjects received a total of 2264 weekly infusions of Hizentra.
No deaths or serious ARs occurred during the study. Two subjects withdrew from the study due to ARs. One subject experienced a severe injection-site reaction one day after the third weekly infusion, and the other subject experienced moderate myositis. Both reactions were judged to be “at least possibly related” to the administration of Hizentra.
Table 2 summarizes the most frequent adverse events (AEs) (experienced by at least 4 subjects), irrespective of causality. Included are all AEs and those considered temporally associated with the Hizentra infusion, i.e., occurring during or within 72 hours after the end of an infusion. Local reactions were the most frequent AEs observed, with injection-site reactions (i.e., swelling, redness, heat, pain, and itching at the site of injection) comprising 98% of local reactions.
Table 2: Incidence of Subjects With Adverse Events (AEs)* (Experienced by 4 or More Subjects) and Rate per Infusion, Irrespective of Causality (ITT Population)
AE ( 4 Subjects)
All AEs*AEs* Occurring During or Within 72 Hours of
Infusion
Number (%) of
Subjects(n=49)
Number (Rate†)of AEs
(n=2264 Infusions)
Number (%)
of Subjects(n=49)
Number (Rate†)of AEs
(n=2264 Infusions)
Local reactions‡ 49 (100) 1340 (0.592) 49 (100) 1322 (0.584)
CSL BehringBRIEF SUMMARY OF PRESCRIBING INFORMATION
Hizentra,Immune Globulin Subcutaneous(Human), 20% LiquidBefore prescribing, please consult full prescribing information, a brief summary of which follows. Some text and references refer to full prescribing information.
1 INDICATIONS AND USAGEHizentra is an Immune Globulin Subcutaneous (Human) (IGSC), 20% Liquid indicated as replacement therapy for primary humoral immunodeficiency (PI). This includes, but is not limited to, the humoral immune defect in congenital agammaglobulinemia, common variable immunodeficiency, X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
4 CONTRAINDICATIONSHizentra is contraindicated in patients who have had an anaphylactic or severe systemic reaction to the administration of human immune globulin or to components of Hizentra, such as polysorbate 80.
Hizentra is contraindicated in patients with hyperprolinemia because it contains the stabilizer L-proline (see Description [11]).
Hizentra is contraindicated in IgA-deficient patients with antibodies against IgA and a history of hypersensitivity (see Description [11]).
5 WARNINGS AND PRECAUTIONS
5.1 HypersensitivitySevere hypersensitivity reactions may occur to human immune globulin or components of Hizentra, such as polysorbate 80. In case of hypersensitivity, discontinue the Hizentra infusion immediately and institute appropriate treatment.
Individuals with IgA deficiency can develop anti-IgA antibodies and anaphylactic reactions (including anaphylaxis and shock) after administration of blood components containing IgA. Patients with known antibodies to IgA may have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions with administration of Hizentra. Hizentra contains 50 mcg/mL IgA (see Description [11]).
5.2 Reactions Reported to Occur With IGIV TreatmentThe following reactions have been reported to occur with IGIV treatment and may occur with IGSC treatment.
Renal Dysfunction/FailureRenal dysfunction/failure, osmotic nephropathy, and death may occur with use of human immune globulin products. Ensure that patients are not volume depleted and assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of Hizentra and at appropriate intervals thereafter.
Periodic monitoring of renal function and urine output is particularly important in patients judged to have a potential increased risk of developing acute renal failure.1 If renal function deteriorates, consider discontinuing Hizentra. For patients judged to be at risk of developing renal dysfunction because of pre-existing renal insufficiency or predisposition to acute renal failure (such as those with diabetes mellitus or hypovolemia, those who are overweight or use concomitant nephrotoxic medicinal products, or those who are over 65 years of age), administer Hizentra at the minimum rate practicable.
Thrombotic EventsThrombotic events may occur with use of human immune globulin products2-4. Patients at increased risk may include those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, hypercoagulable disorders, prolonged periods of immobilization, and/or known or suspected hyperviscosity. Because of the potentially increased risk of thrombosis, consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients judged to be at risk of developing thrombotic events, administer Hizentra at the minimum rate practicable.
Aseptic Meningitis Syndrome (AMS)AMS may occur with use of human immune globulin products.5 The syndrome usually begins within several hours to 2 days following IGIV treatment. AMS is characterized by signs and symptoms including severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting. Cerebrospinal fluid (CSF) studies frequently show pleocytosis up to several thousand cells per cubic millimeter, predominantly from the granulocytic series, with elevated protein levels up to several hundred mg/dL. AMS may occur more frequently in association with high doses (2 g/kg) and/or rapid infusion of IGIV.
Conduct a thorough neurological examination, including CSF studies, to rule out other causes of meningitis in patients exhibiting signs and symptoms of AMS. Discontinuation
42 i n s i d e o u t Spring 2011 43
Table 3: Incidence of Subjects With Adverse Events (AEs)* (Experienced by >5% of Subjects) and Rate† per Infusion, Irrespective of Causality, in the US-Canada Study
AEs* (>5% of Subjects)
All AEsAEs Occurring During or Within 72 Hours of
Infusion
Number (%) of
Subjects(n=65)
Number (Rate†) of AEs per Infusion(n=3656)
Number (%) of
Subjects(n=65)
Number (Rate†) of AEs Per Infusion
(n=3656)
AEs at the injection site‡ 60 (92%) 1789 (0.49) 60 (92%) 1767 (0.4848)
Other AEsHeadacheGastrointestinal disorderFeverNauseaRashSore throatAllergic reactionPainDiarrheaCough increasedGastrointestinal painMigraineSkin disorderAsthmaArthralgiaAstheniaMalaise
31 (48%)24 (37%)16 (25%)12 (18%)11 (17%)10 (15%)7 (11%) 6 (9%) 6 (9%) 6 (9%)5 (8%)5 (8%)5 (8%)5 (8%)4 (6%)4 (6%)4 (6%)
159 (0.04)35 (0.01)28 (0.008)18 (0.005)22 (0.006)17 (0.005)8 (0.002)8 (0.002)6 (0.002)6 (0.002)6 (0.002)5 (0.001)7 (0.002)8 (0.002)4 (0.001)4 (0.001)5 (0.001)
30 (46%)18 (28%)12 (8%)11 (17%)10 (15%)8 (12%)5 (8%)4 (6%)5 (8%)5 (8%)4 (6%)2 (3%)3 (5%)3 (5%)3 (5%)2 (3%)2 (3%)
104 (0.033)24 (0.007)20 (0.005)15 (0.004)16 (0.004)11 (0.003)5 (0.001)4 (0.001)5 (0.001)5 (0.001)5 (0.001)2 (0.001)5 (0.001)4 (0.001)3 (0.001)2 (0.001)2 (0.001)
* Excluding infections. † Rate, number of AEs per infusion. ‡ Includes injection-site inflammation.
The total number of AEs, irrespective of causality, including injection-site reactions, that began during or within 72 hours after the end of an infusion was 2262 (a rate of 0.62 AEs per infusion); excluding injection-site reactions, the rate of AEs per infusion was 0.14.
Table 4 summarizes the severity of local AEs by infusion, irrespective of causality.
Table 4: Severity of Local Adverse Events (AEs) by Infusion, Irrespective of Causality, in the US-Canada Study
AEs(Number of infusions: 3656)
Number (Rate*) of AEs
Number (Rate*) of AEs Occurring During or Within 72 Hours of
InfusionAEs at the injection site
Mild†
Moderate‡
Severe§
Unknown severity
1789 (0.49)1112 (0.30)601 (0.16)65 (0.02)
11 (<0.01)
1767 (0.48)1100 (0.30)593 (0.16)64 (0.02)
10 (<0.01)Discontinuations due to AEs at the injection site 3 subjects
* Rate, number of AEs per infusion.† Defined as those reactions that did not interfere with routine activities. ‡ Defined as those reactions that interfered with routine activities.§ Defined as those reactions that made it impossible to perform routine activities.
Of the three subjects who discontinued the study due to injection-site reactions, one withdrew on Day 1 (Infusion 1) of the wash-in/wash-out period after a moderate injection-site reaction and a mild headache; one withdrew on Day 22 (Infusion 4) of the wash-in/wash-out period following severe injection-site reactions for two weeks; and one withdrew on Day 78 following a mild injection-site reaction.Local reactions decreased substantially after repeated use.Table 5 summarizes the most frequent adverse reactions (experienced by at least 3% of subjects) and considered by the investigator to be at least possibly related to Vivaglobin administration.Table 5: Incidence of Subjects With Adverse Reactions (Experienced in 3% of Subjects) and Rate* Per Infusion in the US-Canada Study
Related Adverse Reactions( 3% Subjects)
Number (%) of Subjects (n=65)
Number (Rate*) of Adverse Reactions
per Infusion (n=3656)
Adverse reactions at the injection site† 60 (92%) 1787 (0.49)Other Adverse reactions
HeadacheNausea RashAsthenia Gastrointestinal disorderFeverSkin disorderTachycardiaUrine abnormality
21 (32%)7 (11%)4 (6%)3 (5%)3 (5%)2 (3%)2 (3%)2 (3%)2 (3%)
59 (0.016)9 (0.002)9 (0.002)3 (0.001)3 (0.001)2 (0.001)3 (0.001)2 (0.001)3 (0.001)
* Rate, number of adverse reactions per infusion. † Includes injection-site inflammation.
Europe-Brazil StudyIn a clinical study conducted in Europe and Brazil, the efficacy and safety of Vivaglobin were evaluated for 10 months in 60 subjects with PI. Subjects were treated weekly with Vivaglobin at a mean dose of 89 mg/kg body weight (range: 51 to 147 mg/kg), which was 101% of their previous weekly IGIV or IGSC dose (see Clinical Studies [14.2]). Study subjects received a total of 2,297 infusions of Vivaglobin.
The AEs and their rates reported in this study were similar to those reported in the US-Canada study, with two exceptions: no episodes of headache were reported; and 18 (a rate of 0.008 per infusion) episodes of fever were judged to be related to the administration of Vivaglobin. One subject discontinued due to repeated local reactions of moderate severity.
6.2 Postmarketing ExperienceBecause postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
VivaglobinAdverse reactions identified during worldwide postmarketing use of Vivaglobin for treatment of PI are allergic-anaphylactic reactions (including dyspnea, pruritus, urticaria, rash, edema and other cutaneous reactions, wheezing, syncope, hypotension, and throat swelling), generalized reactions (including flu-like symptoms, myalgia, chills, fever, tachycardia, arthralgia, nausea and vomiting, diarrhea, gastrointestinal cramping, stomach pain, back pain, headache, headache possibly caused by increased blood pressure, and chest tightness), migraine, and injection-site reactions.
GeneralThe following adverse reactions have been identified and reported during the postmarketing use of IGIV products11:
Renal: Acute renal dysfunction/failure, osmotic nephropathyRespiratory: Apnea, Acute Respiratory Distress Syndrome (ARDS), TRALI, cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasmCardiovascular: Cardiac arrest, thromboembolism, vascular collapse, hypotensionNeurological: Coma, loss of consciousness, seizures, tremor, aseptic meningitis syndromeIntegumentary: Stevens-Johnson syndrome, epidermolysis, erythema multiforme, bullous dermatitisHematologic: Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs’) testGeneral/Body as a Whole: Pyrexia, rigorsMusculoskeletal: Back painGastrointestinal: Hepatic dysfunction, abdominal pain
7 DRUG INTERACTIONS
7.1 Live Virus VaccinesThe passive transfer of antibodies with immunoglobulin administration may interfere with the response to live virus vaccines such as measles/mumps/rubella and varicella (see Patient Counseling Information [17.2]).
7.2 Serological TestingVarious passively transferred antibodies in immunoglobulin preparations may lead to misinterpretation of the results of serological testing.
8 USE IN SPECIFIC POPULATIONS
8.1 PregnancyPregnancy Category C. Animal reproduction studies have not been conducted with Vivaglobin. It is also not known whether Vivaglobin can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Vivaglobin should be given to a pregnant woman only if clearly needed.
8.3 Nursing MothersVivaglobin has not been evaluated in nursing mothers.
8.4 Pediatric UseIn the US-Canada study, Vivaglobin was evaluated in 6 children (ages 5 through 11) and 4 adolescents (ages 13 through 16). In the Europe-Brazil study, Vivaglobin was evaluated in 16 children (ages 3 through 11) and 6 adolescents (ages 13 through 16).The safety and efficacy of Vivaglobin were not studied in pediatric subjects under 2 years of age.There were no differences in the safety and efficacy profiles as compared with adult subjects. No pediatric-specific dosing requirements were necessary to achieve the desired serum IgG levels.For recommendations on the number of simultaneous injection sites for pediatric patients who weigh less than 45 kg (99 pounds), see Administration (2.4).
8.5 Geriatric UseThe clinical studies of Vivaglobin did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently from younger subjects. For recommendations on the number of simultaneous injection sites for geriatric patients, see Administration (2.4).
Manufactured by: Distributed by: CSL Behring GmbH CSL Behring LLC Marburg, Germany Kankakee, IL 60901 USAUS License No. 1765 Based on April 2010 Revision.
CSL BehringBRIEF SUMMARY OF PRESCRIBING INFORMATION
Vivaglobin®
Immune Globulin Subcutaneous (Human) 16% Liquid
Before prescribing, please consult prescribing information, a brief summary of which follows. Some text and references refer to full prescribing information.
1 INDICATIONS AND USAGEVivaglobin is an Immune Globulin Subcutaneous (Human) (IGSC), 16% Liquid indicated as replacement therapy for primary humoral immunodeficiency (PI). This includes, but is not limited to, the primary immunodeficiency in common variable immunodeficiency (CVID), X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.
4 CONTRAINDICATIONS
Vivaglobin is contraindicated in patients who have had an anaphylactic or severe systemic reaction to the administration of Immune Globulin (Human).
Vivaglobin is contraindicated in IgA-deficient patients with antibodies against IgA or a history of hypersensitivity (see Description [11]).
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity ReactionsSevere hypersensitivity reactions may occur (see Patient Counseling Information [17.2]). In case of hypersensitivity, discontinue the Vivaglobin infusion immediately and institute appropriate treatment. Epinephrine should be immediately available to treat any acute severe hypersensitivity reactions.
Individuals with IgA deficiency can develop anti-IgA antibodies and anaphylactic reactions (including anaphylaxis and shock) after administration of blood components containing IgA. Patients with known antibodies to IgA may have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions. Vivaglobin contains
1.7 mg/mL IgA (see Description [11]). The minimum concentration of IgA that will provoke a hypersensitivity reaction is not known; therefore all IgG preparations carry the risk of inducing an anaphylactic reaction to IgA.
5.2 Aseptic Meningitis Syndrome (AMS)AMS has been reported to occur infrequently with IGIV treatment5 and with Vivaglobin treatment. The syndrome usually begins within several hours to 2 days following IGIV treatment. AMS is characterized by signs and symptoms including severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting. Cerebrospinal fluid (CSF) studies frequently show pleocytosis up to several thousand cells per cubic millimeter, predominantly from the granulocytic series, and with elevated protein levels up to several hundred mg/dL. AMS may occur more frequently in association with high doses (2 g/kg) and/or rapid infusion of IGIV.
Patients exhibiting such signs and symptoms should receive a thorough neurological examination, including CSF studies, to rule out other causes of meningitis. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae.
5.3 Reactions Reported with IGIV TreatmentThe following reactions have been reported to occur with IGIV treatment and may occur with IGSC treatment.
Renal Dysfunction/FailureRenal dysfunction/failure, osmotic nephropathy, and death may occur with use of human immune globulin products. Ensure that patients are not volume depleted and assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of Vivaglobin and at appropriate intervals thereafter.
Periodic monitoring of renal function and urine output is particularly important in patients judged to have a potential increased risk of developing acute renal failure.1 If renal function deteriorates, consider discontinuing Vivaglobin. For patients judged to be at risk of developing renal dysfunction because of pre-existing renal insufficiency or predisposition to acute renal failure (such as those with diabetes mellitus or hypovolemia, those who are overweight or use concomitant nephrotoxic medicinal products, or those who are over 65 years of age), administer Vivaglobin at the minimum rate practicable.
Thrombotic EventsThrombotic events may occur with use of human immune globulin products.2-4 Patients at risk may include those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, hypercoagulable disorders, prolonged periods of immobilization, and/or known or suspected hyperviscosity. Because of the potentially increased risk of thrombosis, consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients judged to be at risk of developing thrombotic events, administer Vivaglobin at the minimum rate practicable.
HemolysisVivaglobin may contain blood group antibodies that may act as hemolysins and induce in vivo coating of red blood cells (RBCs) with immunoglobulin, causing a positive direct antiglobulin (Coombs’) test result and hemolysis.6-8 Delayed hemolytic anemia can develop subsequent to immune globulin therapy due to enhanced RBC sequestration, and acute hemolysis, consistent with intravascular hemoylysis, has been reported.9
Monitor recipients of Vivaglobin for clinical signs and symptoms of hemolysis. If these are present after Vivaglobin infusion, perform appropriate confirmatory laboratory testing. If transfusion is indicated for patients who develop hemolysis with clinically compromising anemia after receiving Vivaglobin, perform adequate cross-matching to avoid exacerbating on-going hemolysis.
Transfusion-Related Acute Lung Injury (TRALI)Noncardiogenic pulmonary edema may occur in patients administered human immune globulin products.10 TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Typically, it occurs within 1 to 6 hours following transfusion. Patients with TRALI may be managed using oxygen therapy with adequate ventilatory support.
Monitor recipients of Vivaglobin for pulmonary adverse reactions. If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies in both the product and patient’s serum.
5.4 Transmissible Infectious AgentsBecause Vivaglobin is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses and, theoretically, the Creutzfeldt-Jakob (CJD) agent. No cases of transmission of viral diseases or CJD have been associated with the use of Vivaglobin. Report all infections thought possibly to have been transmitted by Vivaglobin to the CSL Behring Pharmacovigilance Department at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. The physician should discuss the risks and benefits of this product with the patient before prescribing or administering it to the patient (see Patient Counseling Information [17.2]).
5.5 Laboratory TestsAfter infusion of IgG, the transitory rise of the various passively transferred antibodies in the patient’s blood may yield positive serological testing results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g., A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs’) test.
6 ADVERSE REACTIONSThe most common adverse reactions (those AEs considered by the investigator to be at least possibly related to Vivaglobin administration) observed in 5% of study subjects receiving Vivaglobin were local injection-site reactions (swelling, redness, and itching), headache, nausea, rash, asthenia, and gastrointestinal disorder.
6.1 Clinical Studies ExperienceBecause clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not re�ect the rates observed in practice.
US-Canada StudyThe safety of Vivaglobin was evaluated in a clinical study in the US and Canada for 12 months in 65 subjects with PI who had been previously treated with IGIV every 3 or 4 weeks (see Clinical Studies [14.1]). After 3 months, subjects were switched from IGIV to weekly subcutaneous administration of Vivaglobin for 12 months. Subjects were treated weekly with Vivaglobin at a mean dose of 158 mg/kg body weight (range: 34 to 352 mg/kg). The 65 subjects received a total of 3,656 infusions of Vivaglobin.
Table 2 shows the number of subjects who withdrew from the US-Canada study due to adverse events (AEs) and the AEs leading to discontinuation.
Table 2: Subjects with Adverse Events (AEs) Leading to Discontinuation, US-Canada Study
AEsSubjects with AEs At Least
Possibly Related
Subjects with AEs Irrespective of
Causality
Total Number (%) of Subjects
Subjects with at least 1 AE leading to discontinuation
4 1 5 (8%)
Injection-site reaction 3 – 3 (5%)Intestinal obstruction – 1 1 (2%)Hyperventilation 1* – 1 (2%)Tachycardia 1* – 1 (2%)
* One subject experienced hyperventilation and tachycardia.
Table 3 summarizes the most frequent AEs (experienced by more than 5% of subjects), irrespective of causality. It includes all AEs and those considered temporally associated with the Vivaglobin infusion, i.e., occurring during the infusion or within 72 hours after the end of the infusion.

in the community
44 i n s i d e o u t Spring 2011 45
care, this is crucial. “We’ve found that most of the sponsorship money, regardless of the country, goes to school fees to help the child get a good education and lift the family out of poverty, and transportation to a hospital when the child has a bleed,” Kelley notes.
Along with its dedication to the bleeding disorders community, Save One Life knows the importance of using its funds carefully and diligently. To achieve this, the foundation builds strong personal relationships with the non-governmental organizations (NGOs) that manage its pro-grams in developing countries. This helps define Save One Life as, not just a charity, but a development tool that helps promote long-term care and capacity-building with NGOs through training, progress monitor-ing and strict accountability.
Today the organization provides sponsorships to more than 758 individuals with bleeding disorders in 11 developing nations (see sidebar). Sponsors receive an attractive photo and biography of their sponsored child and are encouraged to write them.
ASD Healthcare is proud to support Save One Life and its mission. Neil Herson, president of ASD Healthcare said, “The contributions Save One Life makes to the hemophilia community are invalu-able in raising awareness of the challenges patients face around the world and in saving lives.”
Individuals, groups and companies interested in supporting Save One Life are encouraged to participate by visiting www.saveonelife.net. Save One Life offers a variety of ways to support their mission, including the Mount Kilimanjaro Climb for a Cause in August (see below).
Imagine, for a moment, living with hemo-philia. Consider the uncertainties – how an ordinary day can turn into a life-or-death emergency. Imagine the resources needed to improve the quality of your life or to save it. And imagine the cost – the price of life-saving factor, refrigeration, electric-ity, clean water – all things we take for granted.
Now, imagine living that life in a poor country. No money for medications. No refrigeration to keep it viable. No clean water. And no local treatment center.
Those are the life-and-death chal-lenges patients with hemophilia face every
day in countries like Zimbabwe, India and Honduras. They’re the life-and-death chal-lenges Laurie Kelley saw during her many trips to educate patients about hemophilia in developing countries. As the mother of a child with hemophilia, she knew first-hand the daily struggles. But in poor countries, where most hemophilia patients live in households earning about $1 a day, those challenges become staggering.
Kelley explains, “Hemophilia is a devastating inherited blood disorder that cause severe pain, crippling and even death when left untreated. In developing countries there are usually no government budgets to purchase the blood-clotting
medicine needed to sustain life. Government and social agencies
must use scarce resources on other pressing
problems – infectious diseases, clean
water, natural disasters,
high un-employ-
ment – not on a rare chronic disorder.” To address this need, Kelley founded
Save One Life in 2000, a registered international nonprofit organization. Her vision was to build a humanitarian bridge by raising awareness and encouraging people in developed countries to give back to the rest of the world in a personal way. “I believed that families with hemophilia in wealthier countries, with vast resources and adequate healthcare, would be willing to help once they understood the suffering of people with hemophilia in developing countries,” Kelley said.
Save One Life provides sponsorships for individual hemophilia patients. People in developed countries can choose a child or young adult from a poor country to support for only $20 a month, less than a dollar a day. These funds provide vita-mins to anemic children with hemophilia, pain medication, school fees, clothing and other daily living needs. But most of all, these funds provide transportation to the clinic when a child has a bleed. Since these families live in urban slums or distant, rural villages far from medical
Save One Life
This August, eleven Save One Life supporters, including founder Laurie Kelley and ASD Healthcare president Neil Herson, will be climbing for a cause as they scale the highest mountain in Africa – 19,340 foot Mt. Kilimanjaro.
The climbers will pay their own expenses for the trip so that all funds raised will go directly to Save One Life programs in Africa. In addition to the financial goals, this climb also will put a spotlight on hemophilia care in Africa. It’s an event that will inspire people everywhere that they can make a difference in the lives of those affected by hemophilia.
To support your favorite climber, go to http://saveonelife.net/mt-kilimanjaro-climb.php Read about the event, learn about the climbers and join in by supporting your favorite climber.
“I believed that families with hemophilia in wealthier countries, with vast resources and adequate healthcare, would be willing to help once they understood the suffering of people with hemophilia in developing countries.”
– Laurie Kelley Founder and President of Save One Life
Join the Adventure Mount Kilimanjaro Climb for a Cause
Save One Life was founded in 2000 by Laurie Kelley as a registered inter-national nonprofit organization. The foundation offers individuals, families, companies and organizations the oppor-tunity to sponsor a child or adult with a bleeding disorder in developing coun-try. Today, more than 758 hemophilia patients are being sponsored in 11 coun-tries – Romania, Philippines, India, Nepal, Pakistan, Dominican Republic, Belize, Honduras, Egypt, Zimbabwe and Kenya.
The organization also works in part-nership with the hemophilia foundations in each country where their members vol-unteer their care, time and resources to improve the lives of impoverished people with bleeding disorders. As a result, 100% of sponsorship donations go directly to the sponsored child and their local hemo-philia chapter.
Our goals: • Identify people with bleeding disorders
who need aid in the developing world • Match them with sponsors who offer
long-term friendship and financial support
• Partner with and mentor local hemo-philia organizations through program funding
• Raise public awareness about people with hemophilia in the developing world
about save one life
A Bridge of Hope for Hemophilia Patients in Developing Countries
www.saveonelife.net

46 i n s i d e o u t Spring 2011 47
Devetta Coulter shares
a moment with her fiancé
Dwayne James and her thoughts
on True Blue service with you.
“ For me, delivering exceptional customer support is personal.
As Vice President of Business Operations, I know how my
teams make a difference in the lives of patients. Relationships
become personal when you know people depend on your
actions. That’s why our operating systems – whether it’s
customer service, project management, administration, internal
or external – are built to offer proactive solutions, to think
ahead, to plan ahead and to utilize the most effective
technologies in supporting our customers and the patients
who depend on them. That’s True Blue.”
Our True Blue culture encompasses all aspects of our business.
From technology offerings, to customer support, to patient advocacy,
to community involvement – ASD Healthcare is here for you. Trust us
to provide the products and support you need in an ever-changing
landscape of medicine, policy and patient demographics.
Call us. Challenge us. Experience True Blue.
Learn more about True Blue by going to www.asdtrueblue.com.
Call us at 800.746.6273
ASD_Ad_Q2 InsideOut_Devetta.indd 1 6/21/11 11:36 AM
Devetta Coulter shares
a moment with her fiancé
Dwayne James and her thoughts
on True Blue service with you.
“ For me, delivering exceptional customer support is personal.
As Vice President of Business Operations, I know how my
teams make a difference in the lives of patients. Relationships
become personal when you know people depend on your
actions. That’s why our operating systems – whether it’s
customer service, project management, administration, internal
or external – are built to offer proactive solutions, to think
ahead, to plan ahead and to utilize the most effective
technologies in supporting our customers and the patients
who depend on them. That’s True Blue.”
Our True Blue culture encompasses all aspects of our business.
From technology offerings, to customer support, to patient advocacy,
to community involvement – ASD Healthcare is here for you. Trust us
to provide the products and support you need in an ever-changing
landscape of medicine, policy and patient demographics.
Call us. Challenge us. Experience True Blue.
Learn more about True Blue by going to www.asdtrueblue.com.
Call us at 800.746.6273
ASD_Ad_Q2 InsideOut_Devetta.indd 1 6/21/11 11:36 AM

48 i n s i d e o u t Spring 2011 49
The goals of pharmacotherapy are to neutralize the toxin of snake bites, to reduce morbidity and to prevent complications.
A neutralizing antibody gives antivenin efficacy. The current product approved by the U.S. Food and Drug Administration (FDA) in 2000 (CroFab®, BTG International Inc.), is a mixed monovalent immunoglobulin fragment derived from sheep but purified to avoid other antigenic proteins.
While rare, immediate hypersensitivity reactions (symptoms of acute anaphylactoid reactions such as pruritus, urticaria or wheezing occurred in roughly 6% of patients in prospective studies and were generally mild) and delayed hypersensitivity (serum sickness occurred in 5-10% of patients in prospective studies) reactions have been reported.1 To achieve maximum efficacy, antivenin should be administer within six hours of the snake bite.
A local expert or poison control center should be consulted prior to giving antivenin (1-800-222-1222).
CroFab is made specifically from venom of the Eastern and Western Diamondback rattlesnakes, Mohave rattlesnakes and the cottonmouth or water moccasin snakes. The purpose of any antivenin is to bind the toxins in the venom and prevent both local and systemic results.
CroFab has been used in Crotalid bites with good effect (reduced fasciotomy) and reductions in antivenin toxicity.
To read an expanded discussion on snakebite treatments, go to www.biomedcentral.com/bmcemergmed, then select, “Unified Treatment Algorithm for the Management of Crotaline Snakebite in the United States: Results of an Evidence-Informed Consensus Workshop.”
source: lavonas eJ. unified treatment algorithm for the Management of Crotaline snakebite in the united states: results of an evidence-informed Consensus workshop. biomed Central. 2011; 11:2.
Most venomousThe most venomous of these vipers would be the rattlesnake. They are responsible for the most fatalities in the U.S. annually.
Silent dangerWater moccasins (commonly referred to as cottonmouths) are less common and less aggressive than rattlesnakes. They are dangerous and don’t give any type of warning like that of a rattlesnake. Victims are often caught off guard.
More but lessCopperheads are responsible for the most reported bites, but their venom (in general) is considered to be sub-lethal. However, their bite is a medical emergency. You should seek immediate medical attention for a bite regardless of the type of snake.
It’s the time of year when people like to be outdoors. Whether we are camping, picnicking, hiking or just gazing upon some beautiful scenery, a snake bite can bring our enjoyment of Mother Nature to an abrupt end!
Snakes bite about 8,000 people each year in the United States. Only 12 people each year die from snake bite. Children are at greater risk of dying or having serious complications from a snake bite because of their smaller size. Most snakes will avoid
people and bite only when threatened or surprised. A bite from a poisonous snake is considered a medical emergency.
There are two types of venomous (or poisonous) snakes found in the United States: pit vipers – rattlesnakes, copperheads and cottonmouths – and elapids – coral snakes. Pit vipers are responsible for 98% of all poisonous snake bites each year. However, more and more exotic and foreign snakes are brought into the country illegally. For that reason, all snake bites are considered serious. Knowing what to do in case of a snake bite is important.
Symptoms depend upon the type of snake causing the bite. Each individual may experience different symptoms. Pit viper bites are generally painful when they occur, symptoms usually begin right away and fang marks are seen. Pit viper venom contains peptides and proteins that damage the red blood cells. A bite from a coral snake may be painless at first and symptoms may not develop right away. Coral snake venom is primarily a neurotoxin.
Play it Safe.Avoiding snake bites is the best way to keep safe outdoors. Leave snakes alone. Don’t pick up any snake – even though you believe it’s not poisonous. When hiking, wear thick leather boots and long pants. Stay on the trails and out of tall, grassy areas. Use a walking stick to test ahead of you when entering any areas where you can’t see your feet. Avoid areas under rocks or logs and woodpiles.
Remember, most snakes are harmless and most snake bites are not life threaten-ing. Don’t try to determine if the snake is poisonous or not. Once bitten, stay calm and always seek medical assistance. The good news is that the majority of snake bites can be treated effectively when the victim is transported as soon as possible to a medical facility that has antivenin.
references: paul s. auerbach, wilderness Medicine, http://www.nim.nih.gov/medlineplus/ency/article/000031.htm; accessed June 12, 2009 tintinalli and stapcynski, editors, emergency Medicine: a Comprehensive guide, 6th edition.
staying Safe this Snake Bite Season
Most snake bites in the U.S. (Texas has the highest number of reported bites annually) come from members of the Crotalidae family, also known as pit vipers. This includes varieties such as rattlesnakes, water moccasins and copperheads. Here’s how they compare:
The
Deadliest Bite
Life-Saving Antivenins

Spring 2011 5150 i n s i d e o u t
Snakes aren’t the only things that can take a bite out of your summer fun. Fido can create his share of havoc, too.
Young children are at the greatest risk for dog bites in the summer and are especially vulnerable to severe bites in the head and neck areas, according to a study quoted in U.S. News and World Report.
Researchers analyzed 84 cases of dog bites in children. The findings were published in the March issue of Otolaryngology (head and neck surgery).
It’s not clear why children are more likely to be bitten in the summer, but it may be because children spend more time outdoors playing with dogs in warmer months, the researchers suggested. Or it may be that dogs are generally more irritable in hot weather.
The study found that 27 percent of dog bite injuries were caused by family pets. The most common sites of bites to the head and neck were the cheeks (34 percent), lips (21 percent) and nose and ears (both 8 percent).
source: “dog bite season is around the Corner,” 12 apr. 2009, John davidson, http://blogs.denverpost.com/fetch
Beware: Dogs bite too
Rabies Risk ❉ Today, the risk of rabies in the U.S. and Canada doesn’t come from Fido. It comes from wild animal – raccoons, skunks, foxes, coy-otes – and mostly bats. So if you’re going into the wild, be aware. Bat bites or scratches can be small and go unnoticed. Call your doctor if you’ve been in close contact with a bat or wild animal. Bites are an urgent reason to seek medical atten-tion, immediately. reference: “rabies” pubMed, www.ncbi.nlm.nih.gov/pubmedhealth
In the United States,
dog bites account for about
1 percent of all emergency
room visits, including
44,000 cases per year of
facial injuries.
Do • Do stay calm! Call for medical assistance
immediately! • Do get away from the snake – many snakes
can strike from quite a distance. • Do minimize movement particularly of the
affected extremity. A victim may need to be carried to a safe area.
• Do splint the affected area to keep it immobile. • Do remove any rings, jewelry or potentially
constricting clothing because swelling can occur.
• Do keep the affected area below the level of the heart.
• Do wash the area of the bite with soap and water.
EMS personnel should monitor the patient’s vital signs. A cool compress or moist dressing can minimize swelling and discomfort. Treat pain according to local protocols as necessary. Transport as soon as possible to the closest hospital that is able to treat the patient with antivenin. If access to medical care cannot be accomplished quickly, a light pressure dressing, such as an elastic bandage, may be applied. If necessary, contact the National Poison Control Center at (800) 222-1222 for additional instructions.
Don’t• Don’t allow the snake bite victim to become
excited and move around. • Don’t apply a tourniquet.• Don’t apply ice directly to the bite. • Don’t cut the bite or try to suck out the
venom by mouth. • Don’t give the victim anything to eat or drink.• Don’t raise the site of the bite above the
level of the victim’s heart. • Above all, don’t waste time hunting for the
snake. If the snake has been killed, be care-ful. The head can still bite reflexively for up to an hour after the snake’s death.
Once Bitten:
Pain, bruising, swelling and necrosis of tissue at or around the bite site
Blood clotting defects (presence of mini hemorrhages on the mucous membranes)
Decreased number of platelets in the blood (affects clotting times)
Cardiovascular and respiratory distress
Snake Bite Symptoms
Low blood pressure and high heart rate, which together equals shock
Depression and/or lethargy
Nausea and excessive salivation
For further information call: Grifols USA, LLC Professional Service: 888 GRIFOLS (888 474 3657)Customer Service: 888 325 8579 Fax: 323 441 7968 www.grifols.com
Grifols Biologicals Inc.5555 Valley Boulevard, Los Angeles, California 90032, USA
10 mL
10 mL
10 mL
Diluent SizePotency
500 IU FIX Range
1000 IU FIX Range
1500 IU FIX Range
Available in the following potencies and color coded assay ranges
A9L1
4-15
-US-
10
Packaged with Mix2Vial® Filter Transfer Set

in our catalog
Spring 2011 53
Our updated catalog now includes not
only our most popular plasma products,
but also products for contrast media,
nephrology, and more!
This revamped section is color-coded
for easy identification and quick reference.
Flip through and see what ASD Healthcare
has to offer, and remember that the product
you are looking for may be found in a
different section of the catalog, depending
on its use.
Is there a product you need, but don’t see
in this catalog? An item you’ve been
ordering that you can’t find here? Don’t
worry – our product offerings expand well
beyond what is listed here. Call us today for
more information and personalized service.
Customer Service 800.746.6273
or www.asdhealthcare.com
Plasma Derivatives
Albumin 54
Antihemophelic 54
Immunologic 56
Hyper-Immune Globulin 57
Pharmaceuticals
Vaccines 58
Specialty 58
Biosurgery 60
Contrast Media
Astellas Pharma US 61
Bayer 61
Bracco 62
Covidien 65
GE 70
Nephrology
Rx 72
Medsurge 75
KOGP-41854_M03_US_Remind_Ad.indd12-22-2010 3:01 PM Suke Yawata / Suke Yawata
Client CodeClient
LiveOverall TrimBleed
# of Colors
KN0000810Bayer/Kogenate
7” x 10”8.25” x 10.875”8.75” x 11.375”
CMYK
Job info
NoneNotes Fonts
Myriad Pro (Semibold, Regular, Bold)
ImagesKogenateFS_BIOSET_4C.eps, BayerHC_Pharm_4C_mini.eps
Inks Cyan, Magenta,
Yellow, Black
Fonts & Images
Saved at
None
from syawata2976 by
Printed At
Kogenate® FS with BIO-SET®
Free Trial Program
For more information, visit kogenatefs.com or call 1-866-329-3449 (Monday through Friday, 8:30 am to 5:30 pm, ET).
Ask your doctor if Kogenate® FS with BIO-SET® is right for you.
BAYER, the Bayer Cross, and KOGENATE are registered trademarks of Bayer.BIO-SET is a registered trademark of Biodome SAS.
©2011 Bayer HealthCare Pharmaceuticals Inc. All rights reserved 01/11 KN10000810
6 free doses*
Enroll today for up to
* Subject to approval. Terms and conditions apply.
S:7”
S:10”
T:8.25”
T:10.875”
B:8.75”
B:11.375”

54 i n s i d e o u t Spring 2011 55
J7192 Kogenate® FS 500 iu BYR 00026-3783-30 vial
J7192 Kogenate® FS 1000 iu BYR 00026-3785-50 vial
J7192 Kogenate® FS 2000 iu BYR 00026-3786-60 vial
J7192 Kogenate® FS 3000 iu BYR 00026-3787-70 vial
J7192 Kogenate® FS w/BIO-SET® 250 iu BYR 00026-3792-20 vial
J7192 Kogenate® FS w/BIO-SET® 500 iu BYR 00026-3793-30 vial
J7192 Kogenate® FS w/BIO-SET® 1000 iu BYR 00026-3795-50 vial
J7192 Kogenate® FS w/BIO-SET® 2000 iu BYR 00026-3796-60 vial
J7192 Kogenate® FS w/BIO-SET® 3000 iu BYR 00026-3797-70 vial
J7192 Recombinate® 250 iu BAX 00944-2831-15 vial
J7192 Recombinate® 500 iu BAX 00944-2832-15 vial
J7192 Recombinate® 1000 iu BAX 00944-2833-15 vial
J7185 Xyntha™ 250 iu WYE 58394-0012-01 vial
J7185 Xyntha™ 500 iu WYE 58394-0013-01 vial
J7185 Xyntha™ 1000 iu WYE 58394-0014-01 vial
J7185 Xyntha™ 2000 iu WYE 58394-0015-01 vial
Factor VIII (Immunoaffinity Purified. Derived from Human Plasma.)
J7190 Hemofil® M 250 iu BAX 00944-2930-01 vial
J7190 Hemofil® M 500 iu BAX 00944-2931-01 vial
J7190 Hemofil® M 1000 iu BAX 00944-2932-01 vial
J7190 Hemofil® M 1700 iu BAX 00944-2933-01 vial
J7190 Monoclate-P® 250 iu CSL 00053-7656-01 vial
J7190 Monoclate-P® 500 iu CSL 00053-7656-02 vial
J7190 Monoclate-P® 1000 iu CSL 00053-7656-04 vial
J7190 Monoclate-P® 1500 iu CSL 00053-7656-05 vial
Factor VIII (Derived from Human Plasma. Contains von Willebrand Factor.)
J7186 Alphanate® 250 iu GFS 68516-4601-01 vial
J7186 Alphanate® 500 iu GFS 68516-4602-01 vial
J7186 Alphanate® 1000 iu GFS 68516-4603-02 vial
J7186 Alphanate® 1500 iu GFS 68516-4604-02 vial
J7187 Humate-P® 250 RCoF CSL 00053-7615-05 vial
J7187 Humate-P® 500 RCoF CSL 00053-7615-10 vial
J7187 Humate-P® 1000 RCoF CSL 00053-7615-20 vial
J7190 Koate®-DVI 250 iu TAL 13533-0665-20 vial
J7190 Koate®-DVI 500 iu TAL 13533-0665-30 vial
J7190 Koate®-DVI 1000 iu TAL 13533-0665-50 vial
J3590 Wilate® 450iu RCo/5ml OCT 67467-181-01 vial
J3590 Wilate® 900iu RCo/10ml OCT 67467-181-02 vial
Factor IX (Recombinant)
J7195 BeneFIX® 250 iu WYE 58394-0003-06 vial
J7195 BeneFIX® 500 iu WYE 58394-0002-06 vial
J7195 BeneFIX® 1000 iu WYE 58394-0001-06 vial
J7195 BeneFIX® 2000 iu WYE 58394-0008-02 vial
Factor IX (Prothrombin Concetrates. Derived from Human Plasma. Contains Factor II, VII, IX and X.)
J7194 Bebulin® VH 500-700 iu BAX 64193-0244-02 vial
J7194 Profilnine® SD 500 iu GFS 68516-3201-1 vial
J7194 Profilnine® SD 1000 iu GFS 68516-3202-2 vial
J7194 Profilnine® SD 1500 iu GFS 68516-3203-2 vial
plasma d
erivativespl
asm
a d
eriv
ati
ves
AlbuminCODE PRODUCT SIZE EQ. UNIT MFR. NDC # PK/CS
P9045 Albumin 5% 250 ml 1 OCT 67467-0623-02 vial
P9047 Albumin 25% 50 ml 1 OCT 67467-0643-01 vial
P9047 Albuminar® 25% 50 ml 1 CSL 00053-7680-02 vial
P9047 Albuminar® 25% 100 ml 2 CSL 00053-7680-03 vial
P9045 Albutein® 5% (Albumin) 250 ml 1 GFS 68516-5214-01 vial
P9045 Albutein® 5% (Albumin) 500 ml 2 GFS 68516-5214-02 vial
P9047 Albutein® 25% (Albumin) 50 ml 1 GFS 68516-5216-01 vial
P9047 Albutein® 25% (Albumin) 100 ml 2 GFS 68516-5216-02 vial
P9047 AlbuRx® 25% 50 ml 1 CSL 44206-0251-05 vial
P9047 AlbuRx® 25% 100 ml 2 CSL 44206-0251-10 vial
P9045 AlbuRx® 5% 250 ml 1 CSL 44206-0310-25 vial
P9045 AlbuRx® 5% 500 ml 2 CSL 44206-0310-50 vial
J3490 Buminate 5% (Albumin) 6 x 250 ml 1 BAX 00944-0491-01 case
J3490 Buminate 5% (Albumin) 6 x 500 ml 2 BAX 00944-0491-02 case
J3490 Buminate 25% (Albumin) 10 x 20 ml 0.4 BAX 00944-0490-01 case
J3490 Flexbumin 25% 24 x 50 ml 1 BAX 00944-0493-01 case
J3490 Flexbumin 25% 12 x 100 ml 2 BAX 00944-0493-02 case
P9041 Plasbumin® 5% (Albumin) 50 ml 0.2 TAL 13533-0685-20 vial
P9045 Plasbumin® 5% (Albumin) 250 ml 1 TAL 13533-0685-25 vial
P9046 Plasbumin® 25% (Albumin) 20 ml 0.4 TAL 13533-0684-16 vial
P9047 Plasbumin® 25% (Albumin) 50 ml 1 TAL 13533-0684-20 vial
P9047 Plasbumin® 25% (Albumin) 100 ml 2 TAL 13533-0684-71 vial
P9043 Plasmanate® 5% (PPF) 50 ml 0.2 TAL 13533-0613-20 vial
P9048 Plasmanate® 5% (PPF) 250 ml 1 TAL 13533-0613-25 vial
AntihemophilicCODE PRODUCT SIZE MFR. NDC # PK/CS
Factor VIIa (Recombinant)
J7189 NovoSeven® RT 1000 mcg NOVO 00169-7010-01 vial
J7189 NovoSeven® RT 2000 mcg NOVO 00169-7020-01 vial
J7189 NovoSeven® RT 8 mg NOVO 00169-7040-01 vial
J7189 NovoSeven® RT 5000 mcg NOVO 00169-7050-01 vial
Factor VIII (Recombinant)
J7192 Advate® w/Baxject II 250 iu BAX 00944-2941-10 vial
J7192 Advate® w/Baxject II 500 iu BAX 00944-2942-10 vial
J7192 Advate® w/Baxject II 1000 iu BAX 00944-2943-10 vial
J7192 Advate® w/Baxject II 1500 iu BAX 00944-2944-10 vial
J7192 Advate® w/Baxject II 2000 iu BAX 00944-2945-10 vial
J7192 Advate® w/Baxject II 3000 iu BAX 00944-2946-10 vial
J7192 Helixate® FS 250 iu CSL 00053-8131-02 vial
J7192 Helixate® FS 500 iu CSL 00053-8132-02 vial
J7192 Helixate® FS 1000 iu CSL 00053-8133-02 vial
J7192 Helixate® FS 2000 iu CSL 00053-8134-02 vial
J7192 Helixate® FS 3000 iu CSL 00053-8135-02 vial
J7192 Kogenate® FS 250 iu BYR 00026-3782-20 vial

56 i n s i d e o u t Spring 2011 57
contrast med
iaJ1459 Privigen® 5 gm CSL 44206-0436-05 vial
J1459 Privigen® 10 gm CSL 44206-0437-10 vial
J1459 Privigen® 20 gm CSL 44206-0438-20 vial
Immune Globulin Subcutaneous (Human) Liquid
J3490 Hizentra™ 5 ml CSL 44206-451-01 vial
J3490 Hizentra™ 10 ml CSL 44206-452-02 vial
J3490 Hizentra™ 20 ml CSL 44206-454-04 vial
J1562 Vivaglobin® 3 ml CSL 00053-7596-01 vial
J1562 Vivaglobin® 10 ml CSL 00053-7596-10 vial
J1562 Vivaglobin® 20 ml CSL 00053-7596-20 vial
Hyper-Immune Globulin Hepatitis B Immune Globulin
CODE PRODUCT SIZE MFR. NDC # PK/CS
Intramuscular Immune Globulins (Human)
J1470 GamaSTAN® S/D 2 ml TAL 13533-0635-04 vial
J1550 GamaSTAN® S/D 10 ml TAL 13533-0635-12 vial
Rho(D) Immune Globulin
J2788 HyperRHO® S/D mini-dose 50 mcg TAL 13533-0631-06 10/pk
J2790 HyperRHO® S/D 300 mcg TAL 13533-0631-02 syringe
J2790 RhoGAM® UF Plus* 300 mcg OCD 0562-7805-01 syringe
J2788 MICRhoGAM® UF Plus* 50 mcg OCD 0562-7806-01 syringe
J2791 Rhophylac® 300 mcg CSL 44206-0300-01 syringe
J2791 Rhophylac® 300 mcg CSL 44206-0300-10 10/pk
J2792 WinRho® SDF Liguid 1500 iu 300 mcg CAN 53270-3300-01 vial
J2792 WinRho® SDF Liguid 2500 iu 500 mcg CAN 53270-3500-01 vial
J2792 WinRho® SDF Liguid 5000 iu 1000 mcg CAN 53270-3100-01 vial
J2792 WinRho® SDF Liguid 15000 iu 3000 mcg CAN 53270-3000-01 vial
*Not sold in state of Florida.
Hepatitis B Immune Globulin
J1573 HepaGam B® 1 ml APO 60505-6071-00 vial
J1573 HepaGam B® 5 ml APO 60505-6072-00 vial
CPT-90371 HyperHEP B® S/D 0.5 ml TAL 13533-0636-03 syringe
CPT-90371 HyperHEP B® S/D 1 ml TAL 13533-0636-02 syringe
CPT-90371 HyperHEP B® S/D 1 ml TAL 13533-0636-01 vial
CPT-90371 HyperHEP B® SD 5 ml TAL 13533-0636-05 vial
C9105 Nabi-HB® 1 ml BIOTEST 59730-4202-01 vial
C9105 Nabi-HB® 5 ml BIOTEST 59730-4203-01 vial
Rabies Immune Globulin
CPT-90375 HyperRAB® S/D 2 ml TAL 13533-0618-02 vial
CPT-90375 HyperRAB® S/D 10 ml TAL 13533-0618-10 vial
CPT-90376 IMOGAM® Rabies-HT 300 iu/2 ml SAN 49281-0190-20 vial
CPT-90376 IMOGAM® Rabies-HT 1500 iu/10 ml SAN 49281-0190-10 vial
Tetanus Immune Globulin
J1670 HyperTET® S/D 1 ml TAL 13533-0634-02 syringe
Cytomegalovirus Immune Globulin Intravenous (Human)
J0850 CytoGam® 2.5 gm/50 ml CSL 44206-3101-01 vial
plasma d
erivativespl
asm
a d
eriv
ati
ves
Factor IX (Derived from Human Plasma)
J7193 AlphaNine® SD 500 iu GFS 68516-3601-2 vial
J7193 AlphaNine® SD 1000 iu GFS 68516-3602-2 vial
J7193 AlphaNine® SD 1500 iu GFS 68516-3603-2 vial
J7193 Mononine® 500 iu CSL 00053-7668-02 vial
J7193 Mononine® 1000 iu CSL 00053-7668-04 vial
Factor XIII (Derived from Human Plasma)
J7199 Corifact PDS 1380 iu CSL 63833-518-02 vial
Fibrinogen Concentrate
J1680 RiaSTAP™ 1 gm CSL 63833-8915-01 vial
Anti-Inhibitor Coagulation Complexes (Derived from Human Plasma)
J7198 Feiba® VH 500 iu BAX 64193-0222-03 vial
J7198 Feiba® VH 1000 iu BAX 64193-0222-04 vial
J7198 Feiba® VH 2500 iu BAX 64193-0222-05 vial
Anti Thrombin III (Derived from Human Plasma)
J7197 Thrombate III® 500 iu TAL 13533-0603-20 vial
Anti Thrombin (Recombinant)
J3590 ATryn® 1750 iu GTC 67386-521-51 vial
Desmopressin Acetate (Useful in Disorders of Hemostasis)
J2597 Stimate® (Nasal Spray) 2.5 ml CSL 00053-2453-00 vial
ImmunologicCODE PRODUCT SIZE MFR. NDC # PK/CS
Immune Globulin Intravenous (Human) Lyophilized
J1566 Carimune® NF 3 gm CSL 44206-0416-03 vial
J1566 Carimune® NF 6 gm CSL 44206-0417-06 vial
J1566 Carimune® NF 12 gm CSL 44206-0416-12 vial
Immune Globulin Intravenous (Human) Liquid
J1572 Flebogamma® DIF 5% 2.5 gm GFS 61953-0004-02 vial
J1572 Flebogamma® DIF 5% 5 gm GFS 61953-0004-03 vial
J1572 Flebogamma® DIF 5% 10 gm GFS 61953-0004-04 vial
J1572 Flebogamma® DIF 5% 20 gm GFS 61953-0004-05 vial
Flebogamma® DIF 10% 5 gm GFS 61953-0005-01 vial
Flebogamma® DIF 10% 10 gm GFS 61953-0005-02 vial
Flebogamma® DIF 10% 20 gm GFS 61953-0005-03 vial
J1569 Gammagard Liquid 1 gm BAX 00944-2700-02 vial
J1569 Gammagard Liquid 2.5 gm BAX 00944-2700-03 vial
J1569 Gammagard Liquid 5 gm BAX 00944-2700-04 vial
J1569 Gammagard Liquid 10 gm BAX 00944-2700-05 vial
J1569 Gammagard Liquid 20 gm BAX 00944-2700-06 vial
J1561 Gamunex® 1 gm TAL 13533-0645-12 vial
J1561 Gamunex® 2.5 gm TAL 13533-0645-15 vial
J1561 Gamunex® 5 gm TAL 13533-0645-20 vial
J1561 Gamunex® 10 gm TAL 13533-0645-71 vial
J1561 Gamunex® 20 gm TAL 13533-0645-24 vial
NEW

58 i n s i d e o u t Spring 2011 59
contrast med
iaJ1931 Aldurazyme® 2.9 mg/5 ml GNZ 58468-0070-01 vial
J9215 Alferon N® 5 mm iu (1 ml) HEMI 54746-0001-01 vial
J3490 Aloprim® 500 mg/30 ml NABI 59730-0560-11 vial
Ammonul® 50 ml MEDI 62592-0720-50 vial
J0583 Angiomax® 250 mg MEDCO 65293-0001-01 10/pk
J3490 Berinert® 500 unit vial CSL 63833-825-02 kit
Buphenyl® 500 mg tabs MEDI 62592-0496-03 btl
Buphenyl® 250 g MEDI 62592-0188-64 btl
J9010 Campath® 30 mg/ml BYR 50419-0357-03 3/pk
J1785 Cerezyme® 200u GNZ 58468-1983-01 vial
J1785 Cerezyme® 400u GNZ 58468-4663-01 vial
C9274 CroFab® 2 x 2 ml BTG 00281-0330-10 pk
C9274 CroFab® 2 x 2 ml BTG 50633-0110-12 vial
J9878 Cubicin® 500 mg CUB 67919-0011-01 10/pk
J0894 Dacogen® 50 mg MGI 62856-0600-01 vial
J1162 DigiFab 40 mg BTG 50633-0120-11 vial
J7323 Euflexxa® 2.25 ml FERR 55566-4100-01 3 syr/pk
J8499 Exjade® 125 mg/30 tabs NOV 00078-0468-15 btl
J8499 Exjade® 250 mg/30 tabs NOV 00078-0469-15 btl
J8499 Exjade® 500 mg/30 tabs NOV 00078-0470-15 btl
J0180 Fabrazyme® 35 mg GNZ 58468-0040-01 vial
J0180 Fabrazyme® 5 mg GNZ 58468-0041-01 vial
J9395 Faslodex® 125 mg/2 x 2.5 ml AZ 00310-0720-25 pk
J9395 Faslodex® 250 mg/1 x 5 ml AZ 00310-0720-50 pk
J9155 Firmagon 240 mg injection FERR 55566-840-01
J9155 Firmagon 5 mg diskhaler FERR 55566-8301-01
J9999 Folotyn™ 20 mg ALLOS 48818-0001-01 vial
J9999 Folotyn™ 40 mg ALLOS 48818-0001-02 vial
Q0166 Granisol™ Oral 2 mg/10 ml 30 ml PediatRx 52547-0801-30 btl
J9999 Halaven 1 mg/2 ml SDV EISAI 62856-0389-01 vial
J9315 Istodax® 10 mg CEL 46026-983-01 1 kit
Kalbitor® 10 mg 47783-0101-01 vial
J3590 Krystexxa™ 8 mg SDV 2 ml SAVIENT 54396-0801-01 vial
J9300 Mylotarg® 5 mg/20 ml HEM GRP 00008-4510-01 vial
J0220 Myozyme® 50 mg GNZ 58468-0150-01 vial
J8999 Nexavar® 200 mg/120 tabs BYR 00026-8488-58 btl
J1640 Panhematin® 100 ml OVA 67386-0701-54 vial
Panretin® 0.1% Gel 60 g EISAI 64365-501-01 tube
J7335 Qutenza® 1 patch/50 g gel NEURO 49685-928-01 kit
J7335 Qutenza® 2 patch/50 g gel NEURO 49685-928-02 kit
J1300 Soliris® 300 mg/30 ml ALX 25682-0001-01 vial
J7321 Supartz® 10 mg/ml S&N 08363-7765-01 pk
J8999 Targretin® 75 mg SGC/100 tabs EISAI 64365-502-01 btl
J8999 Targretin® 1% Gel 60 g EISAI 64365-504-01 tube
J8999 Tasigna® 150 mg/28 caps NOV 00078-0592-51 pk
CPT-90586 TheraCys® w/diluent SAN 49281-0880-01 vial
J8999 Tasigna® 200 mg/28 caps NOV 00078-0526-51 pk
J1190 Totect® Non-Returnable 5000 mg TOPO 38423-0110-01 1 kit
J2323 Tysabri® 300 mg (15 ml) ELAN 59075-0730-15 vial
J9025 Vidaza® 100 mg PhM 59572-0102-01 vial
J2357 Xolair® 150 mg GNT 50242-0040-62 vial
J3490 VPRIV 400 units/vial SDV Shire 54092-0701-04 vial
pharmaceuticalsph
arm
aceu
tica
lsVaccines
CODE PRODUCT SIZE MFR. NDC # PK/CS
Flu
Q2035 Afluria® syringe 10 x 0.5 ml MER 33332-0011-01 syringe
Q2035 Afluria® vial 10 x 5 ml MER 3333-0111-10 vial
FluMist® 10 x 0.2 ml MEDI 66019-0109-10 sprayers
Q2037 Fluvirin® (10 dose vial) 5 ml NOV 66521-0114-10 vial
Q2037 Fluvirin® syringe 10 x 0.5 ml NOV 66521-0114-02 syringe
Q2038 FluZone® pediatric syringe 10 x 0.25 ml SAN 49281-0010-25 syringe
Q2038 FluZone® 10 x .5 ml SAN 49281-0111-25 syringe
Q2038 FluZone® 10 x 5 ml SAN 49281-0011-50 vial
Q2038 FluZone® HD 5 ml SAN 49281-0387-65 syringe
G9035 Relenza 5 mg Diskhaler GSK 00173-0681-01
G9035 Tamiflu 12 mg/ml 25 ml 00004-0810-95 suspension
G9035 Tamiflu 75 mg 10/bx 00004-0800-85 capsules
Haemophilus Inflenzae B
CPT-90648 ActHIB® SAN 49281-0545-05 vial
Meningitis
CPT-90734 Menactra® S/D SAN 49281-0589-05 5/pk
Polio
CPT-90713 IPOL® 10 x 1 mdv SAN 49281-0860-10 vial
Rabies
CPT-90675 RabAvert® 1 ml NOV 00078-0566-51 pk
Tetanus
CPT-90715 Adacel® 10 x 0.5 ml SAN 49281-0400-10 vial
CPT-90700 DAPTACEL® 10 x 1 mdv SAN 49281-0286-10 vial
CPT-90714 DECAVAC™ 10 x 0.5 ml SAN 49281-0291-10 syringe
CPT-90702 Tet Dip Toxoid Adsorbed 10 x 0.5 ml AKN 17478-0131-01 vial
CPT-90702 Dip Tet Tox™ Adsorbed 10 x 1 mdv SAN 49281-0278-10 vial
CPT-90703 Tet Tox™ Adsorbed 10 x 1 mdv SAN 49281-0800-83 vial
CPT-90749 Tet Tox™ Plain 7.5 ml SAN 49281-0812-84 vial
CPT-86700 Tripedia® 0.5 ml SAN 49281-0298-10 vial
Tuberculosis
J3490 Aplisol® 5TU 1 ml KING 42023-0104-01 vial
J3490 Aplisol® 5TU 5 ml KING 42023-0104-05 vial
CPT-86580 Tubersol® 5 TU 1 ml SAN 49281-0752-21 pk
CPT-86580 Tubersol® 5 TU 5 ml SAN 49281-0752-22 pk
Typhoid
CPT-90691 Typhim Vi® .05 ml SAN 49281-0790-51 syringe
CPT-90691 Typhim Vi® 20 dose vial SAN 49281-0790-20 vial
SpecialtyCODE PRODUCT SIZE MFR. NDC # PK/CS
J9264 Abraxane® ABR 68817-0134-50 vial
J2997 Activase® Cathflo 2 mg GNT 50242-0041-64 vial
J8999 Afinitor® 2.5/28 tabs mg NOV 00078-0594-51 pk
J8999 Afinitor® 5 mg/28 tabs NOV 00078-0566-51 pk
J8999 Afinitor® 10 mg/28 tabs NOV 00078-0567-51 pk
NEW

60 i n s i d e o u t Spring 2011 61
contrast med
iaph
arm
aceu
tica
lsBiosurgery Products
CODE PRODUCT SIZE MFR. NDC # PK/CS
921028 Tisseel VH 2 ml BAX 00944-4201-04 vial
921029 Tisseel VH 4 ml BAX 00944-4201-08 vial
921030 Tisseel VH 10 ml BAX 00944-4201-12 vial
921051VP ValuPak 2 ml (1 cs tisseel biologic-only and 1 cs duploJeCt easy prep)
6 x 1 ml BAX 00944-4201-03 cs
921052VP ValuPak 4 ml (1 cs tisseel biologic-only and 1 cs duploJeCt easy prep)
6 x 2 ml BAX 00944-4201-07 cs
921053VP ValuPak 10 ml (1 cs tisseel biologic-only and 1 cs duploJeCt easy prep)
6 x 5 ml BAX 00944-4201-11 cs
921063 Easy Prep 6 x 1 ml BAX cs
921064 Easy Prep 6 x 2 ml BAX cs
921065 Easy Prep 6 x 5 ml BAX cs
921020 Duplocath 35 M.I.S. BAX ea
921021 Duplocath 180 BAX ea
921022 Duplocath 25 BAX ea
921023 DuoFlo Dispenser Kit BAX ea
921031 Spray Set for the Tissomat Spray Device BAX 10/cs
600012 EasySpray Pressure Regulator BAX ea
600013 EasySpray Set BAX 10/cs
921042 FibriJet 57 Duplotip BAX ea
921043 FibriJet 83 Duplotip BAX ea
921044 FibriJet 51 Duplotip BAX ea
921045 FibriJet 102 Duplotip BAX ea
921046 FibriJet 267 Duplotip BAX ea
921047 FibriJet 318 Duplotip BAX ea
921050 Duploreach 35 Extended Spray Applicator BAX 6/cs
921134 DuploGrip Accessory Grip 6 x 2 ml BAX cs
921135 DuploGrip Accessory Grip 6 x 5 ml BAX cs
600029 DuploSpray MIS Applicators 30 cm BAX 5/cs
600030 DuploSpray MIS Applicators 40 cm BAX 5/cs
600033 DuploSpray MIS Applicators 20 cm BAX 5/cs
600031 DuploSpray MIS Replacement Tips BAX 10/cs
600036 Cannula DuploTip 20g x 10 cm BAX 10/cs
600037 Cannula DuploTip 20g x 26 cm BAX 10/cs
600038 Cannula DuploTip 20g x 32 cm BAX 10/cs
934057 FloSeal Matrix Hemostatic Sealant 6 x 5 ml BAX cs
934050 FloSeal NT Matrix Hemostatic Sealant 6 x 5 ml BAX cs
934055 FloSeal Reusable Endoscopic Applicator BAX ea
1500181 FloSeal Endoscopic Applicator BAX 6/cs
934208 FloSeal Curved Applicator Tip 8 cm BAX ea
934210 FloSeal Curved Applicator Tip 10 cm BAX ea
934070 CoSeal™ 2 ml BAX vial
934071 CoSeal™ 4 ml BAX vial
934072 CoSeal™ 8 ml BAX vial
934033 CoSeal™ Applicator tips BAX 5/cs
934034 CoSeal™ Extended Applicator BAX 10/cs
934500 CoSeal™ Spray Accessory Kit BAX 5/cs
600021 CoSeal™ SpraySet BAX 10/cs
600041 Adept ADH Reduction Sol BAX 5/cs
Bayer*
ASD # MFR # DESCRIPTION SIZE NDC # PK/CS
MR
36563 3278959 Eovist Vial 181.43 mg/ml 10 ml 5041932005 5 vl/cs
37941 1658772 Gadavist 7.5 ml vial 7.5 ml 50419032511 20 vl/cs
37942 1641851 Gadavist 10 ml vial 10 ml 50419032512 20 vl/cs
37943 1665637 Gadavist 15 ml vial 15 ml 50419032513 20 vl/cs
37944 Gadavist 604.72 mg/ml vial 65 ml 50419032515 10 vl/cs
31349 1213321 Magnevist Inj syr 10 ml 5041918836 5 syr/cs
30512 1213347 Magnevist Inj syr 15 ml 5041918837 5 syr/cs
30513 1213727 Magnevist Inj syr 20 ml 5041918838 5 syr/cs
31350 1973676 Magnevist Vial 5 ml 5041918805 20 vl/cs
31351 1311935 Magnevist Vial 10 ml 5041918801 20 vl/cs
35324 1311745 Magnevist Vial 15 ml 5041918815 20 vl/cs
35323 3218450 Magnevist Vial 20 ml 5041918802 20 vl/cs
31352 1242668 Magnevist Vial pbp 50 ml 5041918858 10 vl/cs
31353 1240340 Magnevist Vial pbp 100 ml 5041918811 10 vl/cs
CT
36896 1712603 Ultravist Vial 240 mg/ml 100 ml 5041934210 10 vl/cs
36897 1333343 Ultravist Vial 240 mg/ml pbp 200 ml 5041934221 10 vl/cs
36553 1731355 Ultravist Vial 300 mg/ml 50 ml 5041934405 10 vl/cs
32054 1732957 Ultravist Vial 300 mg/ml 100 ml 5041934410 10 vl/cs
36554 1288075 Ultravist Vial 300 mg/ml 125 ml 5041934412 10 vl/cs
36555 1737550 Ultravist Vial 300 mg/ml 150 ml 5041934415 10 vl/cs
36556 1288802 Ultravist Vial 300 mg/ml pbp 200 ml 5041934421 10 vl/cs
36898 1289578 Ultravist Vial 300 mg/ml pbp 500 ml 5041934458 8 vl/cs
36557 1761378 Ultravist Vial 370 mg/ml 50 ml 5041934605 10 vl/cs
36558 1761600 Ultravist Vial 370 mg/ml 100 ml 5041934610 10 vl/cs
36559 1761808 Ultravist Vial 370 mg/ml 150 ml 5041934615 10 vl/cs
36560 1762293 Ultravist Vial 370 mg/ml 200 ml 5041934620 10 vl/cs
36561 1292630 Ultravist Vial 370 mg/ml pbp 250 ml 5041934625 10 vl/cs
36562 1332535 Ultravist Vial 370 mg/ml pbp 500 ml 5041934658 8 vl/cs
Astellas Pharma USASD # MFR # DESCRIPTION SIZE NDC # PK/CS
MR
33195 6501-89 Lexiscan 0.4 mg/ 5 ml syr 5 ml 00469-6501-89 1 each
* No Bayer products shipped to Florida or Arizona.
NEW
NEW
NEW
NEW
NEW

62 i n s i d e o u t Spring 2011 63
contrast med
ia
BraccoCT / X-RayASD # MFR # DESCRIPTION SIZE NDC # PK/CS
21146 1411-11 Isovue M 200 41% vl 10 x 10 ml 10 ml 00270-1411-11 10/cs
26183 1411-25 Isovue M 200 41% 10 x 20 ml 20 ml 00270-1411-25 10/cs
21961 1412-15 Isovue M 300 61% 10 x 15 ml 15 ml 00270-1412-15 10/cs
17752 1314-30 Isovue 200 vl 10 x 50 ml 50 ml 00270-1314-30 10/cs
36668 1314-15 Isovue 200 vl 10 x 200 ml 200 ml 00270-1314-15 10/cs
36669 1317-05 Isovue 250 vl 10 x 50 ml 50 ml 00270-1317-05 10/cs
36670 1317-02 Isovue 250 btl 10 x 100 ml 100 ml 00270-1317-02 10/cs
36671 1317-09 Isovue 250 btl 10 x 150 ml 150 ml 00270-1317-09 10/cs
36672 1317-41 Isovue multi 250 10 x 200 ml 200 ml 00270-1317-41 10/cs
36673 1315-25 Isovue 300 61% 10 x 30 ml 30 ml 00270-1315-25 10/cs
21881 1315-30 Isovue 300 61% 10 x 50 ml 50 ml 00270-1315-30 10/cs
36614 1315-47 Isovue 300 61% btl 10 x 75 ml 75 ml 00270-1315-47 10/cs
32427 1315-35 Isovue 300 61% btl 10 x 100 ml 100 ml 00270-1315-35 10/cs
36615 1315-50 Isovue 300 61% btl 10 x 150 ml 150 ml 00270-1315-50 10/cs
36677 1315-41 Isovue multi 300 btl 10 x 200 ml 200 ml 00270-1315-41 10/cs
36678 1315-98 Isovue multi 300 btl 6 x 500 ml 500 ml 00270-1315-98 6/cs
36679 1316-01 Isovue 370 btl 10 x 50 ml 50 ml 00270-1316-01 10/cs
36680 1316-52 Isovue 370 btl 10 x 75 ml 75 ml 00270-1316-52 10/cs
36681 1316-35 Isovue 370 btl 10 x 100 ml 100 ml 00270-1316-35 10/cs
36682 1316-04 Isovue 370 btl 10 x 125 ml 125 ml 00270-1316-04 10/cs
36683 1316-37 Isovue 370 btl 10 x 150 ml 150ml 00270-1316-37 10/cs
36688 1316-41 Isovue multi 370 10 x 200 ml 200 ml 00270-1316-41 10/cs
36689 1316-98 Isovue multi 370 6 x 500 ml 500 ml 00270-1316-98 6/cs
CT / X-Ray
36690 0265-20 Cholografin 52% vl 1 x 20 ml 20 ml 00270-0265-20 1/cs
31844 1410-30 Cystografin 18% btl 10 x 300 ml 300 ml 00270-1410-30 10/cs
36691 0149-60 Cystografin 30% 10 x 100 ml 100 ml 30270-0149-60 10/cs
29448 0149-57 Cystografin 30% btl 10 x 300 ml 300 ml 00270-0149-57 10/cs
36692 0523-30 Sinografin 10 ml 10/pk 10 ml 30270-0523-30 10/cs
Adminstration Sets and Transfer Devices
36693 0004-75 Fluid Admin Set - Large Bore 00270-0004-75 10/cs
36694 0051-10 Solution Transfer Device 00270-0051-10 10/cs
Contrast Media Products - MR
32442 5164-13 Multihance 529 mg 5 x 10 ml 10 ml 00270-5164-13 5/cs
32441 5164-14 Multihance 529 mg 5 x 15 ml 15 ml 00270-5164-14 5/cs
36622 5164-15 Multihance 529 mg/ml vl 5 x 20 ml 20 ml 00270-5164-15 5/cs
36621 5164-12 Multihance 529 mg/ml vl 5 x 5 ml 5 ml 00270-5164-12 5/cs
36623 5264-16 Multihance MP 529 mg/ml btl 5 x 50 50 ml 00270-5264-16 5/cs
36624 5264-17 Multihance MP 529 mg/ml btl 5 x 100 100 ml 00270-5264-17 5/cs
36618 1111-16 Prohance 279.3 mg/ml PFS 5 x 10 ml 10 ml 00270-1111-16 5/cs
36619 1111-45 Prohance 279.3 mg/ml PFS 5 x 17 ml 17 ml 00270-1111-45 5/cs
36617 1111-01 Prohance 279.3 mg/ml vl 5 x 10 ml 10 ml 00270-1111-01 5/cs
cont
rast
med
ia36616 1111-04 Prohance 279.3 mg/ml vl 5 x 5 ml 5 ml 00270-1111-04 5/cs
36276 1111-03 Prohance 279.3 mg/ml 5 x 20ml 20 ml 00270-1111-03 5/cs
22820 1111-02 Prohance 279.3 mg/ml vl 5 x 15 ml 15 ml 00270-1111-02 5/cs
36620 1111-70 Prohance MP279.3 mg/ml btl 5 x 50 ml 50 ml 00270-1111-70 5/cs
Oral Contrast Agents
22242 0455-40 Gastrografin 37% sol 120 ml 12/cs 120 ml 00270-0445-40 12/cs
35353 44535 Gastrografin Lemon sol 24 x 30 ml 30 ml 00270-0445-35 24/cs
Oral Contrast Agents - CT Barium
35356 7350 Readi-Cat Apple 24 x 450 ml 450 ml 32909-0735-03 24/cs
35355 7250 Readi-Cat Banana 24 x 250 ml 250 ml 32909-0725-07 24/cs
35357 7450 Readi-Cat Banana 24 x 450 ml 450 ml 32909-0725-03 24/cs
35366 721 Readi-Cat Barium1.3% 12 x 900 ml 900 ml 32909-4501-03 12/cs
35364 728 Readi-Cat Barium1.3% 24 x 450 ml 450 ml 32909-0728-01 24/cs
35365 724 Readi-Cat Barium1.3% 4 x 1900 ml 1900 ml 32909-4501-05 4/cs
35354 7150 Readi-Cat Berry 24 x 450 ml 450 ml 32909-0715-03 24/cs
35359 7650 Readi-Cat Creamy Van 24 x 250 ml 250 ml 32909-0755-07 24/cs
35358 7550 Readi-Cat Creamy Van 24 x 450 ml 450 ml 32909-0755-03 24/cs
35360 4503-07 Readi-Cat Mochaccino 24 x 450 ml 450 ml 32909-0775-03 24/cs
35363 729 Readi-Cat2 Barium 2.1% 12 x 900 ml 900 ml 32909-0723-03 12/cs
35361 723 Readi-Cat2 Barium 2.1% 24 x 450 ml 450 ml 32909-0723-01 24/cs
35362 726 Readi-Cat2 Barium 2.1% 4 x 1900 ml 1900 ml 32909-0723-02 4/cs
35370 4501-01 E-Z Cat Barium Conc 24 x 225 ml 225 ml 32909-0720-01 24/cs
35371 4501-07 E-Z Cat Barium Dry 50 x 23 gm 23 gm 00270-4501-07 50/cs
35372 4501-11 Esopho-Cat Barium 24 x 30 gm 30 gm 32909-0738-01 24/cs
35369 4507-01 Volumen 0.1% 24 x 450 ml 450 ml 32909-0945-03 24/cs
35373 4500-02 CT Enema Kit 00270-4500-02 12/cs
Fluoroscopy - Routine Filled Colon Examination
36270 AP14 Polibar ACB Barium lq 14 oz 24/cs 14 oz 00270-9005-04 24/cs
36271 AP16 Polibar ACB Barium lq 16 oz 24/s 16 oz 00270-9005-06 24/cs
36759 816 E-Z Paque Enema Bag 454 g 454 g 00270-9005-03 24/cs
36701 920 Empty Enema Bag 00270-9003-02 48/cs
36702 935 Empty Enema Exacta Bag 00270-9003-04 4/cs
Double Contrast Colon Examination
36513 9012-03 Super XL Empty Enema Kit 24/cs 00270-9012-03 24/cs
36375 9001-01 E-Z Dose Polibar Kit 650 ml 6/cs 650 ml 00270-9001-01 6/cs
36269 L164 Polibar Liq 100% w/v 1900 ml4/cs 1900 ml 00270-9002-02 4/cs
36703 L168 Polibar Liq 105% w/v 1900 ml 4/cs 1900 ml 00270-9002-03 4/cs
Routine Upper GI Examination
36061 9019-01 E-Z Paque 6.2 oz 24/cs 6.2 oz 32909-0750-03 24/cs
35700 9019-02 Ultra-R UD cups 24 x 6 oz 6 oz 32909-0753-01 24/cs
36915 9031-02 Flexible plastic straws 00270-9031-02 144/cs
36716 9030-01 E-Z Paque 10 kg 10 kg 00270-9030-01 1/cs
36717 9030-02 Sensatrast 4.2 oz 24/cs 4.2 oz 00270-9030-02 24/cs
36056 9026-01 E-Z Paque 1200 g 8/cs 1200 g 32909-0750-01 8/cs
36060 9029-01 E-Z Paque 1900 ml 4/cs 1900 ml 32909-0186-01 4/cs
36062 9028-01 E-Z Paque 12 oz 24/cs 12 oz 32909-0186-02 24/cs
36063 9021-01 E-Z Paste 16 oz 12/cs 1 lb 32909-0770-01 12/cs

64 i n s i d e o u t Spring 2011 65
contrast med
iaDouble Contrast Examination of Esophagus, Stomach and Duodenum
35699 9017-02 E-Z HD 250% w/v susp 24 x 12 oz 12 oz 32909-0764-01 24/cs
36059 9018-01 E-Z HD Pail 25 lb 25 lb 32909-0764-02 1/cs
Fluoroscopy
36305 2700 Digibar 190 susp 232 gmud 24/cs 232 gm 33609-0270-19 24/cs
36377 9027-01 Maxibar Suspension 120 ml 24/cs 120 ml 00270-9027-01 24/cs
35997 9020-01 E-Z Gas II Granules 10361-0793-01
36128 9021-02 E-Z Disc 10 Grain Tabs 778 btl 00270-9021-02 btl
Modified Barium Swallowing Studies
36719 9000-01 Varibar Barium Thin liq 148 g 24/cs 148 g 00270-9000-01 24/cs
36720 9000-02 Varibar Barium Nectar 240 ml 24/cs 240 ml 00270-9000-02 24/cs
36721 9000-04 Varibar Barium Thin Honey 250 ml 12/cs 250 ml 00270-9000-04 12/cs
36722 9000-05 Varibar Barium Honey 250 ml 12/cs 250 ml 00270-9000-05 12/cs
36723 9000-06 Varibar Barium Pudding 230 ml 12/cs 230 ml 00270-9000-06 12/cs
Small Bowel/Enteroclysis Studies
36724 9014-01 Entero Vu 13% 100 g Packets 12/cs 100 g 00270-9014-01 12/cs
36725 9014-03 Enteroclysis Admin Kit 12/cs 00270-9014-03 12/cs
36374 9014-06 Liquid Entero Vu13% 600 ml 12/cs 600 ml 00270-9014-06 12/cs
36734 9014-07 Entero Vu 24% 600 ml 12/cs 600 ml 00270-9014-07 12/cs
Virtual Colonoscopy - Patient Prepping and Laxatives
36613 3902-01 Loso Prep Cleansing Kit 24/cs 10361-0306-44 24/cs
36738 3902-02 Loso Prep Bowel Cleansing System 00270-3902-02 50/cs
Ultrasound Gels and Disinfecting Towelettes
36307 6010-01 E-Z Gel 8 oz 12/cs 8 oz 30270-6010-01 12/cs
36150 6010-04 E-Z Gel in 5L Container 4/cs 30270-6010-04 4/cs
36261 6045 E-Z Scanning Gel Clear 5L 4/cs 00270-6010-05 4/cs
36762 6010-02 E-Z Scan .25L Bottle 12/cs 00270-6010-02 12/cs
36763 6010-03 E-Z Scan in 5L Container 12/cs 00270-6010-03 12/cs
36764 Q89072 Sani-Coth Plus Towelettes 160 count 12/cs
cont
rast
med
ia
CovidienMR (Gadolinium)
ASD # MFR # DESCRIPTION SIZE NDC # PK/CS
Gastromark Bottles
31115 112006 GastroMARK® 300 ml 00019-1120-06 12 btl/cs
Optimark Bottles
31116 117702 OptiMARK® 5 ml 00019-1177-02 10 vl/cs
31117 117704 OptiMARK® 10 ml 00019-1177-04 10 vl/cs
31118 117706 OptiMARK® 15 ml 00019-1177-06 10 vl/cs
31119 117708 OptiMARK® 20 ml 00019-1177-08 10 vl/cs
31124 117750 OptiMARK® 50 ml 00019-1177-50 5 vl/cs
Optimark Syringes
31120 117710 OptiMARK® Syringe 10 cc 00019-1177-10 10 syr/cs
31121 117715 OptiMARK® Syringe 15 cc 00019-1177-15 10 syr/cs
31122 117720 OptiMARK® Syringe 20 cc 00019-1177-20 10 syr/cs
31123 117730 OptiMARK® Syringe 30 cc 00019-1177-30 10 syr/cs
Tubing / Syringes
31485 801018 Optistar MR® Mpak 2 x 60 ml Y 60" 50/cs
31486 801019 Optistar MR® Mpak 2 x 60 ml Y 90" 50/cs
31483 801020 Optistar MR® Mpak 60 ml/25 ml Y 60" 50/cs
31484 801021 Optistar MR® Mpak 60 ml/25 ml Y 90" 50/cs
31481 801103 Optistar MR® Syringe 25 ml 100 syr/cs
31482 801104 Optistar MR® Syringe 60 ml 50 syr/cs
Disposable Tubing
31488 801106 Optistar MR® Coiled Y-Tubing 60" 100/cs
31489 801107 Optistar MR® Coiled Y-Tubing 90" 100/cs
Disposable Syringes
31487 801800 Optistar® LE Multipak 2 x 60 ml/90” Y 90" 50/cs
31497 801801 Optistar® LE Syringe 60 ml 50 syr/cs
Prefilled Sodium Chloride
31436 118875 Sodium Chloride Inj, USP 0.9% 50 ml 00019-1188-75 10 syr/cs
Miscellaneous
31266 1550CW Ready-Box Media Warmer LF MDL 1 each
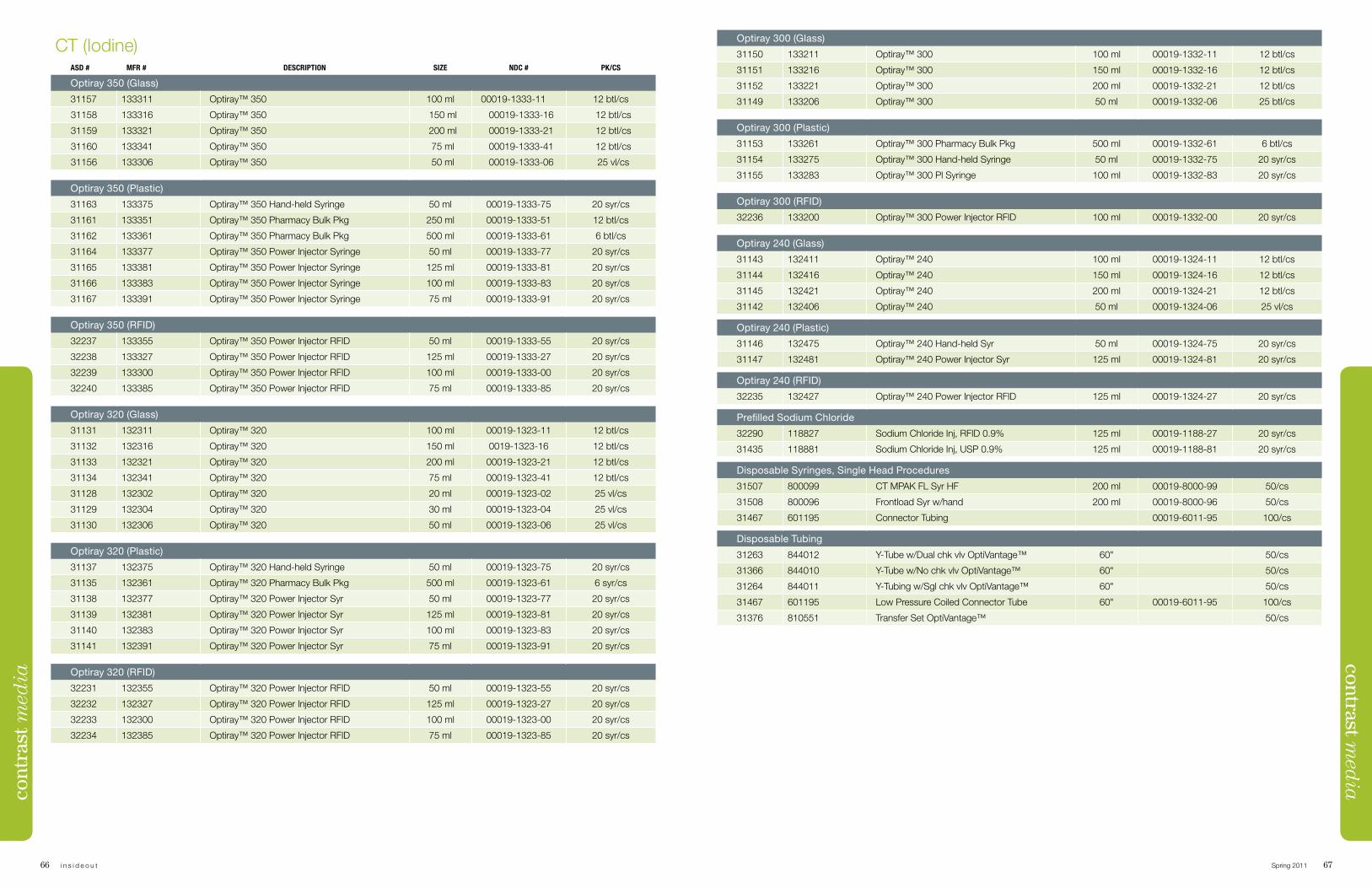
66 i n s i d e o u t Spring 2011 67
contrast med
iaCT (Iodine)
ASD # MFR # DESCRIPTION SIZE NDC # PK/CS
Optiray 350 (Glass)
31157 133311 Optiray™ 350 100 ml 00019-1333-11 12 btl/cs
31158 133316 Optiray™ 350 150 ml 00019-1333-16 12 btl/cs
31159 133321 Optiray™ 350 200 ml 00019-1333-21 12 btl/cs
31160 133341 Optiray™ 350 75 ml 00019-1333-41 12 btl/cs
31156 133306 Optiray™ 350 50 ml 00019-1333-06 25 vl/cs
Optiray 350 (Plastic)
31163 133375 Optiray™ 350 Hand-held Syringe 50 ml 00019-1333-75 20 syr/cs
31161 133351 Optiray™ 350 Pharmacy Bulk Pkg 250 ml 00019-1333-51 12 btl/cs
31162 133361 Optiray™ 350 Pharmacy Bulk Pkg 500 ml 00019-1333-61 6 btl/cs
31164 133377 Optiray™ 350 Power Injector Syringe 50 ml 00019-1333-77 20 syr/cs
31165 133381 Optiray™ 350 Power Injector Syringe 125 ml 00019-1333-81 20 syr/cs
31166 133383 Optiray™ 350 Power Injector Syringe 100 ml 00019-1333-83 20 syr/cs
31167 133391 Optiray™ 350 Power Injector Syringe 75 ml 00019-1333-91 20 syr/cs
Optiray 350 (RFID)
32237 133355 Optiray™ 350 Power Injector RFID 50 ml 00019-1333-55 20 syr/cs
32238 133327 Optiray™ 350 Power Injector RFID 125 ml 00019-1333-27 20 syr/cs
32239 133300 Optiray™ 350 Power Injector RFID 100 ml 00019-1333-00 20 syr/cs
32240 133385 Optiray™ 350 Power Injector RFID 75 ml 00019-1333-85 20 syr/cs
Optiray 320 (Glass)
31131 132311 Optiray™ 320 100 ml 00019-1323-11 12 btl/cs
31132 132316 Optiray™ 320 150 ml 0019-1323-16 12 btl/cs
31133 132321 Optiray™ 320 200 ml 00019-1323-21 12 btl/cs
31134 132341 Optiray™ 320 75 ml 00019-1323-41 12 btl/cs
31128 132302 Optiray™ 320 20 ml 00019-1323-02 25 vl/cs
31129 132304 Optiray™ 320 30 ml 00019-1323-04 25 vl/cs
31130 132306 Optiray™ 320 50 ml 00019-1323-06 25 vl/cs
Optiray 320 (Plastic)
31137 132375 Optiray™ 320 Hand-held Syringe 50 ml 00019-1323-75 20 syr/cs
31135 132361 Optiray™ 320 Pharmacy Bulk Pkg 500 ml 00019-1323-61 6 syr/cs
31138 132377 Optiray™ 320 Power Injector Syr 50 ml 00019-1323-77 20 syr/cs
31139 132381 Optiray™ 320 Power Injector Syr 125 ml 00019-1323-81 20 syr/cs
31140 132383 Optiray™ 320 Power Injector Syr 100 ml 00019-1323-83 20 syr/cs
31141 132391 Optiray™ 320 Power Injector Syr 75 ml 00019-1323-91 20 syr/cs
Optiray 320 (RFID)
32231 132355 Optiray™ 320 Power Injector RFID 50 ml 00019-1323-55 20 syr/cs
32232 132327 Optiray™ 320 Power Injector RFID 125 ml 00019-1323-27 20 syr/cs
32233 132300 Optiray™ 320 Power Injector RFID 100 ml 00019-1323-00 20 syr/cs
32234 132385 Optiray™ 320 Power Injector RFID 75 ml 00019-1323-85 20 syr/cs
Optiray 300 (Glass)
31150 133211 Optiray™ 300 100 ml 00019-1332-11 12 btl/cs
31151 133216 Optiray™ 300 150 ml 00019-1332-16 12 btl/cs
31152 133221 Optiray™ 300 200 ml 00019-1332-21 12 btl/cs
31149 133206 Optiray™ 300 50 ml 00019-1332-06 25 btl/cs
Optiray 300 (Plastic)
31153 133261 Optiray™ 300 Pharmacy Bulk Pkg 500 ml 00019-1332-61 6 btl/cs
31154 133275 Optiray™ 300 Hand-held Syringe 50 ml 00019-1332-75 20 syr/cs
31155 133283 Optiray™ 300 Pl Syringe 100 ml 00019-1332-83 20 syr/cs
Optiray 300 (RFID)
32236 133200 Optiray™ 300 Power Injector RFID 100 ml 00019-1332-00 20 syr/cs
Optiray 240 (Glass)
31143 132411 Optiray™ 240 100 ml 00019-1324-11 12 btl/cs
31144 132416 Optiray™ 240 150 ml 00019-1324-16 12 btl/cs
31145 132421 Optiray™ 240 200 ml 00019-1324-21 12 btl/cs
31142 132406 Optiray™ 240 50 ml 00019-1324-06 25 vl/cs
Optiray 240 (Plastic)
31146 132475 Optiray™ 240 Hand-held Syr 50 ml 00019-1324-75 20 syr/cs
31147 132481 Optiray™ 240 Power Injector Syr 125 ml 00019-1324-81 20 syr/cs
Optiray 240 (RFID)
32235 132427 Optiray™ 240 Power Injector RFID 125 ml 00019-1324-27 20 syr/cs
Prefilled Sodium Chloride
32290 118827 Sodium Chloride Inj, RFID 0.9% 125 ml 00019-1188-27 20 syr/cs
31435 118881 Sodium Chloride Inj, USP 0.9% 125 ml 00019-1188-81 20 syr/cs
Disposable Syringes, Single Head Procedures
31507 800099 CT MPAK FL Syr HF 200 ml 00019-8000-99 50/cs
31508 800096 Frontload Syr w/hand 200 ml 00019-8000-96 50/cs
31467 601195 Connector Tubing 00019-6011-95 100/cs
Disposable Tubing
31263 844012 Y-Tube w/Dual chk vlv OptiVantage™ 60" 50/cs
31366 844010 Y-Tube w/No chk vlv OptiVantage™ 60" 50/cs
31264 844011 Y-Tubing w/Sgl chk vlv OptiVantage™ 60" 50/cs
31467 601195 Low Pressure Coiled Connector Tube 60" 00019-6011-95 100/cs
31376 810551 Transfer Set OptiVantage™ 50/cs
cont
rast
med
ia

68 i n s i d e o u t Spring 2011 69
contrast med
iaConventional CM
ASD # MFR # DESCRIPTION SIZE NDC # PK/CS
Conray 30
31110 95211 Conray™ 30 150 ml 00019-0952-11 12 btl/cs
Conray 43
31169 318309 Conray™ 43 250 ml 00019-3183-09 12 btl/cs
31170 318315 Conray™ 43 50 ml 00019-3183-15 50 vl/cs
Conray 60
31111 95309 Conray™ 100 ml 00019-0953-09 12 btl/cs
31112 95311 Conray™ 150 ml 00019-0953-11 12 btl/cs
31113 95313 Conray™ 30 ml 00019-0953-13 50 vl/cs
31114 95315 Conray™ 50 ml 00019-0953-15 50 vl/cs
Cysto Conray
31108 86207 Cysto-Contray™ II 250 ml 00019-0862-07 12 btl/cs
MD Gastroview
31237 481605 MD-Gastroview™ 120 ml 00019-4816-05 12 btl/cs
31239 481606 MD-Gastroview™ 240 ml 00019-4816-06 12 btl/cs
31238 481604 MD-Gastroview™ 30 ml 00019-4816-04 25 btl/cs
MD-76R
31125 131707 MD-76R™ 100 ml 00019-1317-07 12 btl/cs
31442 131711 MD-76R™ 150 ml 00019-1317-11 12 btl/cs
31126 131709 MD-76R™ 200 ml 00019-1317-09 12 btl/cs
31127 131715 MD-76R™ 50 ml 00019-1317-15 50 vl/cs
Hexabrix
31172 550508 Hexabrix™ 100 ml 00019-5505-08 12 btl/cs
31441 550510 Hexabrix™ 150 ml 00019-5505-10 12 btl/cs
31173 550521 Hexabrix™ 200 ml 00019-5505-21 12 btl/cs
31174 550551 Hexabrix™ 20 ml 00019-5505-51 10 vl/cs
31171 550506 Hexabrix™ 50 ml 00019-5505-06 25 vl/cs
Miscellaneous
31542 14403 Fluid Admin Set, Large Bore 12 pk
31544 265703 Sol Admin RX Burron Set 00019-2657-03 12 pk
UrologyASD # MFR # DESCRIPTION SIZE NDC # PK/CS
Urology
31227 269004 Luer Locknuts (Ultraject™) 10 pk
32377 50012M Tad .035-.018 GW Sys 145 cm 1 each
31258 601278 High Pressure Ext Tubing w/RAT Adapter 10" 25/cs
31259 601279 High Pressure Ext Tubing w/RAT Adapter 20" 25/cs
31260 601280 High Pressure Ext Tubing w/RAT Adapter 30" 25/cs
31503 335563 Uro Drain Bags Sterile 10/cs
31504 337205 Uro Drain Extension Tubing 10/cs
31505 810555 Y Tube Adaptor CT9000® ADV/OptiStat™ 00019-8105-55 50/cs
31506 337200 Uro Drain Y-Connector 10/cs
31476 302050 Handifil 100/cs
31474 337210 FluiDrain LSS 10 10/cs
31475 337220 FluiDrain LSS 20 10/cs
31472 601281 High Pressure Ext Tubing w/ROT Adapter 25/cs
31473 601277 Low Pressure Ext Tube 00019-6012-77 100/cs
31471 335825 Disp Drainage Bag Non-sterile 10/cs
MiscellaneousASD # MFR # DESCRIPTION SIZE. NDC # PK/CS
Miscellaneous Syringes
31491 302100 Syringe w/Handifil 100 ml 50 syr/cs
31492 600172 Syringe w/Handifil 130 ml 50 syr/cs
31493 600269 Syringe w/Handifil 150 ml 50 syr/cs
31494 601360 Syringe w/Handifil for Medrad Inj 150 ml 50 syr/cs
31495 601350 Syringe w/Handifil for Medrad Inj 200 ml 50 syr/cs
31496 600169 Syringe w/Handifil 260 ml 50 syr/cs
31498 900105 Illumena™ Syringe w/Handifil Disp 200 ml 50 syr/cs
31499 601590 Tripack Syringe 200 ml 50 syr/cs
cont
rast
med
ia
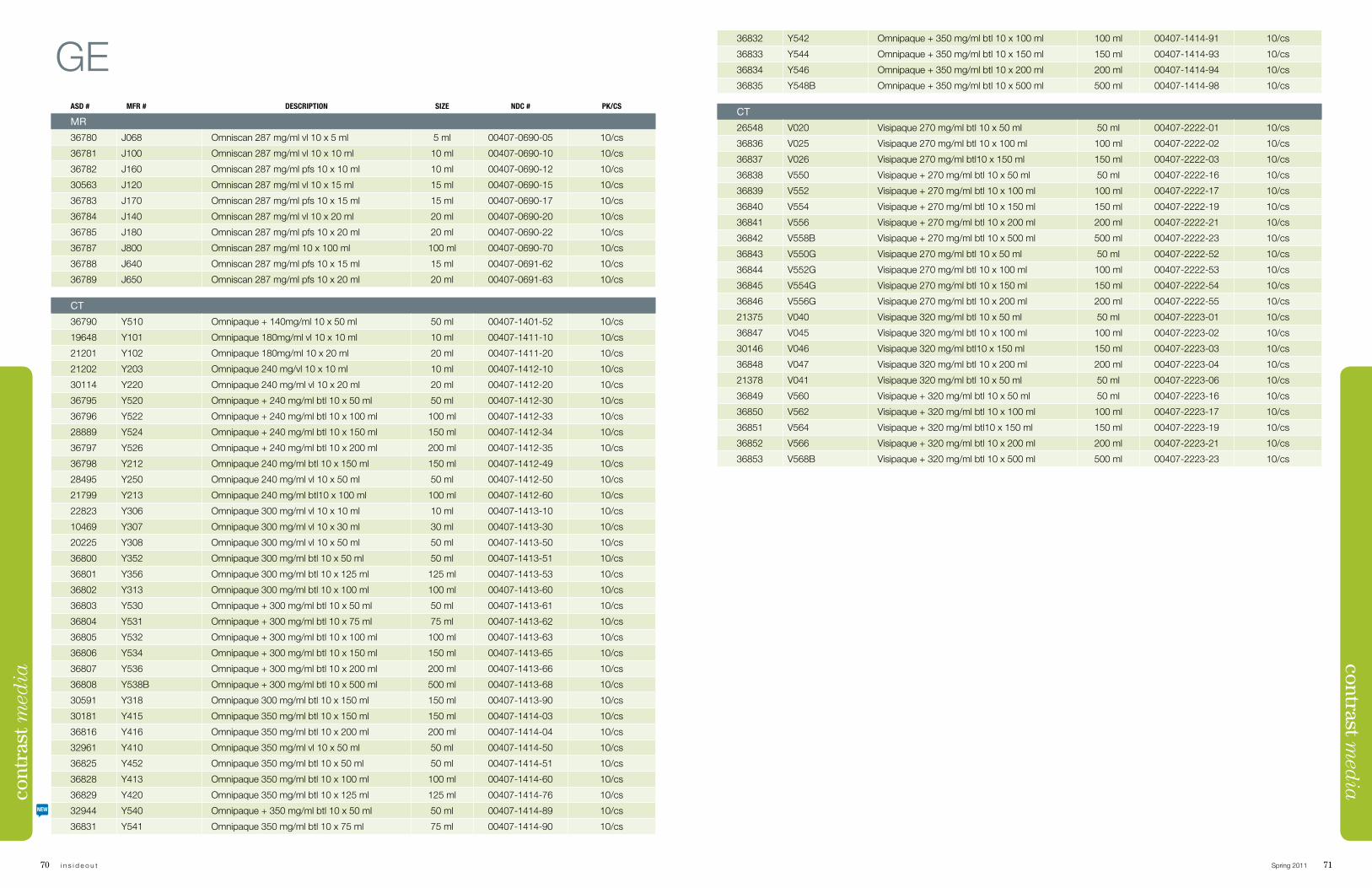
70 i n s i d e o u t Spring 2011 71
contrast med
ia36832 Y542 Omnipaque + 350 mg/ml btl 10 x 100 ml 100 ml 00407-1414-91 10/cs
36833 Y544 Omnipaque + 350 mg/ml btl 10 x 150 ml 150 ml 00407-1414-93 10/cs
36834 Y546 Omnipaque + 350 mg/ml btl 10 x 200 ml 200 ml 00407-1414-94 10/cs
36835 Y548B Omnipaque + 350 mg/ml btl 10 x 500 ml 500 ml 00407-1414-98 10/cs
CT
26548 V020 Visipaque 270 mg/ml btl 10 x 50 ml 50 ml 00407-2222-01 10/cs
36836 V025 Visipaque 270 mg/ml btl 10 x 100 ml 100 ml 00407-2222-02 10/cs
36837 V026 Visipaque 270 mg/ml btl10 x 150 ml 150 ml 00407-2222-03 10/cs
36838 V550 Visipaque + 270 mg/ml btl 10 x 50 ml 50 ml 00407-2222-16 10/cs
36839 V552 Visipaque + 270 mg/ml btl 10 x 100 ml 100 ml 00407-2222-17 10/cs
36840 V554 Visipaque + 270 mg/ml btl 10 x 150 ml 150 ml 00407-2222-19 10/cs
36841 V556 Visipaque + 270 mg/ml btl 10 x 200 ml 200 ml 00407-2222-21 10/cs
36842 V558B Visipaque + 270 mg/ml btl 10 x 500 ml 500 ml 00407-2222-23 10/cs
36843 V550G Visipaque 270 mg/ml btl 10 x 50 ml 50 ml 00407-2222-52 10/cs
36844 V552G Visipaque 270 mg/ml btl 10 x 100 ml 100 ml 00407-2222-53 10/cs
36845 V554G Visipaque 270 mg/ml btl 10 x 150 ml 150 ml 00407-2222-54 10/cs
36846 V556G Visipaque 270 mg/ml btl 10 x 200 ml 200 ml 00407-2222-55 10/cs
21375 V040 Visipaque 320 mg/ml btl 10 x 50 ml 50 ml 00407-2223-01 10/cs
36847 V045 Visipaque 320 mg/ml btl 10 x 100 ml 100 ml 00407-2223-02 10/cs
30146 V046 Visipaque 320 mg/ml btl10 x 150 ml 150 ml 00407-2223-03 10/cs
36848 V047 Visipaque 320 mg/ml btl 10 x 200 ml 200 ml 00407-2223-04 10/cs
21378 V041 Visipaque 320 mg/ml btl 10 x 50 ml 50 ml 00407-2223-06 10/cs
36849 V560 Visipaque + 320 mg/ml btl 10 x 50 ml 50 ml 00407-2223-16 10/cs
36850 V562 Visipaque + 320 mg/ml btl 10 x 100 ml 100 ml 00407-2223-17 10/cs
36851 V564 Visipaque + 320 mg/ml btl10 x 150 ml 150 ml 00407-2223-19 10/cs
36852 V566 Visipaque + 320 mg/ml btl 10 x 200 ml 200 ml 00407-2223-21 10/cs
36853 V568B Visipaque + 320 mg/ml btl 10 x 500 ml 500 ml 00407-2223-23 10/cs
GEASD # MFR # DESCRIPTION SIZE NDC # PK/CS
MR
36780 J068 Omniscan 287 mg/ml vl 10 x 5 ml 5 ml 00407-0690-05 10/cs
36781 J100 Omniscan 287 mg/ml vl 10 x 10 ml 10 ml 00407-0690-10 10/cs
36782 J160 Omniscan 287 mg/ml pfs 10 x 10 ml 10 ml 00407-0690-12 10/cs
30563 J120 Omniscan 287 mg/ml vl 10 x 15 ml 15 ml 00407-0690-15 10/cs
36783 J170 Omniscan 287 mg/ml pfs 10 x 15 ml 15 ml 00407-0690-17 10/cs
36784 J140 Omniscan 287 mg/ml vl 10 x 20 ml 20 ml 00407-0690-20 10/cs
36785 J180 Omniscan 287 mg/ml pfs 10 x 20 ml 20 ml 00407-0690-22 10/cs
36787 J800 Omniscan 287 mg/ml 10 x 100 ml 100 ml 00407-0690-70 10/cs
36788 J640 Omniscan 287 mg/ml pfs 10 x 15 ml 15 ml 00407-0691-62 10/cs
36789 J650 Omniscan 287 mg/ml pfs 10 x 20 ml 20 ml 00407-0691-63 10/cs
CT
36790 Y510 Omnipaque + 140mg/ml 10 x 50 ml 50 ml 00407-1401-52 10/cs
19648 Y101 Omnipaque 180mg/ml vl 10 x 10 ml 10 ml 00407-1411-10 10/cs
21201 Y102 Omnipaque 180mg/ml 10 x 20 ml 20 ml 00407-1411-20 10/cs
21202 Y203 Omnipaque 240 mg/vl 10 x 10 ml 10 ml 00407-1412-10 10/cs
30114 Y220 Omnipaque 240 mg/ml vl 10 x 20 ml 20 ml 00407-1412-20 10/cs
36795 Y520 Omnipaque + 240 mg/ml btl 10 x 50 ml 50 ml 00407-1412-30 10/cs
36796 Y522 Omnipaque + 240 mg/ml btl 10 x 100 ml 100 ml 00407-1412-33 10/cs
28889 Y524 Omnipaque + 240 mg/ml btl 10 x 150 ml 150 ml 00407-1412-34 10/cs
36797 Y526 Omnipaque + 240 mg/ml btl 10 x 200 ml 200 ml 00407-1412-35 10/cs
36798 Y212 Omnipaque 240 mg/ml btl 10 x 150 ml 150 ml 00407-1412-49 10/cs
28495 Y250 Omnipaque 240 mg/ml vl 10 x 50 ml 50 ml 00407-1412-50 10/cs
21799 Y213 Omnipaque 240 mg/ml btl10 x 100 ml 100 ml 00407-1412-60 10/cs
22823 Y306 Omnipaque 300 mg/ml vl 10 x 10 ml 10 ml 00407-1413-10 10/cs
10469 Y307 Omnipaque 300 mg/ml vl 10 x 30 ml 30 ml 00407-1413-30 10/cs
20225 Y308 Omnipaque 300 mg/ml vl 10 x 50 ml 50 ml 00407-1413-50 10/cs
36800 Y352 Omnipaque 300 mg/ml btl 10 x 50 ml 50 ml 00407-1413-51 10/cs
36801 Y356 Omnipaque 300 mg/ml btl 10 x 125 ml 125 ml 00407-1413-53 10/cs
36802 Y313 Omnipaque 300 mg/ml btl 10 x 100 ml 100 ml 00407-1413-60 10/cs
36803 Y530 Omnipaque + 300 mg/ml btl 10 x 50 ml 50 ml 00407-1413-61 10/cs
36804 Y531 Omnipaque + 300 mg/ml btl 10 x 75 ml 75 ml 00407-1413-62 10/cs
36805 Y532 Omnipaque + 300 mg/ml btl 10 x 100 ml 100 ml 00407-1413-63 10/cs
36806 Y534 Omnipaque + 300 mg/ml btl 10 x 150 ml 150 ml 00407-1413-65 10/cs
36807 Y536 Omnipaque + 300 mg/ml btl 10 x 200 ml 200 ml 00407-1413-66 10/cs
36808 Y538B Omnipaque + 300 mg/ml btl 10 x 500 ml 500 ml 00407-1413-68 10/cs
30591 Y318 Omnipaque 300 mg/ml btl 10 x 150 ml 150 ml 00407-1413-90 10/cs
30181 Y415 Omnipaque 350 mg/ml btl 10 x 150 ml 150 ml 00407-1414-03 10/cs
36816 Y416 Omnipaque 350 mg/ml btl 10 x 200 ml 200 ml 00407-1414-04 10/cs
32961 Y410 Omnipaque 350 mg/ml vl 10 x 50 ml 50 ml 00407-1414-50 10/cs
36825 Y452 Omnipaque 350 mg/ml btl 10 x 50 ml 50 ml 00407-1414-51 10/cs
36828 Y413 Omnipaque 350 mg/ml btl 10 x 100 ml 100 ml 00407-1414-60 10/cs
36829 Y420 Omnipaque 350 mg/ml btl 10 x 125 ml 125 ml 00407-1414-76 10/cs
32944 Y540 Omnipaque + 350 mg/ml btl 10 x 50 ml 50 ml 00407-1414-89 10/cs
36831 Y541 Omnipaque 350 mg/ml btl 10 x 75 ml 75 ml 00407-1414-90 10/cs
cont
rast
med
ia
NEW

72 i n s i d e o u t Spring 2011 73
nephrology
RxASD # DESCRIPTION NDC # MANUFACTURER
Ancillary RX
19920 Cal Gluc 10% SDV 10 ml each 63323-0311-10 APP
20490 Carnitor 1 gm/5 ml SDV 5 x 5 ml 54482-0147-01 Sigma-Tau Pharmaceuticals
20490 Carnitor 1 gm/5 ml SDV 5 x 5 ml 54482-0147-01 Sigma -Tau Pharmaceuticals
11153 Diphenhydramne 50 mg Vl 25 x 1 ml 00641-0376-25 Baxter PPI
32986 Hydrogen Peroxide 3% Liq 16 oz each 00869-0871-43 Amerisourcebergen Drug Corporation
29417 Imodium Liq 4 oz 50580-0134-04 Johnson & Johnson
18679 Sod Thiosulfate 25% 50ml SDV 00517-5019-01 Amer Regent Lab
26656 Water Sterile FTV 25 x 10 ml 00409-4887-10 Hospira Worldwide
Antibiotics
28516 Ciprofloxacin 400 mg SDV 40 ml each 00409-4778-86 Hospira Worldwide
34185 Cefazolin 1 gm SDV 25/10 ml 00409-0805-01 Hospira Worldwide
32428 Cubicin Daptomycin 500mg vl 67919-0011-01 Cubist Pharmaceutical
24631 Cubicin Daptomycin 500mg 10/pk 67919-0011-01 Cubist Pharmaceutical
26086 Fortaz 1 gm vl pwd 10 x 25 ml 00173-0378-10 Glaxosmithkline
26708 Gentamicin 40 mg/ml FTV 25 x 2 ml 00409-1207-03 Hospira Worldwide
23421 Gentamicin Top 0.1% crm 15 gm 00168-0071-15 Fougera E and Co.
14181 Rocephin 1 gm vl each 00004-1964-04 Genentech USA
26216 Vancomycin 500 mg FTV 10/bx 00409-4332-01 Hospira Worldwide
26111 Vancomycin 1 gm FTV 10/bx 00409-6533-01 Hospira Worldwide
ESAs
28203 Aranesplysb 200 mcg SDV 1.0 ml 55513-0006-01 Amgen Inc.
28198 Aranesplysb 25 mcg SDV 4 x 1 ml 55513-0002-04 Amgen Inc.
28201 Aranesplysb 100 mcg SDV 4 x 1 ml 55513-0005-04 Amgen Inc.
28200 Aranesplysb 60 mcg SDV 4 x 1 ml 55513-0004-04 Amgen Inc.
28199 Aranesplysb 40 mcg SDV 4 x 1 ml 55513-0003-04 Amgen Inc.
28194 Aranesplysb 100 mcg PFS 4 x .5 ml 55513-0025-04 Amgen Inc.
28196 Aranesplysb 200 mcg PFS 0.4 ml 55513-0028-01 Amgen Inc.
28197 Aranesplysb 300 mcg PFS 0.6 ml 55513-0111-01 Amgen Inc.
28193 Aranesplysb 60 mcg PFS 4 x 0.3 ml 55513-0023-04 Amgen Inc.
28191 Aranesplysb 25 mcg PFS 4 x .42 ml 55513-0057-04 Amgen Inc.
28192 Aranesplysb 40 mcg PFS 4 x .4 ml 55513-0021-04 Amgen Inc.
28204 Aranesplysb 300 mcg SDV 1.0 ml 55513-0110-01 Amgen Inc.
28195 Aranesplysb 150 mcg PFS 4 x 0.3 ml 55513-0027-04 Amgen Inc.
28202 Aranesplysb 150 mcg SDV 4 x 0.75 ml 55513-0053-04 Amgen Inc.
28140 Aranesp Plysb 500 mcg Pfs 1 ml 55513-0032-01 Amgen Inc.
11230 Epogen S3 3 M un/ml vl 10 x 1 ml 55513-0267-10 Amgen Inc.
11295 Epogen S4 4 M Un/ml Vl 10 x 1 ml 55513-0148-10 Amgen Inc.
11294 Epogen S2 2 M Un/ml Vl 10 x 1 ml 55513-0126-10 Amgen Inc.
11296 Epogen S10 10 M un/ml vl 10 x 1 ml 55513-0144-10 Amgen Inc.
11177 Epogen M10 20 mun/2ml Mdv10 x 2 ml 55513-0283-10 Amgen Inc.
11508 Epogen M20 20 mun/ml Mdv 10 x 1 ml 55513-0478-10 Amgen Inc.neph
rolo
gy35151 Ferrlecit 62.5 mg/5 ml amp 10 x 5 ml 00024-2791-50 Aventis Pharmaceuticals (Hmr)
37769 Ferrlecit 62.5 mg/5 ml vl 10 x 5 ml 00024-2792-10 Aventis Pharmaceuticals (Hmr)
34408 Feraheme 510 mg/17 ml SDV 59338-0775-01 Amag Pharmaceuticals
34409 Feraheme 510 mg/17 ml SDV 10/pk 59338-0775-10 Amag Pharmaceuticals
22668 Infed 50 mg/ml vl 10 x 2 ml 52544-0931-02 Watson Pharm
30696 Procrit 20 M un/ml Mdv 4 x 1 ml 59676-0320-04 Ortho Biotech Products
10982 Procrit 40 M un/ml vl 4 x 1 ml 59676-0340-01 Ortho Biotech Products
31310 Procrit 10 M un/ml 2ml MDV 4/pk 59676-0312-04 Ortho Biotech Products
10510 Procrit 10 M un/ml vl 6 x 1 ml 59676-0310-01 Ortho Biotech Products
37763 Nulecit Inj 62.5 mg/10 x 5 ml SDV 52544-0149-87 Watson Pharm
33100 Venofer SDV 10 x 5 ml 49230-0534-10 Fresenius USA Marketing
29962 Venofer 20 mg/ml SDV 25 x 5 ml 00517-2340-25 Amer Regent Lab
20686 Venofer 20 mg/ml vl 10 x 5ml (CKD) 00517-2340-10 Amer Regent Lab
29962 Venofer 20 mg/ml SDV 2 x 5 ml (CKD) 00517-2340-25 Amer Regent Lab
37214 Venofer 50 mg SDV 10 x 2.5 ml (Dialysis) 49230-0530-10 Fresenius USA Marketing
33100 Venofer 100 mg SDV 10 x 5 ml (Dialysis) 49230-0534-10 Fresenius USA Marketing
Vaccines, Hep. B
30326 Engerix adt 20 mcg p/f SDV 10 x 1 ml 58160-0821-11 Glaxosmithkline Vaccines
30218 Engerix ped p/f sdv 10x0.5 ml 58160-0820-11 Glaxosmithkline Vaccines
36216 Engerix adult 20 mcg tiplok 5/pk 58160-0821-48 Glaxosmithkline Vaccines
22353 Recombivax df 40 mcg/ml pf 1 ml 00006-4992-00 Merck & Co.
Heparin
11778 Heparin 1000 u/ml MDV 25 x 30 ml 63323-0540-31 APP
17773 Heparin sod 5 M un MDV 25 x 10 ml 63323-0047-10 APP
36901 Heparin 1000 u/ml MDV 25 x 30 ml 25021-0400-30 Sagent Pharmaceuticals
36906 Heparin sod 5 M MDV vl 25 x 10 ml 25021-0402-10 Sagent Pharmaceuticals
11480 Heparin sod 10V un MDV 25 x 1 ml 63323-0542-01 APP
11479 Heparin sod 5 M un MDV 25 x 1 ml 63323-0262-01 APP
11782 Heparin sod 1 mun/ml MDV 25 x 10 ml 63323-0540-11 APP
Inj. Vitamin D
37547 Calcitriol 0.25 mcg caps 100/btl 63304-0239-01 Ranbaxy
37548 Calcitriol 0.5 mcg caps 100/btl 63304-240-01 Ranbaxy
37813 Calcitriol Oral Solution 1 mcg 15 ml 00054-3120-41 Roxanne
31639 Calcitriol 1 mcg/ml amp 25 x 1 ml 00517-0132-25 Amer Regent Lab
23349 Calcitriol 1 mcg inj 50 x 1 ml 63323-0731-01 APP
23349 Calcitriol 1 mcg amp 50 x 1 ml 63323-0731-01 App
29151 Hectorol 0.5 mcg caps 50/btl 58468-0120-01 Genzyme Renal
37234 Hectorol 1 mcg caps 50/btl 58468-0124-01 Genzyme Renal
37299 Hectorol 2 mcg SDV 50 x 1 ml 58468-0126-01 Genzyme Renal
33635 Hectorol 2.5 mcg caps 50 58468-0121-01 Genzyme Renal
33705 Hectorol 4 mcg 50 x 2 ml 58468-0123-01 Genzyme Renal
37232 Zemplar 1 mcg caps 30/btl 00074-4317-30 Abbott Labs/Onc
37233 Zemplar 2 mcg caps 30/btl 00074-4314-30 Abbott Labs/Onc
20967 Zemplar 2 mcg/ml vl 25 x 1 ml 00074-4637-01 Abbott Labs/Onc
32194 Zemplar 4 mcg caps 30/btl 00074-4315-30 Abbott Labs/Onc
10001 Zemplar 5 mcg/ml 25 x 1 ml FTV 00074-1658-01 Abbott Labs/Onc
10002 Zemplar 5 mcg/ml 25 x 2 ml FTV 00074-1658-02 Abbott Labs/Onc
37381 Zemplar 10 mcg/2 ml mdv 25 x 2 ml 00074-1658-05 Abbott Labs/Onc
NEW
NEW
NEW
NEW
NEW
NEW
NEW
NEW
NEW
NEW
NEW
NEW
NEW

74 i n s i d e o u t Spring 2011 75
Other Rx
17674 Acetamin 325mg tab ud 100 51079-0002-20 Udl Laboratories
20363 Activase cathflo 2 mg SDV 50242-0041-64 Genentech USA
31842 Aplisol 5tu 10test vl 1 ml 42023-0104-01 Jhp Pharmaceuticals
32464 Atropine lfs syr 10 ml 0.1 mg/ml 00409-4911-34 Hospira Worldwide
10093 Clonidine 0.1 mg tab 100/btl 00228-2127-10 Actavis
27537 Dextrose 25 gm ansyr 50 ml each 00409-7517-16 Hospira Worldwide
26223 Dextrose 50% vl 25 x 50 ml 00409-6648-02 Hospira Worldwide
21538 Diphenhydramine 25 mg tab ud 100 51079-0967-20 Udl Laboratories
10704 Diphenhydramin 25 mg cap 100/btl 00185-0648-01 Sandoz
27730 Epinep lfshld syg 10 ml nondl 00409-4921-34 Hospira Worldwide
27229 Epinep 1:10000 18g abjct 10ml 00409-4901-18 Hospira Worldwide
27906 Gelfoam 12-7 mm spg 12/pk 00009-0315-08 Pfizer Pharm
34930 Glucose tab orange 10/pk 38396-0543-64 Amerisourcebergen Drug Corporation
10063 Insulin Hum R 100 u/ml vl 10 ml 00002-8215-01 Eli Lilly & Co
34660 Levaquin 500 mg SDV 20 ml 50458-0164-20 Jom
32794 Lido 0.5% FTV 50 ml each 00409-4275-01 Hospira Worldwide
27029 Lidocaine 1% ansyr syg 5 ml each 00409-9137-05 Hospira Worldwide
26662 Lidocaine 1% ftv 50ml each 00409-4276-02 Hospira Worldwide
26662 Lidocaine 1% ftv 50ml each 00409-4276-02 Hospira Worldwide
14719 Loperamide 2 mg 100 cap 00378-2100-01 Mylan Pharmaceutical
36031 Mannitol 25% FTV 25 x 50 ml 00409-4031-01 Hospira Worldwide
17448 Mannitol 25% SDV 25 x 50 ml 00517-4050-25 Amer Regent Lab
32324 Metoclopram 10 mg/2ml FTV 2 00409-3414-01 Hospira Worldwide
32320 Pain Ease Spray mist 3. oz 00386-0008-02 Gebauer Company
26843 Pain ease 3.5 oz spray 00386-0008-03 Gebauer Company
10486 Pneumovax 23 5dose vl 2.5 ml 00006-4739-00 Merck & Co.
11151 Promethazine 25 mg/ml amp 25 x 1 ml 00641-1495-35 Baxter PPI
18782 Sod Chl .9% 1000 cc bag 12/cs 00264-7800-00 B. Braun Medical
10032 Sod Chl 0.9 Excl 24 x 500 ml sol 00264-7800-10 B.Braun Medical
11199 Sod Chl Conc 23.4% SDV 25 x 30 ml 00517-2930-25 Amer Regent Lab
27615 Sod Bic 8.4% Ls syg 50 ml each 00409-6637-34 Hospira Worldwide
34835 Triple Anti Oint Foil 144 x .9 gm Dynarex Corp
20609 Tubersol 5tu 10 test vl 1 ml 49281-0752-21 Sanofi Pasteur
11062 Tums e-x x/s fruit tab 96/pk 00766-0739-66 Glaxosmithkline
12598 Water Ster SDV 25 x 20 ml 00517-3020-25 Amer Regent Lab
26552 Water Ster FTV 25 x 20 ml 00409-4887-20 Hospira Worldwide
nephrologyneph
rolo
gy
MedsurgeASD # DESCRIPTION MANUFACTURER PK/CS
General
27969 Alcohol Swabticks 3'S Dynarex 25/box
14819 Alcohol Prep Pad (non-sterile) Dynarex 200/box
35009 Alcohol Prep Pad Med. (sterile) Dynarex 200/box
32474 Apron Gowns Disposable 28" x 46" (Plastic) Busse 144/case
32710 Catheter Cap (injectable) Latex Free Molded 100/box
18784 Catheter Cap (dead end / non-injectable) B. Braun 100/box
26860 Chloraprep Single Swabstick Tacy Med 48/box
32684 Cotton Tip Applicators 6" Sterile 2’s Solon 100/box
Dialysis Recliner – 6 Position Winco 1/ea.
32475 Drape Sheet 18 x 26 (other sizes available) Busse 50/bx
26857 Drape Sheet 40" x 48" 2 ply Tidi 100/case
36048 Drape Sheet 40" x 90" 3 ply Tidi 50/cs
34089 Emesis Basin 9" Medline 250/case
35908 Hemastix Test Strips Siemens 50/box
32709 Hemoband (non-sterile) Hemoband 1/ea.
32761 Luer Lock End Caps – Blue Molded 100/box
18318 Medicine Cups 1 oz Dynarex 100/box
32538 Oxygen Mask Rebreather – Adult Allied 1/ea.
15184 Oxygen Nasal Cannula – Adult Allied 1/ea.
14844 Povidone/Iodine Prep Pads (PVP Pads) Dynarex 100/box
14840 Povidone/ Iodine Swabsticks (1 per pack) Dynarex 50/box
28053 Povidone/Iodine Swabsticks (3 per pack) Dynarex 75/box
32754 Recirculating Connector (Male to Male) Molded 100/box
32414 Shoe Covers – Blue (one size fits all) Dynarex 150/pairs
26355 Skin Staple Remover Kits Busse 1/ea.
20941 Suture Removal Kits Busse 1/ea.
15598 Table Paper 21" x 225' Smooth Tidi 12/case
14815 Tape Remover Pads (Sterile) Dynarex 100/box
32643 Transducer Protector (w/luer lock) Molded 100/case
32755 Tube Occluding Clamp (hemostats) Blue Molded 100/bag
20568 Towel 3 ply 13.5" x 18" White 500/CS Graham 500/case
25875 Underpad 17" x 24" (blue) Chux Dynarex 300/case
32605 Urinals w/Cover – Plastic (disposable) Perigon 50/case
33864 Urinals w/Cover – Plastic (disposable) Medline Each
15601 Wash Cloths 10" x 13.5" (blue/disposable) Graham 500/case

76 i n s i d e o u t Spring 2011 77
Sharps containers
17710 Sharps Container 2 gallon - Red (# 8961) Kendall 20/case
22352 Sharps Container 2 gallon - Red (# 8970) Square Kendall 20/case
36603 Sharps Container 3 gallon - Red (# 852221) Square Kendall 10/case
32727 Sharps Container 3 gallon - Red (#8964) Kendall 20/case
23163 Sharps Container 8 gallon - Red (# 8980) Square Kendall 10/case
32042 Sharps Container 8 gallon - Red (#8980S) Slide Lid Kendall 10/case
11973 Sharps Container 18 gallon - Red Kendall 1/ea.
32623 Sharps Container – Brackets 2 and 3 gallon Kendall 1/ea.
Thermometers
13281 Filac Probe Covers Kendall 500/box
32742 Genius Probe Covers Kendall 2100/case
14167 Tempa Dot Thermometers (Oral and Axil) 3M 100/box
24456 Thermoscan Sure Temp Plus 690 Welch Allyn Each
32765 Thermoscan Sure Temp Probe Covers Welch Allyn 1000/cs
25572 Thermoscan Probe Covers Welch Allyn 800/box
Tape
14137 Durapore Tape 1" x 10 yards (Cloth / Silk) 3M 12/rolls
14138 Durapore Tape 2" x 10 yards (Cloth / Silk) 3M 6/rolls
33234 Durapore / Single Use 1" x 1.5 yards (Cloth/Silk) 3M 100/rolls
32338 Hypo-Pore Tape 1" x 10 yards (Paper) Dukal 12/rolls
32577 Hypo-Silk Cloth Tape 1" x 10 yards Dukal 12/rolls
19140 Micropore Tape 1" x 10 yards (Paper) 3M 12/rolls
32535 Micropore / Single Use 1" x 1.5 yards (Paper) 3M 100/rolls
32536 Transpore Tape 1" x 1.5 yards (Clear Plastic) 3M 500/rolls
14151 Transpore Tape 1" x 10 yards (Clear Plastic) 3M 12/rolls
Bandages
34440 Bandage Strip 1" x 3 " (plastic) Dukal 100/box
32553 Bandage Strip 3/4" x 3" (plastic) Dukal 100/box
19216 Bandage Flex Strip 1" x 3" Dynarex 100/box
19204 Bandage Sheer Strip 1" x 3" Dynarex 100/box
32594 Gauze 2" x 2" (non-sterile) 8 Ply Dukal 5000/case
32595 Gauze Pads 2" x 2" 8 Ply (sterile) 2's Dukal 1500/case
32597 Gauze 4" x 4" 12 ply N/S 2000/CS Dukal 2000/case
32693 Gauze 4" x 4" 12 ply (sterile) Dukal 100/box
32796 Gauze 4 x 4 8 Ply (sterile) 2's Dukal 1200/box
24274 Gauze 2" x 2" 8 Ply (sterile) 2's Dynarex 100/box
13275 Telfa Pads 2" x 3" Sterile Kendall 100/box
32344 Op-Site IV3000 2 3/8" x 2 3/4" S/N 100/box
32346 Op-Site IV3000 4" x 4 3/4" S/N 50/box
32345 Op-Site Flexgrid 4" x 4 3/4" S/N 50/box
32578 Tegaderm 2 3/8" x 2 3/4" (compare to 3M brand) Dukal 100/box
32741 Tegaderm 4" x 4 3/4" (compare to 3M brand) Dukal 50/box
14163 Tegaderm 2 3/8" x 2 3/4" 3M Brand 3M 100/box
18528 Tegaderm 4" x 4 3/4" 3M Brand 3M 50/box
32743 Topper Sponge Sterile 4" x 3" Dukal 50/box
32357 Zorband (Large) 1" x 2 3/4" (compare to Sureseal) Exel 100/box
32358 Zorband ( XL) 1 1/4" x 2 3/4"(compare to Sureseal) Exel 100/box
nephrology
Face masks and shields
34204 Face Shield – Full Length (no visor) Precept 1/ea.
18534 Mask (ear loop) Blue Dynarex 50/box
14837 Mask (molded) Blue Dynarex 50/box
18544 Mask (ear loop) w/Face Shield Blue Dynarex 50/bx
Test strips
11262 Accuchek Comfort Curve Test Strip Roche 100 strips
11265 Accuchek Comfort Curve Control Solution Roche 1/box
32328 Ascensia Breeze Gluc. Blood Strips Bayer 50 strips
34128 Bicarb PH II Test Strips (100 per bottle) SerimResearch 5/btls
34124 Chlorine Test Strips (100 per bottle) SerimResearch 6/btls
23596 One Touch Ultra Control Solution 4ml Lifescan 2/v
32640 Peracetic Acid Reagent Strips (100 per bottle) ETS HACH 6/btls
32641 Residual Peroxide Reagent Strips (100 per bottle) ETS HACH 5/btls
32628 Total Chlorine Test Kit ETS HACH 1/kit
32642 Total Chlorine Refill 5 x 100 ETS HACH 1/kit
34130 Water Hardness Reagent Strips (50 per bottle) SerimResearch 6/btls
Gloves
19089 Gloves Powder Free – Vinyl (Small) Sempermed 100/box
22274 Gloves Powder Free – Vinyl Exam (Medium) Dynarex 100/box
19091 Gloves Powder Free – Vinyl (Large) Sempermed 100/box
28282 Gloves SFE Touch Latex PF (Small) Dynarex 100/box
28283 Gloves SFE Touch Latex PF (Medium) Dynarex 100/box
28284 Gloves SFE Touch Latex PF (Large) Dynarex 100/box
28285 Gloves SFE Touch Latex PF ( X-Large) Dynarex 100/box
32407 Gloves Sterile – Latex (Medium) Sm and Lg available Dynarex 50/pairs
32413 Gloves- Nitrile- powder free-small Dynarex 100/box
32412 Gloves- Nitrile- powder free-medium Dynarex 100/box
32411 Gloves-Nitrile-powder free- large Dynarex 100/box
32410 Gloves- Nitrile- powder free- x-large Dynarex 100/box
Needles
10993 Needles 18g x 1" Terumo 100/box
18984 Needles 18g x 1.5" Terumo 100/box
11072 Needles 20g x 1" Terumo 100/box
18985 Needles 21g x 1.5" Terumo 100/box
11065 Needles 23g x 1" Terumo 100/box
11058 Needles 25g x 5/8" Terumo 100/box
Fistula needles
32361 Fistula Needles 15g x 1" w/Back Eye 12" TB Exel 50/box
32362 Fistula Needles 16g x 1" w/Back Eye 12" TB Exel 50/box
32360 Fistula Needles 17g x 1" w/Back Eye 12" TB Exel 50/box
32560 Fistula Needles 17g x 1.25" w/Back Eye 12" TB Exel 50/box
32359 Fistula Needles 15g x 1" w/o B/E 12" TB Exel 50/box
neph
rolo
gy

78 i n s i d e o u t Spring 2011 79
Our Customer Service Representatives are here for you.
12789 Safety Syringe 3 cc 22 g x 1.5" (BD# 309593) B/D 100/box
12811 Safety Syringe 3 cc 25 g x 5/8" (BD# 309592) B/D 100/box
18734 Safety Syringe 5 cc Luer Lock (BD# 305558) B/D 50/box
19561 Safety Syringe 5 cc 21 g x 1.5" (BD# 305561) B/D 50/box
12784 TB Safety Syringe 1 cc 25 g x 5/8" (BD#305554) B/D 100/box
12785 TB Safety Syringe 1 cc 27 g x 1/2" (BD# 305553) B/D 100/box
Kendall – Safety syringes
19521 Safety Syringe 1 cc Insulin 29 g x 1.5" Kendall 100/box
19519 Safety Syringe 1 cc 28 g x 1/2" Kendall 100/box
19518 Safety Syringe 1 cc 25 g x 5/8" Kendall 100/box
19525 Safety Syringe 3 cc 20 g x 1 x 1.5" Kendall 100/box
19302 Safety Syringe 3 cc 21 g x 1" Kendall 100/box
19303 Safety Syringe 3 cc 21 g x 1 x 1.5" Kendall 100/box
32367 Safety Syringe 3 cc 22 g x 1" Kendall 100/box
19528 Safety Syringe 3 cc 22 g x 1.5" Kendall 100/box
19304 Safety Syringe 3 cc 23 g x 1" Kendall 100/box
19530 Safety Syringe 3 cc 25 g x 5/8" Kendall 100/box
19536 Safety Syringe 6 cc 20 g x 1.5" Kendall 50/box
19308 Safety Syringe 6 cc 21 g x 1 x 1.5" Kendall 50/box
19531 Safety Syringe 6 cc Luer Lock Kendall 50/box
19300 Safety Syringe 12 cc 20 g x 1 x 1.5" Kendall 50/box
19301 Safety Syringe 12 cc 21 g x 1 x 1.5" Kendall 50/box
19535 Safety Syringe 12 cc Luer Lock Kendall 50/box
Duopross syringes
32575 3cc 21g x 1.5" Low Dead Space safety syringe Duopross 100/box
32571 1cc 22g x 1 x 1.5" Renal Max Duopross 100/box
32570 1cc 22g x 1.5" Renal Safe safety syringe Duopross 100/box
32576 1cc 25g x 5/8" Safety TB Syringe Duopross 100/box
34418 Insulin Syringe 1cc 29g x 1/2" Duopross 100/box
34915 Safety Syringe 3cc 23g x 1" Duopross 100/box
Syringes
32680 Insulin Syringe 1 cc 27g x 1/2" Terumo 100/box
32562 Insulin Syringe 1 cc 29g x 1/2" Terumo 100/box
22686 Syringe 1 cc 27g x 1/2" Allergy (Low Dead Space) Terumo 100/box
18993 TB Syringe 1 cc 25G x 5/8" Terumo 100/box
32352 TB Syringe 1 cc 25g x 5/8" (Zero Dead Space) Exel 100/box
11033 Syringe 1 cc w/Luer Lock Terumo 100/box
18995 Syringe 3 cc w/Luer Lock Terumo 100/box
18996 Syringe 3 cc 20g x 1" w/Luer Lock Terumo 100/box
18997 Syringe 3 cc 21g x 1" w/Luer Lock Terumo 100/box
18998 Syringe 3 cc 22g x 1" w/Luer Lock Terumo 100/box
11053 Syringe 3 cc 23g x 1" Terumo 100/box
11049 Syringe 3 cc 25g x 5/8" Terumo 100/box
19000 Syringe 5 cc w/Luer Lock Terumo 100/box
11031 Syringe 10 cc w/Luer Lock Terumo 100/box
19003 Syringe 10 cc 21g x 1" Terumo 100/box
33579 Syringe 20 cc w/Luer Lock Terumo 50/box
11403 Syringe 30 cc w/Luer Lock Terumo 25/box
11407 Syringe 60 cc w/Luer Lock Terumo 25/box
B/D – Syringes
12786 TB Syringe 1 cc 25g x 5/8" (BD# 309626) B/D 100/box
12881 TB Syringe 1 cc (w/o needle) (BD# 309602) B/D 100/box
10645 Syringe 10 cc Luer Lock (BD# 309604) B/D 100/box
10638 Syringe 10 cc 21g x 1" (BD# 309642) B/D 100/box
37116 Syringe 3 cc Luer Lock (BD# 309657) B/D 200/bx
12791 Syringe 3 cc 20g x 1.5" (BD# 309579) B/D 100/box
12792 Syringe 3 cc 20g x 1" (BD# 309578) B/D 100/box
12794 Syringe 3 cc 21g x 1" (BD# 309575) B/D 100/box
12795 Syringe 3 cc 22g x 1.5" (BD# 309574) B/D 100/box
12796 Syringe 3 cc 22g x 1" (BD# 309572) B/D 100/box
12797 Syringe 3 cc 23g x 1" (BD# 309571) B/D 100/box
12799 Syringe 3 cc 25g x 1" (BD# 309581) B/D 100/box
12882 Syringe 30 cc Luer Lock (BD# 309650) B/D 40/box
12814 Syringe 5 cc 21g x 1.5" (BD# 309633) B/D 100/box
B/D – Safety syringes
23970 BD Integra 3 cc 22 g x 1.5" Low Dead Space B/D 100/box
23969 BD Integra 3 cc 23 g x 1" Low Dead Space B/D 100/box
12782 Insulin Safety Syringe 1 cc 29 g x 1.5" (BD# 329464) B/D 500/case
18735 Safety Syringe 10 cc Luer Lock (BD# 305559) B/D 50/box
19562 Safety Syringe 10 cc 21 g x 1.5" (BD# 305564) B/D 50/box
12884 Safety Syringe 3 cc Luer Lock (BD# 309606) B/D 100/box
12808 Safety Syringe 3 cc 21 g x 1.5" (BD# 309595) B/D 100/box
nephrologyneph
rolo
gy
During our regular business hours, or during
an after-hours emergency, you can count on
ASD Healthcare to provide you the products
and the service you need, when you need them.
Call ASD Healthcare today to experience our superior service, get connected with a dedicated Account Manager, and start enjoying the benefits of working with ASD Healthcare.
For more information, please visit www.asdhealthcare.com or call us at 866.746.6273.
Customer Service 800.746.6273 Call 8 am – 6:30 pm CsT

in the works
Spring 2011 8180 i n s i d e o u t
COL0
7-4-
US-1
0
Inspired by nature
Grifols provides the following product choices:
For further information call: Grifols USA, LLC Professional Service: 888 GRIFOLS (888 474 3657)Customer Service: 888 325 8579 Fax: 323 441 7968 www.grifols.com
Grifols Biologicals Inc.5555 Valley Boulevard, Los Angeles, California 90032, USA
The Hospital for Sick Children in Montreal Canada is sponsoring an observational study to compare the burden of hemophilia patents in developing countries with that of patients living in developed countries. The proposed study will quantify the burden of arthropathy, physical disability and quality of life in boys with hemophilia in Brazil. This study will also provide an opportunity to compare these outcomes to those observed in Canada where the dominant therapy has become life-long prophylaxis. This study is not yet open for participant recruitment. Estimated comple-tion date is December 2013.
The psychosocial effect of living with hemophilia is the topic of a survey started in Africa, Asia, Europe, North America and South America in May 2011. Sponsored by Novo Nordisk, this observational survey seeks to identify and quantify the key psychosocial issues affecting hemo-philia patients in their daily lives and how these factors affect treatment outcomes, compliance, health and general well-being.
An 18-month interventional study is recruit-ing participants globally to evaluate the efficacy and safety of a prophylactic treat-ment with recombinant activated FVII in reducing the frequency of joint bleeds and the development of joint damage in children with hemophilia A who develop high-titer inhibitors. This study is de-signed to gain evidence of the advantage of the prophylactic, daily treatment with recombinant activated FVII as compared to the conventional on-demand therapy in reducing the bleeding frequency and
preserving the orthopedic status in hemophilic children with inhibitors.
Trials sponsored by Gambro Lundia AB and conducted globally will study the use of Evodial ® in patients requiring heparin-free dialysis. Estimated study completion date is January 2012. This study is not yet open for participant recruitment.
Massachusetts General Hospital seeks to determine safe and effective ways to raise vitamin D levels while monitoring effects on the immune system in a study it is sponsoring. Infection is the second-leading cause of death in individuals requiring dialysis treatment for kidney failure. The study seeks to confirm new research that suggests this high risk may be due in part to low levels of vitamin D.
MD Scientific is conducting an interven-tional study to evaluate the use of sodium bicarbonate to reduce the incidence of contrast induced chronic kidney injury in patients with kidney disease. The study is currently recruiting participants 18 years and older in 13 states throughout the U.S.
An observational study focusing on hospital-ized patients with H1N1 virus is scheduled to be completed this September. One of the study’s goals is to help understand the course of the illness and the characteristics of people who do not do well. The study will also try to learn more about how different treatments and prior vaccination for the flu affects the course of H1N1 virus.
source: Clinical trials.gov, u.s. national institutes of health, web 23 May 2011

von Willebrand Factor/Coagulation Factor VIII Complex (Human)
Developed Specifically for the Treatment of von Willebrand Disease
■ High purity VWF/FVIII complex
■ Double virus inactivated
■ Physiologic 1:1 ratio of VWF and FVIII
■ Parallel pharmacokinetic profiles for FVIII and VWF
■ Clinical efficacy, safety, and tolerability proven in adult and pediatric populations
■ Rapidly dissolved in a small volume
■ Convenient dosing
wilate® is a von Willebrand Factor/Coagulation Factor VIII Complex (Human) indicated for the treatment of spontaneous and trauma-induced bleeding episodes in patients with severe von Willebrand disease (VWD), as well as patients with mild or moderate VWD in whom the use of desmopressin is known or suspected to be ineffective or contraindicated.
Two convenient vial sizes
■ 450 IU VWF:RCo and 450 IU FVIII activities in 5 mL
■ 900 IU VWF:RCo and 900 IU FVIII activities in 10 mL
■ Includes Mix2Vial™ transfer device
rapidly dissolved in a small injection volume, may help save time during administration.
12
6
9
Important safety information:wilate® is contraindicated for individuals with a history of anaphylactic or severe systemic reaction to human plasma-derived products, any ingredient in the formulation, or components of the container. Thromboembolic events have been reported in VWD patients receiving coagulation factor replacement therapies. FVlll activity should be monitored to avoid sustained excessive FVlll levels. wilate® is made from human plasma. The risk of infectious agents, including viruses and, theoretically, the Creutzfeldt-Jakob disease agent, cannot be completely eliminated. The most common adverse reactions to treatment with wilate® in patients with VWD have been urticaria and dizziness. The most serious adverse reactions to treatment with wilate® in patients with VWD have been hypersensitivity reactions.
Octapharma USA, Inc.121 River StreetSuite 1201Hoboken, NJ 07030201-604-1130www.octapharma.com
Customer Service:[email protected]
Medical Affairs:[email protected]
Reimbursement:[email protected] Tel: 800-554-4440Fax: 800-554-6744
www.wilateusa.com
For More Information, please Contact Us
h215_Ad_v1.indd 2 5/23/11 1:43 PMh215_Ad_v1.indd 3 5/23/11 1:43 PM
Related Documents











