Communication Inhibition of mitochondrial bioenergetics: the effects on struc- ture of mitochondria in the cell and on apoptosis *. Konstantin G. Lyamzaev 1 , Denis S. Izyumov 1 , Armine V. Avetisyan 1 , Fuyu Yang 2 , Olga Yu. Pletjushkina 1 and Boris V. Chernyak 1½ 1 A.N. Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia, 2 Institute of Biphysics, Beijing, China Received: 30 April, 2004; accepted: 07 May, 2004 Key words: mitochondria, oxidative phosphorylation, inhibitors, apoptosis The effects of specific inhibitors of respiratory chain, F o F 1 ATP synthase and uncouplers of oxidative phosphorylation on survival of carcinoma HeLa cells and on the structure of mitochondria in the cells were studied. The inhibitors of respiration (piericidin, antimycin, myxothiazol), the F 1 -component of ATP synthase (aurovertin) and uncouplers (DNP, FCCP) did not affect viability of HeLa cells, apoptosis induced by TNF or staurosporin and the anti-apoptotic action of Bcl-2. Apoptosis was induced by combined action of respiratory inhibitors and uncouplers indicating possible pro-apoptotic action of reactive oxygen species (ROS) generated by mitochondria. Short-term incubation of HeLa cells with the mitochondrial inhibitors and 2-deoxyglucose followed by 24–48 h recovery resulted in massive apoptosis. Apoptosis correlated to transient (3–4 h) and limited (60–70%) depletion of ATP. More prolonged or more complete transient ATP depletion induced pronounced ne- Vol. 51 No. 2/2004 553–562 QUARTERLY * This work was presented in poster form at the 29 th Congress of the Federation of European Biochemi- cal Societies, Warsaw, Poland, 26 June–1 July 2004. . This work was supported by grants from The Ludwig Cancer Research Institute, RFBR-China 02-04-39005 and by RFBR Grants No. 04-04-49484 and 02-04-48843. ½ Correspondence to: B.V. Chernyak, A.N. Belozersky Institute, Moscow State University, 119899 Mos- cow, Russia; tel.: (70 95) 939 5549; fax: (70 95) 939 3181; e-mail: [email protected] Abbreviations: AMPK, AMP activated kinase; DMEM, Dulbecco’s modified Eagle’s medium; DNP, 2,4-dinitrophenol; DOG, 2-deoxyglucose; Drp1, dynamin-related protein; FCCP, trifluoromethoxy- carbonylcyanide phenylhydrazone; FCS, fetal calf serum; JNK, c-Jun N-terminal kinase; mTOR, mam- malian target of rapamycin; PTP, permeability transition pore; ROS, reactive oxygen species; STS, staurosporine; TNF, tumor necrosis factor a; zVADfmk, carbobenzoxy-Val-Ala-Asp-fluoromethyl ketone; YFP, yellow fluorescent protein.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Communication
Inhibition of mitochondrial bioenergetics: the effects on struc-ture of mitochondria in the cell and on apoptosis��
Konstantin G. Lyamzaev1, Denis S. Izyumov1, Armine V. Avetisyan1, Fuyu Yang2,
Olga Yu. Pletjushkina1 and Boris V. Chernyak1�
1A.N. Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow,Russia, 2Institute of Biphysics, Beijing, China
Received: 30 April, 2004; accepted: 07 May, 2004
Key words: mitochondria, oxidative phosphorylation, inhibitors, apoptosis
The effects of specific inhibitors of respiratory chain, FoF1ATP synthase anduncouplers of oxidative phosphorylation on survival of carcinoma HeLa cells and onthe structure of mitochondria in the cells were studied. The inhibitors of respiration(piericidin, antimycin, myxothiazol), the F1-component of ATP synthase (aurovertin)and uncouplers (DNP, FCCP) did not affect viability of HeLa cells, apoptosis inducedby TNF or staurosporin and the anti-apoptotic action of Bcl-2. Apoptosis was inducedby combined action of respiratory inhibitors and uncouplers indicating possiblepro-apoptotic action of reactive oxygen species (ROS) generated by mitochondria.Short-term incubation of HeLa cells with the mitochondrial inhibitors and2-deoxyglucose followed by 24–48 h recovery resulted in massive apoptosis.Apoptosis correlated to transient (3–4 h) and limited (60–70%) depletion of ATP.More prolonged or more complete transient ATP depletion induced pronounced ne-
Vol. 51 No. 2/2004
553–562
QUARTERLY
�This work was presented in poster form at the 29th Congress of the Federation of European Biochemi-cal Societies, Warsaw, Poland, 26 June–1 July 2004.�This work was supported by grants from The Ludwig Cancer Research Institute, RFBR-China02-04-39005 and by RFBR Grants No. 04-04-49484 and 02-04-48843.
�Correspondence to: B.V. Chernyak, A.N. Belozersky Institute, Moscow State University, 119899 Mos-cow, Russia; tel.: (70 95) 939 5549; fax: (70 95) 939 3181; e-mail: [email protected]
Abbreviations: AMPK, AMP activated kinase; DMEM, Dulbecco’s modified Eagle’s medium; DNP,2,4-dinitrophenol; DOG, 2-deoxyglucose; Drp1, dynamin-related protein; FCCP, trifluoromethoxy-carbonylcyanide phenylhydrazone; FCS, fetal calf serum; JNK, c-Jun N-terminal kinase; mTOR, mam-malian target of rapamycin; PTP, permeability transition pore; ROS, reactive oxygen species; STS,staurosporine; TNF, tumor necrosis factor �; zVADfmk, carbobenzoxy-Val-Ala-Asp-fluoromethylketone; YFP, yellow fluorescent protein.

crosis. The inhibitors of respiration and uncouplers caused fragmentation of tubularmitochondria and formation of small round bodies followed by swelling. These tran-sitions were not accompanied with release of cytochrome c into the cytosol and werefully reversible. The combined effect of respiratory inhibitors and uncouplers devel-oped more rapidly indicating possible involvement of ROS generated by mitochon-dria. More prolonged (48–72 h) incubation with this combination of inhibitorscaused clustering and degradation of mitochondria.
In the past decade the public view on mito-chondria has changed dramatically. The cleardescription of “power plants” supplying en-ergy (ATP) in the text-books was displacedwith the mysterious image of “Pandora’s box”determining the fate of a cell. In the previousparadigm involvement of mitochondria in pa-thology was limited to impairment of cellularenergetics in genetic diseases, hypoxic andtoxic insults. The recent explosion of experi-mental work demonstrated that various casesof apoptosis critically depend on the releaseof specific mitochondrial proteins into thecytosol (Zamzami & Kroemer, 2001). Themost important usually is the release ofcytochrome c (one of the components of therespiratory chain) catalyzing assembly of alarge cytosolic complex “apoptosome” in-volved in activation of caspases, the major ex-ecutioners of cell death (Adams & Cory,2002). In contrast to the basic bioenergeticprinciples, the mechanisms of sensing ofapoptotic signals by mitochondria are notwell understood.Almost simultaneously the traditional small
round-shaped organelles appeared to be arti-facts of preparations (thin slices) for electronmicroscopy originated from the variable anddynamic mitochondrial reticulum (Amchen-kova et al., 1988). These cable-like structureswere suggested to be an energy (trans-mem-brane electric potential) transporting systemof the cell (Skulachev, 2001). In agreementwith this idea fragmentation of the mitochon-drial reticulum (observed under variousstressful conditions) was attributed to asafety fuse preventing possible short-circuitcollapse of the whole network (Severina et al.,1998). Recently (Frank et al., 2002) it wasshown that fragmentation of mitochondria
not only accompanied apoptosis but was anecessary event preceding the release ofcytochrome c into the cytosol. The molecularmechanisms of mitochondrial fragmentationwere studied for a long time in yeast as amodel of organelle division (Rube & van derBliek, 2004) but application of these results tothe dynamics of mitochondria related to apo-ptosis needs very careful studies.The change in the paradigm of mitochon-
drial structure and function in the cell coin-cided with the change in the basic methods inthis field. The bioenergetics of mitochondriawas investigated using a wonderful arsenal ofspecific inhibitors. Starting with the pioneer-ing work by Lardy and colleagues (Lardy etal., 1964) the inhibitors (usually antibiotics)of almost every component of oxidativephosphorylation were found and the mecha-nisms of inhibition were deciphered. Laterstudies on chemiosmotic mechanisms becamepossible due to discoveries of differentionophores and membrane-permeable indica-tors (Skulachev, 1988). The new era in mito-chondriology coincided with the great suc-cesses in genetic engineering and genomics.Hundreds of new proteins involved in mito-chondria-related signaling were discovered ina very short time. The studies of their func-tions are based on powerful approaches usingknock-out, siRNA or dominant-negative con-structs. For a number of reasons these ap-proaches are very difficult to apply to mito-chondrial proteins involved in bioenergeticfunctioning. Even the nearly successful at-tempts, such as knock-out of cytochrome c (Liet al., 2000), did not help to fill the gapbetween the two areas of research onmitochondria in energy transformation andin apoptosis.
554 K.G. Lyamzaev and others 2004

An attempt for systematic application of theinhibitors of bioenergetic functions of mito-chondria (referred below as “mitochondrialinhibitors”) to studies on apoptosis ispresented here.
MATERIALS AND METHODS
Cell culture. Human carcinoma cells HeLaand green monkey kidney epithelial cells CV-1were grown in Dulbecco’s modified Eagle’smedium (DMEM) medium containing a highlevel of glucose (25 mM), gentamycin sulfate(0.08 mg/ml) and 10% fetal calf serum (FCS)(Gibco) at 37°C and 5% CO2. To monitor themitochondrial structure the cytochromeoxidase subunit VIII was expressed as a fu-sion with yellow fluorescent protein(Mito-YFP, Clontech, U.S.A.) in CV-1 cells.This cell line was a kind gift of Drs. F.K.Gioeva and A.A. Minin (Institute for ProteinResearch, Pushino, Moscow Region, Russia).HeLa cells were stained with mitochondrialspecific dye Mitotracker Green (MolecularProbes). Cells were incubated with inhibitorsat the following concentrations: 2 �Mrotenone (Sigma), 2 �M myxothiazol (Sigma),2 �M antimycin (Sigma), 0.4 mM2,4-dinitrophenol (DNP) (Sigma), 10 �Mtrifluoromethoxycarbonylcyanide phenyl-hydrazone (FCCP) (Sigma).ATP depletion and recovery. For ATP de-
pletion cells were incubated in DMEM with 5mM glucose, 5 mM 2-deoxyglucose (DOG),and alternatively oligomycin (5 �g/ml), 2 �Mmyxothiazol, or 10 �M FCCP for 3 h. Thenthe medium was changed to DMEM with 25mM glucose and the same inhibitors withoutDOG. Cell death was estimated after 24 and48 h.The ATP level was measured by the
luciferin–luciferase method with LKB re-agent according to the manufacturer instruc-tions. In these experiments bicinchoninicacid was used to measure the proteinconcentration.
Cell viability measurements. Cell viabilitywas determined by counting apoptotic and ne-crotic cells. After incubation, cells werestained with fluorescent dyes Hoechst 33342(1 �g/ml, 25 min) and propidium iodide(2 �g/ml, 5 min). The percentage of apoptosiswas calculated by counting cells with con-densed and fragmented nuclei. Necrotic cellswere detected by assessment of propidium io-dide penetrability. In some samples a smallfraction of cells with mixed staining(apoptotic nuclei and propidium iodide posi-tive) was detected. These cases were attrib-uted to apoptosis.
RESULTS
Mitochondrial inhibitors do not induce oraffect apoptosis: the rule and exceptions
Inhibitors of respiration (piericidin, anti-mycin, myxothiazol), ATP synthase (oligo-mycin, aurovertin) and uncouplers (DNP,FCCP) did not cause any loss in viability ofHeLa cells during 48 h in the traditional cellculture medium DMEM supplemented withfetal serum (10%) and glucose (25 mM)(Fig. 1). An important exception was found inexperiments with rotenone. This classic inhib-itor of Complex I of the respiratory chain in-duced cell cycle arrest and following apo-ptosis in HeLa at 2 �M, a concentration whichwas necessary for complete inhibition of un-coupled respiration of these cells in the cul-ture medium. The effect of rotenone was notrelated to inhibition of respiration or a spe-cific effect on Complex I, since another inhibi-tor with identical specificity, piericidin, didnot affect the cell cycle or kill the cells. The ef-fect of rotenone was probably targeted on thecytoskeleton, as it was reported earlier(Brinkley et al., 1974).Tumor necrosis factor (TNF) and the gen-
eral inhibitor of protein kinases stauro-sporine (STS) induced different apoptotic sig-naling pathways and both programs were
Vol. 51 Mitochondrial bioenergetics 555

completely executed and resulted in orderedDNA cleavage (“ladder” formation), fragmen-tation of nuclei and formation of apoptoticbodies in the presence of the mitochondrialinhibitors (Shchepina et al., 2002a; 2002b).The release of cytochrome c into the cytosol,which is a central mitochondria-related eventin both cases, also was not affected. The im-portant anti-apoptotic mitochondria-locatedprotein Bcl-2 inhibited the release ofcytochrome c in the presence of the inhibitorsas well as in control.One interesting exception was represented
by oligomycin. This inhibitor of the protonchannel (Fo) in mitochondrial FoF1ATPasewas found to inhibit TNF-induced release ofcytochrome c and apoptosis (Shchepina et al.,2002b). STS-induced apoptosis in HeLa wasnot affected by oligomycin (Shchepina et al.,
2002b), but in other models this pathway wasalso oligomycin-sensitive (Matsuyama et al.,1998). The effect of oligomycin was not re-lated to inhibition of oxidative phospho-rylation (since inhibitors of respiration anduncouplers were ineffective) or to hyper-polarization of the membrane (since depolar-ization with uncouplers did not relieve the in-hibition). Moreover, the effect was not di-rectly linked to inhibition of ATPase since an-other specific inhibitor aurovertin B did notaffect the release of cytochrome c andapoptosis. In contrast to oligomycin this in-hibitor was targeted to the catalytic (F1) com-ponent of the enzyme. The well-knownnon-mitochondrial target of oligomycinNa+/K+-ATPase of the plasma membrane wasnot responsible for the effects described. Itwas shown that a selective inhibitor of this en-zyme, ouabain did not inhibit apoptosis. In-terestingly, the selective effect of oligomycinon TNF-induced apoptosis correlated with theeffect of cyclosporine A, an inhibitor of thepermeability transition pore (PTP). Thisagent (in combination with trifluoperazinewhich enhance the effect) inhibited TNF-in-duced release of cytochrome c and apoptosiswhile it did not affect STS-induced apoptosisin HeLa. These data allow one to suggest thatoligomycin inhibited the concerned action ofFo and PTP in release of cytochrome c fromthe intermembrane space of mitochondriaduring TNF-induced apoptosis.The effective inhibition of oxidative phos-
phorylation with the inhibitors in use wasconfirmed with measurements of cellularrespiration (Shchepina et al., 2002a; 2002b),so these data indicated that glycolysis inHeLa cells can completely satisfy the de-mand in energy supply for survival of the cellor apoptotic cell death. It was suggested alsothat mitochondrial bioenergetic functioning(respiration, ATP synthesis, generation ofhigh membrane potential) is not critical forapoptosis in general and for the mitochon-dria-related events of the program in particu-lar.
556 K.G. Lyamzaev and others 2004
Figure 1. Cell death induced by mitochondrial in-hibitors in HeLa cells.
Cells were incubated for 24 h in the presence (A) or ab-sence (B) of 10% FCS with the following inhibitors:10 �M FCCP, 5 �g/ml oligomycin (oligo), 2 �Mmyxothiazol (myx) or their combinations as indicated.Apoptosis and necrosis were determined as describedin Materials and Methods.
To reveal possible specific induction of apop-tosis with the mitochondrial inhibitors wehave excluded serum from the culture me-dium suppressing growth factor dependentanti-apoptotic systems (Krasilnikov, 2000).Apoptosis was increased only slightly during24 h and more prolonged incubation resultedin significant necrosis. Significant apoptosiswas observed when a respiratory inhibitor(myxothiazol) was combined with uncoupler(FCCP) or with oligomycin in serum-free me-dium (Fig. 1B). This effect was probablycaused by excessive production of reactive ox-ygen species (ROS) in the initial segments ofthe respiratory chain. ROS generation byComplex I was suggested earlier to be impor-tant for cell death in various pathologies in-cluding ischemia/reperfusion, Parkinson dis-ease, etc. (Fleury et al., 2002).
Programmed cell death induced by energydeprivation
The high concentration of glucose used inthe cell culture medium was significantlyhigher than the physiological level. Presum-ably the limited supply of glycolytic sub-strates is especially important for rapidlygrowing solid tumors. To model these condi-tions we decreased the concentration of glu-cose to 5 mM and supplied the medium with 5mM 2-deoxyglucose (DOG), a non-metabo-lized analog. These conditions did not causeany decrease in viability of HeLa cells indicat-ing the high capacity of oxidative phospho-rylation. As expected, mitochondrial inhibi-tors caused almost complete necrotic celldeath during 24 h in this model. A generalcaspase inhibitor carbobenzoxy-Val-Ala-Asp-fluoromethyl ketone (zVADfmk) or over-expression of Bcl-2 did not inhibit the cell kill-ing indicating that the necrotic morphologydid not appear as a result of incomplete exe-cution of apoptosis. However, if combinedtreatment with DOG and mitochondrial inhib-itors for 3 h was followed by 24 h recovery inhigh-glucose medium (without DOG) signifi-
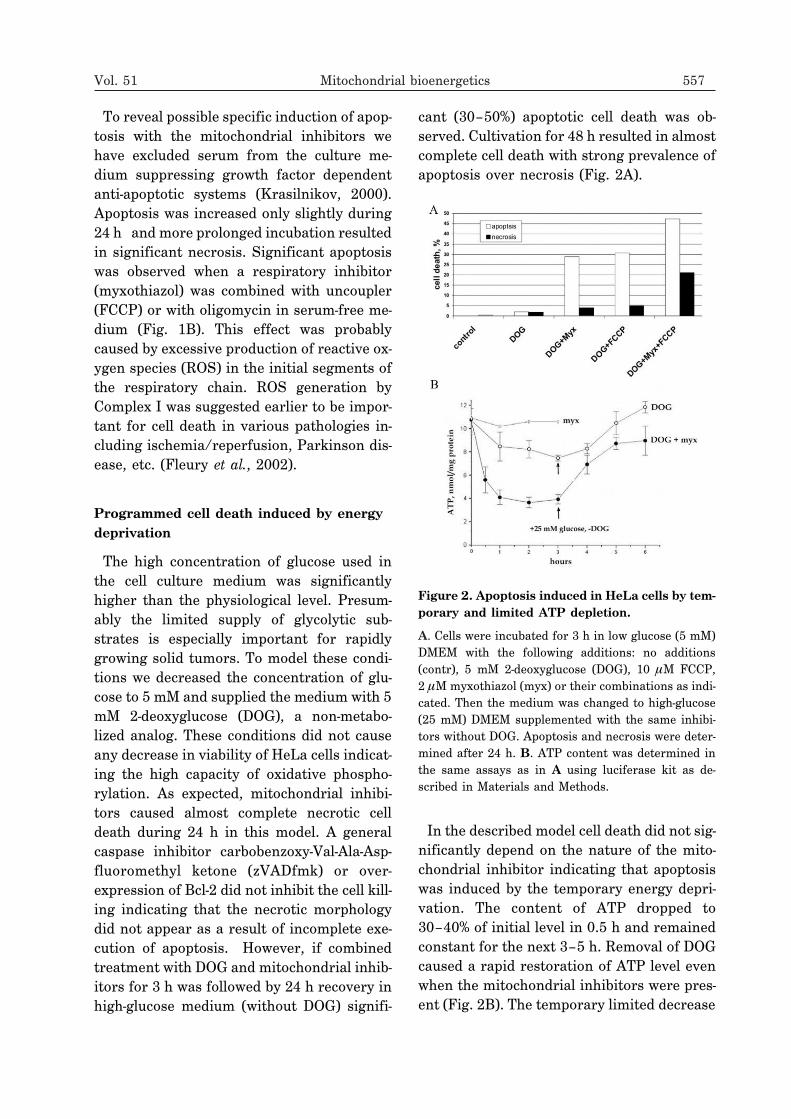
cant (30–50%) apoptotic cell death was ob-served. Cultivation for 48 h resulted in almostcomplete cell death with strong prevalence ofapoptosis over necrosis (Fig. 2A).
In the described model cell death did not sig-nificantly depend on the nature of the mito-chondrial inhibitor indicating that apoptosiswas induced by the temporary energy depri-vation. The content of ATP dropped to30–40% of initial level in 0.5 h and remainedconstant for the next 3–5 h. Removal of DOGcaused a rapid restoration of ATP level evenwhen the mitochondrial inhibitors were pres-ent (Fig. 2B). The temporary limited decrease
Vol. 51 Mitochondrial bioenergetics 557
Figure 2. Apoptosis induced in HeLa cells by tem-porary and limited ATP depletion.
A. Cells were incubated for 3 h in low glucose (5 mM)DMEM with the following additions: no additions(contr), 5 mM 2-deoxyglucose (DOG), 10 �M FCCP,2 �M myxothiazol (myx) or their combinations as indi-cated. Then the medium was changed to high-glucose(25 mM) DMEM supplemented with the same inhibi-tors without DOG. Apoptosis and necrosis were deter-mined after 24 h. B. ATP content was determined inthe same assays as in A using luciferase kit as de-scribed in Materials and Methods.

in ATP appears to be insufficient to cause sig-nificant damage to the cellular structures.Even the lowest concentration of ATP re-mained in the millimolar range, which is wellabove the saturation of the majority ofATP-consuming systems indicating the exis-tence of specific ATP-meter(s). In this modelcombined action of myxothiazol and anuncoupler was significantly stronger (Fig. 2A)in agreement with the results obtained in se-rum-free glucose-rich medium (Fig. 1B). Itcould be suggested that generation of ROS bythe respiratory chain improved apoptotic sig-naling triggered by temporary ATP depletion.When the ATP depletion procedure was pro-
longed to 5 h the following recovery still re-stored the ATP level but resulted mostly innecrotic cell death after 24 or 48 h. A similarswitch to necrosis was observed when ATPdepletion was improved by more complete in-hibition of glycolysis. The mitochondrial in-hibitors in combination with DOG added tothe medium depleted of glucose caused arapid fall in cellular ATP to less then 10% ofinitial level. After 3 h the medium waschanged to complete glucose-rich DMEM andcellular ATP was restored almost completely;the following 24 or 48 h cultivation resultedin massive necrotic death (not shown). Thesedata indicate that temporary ATP depletioncan be a trigger of programmed cell deathwith both necrotic and apoptotic features.Apoptosis induced by ATP depletion was
prevented by inhibition of caspases withzVADfmk or by overexpression of Bcl-2.Translocation of Bax from the cytosol to mito-chondria and release of cytochrome c frommitochondria into the cytosol were observedin this model resembling the typical stress-in-duced apoptosis. Interestingly, zVADfmkstimulated necrosis under the same condi-tions suggesting that caspases not only cata-lyzed apoptosis but also inhibited necrosis. Incontrast to apoptosis, the signaling resultingin necrosis remained poorly characterized.An example of necrosis induced by rotenoneand glucose deprivation in myogenic cells was
recently found to be dependent on activationof the stress-activated protein kinase (JNK)(Gabai et al., 2000; Yaglom et al., 2003). InHeLa cells neither apoptosis nor necrosis wassensitive to inhibitors of JNK or p38 (anotherstress-activated protein kinase). The natureof the putative ATP-meter(s) also remainedmysterious. The candidate sensors includesmammalian target of rapamycin (mTOR)(Dennis et al., 2001), a protein kinase with ex-ceptionally high Km (ATP) and AMPK (Rutteret al., 2003), a kinase that is allosterically acti-vated by AMP. The both kinases are involvedin regulation of gene expression in responseto nutrient starvation but their role in induc-tion of apoptosis is purely speculative.
Inhibition of the bioenergetic functionscauses morphological changes and degrada-tion of mitochondria
Mitochondrial inhibitors in the presence ofglucose induced dramatic changes in mito-chondrial structure independently of ATP de-pletion or apoptotic events. The details of thisprocess were visualized using CV-1 cells ex-pressing yellow fluorescent protein (YFP)fused to subunit VIII of cytochrome oxidase(Fig. 3). It was shown that rotenone,myxothiazol or the uncouplers induced fis-sion of the long mitochondrial filaments aftera significant lag phase (2–3 h). At the nextstep the short fragments were transformed tosmall round bodies. This process included fur-ther fission combined with the change of theshape of the organelles. The following swell-ing of the mitochondria was clearly visible inthe case of uncouplers. A similar sequence ofevents was observed in HeLa cells where mi-tochondria were stained with MitotrackerGreen (not shown). The wash out of theuncouplers caused slow (24 h) complete resto-ration of the original mitochondrial network(Fig. 3A). Exceptionally rapid and intensechanges were induced in mitochondria ofCV-1 by oligomycin. This effect was mostly re-lated to inhibition of Na+/K+-ATPase of the
558 K.G. Lyamzaev and others 2004

plasma membrane (since ouabain caused sim-ilar changes) and was not observed in HeLa.The most rapid fission and the following
transitions of the mitochondrial networkwere induced by combined treatment with re-spiratory inhibitors and uncouplers indicat-ing a possible role of ROS produced by the re-
spiratory chain (Fig. 3B). This suggestion isin good agreement with the observations ofmitochondrial transitions induced by hydro-gen peroxide (0.1–0.4 mM) in the same celllines. Similar steps of mitochondrial fragmen-tation were described (Skulachev et al., 2004)but the initial lag-phase was significantly
Vol. 51 Mitochondrial bioenergetics 559
Figure 3. Fragmentation of mitochondria in CV-1 cells incubated with mitochondrial inhibitors.
A. Cells were incubated with 0.4 mM DNP for 0 h (a), 2 h (b) and 6 h (c). After incubation with DNP (6 h) cells werewashed and cultivated for 24 h (d). Bar 10 �m. B. Cells were incubated with 2 �M myxothiazol (myx) for 6 h (a), orwith 0.4 mM DNP and 2 �M myxothiazol (DNP + myx) for 45 min (b) or 6 h (c). Bar 10 �m.
A
B
shorter. In both models no visible signs ofapoptosis were observed until the final stepsof mitochondrial transitions. The release ofcytochrome c from mitochondria was notobserved even when mitochondria wereswollen.Similar changes in morphology of mitochon-
dria were described in various models ofapoptosis (Desagher & Martinou, 2000). A de-tailed study of staurosporine-induced apo-ptosis in CV-1 revealed a clear time gap be-tween fission of mitochondria and release ofcytochrome c (Skulachev et al., 2004). Inter-
estingly both events were well synchronizedin mitochondrial population of a single celland proceeded very rapidly in comparisonwith the lag phase. Recently, it was foundthat fission of mitochondria during apoptosisdepended on translocation of dynamin-re-lated protein (Drp1) from the cytosol to somelocal sites at the surface of mitochondria(Frank et al., 2002). Bax targeted the samesites and Drp1 dominant-negative mutantprotein not only prevented fission but also in-
hibited apoptosis (Karbowski et al., 2002).There is no evidence that mitochondrial tran-sitions induced by the inhibitors or by hydro-gen peroxide included the same molecularmechanisms; however, it appears that fissionof mitochondria can be necessary but is notsufficient for apoptosis.At the final steps of the treatment with
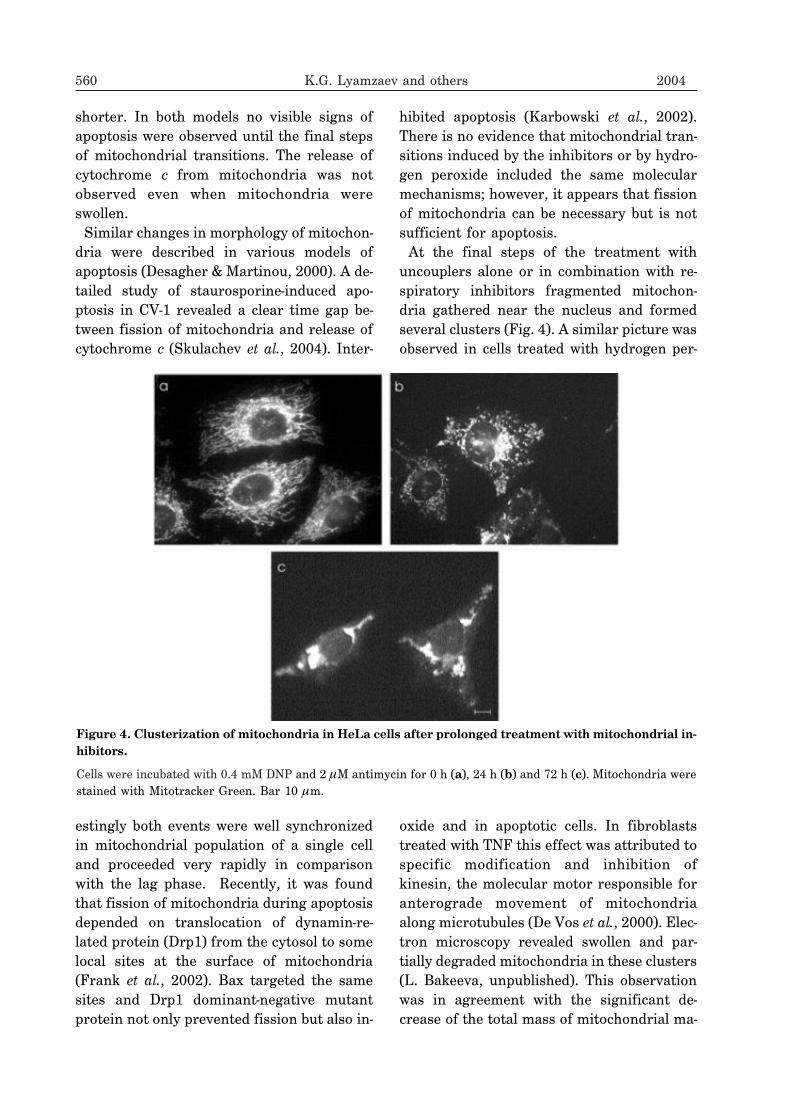
uncouplers alone or in combination with re-spiratory inhibitors fragmented mitochon-dria gathered near the nucleus and formedseveral clusters (Fig. 4). A similar picture wasobserved in cells treated with hydrogen per-
oxide and in apoptotic cells. In fibroblaststreated with TNF this effect was attributed tospecific modification and inhibition ofkinesin, the molecular motor responsible foranterograde movement of mitochondriaalong microtubules (De Vos et al., 2000). Elec-tron microscopy revealed swollen and par-tially degraded mitochondria in these clusters(L. Bakeeva, unpublished). This observationwas in agreement with the significant de-crease of the total mass of mitochondrial ma-
560 K.G. Lyamzaev and others 2004
Figure 4. Clusterization of mitochondria in HeLa cells after prolonged treatment with mitochondrial in-hibitors.
Cells were incubated with 0.4 mM DNP and 2 �M antimycin for 0 h (a), 24 h (b) and 72 h (c). Mitochondria werestained with Mitotracker Green. Bar 10 �m.

terial and content of specific mitochondrialproteins (not shown). Selective elimination ofmitochondria from apoptotic cells was re-cently described (Xue et al., 2001) and the ma-jor role of autophagy in this process was sug-gested. We did not observe accumulation ofautophagosomes in our model, so probablythe mechanism of depletion of the cells of mi-tochondria was different. It should bestressed that in apoptotic models inhibitionof caspases was necessary to prevent thecellular collapse and to observe elimination ofmitochondria.When HeLa cells were treated for 48–72 h
with uncouplers in combination withantimycin or myxothiazol a significant(50–70%) fraction of the cells died but the restof the population was viable, without anysigns of apoptosis and with very low mito-chondrial content (Fig. 4). The cell death wasnot related to energy deprivation, since thesame respiratory inhibitors caused completecessation of oxidative phosphorylation butdid not cause cell killing. Complete depolar-ization of the mitochondrial membrane and(or) hyperproduction of ROS were the mostprobable reasons for cell death. The viablecells depleted of mitochondria probably had aselective advantage due to low content ofpro-apoptotic mitochondrial proteins andelimination of the major source of ROS pro-duction. Thus both the induction of an un-known mechanism(s) of mitochondrial elimi-nation and the selective pressure could be re-sponsible for the observed phenomena. Itcould be suggested that similar processes areresponsible for depletion of mitochondria insome rapidly growing tumors (Cuezva et al.,2002).
The authors are grateful to Professors V.P.Skulachev and Y.M. Vasiliev for support andhelpful discussions, Drs. E.K. Fetisova,L.V. Domnina and O.Yu. Ivanova for help insome experiments, Drs. L.E. Bakeeva andV.B. Saprunova for electron microscopic anal-
ysis, Drs. F.K. Gioeva and A.A. Minin forkind gift of CV-1 cells expressing Mito-YFP.
R E F E R E N C E S
Adams JM, Cory S. (2002) Curr Opin Cell Biol.;14: 715–20.
Amchenkova AA, Bakeeva LE, Chentsov YS,Skulachev VP, Zorov DB. (1988) J Cell Biol.;107: 481–95.
Brinkley BR, Barham SS, Barranco SC, FullerGM. (1974) Exp Cell Res.; 85: 41–6.
Cuezva JM, Krajewska M, de Heredia ML,Krajewski S, Santamaria G, Kim H, ZapataJM, Marusawa H, Chamorro M, Reed JC.(2002) Cancer Res.; 62: 6674–81.
Dennis PB, Jaeschke A, Saitoh M, Fowler B,Kozma SC, Thomas G. (2001) Science.; 294:1102–5.
Desagher S, Martinou JC. (2000) Trends CellBiol.; 10: 369–77.
De Vos K, Severin F, Van Herreweghe F,Vancompernolle K, Goossens V, Hyman A,Grooten J. (2000) J Cell Biol.; 149:1207–14.
Fleury C, Mignotte B, Vayssiere JL. (2002)Biochimie.; 84: 131–41.
Frank S, Gaume B, Bergmann-Leitner ES,Leitner WW, Robert EG, Catez F, Smith CL,Youle RJ. (2001) Dev Cell.; 1: 515–25.
Gabai VL, Meriin AB, Yaglom JA, Wei JY,Mosser DD, Sherman MY. (2000) J BiolChem.; 275: 38088–94.
Karbowski M, Lee YJ, Gaume B, Jeong SY,Frank S, Nechushtan A, Santel A, Fuller M,Smith CL, Youle RJ. (2002) J Cell Biol.;159: 931–8.
Krasilnikov MA. (2000) Biochemistry (Mosc).;65: 59–67.
Lardy HA, Connely JL, Johnson D. (1964) Bio-chemistry.; 19: 1961–8.
Vol. 51 Mitochondrial bioenergetics 561

Li K, Li Y, Shelton JM, Richardson JA,Spencer E, Chen ZJ, Wang X, Williams RS.(2000) Cell.; 101: 389–99.
Matsuyama S, Xu Q, Velours J, Reed JC.(1998) Mol Cell.; 1: 327–36.
Rube DA, van der Bliek AM. (2004) Mol CellBiochem.; 256-257: 331–9.
Rutter GA, Da Silva Xavier G, Leclerc I. (2003)Biochem J.; 375: 1–16.
Severina II, Skulachev VP, Zorov DB. (1988) JCell Biol.; 107: 497–501.
Shchepina LA, Pletjushkina OYu, Avetisyan AV,Bakeeva LE, Fetisova EK, Izyumov DS,Saprunova VB, Vyssokikh MYu, ChernyakBV, Skulachev VP. (2002a) Oncogene.; 21:8149–57.
Shchepina LA, Popova EN, Pletjushkina OYu,Chernyak BV. (2002b) Biochemistry (Mosc).;67: 222–6.
Skulachev VP. (1988) Membrane bioenergetics.Springer-Verlag, New York.
Skulachev VP. (2001) Trends Biochem Sci.; 26:23–9.
Xue L, Fletcher GC, Tolkovsky AM. (2001) CurrBiol.; 11: 361–5.
Yaglom YA, Ekhterae D, Gabai VL, ShermanMY. (2003) J Biol Chem.; 278: 50483–96.
Zamzami N, Kroemer G. (2001) Nat Rev MolCell Biol.; 2: 67–71.
562 K.G. Lyamzaev and others 2004
Related Documents