332 Current Cancer Drug Targets, 2010, 10, 332-342 1568-0096/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd. Inhibition of c-Met with the Specific Small Molecule Tyrosine Kinase In- hibitor SU11274 Decreases Growth and Metastasis Formation of Experi- mental Human Melanoma I. Kenessey 1,2 , M. Keszthelyi 3 , Z. Krámer 1,2 , J. Berta 3,8 , A. Ádám 1 , J. Dobos 2 , M. Mildner 4 , B. Flachner 5 , S. Cseh 5 , G. Barna 6 , B. Szokol 7 , L. rfi 7 , G. Kéri 7 , B. Döme 3,8 , W. Klepetko 8 , J. Tímár 1,2 and J. Tóvári* ,2,3 1 2 nd Institute of Pathology, Semmelweis University, Budapest, Hungary; 2 Department of Experimental Pharmacology, National Institute of Oncology, Budapest, Hungary; 3 Department of Tumor Biology, National Korányi Institute of Pul- monology, Budapest, Hungary; 4 Department of Dermatology, Medical University of Vienna, Austria; 5 TargetEx Ltd., Dunakeszi, Hungary; 6 1 st Institute of Pathology and Experimental Cancer Research, Semmelweis University, Budapest, Hungary; 7 Vichem Chemie Ltd., Budapest, Hungary; 8 Department of Thoracic Surgery, Medical University of Vienna, Vienna, Austria Abstract: The hepatocyte growth factor/scatter factor (HGF/SF) tyrosine kinase (TK) receptor c-Met plays a crucial role in the development of the invasive phenotype of tumors and thus represents an attractive candidate for targeted therapies in a variety of malignancies, including human malignant melanoma (MM). In contrast to what has been shown previously, we were not able to detect any genetic alterations, either in the juxtamembrane- or in the TK-domain of c-Met, in the stud- ied MM cell lines. Nevertheless, c-Met was constitutively active in these cell lines without exogenous HGF/SF stimula- tion. The active receptor was localized to the adhesion sites of the cells. Addition of the c-Met TK inhibitor SU11274 spe- cifically decreased the phosphotyrosine signal at the focal adhesion sites, which was accompanied by a decrease in cell proliferation as well as an increase in apoptotic cells. In addition, non-apoptotic concentrations of SU11274 significantly reduced the in vitro migratory capacity of MM cells in the modified Boyden-chamber assay. Administration of SU11274 significantly decreased primary tumor growth as well as the capacity for liver colony formation of MM cells in SCID mice. Our study provides the first evidence for an in vivo antitumor activity of SU11274 in a human melanoma xenograft model, and suggests c-Met as a valid target for the therapy of MM. Consequently, SU11274 treatment might represent a useful strategy for controlling melanoma progression and metastasis in patients with MM. Keywords: c-Met, tyrosine kinase inhibitor, SU11274, metastasis, human malignant melanoma. INTRODUCTION Aberrant activation of tyrosine kinase (TK) pathways was demonstrated in several common solid tumors, resulting in increased proliferation, survival, invasiveness and metas- tasis [1, 2]. The c-Met oncogene, encoding the receptor-TK for hepatocyte growth factor/scatter factor (HGF/SF) [3], controls genetic programs leading to cell growth, invasion and protection from apoptosis [4, 5]. This heterodimer recep- tor (190 kDa) consists of an - and a -polypeptide chain with disulfide-bounds [6]. The intracellular part of the 140 kDa beta-chain contains several tyrosine residues (Tyr 1003, 1230, 1234, 1235, 1349, 1356 ) for phosphorylation [7]. The major clus- ter is the Tyr 1230/1234/1235 , localized in the signal-regulating ATP-binding site [7]. The deregulated activation of c-Met is crucial not only for the acquisition of tumorigenic properties but also for achieving an invasive phenotype [8]. Aberrant c- Met expression (usually overexpression) has been described in many types of solid tumors (e.g. gastric, head and neck, lung and hereditary papillary renal cancers) and has been shown to correlate with poor prognosis in these malignancies [9]. Overexpression of c-Met is frequently due to gene *Address correspondence to this author at the Department of Experimental Pharmacology, National Institute of Oncology, Ráth György u. 7-9, Buda- pest, H-1122, Hungary; Tel: +36-1-224-8786; Fax: +36-1-224-8706; E-mail: [email protected], [email protected] amplification (such as in uveal melanoma and colorectal-, non small cell lung- and gastric cancers [10-12]), mutations in the TK (hereditary papillary renal cancer [13]) or in the juxtamembrane domains (non-small cell lung cancer [14]) or formation of the TRP-Met fusion gene (gastric cancer [15]). Therapeutic inhibition of the tyrosine kinase activity by small molecule substrates is a possible approach to interfer- ing with such an aberrant activation of TK-type oncogenes, including c-Met. Small molecule tyrosine kinase inhibitors (TKI) bind to the ATP cleft of the TK receptor and selec- tively block growth factor-stimulated signal activation via dimerization and autophosphorylation [16]. Inhibition of phosphorylation leads to depletion of the activated down- stream effectors resulting in attenuation of tumor progres- sion. Malignant melanoma (MM), whose incidence is increas- ing worldwide [17], is resistant to common cytotoxic thera- pies. Improvement in survival can only be achieved with early detection and complete surgical removal. However, MMs have a potential to form organ metastases in the very early phase of primary growth. For this reason, a better un- derstanding of the mechanisms involved in their progression is urgently needed. c-Met is present on normal epithelial cells and melano- cytes as well, and its ligand is expressed by mesenchymal cells of the skin [18]. Moreover, melanoma cells produce

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

332 Current Cancer Drug Targets, 2010, 10, 332-342
1568-0096/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd.
Inhibition of c-Met with the Specific Small Molecule Tyrosine Kinase In-
hibitor SU11274 Decreases Growth and Metastasis Formation of Experi-
mental Human Melanoma
I. Kenessey1,2, M. Keszthelyi3, Z. Krámer1,2, J. Berta3,8, A. Ádám1, J. Dobos2, M. Mildner4, B. Flachner5, S. Cseh5, G. Barna6, B. Szokol7, L. rfi7, G. Kéri7, B. Döme3,8, W. Klepetko8, J. Tímár1,2 and J. Tóvári*,2,3
12
nd Institute of Pathology, Semmelweis University, Budapest, Hungary;
2Department of Experimental Pharmacology,
National Institute of Oncology, Budapest, Hungary; 3Department of Tumor Biology, National Korányi Institute of Pul-
monology, Budapest, Hungary; 4Department of Dermatology, Medical University of Vienna, Austria;
5TargetEx Ltd.,
Dunakeszi, Hungary; 61
st Institute of Pathology and Experimental Cancer Research, Semmelweis University, Budapest,
Hungary; 7Vichem Chemie Ltd., Budapest, Hungary;
8Department of Thoracic Surgery, Medical University of Vienna,
Vienna, Austria
Abstract: The hepatocyte growth factor/scatter factor (HGF/SF) tyrosine kinase (TK) receptor c-Met plays a crucial role in the development of the invasive phenotype of tumors and thus represents an attractive candidate for targeted therapies in a variety of malignancies, including human malignant melanoma (MM). In contrast to what has been shown previously, we were not able to detect any genetic alterations, either in the juxtamembrane- or in the TK-domain of c-Met, in the stud-ied MM cell lines. Nevertheless, c-Met was constitutively active in these cell lines without exogenous HGF/SF stimula-tion. The active receptor was localized to the adhesion sites of the cells. Addition of the c-Met TK inhibitor SU11274 spe-cifically decreased the phosphotyrosine signal at the focal adhesion sites, which was accompanied by a decrease in cell proliferation as well as an increase in apoptotic cells. In addition, non-apoptotic concentrations of SU11274 significantly reduced the in vitro migratory capacity of MM cells in the modified Boyden-chamber assay. Administration of SU11274 significantly decreased primary tumor growth as well as the capacity for liver colony formation of MM cells in SCID mice. Our study provides the first evidence for an in vivo antitumor activity of SU11274 in a human melanoma xenograft model, and suggests c-Met as a valid target for the therapy of MM. Consequently, SU11274 treatment might represent a useful strategy for controlling melanoma progression and metastasis in patients with MM.
Keywords: c-Met, tyrosine kinase inhibitor, SU11274, metastasis, human malignant melanoma.
INTRODUCTION
Aberrant activation of tyrosine kinase (TK) pathways was demonstrated in several common solid tumors, resulting in increased proliferation, survival, invasiveness and metas-tasis [1, 2]. The c-Met oncogene, encoding the receptor-TK for hepatocyte growth factor/scatter factor (HGF/SF) [3], controls genetic programs leading to cell growth, invasion and protection from apoptosis [4, 5]. This heterodimer recep-tor (190 kDa) consists of an - and a -polypeptide chain with disulfide-bounds [6]. The intracellular part of the 140 kDa beta-chain contains several tyrosine residues (Tyr1003,
1230, 1234, 1235, 1349, 1356) for phosphorylation [7]. The major clus-ter is the Tyr1230/1234/1235, localized in the signal-regulating ATP-binding site [7]. The deregulated activation of c-Met is crucial not only for the acquisition of tumorigenic properties but also for achieving an invasive phenotype [8]. Aberrant c-Met expression (usually overexpression) has been described in many types of solid tumors (e.g. gastric, head and neck, lung and hereditary papillary renal cancers) and has been shown to correlate with poor prognosis in these malignancies [9]. Overexpression of c-Met is frequently due to gene
*Address correspondence to this author at the Department of Experimental Pharmacology, National Institute of Oncology, Ráth György u. 7-9, Buda-pest, H-1122, Hungary; Tel: +36-1-224-8786; Fax: +36-1-224-8706; E-mail: [email protected], [email protected]
amplification (such as in uveal melanoma and colorectal-, non small cell lung- and gastric cancers [10-12]), mutations in the TK (hereditary papillary renal cancer [13]) or in the juxtamembrane domains (non-small cell lung cancer [14]) or formation of the TRP-Met fusion gene (gastric cancer [15]).
Therapeutic inhibition of the tyrosine kinase activity by small molecule substrates is a possible approach to interfer-ing with such an aberrant activation of TK-type oncogenes, including c-Met. Small molecule tyrosine kinase inhibitors (TKI) bind to the ATP cleft of the TK receptor and selec-tively block growth factor-stimulated signal activation via dimerization and autophosphorylation [16]. Inhibition of phosphorylation leads to depletion of the activated down-stream effectors resulting in attenuation of tumor progres-sion.
Malignant melanoma (MM), whose incidence is increas-ing worldwide [17], is resistant to common cytotoxic thera-pies. Improvement in survival can only be achieved with early detection and complete surgical removal. However, MMs have a potential to form organ metastases in the very early phase of primary growth. For this reason, a better un-derstanding of the mechanisms involved in their progression is urgently needed.
c-Met is present on normal epithelial cells and melano-cytes as well, and its ligand is expressed by mesenchymal cells of the skin [18]. Moreover, melanoma cells produce

c-Met TKI Decreases Human Melanoma Metastasis in Mice Current Cancer Drug Targets, 2010, Vol. 10, No. 3 333
HGF themselves, and the correlation of c-Met overexpres-sion with the growth of tumor cells suggests that the c-Met/HGF pathway has a pivotal role in melanoma progres-sion in autocrine and paracrine manners as well [19, 20]. c-Met is regulated by MITF (microphthalmia-associated tran-scription factor), a linage-specific transcription factor of melanocytes and melanoma cells [21]. Overexpression of MET in human MM was found both at mRNA level in ge-nomic studies [22, 23] and at protein level in pathological samples [24]. c-Met mutation was described in human MM cells in the juxtamembrane domain [25], but not at the TK domain [26]. These genetic data suggest that MET is a poten-tial target for molecular therapy in human MM. This is cor-roborated by the recent in vitro observation by Puri and col-leagues that the novel c-Met specific tyrosine kinase inhibi-tor SU11274 inhibited the proliferation and differentiation of human melanoma cells and increased their apoptosis at a micromolar concentration [25].
In the current experimental study, we examined the TK status of the c-Met oncogene in several human melanoma cell lines at both gene and protein levels. Moreover, we stud-ied the effect of the specific c-Met TK inhibitor SU11274 on c-Met phosphorylation and on proliferation, apoptosis and migration of human melanoma cells in vitro as well as on the growth and colonization of human melanoma xenografts in vivo.
MATERIAL AND METHODS
Cell Lines and Culture Conditions
The HT168 and HT168-M1 human melanoma lines are derivatives from the A2058 cell line [27]. The HT199 mela-noma line was established by our group. WM35, WM983A and WM983B melanoma cell lines were gifts from M. Her-lyn (Wistar Institute, Philadelphia, PA). The M24met mela-noma line was kindly provided by B. M. Mueller (Scripps Research Institute, La Jolla, CA). Human melanoma cell lines were grown in medium RPMI-1640, while the A431 epidermoid carcinoma cells (which served as a positive con-trol for c-Met) were cultured in DMEM containing 4500 mg/l glucose (Sigma Chemical Co., St. Louis, MO), supple-mented with 5% fetal bovine serum (Sigma) and 1% penicil-lin-streptomycin (Sigma) at 37°C in a humidified atmos-phere of 5% CO2. All of the 8 melanoma cell lines were used for sequence analysis, and HT168-M1, HT199, WM983B and M24met were used for in vitro experiments.
Immunocytochemistry
Melanoma cells were fixed in paraformaldehyde for 10 min and then permeabilized with 0.1% Triton X-100 (Sigma) in phosphate-buffered saline (PBS) for 1 min. After washing in PBS for 3x5 min, slides were blocked with 1% bovine serum albumine (BSA; Sigma) and goat serum (9:1) for 30 min at room temperature, and incubated with the following primary antibodies: mouse monoclonal antibody recognizing the extracellular part of c-Met protein (1:50 in PBS, clone DL-21, Upstate, Charlottesville, VA), rabbit polyclonal anti-body against the intracellular domain of c-Met (1:50 in PBS, C-12, Santa Cruz Biotechnology, CA) or rabbit anti-c-Met[pYpYpY1230/1234/1235], and phosphospecific antibody
(1:20 in PBS, Biosource, Nivelles, Belgium). Cells were washed in PBS for 3x10 min, and then incubated with biotin-conjugated anti-mouse or anti-rabbit IgGs (Amersham, Buckinghamshire, UK) for 40 min at 37ºC (dilution 1:100). After washing, c-Met protein was visualized by streptavidin-FITC (dilution 1:100, Vector Laboratories, Burlingame, CA). Negative controls were prepared by replacing the pri-mary antibody with isotype-matched non-immune IgG (Sigma). Cell nuclei were stained with propidium iodide (PI, Sigma). Slides were covered with Vectashield (Vector Labo-ratories), and cells were examined with a confocal micro-scope (Eclipse C1 Plus, Nikon Optoteam, Vienna, Austria).
On the microscopic sections the FITC-labeled fluorescent plaques by phosphospecific anti-c-Met[pYpYpY1230/1234/1235] were counted manually.
Tyrosine Kinase Inhibitor
The c-Met-specific inhibitor [28], SU11274 [(3Z)-N- (3-chlorophenyl)-3-({3,5-dimethyl-4-[(4-methylpiperazin-1-yl)carbonyl]-1H-pyrrol-2-yl}methylene)-N-methyl-2-oxo-2, 3-dihydro-1H-indole-5-sulfonamid] (Pfizer Inc., San Diego, CA; synthesized by Vichem Chemie Ltd., Budapest, Hun-gary) was suspended in DMSO (Sigma) and used at 0.1-10 μM concentrations in 0.5% DMSO-RPMI for in vitro studies and 0.5 mg/kg for in vivo metastasis assays.
RNA Isolation and cDNA Synthesis
Total RNA was prepared from human melanoma cell lines with different genetic background using RNeasy Mini Kit (Qiagen, Hilden, Germany) or the TRI Reagent (Sigma) according to the manufacturers’ instructions.
Reverse transcription reaction mixture was set up by add-ing 1 μl dNTP mix (10 mM each, Finnzyme, Espoo, Finland) and 1 μl of random primer-oligo(dT) mix (final concentra-tion 2.5 μM each) to 1 μg (in 8 μl DEPC-treated water) of the isolated total RNA. After incubating at 70°C for 10 min the following components were added: 2 μl of 10x M-MLV Reverse Transcriptase Buffer (Sigma), 1 μl of M-MLV Re-verse Transcriptase (200 U/μl, Sigma), 0.5 μl RNase Inhibi-tor (40 U/μl, Promega, Madison, WI), and 6.5 μl DEPC-treated water for a final reaction volume of 20 μl. The reac-tion was run at 37°C for 50 min, and then the enzyme was stopped by incubating at 85°C for 10 min. The efficiency and quality of the reverse transcription of the different sam-ples was checked by a PCR for -actin.
Verification of the Expression of c-Met by PCR and Se-quencing
Expression of c-Met was verified with PCR and DNA sequence analysis of the isolated amplicons. Primers (listed in Table 1) were designed by the Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) or by the Array Designer software (PREMIER Biosoft International, Palo Alto, CA) using the GeneBank RefSeq (Acc: NM_000245). The specificity of the chosen primer pairs was checked by BLAST (http://www.ncbi.nlm.nih.gov/ BLAST/) on GenBank sequences. The PCR was carried out on a Palm-Cycler (Corbett Research, Sydney, Australia) thermal cycler with the following parameters: 94°C 3 min,

334 Current Cancer Drug Targets, 2010, Vol. 10, No. 3 Kenessey et al.
with the following parameters: 94°C 3 min, [94°C 1 min, 59°C 1:10 min, 72°C 1:20 min]x35, 72°C 5 min. The reac-tion mixture contained the following components in a total of 25 μl reaction volume in each tube: 2 μl of the reverse transcription reaction mixture as template (or water for no-template controls), 2.5 μl of 10x PCR Buffer (final Mg2+ concentration 1.5 mM, DyNAzyme, Finnzyme), 2 μl dNTP mix (2.5 mM each), 2.5 μl each of forward and reverse prim-ers (1 μM final concentration for each), 0.4 μl of DNA Po-lymerase (DyNAzyme), and distilled water up to the final reaction volume. The PCR products were electrophoresed on 2% agarose gel, stained with EtBr, and isolated with High Pure PCR Product Purification Kit (Roche, Mannheim, Germany) or MEGA-Spin Agarose Gel Extraction Kit (In-tron Biotechnology Inc., Korea) according to the manufac-turers’ protocol. PCR-based dideoxy dye-terminator DNA sequencing was performed from both directions and the se-quence was analyzed on an ABI PRISM 3100 Genetic Ana-lyzer (Applied Biosystems, Foster City, CA).
Table 1. c-Met-Specific Primers Used in the Study
Primer´s name sequence (5’-3’)
c-Met-1263+20F
c-Met-1568-20R
GATCTGCCATGTGTGCATTC
GACCCTCTGATGTCCCAAGA
c-Met-2182+20F
c-Met-2404-22R
ATACGGTCCTATGGCTGGTG
ACGGTAACTGAAGATGCTTGTC
c-Met-PTK1F
c-Met-PTK1R
GAATCTGCCTGCGAAGTGAAG
ATCAGCAACCTTGACTGTGAATT
c-Met-3340+20F
c-Met-3867-20R
CCACATTGACCTCAGTGCTC
CTGGCAAGACCAAAATCAGC
c-Met-3558+20F
c-Met-4093-21R
TTCTGACCGAGGGAATCATC
GAGTCTTCTCCCTTGCAACAA
c-Met-3793+20F
c-Met-4284-20R
CAGAGACTTGGCTGCAAGAA
GGATACGGAGCGACACATTT
Flow Cytometric Measurement of c-Met Protein Expres-
sion
Cells from monolayer cultures were detached with 0.02% EDTA (Sigma), washed twice with serum-free medium, and then fixed and permeabilized in 1% methanol for 15 min. After blocking nonspecific binding sites with 3% BSA for 15 min, cells were labeled with the specific anti-c-Met primary antibodies detailed above for 45 min at 37 ºC. After washing three times for 5 min with PBS, we used RPE-conjugated goat polyclonal anti-mouse antibody (DakoCytomation, Glostrup, Denmark), biotinylated swine polyclonal anti-rabbit antibody (DakoCytomation) (45 min, 37 ºC) and RPE-conjugated streptavidin (DakoCytomation), as secondary antibodies. Between each step, samples were washed with PBS three times for 5 min. Fluorescence was assayed and analyzed by a flow cytometer (CyFlow SL-Green, Partec, Munster, Germany) using FlowMax software (Partec). Posi-tive events from a total of 104 cells were counted. Negative controls were prepared by replacing the primary antibody with isotype-matched nonimmune IgG (Sigma).
Flow Cytometric Measurement for Apoptosis
Double staining with FITC-Annexin V and cellular DNA using PI was performed as follows. Cells previously treated for 48 h with different concentrations (1, 5 μM) of SU11274 were detached with 0.02% EDTA, washed twice with PBS. Cells were resuspended in binding buffer (10 mM NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl). FITC-Annexin V (Alexis Biochemicals, Switzerland) was added to a final concentra-tion of 1 pg/ml Annexin V and 1 pg/ml PI (Partec, Germany) was added to the resulting cell suspension. The mixture was incubated for 15 min in the dark at room temperature and then measured by FACSCalibur flow cytometer (Becton Dickinson, Sunnyvale, CA) [29].
SDS–PAGE and Western Blot
Previously treated HT168-M1 human melanoma cells were lysed with 1% NP40 (Sigma) in PBS containing 1 mM Na3VO4, centrifuged, and denatured before loading. SDS–PAGE was conducted on 8–18% gradient gels (GE Amer-sham Pharmacia Biotech, Uppsala, Sweden) under non-reducing conditions. The proteins were electro-transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) and immunodetected with a phosphospecific anti-c-Met antibody (p-Tyr1234/1235, Cell Signaling Technology, Danvers, MA, USA; dilution 1:1000), an anti-c-Met anti-body corresponding to the carboxyl-terminal sequence of c-Met (Cell Signaling Technology, Danvers, MA, USA; dilu-tion 1:1000) and an anti-glyceraldehyde-3-phosphate-dehydrogenase antibody (Biogenesis, Poole, UK; dilution 1:5000). Reaction products were detected by chemilumines-cence with the ChemiGlow reagent (Biozyme Laboratories Limited, South Wales, UK) according to the manufacturer’s instructions.
Cell Proliferation Assay
Cell suspensions containing 5x104 viable cells/ml were plated in 96-well dishes (Greiner, Frickenhausen, Germany) and after 24 hours of incubation were treated with SU11274 TKI at concentrations of 0.1-5 μM for 48 hours in 200 μl serum-containing or serum-free medium. At the end of incu-bation, 20 μl of 5 mg/ml thiazolyl blue tetrazolium bromide (MTT, Sigma) were added to the cell medium for 4 hours at 37°C, then the medium was removed and tetrazolium crys-tals were dissolved in 100 μl DMSO (Sigma). Absorbance was measured at 570 nm using ELISA Microplate Reader (BioRad, Hercules, CA). The 50% inhibitory concentrations (IC50) were calculated by Dose-Effect Analysis with Micro-computers software (Elsevier-Biosoft, Cambridge, UK).
siRNA Transfection
siRNA against c-Met was obtained from Santa Cruz Bio-technology (Santa Cruz, CA). MOCK transfection was done simultaneously using control siRNA (Santa Cruz Biotech-nology) as negative control. The transfection was performed according to the manufacturer’s instructions. Briefly, 2x105 cells per well were incubated in a 6-well plate, in 2 ml anti-biotic-free normal growth medium supplemented with serum for 24 hours, until 60% of confluence was reached. siRNA duplex solution was added (6 μl of siRNA into 100 μl of

c-Met TKI Decreases Human Melanoma Metastasis in Mice Current Cancer Drug Targets, 2010, Vol. 10, No. 3 335
Transfection Medium – Solution A) directly to the dilute Transfection Reagent (6 μl of Transfection Reagent into 100 μl of Transfection Medium – Solution B). The solution was mixed and incubated for 45 minutes at room temperature. The cells were washed once with 2 ml of siRNA Transfec-tion Medium and 0.8 ml siRNA Transfection Medium con-taining Solution A + Solution B was immediately added to each well and mixed gently. The cells were incubated for 16 hours. The transfected cells were used in cell proliferation assay described above.
Verification of the Transfection by Real-Time PCR (RT-PCR)
Total RNA was extracted from the transfected (MOCK or siRNA against c-Met) HT168-M1 human melanoma cells, using Trizol –reagent (Invitrogen) and purified with DNA-free DNase kit (Ambion) according to the manufacturer’s protocol. Then RNA was used for reverse transcription, which was performed using deoxy-NTPs (0.5 mM each), a mixture of random primer and oligo dT (final concentration 3 μM), RNasin ribonuclease inhibitor (Promega), reverse transcription buffer and M-MLV Reverse Transcriptase (Sigma). RNA solutions were incubated for 50 min at 37°C, then for 10 min at 85°C.
The quality of cDNA and the purity of the RNA was monitored by the amplification of -actin housekeeping gene (primers: 5’-GTG GGG CGC CCC AGG CAC CCA-3’ és 5’-CTC CTT AAT GTC ACG CAC GAT TTC-3’). All sam-ples were clean of genomic DNA, and good enough for fur-ther examinations.
Quantitative real-time PCR was performed using the cDNA as template with TaqMan Universal PCR Master mix (Applied Biosystems) and TaqMan premade gene expression assays (Applied Biosystems) to amplify cMET (Hs00179845_m1). All reactions were conducted as follows: 50ºC for 2 min and 40 cycles of 10 s at 95ºC and 1 min at 60ºC in an Applied Biosystems 7500 Real-time PCR System. All samples were assayed in triple and control water samples were included in each experiment. The endogenous expres-sion reference was the -actin gene (Hs03023880_g1).
Modified Boyden-Chamber Migration Assay
Cell migration was assayed by a method reported previ-ously by Albini et al. [30]. We used 96-well CXF8 plates (polycarbonate filter with 8 μm pore size, Neuroprobe Inc., Cabin John, MD) without coating. HT168-M1 human mela-noma cells were harvested with 0.02% EDTA, washed twice with serum-free medium, and resuspended at a density of 106 cells/ml in medium with 0.1% BSA. 20 l of the cell suspen-sion was placed on top of the membrane with or without SU11274 TKI at concentrations of 1, 5, and 10 μM, and the lower compartment was filled with 30 l of fibronectin in RPMI (100 g/ml, Sigma). Cells were allowed to migrate for 6 h at 37 ºC in a humidified atmosphere of 5% CO2. The cells on the upper surface of the filter were then removed mechanically and the membranes were stained with toluidine blue. The migrated cells were counted manually under a light microscope.
Animal Experiments for Liver Metastasis
SCID mice were bred and maintained in our specific pathogen-free mouse colony and housed 10 to a cage. Previ-ously cultured human melanoma HT168-M1 cells from monolayer were detached with 0.02% EDTA (Sigma), washed twice with serum-free medium, and single cell sus-pension was inoculated into the spleen of SCID mice with the number of 106 cells/animal. Tyrosine kinase c-Met spe-cific inhibitor SU11274 was suspended in physiologic saline containing 1% DMSO. Twenty days following intrasplenic injection of the tumor cells, animals were treated intraperito-neally with 0.5 mg/kg SU11274 or solvent control daily for 21 days (10 animals per group). At the endpoint, the weight of the primary tumors was measured and the number of liver colonies was counted under a stereomicroscope. The SU11274-treated group was compared to the solvent-treated controls; the effects of the treatment were calculated in per-cent of control group. All animal experiments were con-ducted following standards and procedures approved by the Animal Care and Use Committee of the National Institute of Oncology, Budapest.
Statistics
To determine statistical differences between two groups t-test were used. Statistical differences between more than two groups, ANOVA were used with the post hoc Scheffé-test, where parametric methods were available. For the ani-mal experiments we used the non-parametric Mann-Whitney U-test. Statistical significance was determined when P val-ues were <0.05. Statistical analysis was performed by Statis-tica 6.0 software (StatSoft, Tulsa, OK).
RESULTS
Human Melanoma Cell Lines Express the Wild-Type
c-Met Gene
Six different regions of the c-Met gene were amplified by RT-PCR (Table 1). Two primer pairs targeted the extracellu-lar region (Sema domain and plexin-like domain), and four others the intracellular region of c-Met, including one primer pair for the TK region. The product of -actin served as the positive and H2O as the negative control for the PCR me-thod. Compared to the A431 squamous cancer cell line, melanoma lines had no genetic alterations in c-Met either in the extracellular or in the intracellular domains. Sequencing of the PCR fragments revealed the presence of authentic c-Met gene products in our human melanoma cell lines. Moreover, our study detected wild type c-Met gene variants at the TK and juxtamembrane domains in all the investigated melanoma cell lines (Fig. 1), whereas previous findings have shown missense mutations in human melanoma cell lines occurring in the juxtamembrane region at the position of 2843 (A>G) or 2962 (C>T) [25].
Human Melanoma Cell Lines Express Active c-Met Protein
Fixed and permeabilized cells were labeled with antibod-ies specific for the extracellular or the intracellular regions of

336 Current Cancer Drug Targets, 2010, Vol. 10, No. 3 Kenessey et al.
the c-Met receptor, and the ratio of positive cells was evalu-ated by flow cytometry. All human melanoma cell lines used were positive for both the extra- and intracellular domains (in the range of 21.5-51.6%; data not shown). Immunofluo-rescence microscopy yielded similar results: melanoma cells expressed the entire c-Met protein (Fig. 2a-b). Moreover, immunolabeling with a phosphospecific antibody (Tyr1230/1234/1235) showed that the c-Met protein was constitu-tively phosphorylated even without exogenous HGF stimula-tion, and that the active receptors were localized at the adhe-sion sites of the cells (Fig 2c).
SU11274 Inhibited the Phosphorylation of c-Met Protein
in HT168-M1 Cells
Among the studied MM cell lines, the highly metastatic, liver-specific HT168-M1 cells expressed the highest amount of c-Met protein as determined by flow cytometry (51.6±3.9%). Treatment of HT168-M1 cells with SU11274 at a concentration of 5 μM significantly (P<0.05) decreased c-Met protein phosphorylation compared to that of control cells (mean fluorescence intensities were 6.21±1.27 vs. 12.25±3.56, respectively), as measured by flow cytometry using the phosphospecific antibody (p-Tyr1230/1234/1235).
Fig. (1). Representative sequence data from human melanoma cell lines in the juxtramembrane region. Sequence analysis did not re-veal mutations neither at the position 2843 (arrow) (a) nor at the position 2962 (arrow) (b), as it was described previously in other melanoma cell lines [25].
Fig. (2). Expression of phosphorylated c-Met protein in HT168-M1 human melanoma cells. Immunofluorescent detection of c-Met pro-tein (green) using antibodies against the intracellular (a) and extracellular (b) domain of the protein, or phosphospecific sites of the tyrosine kinase domain (c,d). Administration of the c-Met-specific small TK inhibitor (d) significantly decreased the protein phosphorylation in the adhesion site when compared to untreated cells (c). Arrows: constitutively active phospho-c-Met is localized in the adhesion sites. Nuclei were labeled by PI (red). Bars: 20 m.

c-Met TKI Decreases Human Melanoma Metastasis in Mice Current Cancer Drug Targets, 2010, Vol. 10, No. 3 337
The decreased c-Met phosphorylation at the adhesion sites after SU11274 treatment was detected by immunohisto-chemical analysis as well (Fig. 2d). On the microscopic sec-tions, the fluorescent plaques labeled with anti-phospho-c-Met(p-Tyr1230/1234/1235) antibody was counted manually and compared to untreated control cells. SU11274 treatment sig-nificantly reduced the numbers of active c-Met containing plaques (4.04±3.78 vs. 16.44±7.74; SU11274 vs. Control; mean±SD, P<0.05).
The time-dependent inhibition of c-Met phosphory-latin/activation in HT168-M1 human melanoma cells by SU11274 was further analyzed by Western-blot analysis. Incubation of HT168-M1 cells with 5 M SU11274 led to a time-dependent decrease in c-Met phosphorylation (Fig. 3a). As shown in Fig. (3b), inhibition of c-Met-phosphorylation by SU11274 was concentration dependent and 5 M was already sufficient to inhibit phosphorylation by more than 90%. However, SU11274-treatment caused a minimal altera-tion in the expression of c-Met protein in HT168-M1 mela-noma cells (Fig. 3a-b).
Fig. (3). Time and dose-dependent inhibition of c-Met phos-
phorylation by SU11274. The time-dependent (a) and dose-dependent (b) inhibitory effects of SU11274 were detected by Western-blot analysis using a phosphospecific antibody against c-Met (p-Tyr1234/1235). For the detection of p-c-Met alteration after TKI treatments, antibodies against c-Met and the house-keeping molecule GAPDH were used. After quantification of the bands, the ratios of p-c-Met over total c-Met were calculated (numbers in pa-renthesis below the blots refer to these ratios).
Effect of SU11274 and siRNA Against c-Met on the In
Vitro Proliferation of Human Melanoma Cells
The TK-inhibitor SU11274 significantly decreased the in vitro proliferative capacity of the human melanoma cells both in serum-containing and in serum-free media, with IC50 values between 2.20-3.08 and 0.50-2.01 M, respec-tively (Table 2).
Table 2. Calculated 50% Inhibitory Concentration Values
(IC50, M) of SU11274 on the Proliferation of Hu-
man Melanoma Cell Lines In Vitro in the Absence
(Se-) or in the Presence (Se+) of Fetal Bovine Serum.
The A431 Human Squamous Cell Line Served as a
Reference
Se+ Se-
A431 1.675 1.455
HT168-M1 2.204 0.811
HT199 2.429 1.552
WM983B 2.51 0.498
M24met 3.078 2.006
To confirm that this activity of SU11274 is specific for c-
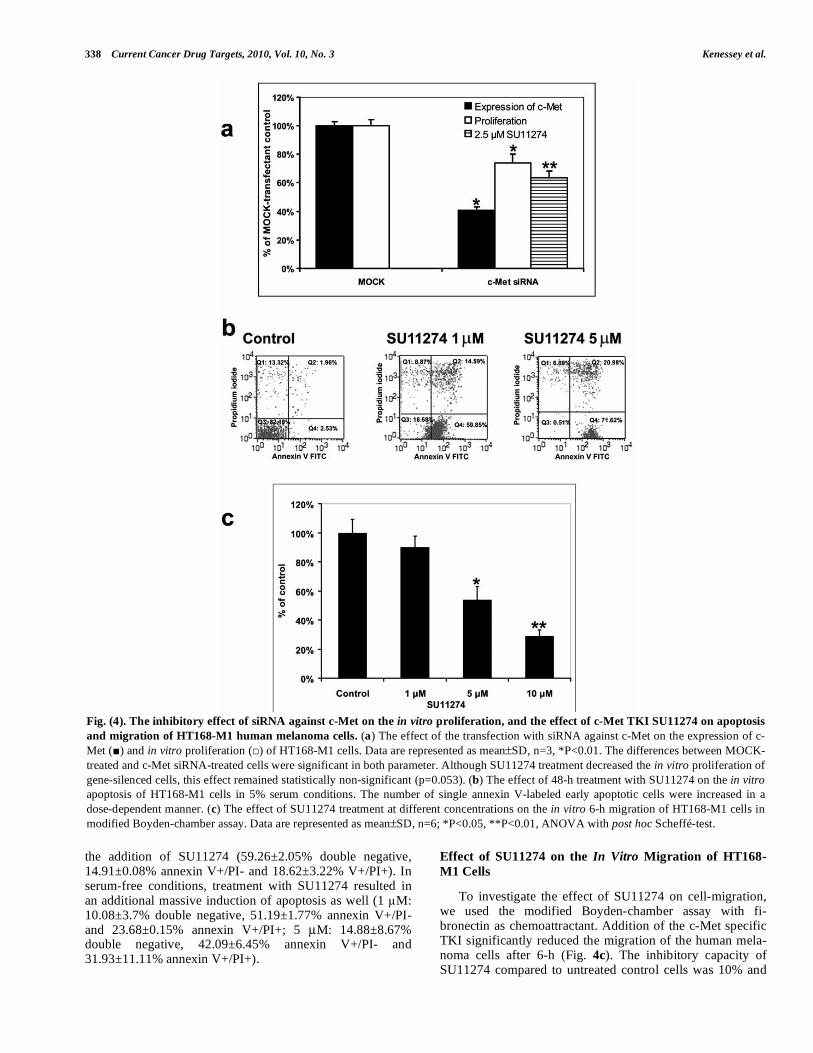
Met inactivation, we performed knock-down experiments using siRNA technology. Inhibition of c-Met expression was analyzed by RT-PCR. Compared to MOCK-transfected con-trol cells (100%), c-Met siRNA-treated cells expressed 41±2.05% percent of c-Met (Fig. 4a). As shown in Fig. (4a), transfection of HT168-M1 cells with c-Met specific siRNA led to a significant inhibition of proliferation in these cells (74.16±5.83% of MOCK-transfected control, P<0.01). Fol-lowing transfection, gene-silenced cells were treated with SU11274, which resulted additional, although statistically non-significant anti-proliferative effect (p=0.053).
Effects of SU11274 on the In vitro Apoptosis of HT168-M1 Cells
To investigate the effect of c-Met inactivation by SU11274 on cell survival/apoptosis, we treated HT168-M1 cells with this inhibitor for 48 hours, and analyzed the cells by FACS-analysis. Double staining with annexin V and propidium revealed a strong induction of apoptosis in HT168-M1 cells after treatment with SU11274 in both se-rum-containing (Fig. 4b) and serum-free conditions (data not shown). Without treatment, most of the cells were viable in the serum-containing medium (82.14±0.07% double nega-tive, 2.46±0.1% annexin V+/PI- and 1.71±0.36% annexin V+/PI+). Treatment with SU11274 resulted in a strong in-crease in annexin V and PI positive cells (1 M: 14.45±3.14% double negative, 59.81±0.06% annexin V+/PI- and 14.01±0.83% annexin V+/PI+; 5 μM: 0.57±0.04% dou-ble negative, 68.09±4.99% annexin V+/PI- and 24.22±4.58% annexin V+/PI+). Higher concentrations of SU11274 led to an increase in double positive cells indicative of necrotic or late apoptotic cells (data not shown). Similar effects were observed under serum-free conditions. Compared to cells in the serum-containing medium, the apoptotic cells under se-rum-free conditions showed an increase in number without
338 Current Cancer Drug Targets, 2010, Vol. 10, No. 3 Kenessey et al.
the addition of SU11274 (59.26±2.05% double negative, 14.91±0.08% annexin V+/PI- and 18.62±3.22% V+/PI+). In serum-free conditions, treatment with SU11274 resulted in an additional massive induction of apoptosis as well (1 M: 10.08±3.7% double negative, 51.19±1.77% annexin V+/PI- and 23.68±0.15% annexin V+/PI+; 5 μM: 14.88±8.67% double negative, 42.09±6.45% annexin V+/PI- and 31.93±11.11% annexin V+/PI+).
Effect of SU11274 on the In Vitro Migration of HT168-
M1 Cells
To investigate the effect of SU11274 on cell-migration, we used the modified Boyden-chamber assay with fi-bronectin as chemoattractant. Addition of the c-Met specific TKI significantly reduced the migration of the human mela-noma cells after 6-h (Fig. 4c). The inhibitory capacity of SU11274 compared to untreated control cells was 10% and
Fig. (4). The inhibitory effect of siRNA against c-Met on the in vitro proliferation, and the effect of c-Met TKI SU11274 on apoptosis
and migration of HT168-M1 human melanoma cells. (a) The effect of the transfection with siRNA against c-Met on the expression of c-Met ( ) and in vitro proliferation ( ) of HT168-M1 cells. Data are represented as mean±SD, n=3, *P<0.01. The differences between MOCK-treated and c-Met siRNA-treated cells were significant in both parameter. Although SU11274 treatment decreased the in vitro proliferation of gene-silenced cells, this effect remained statistically non-significant (p=0.053). (b) The effect of 48-h treatment with SU11274 on the in vitro apoptosis of HT168-M1 cells in 5% serum conditions. The number of single annexin V-labeled early apoptotic cells were increased in a dose-dependent manner. (c) The effect of SU11274 treatment at different concentrations on the in vitro 6-h migration of HT168-M1 cells in modified Boyden-chamber assay. Data are represented as mean±SD, n=6; *P<0.05, **P<0.01, ANOVA with post hoc Scheffé-test.
c-Met TKI Decreases Human Melanoma Metastasis in Mice Current Cancer Drug Targets, 2010, Vol. 10, No. 3 339
70% with concentrations of SU11274 of 1 μM and 10 μM, respectively (Fig. 4c). No effect on the cell viability was observed after this short incubation period. Compared to untreated control cells, the viability after addition of 10 M SU11274 was 115.57±12.96%, as determined by MTT-assay (P=0.32).
SU11274 Inhibited Intrasplenic Growth and Liver Colo-nization of HT168-M1 Xenograft
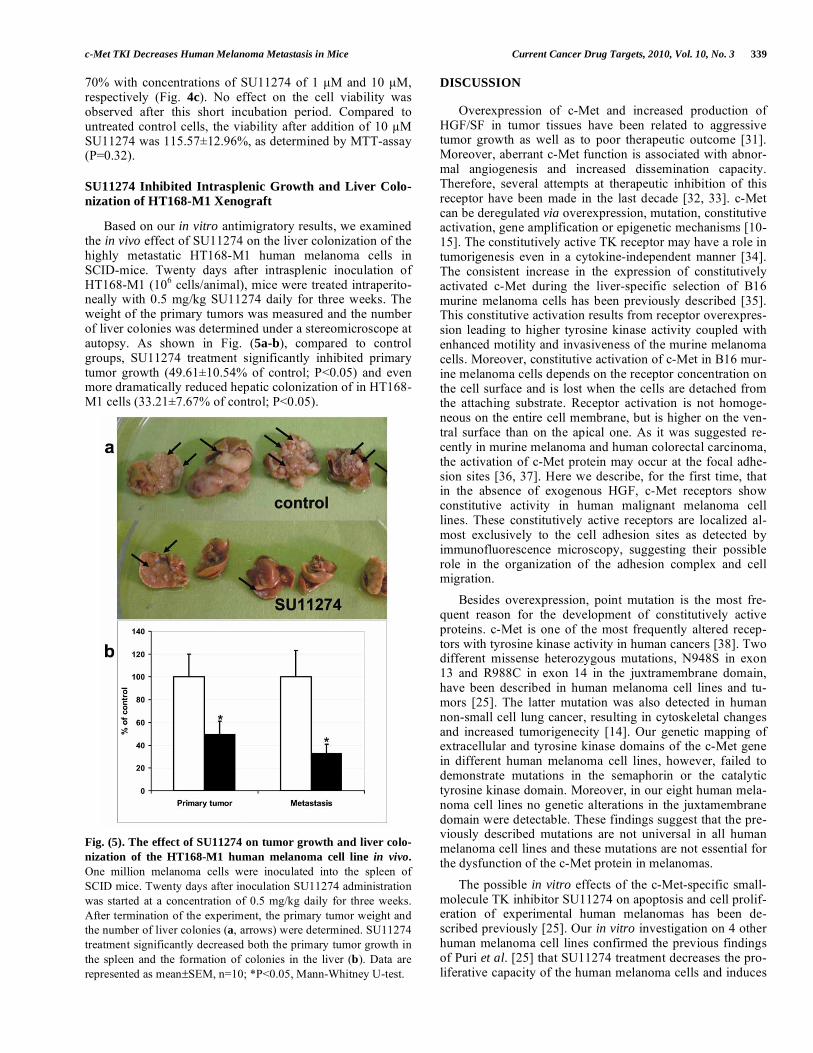
Based on our in vitro antimigratory results, we examined the in vivo effect of SU11274 on the liver colonization of the highly metastatic HT168-M1 human melanoma cells in SCID-mice. Twenty days after intrasplenic inoculation of HT168-M1 (106 cells/animal), mice were treated intraperito-neally with 0.5 mg/kg SU11274 daily for three weeks. The weight of the primary tumors was measured and the number of liver colonies was determined under a stereomicroscope at autopsy. As shown in Fig. (5a-b), compared to control groups, SU11274 treatment significantly inhibited primary tumor growth (49.61±10.54% of control; P<0.05) and even more dramatically reduced hepatic colonization of in HT168-M1 cells (33.21±7.67% of control; P<0.05).
Fig. (5). The effect of SU11274 on tumor growth and liver colo-
nization of the HT168-M1 human melanoma cell line in vivo.
One million melanoma cells were inoculated into the spleen of SCID mice. Twenty days after inoculation SU11274 administration was started at a concentration of 0.5 mg/kg daily for three weeks. After termination of the experiment, the primary tumor weight and the number of liver colonies (a, arrows) were determined. SU11274 treatment significantly decreased both the primary tumor growth in the spleen and the formation of colonies in the liver (b). Data are represented as mean±SEM, n=10; *P<0.05, Mann-Whitney U-test.
DISCUSSION
Overexpression of c-Met and increased production of HGF/SF in tumor tissues have been related to aggressive tumor growth as well as to poor therapeutic outcome [31]. Moreover, aberrant c-Met function is associated with abnor-mal angiogenesis and increased dissemination capacity. Therefore, several attempts at therapeutic inhibition of this receptor have been made in the last decade [32, 33]. c-Met can be deregulated via overexpression, mutation, constitutive activation, gene amplification or epigenetic mechanisms [10-15]. The constitutively active TK receptor may have a role in tumorigenesis even in a cytokine-independent manner [34]. The consistent increase in the expression of constitutively activated c-Met during the liver-specific selection of B16 murine melanoma cells has been previously described [35]. This constitutive activation results from receptor overexpres-sion leading to higher tyrosine kinase activity coupled with enhanced motility and invasiveness of the murine melanoma cells. Moreover, constitutive activation of c-Met in B16 mur-ine melanoma cells depends on the receptor concentration on the cell surface and is lost when the cells are detached from the attaching substrate. Receptor activation is not homoge-neous on the entire cell membrane, but is higher on the ven-tral surface than on the apical one. As it was suggested re-cently in murine melanoma and human colorectal carcinoma, the activation of c-Met protein may occur at the focal adhe-sion sites [36, 37]. Here we describe, for the first time, that in the absence of exogenous HGF, c-Met receptors show constitutive activity in human malignant melanoma cell lines. These constitutively active receptors are localized al-most exclusively to the cell adhesion sites as detected by immunofluorescence microscopy, suggesting their possible role in the organization of the adhesion complex and cell migration.
Besides overexpression, point mutation is the most fre-quent reason for the development of constitutively active proteins. c-Met is one of the most frequently altered recep-tors with tyrosine kinase activity in human cancers [38]. Two different missense heterozygous mutations, N948S in exon 13 and R988C in exon 14 in the juxtramembrane domain, have been described in human melanoma cell lines and tu-mors [25]. The latter mutation was also detected in human non-small cell lung cancer, resulting in cytoskeletal changes and increased tumorigenecity [14]. Our genetic mapping of extracellular and tyrosine kinase domains of the c-Met gene in different human melanoma cell lines, however, failed to demonstrate mutations in the semaphorin or the catalytic tyrosine kinase domain. Moreover, in our eight human mela-noma cell lines no genetic alterations in the juxtamembrane domain were detectable. These findings suggest that the pre-viously described mutations are not universal in all human melanoma cell lines and these mutations are not essential for the dysfunction of the c-Met protein in melanomas.
The possible in vitro effects of the c-Met-specific small-molecule TK inhibitor SU11274 on apoptosis and cell prolif-eration of experimental human melanomas has been de-scribed previously [25]. Our in vitro investigation on 4 other human melanoma cell lines confirmed the previous findings of Puri et al. [25] that SU11274 treatment decreases the pro-liferative capacity of the human melanoma cells and induces

340 Current Cancer Drug Targets, 2010, Vol. 10, No. 3 Kenessey et al.
apoptosis in the 1-5 μM concentration range. However, we also demonstrated that SU11274 decreased the phosphoryla-tion of c-Met proteins at the adhesion sites of the fibronectin-attached human melanoma cells. This special localization of active c-Met protein and the fact that c-Met is the receptor of the motility inducer HGF/SF suggests that at concentrations showing no effect on cell viability, SU11274 may influence the migratiory capacity of the human melanoma cells. In line with that, our in vitro motility assay results indicate that the small-molecule inhibitor specifically targeting c-Met indeed decreases the migration of the HT168-M1 human melanoma cells. It is well documented that HGF/SF stimulation induces several different biological responses, including an invasive phenotype of cancer cells. Major components of this phe-nomenon are the alterations in spreading, attachment and migration of the tumor cells [4]. Through binding to its re-ceptor, HGF/SF can induce several different signaling path-ways including RAS, STAT3, FAK, SRC and PI3K [12]. Among these, PI3K is thought to have a central role in the signalization of c-Met. Stimulation of the PI3K pathways is associated with the regulation of cell proliferation, apoptosis and particularly, migration [39, 40]. Activation of PI3K re-sulted in anchorage independence [41], adhesion remodeling [42], and recruitment of molecules responsible for organiza-tion of the cytoskeleton [43]. Moreover, specific exogenous miRNAs leading to suppression of c-Met expression in pri-mary melanoma cells, have been shown to decrease c-Met-mediated cell migration [44]. These data, together with our in vitro motility assay, suggest that SU11274 may influence the in vivo metastasis formation capacity of the human mela-noma cells by decreasing their ability to migrate. In our in vivo model system, where human melanoma cells were in-oculated into the spleen of SCID mice and tumorcolonies were formed in the liver, we were able to test the effect of SU11274 not only directly on primary tumor growth, but also on the development of liver colonies. We found that in vivo administration of SU11274 significantly delayed the primary tumor growth in SCID mice, which corroborates our in vitro data showing that the specific c-Met inhibitor de-creased the proliferation of human melanoma cells. Moreo-ver, SU11274 treatment at 0.5 mg/kg concentration dramati-cally reduced the number of the HT168-M1 human mela-noma colonies in the liver.
In summary, our preclinical study demonstrates that c-Met is constitutively active without exogenous HGF stimula-tion in human melanoma cell lines. To the best of our knowledge, our data provides the first report on the in vivo efficacy of SU11274 in a human melanoma xenograft model. The c-Met-specific small tyrosine kinase inhibitor success-fully inhibited not only primary tumor growth, but also liver colony formation by human melanoma cells in SCID mice. These data may promote the clinical development of this c-Met inhibitor targeting human melanoma.
ABBREVIATIONS
A = adenosine
ATP = adenosine triphosphate
BSA = bovine serum albumin
C = cytosine
EDTA = ethylenediaminetetraacetic acid
FAK = focal adhesion kinase
G = guanine
GAPDH = glyceraldehyde-3-phosphate
HGF = hepatocyte growth factor
IC50 = half maximal inhibitory concentration
ip = intraperitoneally
MITF = microphthalmia-associated transcription factor
MM = malignant melanoma
MTT = thiazolyl blue tetrazolium bromide
P = probability
p-Tyr = phosphorylated tyrosine
PBS = phosphate buffered saline
PCR = polymerase chain reaction
PI = propidium iodide
PI3K = phosphatidylinositol-3 kinase
RNA = ribonucleic acid
RT-PCR = real time PCR
SCID = severe combined immunodeficiency
SDS-PAGE = sodium dodecyl sulfate polyacrylamide gel electrophoresis
Se- = in the absence fetal bovine serum
Se+ = in the presence of fetal bovine serum
SF = scatter factor
T = thymine
TK = tyrosine kinase
TKI = tyrosine kinase inhibitor
GRANT SUPPORT
This work was supported by the following grants: Na-tional Science Foundation-OTKA-F68916; OTKA-NK73082 (B.D.); OTKA-D48519, OTKA-F46501, OTKA-K76293 (J.Tó.), and OTKA-NK-72595 (J.Ti.); European Commis-sion, Framework Programme 6, LSHC-CT-2006-037559 (S.Cs). EGT/Norwegian Financial Mechanism-HU0125 (B.D., J.Ti., J.Tó.); European Commission, Framework Pro-gramme 6, LSHC-CT-2006-037559 (S.Cs); ESMO Transla-tional Research Fellowship (B.D.); RegIonCo-L00052 Cross-border Co-operation Program Hungary–Austria 2007-2013 (B.D., J.Tó.); further support: J. Tóvári is a recipient of the Eötvös Hungarian State Scholarship. Judit Berta is a re-cipient of the Ernst Mach fellowship of the Austrian Agency for International Cooperation in Education and Research.
ACKNOWLEDGEMENTS
We kindly thank Katalin Derecskei for her excellent technical assistance and Andrea Ladányi and Lara M. Strong for critical reviewing the manuscript.

c-Met TKI Decreases Human Melanoma Metastasis in Mice Current Cancer Drug Targets, 2010, Vol. 10, No. 3 341
REFERENCES
[1] Tamura, K.; Fukuoka, M. Molecular target-based cancer therapy: tyrosine kinase inhibitors. Int. J. Clin. Oncol. 2003, 8, 207-211.
[2] Press, M. F.; Lenz, H. J. EGFR, HER2 and VEGF pathways: vali-dated targets for cancer treatment. Drugs 2007, 67, 2045-2075.
[3] Bottaro, D. P.; Rubin, J. S.; Faletto, D. L.; Chan, A. M.; Kmiecik, T. E.; Vande Woude G. F.; Aaronson, S. A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802-804.
[4] Comoglio, P. M.; Trusolino L. Invasive growth: from development to metastasis. J. Clin. Invest. 2002, 109, 857-862.
[5] Trusolino, L.; Comoglio, P. M. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289-300.
[6] Faletto, D. L.; Tsarfaty, I.; Kmiecik, T. E.; Gonzatti, M.; Suzuki, T.; Vande Woude, G. F. Evidence for non-covalent clusters of the c-met proto-oncogene product. Oncogene 1992, 7, 1149-1157.
[7] Ma, P. C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metasta-
sis Rev. 2003, 22, 309-325. [8] Corso, S.; Comoglio, P. M.; Giordano, S. Cancer therapy: can the
challenge be MET? Trends Mol. Med. 2005, 11, 284-292. [9] Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G. F.
Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915-925.
[10] Di Renzo, M. F.; Olivero, M.; Giacomini, A.; Porte, H.; Chastre, E.; Mirossay, L.; Nordlinger, B.; Bretti, S.; Bottardi, S.; Giordano, S.; et al. Overexpression and amplification of the met/HGF recep-tor gene during the progression of colorectal cancer. Clin. Cancer
Res. 1995, 1, 147-154. [11] Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.;
Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary re-sected lung cancers. J. Thorac. Oncol. 2009, 4, 5-11.
[12] Migliore, C.; Giordano, S. Molecular cancer therapy: can our ex-pectation be MET? Eur. J. Cancer 2008, 44, 641-651.
[13] Linehan, W. M.; Pinto, P. A.; Srinivasan, R.; Merino, M.; Choyke, P.; Choyke, L.; Coleman, J.; Toro, J.; Glenn, G.; Vocke, C.; Zbar, B.; Schmidt, L. S.; Bottaro, D.; Neckers, L. Identification of the genes for kidney cancer: opportunity for disease-specific targeted therapeutics. Clin. Cancer Res. 2007, 13, 671s-679s.
[14] Ma, P. C.; Kijima, T.; Maulik, G.; Fox, E. A.; Sattler, M.; Griffin, J. D.; Johnson, B. E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272-6281.
[15] Yu, J.; Miehlke, S.; Ebert, M. P.; Hoffmann, J.; Breidert, M.; Al-pen, B.; Starzynska, T.; Stolte, Prof. M.; Malfertheiner, P.; Bayer-dorffer, E. Frequency of TPR-MET rearrangement in patients with gastric carcinoma and in first-degree relatives. Cancer 2000, 88, 1801-1806.
[16] Morotti, A.; Mila, S.; Accornero, P.; Tagliabue, E.; Ponzetto, C. K252a inhibits the oncogenic properties of Met, the HGF receptor. Oncogene 2002, 21, 4885-4893.
[17] Garbe, C.; Leiter, U. Melanoma epidemiology and trends. Clin.
Dermatol. 2009, 27, 3-9. [18] Hsu, M. Y.; Meier, F.; Herlyn, M. Melanoma development and
progression: a conspiracy between tumor and host. Differentiation 2002, 70, 522-536.
[19] Natali, P. G.; Nicotra, M. R.; Di Renzo, M. F.; Prat, M.; Bigotti, A.; Cavaliere, R.; Comoglio, P. M. Expression of the c-Met/HGF re-ceptor in human melanocytic neoplasms: demonstration of the rela-tionship to malignant melanoma tumour progression. Br. J. Cancer 1993, 68, 746-750.
[20] Li, G.; Schaider, H.; Satyamoorthy, K.; Hanakawa, Y.; Hashimoto, K.; Herlyn, M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene 2001, 20, 8125-8135.
[21] McGill, G. G.; Haq, R.; Nishimura, E. K.; Fisher, D. E. c-Met expression is regulated by Mitf in the melanocyte lineage. J. Biol.
Chem. 2006, 281, 10365-10373. [22] Riker, A. I.; Enkemann, S. A.; Fodstad, O.; Liu, S.; Ren, S.; Mor-
ris, C.; Xi, Y.; Howell P.; Metge, B.; Samant, R. S.; Shevde, L. A.;
Li, W.; Eschrich, S.; Daud, A.; Ju, J.; Matta, J. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genomics 2008, 1, 13.
[23] Nambiar, S.; Mirmohammadsadegh, A.; Doroudi, R.; Gustrau, A.; Marini, A.; Roeder, G.; Ruzicka, T.; Hengge, U. R. Signaling net-works in cutaneous melanoma metastasis identified by complemen-tary DNA microarrays. Arch. Dermatol. 2005, 141, 165-173.
[24] Cruz, J.; Reis-Filho, J. S.; Silva, P.; Lopes, J. M. Expression of c-met tyrosine kinase receptor is biologically and prognostically rele-vant for primary cutaneous malignant melanomas. Oncology 2003, 65, 72-82.
[25] Puri, N.; Ahmed, S.; Janamanchi, V.; Tretiakova, M.; Zumba, O.; Krausz, T.; Jagadeeswaran, R.; Salgia, R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin.
Cancer Res. 2007, 13, 2246-2253. [26] Seidl, H.; Weger, W.; Wolf, P.; Kerl, H.; Schaider, H. Lack of
oncogenic mutations in the c-Met catalytic tyrosine kinase domain in acral lentiginous melanoma. Int. J. Dermatol. 2008, 47, 1327-1329.
[27] Ladanyi, A.; Timar, J.; Paku, S.; Molnar, G.; Lapis, K. Selection and characterization of human melanoma lines with different liver-colonizing capacity. Int. J. Cancer 1990, 46, 456-461.
[28] Sattler, M.; Pride, Y. B.; Ma, P.; Gramlich, J. L.; Chu, S. C.; Quin-nan, L. A.; Shirazian, S.; Liang, C.; Podar, K.; Christensen, J. G.; Salgia, R. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003, 63, 5462-5469.
[29] Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutelingsperger, C. A novel assay for apoptosis. Flow cytometric detection of phos-phatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39-51.
[30] Albini, A.; Iwamoto, Y.; Kleinman, H. K.; Martin, G. R.; Aaron-son, S. A.; Kozlowski, J. M.; McEwan, R. N. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res 1987, 47, 3239-3245.
[31] Jiang, W.; Hiscox, S.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor/scatter factor, its molecular, cellular and clinical im-plications in cancer. Crit. Rev. Oncol. Hematol. 1999, 29, 209-48.
[32] Haddad, R.; Lipson, K. E.; Webb, C. P. Hepatocyte growth factor expression in human cancer and therapy with specific inhibitors. Anticancer Res. 2001, 21, 4243-4252.
[33] Longati, P.; Comoglio, P. M.; Bardelli, A. Receptor tyrosine kinases as therapeutic targets: the model of the MET oncogene. Curr. Drug Targets 2001, 2, 41-55.
[34] Lengyel, E.; Sawada, K.; Salgia, R. Tyrosine kinase mutations in human cancer. Curr. Mol. Med. 2007, 7, 77-84.
[35] Rusciano, D.; Lorenzoni, P.; Burger, M. M. Expression of constitu-tively activated hepatocyte growth factor/scatter factor receptor (c-met) in B16 melanoma cells selected for enhanced liver coloniza-tion. Oncogene 1995, 11, 1979-1987.
[36] Rusciano, D.; Lorenzoni, P.; Burger, M. M. Constitutive activation of c-Met in liver metastatic B16 melanoma cells depends on both substrate adhesion and cell density and is regulated by a cytosolic tyrosine phosphatase activity. J. Biol. Chem. 1996, 271, 20763-20769.
[37] Fazekas, K.; Csuka, O.; Koves, I.; Raso, E.; Timar, J. Experimental and clinicopathologic studies on the function of the HGF receptor in human colon cancer metastasis. Clin. Exp. Metastasis 2000, 18, 639-649.
[38] Danilkovitch-Miagkova, A.; Zbar, B. Dysregulation of Met recep-tor tyrosine kinase activity in invasive tumors. J. Clin. Invest. 2002, 109, 863-867.
[39] Graziani, A.; Gramaglia, D.; Cantley, L. C.; Comoglio, P. M. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor re-ceptor associates with phosphatidylinositol 3-kinase. J. Biol. Chem. 1991, 266, 22087-22090.
[40] Xiao, G. H.; Jeffers, M.; Bellacosa, A.; Mitsuuchi, Y.; Vande Woude, G. F.; Testa, J. R. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci.
USA 2001, 98, 247-252. [41] Toker, A. Protein kinases as mediators of phosphoinositide 3-
kinase signaling. Mol. Pharmacol. 2000, 57, 652-658.

342 Current Cancer Drug Targets, 2010, Vol. 10, No. 3 Kenessey et al.
[42] Trusolino, L.; Cavassa, S.; Angelini, P.; Ando, M.; Bertotti, A.; Comoglio, P. M.; Boccaccio, C. HGF/scatter factor selectively promotes cell invasion by increasing integrin avidity. FASEB J. 2000, 14, 1629-1640.
[43] Wells, C. M.; Abo, A.; Ridley, A. J. PAK4 is activated via PI3K in HGF-stimulated epithelial cells. J. Cell Sci. 2002, 115, 3947-3956.
[44] Migliore, C.; Petrelli, A.; Ghiso, E.; Corso, S.; Capparuccia, L.; Eramo, A.; Comoglio, P. M.; Giordano, S. MicroRNAs impair MET-mediated invasive growth. Cancer Res. 2008, 68, 10128-10136.
Received: May 01, 2009 Revised: February 26, 2010 Accepted: March 27, 2010
PMID: 20370683
Related Documents











