Inhibition of brain CYP2D lowers codeine-induced analgesia in rats by Kaidi Zhou A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Pharmacology and Toxicology University of Toronto © Copyright by Kaidi Zhou 2012

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Inhibition of brain CYP2D lowers codeine-induced
analgesia in rats
by
Kaidi Zhou
A thesis submitted in conformity with the requirements
for the degree of Master of Science
Graduate Department of Pharmacology and Toxicology
University of Toronto
© Copyright by Kaidi Zhou 2012

ii
Inhibition of brain CYP2D lowers codeine-induced analgesia in
rats
Kaidi Zhou
Master of Science
Graduate Department of Pharmacology and Toxicology
University of Toronto
2012
Abstract
CYP2D6 metabolizes codeine to morphine, the active analgesic metabolite.
Variation in brain CYP2D6 activity may affect brain morphine levels after codeine
administration and thereby influence analgesia. We investigate the effect of
inhibiting brain CYP2D on codeine-induced analgesia. METHODS: Rats received
intracerebroventricular (i.c.v.) injections of CYP2D inhibitors or vehicle controls.
Rats were then given subcutaneous codeine injections and analgesia was measured
with the tail-flick test. Morphine and codeine concentrations in brain and plasma
were measured. CYP2D activity in brain and liver were assessed in vitro. RESULTS:
Compared to vehicle treatment, i.c.v. inhibitor treatments resulted in lower codeine-
induced analgesia, lower morphine levels in brain but not in plasma after codeine
injections, and lower CYP2D activity in brain membranes but not in liver
microsomes. CONCLUSIONS: Inhibiting brain CYP2D reduces codeine’s metabolism
to morphine, resulting in less analgesia. Variation in brain CYP2D6 activity may
influence response to codeine and other CYP2D6 substrates.

iii
Acknowledgements
First and foremost, I would like to thank my supervisor Dr. Rachel F. Tyndale for all
of her support and guidance throughout the course of my graduate study. I am
grateful to her for providing me with this amazing opportunity to learn and grow
intellectually and professionally. Her ambition, hard work and professionalism are
inspiring and have encouraged me to be a more effective student and scientist.
I would like to thank Dr. Jose N. Nobrega for being my M.Sc. advisor, as well
as Dr. Ali Salahpour, Dr. Daniel J. Mueller and Dr. John W. Semple for serving on my
defense committee. Their sharing of time, knowledge and constructive criticism are
greatly appreciated.
I am thankful to each and every member of Dr. Tyndale’s lab for generously
providing their advice, time, knowledge, and technical expertise during my M.Sc. I
am truly fortunate to have been part of a group with such intelligent, caring,
encouraging and supportive people. Special thanks to Dr. Sharon Miksys for her
advice and teaching expertise and for fostering a positive and encouraging lab
environment, to Jibran Khokhar for his enthusiasm, knowledge and help on multiple
aspects of this project, and to Dr. Bin Zhao and Steven Lo for their excellent technical
assistance and time contributions.
Last but not least, I would like to thank my family and friends for their ongoing
love, care and support. I am especially indebted to my parents for their hard work,
patience and dedication, which made all of my opportunities possible.

iv
Table of Contents
Abstract ...................................................................................................................... ii
Acknowledgements ................................................................................................... iii
Table of Contents ....................................................................................................... iv
List of Figures............................................................................................................ vii
Summary of Abbreviations ........................................................................................ ix
Section 1: Introduction ................................................................................................ 1
Statement of Research Problem .................................................................................. 1
Purpose of the Study and Objective ............................................................................ 2
Statement of Research Hypotheses and Rationale ....................................................... 3
Review of the Literature .............................................................................................. 5
1.1 Cytochrome P450 2D6 (CYP2D6) .......................................................................... 5
1.1.1 Cytochromes P450 ............................................................................................. 5
1.1.2 CYP2D6 substrates ............................................................................................. 6
1.1.3 CYP2D6 inhibitors .............................................................................................. 6
1.1.4 CYP2D6 regulation ............................................................................................. 7
1.1.5 CYP2D6 genetic variation ................................................................................... 8
1.1.5a Interethnic variability in CYP2D6 ...................................................................... 9
1.1.6 CYP2D expression in different species ............................................................ 10
1.2 Brain Cytochromes P450 ..................................................................................... 11
1.2.1 Brain CYP expression ....................................................................................... 12
1.2.2 Brain CYP activity ............................................................................................. 14
1.2.3 Brain CYP regulation ........................................................................................ 16
1.2.4 Brain CYP2D6 ................................................................................................... 17
1.2.4a Brain CYP2D expression ................................................................................. 18
1.2.4b Brain CYP2D function and activity .................................................................. 19
1.2.4c Brain CYP2D regulation .................................................................................. 24
1.3 Opioid Analgesics ............................................................................................... 26
1.3.1 Codeine ........................................................................................................... 26
1.3.1a Codeine metabolism in humans ..................................................................... 27
1.3.1b Codeine metabolism in rats ........................................................................... 31
1.3.2 Morphine .......................................................................................................... 33
1.3.2a Spinal mechanisms of morphine-induced analgesia ...................................... 33
1.3.2b Supraspinal mechanisms of morphine-induced analgesia ............................. 34

v
1.4 Rat Tail-Flick Test: Animal Model of Nociception ................................................ 35
1.4.1 Tail-flick reflex ................................................................................................. 35
1.4.2 Effect of opioid analgesics in the tail-flick test .................................................. 37
1.5 Study design ....................................................................................................... 38
Section 2: Materials and Methods ............................................................................. 39
Section 3: Results ...................................................................................................... 50
3.1 Inhibition of brain CYP2D reduced codeine-induced analgesia ......................... 50
3.2 Inhibition of brain CYP2D lowered codeine-induced area under the analgesia
time curve ................................................................................................................. 53
3.3 Inhibiting brain CYP2D did not affect baseline tail-flick latency ......................... 58
3.4 Inhibiting brain CYP2D did not affect morphine-induced analgesia ................... 60
3.5 Inhibiting brain CYP2D did not alter morphine-induced area under the analgesia
time curve ................................................................................................................. 62
3.6 There was no tolerance to the analgesic effects of codeine or morphine ............ 65
3.7 Codeine and morphine doses used resulted in similar levels of analgesia ......... 70
3.8 Inhibitor-treated rats had lower morphine levels in the brain but not plasma at 30
min after codeine injection ....................................................................................... 73
3.9 Analgesia correlated with brain, and not plasma, morphine levels .................... 81
3.10 Inhibitor-treated rats did not have lower morphine levels in the brain at 60 or 90
min after codeine injection ....................................................................................... 86
3.11 Inhibiting brain CYP2D in vivo lowered in vitro codeine metabolism in the brain
but not liver .............................................................................................................. 91
3.12 Inhibiting brain CYP2D in vivo lowered in vitro dextromethorphan metabolism
in the brain but not liver ........................................................................................... 93
Section 4: Discussion, Conclusions, Future Directions .............................................. 95
4.1 Summary and further implications ...................................................................... 95
4.1.1 Rat model of reduced brain CYP2D activity ..................................................... 96
4.1.2 Inhibition of brain CYP2D lowers codeine-induced analgesia ......................... 98
4.1.3 Analgesia correlates with morphine levels in the brain and not plasma ........ 100
4.1.4 Inhibiting brain CYP2D in vivo lowers in vitro enzyme activity in brain
membranes and not liver microsomes .................................................................... 102
4.1.5 Limitations ...................................................................................................... 102
4.2. Clinical relevance of brain CYP2D activity ...................................................... 105
4.2.1 Centrally-acting drugs ................................................................................... 106
4.2.2 Drugs of abuse ............................................................................................... 108
4.2.3 Endogenous substrates .................................................................................. 109

vi
4.2.4 Disease ........................................................................................................... 111
4.3 Other brain CYPs .............................................................................................. 114
4.4 Future directions ............................................................................................... 116
4.4.1 Other uses of rat models of differing levels of brain CYP2D activity .............. 116
4.4.1a Microdialysis ................................................................................................ 116
4.4.1b Different pain model .................................................................................... 116
4.4.1c Role of rat brain CYP2D in meditating drug inactivation .............................. 117
4.4.1d Effect of rat brain CYP2D induction on drug response ................................. 117
4.4.1e Role of rat brain CYP2D in neurotoxin inactivation ....................................... 118
4.4.2 Therapeutic uses of brain CYP2D induction ................................................... 118
4.5 Conclusions ....................................................................................................... 119
References .............................................................................................................. 120
List of Abstracts ....................................................................................................... 148

vii
List of Figures
Figure 1. Morphine levels in rat plasma and brain 30 min after peripheral injection of
codeine or morphine ................................................................................................ 22
Figure 2. Metabolic pathways of codeine in humans ................................................ 29
Figure 3. Inhibition of brain CYP2D reduced codeine-induced analgesia ................ 52
Figure 4. Inhibiting brain CYP2D with propranolol lowered codeine-induced area
under the analgesia time curve between 0-60 min after codeine injection ............... 54
Figure 5. Inhibiting brain CYP2D with propranolol did not lower codeine-induced
area under the analgesia time curve at 60-120 min or 0-120 min after codeine
injection .................................................................................................................... 55
Figure 6. Inhibiting brain CYP2D with propafenone lowered codeine-induced area
under the analgesia time curve between 0-60 min after codeine injection ............... 56
Figure 7. Inhibiting brain CYP2D with propafenone did not lower codeine-induced
area under the analgesia time curve at 60-120 min or 0-120 min after codeine
injection .................................................................................................................... 57
Figure 8. Inhibiting brain CYP2D did not affect baseline tail-flick latency ................ 59
Figure 9. Inhibiting brain CYP2D did not affect morphine-induced analgesia .......... 61
Figure 10. Inhibiting brain CYP2D with propranolol did not alter morphine-induced
area under the analgesia time curve ......................................................................... 63
Figure 11. Inhibiting brain CYP2D with propafenone did not alter morphine-induced
area under the analgesia time curve ......................................................................... 64
Figure 12. Rats treated with propranolol or vehicle did not develop tolerance to
codeine ..................................................................................................................... 66
Figure 13. Rats treated with propafenone or vehicle did not develop tolerance to
codeine ..................................................................................................................... 67
Figure 14. Rats treated with propranolol or vehicle did not develop tolerance to
morphine .................................................................................................................. 68
Figure 15. Rats treated with propafenone or vehicle did not develop tolerance to
morphine .................................................................................................................. 69
Figure 16. Codeine and morphine doses used resulted in similar levels of analgesia
after ACSF (i.c.v. vehicle) treatment ......................................................................... 71
Figure 17. Codeine and morphine doses used resulted in similar levels of analgesia
after cyclodextrin (i.c.v. vehicle) treatment .............................................................. 72
Figure 18. Inhibitor-treated rats had lower morphine levels in the brain but not in
plasma at 30 min after codeine injection ................................................................... 75
Figure 19. Inhibitor-treated rats had lower morphine to codeine ratios in the brain
but not in plasma at 30 min after codeine injection ................................................... 76

viii
Figure 20. Inhibitor-treated rats had lower morphine to total drug ratios in the brain
but not in plasma at 30 min after codeine injection ................................................... 77
Figure 21. Propranolol-treated rats did not have lower codeine levels or total drug
levels in the brain or in plasma at 30 min after codeine injection ............................. 78
Figure 22. Propafenone-treated rats did not have lower codeine levels or total drug
levels in the brain or in plasma at 30 min after codeine injection ............................. 79
Figure 23. Inhibitor-treated rats had similar morphine levels and morphine to
codeine ratios between the anterior and the posterior parts of the brain. ................ 80
Figure 24. Analgesia correlated with brain, and not plasma, morphine levels ......... 82
Figure 25. Analgesia correlated with brain, and not plasma, morphine to codeine
ratios ......................................................................................................................... 83
Figure 26. Analgesia correlated with brain, and not plasma, morphine to total drug
ratios ......................................................................................................................... 84
Figure 27. Analgesia did not correlate with codeine levels or total drug levels in
brain or plasma ......................................................................................................... 85
Figure 28. Inhibitor-treated rats did not have lower morphine levels in the brain at
60 or 90 min after codeine injection .......................................................................... 87
Figure 29. Inhibitor-treated rats did not have lower morphine to codeine ratios in the
brain or plasma at 60 or 90 min after codeine injection ............................................ 88
Figure 30. Inhibitor-treated rats did not have lower morphine to total drug ratios in
the brain or plasma at 60 or 90 min after codeine injection ...................................... 89
Figure 31. Inhibitor-treated rats did not have lower codeine levels or total drug
levels in the brain or plasma at 60 or 90 min after codeine injection ........................ 90
Figure 32. Inhibiting brain CYP2D in vivo lowered in vitro codeine metabolism to
morphine in brain membranes but not in liver microsomes ..................................... 92
Figure 33. Inhibiting brain CYP2D in vivo lowered in vitro dextromethorphan
metabolism to dextrorphan in brain membranes but not in liver microsomes ......... 94

ix
Summary of Abbreviations
ACSF Artificial cerebrospinal fluid
BBB Blood-brain barrier
CNS Central nervous system
COD Codeine
CYP Cytochrome P450
CYP2D Cytochrome P450 2D
CYP2D1 Cytochrome P450 2D1
CYP2D6 Cytochrome P450 2D6
EM Extensive metabolizer
h Hour
HPLC High-performance liquid chromatography
i.c.v. Intracerebroventricular
i.p. Intraperitoneal
MBI Mechanism-based inhibitor
min Minute
MOR Morphine
PM Poor metabolizer
s.c. Subcutaneous
sec Second
UM Ultra-rapid metabolizer

1
Section 1: Introduction
Statement of Research Problem
Cytochrome P450 2D6 (CYP2D6) is an oxidative enzyme that metabolizes many
centrally-acting drugs, including clinically prescribed drugs (e.g. risperidone,
fluoxetine, codeine) as well as drugs of abuse (e.g. amphetamine, MDMA) (Zanger,
Raimundo et al. 2004). CYP2D6 is primarily expressed in the liver but is also
expressed in the brain. Brain CYPs are active in vivo (Miksys and Tyndale 2009), and
in some cell types (e.g. frontal cortex pyramidal neurons) brain CYPs are expressed
at levels as high as those in the liver (Miksys, Hoffmann et al. 2000). Therefore,
CYP2D6 may metabolize centrally-acting drugs locally in the brain and have a
significant impact on drug effect. Examining the role of brain CYPs in drug
metabolism and response will help elucidate the function and importance of brain
CYP activity.
There is large interindividual variation in the response to centrally-acting
drugs, which does not always correlate with plasma drug levels (Michels and
Marzuk 1993a). This may be caused by variation in the degree of metabolism by
brain CYPs, which may affect local drug and metabolite levels in the brain, and in
turn influence drug response.
Brain CYP2D6 levels can vary independently from hepatic CYP2D6 levels.
Brain CYP2D6, unlike hepatic CYP2D6, is induced in animals by nicotine and ethanol

2
(Warner and Gustafsson 1994, Mann, Miksys et al. 2008, Yue, Miksys et al. 2008), and
its levels are higher in smokers and alcoholics (Miksys, Rao et al. 2002, Miksys and
Tyndale 2004, Mann, Miksys et al. 2008). Brain CYP2D6 levels also increase with age
while hepatic CYP2D6 remain the same or even decrease with age (Parkinson,
Mudra et al. 2004, Mann, Miksys et al. 2012). Therefore, environmental factors and
age may result in variation in brain CYP2D6 expression and activity, and thereby
alter the metabolism of, and response to, centrally-acting drugs.
While studies have suggested that brain CYP2D can metabolize drugs in vitro
and in vivo, no studies have examined whether brain CYP2D-mediated drug
metabolism can affect drug response. We thus seek to elucidate the effects of
altering brain CYP2D activity (without affecting liver CYP2D) on drug response.
Clarifying the role of brain CYP-mediated metabolism in drug response may help
explain, at least in part, the interindividual variation in the response to centrally-
acting drugs and the poor correlation between the plasma levels and effects of these
drugs.
Purpose of the Study and Objective
Brain CYPs have been shown to be active in vitro (Albores, Ortega-Mantilla et al.
2001) and in vivo (Miksys and Tyndale 2009), yet it is not clear what their precise
function is in drug metabolism and effect. Thus, the purpose of this study is to
elucidate the impact of brain CYPs on drug biotransformation and response.
CYP2D’s many centrally-acting substrates and in vitro activity in rat brain make it a

3
suitable enzyme for examining the role of brain CYP-mediated metabolism in drug
action. Our objective is to examine the influence of brain CYP2D on the metabolic
activation of a centrally-acting drug (codeine) and on the drug’s subsequent effect
(codeine-induced analgesia).
The role of brain CYP2D could be investigated by manipulating its activity
while leaving hepatic CYP2D activity unchanged. Rat brain CYP2D could be
selectively inhibited in vivo by intracerebroventricular (i.c.v.) injection of CYP2D
inhibitors, without affecting hepatic CYP2D. This provides an animal model of
reduced brain CYP2D activity that could be used to assess the contribution of brain,
as opposed to liver, codeine metabolism to codeine-induced analgesia. Clarifying
the impact of brain CYP2D-mediated metabolism on a drug effect will help us
understand the possible role brain CYPs have in interindividual variation in drug
response.
Statement of Research Hypotheses and Rationale
CYP2D metabolizes the opioid analgesic codeine (prodrug) to morphine (active
metabolite) (Adler, Fujimoto et al. 1955). Since morphine has a 3000-fold greater
affinity for the mu-opioid receptor than does codeine (Pert and Snyder 1973), and
CYP2D6 poor metabolizers produce no morphine from codeine and experience no
analgesia (Sindrup, Brosen et al. 1990, Chen, Somogyi et al. 1991), analgesia from
codeine is dependent on its metabolism to morphine. Codeine is metabolized to
morphine mainly by hepatic CYP2D; morphine then crosses into the brain where it

4
can interact with mu-opioid receptors to elicit analgesia. However, because
morphine is less permeable across the blood-brain barrier than is codeine, there is
a delay in morphine’s entry into the brain compared to codeine’s entry (Oldendorf,
Hyman et al. 1972). Thus, the initial morphine present in the brain after codeine
administration may be solely due to local metabolism of codeine in the brain. This is
supported by rat brain CYP2D’s ability to metabolize codeine in vitro (Chen, Irvine
et al. 1990), and the finding that morphine can be detected in rat brain at 30 min after
intraperitoneal codeine, but not morphine, injections. This implies that at 30 min
after codeine injection, morphine formed from hepatic metabolism has not yet
crossed into the brain, and that morphine found in the brain at this time is due to
brain CYP2D-mediated codeine metabolism.
Hypothesis: We hypothesize that inhibiting rat brain CYP2D will reduce
analgesia during the initial period after codeine administration by decreasing the
metabolism of codeine to morphine in the brain. More specifically, we hypothesize
that i.c.v. injection of CYP2D inhibitors will result in 1) lower brain morphine
concentrations and 2) shorter tail-flick latencies (a measure of analgesia) during the
first 30 min after peripheral codeine injection, compared to i.c.v. injection of vehicle.

5
Review of the Literature
1.1 Cytochrome P450 2D6 (CYP2D6)
1.1.1 Cytochromes P450
Cytochromes P450 (CYPs) are a superfamily of heme-containing enzymes that
oxidize a wide range of substrates (Estabrook 1999, Coon 2005), including drugs,
toxins, and endogenous substances (Rendic and Di Carlo 1997). Most drug-
metabolizing CYPs of the CYP2 family are found mainly in the liver, but also in other
organs such as the brain, intestines and lungs. The liver is thought to be responsible
for systemic drug metabolism, while the other organs may take part in localized, in
situ substrate metabolism (Ding and Kaminsky 2003). The expression and activity
level of a CYP may differ between individuals because of genetic variation and/or
exposure to environmental inducers or inhibitors (Lee, Miksys et al. 2006b, Ai, Li et
al. 2009).
Cytochrome P450 2D6 (CYP2D6), one of the 57 members of the CYP
superfamily in humans (Nelson 2006), makes up only 5% of total CYP content in the
liver (Guengerich 2003, Emoto, Murase et al. 2006) yet is involved in the metabolism
of ~30% of clinically used drugs (Zanger, Raimundo et al. 2004). Its extensive role in
drug metabolism makes it important to understand the function of this enzyme.

6
1.1.2 CYP2D6 substrates
CYP2D6 is capable of metabolically activating or inactivating a wide variety of both
exogenous and endogenous substances (Zanger, Raimundo et al. 2004). These
include clinically used drugs (analgesics, antidepressants, antipsychotics, -
adrenergic blockers, antiarrhythmics), recreational drugs (amphetamine, MDMA) as
well as neurotoxins, neurosteroids, and biogenic amines.
When a CYP is the main contributor (>80%) to the metabolic reaction of a
substrate, that substrate can be used as a probe drug (Frank, Jaehde et al. 2007).
That is, the ratio of parent compound to metabolite resulting from the enzymatic
reaction may be used as an indicator of that CYP’s activity. One such probe drug is
dextromethorphan, which is oxidized predominantly by CYP2D6 to dextrorphan
(Frank, Jaehde et al. 2007, Zhou 2009).
1.1.3 CYP2D6 inhibitors
The activity level of CYPs can be reduced by inhibitors. This can affect drug efficacy
or cause adverse drug reactions. For example, individuals pretreated with the
CYP2D6 inhibitor quinidine produce little morphine from codeine and experience
reduced analgesia (Sindrup, Arendt-Nielsen et al. 1992).
There are different types of CYP inhibitors, including competitive inhibitors
and mechanism-based inhibitors (MBIs; also known as suicide or irreversible
inhibitors). A competitive inhibitor binds at, and thereby blocks, the CYP’s
substrate-binding site (de Groot, Wakenhut et al. 2009). A MBI is a CYP substrate

7
that forms a reactive metabolite which binds covalently to the enzyme (Bertelsen,
Venkatakrishnan et al. 2003, Van, Heydari et al. 2006). MBIs cause the irreversible
loss of enzyme function, requiring the synthesis of new enzyme before activity is
restored.
The antiarrhythmic propafenone is a CYP2D6 competitive inhibitor with a Ki of
2.9 M (Kroemer, Fischer et al. 1991). It also blocks sodium channels and is used to
treat cardiac arrhythmias (Dukes and Vaughan Williams 1984).
The -adrenergic receptor blocker, propranolol, is a selective substrate with
a high affinity for CYP2D6 (Yamamoto, Suzuki et al. 2003). It is used to treat
hypertension, angina pectoris, and cardiac arrhythmias (Komura and Iwaki 2005). It
is also a potent MBI of CYP2D6 (Masubuchi, Narimatsu et al. 1994), with a Ki of 1 μM
(Rowland, Yeo et al. 1994). Propranolol undergoes 4-hydroxylation by CYP2D which
is associated with the formation of a reactive metabolite in the active site of the
enzyme. This reactive species covalently binds to the active site, thereby
inactivating the enzyme (Rowland, Yeo et al. 1994, Narimatsu, Arai et al. 2001).
CYP2D6 inactivates the neurotoxin 1-methyl-4-phenylpyridinium (MPP+); treating
cells with 0.1–30 M propranolol significantly increased the neurotoxicity and cell
death caused by MPP+ (Mann and Tyndale 2010).
1.1.4 CYP2D6 regulation
Hepatic CYP2D6 is constitutively expressed during adulthood (Transon, Lecoeur et
al. 1996, Stevens, Marsh et al. 2008) and is uninducible by common CYP inducers

8
such as phenobarbital (Rae, Johnson et al. 2001, Edwards, Price et al. 2003). Even so,
the promoter of CYP2D6 contains binding sites for positively and negatively acting
transcription factors such as Oct-1, YY-1, heterogeneous nuclear ribonucleoprotein
K and GABP (Yokomori, Kobayashi et al. 1995, Mizuno, Takahashi et al. 2003, Sakai,
Sakamoto et al. 2009). Hepatic CYP2D6 is transcriptionally regulated in large part by
the hepatocyte nuclear factor-4α (HNF-4α) transcription factor (Cairns, Smith et al.
1996, Jover, Bort et al. 1998, Corchero, Granvil et al. 2001). Expression of this
transcription factor is correlated with CYP2D6 expression (Cairns, Smith et al. 1996,
Corchero, Granvil et al. 2001). Expression of HNF-4α is highest in the liver and
kidneys, which is also where CYP2D6 expression is highest (Gonzalez 1990, Xie,
Liao et al. 2009).
1.1.5 CYP2D6 genetic variation
CYP2D6’s role in metabolizing an extensive range of commonly prescribed drugs
makes the wide interindividual variation in its functional levels important. There are
more than 80 known CYP2D6 allelic variants
(http://www.cypalleles.ki.se/cyp2d6.htm) which include gene deletions, frameshift
mutations, insertions, synonymous and non-synonymous substitutions, and copy
number variants (Gaedigk, Simon et al. 2008). These variants can be grouped into
null, reduced, normal, and increased function alleles. Individuals can be grouped
based on CYP2D6 genotype into four CYP2D6 phenotypic categories: poor
metabolizer (PM) (Rae, Johnson et al. 2001), intermediate metabolizer (IM),

9
extensive metabolizer (EM), and ultra-rapid metabolizer (UM) (Gaedigk, Simon et al.
2008).
Genetic variation in CYP2D6 is a major contributor to the interethnic and
interindividual differences in CYP2D6 activity. This variation in CYP2D6 activity can
then affect an individual’s response to the numerous drugs which are metabolically
activated or inactivated by CYP2D6. For example, PMs experience no analgesia
from codeine, which is activated by CYP2D6 (Sindrup, Brosen et al. 1990, Chen,
Somogyi et al. 1991). PMs experience increased side effects from the antipsychotics
haloperidol and risperidone, which are metabolized by CYP2D6 (de Leon, Susce et
al. 2005, Ingelman-Sundberg, Sim et al. 2007). UMs have lower drug efficacy from
the antidepressant imipramine, which is inactivated by CYP2D6 (Schenk, van
Fessem et al. 2008).
1.1.5a Interethnic variability in CYP2D6
The frequencies of CYP2D6 alleles that result in different levels of enzyme activity
vary substantially between ethnic groups, and this contributes to the interethnic
variability in CYP2D6 activity. Caucasians have the highest prevalence of PMs (5-
10%) (Zanger, Raimundo et al. 2004), which is largely (70-90%) due to the high
frequency (20-25%) of the CYP2D6*4 allele in this population (Zanger, Raimundo et
al. 2004, Neafsey, Ginsberg et al. 2009, Abraham, Maranian et al. 2010). Individuals
of African descent have the highest frequency (20-34%) of the CYP2D6*17 reduced-
function allele, which results in much lower CYP2D6 activity than wild-type

10
(Gaedigk, Bhathena et al. 2005). The most prevalent (37-70%) variant among Asians
is the reduced-function allele CYP2D6*10, which results in lower CYP2D6 activity
than wild-type (Garcia-Barcelo, Chow et al. 2000, Neafsey, Ginsberg et al. 2009).
CYP2D6 copy number variants, which result in a UM phenotype, are the most
frequent in North African (28-56%) and Middle Eastern (3-10%) populations.
Ethiopians, Saudi Arabians, and Spaniards have the highest occurrence CYP2D6
UMs, which comprise ~29%, ~20% and ~10% of the population, respectively
(Agundez, Ledesma et al. 1995, Dahl, Johansson et al. 1995, Aklillu, Persson et al.
1996, McLellan, Oscarson et al. 1997, Sachse, Brockmoller et al. 1997). This
interethnic variation in CYP2D6 may make some populations more susceptible to
adverse drug reactions or altered drug efficacy (De Gregori, Allegri et al. 2010).
1.1.6 CYP2D expression in different species
Members of the CYP2D subfamily have been identified among many mammalian
species including human, monkey, rat and mouse. While CYPs are mostly well
conserved across species (Lin 1995), small genetic differences can cause
interspecies differences in CYP expression, activity, substrate specificity and
regulation. Such is the case for CYP2D.
Whereas CYP2D6 is the only functional CYP2D isozyme in humans, mice have
nine different isozymes of Cyp2d. While Cyp2d22 is the most similar out of these
nine to CYP2D6 in terms of amino acid identity (Yu and Haining 2006, McLaughlin,
Dickmann et al. 2008), it differs from CYP2D6 in substrate specificity (Blume,

11
Leonard et al. 2000, Yu and Haining 2006). In contrast to human CYP2D6, Cyp2d22
and other mouse Cyp2ds have only a weak ability to 4-hydroxylate debrisoquine
and O-demethylate dextromethorphan (Lofgren, Hagbjork et al. 2004, Yu, Idle et al.
2004, Yu and Haining 2006, McLaughlin, Dickmann et al. 2008, Shen and Yu 2009).
Rats have six different CYP2D isozymes: CYP2D1, 2, 3, 4, 5 and 18. These vary
in substrate specificity, metabolism, and inhibition profiles (Strobl, von Kruedener et
al. 1993, Hiroi, Chow et al. 2002); they also have different tissue-specific expression
patterns, with CYP2D1 and CYP2D2 being the isozymes most abundant in the liver
(Wyss, Gustafsson et al. 1995, Haduch, Bromek et al. 2011). CYP2D1 has 71% amino
acid identity to human CYP2D6 (Funae, Kishimoto et al. 2003) and is believed to be
the rat homologue of human CYP2D6 (Miksys, Rao et al. 2000). CYP2D1 is capable of
performing many CYP2D6-mediated reactions, such as codeine and
dextromethorphan O-demethylation, and debrisoquine 4-hydroxylation (Matsunaga,
Zanger et al. 1989, Xu, Aasmundstad et al. 1997, Miksys, Rao et al. 2000). These
similarities between rat and human CYP2D enzymes make the rat a useful model of
human CYP2D6-mediated drug metabolism.
1.2 Brain Cytochromes P450
Most CYPs are expressed mainly in the liver, where they are responsible for the
majority of drug metabolism and drug clearance in the body. However, metabolism
by CYPs in extrahepatic tissues may significantly affect drug efficacy by changing
the local, target-site drug concentrations. Moreover, because of the tissue-specific

12
ways that CYPs are regulated, different tissues may respond differently to the same
drug. For example, many centrally-acting drugs are both substrates and inducers of
brain CYPs (e.g. nicotine, phenytoin, ethanol (Miksys, Hoffmann et al. 2000,
Schoedel, Sellers et al. 2001, Howard, Miksys et al. 2003, Meyer, Gehlhaus et al.
2007)), and thus may alter the brain’s sensitivity to these drugs and other brain CYP
substrates. Since the total CYP content in the brain is only a fraction of that in the
liver (Hedlund, Gustafsson et al. 2001, Gervasini, Carrillo et al. 2004), it is unlikely
that metabolism by brain CYPs affects plasma drug levels (Hedlund, Gustafsson et
al. 2001). However, their highly localized expression in different brain regions might
produce microenvironments in which brain CYP-mediated metabolism has a
significant impact on local drug levels and effect (Miksys and Tyndale 2002, Ghosh,
Gonzalez-Martinez et al. 2010). Metabolism by brain CYPs may be particularly
important for centrally-acting substrates which have active metabolites that are not
able to cross the blood-brain barrier. In such cases, the local production of
metabolites in brain may be crucial to the effect of drugs, toxins, and endogenous
neurochemicals.
1.2.1 Brain CYP expression
Of the 57 human CYP transcripts, 41 have been identified in the brain so far (Dauchy,
Dutheil et al. 2008, Dutheil, Dauchy et al. 2009). Only a fraction of these (i.e., CYP1A,
CYP1B, CYP2B, CYP2C, CYP2D, CYP2E, and CYP3A families) have been examined
in the brain at the transcript, protein, and/or activity level (Haining 2007, Dauchy,

13
Dutheil et al. 2008, Dutheil, Dauchy et al. 2009). In the brain, CYPs are expressed in
various cellular membranes including the plasma membrane, endoplasmic
reticulum, and mitochondrial membrane (Miksys, Rao et al. 2000, Howard, Miksys et
al. 2003, Miksys, Lerman et al. 2003, Haining 2007, Woodland, Huang et al. 2008,
Dutheil, Dauchy et al. 2009).
The expression of brain CYPs varies greatly depending on region and cell-
type (Dutheil, Dauchy et al. 2009). Within brain regions, CYPs are expressed at
different levels in pyramidal, Purkinje, granular, neuronal, astrocytic, and glial cells
(Miksys, Hoffmann et al. 2000, Howard, Miksys et al. 2003, Miksys, Lerman et al.
2003, Dutheil, Beaune et al. 2008). While the level of CYPs in the brain has been
estimated to be 1-10% of that in the liver (Hedlund, Wyss et al. 1996, Gervasini,
Carrillo et al. 2004), brain tissue is not homogenous and CYPs are not uniformly
expressed across regions, so this percentage range is unlikely to reflect all brain
regions or all CYPs. For example, there is a ~2.5-fold difference in CYP2B
expression between brain regions of highest and lowest expression in both humans
and rats (Miksys, Hoffmann et al. 2000, Miksys, Lerman et al. 2003). In fact,
expression levels of CYPs in brain cells (e.g., CYP2B in frontal cortex pyramidal
neurons) can be equal to, or higher than, levels in hepatocytes (Miksys, Hoffman et
al. 2000). The highly localized expression of brain CYPs is thought to create
microenvironments in which local, in situ drug metabolism occurs in the brain (Britto
and Wedlund 1992, Miksys and Tyndale 2009). This may in turn alter the local
pharmacokinetics and effect of these drugs.
CYPs are present at the BBB (Miksys, Rao et al. 2000, Miksys, Lerman et al.
2003, Dauchy, Dutheil et al. 2008, Ghosh, Gonzalez-Martinez et al. 2010) as well as in

14
areas lacking BBB such as the choroid plexus and posterior pituitary (Volk,
Hettmannsperger et al. 1991, Ghersiegea, Perrin et al. 1993, Miksys, Rao et al. 2000).
This suggests that brain CYPs may play a role in assisting the BBB in preventing
drugs and toxins from entering the brain. Because some metabolites made in the
periphery are less permeable across the BBB, the formation of metabolites within the
brain can be crucial to the effect of centrally-acting drugs.
1.2.2 Brain CYP activity
Studies of in vitro brain CYP activity using brain homogenates have suggested that
brain CYPs are able to carry out the same reactions as hepatic CYPs. This has been
shown using different substrates including nicotine, chlorpyrifos and codeine
(Chambers and Chambers 1989, Chen, Irvine et al. 1990, Jacob, Ulgen et al. 1997).
Other in vitro studies have revealed brain CYPs to have similar substrate
specificities and affinities (Km) as their hepatic forms (Forsyth and Chambers 1989,
Lin, Kumagai et al. 1992, Bhamre, Anandatheerathavarada et al. 1993, Ghersiegea,
Perrin et al. 1993, Narimatsu, Yamamoto et al. 1999, Tyndale, Li et al. 1999, Bhagwat,
Boyd et al. 2000, Voirol, Jonzier-Perey et al. 2000). However, as cofactors were
added to these reactions, it is possible that there are insufficient endogenous levels
of these cofactors in the brain to carry out these reactions in vivo.
Studying in vivo brain CYP activity is challenging since peripheral metabolites
formed from hepatic metabolism can enter into the brain, thus making it hard to
distinguish metabolites formed from hepatic versus brain metabolism. Also, heme

15
levels in the brain are rate-limiting for at least some CYP functions, suggesting that
brain CYPs may not all be functional in vivo (Meyer, Lindberg et al. 2005). Another
rate-limiting factor may be a potential lack of the coenzyme NADPH-cytochrome
P450 oxidoreductase (POR), which is required for CYP function, near brain CYPs
(Miksys and Tyndale 2009). These factors contribute to the shortage of evidence for
in situ brain CYP activity.
The finding that the coenzyme POR is expressed in the same regions as brain
CYPs (Haglund, Kohler et al. 1984, Ghersiegea, Minn et al. 1989, Bergh and Strobel
1992, Bergh and Strobel 1996, Riedl, Watts et al. 1996, Conroy, Fang et al. 2010)
lends support to the feasibility of in situ brain CYP activity. It has recently been
shown that brain CYP2B protein is active in situ in living rats (Miksys and Tyndale
2009). Rats were pretreated on one side of the brain with a CYP2B MBI before
receiving bilateral intracerebral injections of a different radiolabeled CYP2B MBI.
This radiolabeled MBI becomes bound upon being metabolized by CYP2B. There
was significantly lower radiolabel binding on the inhibitor-treated side of the brain
compared to the untreated side (Miksys and Tyndale 2009). This demonstrated that
rat brain CYP2B is active in vivo, without the addition of cofactors.
The function of brain CYPs may be to protect against exogenous drugs and
toxins and/or to metabolize or catalyze the formation of endogenous compounds
such as neurosteroids and biogenic amines (Haining 2007). Certain CYPs can
metabolize or catalyze the formation of serotonin, dopamine, arachidonic acid,
pregnenolone, estradiol, androstenedione, testosterone, and melatonin (Rifkind, Lee
et al. 1995, Doostzadeh and Morfin 1997, Rosenbrock, Hagemeyer et al. 1999, Ohe,
Hirobe et al. 2000, Wang, Napoli et al. 2000, Fradette, Yamaguchi et al. 2004, Ma,

16
Idle et al. 2005, Bromek, Haduch et al. 2010). CYP2B metabolically inactivates the
anaesthetic propofol; inhibiting brain (but not hepatic) CYP2B resulted in longer
propofol-induced sleep times in rats (Khokhar and Tyndale 2011), suggesting that
brain CYP activity can have a meaningful impact on drug response.
1.2.3 Brain CYP regulation
Brain CYPs are induced in different ways depending on the CYP, brain region, cell
type, and inducer, and are also regulated differently from their hepatic forms
(Miksys and Tyndale 2002, Miksys and Tyndale 2004). Inducers of hepatic CYPs do
not always induce the corresponding CYPs in the brain, and vice versa. For
example, nicotine and ethanol can induce CYPs in vivo in an organ- and CYP-specific
way. Nicotine induces CYP2E1 in both the brain and liver in monkeys and rats, but it
induces CYP2B and CYP2D only in the brain (Miksys, Hoffmann et al. 2000, Joshi and
Tyndale 2006, Lee, Miksys et al. 2006a, Mann, Miksys et al. 2008, Yue, Miksys et al.
2008, Yue, Khokhar et al. 2009). Ethanol induces CYP2E1 in both the brain and liver
in rats, but it induces CYP2B and CYP2D in the liver only (Warner and Gustafsson
1994, Schoedel, Sellers et al. 2001, Howard, Miksys et al. 2003, Schoedel and
Tyndale 2003). In humans, cigarette smoking and alcohol use are both associated
with higher levels of CYP2B6, CYP2D6 and CYP2E1 in certain brain regions (Miksys,
Rao et al. 2002, Howard, Miksys et al. 2003, Miksys, Lerman et al. 2003, Miksys and
Tyndale 2004).

17
The induction of brain CYPs can alter drug effect and contribute to the
interindividual variation in response to centrally-acting drugs (Chimbira and
Sweeney 2000, Jabs, Bartsch et al. 2003, Funck-Brentano, Boelle et al. 2005,
Lysakowski, Dumont et al. 2006, George, Sacco et al. 2008). Drug plasma levels do
not always correlate well with drug response, and this is particularly the case for
certain centrally-acting drugs such as antipsychotics and antidepressants (Michels
and Marzuk 1993a, Nelson, Mazure et al. 1995, Lane, Chiu et al. 2000, Spina, Avenoso
et al. 2001, Riedel, Schwarz et al. 2005 ). Even at plasma levels of these drugs that
are expected to produce maximal therapeutic effects and minimal adverse effects,
there can be either no therapeutic effect or adverse side effects (Michels and Marzuk
1993a, Michels and Marzuk 1993b). This phenomenon may be partly accounted for
by drug metabolism occurring in the brain. Induction of brain CYPs as a result of
exposure to nicotine or alcohol may magnify this effect and contribute to
interindividual variation in drug response. In support of this, smokers require
higher doses of the anaesthetic propofol, which is inactivated by CYP2B6, in order to
achieve loss of consciousness, consistent with smokers having increased CYP2B6
levels and experiencing less adverse side effects from propofol (Chimbira and
Sweeney 2000, Lysakowski, Dumont et al. 2006).
1.2.4 Brain CYP2D6
CYP2D6 may be an important enzyme in the brain as it metabolizes many centrally-
acting substrates which include clinically prescribed drugs (Zanger, Raimundo et al.

18
2004) as well as drugs of abuse, neurotoxins and endogenous neurochemicals
(Miksys, Rao et al. 2000, Mann, Miksys et al. 2008).
1.2.4a Brain CYP2D expression
CYP2D expression has been detected in the brain of rat, mouse, dog, monkey, and
human (Fonne-Pfister, Bargetzi et al. 1987, Niznik, Tyndale et al. 1990, Tyndale,
Sunahara et al. 1991, Tyndale, Li et al. 1999, Siegle, Fritz et al. 2001, Miksys, Cheung
et al. 2005). In rats, CYP2D1, CYP2D4, CYP2D5 and CYP2D18 have been detected in
the brain (Komori 1993, Wyss, Gustafsson et al. 1995, Coleman, Spellman et al. 2000,
Miksys, Rao et al. 2000). CYP2D4 is mainly expressed in the brain and is the most
abundant CYP2D isozyme in the rat brain (Komori 1993, Wyss, Gustafsson et al.
1995), while CYP2D18 is thought to be expressed only in the brain (Coleman,
Spellman et al. 2000). Rat CYP2D2 and CYP2D3 have yet to be found in the brain
(Miksys, Rao et al. 2000).
In humans, CYP2D6 is expressed in most brain regions, including the
neocortex, caudate, putamen, globus pallidus, nucleus accumbens, hippocampus,
hypothalamus, thalamus, substantia nigra, cerebellum, and medulla oblongata
(Gilham, Cairns et al. 1997, McFayden, Melvin et al. 1998, Siegle, Fritz et al. 2001,
Miksys, Rao et al. 2002). CYP2D6 protein levels are highest in the caudate, putamen,
cortex, and cerebellum (Miksys, Rao et al. 2002).
In rats, CYP2D protein is also expressed in most brain regions, and moderate
to high levels are found in the cerebellum, hippocampus, medulla oblongata, pons,

19
cerebral cortex, striatum (caudate/putamen), thalamus, substantia nigra, choroid
plexus, and amygdaloid complex (Miksys, Rao et al. 2000).
Human CYP2D6 protein is expressed in a cell type-specific manner in the
brain. High levels are found in pigmented neurons of the substantia nigra, pyramidal
cells of the hippocampus and frontal cortex, Purkinje cells of the cerebellum, glial
cells, astrocytes, and endothelial cells at the BBB (Gilham, Cairns et al. 1997, Siegle,
Fritz et al. 2001, Miksys, Rao et al. 2002, Dauchy, Miller et al. 2009, Dutheil, Jacob et
al. 2010), similar to the cell types in which rat brain CYP2D is expressed (Michels
and Marzuk 1993a, Michels and Marzuk 1993b, Watts, Riedl et al. 1998, Riedl, Watts
et al. 1999).
1.2.4b Brain CYP2D function and activity
CYP2D6 activity in the brain may be important as many of CYP2D6’s substrates act
within the CNS. These include clinically prescribed drugs such as the
antidepressants fluoxetine and paroxetine, the analgesics codeine and oxycodone,
and the antipsychotics risperidone and haloperidol (Zanger, Raimundo et al. 2004).
Some CYP2D6 substrates are also commonly abused drugs such as codeine,
oxycodone, MDMA and dextromethorphan (Zanger, Raimundo et al. 2004). Brain
CYP2D6-mediated metabolism of these drugs could alter their disposition within the
brain and thereby alter their efficacy, side-effect profile and abuse liability. We are
thus interested in studying brain CYP2D activity and its role in drug metabolism and
response.

20
Brain CYP2D activity has been demonstrated in vitro using hydroxylation of
bufuralol and MDMA and demethylation of codeine and dextromethorphan in rats,
and sparteine demethylation in dogs (Chen, Irvine et al. 1990, Tyndale, Sunahara et
al. 1991, Lin, Kumagai et al. 1992, Jolivalt, Minn et al. 1995, Tyndale, Li et al. 1999,
Coleman, Spellman et al. 2000, Voirol, Jonzier-Perey et al. 2000). CYP2D substrate
affinities (Km) in the brain are comparable to those in the liver; however, because
CYP expression is lower in the brain, the maximal velocity (Vmax) and substrate
turnover (Vmax/Km) are lower (Tyndale, Sunahara et al. 1991, Coleman, Spellman et
al. 2000).
Cultured SH-SY5Y (a neuron-like cell line) cells can metabolize codeine to
morphine, a reaction catalyzed by CYP2D6 (Poeaknapo, Schmidt et al. 2004). These
cells can also metabolize the CYP2D probe drug 3-[2-(N,N-diethyl-N-
methylammonium)-ethyl]-7-methoxy-4-methylcoumarin, and this reaction was
inhibited by CYP2D inhibitors (Mann and Tyndale 2010). These findings suggest that
brain CYP2D is active and carries out these enzymatic reactions.
The formation of dextrorphan from dextromethorphan has been demonstrated
in rat brain membranes (Tyndale, Li et al. 1999). This reaction was inhibited by
classic CYP2D inhibitors and by antibodies raised against CYP2D1, but not by
inhibitors or antibodies against CYP2B, CYP2C or CYP3A. Rat CYP2D activity varies
across brain regions, and there was also a strong correlation of dextromethorphan
O-demethylation with brain CYP2D1 mRNA levels, as well as with brain CYP2D
protein levels. The rat cerebellum displayed the highest dextromethorphan
metabolism and CYP2D protein levels. These findings suggest that CYP2D1 is
responsible for this reaction in rat brain. Because dextromethorphan is a CYP2D6

21
probe drug and its O-demethylation is measure of human CYP2D6 activity, this
suggests that CYP2D1 is the rat homologue of human CYP2D6 (Miksys, Rao et al.
2000, Frank, Jaehde et al. 2007). CYP2D1 has been shown to perform other CYP2D6-
mediated reactions as well, such as codeine O-demethylation and debrisoquine 4-
hydroxylation (Matsunaga, Zanger et al. 1989, Xu, Aasmundstad et al. 1997).
The activity of brain CYP2D in vivo has been suggested by the local
metabolism of codeine to morphine in rat brain during the first 30 minutes after
peripheral codeine injection, which is believed to be the responsible for brain
morphine levels and analgesia at this time point (Chen, Irvine et al. 1990). Rats were
injected with either codeine (20 mg/kg, i.p.) or morphine (1 mg/kg, i.p.) at doses
that produced equal plasma morphine levels between the two drugs. At 30 min after
injection, while plasma morphine levels were the same between codeine- and
morphine-treated rats, morphine in the brain was found in the codeine-treated rats
but could not be detected in the morphine-treated rats (Figure 1). This strongly
suggests that at this time point, morphine formed from hepatic metabolism has not
yet crossed into the brain.

22
1.a) Plasma b) Brain
Figure 1. Morphine levels in rat plasma and brain 30 min after peripheral
injection of codeine or morphine. Morphine concentrations in (a) plasma and (b)
brain after i.p. injection of 20 mg/kg codeine phosphate (white bar) or 1 mg/kg
morphine sulphate (grey bar) (n=4/group). # no morphine detected. This figure is
adapted from Chen, Irvine et al. (1990).
0
400
800
1200[M
orp
hin
e] (n
g/m
l, m
ean
+ S
EM)
Codeine Morphine 0
12
24
36
[Mo
rph
ine]
(ng
/g, m
ean
+ S
EM)
Codeine Morphine
#

23
These findings are in concordance with the fact that morphine has one less
methyl group than codeine, which is expected to make morphine less lipid soluble
and therefore less able to cross the BBB. In one study (Oldendorf, Hyman et al.
1972), the brain uptake of codeine or morphine (i.e., the brain content of the drug as
a percentage of a highly diffusible reference substance injected simultaneously) was
measured in rats. At 15 sec after intra-arterial injection, the uptake of codeine was
24%, whereas the uptake of morphine was below quantification. At 30 sec after
intravenous injection of codeine or morphine, the uptake of codeine was nearly
complete whereas this was much lower with morphine. These findings indicate that
codeine enters the brain faster than morphine does. Morphine is also transported
out of the brain by efflux transporters at the BBB, and this could contribute to the
delay in antinociception after morphine administration in rats (Bouw, Gardmark et
al. 2000). These findings suggests that morphine found in the brain during the first 30
min after codeine administration may be due to local morphine formation in the
brain (Chen, Irvine et al. 1990). In addition, in rat brains infused with the neurotoxin
MPTP, there was local inactivation of MPTP to PTP in the striatum, a reaction
catalyzed by CYP2D (Vaglini, Pardini et al. 2004). Thus, while the study by Chen,
Irvine et al. (1990) did not explicitly determine whether morphine found in the brain
after codeine injection was formed by brain CYP2D-mediated metabolism, together
these studies suggest that CYP2D is capable of metabolizing substrates in situ in the
brain.
In addition to metabolizing many centrally-acting drugs and inactivating
neurotoxins, CYP2D6 can also metabolize endogenous compounds found in the
brain, such as biogenic amines (Yu, Idle et al. 2003a). For example, cDNA expressed

24
CYP2D6 and CYP2D6 transgenic mice can metabolize 5-methoxytryptamine to
serotonin (Yu, Idle et al. 2003a). Rat brain membranes, as well as CYP2D6 expressed
in yeast cells, can convert tyramine to dopamine (Hiroi, Imaoka et al. 1998, Bromek,
Haduch et al. 2010). These findings suggest that brain CYP2D6 activity may have an
impact on behaviour or personality (Hiroi, Imaoka et al. 1998, Yu, Idle et al. 2003a),
which is further supported by the association of CYP2D6 genotype with personality
traits (Gan, Ismail et al. 2004, Roberts, Luty et al. 2004, Kirchheiner, Lang et al. 2006,
Penas-Lledo, Dorado et al. 2009, Gonzalez, Penas-Lledo et al. 2008).
1.2.4c Brain CYP2D regulation
Brain CYP2D, unlike hepatic CYP2D, is inducible by various centrally-acting drugs
in a drug- and brain region-specific way (Rae, Johnson et al. 2001, Edwards, Price et
al. 2003). For example, the antipsychotic clozapine increased levels of rat CYP2D
protein in neurons of the substantia nigra, ventral tegmental area, olfactory bulb,
and cerebellum but it did not alter hepatic levels (Hedlund, Wyss et al. 1996).
CYP2D mRNA levels in the brain were unaltered, which suggests that brain CYP2D is
induced at the posttranscriptional level. The antidepressant fluoxetine produced an
increase in CYP2D protein and activity in rat cerebellum (Haduch, Bromek et al.
2011). Rats treated with thioridazine had an increase in CYP2D protein and activity in
the substantia nigra and cerebellum (Haduch, Bromek et al. 2011). Toluene caused
an increase in CYP2D4 mRNA, protein, and activity in rat brain (Mizuno, Hiroi et al.
2003).

25
Nicotine and ethanol have been shown to induce brain CYP2D in multiple
species. In rats, chronic (7 day) nicotine treatment increased CYP2D protein in the
cerebellum, hippocampus, and striatum (Yue, Miksys et al. 2008). The CYP2D mRNA
was unchanged, suggesting that induction is due to posttranscriptional modification.
In addition, hepatic CYP2D was unaltered. Acute ethanol treatment induced rat brain
CYP2D (Warner and Gustafsson 1994). In monkeys, nicotine treatment increased
CYP2D protein in the brain when compared to saline-treated animals; there was no
change in hepatic CYP2D (Mann, Miksys et al. 2008). Ethanol self-administration in
monkeys increased brain CYP2D without altering hepatic CYP2D (Miller, Miksys et
al. 2012). In humans, we have observed elevated brain CYP2D6 levels in alcoholics
compared to non-alcoholics and smokers compared to non-smokers (Miksys, Rao et
al. 2002, Miksys and Tyndale 2004).
In addition to regulation by environmental inducers, brain CYP2D is also
under developmental and hormonal regulation. Brain CYP2D6 levels increase with
age (Mann, Miksys et al. 2012). Estrogen and testosterone can change brain CYP2D
mRNA levels in ovariectomized rats (Bergh and Strobel 1996). Testosterone induced
brain CYP2D, whereas testosterone combined with estrogen treatment reduced
brain CYP2D induction, suggesting that estrogen may block the induction of brain
CYP2D levels.
The mechanism(s) of brain CYP2D induction is as of yet unknown. Because
nicotine does not increase CYP2D mRNA levels (Hedlund, Wyss et al. 1996, Mann,
Miksys et al. 2008, Yue, Miksys et al. 2008), this implies that CYP2D induction by
nicotine occurs via post-transcriptional events. These could potentially include
decreased splicing, increased translation, increased enzyme stabilization, and

26
decreased protein degradation. In rats, nicotine regulates certain ubiquitinating
proteins in the brain (Kane, Konu et al. 2004), which may alter CYP2D levels by
affecting degradation.
Increased levels of brain CYP2D6 may result in increased substrate
metabolism, which in turn may lead to altered efficacy of clinical drugs, as well as
altered susceptibility to adverse drug reactions. Evidence for this comes from the
observations that smokers have less extrapyramidal side effects from antipsychotics
(inactivated by CYP2D6) than nonsmokers (Jabs, Bartsch et al. 2003), and that
smokers and seniors experience less efficacy from antidepressants (inactivated by
CYP2D6) than nonsmokers and younger patients (Nelson, Mazure et al. 1995,
George, Sacco et al. 2008). The higher levels of brain CYP2D6 in smokers and older
individuals may increase the inactivation of these drugs in the brain, resulting in
reduced therapeutic and/or adverse effects.
1.3 Opioid Analgesics
1.3.1 Codeine
CYP2D6 metabolizes the opioid analgesic codeine (prodrug) to morphine (active
metabolite) (Adler, Fujimoto et al. 1955). Opioids confer their analgesic effects
through interacting with mu-opioid receptors, which are ‘Gi/Go-coupled’ receptors
(Law, Wong et al. 2000). Morphine has much greater (3000-fold) affinity for mu-
opioid receptors than does codeine (Pert and Snyder 1973), so even though

27
morphine is a minor metabolite, codeine-induced analgesia is dependent on its
metabolism to morphine. Codeine must be metabolized to morphine to produce
analgesia in both humans (Chen, Somogyi et al.1991) and rats (Mikus, Somogyi et al.
1991, Cleary, Mikus et al. 1994), and this reaction is performed solely by CYP2D
(Thorn, Klein et al. 2009). In humans, CYP2D6 PMs and individuals pretreated with
the CYP2D6 inhibitor quinidine produce little to no morphine from codeine and
experience no analgesia (Sindrup, Brosen et al. 1990, Chen, Somogyi et al. 1991,
Sindrup, Arendt-Nielsen et al. 1992). In rats pretreated with i.p. injections of the
CYP2D1 inhibitor quinine, there was a substantial reduction in codeine-induced
analgesia compared to untreated rats (Cleary, Mikus et al. 1994). Furthermore,
female Dark-Agouti rats, which lack CYP2D1 and are an animal model of CYP2D6
PMs, experienced no analgesia from codeine (Cleary, Mikus et al. 1994). Therefore,
analgesia from codeine requires its conversion to morphine by CYP2D. Variation in
brain CYP2D activity may affect morphine levels in the brain after codeine
administration, which in turn may affect the analgesic response to codeine.
1.3.1a Codeine metabolism in humans
In the human liver, 50-70% of codeine is glucuronidated by UGT2B7 (Coffman, Rios
et al. 1997) and UGT2B4 (Court, Krishnaswamy et al. 2003) to codeine-6-glucuronide,
10-15% is N-demethylated by CYP3A4 to norcodeine (Caraco, Tateishi et al. 1996,
Yue and Sawe 1997), 0-15% is O-demethylated by CYP2D6 to morphine (Thorn,
Klein et al. 2009), and 5-15% is excreted unchanged. About 60% of morphine is

28
glucuronidated to morphine-3-glucuronide, and 5-10% is glucuronidated to
morphine-6-glucuronide (Lotsch, Stockmann et al. 1996, Ohno, Kawana et al. 2008).
Both of these conjugations are performed mainly by UGT2B7, with a small
contribution by UGT1A1 (Holthe, Klepstad et al. 2002). Morphine is also N-
demethylated to normorphine, mainly by CYP3A4 with CYP2C8 playing a smaller
role (Projean, Morin et al. 2003). Normorphine can also be formed by the O-
demethylation of norcodeine by CYP2D6 (Yue, Hasselstrom et al. 1991). Norcodeine
can be glucuronidated to norcodeine-6-glucuronide (Yue, Hasselstrom et al. 1991).
Besides morphine, the other metabolites of codeine that have an analgesic effect are
normorphine and morphine-6-glucuronide (Lasagna and De Kornfeld 1958,
Osborne, Joel et al. 1988)). Both of these first require the O-demethylation of codeine
(or of norcodeine) by CYP2D6 in order to be formed (Yue, Hasselstrom et al. 1991).
Therefore, O-demethylation by CYP2D6 is necessary in the formation of all of
codeine’s analgesic metabolites (Figure 2). The difference in the O-demethylation
of codeine can be as large as 25-fold between EMs and PMs and 45-fold between
UMs and the PMs (Yue, Alm et al. 1997).

29
Figure 2. Metabolic pathways of codeine in humans. Analgesic metabolites are
in uppercase.
Codeine
Codeine-6-glucuronide MORPHINE Norcodeine Unchanged codeine
MORPHINE- 6-GLUCURONIDE
Morphine- 3-glucuronide
NORMORPHINE Norcodeine- 6-glucuronide

30
In the human brain, CYP2D6 expression and activity are much higher than
those of CYP3A, which are undetectable in some areas (Voirol, Jonzier-Perey et al.
2000, Dutheil, Dauchy et al. 2009). Therefore, CYP2D6 may play a larger role in
codeine metabolism than CYP3A in the brain.
In addition to altering codeine-induced analgesia, variation in CYP2D6
activity also affects susceptibility to codeine toxicity and abuse. For example, when
a CYP2D6 UM who had 3 or more functional alleles received a small dose of codeine,
this lead to serious toxicity as a result of the high levels and fast rates of morphine
and morphine-6-glucuronide formed (Gasche, Daali et al. 2004). CYP2D6 PMs are
underrepresented among individuals dependent on oral opioid drugs, which
suggests that the O-demethylated metabolites of codeine confer its reinforcing
effects, and that low CYP2D6 activity may reduce susceptibility to codeine abuse
(Tyndale, Droll et al. 1997). This is further supported by studies which have
examined the effect of CYP2D6 inhibitors on codeine abuse liability. Individuals
pretreated with quinidine had lower plasma levels of O-demethylated metabolites
and experienced fewer positive subjective effects from codeine
(Kathiramalainathan, Kaplan et al. 2000). Fourteen long-term users of oral opioid
drugs (primarily codeine) who were treated with fluoxetine, a CYP2D6 inhibitor, had
a decrease in CYP2D6 activity as well as a 30% to 100% decrease in opioid use
(Romach, Otton et al. 2000). Therefore, variation in brain CYP2D6 activity due to
genetics, environmental inducers or age may lead to differences in brain morphine
levels after codeine administration, which may have implications for the analgesia as
well as abuse liability of codeine.

31
1.3.1b Codeine metabolism in rats
At 24 h after rats were given 10 mg/kg s.c. codeine, 23.9% of the dose was excreted
in the urine as morphine-3-glucuronide, 4.3% as free morphine, 1.6% as unchanged
codeine, and 0.2% as codeine glucuronide; morphine-6-glucuronide was not
detected (Oguri, Hanioka et al. 1990). Rat UGT2B1 only forms the 3-glucuronide, and
whereas rat UGT2B7 can glucuronidate at both the 3- and 6-positions, rat UGT2B7 is
ten times more efficient at catalyzing the 3-glucuronidation (Ritter 2000).
CYP2D1 is responsible for the O-demethylations of codeine to morphine and
norcodeine to normorphine in rats. When antibodies for different rat CYPs were
tested in rat liver microsomes, only the anti-CYP2D1 antibody significantly inhibited
the O-demethylation of these substrates (Xu, Aasmundstad et al. 1997). Also, the
specific CYP2D1 inhibitor quinine inhibited the codeine and norcodeine O-
demethylations, whereas the CYP3A inhibitor did not (Xu, Aasmundstad et al. 1997).
Furthermore, these O-demethylations were impaired in the liver microsomes of
female Dark Agouti rats, which are known to have reduced CYP2D activity (Xu,
Aasmundstad et al. 1997). These findings provide evidence that the metabolism of
codeine to morphine and norcodeine to normorphine in rats is mediated by
CYP2D1.
Codeine can be O-demethylated in rat brain in vitro as demonstrated by the
formation of morphine from codeine by rat brain homogenates (Chen, Irvine et al.
1990). There is also in vivo evidence of codeine metabolism in rat brain. As
described previously in Section 2.4.2, in rats that received peripheral injection of
either codeine or morphine, morphine was found in codeine-treated rats but could

32
not be detected in morphine-treated at 30 min after drug injection, even though
plasma morphine levels were similar from the two drugs. This indicates that at this
time point, morphine formed from hepatic metabolism has not yet crossed into the
brain. This is consistent with the lower lipophilicity (and therefore lesser ease of
crossing the BBB) of morphine compared to codeine and the quicker uptake of
codeine than morphine into the brain (Oldendorf, Hyman et al. 1972). Thus, during
the first 30 min after codeine administration, morphine found in the brain may be
due to local conversion of codeine to morphine in the brain (Chen, Irvine et al.
1990).
In further support of the impact of local codeine metabolism in the brain, both
mu-opioid receptors and CYP2D are widely distributed throughout the brain, and
many brain regions where rat CYP2D levels are moderate to high (cerebral cortex,
striatum (caudate/putamen), hippocampus, brainstem) also are dense with mu-
opioid receptors (Arvidsson, Riedl et al. 1995; Miksys, Rao et al. 2000). Thus,
morphine formed in the brain can immediately bind to the proximate mu-opioid
receptors. Variation in brain CYP2D6 activity may therefore affect morphine levels
in the brain after codeine administration, which in turn may affect response to
codeine.

33
1.3.2 Morphine
1.3.2a Spinal mechanisms of morphine-induced
analgesia
Nociception is the process by which stimuli capable of causing tissue damage
(noxious thermal, mechanical, or chemical stimuli) are detected by primary sensory
neurons called nociceptors (Basbaum and Jessell 2000). Nociceptors convey this
noxious information by projecting to the dorsal horn of the spinal cord, where they
release glutamate and peptides to excite second order neurons (Fields 2004). A
subset of these second order neurons, in turn, project and transmit pain messages to
areas in the brain including the thalamus, brainstem, and ultimately the cerebral
cortex (Basbaum, Bautista et al. 2009).
Because the dorsal horn of the spinal cord is where the first synapse in pain
transmission is located, it is an effective target for the inhibition of pain transmission
by opioids (Heinricher, Tavares et al. 2009). Morphine’s interaction with mu opioid
receptors within the dorsal horn results in the suppression of the release of
neurotransmitters by nociceptors, as well as the hyperpolarization of second order
neurons, thereby reducing the transmission of pain information to higher centres
(McFadzean 1988, Simonds 1988, Lipp 1991).

34
1.3.2b Supraspinal mechanisms of morphine-induced analgesia
Supraspinal (or descending) control of pain transmission at the dorsal horn of the
spinal cord is mediated by several brain areas, all of which have mu-opioid
receptors (Mansour, Khachaturian et al. 1988, Arvidsson, Riedl et al. 1995, Mansour,
Fox et al. 1995, Akil, Owens et al. 1998). The most studied of these areas is the
periaqueductal gray (PAG) - rostral ventromedial medulla (RVM) system, which is
regarded as the principal site of action of opioids (Yaksh, Yeung et al. 1976,
Hohmann, Suplita et al. 2005, Leith, Wilson et al. 2007). The PAG receives input from
the hypothalamus and limbic forebrain structures including the amygdala, as well as
from the spine. The PAG synapses with the RVM, which in turn terminates in the
dorsal horn (Heinricher, Tavares et al. 2009). There is evidence that activation of the
PAG-RVM system results in the release of serotonin and norepinephrine at the spinal
level, and that this mediates its pain-modulatory effects (Proudfit and Hammond
1981, Jensen and Yaksh 1986, Pang and Vasko 1986).
The PAG-RVM system can exert both inhibitory and facilitatory effects on pain
transmission (Heinricher, Tavares et al. 2009). These two opposing effects result
from the activity of two cell classes found in the PAG and RVM called ON-cells and
OFF-cells (Heinricher, Cheng et al. 1987, Fields, Heinricher et al. 1991). In keeping
with their role in pain regulation, RVM ON- and OFF-cells project specifically to
dorsal horn laminae involved in nociceptive transmission (Fields, Malick et al. 1995).
It is the OFF-cells that function as the pain-inhibiting output from the PAG-RVM
system and the ON-cells that are the pain-facilitating output (Heinricher and Ingram

35
2008). Pain threshold is lowest when ON-cells are active and OFF-cells are silent
(Heinricher, Barbaro et al. 1989, Heinricher, Haws et al. 1991).
Opioids analgesics produce their effects by modulating this system
(Heinricher, Morgan et al. 1994, Heinricher, McGaraughty et al. 1999). Opioids, by
acting on mu-opioid receptors, directly inhibit ON-cells. Opioids indirectly activate
OFF-cells, which do not express mu-opioid receptors (Fields, Heinricher et al. 1991,
Heinricher, Morgan et al. 1992, Heinricher, Morgan et al. 1994, Heinricher,
McGaraughty et al. 1999). OFF cells are inhibited by GABA (γ-aminobutyric acid)-
releasing cells (Heinricher, Morgan et al. 1992). These cells have mu-opioid
receptors, and opioids act on them to inhibit the release of GABA, and thereby
activate (disinhibit) OFF-cells (Vaughan and Christie 1997, Vaughan, Bagley et al.
2003).
1.4 Rat Tail-Flick Test: Animal Model of Nociception
1.4.1 Tail-flick reflex
The rat tail-flick test is one of the most widely used models of nociception (Hardy
1953, Hardy, Stoll et al. 1957, Le Bars, Gozariu et al. 2001). It consists of applying a
thermal stimulus in the form of an infrared heat beam to a rat’s tail, which triggers
the withdrawal of the tail by a quick, abrupt movement called the tail-flick reflex
(D'Amour and Smith 1941, Smith, D'Amour et al. 1943). The measured parameter is

36
the time from start of heat exposure to tail-flick reflex, and this is referred to as “tail-
flick latency” (TFL). A prolonging of TFL is an indication of analgesia.
The advantages of this method are as follows. It requires a simple apparatus
and is easy to perform on rats that have been habituated to manipulation. The tail-
flick reflex is easily observed and there is small interanimal variability in baseline
TFL (Le Bars, Gozariu et al. 2001). TFL stays the same with repeated testing if heat
intensity is kept constant and if tissue damage is avoided (Grossman, Basbaum et al.
1982). Tail flicks rarely occur spontaneously (Grossman, Basbaum et al. 1982).
Furthermore, this test is very sensitive to opioids (Le Bars, Gozariu et al. 2001).
Opioids are the only drugs that at nontoxic doses can inhibit tail-flick reflex during
extended (20-30 seconds) exposure to noxious heat (Grumbach 1975). This test is
able to predict the analgesic effects of opioids in humans (Archer and Harris 1965,
Grumbach 1966).
The nociceptors mediating the tail-flick reflex project to the superficial
laminae of the dorsal horn of the spinal cord (Grossman, Basbaum et al. 1982). There
is a high concentration of opioid receptors at this site (Pert, Kuhar et al. 1975, Atweh
and Kuhar 1977) and many of these are located on nociceptor terminals from the tail
(Lamotte, Pert et al. 1976, Fields, Emson et al. 1980). This is in line with the
observation that opioid drugs inhibit tail-flick reflex.
The tail-flick reflex is a spinal reflex, but it is under the influence of
supraspinal structures (Yaksh and Rudy 1978, Millan 2002). The tail-flick reflex can
be completely inhibited by electrical stimulation of brainstem regions (Grossman,
Basbaum et al. 1982). Microinjection of morphine or other opioids into the PAG or
RVM of the rat increases TFL (Jacquet and Lajtha 1973, Yaksh, Yeung et al. 1976,

37
Lewis and Gebhart 1977). When descending pathways are disrupted, such as after
spinal cord transection or cold-block, systemically administered morphine is less
effective in increasing TFL (Irwin, Houde et al. 1951, Basbaum, Clanton et al. 1976,
Basbaum, Marley et al. 1977, Sinclair, Main et al. 1988). These findings suggest that
the tail-flick test is a useful model for assessing supraspinally-mediated analgesia.
1.4.2 Effect of opioid analgesics in the tail-flick test
During instances of pain such as the tail-flick reflex, ON-cells become active and
OFF-cells are silenced (Barbaro, Heinricher et al. 1989, Heinricher, Barbaro et al.
1989). When opioids are microinjected into the PAG or RVM, or administered
systemically, OFF-cells fire continuously and more rapidly, and ON cells become
silent, with concurrent inhibition of tail-flick reflex (Fields 2004). When OFF-cell
activation is selectively blocked, morphine-induced analgesia is abolished
(Heinricher, McGaraughty et al. 1999). This indicates that OFF-cell activation is
necessary for morphine-induced analgesia to occur whether given systemically or
supraspinally (Heinricher, McGaraughty et al. 1997, Heinricher, McGaraughty et al.
2001).
Following electrical stimulation or microinjection of morphine in the PAG or
NRM, there is a release of serotonin in the spinal cord (Yaksh and Tyce 1979,
Hammond, Tyce et al. 1985). The analgesic effect on tail-flick reflex of morphine
microinjected into the PAG or NRM was diminished by intrathecal administration of
antagonists of serotonin and -adrenergic, but not opioid, receptors (Yaksh 1979,

38
Yaksh and Wilson 1979, Camarata and Yaksh 1985, Jensen and Yaksh 1986). These
findings suggest that supraspinal morphine activates downstream serotonergic and
-adrenergic systems which regulate spinal pain transmission (Jones and Gebhart
1988).
1.5 Study design
1. Rats received i.c.v. injections of either propranolol (mechanism-based CYP2D
inhibitor), propafenone (competitive CYP2D inhibitor) or the respective
vehicle.
2. Rats were used in one of five different types of experiments, to assess the
effect of i.c.v. CYP2D inhibitor treatment on:
a) Analgesia after codeine injection, as measured by tail-flick latency (TFL).
b) In vivo codeine metabolism, as measured by morphine and codeine levels
in brain and plasma after codeine injection.
c) In vitro CYP2D activity, as measured by morphine or dextrorphan
formation by brain membranes and liver microsomes incubated with
codeine or dextromethorphan.
d) Baseline nociception, as measured by TFL.
e) Analgesia after morphine injection, as measured by TFL.

39
Section 2: Materials and Methods
Animals
Male adult Wistar rats (250–300 g; Charles River, St-Constant, QC, Canada) were
kept in pairs or triplets under a 12 h artificial light/dark cycle (lights on at 6:00 AM).
Rats were handled, towel-restrained, and placed on the tail-flick meter daily to
acclimate them to testing procedures. All procedures were approved by the Animal
Care Committee at the University of Toronto.
Drug treatment
Propranolol hydrochloride (Sigma-Aldrich), a CYP2D mechanism-based inhibitor
(MBI) (Masubuchi, Narimatsu et al. 1994), was dissolved in artificial cerebrospinal
fluid (ACSF; 126 mM NaCl, 2.68 mM KCl, 1 mM Na2HPO4, 0.88 mM MgSO4, 22 mM
NaHCO3, 1.45 mM CaCl2, 11 mM D-glucose, pH 7.4), and 20-40 μg of the base was
injected intracerebroventricularly (i.c.v.) in a 1-4 μl volume. Propafenone
hydrochloride (Sigma-Aldrich), a CYP2D competitive inhibitor (Xu, Aasmundstad et
al. 1995), was dissolved in a 20% w/v solution of 2-hydroxypropyl-β-cyclodextrin
(cyclodextrin; Sigma-Aldrich) in water, and 40 μg of the base was given i.c.v. in a 4
µl volume. These two inhibitors were chosen for a variety of reasons as follows. They
inhibit CYP2D through different mechanisms, propranolol through mechanism-
based inhibition and propafenone through competitive inhibition (Kroemer, Fischer
et al. 1991; Masubuchi, Narimatsu et al. 1994). They selectively inhibit CYP2D
(Masubuchi, Fujita et al. 1991, Turpeinen, Korhonen et al. 2006, McGinnity, Waters et

40
al. 2008). They have different pharmacological actions and neither is expected to
have any central effects on its own that would influence analgesia or nociception
(Dukes and Vaughan Williams 1984, Komura and Iwaki 2005). The inhibitor doses
were chosen based on pilot studies showing that these doses did not inhibit hepatic
CYP2D (whereas higher doses did). The i.c.v. route of administration was used to
selectively target CYP2D in the brain (and not liver). Also, i.c.v. injection allows the
inhibitors to distribute across the entire brain and inhibit the total CYP2D content in
the brain.
Codeine phosphate (PCCA) was dissolved in sterile saline (0.9% NaCl; pH 7)
and injected subcutaneously (s.c.) at 30 mg base/kg body weight. This dose was
chosen based on a previous study that showed that this dose induced analgesia in
the tail-flick test in all animals tested over a 2 h time period (Cleary, Mikus et al.
1994). The s.c. route of administration was used because it avoids first-pass
metabolism by the liver, thus enhancing our ability to examine the potential role of
brain CYP2D-mediated codeine metabolism. Morphine sulfate (PCCA) was
dissolved in sterile distilled water and injected s.c. at 5 mg base/kg body weight.
This dose was chosen as it is the dose that produces equivalent analgesia in the tail-
flick test as the 30 mg/kg codeine dose used (Lewis, Sherman et al. 1981).
Inhibition of rat brain CYP2D via i.c.v. injection of CYP2D inhibitor
Rats were anesthetized with isoflurane and placed in a stereotaxic frame. In rats that
were to receive propranolol and be used for in vivo codeine metabolism
experiments or in vitro CYP2D activity experiments, rats received an i.c.v. injection
into the right lateral ventricle (Bregma coordinates: dorsal-ventral, -3.6; anterior-

41
posterior, -0.9; lateral, -1.4) (Paxinos and Watson 1986) of either 20 μg propranolol
(CYP2D MBI) dissolved in 1 μl ACSF, or 1 μl ACSF (vehicle control). All i.c.v.
injections were made over 120 sec, and the Hamilton syringe was left in place for
120 sec after injection.
In all rats that were to receive propafenone, as well as in rats that were to
receive propranolol and undergo tail-flick testing, i.c.v. cannulation was required to
allow for i.c.v. inhibitor injection immediately before injection of opioid drug (as in
the case of propafenone) or to allow for more than one i.c.v. injection per rat (as in
the case for tail-flick testing). Intracerebroventricular cannulas were inserted into
the right lateral ventricle (same coordinates as above). After a one week recovery
period, rats received an i.c.v. injection into the cannula of 20 or 40 μg propranolol
(CYP2D MBI) dissolved in 4 μl ACSF, 4 μl ACSF (vehicle control), 40 μg propafenone
(CYP2D competitive inhibitor) dissolved in 4 μl cyclodextrin, or 4 μl cyclodextrin
(vehicle control). All i.c.v. injections were made over 60 sec, and the injector was
left in place in the cannula for 60 sec after injection. To confirm the patency and
correct placement of the cannulas in the lateral ventricle, dipsogenic response to
i.c.v. administration of angiotensin II (Sigma-Aldrich) was tested before each i.c.v.
inhibitor injection (Vento and Daniels 2010).
Effect of i.c.v. CYP2D inhibitor injection on codeine-induced analgesia
In drug-naïve rats, TFL was measured three times in each rat, and the mean of the
three TFLs was used as that rat’s baseline TFL. All TFLs were taken by restraining the
rat with a towel and positioning it on the tail-flick meter so that the heat beam was
~2-3 cm from the end of its tail. Heat intensity was adjusted to produce baseline TFLs

42
of ~2-4 sec, and the same heat intensity was used for all rats for all TFL
measurements. Codeine was injected at 30 mg/kg s.c. 24 h after i.c.v. injection of a
CYP2D MBI (20 μg propranolol) or vehicle control (ACSF), or 5 min after i.c.v.
injection of a CYP2D competitive inhibitor (40 μg propafenone) or vehicle control
(cyclodextrin). The reason for the difference in timing is that a MBI results in a longer
lasting inhibition and can be given ahead of time before the opioid test drug, as
opposed to a competitive inhibitor which has to be given during the same time as
the test drug. This is because MBIs result in permanent loss of enzyme function that
can only be restored by synthesis of new enzyme (Bertelsen, Venkatakrishnan et al.
2003, Van, Heydari et al. 2006). Thus, an additional advantage of using propranolol is
that it is cleared from the body before codeine is given and therefore not expected
to have an effect on analgesia.
TFL was measured for 2 h after codeine injection. This length of time was
chosen because, while analgesia from codeine can last longer than this in rats
(Cleary, Mikus et al. 2004), we are interested in the earlier period after codeine
administration during which brain CYP2D is expected to play a larger role in
analgesia. Also, previous studies have shown this 2 h period to include the rise in the
analgesia time curve during the tail-flick test, as well as the plateau (Cleary, Mikus et
al. 2004, Lewis, Sherman et al. 1981). A cut-off of 10 sec was used to avoid damaging
the tail skin. Each rat’s baseline TFL was subtracted from its TFLs after opioid
injection, and these are the values used in the results. A within-animal design was
used in which, after a 2 week washout period, rats were crossed over (i.c.v. inhibitor
vs. vehicle) and retested with codeine. Thus, each rat acted as its own control, which

43
accounts for possible genetic variation between rats. In both phases, half the rats
received inhibitor and the other half received vehicle.
As a control, TFL was measured 24 h after 40 μg i.c.v. propranolol and 5 min
after 40 μg i.c.v. propafenone injection in the absence of codeine, and this was
compared with baseline TFL. This dose of propranolol was chosen to confirm that,
even at higher doses than used before codeine, propranolol would not have any
effect on nociception on its own. As another control, rats received morphine at 5
mg/kg, s.c. (Lewis, Sherman et al. 1981) 24 h after i.c.v. injection of 40 μg
propranolol or ACSF, or 5 min after i.c.v. injection of 40 μg propafenone or
cyclodextrin. This dose of propranolol was chosen to verify that, even at higher
doses than used before codeine, propranolol would not have any effect on
morphine’s analgesic actions. TFL was measured for 2 h after morphine injection. A
within-animal design was used in which after a 2 week washout period, rats were
crossed over (inhibitor vs. vehicle) and retested with morphine. In this way, each rat
could act as its own control. In both phases, half the rats received inhibitor and the
other half received vehicle.
Measurement of codeine and morphine levels in brain and plasma
Rats received codeine (30 mg/kg, s.c.) 24 h after propranolol (20 μg, i.c.v.) or ACSF
injection, or 5 min after propafenone (40 μg, i.c.v.) or cyclodextrin injection.
Propranolol-treated rats were decapitated at 30, 60 or 90 min after codeine injection,
and propafenone-treated rats were decapitated at 30 min after codeine injection.
Trunk blood and brains were collected, and brains were halved into hemispheres,
and the right hemisphere was further split into anterior and posterior portions. Blood

44
was centrifuged at 5000 g for 10 min, and supernatants were kept. Based on previous
methods (Kudo, Ishida et al. 2006), half brains were homogenized 1:3 (w/v) in 0.01 M
HCl, centrifuged at 5000 g for 10 min, and supernatants were kept. Morphine and
codeine concentrations in plasma and brain samples were measured using HPLC as
described below (Freiermuth and Plasse 1997, He, Shay et al. 1998).
In vitro codeine and dextromethorphan oxidation in brain and liver
Brain and hepatic CYP2D activity were measured using in vitro oxidation of codeine
as the drug of interest as well as dextromethorphan which is a typical CYP2D probe
drug (Von Moltke, Greenblatt et al. 1998, Frank, Jaehde et al. 2007). Rat brain
membranes were prepared as previously described (Tyndale, Li et al. 1999) from
rats that received an i.c.v. injection of MBI (20 μg propranolol) or vehicle control
(ACSF) 24 h prior to sacrifice. Briefly, immediately after sacrifice, each whole brain
was homogenized in 10 ml ACSF (approximately 1:5 (w:v)), centrifuged at 3000 g for
5 min, and the supernatant was kept. The pellet was resuspended in 10 ml ACSF and
centrifuged at 3000 g for 5 min, and the supernatant was combined with the initial
supernatant. The combined supernatant was centrifuged at 100,000 g for 60 min, and
the pellet was resuspended in ACSF. Protein concentration was measured using the
Bradford assay with a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Mississauga,
Canada). Because brain CYPs are more labile than their hepatic forms and their
activity is reduced by freezer storage (Tyndale, Li et al. 1999, Voirol, Jonzier-Perey
et al. 2000), membranes were prepared immediately after sacrifice and freshly
prepared (i.e., never frozen) membranes were used for all incubations.

45
For brain codeine oxidation, freshly prepared membranes (6 mg protein/ml)
were incubated with 500 μM (10 x Km (Mikus, Somogyi et al. 1991)) codeine, 5 mM
MgCl2 and 1 mM NADPH in ACSF (pH 7.4) for 120 min at 37 °C and 95% O2/5% CO2
in a final volume of 1 ml. The reaction was stopped using 200 μl bicarbonate buffer
solution (1 M, pH 9.7).
For brain dextromethorphan oxidation, as before (Tyndale, Li et al. 1999),
freshly prepared membranes (3 mg protein/ml) were incubated with 25 μM (10 x
Km) dextromethorphan and 1 mM NADPH in ACSF (pH 7.4) for 120 min at 37 °C and
95% O2/5% CO2 in a final volume of 1 ml. The reaction was stopped using 200 μl
bicarbonate buffer solution (1 M, pH 9.7). Linearity of dextrorphan formation at this
protein concentration and incubation time was demonstrated previously (Tyndale, Li
et al. 1999).
Rat liver microsomes were prepared as previously described (Siu,
Wildenauer et al. 2006) from rats that had received an i.c.v. injection of MBI (20 μg
propranolol) or vehicle control (ACSF) 24 h prior to sacrifice. Briefly, liver tissue was
homogenized 1:4 (w/v) in 1.15% KCl, centrifuged at 9000 g for 20 min, and the
supernatants were further centrifuged at 100,000 g for 60 min. The pellets were
resuspended in KCl. Protein concentrations were measured using the Bradford assay
using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Mississauga, Canada).
For hepatic codeine oxidation, as before (Xu, Aasmundstad et al. 1997),
microsomes (0.25 mg protein/ml) were incubated with 500 μM codeine (10 x Km
(Mikus, Somogyi et al. 1991)), 5 mM MgCl2 and 1 mM NADPH in 100 mM
NaH2PO4*H2O buffer (pH 7.4) for 20 min at 37 °C in a final volume of 0.5 ml. The
reaction was stopped using 100 μl acetonitrile. Linearity of morphine formation at

46
this protein concentration and incubation time was demonstrated previously (Xu,
Aasmundstad et al. 1997).
For hepatic dextromethorphan oxidation, as before (Kerry, Somogyi et al.
1993, Vuppugalla and Mehvar 2005), microsomes (0.5 mg protein/ml) were
incubated with 25 μM dextromethorphan (10 x Km) and 1 mM NADPH in 100 mM
NaH2PO4*H2O buffer (pH 7.4) for 5 min at 37 °C in a final volume of 0.5 ml. The
reaction was stopped using 100 μl acetonitrile. Linearity of dextrorphan formation at
this protein concentration and incubation time was demonstrated previously (Kerry,
Somogyi et al. 1993, Vuppugalla and Mehvar 2005).
For all liver and brain incubations, controls were used in which there was one
incubation mixture without substrate and one incubation mixture without protein.
Substrate and metabolite levels after incubation were measured using HPLC.
HPLC
HPLC methods were based on modifications to previous techniques for measuring
codeine and morphine (Freiermuth and Plasse 1997, He, Shay et al. 1998), and
dextromethorphan and dextrorphan (Hendrickson, Gurley et al. 2003, Flores-Perez,
Flores-Perez et al. 2004).
Sample preparation
For all brain and plasma samples containing codeine, the internal standard 2-
benzoxazolinone (1 μg) was added. Extractions were performed using Bond Elut C18
(3 ml, 200 mg) solid phase extraction cartridges (Agilent, USA). Cartridges were first
conditioned with methanol (2 ml), followed by Milli-Q water (2 ml). The sample was

47
then applied to the cartridge and washed with 2 x 1 ml of Milli-Q water followed by 2
x 0.5 ml of a 40% acetonitrile solution in water. The drugs were eluted with 3 x 0.5 ml
of a 0.05 M HCl solution at 10% in acetonitrile. The eluate was evaporated to dryness
at 37ºC under a nitrogen stream and reconstituted with 110 μl of mobile phase and
100 μl of the solution was injected into the HPLC system.
For all brain samples containing dextromethorphan, the internal standard 2-
benzoxazolinone (50 ng) was added with 5 ml hexane-butanol (95:5 v/v). The
mixture was vortexed for 10 sec, mechanically shaken for 10 min and centrifuged at
1400 g for 10 min. The organic layer was then transferred to a 10 ml tube and
evaporated to dryness at 37ºC under a nitrogen stream. The residue was redissolved
in 110 μl of mobile phase and 100 μl of the solution was injected into the HPLC
system.
For all liver samples containing codeine or dextromethorphan, based on
previous methods (Mikus, Somogyi et al. 1991, Kerry, Somogyi et al. 1993), after
stopping the incubation reaction, samples were centrifuged at 13,250 g for 10 min
and 90 μl of the supernantant was directly injected into the HPLC system.
Chromatographic conditions
All samples containing codeine were analyzed by HPLC with ultraviolet detection
(HPLC-UV) (Agilent 1200 Separation Module). The limits of quantification were 25
ng/ml for morphine and 250 ng/ml for codeine from plasma as well as liver and
brain in vitro incubation mixtures, and 5 ng/g for morphine and 50 ng/g for codeine
assessed from brain homogenates. For plasma and the liver and brain in vitro
incubation mixtures, the assay was linear from 25 to 500 ng/ml for morphine and 250

48
to 2000 ng/ml for codeine, with an extraction efficiency of 76.9% for morphine and
83.5% for codeine. For brain homogenates, the assay was linear from 5 to 500 ng/g
for morphine and 50 to 1000 ng/g for codeine, with an extraction efficiency of 82.7%
for morphine and 90.1% for codeine. The HPLC-UV system was set for detection at
214 nm and morphine, codeine and internal standard were separated on an Agilent
ZORBAX SB-C18 Column (250 x 4.6 mm I.D.; particle size, 5 um). The mobile phase
used was methanol - 0.05M phosphate buffer, pH 5.8 (29.3/70.7, v/v) and the flow
rate was 1 ml/min. The retention times were 4.1 min for morphine, 9.4 min for
codeine, and 16.7 min for the internal standard.
All samples containing dextromethorphan were analyzed by HPLC with
fluorescence detection (HPLC-FLD) (Agilent 1200 Separation Module), with a limit of
quantification of 5 ng/ml for both dextromethorphan and dextrorphan. The assay
was linear from 5 to 150 ng/ml with an extraction efficiency of 80.3% for
dextromethorphan and 72.8% for dextrorphan. The HPLC-FLD system was set for
detection at 230 excitation wavelength and 330 nm emission wavelength, and
dextromethorphan, dextrorphan and internal standard were separated on an
Agilent ZORBAX Bonus-RP Column (150 x 4.6 mm I.D.; particle size, 5 um). The
mobile phase used was acetonitrile/potassium phosphate buffer (22.3:77.7 v/v, pH
5.07) containing 34 mM potassium phosphate monobasic, 34 mM citric acid, 3.3 mM
heptane sulfonic acid and 0.5% triethylamine, and the flow rate was 1.2 ml/min. The
retention times were 5.1 min for dextrorphan, 6.6 min for internal standard, and 12.5
min for dextromethorphan.

49
Statistical analyses
Paired t-tests (because of the within-animal design) were used for comparisons of
TFLs and AUCs following codeine or morphine injection after inhibitor treatment
versus after vehicle treatment, of TFLs and AUCs between the two phases of codeine
or morphine injection, of morphine concentrations and morphine to codeine ratios in
the anterior versus the posterior part of the brain, and of baseline TFLs versus TFLs
after inhibitor treatment. Unpaired t-tests (because of the between-animal design)
were used for comparisons of TFLs and AUCs after codeine injection versus after
morphine injection, of morphine concentrations, morphine to codeine ratios,
morphine to total drug ratios, codeine concentrations and total drug concentrations
in brain and plasma between inhibitor- and vehicle-treated rats, and of velocities
from brain membrane and liver microsome in vitro metabolism studies between
inhibitor- and vehicle-treated rats.

50
Section 3: Results
3.1 Inhibition of brain CYP2D reduced codeine-induced
analgesia
Codeine is metabolized by CYP2D to the active morphine metabolite which confers
analgesia. To test the behavioural effects of altering brain CYP2D activity, we
examined whether inhibiting brain CYP2D would reduce analgesia following
codeine administration. We used two different CYP2D inhibitors: propranolol – a
mechanism-based inhibitor (MBI) and propafenone – a competitive inhibitor.
Rats were injected with either 20 μg i.c.v. propranolol, 40 μg i.c.v.
propafenone, or their respective vehicles. Codeine was injected at 30 mg/kg s.c. 24
h after propranolol injection and 5 min after propafenone injection. Tail-flick latency
(TFL) was measured for 2 h after codeine injection. After a two week washout period,
rats were crossed over so that those that had received inhibitor in the first phase
received vehicle in the second phase, and vice versa (the same number of rats
received each treatment in each phase). They were then retested with codeine.
Because each rat received both inhibitor and vehicle, TFL after inhibitor treatment
could be compared with TFL after vehicle treatment within the same animal, with
each rat acting as its own control.
Compared to vehicle treatment, propranolol treatment resulted in
significantly shorter TFL at 15 (p<0.02; n=4 total), 20 (p<0.02; n=16 total), 30
(p<0.004; n=16 total), and 40 (p<0.005; n=16 total) min after codeine injection

51
(Figure 3.a). Compared to vehicle treatment, propafenone treatment resulted in
significantly shorter TFL at 20 (p<0.03; n=12 total), 25 (p<0.05; n=7 total), 30 (p<0.03;
n=12 total), and 40 (p<0.04; n=12 total) min after codeine injection (Figure 3.b). The
n-values are not the same at all each time point because two different sets of rats
were used, and TFL was not measured at the 5, 15 and 25 min time points in the first
set of rats. In the second set of experiments TFL was measured at these additional
time points.

52
3.a)
b)
Figure 3. Inhibition of brain CYP2D reduced codeine-induced analgesia. (a)
Compared to vehicle treatment, propranolol treatment resulted in significantly
shorter tail-flick latency (TFL) at 15 (p<0.02), 20 (p<0.02), 30 (p<0.004) and 40
(p<0.005) min after codeine injection (n=4-16 total/time point). (b) Compared to
vehicle treatment, propafenone treatment resulted in significantly shorter TFL at 20
(p<0.03), 25 (p<0.05), 30 (p<0.03), and 40 (p<0.04) min after codeine injection (n=7-
12 total/time point). *p<0.05, **p<0.01.
-1
1
3
5
7
0 30 60 90 120
TFL
(sec
, mea
n +
SEM
)
Time after codeine injection (min)
Vehicle
Propranolol
* *
** **
-1
1
3
5
7
0 30 60 90 120
TFL
(sec
, mea
n +
SEM
)
Time after codeine injection (min)
Vehicle
Propafenone
* *
* *

53
3.2 Inhibition of brain CYP2D lowered codeine-induced area
under the analgesia time curve
Another way to assess the behavioural effects of brain CYP2D inhibition is to
investigate the area under the analgesia time curve (AUC), which is a measure of
total analgesia over the time period examined. We chose to examine the period up
to 30 min after codeine injection as this was hypothesized based on Chen’s data
(1990) to be the time period during which inhibitor treatment would result in lower
analgesia. However, based on our tail-flick data, analgesia appeared to be lower
after inhibitor treatment up to 60 min after codeine injection, so we also analyzed
analgesia AUC for 30-60 min and 0-60 min after codeine injection. We also measured
AUC for 60-120 min after codeine injection during which no difference in analgesia
after inhibitor treatment was expected. Additionally, we measured AUC for 0-120
min after codeine injection to investigate the effect of inhibitor treatment on
analgesia over the total time period examined.
Compared to vehicle treatment, 20 μg i.c.v. propranolol treatment resulted in
significantly lower AUC for 0-30, 30-60 and 0-60 min after 30 mg/kg s.c. codeine
injection (p<0.01, p<0.05, p<0.02, respectively; Figure 4.a-c), but not for 60-120
min or 0-120 min after codeine injection (p>0.8, p>0.5, respectively; Figure 5.a,b)
(n=16 total). Compared to vehicle treatment, 40 μg i.c.v. propafenone treatment
resulted in significantly lower AUC for 0-30, 30-60 and 0-60 min after codeine
injection (p<0.01, p<0.05, p<0.02, respectively; Figure 6.a-c), but not for 60-120
min or 0-120 min after codeine injection (p>0.2, p>0.07, respectively; Figure 7.a,b)
(n=12 total).

54
4.a) 0-30 min b) 30-60 min
c) 0-60 min
Figure 4. Inhibiting brain CYP2D with propranolol lowered codeine-induced
area under the analgesia time curve between 0-60 min after codeine injection.
Compared to vehicle treatment, propranolol treatment resulted in significantly
lower area under the analgesia time curve (AUC) for (a) 0-30 min (p<0.01), (b) 30-60
min (p<0.05) and (c) 0-60 min (p<0.02) after codeine injection (n=16 total). *p<0.05,
**p<0.01.
0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Vehicle Propranolol
*
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Vehicle Propranolol
**
0
70
140
210
AU
C 3
0-6
0
min
*se
c, m
ean
+ S
EM
Vehicle Propranolol
*

55
5.a) 60-120 min b) 0-120 min
Figure 5. Inhibiting brain CYP2D with propranolol did not lower codeine-
induced area under the analgesia time curve at 60-120 min or 0-120 min after
codeine injection. Compared to vehicle treatment, propranolol treatment did not
result in significantly different area under the analgesia time curve (AUC) for (a) 60-
120 min (p>0.8) or (b) 0-120 min (p>0.5) after codeine injection (n=16 total).
0
120
240
360A
UC
60
-12
0
min
*sec
, mea
n +
SEM
Vehicle Propranolol 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Vehicle Propranolol

56
6.a) 0-30 min b) 30-60 min
c) 0-60 min
Figure 6. Inhibiting brain CYP2D with propafenone lowered codeine-induced
area under the analgesia time curve between 0-60 min after codeine injection.
Compared to vehicle treatment, propafenone treatment resulted in significantly
lower area under the analgesia time curve (AUC) for (a) 0-30 min (p<0.01), (b) 30-60
min (p<0.04) and (c) 0-60 min (p<0.02) after codeine injection (n=12 total). *p<0.05,
**p<0.01.
0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Vehicle Propafenone
*
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Vehicle Propafenone
**
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Vehicle Propafenone
*

57
7.a) 60-120 min b) 0-120 min
Figure 7. Inhibiting brain CYP2D with propafenone did not lower codeine-
induced area under the analgesia time curve at 60-120 min or 0-120 min after
codeine injection. Compared to vehicle treatment, propafenone treatment did not
result in significantly different area under the analgesia time curve (AUC) for (a) 60-
120 min (p>0.2) or (b) 0-120 min (p>0.07) after codeine injection (n=12 total).
0
120
240
360A
UC
60
-12
0
min
*sec
, mea
n +
SEM
Vehicle Propafenone 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Vehicle Propafenone

58
3.3 Inhibiting brain CYP2D did not affect baseline tail-flick
latency
As a control we assessed whether either of the two CYP2D inhibitors on their own
would have an effect on nociception and cause tail-flick latency (TFL) to deviate from
baseline. In drug-naïve rats, TFL was measured three times in each rat within a 15
min period. The mean of the three TFLs in each rat was used as that rat’s own
baseline TFL. Baseline TFLs were similar in all the rats (mean+SEM=3.09+0.09 sec,
range=2.6-3.64 sec) and were also similar within each rat (within-animal SEM
ranged from 0.009-0.14). Immediately after measuring baseline TFL, rats were
injected with either 40 μg i.c.v. propranolol or 40 μg i.c.v. propafenone.
At 24 h after propranolol injection or 5 min after propafenone injection, TFLs
were measured three times in each rat within a 15 min period. The mean of the three
TFLs in each rat was used as that rat’s own TFL after propranolol or propafenone
treatment. There was no significant difference between baseline TFL and TFL after
propranolol treatment (p>0.3; n=10 total; Figure 8.a). There was also no significant
difference between baseline TFL and TFL after propafenone treatment (p>0.1; n=8
total; Figure 8.b).
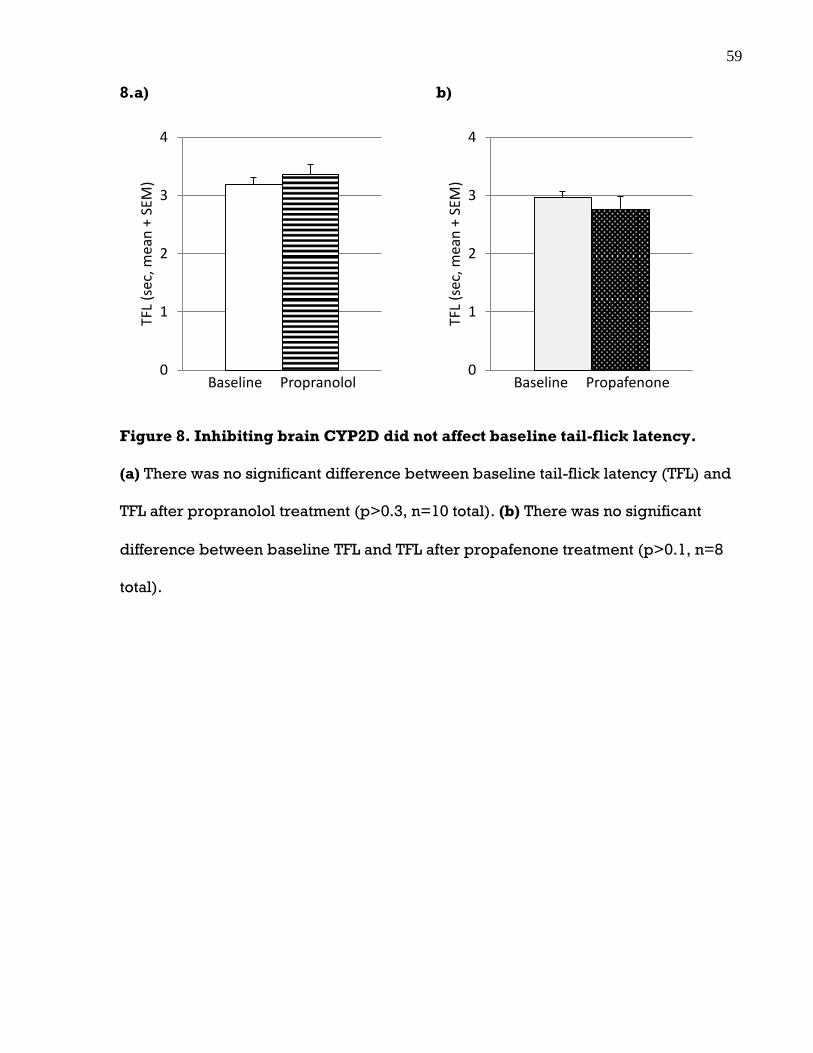
59
8.a) b)
Figure 8. Inhibiting brain CYP2D did not affect baseline tail-flick latency.
(a) There was no significant difference between baseline tail-flick latency (TFL) and
TFL after propranolol treatment (p>0.3, n=10 total). (b) There was no significant
difference between baseline TFL and TFL after propafenone treatment (p>0.1, n=8
total).
0
1
2
3
4TF
L (s
ec, m
ean
+ S
EM)
Baseline Propranolol 0
1
2
3
4
TFL
(sec
, mea
n +
SEM
)
Baseline Propafenone

60
3.4 Inhibiting brain CYP2D did not affect morphine-induced
analgesia
As another control, we assessed whether either of the two CYP2D inhibitors would
affect analgesia following morphine injection. Since morphine is the active analgesic
compound, and it is not metabolized by CYP2D, its analgesic effect should not be
altered by changes in CYP2D activity.
Rats were injected with either 40 μg i.c.v. propranolol, 40 μg i.c.v.
propafenone, or their respective vehicles. Morphine was injected at 5 mg/kg s.c. 24
h after propranolol injection and 5 min after propafenone injection. Tail-flick latency
(TFL) was measured for 2 h after morphine injection. After a two week washout
period, rats were crossed over so that those that had received inhibitor in the first
phase received vehicle in the second phase, and vice versa (the same number of
rats received each treatment in each phase). They were then retested with
morphine. Because each rat received both inhibitor and vehicle, TFL after inhibitor-
treatment could be compared with TFL after vehicle treatment within the same
animal, with each rat acting as its own control.
Compared to vehicle treatment, propranolol treatment did not result in
significantly different TFL at any time point after morphine injection (p>0.08 at all
time points; n=12 total; Figure 9.a). Compared to vehicle treatment, propafenone
treatment did not result in significantly different TFL at any time point after morphine
injection (p>0.1 at all time points; n=6 total; Figure 9.b).

61
9.a)
b)
Figure 9. Inhibiting brain CYP2D did not affect morphine-induced analgesia.
(a) Compared to vehicle treatment, propranolol treatment did not result in
significantly different TFL after morphine injection (p>0.08 at all time points, n=12
total). (b) Compared to vehicle treatment, propafenone treatment did not result in
significantly different TFL after morphine injection (p>0.1 at all time points, n=6
total).
-1
1
3
5
7
0 30 60 90 120
TFL
(sec
, mea
n +
SEM
)
Time after morphine injection (min)
Vehicle
Propranolol
-1
1
3
5
7
0 30 60 90 120
TFL
(sec
, mea
n +
SEM
)
Time after morphine injection (min)
Vehicle
Propafenone

62
3.5 Inhibiting brain CYP2D did not alter morphine-induced
area under the analgesia time curve
As another way to assess whether either of the two CYP2D inhibitors would affect
analgesia following morphine injection, we investigated the area under the
analgesia time curve (AUC) for morphine. We chose to examine the AUCs for the
same periods of time following morphine injection as we did for codeine, so we
could compare the AUCs of these two opioids.
Compared to vehicle treatment, 40 μg i.c.v. propranolol treatment did not
result in significantly different AUC for 0-30, 30-60, 0-60, 60-120 or 0-120 min after 5
mg/kg s.c. morphine injection (p>0.4 for all time periods; n=12 total; Figure 10.a-
e). Compared to vehicle treatment, 40 μg i.c.v. propafenone treatment also did not
result in significantly different AUC for 0-30, 30-60, 0-60, 60-120 or 0-120 min after
morphine injection (p>0.2 for all time periods; n=6 total; Figure 11.a-e).

63
10.a) 0-30 min b) 30-60 min
c) 0-60 min d) 60-120 min
e) 0-120 min
Figure 10. Inhibiting brain CYP2D with propranolol did not alter morphine-
induced area under the analgesia time curve. Compared to vehicle treatment,
propranolol treatment did not result in significantly different AUC for (a) 0-30 min,
(b) 30-60, (c) 0-60 min, (d) 60-120 min or (e) 0-120 min after morphine injection
(p>0.4 for all time periods, n=12 total).
0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Vehicle Propranolol
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Vehicle Propranolol 0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Vehicle Propranolol
0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Vehicle Propranolol 0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Vehicle Propranolol

64
11.a) 0-30 min b) 30-60 min
c) 0-60 min d) 60-120 min
e) 0-120 min
Figure 11. Inhibiting brain CYP2D with propafenone did not alter morphine-
induced area under the analgesia time curve. Compared to vehicle treatment,
propafenone treatment did not result in significantly different AUC for (a) 0-30 min,
(b) 30-60, (c) 0-60 min, (d) 60-120 min or (e) 0-120 min after morphine injection
(p>0.2 for all time periods, n=6 total).
0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Vehicle Propafenone
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Vehicle Propafenone 0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Vehicle Propafenone
0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Vehicle Propafenone 0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Vehicle Propafenone

65
3.6 There was no tolerance to the analgesic effects of codeine
or morphine
We compared the tail-flick latencies (TFLs) and areas under the analgesia time
curves (AUCs) between the two phases of codeine or morphine treatment (i.e., the
first phase of opioid testing and the crossover phase two weeks later) to confirm that
the rats did not develop tolerance to either of the opioids within this testing
schedule. In rats treated with propranolol or vehicle and tested with codeine, there
was no significant difference between the two phases in TFL (p>0.1 at all time points;
Figure 12.a) or AUC (p>0.6 at all time periods; Figure 12.b-f) (n=4-16 total). In rats
treated with propafenone or vehicle and tested with codeine, there was no
significant difference between the two phases in TFL (p>0.1 at all time points; Figure
13.a) or AUC (p>0.2 at all time periods; Figure 13.b-f) (n=7-12 total). In rats treated
with propranolol or vehicle and tested with morphine, there was no significant
difference between the two phases in TFL (p>0.08 at all time points; Figure 14.a) or
AUC (p>0.4 at all time periods; Figure 14.b-f) (n=12 total). In rats treated with
propafenone or vehicle and tested with morphine, there was no significant
difference between the two phases in TFL (p>0.08 at all time points; Figure 15.a) or
AUC (p>0.1 at all time periods; Figure 15.b-f) (n=6 total).

66
12.a) b) 0-30 min
c) 30-60 min d) 0-60 min
e) 60-120 min f) 0-120 min
Figure 12. Rats treated with propranolol or vehicle did not develop tolerance to
codeine. In rats treated with propranolol or vehicle and tested with codeine, there
was no significant difference between the two phases in (a) tail-flick latency (TFL)
(p>0.1 at all time points) or (b-f) area under the analgesia time curve (AUC) (p>0.6
at all time periods) (n=4-16 total).
-1
1
3
5
7
0 30 60 90 120TFL
(sec
, mea
n +
SEM
)
Time after codeine injection (min)
Phase 1
Phase 2
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2 0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2 0
230
460
690
AU
C 0
-12
0
min
*se
c, m
ean
+ S
EM
Phase 1 Phase 2

67
13.a) b) 0-30 min
c) 30-60 min d) 0-60 min
e) 60-120 min f) 0-120 min
Figure 13. Rats treated with propafenone or vehicle did not develop tolerance to
codeine. In rats treated with propafenone or vehicle and tested with codeine, there
was no significant difference between the two phases (a) in tail-flick latency (TFL)
(p>0.1 at all time points) or (b-f) area under the analgesia time curve (AUC) (p>0.2
at all time periods) (n=7-12 total).
-1
1
3
5
7
0 30 60 90 120TFL
(sec
, mea
n +
SEM
)
Time after codeine injection (min)
Phase 1
Phase 2
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2 0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2

68
14.a) b) 0-30 min
c) 30-60 min d) 0-60 min
e) 60-120 min f) 0-120 min
Figure 14. Rats treated with propranolol or vehicle did not develop tolerance to
morphine. In rats treated with propranolol or vehicle and tested with morphine,
there was no significant difference between the two phases in (a) tail-flick latency
(TFL) (p>0.08 at all time points) or (b-f) area under the analgesia time curve (AUC)
(p>0.4 at all time periods) (n=12 total).
-1
1
3
5
7
0 30 60 90 120TFL
(sec
, mea
n +
SEM
)
Time after morphine injection (min)
Phase 1
Phase 2
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2 0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2
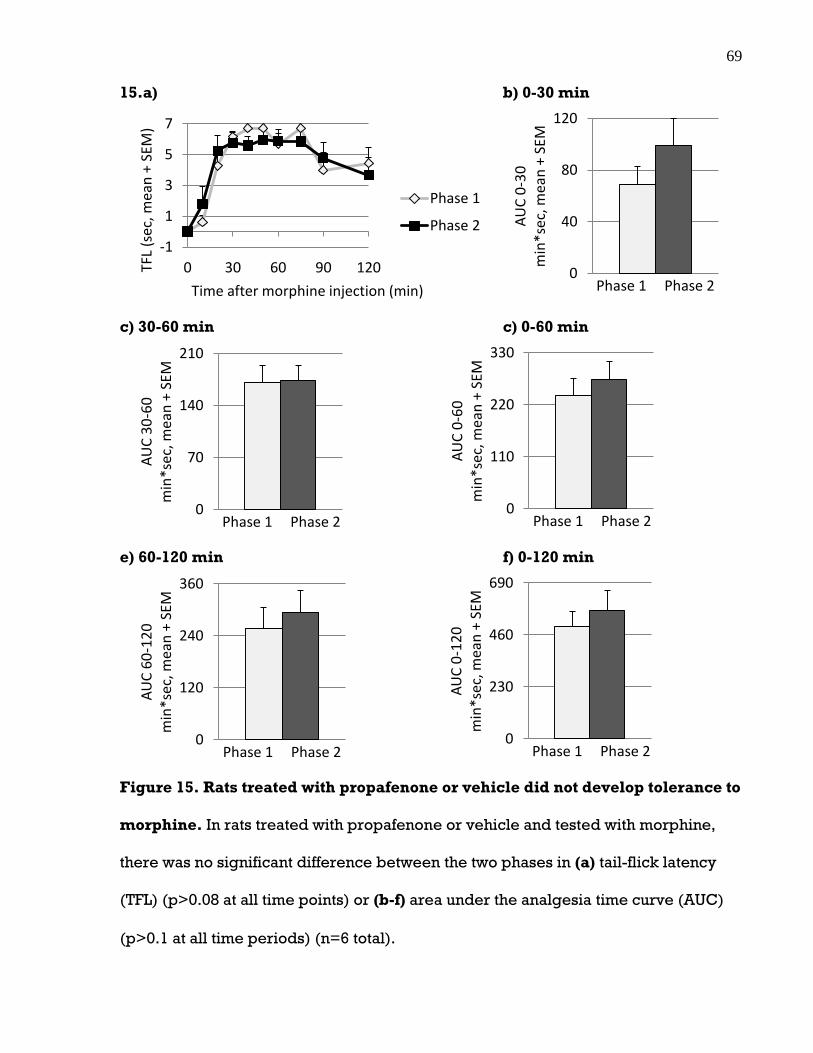
69
15.a) b) 0-30 min
c) 30-60 min c) 0-60 min
e) 60-120 min f) 0-120 min
Figure 15. Rats treated with propafenone or vehicle did not develop tolerance to
morphine. In rats treated with propafenone or vehicle and tested with morphine,
there was no significant difference between the two phases in (a) tail-flick latency
(TFL) (p>0.08 at all time points) or (b-f) area under the analgesia time curve (AUC)
(p>0.1 at all time periods) (n=6 total).
-1
1
3
5
7
0 30 60 90 120TFL
(sec
, mea
n +
SEM
)
Time after morphine injection (min)
Phase 1
Phase 2
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2 0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2
0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Phase 1 Phase 2 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Phase 1 Phase 2

70
3.7 Codeine and morphine doses used resulted in similar
levels of analgesia
To validate our choice of codeine and morphine doses, we compared the tail-flick
latencies (TFLs) and areas under the analgesia curves (AUCs) resulting from each
opioid after i.c.v. vehicle treatment to check that they produced equivalent
analgesia. Following ACSF (vehicle for propranolol) treatment, there was no
significant difference in TFL (p>0.3 for all time points; Figure 16.a) or AUC (p>0.1
for all time periods; Figure 16.b-f) after 30 mg/kg s.c. codeine (n=16) compared to
after 5 mg/kg s.c. morphine (n=12). Following cyclodextrin (vehicle for
propafenone) treatment, there was no significant difference in TFL (p>0.1 for all time
points; Figure 17.a) or AUC (p>0.5 for all time periods; Figure 17.b-f) after 30
mg/kg s.c. codeine (n=12) compared to after 5 mg/kg s.c. morphine (n=6).

71
16.a) b) 0-30 min
b) 30-60 min c) 0-60 min
d) 60-120 min e) 0-120 min
Figure 16. Codeine and morphine doses used resulted in similar levels of
analgesia after ACSF (i.c.v. vehicle) treatment. Following ACSF (vehicle for
propranolol) treatment, there was no significant difference in (a) tail-flick latency
(TFL) (p>0.3) or (b-e) area under the analgesia time curve (AUC) (p>0.1) after 30
mg/kg s.c. codeine (n=16) compared to after 5 mg/kg s.c. morphine (n=12).
-1
1
3
5
7
0 30 60 90 120 TFL
(se
c, m
ean
+ S
EM)
Time after opioid injection (min)
Codeine
Morphine
0
40
80
120
AU
C 0
-30
m
in*
sec,
mea
n +
SEM
Codeine Morphine
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Codeine Morphine 0
110
220
330
AU
C 0
-60
m
in*s
ec, m
ean
+ S
EM
Codeine Morphine
0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Codeine Morphine 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Codeine Morphine

72
17.a) b) 0-30 min
b) 30-60 min c) 0-60 min
d) 60-120 min e) 0-120 min
Figure 17. Codeine and morphine doses used resulted in similar levels of
analgesia after cyclodextrin (i.c.v. vehicle) treatment. Following cyclodextrin
(vehicle for propafenone) treatment, there was no significant difference in (a) tail-
flick latency (TFL) (p>0.1) or (b-e) area under the analgesia time curve (AUC)
(p>0.5) after 30 mg/kg s.c. codeine (n=12) compared to after 5 mg/kg s.c. morphine
(n=6).
-1
1
3
5
7
0 30 60 90 120 TFL
(se
c, m
ean
+ S
EM)
Time after opioid injection (min)
Codeine
Morphine
0
40
80
120
AU
C 0
-30
m
in*s
ec, m
ean
+ S
EM
Codeine Morphine
0
70
140
210
AU
C 3
0-6
0
min
*sec
, mea
n +
SEM
Codeine Morphine 0
110
220
330
AU
C 0
-60
m
in*
sec,
mea
n +
SEM
Codeine Morphine
0
120
240
360
AU
C 6
0-1
20
m
in*s
ec, m
ean
+ S
EM
Codeine Morphine 0
230
460
690
AU
C 0
-12
0
min
*sec
, mea
n +
SEM
Codeine Morphine

73
3.8 Inhibitor-treated rats had lower morphine levels in the
brain but not plasma at 30 min after codeine injection
To assess the pharmacokinetic effects of the CYP2D inhibitors and to see if these are
consistent with differences in analgesia, we examined morphine and codeine levels
in brain and plasma at 30 min after codeine injection in inhibitor- and vehicle-
treated rats. This time point was chosen because it was when we hypothesized,
based on Chen’s data (1990), that brain morphine levels would be lower in inhibitor-
treated rats. This is also when the largest difference in behaviour (i.e., TFL) after
inhibitor treatment, compared to after vehicle treatment, occurred (Figure 3).
Morphine concentration was measured because morphine is the active metabolite
which confers analgesia. The ratio of morphine concentration to codeine
concentration (i.e., ratio of parent drug to metabolite) was calculated because this is
a measure of metabolism. Codeine concentration was measured to verify that it does
not correlate with analgesia. Total drug concentration (morphine plus codeine) was
calculated to ensure that it was the same after inhibitor treatment compared to after
vehicle treatment. The ratio of morphine concentration to total drug concentration
was calculated to confirm that morphine levels correlate with analgesia. These
parameters were examined in both brain and plasma to check that the inhibitors
were selectively affecting brain and not hepatic metabolism.
Rats were injected with either 20 μg i.c.v. propranolol, 40 μg i.c.v.
propafenone, or their respective vehicles. Codeine was injected at 30 mg/kg s.c. 24
h after propranolol injection and 5 min after propafenone injection. Rats were
sacrificed 30 min after codeine injection.

74
Compared to vehicle-treated rats, propranolol-treated rats had significantly
lower morphine concentrations, morphine to codeine ratios and morphine to total
drug ratios in the brain (p<0.02, p<0.05, p<0.05, respectively) but not in plasma
(p>0.6, p>0.7, p>0.7, respectively) (n=11/group; Figure 18-20). Codeine
concentrations and total drug concentrations were not significantly different
between propranolol- and vehicle-treated rats in brain (p>0.6, p>0.6, respectively)
or plasma (p>0.6, p>0.6, respectively) (n=11/group; Figure 21). Compared to
vehicle-treated rats, propafenone-treated rats also had significantly lower morphine
concentrations, morphine to codeine ratios, and morphine to total drug ratios in the
brain (p<0.006, p<0.03, p<0.03, respectively) but not in plasma (p>0.6, p>0.8,
p>0.8, respectively) (n=8/group; Figure 18-20). Codeine concentrations and total
drug concentrations were not significantly different between propafenone- and
vehicle-treated rats in brain (p>0.7, p>0.7, respectively) or plasma (p>0.9, p>0.9,
respectively) (n=8/group; Figure 22). As well, morphine levels and morphine to
codeine ratios were not significantly different between the anterior and the posterior
portion of the brains of propranolol-treated rats (p>0.6, p>0.4, respectively; n=8
total; Figure 23.a,c) or propafenone-treated rats (p>0.3, p>0.3, respectively; n=4
total; Figure 23.b,d), suggesting that the inhibitors distributed and inhibited CYP2D
throughout the brain.

75
18.a) Brain b) Plasma
c) Brain d) Plasma
Figure 18. Inhibitor-treated rats had lower morphine levels in the brain but not
in plasma at 30 min after codeine injection. Compared to vehicle-treated rats,
propranolol-treated rats had significantly lower morphine concentrations in (a) the
brain (p<0.02) but not in (b) plasma (p>0.6) at 30 min after codeine injection
(n=11/group). Compared to vehicle-treated rats, propafenone-treated rats had
significantly lower morphine concentrations in (c) the brain (p<0.006) but not in (d)
plasma (p>0.6) at 30 min after codeine injection (n=8/group). *p<0.05, **p<0.01.
MOR=morphine.
0
15
30
45
MO
R (
ng
/ml,
mea
n +
SEM
)
Vehicle Propranolol 0
15
30
45
MO
R (
ng
/g, m
ean
+ S
EM)
Vehicle Propranolol
*
0
15
30
45
MO
R (
ng
/ml,
mea
n +
SEM
)
Vehicle Propafenone 0
15
30
45
MO
R (
ng
/g, m
ean
+ S
EM)
Vehicle Propafenone
**

76
19.a) Brain b) Plasma
c) Brain d) Plasma
Figure 19. Inhibitor-treated rats had lower morphine to codeine ratios in the
brain but not in plasma at 30 min after codeine injection. Compared to vehicle-
treated rats, propranolol-treated rats had significantly lower morphine to codeine
ratios in (a) the brain (p<0.05) but not in (b) plasma (p>0.7) at 30 min after codeine
injection (n=11/group). Compared to vehicle-treated rats, propafenone-treated rats
had significantly lower morphine to codeine ratios in (c) the brain (p<0.03) but not in
(d) plasma (p>0.8) at 30 min after codeine injection (n=8/group). *p<0.05.
MOR=morphine, COD=codeine.
0
0.01
0.02
0.03
MO
R/C
OD
(m
ean
+ S
EM)
Vehicle Propranolol
*
0
0.01
0.02
0.03
MO
R/C
OD
(m
ean
+ S
EM)
Vehicle Propranolol
0
0.01
0.02
0.03
MO
R/C
OD
(m
ean
+ S
EM)
Vehicle Propafenone
*
0
0.01
0.02
0.03
MO
R/C
OD
(m
ean
+ S
EM)
Vehicle Propafenone

77
20.a) Brain b) Plasma
c) Brain d) Plasma
Figure 20. Inhibitor-treated rats had lower morphine to total drug ratios in the
brain but not in plasma at 30 min after codeine injection. Compared to vehicle-
treated rats, propranolol-treated rats had significantly lower morphine to total drug
ratios in (a) the brain (p<0.05) but not in (b) plasma (p>0.7) at 30 min after codeine
injection (n=11/group). Compared to vehicle-treated rats, propafenone-treated rats
had significantly lower morphine to total drug ratios in (c) the brain (p<0.03) but not
in (d) plasma (p>0.8) at 30 min after codeine injection (n=8/group). *p<0.05.
MOR=morphine, COD=codeine.
0
0.01
0.02
0.03M
OR
/(M
OR
+CO
D)
m
ean
+ S
EM
Vehicle Propranolol 0
0.01
0.02
0.03
MO
R/(
MO
R+C
OD
) m
ean
+ S
EM
Vehicle Propranolol
0
0.01
0.02
0.03
MO
R/(
MO
R+C
OD
) m
ean
+ S
EM
Vehicle Propafenone 0
0.01
0.02
0.03
MO
R/(
MO
R+C
OD
) m
ean
+ S
EM
Vehicle Propafenone
*
*

78
21.a) Brain b) Plasma
c) Brain d) Plasma
Figure 21. Propranolol-treated rats did not have lower codeine levels or total
drug levels in the brain or in plasma at 30 min after codeine injection. Codeine
concentrations (a,b) and total drug concentrations (c,d) were not significantly
different between propranolol- and vehicle-treated rats in brain or plasma (p>0.6 for
each measurement, n=11/group). COD=codeine, MOR=morphine.
0
1500
3000
4500
CO
D (
ng
/ml,
me
an +
SEM
)
Vehicle Propranolol 0
1500
3000
4500C
OD
(n
g/g
, mea
n +
SEM
)
Vehicle Propranolol
0
1500
3000
4500M
OR
+CO
D (
ng
/ml,
mea
n +
SEM
)
Vehicle Propranolol 0
1500
3000
4500
MO
R+C
OD
(n
g/g
, mea
n +
SEM
)
Vehicle Propranolol

79
22.a) Brain b) Plasma
c) Brain d) Plasma
Figure 22. Propafenone-treated rats did not have lower codeine levels or total
drug levels in the brain or in plasma at 30 min after codeine injection. Codeine
concentrations (a,b) and total drug concentrations (c,d) were not significantly
different between propafenone- and vehicle-treated rats in brain (p>0.7 for both
measurements) or plasma (p>0.9 for both measurements) (n=8/group).
COD=codeine, MOR=morphine.
0
1500
3000
4500
CO
D (
ng
/ml,
mea
n +
SEM
)
Vehicle Propafenone 0
1500
3000
4500
CO
D (
ng
/g, m
ean
+ S
EM)
Vehicle Propafenone
0
1500
3000
4500
MO
R+C
OD
(n
g/m
l, m
ean
+ S
EM)
Vehicle Propafenone 0
1500
3000
4500
MO
R+C
OD
(n
g/g
, mea
n +
SEM
)
Vehicle Propafenone

80
23.a) Propranolol b) Propafenone
c) Propranolol d) Propafenone
Figure 23. Inhibitor-treated rats had similar morphine levels and morphine to
codeine ratios between the anterior and the posterior parts of the brain.
Morphine levels and morphine to codeine ratios were not significantly different
between the anterior and the posterior portion of the brains of (a,c) propranolol-
treated rats (p>0.6, p>0.4, respectively; n=8 total) or (b,d) propafenone-treated rats
(p>0.3, p>0.3, respectively; n=4 total).
0
11
22
33
MO
R (
ng
/g, m
ean
+ S
EM)
Anterior Posterior 0
11
22
33
MO
R (
ng
/g, m
ean
+ S
EM)
Anterior Posterior
0
0.005
0.01
0.015
MO
R/C
OD
(m
ean
+ S
EM)
Anterior Posterior 0
0.005
0.01
0.015
MO
R/C
OD
(m
ean
+ S
EM)
Anterior Posterior

81
3.9 Analgesia correlated with brain, and not plasma, morphine
levels
For rats in which plasma and brain morphine and codeine levels were measured at
30 min after codeine injection, tail-flick latency (TFL) was measured right before
sacrifice. This allowed us to assess the correlations between TFL and the various
pharmacokinetic parameters measured, and to confirm which of these parameters is
responsible for analgesia.
TFL trended toward correlating with brain morphine concentration (p=0.054,
Figure 24.a), and correlated significantly with brain morphine to codeine ratio
(p<0.006, Figure 25.a) and brain morphine to total drug ratio (p<0.006, Figure
26.a) There was no correlation between TFL and brain or plasma codeine
concentration (p>0.5, Figure 27.a; p>0.8, Figure 27.b), brain or plasma total drug
concentration (p>0.5, Figure 27. c; p>0.8; Figure 27.d), plasma morphine
concentration (p>0.8, Figure 24.b), plasma morphine to codeine ratio (p>0.7,
Figure 25.b), or plasma morphine to total drug ratio (p>0.7, Figure 26.b) (n=20
total for all brain measurements, n=21 total for all plasma measurements).

82
24.a) Brain b) Plasma
Figure 24. Analgesia correlated with brain, and not plasma, morphine levels.
Tail-flick latency (TFL) trended toward correlating with morphine concentration in
(a) the brain (p=0.054, n=20 total) and not in (b) plasma (p>0.8, n=21 total). Dark
diamonds and dark squares represent propranolol- and propafenone-treated rats,
and pale diamonds and pale squares represent the respective vehicle-treated rats.
MOR=morphine.
-2
0
2
4
6
8
0 20 40 60
TFL
(sec
)
Brain MOR (ng/g)
-2
0
2
4
6
8
0 20 40 60
TFL
(sec
)
Plasma MOR (ng/ml)
R = 0.43 R = 0.04

83
25.a) Brain b) Plasma
Figure 25. Analgesia correlated with brain, and not plasma, morphine to
codeine ratios. Tail-flick latency (TFL) correlated significantly with morphine to
codeine ratios in (a) the brain (p<0.006, n=20 total) and not in (b) plasma (p>0.7,
n=21 total). Dark diamonds and dark squares represent propranolol- and
propafenone-treated rats, and pale diamonds and pale squares represent the
respective vehicle-treated rats. COD=codeine, MOR=morphine.
-2
0
2
4
6
8
0 0.01 0.02 0.03 0.04
TFL
(sec
)
Brain MOR/COD
-2
0
2
4
6
8
0 0.01 0.02 0.03 0.04
TFL
(sec
)
Plasma MOR/COD
R = 0.59 R = 0.08

84
26.a) Brain b) Plasma
Figure 26. Analgesia correlated with brain, and not plasma, morphine to total
drug ratios. Tail-flick latency (TFL) correlated significantly with morphine to total
drug ratios in (a) the brain (p<0.006, n=20 total) and not in (b) plasma (p>0.7, n=21
total). Dark diamonds and dark squares represent propranolol- and propafenone-
treated rats, and pale diamonds and pale squares represent the respective vehicle-
treated rats. COD=codeine, MOR=morphine.
R = 0.59
-2
0
2
4
6
8
0 0.01 0.02 0.03 0.04
TFL
(se
c)
Brain MOR/(MOR+COD)
R = 0.08
-2
0
2
4
6
8
0 0.01 0.02 0.03 0.04
TFL
(se
c)
Plasma MOR/(MOR+COD)

85
27.a) Brain b) Plasma
c) Brain d) Plasma
Figure 27. Analgesia did not correlate with codeine levels or total drug levels in
brain or plasma. There was no correlation between tail-flick latency (TFL) and
codeine concentration (a,b) or total drug concentration (c,d) in brain (p>0.5, p>0.5,
respectively; n=20 total) or plasma (p>0.8, p>0.8, respectively; n=21 total). Dark
diamonds and dark squares represent propranolol- and propafenone-treated rats,
and pale diamonds and pale squares represent the respective vehicle-treated rats.
COD=codeine, MOR=morphine.
-2
0
2
4
6
8
0 2000 4000 6000
TFL
(sec
)
Brain COD (ng/g)
-2
0
2
4
6
8
0 2000 4000 6000
TFL
(sec
)
Plasma COD (ng/ml)
-2
0
2
4
6
8
0 2000 4000 6000
TFL
(sec
)
Brain MOR+COD (ng/g)
-2
0
2
4
6
8
0 2000 4000 6000
TFL
(sec
)
Plasma MOR+COD (ng/ml)
R = 0.13 R = 0.03
R = 0.13 R = 0.03

86
3.10 Inhibitor-treated rats did not have lower morphine levels
in the brain at 60 or 90 min after codeine injection
To further investigate whether the pharmacokinetic effects of brain CYP2D inhibition
on codeine were consistent with its behavioural effects, we examined brain and
plasma morphine and codeine levels at 60 and 90 min after codeine injection in
inhibitor- and vehicle-treated rats. These time points were chosen because they
were when there was no longer a significant difference in behaviour (i.e., TFL) after
inhibitor compared to after vehicle treatment.
Rats were injected with either 20 μg i.c.v. propranolol or vehicle. Codeine was
injected at 30 mg/kg s.c. 24 h after propranolol injection. Rats were sacrificed 60 or
90 min after codeine injection.
At both 60 and 90 min after codeine injection, there was no significant
difference in morphine concentrations in brain (p>0.1, p>0.9, respectively; Figure
28.a) or plasma (p>0.5, p>0.2, respectively; Figure 28.b), morphine to codeine
ratios in brain (p>0.4, p>0.2, respectively; Figure 29.a) or plasma (p>0.2, p>0.8,
respectively; Figure 29.b), morphine to total drug ratios in brain (p>0.4, p>0.2,
respectively; Figure 30.a) or plasma (p>0.2, p>0.8, respectively; Figure 30.b),
codeine concentrations in brain (p>0.6, p>0.4, respectively; Figure 31.a) or plasma
(p>0.1, p>0.5, respectively; Figure 31.b), or total drug concentrations in brain
(p>0.6, p>0.4, respectively; Figure 31.c) or plasma (p>0.1, p>0.5, respectively;
Figure 31.d) between propranolol- and vehicle-treated rats (n=7/group for 60 min;
n=8/group for 90 min).

87
28.a) Brain b) Plasma
Figure 28. Inhibitor-treated rats did not have lower morphine levels in the brain
at 60 or 90 min after codeine injection. At both 60 min (n=7/group) and 90 min
(n=8/group) after codeine injection, there was no significant difference in morphine
concentrations in (a) brain (p>0.1, p>0.9, respectively) or (b) plasma (p>0.5, p>0.2,
respectively) between propranolol-treated rats and vehicle-treated rats.
COD=codeine, MOR=morphine.
0
15
30
45
60 90
MO
R (
ng
/g, m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol
0
15
30
45
60 90
MO
R (
ng
/g, m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol

88
29. a) Brain b) Plasma
Figure 29. Inhibitor-treated rats did not have lower morphine to codeine ratios
in the brain or plasma at 60 or 90 min after codeine injection. At both 60 min
(n=7/group) and 90 min (n=8/group) after codeine injection, there was no
significant difference in morphine to codeine ratio in (a) brain (p>0.4, p>0.2,
respectively) or (b) plasma (p>0.2, p>0.8, respectively) between propranolol-
treated rats and vehicle-treated rats. COD=codeine, MOR=morphine.
0
0.01
0.02
0.03
60 90
MO
R/C
OD
(m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol
0
0.01
0.02
0.03
60 90
MO
R/C
OD
(m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol
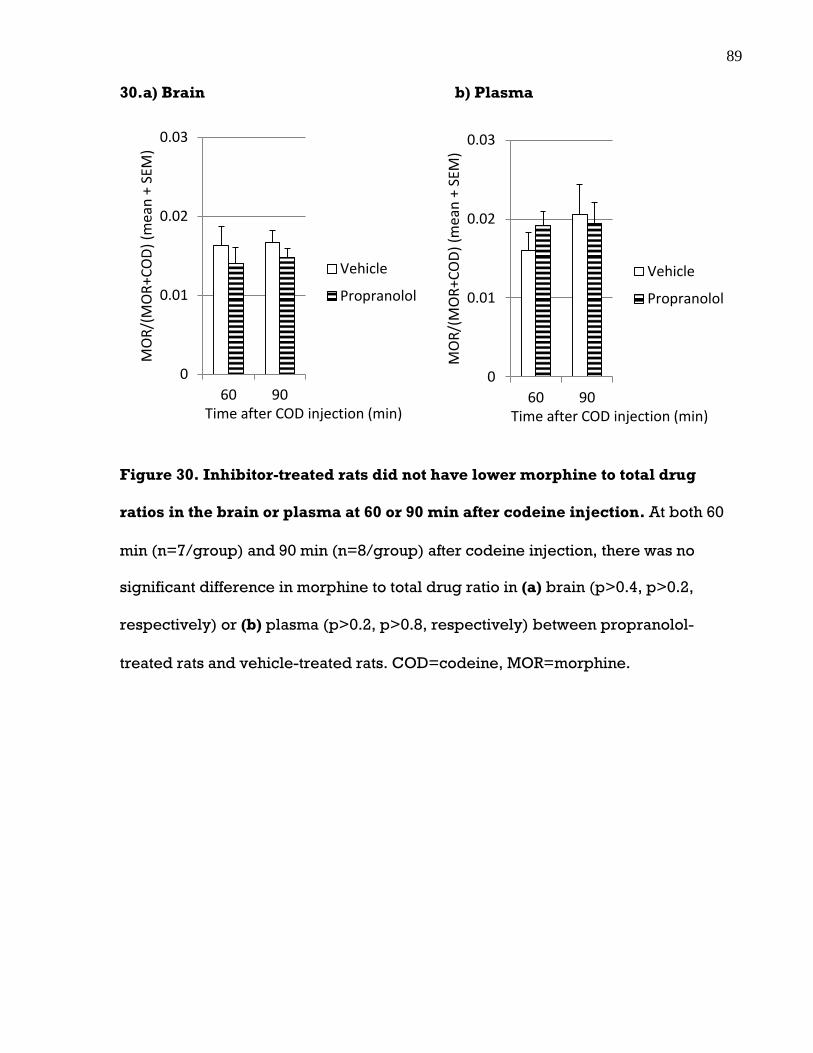
89
30.a) Brain b) Plasma
Figure 30. Inhibitor-treated rats did not have lower morphine to total drug
ratios in the brain or plasma at 60 or 90 min after codeine injection. At both 60
min (n=7/group) and 90 min (n=8/group) after codeine injection, there was no
significant difference in morphine to total drug ratio in (a) brain (p>0.4, p>0.2,
respectively) or (b) plasma (p>0.2, p>0.8, respectively) between propranolol-
treated rats and vehicle-treated rats. COD=codeine, MOR=morphine.
0
0.01
0.02
0.03
60 90
MO
R/(
MO
R+C
OD
) (m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol
0
0.01
0.02
0.03
60 90
MO
R/(
MO
R+C
OD
) (m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol

90
31.a) Brain b) Plasma
c) Brain d) Plasma
Figure 31. Inhibitor-treated rats did not have lower codeine levels or total drug
levels in the brain or plasma at 60 or 90 min after codeine injection. At both 60
min (n=7/group) and 90 min (n=8/group) after codeine injection, there was no
significant difference in codeine concentration (a,b) or total drug concentration (c,d)
in (a,c) brain (p>0.6 at 60 min and p>0.4 at 90 min for both measurements) or (b,d)
plasma (p>0.1 at 60 min and p>0.5 at 90 min for both measurements) between
propranolol-treated rats and vehicle-treated rats. COD=codeine, MOR=morphine.
0
1500
3000
4500
60 90
CO
D (
ng
/g, m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol
0
1500
3000
4500
60 90
CO
D (
ng
/g, m
ean
+ S
EM)
Time after COD injection (min)
Vehicle
Propranolol
0
1500
3000
4500
60 90
MO
R+C
OD
(n
g/g
, mea
n +
SEM
)
Time after COD injection (min)
Vehicle
Propranolol
0
1500
3000
4500
60 90
MO
R+C
OD
(n
g/g
, mea
n +
SEM
)
Time after COD injection (min)
Vehicle
Propranolol

91
3.11 Inhibiting brain CYP2D in vivo lowered in vitro codeine
metabolism in the brain but not liver
To confirm that i.c.v. injections of CYP2D inhibitors could selectively decrease
morphine formation from codeine in the brain, we injected rats with i.c.v.
propranolol or vehicle, sacrificed the animals, and then measured the effect on
codeine metabolism ex vivo in brain membranes and liver microsomes. If
propranolol had irreversibly inhibited CYP2D in the brain and not in the liver, then
brain membranes should have more in vitro CYP2D activity in vehicle- versus
propranolol-treated rats, while in liver microsomes they should be similar. This
would provide further evidence to support that the lower morphine levels in the
brains of inhibitor-treated was indeed due to irreversible brain CYP2D inhibition
and reduced codeine metabolism in the brain.
Rats were injected with either 20 μg i.c.v. propranolol or vehicle and then
sacrificed 24 h later. In brain membranes incubated with codeine (500 μM=10 x Km
(Mikus, Somogyi et al. 1991)), velocity of morphine formation was significantly lower
in propranolol-treated rats than in vehicle-treated rats (p<0.04; n=8/group; Figure
32.a). In liver microsomes incubated with 500 μM codeine, there was no significant
difference in the velocity of morphine formation between propranolol- and vehicle-
treated rats (p>0.9; n=8/group; Figure 32.b).
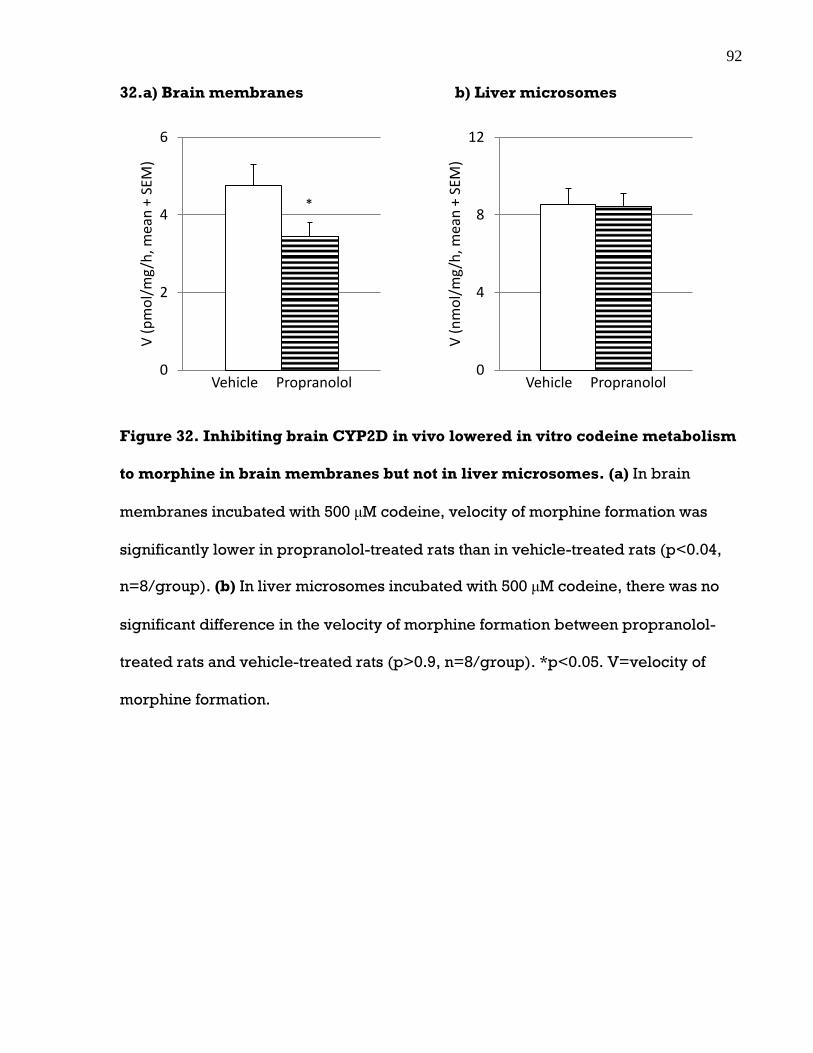
92
32.a) Brain membranes b) Liver microsomes
Figure 32. Inhibiting brain CYP2D in vivo lowered in vitro codeine metabolism
to morphine in brain membranes but not in liver microsomes. (a) In brain
membranes incubated with 500 μM codeine, velocity of morphine formation was
significantly lower in propranolol-treated rats than in vehicle-treated rats (p<0.04,
n=8/group). (b) In liver microsomes incubated with 500 μM codeine, there was no
significant difference in the velocity of morphine formation between propranolol-
treated rats and vehicle-treated rats (p>0.9, n=8/group). *p<0.05. V=velocity of
morphine formation.
0
2
4
6V
(p
mo
l/m
g/h
, mea
n +
SEM
)
Vehicle Propranolol
*
0
4
8
12
V (
nm
ol/
mg
/h, m
ean
+ S
EM)
Vehicle Propranolol

93
3.12 Inhibiting brain CYP2D in vivo lowered in vitro
dextromethorphan metabolism in the brain but not liver
To further confirm that i.c.v. injections of CYP2D inhibitors could reduce brain
CYP2D activity, we measured the effect of i.c.v. propranolol treatment on the ex vivo
metabolism of a CYP2D probe drug, dextromethorphan, in brain membranes and
liver microsomes. If i.c.v. propranolol irreversibly inhibits CYP2D in the brain and
not in the liver, then brain membranes should have more in vitro CYP2D activity in
vehicle- versus propranolol-treated rats, while in liver microsomes they should be
similar. If i.c.v. propranolol can inhibit the metabolism of an additional CYP2D
substrate other than codeine in brain membranes but not in liver microsomes ex
vivo, this would provide further evidence that i.c.v. inhibitor treatment inhibits brain,
but not hepatic, CYP2D activity.
Rats were injected with either 20 μg i.c.v. propranolol or vehicle and then
sacrificed 24 h later. In brain membranes incubated with dextromethorphan (25
μM=10x Km (Kerry, Somogyi et al. 1993)), velocity of dextrorphan formation trended
toward being lower in propranolol-treated rats than in vehicle-treated rats (p=0.095;
n=12/group; Figure 33.a). In liver microsomes incubated with 25 μM
dextromethorphan, there was no significant difference in the velocity of dextrorphan
formation between propranolol- and vehicle-treated rats (p>0.8; n=12/group;
Figure 33.b).
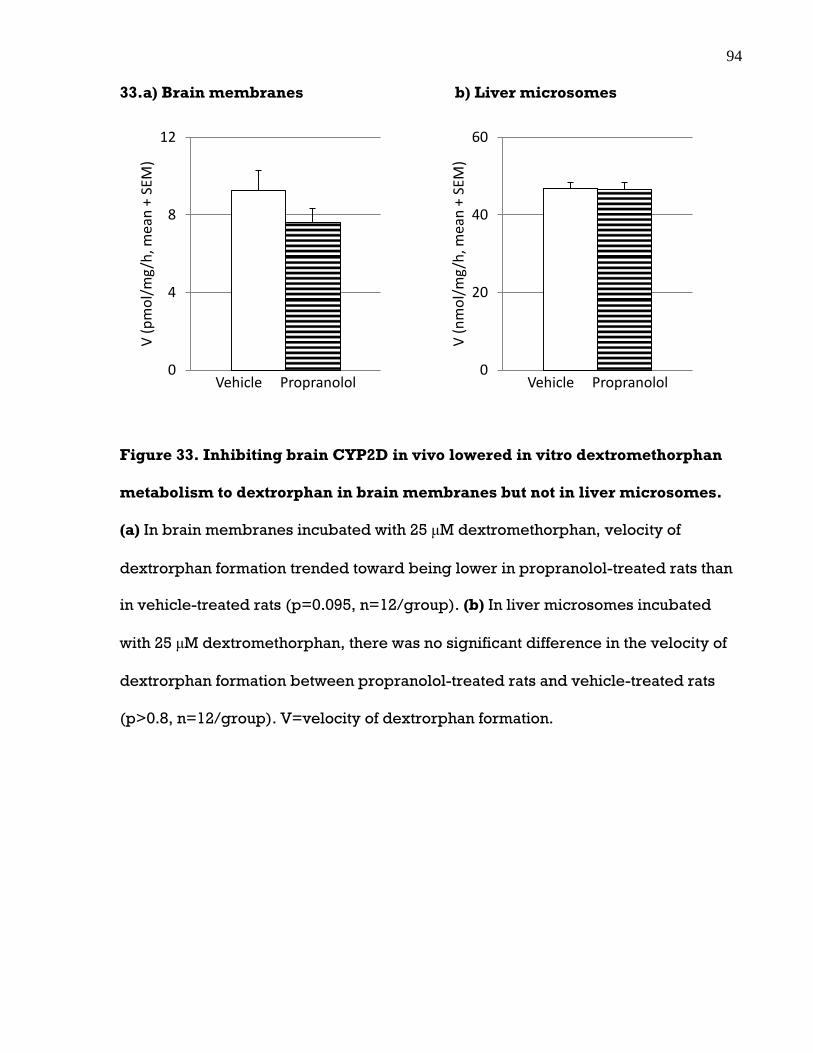
94
33.a) Brain membranes b) Liver microsomes
Figure 33. Inhibiting brain CYP2D in vivo lowered in vitro dextromethorphan
metabolism to dextrorphan in brain membranes but not in liver microsomes.
(a) In brain membranes incubated with 25 μM dextromethorphan, velocity of
dextrorphan formation trended toward being lower in propranolol-treated rats than
in vehicle-treated rats (p=0.095, n=12/group). (b) In liver microsomes incubated
with 25 μM dextromethorphan, there was no significant difference in the velocity of
dextrorphan formation between propranolol-treated rats and vehicle-treated rats
(p>0.8, n=12/group). V=velocity of dextrorphan formation.
0
4
8
12V
(p
mo
l/m
g/h
, mea
n +
SEM
)
Vehicle Propranolol 0
20
40
60
V (
nm
ol/
mg
/h, m
ean
+ S
EM)
Vehicle Propranolol

95
Section 4: Discussion, Conclusions, Future Directions
4.1 Summary and further implications
While previous studies have demonstrated the activity (Tyndale, Li et al. 1999) and
inducibility (Mann, Miksys et al. 2008, Yue, Miksys et al. 2008) of brain CYP2D, they
did not indicate whether brain CYP2D expression or activity levels were high
enough to influence drug response. This is the first study to show that brain CYP2D-
mediated metabolism can alter the effect of a centrally-acting drug. We decreased
rat brain CYP2D activity through the use of CYP2D inhibitors, which decreased
codeine-induced analgesia. We also demonstrated that morphine concentrations in
the brain correlated with analgesia. Our results reveal that brain CYP2D activity in
the rat plays a significant role in the metabolism and effect of codeine, suggesting
that brain CYP2D-mediated metabolism may have an important impact on the
response to the numerous other centrally-acting CYP2D substrates as well. The
response to centrally-acting drugs can display large interindividual variation as well
as poor correlations with plasma drug levels (Michels and Marzuk 1993a).
Differences in the level of drug metabolism in the brain, which does not affect
plasma drug levels as shown here, may contribute to this variation in drug response.

96
4.1.1 Rat model of reduced brain CYP2D activity
A factor that has made it difficult in the past to ascertain the contribution of brain
CYP2D to drug metabolism is the challenge of differentiating between metabolites
that were formed in the brain versus those that were formed in the liver and crossed
into the brain. Establishing the role of brain CYP2D in local drug metabolism and
response requires a model in which CYP2D activity can be altered in the brain
without changing hepatic activity. Using i.c.v. injection of CYP2D inhibitors, we were
able to selectively inhibit rat brain CYP2D activity. The decrease in brain but not
plasma morphine concentrations, and brain but not hepatic enzyme activity, after
i.c.v. injections of CYP2D inhibitors offers convincing evidence that the decrease in
codeine-induced analgesia was due to a decrease in brain, and not liver, CYP2D-
mediated metabolism of codeine to morphine.
To assess the effect of inhibiting brain CYP2D on the analgesic response to
codeine, the use of propranolol and propafenone has several advantages over other
CYP2D inhibitors. Propranolol, a -adrenergic receptor blocker, and propafenone, a
sodium channel blocker, are not expected to affect the CNS in a way that would
affect behaviour in the tail-flick test, and there have been no reports of either of
these drugs having an effect on nociception or analgesia. This attribute makes them
more suitable for our purposes than other CYP2D inhibitors. For example, the
antidepressant paroxetine is a potent CYP2D6 MBI (Bertelsen, Venkatakrishnan et al.
2003), but it has antinociceptive effects, possibly through opioidergic mechanisms
(Duman, Kesim et al. 2004). Quinidine, a commonly used CYP2D competitive
inhibitor (Brosen, Gram et al. 1987), enhances morphine-induced analgesia by

97
inhibiting efflux transporters (Okura, Morita et al. 2009). Quinine, a potent CYP2D
competitive inhibitor in rats (Kobayashi, Murray et al. 1989, Kerry, Somogyi et al.
1993, Xu, Aasmundstad et al. 1995), has antinociceptive effects possibly through
dopaminergic mechanisms (Amabeoku, Ewesuedo et al. 1992). Fluoxetine, another
CYP2D competitive inhibitor, has its own antinociceptive effects in the tail-flick test,
possibly through central opioid pathways (Schreiber, Backer et al. 1996, Singh, Jain
et al. 2001), and it potentiates morphine-induced analgesia (Erjavec, Coda et al.
2000, Nayebi, Hassanpour et al. 2001). Furthermore, propranolol and propafenone
are relatively selective inhibitors of CYP2D (Masubuchi, Fujita et al. 1991,
Turpeinen, Korhonen et al. 2006, McGinnity, Waters et al. 2008), unlike paroxetine
which is also a MBI of CYP3A (Obach, Walsky et al. 2007) and fluoxetine which also
potently inhibits CYP2C19 (Turpeinen, Korhonen et al. 2006, McGinnity, Waters et
al. 2008). Thus, propranolol and propafenone were better choices than many other
CYP2D inhibitors.
An advantage of using a MBI, propranolol, was that it could be administered
well ahead of the test drug. Covalent modifications of enzymes by MBIs are
permanent and activity can only be recovered via synthesis of new enzyme, which
therefore results in a longer lasting inhibition that is not reversed by the presence of
a higher affinity substrate. Propranolol’s elimination half-life in rat blood and brain
are both approximately 1 h (Bianchetti, Elghozi et al. 1980). Therefore, there was
virtually no propranolol left in the body by the time codeine was given 24 h later,
which limits the chance of off-target effects. Showing that brain morphine levels and
analgesia from codeine were reduced by two CYP2D inhibitors with different
mechanisms of inhibition and different pharmacological actions supports the notion

98
that these results were indeed due to brain CYP2D inhibition as opposed to some
other property or action specific to either inhibitor.
4.1.2 Inhibition of brain CYP2D lowers codeine-induced
analgesia
We showed that inhibitor treatment resulted in lower tail-flick latency (TFL) and area
under the analgesia time curve (AUC) compared to vehicle treatment during the first
30 min after codeine injection, which is consistent with our hypothesis that inhibiting
brain CYP2D would reduce analgesia during the initial period after codeine
injection. However, TFL at 40 min after codeine injection and AUC from 30-60 min
and 0-60 min after codeine injection were also significantly lower after inhibitor
treatment, suggesting that the effects of brain CYP2D inhibition last longer than
estimated from Chen, Irvine et al. (1990). TFLs later than 40 min after codeine
injection and AUC for 60-120 min and 0-120 min after codeine injection were not
significantly different after inhibitor treatment compared to after vehicle treatment,
suggesting that inhibiting brain CYP2D only affects the earlier period after codeine
injection. We interpret these results as such: during the initial 40 min period after
codeine injection in rats, analgesia is mediated mainly by morphine formed by brain
CYP2D-mediated codeine metabolism, as relatively less morphine formed by
hepatic CYP2D has crossed into the brain at this time. Thus, during this period,
inhibition of brain CYP2D activity and the ensuing reduction in brain morphine
levels results in lower analgesia. Over time, morphine formed by hepatic CYP2D

99
accumulates in the periphery and crosses the BBB, and by 60 min after codeine
injection, sufficient amounts of morphine have entered the brain to offset the initial
differences in brain morphine levels that were due to differences in brain CYP2D
activity. Because more morphine is formed by the liver than by the brain, analgesia
at these later time points is mediated mainly by morphine formed by hepatic CYP2D
that crosses into the brain. Inhibition of brain CYP2D did not affect hepatic CYP2D
activity, and thus did not result in lower analgesia past 60 min after codeine
injection. Our interpretation is supported by the finding that morphine could be
detected in the brains of codeine- but not morphine-injected rats at 30 min after
peripheral injection (Chen, Irvine et al. 1990), implying that morphine in the brain at
this time is due to local codeine metabolism as morphine from the periphery had not
yet entered the brain. Also in line with our interpretation are findings that brain
uptake of codeine is faster than that of morphine (Oldendorf, Hyman et al. 1972), that
morphine is transported out of the brain by efflux transporters (Bouw, Gardmark et
al. 2000), and the fact that morphine has one less methyl group than codeine, which
is expected to make morphine less lipid soluble and therefore less able to cross the
BBB. Altogether, our findings indicate that inhibiting brain CYP2D reduces analgesia
during the first hour after codeine injection in rats, and that variation in brain CYP2D
activity may influence the onset of analgesia from codeine.
We did not observe a difference between baseline TFL and TFL after inhibitor
treatment, which indicates that the inhibitors by themselves do not affect
nociception. We also saw that inhibitor treatment did not result in different TFL or
AUC compared to vehicle treatment during any time after morphine injection, which
was expected as morphine, being the active drug, does not depend on CYP2D

100
activity in order to confer analgesia. This finding also indicates that the inhibitors did
not lower codeine-induced analgesia by way of interfering with morphine’s actions
(e.g., by affecting mu-opioid receptors or drug transporters). TFL and AUC did not
differ between the two phases of codeine or morphine testing, indicating that the
rats did not develop tolerance to either of the opioids. Following vehicle treatment,
there was no difference in TFL or AUC after codeine injection compared to after
morphine injection, which verifies that the codeine and morphine doses we used
produce equivalent analgesia. Altogether, these findings indicate that inhibiting
brain CYP2D reduces analgesia from codeine but not from an equianalgesic dose of
morphine.
4.1.3 Analgesia correlates with morphine levels in the brain
and not plasma
We showed that at 30 min after codeine injection, which is when the largest
difference in TFL after inhibitor treatment compared to after vehicle treatment
occurred, inhibitor-treated rats had lower brain morphine concentrations,
corresponding to the lower TFLs. We also showed that at this time, inhibitor-treated
rats had lower morphine to codeine ratios in the brain, consistent with the notion that
less codeine was metabolized to morphine in the brains of these animals. The ratio of
morphine to total (morphine plus codeine) drug concentration in the brain was also
lower in inhibitor-treated rats at this time, indicating that a smaller portion of the
total drug was found as morphine in the inhibited animals. There was no difference

101
between inhibitor- and vehicle-treated rats in codeine levels (as morphine is a minor
metabolite) or in total drug levels in brain or plasma at this time, indicating that
differences in analgesia were not due to differences in either of these two
parameters. There was also no difference between inhibitor- and vehicle-treated
rats in any plasma parameters at this time, indicating that hepatic CYP2D activity was
not affected by inhibitor treatment and that differences in analgesia were not due to
plasma drug levels. At this time, TFL correlated with brain morphine levels, brain
morphine to codeine ratios, and brain morphine to total drug ratios, but not with
brain codeine levels, brain total drug levels or any parameter in the plasma.
At 60 or 90 min after codeine injection, when there was no difference in TFL
after inhibitor treatment compared to after vehicle treatment, there was
correspondingly no difference in any of the pharmacokinetic parameters between
inhibitor- and vehicle-treated rats. This is in line with the belief that at these later
time points, analgesia is mediated by morphine formed in the liver that crosses into
the brain, and that initial differences in brain morphine levels between inhibitor-
and vehicle-treated rats seen at 30 min are now masked by the large amount of
morphine that has entered the brain from the periphery. Overall, these results
suggest that analgesia is mediated by morphine in the brain, and that the differences
in codeine-induced analgesia we observed after inhibitor treatment compared to
after vehicle treatment were due to differences in brain morphine levels.

102
4.1.4 Inhibiting brain CYP2D in vivo lowers in vitro enzyme
activity in brain membranes and not liver microsomes
CYP2D velocity, measured using codeine and dextromethorphan as substrates, was
lower in brain membranes of rats that had received i.c.v. inhibitor injections
compared to rats that had received i.c.v. vehicle injections, indicating that inhibitor
treatment did indeed result in the inhibition of brain CYP2D activity. In liver
microsomes, there was no difference in CYP2D velocity between inhibitor- and
vehicle-treated rats, indicating that hepatic CYP2D was not inhibited by i.c.v.
inhibitor injection. These findings provide evidence that the lower brain morphine
levels seen in inhibitor-treated rats were indeed due to reduced brain CYP2D-
mediated metabolism of codeine to morphine. Therefore, while hepatic CYP2D may
be responsible for the bulk of morphine formed from the systemic codeine injection,
localized, in situ codeine metabolism in the brain can meaningfully impact brain
morphine levels and, in turn, response to codeine.
4.1.5 Limitations
A limitation of this study was that propafenone, being a competitive inhibitor, had to
be administered during the same time period as codeine, which could potentially
put more stress on the animal and interfere with the response that is being
observed. It is also not known whether propafenone interacts with codeine in a way
that would reduce analgesia. However, given the very similar responses seen
following the two different inhibitors, this does not appear to have been an issue.

103
Another limitation was that propranolol, being lipophilic, is able to cross the
BBB (Rowland, Yeo et al. 1994, Komura and Iwaki 2005). Therefore, higher doses of
i.c.v. administered propranolol can cross into the periphery and inhibit hepatic
CYP2D, as was the case with the 40 g dose used in our pilot study (but not with the
lower 20 g dose used in our subsequent experiments). This put an upper limit to the
inhibitor dose we could use and thus we may not have completely inhibited CYP2D
in the brain; after inhibitor treatment there was still morphine in the brain at 30 min
following codeine injection and ex vivo CYP2D activity in the brain membranes.
Identifying a CYP2D inhibitor that does not cross the BBB would be advantageous as
it would allow higher doses to be used which may achieve more complete brain
CYP2D inhibition and result in an even larger effect on drug metabolism and
response, more analogous to those who are CYP2D6 PMs.
An additional limitation was that the heat exposure cut-off of 10 sec (which was
to prevent damage to tail skin) did not allow us to detect differences in analgesia in
the period when analgesia from codeine peaked, during which TFLs of 10 sec were
reached both after inhibitor treatment as well as after vehicle treatment. Therefore, it
is unclear whether the lack of difference seen in TFL between inhibitor versus
vehicle treatment after 40 min following codeine injection was due to the cut-off or to
an actual lack of difference in analgesia. It is therefore possible that inhibition of
brain CYP2D had a larger or longer effect on codeine-induced analgesia than what
we were able to detect with this model at the dose of codeine used. This may have
more likely been the case for the propafenone-treated rats, in which 8 out of 12 rats
reached TFLs of 10 sec after both vehicle and inhibitor treatment, compared to the
propranolol-treated rats in which only 6 out of 16 did.

104
We used a subcutaneous route of administration for codeine instead of the
oral route which is typically used in humans. This route was chosen because it
minimizes first pass metabolism by the liver, thereby allowing us to better detect the
effects of brain-mediated metabolism. However, a disadvantage of this method is
that it may not accurately represent the impact of brain CYP2D on the response to
codeine when it is administered by its typical route. Oral codeine is subject to first
pass effects, so even though we have shown that brain CYP2D-mediated metabolism
influences response to subcutaneously injected codeine, the same may not hold
when codeine is given orally. It would be useful to verify whether the impact of brain
CYP2D on codeine response will remain in the presence of more extensive hepatic
metabolism from the oral mode of administration. This is suggested to be the case
by the finding that, inhibiting brain, but not hepatic, CYP2B altered the sleep-times
induced by intaperitoneal injection of the anaesthetic propofol (metabolized by
CYP2B) (Khokhar and Tyndale 2011). Since intraperitoneal injection, like oral
administration, is subject to first pass metabolism, this finding suggests that brain
CYP activity can have a meaningful impact on drug response even in the presence of
hepatic metabolism.
In summary, by using i.c.v. injections of CYP2D inhibitors, we inhibited
CYP2D in rat brain and not liver, and established an animal model of reduced brain
CYP2D activity. We used this model to evaluate the role of brain CYP2D in the
metabolism and effect of codeine, a centrally-acting drug. We found that inhibition
of brain CYP2D could decrease the metabolic activation of codeine to morphine in
the brain and reduce codeine-induced analgesia. Differences in brain CYP activity
may contribute to interindividual variation in the response to centrally-acting drugs.

105
For example, the higher CYP2D6 levels in smokers and seniors might help explain
the altered efficacy or side-effect profiles of centrally-acting drugs seen in these
individuals (Nelson, Mazure et al. 1995, Jabs, Bartsch et al. 2003, Mann, Miksys et al.
2008, Mann, Miksys et al. 2012).
4.2. Clinical relevance of brain CYP2D activity
We have demonstrated that rat brain CYP2D is functional in vivo and can have a
significant contribution to drug effect. While our findings may not have definite
clinical implications in codeine-induced analgesia in humans, they do point to the
functionality of rat brain CYP2D and its ability to affect drug response through local
metabolism. Brain CYP2D-mediated metabolism may not only have a bearing on
drug efficacy, but the involvement of CYP2D in the formation or metabolism of
endogenous compounds such as serotonin (Yu, Idle et al. 2003a) and dopamine
(Bromek, Haduch et al. 2010), indicates that these CYPs may also play a role in
normal brain function. This notion is supported by associations of CYP2D6 genotype
with resting brain activity (Kirchheiner, Seeringer et al. 2011) and personality traits
(Bijl, Luijendijk et al. 2009). CYP2D also inactivates neurotoxins such as 1-methyl-4-
phenylpyridinium (MPP+) (Mann and Tyndale 2010). Inhibition of CYP2D increased
the neurotoxicity induced by MPP+ in a human neuronal cell line (Mann and Tyndale
2010). The induction of brain CYP2D by commonly used drugs such as alcohol and
nicotine (Warner and Gustafsson 1994, Miksys, Rao et al. 2002, Miksys and Tyndale
2004, Mann, Miksys et al. 2008, Yue, Miksys et al. 2008, Miller, Miksys et al. 2012)

106
may affect the likelihood of neurotoxicity following exposure to, and subsequent
metabolism of, neurotoxic substances. The multiple families of drug-metabolizing
CYPs in the brain, including CYP2B, CYP2D, CYP2E1, CYP3A and CYP4 (Strobel,
Thompson et al. 2001, Meyer, Gehlhaus et al. 2007) and their diverse range of
centrally-acting substrates, lends support to the brain being an important organ in
drug metabolism.
4.2.1 Centrally-acting drugs
Brain CYP2D expression and activity can vary among individuals based on genetics,
exposure to environmental inducers or differences in age. Differences in brain
CYP2D levels may affect the local pharmacokinetics of its many centrally-acting
substrates, which include clinical drugs, drugs of abuse, neurotoxins and
endogenous substances. The induction of brain CYP2D6 may result in increased
substrate metabolism, which in turn may lead to altered efficacy of clinical drugs, as
well as altered susceptibility to adverse drug reactions. Smokers respond differently
to centrally-acting CYP2D6 substrates such as antipsychotics and antidepressants
compared to nonsmokers (Jabs, Bartsch et al. 2003, George, Sacco et al. 2008).
Smokers also have elevated CYP2D6 levels in the brain, and not liver (Miksys and
Tyndale 2004, Mann, Miksys et al. 2008). We can therefore speculate that these
differences in drug response are due to differences in the level of brain CYP2D6-
mediated metabolism.

107
CYP2D6 metabolizes various antipsychotics including haloperidol,
perphenazine, thioridazine and risperidone (Ingelman-Sundberg 2005). Plasma
levels of risperidone and its active metabolite do not correlate with drug response
(Spina, Avenoso et al. 2001) or extrapyramidal side effects (EPS) (Lane, Chiu et al.
2000, Riedel, Schwarz et al. 2005) in schizophrenia patients, and this may be due to
variation in drug metabolism in the brain. Smokers have less EPS from
antipsychotics than non-smokers (Jabs, Bartsch et al. 2003). CYP2D in the human
brain is elevated in smokers, particularly in the basal ganglia which is involved in
EPS (Jabs, Bartsch et al. 2003, Mann, Miksys et al. 2008). Higher brain CYP2D6
activity, and therefore increased clearance of antipsychotics, may contribute to the
reduced EPS in smokers (Funck-Brentano, Boelle et al. 2005). Moreover,
antidepressants (inactivated by CYP2D6) were less effective in smokers compared
to non-smokers (George, Sacco et al. 2008). This may be attributable to the higher
levels of brain CYP2D6 in smokers which may increase inactivation of
antidepressants in the brain.
Because brain CYP2D6 levels increase with age, the impact of drug
metabolism in the brain by CYP2D6 may be augmented in seniors (Mann, Miksys et
al. 2012). Desipramine, an antidepressant inactivated by CYP2D6, was less effective
in older (>75 years) individuals than in younger patients when controlling for drug
dose and plasma levels (Nelson, Mazure et al. 1995). The increased levels of brain
CYP2D6 in individuals 60 to 80 years of age may increase rates of desipramine
inactivation and contribute to its reduced efficacy. In sum, variation in brain CYP2D6
levels, whether due to genetics, environmental induction or age may alter the
therapeutic and/or adverse effects of centrally-acting drugs.

108
4.2.2 Drugs of abuse
Codeine, along with being commonly prescribed for pain relief, is also widely
abused. Our observation that brain CYP2D activity influences the early analgesic
effects of codeine also has implications for codeine’s abuse liability. The abuse
liability of a drug depends, at least partly, on the quickness of onset of its reinforcing
effects (Griffiths and Wolf 1990, Farre and Cami 1991). As previously suggested
(Tyndale, Droll et al. 1997, Kathiramalainathan, Kaplan et al. 2000), the reinforcing
qualities of codeine come from its O-demethylated metabolites (morphine,
morphine-6-glucuronide), which are formed by CYP2D. Therefore, analogous to the
analgesic effects of codeine, the initial reinforcing effects of codeine should be
mediated by its metabolism to morphine in the brain, before morphine from the
periphery has crossed into the brain. Since the formation of morphine in the brain
should occur at a higher rate in those with elevated brain CYP2D activity (smokers,
alcoholics, seniors), these individuals are expected to experience a quicker onset of
reinforcing effects from codeine and may be more prone to codeine abuse. Higher
brain CYP2D6 activity can also be speculated to be a risk factor for the abuse of
recreational drugs that are inactivated by CYP2D6 (e.g., amphetamine), as the
abuse liability of a drug also depends partly on the quickness of offset of its
reinforcing effects (Griffiths and Wolf 1990, Farre and Cami 1991). Overall, altered
brain CYP2D levels, due to genetics, environmental induction or aging, could affect
the disposition of drugs of abuse metabolized by CYP2D (e.g. amphetamine, MDMA,
dextromethorphan), and thus lead to interindividual differences in the susceptibility
to abusing these drugs.

109
4.2.3 Endogenous substrates
CYP2D is involved in the synthesis and metabolism of a range of centrally-acting
endogenous substances (Funae, Kishimoto et al. 2003), which suggests a role for this
enzyme in basal brain function. Because CYP2D6 can catalyze the formation of
dopamine, serotonin, epinephrine and norepinephrine, differences in CYP2D6
levels are believed to have an impact on personality or mood (Funae, Kishimoto et
al. 2003, Yu, Idle et al. 2003a, Bromek, Haduch et al. 2010). Genetic variation in
CYP2D6 is associated with various personality and behavioural traits (Roberts, Luty
et al. 2004, Kirchheiner, Lang et al. 2006, Gonzalez, Penas-Lledo et al. 2008, Ahlner,
Zackrisson et al. 2010). For example, CYP2D6 PMs have been reported to have
higher anxiety (Llerena, Edman et al. 1998, Gonzalez, Penas-Lledo et al. 2008), less
success in being socialized (Llerena, Edman et al. 1998), higher impulsivity
(Gonzalez, Penas-Lledo et al. 2008, Penas-Lledo, Dorado et al. 2009), higher ease of
decision making (Bertilsson, Alm et al. 1989), lower harm avoidance (Roberts, Luty
et al. 2004), more novelty seeking behaviours (Roberts, Luty et al. 2004), and lower
competitiveness (Kirchheiner, Lang et al. 2006, Gonzalez, Penas-Lledo et al. 2008).
In addition to catalyzing the conversion of endogenous precursor compounds
to neurotransmitters, CYP2D6 also metabolizes neurosteroids such as progesterone
and allopregnanolone (Hiroi, Kishimoto et al. 2001, Kishimoto, Hiroi et al. 2004,
Niwa, Okada et al. 2008). Progesterone can regulate the synthesis and release of
neurotransmitters and neuropeptides (Pluchino, Luisi et al. 2006). Progesterone
increases serotonin turnover, catechol-O-methyltransferase activity, and MAO
activity in rats (Genazzani, Stomati et al. 2000). In women, progesterone treatment

110
changes mood (Hlatky, Boothroyd et al. 2002). Allopregnanolone, a metabolite of
progesterone, acts on γ-aminobutyric acid receptors and thus induces anxiolysis and
sedation (Pluchino, Luisi et al. 2006). Changes in allopregnanolone levels have been
linked with depression, anxiety, and irritability (Pluchino, Luisi et al. 2006). CYP2D6
also metabolizes anandamide (Snider, Sikora et al. 2008), an endocannabinoid that is
implicated in mood, anxiety, and emotional processing (Bambico and Gobbi, 2008).
In summary, variation in brain CYP2D6 may alter levels of these endogenous
neuromodulators and consequently influence personality, behaviour and mood.
A further function of brain CYP2D6 may be the production of endogenous
morphine. Various studies have shown that humans (Zhu, Cadet et al. 2005) and
animals (Kodaira and Spector 1988, Amann, Roos et al. 1995, Zhu, Mantione et al.
2005) are able to synthesize morphine, and that CYP2D is important in this process
as it metabolizes multiple morphine precursors (Zhu, Cadet et al. 2005, Zhu,
Mantione et al. 2005, Kream, Stefano et al. 2006). The formation of morphine from
tyramine, tyrosine and codeine was demonstrated both in vitro and in vivo in the
marine invertebrate Mytilus edulis; morphine formation was substantially reduced by
the CYP2D inhibitor quinidine (Zhu, Mantione et al. 2005, Zhu 2008). Human white
blood cells express CYP2D6 which can synthesize morphine from tyramine,
norlaudansoline and codeine; morphine production was reduced by the CYP2D6
inhibitors bufuralol, quinidine and paroxetine (Zhu, Cadet et al. 2005). Together,
these findings suggest that CYP2D is involved in endogenous morphine synthesis.
The potential functional significance of this was demonstrated in CYP2D6 PMs and
EMs who were assessed for pain thresholds with the cold pressor test (Sindrup,
Poulsen et al. 1993). Peak pain ratings and area under the pain rating-time curve

111
were significantly higher in CYP2D6 PMs than in EMs (Sindrup, Poulsen et al. 1993).
PMs may therefore have lower pain tolerance than EMs, and this is speculated to be
due to a lack of endogenous morphine production by brain CYP2D6 (Sindrup,
Poulsen et al. 1993). Levels of endogenous morphine excreted in urine were not
found to differ between CYP2D6 PMs and EMs, suggesting that other CYPs may also
be involved in morphine biosynthesis (Mikus, Bochner et al. 1994). However, brain
CYP2D6 may synthesize endogenous morphine locally in the brain, which may
modulate nociception and suggests an additional role that brain CYP2D6 may have
in analgesia (Zhu 2008). In further support of this, there is high expression of
CYP2D6 in the thalamus, a brain region that is involved in nociception (Mann, Miksys
et al. 2008, Wilson, Uhelski et al. 2008). Interindividual variability in thalamus
activity correlates with pain threshold (Dostrovsky 2000, Lenz, Weiss et al. 2004,
Ochsner, Ludlow et al. 2006), and thalamus activity (as measured by resting brain
perfusion) also correlates with CYP2D6 genotype (Kirchheiner, Seeringer et al.
2011). Altogether, variation in brain CYP2D may alter analgesia by affecting the
formation of morphine from exogenous compounds such as codeine as well by
influencing the synthesis of endogenous morphine.
4.2.4 Disease
CYP2D may have a protective role against Parkinson’s disease (PD). PD is a
neurodegenerative disorder characterized by the loss of dopaminergic neurons in
the substantia nigra that project to the striatum (Fahn 2010). A risk factor for PD is
exposure to toxins such as pesticides (Di Monte 2003, Olanow 2007). CYP2D6 can

112
inactivate numerous neurotoxins including 1-methyl-4-phenylpyridinium (MPP+)
(Mann and Tyndale 2010), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
(Modi, Gilham et al. 1997), tetrahydroisoquinolines (Suzuki, Fujita et al. 1992),
harmaline, harmine (Yu, Idle et al. 2003b) and N-methyl-β-carbolines (Herraiz,
Guillen et al. 2006). CYP2D6 PM status is associated with an increased risk for PD
(McCann, Pond et al. 1997), which is further increased when these individuals are
exposed to pesticides (Deng, Newman et al. 2004, Elbaz, Levecque et al. 2004). This
suggests that a lack of CYP2D6 activity, and thus an inability to inactivate
environmental neurotoxins, makes individuals more vulnerable to their effects.
Among PD cases, CYP2D6 PMs were overrepresented, and EMs had ~40-50% lower
levels of brain CYP2D6 compared to control EMs (Mann, Miksys et al. 2012).
Therefore, low levels of brain CYP2D6 may increase the risk of PD, while high levels
may protect against it.
The observation that PD cases had lower brain CYP2D6 levels compared to
controls (Mann, Miksys et al. 2012) is in line with the findings that inhibiting CYP2D
increased toxicity of MPP+ (a Parkinsonian neurotoxin inactivated by CYP2D) in
human neuronal cells (Mann and Tyndale 2010) and that over-expressing CYP2D6
protected against MPP+ toxicity in PC12 cells (Matoh, Tanaka et al. 2003).
Furthermore, brain CYP2D6 is located in areas that are ideal for the inactivation of
Parkinsonian neurotoxins (e.g., basal ganglia, BBB, dopaminergic neurons) (Gilham,
Cairns et al. 1997, Miksys, Rao et al. 2002, Dutheil, Jacob et al. 2010). Thus, brain
CYP2D6 may protect against specific neurotoxins and thereby reduce the risk for
PD.

113
Smokers have high levels of brain CYP2D6, especially in the basal ganglia
including the substantia nigra (Mann, Miksys et al 2008), and smokers also have a
reduced risk for PD (Alves, Kurz et al. 2004). Nicotine has been shown to protect
against Parkinsonian neurotoxins both in vitro and in vivo (Khwaja, McCormack et al.
2007, Quik, O’Neill et al. 2007). For example, pretreating monkeys with nicotine
protected against nigrostriatal damage from MPTP (Quik, Parameswaran et al. 2006).
Moreover, nicotine significantly induced monkey CYP2D in the basal ganglia
including the substantia nigra (Mann, Miksys et al 2008). Therefore, the increased
levels of brain CYP2D resulting from induction by smoking and nicotine may protect
against PD.
Brain CYP2D6, through its ability to synthesize dopamine from tyramine, may
help offset the loss of dopamine in PD and thereby alleviate Parkinsonian symptoms.
Brain CYP2D6 is ideally located for this purpose in the basal ganglia and in
dopaminergic neurons (Gilham, Cairns et al. 1997, Riedl, Watts et al. 1999, Bromek,
Haduch et al. 2010, Gonzalez-Hernandez, Cruz-Muros et al. 2010). Nicotine treatment
has been demonstrated to improve dopaminergic tone and motor symptoms in PD
cases, and this may partly be due to increased dopamine formation by nicotine-
induced brain CYP2D6 (Fagerstrom, Pomerleau et al. 1994, Kelton, Kahn et al. 2000,
Quik, Cox et al. 2007). Altogether, variation in brain CYP2D6, due to genetics or
environmental inducers, may alter the risk for and symptom severity of PD. Genetic
variation in CYP2D6 may also be linked with predispositions to psychiatric illnesses
such as schizophrenia, eating disorders, and major depressive disorder (Dorado,
Penas-Lledo et al. 2007, Llerena, Dorado et al. 2007, Gonzalez, Penas-Lledo et al.
2008, Ahlner, Zackrisson et al. 2010, Penas-Lledo, Dorado et al. 2012).

114
4.3 Other brain CYPs
Our data suggests that other brain CYPs may also be functional in vivo and have an
important role in local substrate metabolism. This has been demonstrated to be the
case with brain CYP2B in rats. CYP2B metabolically inactivates the anaesthetic
propofol (Court, Duan et al. 2001). Inhibiting rat brain, and not liver, CYP2B resulted
in an increase in propofol-induced sleep times, while inducing brain CYP2B
decreased sleep times (Khokhar and Tyndale 2011). CYP2B is induced by nicotine
in the brain but not in the liver (Miksys, Hoffmann et al. 2000, Lee, Miksys et al.
2006a, Yue, Khokhar et al. 2009), and smokers have higher CYP2B levels in the
brain, and not liver compared to nonsmokers (Miksys, Lerman et al. 2003, Miksys
and Tyndale 2004). Accordingly, case reports have suggested that smokers require
higher propofol doses to achieve loss of consciousness, and also have fewer side
effects from propofol, compared to nonsmokers (Chimbira and Sweeney 2000,
Lysakowski, Dumont et al. 2006), possibly due to higher brain CYP2B-mediated
propofol inactivation (Tate and Cook 1996, Trapani, Altomare et al. 2000). Therefore,
variation in brain CYP2B activity may alter response to centrally-acting drugs.
CYP2B can also metabolize neurotoxins such as the insecticide, chlorpyrifos, which
is activated to a neurotoxic metabolite. Inhibition of rat brain CYP2B blocked
chlorpyrifos activation and reduced the neurotoxic effects of chlorpyrifos treatment
(Khokhar and Tyndale 2012). Chronic nicotine treatment in rats increased the
neurotoxic effects of chlorpyrifos (Abou-Donia, Abdel-Rahman et al. 2003), possibly
due to induction of brain CYP2B by nicotine and a resultant increase in chlorpyrifos
activation. Together, these findings indicate that differences in brain CYP2B activity

115
impact the metabolism and effects of propofol, chlorpyrifos, and probably the
numerous other centrally-acting CYP2B substrates as well (e.g. diazepam,
methadone, nicotine, cocaine, phencyclidine).
CYP2E1 is also expressed in the brain and can metabolize a variety of
centrally-acting substrates including ethanol, anaesthetics, and neurotoxins (Caro
and Cederbaum 2004). While ethanol metabolism in the brain is mainly mediated by
catalase, CYP2E1 also plays a significant role (Vasiliou, Ziegler et al. 2006). CYP2E1
knockout mice had longer ethanol-induced sleep-times than did wild-type mice,
suggesting decreased ethanol metabolism in the brains of knockout mice (Vasiliou,
Ziegler et al. 2006). However, these animals had lower CYP2E1 activity the liver as
well, thereby making it unclear how much of the effect on sleep-times can be
attributed to brain versus hepatic CYP2E1. Brain CYP2E1 is induced by ethanol, and
this induction is associated with higher oxidative damage in astrocytes (Montoliu,
Sancho-Tello et al. 1995). There are also higher brain CYP2E1 levels in smokers and
in animals treated with nicotine and ethanol (Howard, Miksys et al. 2003, Schoedel
and Tyndale 2003, Miksys and Tyndale 2004, Joshi and Tyndale 2006, Yue, Khokhar
et al. 2009). Thus, variable levels of brain CYP2E1 could affect the local metabolism
of CYP2E1 substrates as well the health of brain cells.

116
4.4 Future directions
4.4.1 Other uses of rat models of differing levels of brain
CYP2D activity
4.4.1a Microdialysis
Microdialysis could be used to measure codeine and morphine levels in the brain in
vivo after peripheral codeine injection in rats that had received i.c.v. CYP2D
inhibitor treatment versus i.c.v. vehicle treatment. These drug levels can then be
correlated with analgesia in the tail-flick test. Microdialysis allows drug levels to be
measured at multiple time points in the same rat (whereas with the method used in
our study, rats had to be sacrificed at the time point being analyzed). Therefore,
microdialysis may provide more comprehensive data on the pharmacokinetics of
codeine and morphine in the brain and plasma over time, and may allow a more
precise understanding of the relative importance of brain versus hepatic CYP2D-
mediated metabolism in analgesia at different times after codeine injection.
4.4.1b Different pain model
It would be useful to determine if our results can be replicated using a different pain
model, such as the warm plate test. This consists of placing a rat onto a metallic plate
that is heated to a constant temperature by a thermode or hot liquid (Woolfe and
MacDonald 1944, Eddy and Leimbach 1953, O'Callaghan and Holtzman 1975). The

117
reaction times of two behaviours, paw licking and jumping, are measured, with an
increase in reaction time indicating analgesia. Both behaviours are under
supraspinal control, and both are modulated by opioids (Le Bars, Gozariu et al.
2001).
4.4.1c Role of rat brain CYP2D in meditating drug inactivation
While we have demonstrated that inhibiting brain CYP2D decreases the metabolic
activation of a prodrug and thereby decreases drug efficacy, future studies should
determine whether inhibiting brain CYP2D also decreases the metabolic inactivation
of a drug and results in enhanced drug effects. Examples of drugs that are
inactivated by CYP2D include amphetamine (stimulant, drug of abuse), desipramine
and imipramine (antidepressants).
4.4.1d Effect of rat brain CYP2D induction on drug response
While we have demonstrated the effects of inhibiting brain CYP2D on drug
metabolism and response, future studies should also determine whether inducing
brain CYP2D results in an increase in brain CYP2D-mediated drug activation or
inactivation and the corresponding increase or decrease in drug effect. Induction of
brain CYP2D (without altering hepatic CYP2D) can be achieved through chronic
nicotine treatment in rats as has been shown previously (Yue, Miksys et al. 2008),
and drug response can be tested in the resultant rat model of increased CYP2D

118
activity. This model may reflect the elevated levels of brain CYP2D in human
smokers and be used to elucidate the altered response to centrally-acting CYP2D
substrates observed in these individuals.
4.4.1e Role of rat brain CYP2D in neurotoxin inactivation
While studies suggest that brain CYP2D protects against Parkinson’s disease (PD), it
is not known how important a role brain CYP2D plays in this function in vivo.
Manipulating brain CYP2D levels using inhibitors and inducers in an in vivo rat
model of PD (e.g., neurotoxicity caused by MPTP or MPP+ exposure) could be used
to determine the influence of brain CYP2D on the metabolism and effects of
Parkinsonian neurotoxins.
4.4.2 Therapeutic uses of brain CYP2D induction
Future studies should evaluate the efficacy of increasing brain CYP2D6 activity as a
new therapeutic technique for protecting against neurotoxicity (e.g. from MPTP or
MPP+). The induction of brain CYP2D6 would be valuable in circumstances where
low CYP2D6 levels may increase the risk of disease, such as the case with PD.
Nicotine may be used as a preventative measure to induce brain CYP2D6 and
increase local neurotoxin inactivation in brain regions affected by PD (e.g., striatum
and substantia nigra). This may lower the risk for, or delay the onset of, PD. Nicotine
may also be used after the onset of PD to help ameliorate symptoms (Kelton, Kahn et

119
al. 2000, Vieregge, Sieberer et al. 2001). Nicotine’s induction of brain CYP2D6 may
increase the CYP2D6-mediated synthesis of dopamine, which may help compensate
for the loss of dopamine in PD.
4.5 Conclusions
CYP2D6 is an important enzyme that metabolizes a wide variety of drugs, toxins and
endogenous substrates. CYP2D6 is genetically polymorphic, and the expression and
activity of CYP2D in the brain can be altered by environmental inducers and
increases in age, as demonstrated in animal and human studies. Using a rat model of
reduced brain CYP2D activity, we have shown that inhibiting brain CYP2D results in
lower brain morphine levels and less analgesia after codeine administration,
suggesting that brain CYP2D has an important role in the metabolism and effect of
codeine, a centrally-acting CYP2D substrate. Therefore, brain CYP2D6 may have a
significant impact on response to codeine (including analgesia and abuse liability)
and other drugs metabolized by CYP2D6 (e.g. antidepressants, antipsychotics).
Differences in brain CYP2D6 activity, whether through genetic variation, exposure to
alcohol or nicotine, or age, may contribute to the interindividual variation in
therapeutic efficacy and side effect profiles of centrally-acting drugs metabolized by
CYP2D6. The expression of multiple families of CYPs in the brain, and their diverse
array of centrally-acting substrates, supports the brain as being an important organ
in the metabolism of drugs, toxins and endogenous substrates.

120
References
Abou-Donia, M. B., A. Abdel-Rahman, L.B. Goldstein, A.M. Dechkovskaia, D.U. Shah,
S.L. Bullman and W.A. Khan (2003). "Sensorimotor deficits and increased brain
nicotinic acetylcholine receptors following exposure to chlorpyrifos and/or nicotine
in rats." Arch Toxicol 77(8): 452-458.
Abraham, J. E., M. J. Maranian, K. E. Driver, R. Platte, B. Kalmyrzaev, C. Baynes, C.
Luccarini, M. Shah, S. Ingle, D. Greenberg, H. M. Earl, A. M. Dunning, P. D. P.
Pharoah and C. Caldas (2010). "CYP2D6 gene variants: association with breast
cancer specific survival in a cohort of breast cancer patients from the United
Kingdom treated with adjuvant tamoxifen." Breast Cancer Res 12(4): R64.
Adler, T. K., J. M. Fujimoto, E. L. Way and E. M. Baker (1955). "The metabolic fate of
codeine in man." J Pharmacol Exp Ther 114(3): 251-262.
Agundez, J. A. G., M. C. Ledesma, J. M. Ladero and J. Benitez (1995). "Prevalence of
CYP2D6 gene duplication and its repercussion on the oxidative phenotype in a white
population." Clin Pharmacol Ther 57(3): 265-269.
Ahlner, J., A. L. Zackrisson, B. Lindblom and L. Bertilsson (2010). "CYP2D6, serotonin
and suicide." Pharmacogenomics 11(7): 903-905.
Ai, C. Z., Y. Li, Y. H. Wang, Y. D. Chen and L. Yang (2009). "Insight into the effects of
chiral isomers quinidine and quinine on CYP2D6 inhibition." Bioorg Med Chem Lett
19(3): 803-806.
Akil, H., C. Owens, H. Gutstein, L. Taylor, E. Curran and S. Watson (1998).
"Endogenous opioids: overview and current issues." Drug Alcohol Depend 51(1-2):
127-140.
Aklillu, E., I. Persson, L. Bertilsson, I. Johansson, F. Rodrigues and M.
IngelmanSundberg (1996). "Frequent distribution of ultrarapid metabolizers of
debrisoquine in an Ethiopian population carrying duplicated and multiduplicated
functional CYP2D6 alleles." J Pharmacol Exp Ther 278(1): 441-446.
Albores, A., G. Ortega-Mantilla, A. Sierra-Santoyo, M. E. Cebrian, J. L. Munoz-
Sanchez, J. V. Calderon-Salinas and M. Manno (2001). "Cytochrome P450 2B
(CYP2B)-mediated activation of methyl-parathion in rat brain extracts." Toxicol Lett
124(1-3): 1-10.
Alves, G., M. Kurz, S.A. Lie and J.P. Larsen (2004). "Cigarette smoking in Parkinson's
disease: influence on disease progression." Mov Disord 19(9): 1087-1092.
Amabeoku, G., R. Ewesuedo and F. Abuh (1992). "Influence of some dopaminergic
agents on antinociception produced by quinine in mice." Prog
Neuropsychopharmacol Biol Psychiatry 16(3): 351-360.

121
Amann, T., P.H. Roos, H. Huh and M.H. Zenk (1995). "Purifi cation and
characterization of a cytochrome P450 enzyme from pig liver, catalyzing the phenol
oxidative coupling of (R)-reticuline to salutaridine, the critical step in morphine
biosynthesis." Heterocycles 40: 425–440
Archer, S. and L. S. Harris (1965). "Narcotic antagonists." Fortschr Arzneimittelforsch
8: 261-320.
Arvidsson, U., M. Riedl, S. Chakrabarti, J. H. Lee, A. H. Nakano, R. J. Dado, H. H. Loh,
P. Y. Law, M. W. Wessendorf and R. Elde (1995). "Distribution and targeting of a mu-
opioid receptor (MOR1) in brain and spinal cord." J Neurosci 15(5 Pt 1): 3328-3341.
Atweh, S. F. and M. J. Kuhar (1977). "Autoradiographic localization of opiate
receptors in rat brain. I. Spinal cord and lower medulla." Brain Res 124(1): 53-67.
Bambico, F.R. and G. Gobbi (2008). "The cannabinoid CB1 receptor and the
endocannabinoid anandamide: possible antidepressant targets." Expert Opin Ther
Targets 12(11): 1347-1366.
Barbaro, N. M., M. M. Heinricher and H. L. Fields (1989). "Putative nociceptive
modulatory neurons in the rostral ventromedial medulla of the rat display highly
correlated firing patterns." Somatosens Mot Res 6(4): 413-425.
Basbaum, A. I., D. M. Bautista, G. Scherrer and D. Julius (2009). "Cellular and
Molecular Mechanisms of Pain." Cell 139(2): 267-284.
Basbaum, A. I., C. H. Clanton and H. L. Fields (1976). "Opiate and stimulus produced
analgesia: Functional anatomy of a medullospinal pathway." Proc Natl Acad Sci USA
73(12): 4685-4688.
Basbaum, A. I. and T. Jessell (2000). The perception of pain. Principles of
Neuroscience. E. R. Kandel, J. Schwartz and T. Jessell. New York, Appleton and
Lange 472–491.
Basbaum, A. I., N. J. E. Marley, J. Okeefe and C. H. Clanton (1977). "Reversal of
morphine and stimulus-produced analgesia by subtotal spinal cord lesions." Pain
3(1): 43-56.
Bergh, A. F. and H. W. Strobel (1992). "Reconstitution of the brain mixed function
oxidase system: purification of NADPH-cytochrome P450 reductase and partial
purification of cytochrome P450 from whole rat brain." J Neurochem 59(2): 575-581.
Bergh, A. F. and H. W. Strobel (1996). "Anatomical distribution of NADPH-
cytochrome P450 reductase and cytochrome P4502D forms in rat brain: Effects of
xenobiotics and sex steroids." Mol Cell Biochem 162(1): 31-41.
Bertelsen, K. M., K. Venkatakrishnan, L. L. Von Moltke, R. S. Obach and D. J.
Greenblatt (2003). "Apparent mechanism-based inhibition of human CYP2D6 in vitro

122
by paroxetine: Comparison with fluoxetine and quinidine." Drug Metab Dispos
31(3): 289-293.
Bertilsson, L., C. Alm, C. De Las Carreras, J. Widen, G. Edman and D. Schalling
(1989). "Debrisoquine hydroxylation polymorphism and personality." Lancet
1(8637): 555.
Bhagwat, S. V., M. R. Boyd and V. Ravindranath (2000). "Multiple forms of
cytochrome P450 and associated monooxygenase activities in human brain
mitochondria." Biochem Pharmacol 59(5): 573-582.
Bhamre, S., H. K. Anandatheerathavarada, S. K. Shankar, M. R. Boyd and V.
Ravindranath (1993). "Purification of multiple forms of cytochrome P450 from a
human brain and reconstitution of catalytic activities." Arch Biochem Biophys 301(2):
251-255.
Bianchetti, G., J. L. Elghozi, R. Gomeni, P. Meyer and P. L. Morselli (1980). "Kinetics
of distribution of dl-propranolol in various organs and discrete brain areas of the
rat." J Pharmacol Exp Ther 214(3): 682-687.
Bijl, M. J., H. J. Luijendijk, J.F. van den Berg, L.E. Visser, R.H. van Schaik, A. Hofman,
A.G. Vulto, T. van Gelder, H. Tiemeier and B.H. Stricker. (2009). "Association
between the CYP2D6*4 polymorphism and depression or anxiety in the elderly."
Pharmacogenomics 10(4): 541-547.
Blume, N., J. Leonard, Z. J. Xu, O. Watanabe, H. Remotti and J. Fishman (2000).
"Characterization of Cyp2d22, a novel cytochrome P450 expressed in mouse
mammary cells." Arch Biochem Biophys 381(2): 191-204.
Bouw, M. R., M. Gardmark and M. Hammarlund-Udenaes (2000). "Pharmacokinetic-
pharmacodynamic modelling of morphine transport across the blood-brain barrier
as a cause of the antinociceptive effect delay in rats - A microdialysis study." Pharm
Res 17(10): 1220-1227.
Britto, M. R. and P. J. Wedlund (1992). "Cytochrome P-450 in the brain. Potential
evolutionary and therapeutic relevance of localization of drug-metabolizing
enzymes." Drug Metab Dispos 20(3): 446-450.
Bromek, E., A. Haduch and W. A. Daniel (2010). "The ability of cytochrome P450 2D
isoforms to synthesize dopamine in the brain: An in vitro study." Eur J Pharmacol
626(2-3): 171-178.
Brosen, K., L. F. Gram, T. Haghfelt and L. Bertilsson (1987). "Extensive metabolizers
of debrisoquine become poor metabolizers during quinidine treatment." Pharmacol
Toxicol 60(4): 312-314.
Cairns, W., C. A. D. Smith, A. W. McLaren and C. R. Wolf (1996). "Characterization of
the human cytochrome P4502D6 promoter - A potential role for antagonistic

123
interactions between members of the nuclear receptor family." J Biol Chem 271(41):
25269-25276.
Camarata, P. J. and T. L. Yaksh (1985). "Characterization of the spinal adrenergic
receptors mediating the spinal effects produced by the microinjection of morphine
into the periaqueductal gray." Brain Res 336(1): 133-142.
Caraco, Y., T. Tateishi, F. P. Guengerich and A. J. Wood (1996). "Microsomal codeine
N-demethylation: cosegregation with cytochrome P4503A4 activity." Drug Metab
Dispos 24(7): 761-764.
Caro, A. A. and A. I. Cederbaum (2004). "Oxidative stress, toxicology, and
pharmacology of CYP2E1." Ann Rev Pharmacol Toxicol 44: 27-42.
Chambers, J. E. and H. W. Chambers (1989). "Oxidative desulfuration of
chlorpyrifos, chlorpyrifos-methyl, and leptophos by rat brain and liver." J Biochem
Toxicol 4(3): 201-203.
Chen, Z. R., R. J. Irvine, F. Bochner and A. A. Somogyi (1990). "Morphine formation
from codeine in rat brain: a possible mechanism of codeine-induced analgesia." Life
Sci 46(15): 1067-1074.
Chen, Z. R., A. A. Somogyi, G. Reynolds and F. Bochner (1991). "Disposition and
metabolism of codeine after single and chronic doses in one poor and seven
extensive metabolisers." Br J Clin Pharmacol 31(4): 381-390.
Chimbira, W. and B. P. Sweeney (2000). "The effect of smoking on postoperative
nausea and vomiting." Anaesthesia 55(6): 540-544.
Cleary, J., G. Mikus, A. Somogyi and F. Bochner (1994). "The influence of
pharmacogenetics on opioid analgesia: studies with codeine and oxycodone in the
Sprague-Dawley/Dark Agouti rat model." J Pharmacol Exp Ther 271(3): 1528-1534.
Coffman, B. L., G. R. Rios, C. D. King and T. R. Tephly (1997). "Human UGT2B7
catalyzes morphine glucuronidation." Drug Metab Dispos 25(1): 1-4.
Coleman, T., E. F. Spellman, A. Rostami-Hodjegan, M. S. Lennard and G. T. Tucker
(2000). "The 1 '-hydroxylation of rac-bufuralol by rat brain microsomes." Drug Metab
Dispos 28(9): 1094-1099.
Conroy, J. L., C. Fang, J. Gu, S. O. Zeitlin, W. Z. Yang, J. Yang, M. A. VanAlstine, J. W.
Nalwalk, P. J. Albrecht, J. E. Mazurkiewicz, A. Snyder-Keller, Z. X. Shan, S. Z. Zhang,
M. P. Wentland, M. Behr, B. I. Knapp, J. M. Bidlack, O. P. Zuiderveld, R. Leurs, X. X.
Ding and L. B. Hough (2010). "Opioids activate brain analgesic circuits through
cytochrome P450/epoxygenase signaling." Nat Neurosci 13(3): 284-286.
Coon, M. J. (2005). Cytochrome P450: Nature's most versatile biological catalyst.
Annu Rev Pharmacol Toxicol. 45: 1-25.

124
Corchero, J., C. P. Granvil, T. E. Akiyama, G. P. Hayhurst, S. Pimprale, L.
Feigenbaum, J. R. Idle and F. J. Gonzalez (2001). "The CYP2D6 humanized mouse:
effect of the human CYP2D6 transgene and HNF4alpha on the disposition of
debrisoquine in the mouse." Mol Pharmacol 60(6): 1260-1267.
Court, M. H., S. X. Duan, L.M. Hesse, K. Venkatakrishnan and D.J. Greenblatt (2001).
"Cytochrome P-450 2B6 is responsible for interindividual variability of propofol
hydroxylation by human liver microsomes." Anesthesiology 94(1): 110-119.
Court, M. H., S. Krishnaswamy, Q. Hao, S. X. Duan, C. J. Patten, L. L. Von Moltke and
D. J. Greenblatt (2003). "Evaluation of 3'-azido-3'-deoxythymidine, morphine, and
codeine as probe substrates for UDP-glucuronosyltransferase 2B7 (UGT2B7) in
human liver microsomes: specificity and influence of the UGT2B7*2 polymorphism."
Drug Metab Dispos 31(9): 1125-1133.
D'Amour, F. E. and D. L. Smith (1941). "A method for determining loss of pain
sensation." J Pharmacol Exp Ther 72(1): 74-79.
Dahl, M. L., I. Johansson, L. Bertilsson, M. Ingelmansundberg and F. Sjoqvist (1995).
"Ultrarapid hydroxylation of debrisoquine in a Swedish population. Analysis of the
molecular genetic basis." J Pharmacol Exp Ther 274(1): 516-520.
Dauchy, S., F. Dutheil, R. J. Weaver, F. Chassoux, C. Daumas-Duport, P. O. Couraud,
J. M. Scherrmann, I. De Waziers and X. Decleves (2008). "ABC transporters,
cytochromes P450 and their main transcription factors: expression at the human
blood-brain barrier." J Neurochem 107(6): 1518-1528.
Dauchy, S., F. Miller, P. O. Couraud, R. J. Weaver, B. Weksler, I. A. Romero, J. M.
Scherrmann, I. De Waziers and X. Decleves (2009). "Expression and transcriptional
regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral
microvascular endothelial cells." Biochem Pharmacol 77(5): 897-909.
De Gregori, M., M. Allegri, S. De Gregori, G. Garbin, C. Tinelli, M. Regazzi, S.
Govoni and G. N. Ranzani (2010). "How and Why to Screen for CYP2D6
Interindividual Variability in Patients Under Pharmacological Treatments." Curr
Drug Metab 11(3): 276-282.
de Groot, M. J., F. Wakenhut, G. Whitlock and R. Hyland (2009). "Understanding
CYP2D6 interactions." Drug Discov Today 14(19-20): 964-972.
de Leon, J., M. T. Susce, R. M. Pan, M. Fairchild, W. H. Koch and P. J. Wedlund (2005).
"The CYP2D6 poor metabolizer phenotype may be associated with risperidone
adverse drug reactions and discontinuation." J Clin Psychiatry 66(1): 15-27.
Deng, Y., B. Newman, M.P. Dunne, P.A. Silburn and G.D. Mellick (2004). "Further
evidence that interactions between CYP2D6 and pesticide exposure increase risk for
Parkinson's disease." Ann Neurol 55(6): 897.

125
Di Monte, D.A. (2003). "The environment and Parkinson's disease: is the nigrostriatal
system preferentially targeted by neurotoxins?" Lancet Neurol 2(9): 531-538.
Ding, X. X. and L. S. Kaminsky (2003). "Human extrahepatic cytochromes P450:
Function in xenobiotic metabolism and tissue-selective chemical toxicity in the
respiratory and gastrointestinal tracts." Ann Rev Pharmacol Toxicol 43: 149-173.
Doostzadeh, J. and R. Morfin (1997). "Effects of cytochrome P450 inhibitors and of
steroid hormones on the formation of 7-hydroxylated metabolites of pregnenolone in
mouse brain microsomes." J Endocrinol 155(2): 343-350.
Dorado, P., E. M. Penas-Lledo and A. Llerena (2007). "CYP2D6 polymorphism:
implications for antipsychotic drug response, schizophrenia and personality traits."
Pharmacogenomics 8(11): 1597-1608.
Dostrovsky, J.O. (2000). "Role of thalamus in pain." Prog Brain Res 129: 245-257.
Dukes, I. D. and E. M. Vaughan Williams (1984). "The multiple modes of action of
propafenone." Eur Heart J 5(2): 115-125.
Duman, E. N., M. Kesim, M. Kadioglu, E. Yaris, N. I. Kalyoncu and N. Erciyes (2004).
"Possible involvement of opioidergic and serotonergic mechanisms in
antinociceptive effect of paroxetine in acute pain." J Pharmacol Sci 94(2): 161-165.
Dutheil, F., P. Beaune and M. A. Loriot (2008). "Xenobiotic metabolizing enzymes in
the central nervous system: Contribution of cytochrome P450 enzymes in normal and
pathological human brain." Biochimie 90(3): 426-436.
Dutheil, F., S. Dauchy, M. Diry, V. Sazdovitch, O. Cloarec, L. Mellottee, I. Bieche, M.
Ingelman-Sundberg, J. P. Flinois, I. de Waziers, P. Beaune, X. Decleves, C.
Duyckaerts and M. A. Loriot (2009). "Xenobiotic-Metabolizing Enzymes and
Transporters in the Normal Human Brain: Regional and Cellular Mapping as a Basis
for Putative Roles in Cerebral Function." Drug Metab Dispos 37(7): 1528-1538.
Dutheil, F., A. Jacob, S. Dauchy, P. Beaune, J. M. Scherrmann, X. Decleves and M. A.
Loriot (2010). "ABC transporters and cytochromes P450 in the human central nervous
system: influence on brain pharmacokinetics and contribution to neurodegenerative
disorders." Expert Opin Drug Metab Toxicol 6(10): 1161-1174.
Eddy, N. B. and D. Leimbach (1953). "Synthetic analgesics. II. Dithienylbutenyl- and
dithienylbutylamines." J Pharmacol Exp Ther 107(3): 385-393.
Edwards, R. J., R. J. Price, P. S. Watts, A. B. Renwick, J. M. Tredger, A. R. Boobis and
B. G. Lake (2003). "Induction of cytochrome P450 enzymes in cultured precision-cut
human liver slices." Drug Metab Dispos 31(3): 282-288.
Elbaz, A., C. Levecque, J. Clavel, J.S. Vidal, F. Richard, P. Amouyel, A. Alperovitch,
M.C. Chartier-Harlin and C. Tzourio (2004). "CYP2D6 polymorphism, pesticide
exposure, and Parkinson's disease." Ann Neurol 55(3): 430-434.

126
Emoto, C., S. Murase and K. Iwasaki (2006). "Approach to the prediction of the
contribution of major cytochrome P450 enzymes to drug metabolism in the early
drug-discovery stage." Xenobiotica 36(8): 671-683.
Erjavec, M. K., B. A. Coda, Q. Nguyen, G. Donaldson, L. Risler and D. D. Shen (2000).
"Morphine-fluoxetine interactions in healthy volunteers: Analgesia and side effects."
J Clin Pharmacol 40(11): 1286-1295.
Estabrook, R. (1999). "An introduction to the cytochrome P450s." Mol Aspects Med
20(1-2): 5-12, 13-137.
Fagerstrom, K.O., O. Pomerleau, B. Giordani and F. Stelson (1994). "Nicotine may
relieve symptoms of Parkinson's disease." Psychopharmacology (Berl) 116(1): 117-
119.
Fahn, S. (2010). "Parkinson's disease: 10 years of progress, 1997-2007." Mov Disord
25 Suppl 1: S2-S14.
Farre, M. and J. Cami (1991). "Pharmacokinetic considerations in abuse liability
evaluation." Br J Addict 86(12): 1601-1606.
Fields, H. (2004). "State-dependent opioid control of pain." Nat Rev Neurosci 5(7):
565-575.
Fields, H. L., P. C. Emson, B. K. Leigh, R. F. T. Gilbert and L. L. Iversen (1980).
"Multiple opiate receptor sites on primary afferent fibers." Nature 284(5754): 351-
353.
Fields, H. L., M. M. Heinricher and P. Mason (1991). "Neurotransmitters in
nociceptive modulatory circuits." Annu Rev Neurosci 14: 219-245.
Fields, H. L., A. Malick and R. Burstein (1995). "Dorsal horn projection targets of on
and off cells in the rostral ventromedial medulla." J Neurophysiol 74(4): 1742-1759.
Flores-Perez, J., C. Flores-Perez, H. Juarez-Olguin, I. Lares-Asseff and M. Sosa-
Macias (2004). "Determination of dextromethorphan and dextrorphan in human
urine by high performance liquid chromatography for pharmacogenetic
investigations." Chromatographia 59(7-8): 481-485.
Fonne-Pfister, R., M. J. Bargetzi and U. A. Meyer (1987). "MPTP, the neurotoxin
inducing Parkinson's disease, is a potent competitive inhibitor of human and rat
cytochrome P450 isozymes (P450bufI, P450db1) catalyzing debrisoquine 4-
hydroxylation." Biochem Biophys Res Commun 148(3): 1144-1150.
Forsyth, C. S. and J. E. Chambers (1989). "Activation and degradation of the
phosphorothionate insecticides parathion and EPN by rat brain." Biochem Pharmacol
38(10): 1597-1603.

127
Fradette, C., N. Yamaguchi and P. du Souich (2004). "5-hydroxytryptamine is
biotransformed by CYP2C9, 2C19 and 2B6 to hydroxylamine, which is converted
into nitric oxide." Br J Pharmacol 141(3): 407-414.
Frank, D., U. Jaehde and U. Fuhr (2007). "Evaluation of probe drugs and
pharmacokinetic metrics for CYP2D6 phenotyping." Eur J Clin Pharmacol 63(4): 321-
333.
Freiermuth, M. and J. C. Plasse (1997). "Determination of morphine and codeine in
plasma by HPLC following solid phase extraction." J Pharm Biomed Anal 15(6): 759-
764.
Funae, Y., W. Kishimoto, T. Cho, T. Niwa and T. Hiroi (2003). "CYP2D in the brain."
Drug Metab Pharmacokinet 18(6): 337-349.
Funck-Brentano, C., P. Y. Boelle, C. Verstuyft, C. Bornert, L. Becquemont and J. M.
Poirier (2005). "Measurement of CYP2D6 and CYP3A4 activity in vivo with
dextromethorphan: sources of variability and predictors of adverse effects in 419
healthy subjects." Eur J Clin Pharmacol 61(11): 821-829.
Gaedigk, A., A. Bhathena, L. Ndjountche, R. E. Pearce, S. M. Abdel-Rahman, S. W.
Alander, L. D. Bradford and J. S. Leeder (2005). "Identification and characterization of
novel sequence variations in the cytochrome P4502D6 (CYP2D6) gene in African
Americans." Pharmacogenomics J 5(3): 173-182.
Gaedigk, A., S. D. Simon, R. E. Pearce, L. D. Bradford, M. J. Kennedy and J. S. Leeder
(2008). "The CYP2D6 activity score: Translating genotype information into a
qualitative measure of phenotype." Clin Pharmacol Ther 83(2): 234-242.
Gan, S.H., R. Ismail, W.A. Wan Adnan, W. Zulmi, N. Kumaraswamy and E.T. Larmie
(2004) "Relationship between Type A and B personality and debrisoquine
hydroxylation capacity." Br J Clin Pharmacol 57(6): 785-789.
Garcia-Barcelo, M., L. Y. Chow, H. F. K. Chiu, Y. K. Wing, D. T. S. Lee, K. L. Lam and
M. M. Y. Waye (2000). "Genetic analysis of the CYP2D6 locus in a Hong Kong
Chinese population." Clin Chem 46(1): 18-23.
Gasche, Y., Y. Daali, M. Fathi, A. Chiappe, S. Cottini, P. Dayer and J. Desmeules
(2004). "Codeine intoxication associated with ultrarapid CYP2D6 metabolism." N
Engl J Med 351(27): 2827-2831.
Genazzani, A.R., M. Stomati, A. Morittu, F. Bernardi, P. Monteleone, E. Casarosa, R.
Gallo, C. Salvestroni and M. Luisi (2000). "Progesterone, progestagens and the
central nervous system." Hum Reprod 15 Suppl 1: 14-27.
George, T. P., K. A. Sacco, J. C. Vessicchio, A. H. Weinberger and R. D. Shytle
(2008). "Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-

128
refractory major depressive disorder - A preliminary study." J Clin
Psychopharmacol 28(3): 340-344.
Gervasini, G., J. A. Carrillo and J. Benitez (2004). "Potential role of cerebral
cytochrome P450 in clinical pharmacokinetics - Modulation by endogenous
compounds." Clin Pharmacokinet 43(11): 693-706.
Ghersiegea, J. F., A. Minn, J. L. Daval, Z. Jayyosi, V. Arnould, H. Souhailielamri and
G. Siest (1989). "NADPH:cytochrome P-450(c) reductase: biochemical
characterization in rat brain and cultured neurons and evolution of activity during
development." Neurochem Res 14(9): 883-888.
Ghersiegea, J. F., R. Perrin, B. Leiningermuller, M. C. Grassiot, C. Jeandel, J. Floquet,
G. Cuny, G. Siest and A. Minn (1993). "Subcellular localization of cytochrome P450,
and activities of several enzymes responsible for drug metabolism in the human
brain." Biochem Pharmacol 45(3): 647-658.
Ghosh, C., J. Gonzalez-Martinez, M. Hossain, L. Cucullo, V. Fazio, D. Janigro and N.
Marchi (2010). "Pattern of P450 expression at the human blood-brain barrier: roles of
epileptic condition and laminar flow." Epilepsia 51(8): 1408-1417.
Gilham, D. E., W. Cairns, M. J. I. Paine, S. Modi, R. Poulsom, G. C. K. Roberts and C.
R. Wolf (1997). "Metabolism of MPTP by cytochrome P4502D6 and the demonstration
of 2D6 mRNA in human foetal and adult brain by in situ hybridization." Xenobiotica
27(1): 111-125.
Gonzalez, F. J. (1990). "Molecular genetics of the P-450 superfamily." Pharmacology
& Therapeutics 45(1): 1-38.
Gonzalez, I., E. M. Penas-Lledo, B. Perez, P. Dorado, M. Alvarez and L. L. A (2008).
"Relation between CYP2D6 phenotype and genotype and personality in healthy
volunteers." Pharmacogenomics 9(7): 833-840.
Gonzalez-Hernandez, T., I. Cruz-Muros, D. Afonso-Oramas, J. Salas-Hernandez and J.
Castro-Hernandez (2010). "Vulnerability of mesostriatal dopaminergic neurons in
Parkinson's disease." Front Neuroanat 4: 140.
Griffiths, R. R. and B. Wolf (1990). "Relative abuse liability of different
benzodiazepines in drug abusers." J Clin Psychopharmacol 10(4): 237-243.
Grossman, M. L., A. I. Basbaum and H. L. Fields (1982). "Afferent and efferent
connections of the rat tail-flick reflex (a model used to analyze pain control
mechanisms)." J Comp Neurol 206(1): 9-16.
Grumbach, L. (1966). The prediction of analgesic activity in man by animal testing. .
Pain, 15th International Symposium, Detroit, 1964. R. S. Knighton and P. R. Dumke.
Boston, Little Brown: 163–182.

129
Grumbach, L. (1975). Pain: Research and Treatment Further Observations From City
of Hope National Medical Centre. B. L. Crue. New York Academic Press: 163-182.
Guengerich, F. P. (2003). "Cytochromes P450, drugs, and diseases." Mol Interv 3(4):
194-204.
Haduch, A., E. Bromek and W. A. Daniel (2011). "The effect of psychotropic drugs on
cytochrome P450 2D (CYP2D) in rat brain." Eur J Pharmacol 651(1-3): 51-58.
Haglund, L., C. Kohler, T. Haaparanta, M. Goldstein and J. A. Gustafsson (1984).
"Presence of NADPH-cytochrome P450 reductase in central catecholaminergic
neurones." Nature 307(5948): 259-262.
Haining, R. L. (2007). Cytochrome P450 Reactions in the Human Brain. Handbook of
Neurochemistry and Molecular Neurobiology. A. Lajtha. New York, NY, Springer
Science 43-91.
Hammond, D. L., G. M. Tyce and T. L. Yaksh (1985). "Efflux of 5-hydroxytryptamine
and noradrenaline into spinal cord superfusates during stimulation of the rat
medulla." J Physiol 359: 151-162.
Hardy, J. D. (1953). "Thresholds of Pain and Reflex Contraction as Related to Noxious
Stimulation." J Appl Physiol 5(12): 725-739.
Hardy, J. D., A. M. Stoll, D. Cunningham, W. M. Benson and L. Greene (1957).
"Responses of the rat to thermal radiation." Am J Physiol 189(1): 1-5.
He, H., S. D. Shay, Y. Caraco, M. Wood and A. J. Wood (1998). "Simultaneous
determination of codeine and its seven metabolites in plasma and urine by high-
performance liquid chromatography with ultraviolet and electrochemical detection."
J Chromatogr B Biomed Sci Appl 708(1-2): 185-193.
Hedlund, E., J. A. Gustafsson and M. Warner (2001). "Cytochrome P450 in the brain;
a review." Curr Drug Metab 2(3): 245-263.
Hedlund, E., A. Wyss, T. Kainu, M. Backlund, C. Kohler, M. PeltoHuikko, J. A.
Gustafsson and M. Warner (1996). "Cytochrome P4502D4 in the brain: Specific
neuronal regulation by clozapine and toluene." Mol Pharmacol 50(2): 342-350.
Heinricher, M. M., N. M. Barbaro and H. L. Fields (1989). "Putative nociceptive
modulating neurons in the rostral ventromedial medulla of the rat: firing of on- and
off-cells is related to nociceptive responsiveness." Somatosens Mot Res 6(4): 427-
439.
Heinricher, M. M., Z. F. Cheng and H. L. Fields (1987). "Evidence for two classes of
nociceptive modulating neurons in the periaqueductal gray." J Neurosci 7(1): 271-
278.

130
Heinricher, M. M., C. M. Haws and H. L. Fields (1991). "Evidence for GABA-mediated
control of putative nociceptive modulating neurons in the rostral ventromedial
medulla: iontophoresis of bicuculline eliminates the off-cell pause." Somatosens Mot
Res 8(3): 215-225.
Heinricher, M. M. and S. L. Ingram (2008). The brainstem and nociceptive
modulation. The Senses, a Comprehensive Reference. Pain, Vol. 5. M. C. Bushnell
and A. I. Basbaum. San Diego, Academic Press: 593–626.
Heinricher, M. M., S. McGaraughty and D. A. Farr (1999). "The role of excitatory
amino acid transmission within the rostral ventromedial medulla in the
antinociceptive actions of systemically administered morphine." Pain 81(1-2): 57-65.
Heinricher, M. M., S. McGaraughty and D. K. Grandy (1997). "Circuitry underlying
antiopioid actions of orphanin FQ in the rostral ventromedial medulla." J
Neurophysiol 78(6): 3351-3358.
Heinricher, M. M., S. McGaraughty and V. Tortorici (2001). "Circuitry underlying
antiopioid actions of cholecystokinin within the rostral ventromedial medulla." J
Neurophysiol 85(1): 280-286.
Heinricher, M. M., M. M. Morgan and H. L. Fields (1992). "Direct and indirect actions
of morphine on medullary neurons that modulate nociception." Neuroscience 48(3):
533-543.
Heinricher, M. M., M. M. Morgan, V. Tortorici and H. L. Fields (1994). "Disinhibition
of off-cells and antinociception produced by an opioid action within the rostral
ventromedial medulla." Neuroscience 63(1): 279-288.
Heinricher, M. M., I. Tavares, J. L. Leith and B. M. Lumb (2009). "Descending control
of nociception: Specificity, recruitment and plasticity." Brain Res Rev 60(1): 214-225.
Hendrickson, H. P., B. J. Gurley and W. D. Wessinger (2003). "Determination of
dextromethorphan and its metabolites in rat serum by liquid-liquid extraction and
liquid chromatography with fluorescence detection." J Chromatogr B Analyt Technol
Biomed Life Sci 788(2): 261-268.
Herraiz, T., H. Guillen, V.J. Aran, J.R. Idle and F.J. Gonzalez (2006). "Comparative
aromatic hydroxylation and N-demethylation of MPTP neurotoxin and its analogs, N-
methylated betacarboline and isoquinoline alkaloids, by human cytochrome P450
2D6." Toxicol Appl Pharmacol 216(3): 387-398.
Hiroi, T., T. Chow, S. Imaoka and Y. Funae (2002). "Catalytic specificity of CYP2D
isoforms in rat and human." Drug Metab Dispos 30(9): 970-976.
Hiroi, T., S. Imaoka and Y. Funae (1998). "Dopamine formation from tyramine by
CYP2D6." Biochem Biophys Res Commun 249(3): 838-843.

131
Hiroi, T., W. Kishimoto, T. Chow, S. Imaoka, T. Igarashi, and Y. Funae (2001).
"Progesterone oxidation by cytochrome P450 2D isoforms in the brain."
Endocrinology, 142(2): 3901-3908.
Hlatky, M.A., D. Boothroyd, E. Vittinghoff, P. Sharp and M.A. Whooley (2002).
"Quality-of life and depressive symptoms in postmenopausal women after receiving
hormone therapy: results from the Heart and Estrogen/Progestin Replacement Study
(HERS) trial." JAMA, 287(5) 591-597.
Hohmann, A. G., R. L. Suplita, N. M. Bolton, M. H. Neely, D. Fegley, R. Mangieri, J. F.
Krey, J. M. Walker, P. V. Holmes, J. D. Crystal, A. Duranti, A. Tontini, M. Mor, G.
Tarzia and D. Piomelli (2005). "An endocannabinoid mechanism for stress-induced
analgesia." Nature 435(7045): 1108-1112.
Holthe, M., P. Klepstad, K. Zahlsen, P. C. Borchgrevink, L. Hagen, O. Dale, S. Kaasa,
H. E. Krokan and F. Skorpen (2002). "Morphine glucuronide-to-morphine plasma
ratios are unaffected by the UGT2B7 H268Y and UGT1A1*28 polymorphisms in
cancer patients on chronic morphine therapy." Eur J Clin Pharmacol 58(5): 353-356.
Howard, L. A., S. Miksys, E. Hoffmann, D. Mash and R. F. Tyndale (2003). "Brain
CYP2E1 is induced by nicotine and ethanol in rat and is higher in smokers and
alcoholics." Br J Pharmacol 138(7): 1376-1386.
Ingelman-Sundberg, M. (2005). "Genetic polymorphisms of cytochrome P450 2D6
(CYP2D6): clinical consequences, evolutionary aspects and functional diversity."
Pharmacogenomics J 5(1): 6-13.
Ingelman-Sundberg, M., S. C. Sim, A. Gomez and C. Rodriguez-Antona (2007).
"Influence of cytochrome P450 polymorphisms on drug therapies: Pharmacogenetic,
pharmacoepigenetic and clinical aspects." Pharmacol Ther 116(3): 496-526.
Irwin, S., R. W. Houde, D. R. Bennett, L. C. Hendershot and M. H. Seevers (1951).
"The effects of morphine, methadone and meperidine on some reflex responses of
spinal animals to nociceptive stimulation." J Pharmacol Exp Ther 101(2): 132-143.
Jabs, B. E., A. J. Bartsch and B. Pfuhlmann (2003). "Susceptibility to neuroleptic-
induced parkinsonism - age and increased substantia nigra echogenicity as putative
risk factors." Eur Psychiatry 18(4): 177-181.
Jacob, P., 3rd, M. Ulgen and J. W. Gorrod (1997). "Metabolism of (-)-(S)-nicotine by
guinea pig and rat brain: identification of cotinine." Eur J Drug Metab Pharmacokinet
22(4): 391-394.
Jacquet, Y. F. and A. Lajtha (1973). "Morphine action at central nervous system sites
in rat: analgesia or hyperalgesia depending on site and dose." Science 182(4111):
490-492.

132
Jensen, T. S. and T. L. Yaksh (1986). "Examination of spinal monoamine receptors
through which brainstem opiate-sensitive systems act in the rat." Brain Res 363(1):
114-127.
Jolivalt, C., A. Minn, M. Vincentviry, M. M. Galteau and G. Siest (1995).
"Dextromethorphan O-demethylase activity in rat brain microsomes." Neurosci Lett
187(1): 65-68.
Jones, S. L. and G. F. Gebhart (1988). "Inhibition of spinal nociceptive transmission
from the midbrain, pons and medulla in the rat: activation of descending inhibition
by morphine, glutamate and electrical stimulation." Brain Res 460(2): 281-296.
Joshi, M. and R. F. Tyndale (2006). "Induction and recovery time course of rat brain
CYP2E1 after nicotine treatment." Drug Metab Dispos 34(4): 647-652.
Jover, R., R. Bort, M. J. Gomez-Lechon and J. V. Castell (1998). "Re-expression of
C/EBP alpha induces CYP2B6, CYP2C9 and CYP2D6 genes in HepG2 cells." FEBS
Lett 431(2): 227-230.
Kane, J. K., O. Konu, J. Z. Ma and M. D. Li (2004). "Nicotine coregulates multiple
pathways involved in protein modification/degradation in rat brain." Mol Brain Res
132(2): 181-191.
Kathiramalainathan, K., H. L. Kaplan, M. K. Romach, U. E. Busto, N. Y. Li, J. Sawe, R. F.
Tyndale and E. M. Sellers (2000). "Inhibition of cytochrome P450 2D6 modifies
codeine abuse liability." J Clin Psychopharmacol 20(4): 435-444.
Kelton, M.C., H.J. Kahn, C.L. Conrath and P.A. Newhouse (2000). "The effects of
nicotine on Parkinson's disease." Brain Cogn 43(1-3): 274-282.
Kerry, N. L., A. A. Somogyi, G. Mikus and F. Bochner (1993). "Primary and secondary
oxidative metabolism of dextromethorphan. In vitro studies with female Sprague-
Dawley and Dark Agouti rat liver microsomes." Biochem Pharmacol 45(4): 833-839.
Khokhar, J. Y. and R. F. Tyndale (2011). "Drug metabolism within the brain changes
drug response: selective manipulation of brain CYP2B alters propofol effects."
Neuropsychopharmacology 36(3): 692-700.
Khokhar, J. Y. and R. F. Tyndale (2012). "Rat brain CYP2B-enzymatic activation of
chlorpyrifos to the oxon mediates cholinergic neurotoxicity." Toxicol Sci 126(2): 325-
335.
Khwaja, M., A. McCormack, J.M. McIntosh, D.A. Di Monte and M. Quik (2007).
"Nicotine partially protects against paraquat-induced nigrostriatal damage in mice;
link to alpha6beta2*nAChRs." J Neurochem 100(1): 180-190.
Kirchheiner, J., U. Lang, T. Stamm, T. Sander and J. Gallinat (2006). "Association of
CYP2D6 genotypes and personality traits in healthy individuals." J Clin
Psychopharmacol 26(4): 440-442.

133
Kirchheiner, J., A. Seeringer, A. L. Godoy, B. Ohmle, C. Maier, P. Beschoner, E. J. Sim
and R. Viviani (2011). "CYP2D6 in the brain: genotype effects on resting brain
perfusion." Mol Psychiatry 16(3): 333-341.
Kishimoto, W., T. Hiroi, M. Shiraishi, M. Osada, S. Imaoka, S. Kominami, T. Igarashi
and Y. Funae (2004). "Cytochrome P450 2D catalyze steroid 21-hydroxylation in the
brain." Endocrinology 145(2): 699-705.
Kobayashi, S., S. Murray, D. Watson, D. Sesardic, D. S. Davies and A. R. Boobis
(1989). "The specificity of inhibition of debrisoquine 4-hydroxylase activity by
quinidine and quinine in the rat is the inverse of that in man." Biochem Pharmacol
38(17): 2795-2799.
Kodaira, H. and S. Spector (1988). "Transformation of thebaine to oripavine, codeine,
and morphine by rat liver, kidney, and brain microsomes." Proc Natl Acad Sci USA
85(4): 1267-1271.
Komori, M. (1993). "A novel P450 expressed at high level in rat brain." Biochem
Biophys Res Commun 196(2): 721-728.
Komura, H. and M. Iwaki (2005). "Pharmacokinetics and metabolism of metoprolol
and propranolol in the female DA and female Wistar rat: the female DA rat is not
always an animal model for poor metabolizers of CYP2D6." J Pharm Sci 94(2): 397-
408.
Kream, R. M. and G. B. Stefano (2006). "De novo biosynthesis of morphine in animal
cells: an evidence-based model." Med Sci Monit 12(10): RA207-219.
Kroemer, H. K., C. Fischer, C. O. Meese and M. Eichelbaum (1991).
"Enantiomer/enantiomer interaction of (S)- and (R)-propafenone for cytochrome
P450IID6-catalyzed 5-hydroxylation: in vitro evaluation of the mechanism." Mol
Pharmacol 40(1): 135-142.
Kudo, K., T. Ishida, N. Nishida, N. Yoshioka, H. Inoue, A. Tsuji and N. Ikeda (2006).
"Simple and sensitive determination of free and total morphine in human liver and
kidney using gas chromatography-mass spectrometry." J Chromatogr B Analyt
Technol Biomed Life Sci 830(2): 359-363.
Lamotte, C., C. B. Pert and S. H. Snyder (1976). "Opiate receptor binding in primate
spinal cord: Distribution and changes after dorsal root section." Brain Res 112(2):
407-412.
Lane, H. Y., W. C. Chiu, J. C. Y. Chou, S. T. Wu, M. H. Su and W. H. Chang (2000).
"Risperidone in acutely exacerbated schizophrenia: Dosing strategies and plasma
levels." J Clin Psychiatry 61(3): 209-214.
Lasagna, L. and T. J. De Kornfeld (1958). "Analgesic potency of normorphine in
patients with postoperative pain." J Pharmacol Exp Ther 124(3): 260-263.

134
Law, P. Y., Y. H. Wong and H. H. Loh (2000). "Molecular mechanisms and regulation
of opioid receptor signaling." Annu Rev Pharmacol Toxicol 40: 389-430.
Le Bars, D., M. Gozariu and S. W. Cadden (2001). "Animal models of nociception."
Pharmacol Rev 53(4): 597-652.
Lee, A. M., S. Miksys, R. Palmour and R. F. Tyndale (2006a). "CYP2B6 is expressed in
African Green monkey brain and is induced by chronic nicotine treatment."
Neuropharmacology 50(4): 441-450.
Lee, A. M., S. Miksys and R. F. Tyndale (2006b). "Phenobarbital increases monkey in
vivo nicotine disposition and induces liver and brain CYP2B6 protein." Br J
Pharmacol 148(6): 786-794.
Leith, J. L., A. W. Wilson, L. F. Donaldson and B. M. Lumb (2007). "Cyclooxygenase-
1-derived prostaglandins in the periaqueductal gray differentially control C-versus
A-fiber-evoked spinal nociception." J Neurosci 27(42): 11296-11305.
Lenz, F.A., N. Weiss, S. Ohara, C. Lawson and J.D. Greenspan (2004). "The role of the
thalamus in pain." Suppl Clin Neurophysiol 57: 50-61.
Lewis, J. W., J. E. Sherman and J. C. Liebeskind (1981). "Opioid and non-opioid stress
analgesia: assessment of tolerance and cross-tolerance with morphine." J Neurosci
1(4): 358-363.
Lewis, V. A. and G. F. Gebhart (1977). "Evaluation of the periaqueductal central gray
(PAG) as a morphine-specific locus of action and examination of morphine-induced
and stimulation-produced analgesia at coincident PAG loci." Brain Res 124(2): 283-
303.
Lin, J. H. (1995). "Species similarities and differences in pharmacokinetics." Drug
Metab Dispos 23(10): 1008-1021.
Lin, L. Y., Y. Kumagai and A. K. Cho (1992). "Enzymatic and chemical
demethylenation of (methylenedioxy)amphetamine and
(methylenedioxy)methamphetamine by rat brain microsomes." Chem Res Toxicol
5(3): 401-406.
Lipp, J. (1991). "Possible mechanisms of morphine analgesia." Clin Neuropharmacol
14(2): 131-147.
Llerena, A., P. Dorado, E. M. Penas-Lledo, M. C. Caceres and A. De la Rubia (2007).
"Low frequency of CYP2D6 poor metabolizers among schizophrenia patients."
Pharmacogenomics J 7(6): 408-410.
Llerena, A., G. Edman, J. Cobaleda, J. Benitez, D. Schalling and L. Bertilsson (1993).
"Relationship between personality and debrisoquine hydroxylation capacity -
suggestion of an endogenous neuroactive substrate or product of the cytochrome-
P4502D6." Acta Psychiatr Scand 87(1): 23-28.

135
Lofgren, S., A. L. Hagbjork, S. Ekman, R. Fransson-Steen and Y. Terelius (2004).
"Metabolism of human cytochrome P450 marker substrates in mouse: a strain and
gender comparison." Xenobiotica 34(9): 811-834.
Lotsch, J., A. Stockmann, G. Kobal, K. Brune, R. Waibel, N. Schmidt and G.
Geisslinger (1996). "Pharmacokinetics of morphine and its glucuronides after
intravenous infusion of morphine and morphine-6-glucuronide in healthy
volunteers." Clin Pharmacol Ther 60(3): 316-325.
Lysakowski, C., L. Dumont, C. Czarnetzki, D. Bertrand, E. Tassonyi and M.R. Tramer
(2006). "The effect of cigarette smoking on the hypnotic efficacy of propofol."
Anaesthesia 61(9): 826-831.
Ma, X. C., J. R. Idle, K. W. Krausz and F. J. Gonzalez (2005). "Metabolism of melatonin
by human cytochromes P450." Drug Metab Dispos 33(4): 489-494.
Mann, A., S. Miksys, A. Lee, D. C. Mash and R. F. Tyndale (2008). "Induction of the
drug metabolizing enzyme CYP2D in monkey brain by chronic nicotine treatment."
Neuropharmacology 55(7): 1147-1155.
Mann, A., S. L. Miksys, A. Gaedigk, S. J. Kish, D. C. Mash and R. F. Tyndale (2012).
"The neuroprotective enzyme CYP2D6 increases in the brain with age and is lower in
Parkinson's disease patients." Neurobiol Aging 33(9): 2160-2171.
Mann, A. and R. F. Tyndale (2010). "Cytochrome P450 2D6 enzyme neuroprotects
against 1-methyl-4-phenylpyridinium toxicity in SH-SY5Y neuronal cells." Eur J
Neurosci 31(7): 1185-1193.
Mansour, A., C. A. Fox, H. Akil and S. J. Watson (1995). "Opioid-receptor mRNA
expression in the rat CNS: anatomical and functional implications." Trends Neurosci
18(1): 22-29.
Mansour, A., H. Khachaturian, M. E. Lewis, H. Akil and S. J. Watson (1988). "Anatomy
of CNS opioid receptors." Trends Neurosci 11(7): 308-314.
Masubuchi, Y., S. Fujita, M. Chiba, N. Kagimoto, S. Umeda and T. Suzuki (1991).
"Impairment of debrisoquine 4-hydroxylase and related monooxygenase activities
in the rat following treatment with propranolol." Biochem Pharmacol 41(6-7): 861-
865.
Masubuchi, Y., S. Narimatsu, S. Hosokawa and T. Suzuki (1994). "Role of the CYP2D
subfamily in metabolism-dependent covalent binding of propranolol to liver
microsomal protein in rats." Biochem Pharmacol 48(10): 1891-1898.
Matoh, N., S. Tanaka, M. Takehashi, M. Banasik, T. Stedeford, E. Masliah, S. Suzuki, Y.
Nishimura and K. Ueda (2003). "Overexpression of CYP2D6 attenuates the toxicity of
MPP+ in actively dividing and differentiated PC12 cells." Gene Expr 11(3-4): 117-
124.

136
Matsunaga, E., U. M. Zanger, J. P. Hardwick, H. V. Gelboin, U. A. Meyer and F. J.
Gonzalez (1989). "The CYP2D gene subfamily: analysis of the molecular basis of the
debrisoquine 4-hydroxylase deficiency in DA rats." Biochemistry 28(18): 7349-7355.
McCann, S.J., S.M. Pond, K.M. James and D.G. Le Couteur (1997). "The association
between polymorphisms in the cytochrome P-450 2D6 gene and Parkinson's disease:
a case-control study and meta-analysis." J Neurol Sci 153(1): 50-53.
McFadzean, I. (1988). "The ionic mechanisms underlying opioid actions."
Neuropeptides 11(4): 173-180.
McFayden, M. C., W. T. Melvin and G. I. Murray (1998). "Regional distribution of
individual forms of cytochrome P450 mRNA in normal adult human brain." Biochem
Pharmacol 55(6): 825-830.
McGinnity, D. F., N. J. Waters, J. Tucker and R. J. Riley (2008). "Integrated in vitro
analysis for the in vivo prediction of cytochrome P450-mediated drug-drug
interactions." Drug Metab Dispos 36(6): 1126-1134.
McLaughlin, L. A., L. J. Dickmann, C. R. Wolf and C. J. Henderson (2008). "Functional
expression and comparative characterization of nine murine cytochromes P450 by
fluorescent inhibition screening." Drug Metab Dispos 36(7): 1322-1331.
McLellan, R. A., M. Oscarson, J. Seidegard, D. A. Evans and M. Ingelman-Sundberg
(1997). "Frequent occurrence of CYP2D6 gene duplication in Saudi Arabians."
Pharmacogenetics 7(3): 187-191.
Meyer, R. P., M. Gehlhaus, R. Knoth and B. Volk (2007). "Expression and function of
cytochrome p450 in brain drug metabolism." Curr Drug Metab 8(4): 297-306.
Meyer, R. P., R. L. Lindberg, F. Hoffmann and U. A. Meyer (2005). "Cytosolic
persistence of mouse brain CYP1A1 in chronic heme deficiency." Biol Chem 386(11):
1157-1164.
Michels, R. and P. M. Marzuk (1993a). "Progress in psychiatry (1)." N Engl J Med
329(8): 552-560.
Michels, R. and P. M. Marzuk (1993b). "Progress in psychiatry (2)." N Engl J Med
329(9): 628-638.
Miksys, S., E. Hoffmann and R. F. Tyndale (2000). "Regional and cellular induction of
nicotine-metabolizing CYP2B1 in rat brain by chronic nicotine treatment." Biochem
Pharmacol 59(12): 1501-1511.
Miksys, S., C. Lerman, P. G. Shields, D. C. Mash and R. F. Tyndale (2003). "Smoking,
alcoholism and genetic polymorphisms alter CYP2B6 levels in human brain."
Neuropharmacology 45(1): 122-132.

137
Miksys, S., Y. Rao, E. Hoffmann, D. C. Mash and R. F. Tyndale (2002). "Regional and
cellular expression of CYP2D6 in human brain: higher levels in alcoholics." J
Neurochem 82(6): 1376-1387.
Miksys, S., Y. Rao, E. M. Sellers, M. Kwan, D. Mendis and R. F. Tyndale (2000).
"Regional and cellular distribution of CYP2D subfamily members in rat brain."
Xenobiotica 30(6): 547-564.
Miksys, S. and R. F. Tyndale (2004). "The unique regulation of brain cytochrome
P450 2 (CYP2) family enzymes by drugs and genetics." Drug Metab Rev 36(2): 313-
333.
Miksys, S. and R. F. Tyndale (2009). "Brain drug-metabolizing cytochrome P450
enzymes are active in vivo, demonstrated by mechanism-based enzyme inhibition."
Neuropsychopharmacology 34(3): 634-640.
Miksys, S. L., C. Cheung, F. J. Gonzalez and R. F. Tyndale (2005). "Human CYP2D6
and mouse CYP2Ds: organ distribution in a humanized mouse model." Drug Metab
Dispos 33(10): 1495-1502.
Mikus, G., F. Bochner, M. Eichelbaum, P. Horak, A. A. Somogyi and S. Spector
(1994). "Endogenous codeine and morphine in poor and extensive metabolisers of
the CYP2D6 (debrisoquine/sparteine) polymorphism." J Pharmacol Exp Ther 268(2):
546-551.
Mikus, G., A. A. Somogyi, F. Bochner and M. Eichelbaum (1991). "Codeine O-
demethylation: rat strain differences and the effects of inhibitors." Biochem
Pharmacol 41(5): 757-762.
Miksys, S. L. and R. F. Tyndale (2002). "Drug-metabolizing cytochrome P450s in the
brain." J Psychiatry Neurosci 27(6): 406-415.
Millan, M. J. (2002). "Descending control of pain." Prog Neurobiol 66(6): 355-474.
Miller, R.T., S. Miksys and R.F. Tyndale (2012). "An investigation of the expression of
brain CYP2D6 in a monkey model of nicotine and ethanol exposure." Society for
Research on Nicotine and Tobacco, Houston, TX.
Mizuno, D., T. Hiroi, P. Ng, W. Kishimoto, S. Imaoka and Y. Funae (2003). "Regulation
of CYP2D expression in rat brain by toluene." Osaka City Med J 49(1): 49-56.
Mizuno, D., Y. Takahashi, T. Hiroi, S. Imaoka, T. Kamataki and Y. Funae (2003). "A
novel transcriptional element which regulates expression of the CYP2D4 gene by
Oct-1 and YY-1 binding." Biochim Biophys Acta 1627(2-3): 121-128.
Modi, S., D.E. Gilham, M.J. Sutcliffe, L.Y. Lian, W.U. Primrose, C.R. Wolf and G.C.
Roberts (1997). "1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine as a substrate of
cytochrome P450 2D6: allosteric effects of NADPH-cytochrome P450 reductase."
Biochemistry 36(15): 4461-4470.

138
Montoliu, C., M. Sancho-Tello, I. Azorin, M. Burgal, S. Valles, J. Renau-Pigueras and
C. Guerri (1995). "Ethanol increases cytochrome P4502E1 and induces oxidative
stress in astrocytes." J Neurochem 65(6): 2561-2570.
Narimatsu, S., T. Arai, Y. Masubuchi, T. Horie, M. Hosokawa, K. Ueno, H. Kataoka, S.
Yamamoto, T. Ishikawa and A. K. Cho (2001). "Inactivation of rat cytochrome P450 2D
enzyme by a further metabolite of 4-hydroxypropranolol, the major and active
metabolite of propranolol." Biol Pharm Bull 24(9): 988-994.
Narimatsu, S., S. Yamamoto, T. Koitabashi, R. Kato, Y. Masubuchi, T. Suzuki and T.
Horie (1999). "Biphasic kinetics of imipramine N-oxidation in rat brain microsomes."
Biol Pharm Bull 22(3): 253-256.
Nayebi, A. R. M., M. Hassanpour and H. Rezazadeh (2001). "Effect of chronic and
acute administration of fluoxetine and its additive effect with morphine on the
behavioural response in the formalin test in rats." J Pharm Pharmacol 53(2): 219-225.
Neafsey, P., G. Ginsberg, D. Hattis and B. Sonawane (2009). "Genetic polymorphism
in cytochrome P450 2D6 (CYP2D6): Population distribution of CYP2D6 activity." J
Toxicol Environ Health B Crit Rev 12(5-6): 334-361.
Nelson, D. R. (2006). "Cytochrome P450 nomenclature, 2004." Methods Mol Biol 320:
1-10.
Nelson, J. C., C. M. Mazure and P. I. Jatlow (1995). "Desipramine treatment of major
depression in patients over 75 years of age." J Clin Psychopharmacol 15(2): 99-105.
Niwa, T., K. Okada, T. Hiroi, S. Imaoka, S. Narimatsu and Y. Funae (2008). "Effect of
psychotropic drugs on the 21-hydroxylation of neurosteroids, progesterone and
allopregnanolone, catalyzed by rat CYP2D4 and human CYP2D6 in the brain." Biol
Pharm Bull 31(3): 348-351.
Niznik, H. B., R. F. Tyndale, F. R. Sallee, F. J. Gonzalez, J. P. Hardwick, T. Inaba and
W. Kalow (1990). "The dopamine transporter and cytochrome P45OIID1
(debrisoquine 4-hydroxylase) in brain: resolution and identification of two distinct
[3H]GBR-12935 binding proteins." Arch Biochem Biophys 276(2): 424-432.
Obach, R. S., R. L. Walsky and K. Venkatakrishnan (2007). "Mechanism-based
inactivation of human cytochrome p450 enzymes and the prediction of drug-drug
interactions." Drug Metab Dispos 35(2): 246-255.
O'Callaghan, J. P. and S. G. Holtzman (1975). "Quantification of the analgesic activity
of narcotic antagonists by a modified hot-plate procedure." J Pharmacol Exp Ther
192(3): 497-505.
Ochsner, K.N., D.H. Ludlow, K. Knierim, J. Hanelin, T. Ramachandran, G.C. Glover
and S.C. Mackey (2006). "Neural correlates of individual differences in pain-related
fear and anxiety." Pain 120(1-2): 69-77.

139
Oguri, K., N. Hanioka and H. Yoshimura (1990). "Species differences in metabolism
of codeine: urinary excretion of codeine glucuronide, morphine-3-glucuronide and
morphine-6-glucuronide in mice, rats, guinea pigs and rabbits." Xenobiotica 20(7):
683-688.
Ohe, T., M. Hirobe and T. Mashino (2000). "Novel metabolic pathway of estrone and
17beta-estradiol catalyzed by cytochrome P-450." Drug Metab Dispos 28(2): 110-
112.
Ohno, S., K. Kawana and S. Nakajin (2008). "Contribution of UDP-
glucuronosyltransferase 1A1 and 1A8 to morphine-6-glucuronidation and its kinetic
properties." Drug Metab Dispos 36(4): 688-694.
Okura, T., Y. Morita, Y. Ito, Y. Kagawa and S. Yamada (2009). "Effects of quinidine on
antinociception and pharmacokinetics of morphine in rats." J Pharm Pharmacol
61(5): 593-597.
Olanow, C.W. (2007). "The pathogenesis of cell death in Parkinson's disease--2007."
Mov Disord 22 Suppl 17: S335-342.
Oldendorf, W. H., S. Hyman, L. Braun and S. Z. Oldendorf (1972). "Blood-brain
barrier: penetration of morphine, codeine, heroin, and methadone after carotid
injection." Science 178(4064): 984-986.
Osborne, R., S. Joel, D. Trew and M. Slevin (1988). "Analgesic activity of morphine-6-
glucuronide." Lancet 1(8589): 828.
Pang, I. H. and M. R. Vasko (1986). "Effect of depletion of spinal cord norepinephrine
on morphine-induced antinociception." Brain Res 371(1): 171-176.
Parkinson, A., D. R. Mudra, C. Johnson, A. Dwyer and K. M. Carroll (2004). "The
effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme
activity in human liver microsomes and inducibility in cultured human hepatocytes."
Toxicol Appl Pharmacol 199(3): 193-209.
Paxinos, G. and C. Watson (1986). The Rat Brain in Stereotaxic Coordinates, 2nd edn.
San Diego, Academic Press.
Penas-Lledo, E. M., P. Dorado, Z. Aguera, M. Gratacos, X. Estivill, F. Fernandez-
Aranda and A. Llerena (2012). "CYP2D6 polymorphism in patients with eating
disorders." Pharmacogenomics J 12(2): 173-175.
Penas-LLedo, E. M., P. Dorado, R. Pacheco, I. Gonzalez and L. L. A (2009). "Relation
between CYP2D6 genotype, personality, neurocognition and overall
psychopathology in healthy volunteers." Pharmacogenomics 10(7): 1111-1120.
Pert, C. B., M. J. Kuhar and S. H. Snyder (1975). "Autoradiographic localization of the
opiate receptor in rat brain." Life Sciences 16(12): 1849-1853.

140
Pert, C. B. and S. H. Snyder (1973). "Properties of opiate-receptor binding in rat
brain." Proc Natl Acad Sci U S A 70(8): 2243-2247.
Pluchino, N., M. Luisi, E. Lenzi, M. Centofanti, S. Begliuomini, L. Freschi, F. Ninni and
A.R. Genazzani (2006). "Progesterone and progestins: effects on brain,
allopregnanolone and beta-endorphin." J Steroid Biochem Mol Biol 102(1-5): 205-
213.
Poeaknapo, C., J. Schmidt, M. Brandsch, B. Drager and M. H. Zenk (2004).
"Endogenous formation of morphine in human cells." Proc Natl Acad Sci U S A
101(39): 14091-14096.
Projean, D., P. E. Morin, T. M. Tu and J. Ducharme (2003). "Identification of CYP3A4
and CYP2C8 as the major cytochrome P450 s responsible for morphine N-
demethylation in human liver microsomes." Xenobiotica 33(8): 841-854.
Proudfit, H. K. and D. L. Hammond (1981). "Alterations in nociceptive threshold and
morphine-induced analgesia produced by intrathecally administered amine
antagonists." Brain Res 218(1-2): 393-399.
Quik, M., H. Cox, N. Parameswaran, K. O'Leary, J.W. Langston and D. Di Monte
(2007). "Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys." Ann
Neurol 62(6): 588-596.
Quik, M., M. O'Neill and X.A. Perez (2007). "Nicotine neuroprotection against
nigrostriatal damage: importance of the animal model." Trends Pharmacol Sci 28(5):
229-235.
Quik, M., N. Parameswaran, S.E. McCallum, T. Bordia, S. Bao, A. McCormack, A. Kim,
R.F. Tyndale, J.W. Langston and D.A. Di Monte (2006). "Chronic oral nicotine
treatment protects against striatal degeneration in MPTP-treated primates." J
Neurochem 98(6): 1866-1875.
Rae, J. M., M. D. Johnson, M. E. Lippman and D. A. Flockhart (2001). "Rifampin is a
selective, pleiotropic inducer of drug metabolism genes in human hepatocytes:
studies with cDNA and oligonucleotide expression arrays." J Pharmacol Exp Ther
299(3): 849-857.
Rendic, S. and F. J. Di Carlo (1997). "Human cytochrome P450 enzymes: a status
report summarizing their reactions, substrates, inducers, and inhibitors." Drug
Metab Rev 29(1-2): 413-580.
Riedel, M., M. J. Schwarz, M. Strassnig, I. Spellmann, A. Muller-Arends, K. Weber, J.
Zach, N. Muller and H. J. Moller (2005). "Risperidone plasma levels, clinical response
and side-effects." Eur Arch Psychiatry Clin Neurosci 255(4): 261-268.

141
Riedl, A. G., P. M. Watts, R. J. Edwards, A. R. Boobis, P. Jenner and C. D. Marsden
(1996). "Selective localisation of P450 enzymes and NADPH-P450 oxidoreductase in
rat basal ganglia using anti-peptide antisera." Brain Res 743(1-2): 324-328.
Riedl, A. G., P. M. Watts, R. J. Edwards, T. Schulz-Utermoehl, A. R. Boobis, P. Jenner
and C. D. Marsden (1999). "Expression and localisation of CYP2D enzymes in rat
basal ganglia." Brain Res 822(1-2): 175-191.
Rifkind, A. B., C. Lee, T. K. Chang and D. J. Waxman (1995). "Arachidonic acid
metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: regioselective
oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid
epoxygenation in human liver microsomes." Arch Biochem Biophys 320(2): 380-389.
Ritter, J. K. (2000). "Roles of glucuronidation and UDP-glucuronosyltransferases in
xenobiotic bioactivation reactions." Chem Biol Interact 129(1-2): 171-193.
Roberts, R. L., S. E. Luty, R. T. Mulder, P. R. Joyce and M. A. Kennedy (2004).
"Association between cytochrome P450 2D6 genotype and harm avoidance." Am J
Med Genet B Neuropsychiatr Genet 127B(1): 90-93.
Romach, M. K., S. V. Otton, G. Somer, R. F. Tyndale and E. M. Sellers (2000).
"Cytochrome P450 2D6 and treatment of codeine dependence." J Clin
Psychopharmacol 20(1): 43-45.
Rosenbrock, H., C. E. Hagemeyer, I. Singec, R. Knoth and B. Volk (1999).
"Testosterone metabolism in rat brain is differentially enhanced by phenytoin-
inducible cytochrome P450 isoforms." J Neuroendocrinol 11(8): 597-604.
Rowland, K., W. W. Yeo, S. W. Ellis, I. G. Chadwick, I. Haq, M. S. Lennard, P. R.
Jackson, L. E. Ramsay and G. T. Tucker (1994). "Inhibition of CYP2D6 activity by
treatment with propranolol and the role of 4-hydroxy propranolol." Br J Clin
Pharmacol 38(1): 9-14.
Sachse, C., J. Brockmoller, S. Bauer and I. Roots (1997). "Cytochrome P450 2D6
variants in a Caucasian population: allele frequencies and phenotypic
consequences." Am J Hum Genet 60(2): 284-295.
Sakai, N., K. Q. Sakamoto, S. Fujita and M. Ishizuka (2009). "The importance of
heterogeneous nuclear ribonucleoprotein K on cytochrome P450 2D2 gene
regulation: its binding is reduced in Dark Agouti rats." Drug Metab Dispos 37(8):
1703-1710.
Schenk, P. W., M. A. van Fessem, S. Verploegh-Van Rij, R. A. Mathot, T. van Gelder,
A. G. Vulto, M. van Vliet, J. Lindemans, J. A. Bruijn and R. H. van Schaik (2008).
"Association of graded allele-specific changes in CYP2D6 function with imipramine
dose requirement in a large group of depressed patients." Mol Psychiatry 13(6): 597-
605.

142
Schoedel, K. A., E. M. Sellers and R. F. Tyndale (2001). "Induction of CYP2B1/2 and
nicotine metabolism by ethanol in rat liver but not rat brain." Biochem Pharmacol
62(8): 1025-1036.
Schoedel, K. A. and R. F. Tyndale (2003). "Induction of nicotine-metabolizing CYP2B1
by ethanol and ethanol-metabolizing CYP2E1 by nicotine: summary and
implications." Biochim Biophys Acta 1619(3): 283-290.
Schreiber, S., M. M. Backer, R. Weizman and C. G. Pick (1996). "The antinociceptive
effects of fluoxetine." Pain Clinic 9(3): 349-356.
Shen, H. W. and A. M. Yu (2009). "Difference in desipramine metabolic profile
between wild-type and CYP2D6-humanized mice." Drug Metab Lett 3(4): 234-241.
Siegle, I., P. Fritz, K. Eckhardt, U. M. Zanger and M. Eichelbaum (2001). "Cellular
localization and regional distribution of CYP2D6 mRNA and protein expression in
human brain." Pharmacogenetics 11(3): 237-245.
Simonds, W. F. (1988). "The molecular basis of opioid receptor function." Endocr Rev
9(2): 200-212.
Sinclair, J. G., C. D. Main and G. F. Lo (1988). "Spinal vs. supraspinal actions of
morphine on the rat tail-flick reflex." Pain 33(3): 357-362.
Sindrup, S. H., L. Arendt-Nielsen, K. Brosen, P. Bjerring, H. R. Angelo, B. Eriksen and
L. F. Gram (1992). "The effect of quinidine on the analgesic effect of codeine." Eur J
Clin Pharmacol 42(6): 587-591.
Sindrup, S. H., K. Brosen, P. Bjerring, L. Arendt-Nielsen, U. Larsen, H. R. Angelo and
L. F. Gram (1990). "Codeine increases pain thresholds to copper vapor laser stimuli
in extensive but not poor metabolizers of sparteine." Clin Pharmacol Ther 48(6): 686-
693.
Sindrup, S. H., L. Poulsen, K. Brosen, L. Arendt-Nielsen and L. F. Gram (1993). "Are
poor metabolisers of sparteine/debrisoquine less pain tolerant than extensive
metabolisers?" Pain 53(3): 335-339.
Siu, E. C., D. B. Wildenauer and R. F. Tyndale (2006). "Nicotine self-administration in
mice is associated with rates of nicotine inactivation by CYP2A5."
Psychopharmacology (Berl) 184(3-4): 401-408.
Smith, D. L., M. C. D'Amour and F. E. D'Amour (1943). "The analgesic properties of
certain drugs and drug combinations." Journal of Pharmacology and Experimental
Therapeutics 77(2): 184-193.
Snider, N.T., M.J. Sikora, C. Sridar, T.J. Feuerstein, J.M. Rae and P.F. Hollenberg
(2008). "The endocannabinoid anandamide is a substrate for the human polymorphic
cytochrome P450 2D6." J Pharmacol Exp Ther, 327(2): 538-545.

143
Spina, E., A. Avenoso, G. Facciola, M. Salemi, M. G. Scordo, M. Ancione, A. G. Madia
and E. Perucca (2001). "Relationship between plasma risperidone and 9-
hydroxyrisperidone concentrations and clinical response in patients with
schizophrenia." Psychopharmacology (Berl) 153(2): 238-243.
Stevens, J. C., S. A. Marsh, M. J. Zaya, K. J. Regina, K. Divakaran, M. Le and R. N.
Hines (2008). "Developmental changes in human liver CYP2D6 expression." Drug
Metab Dispos 36(8): 1587-1593.
Strobel, H. W., C. M. Thompson and L. Antonovic. (2001). "Cytochromes P450 in
brain: function and significance." Curr Drug Metab 2(2): 199-214.
Strobl, G. R., S. von Kruedener, J. Stockigt, F. P. Guengerich and T. Wolff (1993).
"Development of a pharmacophore for inhibition of human liver cytochrome P-450
2D6: molecular modeling and inhibition studies." J Med Chem 36(9): 1136-1145.
Suzuki, T., S. Fujita, S. Narimatsu, Y. Masubuchi, M. Tachibana, S. Ohta and M. Hirobe
(1992). "Cytochrome P450 isozymes catalyzing 4-hydroxylation of parkinsonism-
related compound 1,2,3,4-tetrahydroisoquinoline in rat liver microsomes." Faseb J
6(2): 771-776.
Tate, S. and H. Cook (1996). "Postoperative nausea and vomiting. 1: Physiology and
aetiology." Br J Nurs 5(16): 962, 964, 966 passim.
Thorn, C. F., T. E. Klein and R. B. Altman (2009). "Codeine and morphine pathway."
Pharmacogenet Genomics 19(7): 556-558.
Trapani, G., C. Altomare, G. Liso, E. Sanna and G. Biggio (2000). "Propofol in
anaesthesia. Mechanism of action, structure-activity relationships, and drug
delivery." Curr Med Chem 7(2): 249-271.
Transon, C., S. Lecoeur, T. Leemann, P. Beaune and P. Dayer (1996). "Interindividual
variability in catalytic activity and immunoreactivity of three major human liver
cytochrome P450 isozymes." Eur J Clin Pharmacol 51(1): 79-85.
Turpeinen, M., L. E. Korhonen, A. Tolonen, J. Uusitalo, R. Juvonen, H. Raunio and O.
Pelkonen (2006). "Cytochrome P450 (CYP) inhibition screening: comparison of three
tests." Eur J Pharm Sci 29(2): 130-138.
Tyndale, R. F., K. P. Droll and E. M. Sellers (1997). "Genetically deficient CYP2D6
metabolism provides protection against oral opiate dependence."
Pharmacogenetics 7(5): 375-379.
Tyndale, R. F., Y. Li, N. Y. Li, E. Messina, S. Miksys and E. M. Sellers (1999).
"Characterization of cytochrome P-450 2D1 activity in rat brain: high-affinity kinetics
for dextromethorphan." Drug Metab Dispos 27(8): 924-930.
Tyndale, R. F., R. Sunahara, T. Inaba, W. Kalow, F. J. Gonzalez and H. B. Niznik
(1991). "Neuronal cytochrome P450IID1 (debrisoquine/sparteine-type): potent

144
inhibition of activity by (-)-cocaine and nucleotide sequence identity to human
hepatic P450 gene CYP2D6." Mol Pharmacol 40(1): 63-68.
Vaglini, F., C. Pardini, C. Viaggi, C. Bartoli, D. Dinucci and G. U. Corsini (2004).
"Involvement of cytochrome P450 2E1 in the 1-methyl-4-phenyl-1,2,3,6-
tetrahydropyridine-induced mouse model of Parkinson's disease." J Neurochem
91(2): 285-298.
Van, L. M., A. Heydari, J. Yang, J. Hargreaves, K. Rowland-Yeo, M. S. Lennard, G. T.
Tucker and A. Rostami-Hodjegan (2006). "The impact of experimental design on
assessing mechanism-based inactivation of CYP2D6 by MDMA (Ecstasy)." J
Psychopharmacol 20(6): 834-841.
Vasiliou, V., T. L. Ziegler, P. Bludeau, D.R. Petersen, F.J. Gonzalez and R.A. Deitrich
(2006). "CYP2E1 and catalase influence ethanol sensitivity in the central nervous
system." Pharmacogenet Genomics 16(1): 51-58.
Vaughan, C.W., E. E. Bagley, G. M. Drew, A. Schuller, J. E. Pintar, S. P. Hack and M. J.
Christie (2003). "Cellular actions of opioids on periaqueductal grey neurons from
C57B16/J mice and mutant mice lacking MOR-1." Br J Pharmacol 139(2): 362-367.
Vaughan, C.W. and M. J. Christie (1997). "Presynaptic inhibitory action of opioids on
synaptic transmission in the rat periaqueductal grey in vitro." J Physiol 498 ( Pt 2):
463-472.
Vento, P.J. and D. Daniels (2010). "Repeated administration of angiotensin II reduces
its dipsogenic effect without altering saline intake." Exp Physiol 95(6): 736-745.
Vieregge, A., M. Sieberer, H. Jacobs, J.M. Hagenah and P. Vieregge (2001).
"Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled
study." Neurology 57(6): 1032-1035.
Voirol, P., M. Jonzier-Perey, F. Porchet, M. J. Reymond, R. C. Janzer, C. Bouras, H. W.
Strobel, M. Kosel, C. B. Eap and P. Baumann (2000). "Cytochrome P-450 activities in
human and rat brain microsomes." Brain Res 855(2): 235-243.
Volk, B., U. Hettmannsperger, T. Papp, Z. Amelizad, F. Oesch and R. Knoth (1991).
"Mapping of phenytoin-inducible cytochrome P450 immunoreactivity in the mouse
central nervous system." Neuroscience 42(1): 215-235.
Von Moltke, L.L., D.J. Greenblatt, J.M. Grassi, B.W. Granda, K. Venkatakrishnan, J.
Schmider, J.S. Harmatz and R.I. Shader (1998). "Multiple human cytochromes
contribute to biotransformation of dextromethorphan in-vitro: role of CYP2C9,
CYP2C19, CYP2D6, and CYP3A." J Pharm Pharmacol 50(9): 997-1004.
Vuppugalla, R. and R. Mehvar (2005). "Enzyme-selective effects of nitric oxide on
affinity and maximum velocity of various rat cytochromes P450." Drug Metab Dispos
33(6): 829-836.

145
Wang, H., K. L. Napoli and H. W. Strobel (2000). "Cytochrome P450 3A9 catalyzes the
metabolism of progesterone and other steroid hormones." Mol Cell Biochem 213(1-
2): 127-135.
Warner, M. and J. A. Gustafsson (1994). "Effect of ethanol on cytochrome P450 in the
rat brain." Proc Natl Acad Sci U S A 91(3): 1019-1023.
Watts, P. M., A. G. Riedl, D. C. Douek, R. J. Edwards, A. R. Boobis, P. Jenner and C. D.
Marsden (1998). "Co-localization of P450 enzymes in the rat substantia nigra with
tyrosine hydroxylase." Neuroscience 86(2): 511-519.
Wilson, H.D., M.L. Uhelski, and P.N. Fuchs (2008). "Examining the role of the medial
thalamus in modulating the affective dimension of pain." Brain Res 1229: 90-99.
Woodland, C., T. T. Huang, E. Gryz, R. Bendayan and J. P. Fawcett (2008).
"Expression, activity and regulation of CYP3A in human and rodent brain." Drug
Metab Rev 40(1): 149-168.
Woolfe, G. and A. D. MacDonald (1944). "The evaluation of the analgesic action of
pethidine hydrochloride (Demerol)." J Pharmacol Exp Ther 80(3): 300-307.
Wyss, A., J. A. Gustafsson and M. Warner (1995). "Cytochromes P450 of the 2D
subfamily in rat brain." Mol Pharmacol 47(6): 1148-1155.
Xie, X., H. Liao, H. Dang, W. Pang, Y. Guan, X. Wang, J. Y. Shyy, Y. Zhu and F. M.
Sladek (2009). "Down-regulation of hepatic HNF4alpha gene expression during
hyperinsulinemia via SREBPs." Mol Endocrinol 23(4): 434-443.
Xu, B. Q., T. A. Aasmundstad, A. Bjorneboe, A. S. Christophersen and J. Morland
(1995). "Ethylmorphine O-deethylation in isolated rat hepatocytes. Involvement of
codeine O-demethylation enzyme systems." Biochem Pharmacol 49(4): 453-460.
Xu, B. Q., T. A. Aasmundstad, A. S. Christophersen, J. Morland and A. Bjorneboe
(1997). "Evidence for CYP2D1-mediated primary and secondary O-dealkylation of
ethylmorphine and codeine in rat liver microsomes." Biochem Pharmacol 53(4): 603-
609.
Yaksh, T. L. (1979). "Direct evidence that spinal serotonin and noradrenaline
terminals mediate the spinal antinociceptive effects of morphine in the
periaqueductal gray." Brain Res 160(1): 180-185.
Yaksh, T. L. and T. A. Rudy (1978). "Narcotic analgestics: CNS sites and mechanisms
of action as revealed by intracerebral injection techniques." Pain 4(4): 299-359.
Yaksh, T. L. and G. M. Tyce (1979). "Microinjection of morphine into the
periaqueductal gray evokes the release of serotonin from spinal cord." Brain Res
171(1): 176-181.

146
Yaksh, T. L. and P. R. Wilson (1979). "Spinal serotonin terminal system mediates
antinociception." J Pharmacol Exp Ther 208(3): 446-453.
Yaksh, T. L., J. C. Yeung and T. A. Rudy (1976). "Systematic examination in the rat of
brain sites sensitive to the direct application of morphine: observation of differential
effects within the periaqueductal gray." Brain Res 114(1): 83-103.
Yamamoto, T., A. Suzuki and Y. Kohno (2003). "High-throughput screening to
estimate single or multiple enzymes involved in drug metabolism: microtitre plate
assay using a combination of recombinant CYP2D6 and human liver microsomes."
Xenobiotica 33(8): 823-839.
Yokomori, N., R. Kobayashi, R. Moore, T. Sueyoshi and M. Negishi (1995). "A DNA
methylation site in the male-specific P450 (Cyp 2d-9) promoter and binding of the
heteromeric transcription factor GABP." Mol Cell Biol 15(10): 5355-5362.
Yu, A. M. and R. L. Haining (2006). "Expression, purification, and characterization of
mouse CYP2d22." Drug Metab Dispos 34(7): 1167-1174.
Yu, A. M., J. R. Idle and F. J. Gonzalez (2004). "Polymorphic cytochrome P450 2D6:
humanized mouse model and endogenous substrates." Drug Metab Rev 36(2): 243-
277.
Yu A.M., J.R. Idle, L.G. Byrd, K.W. Krausz, A. Kupfer and F.J. Gonzalez (2003a).
"Regeneration of serotonin from 5-methoxytryptamine by polymorphic human
CYP2D6." Pharmacogenetics 13(3):173–181.
Yu, A.M., J.R. Idle, K.W. Krausz, A. Kupfer and F.J. Gonzalez (2003b). "Contribution of
individual cytochrome P450 isozymes to the O-demethylation of the psychotropic
betacarboline alkaloids harmaline and harmine." J Pharmacol Exp Ther 305(1): 315-
322.
Yue, Q. Y., J. Hasselstrom, J. O. Svensson and J. Sawe (1991). "Pharmacokinetics of
codeine and its metabolites in Caucasian healthy volunteers: comparisons between
extensive and poor hydroxylators of debrisoquine." Br J Clin Pharmacol 31(6): 635-
642.
Yue, J., J. Khokhar, S. Miksys and R. F. Tyndale (2009). "Differential induction of
ethanol-metabolizing CYP2E1 and nicotine-metabolizing CYP2B1/2 in rat liver by
chronic nicotine treatment and voluntary ethanol intake." Eur J Pharmacol 609(1-3):
88-95.
Yue, J., S. Miksys, E. Hoffmann and R. F. Tyndale (2008). "Chronic nicotine treatment
induces rat CYP2D in the brain but not in the liver: an investigation of induction and
time course." J Psychiatry Neurosci 33(1): 54-63.
Yue, Q. Y., C. Alm, J. O. Svensson and J. Sawe (1997). "Quantification of the O- and N-
demethylated and the glucuronidated metabolites of codeine relative to the

147
debrisoquine metabolic ratio in urine in ultrarapid, rapid, and poor debrisoquine
hydroxylators." Ther Drug Monit 19(5): 539-542.
Yue, Q. Y. and J. Sawe (1997). "Different effects of inhibitors on the O- and N-
demethylation of codeine in human liver microsomes." Eur J Clin Pharmacol 52(1):
41-47.
Zanger, U. M., S. Raimundo and M. Eichelbaum (2004). "Cytochrome P450 2D6:
overview and update on pharmacology, genetics, biochemistry." Naunyn
Schmiedebergs Arch Pharmacol 369(1): 23-37.
Zhou, S. F. (2009). "Polymorphism of human cytochrome P450 2D6 and its clinical
significance: Part I." Clin Pharmacokinet 48(11): 689-723.
Zhu, W. (2008). "CYP2D6: a key enzyme in morphine synthesis in animals." Med Sci
Monit 14(11): SC15-18.
Zhu, W., P. Cadet, G. Baggerman, K.J. Mantione and G.B. Stefano (2005). "Human
white blood cells synthesize morphine: CYP2D6 modulation." J Immunol 175(11):
7357-7362.
Zhu, W., K. J. Mantione, L. Shen, P. Cadet, T. Esch, Y. Goumon, E. Bianchi, D. Sonetti
and G. B. Stefano (2005). "Tyrosine and tyramine increase endogenous ganglionic
morphine and dopamine levels in vitro and in vivo: cyp2d6 and tyrosine
hydroxylase modulation demonstrates a dopamine coupling." Med Sci Monit 11(11):
BR397-404.

148
List of Abstracts
Zhou, K., J.Y. Khokhar and R.F. Tyndale (2012). “The role of brain CYP2D metabolism
in codeine analgesia.” Visions in Pharmacology, Toronto.
Zhou, K., J.Y. Khokhar and R.F. Tyndale (2012). “The role of brain CYP2D metabolism
in codeine analgesia.” Canadian College of Neuropsychopharmacology, Vancouver.
Zhou, K., J.Y. Khokhar and R.F. Tyndale (2012). “The role of brain CYP2D metabolism
in codeine analgesia.” Southern Ontario Neuroscience Association, Toronto.
Related Documents




![Fioricet with Codeine (butalbital, acetaminophen, … · Fioricet with Codeine (butalbital, acetaminophen, caffeine, and codeine phosphate) capsule [Watson Pharmaceuticals, Inc.]](https://static.cupdf.com/doc/110x72/5b0089f27f8b9a89598cc123/fioricet-with-codeine-butalbital-acetaminophen-with-codeine-butalbital.jpg)






