The FASEB Journal express article 10.1096/fj.00-0676fje. Published online March 20, 2001. Inhibition of Alzheimer’s disease β-amyloid aggregation, neurotoxicity, and in vivo deposition by nitrophenols: implications for Alzheimer’s therapy Fernanda G. De Felice*, Jean-Christophe Houzel † , José Garcia-Abreu † , Paulo Roberto F. Louzada, Jr.*, Rosenilde C. Afonso † , M. Nazareth L. Meirelles ‡ , Roberto Lent † , Vivaldo Moura Neto † , and Sérgio T. Ferreira* *Departamento de Bioquímica Médica and † Departamento de Anatomia, Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21944-590, Brazil; ‡ Departamento de Ultraestrutura e Biologia Celular, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, RJ, Brazil Corresponding author: Sérgio T. Ferreira, Depto. de Bioquímica Médica, Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21944-590, Brazil. E-mail: [email protected] ABSTRACT Alzheimer’s disease (AD) is a major public health problem, and there is currently no clinically accepted treatment to cure it or to stop its progression. Fibrillar aggregates of the β−amyloid peptide (Aβ) are major constituents of the senile plaques found in the brains of AD patients and have been related to AD neurotoxicity. Here it is shown that nitrophenols prevent aggregation and cause disaggregation of Aβ fibrils and that they strongly prevent the neurotoxicity of Aβ to rat hippocampal neurons in culture. Furthermore, by using an in vivo model system of cerebral amyloid deposition, it is shown that nitrophenols cause a marked reduction in the volume occupied by amyloid deposits in the hippocampi of rats. These results raise the possibility that nitrophenols or their derivatives may be useful lead compounds for the development of drugs to prevent the neurotoxicity and deposition of Aβ in AD. Key words: amyloidosis • fibrillogenesis • drug development lzheimer’s disease (AD) affects a large fraction of the elderly population worldwide. It is estimated that about 20 million people suffer from AD-attributable dementia (1), with 20% of the individuals above 75 years old (and ~50% of those above 85) at risk of developing the disease. Despite considerable efforts aiming at understanding the molecular basis and physiopathology of this devastating disease, currently no clinically accepted treatments to cure it or to stop its progression have been found. The β-amyloid peptide (Aβ) is produced by proteolytic cleavage of the amyloid precursor protein (APP), and Aβ plays a central role in the neuropathology of AD (2–4). Aβ undergoes aggregation into amyloid fibrils, which are the major protein constituents of the senile plaques found in the brains of AD patients (5, 6). Although β-amyloid peptides are the primary components of the AD plaques, it is uncertain whether Aβ aggregation is mandatory for toxicity. On the one hand, previous studies have shown that free, nonpolymerized Aβ is neurotoxic (reviewed in 7). Nonpolymerized Aβ peptides insert themselves into natural and artificial lipid bilayer membranes and form nonselective cation channels that may trigger cellular death (8– 15). On the other hand, considerable evidence indicates that fibrillar aggregation of Aβ and amyloid deposition are related to AD neurotoxicity (for example, see 16–18). Thus, agents capable of interfering with aggregation might be potentially useful in preventing or diminishing the toxicity of Aβ. In this regard, numerous recent studies have aimed at interfering with amyloid aggregation as a possible route for prevention or treatment of AD. For example, the pineal hormone, melatonin, has been reported to interact with Aβ, inhibiting fibrillogenesis and neurotoxicity (19, 20). Monoclonal antibodies raised against the N-terminal region of Aβ have been shown to cause fibril disaggregation and inhibition of neurotoxicity (21–23). Beta- sheet breaker peptides inhibit amyloid aggregation and prevent neurotoxicity and in vivo cerebral Aβ deposition (24, 25). Finally, recent studies showed that immunization with Aβ prevented or reduced the development of amyloid deposition in the brains of transgenic mice that express human APP and develop plaque pathology (26–30). We now report the effects of nitrophenols on amyloid aggregation, in vitro neurotoxicity, and in vivo deposition in rat brains. The present results indicate that nitrophenols can be useful lead compounds for the design of small molecule inhibitors of β-amyloid toxicity and deposition in AD. METHODS Aβ solutions and amyloid aggregation Synthetic Aβ peptides of different chain-lengths (Bachem Inc., Torrance, Calif.) were freshly dissolved from lyophilized powder in 50% (v/v) trifluoroethanol (TFE) in phosphate-buffered saline (PBS). Aggregation was triggered by dilution of aliquots from the stock solution into PBS (resulting in ≤0.5% residual TFE) and was followed as a function of time by right-angle light scattering measurements. Light scattering was measured in sealed cuvettes at 500 nm on ISS Inc. (Champaign, Ill.) PC1 or Hitachi F-4500 spectrofluorometers. Except as indicated in Figure 1B, all measurements were carried out at 23 o C. Low- temperature experiments were performed by using a thermostated cell holder and by flushing the cell compartment with N 2 to avoid condensation. All results shown represent equilibrium light scattering values obtained for each sample. A

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

The FASEB Journal express article 10.1096/fj.00-0676fje. Published online March 20, 2001.
Inhibition of Alzheimer’s disease β-amyloid aggregation, neurotoxicity, and in vivo deposition by nitrophenols: implications for Alzheimer’s therapy Fernanda G. De Felice*, Jean-Christophe Houzel†, José Garcia-Abreu†, Paulo Roberto F. Louzada, Jr.*, Rosenilde C. Afonso†, M. Nazareth L. Meirelles‡, Roberto Lent†, Vivaldo Moura Neto†, and Sérgio T. Ferreira* *Departamento de Bioquímica Médica and †Departamento de Anatomia, Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21944-590, Brazil; ‡Departamento de Ultraestrutura e Biologia Celular, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, RJ, Brazil Corresponding author: Sérgio T. Ferreira, Depto. de Bioquímica Médica, Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21944-590, Brazil. E-mail: [email protected] ABSTRACT Alzheimer’s disease (AD) is a major public health problem, and there is currently no clinically accepted treatment to cure it or to stop its progression. Fibrillar aggregates of the β−amyloid peptide (Aβ) are major constituents of the senile plaques found in the brains of AD patients and have been related to AD neurotoxicity. Here it is shown that nitrophenols prevent aggregation and cause disaggregation of Aβ fibrils and that they strongly prevent the neurotoxicity of Aβ to rat hippocampal neurons in culture. Furthermore, by using an in vivo model system of cerebral amyloid deposition, it is shown that nitrophenols cause a marked reduction in the volume occupied by amyloid deposits in the hippocampi of rats. These results raise the possibility that nitrophenols or their derivatives may be useful lead compounds for the development of drugs to prevent the neurotoxicity and deposition of Aβ in AD. Key words: amyloidosis • fibrillogenesis • drug development
lzheimer’s disease (AD) affects a large fraction of the elderly population worldwide. It is estimated that about 20 million people suffer from AD-attributable dementia
(1), with 20% of the individuals above 75 years old (and ~50% of those above 85) at risk of developing the disease. Despite considerable efforts aiming at understanding the molecular basis and physiopathology of this devastating disease, currently no clinically accepted treatments to cure it or to stop its progression have been found. The β-amyloid peptide (Aβ) is produced by proteolytic cleavage of the amyloid precursor protein (APP), and Aβ plays a central role in the neuropathology of AD (2–4). Aβ undergoes aggregation into amyloid fibrils, which are the major protein constituents of the senile plaques found in the brains of AD patients (5, 6). Although β-amyloid peptides are the primary components of the AD plaques, it is uncertain whether Aβ aggregation is mandatory for toxicity. On the one hand, previous studies have shown that free, nonpolymerized Aβ is neurotoxic (reviewed in 7). Nonpolymerized Aβ peptides insert themselves into natural and artificial lipid bilayer membranes and form nonselective cation channels that may trigger cellular death (8–15). On the other hand, considerable evidence indicates that fibrillar aggregation of Aβ and amyloid deposition are related to
AD neurotoxicity (for example, see 16–18). Thus, agents capable of interfering with aggregation might be potentially useful in preventing or diminishing the toxicity of Aβ. In this regard, numerous recent studies have aimed at interfering with amyloid aggregation as a possible route for prevention or treatment of AD. For example, the pineal hormone, melatonin, has been reported to interact with Aβ, inhibiting fibrillogenesis and neurotoxicity (19, 20). Monoclonal antibodies raised against the N-terminal region of Aβ have been shown to cause fibril disaggregation and inhibition of neurotoxicity (21–23). Beta-sheet breaker peptides inhibit amyloid aggregation and prevent neurotoxicity and in vivo cerebral Aβ deposition (24, 25). Finally, recent studies showed that immunization with Aβ prevented or reduced the development of amyloid deposition in the brains of transgenic mice that express human APP and develop plaque pathology (26–30). We now report the effects of nitrophenols on amyloid aggregation, in vitro neurotoxicity, and in vivo deposition in rat brains. The present results indicate that nitrophenols can be useful lead compounds for the design of small molecule inhibitors of β-amyloid toxicity and deposition in AD. METHODS Aβ solutions and amyloid aggregation Synthetic Aβ peptides of different chain-lengths (Bachem Inc., Torrance, Calif.) were freshly dissolved from lyophilized powder in 50% (v/v) trifluoroethanol (TFE) in phosphate-buffered saline (PBS). Aggregation was triggered by dilution of aliquots from
the stock solution into PBS (resulting in ≤0.5% residual TFE) and was followed as a function of time by right-angle light scattering measurements. Light scattering was measured in sealed cuvettes at 500 nm on ISS Inc. (Champaign, Ill.) PC1 or Hitachi F-4500 spectrofluorometers. Except as indicated in Figure 1B, all measurements were carried out at 23oC. Low-temperature experiments were performed by using a thermostated cell holder and by flushing the cell compartment with N2 to avoid condensation. All results shown represent equilibrium light scattering values obtained for each sample.
A

Aggregated samples were also examined by transmission electron microscopy. In this case, Aβ (22 µM) was incubated in PBS in the absence or in the presence of nitrophenols, as indicated in Results. After 48 h, samples were stained with 1% uranyl acetate. Cell culture experiments, immunostaining, and viability assay Hippocampi from 18-day-old rat embryos were dissected and cultured as previously described (31) with minor modifications. Cells were plated on glass coverslips previously coated with 1.5 µg/ml polyornithine (Sigma, St. Louis, Mo.) in Basal Eagle’s Medium (Gibco, Rockville, Md.) enriched with 10% fetal calf serum (Hyclone, Logan, UT) for the first 24 h of culture. After
that, proliferation of nonneuronal cells was inhibited with 10 µM arabinosyl cytoside and the serum concentration was lowered to 2%. Aβ1-42 (44 µM), in the absence or in the presence of nitrophenols, was added after 48 h of culture and kept for 3 days. All Aβ solutions used in cell culture experiments were prepared and kept at all times under sterile conditions. In addition, to further exclude the possibility of bacterial contamination of Aβ preparations, lipopolysaccharide (LPS) levels were determined in aliquots from different preparations by using the QCL 1000 kit (Biowhittaker, Walkersville, Md.). LPS levels in the samples were found to be below the detection limit of the method (<0.06 IU/ml or <12 pg/ml). Control cultures consisting of neurons cultured in growth medium alone or in the presence of residual TFE (0.5% v/v) were also prepared. Daily observations were carried out during this period. The overall morphology of the neurons in culture was examined by immunostaining with an anti-Tau polyclonal antibody (DAKO Corp., Carpinteria, Calif.), as previously described (32). Briefly, cells were washed twice with fresh medium, fixed with 4% paraformaldehyde, 4% sucrose in PBS, permeabilized with 0.1% Triton X-100, and incubated for 1 h with anti-Tau antibody (1:200 dilution). Staining was performed by incubation with Cy3-conjugated anti-rabbit IgG (Gibco; 1:600 dilution). Fluorescence microscopy was carried out on a Zeiss Axioplan microscope. Cell viability in cultures incubated with or without Aβ was assessed by trypan blue exclusion. Immediately before counting, the medium was removed, and cultures were washed once with PBS and incubated for 5 min with 0.4% trypan blue. Randomly chosen fields were counted in a Zeiss Televal microscope. Percentages of live neurons are expressed relative to the total number of neurons observed in each field. Five independent fields were counted for each experimental condition (which were carried out in triplicate). Essentially identical results were obtained in a repeat experiment using neurons from another animal. In vivo cerebral amyloid deposition assay Male adult Wistar rats (280–320 g) were anesthetized with chloropent (3.3 ml/kg) and placed into a stereotaxic frame. The left hippocampus (standard coordinates: A 5.0; L 2.0; H 6.8) was injected with 3 nmol of Aβ1−42 (from a previously diluted stock in 55 µl of PBS containing 9% dimethylsulfoxide). A volume of 1.5 µl was administered at a constant flow rate during a 15-min period. The micropipette was left in situ for 15 min after
injection, withdrawn 0.2 mm, left for 3 min, and then slowly withdrawn completely. The right hippocampus of each rat received a mixture of Aβ and 2,4-dinitrophenol (DNP; 0.76 mM). In this case, the concentration of DNP was increased so as to maintain approximately the same ratio of Aβ/DNP concentrations used in cell culture experiments. The animals were kept on a heating pad during surgery and until they regained their righting reflex. Animals did not receive further medication and were killed 8 days later. After transcardiac perfusion with saline followed by 4% paraformaldehyde, brains were cryoprotected and frozen-cut into 20-µm-thick coronal sections. Alternate sections were stained with Thioflavine-S (to reveal amyloid plaques) or cresyl violet (for inspection of cytoarchitecture) or were simply dehydrated for autofluorescence examination, which provides another means for visualizing amyloid plaques. Sections were observed by using an epifluorescence microscope (Zeiss Axioplan) and digitized with a CCD camera (Zeiss, ZVS-47EC). Images were analyzed with ScionImage to measure the areas occupied by amyloid aggregates. Thioflavin-stained sections used for cresyl violet staining were interpolated in the image analysis. Total volumes occupied by amyloid deposits were integrated over all sections spanning the deposition region. RESULTS Stability of β-amyloid fibrils We first investigated the stability of fibrillar Aβ towards chaotropic agents and temperature. Aβ peptides of 40 and 42 amino acid residues (Aβ1-40 and Aβ1-42, respectively) are the main constituents of senile plaques, but other peptides of varying lengths are also present (4, 33). Previous studies have shown that the kinetics of aggregation of Aβ are sharply dependent on peptide length (34). In our studies, we have used both full-length Aβ (Aβ1−42 and Aβ1−43) and the C-terminally truncated Aβ1-28 peptide. Peptide aggregation was triggered by dilution in PBS and was followed by light-scattering measurements (Fig. 1), thioflavin T fluorescence (not shown) and electron microscopy (Fig. 2). Equilibrium light scattering values were used as an index of the extent of aggregation under different experimental conditions. In accordance with previous reports (34), aggregation of Aβ1-42 and Aβ1-43 was very fast (i.e., complete within a few minutes after dilution in PBS), whereas aggregation of Aβ1-28 was quite slow (i.e., 10–12 days). We found that the stability of fibrillar Aβ in guanidine hydrochloride (GdnHCl) solutions was markedly dependent on peptide chain length (Fig. 1A). For Aβ1-
28, complete disaggregation was observed at 3 M GdnHCl, whereas full disaggregation of Aβ1-42 required 5-6 M GdnHCl. All polar and charged amino acid residues of Aβ are located in the 28 amino acid-long N-terminal portion of the peptide. Residues 29-42 comprise a cluster of nonpolar amino acids contained in a transmembrane sequence of the amyloid precursor protein (35). Thus, the higher stabilities of Aβ1-42 and Aβ1-43 relative to Aβ1-28 (Fig. 1A) suggest that the C-terminal nonpolar amino acid sequence in the former two peptides mediates hydrophobic interactions that are important for fibril stability. Hydrophobic interactions are known to be destabilized by low temperatures, due to the decrease in the entropic contribution to the hydrophobic effect. In line with this, we found that

decreasing temperature from 25oC to 1oC caused reversible and nearly complete disaggregation of Aβ (Fig. 1B). Thus, if we extend previous suggestions from the literature (36–39), these results indicate that a significant contribution to the stability of Aβ aggregates comes from entropy-driven hydrophobic interactions and lead to the hypothesis that hydrophobic compounds could be effective in destabilizing and disaggregating amyloid fibrils. Inhibition of amyloid aggregation by nitrophenols Based on the results described above, our investigations focused on the effects of a number of hydrophobic compounds on amyloid stability. Our choice of candidate compounds was guided by the following criteria: 1) the compounds should be sufficiently hydrophobic to interfere with key hydrophobic interactions in amyloid fibrils; 2) they should still be sufficiently water-soluble to make their utilization straightforward in in vitro and in vivo assays. In addition, other desirable characteristics included low molecular weight and lack of neurotoxicity at the concentrations required for effective inhibition of amyloid aggregation. Some of the compounds we tested were nitrophenols. Addition of micromolar concentrations of 2,4-dinitrophenol (DNP) or 3-nitrophenol (NP) caused marked disaggregation of previously formed Aβ fibrils (Fig. 1C). IC50 values of approximately 7 µM and 80 µM were found for DNP and NP, respectively. DNP (20 µM) completely abolished light scattering from Aβ suspensions, which indicated complete disaggregation of fibrillar amyloid. Direct demonstration that DNP and NP inhibited the aggregation of fibrillar amyloid was obtained by transmission electron microscopy (Fig. 2). Abundant fibrils were observed in control samples of Aβ1-42 (Fig. 2A), whereas samples in which Aβ was added to the medium in the presence of NP or DNP were completely devoid of fibrils and contained only occasional scattered amorphous aggregates (as shown in Figs. 2B and C). Nitrophenols block the neurotoxicity of Aβ The possible protective effects of nitrophenols against Aβ-induced neurotoxicity were investigated. To this end, 48-h primary cultures of E18 rat hippocampal neurons were used (31). Aβ1−42 (44 µM) was added to the medium and incubation was continued for 72 h. Whereas control hippocampal neurons exhibited large cell bodies and long, branched neurites (Fig. 3A), significant neuronal degeneration and death was observed after 72 h of culture in the presence of Aβ1−42 (Fig. 3B). Large numbers of Aβ-treated neurons became detached from the plate during the immunostaining washes (Fig. 3B), which suggests that neuronal adhesion was impaired. Furthermore, the remaining cell bodies of Aβ-treated neurons were attached to the plate, but their neurites were retracted and thin and sometimes detached from the plate. When incubation with Aβ was carried out in the presence of NP or DNP, a marked protection against neurotoxicity was observed (Figs. 3C and D, respectively). In the presence of nitrophenols, neurons treated with Aβ showed large cell bodies and long neurites with good adhesion properties, and the morphological aspect of the cultures was similar to a control 5-day hippocampal culture.
The results obtained in cell culture experiments were quantitated by measuring neuronal survival by trypan blue exclusion and are summarized in Figure 4. Incubation with Aβ1−42 caused significant cell death, with only 25% neuronal survival vs. 86% survival in control cultures. Remarkably, addition of nitrophenols to the incubation medium almost completely blocked Aβ-induced cell death (74% and 65% neuronal survival in the presence of Aβ plus DNP or NP, respectively). In vivo inhibition of amyloid deposition To evaluate the effects of nitrophenols as inhibitors of amyloid deposition in vivo, we have used a rat model of cerebral Aβ deposition, as previously described (18, 24). Aβ was injected alone or in the presence of DNP into the left or right hippocampi of rats, respectively, and the areas occupied by amyloid deposits on either side were measured on consecutive sections stained with thioflavin S. The experimental protocol used was designed to minimize the influence of individual variability in animal response by injecting Aβ into one hemisphere and an identical amount of Aβ plus DNP into the other hemisphere for each individual rat. Figure 5A shows representative hippocampal sections stained with thioflavin S or directly visualized by using the autofluorescence of amyloid, as indicated. The total volume occupied by amyloid deposits was integrated by image analysis over all consecutive sections spanning the site of deposition. Figure 5B–E shows results of the integration for four different
rats. Coinjection of Aβ and DNP caused a reduction of 86 ± 17% in the volume of amyloid deposits in rat brains relative to the volume occupied when Aβ was injected alone (p = 0.05, n = 4; one-tailed paired t-test). DISCUSSION The present results show that nitrophenols inhibit amyloid aggregation and cause disaggregation of previously formed amyloid fibrils. These effects are accompanied by marked protection against Aβ toxicity in primary cultures of hippocampal neurons. Furthermore, microinjection experiments showed that nitrophenols inhibit amyloid deposition in rat brains. Nitrophenols are small water-soluble compounds, yet are presumably sufficiently hydrophobic to cross the blood-brain barrier and gain access to the CNS. DNP and other nitrophenols are known to be toxic at high concentrations, an effect that may be related to interference with cellular energy metabolism due to uncoupling of oxidative phosphorylation. For rats, the LD50 values for oral administration of NP and DNP are 933 and 30 mg/kg body weight, respectively (40). In the 1930s, DNP was used clinically in humans as a treatment for obesity. However, its use was subsequently discontinued due to the occurrence of numerous deleterious side-effects at relatively high dosages (3–5 mg/kg body weight) (41, 42). Such doses are ca. 35- to 60-fold higher than the IC50 value reported here for inhibition of Aβ fibril formation by DNP. Furthermore, it is important to note that the nitrophenols by themselves had no detectable toxic effects to neuronal primary cultures at the concentrations used in our studies and that the cytoarchitecture of rat brains (as revealed by staining with cresyl violet) also appeared normal in hemispheres injected with DNP. Finally, it is important to consider that at present there is no effective treatment available for amyloidoses,

including Alzheimer’s disease, Type II diabetes, and prion-related spongiform encephalopathies. Thus, although clinical use of DNP was discontinued due to its side-effects in the treatment of a relatively benign condition such as obesity, the lack of effective treatments for such devastating diseases as the amyloidoses may justify a reappraisal of the possible clinical use of nitrophenols at lower, subtoxic concentrations. Furthermore, an attractive possibility lies in the use of nitrophenols as lead compounds for the development of small molecule inhibitors of amyloidogenesis active at lower concentrations or with fewer undesirable side-effects. A structure-based drug design approach has recently been used in the development of drugs that prevent amyloid formation from transthyretin, which is involved in familial amyloid polyneuropathy, amyloid cardiomyopathy, and senile systemic amyloidosis (43). This finding clearly represents a major advance in the development of anti-amyloidogenic drugs. However, the feasibility of structure-based drug design is evidently dependent on knowledge of the structure of the aggregating species, which is not yet available for Alzheimer’s disease. When this condition is met, we may be in a better position to understand how nitrophenols (or their derivatives) interact with Aβ, and the mechanism of inhibition of amyloid formation and neurotoxicity. In conclusion, we propose that nitrophenols and their derivatives should be explored as possible drug candidates or lead compounds for the development of drugs to prevent amyloid aggregation and neurotoxicity in Alzheimer’s disease. ACKNOWLEDGMENTS We thank M. M. Sorenson for critical reading of the manuscript and P. T. Bozza for determination of LPS levels in Aβ samples. FGDF, PRFL, and JCH are supported by fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico and Coordenação de Aperfeiçoamento de Pessoal Docente de Nível Superior. This work was supported by a Howard Hughes Medical Institute International Research Scholar grant (to STF). REFERENCES 1. Haass, C. and De Strooper, B. (1999) The presenilins in Alzheimer’s
disease—Proteolysis holds the key. Science 286, 916–919 2. Selkoe, D.J. (1994) Alzheimer's disease: a central role for amyloid. J.
Neuropathol. Exp. Neurol. 53, 438–447 3. Yankner, B. A. (1996) Mechanisms of neuronal degeneration in
Alzheimer’s disease. Neuron 16, 921–932 4. Verbeek M. M., Ruiter, D. J., and de Waal, R. M. W. (1997) The role
of amyloid in the pathogenesis of Alzheimer’s Disease. Biol. Chem. 378, 937–950
5. Glenner, G. G., and Wong, C. W. (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890
6. Masters, C. L, Simms, G., Weinman, N. A., Multhaup, G., McDonald, B. L., and Beyreuther, K. (1985) Amyloid plaque core protein in Alzheimer’s disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 82, 4245–4249
7. Kourie, J. I., and Shorthouse, A. A. (2000) Properties of cytotoxic peptide-formed ion channels. Am. J. Physiol. Cell Physiol. 278, C1063–C1087
8. Arispe, N., Rojas, E., and Pollard, H. D. (1993) Alzheimer’s disease amyloid β protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 90, 567–571
9. Arispe, N., Pollard, H.D., and Rojas, E. (1993) Giant multilevel cation channels formed by Alzheimer’s disease amyloid β-protein [AβP-(1-40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA 90, 10573–10577
10. Arispe, N., Pollard, H. B., and Rojas, E. (1996) Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc. Natl. Acad. Sci. USA 93,1710–1715
11. Kawahara, M., Kuroda, Y., Arispe, N., and Rojas, E. (2000) Alzheimer’s β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 275, 14077–14083
12. Vargas, J., Alarcón, J. M., and Rojas, E. (2000) Displacement currents associated with the insertion of Alzheimer’s disease amyloid β-peptide into planar bilayer membranes. Biophys. J. 79, 934–944
13. Pallitto, M. M., Ghanta, J., Heinzelman, P., Kiessling, L. L., and Murphy, R. M. (1999) Recognition sequence design for peptidyl modulators of beta-amyloid aggregation and toxicity. Biochemistry 38, 3570–3578
14. Bhatia, R., Lin, H., and Lal, R. (2000) Fresh and globular amyloid β protein (1–42) induces rapid cellular degeneration: evidence for AβP channel-mediated cellular toxicity. FASEB J. 14, 1233–1243
15. Zhu, Y.J., Lin, H., and Lal, R. (2000) Fresh and nonfibrillar amyloid β protein(1–40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J. 14, 1244–1254
16. Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G., and Cotman, C. W. (1993) Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 13, 1676–1687
17. Lorenzo, A., and Yankner, B. A. (1994) Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. USA 91, 12243–12247
18. Geula, C., Wu, C.K., Saroff, D., Lorenzo, A., Yuan, M., and Yankner, B.A. (1998) Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat. Med. 4, 827–831
19. Pappolla, M. A., Sos, M., Omar, R.A., Bick, R.J., Hickson-Bick, D.L., Reiter, R.J., Efthimiopoulos, S., and Robakis, N.K. (1997) Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 17, 1683–1690
20. Pappolla, M., Bozner, P., Soto, C., Shao, H., Robakis, N. K., Zagorski, M., Frangione. B., and Ghiso, J. (1998) Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J. Biol. Chem. 273, 7185–7188
21. Solomon, B., Koppel, R., Hanan, E., and Katzav, T. (1996) Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 93, 452–455
22. Solomon, B., Koppel, R., Frenkel D., and Hanan-Aharon, E. (1997) Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc. Natl. Acad. Sci. USA 94, 4109–4112
23. Frenkel, D., Balass, M., Katchalski-Katzir, E., and Solomon, B. (1999) High-affinity binding of monoclonal antibodies to the sequential epitope EFRH of beta-amyloid peptide is essential for modulation of fibrillar aggregation. J. Neuroimmunol. 95, 136–142
24. Soto, C., Sigurdsson, E. M., Morelli, L., Kumar, R. A., Castano, E. M., and Frangione, B. (1998) Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat. Med. 4, 822–826
25. Poduslo, J.F., Curran, G.L., Kumar, A., Frangione, B., and Soto, C. (1999) Beta-sheet breaker peptide inhibitor of Alzheimer's amyloidogenesis with increased blood-brain barrier permeability

and resistance to proteolytic degradation in plasma. J. Neurobiol. 39, 371–382
26. Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., Kholodenko, D., Lee, M., Liao, Z., Lieberburg, I., Motter, R., Mutter, L., Soriano, F., Shopp, G., Vasquez, N., Vandevert, C., Walker, S., Wogulis, M., Yednock, T., Games, D., and Seubert, P. (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature (London) 400, 173–177
27. Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., Kholodenko, D., Lee, M., Lieberburg, I., Motter, R., Nguyen, M., Soriano, F., Vasquez, N., Weiss, K., Welch, B., Seubert, P., Schenk, D., and Yednock, T. (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer’s disease. Nat. Med. 6, 916–919
28. Chen, G., Chen, K. S., Knox, J., Inglis, J., Bernard, A., Martin, S. J., Justice, A., McConlogue, L., Games, D., Freedman, S. B., and Morris, R. G. M. (2000) A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature (London) 408, 975–979
29. Janus, C., Pearson, J., McLaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., Chishti, M. A., Horne, P., Heslin, D., French, J., Mount, H. T., Nixon, R. A., Mercken, M., Bergeron, C., Fraser, P. E., ST George-Hyslop, P., and Westaway, D. (2000) Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature (London) 408, 979–982
30. Morgan, D., Diamond, D. M., Gottschall, P. E., Ugen, K. E., Dickey, C., Hardy, J., Duff, K., Jantzen, P., Dicarlo, G., Wilcock, D., Connor, K., Hatcher, J., Hope, C., Gordon, M., and Arendash, G. W. (2000) Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature (London) 408, 982–985
31. Martins, V. R., Graner, E., Garcia-Abreu, J., de Souza, S. J., Mercadante, A. F., Veiga, S. S., Zanata, S. M., Neto, V. M., and Brentani, R. R. (1997) Complementary hydropathy identifies a cellular prion protein receptor. Nat. Med. 3, 1376–1382
32. Garcia-Abreu, J., Moura Neto, V., Carvalho, S.L., and Cavalcante, L.A. (1995) Regionally specific properties of midbrain glia: I. Interactions with midbrain neurons. J. Neurosci. Res. 40, 471–477
33. Selkoe, D. J. (1999) Translating cell biology into therapeutic advances in Alzheimer's disease. Nature (London) 399(Suppl), A23–31
34. Jarrett, J.T., Berger, E.P., and Lansbury, P.T., Jr. (1993) The C-terminus of the beta protein is critical in amyloidogenesis. Ann. N. Y. Acad. Sci. 695, 144–148
35. Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., Multhaup, G., Beyreuther, K., and Muller-Hill, B. (1987) The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature (London) 325, 733–736
36. Halverson, K., Fraser, P.E., Kirschner, D.A., and Lansbury, P.T., Jr. (1990) Molecular determinants of amyloid deposition in Alzheimer's disease: conformational studies of synthetic beta-protein fragments. Biochemistry 29, 2639–2644
37. Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C.L., and Beyreuther, K. (1991) Aggregation and secondary structure of synthetic amyloid beta A4 peptides of Alzheimer's disease. J. Mol. Biol. 218,149–163
38. Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C.L., and Beyreuther, K. (1992) Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer's disease beta A4 peptides. J. Mol. Biol. 228, 460–473
39. Snyder, S. W., Ladror, U. S., Wade, W. S., Wang, G. T., Barrett, L. W., Matayoshi, E. D., Huffaker, H. J., Krafft, G. A., and Holzman, T. F. (1994) Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys. J. 67, 1216–1228
40. The Merck Index, Eleventh Edition; 1989. Merck and Co., Inc.: Rahway, N.J.
41. MacBryde, C.M., and Taussig, B.L. (1935) Functional changes in liver, heart and muscles, and loss of dextrose tolerance resulting from dinitrophenol. JAMA 105, 13–17
42. Simon, E.W. (1953) Mechanisms of dinitrophenol toxicity. Biol. Rev. 28, 453–479
43. Klabunde, T., Petrassi, H. M., Oza, V. B., Raman, P., Kelly, J. W., and Sacchettini, J. C. (2000) Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 7, 312–321
Received October 23, 2000; revised January 8, 2001.

Fig. 1
Figure 1. Amyloid stability and disaggregation by nitrophenols. (a) Disaggregation of Aβ (10 µM) by GdnHCl; samples were incubated 1 h in the presence of the indicated concentrations of GdnHCl before measurements. Different symbols correspond to Aβ1-28 (�), Aβ1-42 (O) and Aβ1-43 (∆), and represent averages ± SD from 3–5 determinations. (b) Cold disaggregation of Aβ1-42. The sample was initially aggregated at 25oC and was cooled progressively to the indicated temperatures (circles). Light-scattering intensities were acquired after 20 min equilibration at each temperature. Triangles represent scattering intensities measured on reheating the sample to room temperature and indicate the reversibility of the cold-disaggregation process. (c) Disaggregation of Aβ1-42 by nitrophenols. NP (O) or DNP (�) were added to previously aggregated Aβ samples. Light-scattering intensities were measured 1.5 h after addition of the drugs. Symbols represent averages ± SD from three experiments.

Fig. 2
Figure 2. Inhibition of Aββββ1-42 aggregation by nitrophenols. (A) Control amyloid fibrils (9–10 nm diameter) obtained after aggregation of Aβ1-42 for 2 days in PBS. Magnification: 78,750×. (B) Aβ1-42 Sample incubated for 2 days in the presence of 100 µM NP. Inspection of a large number of EM fields failed to reveal fibrillar aggregates, with only occasional amorphous deposits present as shown in the micrograph. Magnification: 52,500×. (C) Aβ1-42 Sample incubated for 2 days in the presence of 20 µM DNP. Magnification: 52,500×.
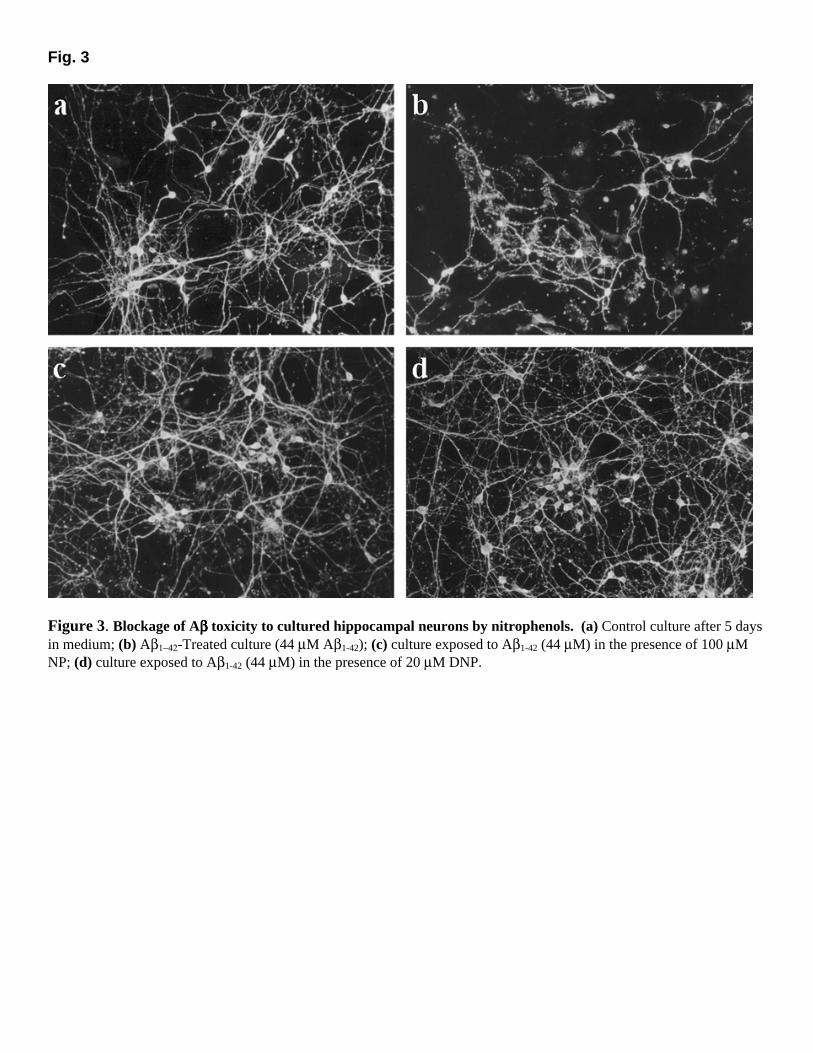
Fig. 3
Figure 3. Blockage of Aββββ toxicity to cultured hippocampal neurons by nitrophenols. (a) Control culture after 5 days in medium; (b) Aβ1−42-Treated culture (44 µM Aβ1-42); (c) culture exposed to Aβ1-42 (44 µM) in the presence of 100 µM NP; (d) culture exposed to Aβ1-42 (44 µM) in the presence of 20 µM DNP.
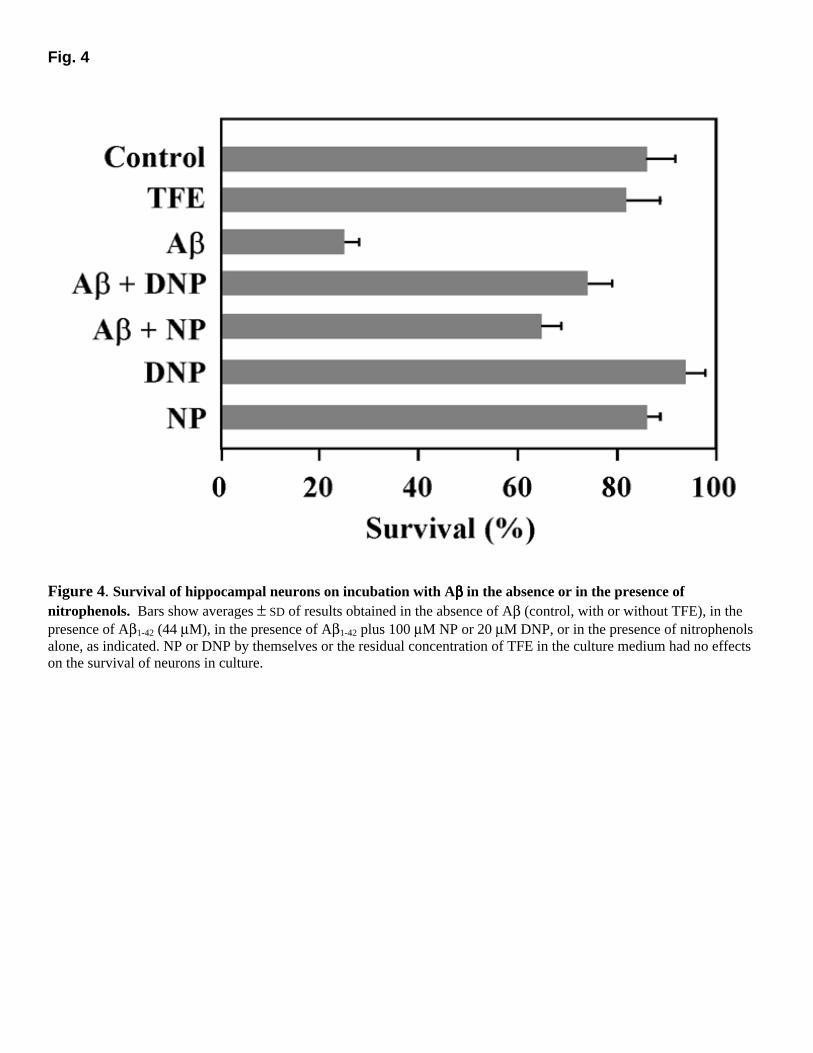
Fig. 4
Figure 4. Survival of hippocampal neurons on incubation with Aββββ in the absence or in the presence of nitrophenols. Bars show averages ± SD of results obtained in the absence of Aβ (control, with or without TFE), in the presence of Aβ1-42 (44 µM), in the presence of Aβ1-42 plus 100 µM NP or 20 µM DNP, or in the presence of nitrophenols alone, as indicated. NP or DNP by themselves or the residual concentration of TFE in the culture medium had no effects on the survival of neurons in culture.

Fig. 5
Figure 5. Reduction of cerebral Aββββ deposition and prevention of amyloid fibril formation in vivo by DNP. (a) Schematic diagram of the injection protocol and representative hippocampal sections visualized by thioflavine-S staining or autofluorescence, as indicated. (b–e) Quantitative analysis of the area of amyloid deposits in consecutive rat hippocampal sections (see Materials and Methods). Blue bars correspond to injection of Aβ alone (left hippocampus), and red bars correspond to coinjection of Aβ with DNP (right hippocampus). Dashed bars correspond to interpolated values. Different panels represent results obtained with different animals.
Related Documents











