REVIEW ARTICLE Inherited neuromuscular disorders: Pathway to diagnosisManoj P Menezes 1 and Kathryn N North 1,2 1 Institute for Neuroscience and Muscle Research, The Children’s Hospital at Westmead, Sydney and 2 Discipline of Paediatrics and Child Health, Faculty of Medicine, University of Sydney, New South Wales, Australia Abstract: Muscle weakness in childhood can be caused by a lesion at any point extending from the motor cortex, brainstem and spinal cord to the anterior horn cell, peripheral nerve, neuromuscular junction and muscle. A comprehensive history and physical examination is essential to aid classification of the neuromuscular disorder and direct gene testing. The more common disorders such as spinal muscular atrophy, Duchenne muscular dystrophy, myotonic dystrophy and facioscapulohumeral dystrophy may be diagnosed on direct gene testing based on the history and clinical examination. The congenital myopathies are classified based on structural abnormalities on muscle biopsy, while protein abnormalities on immunohistochemistry and immunoblotting aid classification of the muscular dystrophies. In this review, we provide an approach to diagnosis of a child with weakness, with a focus on the inherited neuromuscular disorders, and the features on history, examination and investigation that help to distinguish between them. Key words: child; Duchenne; hypotonia; muscle weakness; muscular dystrophy; neuromuscular disease. Muscle weakness in childhood can be caused by a lesion at any point extending from the motor cortex, brainstem and spinal cord to the anterior horn cell, peripheral nerve, neuromuscular junction and muscle. The clinical presentation can occur at any age; as a ‘floppy infant’, with delayed motor milestones in the first 2 years of life, or in childhood with abnormal gait, difficulty with running and climbing stairs and frequent falls. There is a very wide range of possible causes, both inherited and acquired. Acquired disorders can often be distinguished by acute or sub- acute onset in a child with normal developmental milestones and a history or associated features consistent with infectious, autoimmune or vascular pathology. In this review, we provide an approach to diagnosis of a child with weakness, with a focus on the inherited neuromuscular disorders, and the features on history, examination and investigation that help to distinguish between them. Approach to Diagnosis of the Child with Weakness Clinical history and physical examination The clinical history (Table 1) helps identify the age of onset, distribution and progress of weakness, and mode of inheritance. Key Points 1 A comprehensive history and physical examination is essential to aid classification of the neuromuscular disorder and direct gene testing. 2 Most cases of spinal muscular atrophy, Duchenne muscular dystrophy, myotonic dystrophy and facioscapulohumeral dys- trophy are diagnosed on gene testing based on a suggestive clinical phenotype without the need for invasive tests. 3 The congenital myopathies are classified based on structural abnormalities on muscle biopsy, while protein abnormalities on immunohistochemistry and immunoblotting aid classifica- tion of the muscular dystrophies. Correspondence: Professor Kathryn North, Institute for Neuroscience and Muscle Research, The Children’s Hospital at Westmead, Sydney, NSW 2145, Australia. Fax: +61 2 9845 3389; email: [email protected] Accepted for publication 29 May 2011. Table 1 Salient features in the history • Pregnancy – Polyhydramnios, intrauterine movements, intrauterine lie • Birth – Breech presentation, congenital hip dislocation, contractures, absent flexion creases – Medications given to mother (magnesium sulphate, opioids, anaesthetic agents) • Neonatal period and infancy – Respiratory distress – Hypotonia and floppiness – Sucking and swallowing difficulties • Distribution of the weakness – Proximal, distal or global – Facial or bulbar weakness – Ophthalmoplegia • Family history – Consanguinity – Family history of a neuromuscular disorder and pedigree doi:10.1111/j.1440-1754.2011.02210.x Journal of Paediatrics and Child Health 48 (2012) 458–465 © 2011 The Authors Journal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians) 458

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

REVIEW ARTICLE
Inherited neuromuscular disorders: Pathway to diagnosisjpc_2210 458..465
Manoj P Menezes1 and Kathryn N North1,2
1Institute for Neuroscience and Muscle Research, The Children’s Hospital at Westmead, Sydney and 2Discipline of Paediatrics and Child Health, Faculty of
Medicine, University of Sydney, New South Wales, Australia
Abstract: Muscle weakness in childhood can be caused by a lesion at any point extending from the motor cortex, brainstem and spinal cordto the anterior horn cell, peripheral nerve, neuromuscular junction and muscle. A comprehensive history and physical examination is essentialto aid classification of the neuromuscular disorder and direct gene testing. The more common disorders such as spinal muscular atrophy,Duchenne muscular dystrophy, myotonic dystrophy and facioscapulohumeral dystrophy may be diagnosed on direct gene testing based on thehistory and clinical examination. The congenital myopathies are classified based on structural abnormalities on muscle biopsy, while proteinabnormalities on immunohistochemistry and immunoblotting aid classification of the muscular dystrophies. In this review, we provide anapproach to diagnosis of a child with weakness, with a focus on the inherited neuromuscular disorders, and the features on history, examinationand investigation that help to distinguish between them.
Key words: child; Duchenne; hypotonia; muscle weakness; muscular dystrophy; neuromuscular disease.
Muscle weakness in childhood can be caused by a lesion at anypoint extending from the motor cortex, brainstem and spinalcord to the anterior horn cell, peripheral nerve, neuromuscularjunction and muscle. The clinical presentation can occur at anyage; as a ‘floppy infant’, with delayed motor milestones in thefirst 2 years of life, or in childhood with abnormal gait, difficultywith running and climbing stairs and frequent falls. There is avery wide range of possible causes, both inherited and acquired.Acquired disorders can often be distinguished by acute or sub-acute onset in a child with normal developmental milestonesand a history or associated features consistent with infectious,autoimmune or vascular pathology. In this review, we providean approach to diagnosis of a child with weakness, with a focuson the inherited neuromuscular disorders, and the features on
history, examination and investigation that help to distinguishbetween them.
Approach to Diagnosis of the Childwith Weakness
Clinical history and physical examination
The clinical history (Table 1) helps identify the age of onset,distribution and progress of weakness, and mode of inheritance.
Key Points
1 A comprehensive history and physical examination is essentialto aid classification of the neuromuscular disorder and directgene testing.
2 Most cases of spinal muscular atrophy, Duchenne musculardystrophy, myotonic dystrophy and facioscapulohumeral dys-trophy are diagnosed on gene testing based on a suggestiveclinical phenotype without the need for invasive tests.
3 The congenital myopathies are classified based on structuralabnormalities on muscle biopsy, while protein abnormalitieson immunohistochemistry and immunoblotting aid classifica-tion of the muscular dystrophies.
Correspondence: Professor Kathryn North, Institute for Neuroscience andMuscle Research, The Children’s Hospital at Westmead, Sydney, NSW 2145,Australia. Fax: +61 2 9845 3389; email: [email protected]
Accepted for publication 29 May 2011.
Table 1 Salient features in the history
• Pregnancy
– Polyhydramnios, intrauterine movements, intrauterine lie
• Birth
– Breech presentation, congenital hip dislocation, contractures,
absent flexion creases
– Medications given to mother (magnesium sulphate, opioids,
anaesthetic agents)
• Neonatal period and infancy
– Respiratory distress
– Hypotonia and floppiness
– Sucking and swallowing difficulties
• Distribution of the weakness
– Proximal, distal or global
– Facial or bulbar weakness
– Ophthalmoplegia
• Family history
– Consanguinity
– Family history of a neuromuscular disorder and pedigree
doi:10.1111/j.1440-1754.2011.02210.x
bs_bs_banner
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The Authors
Journal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
458
Decreased fetal movements in utero may indicate a prenatalonset of weakness and a significantly affected fetus with lack ofantigravity movement due to a neuromuscular disorder.1
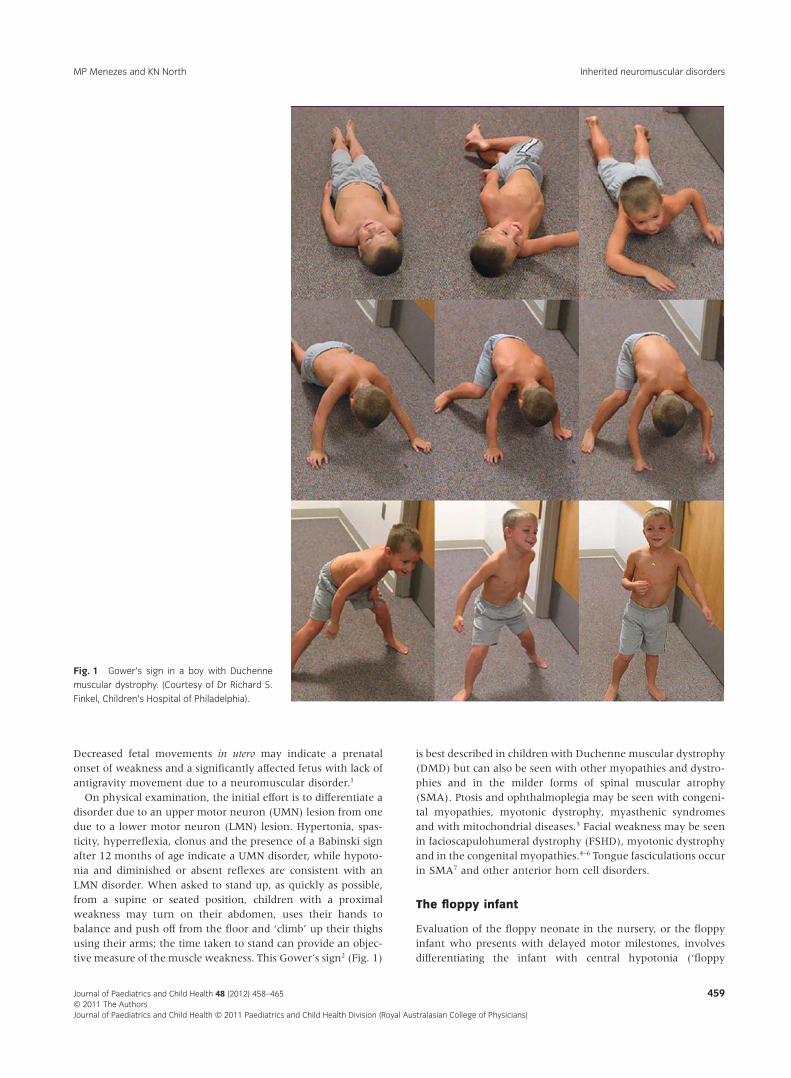
On physical examination, the initial effort is to differentiate adisorder due to an upper motor neuron (UMN) lesion from onedue to a lower motor neuron (LMN) lesion. Hypertonia, spas-ticity, hyperreflexia, clonus and the presence of a Babinski signafter 12 months of age indicate a UMN disorder, while hypoto-nia and diminished or absent reflexes are consistent with anLMN disorder. When asked to stand up, as quickly as possible,from a supine or seated position, children with a proximalweakness may turn on their abdomen, uses their hands tobalance and push off from the floor and ‘climb’ up their thighsusing their arms; the time taken to stand can provide an objec-tive measure of the muscle weakness. This Gower’s sign2 (Fig. 1)
is best described in children with Duchenne muscular dystrophy(DMD) but can also be seen with other myopathies and dystro-phies and in the milder forms of spinal muscular atrophy(SMA). Ptosis and ophthalmoplegia may be seen with congeni-tal myopathies, myotonic dystrophy, myasthenic syndromesand with mitochondrial diseases.3 Facial weakness may be seenin facioscapulohumeral dystrophy (FSHD), myotonic dystrophyand in the congenital myopathies.4–6 Tongue fasciculations occurin SMA7 and other anterior horn cell disorders.
The floppy infant
Evaluation of the floppy neonate in the nursery, or the floppyinfant who presents with delayed motor milestones, involvesdifferentiating the infant with central hypotonia (‘floppy
Fig. 1 Gower’s sign in a boy with Duchenne
muscular dystrophy. (Courtesy of Dr Richard S.
Finkel, Children’s Hospital of Philadelphia).
MP Menezes and KN North Inherited neuromuscular disorders
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The AuthorsJournal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
459

strong’) from one with a disorder of the LMN (‘floppy weak’). Aguide to the physical examination of a floppy infant is listed inTable 2. A floppy infant will have prominent head lag on the‘pull to sit’ manoeuvre, slip through on shoulder suspension,have an abnormal ‘scarf sign’ with the elbow crossing themidline when the hand is pulled towards the contralateralshoulder and be unable to maintain the head in the horizontalposition on ventral suspension.8 If the deep tendon reflexes areeasily elicited, then a primary neuromuscular disorder is lesslikely.
The most common cause of the floppy infant is birthasphyxia, and infants with weakness due to a primary neuro-muscular disorder are more susceptible to difficulties at deliv-ery and may present with a combination of hypoxic ischaemicencephalopathy and peripheral weakness. The list of causesfor central hypotonia8 (syndromes like Down syndrome andPrader–Willi syndrome, structural brain abnormalities, delayedand abnormal myelination and spinal cord disorders) is longand varied and beyond the scope of this review. Initial evalu-ation for presumed central hypotonia usually includes thyroid
function tests, urine metabolic screen, serum lactate, cranialand spinal imaging, karyotype and a comparative genomichybridisation array.
The floppy infant with a primary neuromuscular disordertypically presents as an alert baby who has paucity of move-ment, particularly proximal antigravity movement, and dimin-ished or absent reflexes. The floppy weak infant, and the weakolder child, may have a lesion at the anterior horn cell, periph-eral nerve, neuromuscular junction or muscle. The commondiseases associated with each of the lesions are listed in Table 3.
Investigations
The challenge for the clinician is first to determine the level ofthe lesion and then to proceed to definitive genetic diagnosis.This is usually achieved by a combination of clinical assess-ment and relevant investigations. The serum creatine kinase(CK) may be normal to moderately raised (5¥ normal) in SMAand the congenital myopathies, with a more significant rise(often greater than 10¥ normal) in the muscular dystrophies.9
Electromyography (EMG) and nerve conduction studies helpto differentiate neurogenic from myopathic weakness and helpclassify the neuropathy as demyelinating, axonal or interme-diate and as sensorimotor or primarily motor or sensory neu-ropathy.10 The presence of a decrement on repetitive nervestimulation at low rates (2–5 Hz) is useful in identifying thosewith congenital or acquired myasthenia. Prolonged runs ofmotor unit potentials with a waxing and waning frequencyand amplitude, described as resembling the sound of amotorcycle or chainsaw, may be heard on insertion ofthe needle electrode on EMG in older patients with myotonicdystrophy.11
The muscle biopsy (Fig. 2) may show evidence of chronicdenervation, dystrophic change or structural abnormalities. Thepresence of small angular fibres, fibre-type grouping and type 2fibre atrophy indicates chronic denervation as seen in SMA andCharcot-Marie-Tooth disease (CMT).12,13 In the congenital andlimb-girdle dystrophies, the muscle biopsy is characterised bydiffuse variation in fibre size, necrotic and regenerating fibresand fibrosis.14 The nerve biopsy sections and teased fibre prepa-rations are useful in the evaluation of neuropathies that remain
Table 2 Physical examination in an infant with hypotonia
• Posture and activity at rest
– ‘Frog-leg posture’ versus flexed posture
– Presence of spontaneous and antigravity movement
– Paradoxical respiration
– Deep tendon reflexes
• Posture and activity with specific manoeuvres
– Ventral suspension
– Traction on hands in supine position –‘pull to sit’
– Traction response of flexor muscles of arm and leg
– ‘Scarf sign’
– Posture of a passively elevated limb
– Response to noxious stimuli
• Assessment of cranial nerve function
– Ophthalmoplegia, facial or bulbar weakness
– Fasciculation and atrophy of the tongue
• Other
– Organomegaly (e.g. Pompe disease)
– Bone or joint abnormalities (suggest in utero onset)
• Examination of the parents and siblings
– Facial muscle weakness
– Myotonia
– Dysmorphic features
• Features suggesting a central hypotonia
– Impaired alertness, seizures
– Poor feeding and sucking
– Microcephaly/macrocephaly
– Dysmorphism
– Nystagmus and retinal abnormalities
– Hypotonia with normal strength
– Reflexes normal or brisk
• Features suggesting a lower motor unit disorder
– Alert baby who is ‘not moving’
– Weakness with lack of antigravity movements
– Diminished or absent reflexes
Table 3 Inherited disorders of the lower motor neuron
Level of lesion Common disorders
Anterior horn cell Spinal muscular atrophy
Peripheral nerve Charcot-Marie-Tooth disease
Neuromuscular junction ‘Transient’ neonatal myasthenia
Congenital myasthenic syndromes
Juvenile myasthenia gravis
Muscle Duchenne and Becker muscular dystrophy
Other limb-girdle dystrophies
Congenital myopathies
Congenital muscular dystrophies
Myotonic dystrophy
Inherited neuromuscular disorders MP Menezes and KN North
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The Authors
Journal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
460
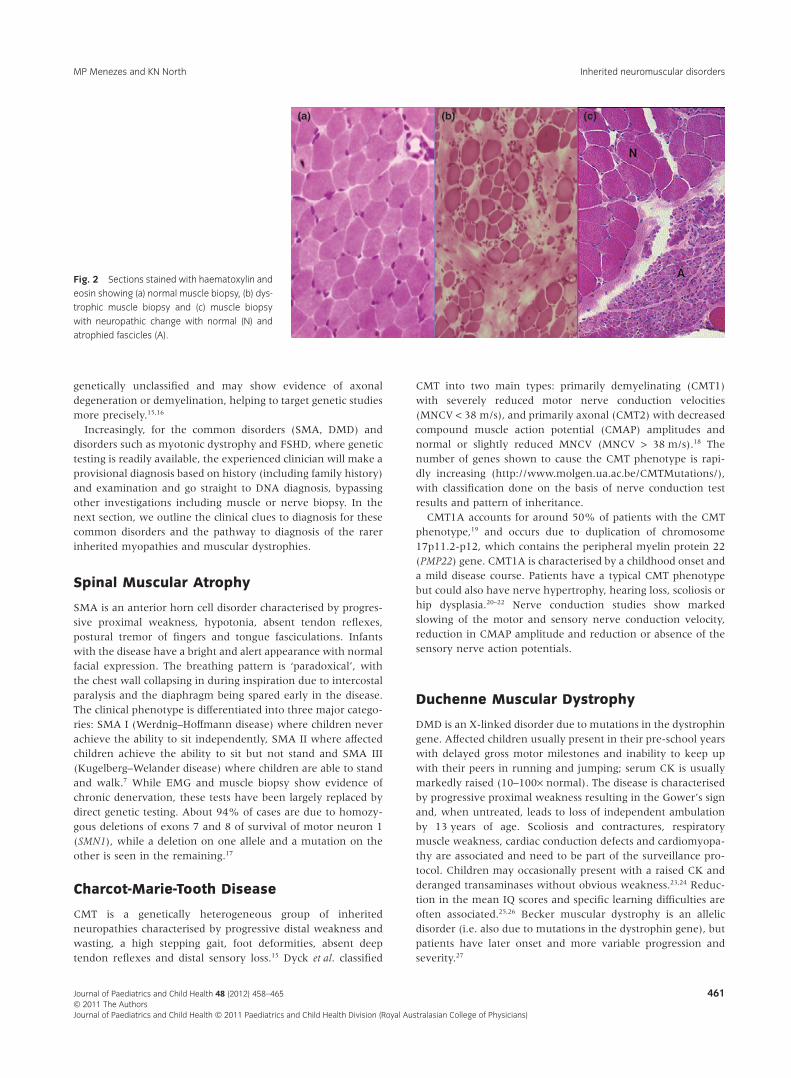
genetically unclassified and may show evidence of axonaldegeneration or demyelination, helping to target genetic studiesmore precisely.15,16
Increasingly, for the common disorders (SMA, DMD) anddisorders such as myotonic dystrophy and FSHD, where genetictesting is readily available, the experienced clinician will make aprovisional diagnosis based on history (including family history)and examination and go straight to DNA diagnosis, bypassingother investigations including muscle or nerve biopsy. In thenext section, we outline the clinical clues to diagnosis for thesecommon disorders and the pathway to diagnosis of the rarerinherited myopathies and muscular dystrophies.
Spinal Muscular Atrophy
SMA is an anterior horn cell disorder characterised by progres-sive proximal weakness, hypotonia, absent tendon reflexes,postural tremor of fingers and tongue fasciculations. Infantswith the disease have a bright and alert appearance with normalfacial expression. The breathing pattern is ‘paradoxical’, withthe chest wall collapsing in during inspiration due to intercostalparalysis and the diaphragm being spared early in the disease.The clinical phenotype is differentiated into three major catego-ries: SMA I (Werdnig–Hoffmann disease) where children neverachieve the ability to sit independently, SMA II where affectedchildren achieve the ability to sit but not stand and SMA III(Kugelberg–Welander disease) where children are able to standand walk.7 While EMG and muscle biopsy show evidence ofchronic denervation, these tests have been largely replaced bydirect genetic testing. About 94% of cases are due to homozy-gous deletions of exons 7 and 8 of survival of motor neuron 1(SMN1), while a deletion on one allele and a mutation on theother is seen in the remaining.17
Charcot-Marie-Tooth Disease
CMT is a genetically heterogeneous group of inheritedneuropathies characterised by progressive distal weakness andwasting, a high stepping gait, foot deformities, absent deeptendon reflexes and distal sensory loss.15 Dyck et al. classified
CMT into two main types: primarily demyelinating (CMT1)with severely reduced motor nerve conduction velocities(MNCV < 38 m/s), and primarily axonal (CMT2) with decreasedcompound muscle action potential (CMAP) amplitudes andnormal or slightly reduced MNCV (MNCV > 38 m/s).18 Thenumber of genes shown to cause the CMT phenotype is rapi-dly increasing (http://www.molgen.ua.ac.be/CMTMutations/),with classification done on the basis of nerve conduction testresults and pattern of inheritance.
CMT1A accounts for around 50% of patients with the CMTphenotype,19 and occurs due to duplication of chromosome17p11.2-p12, which contains the peripheral myelin protein 22(PMP22) gene. CMT1A is characterised by a childhood onset anda mild disease course. Patients have a typical CMT phenotypebut could also have nerve hypertrophy, hearing loss, scoliosis orhip dysplasia.20–22 Nerve conduction studies show markedslowing of the motor and sensory nerve conduction velocity,reduction in CMAP amplitude and reduction or absence of thesensory nerve action potentials.
Duchenne Muscular Dystrophy
DMD is an X-linked disorder due to mutations in the dystrophingene. Affected children usually present in their pre-school yearswith delayed gross motor milestones and inability to keep upwith their peers in running and jumping; serum CK is usuallymarkedly raised (10–100¥ normal). The disease is characterisedby progressive proximal weakness resulting in the Gower’s signand, when untreated, leads to loss of independent ambulationby 13 years of age. Scoliosis and contractures, respiratorymuscle weakness, cardiac conduction defects and cardiomyopa-thy are associated and need to be part of the surveillance pro-tocol. Children may occasionally present with a raised CK andderanged transaminases without obvious weakness.23,24 Reduc-tion in the mean IQ scores and specific learning difficulties areoften associated.25,26 Becker muscular dystrophy is an allelicdisorder (i.e. also due to mutations in the dystrophin gene), butpatients have later onset and more variable progression andseverity.27
(a) (b) (c)
Fig. 2 Sections stained with haematoxylin and
eosin showing (a) normal muscle biopsy, (b) dys-
trophic muscle biopsy and (c) muscle biopsy
with neuropathic change with normal (N) and
atrophied fascicles (A).
MP Menezes and KN North Inherited neuromuscular disorders
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The AuthorsJournal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
461

Diagnosis is confirmed by identifying a deletion or duplicationon multiplex polymerase chain reaction or multiplex ligation-dependent probe amplification. If this is negative, sequencingof the dystrophin gene may identify point mutations or smalldeletion/insertions. A muscle biopsy is required if genetic testingis negative despite a suggestive phenotype and may showabsence or reduction in dystrophin on immunohistochemistryor immunoblotting23 (Fig. 3). Consensus recommendations fordiagnosis and management have recently been published.23,28
Other Forms of Muscular Dystrophy
There are numerous other genetic forms of muscular dystrophy,with X-linked, autosomal dominant and autosomal recessiveinheritance. They are classified according to the age of presen-tation and pattern of weakness and have in common histologi-cal evidence of degeneration and regeneration on muscle biopsyassociated with weakness and a raised serum CK. When chil-dren present at birth with hypotonia and weakness with adystrophic picture on muscle biopsy, they are classified ashaving a congenital muscular dystrophy. Common geneticcauses include mutations in the genes encoding the extracellu-lar matrix proteins collagen VI, merosin (laminin-a2) and pro-teins that glycosylate a-dystroglycan. Childhood- or adult-onsetmuscular dystrophies are classified as a limb-girdle musculardystrophy (LGMD). DMD can be considered as a severe child-hood onset form of X-linked LGMD. Common genetic causesof other LGMDs include mutations in the genes encodingdystrophin-associated proteins at the muscle membrane such asthe sarcoglycans, dysferlin, caveolin-3 and calpain.9
Specific protein analysis of the patient muscle biopsy byimmunohistochemistry and western blot plays an important
role in distinguishing between the different forms of musculardystrophy and in directing genetic testing. In the X-linked andautosomal recessive forms of muscular dystrophy, an absence orreduction of the disease-associated protein may be seen onimmunohistochemistry, while reduction or abnormal proteinmay be identified on western blot.14 In autosomal dominantdisorders, protein studies are usually less helpful since expres-sion of protein from the normal copy of the gene may concealthe effects of the mutant copy.
Occasionally, clinical findings may help to direct genesequencing in combination with protein findings on musclebiopsy. For example, calf hypertrophy is seen with Duchenneand Becker muscular dystrophy, sarcoglycanopathies, calpain-opathy and the a-dystroglycanopathies, while selective calfatrophy is seen with LGMD due to mutations in the gene encod-ing dysferlin.9 The phenotype with collagen VI-deficient myopa-thy includes distal laxity and hyperextensibility, long fingerflexion contractures, follicular hyperkeratosis on the extensorsurfaces of the limbs and prominent calcanei.29 An extensive listof the monogenic neuromuscular disorders and their causativemutation is available at http://www.musclegenetable.org
Congenital Myopathies
The congenital myopathies usually present at birth or childhoodwith hypotonia and generalised weakness and a static or slowlyprogressive course, and are classified into subgroups based onthe presence of distinct structural abnormalities in the musclebiopsy. These include distinctive protein accumulations in theform of rods or nemaline bodies (nemaline myopathy), ‘cores’devoid of oxidative activity (central core and multi-minicoredisease), centrally, rather than peripherally, placed nuclei
(a) (b)
Fig. 3 Absence of dystrophin in a patient (P)
with Duchenne muscular dystrophy, when com-
pared with control (C), on immunohistochemis-
try (Panel a) and western blot (Panel b).
Inherited neuromuscular disorders MP Menezes and KN North
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The Authors
Journal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
462

(centronuclear myopathies) and selective atrophy of type 1(slow) fibres (congenital fibre-type disproportion).30,31 Genesthat cause congenital myopathies often encode protein compo-nents of the muscle contractile apparatus (the sarcomere) andproteins involved in Ca2+ handling,32 resulting in inefficientmuscle contraction. CK is usually normal or only mildlyelevated. The presence of ophthalmoplegia, facial and bulbarinvolvement may help differentiate congenital myopathies fromcongenital muscular dystrophies, anterior horn cell disordersand early-onset peripheral neuropathies.32
Other Inherited DisordersInvolving Muscle
Myotonic dystrophy is an autosomal dominant disorder charac-terised by hypotonia, weakness that may be proximally or dis-tally predominant, bilateral facial weakness, learning difficultiesand myotonia on clinical examination and EMG. There are twogenetic subtypes: myotonic dystrophy type 1 (DM1) is caused byan expanded CTG repeat in the dystrophia myotonica-proteinkinase gene,33 while type 2 (DM2) is caused by a CCTG expan-sion in the zinc finger protein 9 gene.34 There is a wide range ofclinical severity and age on onset that roughly correlates withthe size of the repeat.
Clinical variability between parent and offspring and amongfamily members may be due to an increase in the size of theexpansion; this most often occurs when the mutant gene isinherited from an affected mother. Congenital myotonic dystro-phy only occurs in DM1 and is associated with maternal trans-mission of an expanded trinucleotide repeat (usually with>1500 repeats). Patients present in the newborn period withhypotonia, talipes, bilateral facial weakness with a tented upperlip, open mouth and high arched palate. There may be a historyof prematurity, reduced fetal movements and polyhydramnios.A weak cry and poor suck are often present, and respiratoryinsufficiency may be prominent and require mechanical venti-lation. Reflexes are absent. The clinical course is characterisedby motor and speech delay and a variable degree of learningdifficulty. Neither clinical nor electrical myotonia is common innewborns and infants with myotonic dystrophy, and clinical andEMG analysis of the mother may be more useful. Cognitivedifficulties are the presenting feature in those with a childhoodonset in whom distal limb weakness, facial weakness and clini-cal myotonia may be present, but respiratory difficulties arenot prominent.5 Cardiac arrhythmias and cardiomyopathymay occur in the second decade in both the congenital andchildhood forms.35,36 DM2 differs from DM1 in having proximalweakness and absence of a congenital onset.
FSHD has a characteristic distribution of muscle involvementthat often leads to targeted genetic testing without the need fora muscle biopsy. Clinical presentation is mostly in the seconddecade, with facial weakness, weakness of the scapular fixatorsleading to scapular winging and biceps and triceps wasting withsparing of the deltoid. Muscle involvement is typically asym-metric and progresses to involve the trunk muscles and then thehip girdle. High-frequency hearing loss and retinal telangiectasiaare common, and cardiac arrhythmias are rarely associated.6 Inthe majority of patients with typical FSHD (>90%), there is a
detectable deletion involving the subtelomeric region of chro-mosome 4q35.37 The specific genes involved have not yet beendetermined.
Newer Diagnostic Modalities inNeuromuscular Disorders
Two exciting new modalities, microarray or ‘chip’ technologyand exome sequencing, promise to greatly improve the rate ofpositive gene diagnosis in neuromuscular disorders. Microarraytechnology involves the use of new generation sequencers tosequence for all known myopathy/muscular dystrophy/neuropathy genes in a single reaction, significantly reducingtime and cost.9 While this technology is useful for identifyingmutations in known genes, targeted sequencing of all protein-coding regions (‘exomes’) holds promise for identifying yetunknown genes that cause muscle diseases, especially in smallkindreds or among sporadic unrelated individuals.38 False-positive and false-negative results remain critical issues withthese emerging technologies, increasing the importance of accu-rately establishing the phenotype through history and clinicalexamination.
Multiple Choice Questions
1. A 4-year-old boy is seen in the outpatients department afterhaving been noticed in pre-school having difficulty in gettingoff the floor and falling more frequently than his peers. Onexamination, he has prominent calves, a Gower’s sign whilerising from the floor, normal facial strength and extra-ocularmovements and a persistently raised creatine kinase (CK)>13 000 U/L. There is no family history of a neuromusculardisorder.The most appropriate next diagnostic test would be:A Nerve conduction tests/electromyogram (EMG).B Deletions/duplication analysis of the dystrophin gene.C Testing for size of CTG repeats in DMPK gene.D Targeted mutational analysis of the SMN1 gene.E Muscle biopsy.
2. An 8-month-old girl presents to the outpatients departmentwith delayed motor milestones, not having learned to roll orsit yet. On examination, she is not dysmorphic and has abright and alert appearance with normal facial expression.She is hypotonic with no proximal antigravity movementand absent reflexes in the upper and lower limbs. She has aparadoxical breathing pattern and tongue fasciculations. HerCK is 300 U/L (normal 0–180 U/L).The test most likely to yield a positive diagnosis is:A Magnetic resonance imaging (MRI) of the brain.B Karyotype.C FISH for Prader–Willi syndrome.D Testing for size of CTG repeats in DMPK gene.E Targeted mutational analysis of the SMN1 gene.
3. A child with an early onset proximal weakness, with oph-thalmoplegia, facial weakness and a static course with anormal CK would be classified as having a:A Congenital myopathy.B Congenital muscular dystrophy.C Limb-girdle muscular dystrophy.
MP Menezes and KN North Inherited neuromuscular disorders
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The AuthorsJournal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
463

D Peripheral neuropathy.E Anterior horn cell disorder.
Answers1 B The presentation with proximal weakness in childhood,with a significantly raised CK is consistent with a limb-girdlemuscular dystrophy, and Duchenne muscular dystrophy (DMD)would be the most common cause in a male child. Calf hyper-trophy is often seen in DMD. A family history would not bepresent in the case of a de novo mutation. A multiplex ligation-dependent probe amplification (MLPA) or multiplex polymerasechain reaction would identify deletions and duplications,present in approximately 70% of cases of DMD, and sequencingof the dystrophin gene identifies small deletions/insertions/mutations in the remaining.A The significantly raised CK and proximal weakness would
suggest a muscular dystrophy. Nerve conduction tests wouldbe normal and EMG may show only non-specific myopathicchanges.
C Children with a childhood onset of myotonic dystro-phy (testing for size of CTG repeats in DMPK gene) usuallypresent with cognitive difficulties and have facial and distallimb weakness. CK is normal or only mildly (¥5 normal)raised.
D While the child with SMA3 (targeted mutational analysis of theSMN1 gene) may present with a proximal weakness andGower’s sign, calf hypertrophy is not seen and the CK is onlymildy (¥5 normal) raised.
E A muscle biopsy would be invasive, but may need to beperformed if MLPA and sequencing of the dystrophin genedo not show a mutation.
2 E The hypotonia, weakness and areflexia would place thischild into the ‘floppy weak’ category. The proximal weakness,areflexia and tongue fasciculations would be consistent with ananterior horn cell disorder, of which spinal muscular atrophy(targeted mutational analysis of the SMN1 gene) would be the mostcommon.A–C MRI Brain, karyotype and FISH for Prader–Willi syndromewould be more appropriate for a child with central hypotonia(floppy strong).D Generalised muscle weakness and facial weakness would bemore likely with congenital myotonic dystrophy (testing for size ofCTG repeats in DMPK gene).3 A The congenital myopathies usually present at birth orchildhood with hypotonia and weakness and a static or slowlyprogressive course. CK is usually normal or only mildlyelevated. The presence of ophthalmoplegia, facial and bulbarinvolvement may help differentiate congenital myopathies fromcongenital muscular dystrophies, anterior horn cell disordersand early onset peripheral neuropathies.B, C Muscular dystrophies are usually associated with a raisedserum creatine kinase. Ophthalmoplegia and facial weaknessare not associated. The course may be progressive with deterio-ration in strength or respiratory function.D A child with an inherited peripheral neuropathy wouldhave distal weakness and no ophthalmoplegia or facialweakness.E Anterior horn cell disorders, characterised by SMA, have aprogressive course and ophthalmoplegia and facial weakness arenot associated, while tongue fasciculations may be seen.
References
1 Vasta I, Kinali M, Messina S et al. Can clinical signs identify newbornswith neuromuscular disorders? J. Pediatr. 2005; 146: 73–9.
2 Tyler KL, McHenry LC Jr. Fragments of neurologic history:pseudohypertrophic muscular dystrophy and Gowers’ sign. Neurology1983; 33: 88–9.
3 Jones KJ, North KN. External ophthalmoplegia in neuromusculardisorders: case report and review of the literature. Neuromuscul.Disord. 1997; 7: 143–51.
4 North KN, Goebel HH. Congenital myopathies. In: Jones HR, De VivoDC, Darras BT, eds. Neuromuscular Disorders of Infancy, Childhood,and Adolescence: A Clinician’s Approach. Philadelphia: ButterworthHeinemann, 2003; 601–32.
5 Schara U, Schoser BG. Myotonic dystrophies type 1 and 2: a summaryon current aspects. Semin. Pediatr. Neurol. 2006; 13: 71–9.
6 Tawil R, Van Der Maarel SM. Facioscapulohumeral musculardystrophy. Muscle Nerve 2006; 34: 1–15.
7 Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008; 371:2120–33.
8 Bodensteiner JB. The evaluation of the hypotonic infant. Semin.Pediatr. Neurol. 2008; 15: 10–20.
9 Tesi-Rocha C, Hoffman E. Limb–girdle and congenital musculardystrophies: current diagnostics, management, and emergingtechnologies. Curr. Neurol. Neurosci. Rep. 2010; 10: 267–76.
10 Johnsen B, Fuglsang-Frederiksen A. Electrodiagnosis ofpolyneuropathy. Neurophysiol. Clin. 2000; 30: 339–51.
11 Kimura J. Types of electromyographic abnormalities. In: Kimura J, ed.Electrodiagnosis in Diseases of Nerve and Muscle: Principles andPractice, 3rd edn. New York: Oxford University Press, 2001;339–69.
12 Anthony DC, De Girolami U, Shapiro F. Muscle biopsy. In: Jones HR,De Vivo DC, Darras BT, eds. Neuromuscular Disorders of Infancy,Childhood, and Adolescence: A Clinician’s Approach. Philadelphia:Butterworth Heinemann, 2003; 75–90.
13 Mishra VN, Kalita J, Kesari A, Mittal B, Shankar SK, Misra UK. A clinicaland genetic study of spinal muscular atrophy. Electromyogr. Clin.Neurophysiol. 2004; 44: 307–12.
14 Sewry C. Muscular dystrophies: an update on pathology anddiagnosis. Acta Neuropathol. 2010; 120: 343–58.
15 Pareyson D, Marchesi C. Diagnosis, natural history, andmanagement of Charcot-Marie-Tooth disease. Lancet Neurol. 2009; 8:654–67.
16 Vallat JM, Sommer C, Magy L. Chronic inflammatory demyelinatingpolyradiculoneuropathy: diagnostic and therapeutic challenges for atreatable condition. Lancet Neurol. 2010; 9: 402–12.
17 Ogino S, Wilson RB. Genetic testing and risk assessment for spinalmuscular atrophy (SMA). Hum. Genet. 2002; 111: 477–500.
18 Dyck PJ, Chance P, Lebo R, Carney JA. Hereditary motor and sensoryneuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF,eds. Peripheral Neuropathy. Philadelphia: W.B. Saunders Company,1993; 1094–136.
19 Boerkoel CF, Takashima H, Lupski JR. The genetic convergence ofCharcot-Marie-Tooth disease types 1 and 2 and the role of genetics insporadic neuropathy. Curr. Neurol. Neurosci. Rep. 2002; 2: 70–7.
20 Shy ME, Lupski JR, Chance PF, Klein CJ, Dyck PJ. Hereditary Motor andSensory Neuropathies: an overview of clinical, genetic,electrophysiologic, and pathologic features. In: Dyck PJ, Thomas PK,eds. Peripheral Neuropathy. Philadelphia: Elsevier Saunders, 2005;1623–58.
21 Birouk N, Gouider R, Le Guern E et al. Charcot-Marie-Tooth diseasetype 1A with 17p11.2 duplication. Clinical and electrophysiologicalphenotype study and factors influencing disease severity in 119cases. Brain 1997; 120: 813–23.
Inherited neuromuscular disorders MP Menezes and KN North
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The Authors
Journal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
464

22 Bamford NS, White KK, Robinett SA, Otto RK, Gospe SM, Jr.Neuromuscular hip dysplasia in Charcot-Marie-Tooth disease type 1A.Dev. Med. Child Neurol. 2009; 51: 408–11.
23 Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management ofDuchenne muscular dystrophy, part 1: diagnosis, andpharmacological and psychosocial management. Lancet Neurol.2010; 9: 77–93.
24 Bushby KM, Hill A, Steele JG. Failure of early diagnosis in symptomaticDuchenne muscular dystrophy. Lancet 1999; 353: 557–8.
25 Cotton S, Voudouris NJ, Greenwood KM. Intelligence and Duchennemuscular dystrophy: full-scale, verbal, and performance intelligencequotients. Dev. Med. Child Neurol. 2001; 43: 497–501.
26 Hinton VJ, De Vivo DC, Nereo NE, Goldstein E, Stern Y. Poor verbalworking memory across intellectual level in boys with Duchennedystrophy. Neurology 2000; 54: 2127–32.
27 Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene,several proteins, multiple phenotypes. Lancet Neurol. 2003; 2:731–40.
28 Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management ofDuchenne muscular dystrophy, part 2: implementation ofmultidisciplinary care. Lancet Neurol. 2010; 9: 177–89.
29 Lampe AK, Bushby KMD. Collagen VI related muscle disorders. J. Med.Genet. 2005; 42: 673–85.
30 Laing NG. Congenital myopathies. Curr. Opin. Neurol. 2007; 20:583–9.
31 Sewry CA, Jimenez-Mallebrera C, Muntoni F. Congenital myopathies.Curr. Opin. Neurol. 2008; 21: 569–75.
32 North K. What’s new in congenital myopathies? Neuromuscul. Disord.2008; 18: 433–42.
33 Mahadevan M, Tsilfidis C, Sabourin L et al. Myotonic dystrophymutation: an unstable CTG repeat in the 3′ untranslated region of thegene. Science 1992; 255: 1253–5.
34 Liquori CL, Ricker K, Moseley ML et al. Myotonic dystrophy type 2caused by a CCTG expansion in intron 1 of ZNF9. Science 2001; 293:864–7.
35 Bhakta D, Lowe MR, Groh WJ. Prevalence of structural cardiacabnormalities in patients with myotonic dystrophy type I. Am. Heart J.2004; 147: 224–7.
36 Groh WJ, Groh MR, Saha C et al. Electrocardiographic abnormalitiesand sudden death in myotonic dystrophy type 1. N. Engl. J. Med.2008; 358: 2688–97.
37 Lemmers RJ, van der Vliet PJ, Klooster R et al. A unifying geneticmodel for facioscapulohumeral muscular dystrophy. Science 2010;329: 1650–3.
38 Ng SB, Turner EH, Robertson PD et al. Targeted capture and massivelyparallel sequencing of 12 human exomes. Nature 2009; 461: 272–6.
MP Menezes and KN North Inherited neuromuscular disorders
Journal of Paediatrics and Child Health 48 (2012) 458–465© 2011 The AuthorsJournal of Paediatrics and Child Health © 2011 Paediatrics and Child Health Division (Royal Australasian College of Physicians)
465
Related Documents











