724 • JID 2004:189 (15 February) • Goerke et al. MAJOR ARTICLE Increased Frequency of Genomic Alterations in Staphylococcus aureus during Chronic Infection Is in Part Due to Phage Mobilization Christiane Goerke, 1 Saskia Matias y Papenberg, 1 Simone Dasbach, 1 Klaus Dietz, 2 Rita Ziebach, 3 Barbara C. Kahl, 4 and Christiane Wolz 1 Instituts fu ¨r 1 Allgemeine Hygiene und Umwelthygiene and 2 Medizinische Biometrie, and 3 Universita ¨tsklinik fu ¨r Kinderheilkunde und Jugendmedizin, Universita ¨t Tu ¨bingen, Tu ¨bingen, and 4 Medizinische Mikrobiologie, Universita ¨t Mu ¨nster, Mu ¨nster, Germany We assessed the nature and frequency of genome alterations in Staphylococcus aureus during chronic lung infection in patients with cystic fibrosis (CF) and during colonization of the nares in healthy individuals. Only individuals harboring the same S. aureus clone on consecutive samplings were included in the present study. Clone definition was based on pulsed-field gel electrophoresis (PFGE) analysis. Minor fragment variations in consecutive clones were interpreted as genome alterations. The frequency of genome alterations was signifi- cantly higher in S. aureus derived from patients with CF (mean time, 1.03 years) than in isolates derived from healthy individuals (mean time, 13.4 years). In total, 19 S. aureus strain pairs showing genome alterations were available for molecular analysis to clarify the nature of recombinational events in the host environment. In 8 cases, genome alteration could be linked to phage mobilization. Phage conversion of b-toxin production was evident in 7 pairs. In 1 strain pair, changes in the PFGE pattern were accompanied by deletion of a phage similar to ETA. Obviously, phage mobilization plays an important role in vivo. During long-term lung infection in patients with CF, the specific host response and/or the regular exposure to antibiotics exercises strong selective pressure on the pathogen. Genome plasticity may facilitate the adaptation to various host conditions. Genetic variation is a requirement for the biological evolution of bacterial pathogens. The extent of variation is determined by the frequencies of mutation and re- combination within a given population. The resulting differences in gene content and allelic variations are pre- sumably advantageous for adaptation to various host conditions. The rate of recombination seems to be spe- cies specific, leading to considerable differences in the population structure of various pathogens [1, 2]. By use of multilocus sequence typing, an intermittent re- combination frequency was assessed for the human pathogen Staphylococcus aureus, compared with Esch- Received 3 June 2003; accepted 15 August 2003; electronically published 29 January 2004. Financial support: Deutsche Forschungsgemeinschaft (grants Wo 578/3-2 and Wo 578/3-3); Mukoviszidose e.V. Reprints or correspondence: Dr. Christiane Goerke, Institut fu ¨r Allgemeine Hygiene und Umwelthygiene, Universita ¨t Tu ¨bingen, Wilhelmstr. 31, 72074 Tu ¨bingen, Germany ([email protected]). The Journal of Infectious Diseases 2004; 189:724–34 2004 by the Infectious Diseases Society of America. All rights reserved. 0022-1899/2004/18904-0022$15.00 erichia coli with very low recombination frequencies representing one end of the scale and Neisseria men- ingitidis with very high frequencies representing the other end [3]. Microarray analysis revealed that genetic variation between S. aureus lineages is extensive, with 22% of the genome comprising strain-specific material [4]. Accordingly, publication of the genome sequence of 3 S. aureus strains revealed the presence of multiple mobile elements—such as bacteriophages, pathogenic- ity islands, and transposons [5, 6]. S. aureus asymptomatically colonizes the anterior nares of humans but also causes a wide spectrum of acute and chronic diseases. Evolution of the species was probably driven by adaptation to the environment of the nose, which is thought to be the primary reservoir for subsequent infection [7]. On the other hand, during the course of infection, the specific host response or subinhibitory antibiotic concentrations may exercise se- lective pressure, resulting in microevolutionary pro- cesses in the pathogen. In patients with cystic fibrosis (CF), S. aureus causes a chronic lung infection, which

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

724 • JID 2004:189 (15 February) • Goerke et al.
M A J O R A R T I C L E
Increased Frequency of Genomic Alterationsin Staphylococcus aureus during Chronic InfectionIs in Part Due to Phage Mobilization
Christiane Goerke,1 Saskia Matias y Papenberg,1 Simone Dasbach,1 Klaus Dietz,2 Rita Ziebach,3 Barbara C. Kahl,4
and Christiane Wolz1
Instituts fur 1Allgemeine Hygiene und Umwelthygiene and 2Medizinische Biometrie, and 3Universitatsklinik fur Kinderheilkunde und Jugendmedizin,Universitat Tubingen, Tubingen, and 4Medizinische Mikrobiologie, Universitat Munster, Munster, Germany
We assessed the nature and frequency of genome alterations in Staphylococcus aureus during chronic lunginfection in patients with cystic fibrosis (CF) and during colonization of the nares in healthy individuals. Onlyindividuals harboring the same S. aureus clone on consecutive samplings were included in the present study.Clone definition was based on pulsed-field gel electrophoresis (PFGE) analysis. Minor fragment variations inconsecutive clones were interpreted as genome alterations. The frequency of genome alterations was signifi-cantly higher in S. aureus derived from patients with CF (mean time, 1.03 years) than in isolates derived fromhealthy individuals (mean time, 13.4 years). In total, 19 S. aureus strain pairs showing genome alterationswere available for molecular analysis to clarify the nature of recombinational events in the host environment.In 8 cases, genome alteration could be linked to phage mobilization. Phage conversion of b-toxin productionwas evident in 7 pairs. In 1 strain pair, changes in the PFGE pattern were accompanied by deletion of a phagesimilar to ETA. Obviously, phage mobilization plays an important role in vivo. During long-term lung infectionin patients with CF, the specific host response and/or the regular exposure to antibiotics exercises strongselective pressure on the pathogen. Genome plasticity may facilitate the adaptation to various host conditions.
Genetic variation is a requirement for the biological
evolution of bacterial pathogens. The extent of variation
is determined by the frequencies of mutation and re-
combination within a given population. The resulting
differences in gene content and allelic variations are pre-
sumably advantageous for adaptation to various host
conditions. The rate of recombination seems to be spe-
cies specific, leading to considerable differences in the
population structure of various pathogens [1, 2]. By
use of multilocus sequence typing, an intermittent re-
combination frequency was assessed for the human
pathogen Staphylococcus aureus, compared with Esch-
Received 3 June 2003; accepted 15 August 2003; electronically published 29January 2004.
Financial support: Deutsche Forschungsgemeinschaft (grants Wo 578/3-2 and Wo578/3-3); Mukoviszidose e.V.
Reprints or correspondence: Dr. Christiane Goerke, Institut fur Allgemeine Hygieneund Umwelthygiene, Universitat Tubingen, Wilhelmstr. 31, 72074 Tubingen, Germany([email protected]).
The Journal of Infectious Diseases 2004; 189:724–34� 2004 by the Infectious Diseases Society of America. All rights reserved.0022-1899/2004/18904-0022$15.00
erichia coli with very low recombination frequencies
representing one end of the scale and Neisseria men-
ingitidis with very high frequencies representing the
other end [3]. Microarray analysis revealed that genetic
variation between S. aureus lineages is extensive, with
22% of the genome comprising strain-specific material
[4]. Accordingly, publication of the genome sequence
of 3 S. aureus strains revealed the presence of multiple
mobile elements—such as bacteriophages, pathogenic-
ity islands, and transposons [5, 6].
S. aureus asymptomatically colonizes the anterior
nares of humans but also causes a wide spectrum of
acute and chronic diseases. Evolution of the species was
probably driven by adaptation to the environment of
the nose, which is thought to be the primary reservoir
for subsequent infection [7]. On the other hand, during
the course of infection, the specific host response or
subinhibitory antibiotic concentrations may exercise se-
lective pressure, resulting in microevolutionary pro-
cesses in the pathogen. In patients with cystic fibrosis
(CF), S. aureus causes a chronic lung infection, which

Genomic Alterations in S. aureus • JID 2004:189 (15 February) • 725
is regularly treated with antibiotics. However, complete erad-
ication of the pathogen is not achieved. An extensive adaptation
to the CF lung has been described for Pseudomonas aeruginosa.
This adaptation is characterized by elevated mutation frequen-
cies [8] and genome alterations [9], resulting in a fixed CF
phenotype. Whether such extensive adaptations occur in S.
aureus during lung infection related to CF is unclear.
In the present study, we assessed the nature and frequency
of genome alterations during chronic lung infection in patients
with CF and during colonization of the nares in healthy in-
dividuals. Genome analysis by pulsed-field gel electrophoresis
(PFGE) enables us to register DNA rearrangements and the ac-
quisition or loss of genetic elements. It also allows high-resolution
discrimination of strains [10] and has been successfully used to
define clonal lineages within the S. aureus species [11–13].
We were able to show that the frequency of genome alter-
ations during lung infection in patients with CF was signifi-
cantly higher than that during colonization of the nares in
healthy individuals. In a subset of strains, genome alterations
could be traced to phage mobilization.
SUBJECTS, MATERIALS, AND METHODS
Study design. S. aureus isolates were obtained from sputum
specimens or throat swabs obtained from 118 patients with CF
attending 3 European CF clinics and from nose swabs obtained
from 208 healthy individuals. Informed consent was obtained
from patients or their parents or guardians, and the human-
experimentation guidelines of the University of Tubingen were
followed in the conduct of clinical research. Standard proce-
dures were used to isolate and identify S. aureus from the
different specimens. Genome types were assigned to each isolate
by typing with PFGE. Only consecutive intervals with the same
S. aureus genome type were included in the study, reducing the
study population to 26 patients with CF and 38 healthy control
subjects. One interval was available from 11 patients with CF,
2 intervals were available from 7 patients, 3 intervals were avail-
able from 4 patients, 4 intervals were available from 2 patients,
and 6 intervals were available from 3 patients. One interval was
available from 23 healthy individuals, 2 intervals were available
from 7 individuals, 3 intervals were available from 7 individuals,
and 4 intervals were available from 1 individual. In the CF
group, intervals varied between 6 and 538 days (median, 91
days), and, in the control group, intervals varied between 31
and 304 days (median, 133 days).
Genome typing with PFGE. PFGE was performed after
restriction endonuclease digestion of whole chromosomal
DNA, with SmaI (Roche Biochemicals) and EagI (EclXI; Roche
Biochemicals), as described elsewhere [14]. The restriction frag-
ments were separated by a contour-clamped homogeneous elec-
tric field (CHEF-DRII system; BioRad) in 0.5� Tris-borate EDTA
buffer at 12�C and 200 V. Running conditions for SmaI-digested
DNA were 1–15 s for 12 h, followed by 30–70 s for 12 h; and
those for EagI-digested DNA were 1–15 s for 12 h, followed by
20–50 s for 12 h. Digested whole chromosomal DNA of S. aureus
strain COL was used as size standard. The gels were evaluated
by use of WinCam3 software (Cybertech). Isolates showing dif-
ferences in the fragment pattern that could be explained by 1 or
2 genetic events (insertions, deletions, point mutations, or trans-
position) were classified as belonging to 1 clonal lineage.
Southern hybridization of PFGE gels. Ethidium bromide–
stained PFGE gels were nicked twice, from both sides, in a UV
chamber with 60 mJ. Depurination was performed in 0.25 mol/
L HCl for 15 min, followed by 2 cycles of denaturation in 3
mol/L NaCl and 0.4 mol/L NaOH for 15 min. Gels were blot-
ted by capillary transfer onto positively charged nylon mem-
branes (Roche Biochemicals) with denaturation buffer. High-
stringency hybridization was performed in accordance with the
instructions given by the manufacturer of the digoxigenin la-
beling and detection kit (Roche Biochemicals); signals were
detected by chemiluminescence.
Phage typing, antibiotic-resistance pattern, and polymor-
phisms of the agr locus. Phage typing and antibiotic-resistance
determination were performed by the reference laboratory (Rob-
ert Koch-Institut, Wernigerode, Germany), by use of standard
methods. The following antibiotics were tested: penicillin, oxa-
cillin, gentamicin, erythromycin, clindamycin, tetracycline, tei-
coplanin, chloramphenicol, vancomycin, ciprofloxacin, trimeth-
oprim-sulfamethoxazole, fusidic acid, mupirocin, linezolid, and
moxifloxacin.
Identification of agr locus restriction fragment–length poly-
morphisms was performed as described elsewhere [15, 16]. In brief,
the variable part of the agr operon was amplified, and the generated
amplicons were digested with DraI (Roche Biochemicals).
Genome stability in vitro. Five S. aureus strains were an-
alyzed for changes after prolonged subculturing in vitro. Two
pairs of clinical isolates showing genome alterations in vivo
(strain pairs i1/i2 and s2/s3) and the prototypic S. aureus strain
RN6390 [17] were chosen. In 1 series of experiments, the 5
isolates were subcultured on sheep blood agar plates daily for
33 days. Every 10th passage was stored for typing. Each plate
was inspected for the occurrence of phenotypic variants (he-
molysis pattern and colony morphology). These variants were
stored and subcultured separately.
In a second approach, liquid medium (CYPG [18]) was in-
oculated with single colonies of isolate i2 and RN6390 and
cultured at 37�C into deep stationary phase (48 h). Both cul-
tures were sequentially subcultured 14 times every other day
by use of 1:100 dilutions in fresh medium. Colony-forming
units and changes in phenotype were determined on sheep
blood agar plates. Phenotypic variants were separately stored

726 • JID 2004:189 (15 February) • Goerke et al.
Table 1. Target sites of fragment-specific hybridization probes and primers used for gen-eration of probes.
Probe N315 coordinates, nt Target site, ORF Primer(s)a
Amplicon 17 1,033,254–1,033,844 fmt AB4
Amplicon 19 1,206,110–1,206,748 cfxE AB4
Amplicon 3 1,971,268–1,971,870 pcrA AB4
Amplicon 4 1,159,978–1,160,530 pbpA AB4
Amplicon 5 2,156,808–2,157,400 atpG AB4
clfA 848,484–851,453 Clumping factor A GGCGTGGCTTCAGTGCTTGTA;CACCAGTTACCGGCGTTTCTTC
Eag7 980,667–982,053 SA0867 GATGAAAATAGACAAAAGAT;CGTGATGATACCAAGCAAATG
hla 1,140,562–1,141,521 a-hemolysin
hlb 2,049,532–2,050,410 b-hemolysin AGCTTCAAACTTAAATGTCA;GCTATCATTATCGAATCCAC
RNAIII 2,079,076–2,079,210 hld
sae 755,280–765,335 saeS CCATTTACGCCTTAACTTTA;TAGTCATATCCCCAAACTT
NOTE. ORF, open-reading frame.a Primer sequence for amplicons are listed in Subjects, Materials, and Methods; primer sequences for hla
and RNAIII are listed in [37].
and typed. After 48 h, a decline in variable counts of ∼1 log
was observed, indicating stress conditions.
Generation of fragment-specific amplicons and probes.
For the molecular characterization of genome alterations, PFGE
was performed with SmaI-digested genomic DNA from isolates
of interest, as described above. Differing fragments were excised
from the agarose gel, were washed with polymerase chain re-
action (PCR)–grade water, and were stored at 4�C. Random
PCR was performed in 50-mL volumes containing small pieces
of the following agarose plugs: 0.2 mmol/L dNTP mix, 2 mmol/
L MgCl2, 2.5 U of HotStar Taq polymerase (Qiagen), and 10
pmol of arbitrary primers tt-AB4 (CAGTTCAAGCTTGTCCA-
GGAATTCNNNNNNNAGATT) or ct-AB4 (CAGTTCAAGCT-
TGTCCAGGAATTCNNNNNNNAGACT). A nested PCR was
performed by use of primer AB4 (CAGTTCAAGCTTGTCCA-
GGAATTC), which consisted of the conserved part of the ar-
bitrary primers. PCR amplicons were cloned in the pCR2.1
TOPO vector (Invitrogen) and were transformed in E. coli
TOP10. Plasmid DNA was purified by use of the Wizard Mini-
preps system (Promega) and were sequenced by use of M13
forward and reverse sequencing primers. Digoxigenin-labeled
fragment-specific probes were generated by PCR labeling
(Roche Biochemicals) with the AB4 primer, by use of the TOPO
vectors containing the cloned PCR amplicons as a template.
Hybridization probes directed against known genes and against
sequences spanning the SmaI and EagI restriction sites were
generated by use of the specific primers listed in table 1.
Suppressive subtractive hybridization (SSH). Genomic
subtraction was performed by use of the PCR-Select Bacterial
Genome Subtraction Kit (Clontech) using strain m47 as the tester
and strain m46 as the driver. In brief, 2 mg of genomic DNA
from each strain was digested with AluI, and 2 different PCR
adaptors were ligated to 2 aliquots of the tester DNA. Two hy-
bridizations in which an excess of driver DNA was added were
then performed at 50�C. PCR products obtained after SSH were
cloned in the pCR2.1 TOPO vector and were sequenced.
PCR. Long-range PCR was performed by use of the
TripleMaster PCR System (Eppendorf), in accordance with the
manufacturer’s instructions for amplification of 10-kb targets.
The following primer pairs were used for amplification of the
region nt 875,220–931,123 in N315: primer 1 (forward, ACTTT-
TATCCTCAGTTTTGT; reverse, TTATGCACTTAGTCGAAGCT),
primer 2 (forward, AATTGAACTAAAATCATTGCAATG; reverse,
AAAACAAGCATCTGCGTCAC), primer 3 (forward, TCGAGGA-
AAATTCCAGATAAATT; reverse, GTGGCATATTACTAAAGTC-
TCTTGC), primer 4 (forward, GGCACCGTTTAAGACGAATT; re-
verse, GTTATCAAACACCCGAAACA), primer 5 (forward, CGTA-
TAGCGTTTTTCAAAATGGCT; reverse, GTGCTTCTTCTTTAC-
CGTATCTTTC), and primer 6 (forward, GCAGACAAAGATTTC-
TCTGT; reverse, GCTTTGCGATCAGTGATATC).
The attB site of phage ETA was amplified by use of standard
PCR with the published primers [19]. The resulting PCR frag-
ment of strain m46 was sequenced (4base lab, Reutlingen, Ger-
many). The attB site of phage L54 was amplified by use of the
primers TGGTCATGATGCAAGAGAAG and CTTCAACACG-
CAACAAGTCA.
DNA sequence analysis. All sequencing was performed by
4base lab by use of the DYNAMIC Sequence Kit (Amersham
Biosciences). Arbitrary primers used for random PCR were
designed by M. Bayer (4base lab).

Genomic Alterations in S. aureus • JID 2004:189 (15 February) • 727
Figure 1. Example of 5 Staphylococcus aureus strain pairs showing genome alterations after pulsed-field gel electrophoresis of SmaI- or EagI-digestedgenomic DNA.
Sequence data were analyzed by use of Vector NTI software
(version 8; InforMax). The sequence of strain N315 was obtained
from the European Molecular Biology Laboratory nucleotide da-
tabase (accession number BA000018), and the sequence of strain
COL was obtained from The Institute of Genomic Research
(TIGR; ftp://ftp.tigr.org/pub/data/s_aureus_COL).
Statistical analysis. The transformation rates (frequency
of genome alterations) were estimated in the 2 groups by the
maximum-likelihood method. This model assumes an expo-
nential (e) distribution until the time of transformation. If a
transformation was observed during an interval of t, then the
contribution to the likelihood was given by the expression
, where l is the transformation rate to be estimated.(�lt)1 � e
The inverse of l is the expected time to transformation. If there
was no transformations during an interval of t, then the con-
tribution to the likelihood was .(�lt)e
To compare the observations with the model, we calculated
Kaplan-Meier curves for the transformation-free time by im-
puting the most likely time of transformation, using a uniformly
distributed time during the intervals for which a transformation
was observed. Intervals without transformation were entered in
the Kaplan-Meier estimates as censored observations. Relative
frequencies were compared by use of Fisher’s exact test.
RESULTS
Frequency of S. aureus genome alterations in vivo. Con-
secutive S. aureus isolates were obtained from sputum speci-
mens or throat swabs from patients with CF and from nose
swabs from healthy control subjects. These isolates were ge-
nome typed by use of PFGE. Only individuals harboring the
same S. aureus clone on consecutive samplings (1 interval) were
included in the study. Clone definition was based on the PFGE
result after restriction digestion with 2 enzymes (SmaI and
EagI). Fragment variations detected in consecutive isolates of
the same clonal lineage, after digestion with both enzymes, were
interpreted as genome alterations (figure 1). A total of 93 iso-
lates from 26 patients with CF were available for analysis of
genome variations over time, representing 59 intervals, with a
mean duration of 91 days (minimum, 6 days; maximum, 538
days). A total of 102 isolates were available from 38 healthy
control subjects, representing 62 intervals, with a mean dura-
tion of 133 days (minimum, 31 days; maximum, 304 days).
Significantly more patients with CF (13/26) harbored S. aureus
clones showing genome alterations than did healthy control sub-
jects (2/38) ( ). The 2 alterations detected in S. aureusP ! .0001
clones from the control group were obtained from 2 different
individuals. In 1 patient with CF, 3 independent genome al-
terations in the infecting S. aureus clones were detected over
time; in 2 patients, 2 independent genome alterations were
found. Multiple intervals from 1 patient were treated as in-
dependent episodes. On the basis of the changes observed in
S. aureus within given intervals, a mean time for the observed
genome alterations was calculated for both groups. The S. au-
reus clones showed changes in the PFGE pattern after a mean
of 1.03 years (95% confidence interval [CI], 0.66–1.74 years) in
the CF group and after a mean of 13.4 years (95% CI, 4.3–80.3
years) in the control group (figure 2). The calculated time dif-
ference for S. aureus genome alterations between patients with
CF and healthy control subjects was significant ( ).P ! .0001
Association between clonal lineages and genome altera-
tions. Isolates from 26 patients with CF and from 38 healthy
control subjects could be differentiated by PFGE typing into
20 distinct S. aureus genome types (table 2). The clonality of

728 • JID 2004:189 (15 February) • Goerke et al.
Figure 2. Kaplan-Meier curves for the observed frequencies of genomealterations in the cystic fibrosis (CF) group and in the group of healthyindividuals. The dotted lines show the expected frequencies. To comparethe observations with the model, we calculated Kaplan-Meier curves forthe transformation-free time by imputing the most likely time of transfor-mation, using a uniformly distributed time during the intervals for which atransformation was observed. Intervals without transformations were en-tered in the Kaplan-Meier estimates as censored observations.
isolates assigned to the same genome type was further con-
firmed by additional typing methods (phage typing and agr
group polymorphism). Consecutive isolates were always iden-
tical by phage and agr group. No variation in antibiotic-resis-
tance patterns was detected between consecutive isolates. In 1
of the strain pairs, both isolates were methicillin resistant (data
not shown).
Several epidemiologically unrelated persons were colonized
with the same S. aureus clone. Eleven of 20 genome types were
obtained from 11 individual, and none was specifically associ-
ated with patients with CF. Genome alterations were observed
in 11 of 20 S. aureus genome types. No association was observed
between the frequency of genome alterations and certain clonal
lineages of S. aureus.
Genome alterations in vitro. To analyze whether genome
alterations can also occur during extended in vitro subculturing,
2 pairs of clones showing alterations in vivo and the laboratory
strain RN6390 were passaged 33 times on sheep blood agar
plates. Although nonhemolytic variants arose during subcul-
tivation in all 5 isolates, this was not accompanied by changes
in the PFGE pattern. In general, none of the descendents of
the original 5 isolates showed genome alterations. To further
evaluate whether starvation induces alterations, isolates were
grown to deep stationary phase in liquid medium and were
subcultured 14 times. Again, although phenotypic variations
occurred, no changes in the PFGE pattern were detected.
Molecular analysis of genome alterations. In total, 19
strain pairs showing alterations in the SmaI and EagI restriction
pattern after PFGE were available for molecular analysis. These
strains are described in more detail in table 3. To map the
affected regions in the genome, fragments of interest were ex-
cised from the PFGE gels and were subjected to random PCR.
Southern hybridization was performed by use of digoxigenin-
labeled probes derived from the generated amplicons. Probes
reacted with the excised PFGE fragment and the altered frag-
ment in the consecutive strain. To identify the altered region,
amplicons were sequenced and mapped to the published N315
genome [5]. In general, amplicons derived from a given frag-
ment clustered within a distinct region of the N315 genome.
The same clustering was observed when the genome sequence
of S. aureus COL (TIGR) was used. Thus, the gene order seems
to be conserved, and we supposed a similar organization in
our strains. Therefore, hybridization probes directed against
known genes and against sequences spanning the SmaI and
EagI restriction sites from the region in question were used to
narrow the sites of variation. We concentrated on the molecular
characterization of genome alterations in selected pairs in which
we could assume that the differences between the strains were
solely due to insertions or deletions.
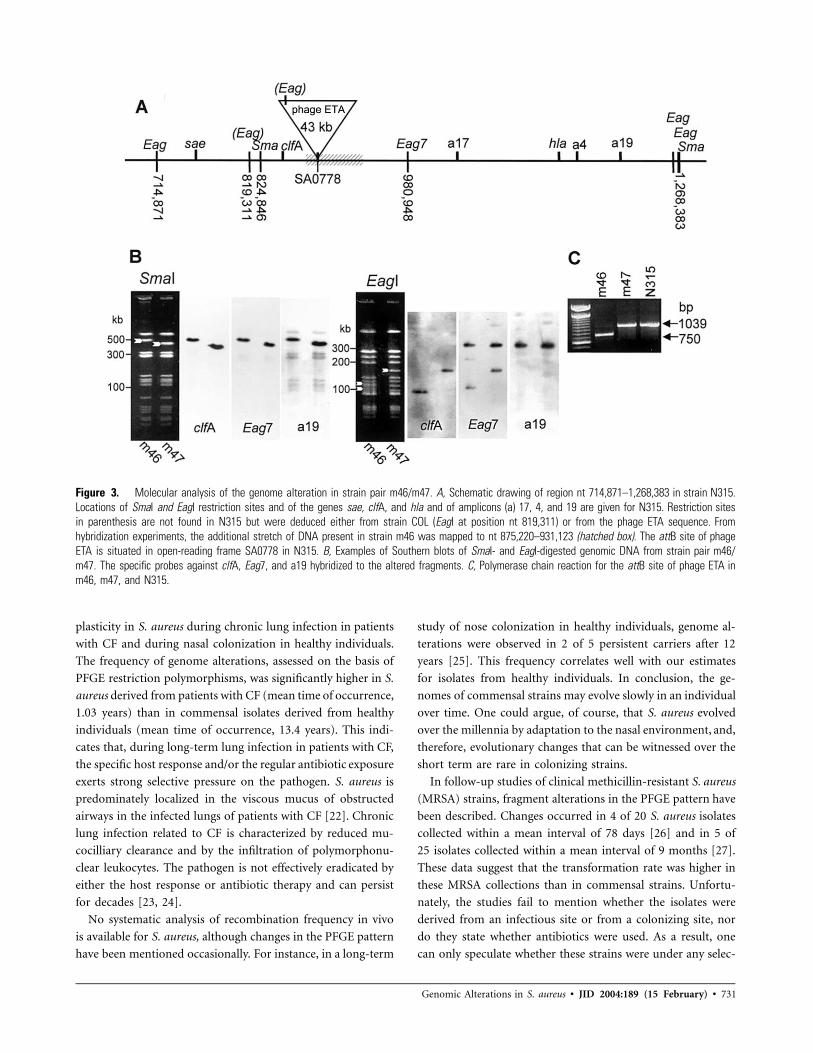
Genome alteration due to deletion of a phage ETA homo-
logue. In 1 strain pair (m46/m47), a deletion of ∼50 kb was
evident in the SmaI restriction pattern of the second isolate, m47
(figure 3B). In the EagI pattern, 2 fragments (110 and 90 kb) in
strain m46 were replaced by a single fragment of 160 kb in strain
m47. After the generation of fragment-specific amplicons, hy-
bridization, and sequencing of the probes (amplicon 4, amplicon
17, and amplicon 19), this alteration was mapped to the SmaI
fragment nt 824,846–1,268,383 in N315, encompassing, for in-
stance, clfA and hla (figure 3A and 3B). On the basis of the
hybridization analysis, we concluded that strain m46 contains
an additional stretch of DNA (40–50 kb long) that has no
counterpart in strain m47 or N315 and that is probably located
in the region corresponding to nt 875,220–931,123 in N315.
Overlapping primers for long-range PCR were generated for
this region. The region corresponding to nt 883,225–886,164
in N315 could not be amplified from strain m46.
To identify the stretch of additional DNA, the strain pair
m46/m47 was subjected to suppressive subtractive hybridiza-
tion. Two resulting clones had a high degree of homology to
ETA, a phage of 43 kb [19]. Thus, the genome alterations in
the strain pair m46/m47 could be explained by insertion of
either phage ETA or an ETA homologue in m46. The attach-
ment site for phage ETA was mapped to an open-reading frame
(ORF) coding for a hypothetical protein on the chromosome
of COL [19]. This ORF corresponds to SA0778 in N315, which
lies in the predicted region of the insertion site in m46. Phage
conversion was further verified by amplification with primers
specific for the attachment site of phage ETA (figure 3C). An
amplicon of the expected size was detected in strain m47 and
in N315. In the phage-positive strain m46, a fragment of 750

Genomic Alterations in S. aureus • JID 2004:189 (15 February) • 729
Table 2. Distribution of 20 distinct Staphylococcus aureus genome types (GT) in patientswith cystic fibrosis (CF) and healthy control subjects and no. of genome alterations in eachclonal lineage.
GT
Patients with CF Healthy control subjects
No. of patientsharboring GT
(no. of intervals)a
No. ofgenome
alterations
No. of controlsubjects harboring GT
(no. of intervals)a
No. ofgenome
alterations
1 2 (7) 3 1 (1) 02 6 (9) 4 8 (10) 07 2 (3) 2 5 (8) 016 3 (8) 2 3 (3) 017 2 (2) 1 0 (0) 019 2 (3) 0 2 (3) 030 0 (0) 0 3 (6) 036 3 (7) 1 10 (20) 048 1 (1) 0 1 (1) 054 1 (3) 0 2 (3) 0109 2 (8) 1 1 (1) 0Rareb (8, 15, 18, 31, 40,
43, 49, 88, 108)5 (8) 3 4 (6) 2
a Two consecutive S. aureus isolates with identical GTs were obtained during 1 interval.b Genome types that were isolated in only 1 individual.
bp was amplified. Sequencing of this fragment revealed that
part of phage ETA (128 bp at the 3′ end), the attR site, and
part of the flanking region were amplified because of false
priming of the upper primer within the phage. The sequences
obtained for the partial phage ETA and the attR site were iden-
tical to the published sequences (GenBank accession numbers
AB046707 and AP001553). Further results obtained by sub-
tractive hybridization indicate that the phage present in m46
has a mosaic structure: 2 of the analyzed clones showed ho-
mology only to phage 11 or 12, respectively. Probes generated
from these clones hybridized to the same EagI fragments in
m46 as did the phage ETA–specific clones.
To further clarify whether phage ETA conversion is respon-
sible for genome alterations in other strain pairs, we amplified
the attB site for phage ETA. In all the strains, amplification
resulted in the predicted fragment, indicating that none is phage
ETA positive.
Genome alteration due to mobilization of phage 42 and its
derivates. In another strain pair (cfs425/cfs555), the alteration
was mapped to 2 neighboring SmaI fragments, nt 1,922,490–
2,031,794 and nt 2,031,794–2,109,683, in phage N315 by use of
amplicon 3, amplicon 5, and RNAIII as probes. The SmaI site
separating these 2 fragments is localized on phage N315. This
phage is homologous to phage 42 and its derivates, which have
their attachment site in the gene for b-hemolysin (hlb). Phage
insertion leads to negative conversion of hlb expression [20].
Strain cfs425 showed no Hlb production on sheep blood agar
plates, whereas strain cfs555 was positive. Thus, a phage 42-like
phage was probably deleted in strain cfs555. Accordingly, hlb was
detected on 2 fragments in strain cfs425 and on 1 fragment in
strain cfs555, by Southern hybridization with an hlb-specific
probe (figure 4A and 4B). As with strain N315, phage insertion
resulted in an additional SmaI restriction site in strain cfs425,
creating 2 fragments of 100 kb and 60 kb, respectively.
Furthermore, we screened for Hlb expression in all 19 strain
pairs showing genome alterations. In 7 cases, conversion of hlb
expression was observed: in 2 cases, the phenotype changed
from Hlb positive to Hlb negative, whereas, in the other 5 cases,
strains were rendered positive. All conversions were accom-
panied by a shift of the fragment reacting with the hlb probe,
indicating phage involvement (figure 4B). Phage insertion re-
sulted in the addition of an SmaI restriction site in 6 cases; in 1
case, the phage carried no SmaI site. In 3 of 7 cases, phage
integration was the sole cause of the detected fragment altera-
tions. In the remaining 4 cases, additional fragment alterations
not linked to hlb conversion were evident from the PFGE pattern.
No genome alteration due to mobilization of phage L54.
Insertion of phage L54 in the lipase gene (geh) inactivates glycerol
ester hydrolase production in S. aureus [21]. To clarify whether
phage L54 conversion is responsible for genome alterations in
our strain pairs, we amplified the attB site for phage L54 (Gen-
Bank accession number STAL54BOB). In all the strains, ampli-
fication resulted in the predicted fragment, indicating that none
is phage L54 positive.
DISCUSSION
To understand the evolution of pathogens, it is essential to
establish the nature and frequency of recombination, as well
as the driving forces behind it. Here, we have analyzed genome
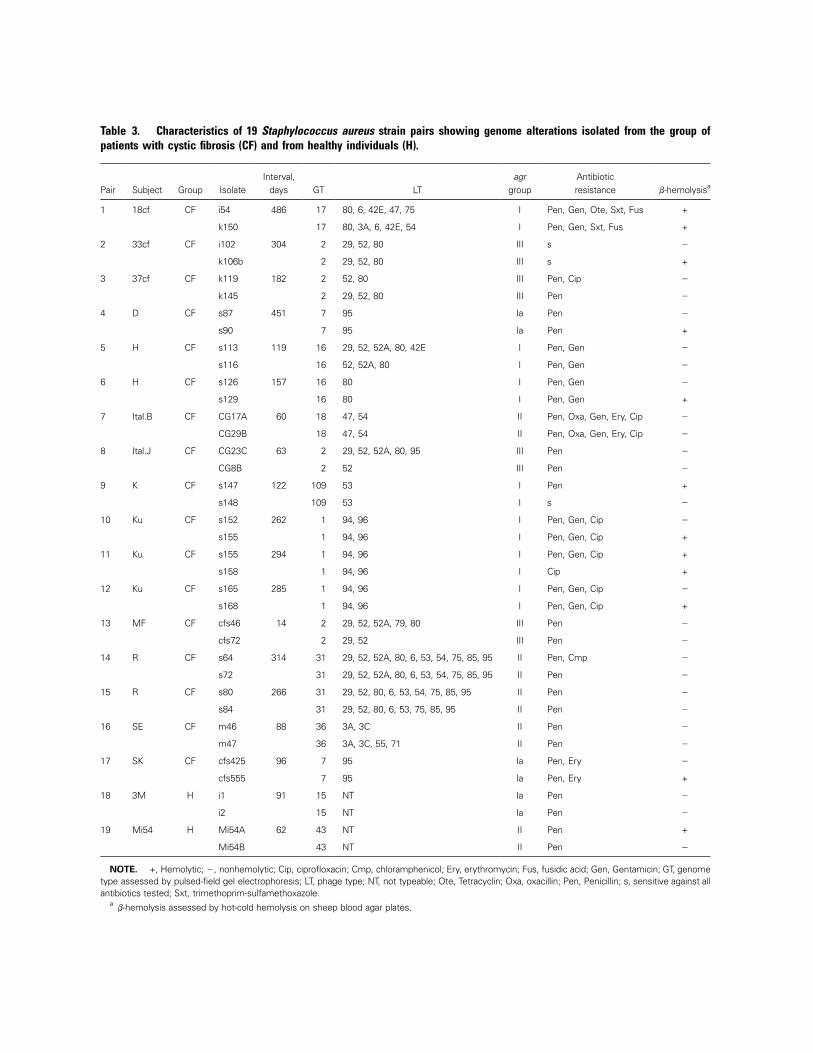
Table 3. Characteristics of 19 Staphylococcus aureus strain pairs showing genome alterations isolated from the group ofpatients with cystic fibrosis (CF) and from healthy individuals (H).
Pair Subject Group IsolateInterval,
days GT LTagr
groupAntibioticresistance b-hemolysisa
1 18cf CF i54 486 17 80, 6, 42E, 47, 75 I Pen, Gen, Ote, Sxt, Fus +
k150 17 80, 3A, 6, 42E, 54 I Pen, Gen, Sxt, Fus +
2 33cf CF i102 304 2 29, 52, 80 III s �
k106b 2 29, 52, 80 III s +
3 37cf CF k119 182 2 52, 80 III Pen, Cip �
k145 2 29, 52, 80 III Pen �
4 D CF s87 451 7 95 Ia Pen �
s90 7 95 Ia Pen +
5 H CF s113 119 16 29, 52, 52A, 80, 42E I Pen, Gen �
s116 16 52, 52A, 80 I Pen, Gen �
6 H CF s126 157 16 80 I Pen, Gen �
s129 16 80 I Pen, Gen +
7 Ital.B CF CG17A 60 18 47, 54 II Pen, Oxa, Gen, Ery, Cip �
CG29B 18 47, 54 II Pen, Oxa, Gen, Ery, Cip �
8 Ital.J CF CG23C 63 2 29, 52, 52A, 80, 95 III Pen �
CG8B 2 52 III Pen �
9 K CF s147 122 109 53 I Pen +
s148 109 53 I s �
10 Ku CF s152 262 1 94, 96 I Pen, Gen, Cip �
s155 1 94, 96 I Pen, Gen, Cip +
11 Ku CF s155 294 1 94, 96 I Pen, Gen, Cip +
s158 1 94, 96 I Cip +
12 Ku CF s165 285 1 94, 96 I Pen, Gen, Cip �
s168 1 94, 96 I Pen, Gen, Cip +
13 MF CF cfs46 14 2 29, 52, 52A, 79, 80 III Pen �
cfs72 2 29, 52 III Pen �
14 R CF s64 314 31 29, 52, 52A, 80, 6, 53, 54, 75, 85, 95 II Pen, Cmp �
s72 31 29, 52, 52A, 80, 6, 53, 54, 75, 85, 95 II Pen �
15 R CF s80 266 31 29, 52, 80, 6, 53, 54, 75, 85, 95 II Pen �
s84 31 29, 52, 80, 6, 53, 75, 85, 95 II Pen �
16 SE CF m46 88 36 3A, 3C II Pen �
m47 36 3A, 3C, 55, 71 II Pen �
17 SK CF cfs425 96 7 95 Ia Pen, Ery �
cfs555 7 95 Ia Pen, Ery +
18 3M H i1 91 15 NT Ia Pen �
i2 15 NT Ia Pen �
19 Mi54 H Mi54A 62 43 NT II Pen +
Mi54B 43 NT II Pen �
NOTE. +, Hemolytic; �, nonhemolytic; Cip, ciprofloxacin; Cmp, chloramphenicol; Ery, erythromycin; Fus, fusidic acid; Gen, Gentamicin; GT, genometype assessed by pulsed-field gel electrophoresis; LT, phage type; NT, not typeable; Ote, Tetracyclin; Oxa, oxacillin; Pen, Penicillin; s, sensitive against allantibiotics tested; Sxt, trimethoprim-sulfamethoxazole.
ab-hemolysis assessed by hot-cold hemolysis on sheep blood agar plates.
Genomic Alterations in S. aureus • JID 2004:189 (15 February) • 731
Figure 3. Molecular analysis of the genome alteration in strain pair m46/m47. A, Schematic drawing of region nt 714,871–1,268,383 in strain N315.Locations of SmaI and EagI restriction sites and of the genes sae, clfA, and hla and of amplicons (a) 17, 4, and 19 are given for N315. Restriction sitesin parenthesis are not found in N315 but were deduced either from strain COL (EagI at position nt 819,311) or from the phage ETA sequence. Fromhybridization experiments, the additional stretch of DNA present in strain m46 was mapped to nt 875,220–931,123 (hatched box). The attB site of phageETA is situated in open-reading frame SA0778 in N315. B, Examples of Southern blots of SmaI- and EagI-digested genomic DNA from strain pair m46/m47. The specific probes against clfA, Eag7, and a19 hybridized to the altered fragments. C, Polymerase chain reaction for the attB site of phage ETA inm46, m47, and N315.
plasticity in S. aureus during chronic lung infection in patients
with CF and during nasal colonization in healthy individuals.
The frequency of genome alterations, assessed on the basis of
PFGE restriction polymorphisms, was significantly higher in S.
aureus derived from patients with CF (mean time of occurrence,
1.03 years) than in commensal isolates derived from healthy
individuals (mean time of occurrence, 13.4 years). This indi-
cates that, during long-term lung infection in patients with CF,
the specific host response and/or the regular antibiotic exposure
exerts strong selective pressure on the pathogen. S. aureus is
predominately localized in the viscous mucus of obstructed
airways in the infected lungs of patients with CF [22]. Chronic
lung infection related to CF is characterized by reduced mu-
cocilliary clearance and by the infiltration of polymorphonu-
clear leukocytes. The pathogen is not effectively eradicated by
either the host response or antibiotic therapy and can persist
for decades [23, 24].
No systematic analysis of recombination frequency in vivo
is available for S. aureus, although changes in the PFGE pattern
have been mentioned occasionally. For instance, in a long-term
study of nose colonization in healthy individuals, genome al-
terations were observed in 2 of 5 persistent carriers after 12
years [25]. This frequency correlates well with our estimates
for isolates from healthy individuals. In conclusion, the ge-
nomes of commensal strains may evolve slowly in an individual
over time. One could argue, of course, that S. aureus evolved
over the millennia by adaptation to the nasal environment, and,
therefore, evolutionary changes that can be witnessed over the
short term are rare in colonizing strains.
In follow-up studies of clinical methicillin-resistant S. aureus
(MRSA) strains, fragment alterations in the PFGE pattern have
been described. Changes occurred in 4 of 20 S. aureus isolates
collected within a mean interval of 78 days [26] and in 5 of
25 isolates collected within a mean interval of 9 months [27].
These data suggest that the transformation rate was higher in
these MRSA collections than in commensal strains. Unfortu-
nately, the studies fail to mention whether the isolates were
derived from an infectious site or from a colonizing site, nor
do they state whether antibiotics were used. As a result, one
can only speculate whether these strains were under any selec-

732 • JID 2004:189 (15 February) • Goerke et al.
Fig. 4. Examples of Staphylococcus aureus strain pairs where genomealterations were linked to conversion of hlb expression. A, Pulsed-field gelelectrophoresis after SmaI digestion of genomic DNA. Fragments hybridizingwith the hlb probe are indicated with white arrows. B, Southern hybridizationwith digoxigenin-labeled hlb-specific probe.
tive pressure. On the other hand, MRSA recently evolved and
spread from a limited number of clonal lineages—so these
clones may be naturally more adaptable than others. We ob-
served genome alterations in methicillin-sensitive strains of dif-
ferent genetic backgrounds. Thus, genome plasticity is not a
trait of certain clonal lineages but instead seems to be a pre-
requisite for a strain’s ability to adapt to hostile environments.
In contrast to the in vivo setting, no genome alterations were
detected after in vitro subculturing under a variety of condi-
tions. The stability of the PFGE patterns is commonly accepted
and is one of the reasons for the widespread application of this
method for strain typing. In Campylobacter jejuni as well, ge-
netic recombination occurred only after intestinal passage in
chickens, but not during growth in vitro [28]. Furthermore, it
has been shown for S. epidermidis examined in human infection
and in a foreign-body model that these in vivo environments
are conducive to genetic exchange [29]. Thus, genomic plas-
ticity is an important characteristic of pathogens, equipping
them for survival in various hosts.
In total, 19 S. aureus strain pairs were available for molecular
analysis to clarify the nature of recombinational events in the
host environment. In at least 8 cases, phage mobilization was
the molecular basis for the genome alterations. In 1 strain pair,
the observed genome alteration was due to the deletion of a
phage similar to ETA. In the parental strain m46, the phage
was integrated at the known attB site for the phage family [19].
The m46 phage was observed to have a mosaic structure, with
homology to the phages ETA, 11, and 12. Mosaic structure has
been described as a general feature of phage genomes [30].
Screening of our isolates showed no other strains with inte-
grated ETA-like phages. This is consistent with the observation
of Yamaguchi et al. [19] that phage ETA preferentially lysog-
enizes S. aureus strains of phage group 3A and 3C. In the strain
collection in the present study, only the pair m46/m47 belonged
to this phage group.
Phage conversion of b-toxin production was evident in 7
pairs. Conversion is mediated by insertion of phage 42 and its
variants into a conserved attachment site in hlb [20, 31]. The
ensuing change in the PFGE pattern results either in the shift
of an SmaI fragment or in the separation into 2 fragments due
to an SmaI restriction site within the phage [31]. Both types
of changes were observed, indicating that different variants of
phage 42 were present in our strain collection.
Obviously, phage mobilization plays an important role dur-
ing infection. S. aureus phages may either carry accessory vir-
ulence factors (e.g., sak, sea, or eta) or interrupt chromosomal
virulence genes (e.g., hlb or geh). Therefore, both insertion and
deletion of phages can be accompanied by the augmentation
of a virulence trait. However, no apparent advantage of any of
these factors is evident in the environment of the CF host.
Alternatively, the observed phage involvement may mirror an
enhanced potency of lateral gene transfer, for which phages are
the primary vehicle between S. aureus strains. Theoretically,
homologous recombination mediated by transduction occurs
all over the chromosome, providing the organism with a chance
for broad genetic variation. In combination with a higher mu-
tation frequency, it may provide advantages under conditions
of severe selective pressure. Hypermutable strains of P. aeru-
ginosa are selected during chronic lung infection related to CF
and persist for years in most patients [8]. However, the oc-
currence of hypermutators in S. aureus strain collections from
patients with CF is controversial [32, 33]. Increasing the re-
combination frequency may be another method of ensuring
variation in this pathogen.
Many prophages are induced by environmental conditions
that lead to DNA damage, including exposure to reactive ox-
ygen species generated by leukocytes or exposure to exogenous
agents such as antibiotics [34]. Both conditions are present in
the infected lungs of patients with CF and could lead to the
phage mobilization detected in our strains. In addition, Broudy

Genomic Alterations in S. aureus • JID 2004:189 (15 February) • 733
et al. discovered a soluble phage-inducing factor elaborated
by human pharyngeal epithelial cells that induces the SpeC-
encoding phages of streptococci [35].
Horizontal gene movements between different strains are
likely to be favored when the donor and recipient strains occupy
the same site. Cocolonization with multiple S. aureus strains
occurs regularly in the CF lung but is rarely observed in the
nose [16]. Alternatively, there might be a changing proportion
over time of lysogenized and phage-free bacterial cells in the
infected lungs of patients with CF. Additionally, free phages
may be present at the site of colonization or infection.
Besides the importance of phage mobilization during infec-
tion, one must keep in mind that genome alterations could be
traced to phages in only a subset of our strain pairs. A prelim-
inary analysis of the remaining pairs leads us to believe that
intrachromosomal alterations, such as inversions or duplica-
tions, might occur and could be mediated by insertion sequence
elements [36] or transposons. The clarification of these events
requires the extensive mapping of the strains in question. This
is an ongoing project.
Acknowledgment
We would like to thank W. Witte (Robert Koch-Institut,
Wernigerode) for phage typing and antibiotic-resistance testing
of isolates.
References
1. Smith JM, Smith NH, O’Rourke M, Spratt BG. How clonal are bacteria?Proc Natl Acad Sci USA 1993; 90:4384–8.
2. Smith JM, Feil EJ, Smith NH. Population structure and evolutionarydynamics of pathogenic bacteria. Bioessays 2000; 22:1115–22.
3. Feil EJ, Holmes EC, Bessen DE, et al. Recombination within naturalpopulations of pathogenic bacteria: short-term empirical estimates andlong-term phylogenetic consequences. Proc Natl Acad Sci USA 2001;98:182–7.
4. Fitzgerald JR, Sturdevant DE, Mackie SM, Gill SR, Musser JM. Evo-lutionary genomics of Staphylococcus aureus: insights into the originof methicillin-resistant strains and the toxic shock syndrome epidemic.Proc Natl Acad Sci USA 2001; 98:8821–6.
5. Kuroda M, Ohta T, Uchiyama I, et al. Whole genome sequencing ofmeticillin-resistant Staphylococcus aureus. Lancet 2001; 357:1225–40.
6. Baba T, Takeuchi F, Kuroda M, et al. Genome and virulence deter-minants of high virulence community-acquired MRSA. Lancet 2002;359:1819–27.
7. von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriageas a source of Staphylococcus aureus bacteremia. N Engl J Med 2001;344:11–6.
8. Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequencyof hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infec-tion. Science 2000; 288:1251–4.
9. Romling U, Schmidt KD, Tummler B. Large genome rearrangementsdiscovered by the detailed analysis of 21 Pseudomonas aeruginosa cloneC isolates found in environment and disease habitats. J Mol Biol 1997;271:386–404.
10. Tenover FC, Arbeit RD, Goering RV, et al. Interpreting chromosomal
DMA restriction patterns produced by pulsed-field gel electrophoresis:criteria for bacterial strain typing. J Clin Microbiol 1995; 33:2233–9.
11. Goerke C, Kraning K, Stern M, Doring G, Botzenhart K, Wolz C.Molecular epidemiology of community-acquired Staphylococcus aureusin families with and without cystic fibrosis patients. J Infect Dis 2000;181:984–9.
12. Oliveira DC, Tomasz A, de Lencastre H. Secrets of success of a humanpathogen: molecular evolution of pandemic clones of meticillin-resis-tant Staphylococcus aureus. Lancet Infect Dis 2002; 2:180–9.
13. Booth MC, Pence LM, Mahasreshti P, Callegan MC, Gilmore MS.Clonal associations among Staphylococcus aureus isolates from varioussites of infection. Infect Immun 2001; 69:345–52.
14. Schlichting C, Branger C, Fournier JM, et al. Typing of Staphylococcusaureus by pulsed-field gel electrophoresis, zymotyping, capsular typing,and phage typing: resolution of clonal relationships. J Clin Microbiol1993; 31:227–32.
15. Papakyriacou H, Vaz D, Simor A, Louie M, McGavin MJ. Molecularanalysis of the accessory gene regulator (agr) locus and balance of vir-ulence factor expression in epidemic methicillin-resistant Staphylococcusaureus. J Infect Dis 2000; 181:990–1000.
16. Goerke C, Kummel M, Dietz K, Wolz C. Evaluation of intraspeciesinterference due to agr polyporphism in Staphylococcus aureus duringinfection and colonization. J Infect Dis 2003; 188:250–6.
17. Novick RP, Projan SJ, Kornblum J, et al. The agr P2 operon: an au-tocatalytic sensory transduction system in Staphylococcus aureus. MolGen Genet 1995; 248:446–58.
18. Novick RP. Genetic systems in staphylococci. Methods Enzymol1991; 204:587–636.
19. Yamaguchi T, Hayashi T, Takami H, et al. Phage conversion of exfoli-ative toxin A production in Staphylococcus aureus. Mol Microbiol 2000;38:694–705.
20. Coleman DC, Sullivan DJ, Russell RJ, Arbuthnott JP, Carey BF, Pom-eroy HM. Staphylococcus aureus bacteriophages mediating the simul-taneous lysogenic conversion of b-lysin, staphylokinase and enterotoxinA: molecular mechanism of triple conversion. J Gen Microbiol 1989;135:1679–97.
21. Lee CY, Iandolo JJ. Lysogenic conversion of staphylococcal lipase iscaused by insertion of the bacteriophage L54a genome into the lipasestructural gene. J Bacteriol 1986; 166:385–91.
22. Ulrich M, Herbert S, Berger J, et al. Localization of Staphylococcusaureus in infected airways of patients with cystic fibrosis and in a cellculture model of S. aureus adherence. Am J Respir Cell Mol Biol 1998;19:83–91.
23. Kahl BC, Herrmann M, Everding AS, et al. Persistent infection withsmall colony variant strains of Staphylococcus aureus in patients withcystic fibrosis. J Infect Dis 1998; 177:1023–9.
24. Branger C, Gardye C, Lambert-Zechovsky N. Persistence of Staphylo-coccus aureus strains among cystic fibrosis patients over extended periodsof time. J Med Microbiol 1996; 45:294–301.
25. VandenBergh MF, Yzerman EP, van Belkum A, Boelens HA, Sijmons M,Verbrugh HA. Follow-up of Staphylococcus aureus nasal carriage after 8years: redefining the persistent carrier state. J Clin Microbiol 1999; 37:3133–40.
26. Hartstein AI, Phelps CL, Kwok RY, Mulligan ME. In vivo stability anddiscriminatory power of methicillin-resistant Staphylococcus aureus typ-ing by restriction endonuclease analysis of plasmid DNA compared withthose of other molecular methods. J Clin Microbiol 1995; 33:2022–6.
27. Maslow JN, Brecher S, Gunn J, Durbin A, Barlow MA, Arbeit RD.Variation and persistence of methicillin-resistant Staphylococcus aureusstrains among individual patients over extended periods of time. EurJ Clin Microbiol Infect Dis 1995; 14:282–90.
28. Hanninen ML, Hakkinen M, Rautelin H. Stability of related humanand chicken Campylobacter jejuni genotypes after passage through chickintestine studied by pulsed-field gel electrophoresis. Appl Environ Mi-crobiol 1999; 65:2272–5.
29. Van Eldere J, Peetermans WE, Struelens M, Deplano A, Bobbaers H.

734 • JID 2004:189 (15 February) • Goerke et al.
Polyclonal staphylococcal endocarditis caused by genetic variability. ClinInfect Dis 2000; 31:24–30.
30. Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. Evolution-ary relationships among diverse bacteriophages and prophages: all theworld’s a phage. Proc Natl Acad Sci USA 1999; 96:2192–7.
31. Smeltzer MS, Hart ME, Iandolo JJ. The effect of lysogeny on the genomicorganization of Staphylococcus aureus. Gene 1994; 138:51–7.
32. O’Neill AJ, Chopra I. Insertional inactivation of mutS in Staphylococcusaureus reveals potential for elevated mutation frequencies, although theprevalence of mutators in clinical isolates is low. J Antimicrob Chemother2002; 50:161–9.
33. Prunier AL, Malbruny B, Laurans M, Brouard J, Duhamel JF, LeclercqR. High rate of macrolide resistance in Staphylococcus aureus strains frompatients with cystic fibrosis reveals high proportions of hypermutable
strains. J Infect Dis 2003; 187:1709–16.34. Wagner PL, Waldor MK. Bacteriophage control of bacterial virulence.
Infect Immun 2002; 70:3985–93.35. Broudy TB, Pancholi V, Fischetti VA. The in vitro interaction of Strep-
tococcus pyogenes with human pharyngeal cells induces a phage-en-coded extracellular DNase. Infect Immun 2002; 70:2805–11.
36. Ziebuhr W, Dietrich K, Trautmann M, Wilhelm M. Chromosomalrearrangements affecting biofilm production and antibiotic resistancein a Staphylococcus epidermidis strain causing shunt-associated ventric-ulitis. Int J Med Microbiol 2000; 290:115–20.
37. Goerke C, Campana S, Bayer MG, Doring G, Botzenhart K, Wolz C.Direct quantitative transcript analysis of the agr regulon of Staphylo-coccus aureus during human infection in comparison to the expressionprofile in vitro. Infect Immun 2000; 68:1304–11.
Related Documents











