Inborn Errors Of Metabolism DR.P.S.K. RAJKUMAR SHANKAR 1 st YEAR MDS DEPT.PUBLIC HEALTH DENTISTRY TNGDC&H. 17/12/2014 1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Inborn Errors Of Metabolism
DR.P.S.K. RAJKUMAR SHANKAR
1st YEAR MDS
DEPT.PUBLIC HEALTH DENTISTRY
TNGDC&H.
17/12/2014 1

Contents
• Introduction
• Epidemiology
• Pathophysiology
• Various Disorders of Metabolism
• Clinical Features of IEMs
• Diagnosis
• Treatment
17/12/2014 2

Introduction6
Inborn errors of metabolism are group of genetically determined
biochemical disorder in which inherited defect in a single specific enzyme
that results in disruption or abnormality in a specific metabolic pathway
• Single gene defects
• Mostly autosomal recessive(exepct hunter’s,fabry’s,lesch-nyhan)
17/12/2014 3

Epidemiology
• According to a study in British Columbia, the overall incidence is approximately 40 cases
per 100,000 live births .
• Approximately 24 children per 100,000 births have a disease involving amino acids (e.g.
PKU), organic acids, primary lactic acidosis, galactosemia, or a urea cycle disease.
• Approximately 2.3 children per 100,000 births have some form of glycogen storage
disease.
• Approximately 8cases per 100,000 births (1 in 12,500) have a lysosomal storage disease;
• Approximately 3cases per 100 000 births have a respiratory chain-based mitochondrial
disease and 3 to 4 cases per 100 000 of births have a peroxisomal disorder.
17/12/2014 4

Epidemiology cont…,
• Mortality/Morbidity
Asymptomatic with slow degeneration over decades can be life-threateningover hours
Episodic with intermittent decompensations
• Race
Incidence varies with different race and ethnic groups
Incidence of Cystic fibrosis,1/1600people of European descent
Incidence of Sickle cell anemia,1/600people of African descent
Incidence of Tay-Sachs,1/3500people of Ashkenazi Jews
• Sex
Male-to-female ratio 1:1 for autosomal recessive transmission and X-linked
recessive transmission.
• AgePresenting at birth- phenylketonuria
Or later life -diabetes mellitus
17/12/2014 5

Pathophysiology
Gene mutations or gene deletions of ,,,,
DNA Enzyme
which code for a Receptor
Transport vehicle
Membrane pump
Structural element
Single gene defects result in abnormalities in the synthesis or catabolism of
Proteins, carbohydrates, fats or complex molecules.
Defect in an enzyme or transport protein, which results in block in a pathway.
Effects are due to toxic accumulations of substances .
17/12/2014 6
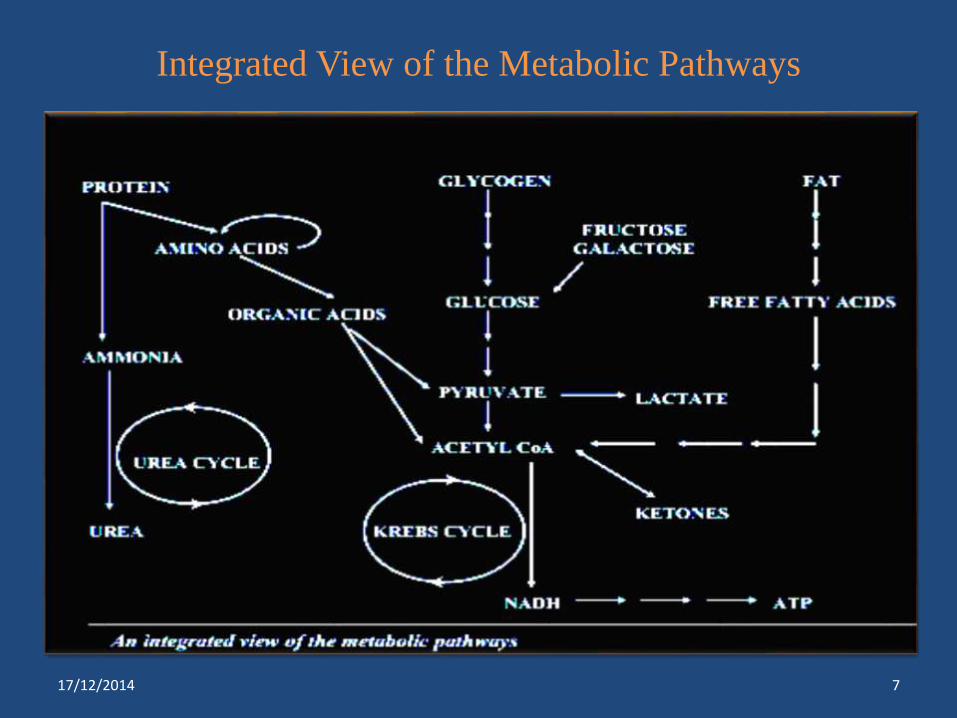
Integrated View of the Metabolic Pathways
17/12/2014 7

IEMs - Categories
Disorders that result in toxic accumulation
• Disorders of protein metabolism
• Disorders of carbohydrate intolerance
• Lysosomal storage disorders
Disorders of energy production, utilization
• Fatty acid oxidation defects
• Disorders of carbohydrate utilization
• Mitochondrial disorders
• Peroxisomal disorders
17/12/2014 8

Various Metabolic Disorders
Disorders of :
• Carbohydrate Metabolism
• Amino acid Metabolism
• Organic acid Metabolism
• Lipid Metabolism
• Nucleotide Metabolism
• Porphyrin Metabolism
• Mineral and Electrolyte Metabolism
• Mitochondrial Function
• Peroxisomal Function
• Steroid Metabolism
• Hereditary Anemias
17/12/2014 9

DISORDERS OF CARBOHYDRATE METABOLISM
17/12/2014 10
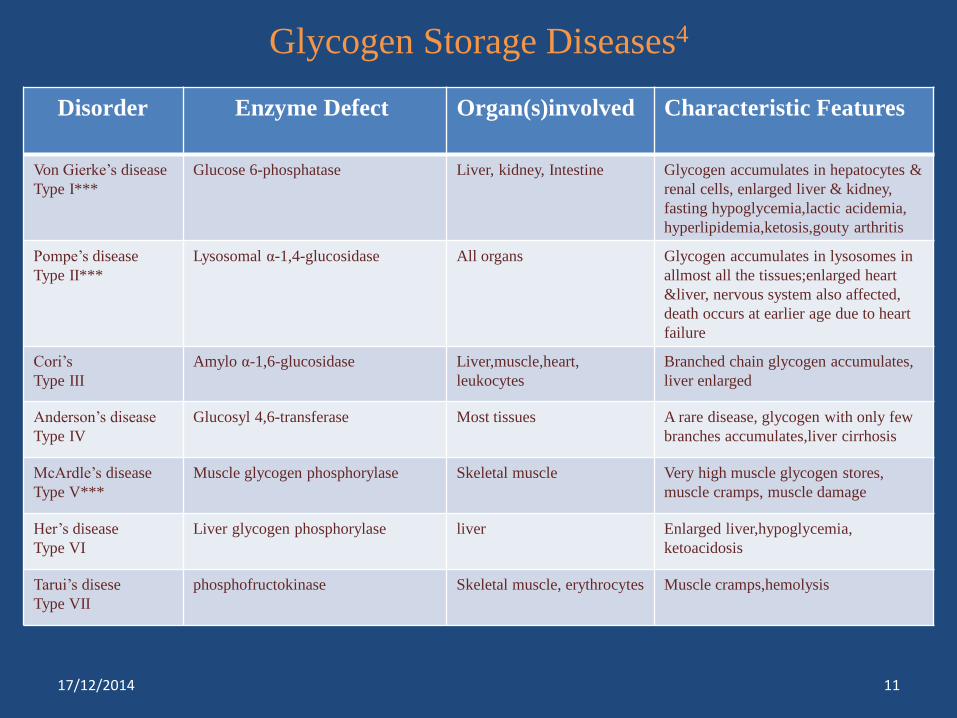
Glycogen Storage Diseases4
Disorder Enzyme Defect Organ(s)involved Characteristic Features
Von Gierke’s disease
Type I***
Glucose 6-phosphatase Liver, kidney, Intestine Glycogen accumulates in hepatocytes &
renal cells, enlarged liver & kidney,
fasting hypoglycemia,lactic acidemia,
hyperlipidemia,ketosis,gouty arthritis
Pompe’s disease
Type II***
Lysosomal α-1,4-glucosidase All organs Glycogen accumulates in lysosomes in
allmost all the tissues;enlarged heart
&liver, nervous system also affected,
death occurs at earlier age due to heart
failure
Cori’s
Type III
Amylo α-1,6-glucosidase Liver,muscle,heart,
leukocytes
Branched chain glycogen accumulates,
liver enlarged
Anderson’s disease
Type IV
Glucosyl 4,6-transferase Most tissues A rare disease, glycogen with only few
branches accumulates,liver cirrhosis
McArdle’s disease
Type V***
Muscle glycogen phosphorylase Skeletal muscle Very high muscle glycogen stores,
muscle cramps, muscle damage
Her’s disease
Type VI
Liver glycogen phosphorylase liver Enlarged liver,hypoglycemia,
ketoacidosis
Tarui’s disese
Type VII
phosphofructokinase Skeletal muscle, erythrocytes Muscle cramps,hemolysis
17/12/2014 11
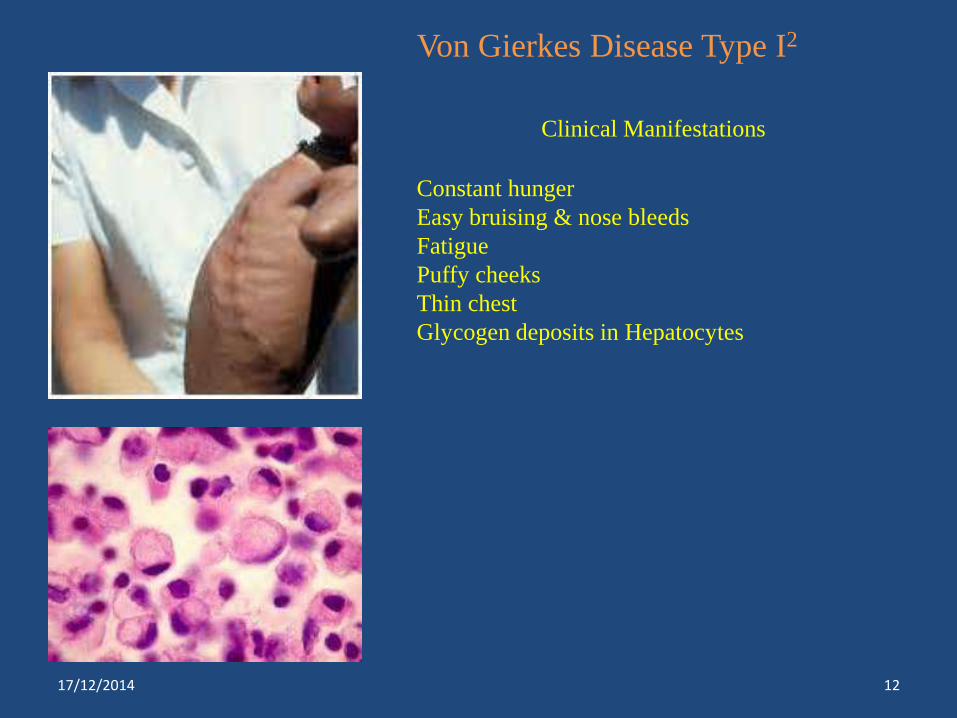
Von Gierkes Disease Type I2
Clinical Manifestations
Constant hunger
Easy bruising & nose bleeds
Fatigue
Puffy cheeks
Thin chest
Glycogen deposits in Hepatocytes
17/12/2014 12

Pompe’s Disease6
17/12/2014 13

McArdle’s Disease6
17/12/2014 14
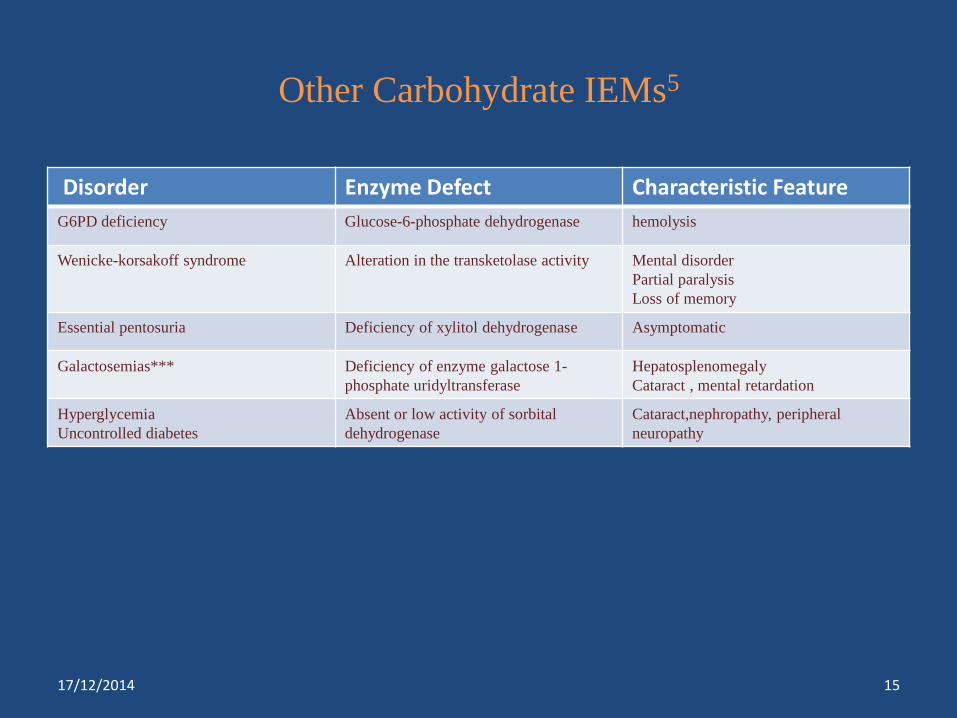
Other Carbohydrate IEMs5
Disorder Enzyme Defect Characteristic Feature
G6PD deficiency Glucose-6-phosphate dehydrogenase hemolysis
Wenicke-korsakoff syndrome Alteration in the transketolase activity Mental disorder
Partial paralysis
Loss of memory
Essential pentosuria Deficiency of xylitol dehydrogenase Asymptomatic
Galactosemias*** Deficiency of enzyme galactose 1-
phosphate uridyltransferase
Hepatosplenomegaly
Cataract , mental retardation
Hyperglycemia
Uncontrolled diabetes
Absent or low activity of sorbital
dehydrogenase
Cataract,nephropathy, peripheral
neuropathy
17/12/2014 15
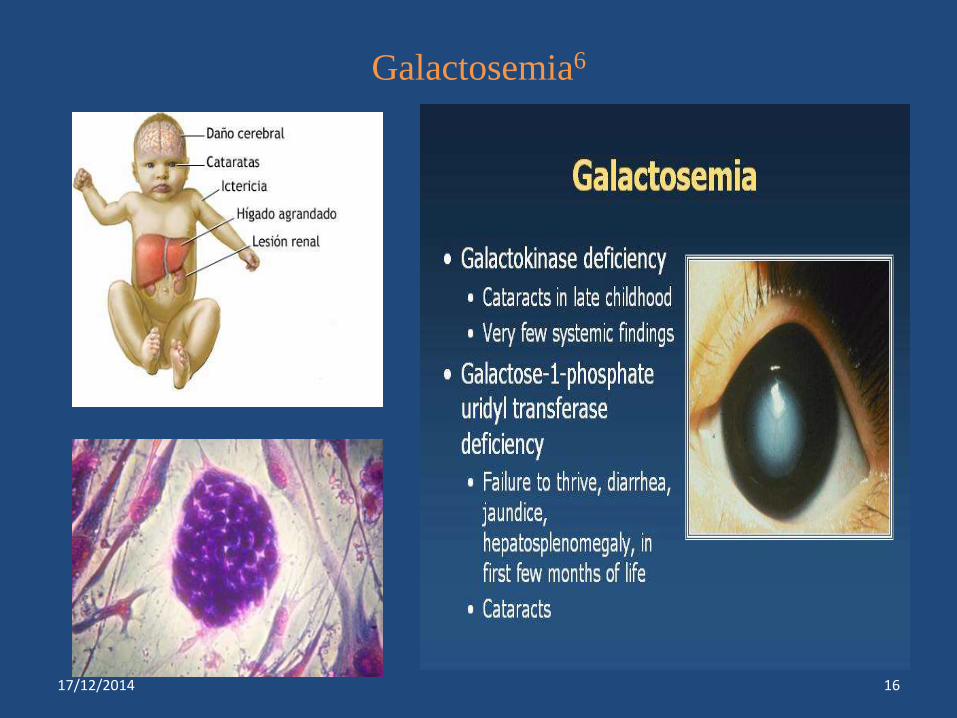
Galactosemia6
17/12/2014 16
Mucopolysaccharidosis1
Accumulation of mucopolysaccharides in tissues like liver, spleen, cornea, heart, blood
vessels and these patients have coarse facial features
Disorders Enzyme Defect Accumulated Product
Hurler’s Syndrome*** α- idouronidase Heparan, dermatan sulfate
Hunter Syndrome*** Idouronate sulfatase Heparan, dermatan sulfate
MaroTeauxlamy N-acetyl galactosamine sulfatase Dermatan sulfate
Mucolipidosis VIII β-glucouronidase Heparan, dermatan sulfate
Sanflippo Type A
Sanflippo Type B
Sanflippo Type C
Sanflippo Type D
Heparan sulfamidase
N-acetyl glucosamine
Α-glucosaminide acetyl tranferase
N-acetyl glucosamine 6-sulfatase
Heparan sulfate
Heparan sulfate
Heparan sulfate
Heparan sulfate
Morquio Type A***
Morquio Type B
Galactose 6-sulfatase
Β-galactosidase
Keratin/chondroitin sulfate
keratin sulfate
17/12/2014 17

Hunter’s Syndrome6
17/12/2014 18
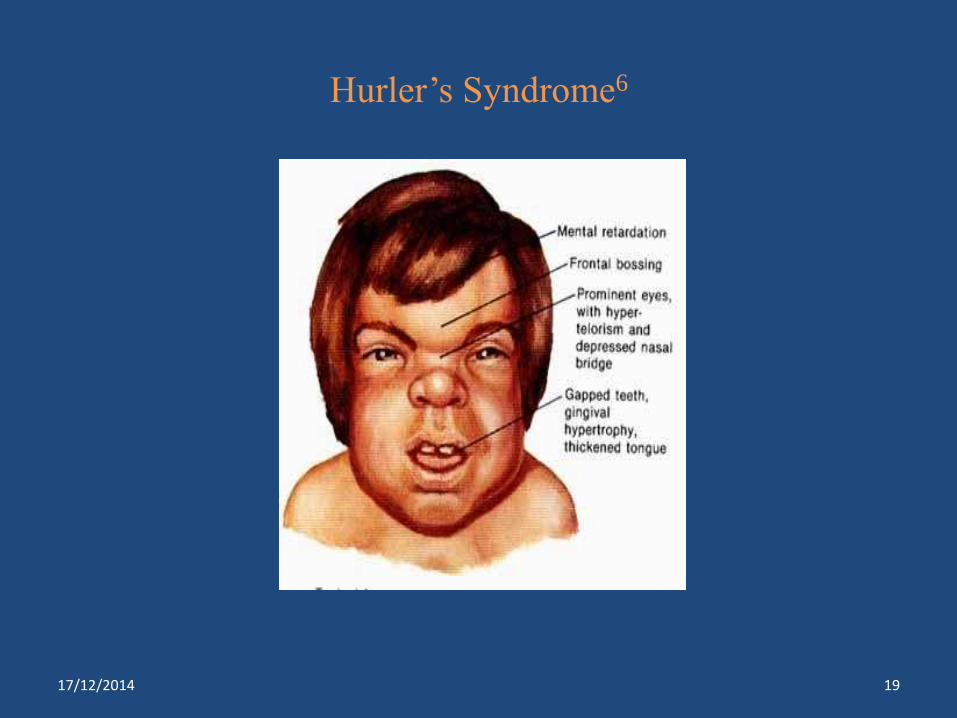
Hurler’s Syndrome6
17/12/2014 19
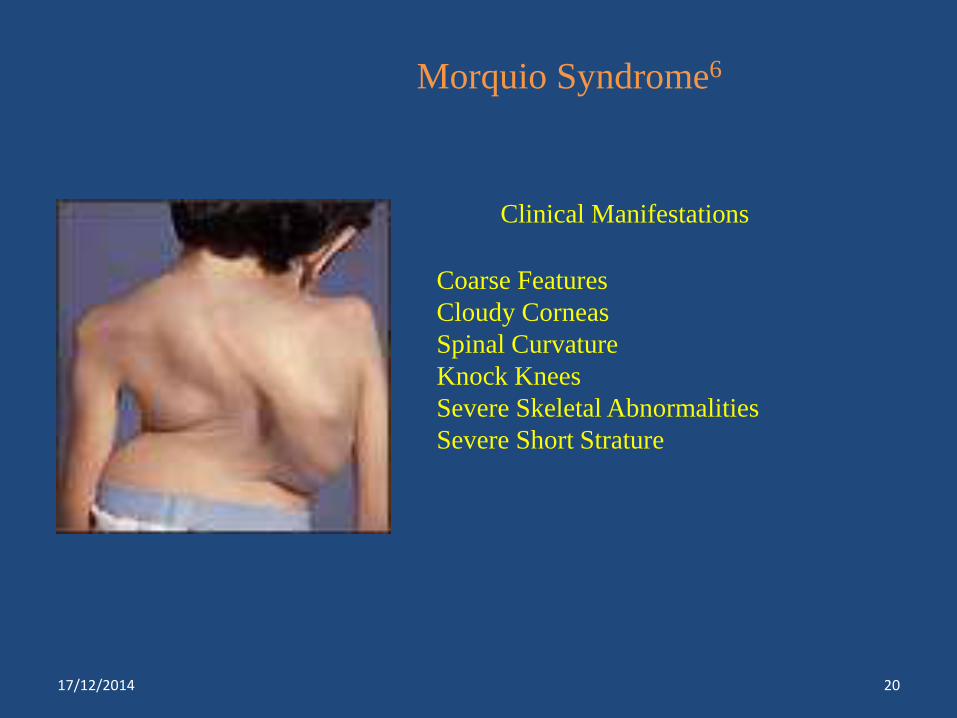
Morquio Syndrome6
Clinical Manifestations
Coarse Features
Cloudy Corneas
Spinal Curvature
Knock Knees
Severe Skeletal Abnormalities
Severe Short Strature
17/12/2014 20

DISORDERS OF AMINO ACID METABOLISM
17/12/2014 21

I. Metabolic Defects in Urea Cycle5
Disorders Enzyme defect
Hyperammonemia type 1 Carbamoyl phosphate synthase I
Hyperammonemia type II Ornithine transcarbamoylase
Citrullinemia Arginosuccinate synthase
Arginosuccinic aciduria Arginosuccinase
Hyperargininemia Arginase
17/12/2014 22
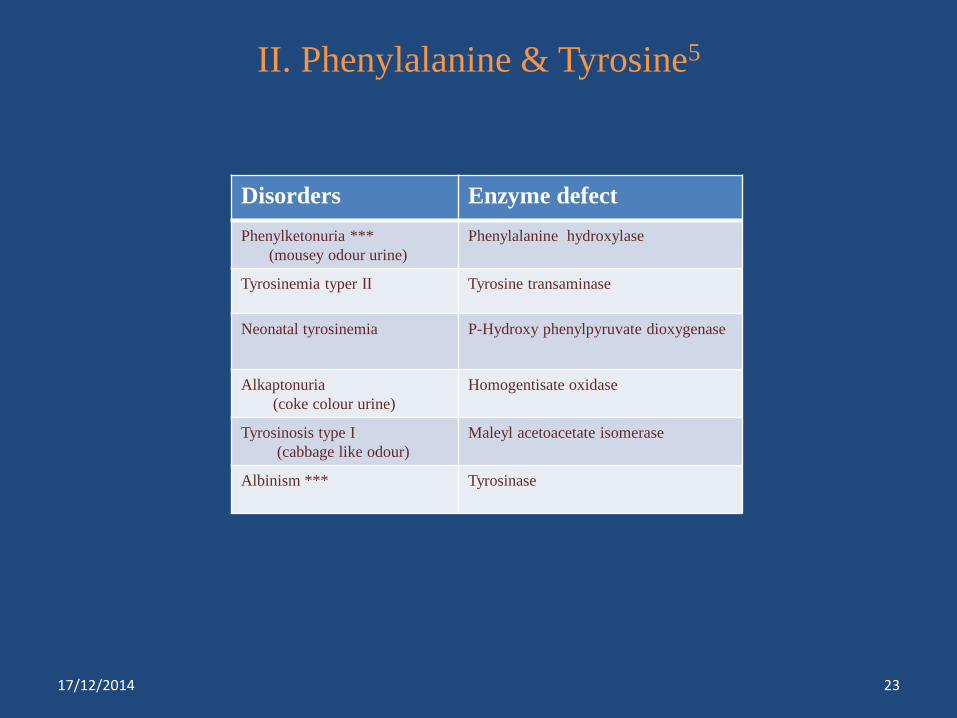
II. Phenylalanine & Tyrosine5
Disorders Enzyme defect
Phenylketonuria ***
(mousey odour urine)
Phenylalanine hydroxylase
Tyrosinemia typer II Tyrosine transaminase
Neonatal tyrosinemia P-Hydroxy phenylpyruvate dioxygenase
Alkaptonuria
(coke colour urine)
Homogentisate oxidase
Tyrosinosis type I
(cabbage like odour)
Maleyl acetoacetate isomerase
Albinism *** Tyrosinase
17/12/2014 23

Phenylketonuria6
17/12/2014 24
Clinical Manifestations
Mental retardation
Defective myelin formation
Hypopigmentation
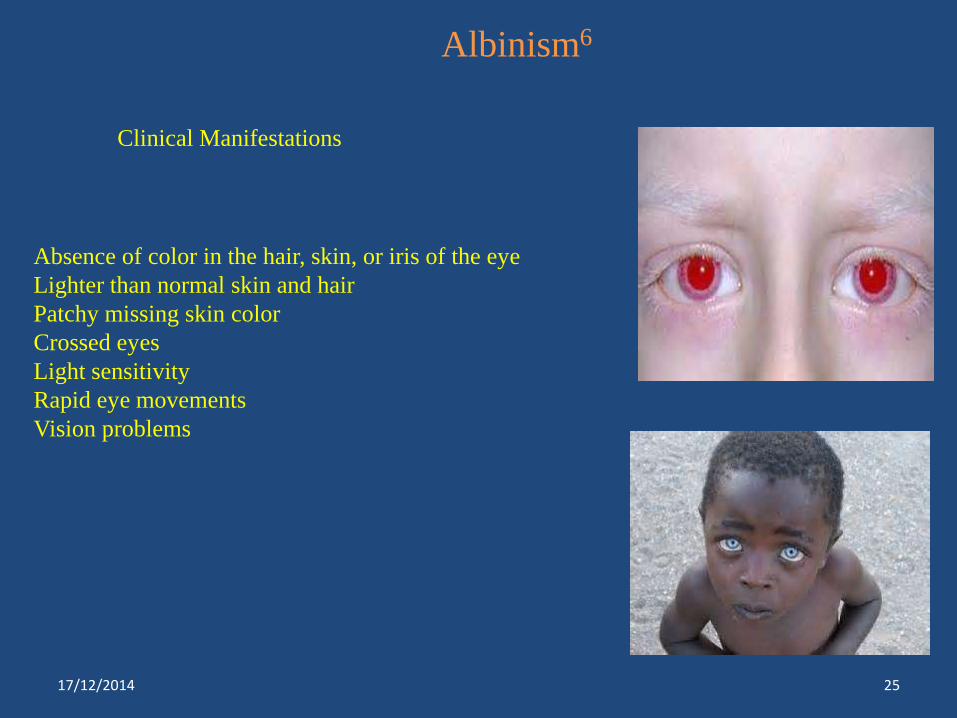
Albinism6
Clinical Manifestations
Absence of color in the hair, skin, or iris of the eye
Lighter than normal skin and hair
Patchy missing skin color
Crossed eyes
Light sensitivity
Rapid eye movements
Vision problems
17/12/2014 25

III. Sulfur Amino acids5
(methionine, cysteine, cystine)
Disorders Enzyme defect
Cystinuria Defect in renal reabsorption
Cystinosis Impairment in cystine utilization
Homocystinuria type I*** Cystathionine synthetase
Homocystinuria type II N 5, N10-Methylene THF reductase
Homocystinuria type III N 5-Methyl THF- homoscysteine
methyltransferase
Cystathionuria Cystathioninase
17/12/2014 26
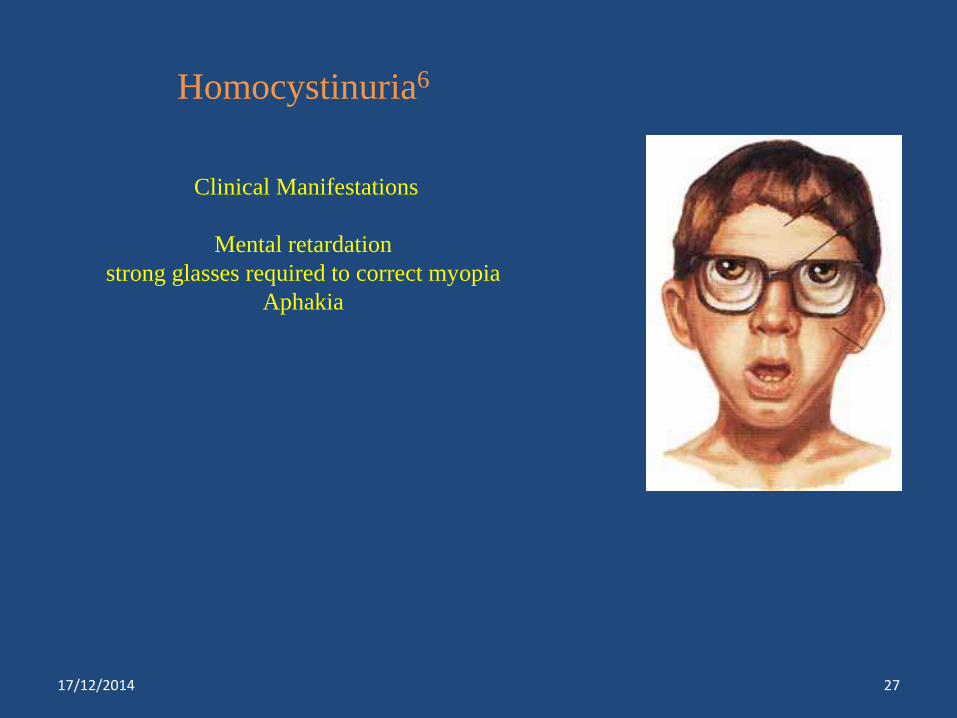
Homocystinuria6
Clinical Manifestations
Mental retardation
strong glasses required to correct myopia
Aphakia
17/12/2014 27

IV. Glycine5
Disorders Enzyme defect
Glycinuria Defect in renal
reabsorption
Primary hyperoxaluria Glycine transaminase
V. Tryptophan5
Disorders Enzyme defect
Hartnup’s disease*** Defective intestinal
absorption
17/12/2014 28
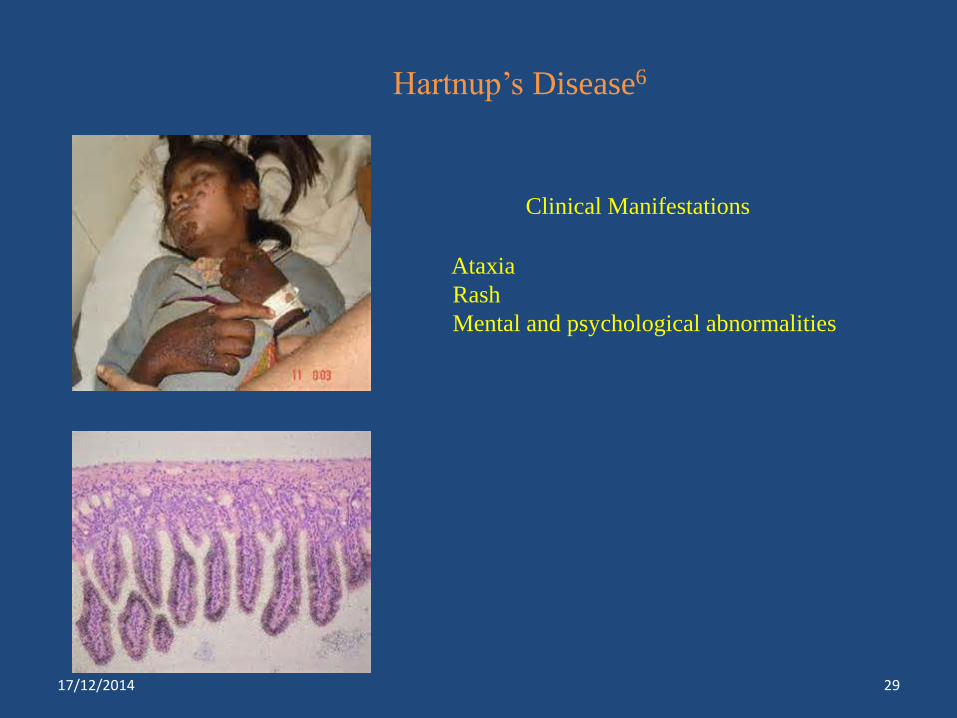
Hartnup’s Disease6
Clinical Manifestations
Ataxia
Rash
Mental and psychological abnormalities
17/12/2014 29

VI. Branched Chain Amino Acids5
(valine,leucine,isoleucine)
Disorders Enzyme defect
Maple syrup urine disease***
(bunrt sugar urine)
Branched chain α-keto acid
dehydrogenase
Intermittent branched chain
ketonuria
Variant of the above enzyme
Hypervalinemia Valine transaminase
Isovaleric acidemia Isovaleryl CoA dehydrogenase
17/12/2014 30
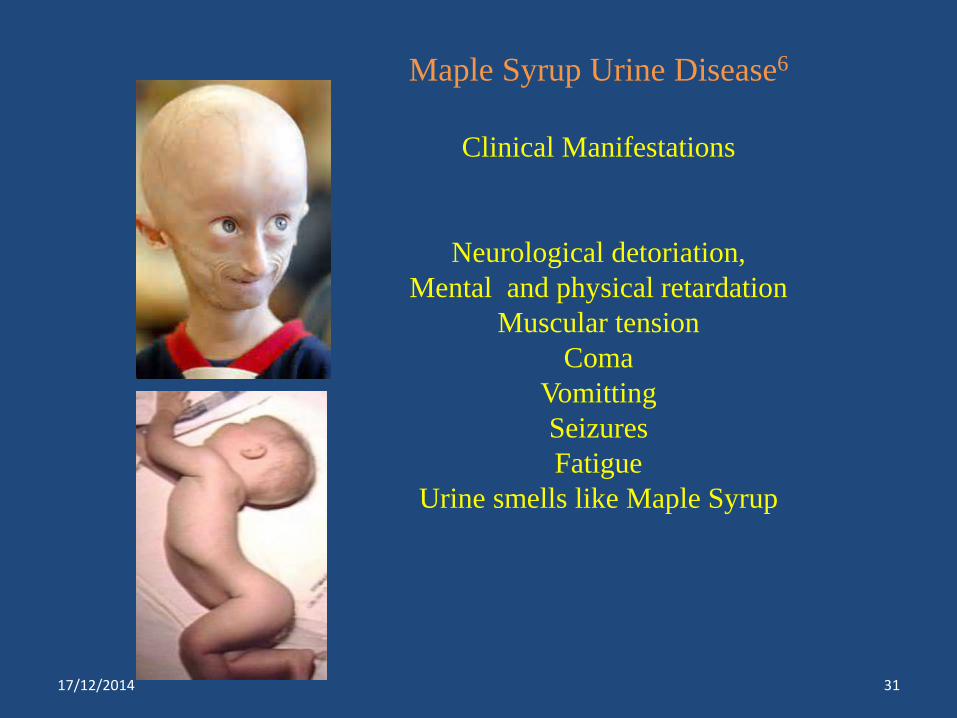
Maple Syrup Urine Disease6
Clinical Manifestations
Neurological detoriation,
Mental and physical retardation
Muscular tension
Coma
Vomitting
Seizures
Fatigue
Urine smells like Maple Syrup
17/12/2014 31

VII. Hystidine5
Disorders Enzyme defect
Histidinemia Histidase
VIII. Proline5
Disorders Enzyme defect
Hyperprolinemiatype I Proline oxidase
17/12/2014 32

DISORDERS OF ORGANIC ACID METABOLISM
17/12/2014 33

Inborn Errors of Organic Acid Metabolism6
Disorders
Methylmalonic acidemia
Proprionic acidemia
Isovaleric acidemia
Glutaric aciduria type II
Dicarboxylic aciduria
17/12/2014 34
DISORDERS OF LIPID METABOLISM
17/12/2014 35
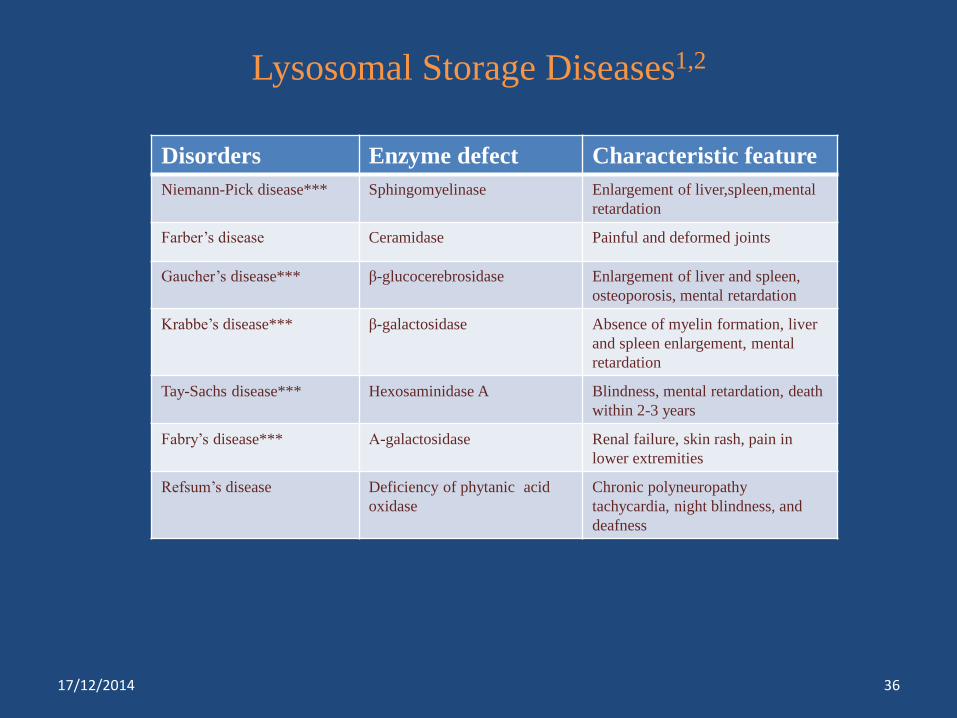
Lysosomal Storage Diseases1,2
Disorders Enzyme defect Characteristic feature
Niemann-Pick disease*** Sphingomyelinase Enlargement of liver,spleen,mental
retardation
Farber’s disease Ceramidase Painful and deformed joints
Gaucher’s disease*** β-glucocerebrosidase Enlargement of liver and spleen,
osteoporosis, mental retardation
Krabbe’s disease*** β-galactosidase Absence of myelin formation, liver
and spleen enlargement, mental
retardation
Tay-Sachs disease*** Hexosaminidase A Blindness, mental retardation, death
within 2-3 years
Fabry’s disease*** Α-galactosidase Renal failure, skin rash, pain in
lower extremities
Refsum’s disease Deficiency of phytanic acid
oxidase
Chronic polyneuropathy
tachycardia, night blindness, and
deafness
17/12/2014 36
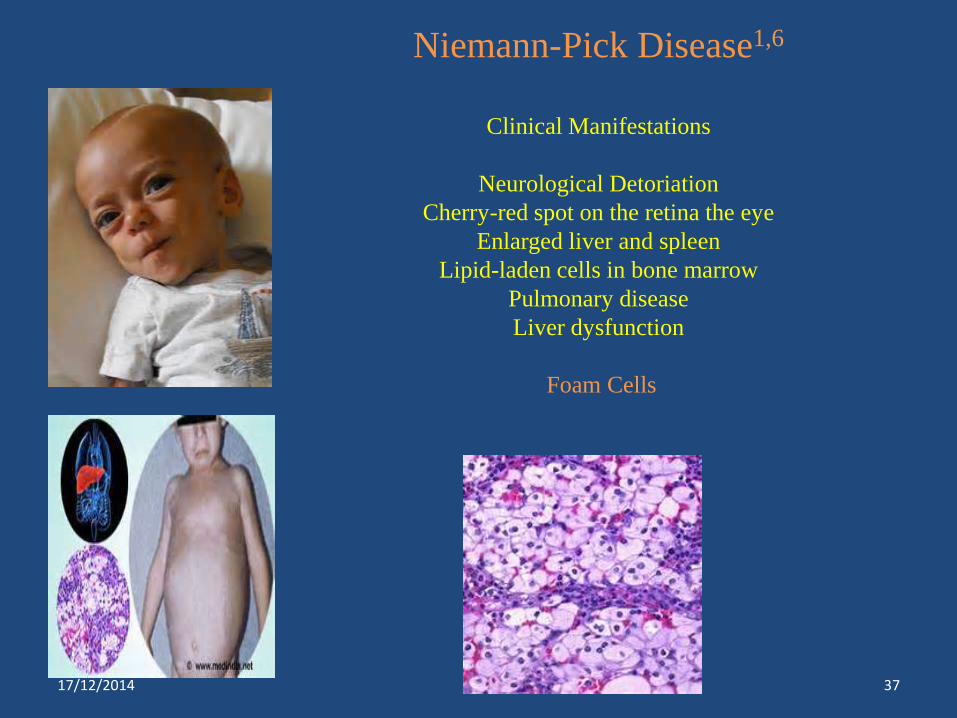
Niemann-Pick Disease1,6
Clinical Manifestations
Neurological Detoriation
Cherry-red spot on the retina the eye
Enlarged liver and spleen
Lipid-laden cells in bone marrow
Pulmonary disease
Liver dysfunction
Foam Cells
17/12/2014 37

Gaucher’s Disease1,6
Gaucher Cell
17/12/2014 38
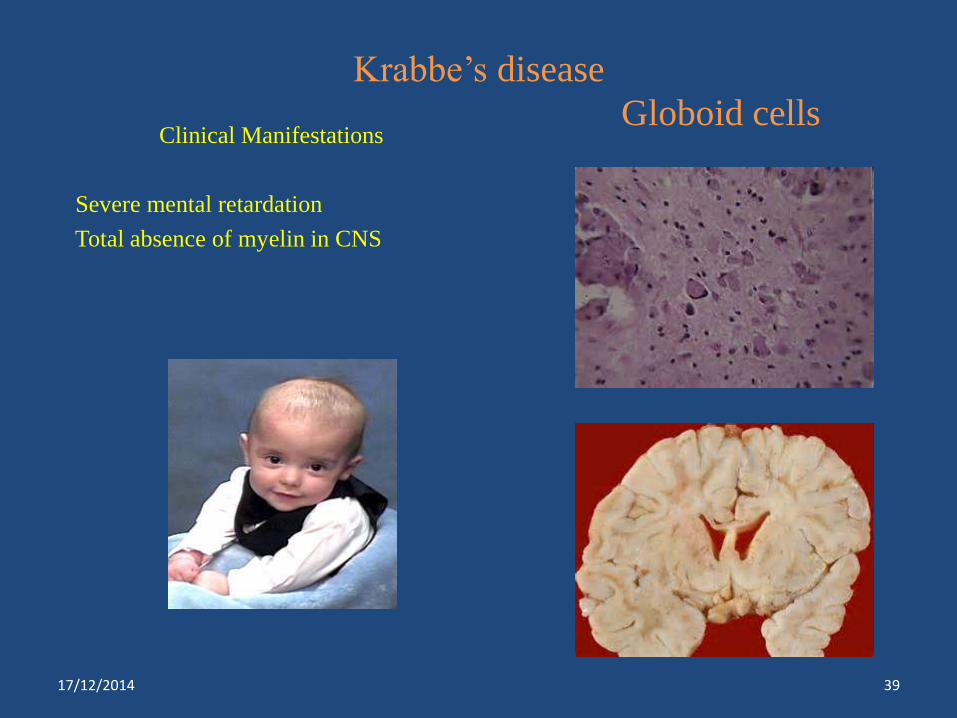
Krabbe’s disease
Globoid cellsClinical Manifestations
Severe mental retardation
Total absence of myelin in CNS
17/12/2014 39
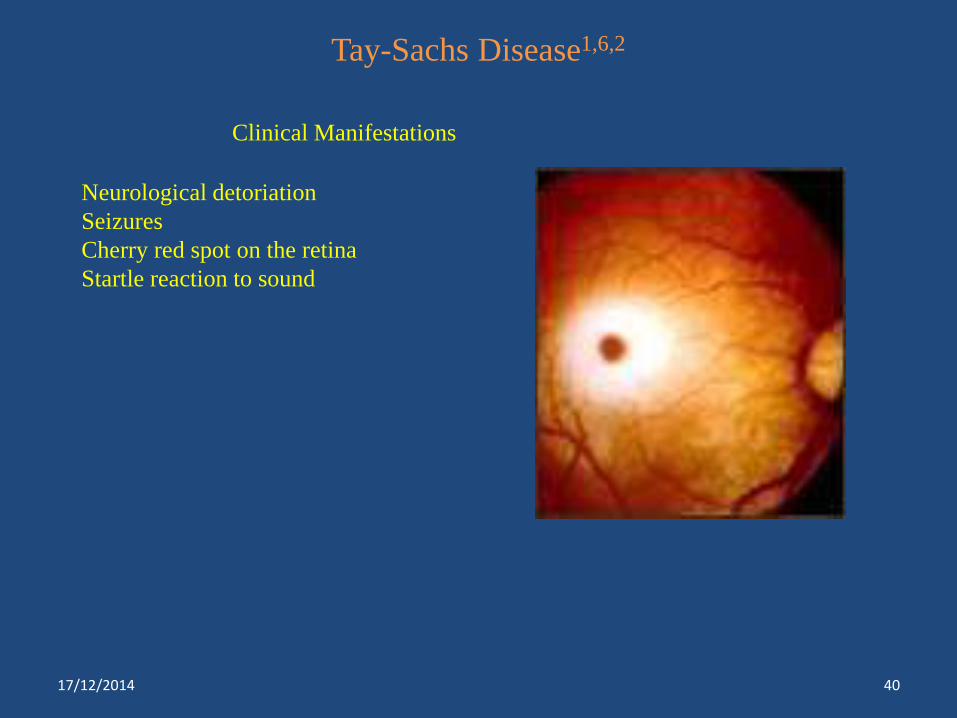
Tay-Sachs Disease1,6,2
Clinical Manifestations
Neurological detoriation
Seizures
Cherry red spot on the retina
Startle reaction to sound
17/12/2014 40
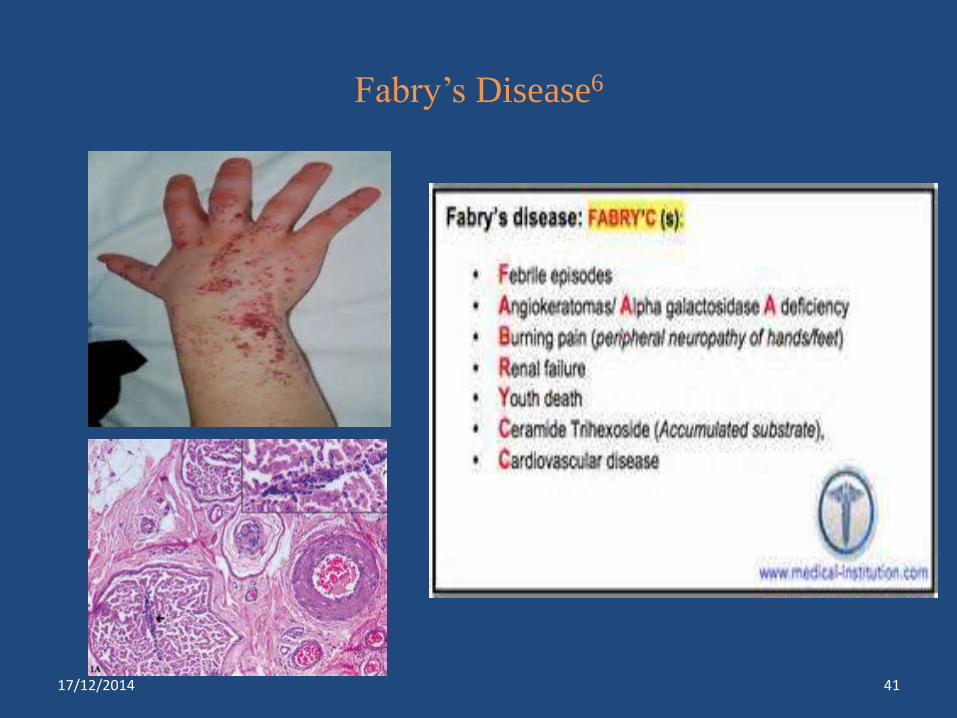
Fabry’s Disease6
17/12/2014 41

DISORDERS OF NUCLEOTIDE METABOLISM
17/12/2014 42

Inborn Errors of Purine Metabolism6
Disorders Enzyme defect
Anabolism
Lesch-Nyhan syndrome***
Catabolism
Gout
Adenylosuccinate lyase deficiency
Adenosine monophosphate deaminase
deficiency,typeI
Adenine phosphoribosyl transferase deficiency
Adenosine deaminase deficiency
Purine nucleotide phosphorylase deficiency
Inborn Errors of Pyrimidine Metabolism6
Disorders Enzyme defect
Anabolism
Orotic aciduria/
Miller’s Syndrome
Orotate phosphoribosyl
transferase
Catabolism Dihydeopyrimidine
dehydeogenase deficiency
17/12/2014 43
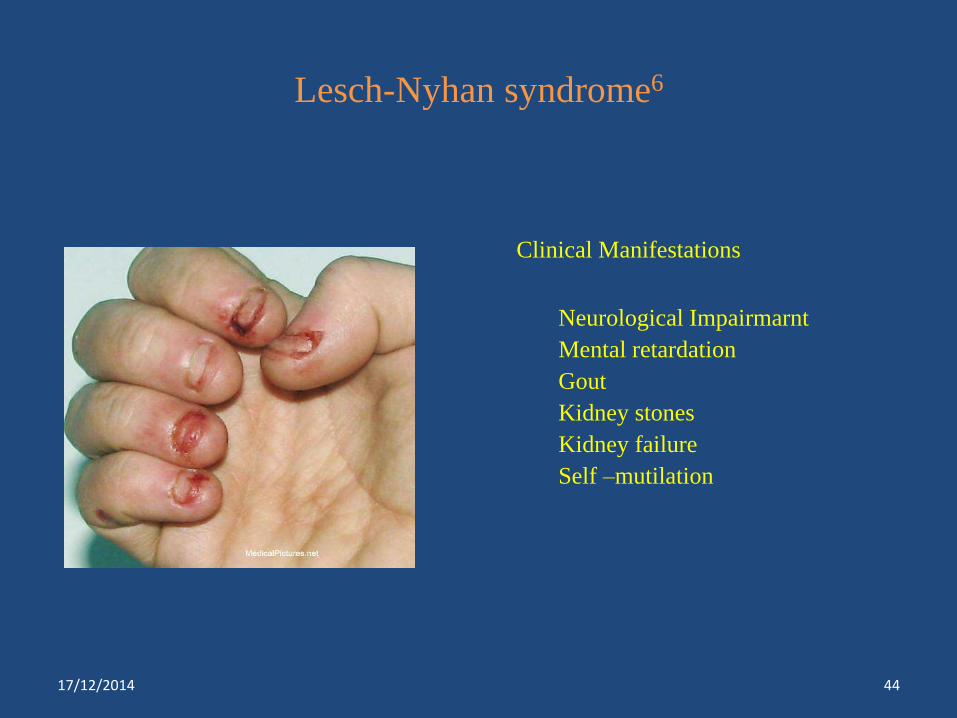
Lesch-Nyhan syndrome6
Clinical Manifestations
Neurological Impairmarnt
Mental retardation
Gout
Kidney stones
Kidney failure
Self –mutilation
17/12/2014 44

Inborn Errors of Porphyrin Metabolism6
Disorders Enzyme defect Characteristic
feature
Acute intermittent porphyria ** Porphobilinogen deaminase
deficiency
Abdominal pain, constipation,
muscle weakness
paresthesias,dysuria,dark
urine,urinary
retention,incontinence,
neuropsychiatric
symptoms,electrolytes
abnormalities SIADH leads to
seizures
17/12/2014 45
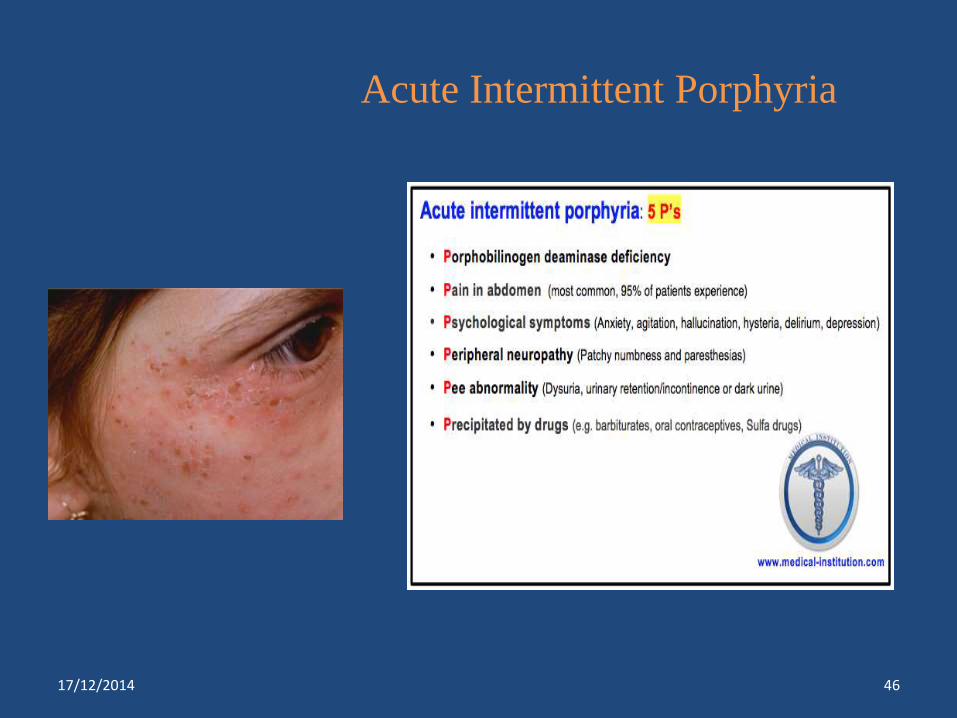
Acute Intermittent Porphyria
17/12/2014 46

Inborn Errors of Mineral and Electrolyte Metabolism6
Transition Metal Electrolyte
Fe
High
Primary iron overload disorder
Hemochromatosis
aceruloplasminemia
Atransferrinemia
Hemosiderosis
Deficiency
Iron deficiency
Na+ & K+
Cl- membrane transport deficiency
Cystic fibrosis
Ca2+
High
Hypercalcemia
Milk alkali syndrome(Burnett’s)
Calcinosis cutis
Dystrophic calcification
Deficiency
Hypocalcemia
Osteomalacia
Albright’s hereditary
osteodystrophy
Pseudohypoparathyroidism
Cu
High
Copper toxicity
Wilson’s disease***
Copper binding ATPase
Deficiency
Menkes disease
Po43-
High -Hyperphosphatemia
Deficiency
Hypophosphatemia
Hypophosphatasia
Zn
High
Zinc toxicity
Acrodermatitis enteropathica
Mg2+
High -Hypermagnesemia
Deficiency
Hypomagnesemia17/12/2014 47
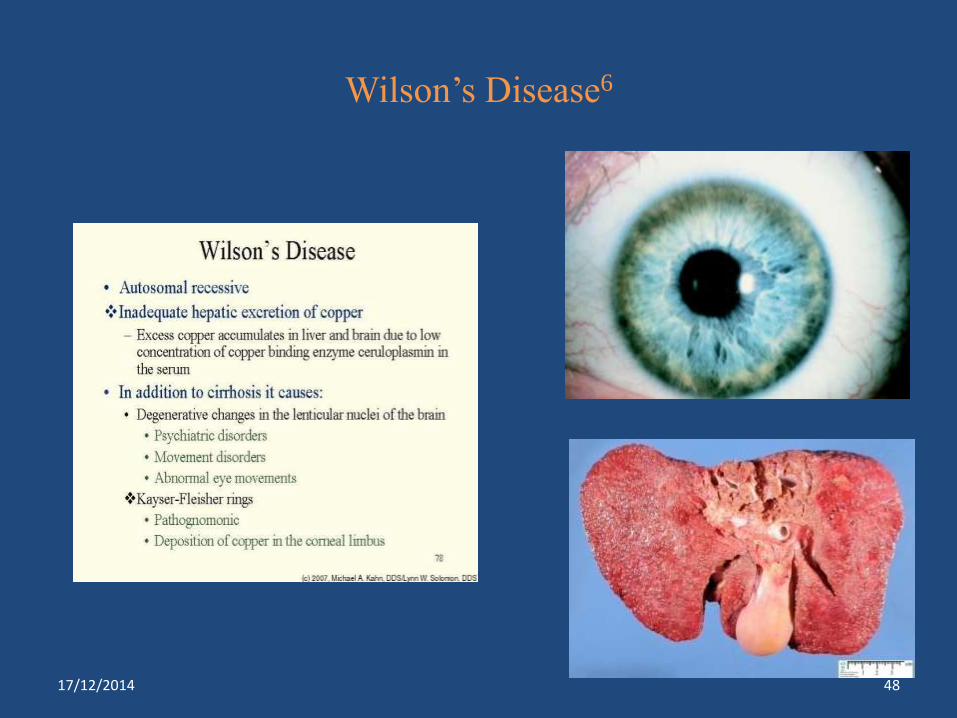
Wilson’s Disease6
17/12/2014 48

Inborn Errors of Mitochondrial Function6
Disorders
Chronic progressive external ophthalmoplegia
Kearns-Sayre Syndrome
Lebers hereditary optic neuropathy
Isolated myopathy
Severe encephalomyopathy
pearsonsyndrome
17/12/2014 49

Inborn Errors of Peroxisomal Function6
Disorders
Zellweger syndrome
Adrenoleukodystrophy
Refsum disease
17/12/2014 50

Inborn Errors of Steroid Metabolism6
Disorders
Lipoid congenital adrenal hyperplasia
Congenital adrenal hyperplasia
Disorders
Sickle cell anemia
Thallasemia
Inborn Errors of Hereditary Anemia3
17/12/2014 51

When to suspect an IEM?6
Every child with unexplained ….
-neurological detoriation
Metabolic acidosis
Hypoglycemia
Inappropriate ketosis
Hypotonia
Cardiomyopathy
Hepatocellular dysfunction
Failure to thrive
Abnormal hair
Odour
…should suspected of having a metabolic disorder
17/12/2014 52

Neonatal Clinical Manifestations of IEMs6
Neurologic Signs:
Poor suck
Lethargy (progressive to coma)
Abnormalities of tone
Loss of reflexes
Seizures
Gastrointestinal Signs:
Poor feeding
Vomitting
Diarrhea
Respiratory Signs
Hyperpnea
Respiratory failure:
Organomegaly
Liver
Heart
17/12/2014 53

DIAGNOSIS
17/12/2014 54

Common Screening Tests6
• Ferric chloride test
• Ninhydrin paper chromatography
• Quantitative measurement of amino acids in plasma and urine
• Ninhydrin post column liquid ion-exchange chromatography
• Urine organic acid analysis by Gas chromatography-mass spectrometry
• Plasma acylcarnitines analysis by mass spectrometry
• Urine purines and pyrimidines analysis by gas chromatography
• Tissue biopsy or necropsy; liver, muscle, brain, bone marrow. Skin biopsy and
fibroblast cultivation for specific enzyme testing
• Specific DNA testing
17/12/2014 55

Diagnosis6
• IEM can be detected in fetus in utero by the examination of blood cells obtained by
amniocentesis and fetoscopy
• New born screening ( must do on all infants in NICU)
PKU
Hemoglobinopathies
MSUD
• Laboratory tests after birth show higher than normal levels of particular metabolites in
the blood and urine
The values are higher in homozygous than in heterozygous carriers
Signs of various defects are usually seen only homozygous carriers
17/12/2014 56

TREATMENT
17/12/2014 57

Management6
• Dietary restriction
• Removal of food in the diet containing the non-degradable metabolite prevent
its accumulation . In those cases of IEM in which the non-degradable
metabolite is endogenous no treatment is available.
• Dietary supplementation or replacement
• Vitamins
• Intermediary metabolites, compounds or drugs that facilitate or retard specific
metabolic pathways
• Dialysis
• Enzyme replacement
• Gene therapy
• Bone marrow or organ transplantation
• Treatment of symptoms and complications
• Prenatal diagnosis and avoidance of pregnancy or abortion of an affected fetus
17/12/2014 58

Treatment of Acutely-Sick Child6
General therapy:
• Maintain vital functions
• Oxygenation
• Hydration
• Acid/base balance
Specific therapy:
• Treat infection
• High dose I.V glucose
• Carnitine supplementation
TO IDENTIFY PRIMARY METABOLIC DISORDER
17/12/2014 59

Therapeutic Measures of IEMs6
• D/C oral intake temporarily
• IVF’s with glucose to give 12-15 mg/kg/min glucose and atleast 60 kcal/kg to
prevent catabolism(may worsen PDH)
• Bicarbonate/citrate carnitine/glycine
• Na benzoate/arginine /citrulline
• Dialysis-not exchange transfusion
• Vitamins-often given in cocktails before dx is known
-biotin, B6, B12, riboflavin, thiamine, folate
17/12/2014 60

Treatment of Genetic Diseases6
• Modify environment e.g., diet , drugs
• Surgical ,correct or repair defect or organ transplantation
• Modify or replace defective gene product, mega dose vitamin therapy or enzyme
replacement
• Replace defective gene
• Correct altered DNA in defective gene
17/12/2014 61

References
• Robbin’s Basic Pathology, Kumar, Abbas, Aster; IXth Edition, pg 218 – 234
• W.A.D. Anderson’s Pathology Vth Edition Chap 31, pg 1041
• Anderson’s Pathology VIIIth Edition chap 3, pg 101 - 104
• Fundamentals of Biochemistry, A.C. Deb
• Text book of Biochemistry, U. Sathyanarayana, U.Chakrapani; Chap 13, 15
• Net resources
17/12/2014 62

THANKYOU
17/12/2014 63