Applied Catalysis B: Environmental 104 (2011) 84–90 Contents lists available at ScienceDirect Applied Catalysis B: Environmental journal homepage: www.elsevier.com/locate/apcatb In situ study of ozone and hybrid plasma Ag–Al catalysts for the oxidation of toluene: Evidence of the nature of the active sites Monica Magureanu a , Daniela Piroi a , Nicolae Bogdan Mandache a , Vasile I. Pârvulescu b,∗ , Viorica Pârvulescu c , Bogdan Cojocaru b , Chris Cadigan d , Ryan Richards d , Helen Daly e , Christopher Hardacre e a Department of Plasma Physics and Nuclear Fusion, National Institute for Lasers, Plasma and Radiation Physics, Magurele-Bucharest, Romania b Department of Chemical Technology and Catalysis, University of Bucharest, Bucharest, Romania c Institute of Physical Chemistry of the Romanian Academy, Bucharest, Romania d Colorado School of Mines, Department of Chemistry and Geochemistry, Golden, CO, USA e School of Chemistry and Chemical Engineering, Queen’s University, Belfast, BT9 5AG, UK article info Article history: Received 8 December 2010 Received in revised form 16 February 2011 Accepted 18 February 2011 Available online 24 February 2011 Keywords: Colloid Silver Oxidation Hydrocarbon Plasma Catalyst abstract Silver colloids have been prepared by reducing AgNO 3 in aqueous solution and embeded in alumina following a sol–gel procedure in the presence of Pluronic 84 ((EO) 19 (PO) 39 (EO) 19 ), as surfactant. Plasma- catalytic experiments aimed at the mineralization of toluene showed that the selectivity to CO 2 was significantly increased in the presence of Ag catalysts compared with results obtained using the plasma alone. In-situ studies of the ozone interaction with catalysts provide an insight into the nature of the active sites of supported silver colloids for mineralization reactions. It is noticeable that when ozone is chemisorbed on embedded Ag colloidal catalysts no change in the silver oxidation state or size is found. The population of the chemisorbed species is higher at lower temperatures, where the non-selective decomposition of ozone is smaller. The catalysts exhibit high stability, preserving the structural and textural properties after the catalytic tests, that is indeed very important in the presence of ozone. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Chemical oxidation through the use of agents such as ozone, hydrogen peroxide, chlorine or chlorine dioxide is currently reported for mineralization of organic compounds [1]. However, the use of chlorine compounds is not environmentally acceptable while both hydrogen peroxide and ozone are, in general, too expen- sive for general use due to the method of production. Additionally, too high a concentration of ozone may lead to supplementary pollu- tion concerns. Methods using microorganism to remove VOCs have been reported. These utilize special reactors, usually referred to as biofilters [2]; however, their operation requires special condi- tions before and during usage. In spite of the drawbacks, ozone has received attention for total mineralization of organic compounds. Among others, benzene, toluene, xylene and ethylbenzene (BTEX’s) are the most common contaminants released from water and soil. Several treatment methods regarding their VOCs elimina- tion have been reported. However, in many cases these require the ∗ Corresponding author. Tel.: +40 21 410 02 41; fax: +40 21 410 02 41. E-mail address: [email protected] (V.I. Pârvulescu). transfer of the VOC from one phase to another via vapour extraction or the adsorption on activated carbon [3], or advanced destruction methods involving catalytic oxidation [4] or incineration. These methods gave reasons to develop new methodologies for the treat- ment of aromatic VOCs in the gas phase. In this context, one possible option is the gas phase reaction between ozone and VOCs, lead- ing to a reduction of emitted contaminants. Practical applications have already been reported [5]. An important advantage of using ozone is the capacity to eliminate the polycyclic aromatic hydro- carbons, which principally originate from incomplete combustion and pyrolytic processes at high temperatures that are recognized as important pollutants with carcinogenic and mutagenic properties [6]. However, air treatment ozonation in gaseous phase has not been explored in detail. Studies carried out with alkenes to elucidate the reaction mechanism showed that the hydroxyl radical (OH • ) forms as a reaction product and reacts ca. 4–5 orders of magnitude faster than ozone and also reacts rapidly with carbonyls, especially aldehydes, that are major products of the ozone–alkene reaction [7]. Plasma-catalytic hybrid systems have been studied extensively in the last years for the oxidation of various VOCs. The main advan- 0926-3373/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.apcatb.2011.02.025

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

It
MVHa
b
c
d
e
a
ARRAA
KCSOHPC
1
hrtwsttbatr
(at
0d
Applied Catalysis B: Environmental 104 (2011) 84–90
Contents lists available at ScienceDirect
Applied Catalysis B: Environmental
journa l homepage: www.e lsev ier .com/ locate /apcatb
n situ study of ozone and hybrid plasma Ag–Al catalysts for the oxidation ofoluene: Evidence of the nature of the active sites
onica Magureanua, Daniela Piroia, Nicolae Bogdan Mandachea, Vasile I. Pârvulescub,∗,iorica Pârvulescuc, Bogdan Cojocarub, Chris Cadigand, Ryan Richardsd,elen Dalye, Christopher Hardacree
Department of Plasma Physics and Nuclear Fusion, National Institute for Lasers, Plasma and Radiation Physics, Magurele-Bucharest, RomaniaDepartment of Chemical Technology and Catalysis, University of Bucharest, Bucharest, RomaniaInstitute of Physical Chemistry of the Romanian Academy, Bucharest, RomaniaColorado School of Mines, Department of Chemistry and Geochemistry, Golden, CO, USASchool of Chemistry and Chemical Engineering, Queen’s University, Belfast, BT9 5AG, UK
r t i c l e i n f o
rticle history:eceived 8 December 2010eceived in revised form 16 February 2011ccepted 18 February 2011vailable online 24 February 2011
a b s t r a c t
Silver colloids have been prepared by reducing AgNO3 in aqueous solution and embeded in aluminafollowing a sol–gel procedure in the presence of Pluronic 84 ((EO)19(PO)39(EO)19), as surfactant. Plasma-catalytic experiments aimed at the mineralization of toluene showed that the selectivity to CO2 wassignificantly increased in the presence of Ag catalysts compared with results obtained using the plasmaalone. In-situ studies of the ozone interaction with catalysts provide an insight into the nature of the
eywords:olloidilverxidationydrocarbonlasma
active sites of supported silver colloids for mineralization reactions. It is noticeable that when ozone ischemisorbed on embedded Ag colloidal catalysts no change in the silver oxidation state or size is found.The population of the chemisorbed species is higher at lower temperatures, where the non-selectivedecomposition of ozone is smaller. The catalysts exhibit high stability, preserving the structural andtextural properties after the catalytic tests, that is indeed very important in the presence of ozone.
© 2011 Elsevier B.V. All rights reserved.
atalyst. Introduction
Chemical oxidation through the use of agents such as ozone,ydrogen peroxide, chlorine or chlorine dioxide is currentlyeported for mineralization of organic compounds [1]. However,he use of chlorine compounds is not environmentally acceptablehile both hydrogen peroxide and ozone are, in general, too expen-
ive for general use due to the method of production. Additionally,oo high a concentration of ozone may lead to supplementary pollu-ion concerns. Methods using microorganism to remove VOCs haveeen reported. These utilize special reactors, usually referred tos biofilters [2]; however, their operation requires special condi-ions before and during usage. In spite of the drawbacks, ozone haseceived attention for total mineralization of organic compounds.
Among others, benzene, toluene, xylene and ethylbenzeneBTEX’s) are the most common contaminants released from waternd soil. Several treatment methods regarding their VOCs elimina-ion have been reported. However, in many cases these require the
∗ Corresponding author. Tel.: +40 21 410 02 41; fax: +40 21 410 02 41.E-mail address: [email protected] (V.I. Pârvulescu).
926-3373/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.apcatb.2011.02.025
transfer of the VOC from one phase to another via vapour extractionor the adsorption on activated carbon [3], or advanced destructionmethods involving catalytic oxidation [4] or incineration. Thesemethods gave reasons to develop new methodologies for the treat-ment of aromatic VOCs in the gas phase. In this context, one possibleoption is the gas phase reaction between ozone and VOCs, lead-ing to a reduction of emitted contaminants. Practical applicationshave already been reported [5]. An important advantage of usingozone is the capacity to eliminate the polycyclic aromatic hydro-carbons, which principally originate from incomplete combustionand pyrolytic processes at high temperatures that are recognized asimportant pollutants with carcinogenic and mutagenic properties[6].
However, air treatment ozonation in gaseous phase has not beenexplored in detail. Studies carried out with alkenes to elucidatethe reaction mechanism showed that the hydroxyl radical (OH•)forms as a reaction product and reacts ca. 4–5 orders of magnitude
faster than ozone and also reacts rapidly with carbonyls, especiallyaldehydes, that are major products of the ozone–alkene reaction[7].Plasma-catalytic hybrid systems have been studied extensivelyin the last years for the oxidation of various VOCs. The main advan-

lysis B
tit[gfhheniu
ttsobce
goatTtb[fws
stydcrbcivaosfr
2
itdra1prstoTp
M. Magureanu et al. / Applied Cata
age of non-thermal plasmas is the fact that the available energys used selectively to accelerate electrons and not spent in heatinghe entire gas stream, which remains close to room temperature4,8]. The high-energy electrons excite, dissociate, and ionize theas molecules, generating chemically active species, which reacturther with the VOC molecules and decompose them. Thus, aighly reactive environment is created at low temperature. Plasmaas been found to be an effective method for VOC decompositionspecially at low VOC concentrations, where the conventional tech-ologies are less suitable for treatment [9]. The drawback consists
n the low selectivity of the process towards mineralization withndesirable by-products being reported in many studies [8,10,11].
By combining non-thermal plasma and heterogeneous catalysis,he high efficiency of non-thermal activation and the high selec-ivity of catalysts can lead to a synergetic effect. Increased CO2electivity in the presence of catalysts as compared with the valuesbtained with plasma in the absence of the catalyst has frequentlyeen reported [11–15]. Improved results were reported in plasma-atalytic systems as compared with thermally activated catalysis,specially at low catalyst temperatures [16].
Transition metal oxides, especially manganese oxides, have, ineneral, been used [17] to promote VOC oxidation using atomicxygen due to their ability to decompose ozone. Furthermore, cat-lysts containing Ag were reported to exhibit a higher activity inhe decomposition of ozone as compared with other oxides [18].he influence of the support and its acidity in ozone decomposi-ion was examined by impregnating different loadings of silver onoth microporous (H-Beta) and mesoporous (H-MCM-41) supports19] and the acidic materials were shown to exhibit a lower activityor the decomposition of ozone. Recently, silver supported on SiO2as reported to be disrupted (particles broken down into smaller
izes) by ozone, leading to supported small Ag oxide particles [20].The aim of this study is to present a new route to generate stable
upported silver catalysts under an ozone atmosphere and examineheir behavior in the oxidation of toluene. Plasma assisted catal-sis was carried out in a two-stage configuration consisting of aielectric barrier discharge (DBD) and the alumina supported Agatalysts placed downstream of the plasma reactor. In this configu-ation, ozone was generated in the plasma in the desired amountsy adjusting plasma operation conditions and was fed through theatalyst bed thus influencing the reaction pathway. In addition, then-situ study of the ozone interaction with these catalysts preparedia embedding of pre-synthesised silver colloids in a surfactantssisted alumina support synthesis in order to examine the naturef the active sites of silver for mineralization reactions. Previoustudies demonstrated that such catalysts may be very effectiveor the selective conversion of NOx to N2 using hydrocarbon SCReactions [21].
. Experimental
Al2O3 support was prepared from 98.5 g aluminum butox-de (Al(OBu)3) which was dissolved in isobutyl alcohol and thenhe mixture 17.2 g Pluronic 84 ((EO)19(PO)39(EO)19), previouslyissolved in isobutyl alcohol, was added as the surfactant. Theesulting mixture was then refluxed at 70 ◦C for 6 h. Water wasdded and the reflux continued at 80 ◦C for 10 h and, thereafter, at00 ◦C for 20 h. The catalysts containing silver used silver colloidsrepared by reducing AgNO3 in an aqueous solution following aecently reported procedure [21]. In a typical preparation, 0.294 g
odium citrate was added to 50 mL of 10 mM AgNO3 aqueous solu-ion in an ice bath. NaBH4 (0.019 g) was added to the solution atnce with strong stirring and a black powder formed in solution.he solution/precipitate, was then filtered, and dried at room tem-erature in a vacuum oven. The Ag colloid dissolved in water was: Environmental 104 (2011) 84–90 85
then added to the aluminum hydroxide gel under vigourous stir-ring, resulting in samples containing 3 and 5 wt% Ag which weredenoted as AlAg23 and AlAg25, respectively. Additionally, a AlAg13sample, containing 3 wt% Ag, was prepared changing the molarratio alkoxide:alcohol:water to 1:10:25. The samples were aged atroom temperature for 48 h, dried under vacuum at 110 ◦C, and thencalcined at 500 ◦C with a slope of 0.5 ◦C min−1.
The Ag content was confirmed by ICP-AES analysis. The pre-pared catalysts were studied ex-situ by BET nitrogen physisorptionusing a Micromeritics ASAP-2010 automated instrument, pow-der X-ray diffraction (XRD) with a Shimadzu XRD-7000 with CuK� radiation (� = 1.5418 A, 40 kV, 40 mA), transmission electronmicroscopy (TEM) with a JEOL JEM-1010 instrument, and X-rayphotoelectron spectroscopy (XPS) (Specs GmbH) as well as in-situunder an oxygen-ozone atmosphere by diffuse reflectance UV–Vis(PerkinElmer Lambda 650S) and infrared spectroscopy (BrukerEquinox 55 spectrometer) (DR-UV–Vis and DRIFTS).
Ozone was generated with a Trio3gen Ozone 1 instrument. Forthe in-situ experiments with ozone a concentration of ozone of360 ppm in a 0.075 L/min flow of He was used. The concentrationof ozone generated in the plasma varied in the range 300–550 ppmover the SIE range used (80–350 J/L). The ozone concentration wasmeasured with an ozone detector Ozomat MP, Anseros.
Time-on-stream experiments were carried out using a concen-tration of toluene of 50 ppmv with a total flow rate of 0.51 L/min.The concentration of ozone at the entrance (1000 ppm) and at theexit of the reactor was and was measured with a an Ozone Moni-tor Model 460 M, from Teledyne Instruments. The effluent gas wasanalysed by gas chromatography, using a Shimadzu GC-2014 withFID detector. A gas analyser (Ultramat 6, Siemens) was coupledon-line to monitor continuously the concentrations of CO2 and COresulting from toluene oxidation.
The non-thermal plasma was generated in a DBD reactor withcoaxial geometry, packed with spherical quartz pellets of 1 mmdiameter. The experiments were carried out in dry air containing50 ppm toluene. The reactor was made of quartz, and had an outerdiameter of 22 mm with a wall thickness of 1.5 mm. The inner elec-trode was a copper rod of 11 mm diameter and was placed on theaxis of the reactor. The outer electrode was painted with silver pasteon the outside of the tube on a length of 10 cm. The electrical circuitis based on a high voltage transformer with a transformation ratioTR = 300, which provides sinusoidal voltage at 50 Hz frequency. Thesinusoidal voltage of 11–23 kV amplitude was applied to the innerelectrode, while the outer electrode was connected with ground.
The discharge voltage was measured by a high voltage probe(Tektronix P6015). The discharge current was determined from thevoltage drop across a shunt resistor (Rs = 3 �) connected in serieswith the earthed electrode. The total charge dissipated in the dis-charge was measured with a non-inductive capacitor (C = 1 �F),placed instead of the shunt resistor. The discharge voltage, currentand total charge were monitored by a digital oscilloscope (Tek-tronix TDS 2022). The average electrical power dissipated in thedischarge was calculated by the Lissajous method [22]. The Lis-sajous figure, obtained by plotting the total charge versus appliedvoltage, is a parallelogram, whose area is equal to the energydeposited in the discharge in a cycle [22]. The average power inthe discharge can be calculated by multiplying this area with thefrequency of the applied voltage.
This method allows the determination of toluene concentrationsbefore and after plasma treatment (and implicitly the calculationof toluene conversion), as well as the detection of eventual organic
reaction products resulting from toluene decomposition in plasma.Toluene conversion was determined by the ratio between theconcentration of toluene in the effluent gas and the initial tolueneconcentration. Since the only gaseous reaction products were COand CO2, the selectivity towards mineralization was defined as the
86 M. Magureanu et al. / Applied Catalysis B: Environmental 104 (2011) 84–90
Ft
ro
1oTwtfs
3
3
drott
bra
ioiftFaowca2r
3
ste
Fig. 2. Average power dissipated in the DBD as a function of the amplitude of theapplied voltage.
Small amounts of polymeric deposits were observed on the innerelectrode and on the walls of the plasma reactor, amounting to lessthan 5% of the toluene converted.
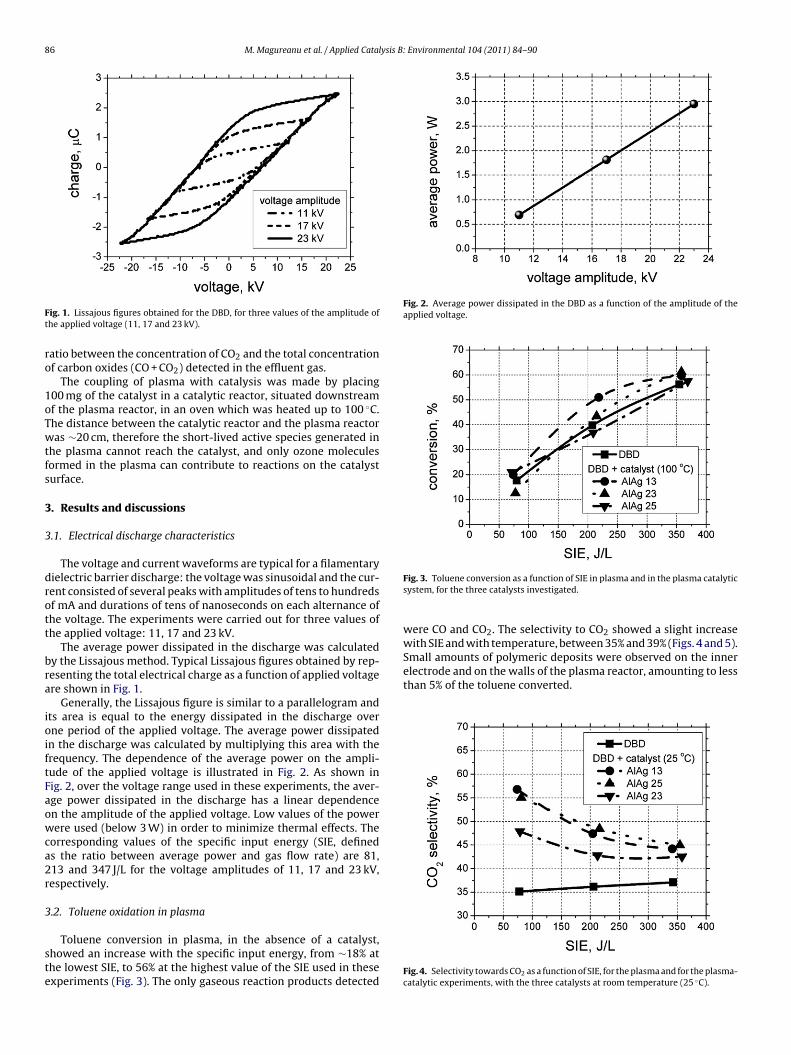
ig. 1. Lissajous figures obtained for the DBD, for three values of the amplitude ofhe applied voltage (11, 17 and 23 kV).
atio between the concentration of CO2 and the total concentrationf carbon oxides (CO + CO2) detected in the effluent gas.
The coupling of plasma with catalysis was made by placing00 mg of the catalyst in a catalytic reactor, situated downstreamf the plasma reactor, in an oven which was heated up to 100 ◦C.he distance between the catalytic reactor and the plasma reactoras ∼20 cm, therefore the short-lived active species generated in
he plasma cannot reach the catalyst, and only ozone moleculesormed in the plasma can contribute to reactions on the catalysturface.
. Results and discussions
.1. Electrical discharge characteristics
The voltage and current waveforms are typical for a filamentaryielectric barrier discharge: the voltage was sinusoidal and the cur-ent consisted of several peaks with amplitudes of tens to hundredsf mA and durations of tens of nanoseconds on each alternance ofhe voltage. The experiments were carried out for three values ofhe applied voltage: 11, 17 and 23 kV.
The average power dissipated in the discharge was calculatedy the Lissajous method. Typical Lissajous figures obtained by rep-esenting the total electrical charge as a function of applied voltagere shown in Fig. 1.
Generally, the Lissajous figure is similar to a parallelogram andts area is equal to the energy dissipated in the discharge overne period of the applied voltage. The average power dissipatedn the discharge was calculated by multiplying this area with therequency. The dependence of the average power on the ampli-ude of the applied voltage is illustrated in Fig. 2. As shown inig. 2, over the voltage range used in these experiments, the aver-ge power dissipated in the discharge has a linear dependencen the amplitude of the applied voltage. Low values of the powerere used (below 3 W) in order to minimize thermal effects. The
orresponding values of the specific input energy (SIE, defineds the ratio between average power and gas flow rate) are 81,13 and 347 J/L for the voltage amplitudes of 11, 17 and 23 kV,espectively.
.2. Toluene oxidation in plasma
Toluene conversion in plasma, in the absence of a catalyst,howed an increase with the specific input energy, from ∼18% athe lowest SIE, to 56% at the highest value of the SIE used in thesexperiments (Fig. 3). The only gaseous reaction products detected
Fig. 3. Toluene conversion as a function of SIE in plasma and in the plasma catalyticsystem, for the three catalysts investigated.
were CO and CO2. The selectivity to CO2 showed a slight increasewith SIE and with temperature, between 35% and 39% (Figs. 4 and 5).
Fig. 4. Selectivity towards CO2 as a function of SIE, for the plasma and for the plasma-catalytic experiments, with the three catalysts at room temperature (25 ◦C).

M. Magureanu et al. / Applied Catalysis B: Environmental 104 (2011) 84–90 87
Fc
3
to
ttitawrtprmacttep
sctiosdahct
dilwt
imC
ig. 5. Selectivity towards CO2 as a function of SIE, for the plasma and for the plasma-atalytic experiments, with the three catalysts heated at 100 ◦C.
.3. Toluene oxidation in plasma-catalytic experiments
Toluene conversion is plotted in Fig. 3 as a function of SIE forhe plasma-catalytic experiments. The results shown here werebtained with the catalysts heated at 100 ◦C.
Toluene conversion was not significantly enhanced by the addi-ion of catalysts, neither at room temperature, not at higheremperature (100 ◦C), as seen in Fig. 3; however, a significantncrease in the selectivity to CO2 was observed. Figs. 4 and 5 showhe selectivity towards CO2 as a function of SIE, for the plasma in thebsence of the catalyst and for the plasma-catalytic experimentsith the three catalysts at room temperature (25 ◦C) and 100 ◦C,
espectively. The concentration of CO2 increased significantly inhe presence of catalysts, as compared with the values obtained bylasma in the absence of the catalyst, while the CO concentrationemained the same as in the plasma-only experiments. As it wasentioned above no other gaseous reaction products except CO
nd CO2 were detected. Although the toluene conversion was nothanged significantly in the presence of the catalysts as comparedo plasma alone, its presence results in the increase in CO2 concen-ration due to an advanced oxidation of toluene on the catalyst. Anvidence of this behavior is the drastic diminution of the condensedolyaromatic compounds.
As shown in Figs. 4 and 5, the selectivity towards CO2 increasedignificantly in the presence of all the investigated catalysts, asompared to the results obtained using plasma alone. At roomemperature AlAg13 and AlAg25 performed equally well, improv-ng the CO2 selectivity by up to 20% as compared with the resultsbtained using the plasma without the catalyst, while AlAg23howed slightly lower values of the selectivity. A decreasing ten-ency was observed for the CO2 selectivity when increasing SIE, forll the three catalysts. However, this is compensated by the muchigher conversion at high SIE, so that the total amount of tolueneonverted into CO2 was ∼3 times higher at 347 J/L SIE as comparedo the one obtained at 81 J/L.
For the AlAg25 catalyst the increase in temperature up to 100 ◦Cid not lead to a further improvement in CO2 selectivity. A slight
ncrease in selectivity was observed when heating the AlAg23 cata-yst up to 100 ◦C, from 43–48% to 45–55%. The highest selectivities
ere obtained using the AlAg13 catalyst. In this case, the CO2 selec-ivity was in the range 50–65%.
The evolution of CO2 selectivity as a function of time is shownn Fig. 6 for the AlAg13 catalyst heated at 80 ◦C and for the inter-
ediate plasma power (1.8 W, corresponding to SIE 210 J/L). TheO2 selectivity was found to decrease slightly with time, while
Fig. 6. Selectivity towards CO2 as a function of treatment time, for the AlAg13catalyst heated at 80 ◦C and for the intermediate plasma power (1.8 W).
the CO selectivity increased slightly, since CO and CO2 were theonly detected reaction products. Toluene conversion remainedunchanged over the investigated time interval.
A lower SIE will correspond to less ozone that instead of the het-erogeneous catalytic route will favor the radicalic route in whichthe carbonaceous deposits will block the catalyst surface. How-ever, unreacted toluene can be ruled out, since its concentrationis higher at lower SIE and decreases with increasing SIE. The role ofozone in the oxidation of hydrocarbons and CO was investigated in[23] using a combination of non-thermal plasma and �-Al2O3. Theauthors found that that in the absence of a catalytic active speciessuch silver in this case, the catalytic function of �-Al2O3 was deac-tivated in time. However, the deactivation of �-Al2O3 did not occurin the absence of hydrocarbons, indicating that the characteristicproducts of a discharge in air (e.g. NOx, OH and O radicals, O3) donot cause a measurable deactivation of the catalyst [23]. Rolandet al. [23] concluded that CO2 resulting from the mineralizationof hydrocarbons is responsible for poisoning the catalytic sites of�-Al2O3 that is very correct for that example.
Regarding the mechanism responsible for pollutants removal intwo-stage plasma-catalytic hybrid systems, it is well known thatthe short-lived active species generated in the plasma disappearbefore they reach the catalyst, due to their high reactivity and shortlifetime. Therefore, the main role played by plasma in this case maybe to transform the toluene into an oxygenated molecule using theactivated ozone which is more tractable towards oxidation on thecatalyst surface [9]. However, in the present case no other com-pounds except un-reacted toluene, CO and CO2 were detected byGC in the effluent gas from the plasma reactor. Another impor-tant possibility is that the ozone generated in the plasma has asufficiently long lifetime to reach the catalyst and react with thepollutants on its surface, resulting either in increased conversionand/or selectivitiy towards the mineralization product, CO2. Theozone influence on the catalyst properties is, therefore, essential inorder to understand the reaction mechanism.
3.4. Catalyst characterization
Textural characterization indicated that these materials aremesoporous with a surface area higher than 400 m2 g−1 and abimodal pore size distribution with pores of 3 and 6 nm (Table 1).XRD patterns recorded at small angles did not indicate the presence
of any mesoporous structural organization. The patterns shown inFig. 7 correspond to the reflection planes of bulk Ag (2� = 38.1◦,44.3◦, 64.4◦ and 77.4◦ corresponding to the (1 1 1), (2 0 0), (2 2 0)and (3 1 1) reflexion planes respectively) as well as of �-alumina
88 M. Magureanu et al. / Applied Catalysis B: Environmental 104 (2011) 84–90
Table 1Textural characterization of the investigated catalysts.
Sample Ag content(wt%)
BET surfacearea (m2 g−1)
Pore volume(cm3 g−1)
Pore size (nm)
AlM1 0 524 1.02 Monomodal, 7.5
((ctos
turfieso3iris
abiathsd
3outc
FA
Fig. 8. XPS spectra of Ag 3d5/2 level for AlAg13: (a) fresh; (b) after 24 h; (c) after 48 h.
AlAg13 3 408 0.97 Monomodal, 7.5AlAg23 3 333 0.68 Bimodal, 3 and 7.5AlAg25 5 336 0.58 Bimodal, 3 and 6
2� = 37.5◦, 39.5◦, 46.1◦, 60.3◦ and 67◦ corresponding to (3 1 1),2 2 2), (4 0 0), (5 1 1) and (4 4 0) reflexion planes, respectively), indi-ated that at least some of the silver is present as large particles ofhe order of 24 nm from the Debye–Scherrer equation. Exposure tozone caused no significant change in the XRD patterns although amall decrease in the silver features was observed.
TEM confirmed the preservation of the mesoporous texture afterhe incorporation of the Ag colloid in the alumina support (Fig-re not shown); however, the individual particles could not beesolved in this system even with the use of high angle annular darkeld (HAADF) experiments confirming these particles are partiallymbedded in the alumina matrix. XPS confirmed the presence ofilver, and the binding energy analysis showed the preservationf the Ag0 state even after calcination. The binding energy of Agd5/2 level was observed at 368.6 eV for both silver loadings which
s typical for Ag0 (Fig. 8) [24]. Spectra collected after 24 and 48 h,espectively, showed no change both in the binding energy andn the XPS Ag/Al atomic ratio. This is a very good evidence of thetability of these catalysts.
Fig. 9a shows DR-UV–Vis spectra collected at 30 ◦C under heliumnd ozone. The main feature of the 3% Ag sample at 30 ◦C underoth helium and ozone is a band at 225 nm which is assigned to
solated Ag+ ions [25]. No change in the band position was detecteds a function of temperature under helium (Fig. 9b). Under ozonehe band at 225 nm increases in intensity and shifts to 218 nm oneating to 100 ◦C (Fig. 9c). Heating the 3% Ag sample under ozonehows no change in species but an increase in intensity of the bandue to Ag+ species.
The UV–DR spectrum of the AlAg25 sample under helium at0 ◦C is also dominated by a band at 225 nm; there is no effect
f loading on the Ag species under helium. However, at 30 ◦Cnder ozone, bands due to Agnı+ (260 nm) and large silver clus-ers (320 nm) are present. Under ozone, Ag species are changedompared to the fresh catalyst even at 30 ◦C for this higher loading
ig. 7. XRD patterns of AlAg23 (a) before and (b) after exposure to ozone and oflAg25 (c) before and (d) after exposure to ozone.
Fig. 9. DR-UV–Vis spectra of (a) 3% and 5% AlAg at 30 ◦C under helium and ozone(0.06% O3 in O2); normalized to the most intense band in the spectrum. The AlAg25(grey line) shows an increase in Agn
ı+ clusters under ozone at 30 ◦C yet under heliumthe spectra resembles the AlAg23 which is comparable under helium and ozone. (b)AlAg23 under helium with increasing temperature and (c) AlAg23 under ozone withincreasing temperature. Spectra were recorded every 10 min.

M. Magureanu et al. / Applied Catalysis B
Fig. 10. In situ O3-toluene-DRIFT spectra on the AlAg23 catalyst: (A)41r3
cahp
opsptAat
tdbicpSnTO
aOm
an enhanced interaction of ozone with the catalyst following the
000–2800 cm−1 region: (a) 80 ◦C in He, (b) 80 ◦C in O3, (c) 80 ◦C + toluene, (d)50 ◦C + toluene, (e) 300 ◦C + toluene, (f) 80 ◦C + toluene + O3; (B) 2500–1000 cm−1
egion: (a) 80 ◦C in He, (b) 80 ◦C in O3, (c) 80 ◦C + toluene, (d) 150 ◦C + toluene, (e)00 ◦C + toluene, (f) 80 ◦C + toluene + O3. Spectra were recorded every 10 min.
atalyst. On heating to 100 ◦C under ozone, these bands are depletednd only Ag+ ions are present as for the AlAg23 sample. This isowever the temperature ozone starts to be decomposed in theresence of a catalyst [26].
The higher loading affects the initial silver species but underzone, it is Ag+ species which are present at the reaction tem-erature of 80 ◦C irrespective of loading which indicates that Ag+
pecies may be important and stable under an ozone feed com-ared to silver clusters or larger particles. It is noteworthy, that forhe impregnated catalysts (also 3% loading of Ag), the interaction ofg with ozone led to the redispersion of small clusters to Ag+ ions,s has been observed for the colloid catalysts and also reported inhe literature [6].
In-situ DRIFTS studies were performed using fresh catalyst ashe reference material. The presence of ozone led to a significantecrease in the bands assigned to surface hydroxyl groups and car-onate species resulting from calcination of the catalysts. Silver
s known to be passivated by chemisorbed oxygen, hydroxyl andarbonate species under ambient conditions [27]; however, in theresence of ozone these species are scavenged from the surface.ome surface hydroxyl remains even after this treatment with aew band found at 3732 cm−1 that increases with time (Fig. 10).his feature is thought to be due to perturbations in the remainingH groups by ozone adsorption (Fig. 10).
This demonstrates that initially the role of ozone is essentiallycleaning of the surface with preserving a certain population ofH species. Similar changes are also found under thermal treat-ent in an inert gas environment; however, these changes are
Fig. 11. Proposed model for the interact
: Environmental 104 (2011) 84–90 89
very slow compared to ozone. The band at 3732 was not observedunder He, indicating the final state of the catalyst is different underthe two different atmospheres, i.e. the thermal treatment in inertatmosphere is leading to a more advanced dehydroxylation.
Fig. 10 shows the in-situ DRIFT spectra following treatment ofthe catalyst in a mixture of O3 and toluene for the AlAg23 cata-lysts. The adsorption of toluene at 80 ◦C on a cleaned surface (priorto adsorption of toluene the catalyst was held at 150 ◦C underO3/O2 feed for 2 h) leads to the appearance of a weak bands at3028, 1604 and 1496 cm−1. The band at 3028 cm−1 is due to C–Hstretch and the bands at 1604 and 1496 cm−1 to ring stretchingvibrations of adsorbed toluene. A negative band at 3745 cm−1 isalso observed which suggests that toluene adsorption is interact-ing with OH of alumina surface. As the temperature is increased to300 ◦C, bands due to adsorbed toluene are depleted. On returningto 80 ◦C, adsorbed toluene is observed again. Upon introduction ofozone, bands due to toluene are depleted and new weak bands areevident and assigned to phenolate C C stretching at 1590 cm−1
(shoulder on 1604 cm−1 ring stretching vibration) and between1460 and 1430 cm−1 and also a new band at 1300 cm−1 which isassigned to the C–O stretching of phenolate [28].
No change in the oxidation state or size of the embedded col-loids have been observed after several catalytic cycles, provingthat this preparation methodology leads to very stable catalystsfor oxidation under harsh conditions using ozone. As mentionedpreviously [18], impregnation leads to precursor catalysts whichafter the exposure to ozone generate, via a disruption mechanism,active and non-active spectator species. This preparation procedureleads only to stable active species in which all the introduced silverparticipates in the catalytic reaction.
Previous catalytic studies with these catalysts in reduction ofNO with propene and decane under real exhaust gas conditionsshowed that AlAg13 and AlAg23 behave differently with AlAg13exhibiting the lowest activity in the conversion of NO and a lossof its oxidative properties after thermal ageing. Conversely, for theAlAg23 it was shown that a subsequent thermal ageing leads toan enhancement of the conversion of NO and a slight alteration ofthe selectivity [21]. It was thus suggested that the balance betweenAg0/Ag+ is the main factor that draws the catalytic performance. Inthat particular case it was demonstrated that the presence of oxidicsilver species do not play a key role in the NO/O2 reaction.
Based on data collected in this study we propose the interac-tion model given in Fig. 11. This is essentially different from thatdescribed previously [20]. It shows a first step representing a clean-ing of the surface and a passivation to Agı+ species (demonstratedby DRIFTS) and an accumulation of ozone demonstrated by DR-UV–Vis spectra. According to XPS and XRD results the core of theembedded silver colloids is constituted from Ag0 species. The cover-age of ozone exhibits no influence on the state of the silver particles.
The AlAg13 catalyst contains a greater extent of Ag0 that allows
way presented in Fig. 11. This Ag0/Ag+ ratio does not depend onthe particle size but does seem to be sensitive to the operating con-ditions as it is demonstrated in the ozone oxidation of toluene andNO reduction.
ion of ozone with catalyst surface.

90 M. Magureanu et al. / Applied Catalysis B:
iHuiiit
tbapttfdmfo
dwhs
Nc
hcsahhticstc
4
vaaOacn
[[
[[
[
[
[
[[[
[
[
[[[
[
[[
[
Scheme 1. Interaction of toluene with ozone.
In the next step the activated ozone interacts with toluene lead-ng to a mineralization according to a reported route (Eq. (1)) [29].owever, although this is different from those generally reportedsing photocatalysis [30] in the sense the activation of ozone occurs
n this case on the heterogeneous surface (Fig. 11 most probably) itnvolves the same intermediates. This mechanism is also explain-ng the almost complete diminution of the carbonaceus deposits inhe presence of the catalyst.
However, plasma is a very complex system, and as we men-ioned above the reaction also occurs in the absence of the catalystut with a lower selectivity. The change in the selectivity mayccount for the model presented in Fig. 11. Actually, we have twoarallel routes, one radicalic in plasma alone and another in whichhe presence of the catalyst induces a heterogeneous oxidation ofoluene. Under plasma alone, due to the radicalic mechanism, theormation of carbonaceous deposits occurs. Under catalytic con-itions (see Scheme 1), the radicalic route is suppressed and theineralization enhanced with an improved selectivity to CO2. In
act, this is one of the examples in which the direct activation ofzone is proved.
A too high energy density is also detrimental since it is pro-ucing more ozone. Under these conditions the surface is coveredith ozone in several layers thus diminishing its activation by theeterogeneous catalyst and under these conditions the reaction istopped at the level of CO.
Ozonation of toluene can occur on a range of materials includingaX, NaY and MCM-41 [31] but in that case the behavior is verylose to that occurring in plasma in the absence of any catalyst.
A very important feature of our Ag-based catalysts is the veryigh stablity. As in situ measurements demonstrate the catalystan recover its initial state after the catalytic tests. XPS addedupplementary arguments in this sense (Fig. 8). Also AlAg cat-lysts prepared by the same procedure as in the present workave been tested for catalytic removal of NO in the presence ofydrocarbons (propene and decane) under real exhaust gas condi-ions [21]. Although the reaction is very different on this case, NOs also a strong oxidant while the investigated hydrocarbons canreate carbonaceous deposits. Catalysts analysis after the reactionhowed that neither the oxidation state of the nanoparticles, norhe relative surface concentration of metal changed under theseonditions.
. Conclusions
In-situ study of ozone interaction with Ag–Al catalysts preparedia embedding of pre-synthesised silver colloids in a surfactantssisted alumina support, provides insight as to the nature of the
ctive sites of supported silver colloids for mineralization reactions.zone is chemisorbed on embedded Ag colloidal catalysts withoutny change in the oxidation state and size. The population of thehemisorbed species is higher at lower temperatures, where theon-selective decomposition of ozone is smaller.[
[[
Environmental 104 (2011) 84–90
Working under hybrid plasma catalyst conditions it was demon-strate that indeed silver participates as a catalyst in this reaction,changing the pathway and in consequence the selectivity to CO2.The catalysts exhibit high stability, preserving the structural andtextural properties after the catalytic tests, that is indeed veryimportant in the presence of ozone.
Acknowledgements
We thank Centacata EU-Transitional for the fellowship of Bog-dan Cojocaru in Queen’s University and Project 21-048/ANCS for thefinancial support. RR would also like to thank the National Renew-able Energy Laboratory for HAADF TEM studies and James Ranvillefor ICP-AES experiments. Some authors (MM, DP, NBM, BC) thankUEFISCSU for financial support (project IDEI-223). HD thanks theEPSRC under the CASTech programme for funding.
References
[1] K. Urashima, J.-S. Chang, IEEE Trans. Diel. Electr. Insul. 7 (2000) 602–614.[2] J.S. Devinny, M.A. Deshusses, T.S. Webster, Biofiltration for Air Pollution Control,
Lewis Publishers, Boca Raton, FL, 1999.[3] Y. Shih, M. Li, J. Hazard. Mater. 154 (2008) 21–28.[4] M. Magureanu, N.B. Mandache, E. Gaigneaux, C. Paun, V.I. Parvulescu, J. Appl.
Phys. 99 (2006) 123301.[5] I. Chairez, R. Fuentes, T. Poznyak, M. Franco, A. Poznyak, Catal. Today 151 (2010)
159–165.[6] Y. Bedjanian, M.L. Nguyen, Chemosphere 79 (2010) 387–393.[7] E. Grosjean, D. Grosjean, Environ. Sci. Technol. 31 (1997) 2421–2427.[8] F. Holzer, F.D. Kopinke, U. Roland, Plasma Chem. Plasma Process. 25 (2005)
595–611.[9] H.L. Chen, H.M. Lee, S.H. Chen, M.B. Chang, S.J. Yu, S.N. Li, Environ. Sci. Technol.
43 (2009) 2216–2227.10] S. Futamura, T. Yamamoto, IEEE Trans. Ind. Appl. 33 (1997) 447–453.11] M. Magureanu, N.B. Mandache, V.I. Parvulescu, C. Subrahmanyam, A. Renken,
L. Kiwi-Minsker, Appl. Catal. B: Environ. 74 (2007) 270–277.12] S. Futamura, H. Einaga, H. Kabashima, L.Y. Hwan, Catal. Today 89 (2004) 89–95.13] S. Delagrange, L. Pinard, J.-M. Tatibouet, Appl. Catal. B: Environ. 68 (2006)
92–98.14] M. Magureanu, N.B. Mandache, J. Hu, R. Richards, M. Florea, V.I. Parvulescu,
Appl. Catal. B: Environ. 76 (2007) 275–281.15] C. Subrahmanyam, M. Magureanu, A. Renken, L. Kiwi-Minsker, Appl. Catal. B:
Environ. 65 (2006) 150–156.16] T. Blackbeard, V. Demidyuk, S.L. Hill, J.C. Whitehead, Plasma Chem. Plasma
Process. 29 (2009) 411–419.17] M.H. Kim, K.-H. Choo, Catal. Commun. 8 (2007) 462–466.18] S. Imamura, M. Ikebata, T. Ito, T. Ogita, Ind. Eng. Chem. Res. 30 (1991) 217–221.19] N. Kumar, P. Konova, A. Naydenov, T. Heikillae, T. Salmi, D.Y. Murzin, Catal. Lett.
98 (2004) 57–60.20] A. Naydenov, P. Konova, P. Nikolov, F. Klingstedt, N. Kumar, D. Kovacheva, P.
Stefanov, R. Stoyanova, D. Mehandjiev, Catal. Today 137 (2008) 471–474.21] V.I. Pârvulescu, B. Cojocaru, V. Pârvulescu, R. Richards, Z. Li, C. Cadigan, P.
Granger, P. Miquel, C. Hardacre, J. Catal. 272 (2010) 92–100.22] Z. Falkenstein, J.J. Coogan, J. Phys. D: Appl. Phys. 30 (1997) 817–825.23] U. Roland, F. Holzer, F.D. Kopinke, Appl. Catal. B: Environ. 58 (2005) 217–226.24] C. Wagner, W. Riggs, L. Davis, J. Moulder, G. Mullenberg (Eds.), Handbook of
X-ray Photoelectron Spectroscopy, PerkinElmer Corp., Eden Prairie, MN, USA,1974.
25] K. Shimizu, M. Tsuzuki, K. Kato, S. Yokota, K. Okumura, A. Satsuma, J. Phys.Chem. C 111 (2007) 950–959.
26] H. Einaga, M. Harada, S. Futamura, Chem. Phys. Lett. 408 (2005) 377–380.27] G.J. Millar, J.B. Metson, G.A. Bowmaker, R.P. Cooney, J. Chem. Soc. Faraday Trans.
91 (1995) 4149–4159.28] A.A. Davydov, E.M. Knyazeva, M.L. Shepotko, J. Appl. Spectrosc. 49 (1988)
731–736.29] A.G. Galstyan, S.G. Galstyan, N.F. Tyupalo, Russ. J. Appl. Chem. 83 (2010)
267–270.30] K.-P. Yu, G.W.M. Lee, Appl. Catal. B: Environ. 75 (2007) 29–38.31] C.W. Kwong, C.Y.H. Chao, K.S. Hui, M.P. Wan, Atmos. Environ. 42 (2008)
2300–2311.
Related Documents











