Research article In Silico Characterization and Molecular Evolutionary Analysis of a Novel Superfamily of Fungal Effector Proteins Ioannis Stergiopoulos,* ,1,2,3,4 Yiannis A.I. Kourmpetis, 5,6 Jason C. Slot, 2 Freek T. Bakker, 7 Pierre J.G.M. De Wit,* , y,3,8 and Antonis Rokas* , y,2 1 Department of Plant Pathology, University of California Davis 2 Department of Biological Sciences, Vanderbilt University 3 Laboratory of Phytopathology, Wageningen University and Research Centrum, Wageningen, The Netherlands 4 Centre for BioSystems Genomics, Wageningen, The Netherlands 5 Laboratory of Bioinformatics, Wageningen University and Research Centrum, Wageningen, The Netherlands 6 Functional Genomics Group, BioAnalytical Science Department, Nestec Ltd, Nestle ´ Research Center 7 Biosystematics Group, Wageningen University and Research Centrum, Wageningen, The Netherlands 8 Department of Botany and Microbiology, King Saud University, Riyadh, Saudi Arabia *Corresponding authors: E-mail: [email protected]; [email protected]; [email protected]. yThese authors contributed equally to this work. Associate Editor: Matthew Hahn Abstract Most fungal plant pathogens secrete effector proteins during pathogenesis to manipulate their host’s defense and promote disease. These are so highly diverse in sequence and distribution, they are essentially considered as species-specific. However, we have recently shown the presence of homologous effectors in fungal species of the Dothideomycetes class. One such example is Ecp2, an effector originally described in the tomato pathogen Cladosporium fulvum but later detected in the plant pathogenic fungi Mycosphaerella fijiensis and Mycosphaerella graminicola as well. Here, using in silico sequence-similarity searches against a database of 135 fungal genomes and GenBank, we extend our queries for homologs of Ecp2 to the fungal kingdom and beyond, and further study their history of diversification. Our analyses show that Ecp2 homologs are members of an ancient and widely distributed superfamily of putative fungal effectors, which we term Hce2 for H omologs of C . fulvum E cp2 . Molecular evolutionary analyses show that the superfamily originated and diversified within the fungal kingdom, experiencing multiple lineage-specific expansions and losses that are consistent with the birth-and-death model of gene family evolution. Newly formed paralogs appear to be subject to diversification early after gene duplication events, whereas at later stages purifying selection acts to preserve diversity and the newly evolved putative functions. Some members of the Hce2 superfamily are fused to fungal Glycoside Hydrolase family 18 chitinases that show high similarity to the Zymocin killer toxin from the dairy yeast Kluyveromyces lactis, suggesting an analogous role in antagonistic interactions. The observed high rates of gene duplication and loss in the Hce2 superfamily, combined with diversification in both sequence and possibly functions within and between species, suggest that Hce2s are involved in adaptation to stresses and new ecological niches. Such findings address the need to rationalize effector biology and evolution beyond the perspective of solely host-microbe interactions. Key words: adaptive evolution, birth-and-death, diversification, effectors, fungi, GH18 chitinases. Introduction Effectors are low molecular weight proteins that are secreted by bacteria, oomycetes, and fungi to manipulate and adapt to their hosts and their environment (Hogenhout et al. 2009). Unlike bacteria and oomycetes from which several effector families have been described (Jiang et al. 2006; Sarkar et al. 2006; Hajri et al. 2009), only very few homologous effector proteins are known from fungi. Indeed, one of the hallmarks of fungal effector proteins so far has been their high sequence divergence and species-specificity (Stergiopoulos and de Wit 2009). So far, Ecp6, a secreted effector protein from the tomato pathogen C. fulvum (class Dothideomycetes) was the only known fungal effector with homologs in several other fungal species. Homology in this case, however, is mainly based on the presence of LysM motifs in this protein, a domain that is widespread among fungi of diverse taxa and lifestyles (de Jonge and Thomma 2009). More recently, a com- parative genomics study in Dothideomycetes showed the presence of highly divergent homologs of the C. fulvum Avr4 and Ecp2 effector proteins in the closely related banana pathogen M. fijiensis (Avr4 and Ecp2) and the wheat pathogen M. graminicola (Ecp2) (Stergiopoulos et al. 2010). Interestingly, both M. fijiensis and M. graminicola con- tain three highly divergent homologs (64–25% identity at the amino acid level) of C. fulvum Ecp2. Phylogenetic analysis of ß The Author 2012. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: [email protected] Mol. Biol. Evol. 29(11):3371–3384 doi:10.1093/molbev/mss143 Advance Access publication May 23, 2012 3371

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Research
articleIn Silico Characterization and Molecular Evolutionary Analysisof a Novel Superfamily of Fungal Effector ProteinsIoannis Stergiopoulos,*,1,2,3,4 Yiannis A.I. Kourmpetis,5,6 Jason C. Slot,2 Freek T. Bakker,7
Pierre J.G.M. De Wit,*,y,3,8 and Antonis Rokas*,y,2
1Department of Plant Pathology, University of California Davis2Department of Biological Sciences, Vanderbilt University3Laboratory of Phytopathology, Wageningen University and Research Centrum, Wageningen, The Netherlands4Centre for BioSystems Genomics, Wageningen, The Netherlands5Laboratory of Bioinformatics, Wageningen University and Research Centrum, Wageningen, The Netherlands6Functional Genomics Group, BioAnalytical Science Department, Nestec Ltd, Nestle Research Center7Biosystematics Group, Wageningen University and Research Centrum, Wageningen, The Netherlands8Department of Botany and Microbiology, King Saud University, Riyadh, Saudi Arabia
*Corresponding authors: E-mail: [email protected]; [email protected]; [email protected].
yThese authors contributed equally to this work.
Associate Editor: Matthew Hahn
Abstract
Most fungal plant pathogens secrete effector proteins during pathogenesis to manipulate their host’s defense and promotedisease. These are so highly diverse in sequence and distribution, they are essentially considered as species-specific. However, wehave recently shown the presence of homologous effectors in fungal species of the Dothideomycetes class. One such example isEcp2, an effector originally described in the tomato pathogen Cladosporium fulvum but later detected in the plant pathogenicfungi Mycosphaerella fijiensis and Mycosphaerella graminicola as well. Here, using in silico sequence-similarity searches against adatabase of 135 fungal genomes and GenBank, we extend our queries for homologs of Ecp2 to the fungal kingdom and beyond,and further study their history of diversification. Our analyses show that Ecp2 homologs are members of an ancient and widelydistributed superfamily of putative fungal effectors, which we term Hce2 for Homologs of C. fulvum Ecp2. Molecular evolutionaryanalyses show that the superfamily originated and diversified within the fungal kingdom, experiencing multiple lineage-specificexpansions and losses that are consistent with the birth-and-death model of gene family evolution. Newly formed paralogsappear to be subject to diversification early after gene duplication events, whereas at later stages purifying selection acts topreserve diversity and the newly evolved putative functions. Some members of the Hce2 superfamily are fused to fungalGlycoside Hydrolase family 18 chitinases that show high similarity to the Zymocin killer toxin from the dairy yeastKluyveromyces lactis, suggesting an analogous role in antagonistic interactions. The observed high rates of gene duplicationand loss in the Hce2 superfamily, combined with diversification in both sequence and possibly functions within and betweenspecies, suggest that Hce2s are involved in adaptation to stresses and new ecological niches. Such findings address the need torationalize effector biology and evolution beyond the perspective of solely host-microbe interactions.
Key words: adaptive evolution, birth-and-death, diversification, effectors, fungi, GH18 chitinases.
IntroductionEffectors are low molecular weight proteins that are secretedby bacteria, oomycetes, and fungi to manipulate and adapt totheir hosts and their environment (Hogenhout et al. 2009).Unlike bacteria and oomycetes from which several effectorfamilies have been described (Jiang et al. 2006; Sarkar et al.2006; Hajri et al. 2009), only very few homologous effectorproteins are known from fungi. Indeed, one of the hallmarksof fungal effector proteins so far has been their high sequencedivergence and species-specificity (Stergiopoulos and de Wit2009). So far, Ecp6, a secreted effector protein from thetomato pathogen C. fulvum (class Dothideomycetes) was
the only known fungal effector with homologs in severalother fungal species. Homology in this case, however, ismainly based on the presence of LysM motifs in this protein,a domain that is widespread among fungi of diverse taxa andlifestyles (de Jonge and Thomma 2009). More recently, a com-parative genomics study in Dothideomycetes showed thepresence of highly divergent homologs of the C. fulvumAvr4 and Ecp2 effector proteins in the closely relatedbanana pathogen M. fijiensis (Avr4 and Ecp2) and thewheat pathogen M. graminicola (Ecp2) (Stergiopoulos et al.2010). Interestingly, both M. fijiensis and M. graminicola con-tain three highly divergent homologs (64–25% identity at theamino acid level) of C. fulvum Ecp2. Phylogenetic analysis of
� The Author 2012. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, pleasee-mail: [email protected]
Mol. Biol. Evol. 29(11):3371–3384 doi:10.1093/molbev/mss143 Advance Access publication May 23, 2012 3371

these seven Ecp2s indicated clustering based on orthologyrather than paralogy, suggesting that gene duplications tookplace prior to speciation (Stergiopoulos et al. 2010). Ecp2 is a165 amino acid (aa) secreted protein that was originally iden-tified as a virulence factor in C. fulvum, since disruption redu-ces virulence of the fungus on tomato plants (Lauge et al.1997). Although the intrinsic function of this effector proteinduring pathogenesis is still unknown, it has been hypothe-sized that Ecp2 interacts with a host virulence target toinduce necrosis and the release of nutrients from the hostcells during infection (Stergiopoulos et al. 2010). Indeed, theclosest homolog of the C. fulvum Ecp2 in M. fijiensis wasshown to trigger necrosis in tomato plants, irrespectively ofthe presence of the cognate Cf-Ecp2 resistance gene that me-diates Ecp2 recognition and subsequent resistance reactionsin tomato (Stergiopoulos et al. 2010).
The identification of homologous effectors within thefungal class of Dothideomycetes suggests that, as in bacteriaand oomycetes, core effector proteins that facilitate basicvirulence functions on a diverse set of hosts are also presentin fungi. Thus, differences in the effector repertoire amongstfungal species could reflect adaptation to specific hosts orcultivars (Stergiopoulos et al. 2010). Theoretically, both con-vergent evolution driven by the need to cope with similarenvironmental challenges, and divergent evolution driven byshared ancestry between different species, could potentiallyshape the effector repertoire of fungal species. In this respect,two studies in oomycete species of Phytophthora spp. showedthat their effector repertoire has been largely shaped by di-vergent evolution, where high levels of genetic diversity aredriven by selection pressure imposed by the host(s) (Jianget al. 2006; Jiang et al. 2008). However, although a few studieshave described the sequence diversity and distribution of ef-fector alleles in populations of a particular species (Schurchet al. 2004; Ma et al. 2006; Stergiopoulos et al. 2007; Hajriet al. 2009; Stukenbrock and McDonald 2009), detailed stud-ies addressing the presence of effector families in fungi andtheir evolutionary paths after speciation are mostly lacking(de Jonge and Thomma 2009; Chuma et al. 2011).
To address this issue, we systematically searched for thepresence of putative Ecp2 effector homologs across the entirefungal kingdom and beyond, and analyzed their history ofevolutionary diversification. We show that Ecp2 is amember of a novel and widely distributed within the fungalkingdom multigene superfamily, which we have now desig-nated Hce2, for Homologs of C. fulvum Ecp2 effector. Detailedevolutionary analysis of this superfamily of putative effectorsin fungi shows that it most likely originated in aPezizomycotina ancestor and has subsequently diverged ex-tensively, experiencing multiple lineage-specific expansionsand losses that are consistent with the birth-and-deathmodel of gene family evolution. We also identified withinthe Hce2 subfamily members with a unique fusion tofungal Glycoside Hydrolase family 18 (GH18) chitinases thatshow highest similarity to the Zymocin �- and �-subunits ofthe heterotrimeric (���) killer toxin from the dairy yeastK. lactis, suggesting a functional linkage between the twodomains. Thus, we propose that in addition to gene
duplication and rapid diversification, new or enhanced spe-cificities of putative effectors can be generated by acquisitionof new protein domains as well. In summary, we identify andcharacterize a novel superfamily in fungi of putative core ef-fector proteins, shedding light on its origin and diversificationby showing that gene duplication, rapid diversification andrecombination with novel protein domains, all contribute increating remarkably diverse putative effector molecules re-quired for plant parasitism and likely adaptation to stressfulenvironments as well. Based on our findings, we further pro-pose that the biology and evolution of effector proteins andvirulence traits in general should be studied in a broaderecological context that will allow us to fully understand mi-crobial pathogenicity beyond the current perspective ofhost–microbe interactions alone.
Materials and MethodsA detailed description of methods applied is provided assupplementary material.
Identification of Putative Ecp2 Homologs
We searched for homologs of Ecp2 against thenon-redundant protein database nr (GenBank) and a localdatabase of 135 fungal genomes that was constructed fromgenomes available in public databases (supplementary fig. S1and table S1, Supplementary Material online). Searches wereperformed using PSI-Blast (e-value cutoff: 10�5) iterated tillsaturation with the previously identified Ecp2 orthologs fromC. fulvum (CAA78401.1), M. fijiensis (Mfij_52972, Mfij_60658,Mfij_198160), and M. graminicola (Mgra_104404,Mgra_107904, Mgra_106176) (Stergiopoulos et al. 2010).The reference set of seven Ecp2 orthologs, together withtheir five best BlastP hits against nr and our local databasewere further exploited as a training set for HMM built andmotif discovery, using the HMMER (v3.0) (http://hmmer.janelia.org) software package and the GLAM2SCAN program(Frith et al. 2008) of the MEME suite (v.4.6.1) (Bailey et al.2006), respectively. These programs were used to filter the listof PSI-Blast hits from false positives that might have beenobtained during the PSI-Blast step. To maximize output, fur-ther BlastP and tBlastN searches were performed using Hce2sequences from close related species as queries. Finally, localsynteny was utilized to carefully examine genomic loci inclosely related species that were likely to hold homologs over-looked or mis-annotated by the automatic annotations pro-vided in public genome databases.
Pairwise similarity scores among retrieved Hce2 homologswere calculated using the Needleman-Wunsch algorithm(Needleman and Wunsch 1970), as implemented in the“Needleall” software package available from EMBOSS, whilepairwise e-value scores were computed from an all-versus-allBlastP analysis, executed using a locally installed BlastALL ap-plication available from NCBI. The query database consistedof all 153 Hce2 proteins identified in this study. Matrices ofpairwise similarity scores and BlastP e-values were generatedusing the MultiExperiment Viewer (MeV: v4.7.1) software(http://www.tm4.org/mev/).
3372
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE

Protein sequence alignments were performed using theGLAM2 (Gapped Local Alignment of Motifs) algorithm(Frith et al. 2008) available by the MEME suite (Bailey et al.2006). The parameters used were z 150, a 120, b 250, w 120 r10, and n 25000, where z is the minimal number of sequencesin the alignment, a, b, and w the minimum, maximum, andinitial number of aligned columns, respectively, r the numberof alignment runs and n the number of iterations withoutimprovement for each run.
Phylogenetic Analyses
Bayesian inference (BI) and Maximum Likelihood (ML) meth-ods as implemented in MrBayes (v3.1.2) (Ronquist,Huelsenbeck 2003) and RAxML (v7.2.8) (Stamatakis 2006),respectively, were used to infer the phylogenetic trees pre-sented in this study and estimate clade support. For each BIanalysis 10 independent runs were performed using a mixedamino acid substitution model. Every run consisted of2,000,000 iterations, a burn-in of 2,000 iterations, fourchains, and a sampling frequency of every 100 generations.Convergence was examined using Tracer (v1.5) (http://tree.bio.ed.ac.uk/software/tracer/) and sampled trees from all runswere concatenated into a single tree using the sumt function.For each ML analysis we performed five independent fastMLbootstrap heuristic searches that begun with parsimony-derived trees (Stamatakis 2006; Stamatakis et al. 2008). Eachrun consisted of 2,000 bootstrap replicates and trees wereinferred under the DAYHOFF+� amino acid substitutionmatrix. A bootstrap consensus tree from all runs was obtainedusing the Extended Majority Rule as implemented in theprogram CONSENSE from the Phylip software package(v3.67) (Felsenstein 2005).
Maximum Likelihood Ancestral State Reconstructionand Reconciled Tree Analyses
Correlation between Hce2 numbers and species ecology wasperformed using pairwise comparisons (Read and Nee 1995;Maddison 2000) as implemented in the Mesquite softwarepackage (v2.73) (Maddison and Maddison 2010). All correla-tion analyses were performed with the “most pairs” option,under which the choice of which pairs of states in the twocharacters are to be compared is aimed to maximize thenumber of pairs compared, regardless of the states in thecharacters.
Reconciliation of the inferred phylogenetic Hce2 trees withthe Ascomycota species tree was performed using Notung(v2.6) (Durand et al. 2006; Vernot et al. 2008). The speciestree was deduced from the available published phylogeneticdata (Fitzpatrick et al. 2006; James et al. 2006; Schoch et al.2009). To limit the impact of error in the Hce2 phylogenies onthe estimation of gain and loss events, all clades with <70%bootstrap support or 99% posterior probability were col-lapsed prior to reconciliation with the species tree. In allcases, a rearrangement step was followed after the initial rec-onciliation of the Hce2 phylogenies with the Ascomycotaspecies tree, in order to minimize the number of duplicationand loss events (Durand et al. 2006).
Detection of Local Synteny
The progressive MAUVE algorithm as implemented in theMAUVE software package (v2.3.1) (Darling et al. 2004) wasused to align genomic regions of 10 kb on either side of Hce2genes (20 kb in total) and identify areas of local syntenyamong them. Default seed weight was set to 11 in order toincrease the sensitivity of the alignment and all other param-eters were left at default.
Tests for Selection and Diversificationin Putative Functions
The pattern of molecular evolution at individual sites of theHce2 sequences was investigated using the site models (M0,M1a, M2a, M3, M7, and M8) implemented in the CODEMLprogram of the PAML software package (v4.4) (Yang 1997,2007). Positive selection was inferred only when the modelsthat allow for positively selected sites (M2a, M8) fitted thedata better than their nested null models (M1a, M7, respec-tively) based on Likelihood Ratio Tests (LRT). If this was thecase then positively selected sites were assigned based onposterior probabilities (>95%) calculated using the BayesEmpirical Bayes (BEB) and Naive Empirical Bayes (NEB) esti-mation methods in models M2a and M8 (Nielsen and Yang1998; Yang et al. 2005).
Functional divergence between groups of orthologousHce2 proteins was detected on the basis of shifts insite-specific evolutionary rates (Type I functional divergence)and group-specific changes in amino acid biochemical prop-erties (Type II functional divergence) after gene duplication,using DIVERGE (v2.0) (Gu 2001). The method uses a maxi-mum likelihood approximation to measure the coefficient offunctional divergence (#) (Gu 1999, 2001, 2006). Rejection ofthe null hypothesis that #= 0 in favor of #> 0 indicatedfunctional divergence (Gu 1999, 2001). Detection of aminoacid residues responsible for Type I and/or Type II functionaldivergence was based on the posterior probabilities calculatedfor each position in the alignment that indicates the likeli-hood of a site to contributing to Type I and/or Type II diver-gence between groups.
Results
In Silico Identification of Ecp2 Homologs
In silico sequence-similarity searching of 135 fungal genomesand the GenBank nr database (supplementary fig. S1 andtable S1, Supplementary Material online), identified 153 pu-tative homologs of Ecp2, including three pseudogenes (sup-plementary table S2, Supplementary Material online).Pairwise sequence comparisons indicated that most homo-logs were highly divergent, sharing �20–40% of amino acidsimilarity. Despite the high sequence divergence among theidentified homologs, pairwise similarities and blast-hite-values computed in an all-versus-all BlastP analysis indicatedan asymmetric but nearly fully connected network ofquery-hit pairs with e-values �10�6 and >40% similarity(supplementary fig. S2, Supplementary Material online). Asexpected, e-values and similarity scores were higher betweenprotein pairs from closely related species but dramatically
3373
Evolution of a Fungal Effector Superfamily . doi:10.1093/molbev/mss143 MBE
decreased as an inverse function of the phylogenetic distancebetween the Ecp2-containing species. Overall, based on thefact that all 153 putative Ecp2 homologs retrieved from theheuristic Blast and HMM searches form a connected networkof significant blast hits (e-values�10�6 and>40% amino acidsimilarity), we infer that they are members of a single super-family, which we name the Hce2 superfamily for Homologs ofCladosporium fulvum Ecp2 effector.
Annotation and Domain Organization of Members ofthe Hce2 Superfamily
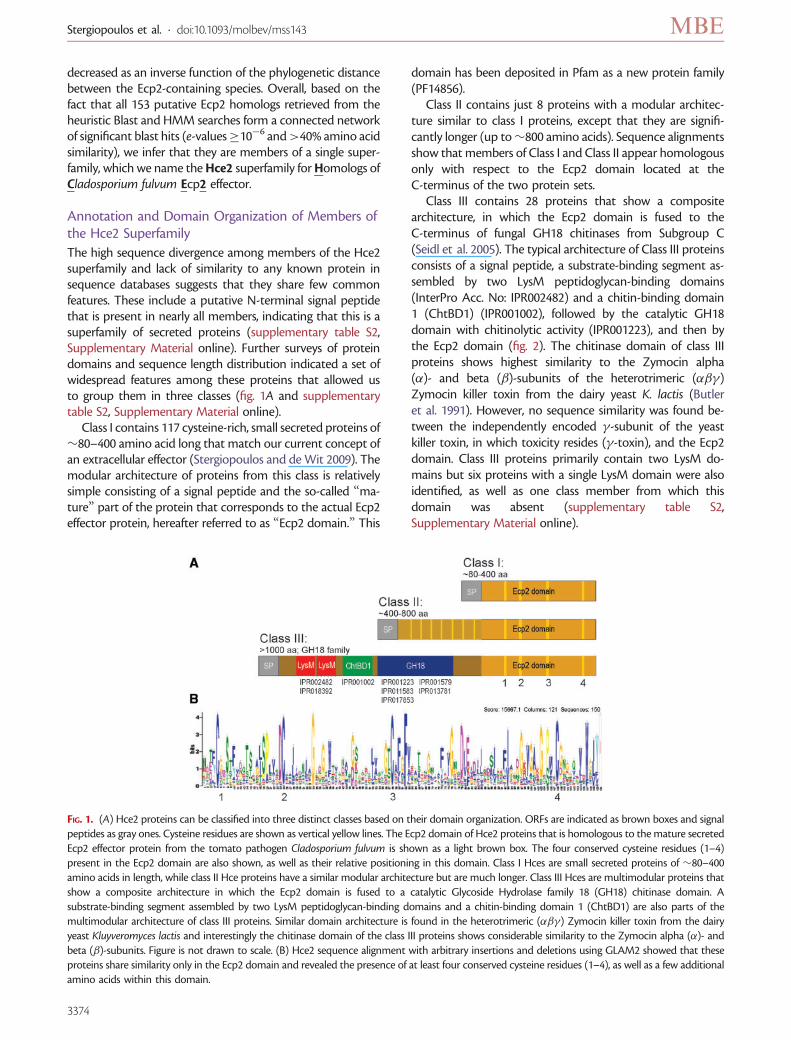
The high sequence divergence among members of the Hce2superfamily and lack of similarity to any known protein insequence databases suggests that they share few commonfeatures. These include a putative N-terminal signal peptidethat is present in nearly all members, indicating that this is asuperfamily of secreted proteins (supplementary table S2,Supplementary Material online). Further surveys of proteindomains and sequence length distribution indicated a set ofwidespread features among these proteins that allowed usto group them in three classes (fig. 1A and supplementarytable S2, Supplementary Material online).
Class I contains 117 cysteine-rich, small secreted proteins of�80–400 amino acid long that match our current concept ofan extracellular effector (Stergiopoulos and de Wit 2009). Themodular architecture of proteins from this class is relativelysimple consisting of a signal peptide and the so-called “ma-ture” part of the protein that corresponds to the actual Ecp2effector protein, hereafter referred to as “Ecp2 domain.” This
domain has been deposited in Pfam as a new protein family(PF14856).
Class II contains just 8 proteins with a modular architec-ture similar to class I proteins, except that they are signifi-cantly longer (up to�800 amino acids). Sequence alignmentsshow that members of Class I and Class II appear homologousonly with respect to the Ecp2 domain located at theC-terminus of the two protein sets.
Class III contains 28 proteins that show a compositearchitecture, in which the Ecp2 domain is fused to theC-terminus of fungal GH18 chitinases from Subgroup C(Seidl et al. 2005). The typical architecture of Class III proteinsconsists of a signal peptide, a substrate-binding segment as-sembled by two LysM peptidoglycan-binding domains(InterPro Acc. No: IPR002482) and a chitin-binding domain1 (ChtBD1) (IPR001002), followed by the catalytic GH18domain with chitinolytic activity (IPR001223), and then bythe Ecp2 domain (fig. 2). The chitinase domain of class IIIproteins shows highest similarity to the Zymocin alpha(�)- and beta (�)-subunits of the heterotrimeric (���)Zymocin killer toxin from the dairy yeast K. lactis (Butleret al. 1991). However, no sequence similarity was found be-tween the independently encoded �-subunit of the yeastkiller toxin, in which toxicity resides (�-toxin), and the Ecp2domain. Class III proteins primarily contain two LysM do-mains but six proteins with a single LysM domain were alsoidentified, as well as one class member from which thisdomain was absent (supplementary table S2,Supplementary Material online).
FIG. 1. (A) Hce2 proteins can be classified into three distinct classes based on their domain organization. ORFs are indicated as brown boxes and signalpeptides as gray ones. Cysteine residues are shown as vertical yellow lines. The Ecp2 domain of Hce2 proteins that is homologous to the mature secretedEcp2 effector protein from the tomato pathogen Cladosporium fulvum is shown as a light brown box. The four conserved cysteine residues (1–4)present in the Ecp2 domain are also shown, as well as their relative positioning in this domain. Class I Hces are small secreted proteins of �80–400amino acids in length, while class II Hce proteins have a similar modular architecture but are much longer. Class III Hces are multimodular proteins thatshow a composite architecture in which the Ecp2 domain is fused to a catalytic Glycoside Hydrolase family 18 (GH18) chitinase domain. Asubstrate-binding segment assembled by two LysM peptidoglycan-binding domains and a chitin-binding domain 1 (ChtBD1) are also parts of themultimodular architecture of class III proteins. Similar domain architecture is found in the heterotrimeric (���) Zymocin killer toxin from the dairyyeast Kluyveromyces lactis and interestingly the chitinase domain of the class III proteins shows considerable similarity to the Zymocin alpha (�)- andbeta (�)-subunits. Figure is not drawn to scale. (B) Hce2 sequence alignment with arbitrary insertions and deletions using GLAM2 showed that theseproteins share similarity only in the Ecp2 domain and revealed the presence of at least four conserved cysteine residues (1–4), as well as a few additionalamino acids within this domain.
3374
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE

Despite the high sequence divergence among Hce2proteins, alignment using the GLAM2 algorithm revealedthe presence of at least four conserved cysteine residueswithin their Ecp2 domain, as well as a few additional aminoacids (fig. 1B).
Taxonomic Distribution of the Hce2 Superfamily
All 153 members of the Hce2 superfamily are distributed onlyamong fungi (52 species; 46 in the phylum Ascomycota and 6in Basidiomycota; supplementary fig. S3 and table S1,Supplementary Material online). Within Ascomycota, Hce2genes were only found in the subphylum Pezizomycotina,and were most abundant within the classes of Sordariomy-cetes (genomes from 25/25 species examined), Dothideomy-cetes (9/13 species examined), and Eurotiomycetes (12/25species examined) (fig. 2). We did not recover any sequencesfrom Leotiomycetes (2 species examined) or Lecanoromy-cetes (1 species examined), the sister classes of Sordariomy-cetes and Eurotiomycetes in Pezizomycotina, respectively.Also, putative Hce2 genes were not recovered from speciesof Taphrinomycotina (6 species examined) or Saccharomyco-tina (25 species examined), the sister subphyla of Pezizomy-cotina in Ascomycota. It is possible that Hce2 genes have beenlost from these classes or, alternatively, this could be due to
incomplete sampling and the considerably lower taxonomicrepresentation of some of these classes in sequence databases.All six members of the Hce2 superfamily identified in Basidio-mycota were from the subphylum of Agaromycotina (29 spe-cies examined), although the absence of Hce2 genes inPucciniomycotina (4 species examined) and Ustilaginomyco-tina (2 species examined) might also be due to the underrep-resentation of genomes from these species in publicdatabases. Finally, the highly discontinuous distribution ofHce2 superfamily members was also evident at the order,family and genus levels (supplementary fig. S3 and table S1,Supplementary Material online).
Within species, the number of Hce2 paralogs per genomevaried from one to as many as 14, suggesting several rounds ofgene duplications and losses (fig. 3). Most species had one(20 species), two (10 species), or three (10 species) Hce2 genesin their genomes, but a substantial increase in gene copynumber was observed in species of Sordariaceae, includingNeurospora discreta (9 genes), N. crassa (10 genes), N. tetra-sperma (10 genes), and Sordaria macrospora (14 genes), aswell as in the Sordariales Podospora anserina (8 genes) and theBasidiomycete Auricularia delicata (7 genes) (supplementaryfig. S3 and tables S1 and S2, Supplementary Material online).Evidence of lineage-specific expansions and contractions ofthe Hce2 superfamily were evident, even when comparingclose related species within the same genus. For example,the distribution of Hce2 genes in species of Trichodermavaried from one (T. reesei), to two (T. atroviride) or three(T. virens) copies per genome.
This highly variable distribution of Hce2 genes in the dif-ferent fungal lineages suggests a complex and dynamic evo-lutionary history consistent with the birth-and-death modelof gene family evolution (Nei et al. 1997; Nei and Rooney2005). To infer ancestral gene copy number and identifykey events in the evolutionary history of the Hce2 superfamily,we performed parsimony based ancestral character state re-construction on a species tree phylogeny of Ascomycota thatwas constructed with reference to available published phy-logenies. Our analysis shows that the Hce2 superfamily mostlikely arose in Pezizomycotina and that the most recentcommon ancestor had a single Hce2 (supplementary fig. S4,Supplementary Material online). The subsequent evolution ofthe superfamily consists of multiple independent episodes ofexpansion and contraction across different fungal lineages.For example, multiple independent rounds of gene duplica-tions have resulted in a large-scale expansion in species ofSordariomycetes, whereas Hce2 genes have been largely lost inEurotiomycetes. Similarly, within Dothideomycetes an expan-sion of the superfamily occurred in species of Capnodiales,whereas a contraction occurred in species of Pleosporales.
To test whether the distribution and lineage-specificexpansions and contractions of the Hce2 superfamily wereassociated with the species ecology, we classified species as1) plant pathogens, 2) pathogens or parasites of animals,human, insects, nematodes, or other fungi, and 3) non-pathogens (saprophytic), and used a phylogenetic correlationtest (Read and Nee 1995; Maddison 2000) to evaluate therelationship between Hce2 numbers and lifestyle (fig. 4 and
FIG. 2. Differential distribution of members of the Hce2 superfamilyacross higher fungal taxonomic categories. The fungal tree was assem-bled manually with reference to available published phylogenies.Phylogenentic uncertainties concerning the placement of specificfungal lineages in the tree are indicated by dotted lines. End nodesare color-coded based on the presence (green) or absence (red) ofHce2 genes in a particular fungal order. Not sampled orders are indi-cated by gray end nodes. Numbers in parentheses above the nodesspecify the number of species in each order that have Hce2 genes rel-atively to the total number of species examined in that order, while thethickness of the green lines is proportional to the ratio of these twonumbers. The distribution shows that Hce2 genes are mostly present inSordariomycetes (25/2), followed by Dothideomycetes (9/13) and finallyEurotiomycetes (12/25). In Basidiomycota, Hce2 members are only pre-sent in Agaromycotina (6/29).
3375
Evolution of a Fungal Effector Superfamily . doi:10.1093/molbev/mss143 MBE

supplementary fig. S4, Supplementary Material online). Nocorrelation could be found between a particular lifestyleand gene expansion or contraction across fungal species(P> 0.05). However, Class III Hce2 proteins were mostly as-sociated with non-plant pathogenic (46.5%) and saprophytic(35.7%) fungi rather than plant pathogens (17.8%), whereas85% of Class I and II proteins were almost equally distributedbetween plant pathogens and saprophytes (supplementarytable S3, Supplementary Material online). This proportionalrepresentation of Hce2 genes from the three classes acrossfungi with different lifestyles could reflect functional diver-gence and lineage-specific expansions and contractions re-lated to the ecological niches that the different speciesoccupy.
Molecular Evolution of the Hce2 Superfamily
To characterize the evolution of the Hce2 superfamily weexamined the phylogenetic relations of the Ecp2 domain be-tween its members, using Bayesian inference (BI) andMaximum Likelihood (ML) approaches. Tree topologies de-rived with BI and ML methods were nearly identical and hadgenerally low clade support, especially for deep internodes(supplementary fig. S5 and S6, Supplementary Materialonline), suggesting that the history of the Hce2 superfamilycannot be confidently resolved beyond the level of closelyrelated species. This lack of support is likely due to the limitedphylogenetic signal contained in the short but highly diver-gent aa sequence alignment of Hce2 superfamily members.Perhaps the only exception is the robust support (100% BIposterior probability and 85% ML bootstrap support) for the
internode connecting Basidiomycota and Ascomycota. Incontrast, the topology of the shallow internodes is largelyconsistent with the consensus fungal species phylogeny andis generally fairly strongly supported (>65% ML bootstrapsupport and >95% BI probability). Shallow internodes showextensive clustering in the Hce2 superfamily based on orthol-ogy rather than paralogy across the entire phylogeny, suggest-ing that gene duplications occurred prior to speciation.Exceptions are a weakly supported clade of three Hce2paralogs from the plant pathogen M. graminicola, a well-supported two-protein clade from S. macrospora, and asix-paralog clade from the Basidiomycete A. delicata. Theseduplications are likely recent, indicating that “birth” is still anongoing process in the evolution of the Hce2 superfamily.
Both duplication and loss were frequently inferred inclades of close relatives, thus supporting a mode of birth-and-death evolution for the Hce2 superfamily. To obtain amore thorough picture of the pattern of birth-and-deathevents, we performed phylogenetic reconciliation analysis ofthe Ecp2 domain topology with the Ascomycota species tree(fig. 5). The analysis identified 25 duplication (D) and 54 lossevents (L) (D/L Score = 91.5), whereas reconciliation afterrearrangement of the Hce2 topology to obtain the minimalscore for duplication and losses, identified 23 duplication and37 loss events (D/L Score = 60). These numbers should beconsidered as approximate since the history of alternativeevents from reconciliation assuming equal weights for dupli-cations and losses indicates a range of 29–37 duplications and23–31 losses (D/L Score = 60.0), respectively. Nevertheless, theanalysis exposed once more the dynamic evolutionary historyof the Hce2 superfamily, consisting of recurrent episodes of
Fig. 3. Differential distributions of Hce2 paralogs across fungal species. Most species (20�) have a single Hce2 gene in their genome, while a largeexpansion of the family is observed in the species of Sordariaceae, Neurospora discreta (9 genes), N. crassa (10 genes), N. tetrasperma (10 genes), andSordaria macrospora (14 genes), as well as the Sordariales Podospora anserina (8 genes) and the Basidiomycete Auricularia delicata (7 genes).
3376
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE

duplications and losses taking place across the entirePezizomycotina clade and which continue to occur even tothe present day. The reconciliation analysis also showed alarge expansion of the Hce2 superfamily in the Sordarialesclade as a result of 11 duplication events, five of which tookplace before the divergence of the Chaetoniaceae,Lasiosphaeriaceae and Sordariaceae families. Since then, theHce2 superfamily has continued to expand in Sordariaceae(N. discreta, N. crassa, N. tetrasperma, and S. macrospora), asindicated by the three recent duplication events suggestedto have taken place in S. macrospora, but has contractedin Chaetoniaceae (Chaetomium globosum, Sporotrichum ther-mophile, Thielavia terrestris). Similar observations can be
made for other fungal lineages as well, as for example theexpansion of the Hce2 superfamily in Glomerellales (includingVerticillium and Colletotrichum spp.).
Despite the fact that the deeper nodes of the Hce2 phy-logeny are not well supported, it appears that there are twomajor groupings, of Class I/Class II proteins on the one handand of class III proteins on the other (supplementary fig. S5and S6, Supplementary Material online). Both BI and MLmethods consistently reproduce the class III protein clade,albeit with weak support, thus suggesting a monophyleticorigin for this clade. A pattern of interspecies clustering wasalso evident within this clade but a general arrangement ac-cording to the consensus fungal species phylogeny was not as
Fig. 4. The number of Hce2s per genome is not correlated with the fungal species ecology. Maximum parsimony-based ancestral character statereconstruction tracing variation in Hce2 numbers in species of Pezizomycotina (A) and their pathogenic lifestyle (B). The Pezizomycotina speciestopology upon which the multistate state characters were traced was assembled manually with reference to available published phylogenies. Braches onthe left panel are color-coded based on the present (end nodes) and inferred (deeper nodes) numbers of Hce2 genes in each species or its ancestor. In asimilar way, branches on the right panel are color-coded according to the species life style as plant pathogens (green nodes), human, and/or animalpathogens (blue nodes) and non-pathogenic or saprophytes (orange nodes). In both panels, branches supported by more than one character states areindicated by two-color patterns. For easier interpretation of the image on the left panel the number of states (range of Hce2 gene numbers) on eachbranch is also indicated, as well as gain (G) and losses (L) along specific lineages. The maximum parsimony reconstruction shows that the Hce2superfamily most likely arose in Pezizomycotina and followed a complex evolutionary path with multiple independent expansions and contractionsacross the different fungal lineages that are consistent with a birth-and-death model of evolution. However, no specific correlation could be foundbetween the fungal species ecology and Hce2 gene numbers in the different fungal species.
3377
Evolution of a Fungal Effector Superfamily . doi:10.1093/molbev/mss143 MBE

evident as with Class I and Class II proteins. This resulted in anumber of apparent, albeit unsupported, differences betweenthe Hce2 and the species phylogeny in this clade that could beeither an artifact of the fairly unresolved phylogenetic rela-tionships or indicative of extensive gene loss in an array offungal lineages.
Birth-and-Death Evolution within Specific FungalLineages
To better understand the tempo and mode of evolutionwithin the Hce2 superfamily, we examined in greater detail
Ecp2 proteins from Sordariaceae (Sordariomycetes) andArthrodermataceae (Eurotiomycetes) (presented in Sup-plementary Material online). We chose these fungal familiesbecause 1) each represented a different order of Ascomycotafrom which Hce2 genes were identified, 2) several differenthighly similar Hce2 ortholog groups were present in at leastfour closely related species of the family that would allow theuse of codon-based models for the detection of changes inevolutionary pressure in members of the Hce2 superfamily,and 3) they exhibited other interesting features, such as aparticularly large lineage-specific expansion (Sordariaceae),
Fig. 5. The evolution of the Hce2 superfamily follows the birth-and-death model of gene family evolution. Reconciliation of the Ecp2 domain genealogywith the Ascomycetes species tree was performed using the Notung software. The Ascomycota species tree was assembled manually with reference toavailable published phylogenies, while the Hce2 genealogy is a maximum likelihood-inferred extended majority-rule consensus bootstrap tree. Hce2 geneduplications are indicated by yellow dots and losses by red dots. When losses occur on specific lineages, then these are specified as blue nodes on thephylogenetic tree. The analysis suggested the occurrence of 23 duplication and 37 loss events in the evolutionary history of the Hce2 superfamily inAscomycota.
3378
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE

and, unlike other fungal families, 4) they comprised mainly ofClass III chitinase-associated proteins (Arthrodermataceae).
SordariaceaeFour species were analyzed from this family, each of whichcontained a large number of Hce2 paralogs in their genome(N. crassa: 10 paralogs, N. discrete: 9 paralogs, N. tetrasperma:10 paralogs, and S. macrospora: 14 paralogs). BI phylogeneticanalysis of the Ecp2 domain of Sordariaceae Hce2 proteinsidentified 10 distinct groups of orthologs (Groups I–X;fig. 6A). All but one of the ortholog groups (Group IV),were supported by very high posterior probability clade sup-port values. Groups I and II contain exclusively class III pro-teins and form a robustly supported (100% BI posteriorprobability) clade. The sister relationship of these twogroups supports our previous assumptions (see supplemen-tary results, Supplementary Material online) on the early ra-diation and diversification of class III proteins. Nine ofthe groups contained orthologs from all four species, withonly group I containing orthologs from N. crassa andN. tetrasperma, indicating the loss of this ortholog inN. discreta and S. macrospora. Indeed, reconciliation of theSordariaceae Hce2 gene tree with the Ascomycota species treeconfirmed this loss (L) and further suggested the occurrenceof 13 duplications (D) in the evolutionary history of the Hce2superfamily in Sordariaceae (D/L Score = 21.5 D = 13, L = 2,99% BI posterior probability Edge Weight Threshold)(fig. 6B). Four of these duplications were associated exclusivelywith S. macrospora, thus contributing to the expansion of thissuperfamily in this species. The other nine duplications alltook place in the four species’ common ancestor.
It should be noted that the placement of sequenceNtet_85717 from N. tetrasperma differed between the phy-logeny constructed using only the sequences from the fourSordariaceae species and the phylogeny using all Hce2 super-family sequences. In the Sordariaceae-specific phylogeny thisgene is placed in group IV, while in the overall phylogeny itappears together with sequences from group II. However, byanalyzing syntenic information within 10 kb regions (supple-mentary fig. S7, Supplementary Material online) of theSordariaceae Hce2 genes it was finally determined thatNtet_85717 should be regarded as orthologous to group IVHce2 proteins. The only genomic loci that were not syntenicto any other were the ones flanking genes CBI57005.1,CBI57631.1, CBI57148.1, and CBI54907.1, which were inferredby the reconciliation analysis to be the products of gene du-plications within S. macrospora, thus confirming these resultsand indicating a rapid dispersal of the Hce2 genes in genomesafter duplication.
Gene duplication can generate new genes with novel oraltered functions and functional divergence of paralog genesis a major factor promoting their retention in the genome. Insuch cases, adaptive evolution or relaxed selection in earlystages after duplication plays a critical role towards the func-tional diversification of the two copies, whereas at later stagespurifying selection can ensure the maintenance of their dis-tinct functions. To investigate changes in evolutionary pres-sure on members of Hce2 superfamily in Sordariaceae that
could be indicative of diversification in putative functions, weconducted a series of likelihood ratio tests between alterna-tive models of codon evolution for each group of orthologs.Because the per-site frequency of synonymous substitutionswas saturated (dS> 1.0) on several internal branches of theSordariaceae Hce2 gene phylogeny, thus preventing accurateestimation of the o (dN/dS) ratio, we restricted our analysiswithin the orthologous groups of Hce2s. Group I was excludedfrom the analysis as it consists of only two members.Estimates of o under the Model M0 showed low overallvalues ranging from 0.06 to 0.23 depending on the groupexamined (table S3). Interestingly, LRT of site-specificmodels showed (irrespective of dS saturation) that for allnine groups examined, model M3 fitted the data significantlybetter than model M0, thus suggesting heterogeneous selec-tion pressure among codon sites that could have promoteddivergence of the Hce2 superfamily members across clades.For groups III, V, VIII, and IX all three classes of o under M3had values <1, suggesting purifying selection. However, forgroups II, IV, VI, VII, and X, a small proportion of sites (1–3%)were predicted to show o> 1. To identify the specific codonsites evolving with o> 1, we further tested whether modelsM2a and M8 that allow for positively selected sites fit the databetter than their null models M1a and M7, respectively. Ineight of the nine groups, LRTs between models M1a and M2aand between M7 and M8 were not significantly different,suggesting it is unlikely that these are sites under strong pos-itive selection. In contrast, in group VII all three LRT testsshowed that models M3, M2a, and M8 fitted the data signif-icantly better than their nested null models M0, M1a, and M7,respectively, leading to the identification of four sites under-going positive selection. Two of these sites, though, were con-sistently detected with significant posterior probabilities(P> 95%), and they involved transitions from Alanine-to-Lysine or vice-versa and from a Tryptophan-to-Leucine andThreonine (supplementary fig. S8, Supplementary Materialonline). These sites were located in highly variable regionsof the overall alignment. Nevertheless, despite the presenceof two positively selected sites in Group VII, purifying selec-tion is the dominant evolutionary force shaping the evolutionof the Hce2 ortholog groups in Sordariaceae.
The codon-based analysis is restricted to within group var-iation and does not provide any information for changes inevolutionary rates between groups. Furthermore, it is unlikelythat positive selection would have operated on large numbersof sites of the duplicated Hce2 genes over prolonged periodsof time. Rather, diversification in putative functions mighthave happened soon after gene duplication. Changes in func-tional constraints at individual sites could reflect a change inrates of evolution (known as Type I divergence) and result inamino acid sites that are highly conserved in one group ofparalogs but highly variable in another (Gu 1999, 2001).Alternatively, these changes in functional constraints at indi-vidual sites could reflect a change in amino acid properties(known as Type II divergence) and result in amino acid sitesthat, although conserved within each group of paralogous,when compared between such groups differ radically in theirbiochemical properties (Gu 2006). To test for evidence of
3379
Evolution of a Fungal Effector Superfamily . doi:10.1093/molbev/mss143 MBE

Type I and/or Type II functional divergence in the paraloggroups of Hce2 proteins in Sordariaceae, we conducted allpossible pairwise comparisons between Groups II-to-X andestimated their coefficient of Type I (#I) and Type II (#II)functional divergence (Gu 1999, 2001, 2006).
Estimates of #I for almost all examined pairs were moder-ate (0.5�#I� 0) to high (1�#I> 0.5), suggesting significantshifts in evolutionary rates between the groups (supplemen-tary table S4, Supplementary Material online). However, allpairwise comparisons of group IV with the rest of the groupsresulted in #I = 0, indicating absence of Type I functional di-vergence. This is surprising given that Group IV is the mostdivergent in sequence when compared to all other groups(supplementary table S4, Supplementary Material online). It ispossible that this lack of significance is an artifact caused bythe high degree of sequence divergence within Group IV,which is almost 2-fold higher (0.43%) as compared to allother groups (0.18% on average). From the 28 pairwise com-parisons, #I was significantly higher than 0 in 24 (P< 0.05).Consideration of the site-specific posterior probabilities didnot identify any specific protein domains with elevated ratesof evolutionary divergence. Rather a large number of aminoacid sites distributed across the entire alignment display high(#I� 0.8) Type I divergence (supplementary fig. S9A,Supplementary Material online). Evidence of Type II func-tional divergence was also present. Of the 36 comparisons28 were statistically significant. Again, comparisons involvinggroup IV were not significant. Type II functional divergencedetected a large number of amino acid sites distributed across
the alignment with radical and statistically significant changes(#II> 1) in their biochemical properties (supplementary fig.S9B, Supplementary Material online). Collectively, the data forType I and Type II functional divergence provide strong evi-dence for early diversification in putative functions, both inevolutionary rates and amino acid properties, between dupli-cated genes in the Hce2 superfamily.
Discussion
Hce2 Is an Ancient Superfamily within Fungi
The taxonomic circumscription of the Hce2 superfamilywithin fungi and the presence of multiple paralogs per speciessuggest that this superfamily originated and diversified withinthe fungal kingdom. Furthermore, the presence of the Hce2superfamily in both Ascomycota and Basidiomycota indicatesthat its origin predates the divergence of Dikarya. However,this scenario requires a complex pattern of gene maintenanceand loss along many intervening lineages and taxa, implying,on one hand, the maintenance of Hce2 genes for long periodsof time in Pezizomycotina (Ascomycota) andAgaricomycotina (Basidiomycota), and, on the other, loss ofHce2 genes on numerous independent lineages (e.g.,Saccharomycotina). Given the clear overrepresentation ofthese genes in Pezizomycotina and the presence of multipleparalogs in species of this subphylum as compared toAgaricomycotina, an alternative hypothesis is that the Hce2superfamily originated prior to, or early in the evolution ofPezizomycotina, from where it was horizontally transferred toAgaricomycotina. This hypothesis relies solely on the taxon
Fig. 6. The majority of duplication events is ancient and predates speciation events in Sordariaceae. (A) The Bayesian Markov Chain Monte Carlo(MCMC) inferred phylogeny of the Hce2 superfamily from species of Sordariaceae using only the Ecp2 domain. Nodes are color-coded according thespecies that the Hce2 proteins were derived from and the groups of orthologous Hce2 proteins are marked with Latin numbers. Bayesian posteriorprobabilities supporting the phylogeny are shown for each node. The phylogeny reveals almost exclusive clustering based on orthology rather thanparalogy, suggesting that the Hce2 superfamily expanded in Sordariaceae prior to the separation of the four species analyzed. Groups I and II consistexclusively from class III proteins that form a monophyletic group, suggesting their early radiation and diversification from the rest of the SordariaceaeHce2 proteins. Within each group, additional instances of gene duplications and losses can be found supporting birth-and-death evolution. (B) Thereconciliation of the Sordariaceae Hce2 genealogy with the Ascomycota species tree. The analysis suggests the occurrence of 13 duplications (yellowdots) and 2 losses (red dots) in the evolutionary history of the Hce2 family within Sordariaceae. Most duplication took place already before the radiationof the four species, while the family seems to have continued expanding in Sordaria macrospora.
3380
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE

distribution of this gene superfamily and it is not supportedby the observed phylogeny, and should therefore be treatedwith caution until more extensive and uniform sampling ofgenomes across fungi has been achieved.
Recurrent Gene Birth-and-Death Defines theEvolution of the Hce2 Superfamily
The Hce2 superfamily is characterized by frequent indepen-dent duplications and losses, even among closely related spe-cies, consistent with the birth-and-death model (Nei et al.1997; Nei and Rooney 2005). Clear evidence ofbirth-and-death evolution in the Hce2 superfamily wasfound by examining lineage-specific variations in gene copynumbers of putative orthologs from closely related specieswithin the same fungal genus or family. In almost all suchcases examined we observed 1) high rates of copy numbervariation even among closely related species, 2) clustering ofHce2 genes based on orthology rather than paralogy, 3) con-servation of gene structure among orthologous Hce2 genes,and in some cases, 4) presence of pseudogenes. All these areconsistent with the birth-and-death model of evolution (Neiet al. 1997; Nei and Rooney 2005).
The most likely scenario for the evolution of the Hce2superfamily in Ascomycota is that a single gene was presentin a common ancestor of Pezizomycotina, which indepen-dently underwent multiple expansions and contractions in alineage-specific manner. In this respect, species of Sordario-mycetes have experienced a large-scale expansion in Hce2numbers since their divergence from Eurotiomycetes. In con-trast, Hce2 genes were mostly lost from species in the order ofEurotiomycetes, whereas in Dothideomycetes they under-went both gains and losses. The ecological pressures drivingsuch lineage-specific expansions and contractions are unclear,since no correlation was found between Hce2 numbers andthe species ecology. The largest lineage-specific expansiontook place in Sordariaceae, which is rather surprising giventhat in N. crassa, and presumably its close relatives as well, thefixation of duplicated genes is greatly reduced by the RepeatInduced Point mutation (RIP) mechanism that largely pre-vents evolution through gene duplication (Galagan et al.2003). The presence of so many paralogs that have escapedthe effects of RIP indicates that these are ancient copies,whose duplication and diversification predates the emer-gence of RIP (Galagan et al. 2003).
Both Divergent Evolution and Purifying SelectionHave Shaped the Evolution of Hce2 Genes
Gene duplication is a major force of genomic innovation,constantly creating new genes, whose retention in thegenome depends on their functional diversification (Lynchand Conery 2000; Conant and Wolfe 2008). In such cases,positive or relaxed selection in one of the two copies inearly stages after duplication drives functional diversification,whereas at later stages purifying selection maintains the newlyacquired function(s) (Lynch and Conery 2000). This seems tobe the case for duplicated Hce2 genes as well. Our analyses onthe Hce2 superfamily support a model where Hce2 duplicates
diversify rapidly early after duplication as a result of positive orrelaxed selection, most likely acquiring new functions as well.Indeed, the high levels of sequence divergence among para-logs and their dispersal in the genome are indicative of anaccelerated rate of evolution after duplication. Moreover,tests for Type I and Type II functional divergence betweenalmost all pairs of paralogous Hce2 groups in Sordariaceaeand Arthrodermataceae indicate that the newly generatedparalogs diversified in their putative functions early in theirevolution. Finally, our molecular evolutionary analyses showthat, once diversified, Hce2 genes within ortholog groups areunder purifying selection. Thus, any changes that contributedto the diversification in putative functions were likely fixedearly in their evolution.
Hce2 Proteins Are Putatively Involved inStress-Responses
The driving force behind the patchy distribution of Hce2genes in different fungal species is unknown but likely relatesto their yet uncharacterized intrinsic function. While genedeletions and duplications are frequently observed in multi-gene families, the diverse and large-scale lineage-specific ex-pansion and losses observed here suggest that Hce2 genesperform taxon-specific roles providing conditional advan-tages in specific environmental niches. One possibility isthat Hce2 genes are gained or lost after speciation eventsaccording to niche-specific selection pressures, thusfine-tuning environmental and/or parasitic fitness (Conantand Wolfe 2008).
Such a dynamic response to the environmental stimuliinvolving high rates of gains and losses is frequently observedin stress-response genes, such as the ones implicated in inter-actions with other organisms, adaptive immunity or pathoa-daptation to new hosts (Ota, Nei 1994; Nei and Rooney 2005;Wapinski et al. 2007; Korbel et al. 2008; Pujol et al. 2008).Effector proteins in particular from fungi, bacteria and oomy-cetes are known for their rapid diversification and acceleratedrates of birth-and-death evolution that lead to highly discon-tinuous distributions, even between taxa belonging to thesame species complex (Jiang et al. 2006; Stergiopoulos et al.2007; Jiang et al. 2008; Stavrinides et al. 2008). The intrinsicfunction of Hce2 genes remains unknown but in the plantpathogenic fungi C. fulvum and M. fijiensis they may functionas effector proteins that both promote virulence in suscepti-ble hosts and induce R-gene-mediated resistance in resistantones (Lauge et al. 1997; Stergiopoulos et al. 2010). It is possi-ble that Hce2 proteins from other plant pathogenic fungihave a similar role in promoting virulence as well. If this isthe case, then the broad distribution of Hce2 genes in fungipathogenic on a variety of hosts suggests that these genes arepart of the pathogenic core that targets broadly conserveddefense components (Stergiopoulos et al. 2010). As such, it isunlikely that they contribute directly to host specializationbut rather facilitate pathogenicity on a wide range of hosts byproviding basic virulence functions. Maintenance or loss ofHce2 genes following speciation and potentially host jumpswould depend both on their role during pathogenesis and the
3381
Evolution of a Fungal Effector Superfamily . doi:10.1093/molbev/mss143 MBE

conservation of their virulence targets in different hosts, butalso on the suite of cognate resistance genes that mediateimmune responses in their hosts (Stergiopoulos et al. 2010).The preservation of multiple highly diverse paralogs per spe-cies would further suggest that the basic function of Hce2proteins can be manipulated to fine-tune interactionswith the host, either by targeting diverse defense components(and acting perhaps even synergistically) or by being ex-pressed at different stages and levels during infection.
The study of fungal effector proteins has so far been ap-proached from a predominately host-microbe interactionsperspective (Morris et al. 2009). Pathogens, however, do notonly have to infect a host in order to be evolutionarily suc-cessful but also need to compete with numerous other mi-crobial species present in their environment and survivesaprophytically for large periods of their life-cycle. Thus,both pathogen biology on the host and the environment inwhich the organism is living-in will strongly influence theevolution of a pathogen’s virulence (Wolinska and King2009; Allen, Little 2011). In this study, a large number ofHce2 genes were identified from fungal species that are par-asitic on human, animals, insects and other fungi or sapro-phytic. In such cases, the function of Hce2s could have beenadapted to meet their demands in these environments, in-cluding activities as toxigenic peptides in antagonistic inter-actions with other microorganisms. The fusion of the Ecp2domain of some Hce2 proteins to fungal GH18 chitinases andtheir association with the Zymocin killer toxin from yeastfavors this hypothesis. Overall, the presence of Hce2s outsideplant pathogens and their implication in possible antagonistinteractions with other microorganisms in their environmentemphasizes the necessity to study the biology and evolutionof virulence traits and of putative effectors beyond the cur-rent perspective of host-microbe interactions and in thebroader context of pathogen ecology. Thus, a more holisticunderstanding of microbial ecology is needed in order to fullyunderstand effector biology and microbial pathogenicity ingeneral. In this respect, studies in human pathogens haveshown that many virulence traits have dual roles in parasiticand environmental fitness and their evolution has beenshaped by forces outside the narrow context ofhuman-pathogen interactions (Pallen and Wren 2007;Morris et al. 2009). The recent characterization of the TypeVI secretion system (T6SS) in bacteria, for example, has led tothe identification of T6SS effectors that are not only impor-tant for pathogenesis but also mediate competitive interac-tions with other bacteria (Jani and Cotter 2010). Similarly, anumber of virulence traits are shared between plant patho-genic fungi and saprophytes (e.g., LysM effectors) or have dualroles in parasitic fitness (e.g. several mycotoxins) and ecolog-ical survival (e.g., melanin) (Nosanchuk and Casadevall 2003;Berestetskiy 2008; de Jonge and Thomma 2009).
Hce2 Genes Show Unique Associations with FungalGH18 Chitinases
GH18 is an ancient family of chitinases, widely distributed inArchaea, Bacteria, and Eukaryota, including humans and
fungi, whose members catalyze the hydrolysis of chitin, astructural component of the cell wall of fungi and the exo-skeleton of arthropods. In fungi, GH18 chitinases are involvedin diverse physiological functions, including mycoparasitismthrough lyses of the antagonist’s cell wall. The chitinasedomain of the class III proteins shows considerable similarityto the Zymocin alpha (�)-subunit of the yeast killer toxin, aheterotrimeric (���) eukaryotic tRNase toxin from the dairyyeast K. lactis that inhibits cell cycle proliferation by arrestingthe G1 phase in Saccharomyces cerevisiae (Stark et al. 1990).Toxicity of Zymocin resides in the intracellularly targeted�-subunit (�-toxin), the import of which into host cells isfacilitated by the larger �- and �-subunits that act from thecell exterior to promote contact with the cell-surface andassociation with the plasma membrane (Stark et al. 1990;Jablonowski et al. 2001). The �-subunit is an exochitinasethat binds to chitin, thereby facilitating docking to and sub-sequent chitinolysis of the cell-wall chitin, whereas the�-subunit is predicted to be associated with thecell-membrane. Based on the presence of similar domainsin class III proteins and Zymocin and the high sequence sim-ilarity of the chitinase domains, it is tempting to speculatethat the Ecp2 domain of fungal GH18 chitinases and the�-subunit of the yeast killer toxin have an analogous role inantagonistic interactions with other microorganisms, despitethe lack of any sequence similarity between these two do-mains. Although testing this hypothesis requires functionalassays, it is intriguing that both toxigenic peptides have re-cruited the same set of proteins for delivery into their hostcells. The fusion of the Ecp2 domain to GH18 chitinases fur-ther suggests that, in addition to gene duplication and rapiddiversification, new effector specificities in fungi might begenerated by the recruitment of unrelated protein domains.This domain recruitment by putative fungal effectors to en-hance or alter their function is a novel finding for fungaleffectors that changes our traditional views on these proteinsand reveals their evolutionary and structural plasticity.
Supplementary MaterialSupplementary tables S1–S5, figures S1–S14, Materials andMethods, and Results are available Molecular Biology andEvolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work used resources of the Advanced Computing Centerfor Research and Education (ACCRE) at Vanderbilt University,Nashville, TN, USA. This work was supported by the SearleScholars Program (to A.R.) and the National ScienceFoundation (DBI-0805625 to J.C.S. and DEB-0844968 toA.R.). I.S. was in part financially supported by the NWO-ERA-PG project ARAPGFP/06.002A entitled “RLP-andRLK-mediated innate immune responses in Arabidopsis andtomato triggered by pathogen-associated molecular patternsand avirulence factors”. This project was co-financed by theCentre for BioSystems Genomics (CBSG), which is part ofthe Netherlands Genomics Initiative, a Dutch organization
3382
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE

for scientific research. P.J.G.M.deW. was co-financed by theRoyal Netherlands Academy of Arts and Sciences.
ReferencesAllen D, Little T. 2011. Dissecting the effect of a heterogeneous environ-
ment on the interaction between host and parasite fitness traits.
Evol Ecol. 25:499–508.
Bailey TL, Williams N, Misleh C, Li WW. 2006. MEME: discovering and
analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34:
W369–W373.
Berestetskiy A. 2008. A review of fungal phytotoxins: from basic studies
to practical use. Appl Biochem Microbiol. 44:453–465.
Butler AR, O’Donnell RW, Martin VJ, Gooday GW, Stark MJ. 1991.
Kluyveromyces lactis toxin has an essential chitinase activity. Eur J
Biochem. 199:483–488.
Chuma I, Isobe C, Hotta Y, et al. (12 co-authors). 2011. Multiple trans-
location of the AVR-Pita effector gene among chromosomes of the
rice blast fungus Magnaporthe oryzae and related species. PLoS
Pathog. 7:e1002147.
Conant GC, Wolfe KH. 2008. Turning a hobby into a job: how duplicated
genes find new functions. Nat Rev Genet. 9:938–950.
Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple align-
ment of conserved genomic sequence with rearrangements.
Genome Res. 14:1394–1403.
de Jonge R, Thomma BP. 2009. Fungal LysM effectors: extinguishers of
host immunity? Trends Microbiol. 17:151–157.
Durand D, Halldorsson BV, Vernot B. 2006. A hybrid micro-
macroevolutionary approach to gene tree reconstruction. J
Comput Biol. 13:320–335.
Felsenstein J. 2005. PHYLIP (Phylogeny Inference Package) version 3.6
Distributed by the author. Available from: http://evolution.genetics.
washington.edu/phylip.html
Fitzpatrick DA, Logue ME, Stajich JE, Butler G. 2006. A fungal phylogeny
based on 42 complete genomes derived from supertree and com-
bined gene analysis. BMC Evol Biol. 6:99.
Frith MC, Saunders NF, Kobe B, Bailey TL. 2008. Discovering sequence
motifs with arbitrary insertions and deletions. PLoS Comput Biol. 4:
e1000071.
Galagan JE, Calvo SE, Borkovich KA, et al. 2003. The genome se-
quence of the filamentous fungus Neurospora crassa. Nature 422:
859–868.
Gu X. 1999. Statistical methods for testing functional divergence after
gene duplication. Mol Biol Evol. 16:1664–1674.
Gu X. 2001. Maximum-likelihood approach for gene family evolution
under functional divergence. Mol Biol Evol. 18:453–464.
Gu X. 2006. A simple statistical method for estimating type-II
(cluster-specific) functional divergence of protein sequences. Mol
Biol Evol. 23:1937–1945.
Hajri A, Brin C, Hunault G, Lardeux F, Lemaire C, Manceau C, Boureau T,
Poussier S. 2009. A “repertoire for repertoire” hypothesis: repertoires
of type three effectors are candidate determinants of host specificity
in Xanthomonas. PLoS One 4:e6632.
Hogenhout SA, Van der Hoorn RA, Terauchi R, Kamoun S. 2009.
Emerging concepts in effector biology of plant-associated organisms.
Mol Plant Microbe In. 22:115–122.
Jablonowski D, Frohloff F, Fichtner L, Stark MJ, Schaffrath R. 2001.
Kluyveromyces lactis zymocin mode of action is linked to RNA po-
lymerase II function via Elongator. Mol Microbiol. 42:1095–1105.
James TY, Kauff F, Schoch CL, et al. (70 co-authors). 2006.
Reconstructing the early evolution of fungi using a six-gene phylog-
eny. Nature 443:818–822.
Jani AJ, Cotter PA. 2010. Type VI secretion: not just for pathogenesis
anymore. Cell Host Microbe 8:2–6.
Jiang RH, Tripathy S, Govers F, Tyler BM. 2008. RXLR effector reservoir in
two Phytophthora species is dominated by a single rapidly evolving
superfamily with more than 700 members. Proc Natl Acad Sci U S A.
105:4874–4879.
Jiang RH, Tyler BM, Whisson SC, Hardham AR, Govers F. 2006. Ancient
origin of elicitin gene clusters in Phytophthora genomes. Mol Biol
Evol. 23:338–351.
Korbel JO, Kim PM, Chen X, Urban AE, Weissman S, Snyder M, Gerstein
MB. 2008. The current excitement about copy-number variation:
how it relates to gene duplications and protein families. Curr Opin
Struct Biol. 18:366–374.
Lauge R, Joosten MHAJ, VandenAckerveken GFJM, VandenBroek HWJ,
De Wit PJGM. 1997. The in planta-produced extracellular proteins
ECP1 and ECP2 of Cladosporium fulvum are virulence factors. Mol
Plant Microbe In. 10:725–734.
Lynch M, Conery JS. 2000. The evolutionary fate and consequences of
duplicate genes. Science 290:1151–1155.
Ma W, Dong FF, Stavrinides J, Guttman DS. 2006. Type III effec-
tor diversification via both pathoadaptation and horizontal
transfer in response to a coevolutionary arms race. PLoS Genet.
2:e209.
Maddison WP. 2000. Testing character correlation using pairwise com-
parisons on a phylogeny. J Theor Biol. 202:195–204.
Maddison WP, Maddison DR. 2010. Mesquite: a modular system
for evolutionary analysis version 2.73. Available from: http://
mesquiteproject.org/mesquite/mesquite.html
Morris CE, Bardin M, Kinkel LL, Moury B, Nicot PC, Sands DC. 2009.
Expanding the paradigms of plant pathogen life history and evolu-
tion of parasitic fitness beyond agricultural bundaries. PLoS Pathog.
5:e1000693.
Needleman SB, Wunsch CD. 1970. A general method applicable to the
search for similarities in the amino acid sequence of two proteins. J
Mol Biol. 48:443–453.
Nei M, Gu X, Sitnikova T. 1997. Evolution by the birth-and-death process
in multigene families of the vertebrate immune system. Proc Natl
Acad Sci U S A. 94:7799–7806.
Nei M, Rooney AP. 2005. Concerted and birth-and-death evolution of
multigene families. Annu Rev Genet. 39:121–152.
Nielsen R, Yang Z. 1998. Likelihood models for detecting positively se-
lected amino acid sites and applications to the HIV-1 envelope gene.
Genetics 148:929–936.
Nosanchuk JD, Casadevall A. 2003. The contribution of melanin to mi-
crobial pathogenesis. Cell Microbiol. 5:203–223.
Ota T, Nei M. 1994. Divergent evolution and evolution by the
birth-and-death process in the immunoglobulin VH gene family.
Mol Biol Evol. 11:469–482.
Pallen MJ, Wren BW. 2007. Bacterial pathogenomics. Nature 449:
835–842.
Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, Jin Y,
Chisholm AD, Ewbank JJ. 2008. Distinct innate immune responses to
infection and wounding in the C. elegans epidermis. Curr Biol. 18:
481–489.
Read AF, Nee S. 1995. Inference from binary comparative data. J Theor
Biol. 173:99–108.
3383
Evolution of a Fungal Effector Superfamily . doi:10.1093/molbev/mss143 MBE

Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian
phylogenetic inference under mixed models. Bioinformatics 19:
1572–1574.
Sarkar SF, Gordon JS, Martin GB, Guttman DS. 2006. Comparative ge-
nomics of host-specific virulence in Pseudomonas syringae. Genetics
174:1041–1056.
Schoch CL, Sung GH, Lopez-Giraldez F, et al. (64 co-authors) 2009. The
Ascomycota tree of life: a phylum-wide phylogeny clarifies the origin
and evolution of fundamental reproductive and ecological traits.
Syst Biol. 58:224–239.
Schurch S, Linde CC, Knogge W, Jackson LF, McDonald BA. 2004.
Molecular population genetic analysis differentiates two virulence
mechanisms of the fungal avirulence gene NIP1. Mol Plant Microbe
In. 17:1114–1125.
Seidl V, Huemer B, Seiboth B, Kubicek CP. 2005. A complete survey of
Trichoderma chitinases reveals three distinct subgroups of family 18
chitinases. FEBS J. 272:5923–5939.
Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylo-
genetic analyses with thousands of taxa and mixed models.
Bioinformatics 22:2688–2690.
Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algo-
rithm for the RAxML Web servers. Syst Biol. 57:758–771.
Stark MJ, Boyd A, Mileham AJ, Romanos MA. 1990. The
plasmid-encoded killer system of Kluyveromyces lactis: a review.
Yeast 6:1–29.
Stavrinides J, McCann HC, Guttman DS. 2008. Host-pathogen inter-
play and the evolution of bacterial effectors. Cell Microbiol. 10:
285–292.
Stergiopoulos I, De Kock MJ, Lindhout P, De Wit PJGM. 2007. Allelic
variation in the effector genes of the tomato pathogen
Cladosporium fulvum reveals different modes of adaptive evolution.
Mol Plant Microbe In. 20:1271–1283.
Stergiopoulos I, De Wit PJGM. 2009. Fungal effector proteins. Annu Rev
Phytopathol. 47:233–263.
Stergiopoulos I, van den Burg HA, Okmen B, Beenen HG, van Liere S,
Kema GH, De Wit PJGM. 2010. Tomato Cf resistance proteins me-
diate recognition of cognate homologous effectors from fungi path-
ogenic on dicots and monocots. Proc Natl Acad Sci U S A. 107:
7610–7615.
Stukenbrock EH, McDonald BA. 2009. Population genetics of fungal and
oomycete effectors involved in gene-for-gene interactions. Mol Plant
Microbe In. 22:371–380.
Vernot B, Stolzer M, Goldman A, Durand D. 2008. Reconciliation with
non-binary species trees. J Comput Biol. 15:981–1006.
Wapinski I, Pfeffer A, Friedman N, Regev A. 2007. Natural history and
evolutionary principles of gene duplication in fungi. Nature 449:
54–61.
Wolinska J, King KC. 2009. Environment can alter selection in host
parasite interactions. Trends Parasitol. 25:236–244.
Yang Z. 1997. PAML: a program package for phylogenetic analysis by
maximum likelihood. Comput Appl Biosci. 13:555–556.
Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood.
Mol Biol Evol. 24:1586–1591.
Yang Z, Wong WS, Nielsen R. 2005. Bayes empirical bayes inference of
amino acid sites under positive selection. Mol Biol Evol. 22:
1107–1118.
3384
Stergiopoulos et al. . doi:10.1093/molbev/mss143 MBE
Related Documents











