Research Collection Doctoral Thesis Versuche zur Synthese von 5-Thiomethyl-D-ribose Author(s): Wyss, Franz Publication Date: 1955 Permanent Link: https://doi.org/10.3929/ethz-a-000089053 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Research Collection
Doctoral Thesis
Versuche zur Synthese von 5-Thiomethyl-D-ribose
Author(s): Wyss, Franz
Publication Date: 1955
Permanent Link: https://doi.org/10.3929/ethz-a-000089053
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2419
Versuche zur Synthese von
5-Thiomethyl-D-ribose
VON DER
EIDGENÖSSISCHEN TECHNISCHEN HOCHSCHULE IN ZÜRICH
ZUR ERLANGUNG DER WÜRDE EINES
DOKTORS DER TECHNISCHEN WISSENSCHAFTEN
GENEHMIGTE
PROMOTIONSARBEIT
VORGELEGT VON
Franz Wyss
Dipl. Ingenieur-Chemiker ETH
von Solothurn und Balm b. Messen (SO)
Referent : Herr Prof. Dr. V. Prelog
Korreferent: Herr P.-D. Dr. E. Hardegger
* /
Zürich 1955
Dissertationsdruckerei Leemami AG.

Leer - Vide - Empty

MEINER LIEBEN MUTTER
IN DANKBARKEIT
GEWIDMET

Leer - Vide - Empty

Meinen verehrten Lehrern
Herrn Prof. Dr. L. Ruzicka
und
Herrn P.-D. Dr. E. Hardegger
danke ich für die grosszügige Leitung dieser Arbeit, das fördernde
Interesse, das sie ihr stets entgegenbrachten und die vielen wert¬
vollen Ratschläge, die mir zuteil wurden.

Leer - Vide - Empty

Inhaltsverzeichnis
Theoretischer Teil 9
Einleitung 9
Geschichte der 5-Thiomethyl-D-ribose 10
Eigene Versuche zur Synthese von 5-Thiomethyl-D-ribose:
a) Aus D-Ribose-mercaptalen 20
b) Aus D-Ribonsäure-y-lacton 23
Experimenteller Teil 32
Zusammenfassung 48
7

Leer - Vide - Empty

Theoretischer Teil
Einleitung
Die vorliegende Dissertation wurde Ende 1950 begonnen, also
zu einem Zeitpunkt, da über die Konfiguration der von J. A. Mandel
und E. K. Dunham aus Hefe isolierten Thiomethyl-aldopentose nur
wenig bekannt war. Kurz vor Beginn meiner Untersuchungenwurde von F. Weygand die endständige Lage der Thiomethyläther-Gruppe in der Hefe-Thiomethyl-pentose wahrscheinlich gemacht.Da die Thiomethyl-pentose der Hefe aus einer Nucleosid-fraktion
stammte und in Nucleosiden als Pentosen bisher nur D-Ribose und
2-Desoxy-D-ribose gefunden worden waren, schien es als Arbeits¬
hypothese aussichtsreich, der natürHchen Hefe-Thiomethyl-pentosezunächst die Konstitution der 5-Thiomethyl-D-ribose zuzuschrei¬
ben.
Die Bestätigung dieser Annahme sollte durch die Synthese der
5-Thiomethyl-D-ribose erbracht werden. Es war naheliegend, zu
diesem Zwecke Verbindungen mit Ribose-Konfiguration herzu¬
stellen, die am C-Atom 5 des Pentosegerüstes eine freie Hydroxyl¬
gruppe aufweisen. Die Darstellung und Konstitutionsbestimmungdieser meist neuen Derivate erwies sich als mühsam und zeitraubend.
Da zudem die weiteren Reaktionen, für die zahlreiche Beispieleaus der Zuckerchemie bekannt sind, unerwartete Schwierigkeitenbrachten, konnte das Endprodukt der Synthese nicht mehr erhal¬
ten werden.
Inzwischen ist sowohl von englischer wie von deutscher Seite
die Synthese der 5-Thiomethyl-D-ribose erfolgreich durchgeführtund die Identität dieser Verbindung mit der natürlichen Thio¬
methyl-pentose aus Hefe bewiesen worden.
9

Geschichte der 5-Thiomethyl-D-ribose
Im Jahre 1912 wurde von J. A. Mandel und E. K. Dunham1)
und zwei Jahre später von U. Suzuki2) bei der Aufarbeitung der
Nucleosidfraktion aus Hefe ein kristallisiertes, schwefelhaltigesAdenosid von der Bruttoformel C11H1503N5S und vom Schmelz¬
punkt 212° isoliert. Erst zehn Jahre später (1924) fanden Suzuki
und Mitarbeiter z), dass bei der unter milden Bedingungen erfolgen¬den sauren Hydrolyse das oben erwähnte Thio-adenosid in 1 Mol
Adenin und 1 Mol eines Thiozuckers von der Zusammensetzung
C6H1204S gespalten wurde.
Die Verbindung C6H1204S ist bis heute der einzige in der Natur
aufgefundene Thiozucker geblieben. Er konnte bisher nicht in kri¬
stallisierter Form erhalten werden, dagegen gab er mehrere gut
kristallisierte Derivate. Er reduzierte Fehlingsche Lösung und ver¬
brauchte bei der Titration nach Willstätter-Schudel 4 Atome Jod4).
Nach der Oxydation mit überschüssigem Blei-tetra-acetat konnte
in den Oxydationsprodukten des Thiozuckers kein Formaldehyd
nachgewiesen werden, obschon in Modellversuchen mit Mannit die¬
ser Nachweis quantitativ gelang. Von Salpetersäure wurde der Thio¬
zucker unter Aufnahme von 2 Atomen Sauerstoff zu einer Mono-
carbonsäure C6H1206S vom Schmelzpunkt 183°—184° oxydiert5).Dieselbe Säure wurde auch bei der bereits erwähnten Titration mit
Hypojodit nach Wülstätter-Schudel erhalten4).Bei der Behandlung mit heisser Salzsäure gab der Thiozucker
Furfurol3). Mit Phenylhydrazin entstand ein kristallisiertes Phenyl -
osazon6) gemäss der Gleichung:
C9H1204S + 3C6H5NHNH2 -> C18H2202N4S + C6H5NH2 + NH3
Acetanhydrid und Pyridin führten ihn in ein bei 66°—67° schmel¬
zendes Triacetylderivat über6).
*) J. A. Mandel, E. K. Dunham, J. Biol. Chem. 11, 85 (1912).
2) U. Suzuki, J. Chem. Soc. Tokio, 34, 1134 (1914).
3) U. Suzuki, S. Odake, T. Mori, Biochem. Z. 154, 278 (1924).
4) G. Wendt, Z. physiol. Chem. 272, 152 (1942).
') U. Suzuki, T. Mori, Biochem. Z. 162, 413 (1925).
6) A. L. Raymond, J. Biol. Chem. 107, 85 (1934).
10

Mit Natriuniamalgam liess sich der Thiozucker C6H1204S unter
Aufnahme von 2 Atomen Wasserstoff zu einem gut kristallisierten,
schwefelhaltigen Zuckeralkohol C6H1404S vom Schmelzpunkt 118°
reduzieren 5). Der Thiozucker-alkohol C6H1404S verbrauchte bei der
stöchiometrisch verlaufenden Titration nach Willstätter-Schvdel mit
zwei Äquivalenten genau halb so viel Jod wie der Thiozucker
C6H1204S4). Nach der Einwirkung von überschüssigem Blei-tetra-
acetat auf den bei 118° schmelzenden Thiozucker-alkohol wurden
in den Oxydationsprodukten 0,7—0,9 Mol Formaldehyd gefunden4).
Monocarbonsäure
C6H12OeS
Adenosid
Thiozucker
/* C6H1204S
\ Adenin
HNO,od.
4HOJ
Phenyl-osazonClgHä!OsN4S
Phenyl¬hydrazin
— kein HCHO
Pb(ac)4
AosO+Pyridin
^ TriacetylderivatC9H180,S
12% HCl
heiss
CHO
O
NaHg
yThiozucker-alkohol
C6H1404SPb(ac),/ \
0,7—0,9HCHO verbraucht 2 HOJ
Die bis hieher beschriebenen, in der Zusammenstellung übersicht¬
lich angeordneten Umsetzungen erlauben folgende Aussagen über
die Konstitution des Thiozuckers C6H1204S:1. Das Vorliegen einer linearen Kette von 5 C-Atomen
11

ergibt sich durch die Überführung des Thiozuckers in Furfurol beim
Erhitzen mit Salzsäure7).2. Mit der Titration des Thiozuckers und seines Reduktionspro¬
duktes C6H1404S mit Hypojodit wird die Anwesenheit einer
Aldehydgruppe bewiesen8)9). In Übereinstimmung damit steht
auch die Oxydation des Thiozuckers mit Salpetersäure, welche
unter Erhaltung sämtlicher Kohlenstoffatome zu einer Mono-
carbonsäure führt7).3. Die Bruttoformel C6H1206S der unter 2. erwähnten Mono-
carbonsäure und deren Bildung aus dem Thiozucker C6H1204S mit
Salpetersäure, bzw. Hypojodit, kann nur an Hand einer Thio-
äthergruppe erklärt werden. Die Oxydation des Thiozucker¬
alkohols mit 2 Mol Hypojodit muss als Umwandlung der Thio-
äthergruppe in das Sulfoxyd interpretiert werden. Der Thiozucker
C6H1204S stellt demnach einen unverzweigten Aldopentose-
thiomethyläther dar.
4. Auf Grund der Umwandlung in ein Phenylosazon C18H2202N4Sbefindet sich benachbart zu der unter 2. bewiesenen Aldehyd¬
gruppe, also am C-Atom 2 der Thiomethyl-pentose ein sekun¬
däres Hydroxyl.Dieser Befund wird gestützt durch den Nachweis von 0,7—0,9
Mol Formaldehyd, bei der Glycolspaltung des Thiozuckeralkohols
C6H1404S mit Blei-tetra-acetat9).5. Die beiden noch nicht erfassten Sauerstoff-Atome müssen auf
Grund der unter 2., 3. und 4. erwähnten Bruttoformeln als Hydro-
xyl-Gruppen vorliegen, in Übereinstimmung mit der Darstellungeines Triacetyl-Derivats aus dem Thiozucker mit Acetanhydrid-
Pyridin.Die Überlegungen 1. bis 5. erlauben es für den Thiozucker
C6H1204S die Strukturformeln I, II und III aufzustellen, wobei zu
berücksichtigen ist, dass von jeder dieser Formeln je acht konfigu-rativ verschiedene Anordnungen der funktionellen Gruppen mög¬lich sind.
') U. Suzuki, T. Mori, Biochem. Z. 162, 413 (1925).
8) Vgl. dagegen P. A. Levene und H. Sobotka, J. Biol. Chem. 59, 465 (1924).
9) G. Wendt, Z. physiol. Chem. 272, 152 (1942).
12

CHO CHO CHO
—OH
SCH3
OH
CH2OH
-OH
-OH
-SCH,
CH2OH
-OH
-OH
-OH
CH^SCHg
h m
Von den Formeln I bis III ist I mit der Thiomethyl-Gruppe am
C3 wenig wahrscheinlich, da bei der Oxydation des Thiozuckers
mit Blei-tetra-acetat kein Formaldehyd nachgewiesen werden
konnte10).Zu Beginn meiner eigenen Arbeiten kam somit auf Grund der
vorstehend besprochenen analytischen Untersuchungen für den
Thiozucker C6H1204S mit grosser Wahrscheinlichkeit eine der acht
stereoisomeren Formen der Verbindungen II und III in Betracht.
Als Ergebnis synthetischer Arbeiten, welche nachfolgend bespro¬chen werden, reduzierte sich die Anzahl der für den Thiozucker
möglichen Konstitutionsformen auf vierzehn.
Seither sind neue Ergebnisse analytischer Untersuchungen be¬
kannt geworden. Um die Stellung der Thiomethyl-Gruppe abzu¬
klären, unterwarfen F. Weygand und Mitarbeiter11) das Thiozucker-
phenylosazon der Oxydation mit Perjodat. Mit einem Überschuss
an Perjodat wurden bis acht „Oxydationsäquivalente"12)13) ver¬
braucht. Bei Verwendung von zwei „Oxydationsäquivalenten"
Perjodat konnte aus den Oxydationsprodukten Mesoxal-dialdehyd-
1,2-bis-phenylhydrazon in über 50-prozentiger Ausbeute isoliert
werden. Dieses Ergebnis lässt sich zwangslos an Hand der Formel
III erklären. Nachdem was über die Perjodspaltung bekannt ist,
kann es aber nicht aus der Strukturformel II abgeleitet werden.
Es bleibt demnach als mögliche Konstitutionsformel des Thio¬
zuckers die Struktur III mit ribo-, arabo-, xylo- oder Iyxo-Kon-
figuration. Von diesen schieden, wie bereits erwähnt, die Verbin-
10) G. Wendt, Z. physiol. Chem. 272, 152 (1942).
") F. Weygand, 0. Trauth und R. Löwenfeld, B. 83, 563 (1950).
12) Gemeint ist vermutlich Mol Perjodat pro Mol Thiozucker.•
13) Vgl. auch: K. Satho und K. Makino, Nature 165, 769 (1950).
13

düngen mit xylo- und lyxo-Konfiguration auf Grund synthetischerArbeiten von A. L. Raymond1*) aus.
A. L. Raymond gelang es, ausgehend von 5-Tosyl-mono-aceton-
xylose IV15), durch Umsetzung mit Kalium-methyl-mercaptid und
anschliessende Hydrolyse des Acetonderivates die kristallisierte
5-Thiomethyl-D-xylose V herzustellen. Ihr Phenylosazon VI, wel¬
ches mit dem der 5-Thiomethyl-D-lyxose identisch ist, erwies sich
als verschieden vom Phenylosazon des natürlichen Thiozuckers.
Für den natürlichen Thiozucker blieb somit nur noch die Konsti¬
tution der 5-Thiomethyl-D-ribose oder der 5-Thiomethyl-D-arabi-nose übrig.
H—C—O CH3
\ /3
/C\-0 CH,
HO-
H-
H—C—O, CH,
KSCH,
-o
in Aceton HO-
H-
O CH,
-0
IV
CHaSCHg
CH=N—NHC6H5 HO—C—H
C=N—NHC6HSHO—
—OH
HO—
H
CH2SCH3
VI
-OH
O
CH2SCH3
V
A.L.Raymond1*) versuchte dann von 2,3-Isopropyliden-5-tosyl-methyl-D-ribofuranosid VII15) ausgehend, in der eben be¬
schriebenen Reaktionsfolge zu 5-Thiomethyl-D-ribose VIII zu ge¬
langen. Er isolierte dabei in schlechter Ausbeute ein kristallisiertes
p-Brom-phenylosazon von welchem er aussagte, es sei mit dem
entsprechenden Osazon des natürlichen Zuckers nicht identisch.
14) A. L. Raymond, J. Biol. Chem. 107, 85 (1934).
lä) P. A. Levene und E. T. Stiller, J. Biol. Chem. 106, 421 (1934).
14

CHO
—OH
—OH
—OH
Aceton+5%CH,OH»
CuSO.+H.SO,
.5)
H—C—OCH3
-Ov /CH3C
/ \—O CH,
H—C—OCH,
TsCl
Pyridin
15)
-o.
—o
CH3
CHS
CH2OH CH2OTs
krist. VII
KSCH,Aceton]
p-Brom-phenylosazonkrist.
nicht identisch mit
entsprechendem Osazon
des nat. Zuckers
CHO
OH
OH
OH
CH-2SCHa
VIII
Hydrolyse
H—C—OCH3
-O CH3\ /c
/ \-O CH,
OidlnSOxio
0
-O
Wir glaubten nicht an die chemischen Argumente, die gegen eine
ribo- bzw. arabo-Konfiguration der natürlichen Thiomethyl-pentose
sprachen. Es schien uns im Gegenteil die Formel III mit der will¬
kürlich angenommenen ribo-Konfiguration am wahrscheinlichsten,
da in allen bisher untersuchten Adenyl-glykosiden aus Nuclein-
säuren pflanzlicher oder tierischer Herkunft nur D-Ribose16) oder
2-Desoxy-D-ribose, nie aber Arabinose aufgefunden wurde.
Unsere Anschauungen wurden durch spätere Untersuchungenbestens bestätigt. F. Weygand, 0. Trauth und R. Löwenfeld1'') bau¬
ten 6-Thiomethyl-D-glucose IX in Form ihres Oxims durch Ein¬
wirkung von Fluordinitrobenzol zur nicht kristallisierenden 5-Thio-
methyl-D-arabinose X ab. Das daraus hergestellte, kristallisierte
Phenylosazon XI war nach Schmelzpunkt, Mischprobe und opti¬scher Drehung mit dem Phenylosazon des natürlichen Thiozuckers
identisch.
16) Vgl. z. B. Advances in carbohydrate-chemistry Vol. VI (1951).
") F. Weygand, 0. Trauth, R. Löwenfeld, B. 83, 563 (1950).
15
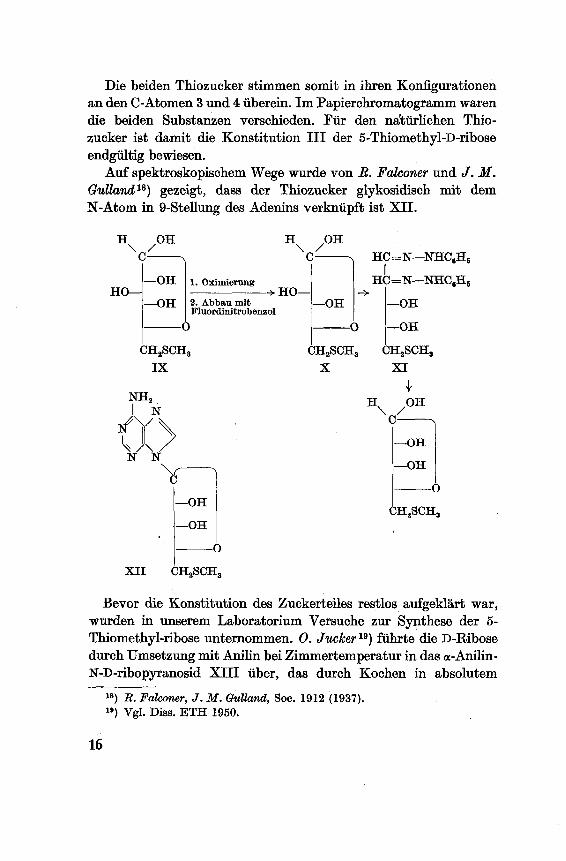
Die beiden Thiozucker stimmen somit in ihren Konfigurationenan den C-Atomen 3 und 4 überein. Im Papierchromatogramm waren
die beiden Substanzen verschieden. Für den natürlichen Thio¬
zucker ist damit die Konstitution III der 5-Thiomethyl-D-ribose
endgültig bewiesen.
Auf spektroskopischem Wege wurde von B. Falconer und J. M.
Ghdland18) gezeigt, dass der Thiozucker glykosidisch mit dem
N-Atom in 9-Stellung des Adenins verknüpft ist XII.
H ÖH
C —
HO—
—OH
—OH
H OH
C
1. Oximiermig
2. Abbau mit
Fluordinltrobenzol
-»HO—
O
CH2SCH3
IX
NH2I N
\ /\N N
c
—OH
—OH
-O
XII CH-SCH
—OH
-O
CH.2SCH3
X
HC=N—NHCeH6
HC=N—NHC6H5
—OH
—OH
CH2SCH3
XI
Hx OH
C
-OH
-OH
O
CHaSCHa
Bevor die Konstitution des Zuckerteiles restlos aufgeklärt war,
wurden in unserem Laboratorium Versuche zur Synthese der 5-
Thiomethyl-ribose unternommen. 0. Jucker19) führte die D-Ribose
durch Umsetzung mit Anilin bei Zimmertemperatur in das a-Anilin-
N-D-ribopyranosid XIII über, das durch Kochen in absolutem
M) R. Falconer, J. M. GuUand, Soc. 1912 (1937).
") Vgl. Diss. ETH 1950.
16

Alkohol in das a-Anilin-N-D-ribofuranosid XIV übergeht20). Durch
Tosylieren und nachfolgendes Acetylieren sollte dieses in das 2,3-
Diacetat-N-5-ditosylat XV bzw. in das 2,3-Diacetat-5-tosylat XVI
umgewandelt werden. Die Umsetzung mit Natriumjodid-Acetanhy-drid bzw. Natrium-methyl-mercaptid und anschliessende Hydrolysesollte dann zur 5-Thiomethyl-D-ribose XVII führen. Es gelang aber
nicht, aus den bei der Tosylierung des N-Furanosids entstandenen
uneinheitlichen Ölen reine Verbindungen abzutrennen. Auch die
Umsetzung der Rohprodukte verlief in letzter Stufe ergebnislos.
HC—NHC6H5
—OH
-OH
-OH
CH,—
HC—NHC6H,
—OH
—OH
O
O
XIII
CH2OH
XIV
HC—NTC6H5
CH2OTs
XVI
oder
S02C7H7
—OAc
—OAc
-0
CHO
—OH
—OH
-OH
C-H2oC-EI3
XVII
XV
Eine weitere Bearbeitung des von 0. Jucker eingeschlagenenWeges zur Synthese der 5-Thiomethyl-ribose schien wegen des
unübersichtlichen Verlaufs der Reaktionen wenig aussichtsreich.
Ich versuchte darnach von Ribose-mercaptalen aus zur 5-Thio¬
methyl-ribose zu gelangen. Es war beabsichtigt die Mercaptale in
Analogie zur partiellen Tosyherung der Glucose in 5-Stellung zu
tosylieren. Der Tosylrest hätte dann in einer oder mehreren Stufen
nach bekannten Vorlagen durch den Thiomethylrest ersetzt werden
sollen. Die Versuche, wie auch analoge Umsetzungen von D-Ribon-
20) Vgl. L. Berger und J. Lee, J. org. ehem. 11, 75 (1946).
17

säure-y-lacton, führten zwar zu einer Anzahl neuer Derivate, die
im folgenden Kapitel beschrieben werden, jedoch nicht zur Syntheseder 5-Thiomethyl-D-ribose.
Die Synthese der 5-Thiomethyl-D-ribose gelang fast gleichzeitigJ. Baddiley, 0. Trauth und F. Weygand sowie W. G. Overend und
L. F. J. Parker21). Den letzteren Autoren gelang es im Gegensatzzu A. L. Raymond22) das 2,3-Isopropyliden-5-tosyl-methyl-D-ribo-furanosid XVIII durch Erhitzen mit Natrium-methylmercaptid in
Acetonlösung in das 5-Methyl-thio-Derivat XIX umzuwandeln und
daraus durch Hydrolyse mit 1-n. Schwefelsäure bei 80° die 5-Thio¬
methyl-D-ribose XX in kristallisierter Form zu gewinnen. Die aus
dem synthetischen Thiozucker hergestellten Derivate erwiesen sich
mit jenen des natürlichen Thiozuckers in allen Eigenschaften als
identisch.
H—C—OCH,
-O CH3
-O CH,
O
CH2OTs
XVIII
H—C—OCH,
-O CH,
x /3
C/
-O CH,
O
CH2SCH3
XIX
H—C—OH I
-OH
—OH
XX
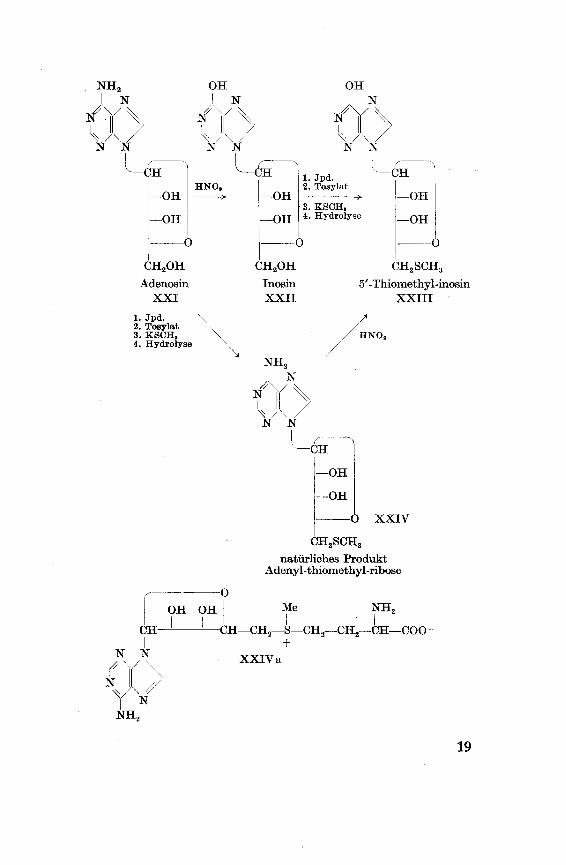
Die Synthese von J. Baddiley, 0. Trauth und F. Weygand23)
geht von Adenosin XXI aus, welches zunächst durch Behandeln
mit salpetriger Säure in Inosin XXII übergeführt wurde. Durch
Einführung des Isopropyhdenrestes, anschliessende Tosyherung in
5-Stellung des Zuckerrestes und Behandlung mit Kaliummethyl-
mercaptid entstand 5'-Thiomethyl-2', 3'-isopropyliden-inosin. Das
nach Abspaltung des Isopropylidenrestes durch Hydrolyse mit
Schwefelsäure in Eisessig hergestellte 5'-Thiomethyl-inosin XXIII
war identisch mit dem Naturprodukt, das auch aus der natürlichen
Adenyl-thiomethyl-pentose XXIV durch Behandlung mit salpetri-
21) Nature, 167, 526 (1951).
**) A. L. Raymond, J. Biol. Chem. 107, 85 (1934).
23) Nature, 167, 359 (1951).
18
NH, OH
N
N
k A /N N
//N
N
CH
-OH
-OH
O
CH2OH
Adenosin
XXI
1. Jpd.2. Tosylat3. KSCH,4. Hydrolyse
N N
HNO,
OH
N/\/N
^ /\ /N N
f
-CH
-OH
-OH
1. Jpd.2. Tosylat
3. KSCH,4. Hydrolyse
CH
'—OH
—OH
-0
CH2OH
Inosin
XXII
NH,
/\/N
N
!..
N N
-0
CH2SCH3
5'-Thiomethyl-inosinXXIII
HNO,
CH
—OH
—OH
O XXIV
0
OH OH
N
N
CH-
N
N
natürliches Produkt
Adenyl-thiomethyl-ribose
CH—CH,
Me
+
NH,
-CH,—CH—COO-
XXIVa
NH,
19

ger Säure gewonnen werden konnte. Die durch Hydrolyse freigesetz¬ten Zucker beider Substanzen waren, wie auch die Nucleoside selbst
und ihre Purinkomponenten, durch Papierchromatographie nicht
voneinander zu unterscheiden. Durch Reduktion des synthetischenThiozuckers mit Natriumamalgam entstand ein 5-Thiomethyl-adonit, der sich in der Mischprobe mit dem Thiomethyl-pentit aus
dem natürlichen Thiozucker als identisch erwies.
Eine Variante der Synthese des 5'-Thiomethyl-inosins XXIII
führt unter Anwendung analoger Zwischenprodukte vom Adenosin
XXI über die Adenyl-thiomethyl-ribose XXIV. Beide Synthesenerlauben der natürlich vorkommenden Adenyl-thiomethyl-ribosebzw. dem 5-Thiomethyl-inosin am glykosidischen C-Atom die
^-Konfiguration zuzuschreiben.
Obwohl eine Diskussion der biologischen Bedeutung der Adenyl-
thiomethyl-ribose den Rahmen meiner Arbeit überschreitet, sei
kurz auf eine soeben erschienene Arbeit von J. Baddiley und G. A.
Jamieson2i) über die Synthese eines „aktiven Methionins" verwie¬
sen. Dieses wichtige Zwischenprodukt verschiedener biologischer
Methylierungsprozesse besitzt die Konstitution XXIV a. Die Syn¬these erfolgte aus Adenyl-thiomethyl-ribose-a-amino-y-brombutter-säure-hydrobromid.
Eigene Versuche zur Synthese von 5-Thiomethyl-D-ribose
a) Aus D-Bibose-mercaptalen
Zu Beginn der eigenen Arbeiten waren zwei missglückte Versuche
zur Synthese der natürlichen 5-Thiomethyl-D-ribose bekannt25)26).In beiden Versuchen — sie sind im vorhergehenden Kapitel kurz
skizziert — wurden Derivate der D-Ribofuranose als Ausgangs¬materialien verwendet. Bei der Durchführung selektiver Umsetzun¬
gen am C-Atom 5 dieser D-Ribofuranose-Derivate traten, ebenfalls
24) Chem. and Ind. No. 13, 375 (1954).
25) A. L. Raymond, J. Biol. Chem. 107, 85 (1934).
2») Vgl. 0. Jucker, Diss. ETH 1950.
20

in beiden Versuchen, unerwartete und scheinbar unüberwindliche
Schwierigkeiten auf27).Es schien deshalb ratsam für die eigenen Untersuchungen zur
- Synthese der 5-Thiomethyl-ribose nicht von D-Ribofuranosiden, son¬
dern von andern Derivaten der D-Ribose auszugehen, welche in
5-Stellung eine freie Oxy-Gruppe aufweisen. Die kurz zuvor in
unserem Laboratorium hergestellten D-Ribose-mercaptale, denen
wie allen übrigen Aldose-mercaptalen eine acyclische Struktur zuzu¬
schreiben ist, schienen zu diesem Zweck besonders geeignet.Es war beabsichtigt die gut kristallisierenden D-Ribose-dibenzyl-
und äthylen-mercaptale XXVI XXVII durch partielle Tosylierungin die 5-Tosyl-mercaptale XXVIII XXVIII a überzuführen und
diese durch Umsetzung mit Methylmercaptiden in die Mercaptaleder natürlichen Thiomethylribose XXIX XXIXa umzuwandeln.
Anschliessend sollte die Zerlegung der Mercaptale nach analogenin der Literatur beschriebenen Beispielen mit Quecksilberchloridund Cadmiumcarbonat vorgenommen werden28). Beim Auftreten
von Schwierigkeiten waren als Varianten vorgesehen:
a) die Isolierung der Mercaptal-5-tosylate als 2,3,4-triacetyl-Deri-vate;
b) die in bekannter Weise vorzunehmende indirekte Umwandlungder Tosylate über die 5-Desoxy-5-jod-mercaptale XXX XXXa
in die Thiomethyl-Derivate, wobei es freistand die Umsetzungenmit den acetylierten, bzw. nicht acetylierten Produkten durch¬
zuführen.
Die Herstellung des Ribose-dibenzyl-mercaptals gelang nach der
Vorschrift von E. Hardegger, E. Schreier und Z. El Heweihi29) mit
überschüssigem Mercaptal in einer Lösung von Chlorwasserstoff in
Dioxan, in ca. 75-prozentiger Ausbeute. Das Dibenzyl-mercaptal
27) Vgl. dazu die Synthese der Thiomethyl-D-ribose nach W. 6. Overend,
L. F. J. Parker, Nature 167, 526 (1951).
28) M.L. Wolfrom, N.R.Newlin, Am. Soc. 52, 3619 (1930). Vgl. dazu:
H. Zinner, B. 83, 418 (1950).
29) E. Hardegger, E. Schreier, Z. El Heweihi, Helv. 33, 1159 (1950). Eine
ähnliche Methode zur Herstellung von Mercaptalen mit Borfluorid-Äther
beschreibt Fieser, Am. Soc. 76, 1945 (1954). Vgl. dazu auch H. Zinner,
B. 83, 275 (1950).
21
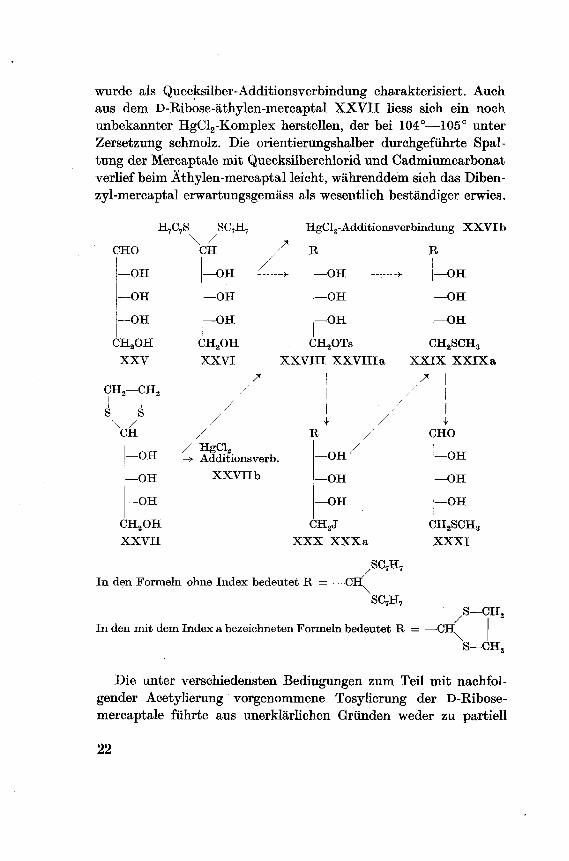
wurde als Quecksilber-Additionsverbindung charakterisiert. Auch
aus dem D-Bibose-äthylen-mercaptal XXVII liess sich ein noch
unbekannter HgCl2-Komplex herstellen, der bei 104°—105° unter
Zersetzung schmolz. Die orientierungshalber durchgeführte Spal¬
tung der Mercaptale mit Quecksilberchlorid und Cadmiumcarbonat
verlief beim Äthylen-mercaptal leicht, währenddem sich das Diben-
zyl-mercaptal erwartungsgemäss als wesentlich beständiger erwies.
H7C7S SC7H7 HgCl2-Additionsverbindung XXVIb
(ÏEÏO (3H -' R R
—OH —OH /__-> —OH y —OH
—OH —OH —OH —OH
—OH —OH —OH —OH
CH2OH
XXV
CH2OH
XXVI
CH2OTs
XXVIII XXVIII a
CH2SCH3
XXIX XXIXa
CHS1S
\
(
—CH21S
/
)H
—OH
/
/
/
/ HgCl2-> Additionsverb
]
1
1
—on
/
/
/
(.
11
1
mo
—OH
—OH XXVIIb —OH —OH
—OH —OH —OH
(M2OH
ÏXVII
(
XXX XXXa
(3H2SCH3
XXXI
SC7H7
In den Formeln ohne Index bedeutet R = —CH\
SC7H7.S—CH»/
In den mit dem Index a bezeichneten Formeln bedeutet R = —CH
0—^Ha
Die unter verschiedensten Bedingungen zum Teil mit nachfol¬
gender Acetylierung vorgenommene Tosylierung der D-Bibose-
mercaptale führte aus unerklärlichen Gründen weder zu partiell
22

noch zu vollständig tosylierten Präparaten. Es wurden zwar aus
den verschiedenen Ansätzen in wechselnden Mengen mehrere scharf
schmelzende prachtvoll kristallisierende Substanzen isoliert, an
deren Einheitlichkeit nicht zu zweifeln war, deren Analysenresultateaber jeder Interpretation widerstanden. Die Versuche zur Um¬
setzung der in ihrer Konstitution nicht erkannten Verbindungenmit Natriumjodid bzw. Natrium-methyl-mercaptiden verliefen
ergebnislos und sind im experimentellen Teil nicht erwähnt. Die
Konstitution der lediglich auf Grund einer Schwefelbestimmung im
experimentellen Teil als 5, X-Ditosyl-D-ribose-dibenzylmercaptalbeschriebenen Verbindung erscheint unsicher. Das nicht weiter
untersuchte Präparat erschien zur Synthese der 5-ThiomethyI-ribose ohnehin wenig geeignet.
b) Aus T)-Riboiisäure-y-lacton
Für die weiteren Versuche zur Synthese der 5-Thiomethyl-D-ribose benützte ich in der Folge das im Handel erhältliche D-Ribon-
säure-y-lacton XXXII als Ausgangsmaterial. Ich beabsichtigtedamit dieselben Umsetzungen durchzuführen, wie mit denD-Ribose-
mercaptalen. Aus D-Ribose-y-lacton sollte demnach das 5-Thio-
methyl-D-ribonsäure-y-lacton XXXVII als Vorstufe zur 5-Thio-
methyl-D-ribose XXXVIII zugänglich sein; die ZwischenprodukteXXXIII bis XXXVI der Synthese sind aus dem Formelschema
ersichtlich.
Die Versuche zur Tosylierung des Ribonsäure-y-lactons lieferten
unter verschiedenen Bedingungen nur ein kristallisiertes Präparatvom Schmelzpunkt 124° und der Zusammensetzung C19H20O9S2eines Ditosylats. Diesem Ditosylat wurde auf Grund der Perjodat-
Spaltung die Konstitution XXXIX eines 3,5-Ditosylats zugewie¬
sen30), trotzdem die erhöhte Reaktionsfähigkeit der in a-Stellungzum Lacton-Carbonyl sich befindlichen Oxy-Gruppe eher ein 2,5-
Ditosylat erwarten liesse.
Ein zweiter Weg um in die vorgesehene Reaktionsfolge zu gelan¬
gen führte zur Umsetzung des Ribonsäure-y-lactons mit Aceton.
30) Vgl. nächstes Kapitel: Oxydation von Derivaten der Ribonsäure
mitHJ04.
23

C=0
—OH
-OH
-O
CHaOH
XXXII
rC=0
—OH
—OH
-O
CH2OTs
XXXIII
CHO
OH
-OH
-OH
XXXVIII
6=0 ^
—OH
—OTs
n
CH20Ts
XXXIX
rc=o
c=o
—OH
—OH
O
CH2J
XXXIVI
I+
>
r ~\C=0 I
-OH
-OH
O
CH2SCIÏ3
XXXVII
(
c=o
-O CH.
\ /s
-O CH3
O
CH2OTs
XLI
X—O CH,
[ | COOK
-O
CH2OH
XL
c7/ \
—O CH3
—OH
CH2OH
XLII
c=o
c/
—0
/CH3\
CH3
CH2J
XXXV
11
o
rc=o
-O /CH3/C\O CH.
-O
CH2SCH3
XXXVI
1
6I0 A
—OH
—0.
YCH20—/ X,
XLa
°,CHa
CH,
24

Diese Umsetzung könnte zum 2,3-Isopropyliden-D-ribonsäure-y-lacton XL führen in dem die cis-Verknüpfung von zwei 5-Ringeneine stabile Anordnung darstellt. Die zweite Möglichkeit der Um¬
setzung des Ribonsäure-y-lactons mit Aceton müsste zum 3,5-Iso-
propyliden-ribonsäure-y-lacton XLa führen. Im 3,5-Isopropyliden-
ribonsäure-y-laeton XLa stehen ein 5- und ein 6-gliedriger Ring in
der etwas gespannten trans-Stellung zueinander. Von den beiden
Isopropylidenverbindungen XL und XLa ist daher wohl eher das
2,3-Isopropyliden-D-ribonsäure-y-lacton XL zu erwarten. Die prak¬tische Durchführung der Umsetzung des Ribonsäure-y-lactons mit
Aceton führte in guter Ausbeute zu einem Präparat vom Schmelz¬
punkt 138° und [a]D = —72°, dessen Konstitution XL durch Abbau
mit Perjodsäure bestätigt wurde30).Beim Versuch zur Oxydation des 2,3-Isopropyliden-ribonsäure-
y-lactons mit Kaliumpermanganat in neutraler, heisser, wässeriger
Lösung, unter Durchleiten von C02, wurde in ca. 35-prozentigerAusbeute nur das Kaliumsalz der 2,3-Isopropyliden-ribonsäureXLII isoliert. Die restlichen 65% Lacton sind wahrscheinlich zu
C02 und H20 oxydiert worden.
Das Isopropyliden-ribonsäure-lacton XXXII wurde bei länge¬rem Kochen mit Thionylchlorid nicht verändert. Auch die Versuche
zur Umsetzung des 2,3-Isopropyliden-ribonsäure-y-lactons mit
Ammoniak, Anilin, Hydrazin und Phenylhydrazin ergaben stets
nur Ausgangsmaterial, obwohl unter denselben Bedingungen das
D-Ribonsäure-y-lacton in das entsprechende Amid vom Smp. 138°,
in das kristallisierte, bei 150° schmelzende Anilid, in das Hydrazidvom Smp. 147° und in das Phenylhydrazid vom Smp. 158° über¬
geführt werden konnte31). An diesen Verbindungen, welche alle in
guter Ausbeute zugänglich waren, wurden die im nächsten Kapitel
besprochenen Versuche zur Oxydation mit Perjodsäure unter¬
nommen.
Die Tosylierung des 2,3-Isopropyliden-ribonsäure-y-lactonsführte nur zu Ölen, deren Zusammensetzung nicht dem 5-Tosyl-
2,3-isopropyliden-ribonsäure-y-lacton C15H1807S XLI entsprach.Auch die Umsetzung der Öle mit Natriumjodid lieferte keine
81) Beilstein, Bd. 3 und Bd. 15.
25

brauchbaren Präparate. Diese Umsetzungen sind im experimen¬tellen Teil nicht beschrieben.
Von D-Ribonsäure-y-lacton aus bestand schliesslich noch eine
dritte Möglichkeit, um in die vorgesehene Reaktionsreihe32) zu ge¬
langen. Die TrityHerung des Lactons XXXII sollte, wie an mehre¬
ren Beispielen bekannt ist33), nur an der primären Oxy-Gruppe
c=o
-OH
-OH
O
CH2OH
XXXII
c=o
—OTr
—OH
O
CH2OTr
XLV
COOTr
-OH
-OH
-OH
CHaOTr
XLIV
r
c=o
—OH
—OH
-O
CH2OTr
XLIII
NHNH,
c=o
—OTr
—OH
—OH
CH2OTr
XLVII
6=0
—0 CH3
\ /3
/\—0 CH3
CH2OTr
XLVI
XL
O
32) Vgl. dazu die Formeln Seite 24.
33) Vgl. z. B. W. W. Pigman und B. M. Goepp jun., Chemistry of the
Carbohydrates (1948).
26

erfolgen und zum 5-Trityl-äther-lacton XLIII führen. Durch die
Umwandlung von XLIII in die Isopropyliden-Verbindung XLVI
und anschliessende Abspaltung der Trityl-Gruppe durch katalytische
Hydrierung sollte das inzwischen auf einfacherem Wege herge¬stellte 2,3-Isopropyliden-D-ribonsäure-y-lacton XL zugänglich sein.
Die praktischen Ergebnisse der Umsetzung des D-Ribonsäure-
y-lactons XXXII mit Tritylchlorid in Pyridin waren aber anders
als erwartet. An Stelle des erwarteten 5-Trityläther-lactons XLIII
waren noch zwei weitere Reaktionsprodukte entstanden. Alle drei
Verbindungen konnten auf Grund verschiedener Löslichkeit von¬
einander getrennt und in reiner Form isoliert werden. Als erstes,
d. h. am schwerstenlösliches Produkt, kristallisierte der 5-Trityl-ätherXLIII vom Smp. 172° ([a]D = +44,8°). Bei weiterem Einengender Chloroformlösung kristallisierte als Hauptprodukt das Mono-
hydrat des D-Ribonsäure-di-trityläther-y-lactons XLV vom Smp.271° ([<x]D = +113,2°), von dem zur weiteren Charakterisierung das
bei 114° schmelzende Hydrazid XLVII hergestellt wurde. Aus der
Mutterlauge des Di-trityläther-lactons XLV wurde noch ein gerin¬
ger Anteil an D-Ribonsäure-tritylester-5-trityläther XLIV (Smp.
180°, [<x]D = +34,6°) isoliert. Die Konstitution der VerbindungenXLIII, XLIV, XLV und XLVII ist .nicht streng bewiesen.
Oxydation von Derivaten der Bibonsäure mit Perjodsäure3i)
Es ist allgemein bekannt, dass freie Zucker sich gegen Perjod-säure wie aüphatische Oxy-aldehyde verhalten, d. h. es tritt Spal¬
tung zwischen allen C-Atomen ein. Mittelständige OH-Gruppenwerden von Perjodsäure zu Ameisensäure oxydiert, während bei
Einwirkung von Bleitetraacetat34)35) C02 entsteht. Bleitetraacetat
greift a-Oxy-säuren zwischen Carboxyl- und a-ständiger Oxy-
Gruppe an, während Perjodsäure im Gegensatz dazu nicht reagiert.In gleicher Weise verhalten sich a-Ketosäuren. Aus Aldonsäuren
entsteht daher mit Perjodsäure die Glyoxylsäure als Spaltstück,wie auch die im experimentellen Teil beschriebenen Versuche an
verschiedenen Derivaten der D-Ribonsäure zeigen. Von den Ver-
M) Vgl. dazu Neuere Methoden der org. Chemie.
36) Organic Reactions, Vol. II.
27

bindungen, die der Oxydation mit Perjodsäure unterworfen wur¬
den, sind das D-Ribonsäure-amid XLVIII und das D-Ribonsäure-
anilid XLIX zu erwähnen. Beide Verbindungen verbrauchten je3 Millimol HJ04 pro Millimol Substanz. Als Spaltstücke wären
demnach 2 Mol Ameisensäure, 1 Mol Formaldehyd und 1 Mol
Glyoxylsäure-amid L bzw. 1 Mol Glyoxylsäure-anilid LI zu erwar¬
ten. Es ist leider nicht gelungen die Glyoxylsäure-Derivate L und
LI in Form ihrer Phenylhydrazone zu isoHeren.
NHR NHR
c=o c=o
L R = H-
-OH HC=03 HJO,
+ LI R = C6H5-OH
"*2 HCOOH
+
-OH CH20
XLVIII R = H—
XLIX R = C6H6
LH R = NH„-
CHOLUI R = C6H5—NH— LIV
Im Gegensatz zum D-Ribonsäure-amid XLVIII bzw. -anilid
XLIX verbrauchten das D-Ribonsäure-hydrazid LH bzw. das
-phenylhydrazid LUI mehr als 3 Mol Perjodsäure. Währenddem
die Oxydationsversuche am Hydrazid LH einen unübersichtlichen
Verlauf nahmen, blieb beim Phenylhydrazid LUI der Verbrauch
bei 4 Mol Perjodsäure stehen. Wahrscheinlich ist die VerbindungLIV unter den Spaltstücken zu finden.
Um das Verhalten von Hydrazinderivaten gegenüber Perjod¬säure genauer zu untersuchen, wurde das Benzoyl-phenylhydrazin
hergestellt LV36) und der Perjodsäure-Oxydation unterworfen. Die
Versuche an diesem Präparat zeigten, dass die Oxydation anfäng¬lich rasch verläuft und dann nach Verbrauch von 1 Mol HJ04stehen bleibt. Das als Oxydationsprodukt erwartete Benzoyl-azo-
36) Vgl. experimenteller Teil.
28
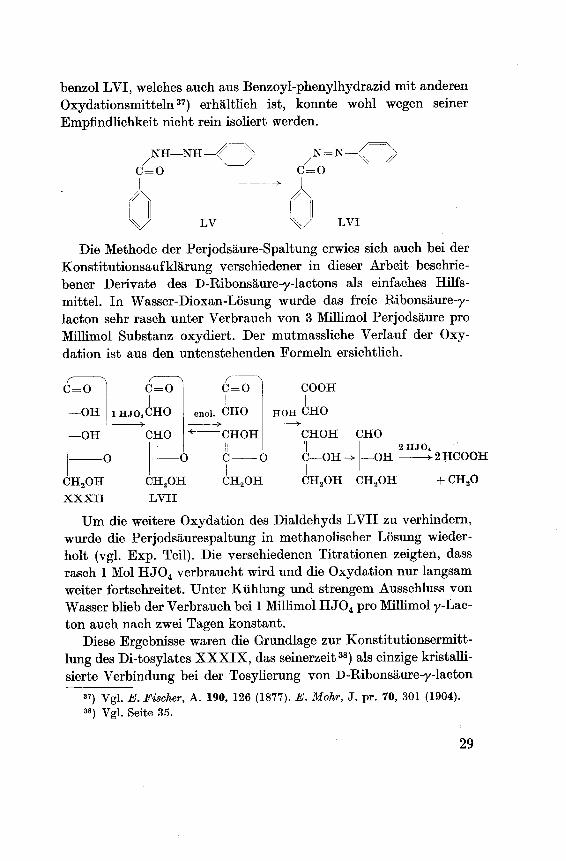
benzol LVI, welches auch aus Benzoyl-phenylhydrazid mit anderen
Oxydationsmitteln37) erhältlich ist, konnte wohl wegen
Empfindlichkeit nicht rein isoliert werden.
seiner
NH—NH-
C=0
LV
Die Methode der Perjodsäure-Spaltung erwies sich auch bei der
Konstitutionsaufklärung verschiedener in dieser Arbeit beschrie¬
bener Derivate des D-Ribonsäure-y-lactons als einfaches Hilfs¬
mittel. In Wasser-Dioxan-Lösung wurde das freie Ribonsäure-y-
lacton sehr rasch unter Verbrauch von 3 Millimol Perjodsäure pro
Millimol Substanz oxydiert. Der mutmassliche Verlauf der Oxy¬
dation ist aus den untenstehenden Formeln ersichtlich.
r
c=o
OH
—OH
fc=o
ihjo.CHO»-
CHO
O
CH2OH
XXXII
c=o
!enol. CHO
*" CHOH
-O c- o
COOH
IHÖH CHO
>
CHOH CHO
2HJ0.
OH S-2HCOOH
+ CH20
C—OH-
LVII
Um die weitere Oxydation des Dialdehyds LVII zu verhindern,
wurde die Perjodsäurespaltung in methanolischer Lösung wieder¬
holt (vgl. Exp. Teil). Die verschiedenen Titrationen zeigten, dass
rasch 1 Mol HJ04 verbraucht wird und die Oxydation nur langsam
weiter fortschreitet. Unter Kühlung und strengem Ausschluss von
Wasser blieb der Verbrauch bei 1 Millimol HJ04 pro Millimol y-Lac-
ton auch nach zwei Tagen konstant.
Diese Ergebnisse waren die Grundlage zur Konstitutionsermitt¬
lung des Di-tosylates XXXIX, das seinerzeit38) als einzige kristalli¬
sierte Verbindung bei der Tosylierung von D-Ribonsäure-y-lacton
37) Vgl. E. Fischer, A. 190, 126 (1877). E. Mohr, J. pr. 70, 301 (1904).
38) Vgl. Seite 35.
29

isoliert worden war. Von den drei für das Di-tosylat möglichenKonstitutionsformeln XXXIX, LVIII und LIX konnten durch die
Perjodsäure-Oxydation39) LVIII und LIX ausgeschaltet werden,
da weder in wasserfreier methanolischer Lösung, noch in wässeriger
Lösung eine Oxydation eintrat. Selbst nach dreiwöchigem Stehen
der Oxydationsmischung bei 0° konnte das unveränderte Aus¬
gangsmaterial in kristalhsierter Form zurückgewonnen werden. Das
2,5-Di-tosylat LVIII, wie auch das 2,3-Di-tosylat LIX hätten in
wässeriger Lösung je 1 HJ04 verbrauchen müssen.
r \
c=o (}=o | c=o
—OH —OTs —OTs
—OTs —OH —OTs
(
—>
1 0
->
}H2OTs (
(
CH2OTs ( ÜHaOH
XXXIX LVIII LIX
In eindeutiger Weise konnten auch auf Grund der Ergebnisse der
Perjodat-Spaltung von den drei möglichen Formeln LX, LXI und
LXII des Isopropyliden-ribonsäure-y-lactons40) zwei ausgeschaltetwerden. Um die hydrolytische Abspaltung des IsopropyHden-Restesdurch die Perjodsäure zu verhindern, war es notwendig die Oxy¬dation erst nach Aufspaltung des Lacton-Ringes in alkalischer
Lösung vorzunehmen.
COONa COONa COONa
—O. CH,
C/
—O
—OH
CH2OH
LX
1HJ04+
CH,0
CH3
—O
—OH
—OH
CH,
CH,
CH20
-OH
°\ /CH*^OH/C
/
CH20CHa
LXI
lHJO„
LXII
kein HJO,
39) Vgl. experimenteller Teil Seite 44.
40) Vgl. Seite 44.
30

Die titrimetrische Verfolgung des Oxydationsverlaufs ergab, dass
insgesamt nur 1 Millimol HJ04 pro Millimol Substanz verbraucht
wurde. 1 Millimol Formaldehyd konnte in quantitativer Ausbeute
als Dimedon-Derivat vom Smp. 187° aus der Oxydationslösungisoliert werden. Das andere Spaltstück, das Natriumsalz der 2,3-
Isopropyliden-L-erithruronsäure, konnte nicht in kristallisierter
Form gefasst werden. Die Interpretation des Oxydationsversuchsbeweist eindeutig, dass die Isopropyliden-Verbindung als 2, 3-Iso-
propyliden-D-ribonsäure-y-lacton XL zu bezeichnen ist.
31

Experimenteller Teil1)
Herstellung von D-Ribose-dibenzyl-mercaptal (XXVI)2)
1,5 g (= 10 Milhmol) D-Ribose wurden in einer Lösung von 1 g
HCl in 10 ccm absolutem Dioxan suspendiert. Die farblose Mischungwurde nach Zugabe von 3,1g Benzylmercaptan im geschlossenenGefäss bei 20° 20—24 Stunden geschüttelt. Nach Abdampfen der
flüchtigen Anteile im Vakuum blieb ein gelblicher, öliger Rückstand,welcher aus Äther-Petroläther kristallisierte. Das aus Methanol-
Petroläther umkristallisierte Präparat wog 2,8 g (75% d. Th.) und
schmolz bei 80°. Das bei 18° 48 Stunden im Hochvakuum getrock¬nete Analysenpräparat enthielt x/2 Mol Kristall-Methanol.
4,675 mg Substanz verbrauchten 2,358 ccm 0,02-n. KJ03
C19H2404S2, i CH3OH Ber. S 16,06%Gef. S 16,17%
Additionsverbindung des D-Ribose-dibenzyl-mercaptals mit
Quecksilber (H)-chlorid (XXVIb)
Eine Lösung von 0,38 g ( = 1,0 Millimol) D-Ribose-dibenzyl-
mercaptal in 15 ccm heissem Alkohol wurde zu einer heissen Lösungvon 0,3g (= ca. 1,1 Millimol) HgCl2 in 20 ccm Alkohol gegeben.NachdemErkalten derMischung wurde die in quantitativerAusbeute
auskristalhsierte Additionsverbindung abfiltriert. Das 48 Stunden
bei 50° im Hochvakuum getrocknete Analysenpräparat vom
Schmelzpunkt 160° enthielt 1 Mol Kristall-Alkohol.
*) Alle Schmelzpunkte sind korrigiert.2) E.Hardegger, E.Schreier, Z. El Heweihi, Helv. 33, 1159 (1950).
H. Zinner, B. 83, 418 (1950).
32

4,776 mg Substanz verbrauchten 1,350 com 0,02-n. KJ03
C19H2404S2,HgCl2,C2H5OH Ber. S 9,2 %Gef. S 9,06%
Additionsverbindung von D-Ribose-äthylen-mercaptal mit
Quecksilber (Il)-chlorid (XXVII b)
Das Präparat wurde in derselben Weise wie die Additionsver¬
bindung von HgCl2 und D-Ribose-dibenzyl-mercaptal (vgl. oben)
hergestellt. Die Additionsverbindung enthielt nach 24 stündigemTrocknen im Hochvakuum bei 18° noch 1/2 Mol Kristall-Alkohol;
sie schmolz unter Zersetzung bei 104°—105°.
4,661 mg Substanz verbrauchten 1,769 ccm 0,02-n. KJ03
C7H1404S2,HgCl2,£C2H5OH Ber. S 12,3 %Gef. S 12,17%
Versuch zur Herstellung von 5-Tosyl-D-ribose-äthylen-mercaptal(XXVIII a) aus D-Ribose-äthylen-mercaptal (XXVII)
2,26 g ( = 10 Millimol) D-Ribose-äthylen-mercaptal vom Schmelz¬
punkt 108°3) und 5g reines Tosylchlorid wurden in 20 ccm abso¬
lutem Pyridin und 5 ccm Chloroform aufgelöst und 120 Stunden
bei Zimmertemperatur stehen gelassen. Die Mischung wurde im
Vakuum auf ca. 15 ccm eingeengt, unter Kühlung mit 1—2 ccm
Wasser versetzt, unter zeitweisem Umschütteln 3 Stunden bei 20°
stehen gelassen und dann in Chloroform aufgenommen. Nach dem
Ausschütteln mit verdünnter Salzsäure, verdünnter Natronlaugeund Wasser, unter jeweiliger Zugabe von Eis, anschliessendem
Trocknen mit Natriumsulfat und Abdampfen des Chloroforms blie¬
ben 3,7 g eines gelblichen Öls zurück, welches beim Stehen teilweise
kristallisierte. Das Präparat wurde zweimal mit je 25 ccm heissem
Methanol extrahiert. Der in Methanol unlösliche Rückstand konnte
3) E. Hardegger, E. Schreier, Z. El Heweihi, Helv. 33, 1163 (1950).
33

aus Chloroform (2 ccm)-Methanol (20 ccm) umkristallisiert werden.
Die mehrmals umkristallisierte, in prachtvollen, glänzenden Nadeln
erhaltene Verbindung schmolz bei 123°—124°. Das Präparat zeigtein der Mischprobe mit D-Ribose-äthylen-mercaptal (Smp. 108°)XXVII eine starke Schmelzpunktserniedrigung. Das Analysenprä¬parat wurde 20 Stunden im Hochvakuum getrocknet.
3,630 mg Substanz gaben 6,442 mg C02 und 1,667 mg H20
3,270 mg Substanz verbrauchten 2,563 ccm 0,02-n. KJ03
C14H20O6S3 Ber. C 44,19 H 5,30 S 25,28%
(5-Tosyl-D-ribose-äthylen-mercaptal)
C21H2608S4 Ber. C 47,2 H 4,87 S 23,95%
(2,5-Ditosyl-D-ribose-äthylen-mercaptal)
Gef. C 48,43 H 5,14 S 25,13%
[a]D = -51° (c = 1,1 in Chloroform)
S, X-Ditosyl-D-ribose-dibenzyl-mercaptal
1,9g (=5 Millimol) D-Ribose-dibenzyl-mercaptal und 2g
( = ca. 10 Millimol) Tosylchlorid wurden in 10 ccm Pyridin und
5 ccm Chloroform aufgelöst und 24 Stunden bei Zimmertemperaturstehen gelassen. Nach Abdampfen des Chloroforms wurde die
Mischung mit 10 ccm Wasser versetzt und eine halbe Stunde bei
20° gehalten. Das Reaktionsprodukt wurde in Chloroform aufge¬nommen und mit 1-n. Salzsäure, 1-n. Natronlauge und Wasser aus¬
geschüttelt. Als Rückstand blieb nach dem Abdampfen des Chloro¬
forms ein gelbliches Öl, das nach langem Stehen und wiederholtem
Anspritzen mit Methanol teilweise kristallisierte. Die Kristalle wur¬
den auf einer Tonplatte von anhaftender Mutterlauge befreit und
mehrmals aus Methanol umkristallisiert. Das 5, X-Ditosylat schmolz
bei 115°; es wurde zur Analyse 20 Stunden bei 60° im Hochvakuum
getrocknet.
4,032 mg Substanz verbrauchten 2,359 ccm 0,02-n. KJ03
C33H3608S4 Ber. S 18,63%Gef. S 18,76%
[a]D = -79° (c = 0,7 in Chloroform)
34

Versuch zur Herstellung von 5-Tosyl-2, 3,4-triacetyl-D-ribose-dibenzyl-mercaptal4)
5,7 g (= 15 Millimol) D-Ribose-dibenzyl-mercaptal und 5,7 g
(= 30 Millimol) reines Tosylchlorid wurden unter Kühlung in
20 ccm absolutem Pyridin und 10 ccm Chloroform aufgelöst. Nach
48 stündigem Stehen bei 20°, Abdampfen des Chloroforms im
Vakuum und Zugabe von 10 ccm Acetanhydrid, wurde die Lösungnochmals 24 Stunden bei 20° aufbewahrt. Zur Aufarbeitung wurde
die Mischung in kaltes Wasser gegossen und mit Benzol ausgeschüt¬telt. Die Benzollösung wurde mit 2-n. Salzsäure, gesättigter Kalium-
hydrogen-carbonat-Lösung undWasser gewaschenundmitNatrium-
sulfat getrocknet. Nach dem Abdampfen des Benzols wurde das
nicht flüchtige gelbliche Öl in Chloroform gelöst und mit Tierkohle
entfärbt. Abdampfen des Chloroforms gab ein farbloses öl, welches
langsam kristalhsierte. Bei mehrmahgem Umkristallisieren des
Präparates aus Methanol entstanden gut ausgebildete, feine Stäb¬
chen. Das Analysenpräparat wurde nochmals aus Benzol-Petrol-
äther umkristallisiert und 24 Stunden bei 60° im Hochvakuum
getrocknet; es zeigte einen Schmelzpunkt von 105°—106° und gabin der Mischprobe mit dem vorstehend beschriebenen Ditosylat(Smp. 115°) eine deutliche Schmelzpunktserniedrigung.
3,677 mg Substanz gaben 8,244 mg C02 und 1,918 mg H204,852 mg Substanz verbrauchten 2,193 ccm 0,02-n. KJ03
C32H3609S3 Ber. C 58,16 H 5,49 S 14,56%(Monotosylat-triacetat)
C37H40Oi0S4 Ber. C 57,5 H 5,18 S 16,58%(Ditosylat-diacetat)
Gef. C 61,19 H 5,84 S 14,49%
[<x]D = - 133° (c = 1,9 in Chloroform)
3, 5-Ditosyl-D-ribonsäure-y-lacton (XXXIX)
4 g ( = 27 Millimol) D-Eibonsäure-y-lacton wurden in 6 ccm
Pyridin und 7,6 g ( = 40 Millimol) Tosylchlorid in 8 ccm Pyridin
4) Ausführung in Anlehnung an die partielle Tosylierung und nachfol¬
gende Acetylierung von Glucose zu |3-Tetraacetyl-D-glucose-6-tosylat nach
E. Hardegger und R. M. Montavon, Helv. 29, 1199 (1946).
35

gelöst. Die zusammengegebenen Lösungen wurden 24 Stunden bei
Zimmertemperatur stehengelassen und dann mit 5 ccm Wasser
versetzt. Nach weiteren 3 Stunden wurden die Reaktionsproduktein Chloroform aufgenommen. Die Chloroform-Lösung wurde mit
2-n. HCl und gesättigter KHCOg-Lösung ausgeschüttelt, mit Was¬
ser neutral gewaschen, mit Natriumsulfat getrocknet und im
Vakuum zur Trockene eingedampft. Der ölige Rückstand wog 2,2 g.
Beim Anspritzen mit Methanol entstanden wenig gelbliche amorphe
Flocken, die abfiltriert wurden. Aus dem Filtrat kristallisierte beim
Stehen das 3,5-Ditosylat. Das Präparat schmolz nach wiederholtem
Umkristallisieren aus Methanol bei 123°—124°. Zur Analyse wurde
das Ditosylat 48 Stunden bei 20° im Hochvakuum getrocknet.
3,704 mg Substanz gaben 6,802 mg C02 und 1,482 mg HaO
4,170 mg Substanz verbrauchten 1,768 com 0,02-n. KJ03
C19H20O9S2 Ber. C 50,0 H 4,39 S 13,6 %
Gef. C 50,12 H 4,48 S 13,59%
2, 3-Isopropyliden-D-ribonsäure-y-Iacton (XL) aus
D-Ribonsäure-y-lacton (XXXII)
10 g D-Ribonsäure-y-lacton wurden mit 20 ccm Aceton und
1 ccm konz. Schwefelsäure unter zeitweisem Umschütteln 24 Stun¬
den stehen gelassen. Die Mischung wurde durch vierstündiges
Schütteln mit überschüssiger, fein pulverisierter, kalzinierter Soda
neutralisiert und dann filtriert. Das klare Filtrat wurde ohne
Erwärmung im Wasserstrahlvakuum eingeengt. Das 2,3-Isopro-
pyliden-D-ribonsäure-y-lacton XL schied sich während des Ein-
dampfens in kristallisierter Form ab. Das mehrmals aus Aceton-
Petroläther umkristallisierte Reaktionsprodukt (9,1 g) schmolz bei
138°.
Zur Analyse wurde die Isopropyliden-Verbindung im Hoch¬
vakuum bei 110° sublimiert.
3,702 mg Substanz gaben 6,906 mg C02 und 2,135 mg H20
C8H1205 Ber. C 51,06 H 6,43%Gef. C 50,90 H 6,45%
[a]D = - 72° (c = 1,4 in Aceton)
36

Einwirkung von Basen: Im Gegensatz zum D-Ribonsäure-y-lacton5) konnte das 2,3-Isopropyliden-D-ribonsäure-y-lacton weder
mit Ammoniak, noch mit Anilin, noch mit Hydrazinhydrat zu kri¬
stallisierten Derivaten umgesetzt werden; aus allen Ansätzen
konnte das Ausgangsmaterial unverändert zurückgewonnen werden.
Umsetzung mit Tosylchlorid: Mehrere Ansätze zur Tosylierungdes 2,3-Isopropyliden-D-ribonsäure-y-lactons mit Tosylchlorid in
Pyridin führten nicht zu kristallisierten Präparaten der Zusammen¬
setzung C15H1807S eines Tosyl-isopropyliden-D-ribonsäure-lactons.
Versuch zur Oxydation des 2, 3-Isopropyliden-D-ribonsäure-y-lactons mit Kaliumpermanganat
Eine Lösung von 1 g Isopropyliden-Verbindung in 40 ccm Was¬
ser wurde mit festem Kaliumpermanganat (1,78 g) versetzt und
unter Durchleiten von Kohlensäuregas auf dem Wasserbade 21/4Stunden erhitzt. Nach Zugabe von wenig Äthanol und nochmaligemkurzen Erhitzen wurde vom Braunstein filtriert und im Wasser¬
strahlvakuum eingedampft. Eine sich als anorganische Substanz
erweisende Fällung, welche beim Anspritzen des entstandenen
Sirups mit Äthanol entstand, wurde verworfen. Aus der alkoholi¬
schen Lösung kristallisierte das Kaliumsalz der 2,3-Isopropyliden-D-ribonsäure (XLII), welches nach Umkristallisieren aus Äthanol-
Wasser bei 217° unter Zersetzung schmolz. Das Analysenpräparatwurde bei 60° 34 Stunden im Hochvakuum getrocknet.
3,844 mg Substanz gaben 5,529 mg C02 und 1,862 mg H20mit K2Cr207 verbrannt.
C8H1306K Ber. C 39,4 H 5,32%Gef. C 39,25 H 5,40%
Versuch zur Umsetzung von 2, 3-Isopropyliden-D-ribonsäure-y-lacton (XL) mit Thionylchlorid
1 g Isopropyliden-Verbindung wurde in 4 ccm Thionylchlorid
gelöst und 6 Stunden am Rückfluss gekocht. Das nicht kristalli-
6) Vgl. Seite 38, 39.
37

sierte Reaktionsprodukt wurde im Kugelrohr bei 130°—150° im
Hochvakuum destilliert, das kristallisierte Destillat im Hoch¬
vakuum bei 120° sublimiert und zur Analyse gegeben. Das Produkt,welches kein Halogen enthielt, erwies sich als Ausgangsmaterial.
3,664 mg Substanz gaben 6,866 mg C02 und 2,100 mg H20
C8H1205 Ber. C 51,06 H 6,43%Gef. C 51,14 H 6,42%
D-Ribonsäure-amid (XLVIII)6)
5 g D-Ribonsäure-y-lacton wurden in 40 ccm Äthanol aufgelöst.In die Lösung wurde während 15 Minuten ein kräftiger Strom von
Ammoniakgas eingeleitet. In der sich erwärmenden Lösung bilde¬
ten sich schon nach einigen Minuten weisse Kristalle. Der Kolben
wurde verschlossen 12 Stunden stehen gelassen. Nach Filtration
des Kristallbreis und Waschen mit Äthanol-Äther wurde das Prä¬
parat aus Wasser-Äthanol umkristallisiert. Das Amid (5,3 g) schmolz
bei 138° unter Zersetzung. Das Analysenpräparat wurde bei 50°
24 Stunden im Hochvakuum getrocknet.
3,616 mg Substanz gaben 4,819 mg C02 und 2,151 mg HaO
C5Hu05N Ber. C 36,36 H 6,71%Gef. C 36,37 H 6,65%
D-Ribonsäure-hydrazid (LH)7)
5 g D-Ribonsäure-y-lacton wurden in 5 ccm Hydrazinhydrat
gelöst und 10 Minuten am Rückfluss gekocht. Nach Anspritzen der
noch heissen Lösung mit Äthanol entstand ein öl, aus dem beim
Abkühlen und Anspritzen mit Äther das Hydrazid auskristalKsierte
(2,5 g). Das Präparat zersetzte sich beim Schmelzpunkt von 149°.
Zur Analyse wurde das Hydrazid aus Äthanol-Wasser umkristalli¬
siert und bei 50° 24 Stunden im Hochvakuum getrocknet.
«) M. Tishler und J. W. Wellmann, TJ.S. 2417143 Mar. 11 (1947).
7) Th. W. J. van Marie, R. 39, 558 (1920).
38

3,562 mg Substanz gaben 4,361 mg C02 und 2,129 mg H20
C6H1206N2 Ber. C 33,33 H 6,71%Gef. C 33,41 H 6,69%
D-Ribonsäure-phenylhydrazid (LUI) s)
3 g D-Ribonsäure-y-lacton wurden in 10 ccm Äthanol aufgelöstund nach Zugabe von 2,5 ccm Phenylhydrazin eine Stunde am
Rüekfluss gekocht. Beim Abkühlen kristallisierte das Phenylhydra¬zid (2,4 g), welches nach Filtration mit wenig Methanol und viel
Äther gewaschen wurde. Nach dem Umkristallisieren aus Methanol-
Benzol schmolz das Produkt bei 158° unter Zersetzung. Bei mehr¬
maligem Umkristallisieren aus Methanol-Benzol zersetzte sich das
Phenylhydrazid. Das Analysenpräparat wurde bei 20° 60 Stunden
im Hochvakuum getrocknet.
3,632 mg Substanz gaben 6,858 mg C02 und 2,043 mg H20
CuH1605N2 Ber. C 51,56 H 6,29%Gef. C 51,53 H 6,29%
D-Ribonsäure-anilid (XLIX) und D-Arabonsäure-anilid9)aus D-Ribonsäure-y-lacton (XXXII)
6 g D-Ribonsäure-y-lacton wurden in 4 g Anilin 6 Stunden am
Rüekfluss gekocht. Die beim Abkühlen entstandenen Kristalle
wurden filtriert und der Reihe nach mit Anilin, Benzol, wenigMethanol und zuletzt mit Äther gewaschen. Nach mehrmaligemUmkristallisieren aus Methanol hatte das D-Arabonsäure-anilid
einen Schmelzpunkt von 197°. Das Analysenpräparat wurde 4 Tagebei 20° im Hochvakuum getrocknet.
3,754 mg Substanz gaben 7,508 mg C02 und 2,130 mg H203,889 mg Substanz gaben 0,209 ccm N2 (24°, 723 mm)
CiiH1506N Ber. C 54,76 H 6,27 N 5,81%Gef. C 54,58 H 6,35 N 5,88%
8) Beilstein, Bd. 15.
>) Th. W. J. van Marie, R. 39, 549 (1920).
39

Aus den zusammengenommenen Mutterlaugen kristallisierte das
D-Ribonsäure-anilid, welches nach mehrmaligem Umkristallisieren
aus Methanol-Benzol einen Schmelzpunkt von 150° aufwies. Das
Analysenpräparat wurde bei 20° 3 Tage im Hochvakuum getrock¬net.
3,756 mg Substanz gaben 7,534 mg COa und 2,134 mg H20
4,357 mg Substanz gaben 0,236 ccm N2 (25°, 726 mm)
CuH1505N Ber. C 54,76 H 6,27 N 5,81%Gef. C 54,77 H 6,36 N 5,93%
Tritylierung von D-Ribonsäure-y-lacton (XXXII)
D-Ribonsäure-y-lacton-5-trityIäther (XLIII)
2 g (= 13,5 Millimol) D-Ribonsäure-y-lacton wurden in 4 ccm
wasserfreiem Pyridin gelöst und ebenso 4,12 g (= 14,8 Millimol)reinstes Tritylchlorid in 9 ccm absolutem Pyridin. Nach dem Zu-
sammengiessen der Lösungen wurde die Mischung 12 Stunden
geschüttelt, anschliessend auf dem Wasserbade 4 Stunden erwärmt
und die Lösung tropfenweise unter starkem Rühren zu 200 ccm
Wasser und Eis gegeben. Die Mischung wurde mit 150 ccm Chloro¬
form ausgezogen. Der Chloroformauszug wurde mit KHS04, gesät¬
tigter KHC03-Lösung und Wasser ausgeschüttelt, mit Na2S04
getrocknet, am Wasserstrahlvakuum auf 100 ccm eingeengt und
stehen gelassen. Aus der Chloroformlösung kristallisierte der 5-Tri-
tyläther des D-Ribonsäure-lactons in farblosen Nadeln (1,9 g),welche nach mehrfachem Umkristallisieren aus Alkohol bei 172°
schmolzen. Das Analysenpräparat wurde 24 Stunden bei 30° im
Hochvakuum getrocknet.
3,558 mg Substanz gaben 9,643 mg C02 und 1,832 mg H20
C24H2205 Ber. C 73,83 H 5,68%Gef. C 73,96 H 5,76%
0]D = +45° (c = 0,8 in Methanol)
D-Ribonsäure-y-lacton-5, X-ditrityläther (XLV)
Bei weiterem Einengen der Chloroformlösung kristallisierte der
5, X-Ditrityläther des D-Ribonsäure-y-lactons als Monohydrat
40

(2,3 g); er schmolz nach mehrmaligem Umkristallisieren aus Chloro¬
form-Alkohol, bzw. Dioxan bei 271°. Die Analysenpräparate wur¬
den bei 30°, bzw. 80° im Hochvakuum 12 Stunden, bzw. 20 Stun¬
den getrocknet.
3,751 mg Substanz gaben 10,918 mg C02 und 1,945 mg H20 (30°)
3,680 mg Substanz gaben 10,901 mg C02 und 1,926 mg H20 (80°)
C43H3605.HaO Ber. C 79,36 H 5,89%Gef. C 79,45 H 5,80%
C43H3606.JH20 Ber. C 80,60 H 5,77%Gef. C 80,84 H 5,86%
[a]D = +113° (c = 0,4 in Dioxan)
D-RUtonsäure-tritylester-5-trityläther (XLIV)
Bei Behandeln des Rückstandes mit Methanol gelang es, als
drittes Präparat den 5-Trityläther des D-Ribonsäure-tritylesters
(XLIV) in kristallisierter Form zu isolieren. Die Mutterlaugen aus
der Gewinnung des 5, X-Ditrityläther-lactons wurden im Vakuum
zur Trockene eingedampft. Das mehrmals aus Methanol umkristalli¬
sierte, 3 Tage bei 30° im Hochvakuum getrocknete Analysenpräpa¬rat schmolz bei 180°.
4,158 mg Substanz gaben 12,077 mg C02 und 2,298 mg H20
C43H3806 Ber. C 79,36 H 5,89%Gef. C 79,26 H 6,18%
[a]D = + 35° (c = 0,8 in Methanol)
Hydrazid des D-Ribonsäure-5, x-ditrityläthers (XLVII)
600 mg D-Ribonsäure-5, X-ditrityläther-y-lactonwurden in 4 ccm
Hydrazinhydrat gelöst, 20 Minuten am Rückfluss gekocht und
anschliessend mit Äthanol angespritzt und stehen gelassen. Die
sich bildenden Kristalle wurden aus Benzol-Äthanol, bzw. aus
Benzol umkristallisiert. Das aus Benzol-Äthanol umkristalhsierte
Analysenpräparat vom Schmelzpunkt 113° wurde bei 20° 3 Tageim Hochvakuum getrocknet.
41

3,686 mg Substanz gaben 10,068 mg C02 und 2,276 mg H20
C43H40O,N2,2C2H5OH Ber. C 74,60 H 6,88%Gef. C 74,54 H 6,91%
Das aus Benzol umkristallisierte Analysenpräparat vom Schmelz¬
punkt 114° wurde bei 50° 2 Tage im Hochvakuum getrocknet.
3,770 mg Substanz gaben 10,806 mg C02 und 2,138 mg H20
6,549 mg Substanz gaben 0,234 ccm N2 (27°, 730 mm)
C43H40OBN2 Ber. C 77,69 H 6,07 N 4,21%
C43H40O6N2-iC6H6 Ber. C 78,75 H 6,14 N 3,75%
C43H40O5N2-iC6He Ber. C 78,3 H 6,08 N 3,81%Gef. C 78,22 H 6,35 N 3,91%
Versuch zur Herstellung von 2,3-Isopropyliden-D-ribonsäure-y-lacton-5-trityläther. 200 mg 5-Trityläther-D-ribonsäure-y-lactonwurden in 15 ccm Aceton aufgelöst und mit 1 g wasserfreiem
Kupfersulfat 24 Stunden bei Zimmertemperatur geschüttelt. Aus
dem eingeengten Filtrat konnte nur kristallisiertes Ausgangs¬material, aber kein Isopropyliden-Derivat isoliert werden.
Oxydation von D-Rihonsäure-y-Iacton mit Perjodsäure
a) Oxydation in Wasser-Dioxan — Verbrauch von 3 Mol HJ04
pro Mol Lacton. Zu einer Lösung von 148 mg (= 1 Millimol)
D-Ribonsäure-y-lacton in 50 ccm Dioxan wurden 9 ccm 0,50-molare
Perjodsäure-Lösung (= 4,5 Millimol HJ04) eingetropft und nach
Zugabe von 25 ccm Wasser mit Dioxan auf 100 ccm aufgefüllt.Nach 5 Minuten wurden zur Titration 10 ccm der Mischung ver¬
wendet. Diese 10 ccm wurden mit 20 ccm Wasser verdünnt, mit
15 ccm gesättigtem, wässrigem Natrium-hydrogencarbonat gepuf¬fert und mit 3 ccm ca. 1-n. Natriumjodid-Lösung versetzt. Das
ausgeschiedene Jod wurde mit 0,1-n. arseniger Säure unter Zusatz
von 2 Tropfen Stärkelösung zur besseren Erkennung des End¬
punktes titriert.
1 ccm 0,1-n. Asa03-Lösung entsprechen 0,05 Millimol Perjod¬säure. Da zur Titration 29 ccm 0,1-n. As203-Lösung nötig waren,
entspricht dies 1,45 Millimol Perjodsäure. Es wurden somit zur
42

Oxydation 4,5 Millimol-1,45 Millimol = 3,05 Millimol Perjodsäure
pro Millimol Substanz verbraucht.
b) Oxydation in methanolischer Lösung. Zu einer Lösung von
148 mg ( = 1 Millimol) D-Ribonsäure-y-lacton in 50 ccm Methanol
wurden 4 ccm 0,47-m. methanolische Perjodsäure zugefügt und mit
Methanol auf 100 ccm aufgefüllt.Je 10 ccm der Oxydationslösung wurden mit 5 ccm ca. 1-n.
methanolischer Natriumjodidlösung versetzt und mit ca. 5 ccm ver¬
dünnter Schwefelsäure angesäuert. Das ausgeschiedene Jod wurde
mit 0,1-n. Natriumthiosulfat-Lösung titriert.
Verbrauch an Perjodsäure
nach pro Millimol Substanz
5 Minuten 0,75 Millimol HJ0415 Minuten 1,15 Millimol HJ04
lVa Std. 1,30 Millimol HJ0418 Std. 1,58 Millimol HJ04
Der Versuch zeigt, dass nach anfänglich raschem Verbrauch von
1 Mol Perjodsäure pro Mol Lacton die Oxydation nur langsamweiter fortschreitet.
c) Oxydation in methanolischer Lösung unter Eis-Kochsalz-
Kühlung. — Verbrauch von 1 Mol HJ04 pro Mol Lacton. Die Oxy¬dation wurde in gleicher Weise, wie unter b) beschrieben ist, aus¬
geführt, nur wurde die Oxydationslösung im Messkolben stets unter
Eis-Kochsalz-Kühlung gehalten.
Verbrauch an Perjodsäure
nach pro Millimol Substanz
15 Minuten 0,15 Millimol HJ04
23/4 Std. 0,38 Millimol HJ0418 Std. 1,03 Millimol HJ0420 Std. 1,04 Millimol HJ04
231/2 Std. 1,10 Millimol HJ0448 Std. 1,15 Millimol HJ04
43

Der Versuch zeigt, dass die Oxydation bei Eis-Kochsalz-Küh¬
lung bei einem Verbrauch von 1 Millimol Perjodsäure pro Millimol
Substanz stehen bleibt.
Oxydation des 3, 5-Ditosyl-D-ribonsäure-y-lactons (XXXIX)mit Perjodsäure
Zu einer mit Eis-Kochsalz gekühlten Lösung von 228 mg ( = 0,5
Millimol) Ditosylat in 50 ccm Methanol wurden 2 ccm 0,47-m.
(= 0;94 Millimol) methanolische Perjodsäure zugefügt und mit
Methanol auf 100 ccm aufgefüllt. Die Titration von 2 Proben à
20 ccm wurde gleich durchgeführt wie im vorherigen Versuch.
Weder nach 15 Minuten noch nach 16 Stunden war eine Oxydationdurch Titration festzustellen.
Nach dem Auffüllen des Messkolbens mit 40 ccm Wasser bis zur
Marke wurde eine weitere Titration ausgeführt. Der Verbrauch an
0,1-n. arseniger Säure betrug nach 15 Minuten 11,3 ccm. Da 1 ccm
0,1-n. arsenige Säure 0,05 Millimol Perjodsäure entspricht, sind
0,56 Millimol Perjodsäure zurücktitriert worden, was der vorgeleg¬ten Menge Perjodsäure, nämlich 3/5 von 0,94 Millimol = 0,56 Milli¬
mol Perjodsäure entspricht. Es ist also auch in Gegenwart von
Wasser, im Gegensatz zum Verhalten des D-Ribonsäure-y-lactons,kein Verbrauch an HJ04 festzustellen.
Nach dreiwöchigem Stehen hatten sich im Messkölbchen Kri¬
stalle abgesetzt, welche nach Filtration und Waschen mit Wasser
bei 124° schmolzen und sich auch in der Mischprobe mit dem Aus¬
gangsmaterial identisch erwiesen. Die klare Lösung zeigte, in der
wie oben durchgeführten Titration, einen Verbrauch von 0,34 Milli¬
mol Perjodsäure pro Millimol Substanz an.
Oxydation von 2, 3-Isopropyliden-D-ribonsäure-y-lacton (XL)mit Perjodsäure
188 mg (= 1 Millimol) Isopropyliden-D-ribonsäure-y-lacton wur¬
den, um den Lactonring zu öffnen, in 12 ccm 0,1-n. Natronlauge auf-
44

gelöst. 2 ccm 0,6-m. ( = 1,2 Millimol) Perjodsäure wurden mit 12 ccm
0,1-n. Natronlauge neutralisiert und dann in die Lösung der Iso-
propyliden-D-ribonsäure eingetropft. Die Mischung wurde mit Was¬
ser auf 50 ccm aufgefüllt. Die Bestimmung der verbrauchten Perjod¬säure erfolgte analog der früher gegebenen Vorschrift10). Je 10 ccm
der Oxydationslösung verbrauchten nach 15 Minuten bzw. nach
3 Stunden 0,4 ccm 0,1-m. As203-Lösung, entsprechend 0,1 Millimol
unverbrauchter Perjodsäure. Die Menge an verbrauchter Perjod¬säure beträgt demnach 1,2 Millimol —0,1 Millimol = 1,1 Millimol
HJ04 pro Millimol Substanz.
Nachweis von Formaldehyd unter den Oxydationsprodukten
Die verbleibenden 30 ccm Oxydationslösung wurden mit einer
Lösung von 140 mg Dimedon in 3 ccm Äthanol versetzt, stark
geschüttelt und zur Kristallisation stehen gelassen. Das aus Ätha¬
nol umkristallisierte Produkt schmolz bei 187°, was mit dem in der
Literatur angegebenen Schmelzpunkt von Dimedon-formaldehydübereinstimmt. Die Überführung des gebildeten Formaldehyds in
das Dimedon-Derivat verlief quantitativ. Von den zu erwartenden
116,8 mg wurden 112,9 mg erhalten.
Oxydation von D-Ribonsäure-amid (XLVIII) mit Perjodsäure
Zu einer Lösung von 165 mg D-Ribonsäure-amid in 50 ccm Was¬
ser wurden 9 ccm 0,6-m. (= 5,4 Millimol) Perjodsäure tropfenweise
zugegeben und anschliessend mit Wasser auf 100 ccm aufgefüllt.Die Bestimmung der verbrauchten Perjodsäure erfolgte analog der
früher gegebenen Vorschrift10). Je 10 ccm der Oxydationslösungverbrauchten nach 15 Minuten, bzw. nach 4 Stunden, je 4,4 ccm
0,1-n. As203-Lösung, was auf den ganzen Ansatz berechnet 2,2
Millimol unverbrauchter Perjodsäure entspricht. Die Menge an
verbrauchter Perjodsäure beträgt demnach 5,4 Millimol — 2,20 Milli¬
mol = 3,2 Millimol HJ04 pro Millimol Substanz.
10) Vgl. Seite 42.
45

In einem Ansatz mit 2 g D-Ribonsäure-amid wurde, nach Ver¬
brauch von 3 Mol HJ04 und Entfernung von Jod- und überschüs¬
siger Perjodsäure, versucht das Glyoxylsäure-amid (L) als Phenyl-hydrazon aus den Oxydationsprodukten zu isolieren. Das Phenyl-hydrazon konnte aber weder in analysenreiner noch in kristalli¬
sierter Form erhalten werden.
Oxydation von D-Ribonsäure-anilid (XLIX) mit Perjodsäure
241 mg D-Ribonsäure-anilid wurden in 20 ccm Methanol aufge¬löst. Nach Eintropfen von 9 ccm 0,6-m. ( = 5,4 Millimol) Perjod¬säure in die mit 50 ccm Wasser verdünnte Lösung wurde mit Was¬
ser zur 100-ccm-Marke aufgefüllt. Die Bestimmung der Perjodsäureerfolgte analog dem vorhergehenden Versuch11). 10 ccm der Oxy¬
dationslösung verbrauchten nach 18 Stunden 4,3 ccm 0,1-n. As203-Lösung, was für die ganze Lösung 2,2 Millimol unverbrauchter
Perjodsäure entspricht. Die Menge an verbrauchter Perjodsäurebeträgt demnach 5,4 Millimol - 2,2 Millimol = 3,2 Millimol HJ04
pro Millimol Substanz.
Benzoyl-phenylhydrazid (LV)12)
Zu einer eisgekühlten Lösung von 10 g Phenylhydrazin in der
fünffachen Menge Äther wurden allmählich 6,5 g Benzoylchlorid
zugetropft. Die von der Reaktionslösung filtrierten Kristalle wurden
mit Wasser aufgekocht und heiss filtriert (6,1 g). Nach mehrmali¬
gem Umkristallisieren aus heissem Äthanol schmolz das Benzoyl-phenylhydrazid bei 168°. Das Analysenpräparat wurde 3 Tage bei
20° im Hochvakuum getrocknet.
3,753 mg Substanz gaben 10,12 mg C02 und 1,922 mg H203,251 mg Substanz gaben 0,395 ccm N2 (26°, 721 mm)
C13H12ON2 Ber. C 73,56 H 5,70 N 13,20%Gef. C 73,59 H 5,73 N 13,16%
u) Vgl. Seite 42.
12) Vgl. E. Fischer, A. 190, 125 (1878); dort. Smp. 168°.
46

Oxydation von Benzoyl-phenylhydrazid (LY) mit Perjodsäure
212 mg ( = 1 Millimol) Benzoyl-phenylhydrazid wurden in 60 ccm
Methanol aufgelöst und tropfenweise mit 4 ccm 0,525-m. (=2,1
Millimol) Perjodsäure versetzt. Nach Auffüllen mit Methanol bis
zur 100-ccm-Marke wurden zur Bestimmung der unverbrauchten
Perjodsäure je 10 ccm verwendet. Die Titration mit arseniger Säure
erfolgte gleich wie in den vorangegangenen Versuchen.
Verbrauch an Perjodsäure
nach pro MiUimol Substanz
15 Minuten 0,75 Millimol HJ04
45 Minuten 0,87 Millimol HJ04
18 Std. 0,95 Millimol HJ0424 Std. 1,07 MiUimol HJ04
47

Zusammenfassung
Zu Beginn meiner Arbeit war die Konfiguration der vor längererZeit aus Hefe isolierten Thiomethyl-pentose noch unbekannt.
Ich beabsichtigte auf synthetischem Wege die 5-Thiomethyl-D-ribose herzustellen und sie mit der natürlichen Thiomethylpentoseaus Hefe zu vergleichen. Die inzwischen von anderer Seite durch¬
geführte Synthese ergab die Identität der beiden Verbindungen.Im theoretischen Teil der vorliegenden Dissertation wurden nach
einer eingehenden Besprechung der in der Literatur beschriebenen
analytischen und synthetischen Untersuchungen über die natürliche
Thiomethyl-pentose der Hefe die eigenen Arbeiten diskutiert. Diese
befassten sich — zum Zwecke der Einführung einer Thiomethyl-Gruppe — mit solchen Derivaten der D-Ribose, bzw. D-Ribonsäure,welche am C-Atom 5 die Durchführung selektiver Umsetzungenerlauben.
Zunächst wurde versucht, die in unserem Laboratorium erst¬
mals hergestellten und von mir als Quecksilberchlorid-Additions¬
verbindungen charakterisierten D-Ribose-äthylen- und -benzyl-
mercaptale partiell zu tosylieren. Die prachtvoll kristallisierten
Reaktionsprodukte gaben aber nicht interpretierbare Analysen¬werte mit hohem Schwefelgehalt und schieden deshalb für die
beabsichtigte Synthese aus.
Erfolgreicher waren die Umsetzungen mit D-Ribonsäure-y-lac-ton. Mit Tosylchlorid entstand daraus das 3,5-Ditosylat, mit Aceton
die 2, 3-Isopropyliden-Verbindung und mit Tritylchlorid der 5-Tri-
tyläther, neben dem 5-Trityl-D-ribonsäure-tritylester und einem
Di-trityläther-lacton. Die Konstitution der ersten drei Verbindun¬
gen konnte durch ihr Verhalten gegenüber Perjodsäure weitgehendgesichert werden. In diesem Zusammenhang wurde auch die Ein¬
wirkung von Perjodsäure auf das Ribonsäure-y-lacton und auf
48

stickstoffhaltige Derivate der Kibonsäure genauer untersucht. Es
gelang erstmals eine Methode zu finden, die es erlaubt, das Ribon-
säure-y-lacton unter Verbrauch von genau 1 Mol Perjodsäure ohne
Öffnung des Lactonringes zu oxydieren. Ferner wurde gefunden,dass Ribonsäure-amid und -anilid normal oxydiert werden; die
Oxydation des Hydrazids nahm einen unübersichtlichen Verlauf,
während jene des Phenylhydrazids nach Aufnahme von 4 Atomen
Sauerstoff zum Stillstand kam. Oxydationsversuche mit dem
Benzoesäure-phenylhydrazid weisen darauf hin, dass die Oxydationvon Ribonsäure-phenylhydrazid mit Perjodsäure und wohl allge¬mein von Carbonsäure-phenylhydraziden zu den unbeständigen
Phenyl-azo-acyl-Derivaten führt.
Obwohl alle weiteren Versuche zur Einführung des Thiomethyl-restes in die oben erwähnten Verbindungen erfolglos verliefen,
dürfte nach den neuesten experimentellen Ergebnissen von W. 0.
Overend und L. F. J. Parker das von mir hergestellte 2,3-Isopro-
pyliden-D-ribonsäure-y-lacton wohl das am leichtesten zugängliche
Ausgangsmaterial für eine ergiebige Synthese der 5-Thiomethyl-D-ribose darstellen.
49

Lebenslauf
Als Sohn des Arnold Joh. Wyss sei., von Solothurn und Balm
b. Messen, Kanton Solothurn und der Anna, geborene Rieh, wurde
ich am 9. Dezember 1924 in Solothurn geboren, wo ich die Primar¬
und Oberrealschule absolvierte. Im Herbst 1943 bestand ich die
eidgenössische Maturitätsprüfung vom Typus C. Anschliessend
arbeitete ich als Laborant in den Ludw. von Roll'sehen Eisenwer¬
ken in Gerlafingen. Nachdem ich die Rekrutenschule bestanden
hatte — während dieser Zeit starb mein Vater —, wurde ich im
Herbst 1944 in die Abteilung für Chemie an der EidgenössischenTechnischen Hochschule in Zürich aufgenommen. Nach mehreren
Unterbrüchen infolge Militärdienstes und Volontariaten in den
Eisenwerken Gerlafingen erhielt ich im Herbst 1950 das Diplomals Ingenieur-Chemiker. Mit der Durchführung der vorliegendenPromotionsarbeit war ich bis im Herbst 1953 im Laboratorium für
organische Chemie (Leitung Prof. Dr. L. Ruzicka) beschäftigt. Seit¬
her habe ich die Stelle eines Hauptlehrers für Chemie und Waren¬
kunde am Lyceum Alpinum in Zuoz (Graubünden) inne.
September 1954.
Related Documents
![2. Das Silizium-Sauerstoff-System - uni-halle.de · 2. DasSilizium-Sauerstoff-System Sauerstoff [gew.%] 1723 °C CRISTOBALITE (Si) Si O L TRIDYMITE α - QUARTZ β - QUARTZ 1414 °C](https://static.cupdf.com/doc/110x72/5f25363fda691d05ae55c121/2-das-silizium-sauerstoff-system-uni-hallede-2-dassilizium-sauerstoff-system.jpg)










