UNIVERSIDADE DE LISBOA FACULDADE DE MEDICINA IL-4 and TAL1 in T-cell acute lymphoblastic leukemia: studies on the participation of microenvironmental cues and cell-autonomous alterations in leukemogenesis Bruno António Caetano Cardoso Doutoramento em Ciências Biomédicas Especialidade em Ciências Morfológicas 2011

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

UNIVERSIDADE DE LISBOA
FACULDADE DE MEDICINA
IL-4 and TAL1 in T-cell acute lymphoblastic
leukemia: studies on the participation of
microenvironmental cues and cell-autonomous
alterations in leukemogenesis
Bruno António Caetano Cardoso
Doutoramento em Ciências Biomédicas
Especialidade em Ciências Morfológicas
2011


iii
UNIVERSIDADE DE LISBOA
FACULDADE DE MEDICINA
IL-4 and TAL1 in T-cell acute lymphoblastic
leukemia: studies on the participation of
microenvironmental cues and cell-autonomous
alterations in leukemogenesis
by
Bruno António Caetano Cardoso
Doutoramento em Ciências Biomédicas
Especialidade em Ciências Morfológicas
Orientador: Doutor João Taborda Barata
Co-orientador: Professora Doutora Leonor Parreira
2011

iv

Preface
v
Preface
This thesis describes the research work under the scope of my PhD project
developed between January of 2006 and July of 2010 at the Instituto de Medicina
Molecular (Lisbon, Portugal) under the supervision of João T. Barata, PhD. During this
period, part of the research work was also carried at the Utrecht Medical Centre
(Utrecht, Netherlands) under the supervision of Prof. Paul J. Coffer.
This thesis is organized in 6 chapters, which are preceded of a summary written in
Portuguese and an abstract briefly describing the work developed. In chapter 1 an
introductory review and the aims of the work are provided. The chapters 2, 3, 4 and 5
the original results are described and discussed. The chapter 6 comprises a generalized
discussion and the biological implications of the data described in this thesis.
The results presented in this thesis are the result of my own research work and it is
clearly acknowledged in the text whenever results or reagents produced by others were
utilized. I was financially supported by a scholarship from Programa SFRH
(SFRH/BD/24722/2005), Fundação para a Ciência e Tecnologia, Portugal. This work
has not been submitted for any degree at this or any university.
The opinions expressed in this publication are from the exclusive responsibility of
the author.
The impression of this thesis was approved by Conselho Científico da Faculdade
de Medicina de Lisboa on the 26th
of October 2011.

Acknowledgements
vi
Acknowledgements
O meu primeiro agradecimento vai para o Doutor João Barata, meu orientador e amigo.
Gostaria de agradecer pela oportunidade que me deu de fazer o que gosto e que sei, pela
experiência e aprendizagem que me proporcionou, pela paciência demonstrada (várias
foram as vezes que vi os seus cabelos levantarem), pela motivação demonstrada mesmo
quando tudo parecia perdido (e não foram poucas as vezes), pela insistência em que me
tornasse um investigador melhor. Sem a sua perseverança este trabalho não seria
possível. Gostaria acima de tudo de agradecer a amizade retribuída ao longo destes
anos, uma amizade sincera que fica e que muito estimo. Mestre, OBRIGADO.
Gostaria também agradecer à minha co-orientadora, a Professora Leonor Parreira pela
supervisão e entusiasmo demonstrado ao longo do meu doutoramento.
I special thank to Prof. Paul Coffer, for receiving in its laboratory during my PhD. I also
like to thank to all the members of Prof. Paul Coffer laboratory, particularly to Jorg van
Loosdregt and to Miranda Buitenhuis for their help and friendship during my stay in
Utrecht.
Os meus colegas de laboratório merecem um agradecimento especial, sem eles não seria
possível conduzir este projecto a bom termo. OBRIGADO pelos momentos fantásticos
na salinha P3C48 e por terem aturado as Brunices diárias, não deve ter sido fácil.
Obrigado jovem Ana Silva pela tua amizade, dedicação e ajuda em tudo o que precisei,
a IP de PTEN foi mesmo o ponto de viragem. Obrigado Cristina, tu de facto és a maior,
sem palavras. Obrigado Leilita, Ana Gírio, Nádia, Daniel, Leonor, Inês, Alice pelo
vosso apoio e pelo vosso entusiasmo. UBCA RULES pessoal. Um agradecimento
especial à Ana Gírio, Inês e Leonor que me puderam ajudar a rever a tese.
Os colegas com quem partilhamos o laboratório do lado também merecem um
agradecimento especial, Isabel, Hélia, Andreia, Zé e Ricardo. OBRIGADO, por tudo,
foi fantástico trabalhar ao vosso lado. Zé… sem palavras. Continua de pé a proposta?
Gostaria também de agradecer ao Prof. João Gonçalves e aos membros do seu
laboratório, em particular à Sylvie e à Mariana, por toda a ajuda e dedicação.
Gostaria de agradecer aos colaboradores do IMM que de alguma forma me ajudaram,
quer nos conselhos, quer naquele pozinho ou solução mágica que faltava na altura da

Acknowledgements
vii
experiência (Marinho eras sempre tu), quer simplesmente naquela pausa para café
(Marco, Jorge, RP). Gostaria de agradecer em especial à Maria Soares, Ana Caetano e
Isabel Pinto pelos Sortings; à Inês Domingues pela ajuda com a Radioactividade e com
os géis grandes e ao Sérgio de Almeida pela ajuda com os Chips. Gostaria também de
agradecer ao GDIMM por proporcionar uns belos jogos de bola e a todos os que neles
participam. Gostaria ainda de agradecer aos rapazes da aldeia: Rp, Marco, Daniel,
Filipe, Malino, Manel. Já vos tinha dito, foi um enorme prazer tocar com vocês, muito
OBRIGADO por aqueles momentos.
Malta, West side coast to coast, ortogonal… o resto já não digo. OBRIGADO pessoal,
Jojó, Mica, Davids, Piu-Piu, Folhini, Tusu, Chico Fresca, Joana e Laranjinha pela vossa
amizade, pela vossa motivação, pela vossa dedicação e por estarem aqui, sempre tão
perto. Um abraço especial ao Jojó que me reviu parte da tese.
Sergito, Pipas, Gentil, vocês não percebem nada disto mas obrigado na mesma, um dia
explico-vos o que fiz. Obrigado pela amizade, pelas jogatanas de bola e pelos concertos
memoráveis daqueles moços que vocês sabem quem são. Sergito obrigado pela
dedicação, pela confiança e pela companhia nos jogos dos nossos moços que tantas
alegrias nos proporcionaram.
Gostaria também de agradecer a uma Instituição muito especial, que prova ano após ano
o verdadeiro significado da palavra trabalho, sacrifício e dedicação sem nunca pedir
nada em troca apenas um mais que justo reconhecimento. Obrigado.
Gostaria de agradecer à minha família por todo o apoio que me prestaram, avós, tios,
primos, sogrinhos e cunhadinha. Aos meus pais devo tudo, por isso não existem
palavras para vos agradecer, mas cá vai OBRIGADO TONINHOS! Este trabalho
também é vosso.
Por fim gostaria de agradecer a uma pessoa incansável em todo este processo, quando as
coisas pareciam sem retorno, quando o acordar era apenas mais um acordar, quando a
ciência deixava de ter piada, ela não me deixava parar. Não é fácil agradecer a uma
pessoa que nos dá tudo. OBRIGADO PUDJI, por estares aqui quando precisei, por não
me deixares desistir quando era o mais fácil, por não me deixares desiludir, pela força
que me fizeste ver que tinha, pelas palavras naqueles dias difíceis e por todos os grandes

Acknowledgements
viii
e fantásticos momentos juntos. OBRIGADO, simplesmente por estares aqui. Este
trabalho é para ti. OBRIGADO PUDJI, POR TUDO.
A TODOS os que tornaram este trabalho possível, o meu sincero OBRIGADO.

Resumo
ix
Resumo
A Leucemia Linfoblástica Aguda (LLA) é o cancro mais frequente em crianças,
resultando da expansão clonal maligna de precursores linfóides. Aproximadamente 15%
dos doentes com LLA apresentam marcadores de células T (LLA-T). Embora os
regimes quimioterápicos actualmente em uso sejam bastante eficazes, existe ainda um
número significativo de doentes que recidivam. Além disso, os regimes intensivos de
quimioterapia estão normalmente associados a efeitos secundários consideráveis a
médio e longo prazo. Para melhor perceber a biologia da LLA-T e determinar novos
alvos terapêuticos é necessário perceber em que medida factores microambientais e
mecanismos intra-celulares influenciam a génese e a progressão da leucemia. A presente
tese procura identificar os mecanismos pelos quais tanto um factor extracelular (IL-4)
como um factor de transcrição celular (TAL1) podem participar no desenvolvimento e
progressão da leucemia.
A Interleucina-4 (IL-4) é uma citocina da cadeia comum-γ, produzida na medula
óssea, que estimula a proliferação in vitro de células LLA-T. No capítulo 2,
demonstramos que a IL-4 induz a progressão do ciclo celular da fase G0/G1 para as
fases S e G2/M em células LLA-T primárias, devido ao aumento da expressão das
ciclinas D2, E e A e à diminuição de expressão do inibidor de cinases dependentes de
ciclinas p27Kip1
. A transfecção de células LLA-T com a proteína de fusão VP22-p27Kip1
,
que é capaz de translocar para o citoplasma e núcleo das células alvo, impede a
proliferação mediada por IL-4. Além disso, a IL-4 estimula a activação de mTOR, como
demonstra o aumento de fosforilação dos seus alvos p70S6K
, S6 e 4E-BP1. A inibição da
sinalização mediada por mTOR com rapamicina impede o crescimento celular, a
progressão do ciclo celular e a proliferação de células LLA-T estimuladas por IL-4.
Estes resultados identificam mTOR como um regulador dos efeitos moleculares e
celulares promovidos por IL-4 em células LLA-T e fortalecem a hipótese do uso de
inibidores farmacológicos de mTOR no tratamento de doentes com LLA-T (Capítulo 2;
Cardoso et al. Leukemia 2009).
O factor de transcrição hélice-volta-hélice TAL1 é aberrantemente expresso em
quase 65% dos doentes com LLA-T. A proteína “Lim-only domain” LMO2, é
geralmente co-expressa com TAL1 neste tipo de leucemia. Estes genes parecem
contribuir para a génese da leucemia, visto que ratinhos transgénicos para TAL1 e
LMO2 desenvolvem leucemia com fenótipo de células T. No entanto, não se confirmou

Resumo
x
até hoje se TAL1 estará envolvido na génese da leucemia em seres humanos, ou apenas
secundariamente activado como resultado do processo de transformação em LLA-T.
Para responder a esta questão, transduzimos progenitores hematopoiéticos com TAL1
e/ou LMO2 e co-cultivamos estes progenitores com células estromais OP9-Dll1, que
têm a capacidade de induzir a diferenciação de células T in vitro. Descobrimos que os
genes TAL1 e LMO2 desregulam a diferenciação de células T em co-cultura com
células estromais. A expressão coordenada destes dois genes leva a um pequeno
aumento de precursores T CD3+CD4
+CD8
+ de tamanho celular aumentado. Esta
observação é particularmente interessante visto que se sabe que os blastos de pacientes
com LLA-T que expressam TAL1 apresentam um imunofenótipo idêntico. Estes
resultados preliminares mostram que TAL1 e LMO2 podem perturbar o normal
desenvolvimento de células T humanas, possivelmente predispondo os timócitos para
transformação maligna (Capítulo 3).
Com o intuito de identificar e caracterizar os eventuais alvos transcricionais
através dos quais TAL1 poderá gerar leucemia de células T, desenvolvemos um sistema
indutível em que a fusão de TAL1 com o domínio de ligação a hormonas (DLH) do
receptor de estrogénio (RE) permite a regulação fina da actividade de TAL1 numa linha
celular T sem expressão de TAL1 endógeno. Após tratamento com 4-Hidróxi-
Tamoxifeno (4HT), a proteína de fusão RE-TAL1 consegue translocar para o núcleo
celular e consequentemente activar o seu programa de trancrição. O perfil de expressão
da linha celular HPB-ALL estavelmente transduzida com a fusão RE-TAL1 e tratada
com 4HT revelou um total de 26 genes cuja expressão aumentou ou diminuiu após
activação de TAL1, em pelo menos 2 experiências independentes. Seleccionámos sete
genes com base na sua função e potencial interesse em cancro e confirmámos a
expressão diferencial de três (CASZ1, DMGDH e OR5M3) por PCR quantitativo em
tempo real. A transfecção de TAL1 numa outra linha celular LLA-T sem expressão
deste gene (P12), resulta igualmente num aumento da expressão destes genes. O
possível envolvimento de CASZ1 nos efeitos anti-apoptóticos e proliferativos mediados
por TAL1 em células LLA-T também foi investigado. A diminuição da expressão de
TAL1 com siRNA na linha celular Jurkat, que expressa TAL1 abundantemente, diminui
significativamente a expressão de CASZ1, e a perda de expressão correlacionacom
perda de viabilidade celular. Acresce que a diminuição da expressão de CASZ1 em
células Jurkat tem efeitos funcionais semelhantes aos que ocorrem após redução da
expressão de TAL1, nomeadamente diminuição da viabilidade celular e proliferação.

Resumo
xi
No geral, estes estudos permitiram a identificação de três novos genes alvo de TAL1,
com possível relevância funcional no contexto do potencial poder oncogénico de TAL1
(Capítulo 4)
TAL1 parece ter não apenas um papel de regulador positivo mas também de
repressor da transcrição. Não é, portanto, de surpreender que tenha sido demonstrado
anteriormente que TAL1 se pode associar a complexos de cromatina repressivos,
nomeadamente HDAC1, e que a incubação com inibidores de HDAC (iHDAC) induz
apoptose em células leucémicas derivadas de ratinhos transgénicos para TAL1. No
capítulo 5, avaliamos o impacto dos iHDAC em TAL1 numa perspectiva diferente,
nomeadamente analisando o seu impacto na expressão de TAL1 e não no impacto na
actividade transcricional. Os nossos estudos revelam que a incubação de células LLA-T
com iHDAC diminui drasticamente a expressão da proteína TAL1. Este efeito é devido
à diminuição da transcrição do gene TAL1 em células que mantêm o locus TAL1 intacto,
mas também devido à diminui da tradução de mRNAs TAL1 em células que contêm a
delecção TAL1d. Igualmente importante é o facto da apoptose induzida pelos iHDAC
ser inibida pela sobre-expressão de TAL1. Os nossos resultados indicam que o
programa apoptótico promovido pelos iHDAC em LLA-T é parcialmente dependente da
diminuição da expressão de TAL1 e sugerem que a integração de iHDAC no protocolo
de tratamento de doentes LLA-T pode trazer benefícios terapêuticos (Cardoso et al,
Leukemia 2011, advance online publication).
O conjunto dos estudos descritos nesta dissertação destacam a importância que os
factores micro-ambientais, como IL-4, podem ter na progressão de LLA-T (por
exemplo, activando mTOR e promovendo a progressão no ciclo celular) e mostram a
importância que factores celulares como TAL1, e também LMO2, podem ter na
predisposição de células T para a transformação maligna e na sobrevivência das células
LLA-T. Finalmente, os resultados desta tese demonstram que tanto factores
extracelulares como lesões intracelulares podem constituir alvos promissores para
intervenção terapêutica em LLA-T.

Abstract
xii
Abstract
Acute lymphoblastic leukemia (ALL) is the most frequent cancer found in
children and results from the clonal expansion of transformed lymphoid precursors.
Approximately 15% of pediatric ALL patients present with a T-cell phenotype (T-
ALL). Despite the recent improvements in the treatment of T-ALL, there are still a high
number of relapses and the intensive chemotherapeutic regiments used are associated
with long-term severe complications. In order to develop new therapeutic strategies that
can further increase efficacy while reducing side effects, one needs to better understand
the pathobiology of T-ALL. In particular, it is necessary to understand how
microenvironmental and cell-autonomous mechanisms influence the initiation and the
progression of leukemia. The present thesis has the preocupation of exploring the
mechanisms by which both an extracellular cue (IL-4) and a cell-intrinsic transcription
factor (TAL1) may partake in leukemia development and maintenance.
Interleukin-4 (IL-4) is a γ-common chain cytokine produced within the bone
marrow microenvironment that is known to promote the in vitro proliferation of T-ALL
cells. In Chapter 2, we present evidence that IL-4 induces primary T-ALL cell cycle
progression from G0/G1 into S and G2/M, by up-regulating cyclin D2, E and A and
down-regulating the cyclin-dependent kinase inhibitor p27kip1
. Transfection of T-ALL
cells with the VP22-p27kip1
fusion protein, which is able to translocate into the
cytoplasm and nucleus of target cells, abrogates IL-4-mediated proliferation. This
indicates that p27kip1
downregulation is mandatory for cell cycle progression of T-ALL
cells stimulated with IL-4. Furthermore, IL-4 stimulates mTOR activation, as
determined by increased phosphorylation of its downstream targets p70S6K
, S6 and 4E-
BP1. Inhibition of mTOR signaling with rapamycin prevents IL-4-induced T-ALL cell
growth, cell cycle progression and proliferation. Our results identify mTOR as a critical
regulator of IL-4-mediated effects in T-ALL cells and support the rationale for using
mTOR pharmacological inhibitors in T-ALL therapy (Cardoso et al. Leukemia 2009).
The basic helix-loop-helix transcription factor TAL1 is aberrantly expressed in up
to 65% of T-ALL patients. LMO2, a Lim-only domain protein, is often co-expressed
ectopically with TAL1 in this malignancy. These genes appear to have leukemogenic
potential, since both TAL1 and LMO2 transgenic mice develop leukemias of T-cell
phenotype. However, it is still unclear whether TAL1 is effectively leukemogenic in
humans, or whether merely participates as a secondary event in the transformation

Abstract
xiii
process in T-ALL. To address this question, we transduced hematopoietic progenitors
with TAL1 and/or LMO2 and co-cultured them with OP9-Dll1 stromal cells, which have
the capacity to induce T-cell differentiation in vitro. We found that TAL1 and LMO2
genes deregulate human T-cell differentiation in stromal cell co-cultures. Interestingly,
the coordinated expression of both TAL1 and LMO2 led to a relative increase in
CD3+CD4
+CD8
+ T-cell precursors with increased cell size. This observation is
particularly interesting given that TAL1-expressing patients normally display a similar
phenotype. These preliminary results show that TAL1 and LMO2 can disrupt normal
human T-cell development, therefore likely predisposing thymocytes to malignant
transformation (Chapter 3).
In our effort to characterize the mechanisms by which TAL1 might promote T-
cell leukemogenesis, we developed a TAL1 inducible system, by fusing TAL1 with the
hormone binding domain (HBD) of the estrogen receptor (ER), which we expressed in a
TAL1-negative T-cell line. Upon 4-Hydroxi-Tamoxifen (4OHT) treatment, ER-TAL1
fusion protein is able to translocate into the nucleus and consequently trigger its
transcriptional program. Gene expression profiling of 4OHT-treated HPB-ALL cells
stably transduced with the ER-TAL1 fusion revealed a total of 26 genes up- or down-
regulated by TAL1 activation, in at least two independent experiments. We selected
seven of those genes on the basis of their function/potential interest in cancer and
confirmed the differential expression of three (CASZ1, DMGDH and OR5M3) by qRT-
PCR. Accordingly, transfection of another TAL1-negative T-ALL cell line, P12, with
TAL1, also led to increased expression of the validated TAL1 target genes. The possible
involvement of CASZ1 in TAL1-mediated anti-apoptotic and proliferative effects in T-
ALL cells was subsequently investigated. Knock-down of TAL1 with siRNA in the
TAL1-positive T-ALL cell line Jurkat decreased the expression of CASZ1, correlating
with loss of cell viability. Moreover, CASZ1 knockdown in Jurkat cells led to
functional effects similar to those of TAL1 knockdown, namely a decrease in survival
and proliferation. Overall, these studies allowed the identification of three novel TAL1
downstream targets, likely with functional relevance for TAL1-mediated leukemogenic
potential (Chapter 4).
TAL1 binds to repressive chromatin complexes, namely involving HDAC1, and
incubation with HDAC inhibitors (HDACis) promotes apoptosis of leukemia cells
derived from TAL1 transgenic mice. In Chapter 5, we evaluated the impact of HDACis
on TAL1 from a somewhat different perspective, namely by analyzing their impact on

Abstract
xiv
TAL1 expression rather than transcriptional activity. We found that incubation of T-
ALL cells with HDACis strikingly down-regulates TAL1 protein expression. This is
due to decreased TAL1 gene transcription in cells with an intact TAL1 locus, and to
impaired TAL1 mRNA translation in cells that harbor the TAL1d deletion. Importantly,
HDACi-induced apoptosis of T-ALL cells is significantly reversed by TAL1 forced
over-expression. Our results indicate that the HDACi-mediated apoptotic program in T-
ALL cells is partially dependent on the down-regulation of TAL1 expression, and
suggest that integration of HDACis into T-ALL treatment protocol may be of potential
therapeutic benefit (Cardoso et al, Leukemia 2011, advance online publication).
Taken together, the results described in this thesis highlight the importance that
microenviromental factors, such as IL-4, might have in the progression of T-ALL (for
instance, by activating mTOR and promoting cell cycle progression), and hint on the
importance that cell-autonomous factors, such as TAL1 and LMO2, may have in
predisposing T-cells for malignant transformation and promoting survival of T-ALL
cells. Importantly, our results further demonstrate that both extracellular cues and
intracellular molecular lesions can constitute targets for therapeutic intervention in T-
ALL.

Abbreviations
xv
Abbreviations
ABC ATP-Binding Cassette
Act D Actinomycin D
ALL Acute Lymphoblastic Leukemia
AML Acute Myeloid Leukemia
APC Allophycocyanin
B-ALL B-cell Acute Lymphoblastic Leukemia
bHLH basic Helix-Loop-Helix
BM Bone Marrow
BrdU Bromodeoxyuridine (5-bromo-2-deoxyuridine)
BSA Bovine Serum Albumin
CALM Clathrin Assembly protein-like Lymphoid-Myeloid
CBHA m-Carboxycinnamic Acid bis-Hydroxamide
CD Cluster of Differentiation
CDK Cyclin Dependent Kinase
cDNA Complementary Deoxyribonucleic Acid
CHIP Carboxyl terminus of Hsc70 Interacting Protein
ChIP Chromatin Immunoprecipitation
CHX Cycloheximide
CK2 Casein Kinase 2
CLP Common Lymphoid Precursor
CML Chronic Myeloid Leukemia
CMV Cytomegalovirus
CNS Central Nervous System
CSF Colony-Stimulating Factor
Dll1 Delta-like protein 1
DMEM Dubelco‟s Modified Eagle Medium

Abbreviations
xvi
DMGDH Dimethylglycine Dehydrogenase
DMSO Dimethyl Sulphoxide
DN Double Negative
DNA Deoxyribonucleic Acid
DNMT DNA methytransferase
DP Double Positive
eGFP Enhanced Green Fluorescent Protein
EGIL European Group for Immunological Characterization of
Leukemias
eIF4E Eukaryotic Initiation Factor 4E
EPO Erythropoietin
ER Estrogen Receptor
ERK Extracellular signal-Regulated Kinase
ETP Early Thymic Progenitors
FACS Flow Activated Cell Sorting
FBS Fetal Bovine Serum
FITC Fluorescein Isothiocyanate
FGF16 Fibroblast Growth Factor 16
FSC Forward Scattered Light
γC Gamma-Common chain
GFP Green Fluorescent Protein
GPA Glycophorin A
GPCRs G-Protein Coupled Receptors
HA Hemaglutinin
HAT Histone Acetyl Transferase
HBD Hormone Binding Domain
HDAC Histone Deacetylase

Abbreviations
xvii
HDACi(s) Histone Deacetylase Inhibitor(s)
HLH Helix Loop Helix
hPGK Human Phosphoglycerate Kinase promoter
HP1 Heterochromatin Protein 1
HRP Horseradish Peroxidase
HSC Hematopoietic Stem Cells
HSP Heat Shock Protein
HSP90 Heat Shock Protein 90
H3K9Ac Acetylated Histone H3 at Lysine 9
ICN Intracellular Notch
IFN Interferon
IL Interleukin
IMDM Iscove‟s Modified Dulbecco‟s Medium
IRES Internal Ribossomal Entry Site
IR4 Insulin-IL-4 Receptor motif
ISP Immature Single Positive
JAK Janus Kinase/ Just Another Kinase
KDa KiloDalton
LCK Lymphoid Cell Kinase
LTR Long Terminal Repeat
MAPK Mitogen-Activated Protein Kinase
MEK Mitogen activated and ERK related Kinase
MEM Minimum Essential Medium
MHC Major Histocompatibility Complex
MLL Mixed Lineage Leukemia
MRD Minimal-Residual-Disease
mTOR Mammalian Target of Rapamycin

Abbreviations
xviii
mTORC1 mTOR Complex 1
mTORC2 mTOR Complex 2
MW Molecular Weight
NAD Nicotinamide Adenine Dinucleotide
NLS Nuclear Localization Signal
ORs Olfactory Receptors
PARP Poly ADP Ribose Polymerase
PB Phenyl Butyrate
PBS Phosphate Buffered Saline
PCR Polimerase Chain Reaction
PDK1 Phosphoinositide-Dependent Kinase 1
PE Phycoerythrin
PerCP Peridinin Chlorophyll Protein
PFA Paraformaldehyde
PGS PBS Gelatin Saponin
PH Plecstrin Homology
PIP2 Phosphatidyl-Inositol-4,5-Biphosphate
PIP3 Phosphatidyl-Inositol-3, 4, 5-Triphosphate
PI3K Phospho-Inositol-3 Kinase
PKA Protein Kinase A
PKB Protein Kinase B (c-Akt)
PKC Protein Kinase C
PTEN Phosphatase and Tensin Homolog
RA Retinoic Acid
RAG Recombination Activation Gene
RALDH2 Retinaldehyde Dehydrogenase 2
RB Retinoblastoma protein

Abbreviations
xix
RNA Ribonucleic Acid
ROS Reactive Oxygen Species
RPMI Roswell Park Memorial Institute medium
RT-PCR Reverse Transcriptase PCR
RTK Receptor Tyrosine Kinase
SAHA Suberoylanilide Hydroxamic Acid
SB Sodium Butyrate
SCID Severe Combined Immunodefficiency
SCL Stem Cell Leukemia (TAL1)
SDS Sodium-Dodecyl-Sulfate
SDS-PAGE SDS Polyacrylamide Gel Electrophoresis
Ser Serine
SIL SCL-Interrupting Locus
SP Single Positive
SPB Sodium Phenyl Butyrate
SSC Side Scattered Light
STAT Signal Transducer and Activator of Transcription
T-ALL T-cell Acute Lymphoblastic Leukemia
TAL1 T-cell Acute Lymphocytic Leukemia protein 1 (SCL)
TAN1 Translocation Associated Notch1
TBP-2 Thioredoxin Binding Protein 2
TCR T-cell Receptor
TCRA/D T-cell Receptor α/δ gene
TCRB T-cell Receptor β gene
TCRG T-cell Receptor γ gene
TEC Thymic Epithelial Cells
TGF-β Transforming Growth Factor –β

Abbreviations
xx
Thr Threonine
TNF Tumor Necrosis Factor
TRX Thioredoxin
TSC Tuberous Sclerosis Complex
TSA Trichostatin A
UCB Umbilical Cord-Blood
UV Ultraviolet light
VPA Valproic Acid
VSVG Vesicular Stomatitis Virus protein G
ZAP70 Zeta-chain Associated Protein kinase 70
4EBP1 eIF4E Binding Protein 1
4OHT 4-Hydroxy-Tamoxifen
7-AAD 7-Amino-Actinomycin D

Table of Contents
xxi
Table of Contents
Preface v
Acknowledgements vi
Resumo ix
Abstract xii
Abbreviations xv
Table of Contents xxi
Index of Figures xxvi
Index of Tables xxx
Chapter 1. Introduction 1
Cancer 2
Leukemia 3
Acute Lymphoblastic Leukemia (ALL) 4
Diagnosis and Treatment 5
T-cell Acute Lymphoblastic Leukemia (T-ALL) 6
Normal T-cell development 7
T-ALL Immunophenotypical Classification 10
Extracellular factors and Microenvironment 10
Cytokine signaling in T-ALL 12
The IL-4/IL-4 receptor signaling axis 12
Genetic abnormalities in T-ALL 14
Cell cycle defects 14

Table of Contents
xxii
Aberrant signaling 15
The PI3K-mTOR pathway 18
An overview 18
Importance in T-ALL 21
Transcription factors 21
The TAL1/SCL oncogene 25
TAL1, more than a key player in leukemia 27
TAL1: structure and function 28
TAL1 target genes 30
Signaling to TAL1 32
TAL1 in normal development 33
Genome Organization 35
Nucleosomes and Histones 35
Histone Acetyl Transferases (HATs) and Histone Deacetylases
(HDACs) 36
HATs 37
HDACs 38
HDAC inhibitors (HDACis) 39
HDACis and gene expression 40
The effect of HDACis on cancer cells 43
Objectives 45
References 46
Chapter 2. Interleukin-4 stimulates proliferation and growth of
T-cell acute lymphoblastic leukemia cells by activating mTOR
signaling 84
Abstract 85
Introduction 85
Materials and Methods 86

Table of Contents
xxiii
Results and Discussion 88
IL-4 signaling promotes proliferation of T-ALL cells by inducing
cell cycle progression 88
IL4 down-regulates p27Kip1
and up-regulates cyclin expression, CDK
activity and Rb hyperphosphorylation 91
Activation of mTOR pathway is mandatory for IL4 induced
proliferation 92
References 94
Chapter 3. TAL1 and LMO2 ectopic expression in human T-
cell progenitors impacts T-cell development in vitro 96
Abstract 97
Introduction 97
Materials and Methods 99
Results 103
Establishment of a system to simultaneously transduce target cells
with three genes 103
Detection of TAL1 and LMO2 in cord-blood CD34+CD38
- cells
105
Forced TAL1 and LMO2 expression in CD34+CD38
- affect human T-
cell differentiation in vitro 106
High TAL1 and LMO2 expression in human thymic progenitors
increases cell proliferation and has a striking effect on T-cell
differentiation 108
Discussion 110
References 112

Table of Contents
xxiv
Chapter 4. Identification of novel TAL1 target genes with
potential impact on T-cell acute lymphoblastic leukemia 118
Abstract 119
Introduction 119
Materials and Methods 120
Results 125
TAL1 inducible system 125
TAL1 activity up-regulates genes associated with cancer 127
CASZ1, DMGDH and OR5M3 are potential TAL1 target genes 129
CASZ1 knock-down decreases T-ALL cell viability and
proliferation 130
Discussion 132
References 134
Chapter 5. TAL1 is down-regulated upon histone deacetylase
inhibition in T-cell acute lymphoblastic leukemia cells 139
Abstract 140
Introduction 140
Materials and Methods 141
Results 145
HDAC inhibition down-regulates TAL1 protein levels in T-ALL
cells 145
HDACi-mediated TAL1 protein down-regulation in T-ALL cells is
not due to increased apoptosis or protein degradation. 146
HDAC inhibition down-regulates TAL1 transcript levels without
affecting TAL1 splicing or mRNA stability 148

Table of Contents
xxv
HDAC inhibition abrogates TAL1 transcription in TAL1-positive
T-ALL cells with an intact TAL1 locus 150
HDAC inhibition up-regulates TAL1 transcripts in T-ALL cells with
TAL1d
151
HDAC inhibition down-regulates TAL1 protein levels by decreasing
translation in T-ALL cells with TAL1d
152
Forced TAL1 expression partially rescues T-ALL cell death induced
by HDAC inhibition 153
Discussion 153
References 156
Chapter 6. Discussion 169
Cytokine signaling in T-ALL: the role of IL-4 170
Is TAL1 a human oncogene? 172
Novel TAL1 target genes in T-ALL and beyond 174
HDAC inhibitors: a novel therapeutic approach in T-ALL? 176
Acetylation, a new clue on TAL1 regulation? 177
Concluding Remarks 178
References 181

Index of Figures
xxvi
Index of Figures
Chapter 1
Figure 1.1. Human T-cell differentiation 8
Figure 1.2. An overview of the PI3K-mTOR signaling pathway.
19
Figure 1.3. Structural comparison of the TAL1 locus and SIL-TAL1
gene rearrangements 27
.
Chapter 2
Figure 2.1. IL-4 stimulates cell cycle progression of primary T-ALL
cells. 90
Figure 2.2. IL-4-mediated activation of mTOR pathway is critical
for cell cycle progression of T-ALL cells. 93
Chapter 3
Figure 3.1.Transduction of MAT vectors into 293T and
CD34+CD38
- cord blood cells. 104
Figure 3.2. Coordinated expression of TAL1 and LMO2 in human
hematopoietic progenitors promotes cell growth and leads to the
differentiation of CD3+CD4
+CD8
+ cells. 107
Figure 3.3. High TAL1 and LMO2 expression in human T-cell
progenitors disrupts normal T-cell differentiation. 109

Index of Figures
xxvii
Chapter 4
Figure 4.1. Overview of the TAL1 inducible system. 126
Figure 4.2. Venn diagram of the number of genes differentially
expressed in each of three independent experiments upon TAL1
activity induction. 127
Figure 4.3. CASZ1, DMGDH and OR5M3 are potential TAL1-target
genes. 129
Figure 4.4 . CASZ1 knock-down decreases T-ALL cell viability and
proliferation 131
Chapter 5
Figure 5.1. HDACis down-regulate TAL1 protein in T-ALL cells.
145
Figure 5.2. HDACi-mediated TAL1 protein down-regulation is not
due to increased apoptosis or increased protein degradation. 147
Figure 5.3. HDACis down-regulate TAL1 through inhibition of
TAL1 gene transcription in TAL1wt
T-ALL cells lines. 149
Figure 5.4. HDACis down-regulate TAL1 by affecting TAL1
protein translation in TAL1d T-ALL cell lines. 151
Figure 5.5. Enforced TAL1 expression partially rescues HDACi-
mediated T-ALL cell death. 154

Index of Figures
xxviii
Supplementary Figure 5.1. HDACi-mediated down-regulation of
TAL1 mRNA is not due to increased apoptosis in TAL1wt
T-ALL cell
lines. 162
Supplementary Figure 5.2. HDACis up-regulate CDKN1A/p21
mRNA expression. 162
Supplementary Figure 5.3. Schematic representation of TAL1 locus
and the primers used to detect total and processed TAL1 mRNA.
163
Supplementary Figure 5.4. TAL1d-expressing T-ALL cells up-
regulated TAL1 mRNA upon HDACi treatment 164
Supplementary Figure 5.5. HDACis induce T-ALL cell death.
165
Supplementary Figure 5.6. Enforced TAL1 expression partially
rescues HDACi-mediated apoptosis of Jurkat cells. 166
Supplementary Figure 5.7 Model for HDACi-mediated TAL1
down-regulation in T-ALL cells. 167
Supplementary Figure 5.8. TAL1 expression is not affect by
inhibition of PI3K and mTOR. 168
Chapter 6
Figure 6.1. IL-4 signaling promotes the proliferation of T-ALL cells
through the activation of the mTOR pathway 171

Index of Figures
xxix
Figure 6.2. Several hypothetical mechanisms could explain TAL1-
mediated up-regulation of CASZ1, DMGDH and OR5M3. 176
Figure 6.3. The role of the extra-cellular cues and cell-autonomous
mechanisms in the progression of T-ALL. 180

Index of Tables
xxx
Index of Tables
Chapter 1
Table 1.1. T-cell receptor genes and their involvement in the
chromosomal translocations in T-ALL. 22
Table 1.2. Chromosomal aberrations involving the TAL1 gene in
T-ALL. 25
Table 1.3. List of the genes whose expression has been shown to be
directly regulated by TAL1. 30
Table 1.4. Charactheristics of human Histone Deacetylases
(HDACs). 38
Table 1.5. Classification of the commonly used HDAC inhibitors
(HDACis) 41
Chapter 2
Table 2.1. Immunophenotype, classification, and response to IL-4 of
primary T-ALL specimens 89

Index of Tables
xxxi
Chapter 3
Table 3.1. List of primers used in the cloning of the MAT plasmids
99
Table 3.2. List of primers used in semi-quantitative-PCR 103
Chapter 4
Table 4.1. List of primers used in the cloning procedures 121
Table 4.2. List of primers for quantitative and semi-quantitative-
PCR. 123
Table 4.3. List of genes identified in the microarray experiments
(similarly regulated in at least 2 of 3 experiments). 128
Chapter 5
Table 5.1. List of primers used in quantitative-PCR. 144
Table 5.2. List of primers used in Chromatin Immunoprecipitation
experiments. 144

Index of Tables
xxxii

Introduction
1
Chapter 1
INTRODUCTION

Introduction
2
Cancer
The first known reports describing cancer in human patients were written on
papyrus in the ancient Egypt. However, the term cancer was introduced later on by the
Greek physician Hippocrates, the father of Medicine. Hippocrates used the term
Karcinus to describe ulcer-forming tumors due to their projections that resemble the
shape of a crab. Later, the Roman physician Celsus translated the word Karcinus to the
latin word Cancer (1).
What exactly is a cancer? To better understand its definition, one should first
realize that it is not a synonym of tumor. The latter is a swelling or mass, and is one of
the four classical signs of inflammation (calor, dolor, rubor, and tumor: heat, pain,
redness, and swelling) as originally recorded by Celsus in the 1st century A.D.
However, a tumor can also arise from the process of neoplasia, which literally means
“new growth”. Neoplasia is the process by which cell proliferation occurs in an
uncontrolled fashion, exceeding normal growth and persisting at the expense of the
host. As a result, for example, of an accumulation of genomic and/or epigenomic
alterations, normal cells can alter their behavior and start an independent program that
does not obey to the rules imposed upon their surrounding neighbours. Consequently, a
population of cells, which started from a single clone, will grow abnormally and form a
mass of cells with an unstructured, simpler architure than that of a normal tissue. Such
an abnormal mass of cells within a normal tissue, not necessarily resulting from an
inflammatory process, is evidently also called a tumor (2, 3). Currently, the term is
often used as a synonym of neoplasia; however, as stated above, tumor refers to the
mass of abnormal cells while neoplasia refers to the process of tumor formation due to
uncontrolled cellular growth (2, 3).
Tumors can be classified as benign or malignant depending on the type of growth
and degree of aggressiveness to the organism. Benign tumors are charactherized by a
slower, localized growth, which occurs by expansion (encapsulated) and therefore does
not result in invasion of surrounding tissues, intravasation or metastasis. This type of
tumors is also generally indolent to their hosts, except when the expansion of benign
tumors interferes with vital organs or tissues. On the other hand, malignant tumors
generally grow rapidly; invade surrounding tissues with very significant impact on
overall architecture, and eventually metastize. Malignant tumors are generally life

Introduction
3
threatening since they colonize vital organs, thereby compromising the normal
physiology of a living organism (2-4).
Knowing what malignant tumors are, one can now properly introduce the
definition of cancer. Cancer is the general term to encompass a high number of diseases
that are caused by malignant tumors (3). Currently, cancer is commonly used as
synonym of malignant tumor (2, 4).
These definitions, useful as they are at the systemic level, do not allow us to fully
grasp the nature of the cells that originate cancer. This relates to the fact that one often
has far more knowledge concerning the consequences than insights into the etiology and
biology of the disease. Nonetheless, strong efforts have been made throughout the years
to identify the essential characteristics of cancer cells. Two seminal reviews by Douglas
Hanahan and Robert Weinberg, summarized decades of research into the proposal that
all cancer cells can be defined as displaying a common set of features that includes
abnormal and uncontrolled growth, high proliferative capacity, insensitivity to death
signals, capacity to promote angiogenesis, deregulated metabolism, genetic instability,
immune evasion capacity and also the ability to spread systemically to other tissues and
organs compromising their normal physiology (5, 6). Whether all cancer cells must
simulateously display all these features is a matter of debate. For some authors the
ability of cancer cells to form metastasis is the only true hallmark of cancer (7).
While cancer progression depends on many factors that extend well beyond the
cancer cell itself, cancer is mainly a genetic disease, caused by mutations in the DNA.
These genetic mutations are associated with the loss of function in tumor-suppressor
genes and gain-of-function in oncogenes (8), occuring in genes that control mechanisms
essential to the normal cellular physiology, such as components of the cell cycle
machinery, apoptosis, metabolism and also signaling pathways (5, 6).
Leukemia
The organism is constantly renewing the pool of blood cells. Hematopoiesis is the
process of formation and development of new blood cells. Leukemia results from the
deregulation of this process by malignant transformation (9, 10).
In 1845, a patient with a massive accumulation of white blood cells and advanced
chronic disease was reported by Dr. Rudolph Virchow, which termed the condition
“weisses blut”, the German expression for “white blood”. Two years later, Virchow

Introduction
4
renamed the term and called it Leukemia, derived from the Greek work “Leukämie”
(11).Leukemia is therefore the designation for blood cell cancer, and it is generally
characterized by the accumulation of immature cells from a particular hematopoietic
lineage. Currently, leukemia is classified according to the lineage of the transformed
cells (lymphoid versus myeloid) and to the proliferation state of the cells (acute versus
chronic) (12). Historically, “acute” and “chronic” referred to the relative time-span of
survival of patients when effective therapy was not available. However, therapeutic
improvements led to the redefinition of these terms, in such way that presently “acute”
is used to characterize leukemias displaying rapid proliferation of blast cells, whereas
“chronic” refers to leukemias with slower proliferation of malignant cells that are in
general relatively well differentiated (12).
Acute Lymphoblastic Leukemia (ALL)
The incidence of leukemia varies with age. In adults, chronic leukemias are more
frequent than acute leukemias (12). In contrast, acute lymphoblastic leukemia (ALL) is
not only the most common form of leukemia but also the most common cancer in
children, accounting for roughly 25% of all the pediatric cancers (12). ALL is a very
heterogeneous disease with variations at the level of the cellular morphology,
immunological markers and cytogenetic abnormalities. The disease results from the
clonal accumulation of immature cells with either B-cell or T-cell markers that are
developing in the bone marrow (BM) or in the thymus (13).
The precise biological mechanisms that lead to the development of ALL are still
largely unknown. Nevertheless, it is generally accepted that ALL malignant
transformation is a multistep process that involves the deregulation of genes that affect
lymphoid homeostasis (via regulation of cell cycle or apoptosis) and normal
hematopoietic development (13, 14).
Chromosomal translocations are the hallmark of ALL (15). The development of
lymphocytes is characterized by sequential gene rearrangements that produce functional
B and T-cell receptors, and the RAG1 and RAG2 enzymes are the proteins responsible
for this process (16). In several cases of ALL, deregulation of the activity of RAG
proteins appears to be responsible for the formation of chimeric proteins, such as TEL-
AML1 (17) but also for the aberrant expression of oncogenes such as TAL1/SCL (18).
These abnormal events are highly associated with ALL (15).

Introduction
5
The occurrence of ALL is higher in industrialized countries (e.g. Italy, USA, and
Switzerland), whereas in developing countries the incidence is significantly lower. The
exact reasons for this epidemiological observation remain a matter of debate. In
addition, the frequency of ALL varies with age. The occurrence is higher at early ages,
peaking at the age of 1 to 4 years with a frequency of 7 cases per 100.000 persons. ALL
frequency declines with time and stabilizes at the occurrence of 1-2 cases per 100.000
persons (19).
Diagnosis and Treatment
The majority of ALL symptoms are associated with the disruption of normal
hematopoiesis. These symptoms include fever, anemia and bone and joint pain. Other
manisfestations such as fatigue, shortness of breath and dizziness are associated with
anemia as a result of the decrease in red blood cell count. Enlargement of organs such as
spleen, liver, lymph nodes and appearance of mediastinal masses are manifestations that
occur upon the progression of the disease. The involvement of the central nervous
system (CNS) is also a common feature, resulting in the appearance of symptoms like
headache, nausea, vomiting, lethargy and cranial nerve dysfunction. The diagnostic of
ALL is achieved when the presence of these symptoms is associated with the molecular,
cytogenetic and immunophenotypic characterization of the leukemic blasts (19).
The treatment of ALL patients has been improving over time with the systematic
testing of new therapies in clinical trials. The use of risk adjusted and intensive
chemotherapy improved dramatically the overall survival rate of ALL patients.
Currently, the overall survival rate of ALL patients is about 80%, however, recent
clinical trials suggest that it can be above 90% (20). Since, as mentioned above, ALL is
a heterogeneous disease, several biological and clinical factors determine the design of
the therapy. The factors that influence the design include the age of the patient,
leukocyte count, genetic characteristics of the leukemic blasts and the early response to
the chemotherapeutic regiment (19).
With few exceptions, the treatment of ALL patients consists in a therapeutic
program that comprises three phases: the remission induction, the
consolidation/intensification phase and elimination of minimal-residual disease (MRD),
also known as continuation therapy. The treatment of ALL also includes therapy
directed to the CNS to prevent the accumulation of leukemic cells in the brain (19, 21).

Introduction
6
The objective of the remission induction phase is to reduce by 99% the initial leukemic
burden and restore normal hematopoiesis. During this initial stage, three drugs are
administrated: a glucocorticoid (prednisone or dexamethasone), vincristine and
asparaginase or an anthracycline such as doxorubicin or daunorubicin. This is a
common protocol for remission induction. However, several adaptations can be made.
Children at very-high risk and adult patients can be treated with additional drugs. ALL
patients of T-cell phenotype (which are frequently included in the high risk group) also
receive treatment with cyclophosphamide. With the current protocols remission is
achieved in 99% of children and 93% of adult ALL patients (21). When normal
hematopoiesis is restored, the intensification phase aims to eradicate the drug-resistant
leukemic cells (and/or leukemic stem cells). There is still no consensus regarding the
duration and the drugs used in the intensification/consolidation-phase. Nonetheless, the
common protocols include the use of several drugs in combination – for example,
combination of methotrexate and 6-mercaptopurine; L-asparginase; dexamethasone;
vincristine; doxorubicin; the combination of thioguanine and cyclophosphamide and
also an epipodophyllotoxin and cytarabine. Stem cell or bone marrow transplantation is
also an option of treatment, but it is usually only applied to high-risk ALL patients that
include patients with the t(9;22)(q34;q11) translocation and cases with initial poor
response to treatment (20).
The remarkable advances in the efficacy of the treatment of ALL are somewhat
hampered by the realization that the intensity of the chemotherapeutic strategies used to
guarantee success are associated with severe long-term side-effects, including
osteonecrosis (22), decreased bone mineral density (23), thrombocytic complications
(24) and cognitive impairment (25). Currently, efforts are still being made to develop
new therapeutic strategies to treat the incurable cases of ALL and to diminish the severe
side effects associated with the intensive treatments regiments.
T-cell Acute Lymphoblastic Leukemia (T-ALL)
Approximately 15% of pediatric ALL patients present with a T-cell phenotype
cases, and in adults the percentage increases up to 25% (15, 26). T-cell acute
lymphoblastic leukemia (T-ALL) is a highly aggressive malignancy (13), and
historically, T-ALL was associated with a poorer prognosis. However, intensive and
risk adjusted chemotherapy led to improved outcome in this disease (27-29). Currently,

Introduction
7
the overall survival rate of T-ALL cases is about 80% in childhood T-ALL and 40-60%
in the adult cases (30).
As described for ALL in general, the main molecular mechanisms behind the
origin of T-ALL are still largely unknown. It is currently accepted that T-cell
leukemogenesis is a stepwise process that culminates in the acquisition of a fully
malignant phenotype. These events include defects in the control of cell cycle
machinery, NOTCH1 mutations that confer self renewal capacity to thymic progenitors,
deregulated expression of pivotal transcription factors and also aberrant activation of
protein kinases (14).
Normal T-cell development
Whatever the exact molecular mechanisms that trigger T-ALL, the malignant
clones originate from precursor cells arrested at a certain T-cell developmental stage.
Thus, to better understand the „framework‟ in which T-ALL occurs, we will briefly
characterize the normal T-cell developmental process.
T-cell development occurs in specialized lymphoid organs. It starts in the bone
marrow and culminates in the thymus, where most of the differentiation occurs (Figure
1.1). The hematopoietic progenitor that gives rise to T-cells arises in the bone marrow
and expresses the CD34 surface marker, indicating that it still maintains some
hematopoietic stem cell (HSC) properties. In fact, evidence suggests that this progenitor
is a Common Lymphoid Progenitor (CLP) with the potential to become a B or a T-cell.
The CLP subsequently migrates to the thymus, the organ where mature T-cells are
produced (31, 32). The commitment to the T-cell lineage depends on the signals derived
from the thymic microenvironment where the Notch signaling has been shown to play
an important role (33-35). During this stage, the early T-cell progenitors also rely on
microenvironmental cues, most notably IL-7, to survive and proliferate (36). After some
rounds of division these early thymocytes start to rearrange the genes that code for the
T-cell receptor (TCR) chains. The TCR is a transmembrane heterodimer composed of
two chains, α and β chains in the αβ T-cell lineage, while T-cells from the δ lineage
contain and δ chains.

Introduction
8

Introduction
9
The δ T-cells are a peculiar and minor subpopulation of T-cells that are important
effectors of innate immunity (37, 38), whereas most T-cells display an αβ TCR and are
involved in adaptive immunity. Each αβ TCR is unique and only recognizes a specific
antigen that is presented by Major Histocompatibility Complex (MHC) molecules (14).
The genomic regions that contain the coding sequence for the different TCR
chains are organized in gene clusters that contain variable (V), diversity (D), joining (J)
and constant (C) gene segments. During T-cell differentiation the RAG1 and RAG2
enzymes are responsible for rearranging the different gene segments to produce a
functional TCR chain by random choice of the V, D and J gene segments. These
recombination events are highly regulated during T-cell differentiation and take place
during specific maturation stages (39). Notably, deregulation of these mechanisms has
been shown to account for the high frequency of translocations involving TCR loci in
T-ALL.
The first chains to be rearranged are the δ, and β (14, 31, 32). Concomitantly
with TCR β gene rearrangement, thymocytes up-regulate the expression of CD4 and
CD8 co-receptors while down-regulating the expression of CD34 (14, 32). Productive
Figure 1.1. Human T-cell differentiation. The majority of T-cell development processes
occur in the thymus. A common lymphoid progenitor with a CD34+ CD1a
- phenotype
migrates from the bone marrow to colonize the thymus. In the initial stage of
differentiation, the thymocyte precursors lack the expression of CD4 and CD8 markers and
are thus called double negative precursors (DN). DN thymocytes receive signals from the
thymic microenvironment to proliferate, particularly from the IL-7 cytokine. The
thymocytes begin the recombination of the genes encoding TCR , δ and β chains. The δ
lineage originates at this stage (Pre-T1). Thymocytes first acquire the expression of CD4
(intermediate single positive, ISP) and start the down-regulation of the CD34 marker, while
subsequently upregulating CD8 to become CD4+CD8
+ double positive (DP) precursors. At
the DP stage the thymocytes assemble the pre-TCR. The signals derived from this receptor
expand this thymocyte population dramatically. The rearrangement of TCR α chain
eventually produces a functional TCR at the cellular membrane. Thymocytes interact with
thymic epithelial cells (TECs) to select functional and competent T-cells. Upon the
selection process, thymocytes keep the expression of either CD4 or CD8, becoming single
positive cells (SP) and move to the peripheral lymphoid organs. Further details are
described in the text.

Introduction
10
rearrangement of the TCRβ gene results in the surface expression of a β-chain that
assembles at the cellular membrane with an invariant pTα chain and CD3 to form the
pre-TCR (40). The formation of a functional pre-TCR complex activates signaling
pathways that promote survival and proliferation of the developing T-cells (41), and
ultimately triggers the rearrangements in the TCRα gene locus, also inhibiting further
rearrangements at the β chain locus (14). The thymocytes with functional αβ TCR
undergo positive and negative selection processes that ultimely result in a pool of T-
cells that only react to foreign antigens presented by MHC molecules. Thymocytes with
an αβ TCR that recognizes with low affinity self antigens presented at MHC molecules
by thymic epithelial cells (TECs) are positively selected and escape apoptosis while
those that do not detect any antigen die by neglect and those with a high affinity are
eliminated by clonal delection (negative selection) to avoid self-reactivity (14). During
the process of positive selection, thymocytes that express both CD4 and CD8 co-
receptors are selected into single positive T-cells that express either CD4 (which
recognizes antigens presented by MHC class II molecules) or CD8 (which recognizes
antigens presented by MHC class I). Functionally mature single-positive thymocytes
subsequently migrate to peripheral lymphoid tissues and, upon recognition of
appropriate antigens, lead to the activation of CD4 helper T-cell and CD8 cytotoxic T-
cell responses (14, 31).
T-ALL Immunophenotypical Classification
Several classifications based on the thymocyte developmental stage at which the
leukemic cells are supposedly blocked have been proposed to divide T-ALL into
discrete immunophenotyic subgroups. In this thesis we adopted the classification by the
European Group for the Immunological Characterization of Leukemias (EGIL) (42),
which is arguably the most generally accepted. This classification divides T-ALL into
four groups according to the following criteria: pro-T-ALL (CD7+ only), pre-T-ALL
(CD7+, CD1
-, CD2 and/or CD5 and/or CD8
+, CD3
-), cortical T-ALL (CD1
+,
independently of the presence of other markers) and mature T-ALL (CD1-, CD3
+) (42).
Extracellular factors and Microenvironment
Cancer cells are not isolated, cells from both solid cancers and leukemias interact
with the surrounding environment. The microenvironment contributes to the proper

Introduction
11
development of the tissues by providing adequate signals to the developing cells,
including proliferation, survival or apoptotic and differentiation signals. The malignant
cells also perceive these signals but, due to their intrinsic proliferative and anti-
apoptotic capacities, do so in a manner that provides a competitive advantage over their
normal counterparts. Moreover, cancer cells can subvert the microenvironment to
produce factors that positively stimulate their growth.
T-ALL arises from the malignant transformation of lymphoid precursors that are
developing in the bone marrow and in the thymus (13). Both of these hematopoietic
niches have a clear role in the supporting T-ALL cells. Stromal support derived from
the BM (43) and the thymus (44) increase the survival and proliferation of T-ALL cells.
Interestingly, it was shown that the in vitro recovery of primary leukemic blasts cultured
with stromal support can predict treatment outcome in the T-ALL patients (43). The
increased survival of T-ALL blasts in the BM stroma is dependent, at least in part, on
the engagement of adhesion molecules and integrins like LFA-1 and ICAM-1(45).
These adhesion molecules were also implicated in resistance to drug-induced apoptosis
in multiple myeloma (46), raising the possibility that the same could occur in T-ALL
patients. Remarkably, the interaction between leukemic cells and specific areas in the
BM can provide a supportive niche that allows the development and proliferation of
malignant blasts (47) while disrupting the normal development of hematopoietic cells
(48).
Chemokines regulate hematopoietic development and lymphocyte biology.
Chemokines are small chemotactic proteins that regulate the homing of leukocytes to
the sites of inflammation, infection and also to the sites where development takes place
(49). Chemokines can, on the other hand, act as key regulators of the homing of cancer
cells to the place of metastasis (50). The chemokine SDF-1/CXCL12 and its receptor
CXCR4 are implicated in the metastatic process of several types of cancers, namely
acute leukemias, breast and lung (51). CXCR4 is expressed in several cancers (52),
including T-ALL (53-55). Interestingly, CXCR4 expression in Acute Myeloid
Leukemia is a prognostic factor associated with low survival rates (56). The SDF-1
/CXCR4 axis was shown to be responsible for the homing of leukemic cells (47) and
normal hematopoietic cells (57) to bone marrow niches, and binding of SDF-1 to
CXCR4 increases the chemotaxis of T-ALL cells (53). Other chemokines, such as
CXCL13 (58) and CCL25 (59), were shown to protect T-ALL cells from apoptosis.

Introduction
12
Furthermore, a recent report showed that the expression of CCR7 in leukemic cells was
essential for their homing to the CNS (60).
Cytokine signaling in T-ALL
Cytokines are another family of soluble proteins that regulate the development of
hematopoietic cells (61, 62). Similar to chemokines, cytokines are produced in the
lymphoid tissues where the hematopoietic cells develop. Cytokines include colony-
stimulating factors (CSFs), interleukins (ILs), interferons (IFNs) and other growth
factors(63). The binding of cytokines to their receptors engages a series of signaling
events that can result in increased proliferation, viability, differentiation and also
apoptosis of the target cells (62). Similar to normal hematopoietic cells, T-ALL blasts
can increase their viability, proliferation and maturation status in response to cytokine
signaling (64-67). Several cytokines were shown increase the proliferation of T-ALL
cells. Interleukin-2 (IL-2) was one of the first cytokines described to promote T-ALL
growth in vitro (64, 68, 69). Likewise, we and others established a clear role for
interleukin-7 (IL-7) in the biology of T-ALL (44, 65, 67, 70, 71). Scupoli and
colleagues demonstrated that IL-7 was the main effector of the T-ALL cell survival
induced by thymic microenviroment in vitro (44). Activation of IL7 signaling in T-ALL
cells in vitro inhibited spontaneous apoptosis through the up-regulation of BCL-2 (65,
67), but also increased the proliferation of the leukemic blasts through the down-
regulation of the cyclin-dependent kinase inhibitor p27Kip1
(65). In T-ALL cells, the PI3-
Kinase (PI3K) pathway is main effector of the IL-7-induced effects (70). In addition, we
showed that IL-4, IL-9 and IL-15 also induce T-ALL cell proliferation and that this
effect was dependent on the maturation status of the T-ALL cells (72). In contrast, other
cytokines can suppress the growth and induce apoptosis of leukemic cells. Interleukin-6
(IL-6) was shown to suppress the growth of T-ALL cells (73) and tumor necrosis factor
–α (TNF-α) was shown to induce apoptosis (74, 75).
The IL-4/IL-4 receptor signaling axis
IL-4 is a γ-common chain cytokine involved in the regulation of the host immune
response against helmintic pathogens (76). IL-4 is produced by several sources, which
include circulating leukocytes like mast cells, basophils and eosinophils (76, 77).

Introduction
13
Moreover, IL-4 expression was also detected in the bone marrow (78), indicating that it
can influence and regulate the development of lymphoid cells.
The IL-4 receptor complex is composed of two subunits, the IL-4 receptor α-chain
and the IL-2 Receptor γ-chain (79). The IL-4 receptor α-chain is a member of the type I
cytokine receptor superfamily (80). This family is characterized by the presence of four
positionally conserved cysteine residues and a conserved W-S-X-W-S motif (W-
tryptophane, S-serine and X-nonconserved aminoacid) in the extracellular regions (81).
In adition, the cytoplasmic tail contains a short conserved aminoacid sequence termed
Box 1. The IL-4 receptor α-chain also contains a region named insulin-IL-4 receptor
motif (IR4) that is responsible for cellular proliferation (82). The IL-2 Receptor γ-chain
also belongs to the type I cytokine receptor superfamily (83). This subunit is shared by
the heteromeric receptor complexes for IL-2 (83), IL-4 (79), IL-7 (84), IL-9 (85) and
also IL-15 (86).
Binding of the IL-4 to its receptor induces heterodimerization of the receptor
complex leading to its phosphorylation by the Janus kinases JAK1 and JAK3 (87).
Receptor phosphorylation by these kinases creates docking sites in the receptor
cytoplasmic tail that lead to the activation of downstream targets. In CD8+ T
lymphocytes, IL-4 stimulation activates several STAT (signal transducer and activator
of transcription) members that included STAT1, STAT3, STAT5 and STAT6 (88).
Furthermore, the PI3K and its downstream targets Protein Kinase B (PKB)/Akt and
p70S6K
were also shown to be activated upon IL-4 stimulation of primary lymphocytes
(88, 89).
IL-4 stimulation was shown to induce the proliferation of primary lymphocytes
(89), as well as pancreatic (90) and prostatic (91) cancer cells. Moreover, IL-4 signaling
also protects a B-cell lymphoma cell line from apoptosis, via PI3K-PKB/Akt pathway
activation (92). Similarly, IL-4 appears to have anti-apoptotic effects on colon cancer
cell lines (93). In contrast, and somewhat surprisingly, it has also been shown that IL-4
can induce apoptosis of B-cell Acute Lymphoblastic Leukemia (B-ALL) patient cells
(94). In T-ALL, IL-4 appears to be mostly pro-tumoral. IL-4 stimulation promotes the
proliferation of T-ALL patient samples (67, 72) and we showed that this effect is
dependent on the maturation stage of the T-ALL blasts (72).

Introduction
14
Genetic abnormalities in T-ALL
T-ALL is characterized by several genetic alterations that disturb normal
hematopoietic development and lead to the malignant transformation. These alterations
occur in genes that control diverse cellular processes, and include point mutations, gene
fusions, translocations, inversions and deletions (14, 95).
Reciprocal translocations between the T-cell receptor (TCR) chain genes (α, β and
δ) and several T-cell oncogenes are quite frequent occurring in up to 35% of the T-ALL
cases (96). The majority of these translocations lead to the aberrant expression of
transcriptional regulators resulting in impaired differentiation and loss of cellular
homeostasis of the developing thymocytes (14, 95). Furthermore, it was demonstrated
that more than 50% of the T-ALL patients display activating mutations in the NOTCH1
gene resulting in increased Notch signaling (97), which is highly associated with T-cell
malignancies (98, 99).
Cell cycle defects
Loss of cell cycle control is a common feature in cancers. Mutations affecting
genes that control cell cycle transitions and DNA damage response, such as RB1 (100)
and TP53 (101), are frequent in cancer. However, mutations affecting these genes are
rare in T-ALL, whereas deletions in the INK4/ARF locus (del 9p21), affecting the
CDKN2A/INK4A and CDKN2B/INK4B genes, are extremely frequent (102-105). The
CDKN2A gene encodes for the p14/p19 and p16 proteins, while the CDKN2B gene
encodes the p15 protein. The p15 and the p16 proteins are inhibitors of cyclin D-CDK4
complexes, thereby maintaining the cells in a quiescent state. The inactivation of these
CDK inhibitors leads to the activation of cyclin D-CDK4 complexes and consequent
phosphorylation of the retinoblastoma protein allowing cells to enter and progress in the
cell cycle. In addition, the INK4/ARF locus can encode for a p14/p19ARF
protein
resulting from transcription of an alternative reading frame (ARF). .p14/p19ARF
interacts
and sequesters the MDM2 protein, which is the negative regulator of the p53 protein,
leading to its up-regulation. The inactivation of the p14/p19ARF
down-regulates p53
protein, which is a critical cell cycle and genotoxic stress response regulator in the cell
(106, 107). Recently, it was also described that the cyclin D2 gene (CCND2), a key
regulator of the G1 to S phase transition, is ectopically expressed in T-ALL patients

Introduction
15
samples as a result of chromosomal translocations that juxtapose this gene to the
regulatory sequences of the TCRA/D or the TCRB loci (108).
Aberrant signaling
Genetic lesions not only lead to aberrant transcriptional activation in T-ALL but
also to aberrant activation of signaling pathways that are crucial for the proliferation and
viability of the leukemic blasts (109).
Tyrosine kinases play a critical role in the signaling events upon TCR
engagement, and are critical in the regulation of the T-cell viability, proliferation and
immune responses. Several of these kinases are aberrantly activated in T-ALL, either by
chromosomal translocations that activate the expression of the kinases or creation of
chimeric fusion genes that code for new proteins with enhanced kinase activity and de-
regulated expression. However, point mutations in key molecules and deletions of
negative regulators are also observed in T-ALL patients (14, 95).
The ABL1 gene codes for a ubiquitously expressed cytoplasmic tyrosine kinase
that plays a role in TCR signaling (110, 111). The t(9;22)(q43;q11) translocation that
creates the BCR-ABL fusion protein is highly common in CML and in B-ALL (15,
112).However, its frequency is very rare in T-ALL (113). In contrast, the NUP214-
ABL1 is found in 6% of the patients as a result of episomal amplification. This gene
fusion is also associated with the expression of other oncogenes, such as HOX11 and
HOX11L2, as well as with the deletion of the CDKN2A locus (114). Other fusions
affecting the ABL1 gene identified in T-ALL are relatively rare, these include the ETV6-
ABL1 gene fusion (115) and the EML1-ABL1 gene fusion that was detected in a single
T-ALL patient (116).
The LCK kinase is a member of SRC family of tyrosine kinases highly expressed
in T-cells which plays a central role in delivering the signals that emanate from TCR-
signaling (117). In rare cases of T-ALL, LCK was found to be ectopically activated as a
result of the t(1;7)(p34;q34) translocation that places the LCK gene under the control of
the TCRB locus (118, 119).
The conserved family of the JAK kinases participates in the coupling of cytokine
receptors to intracellular signaling events thereby regulating important biological
processes such as apoptosis, differentiation, proliferation and immune responses (120).
In T-ALL, the translocation t(9;12)(p24;p13) results in the ETV6-JAK2 gene fusion

Introduction
16
resulting in constitutively active JAK2 signaling (121, 122). The leukemogenic effect of
this gene fusion was demonstrated in a transgenic mouse model that resulted in the
development of fatal leukemia with a selective expansion of CD8+ T-cells (123).
Recently, the occurrence of mutations in the JAK1 gene was reported in 18% of adult
and 2% of pediatric patients with T-ALL. These mutations are associated with poor
response to treatment and overall survival (124).
The FLT3 gene encodes for a receptor tyrosine kinase that is crucial for the
development of hematopoietic stem cells (125). Activating mutations in this gene are
frequent in Acute Myeloid Leukemia (AML) (125), but in contrast, are rare in T-ALL
patients and are restricted to lymphoblasts with a very early phenotype that still
maintain the expression of LYL1, LMO2 and also the KIT receptor (126, 127). The
FLT3 mutations that occur in AML and T-ALL patients are, in both instances, internal
tandem duplications in the juxtamembrane domain or point mutations in the activation
loop of the kinase domain that lead to the activation of the receptor in the absence of the
ligand (125, 126).
The RAS family (N-RAS, K-RAS and H-RAS) of small GTPases is critical to the
transmission of numerous stimuli from the membrane and their integration into
downstream signaling pathways (128). Activating mutations in the genes that code for
these proteins are described in several types of malignancies (129, 130). In T-ALL,
activating mutations in RAS are described in up to 10% of the patients (131-133).
Moreover, in 2% of the T-ALL patients, inactivating mutations were found in NF1, a
negative regulator of the RAS pathway (133). However, there is evidence that RAS
protein activation may occur in around half of the T-ALL patients (134), raising the
possibility that RAS activation (possibly due to stimulation by extracellular cues in the
absence of RAS gene lesions) may play a central role in the pathogenesis of T-ALL.
The Transforming Growth Factor-β (TGF-β) signaling pathway regulates cell
growth, senescence, differentiation and apoptosis (135). Several lines of evidence
implicate TGF-β signaling pathway in cancer, either as a tumor suppressor or tumor
promoter (136). Upon activation of the pathway, the main cytoplasmatic adaptors,
SMAD2 and SMAD3, are translocated to the nucleus where they regulate gene
transcription (137). The link between the TGF-β pathway and T-cell malignancy was
realized not long ago (138, 139). Lucas and colleagues described that decreased TGF-β
signaling in a mouse model results in the expansion of CD8+ memory T-cells leading to
the establishment of T-cell leukemia (138). Moreover, it was demonstrated that T-ALL

Introduction
17
patients display loss of SMAD3 protein in the absence of MADH3 (which encondes for
SMAD3) gene mutations (139). The loss of SMAD3 protein synergized with other
oncogenic events, particularly with the loss of p27Kip1
, to induce T-cell leukemia in
mice (139). The hypothesis that the TGF-β signaling pathway acts to suppress T-cell
leukemogenesis is further supported by the high percentage of T-ALL patients (34%)
that display genomic alterations in members of this pathway (140). These genomic
alterations include deletions in the activators and amplifications in the inhibitors of the
pathway (140).
Recently, LEF1/TCF1, a member of WNT signaling pathway, was shown to be
deleted in a variety of T-ALL patients (141). LEF1 interacts with β-Catenin to promote
gene transcription (142), but also with SMAD4 a pivotal mediator of TGF-β signaling
(143). In T-ALL, Gutierrez and colleages showed that the LEF1 gene is deleted in 11%
of the samples analyzed and identified non-synonymous mutations that produce
premature stop codons in 7% of the cases analyzed. Importantly, LEF1 inactivation in
T-ALL correlates with increased MYC expression, NOTCH1 activating mutations and
early cortical stage of T-cell differentiation (141).
The Notch signaling is critical for the regulation of cell fate decisions in stem cell
maintenance, neurogenesis and T-cell differentiation (35, 144, 145). The Notch
receptors are heterodimeric proteins composed of an extracellular subunit and a
transmembrane subunit that are non-covalently bound through a heterodimerization
domain (109, 146). Activation of the Notch signaling pathway occurs upon binding of
the ligand to the extracellular subunit of the Notch receptor, resulting in serial
proteolytic cleavages. The final cleavage is catalyzed by the γ-secretase complex that
releases the intracellular Notch (ICN) receptor, which activates the transcription of
Notch target genes (109, 146). The involvement of the Notch receptor in T-ALL was
first found in three patients that presented the t(7;9)(q43;q34.4) translocation that
juxtaposed the region that codes for the intracellular NOTCH1 gene (TAN1) to the
regulatory sequence of the TCRB locus (147). Notably, transplantation of bone marrow
transduced with the TAN1 gene into recipient mice led to the development of T-cell
leukemia (98). Recently, it was discovered that the majority of the T-ALL patients
(56%) display NOTCH1 activating mutations. The mutations were found in two distinct
regions of the NOTCH1 gene, in the heterodimerization domain (44%) and in the PEST
domain located in the C-terminus (30%), with a significant percentage of the patients
(17%) displaying mutations in both regions. Increased Notch signaling resulted from

Introduction
18
either the destabilization of the heterodimerization domain or from increased ICN half-
life. Importantly, patients with both types of mutations have synergistic activation of the
Notch signaling pathway (97). Furthermore, another type of NOTCH1 activating
mutations were described by Sulis et al, these mutations are internal duplication
insertions occurring at the vicinity of the exon 28 of the NOTCH1 allele that encode the
extracellular juxtamembrane region of the receptor. These insertations lead to aberrant
NOTCH1 signaling by promoting the final cleavage of the receptor by the γ-secretase
complex. Interstingly, the level of aberrant NOTCH1 signaling depends on the number
of aminoacid residues introduced in the juxtamembrane region of the receptor (148).
The NOTCH1 activating mutations occur in the all the major molecular subtypes of the
T-ALL patients (TAL1+; LYL1
+; HOX11
+; MLL-ENL
+; CALM-AF10
+), which may
indicate that they occur very early in T-cell differentiation (97, 149).
The PI3K-mTOR pathway
The signal transduction pathway involving Phospho-Inositol-3 Kinase (PI3K) and
the mammalian Target of Rapamycin (mTOR) regulates cellular processes such as
viability, proliferation, and differentiation, and have been extensively associated with
hematological malignancies (150-152). In this section, we will briefly describe this
pathway, and discuss the evidence for its involvement in the pathogenesis of T-ALL.
An overview
Surface receptors frequently activate the PI3K-mTOR pathway (Figure 1.2). The
PI3K complex consists of the p110 catalytic subunit and the p85 regulatory subunit. The
activation of this complex leads to the phosphorylation of the Phosphatidyl-Inositol 4,5-
Bisphosphate (PIP2) lipid creating Phosphatidyl-Inositol 3,4,5-Trisphosphate (PIP3).
The PTEN (Phosphatase and Tensin Homologue) tumor suppressor catalyzes the
inverse reaction. Upon formation of PIP3, the PI3K downstream targets Protein Kinase
B (PKB) and PDK1 are recruited to the plasma membrane through their Plecstrin
Homology (PH) domains that anchor them to the PIP3. The PDK1 kinase
phosphorylates PKB in the Thr308 residue. The full activation of PKB is achived upon
phosphorylation in the Ser473 residue by PDK2, which was recently demonstrated to be
the mTORC2 complex (150, 151, 153).

Introduction
19
Activated PKB is known to phosphorylate a variety of cellular targets (154, 155).
PKB downstream targets include the FOXO family of transcription factors (156, 157),
Figure 1.2. An overview of the PI3K-mTOR signaling pathway. Growth factors and
cytokines bind to surface receptor leading to PI3K-mediated signaling. The activation of PI3K
and subsequent phosphorylation of PIP2 to PIP3 are key steps in this signaling pathway.
Activation of PI3K-mTOR results in increased viability, proliferation of cancer cells. Further
details can be found in the main text.

Introduction
20
the GSK-3α/β kinase (158), the pro-apoptotic protein BAD (159, 160) and TSC2, the
negative regulator of the mTORC1 complex (161). FOXOs are critical in the negative
regulation of cell cycle progression and in promoting the apoptotic process (162). The
genes regulated by this family of transcription factors include CDN1A (p21CIP1
),
CDN1B (p27KIP1
), FASL and BIM (163). PKB phosphorylates the FOXO3a transcription
factor in three conserved residues Ser32, Ser215 and Ser315 leading to its inactivation
and subsequent proteasomal degradation (163).
PKB activates mTOR downstream signaling by direct phosphorylation of mTOR
kinase in the Ser 2448 residue (151) and by phosphorylation and inactivation of the
TSC2/TSC1 complex, the negative regulator of the mTORC1 complex (161). The TSC2
protein is phoshorylated by PKB which disrupts the TSC1/TSC2 complex. As a result
the small GTPase Rheb activates the mTORC1 complex (164). The mTORC1 complex
is composed of mTOR, Raptor, PRAS40 and LSt8 proteins and is sensitive to inhibition
by Rapamycin (150, 151, 153). The mTORC1 complex increases protein translation by
directly regulating the p70S6K
kinase and 4EBP1 (165). p70S6K
is activated by direct
phosphorylation mediated by mTORC1 (166, 167). Subsequently, p70S6K
leads to
increased protein translation by phosphorylating the ribosomal protein S6 (168) and
PDCD4 (169). These events result in active translation of 5‟CAP mRNAs (153). In
contrast, the phosphorylation of 4EBP1 by mTORC1 inhibits its activity. Upon
inhibition, 4EBP1 dissociates from eIF4E releasing it to participate in the assembly of
the translation initiation complex (150, 151, 153). Interestingly, it was demonstrated
that the mTORC1 complex in association with p70S6K
kinase also inhibits PI3K
signaling by creating a negative feedback loop to shutdown signaling (170). mTOR is
also a component of the mTORC2 protein complex, which also contains Rictor, SIN1
and mLSt8 (150, 151, 153). This complex is insensitive to Rapamycin and responsible
for the phosphorylation of the PKB kinase in the Ser473 residue, as described above
(165, 171).
Importantly, the PI3K-mTOR axis controls several steps in protein translation and
enhances the production of several oncogenic proteins that include c-Myc, cyclin D1
and Rb. By regulating protein translation, the PI3K-mTOR pathway controls cell
growth and cellular metabolism, which are important processes for tumorigenesis (172).

Introduction
21
Importance in T-ALL
Despite the fact that the PI3K-mTOR pathway and its downstream target PKB are
implicated in several malignancies, until recently very few abnormalities were detected
in this signaling pathway in T-ALL.
Until recently, the notion that the PI3K pathway was implicated in T-ALL came
essentially from the knowledge that T-ALL PTEN-null cell lines exist that display
uncontrolled proliferation (173, 174), and restoration of PTEN activity (175) or
pharmacological blockade of the PI3K pathway (176) resulted in apoptosis of these cell
lines. Avellino and colleagues further extended these analyses to a few T-ALL patient
samples and showed that the PI3K-mTOR axis was crucial for maintenance of viability
of the T-ALL blasts. The incubation with Rapamycin (mTOR inhibitor) and
Wortmannin (PI3K inhibitor) stimulated the apoptosis of the T-ALL samples (177).
Importantly, we subsequently demonstrated that aberrant PI3K signaling is a very
frequent alteration in T-ALL, with 87.5% of the patients tested presenting
hyperactivation of this pathway when compared to normal controls (178). PI3K
pathway constitutive hyperactivation resulted from inactivation of PTEN, by PTEN
gene alterations (present at relatively low frequency) and by PTEN protein inactivation
due to CK2-mediated C-terminal phosphorylation and ROS-dependent oxidation at the
catalytic centre (178). Recently, two additional reports confirmed the relevance of PI3K
signaling in T-ALL, describing genomic abnormalities in members of the PI3K pathway
or in predicted upstream regulators of the pathway in around 40% of the patients
analyzed (133, 140). Importantly, T-ALL patients with mutations in the PTEN gene are
associated with a poorer prognosis than patients that present an intact PTEN locus (133,
179).
Transcription factors
Transcriptional deregulation is a common feature in Acute Leukemias. Unlike
AML and B-ALL, where the formation of chimeric fusion proteins is common, in T-
ALL the most common feature is the aberrant expression of full length transcription
factors and other proteins (180). Recurrent chromosomal translocations are responsible
for the aberrant expression of transcription factors and other key proteins in
differentiating thymocytes altering their transcriptional program (13, 14, 95). The Table
1.1 summarizes the most common chromosomal translocations associated with T-ALL.

Introduction
22
The Homeobox (HOX) genes are a class of transcription factors that are pivotal in
several differentiation processes – including fly patterning (181), axial morphogenesis
(182), neural development (183) and also hematopoiesis (184). These transcription
factors contain a characteristic homeodomain motif of 61 aminoacids that is responsible
for the binding to the DNA (185) and are divided in two classes. The HOX genes that
belong to the class I comprise a complex network of transcriptional regulators that are
organized in four clusters, HOXA-D. A chromosomal inversion [inv(7)(p15q34)] was
reported in 5% of the T-ALL patients that results in the aberrant expression of the
HOXA10 and HOXA11 genes, members of the HOXA gene cluster (186). Another
member of the HOXA gene cluster, the HOXA13 gene was reported to be highly
expressed in T-ALL patients as a result of a complex tranlocation that places it under
the control of the BCL11B locus (187).
The HOX11 (TLX1) and HOX11L2 (TLX3) genes belong to the class II of HOX
genes and have been extensively associated with T-ALL. HOX11 ectopic expression is
mainly due to two chromosomal translocations (see Table 1.1), the t(10;14)(q24;q11)
and the rare variant t(7;10)(q35;q24) that juxtapose the coding sequence of this gene to
the strong regulatory sequences of the TCRA/D and TCRB locus, respectively (188,
189). Furthermore, HOX11 aberrant expression can also occur in the absence of these
chromosomal translocations (149, 190, 191). The HOX11 gene is not normally
T-cell receptor gene Chromosome
location Partner gene
Chromosome
location
T-cell receptor α/δ (TCRA/D) 14q11
HOX11 10q24
TAL1 1p32
LMO1 11p15
LMO2 11p13
CCND2 12p13.3
T-cell receptor β (TCRB) 7q34-35
HOX11 10q24
HOXA cluster 7p15
LYL1 19p13
TAL2 9q32
LCK 1p34
NOTCH1 9q34
CCND2 12p13.3
T-cell receptor У (TCRG) 7p15 no known chromosomal translocations
Table 1.1. T-cell receptor genes and their involvement in the chromosomal translocations in
T-ALL.

Introduction
23
expressed in T-cells (192) and its ectopic expression in hematopoietic precursors and
thymocytes is oncogenic (188, 193). The expression of HOX11 in T-ALL is associated
with an early cortical phenotype (CD3low
/CD4+/CD8
+) and a good prognosis (149, 194).
HOX11 expression in T-ALL correlates with the expression of genes associated with
increased cell cycle progression and proliferation (149, 195). The HOX11L2 gene is
also associated with T-ALL (149) and its expression is detected in up 20% of childhood
T-ALL and 13% in adult cases (14). Ectopic HOX11L2 expression in T-ALL is mainly
due to the cryptic translocation t(5;14)(q53;q32). This translocation places the
HOX11L2 gene under the control of the BCL11B gene, which is highly expressed
during T-cell development (196, 197). Other translocations, namely t(5;14)(q53;q11)
and t(5;7)(q35;q21), also explain the aberrant expression of HOX11L2 in T-ALL (198,
199). Despite the fact that the expression profiles of T-ALL patients with HOX11L2 and
HOX11 aberrant activation share similarities (149), patients with HOX11L2 expression
are associated with a poorer prognosis than patients that express the HOX11 gene (149,
200).
The basic Helix-Loop-Helix (bHLH) family of transcription factors is
characterized by a Helix-Loop-Helix (HLH) motif of 60 aminoacids used to dimerize
with other bHLH transcription factors and by binding to DNA via a basic region N-
terminal to this motif (201). This family is divided in two main classes. The class I (or
A) bHLH transcription factors are ubiquitously expressed and form homo- or
heterodimers. The class II (or B) bHLH transcription factors are expressed in a tissue
specific manner and only form heterodimers with class I bHLH proteins. The E2A gene
codes for two proteins by alternative splicing, E47 and E12, and is the classical class I
bHLH transcription factor. The E2A gene plays a critical role in T-cell differentiation
(202-204). E2A proteins have been shown to activate V(D)J recombination (205) and
regulate the expression of several genes crucial of T-cell differentiation, including the
surrogate pTα chain (206, 207). Importantly, E2A acts as a tumor suppressor gene in T-
cells since down-regulation of E2A expression in T-cells leads to leukemogenesis (208,
209). The class II (or B) bHLH transcription factors were shown to be highly associated
with T-ALL (149, 210-212). The LYL1, TAL1 (its involvement in T-ALL will be
discussed later) and the closely related TAL2 and BHLH1 genes were identified due to
the involvement in recurrent chromosomal translocations in T-ALL (211-215). The
LYL1 gene is down-regulated during T-cell differentiation (190) and is aberrantly
expressed in T-ALL as a result of the chromosomal translocation t(7;19)(q35;p13) (213)

Introduction
24
and other unknown mechanisms (149, 190). The gene expression profile of T-ALL
patients with ectopic LYL1 gene activation correlates with the profile of early T-cell
precursors and its associated with a poor prognosis (190). The TAL2 and BHLH1 genes
are expressed in T-ALL as result of the chromosomal translocations t(7;9)(q34;q32) and
t(14;11)(q11;q22), respectively (211, 212).
The genes that code for the LIM-domain only family of proteins are also
ectopically expressed in T-ALL patients (149). These proteins contain cystiene-rich
motifs in the LIM domains, which are responsible for protein-protein interactions (216),
and act as bridging molecules assembling complexes of transcription factors that
include GATA1, TAL1 and E2A (216, 217). The LMO1 and LMO2 genes are activated
in T-ALL as a result of the chromosomal translocations t(11;14)(p15;q11) and
t(11;14)(p15;q13) that juxtapose the coding regions of these genes to the regulatory
regions of the TCRA and TCRD loci, respectively (218, 219). Ectopic activation of these
genes without known genetic alterations can also occur in T-ALL (149). Recently it was
shown that the deletion del(11)(p12p13) also accounts for LMO2 gene expression in T-
ALL (220). The LMO (LMO1 and LMO2) genes are down-regulated when normal
hematopoietic precursors commit to the lymphoid lineage (190, 221). In contrast, their
expression is detected in up to 45% of the T-ALL patients and is frequently associated
with other oncogenes like the TAL1 and LYL1 (149). Furthermore, LMO1 and LMO2
transgenic mice develop T-cell leukemia (222-225) and synergistically cooperate with
the TAL1 gene (226-228). Recently, it was described that during a gene therapy trial to
correct severe combined immunodeficiency (SCID), several patients developed T-cell
lymphoproliferative disorders similar to T-ALL. The retrovirus that carried the IL2RG
gene integrated near the LMO2 locus leading to its aberrant expression in the
hematopoietic precursors of the transplanted patients (229-233).
The MLL gene (Mixed-lineage leukemia) is known to rearrange with more than 50
partners in lymphoid and in myeloid leukemias (234). The MLL gene is a transcription
regulator functionally homologous to the Drosophila Trithorax gene and regulates the
expression of HOX genes (235, 236). In T-ALL, fusions involving the MLL gene are
described in up to 8% of the patients (237-239). The usual partner in these fusions is the
ENL (MLLT1) gene as a result of the translocation t(11;19)(q23;p13.3) (240). The other
fusion partners of the MLL gene are the AF10, AF6, AF4 and AFX1 genes (14). Gene
expression profile of T-ALL patients with MLL gene fusions shows up-regulated
expression of several HOX genes, such as HOXA10, HOXA9 and HOXC6 and also

Introduction
25
MES1, a HOX gene co-regulator (238). Furthermore, T-ALL patients with MLL gene
fusions represent a clear distinct molecular subtype associated with a specific gene
signature (HOX gene activation) (238).
The translocation t(10;11)(p13;q14) is recurrent in T-ALL, and as a result, the
CALM (Clathrin Assembly protein-like Lymphoid-Myeloid gene, or PICALM) gene is
fused to the AF10 gene (Acute Lymphoblastic Leukemia 1 fused gene on chromosome
10, or MLLT10) (241). The CALM-AF10 fusion occurs in up to 10% of the T-ALL
patients with the γδ phenotype (242) and is associated with poor prognosis (243). T-
ALL patients with the CALM-AF10 fusion protein express a subset of HOX genes that
include HOXA5, HOXA9 and HOXA10 and also the BMI1 oncogene (244).
Interestingly, the MLL fusions and the CALM-AF10 fusion seem to share a common
feature, the up-regulation of several HOX genes, particularly members of the HOXA
gene cluster.
The TAL1/SCL oncogene
TAL1 (T-cell Acute Lymphocytic protein 1) aberrant gene expression is the most
common genetic alteration in T-ALL being detected in up to 65% of the patients (149,
210). T-ALL patients with TAL1 aberrant expression are generally associated with poor
prognosis and with a late cortical phenotype (CD3+CD4
+CD8
+) (149). However, the
first TAL1 positive cases described in the literature presented with early T-cell
phenotype (CD2+CD7
+CD3
-CD4
-CD8
-) (245). TAL1 aberrant gene expression in T-
ALL patients occurs due to several chromosomal aberrations (Table 1.2) such as
translocations and deletions (214, 215, 246-249).
Type Designation Incidence Partner gene
translocation t(1;14)(p32;q11) 3% T-cell receptor δ (TCRA/D)
translocation t(1;7)(p32;q35) n.d. T-cell receptor β (TCRB)
translocation t(1;3)(p32;p21 n.d. TCT1A
translocation t(1;5)(p32;q31) n.d. Unknown
deletion/fusion 1p32 (TAL1d) 26% SIL
Table 1.2. Chromosomal aberrations involving the TAL1 gene in T-ALL.

Introduction
26
Moreover, there is a high number of T-ALL patients with detectable TAL1 gene
expression without rearrangements in the respective locus. The precise molecular
mechanisms leading to ectopic TAL1 expression in these cases remain unknown (190).
The most common translocation t(1;14)(p32;q11) juxtaposes the TAL1 gene to the
strong regulatory elements that control the expression of the TCR δ chain (214) and is
detected in up to 3% of the T-ALL cases.
The other translocations (Table 1.2) are rare and are documented as case-study
reports (247-249). However, the most common chromosomal alteration leading to
aberrant TAL1 gene expression in T-ALL is the SIL-TAL1 gene fusion (246) that is
detected in up to 26% of the T-ALL patients. This gene fusion results from a micro-
deletion of 90 kb in the TAL1 locus removing the regulatory elements that control the
TAL1 gene expression and fusing the TAL1 coding region to the SIL regulatory elements
(Figure 1.3) (246). Although this gene fusion gives rise to three different
rearrangements, the end result at the protein level is always the same (Figure 1.3) (246).
The clear involvement of TAL1 in the pathology of T-ALL is established by the
fact that transgenic mice expressing this oncogene develop T-cell leukemia, although
with a long latency period (250-252). This fact could suggest that an additional
oncogenic event should occur for the establishment of full blown leukemia. Obvious
candidates for the second hit in TAL1-induced leukemogenesis are the LMO genes. The
expression of LMO genes cooperates with TAL1 to accelerate the onset of T-cell
leukemia in transgenic mice (226-228, 253). Importantly, LMO proteins are also co-
expressed with TAL1 in a significant number of T-ALL patients (149) Other oncogenic
events may cooperate with TAL1 in promoting leukemia. For example, expression of
CK2 kinase cooperates with TAL1 to reduce the latency period of T-cell leukemia in
double transgenic mice (250, 251). Despite the fact that TAL1 expression in T-ALL
patients is associated with a differentiated phenotype (149), the establishment of
leukemia in TAL1 transgenic mice is preceded by an early, partial block in T-cell
differentiation (226, 250-252).
TAL1 exerts its leukemogenic activity, at least in part, by interfering with E2A
activity (252), preventing the expression of E2A target genes that include CD4 and pTα
(207, 254). Interestingly, TAL1 overexpression in T-ALL cell lines was shown to
protect them from apoptosis (255, 256), whereas E2A ectopic expression promotes cell
death (257).

Introduction
27
These two facts appear to be linked, as demonstrated by the fact that restoration of
E2A activity in a T-ALL cell line specifically associated with TAL1 and promoted
apoptosis and growth arrest (258).
TAL1, more than a key player in leukemia
As discussed above, TAL1 ectopic expression is extremely frequent in T-ALL
(149) and transgenic expression of TAL1 in mice leads to the development of T-cell
Figure 1.3. Structural comparison of the TAL1 locus and SIL-TAL1 gene
rearrangements. (A) The SIL locus is located at the 5‟ and the MAP17 locus is located at the
3‟ end of the TAL1 locus. Exons are represented as boxes. The white boxes represent exons
that are not translated, while the black boxes show exons that are translated. (B) The three SIL-
TAL1 gene rearrangements are represented as A, B and C. As above, exons are represented as
boxes. The white boxes represent exons that are not translated, while the black boxes show
exons that are translated. The SIL exons are represented in grey and are not translated.

Introduction
28
malignancies (250, 251). However, physiologically, TAL1 is a pivotal transcription
factor in early hematopoiesis and erythropoiesis (259). In this section we will discuss
how TAL1 is regulated and how it exerts its function in normal development.
TAL1: structure and function
The TAL1 oncogene was identified due to its recurrent involvement in
chromosomal translocations that place the TAL1 coding sequences under the control of
the regulatory elements of the genes that code for T-cell receptor chains (214, 215). As
shown in Figure 1.3A, the TAL1 locus is located in the small arm of the chromosome 1
(1p32) surrounded by the SIL locus in the 5‟ end and the MAP17 locus at the 3‟ end
(260, 261). The TAL1 locus contains eight exons (Ia, Ib, IIa, IIb, III, IV, V and VI)
(Figure 1.3A) dispersed over 16 kb, in which exon IV, V and VI code for the TAL1
protein while the others are non-coding exons (260). The splicing events that occur
within the TAL1 gene are highly complex and several TAL1 transcripts have been
described (260). In addition, several regulatory elements have been shown to regulate
TAL1 gene expression, namely three promoters (262, 263), a silencer (264, 265) and
several enhacers (266, 267).
The promoters Ia and Ib are controlled in a tissue-specific manner. The promoter
Ia is located upstream of the Ia exon, it is active in the myeloid lineage (erythroid,
megakayocytes and mast cells), and it is controlled by GATA1 and SP1 (268, 269). The
promoter Ib localizes upstream to the exon Ib, it is active in CD34+ progenitors and in
mast cells, and it is regulated by PU.1 in association with SP1 and SP3 (270, 271).
Interestingly, it was shown that the HTLV1 protein Tax can increase TAL1 gene
expression by activating the Ib promoter through the CREB and NF-κB pathways (272).
The promoter IV is located within the exon IV and is specifically active in human T-
ALL. Transcription driven from this promoter produces a truncated transcript (263),
whose functional impact on leukemia progression has not been characterized, however
it was demonstrated that TAL1 truncated transcripts cooperate with LMO1 to induce
leukemia in transgenic mice (253). The activity of this promoter is regulated by the
binding of PU.1 to a silencer in the 3‟UTR, which is also regulated in a tissue-specific
manner (264, 265).
The expression of the TAL1 gene is also regulated by enhancers (266, 267, 273).
Two enhancers have been identified to direct TAL1 gene expression in endothelial cells

Introduction
29
and during hematopoiesis: the +19 enhancer (3‟) (266) and the -3.9 enhancer (5‟) (267).
The activity of the +19 enhancer was shown to be regulated by GATA2, Fli-1 and Elf-1
(267) (274). A third enhancer that directs TAL1 gene expression in erythroid primitive
precursors is located 40 Kb downstream of the TAL1 Ia exon (273). The regulation of
TAL1 expression is highly complex, due to the existence of these regulatory circuits that
control TAL1 expression in several lineages. Recently, using a genomic tilling array
approach to investigate the regulation of the human TAL1 locus, six more TAL1
regulatory sequences were described dispersed over a genomic area that covers 88 kb,
which included elements with enhancer and (-7, -10, -31) repressor activity (-13), as
well as insulator enhancer-blockers (+53, +57) (261).
The TAL1 gene encodes for two proteins, the full-length protein with 331
aminoacids with approximately 42 KDa (275) and an N-terminal truncated version with
155 aminoacids with approximately 22 KDa (276). The TAL1 protein contains a
proline-rich transactivation domain and a bHLH motif that associates with other bHLH
transcription factors (277). As mentioned above, TAL1 is a class II bHLH transcription
factor, heterodimerizing with class I bHLH transcription factors such as E2A and HEB
to bind DNA (277, 278) in specific sequences called E-boxes (CANNTG), particularly
to the consensus sequence CAGATG (279). TAL1 can act both as transcriptional
repressor and as activator (280). In the context of normal erythropoiesis TAL1 induces
terminal differentiation through the up-regulation of several genes (281-283). TAL1
interacts with transcriptional co-activators like p300 (284) and with the RNA
polymerase II transcriptional machinery, including the p44 subunit of the TFIIH
transcription factor (285). In contrast, in the context of T-cell leukemia TAL1 was
shown to inhibit E2A-mediated transcription (258) and this inhibition appears to be the
trigger for the development of T-cell malignancies in a TAL1 transgenic mouse (252).
Furthermore, TAL1 was shown to interact with repressive chromatin complexes that
include HDAC1 (252, 286), HDAC2 (287) mSIN3A (252, 286), HP1 (288), Suv39h1
(288) and LSD1 (287). Importantly, TAL1 recruits these repressive complexes to inhibit
gene transcription (252, 287). TAL1 was also shown to interact with several binding
partners that include LMO proteins (LMO1 and LMO2), LDB1 and GATA proteins
(289, 290). This multimeric complex recognizes regions in the DNA that contain an E-
box and an adjacent GATA site, which locate in the regulatory regions of genes that are
co-regulated by TAL1 and GATA1 proteins during erythroid development and in
hematopoietic stem cells (290, 291). In T-cell leukemia, TAL1 also interacts with these

Introduction
30
binding partners to up-regulate the expression of TAL1 target genes (292). It is
interesting to note that in a high percentage of T-ALL patients TAL1 gene expression is
associated with LMO genes (LMO1 or LMO2) (149), and these genes also cooperate to
induce aggressive T-cell leukemia in transgenic mice (226, 227).
TAL1 target genes
TAL1 plays a pivotal role in early hematopoiesis (293), erythropoiesis, (281,
294), vasculogenesis (295), and neurogenesis (296). Despite the involvement of TAL1
in all these developmental processes, few TAL1 target genes were so far identified and
clearly validated. Table 1.3 summarizes the TAL1 target genes that have been described
so far.
Gene Context TAL1 action Reference
c-KIT Hematopoiesis up-regulate 291; 297
Runx1 Embryogenesis up-regulate 298
Runx3 Embryogenesis up-regulate 298
GPA Erythropoiesis up-regulate 282
P4.2 Erythropoiesis up-regulate 283
UBE2H Erythropoiesis up-regulate 301
pTα T-cell differentiation down-regulate 207; 254; 302
CD4 T-cell differentiation down-regulate 252
RALDH2 T-ALL up-regulate 292
NFKB1 T-ALL down-regulate 304
NKX3.1 T-ALL up-regulate 305
TALLA1 T-ALL up-regulate 303
TAL1 is fundamental for the biology of HSCs. The receptor tyrosine kinase
encoded by the c-KIT gene is required for proper hematopoietic development and was
identified as a TAL1 target gene (291, 297). It was demonstrated that TAL1 and
interacting partners that include E2A, LMO2, LDB1 and GATA assemble in the
promoter of the KIT gene together with SP1 to activate its expression in hematopoietic
cells (291). During embryonic development, TAL1 positively regulates the expression
of Runx genes, in particular Runx1 and Runx3 genes (298).
The role of TAL1 during red blood cell differentiation is well established. For
instance, enforced expression of TAL1 directs terminal maturation of undifferentiated
MEL cells in the absence of a chemical inducer (281). Moreover, TAL1 was shown to
Table 1.3. List of the genes whose expression has been shown to be directly regulated by TAL1

Introduction
31
regulate the expression of components of the red blood cell membrane, such as
Glycophorin A (GPA) and protein P4.2 (P4.2) (282, 283), during erythropoiesis.
Glycophorin A is highly expressed at the cellular membrane of erythrocytes (299) and
protein P4.2 is part of the erythrocyte membrane cytoskeleton (282, 300). The
expression of these genes is determined by the assembly of a multimeric complex
containing TAL1 and its binding partners in the proximal promoter of these genes
through E-boxes and GATA sites (282, 283). Moreover, during red blood cell
development, TAL1 was also shown to regulate the ubiquitination machinery by
inducing the transcription of the UBE2H gene that encodes for the E2-ubiquitin
conjugase (301).
TAL1 is not expressed in T-cells (190, 221, 254). However, in the context of
TAL1 overexpression in T-cell progenitors and other cell lines, TAL1 has been
associated to the regulation of genes that are important for T-cell development (149,
207, 252, 302). Several lines of evidence suggest that TAL1 regulates the expression of
the pTα gene (207, 254, 302), which is a pivotal protein in normal T-cell differentiation
(41). Reporter assays also show that TAL1 inhibit pTα gene promoter activation in vitro
(302) and in vivo (207, 254). Moreover, TAL1 was also shown to bind to the CD4
enhancer in vivo and inhibit its expression in a TAL1 transgenic mouse model (252).
In the T-ALL context, TAL1 was shown to regulate the expression of several
genes that include TALLA1, RALDH2, NFKB1 and NKX3.1 (292, 303-305). TALLA1 is
considered a highly specific marker of T-ALL (306), whereas RALDH2 is the enzyme
that synthesizes retinoic acid (RA) (307), and NKX3.1 is a homeobox transcription
factor involved in the development of prostate tissue (308) that was shown to mediate
cellular proliferation in T-ALL (305). TAL1 interacts with LMO proteins and GATA3
to up-regulate the expression of these genes in T-ALL (292, 303, 305). Interestingly, it
seems that in the context of T-ALL, TAL1 still interacts with E2A and LMO proteins
but with GATA3 protein instead of GATA1, similar to the regulation of the GPA and
P4.2 genes during terminal erythroid differentiation (282, 283). In contrast to the other
genes described above, NFKB1 expression was shown to be directly repressed by TAL1
(304). The NFKB1 gene encodes p50, a member o the NF-κB/c-Rel family of
transcription factors (309). In T-ALL, the expression levels of p50 are generally low,
since TAL1 together with LMO1 occupies its promoter and recruits HDAC1 to repress
the transcription of the NFKB1 gene (304). The down-regulation of the NFKB1 gene by
TAL1 contributes to the formation of the atypical p65/c-Rel dimer, consequently

Introduction
32
changing the transcriptional program of the NF-κB pathway in T-ALL (304) and
possibly promoting resistance to chemotherapeutic drugs (310).
In the attempt to identify genes that could explain how TAL1 could contribute to
T-cell leukemia, Palomero and colleagues performed a genome wide screen in a human
T-ALL cell line to identify genes whose promoters were bound by TAL1. As one would
expect, TAL1 occupies promoters of genes that are involved in different cellular
functions that include signaling, transcriptional regulation, membrane transport,
vesicular trafficking and metabolism. The authors also identified six genes that changed
their expression upon decrease of TAL1 levels, indicating that they could be directly
regulated by TAL1. TRAF3, RAB40B and EPHB1 were down-regulated, whereas
PTPRU, TTC3 and RPS3A increased their expression upon TAL1 knock-down (311).
Signaling to TAL1
Activation of signaling pathways by external factors or by cell-autonomous
lesions, as described in T-ALL, leads to changes in the phosphorylation status of the
pathway components and consequent alterations in gene expression. Not only TAL1
transcript levels (312), but also the transcriptional activity of TAL1 (313, 314) have
both been shown to be regulated by external signals. So far, three residues in the TAL1
protein were shown to be phosphorylated by different protein kinases: Serines 122
(Ser122) (276) and 172 (Ser172) (313) and Threonine 90 (Thr90) (315).
The Ser122 residue was shown to be phosphorylated in vitro by the extracellular
related kinase 1 (ERK1), a member of the Mitogen-activated Protein Kinase (MAPK)
family (276). Stimulation of proerythroblasts with Erythropoietin (EPO) also appears to
lead to increased phosphorylation of TAL1 Ser122 mediated by ERK1 (316). Talora
and colleagues demonstrated that phosphorylation of this residue by ERK1 kinase, as a
result of increased Pre-TCR signaling, results in increased interaction of TAL1 with
SP1 and subsequent activation of cyclin D1 expression (314). Interestingly, the outcome
of TAL1 protein phosphorylation in this residue is dependent on the cellular
environment. In endothelial cells, hypoxia-induced phosphorylation of TAL1 Ser122
residue drives the ubiquitination and subsequent proteasomal degradation of TAL1
protein (317).
The Ser172 residue was shown to be phosphorylated by Protein kinase A (PKA).
This phosphorylation has no effect on the ability of TAL1 to bind E2A or in its

Introduction
33
subcellular localization. Nevertheless, the phosphorylation in this residue affects the
DNA binding properties of the TAL1/E2A heterodimers in a target-dependent manner.
In other words, phosphorylation of TAL1 Ser172 regulates the binding of TAL1/E2A
heterodimers to specific E-boxes with different core and flanking sequences, thereby
influencing TAL1 transcriptional activity (313).
The Thr90 residue was shown to be phosphorylated by PKB, resulting in TAL1
decreased repressor activity (315). Curiously, Thr90 was also reported to be involved in
TAL1 protein stability. Terme and collaborators showed that treatment of T-ALL cells
with TGF-β activates PKB, which phosphorylates TAL1 in this residue, promoting the
association of TAL1 with the E3-ubiquitin ligase CHIP and consequent TAL1 poly-
ubiquitination and proteasomal degradation. Interestingly, the association between
TAL1 and CHIP is reduced by over-expressing an E2A isoform (E47) indicating that
the formation of TAL1-E2A heterodimers exerts a protective effect (318). Taken
together, these studies may indicate that phosphorylation of Thr90 negatively regulates
the dynamic interaction between TAL1 and E2A proteins, shifting TAL1 away from its
association with E2A (via which TAL1 exerts its transcriptional repressor activity) and
targeting TAL1 for degradation.
As mentioned above, TAL1 cooperates with the ubiquitous protein kinase CK2 to
induce T-cell malignancies in transgenic mice (250, 251). This could suggest that CK2
directly phosphorylates TAL1. However, this has not been demonstrated yet.
Nonetheless, E2A, the main heterodimerization partner of TAL1, was shown to be
phosphorylated (319) by CK2 (320) and p38 kinase (321, 322). The phosphorylation of
E2A by CK2 increases the heterodimerization and transcriptional activity of MyoD
(320), which is another class II bHLH transcription factor highly homologous to TAL1
(323). These reports suggest that CK2 could regulate TAL1 activity indirectly by
interfering with E2A phosphorylation levels.
TAL1 in normal development
As mentioned above, TAL1 in an important player in hematopoiesis (259, 324),
vascular development (295) and, as demonstrated more recently, neurogenesis (296).
The first indication that TAL1 could play a critical role in development came from its
pattern of expression in hematopoietic, vascular and neural tissues (325).

Introduction
34
TAL1 is important for the establishment of early hematopoiesis (326). TAL1
starts to be be expressed very early in embryonic development (327) and it is important
for the differentiation of the hemangioblast (328-330), the progenitor cell that gives rise
to both hematopoietic and endothelial cells (331). A role in the development of the
hemangioblast is attributed to TAL1 due to the fact that ablation of its expression
resulted in complete absence of primitive blood cells in the mouse (326). Furthermore,
the reconstitution of hematopoiesis in TAL1-deficient embryos failed to restore the
angiogenic remodeling that normally occurs in the yolk-sac, implicating TAL1 in the
regulation of primitive vasculogenesis (295). In addition, a role in neo-angiogenesis was
also established using an in vitro vasculature model. TAL1 was shown to regulate the
migration, proliferation and differentiation of adult endothelial cells (332).
TAL1 is also critical in adult hematopoiesis (333, 334). TAL1 is expressed in
HSCs (259) and decreases upon differentiation to most of the hematopoietic lineages
(335). However, erythroid, megakaryocytic and mastocytic lineages still retain its
expression (259) and TAL1 was shown to positively regulate the differentiation of these
hematopoietic lineages (336). The involvement of TAL1 in adult HSC biology is
considerable. Several studies showed that TAL1 is required for HSC function, including
short-term repopulation activity (337), engraftment and self-renewal capacity (338). In
contrast, by deleting TAL1 expression in conditional knock-out mice, Mikkola and
colleagues found that the HSCs still maintained their properties, namely self-renewal,
engraftment and differentiation into lymphoid and myeloid lineages. The authors
attributed this effect to the fact that TAL1 expression is important for the early
specification and development of HSCs but not for their subsequent functions (339).
More recently, it was demonstrated that TAL1 regulates the quiescence of HSCs by
controlling the expression levels of p21 and Id1, thereby preserving HSC long-term
integrity (340).
Several studies highlighted the importance of TAL1 during the development of
erythroid and megakaryocytic lineages (336, 339, 341). Two independent reports
described that specific inactivation of TAL1 gene expression had a severe negative
impact on erythropoiesis and megakaryopoiesis (336, 339). A third report showed that
knock-down of TAL1 expression in human CD34+ HSCs impacted the erythroid and
myeloid lineages (341). Importantly, ectopic TAL1 expression in human CD34+ HSCs
led to an increase in the formation of erythroid and megakaryocytic colonies in culture
(294). A role for TAL1 in red blood cell differentiation is indeed well established (342).

Introduction
35
TAL1 enforced expression was shown to increase red blood cell differentiation (281)
and TAL1 expression and activity is induced by EPO stimulation of erythroblasts (312,
316). Furthermore, TAL1 regulates the expression of P4.2 (282) and GPA (283), genes
that are important for erythrocyte physiology.
TAL1 is also detected in neural tissues (343), namely in neurons of the lateral and
caudal thalamic region, midbrain and hindbrain (344). Inactivation of TAL1 expression
in the brain resulted growth retardation, altered brain morphology and also abnormal
neuron development, implicating TAL1 in the regulation of brain development (296).
Moreover, TAL1 was also described to play a role in differentiation of astrocytes and
V2b interneurons in the p2 domain of the spinal cord (345).
Genome Organization
Tight control of gene expression is necessary to maintain a healthy and
physiological cellular environment. Due to space constrains the genome is tightly
packed inside the cell nucleus. However, this has to be somehow balanced by processes
that allow for access to the transcriptional machinery for gene expression purposes.
Therefore the mechanisms responsible for the genome organization in the nucleus are
also tightly controlled to insure proper gene expression.
Nucleosomes and Histones
In the nucleus, the DNA is tightly condensed, packed and, in conjugation with
proteins such as histones, constitutes the major component of the chromatin. The
chromatin exists in two forms, the heterochromatin which is highly packed and
transcriptionally inactive and euchromatin that is composed of loosely packed DNA and
is a region where active transcription occurs (346). The nucleosomes are protein
complexes that are highly associated with genomic DNA. These structures are
composed of 146 base pair long stretches of DNA that wrap around a histone core
(347). Histones, which are the most abundant proteins in the nucleus, are small weight
proteins (around 11 to 20 KDa) and contain a high percentage of positively charged
residues that are responsible for the strong interaction with the DNA. In eukaryotic cells
five histone proteins were identified. The histone H1 is the linker that binds
nucleosomes together and thereby regulates further packaging of the DNA. The other
histones H2A, H2B, H3 and H4 assemble in the nucleosome core forming an octamer

Introduction
36
composed of two copies of each of these histones. These proteins share a similar
structure.They are composed of a center fold domain and N- and C-terminal tails. The
fold domain participates in the structure of the nucleosome core, whereas the tails are
pivotal for the normal function of cellular processes such as DNA replication and gene
transcription (346, 348-351).
Several post-translational modifications have been identified in the N-terminal
tails of histones, which include acetylation, methylation, phosphorylation,
ubiquitination and ADP-ribosylation (352). Acetylation and methylation of histones are
highly associated with gene expression levels (348). In particular, acetylation of the N-
terminal tail of histones neutralizes the positively charged lysine residues and therefore
reduces the interaction between histones and DNA. This leads to the relaxion of the
packaging of DNA, facilitating the recruitment of transcription factors and the assembly
of RNA polymerase complexes, ultimately promoting gene transcription (346, 349,
350).
Histone Acetyl Transferases (HATs) and Histone Deacetylases (HDACs)
As described above, the level of histone N-terminal acetylation at the nucleosome
core impacts gene transcription. Two classes of enzymes regulate the amount of
acetylation in the N-terminal tail of histones: the Histone Acetyl Transferases (HATs)
and Histone Deacetylases (HDACs). HATs mediate the transfer of acetyl groups from
Acetyl-CoA to lysine residues, decreasing the binding of histones to the DNA (353).
This event opens the chromatin configuration, allowing the recruitment of transcription
factors for activation of gene expression. In opposition, HDACs remove acetyl groups
from the N-terminal tail of histones, resulting in increased DNA-histone interaction.
This closes the chromatin configuration and inhibits the binding of transcription factors
to the DNA. The fine balance between the activities of these enzymes dictates the level
of histone acetylation, thereby regulating gene transcription (346, 350).
Due to its association with gene expression, deregulation of HAT/HDAC
activities can lead to tumorigenesis (354, 355). For example, increased acetylation in
promoters of proto-oncogenes that are normally expressed at low levels would increase
their expression and potentiate the tumorigenic process. In contrast, decreased
acetylation in promoters of tumor suppressor genes would lead to their down-regulation
and possibly contribute to aberrant proliferation of tumor cells (346).

Introduction
37
HATs
The role for acetylation in the regulation of gene expression was suggested 45
years ago (356). However, the molecules responsible for this phenomenon only recently
have been identified (357, 358). HATs regulate gene expression by the mechanisms
described above, but do not bind directly to the DNA. Instead they are recruited by
transcription factors such as TAL1 (286) and other co-factors to acetylate histones that
are in the vicinity of gene promoters (357). The HAT enzymes also participate in DNA
repair (359-361). The p300/CBP complex was shown to acetylate histones at double
strand breaks, thereby facilitating chromatin remodeling and recruitment of non-
homologous end-joining factors such as Ku70 and Ku80 proteins (361).
Several HATs have been identified so far, which are chategorized in two types.
Type A HATs are located in the nucleus and acetylate nucleosomal histones and
chromatin associated proteins, whereas type B HATs are located in the cytoplasm and
acetylate newly translated histones (357). Type A HATs are further divided into three
main groups: the GNAT group includes, among others, the GCN5 and P/CAF members;
the MYST group, and the p300 and CBP acetyltransferases that form their own separate
group (349). It is well established that HATs acetylate histones that form the
nucleosome core (H2A, H2B, H3 and H4). However, it has been shown that HATs can
also acetylate other proteins and modify their activity. Examples of HAT target proteins
include transcription factors such as GATA1, E2F, p53, TAL1 and E2A (349, 362-364).
Of note, TAL1 is acetylated in vivo by P/CAF, increasing its DNA binding activity
(363). E2A proteins were shown to be acetylated by three different HATs: P/CAF, p300
and CBP. Acetylation of E2A increases its transcriptional activity and nuclear
localization (364).
As described above, deregulation of HAT activity can promote the development
of cancer and alterations in the genes that code the HAT enzymes are common in
cancer. These alterations include overexpression, amplification, translocations and also
mutations in these genes. Missense mutations and truncations in the p300 gene were
identified in colon and gastric cancer and other epithelial cancers. Loss of p300
heterozygosity has also been described in 80% of the patients with glioblastomas. Loss
of heterozygosity was also described for the CBP gene in hepatocellular carcinomas. In
AML, the CBP gene was shown to fuse with several partners that include MLL, MOZ,
MYST4 and MORF (349-351).

Introduction
38
HDACs
As described above, Histone Deacetylases (HDACs) enzymes remove acetyl
groups from acetylated histones, and thus are linked to gene repression by creating a
more compact chromatin (346, 349, 350). Similar to HATs, HDACs also do not bind to
DNA and are recruited by large multi-protein complexes that include transcription
factors and general repressors – e.g. Sin3 and N-CoR (346, 365, 366).
Mammalian HDACs are divided in four classes (Table 1.4), based on homology
with yeast HDACs and structure of the catalytic domain. The classe I HDACs (1, 2, 3
and 8) are orthologues of the yeast HDAC Rpd3 protein and are small nuclear proteins
with generalized expression. The HDACs 4, 5, 6, 7, 9 and 10 belong to the class II and
are orthologues of the yeast HDAC Hda1 protein. The class II HDACs are high
molecular weight proteins that shuttle between the cytoplasm and the cellular nucleus
and have tissue specific expression (351, 367, 368). HDAC6 and 10 are classified as IIb
due to the fact that they contain two catalylic domains, whereas HDAC4, 5, 7 and 9 are
IIa. The shuttling of class IIa HDACs was shown to be regulated by N-terminal
phosphorylation and subsequent association with 14-3-3 proteins (369, 370). The
HDAC11 is the only member of the class IV due to the presence of a different catalytic
domain (367). The HDACs that belong to classes I, II and IV contain Zinc in the active
site of the catalytic domain. Contrarily, class III HDACs (Sirtuins 1-7) are homologous
to the yeast Sirt2 and have a unique catalytic domain that is dependent of NAD+
molecules for its activity and lacks Zinc in the catalytic site. This latter characteristic is
highly important, since class III HDACs are not inhibited by any of the conventional
HDAC inhibitors (349, 365, 366).
Similar to HATs, HDACs are also capable of deacetylating other proteins
besides histones, including the transcription factors E2F (371, 372), GATA (373) and c-
Myc (371), the protein chaperone HSP90 (374) and the cytoskeleton protein α-tubulin
(375, 376).
HDACs Class Zn2+
dependency MW (KDa) Localization Expression
HDAC1-3, 8 I Yes 20-55 Nucleus Ubiquitous
HDAC4-7, 9-10 II Yes 70-135 Nucleus Tissue specific
SIRT1-7 III No - Variable Variable
HDAC11 IV Yes 40 Nucleus Ubiquitous
Table 1.4. Charactheristics of Histone Deacetylases (HDACs).

Introduction
39
Although mutations in the genes that code for HDACs are very rare, deregulated
HDAC activity is frequently found in human cancers. Several reports indicate that
HDAC proteins are up-regulated in cancer cells when compared with normal tissues.
Increased HDAC1 expression was demonstrated in prostate (377), gastric (378), colon
(379) and breast carcinomas (380). Moreover, HDAC2 was also shown to have
increased expression in colorectal (76, 379) and gastric (381) carcinomas, whereas
HDAC3 is increased in colon tumors (379). There are also reports indicating that
HDAC4 and HDAC6 activity are up-regulated in breast cancer patients (382, 383).
Recently, Moreno and colleagues found that ALL patients display increased expression
of several HDAC proteins (HDAC2, HDAC3, HDAC6, HDAC7 and HDAC8) and T-
ALL patients showed increased expression of HDAC1 and HDAC4 (384). Cancer cells
not only upregulate HDACs but also show aberrant recruitment of HDACs to promoters
due to the interaction with oncogenic transcription factors (367). For example, the
fusion proteins PML-RARα, PLZF-RARα and AML-ETO1 were shown to induce AML
by recruiting HDAC-containing complexes to repress the expression of specific genes
(385-387). The BCL6 transcription factor is highly associated with B-cell lymphomas
and was shown to recruit HDAC2 to repress the expression of genes that negatively
regulate cell cycle progression (388, 389). As described above, TAL1 interacts with
HDAC1 (286) and was shown recruit HDAC1-containing complexes to repress the
expression of E2A target-genes and induce leukemia in a TAL1 transgenic mouse
model (252).
HDAC inhibitors (HDACis)
The fact that human cancers display abnormal HDAC expression and activity
provides a rationale for the design of inhibitors to target these chromatin regulators in
cancer therapy. In fact, inhibition of HDAC activity was shown to release the repressive
block of genes that lead to cell cycle arrest, apoptosis, differentiation and inhibition of
angiogenesis (346, 351, 353, 367).
Several HDACis have been identified and synthesized. Table 1.5 summarizes the
different HDACis that are currently under use and are involved in clinical trials for
cancer therapy (351, 367). As shown in Table 1.5, HDACis are divided into different
classes according with their chemical structures, which include short-chain fatty acids,
hydroxamic acids, cyclic peptides, benzamides and ketones. Butyrate, Phenylbutyrate,

Introduction
40
Valproic Acid and other short-chain fatty acids are relative weak inhibitors of HDACs.
However, a derivative of Butyrate, the AN-9 compound has a higher in vitro efficacy
than the other fatty acids.
TSA was the first Hydroxamic Acid shown to inhibit HDAC proteins, and it is
highly effective in vitro. However, it was shown to be too toxic to be used in the clinic.
Other Hydroxamic Acids such as SAHA (Vorinostat) and LBH589 (Panobinostat) are
derivatives of TSA that maintain its effectiveness with reduced toxicity. Cyclic peptides
are a complex structurally group of HDACis that includes the naturally occurring
product FK-228, Apicidin and Trapoxin A. These HDACis are highly effective in vitro
and FK-228 is already being tested for clinical purposes. Benzamides are synthetic
products with high in vitro HDAC inhibitory activity that include the compounds MS-
275 and CI-994. Ketones have powerfull in vitro activity but their in vivo toxicity
precludes their use for clinical purposes. Other HDACis include Dupedecin and
MGCD-0103. A common feature to the currently available HDACis is the lack of
specificity. As shown in Table 1.5, most of the HDACis used, inhibit the activity of
several HDACs belonging to one or more classes. Despite displying different chemical
properties, most HDACis contain three structural components that are necessary to
inhibit the activity of HDAC enzymes: a hydrophobic cap, a polar site and a Zinc
binding site. The mechanism of inhibition involves the binding of the HDACi to the
Zinc ion in the HDAC catalytic site, preventing its binding to acetylated proteins (346,
351, 353, 367).
HDACis and gene expression
As mentioned before, incubation with HDACis results in the up-regulation of
genes related to apoptosis and cell cycle arrest (367). A gene that is consistently up-
regulated by HDACis is the CDKN1A which codes for the cyclin-dependent kinase
inhibitor p21CIP1
. Several HDACis described in Table 1.5 (SB; TSA; SAHA;
Depsipetide; MS-275 and Oxamflatin) were shown to induce the expression of this cell
cycle regulator (346, 350, 390). HDACis were also shown to induce the expression of
members of the TNF family and of the mitochondrial apoptotic pathway (367).

Introduction
41
HDAC inhibitors (HDACi) Class Specificity In vitro potency Clinical trials Clinical Trial Reference***
Sodium Butyrate (SB)
Short-chain fatty acids
Class I and IIa mM Phase II -
Phenyl-Butyrate (PB) Class I and IIa mM Phase II -
Valproic Acid (VPA) Class I and IIa mM Phase II NCT00525135
AN9 (Pivanex) n.d. µM Phase II NCT00083473; NCT00087477
Trichostatin A (TSA)
Hydroxamic Acids
Class I and II nM - -
Suberoylanilide Hydroxamic Acid (SAHA) Class I and II µM Phase III NCT00473889; NCT00773747; NCT00128102
M-carboxycinnamic Acid bis-Hydroxamide
(CBHA) n.d. µM - -
NVP-LAQ824 Class I and II nM Phase I -
Panobinostat (LBH589) Class I and II nM Phase III NCT00449761; NCT00425555; NCT00490776
Pyroxamide Class I µM Phase I NCT00042900
PDX101 Class I and II µM Phase I NCT00589290
Depsipeptide (FK228, FR901228)
Cyclic Peptides
Class I nM Phase II NCT00106431; NCT00383565; NCT00077337;
Apicidin Class I** nM - -
Trapoxin A Class I and IIa nM - -
MS-275 Benzamides
Class I µM Phase II NCT00185302; NCT00866333; NCT00828854
N-Acetyldanilide (CI-994) n.d. µM Phase III NCT00005093
Trifluoromethyl Ketone Ketones n.d. µM - -
Dupedecin Other synthetic compounds
Class I µM - -
MGCD-0103 Class I µM Phase II NCT00431873; NCT00358982; NCT00324220
Table 1.5. Classification of the commonly used HDAC inhibitors (HDACis)*
n.d. not determined. * This information was collected from the references 341, 346, 348 and 360. ** inhibits HDAC1, HDAC2 and HDAC3 but not
HDAC8. *** From http://clinicaltrials.gov

Introduction
42
Although several genes have been reported to be up-regulated upon response to HDACi
treatment, microarray studies show that HDACi treatment only affects a slight fraction
of cellular genes. This may, in part, relate to the knowledge that HDACis regulate gene
expression not only by affecting transcription but also by regulating mRNA and protein
stability (346), the latter not being accounted for in microarray analyses.
Interestingly, whereas HDACi treatment up-regulates the expression of genes
associated with apoptosis and cell cycle arrest, it decreases the levels of genes that are
involved in promoting proliferation (391-393). The fact that HDAC inhibition also leads
to down-regulation of gene expression suggests that some of the effects of HDACis are
not due to changes in chromatin conformation, but rather in acetylation levels of other
HDAC targets in the cell (351, 366).
HDACis were also shown to decrease the expression of oncogenes, such as c-Myc
(394, 395), BCL-2 (396), COX-2 (397, 398), DNMT1 (399, 400), Claudin-1 (401), and
BCR-ABL (395, 402). The effect of HDACis does not occur strictly at the level of
transcription. For example, the transcription of DNMT1 (399), which codes for a DNA
methyltransferase associated with cellular transformation (403), was shown to be
inhibited by treatment with Apicidin in cervix cancer cells (399). However, in T-ALL
cells HDAC inhibition induced the degradation of DNMT1 transcripts (400). In fact,
mounting evidence implicates HDACis in the regulation of mRNA stability. Incubation
of endometrial cancer cells with TSA decreased the stability of the DNMT3B transcripts
leading to the down-regulation of this DNA methyltransferase (404). Recently,
Krishnan and colleagues demonstrated that the same mechanism was implicated in the
down-regulation of the tight junction protein Claudin-1 in colon cancer cells
(401).BCR-ABL is the fusion protein created by the t(9;22)(q43;q11) translocation
highly common in CML patients (112). Treatment of CML cells with HDACis induces
the proteasomal degradation of BCR-ABL (402), via acetylation of HSP90 (402) and
activation of the ubiquitin pathway (405). Several other proteins were show to be
degradated upon HDACi exposure, including key regulators of signaling pathways such
as ERK (406) and c-RAF (407). Furthermore, HDACi treatment of renal cancer cells
also led to the proteasomal degradation of Aurora A and B, two kinases that are
involved in the regulation of cell cycle progression in the G2-M stage. The degradation
of these kinases not only induced cell cycle arrest but also increased apoptosis (408).

Introduction
43
HDACi treatment can also affect protein translation. SAHA was shown to
suppress the translation of cyclin D1 in mantle cell lymphoma cells through the direct
inhibition of PI3K (409).
Interestingly, HDACis can also down-regulate the expression of HDACs. In CML
cells, SAHA treatment decreased HDAC3 protein levels (395), whereas VPA induced
proteasomal degradation of HDAC2 (410).
HDACis have a clear pleotropic effect on gene expression and can affect not only
gene transcription, but also mRNA stability, translation and protein stability, which
makes HDACis unique compounds in targeting diseases, such as cancer, that are highly
associated with gene deregulation.
The effect of HDACis on cancer cells
The cytotoxic and cytostatic effects of HDACis seem to be restricted to cancer
cells, since normal tissues are relative resistant to HDACi treatment (411, 412). Two
lines of evidence may explain the resistance of normal tissue to HDAC inhibition.
Warrener and collegues suggest that cancer cells are sensitive to HDACis because they
are defective in cell cycle checkpoints that are triggered upon exposure to HDACis in
normal cells. Normal cells activate these checkpoints, preventing them from further
progressing in the cell cycle, whereas cancer cells without these checkpoints progress
through the cycle with major mitotic defects and eventually die by apoptosis (411, 413).
An alternative line of evidence indicates that it may be the expression and activity of the
ROS scavenger Thioredoxin (TRX) that determines the resistance of normal cells to
HDACi (412). Exposure to HDACis increases ROS to levels that are cytotoxic (414).
However, normal cells up-regulate TRX expression in response to HDACis, which may
protect them from ROS-induced death (412). In contrast, cancer cells decrease TRX
expression upon HDACi exposure, and up-regulate Thioredoxin Binding Protein-2
(TBP-2), which binds TRX and inhibits its reducing activity (415). These two
mechanisms strongly contribute to abrogation of TRX activity in cancer cells and
should make them highly sensitive to HDACis (416). Whatever the exact mechanisms
that make cancer cells particularly sensitive to HDACis, the fact that these drugs
selectively eliminate cancer cells makes them highly appealing to treat cancer patients.
The efficacy of HDACis against tumor cells was demonstrated in vitro in several
cancer cell lines that include neuroblastoma, melanoma, leukemia, breast, ovarian,

Introduction
44
colon, lung, and prostate, amongst others (346, 349, 351, 367). In vivo studies also
showed that HDACis are effective in the treatment of cancer mouse models of breast,
lung, prostate, and gastric cancer, neuroblastoma and leukemia (349). Notably, the few
studies that have addressed the role of HDACis in T-ALL showed that exposure to
HDACis results in leukemic blast apoptosis (417, 418).
Due to their high in vitro efficacy, HDACis were tested in clinical trials for
several types of cancer that included solid tumors and hematological malignancies
(346). One of the most promising HDACis is SAHA, which was tested in phase III
clinical trials for several types of tumors that include lung cancer
(http://clinicaltrials.gov/ NCT00473889), multiple myeloma (NCT00773747) and
malignant pleural mesenthelioma (NCT00128102). Importantly, SAHA was also tested
in phase II clinical trials for cutaneous T-cell lymphoma with a good response in the
treated patients (368). Other promising HDACis are the Hydroxamic Acid LBH589 and
the Benzimide CI-994. LBH589 is currently being tested in phase II clinical trials to
treat hematological malignancies such as Cutaneous T-cell Lymphoma (NCT00425555
and NCT00490776). CI-994 is being tested in phase II clinical trials for pancreatic
cancer (NCT00004861) and multiple myeloma (NCT00005624), and a phase III trial for
lung cancer (NCT00005093).
HDAC inhibitors synergize with several other pharmacological agents resulting in
increased apoptosis of target cells (346, 367, 419). Such drugs include DNA
methyltransferase inhibitors (e.g. 5-Aza- 2‟Deoxycitidine) that activate the transcription
of hyper-methylated genes (420), standard chemotherapeutic drugs (e.g. Doxorubucin)
(421) and BCR-ABL (402), PI3K (406), MEK (422) and PKC (423) kinase inhibitors.
In addition, HDACis induce HSP90 acetylation, promoting degradation of HSP90 client
proteins. Thus, combined incubation with HSP90 inhibitors may also be synergistic
(424). Importantly, combinatorial therapeutic strategies may constitute efficient ways to
overcome the resistance to HDACi that has sometimes been reported. For example, a
mutation in HDAC2 was identified in colon and endometrial tumor cell lines resistant to
TSA (425), and HDAC1 overexpression confers resistance to Butyrate treatment in
melanoma cancer cells (426). Furthermore, it was recently demonstrated that HDACi
treatment in AML cells induces the expression of ABC multidrug transporters,
increasing the efflux of cytotoxic drugs from AML cells and providing a mechanism for
resistance to chemotherapy (427). Despite the occurrence of resistance in cancer cells,
the clinical trials where HDACis participated, either as mono-therapy or in conjunction

Introduction
45
with other drugs, have been so far largely successful, highlighting their promise as
effective tools for the treatment of cancer patients.
Objectives
Despite the significant advances in the treatment of T-ALL (27, 28), around 20%
of the cases remain incurable. In order to define new molecular targets for improved
therapeutic intervention, it is crucial to understand the biology and pathophysiology of
this disease. The fundamental goal of the present thesis was to contribute to a better
understanding of T-ALL and possibly to identify novel targets for therapeutic
intervention. Therefore, we not only analyzed the role of cell-intrinsic mechanisms
(related to TAL1 function) in the biology of T-ALL, but also the possible involvement
of the micro-environment (illustrated by study of IL-4) in the function of leukemia cells.
We had previously shown that c cytokines can increase the proliferation of
primary T-ALL cells in vitro (72). Particularly, IL-7 was able to increase the
proliferation and viability of primary T-ALL cells through the activation of PI3K
pathway. Importantly, pharmacological inhibition of PI3K pathway completely reversed
the effects of IL-7 (70). Here, we used primary T-ALL samples to analyze the
downstream events activated by IL-4. These results have been published (428) and are
described in Chapter 2.
Aberrant expression of the TAL1 gene is considered the most common alteration
in T-ALL, frequently in combination with LMO proteins (LMO1 and LMO2) (149).
However, it is still unknown whether the expression of the TAL1 is the actual cause of
leukemia or, alternatively, is a merely secondary event in the transformation process.
One of the key early steps in leukemogenesis is the blockade of normal T-cell
differentiation that results in the accumulation of malignant T-cell lymphoblasts
arrested at different stages of development (227, 251-253). To test whether TAL1, alone
or in association with LMO2, could impair normal T-cell development, we made use of
in vitro co-culture systems that allow the differentiation of human T-cells from
hematopoietic and thymic progenitors (33, 34, 429). The preliminary results obtained
during this part of the project are described in Chapter 3.
Despite the fact that TAL1 is a transcription factor (246), very few TAL1 target
genes have been identified in the context of T-ALL (292, 303-305). To identify novel
TAL1 targets, we used the 4-OH-Tamoxifen system (430) to induce TAL1 activity in a

Introduction
46
regulated manner, and analyzed the gene expression profile using microarray
technology. The genes identified, validated and partially characterized in this study are
shown in Chapter 4.
TAL1 was shown to interact with HDAC complexes during red blood cell
development (286) and in T-ALL cells (252). Moreover, incubation of tumors derived
from TAL1 transgenic mice with HDACis was reported to selectively kill these tumors
without affecting normal thymocytes or TAL1-negative leukemia cell lines (252).
Interestingly, HDACis decrease the expression of several oncogenes in cancer cells,
either by mRNA or protein degradation or by transcriptional inhibition (399, 401, 402).
To understand whether inhibition of HDAC function could impact on TAL1 expression,
we treated T-ALL cells with different HDACis and analyzed TAL1 expression. The
characterization of the mechanisms by which HDACis affect TAL1 expression are
described in Chapter 5 and were recently published.
References
1. Ackerknecht, E.H. 1958. Historical notes on cancer. Med Hist 2:114-119.
2. Weinberg, R.A. 2007. The Nature of Cancer. In: Weinberg R-A, ed. The Biology of
Cancer. Garland Science 25-57.
3. Dictionary of Cancer Terms at: http://www.cancer.gov/dictionary.
4. Silverstein, A.S., Virginia; Nunn, Laura Silverstein. 2006. The cells gone wild. In:
Silverstein A, ed Cancer. Twenty-First Century Books 7-12.
5. Hanahan, D., and R.A. Weinberg. 2000. The hallmarks of cancer. Cell 100:57-70.
6. Hanahan, D., and R.A. Weinberg. 2011. Hallmarks of cancer: the next generation. Cell
144:646-674.
7. Lazebnik, Y. 2010. What are the hallmarks of cancer? Nat Rev Cancer 10:232-233.
8. Collins, K., T. Jacks, and N.P. Pavletich. 1997. The cell cycle and cancer. Proc Natl
Acad Sci U S A 94:2776-2778.
9. Eaves, C.J.E., A C. 1999. Anatomy and phisiology of hematopoiesis. In: Pui C-H, ed.
Childhood Leukemias. Cambrigde University Press 69-105.
10. Pui, C.H. 1995. Childhood leukemias. N Engl J Med 332:1618-1630.
11. Pinkel, D. 1999. Historical Perspective. In: Pui. C-H, ed. Childhood Leukemias.
Cambrigde University Press 3-18.
12. Onciu M, P.C. 1999. Diagnosis and Classification. In: Pui C-H, ed. Childhood
Leukemias. Cambrigde University Press 21-47.

Introduction
47
13. Uckun, F.M., M.G. Sensel, L. Sun, P.G. Steinherz, M.E. Trigg, N.A. Heerema, H.N.
Sather, G.H. Reaman, and P.S. Gaynon. 1998. Biology and treatment of childhood T-
lineage acute lymphoblastic leukemia. Blood 91:735-746.
14. Graux, C., J. Cools, L. Michaux, P. Vandenberghe, and A. Hagemeijer. 2006.
Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from
thymocyte to lymphoblast. Leukemia 20:1496-1510.
15. Pui, C.H., M.V. Relling, and J.R. Downing. 2004. Acute lymphoblastic leukemia. N
Engl J Med 350:1535-1548.
16. Notarangelo, L.D., S. Santagata, and A. Villa. 2001. Recombinase activating gene
enzymes of lymphocytes. Curr Opin Hematol 8:41-46.
17. Numata, M., S. Saito, and K. Nagata. 2010. RAG-dependent recombination at cryptic
RSSs within TEL-AML1 t(12;21)(p13;q22) chromosomal translocation region.
Biochem Biophys Res Commun 402:718-724.
18. Aplan, P.D., D.P. Lombardi, A.M. Ginsberg, J. Cossman, V.L. Bertness, and I.R.
Kirsch. 1990. Disruption of the human SCL locus by "illegitimate" V-(D)-J
recombinase activity. Science 250:1426-1429.
19. Redaelli, A., B.L. Laskin, J.M. Stephens, M.F. Botteman, and C.L. Pashos. 2005. A
systematic literature review of the clinical and epidemiological burden of acute
lymphoblastic leukaemia (ALL). Eur J Cancer Care (Engl) 14:53-62.
20. Pui, C.H., and W.E. Evans. 2006. Treatment of acute lymphoblastic leukemia. N Engl J
Med 354:166-178.
21. Pui, C.H., L.L. Robison, and A.T. Look. 2008. Acute lymphoblastic leukaemia. Lancet
371:1030-1043.
22. Mattano, L.A., Jr., H.N. Sather, M.E. Trigg, and J.B. Nachman. 2000. Osteonecrosis as
a complication of treating acute lymphoblastic leukemia in children: a report from the
Children's Cancer Group. J Clin Oncol 18:3262-3272.
23. Kaste, S.C., D. Jones-Wallace, S.R. Rose, J.M. Boyett, R.H. Lustig, G.K. Rivera, C.H.
Pui, and M.M. Hudson. 2001. Bone mineral decrements in survivors of childhood acute
lymphoblastic leukemia: frequency of occurrence and risk factors for their
development. Leukemia 15:728-734.
24. Nowak-Gottl, U., C. Wermes, R. Junker, H.G. Koch, R. Schobess, G. Fleischhack, D.
Schwabe, and S. Ehrenforth. 1999. Prospective evaluation of the thrombotic risk in
children with acute lymphoblastic leukemia carrying the MTHFR TT 677 genotype, the
prothrombin G20210A variant, and further prothrombotic risk factors. Blood 93:1595-
1599.

Introduction
48
25. Hill, D.E., K.T. Ciesielski, L. Sethre-Hofstad, M.H. Duncan, and M. Lorenzi. 1997.
Visual and verbal short-term memory deficits in childhood leukemia survivors after
intrathecal chemotherapy. J Pediatr Psychol 22:861-870.
26. Downing, J.R., and K.M. Shannon. 2002. Acute leukemia: a pediatric perspective.
Cancer Cell 2:437-445.
27. Schrappe, M., A. Reiter, W.D. Ludwig, J. Harbott, M. Zimmermann, W. Hiddemann, C.
Niemeyer, G. Henze, A. Feldges, F. Zintl, B. Kornhuber, J. Ritter, K. Welte, H. Gadner,
and H. Riehm. 2000. Improved outcome in childhood acute lymphoblastic leukemia
despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-
BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95:3310-3322.
28. Silverman, L.B., R.D. Gelber, V.K. Dalton, B.L. Asselin, R.D. Barr, L.A. Clavell, C.A.
Hurwitz, A. Moghrabi, Y. Samson, M.A. Schorin, S. Arkin, L. Declerck, H.J. Cohen,
and S.E. Sallan. 2001. Improved outcome for children with acute lymphoblastic
leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97:1211-1218.
29. Goldberg, J.M., L.B. Silverman, D.E. Levy, V.K. Dalton, R.D. Gelber, L. Lehmann,
H.J. Cohen, S.E. Sallan, and B.L. Asselin. 2003. Childhood T-cell acute lymphoblastic
leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium
experience. J Clin Oncol 21:3616-3622.
30. DeAngelo, D.J. 2005. The treatment of adolescents and young adults with acute
lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 123-130.
31. Bommhardt, U., M. Beyer, T. Hunig, and H.M. Reichardt. 2004. Molecular and cellular
mechanisms of T cell development. Cell Mol Life Sci 61:263-280.
32. Staal, F.J., F. Weerkamp, A.W. Langerak, R.W. Hendriks, and H.C. Clevers. 2001.
Transcriptional control of t lymphocyte differentiation. Stem Cells 19:165-179.
33. Jaleco, A.C., H. Neves, E. Hooijberg, P. Gameiro, N. Clode, M. Haury, D. Henrique,
and L. Parreira. 2001. Differential effects of Notch ligands Delta-1 and Jagged-1 in
human lymphoid differentiation. J Exp Med 194:991-1002.
34. Schmitt, T.M., and J.C. Zuniga-Pflucker. 2002. Induction of T cell development from
hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17:749-756.
35. Radtke, F., A. Wilson, and H.R. MacDonald. 2004. Notch signaling in T- and B-cell
development. Curr Opin Immunol 16:174-179.
36. Plum, J., M. De Smedt, G. Leclercq, B. Verhasselt, and B. Vandekerckhove. 1996.
Interleukin-7 is a critical growth factor in early human T-cell development. Blood
88:4239-4245.
37. Bukowski, J.F., C.T. Morita, and M.B. Brenner. 1999. Human gamma delta T cells
recognize alkylamines derived from microbes, edible plants, and tea: implications for
innate immunity. Immunity 11:57-65.

Introduction
49
38. Ferrick, D.A., D.P. King, K.A. Jackson, R.K. Braun, S. Tam, D.M. Hyde, and B.L.
Beaman. 2000. Intraepithelial gamma delta T lymphocytes: sentinel cells at mucosal
barriers. Springer Semin Immunopathol 22:283-296.
39. Tonegawa, S. 1983. Somatic generation of antibody diversity. Nature 302:575-581.
40. von Boehmer, H., and H.J. Fehling. 1997. Structure and function of the pre-T cell
receptor. Annu Rev Immunol 15:433-452.
41. Aifantis, I., M. Mandal, K. Sawai, A. Ferrando, and T. Vilimas. 2006. Regulation of T-
cell progenitor survival and cell-cycle entry by the pre-T-cell receptor. Immunol Rev
209:159-169.
42. Bene, M.C., G. Castoldi, W. Knapp, W.D. Ludwig, E. Matutes, A. Orfao, and M.B.
van't Veer. 1995. Proposals for the immunological classification of acute leukemias.
European Group for the Immunological Characterization of Leukemias (EGIL).
Leukemia 9:1783-1786.
43. Winter, S.S., J. Sweatman, J.J. Shuster, M.P. Link, M.D. Amylon, J. Pullen, B.M.
Camitta, and R.S. Larson. 2002. Bone marrow stroma-supported culture of T-lineage
acute lymphoblastic leukemic cells predicts treatment outcome in children: a Pediatric
Oncology Group study. Leukemia 16:1121-1126.
44. Scupoli, M.T., F. Vinante, M. Krampera, C. Vincenzi, G. Nadali, F. Zampieri, M.A.
Ritter, E. Eren, F. Santini, and G. Pizzolo. 2003. Thymic epithelial cells promote
survival of human T-cell acute lymphoblastic leukemia blasts: the role of interleukin-7.
Haematologica 88:1229-1237.
45. Winter, S.S., J.J. Sweatman, M.B. Lawrence, T.H. Rhoades, A.L. Hart, and R.S.
Larson. 2001. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone
marrow stroma requires involvement of LFA-1 and ICAM-1. Br J Haematol 115:862-
871.
46. Damiano, J.S., A.E. Cress, L.A. Hazlehurst, A.A. Shtil, and W.S. Dalton. 1999. Cell
adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to
apoptosis in human myeloma cell lines. Blood 93:1658-1667.
47. Sipkins, D.A., X. Wei, J.W. Wu, J.M. Runnels, D. Cote, T.K. Means, A.D. Luster, D.T.
Scadden, and C.P. Lin. 2005. In vivo imaging of specialized bone marrow endothelial
microdomains for tumour engraftment. Nature 435:969-973.
48. Colmone, A., M. Amorim, A.L. Pontier, S. Wang, E. Jablonski, and D.A. Sipkins. 2008.
Leukemic cells create bone marrow niches that disrupt the behavior of normal
hematopoietic progenitor cells. Science 322:1861-1865.
49. Olson, T.S., and K. Ley. 2002. Chemokines and chemokine receptors in leukocyte
trafficking. Am J Physiol Regul Integr Comp Physiol 283:R7-28.

Introduction
50
50. Kakinuma, T., and S.T. Hwang. 2006. Chemokines, chemokine receptors, and cancer
metastasis. J Leukoc Biol 79:639-651.
51. Burger, J.A., and T.J. Kipps. 2006. CXCR4: a key receptor in the crosstalk between
tumor cells and their microenvironment. Blood 107:1761-1767.
52. Balkwill, F. 2004. The significance of cancer cell expression of the chemokine receptor
CXCR4. Semin Cancer Biol 14:171-179.
53. Matsumoto, T., S. Jimi, S. Hara, Y. Takamatsu, J. Suzumiya, and K. Tamura. 2010.
Am80 inhibits stromal cell-derived factor-1-induced chemotaxis in T-cell acute
lymphoblastic leukemia cells. Leuk Lymphoma 51:507-514.
54. Dialynas, D.P., M.J. Lee, D.P. Gold, L. Shao, A.L. Yu, M.J. Borowitz, and J. Yu. 2001.
Preconditioning with fetal cord blood facilitates engraftment of primary childhood T-
cell acute lymphoblastic leukemia in immunodeficient mice. Blood 97:3218-3225.
55. Dialynas, D.P., L. Shao, G.F. Billman, and J. Yu. 2001. Engraftment of human T-cell
acute lymphoblastic leukemia in immunodeficient NOD/SCID mice which have been
preconditioned by injection of human cord blood. Stem Cells 19:443-452.
56. Spoo, A.C., M. Lubbert, W.G. Wierda, and J.A. Burger. 2007. CXCR4 is a prognostic
marker in acute myelogenous leukemia. Blood 109:786-791.
57. Sharma, M., F. Afrin, N.K. Satija, R.K. Thripati, and G.U. Gangenahalli. 2011. SDF-
1/CXCR4 Signaling: Indispensable role in Homing and Engraftment of Hematopoietic
Stem Cells in Bone Marrow. Stem Cells Dev 20:933-946.
58. Qiuping, Z., X. Jie, J. Youxin, W. Qun, J. Wei, L. Chun, W. Jin, L. Yan, H. Chunsong,
Y. Mingzhen, G. Qingping, L. Qun, Z. Kejian, S. Zhimin, L. Junyan, and T. Jinquan.
2005. Selectively frequent expression of CXCR5 enhances resistance to apoptosis in
CD8(+)CD34(+) T cells from patients with T-cell-lineage acute lymphocytic leukemia.
Oncogene 24:573-584.
59. Qiuping, Z., L. Qun, H. Chunsong, Z. Xiaolian, H. Baojun, Y. Mingzhen, L.
Chengming, H. Jinshen, G. Qingping, Z. Kejian, S. Zhimin, Z. Xuejun, L. Junyan, and
T. Jinquan. 2003. Selectively increased expression and functions of chemokine receptor
CCR9 on CD4+ T cells from patients with T-cell lineage acute lymphocytic leukemia.
Cancer Res 63:6469-6477.
60. Buonamici, S., T. Trimarchi, M.G. Ruocco, L. Reavie, S. Cathelin, B.G. Mar, A.
Klinakis, Y. Lukyanov, J.C. Tseng, F. Sen, E. Gehrie, M. Li, E. Newcomb, J. Zavadil,
D. Meruelo, M. Lipp, S. Ibrahim, A. Efstratiadis, D. Zagzag, J.S. Bromberg, M.L.
Dustin, and I. Aifantis. 2009. CCR7 signalling as an essential regulator of CNS
infiltration in T-cell leukaemia. Nature 459:1000-1004.
61. Bociek, R.G., and J.O. Armitage. 1996. Hematopoietic growth factors. CA Cancer J
Clin 46:165-184.

Introduction
51
62. Miyajima, A., Y. Ito, and T. Kinoshita. 1999. Cytokine signaling for proliferation,
survival, and death in hematopoietic cells. Int J Hematol 69:137-146.
63. Moqattash, S., and J.D. Lutton. 2004. Leukemia cells and the cytokine network:
therapeutic prospects. Exp Biol Med (Maywood) 229:121-137.
64. Mentz, F., F. Ouaaz, A. Michel, C. Blanc, P. Herve, G. Bismuth, P. Debre, H. Merle-
Beral, and M.D. Mossalayi. 1994. Maturation of acute T-lymphoblastic leukemia cells
after CD2 ligation and subsequent treatment with interleukin-2. Blood 84:1182-1192.
65. Barata, J.T., A.A. Cardoso, L.M. Nadler, and V.A. Boussiotis. 2001. Interleukin-7
promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia
cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood
98:1524-1531.
66. Masuda, M., M. Takanashi, T. Motoji, K. Oshimi, and H. Mizoguchi. 1991. Effects of
interleukins 1-7 on the proliferation of T-lineage acute lymphoblastic leukemia cells.
Leuk Res 15:1091-1096.
67. Karawajew, L., V. Ruppert, C. Wuchter, A. Kosser, M. Schrappe, B. Dorken, and W.D.
Ludwig. 2000. Inhibition of in vitro spontaneous apoptosis by IL-7 correlates with bcl-2
up-regulation, cortical/mature immunophenotype, and better early cytoreduction of
childhood T-cell acute lymphoblastic leukemia. Blood 96:297-306.
68. Touw, I., R. Delwel, G. van Zanen, and B. Lowenberg. 1986. Acute lymphoblastic
leukemia and non-Hodgkin's lymphoma of T lineage: colony-forming cells retain
growth factor (interleukin 2) dependence. Blood 68:1088-1094.
69. Georgoulias, V., M. Allouche, A. Salvatore, C. Clemenceau, and C. Jasmin. 1987.
Interleukin 2 responsiveness of immature T-cell colony-forming cells (T-CFC) from
patients with acute T-cell lymphoblastic leukemias. Cell Immunol 105:317-331.
70. Barata, J.T., A. Silva, J.G. Brandao, L.M. Nadler, A.A. Cardoso, and V.A. Boussiotis.
2004. Activation of PI3K is indispensable for interleukin 7-mediated viability,
proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J
Exp Med 200:659-669.
71. Scupoli, M.T., O. Perbellini, M. Krampera, F. Vinante, F. Cioffi, and G. Pizzolo. 2007.
Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and
human thymocytes on bone marrow stroma. Haematologica 92:264-266.
72. Barata, J.T., T.D. Keenan, A. Silva, L.M. Nadler, V.A. Boussiotis, and A.A. Cardoso.
2004. Common gamma chain-signaling cytokines promote proliferation of T-cell acute
lymphoblastic leukemia. Haematologica 89:1459-1467.
73. Regis, G., L. Icardi, L. Conti, R. Chiarle, R. Piva, M. Giovarelli, V. Poli, and F. Novelli.
2009. IL-6, but not IFN-gamma, triggers apoptosis and inhibits in vivo growth of
human malignant T cells on STAT3 silencing. Leukemia 23:2102-2108.

Introduction
52
74. Jia, L., R.R. Dourmashkin, A.C. Newland, and S.M. Kelsey. 1997. Mitochondrial
ultracondensation, but not swelling, is involved in TNF alpha-induced apoptosis in
human T-lymphoblastic leukaemic cells. Leuk Res 21:973-983.
75. Jia, L., S.M. Kelsey, M.F. Grahn, X.R. Jiang, and A.C. Newland. 1996. Increased
activity and sensitivity of mitochondrial respiratory enzymes to tumor necrosis factor
alpha-mediated inhibition is associated with increased cytotoxicity in drug-resistant
leukemic cell lines. Blood 87:2401-2410.
76. Min, B., M. Prout, J. Hu-Li, J. Zhu, D. Jankovic, E.S. Morgan, J.F. Urban, Jr., A.M.
Dvorak, F.D. Finkelman, G. LeGros, and W.E. Paul. 2004. Basophils produce IL-4 and
accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med 200:507-
517.
77. Gessner, A., K. Mohrs, and M. Mohrs. 2005. Mast cells, basophils, and eosinophils
acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are
sufficient for rapid cytokine production. J Immunol 174:1063-1072.
78. Piccinni, M.P., D. Macchia, P. Parronchi, M.G. Giudizi, D. Bani, R. Alterini, A. Grossi,
M. Ricci, E. Maggi, and S. Romagnani. 1991. Human bone marrow non-B, non-T cells
produce interleukin 4 in response to cross-linkage of Fc epsilon and Fc gamma
receptors. Proc Natl Acad Sci U S A 88:8656-8660.
79. Russell, S.M., A.D. Keegan, N. Harada, Y. Nakamura, M. Noguchi, P. Leland, M.C.
Friedmann, A. Miyajima, R.K. Puri, W.E. Paul, and et al. 1993. Interleukin-2 receptor
gamma chain: a functional component of the interleukin-4 receptor. Science 262:1880-
1883.
80. Idzerda, R.L., C.J. March, B. Mosley, S.D. Lyman, T. Vanden Bos, S.D. Gimpel, W.S.
Din, K.H. Grabstein, M.B. Widmer, L.S. Park, and et al. 1990. Human interleukin 4
receptor confers biological responsiveness and defines a novel receptor superfamily. J
Exp Med 171:861-873.
81. Bazan, J.F. 1990. Structural design and molecular evolution of a cytokine receptor
superfamily. Proc Natl Acad Sci U S A 87:6934-6938.
82. Keegan, A.D., K. Nelms, M. White, L.M. Wang, J.H. Pierce, and W.E. Paul. 1994. An
IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated
IRS-1 phosphorylation and cell growth. Cell 76:811-820.
83. Takeshita, T., H. Asao, K. Ohtani, N. Ishii, S. Kumaki, N. Tanaka, H. Munakata, M.
Nakamura, and K. Sugamura. 1992. Cloning of the gamma chain of the human IL-2
receptor. Science 257:379-382.
84. Noguchi, M., Y. Nakamura, S.M. Russell, S.F. Ziegler, M. Tsang, X. Cao, and W.J.
Leonard. 1993. Interleukin-2 receptor gamma chain: a functional component of the
interleukin-7 receptor. Science 262:1877-1880.

Introduction
53
85. Russell, S.M., J.A. Johnston, M. Noguchi, M. Kawamura, C.M. Bacon, M. Friedmann,
M. Berg, D.W. McVicar, B.A. Witthuhn, O. Silvennoinen, and et al. 1994. Interaction
of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and
XCID. Science 266:1042-1045.
86. Giri, J.G., M. Ahdieh, J. Eisenman, K. Shanebeck, K. Grabstein, S. Kumaki, A. Namen,
L.S. Park, D. Cosman, and D. Anderson. 1994. Utilization of the beta and gamma
chains of the IL-2 receptor by the novel cytokine IL-15. Embo J 13:2822-2830.
87. Miyazaki, T., A. Kawahara, H. Fujii, Y. Nakagawa, Y. Minami, Z.J. Liu, I. Oishi, O.
Silvennoinen, B.A. Witthuhn, J.N. Ihle, and et al. 1994. Functional activation of Jak1
and Jak3 by selective association with IL-2 receptor subunits. Science 266:1045-1047.
88. Acacia de Sa Pinheiro, A., A. Morrot, S. Chakravarty, M. Overstreet, J.H. Bream, P.M.
Irusta, and F. Zavala. 2007. IL-4 induces a wide-spectrum intracellular signaling
cascade in CD8+ T cells. J Leukoc Biol 81:1102-1110.
89. Stephenson, L.M., D.S. Park, A.L. Mora, S. Goenka, and M. Boothby. 2005. Sequence
motifs in IL-4R alpha mediating cell-cycle progression of primary lymphocytes. J
Immunol 175:5178-5185.
90. Prokopchuk, O., Y. Liu, D. Henne-Bruns, and M. Kornmann. 2005. Interleukin-4
enhances proliferation of human pancreatic cancer cells: evidence for autocrine and
paracrine actions. Br J Cancer 92:921-928.
91. Lee, S.O., E. Pinder, J.Y. Chun, W. Lou, M. Sun, and A.C. Gao. 2008. Interleukin-4
stimulates androgen-independent growth in LNCaP human prostate cancer cells.
Prostate 68:85-91.
92. Carey, G.B., E. Semenova, X. Qi, and A.D. Keegan. 2007. IL-4 protects the B-cell
lymphoma cell line CH31 from anti-IgM-induced growth arrest and apoptosis:
contribution of the PI-3 kinase/AKT pathway. Cell Res 17:942-955.
93. Li, B.H., X.Z. Yang, P.D. Li, Q. Yuan, X.H. Liu, J. Yuan, and W.J. Zhang. 2008. IL-
4/Stat6 activities correlate with apoptosis and metastasis in colon cancer cells. Biochem
Biophys Res Commun 369:554-560.
94. Manabe, A., E. Coustan-Smith, M. Kumagai, F.G. Behm, S.C. Raimondi, C.H. Pui, and
D. Campana. 1994. Interleukin-4 induces programmed cell death (apoptosis) in cases of
high-risk acute lymphoblastic leukemia. Blood 83:1731-1737.
95. De Keersmaecker, K., P. Marynen, and J. Coolsi. 2005. Genetic insights in the
pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica 90:1116-1127.
96. Cauwelier, B., N. Dastugue, J. Cools, B. Poppe, C. Herens, A. De Paepe, A.
Hagemeijer, and F. Speleman. 2006. Molecular cytogenetic study of 126 unselected T-
ALL cases reveals high incidence of TCRbeta locus rearrangements and putative new
T-cell oncogenes. Leukemia 20:1238-1244.

Introduction
54
97. Weng, A.P., A.A. Ferrando, W. Lee, J.P.t. Morris, L.B. Silverman, C. Sanchez-Irizarry,
S.C. Blacklow, A.T. Look, and J.C. Aster. 2004. Activating mutations of NOTCH1 in
human T cell acute lymphoblastic leukemia. Science 306:269-271.
98. Pear, W.S., J.C. Aster, M.L. Scott, R.P. Hasserjian, B. Soffer, J. Sklar, and D.
Baltimore. 1996. Exclusive development of T cell neoplasms in mice transplanted with
bone marrow expressing activated Notch alleles. J Exp Med 183:2283-2291.
99. Bellavia, D., A.F. Campese, E. Alesse, A. Vacca, M.P. Felli, A. Balestri, A.
Stoppacciaro, C. Tiveron, L. Tatangelo, M. Giovarelli, C. Gaetano, L. Ruco, E.S.
Hoffman, A.C. Hayday, U. Lendahl, L. Frati, A. Gulino, and I. Screpanti. 2000.
Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in Notch3
transgenic mice. Embo J 19:3337-3348.
100. Knudsen, E.S., and K.E. Knudsen. 2006. Retinoblastoma tumor suppressor: where
cancer meets the cell cycle. Exp Biol Med (Maywood) 231:1271-1281.
101. Greenblatt, M.S., W.P. Bennett, M. Hollstein, and C.C. Harris. 1994. Mutations in the
p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis.
Cancer Res 54:4855-4878.
102. Hebert, J., J.M. Cayuela, J. Berkeley, and F. Sigaux. 1994. Candidate tumor-suppressor
genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous
deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic
leukemias. Blood 84:4038-4044.
103. Bertin, R., C. Acquaviva, D. Mirebeau, C. Guidal-Giroux, E. Vilmer, and H. Cave.
2003. CDKN2A, CDKN2B, and MTAP gene dosage permits precise characterization of
mono- and bi-allelic 9p21 deletions in childhood acute lymphoblastic leukemia. Genes
Chromosomes Cancer 37:44-57.
104. Omura-Minamisawa, M., M.B. Diccianni, A. Batova, R.C. Chang, L.J. Bridgeman, J.
Yu, J. Pullen, W.P. Bowman, and A.L. Yu. 2000. Universal inactivation of both p16
and p15 but not downstream components is an essential event in the pathogenesis of T-
cell acute lymphoblastic leukemia. Clin Cancer Res 6:1219-1228.
105. Cayuela, J.M., A. Madani, L. Sanhes, M.H. Stern, and F. Sigaux. 1996. Multiple tumor-
suppressor gene 1 inactivation is the most frequent genetic alteration in T-cell acute
lymphoblastic leukemia. Blood 87:2180-2186.
106. Sherr, C.J. 2001. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell
Biol 2:731-737.
107. Sherr, C.J., and F. McCormick. 2002. The RB and p53 pathways in cancer. Cancer Cell
2:103-112.

Introduction
55
108. Clappier, E., W. Cuccuini, J.M. Cayuela, D. Vecchione, A. Baruchel, H. Dombret, F.
Sigaux, and J. Soulier. 2006. Cyclin D2 dysregulation by chromosomal translocations to
TCR loci in T-cell acute lymphoblastic leukemias. Leukemia 20:82-86.
109. Cardoso, B.A., A. Girio, C. Henriques, L.R. Martins, C. Santos, A. Silva, and J.T.
Barata. 2008. Aberrant signaling in T-cell acute lymphoblastic leukemia: biological and
therapeutic implications. Braz J Med Biol Res 41:344-350.
110. Zipfel, P.A., W. Zhang, M. Quiroz, and A.M. Pendergast. 2004. Requirement for Abl
kinases in T cell receptor signaling. Curr Biol 14:1222-1231.
111. Wange, R.L. 2004. TCR signaling: another Abl-bodied kinase joins the cascade. Curr
Biol 14:R562-564.
112. Melo, J.V. 1996. The diversity of BCR-ABL fusion proteins and their relationship to
leukemia phenotype. Blood 88:2375-2384.
113. Quentmeier, H., J. Cools, R.A. Macleod, P. Marynen, C.C. Uphoff, and H.G. Drexler.
2005. e6-a2 BCR-ABL1 fusion in T-cell acute lymphoblastic leukemia. Leukemia
19:295-296.
114. Graux, C., J. Cools, C. Melotte, H. Quentmeier, A. Ferrando, R. Levine, J.R.
Vermeesch, M. Stul, B. Dutta, N. Boeckx, A. Bosly, P. Heimann, A. Uyttebroeck, N.
Mentens, R. Somers, R.A. MacLeod, H.G. Drexler, A.T. Look, D.G. Gilliland, L.
Michaux, P. Vandenberghe, I. Wlodarska, P. Marynen, and A. Hagemeijer. 2004.
Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic
leukemia. Nat Genet 36:1084-1089.
115. Van Limbergen, H., H.B. Beverloo, E. van Drunen, A. Janssens, K. Hahlen, B. Poppe,
N. Van Roy, P. Marynen, A. De Paepe, R. Slater, and F. Speleman. 2001. Molecular
cytogenetic and clinical findings in ETV6/ABL1-positive leukemia. Genes
Chromosomes Cancer 30:274-282.
116. De Keersmaecker, K., C. Graux, M.D. Odero, N. Mentens, R. Somers, J. Maertens, I.
Wlodarska, P. Vandenberghe, A. Hagemeijer, P. Marynen, and J. Cools. 2005. Fusion
of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic
t(9;14)(q34;q32). Blood 105:4849-4852.
117. Palacios, E.H., and A. Weiss. 2004. Function of the Src-family kinases, Lck and Fyn, in
T-cell development and activation. Oncogene 23:7990-8000.
118. Burnett, R.C., M.J. Thirman, J.D. Rowley, and M.O. Diaz. 1994. Molecular analysis of
the T-cell acute lymphoblastic leukemia-associated t(1;7)(p34;q34) that fuses LCK and
TCRB. Blood 84:1232-1236.
119. Tycko, B., S.D. Smith, and J. Sklar. 1991. Chromosomal translocations joining LCK
and TCRB loci in human T cell leukemia. J Exp Med 174:867-873.

Introduction
56
120. Rawlings, J.S., K.M. Rosler, and D.A. Harrison. 2004. The JAK/STAT signaling
pathway. J Cell Sci 117:1281-1283.
121. Peeters, P., S.D. Raynaud, J. Cools, I. Wlodarska, J. Grosgeorge, P. Philip, F. Monpoux,
L. Van Rompaey, M. Baens, H. Van den Berghe, and P. Marynen. 1997. Fusion of
TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result
of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood 90:2535-2540.
122. Lacronique, V., A. Boureux, V.D. Valle, H. Poirel, C.T. Quang, M. Mauchauffe, C.
Berthou, M. Lessard, R. Berger, J. Ghysdael, and O.A. Bernard. 1997. A TEL-JAK2
fusion protein with constitutive kinase activity in human leukemia. Science 278:1309-
1312.
123. Carron, C., F. Cormier, A. Janin, V. Lacronique, M. Giovannini, M.T. Daniel, O.
Bernard, and J. Ghysdael. 2000. TEL-JAK2 transgenic mice develop T-cell leukemia.
Blood 95:3891-3899.
124. Flex, E., V. Petrangeli, L. Stella, S. Chiaretti, T. Hornakova, L. Knoops, C. Ariola, V.
Fodale, E. Clappier, F. Paoloni, S. Martinelli, A. Fragale, M. Sanchez, S. Tavolaro, M.
Messina, G. Cazzaniga, A. Camera, G. Pizzolo, A. Tornesello, M. Vignetti, A.
Battistini, H. Cave, B.D. Gelb, J.C. Renauld, A. Biondi, S.N. Constantinescu, R. Foa,
and M. Tartaglia. 2008. Somatically acquired JAK1 mutations in adult acute
lymphoblastic leukemia. J Exp Med 205:751-758.
125. Gilliland, D.G., and J.D. Griffin. 2002. The roles of FLT3 in hematopoiesis and
leukemia. Blood 100:1532-1542.
126. Paietta, E., A.A. Ferrando, D. Neuberg, J.M. Bennett, J. Racevskis, H. Lazarus, G.
Dewald, J.M. Rowe, P.H. Wiernik, M.S. Tallman, and A.T. Look. 2004. Activating
FLT3 mutations in CD117/KIT(+) T-cell acute lymphoblastic leukemias. Blood
104:558-560.
127. Van Vlierberghe, P., J.P. Meijerink, R.W. Stam, W. van der Smissen, E.R. van Wering,
H.B. Beverloo, and R. Pieters. 2005. Activating FLT3 mutations in CD4+/CD8-
pediatric T-cell acute lymphoblastic leukemias. Blood 106:4414-4415.
128. Campbell, S.L., R. Khosravi-Far, K.L. Rossman, G.J. Clark, and C.J. Der. 1998.
Increasing complexity of Ras signaling. Oncogene 17:1395-1413.
129. Bos, J.L. 1989. ras oncogenes in human cancer: a review. Cancer Res 49:4682-4689.
130. Neubauer, A., R.K. Dodge, S.L. George, F.R. Davey, R.T. Silver, C.A. Schiffer, R.J.
Mayer, E.D. Ball, D. Wurster-Hill, C.D. Bloomfield, and et al. 1994. Prognostic
importance of mutations in the ras proto-oncogenes in de novo acute myeloid leukemia.
Blood 83:1603-1611.
131. Yokota, S., M. Nakao, S. Horiike, T. Seriu, T. Iwai, H. Kaneko, H. Azuma, T. Oka, T.
Takeda, A. Watanabe, A. Kikuta, K. Asami, I. Sekine, T. Matsushita, T. Tsuhciya, J.

Introduction
57
Mimaya, S. Koizumi, M. Miyake, K. Nishikawa, Y. Takaue, Y. Kawano, A. Iwai, Y.
Ishida, K. Matsumoto, and T. Fujimoto. 1998. Mutational analysis of the N-ras gene in
acute lymphoblastic leukemia: a study of 125 Japanese pediatric cases. Int J Hematol
67:379-387.
132. Kawamura, M., A. Kikuchi, S. Kobayashi, R. Hanada, K. Yamamoto, K. Horibe, T.
Shikano, K. Ueda, K. Hayashi, T. Sekiya, and et al. 1995. Mutations of the p53 and ras
genes in childhood t(1;19)-acute lymphoblastic leukemia. Blood 85:2546-2552.
133. Gutierrez, A., T. Sanda, R. Grebliunaite, A. Carracedo, L. Salmena, Y. Ahn, S.
Dahlberg, D. Neuberg, L.A. Moreau, S.S. Winter, R. Larson, J. Zhang, A. Protopopov,
L. Chin, P.P. Pandolfi, L.B. Silverman, S.P. Hunger, S.E. Sallan, and A.T. Look. 2009.
High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic
leukemia. Blood 114:647-650.
134. von Lintig, F.C., I. Huvar, P. Law, M.B. Diccianni, A.L. Yu, and G.R. Boss. 2000. Ras
activation in normal white blood cells and childhood acute lymphoblastic leukemia.
Clin Cancer Res 6:1804-1810.
135. Shi, Y., and J. Massague. 2003. Mechanisms of TGF-beta signaling from cell
membrane to the nucleus. Cell 113:685-700.
136. Derynck, R., R.J. Akhurst, and A. Balmain. 2001. TGF-beta signaling in tumor
suppression and cancer progression. Nat Genet 29:117-129.
137. Derynck, R., and Y.E. Zhang. 2003. Smad-dependent and Smad-independent pathways
in TGF-beta family signalling. Nature 425:577-584.
138. Lucas, P.J., N. McNeil, E. Hilgenfeld, B. Choudhury, S.J. Kim, M.A. Eckhaus, T. Ried,
and R.E. Gress. 2004. Transforming growth factor-beta pathway serves as a primary
tumor suppressor in CD8+ T cell tumorigenesis. Cancer Res 64:6524-6529.
139. Wolfraim, L.A., T.M. Fernandez, M. Mamura, W.L. Fuller, R. Kumar, D.E. Cole, S.
Byfield, A. Felici, K.C. Flanders, T.M. Walz, A.B. Roberts, P.D. Aplan, F.M. Balis, and
J.J. Letterio. 2004. Loss of Smad3 in acute T-cell lymphoblastic leukemia. N Engl J
Med 351:552-559.
140. Remke, M., S. Pfister, C. Kox, G. Toedt, N. Becker, A. Benner, W. Werft, S. Breit, S.
Liu, F. Engel, A. Wittmann, M. Zimmermann, M. Stanulla, M. Schrappe, W.D.
Ludwig, C.R. Bartram, B. Radlwimmer, M.U. Muckenthaler, P. Lichter, and A.E.
Kulozik. 2009. High-resolution genomic profiling of childhood T-ALL reveals frequent
copy-number alterations affecting the TGF-beta and PI3K-AKT pathways and deletions
at 6q15-16.1 as a genomic marker for unfavorable early treatment response. Blood
114:1053-1062.
141. Gutierrez, A., T. Sanda, W. Ma, J. Zhang, R. Grebliunaite, S. Dahlberg, D. Neuberg, A.
Protopopov, S.S. Winter, R.S. Larson, M.J. Borowitz, L.B. Silverman, L. Chin, S.P.

Introduction
58
Hunger, C. Jamieson, S.E. Sallan, and A.T. Look. 2010. Inactivation of LEF1 in T-cell
acute lymphoblastic leukemia. Blood 115:2845-2851.
142. van Noort, M., and H. Clevers. 2002. TCF transcription factors, mediators of Wnt-
signaling in development and cancer. Dev Biol 244:1-8.
143. Nawshad, A., D. Medici, C.C. Liu, and E.D. Hay. 2007. TGFbeta3 inhibits E-cadherin
gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1
transcription complex. J Cell Sci 120:1646-1653.
144. Lai, E.C. 2004. Notch signaling: control of cell communication and cell fate.
Development 131:965-973.
145. Duncan, A.W., F.M. Rattis, L.N. DiMascio, K.L. Congdon, G. Pazianos, C. Zhao, K.
Yoon, J.M. Cook, K. Willert, N. Gaiano, and T. Reya. 2005. Integration of Notch and
Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol 6:314-322.
146. Miele, L. 2006. Notch signaling. Clin Cancer Res 12:1074-1079.
147. Ellisen, L.W., J. Bird, D.C. West, A.L. Soreng, T.C. Reynolds, S.D. Smith, and J. Sklar.
1991. TAN-1, the human homolog of the Drosophila notch gene, is broken by
chromosomal translocations in T lymphoblastic neoplasms. Cell 66:649-661.
148. Sulis, M.L., O. Williams, T. Palomero, V. Tosello, S. Pallikuppam, P.J. Real, K.
Barnes, L. Zuurbier, J.P. Meijerink, and A.A. Ferrando. 2008. NOTCH1 extracellular
juxtamembrane expansion mutations in T-ALL. Blood 112:733-740.
149. Ferrando, A.A., D.S. Neuberg, J. Staunton, M.L. Loh, C. Huard, S.C. Raimondi, F.G.
Behm, C.H. Pui, J.R. Downing, D.G. Gilliland, E.S. Lander, T.R. Golub, and A.T.
Look. 2002. Gene expression signatures define novel oncogenic pathways in T cell
acute lymphoblastic leukemia. Cancer Cell 1:75-87.
150. Park, S., N. Chapuis, J. Tamburini, V. Bardet, P. Cornillet-Lefebvre, L. Willems, A.
Green, P. Mayeux, C. Lacombe, and D. Bouscary. 2010. Role of the PI3K/AKT and
mTOR signaling pathways in acute myeloid leukemia. Haematologica 95:819-828.
151. Steelman, L.S., S.L. Abrams, J. Whelan, F.E. Bertrand, D.E. Ludwig, J. Basecke, M.
Libra, F. Stivala, M. Milella, A. Tafuri, P. Lunghi, A. Bonati, A.M. Martelli, and J.A.
McCubrey. 2008. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and
Jak/STAT pathways to leukemia. Leukemia 22:686-707.
152. Panwalkar, A., S. Verstovsek, and F.J. Giles. 2004. Mammalian target of rapamycin
inhibition as therapy for hematologic malignancies. Cancer 100:657-666.
153. Guertin, D.A., and D.M. Sabatini. 2007. Defining the role of mTOR in cancer. Cancer
Cell 12:9-22.
154. Lawlor, M.A., and D.R. Alessi. 2001. PKB/Akt: a key mediator of cell proliferation,
survival and insulin responses? J Cell Sci 114:2903-2910.

Introduction
59
155. Hay, N. 2005. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 8:179-
183.
156. Nakae, J., B.C. Park, and D. Accili. 1999. Insulin stimulates phosphorylation of the
forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive
pathway. J Biol Chem 274:15982-15985.
157. Brunet, A., A. Bonni, M.J. Zigmond, M.Z. Lin, P. Juo, L.S. Hu, M.J. Anderson, K.C.
Arden, J. Blenis, and M.E. Greenberg. 1999. Akt promotes cell survival by
phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857-868.
158. Cross, D.A., D.R. Alessi, P. Cohen, M. Andjelkovich, and B.A. Hemmings. 1995.
Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B.
Nature 378:785-789.
159. del Peso, L., M. Gonzalez-Garcia, C. Page, R. Herrera, and G. Nunez. 1997.
Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science
278:687-689.
160. Scheid, M.P., and V. Duronio. 1998. Dissociation of cytokine-induced phosphorylation
of Bad and activation of PKB/akt: involvement of MEK upstream of Bad
phosphorylation. Proc Natl Acad Sci U S A 95:7439-7444.
161. Inoki, K., Y. Li, T. Zhu, J. Wu, and K.L. Guan. 2002. TSC2 is phosphorylated and
inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648-657.
162. Burgering, B.M., and G.J. Kops. 2002. Cell cycle and death control: long live
Forkheads. Trends Biochem Sci 27:352-360.
163. Greer, E.L., and A. Brunet. 2005. FOXO transcription factors at the interface between
longevity and tumor suppression. Oncogene 24:7410-7425.
164. Long, X., Y. Lin, S. Ortiz-Vega, K. Yonezawa, and J. Avruch. 2005. Rheb binds and
regulates the mTOR kinase. Curr Biol 15:702-713.
165. Sarbassov, D.D., D.A. Guertin, S.M. Ali, and D.M. Sabatini. 2005. Phosphorylation and
regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098-1101.
166. Holz, M.K., B.A. Ballif, S.P. Gygi, and J. Blenis. 2005. mTOR and S6K1 mediate
assembly of the translation preinitiation complex through dynamic protein interchange
and ordered phosphorylation events. Cell 123:569-580.
167. Holz, M.K., and J. Blenis. 2005. Identification of S6 kinase 1 as a novel mammalian
target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem 280:26089-26093.
168. Park, I.H., R. Bachmann, H. Shirazi, and J. Chen. 2002. Regulation of ribosomal S6
kinase 2 by mammalian target of rapamycin. J Biol Chem 277:31423-31429.
169. Dorrello, N.V., A. Peschiaroli, D. Guardavaccaro, N.H. Colburn, N.E. Sherman, and M.
Pagano. 2006. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein
translation and cell growth. Science 314:467-471.

Introduction
60
170. Harrington, L.S., G.M. Findlay, A. Gray, T. Tolkacheva, S. Wigfield, H. Rebholz, J.
Barnett, N.R. Leslie, S. Cheng, P.R. Shepherd, I. Gout, C.P. Downes, and R.F. Lamb.
2004. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of
IRS proteins. J Cell Biol 166:213-223.
171. Hresko, R.C., and M. Mueckler. 2005. mTOR.RICTOR is the Ser473 kinase for
Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem 280:40406-40416.
172. Giles, F.J., and M. Albitar. 2005. Mammalian target of rapamycin as a therapeutic target
in leukemia. Curr Mol Med 5:653-661.
173. Sakai, A., C. Thieblemont, A. Wellmann, E.S. Jaffe, and M. Raffeld. 1998. PTEN gene
alterations in lymphoid neoplasms. Blood 92:3410-3415.
174. Shan, X., M.J. Czar, S.C. Bunnell, P. Liu, Y. Liu, P.L. Schwartzberg, and R.L. Wange.
2000. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the
plasma membrane and hyperresponsiveness to CD3 stimulation. Mol Cell Biol 20:6945-
6957.
175. Xu, Z., D. Stokoe, L.P. Kane, and A. Weiss. 2002. The inducible expression of the
tumor suppressor gene PTEN promotes apoptosis and decreases cell size by inhibiting
the PI3K/Akt pathway in Jurkat T cells. Cell Growth Differ 13:285-296.
176. Uddin, S., A. Hussain, K. Al-Hussein, L.C. Platanias, and K.G. Bhatia. 2004. Inhibition
of phosphatidylinositol 3'-kinase induces preferentially killing of PTEN-null T
leukemias through AKT pathway. Biochem Biophys Res Commun 320:932-938.
177. Avellino, R., S. Romano, R. Parasole, R. Bisogni, A. Lamberti, V. Poggi, S. Venuta,
and M.F. Romano. 2005. Rapamycin stimulates apoptosis of childhood acute
lymphoblastic leukemia cells. Blood 106:1400-1406.
178. Silva, A., J.A. Yunes, B.A. Cardoso, L.R. Martins, P.Y. Jotta, M. Abecasis, A.E.
Nowill, N.R. Leslie, A.A. Cardoso, and J.T. Barata. 2008. PTEN posttranslational
inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell
leukemia viability. J Clin Invest 118:3762-3774.
179. Jotta, P.Y., M.A. Ganazza, A. Silva, M.B. Viana, M.J. da Silva, L.J. Zambaldi, J.T.
Barata, S.R. Brandalise, and J.A. Yunes. 2010. Negative prognostic impact of PTEN
mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia 24:239-242.
180. Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias.
Science 278:1059-1064.
181. Lawrence, P.A., and G. Morata. 1994. Homeobox genes: their function in Drosophila
segmentation and pattern formation. Cell 78:181-189.
182. Deschamps, J., and J. van Nes. 2005. Developmental regulation of the Hox genes
during axial morphogenesis in the mouse. Development 132:2931-2942.

Introduction
61
183. Favier, B., and P. Dolle. 1997. Developmental functions of mammalian Hox genes. Mol
Hum Reprod 3:115-131.
184. van Oostveen, J., J. Bijl, F. Raaphorst, J. Walboomers, and C. Meijer. 1999. The role of
homeobox genes in normal hematopoiesis and hematological malignancies. Leukemia
13:1675-1690.
185. Gehring, W.J., Y.Q. Qian, M. Billeter, K. Furukubo-Tokunaga, A.F. Schier, D.
Resendez-Perez, M. Affolter, G. Otting, and K. Wuthrich. 1994. Homeodomain-DNA
recognition. Cell 78:211-223.
186. Speleman, F., B. Cauwelier, N. Dastugue, J. Cools, B. Verhasselt, B. Poppe, N. Van
Roy, J. Vandesompele, C. Graux, A. Uyttebroeck, M. Boogaerts, B. De Moerloose, Y.
Benoit, D. Selleslag, J. Billiet, A. Robert, F. Huguet, P. Vandenberghe, A. De Paepe, P.
Marynen, and A. Hagemeijer. 2005. A new recurrent inversion, inv(7)(p15q34), leads to
transcriptional activation of HOXA10 and HOXA11 in a subset of T-cell acute
lymphoblastic leukemias. Leukemia 19:358-366.
187. Su, X., H. Drabkin, E. Clappier, E. Morgado, M. Busson, S. Romana, J. Soulier, R.
Berger, O.A. Bernard, and C. Lavau. 2006. Transforming potential of the T-cell acute
lymphoblastic leukemia-associated homeobox genes HOXA13, TLX1, and TLX3.
Genes Chromosomes Cancer 45:846-855.
188. Hatano, M., C.W. Roberts, M. Minden, W.M. Crist, and S.J. Korsmeyer. 1991.
Deregulation of a homeobox gene, HOX11, by the t(10;14) in T cell leukemia. Science
253:79-82.
189. Kennedy, M.A., R. Gonzalez-Sarmiento, U.R. Kees, F. Lampert, N. Dear, T. Boehm,
and T.H. Rabbitts. 1991. HOX11, a homeobox-containing T-cell oncogene on human
chromosome 10q24. Proc Natl Acad Sci U S A 88:8900-8904.
190. Ferrando, A.A., S. Herblot, T. Palomero, M. Hansen, T. Hoang, E.A. Fox, and A.T.
Look. 2004. Biallelic transcriptional activation of oncogenic transcription factors in T-
cell acute lymphoblastic leukemia. Blood 103:1909-1911.
191. Kees, U.R., N.A. Heerema, R. Kumar, P.M. Watt, D.L. Baker, M.K. La, F.M. Uckun,
and H.N. Sather. 2003. Expression of HOX11 in childhood T-lineage acute
lymphoblastic leukaemia can occur in the absence of cytogenetic aberration at 10q24: a
study from the Children's Cancer Group (CCG). Leukemia 17:887-893.
192. Yamamoto, H., M. Hatano, Y. Iitsuka, N.S. Mahyar, M. Yamamoto, and T. Tokuhisa.
1995. Two forms of Hox11 a T cell leukemia oncogene, are expressed in fetal spleen
but not in primary lymphocytes. Mol Immunol 32:1177-1182.
193. Hawley, R.G., A.Z. Fong, M. Lu, and T.S. Hawley. 1994. The HOX11 homeobox-
containing gene of human leukemia immortalizes murine hematopoietic precursors.
Oncogene 9:1-12.

Introduction
62
194. Bergeron, J., E. Clappier, I. Radford, A. Buzyn, C. Millien, G. Soler, P. Ballerini, X.
Thomas, J. Soulier, H. Dombret, E.A. Macintyre, and V. Asnafi. 2007. Prognostic and
oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110:2324-
2330.
195. Riz, I., and R.G. Hawley. 2005. G1/S transcriptional networks modulated by the
HOX11/TLX1 oncogene of T-cell acute lymphoblastic leukemia. Oncogene 24:5561-
5575.
196. Bernard, O.A., M. Busson-LeConiat, P. Ballerini, M. Mauchauffe, V. Della Valle, R.
Monni, F. Nguyen Khac, T. Mercher, V. Penard-Lacronique, P. Pasturaud, L. Gressin,
R. Heilig, M.T. Daniel, M. Lessard, and R. Berger. 2001. A new recurrent and specific
cryptic translocation, t(5;14)(q35;q32), is associated with expression of the Hox11L2
gene in T acute lymphoblastic leukemia. Leukemia 15:1495-1504.
197. Su, X.Y., V. Della-Valle, I. Andre-Schmutz, C. Lemercier, I. Radford-Weiss, P.
Ballerini, M. Lessard, M. Lafage-Pochitaloff, F. Mugneret, R. Berger, S.P. Romana, O.
Bernard, and V. Penard-Lacronique. 2006. HOX11L2/TLX3 is transcriptionally
activated through T-cell regulatory elements downstream of BCL11B as a result of the
t(5;14)(q35;q32). Blood
198. Su, X.Y., M. Busson, V. Della Valle, P. Ballerini, N. Dastugue, P. Talmant, A.A.
Ferrando, D. Baudry-Bluteau, S. Romana, R. Berger, and O.A. Bernard. 2004. Various
types of rearrangements target TLX3 locus in T-cell acute lymphoblastic leukemia.
Genes Chromosomes Cancer 41:243-249.
199. Hansen-Hagge, T.E., M. Schafer, H. Kiyoi, S.W. Morris, J.A. Whitlock, P. Koch, I.
Bohlmann, C. Mahotka, C.R. Bartram, and J.W. Janssen. 2002. Disruption of the
RanBP17/Hox11L2 region by recombination with the TCRdelta locus in acute
lymphoblastic leukemias with t(5;14)(q34;q11). Leukemia 16:2205-2212.
200. Ballerini, P., A. Blaise, M. Busson-Le Coniat, X.Y. Su, J. Zucman-Rossi, M. Adam, J.
van den Akker, C. Perot, B. Pellegrino, J. Landman-Parker, L. Douay, R. Berger, and
O.A. Bernard. 2002. HOX11L2 expression defines a clinical subtype of pediatric T-
ALL associated with poor prognosis. Blood 100:991-997.
201. Jones, S. 2004. An overview of the basic helix-loop-helix proteins. Genome Biol 5:226.
202. Bain, G., M.W. Quong, R.S. Soloff, S.M. Hedrick, and C. Murre. 1999. Thymocyte
maturation is regulated by the activity of the helix-loop-helix protein, E47. J Exp Med
190:1605-1616.
203. Engel, I., C. Johns, G. Bain, R.R. Rivera, and C. Murre. 2001. Early thymocyte
development is regulated by modulation of E2A protein activity. J Exp Med 194:733-
745.

Introduction
63
204. Ikawa, T., H. Kawamoto, A.W. Goldrath, and C. Murre. 2006. E proteins and Notch
signaling cooperate to promote T cell lineage specification and commitment. J Exp Med
203:1329-1342.
205. Bain, G., W.J. Romanow, K. Albers, W.L. Havran, and C. Murre. 1999. Positive and
negative regulation of V(D)J recombination by the E2A proteins. J Exp Med 189:289-
300.
206. Takeuchi, A., S. Yamasaki, K. Takase, F. Nakatsu, H. Arase, M. Onodera, and T. Saito.
2001. E2A and HEB activate the pre-TCR alpha promoter during immature T cell
development. J Immunol 167:2157-2163.
207. Tremblay, M., S. Herblot, E. Lecuyer, and T. Hoang. 2003. Regulation of pT alpha gene
expression by a dosage of E2A, HEB, and SCL. J Biol Chem 278:12680-12687.
208. Bain, G., I. Engel, E.C. Robanus Maandag, H.P. te Riele, J.R. Voland, L.L. Sharp, J.
Chun, B. Huey, D. Pinkel, and C. Murre. 1997. E2A deficiency leads to abnormalities
in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol
Cell Biol 17:4782-4791.
209. Yan, W., A.Z. Young, V.C. Soares, R. Kelley, R. Benezra, and Y. Zhuang. 1997. High
incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Mol
Cell Biol 17:7317-7327.
210. Bash, R.O., S. Hall, C.F. Timmons, W.M. Crist, M. Amylon, R.G. Smith, and R. Baer.
1995. Does activation of the TAL1 gene occur in a majority of patients with T-cell
acute lymphoblastic leukemia? A pediatric oncology group study. Blood 86:666-676.
211. Xia, Y., L. Brown, C.Y. Yang, J.T. Tsan, M.J. Siciliano, R. Espinosa, III, M.M. Le
Beau, and R.J. Baer. 1991. TAL2, a helix-loop-helix gene activated by the
(7;9)(q34;q32) translocation in human T-cell leukemia. Proc Natl Acad Sci U S A
88:11416-11420.
212. Wang, J., S.N. Jani-Sait, E.A. Escalon, A.J. Carroll, P.J. de Jong, I.R. Kirsch, and P.D.
Aplan. 2000. The t(14;21)(q11.2;q22) chromosomal translocation associated with T-cell
acute lymphoblastic leukemia activates the BHLHB1 gene. Proc Natl Acad Sci U S A
97:3497-3502.
213. Mellentin, J.D., S.D. Smith, and M.L. Cleary. 1989. lyl-1, a novel gene altered by
chromosomal translocation in T cell leukemia, codes for a protein with a helix-loop-
helix DNA binding motif. Cell 58:77-83.
214. Chen, Q., J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A. Carroll, W. Crist, B.
Ozanne, M.J. Siciliano, and R. Baer. 1990. The tal gene undergoes chromosome
translocation in T cell leukemia and potentially encodes a helix-loop-helix protein.
Embo J 9:415-424.

Introduction
64
215. Begley, C.G., P.D. Aplan, M.P. Davey, K. Nakahara, K. Tchorz, J. Kurtzberg, M.S.
Hershfield, B.F. Haynes, D.I. Cohen, T.A. Waldmann, and et al. 1989. Chromosomal
translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor
delta-chain diversity region and results in a previously unreported fusion transcript.
Proc Natl Acad Sci U S A 86:2031-2035.
216. Rabbitts, T.H. 1998. LMO T-cell translocation oncogenes typify genes activated by
chromosomal translocations that alter transcription and developmental processes. Genes
Dev 12:2651-2657.
217. Nam, C.H., and T.H. Rabbitts. 2006. The role of LMO2 in development and in T cell
leukemia after chromosomal translocation or retroviral insertion. Mol Ther 13:15-25.
218. McGuire, E.A., R.D. Hockett, K.M. Pollock, M.F. Bartholdi, S.J. O'Brien, and S.J.
Korsmeyer. 1989. The t(11;14)(p15;q11) in a T-cell acute lymphoblastic leukemia cell
line activates multiple transcripts, including Ttg-1, a gene encoding a potential zinc
finger protein. Mol Cell Biol 9:2124-2132.
219. Royer-Pokora, B., U. Loos, and W.D. Ludwig. 1991. TTG-2, a new gene encoding a
cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia
with the t(11;14)(p13;q11). Oncogene 6:1887-1893.
220. Van Vlierberghe, P., M. van Grotel, H.B. Beverloo, C. Lee, T. Helgason, J. Buijs-
Gladdines, M. Passier, E.R. van Wering, A.J. Veerman, W.A. Kamps, J.P. Meijerink,
and R. Pieters. 2006. The cryptic chromosomal deletion del(11)(p12p13) as a new
activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood
108:3520-3529.
221. Pike-Overzet, K., D. de Ridder, F. Weerkamp, M.R. Baert, M.M. Verstegen, M.H.
Brugman, S.J. Howe, M.J. Reinders, A.J. Thrasher, G. Wagemaker, J.J. van Dongen,
and F.J. Staal. 2007. Ectopic retroviral expression of LMO2, but not IL2Rgamma,
blocks human T-cell development from CD34+ cells: implications for leukemogenesis
in gene therapy. Leukemia 21:754-763.
222. McGuire, E.A., C.E. Rintoul, G.M. Sclar, and S.J. Korsmeyer. 1992. Thymic
overexpression of Ttg-1 in transgenic mice results in T-cell acute lymphoblastic
leukemia/lymphoma. Mol Cell Biol 12:4186-4196.
223. Larson, R.C., P. Fisch, T.A. Larson, I. Lavenir, T. Langford, G. King, and T.H.
Rabbitts. 1994. T cell tumours of disparate phenotype in mice transgenic for Rbtn-2.
Oncogene 9:3675-3681.
224. Larson, R.C., H. Osada, T.A. Larson, I. Lavenir, and T.H. Rabbitts. 1995. The
oncogenic LIM protein Rbtn2 causes thymic developmental aberrations that precede
malignancy in transgenic mice. Oncogene 11:853-862.

Introduction
65
225. Neale, G.A., J.E. Rehg, and R.M. Goorha. 1995. Ectopic expression of rhombotin-2
causes selective expansion of CD4-CD8- lymphocytes in the thymus and T-cell tumors
in transgenic mice. Blood 86:3060-3071.
226. Larson, R.C., I. Lavenir, T.A. Larson, R. Baer, A.J. Warren, I. Wadman, K. Nottage,
and T.H. Rabbitts. 1996. Protein dimerization between Lmo2 (Rbtn2) and Tal1 alters
thymocyte development and potentiates T cell tumorigenesis in transgenic mice. Embo
J 15:1021-1027.
227. Chervinsky, D.S., X.F. Zhao, D.H. Lam, M. Ellsworth, K.W. Gross, and P.D. Aplan.
1999. Disordered T-cell development and T-cell malignancies in SCL LMO1 double-
transgenic mice: parallels with E2A-deficient mice. Mol Cell Biol 19:5025-5035.
228. Tremblay, M., C.S. Tremblay, S. Herblot, P.D. Aplan, J. Hebert, C. Perreault, and T.
Hoang. 2010. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and
LMO1 oncogenes. Genes Dev 24:1093-1105.
229. Hacein-Bey-Abina, S., C. Von Kalle, M. Schmidt, M.P. McCormack, N. Wulffraat, P.
Leboulch, A. Lim, C.S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P.
Fraser, J.I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A.
Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E.
Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L.E. Leiva, M. Wissler, C. Prinz, T.H.
Rabbitts, F. Le Deist, A. Fischer, and M. Cavazzana-Calvo. 2003. LMO2-associated
clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science
302:415-419.
230. Hacein-Bey-Abina, S., A. Garrigue, G.P. Wang, J. Soulier, A. Lim, E. Morillon, E.
Clappier, L. Caccavelli, E. Delabesse, K. Beldjord, V. Asnafi, E. MacIntyre, L. Dal
Cortivo, I. Radford, N. Brousse, F. Sigaux, D. Moshous, J. Hauer, A. Borkhardt, B.H.
Belohradsky, U. Wintergerst, M.C. Velez, L. Leiva, R. Sorensen, N. Wulffraat, S.
Blanche, F.D. Bushman, A. Fischer, and M. Cavazzana-Calvo. 2008. Insertional
oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin
Invest 118:3132-3142.
231. Schmidt, M., S. Hacein-Bey-Abina, M. Wissler, F. Carlier, A. Lim, C. Prinz, H. Glimm,
I. Andre-Schmutz, C. Hue, A. Garrigue, F. Le Deist, C. Lagresle, A. Fischer, M.
Cavazzana-Calvo, and C. von Kalle. 2005. Clonal evidence for the transduction of
CD34+ cells with lymphomyeloid differentiation potential and self-renewal capacity in
the SCID-X1 gene therapy trial. Blood 105:2699-2706.
232. Deichmann, A., S. Hacein-Bey-Abina, M. Schmidt, A. Garrigue, M.H. Brugman, J. Hu,
H. Glimm, G. Gyapay, B. Prum, C.C. Fraser, N. Fischer, K. Schwarzwaelder, M.L.
Siegler, D. de Ridder, K. Pike-Overzet, S.J. Howe, A.J. Thrasher, G. Wagemaker, U.
Abel, F.J. Staal, E. Delabesse, J.L. Villeval, B. Aronow, C. Hue, C. Prinz, M. Wissler,

Introduction
66
C. Klanke, J. Weissenbach, I. Alexander, A. Fischer, C. von Kalle, and M. Cavazzana-
Calvo. 2007. Vector integration is nonrandom and clustered and influences the fate of
lymphopoiesis in SCID-X1 gene therapy. J Clin Invest 117:2225-2232.
233. Howe, S.J., M.R. Mansour, K. Schwarzwaelder, C. Bartholomae, M. Hubank, H.
Kempski, M.H. Brugman, K. Pike-Overzet, S.J. Chatters, D. de Ridder, K.C. Gilmour,
S. Adams, S.I. Thornhill, K.L. Parsley, F.J. Staal, R.E. Gale, D.C. Linch, J. Bayford, L.
Brown, M. Quaye, C. Kinnon, P. Ancliff, D.K. Webb, M. Schmidt, C. von Kalle, H.B.
Gaspar, and A.J. Thrasher. 2008. Insertional mutagenesis combined with acquired
somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients.
J Clin Invest 118:3143-3150.
234. Meyer, C., B. Schneider, S. Jakob, S. Strehl, A. Attarbaschi, S. Schnittger, C. Schoch,
M.W. Jansen, J.J. van Dongen, M.L. den Boer, R. Pieters, M.G. Ennas, E. Angelucci,
U. Koehl, J. Greil, F. Griesinger, U. Zur Stadt, C. Eckert, T. Szczepanski, F.K. Niggli,
B.W. Schafer, H. Kempski, H.J. Brady, J. Zuna, J. Trka, L.L. Nigro, A. Biondi, E.
Delabesse, E. Macintyre, M. Stanulla, M. Schrappe, O.A. Haas, T. Burmeister, T.
Dingermann, T. Klingebiel, and R. Marschalek. 2006. The MLL recombinome of acute
leukemias. Leukemia 20:777-784.
235. Schumacher, A., and T. Magnuson. 1997. Murine Polycomb- and trithorax-group genes
regulate homeotic pathways and beyond. Trends Genet 13:167-170.
236. Milne, T.A., S.D. Briggs, H.W. Brock, M.E. Martin, D. Gibbs, C.D. Allis, and J.L.
Hess. 2002. MLL targets SET domain methyltransferase activity to Hox gene
promoters. Mol Cell 10:1107-1117.
237. Hayette, S., I. Tigaud, V. Maguer-Satta, L. Bartholin, X. Thomas, C. Charrin, M.
Gadoux, J.P. Magaud, and R. Rimokh. 2002. Recurrent involvement of the MLL gene
in adult T-lineage acute lymphoblastic leukemia. Blood 99:4647-4649.
238. Ferrando, A.A., S.A. Armstrong, D.S. Neuberg, S.E. Sallan, L.B. Silverman, S.J.
Korsmeyer, and A.T. Look. 2003. Gene expression signatures in MLL-rearranged T-
lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood
102:262-268.
239. Armstrong, S.A., J.E. Staunton, L.B. Silverman, R. Pieters, M.L. den Boer, M.D.
Minden, S.E. Sallan, E.S. Lander, T.R. Golub, and S.J. Korsmeyer. 2002. MLL
translocations specify a distinct gene expression profile that distinguishes a unique
leukemia. Nat Genet 30:41-47.
240. Rubnitz, J.E., B.M. Camitta, H. Mahmoud, S.C. Raimondi, A.J. Carroll, M.J. Borowitz,
J.J. Shuster, M.P. Link, D.J. Pullen, J.R. Downing, F.G. Behm, and C.H. Pui. 1999.
Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and
t(11;19)(q23;p13.3) translocation. J Clin Oncol 17:191-196.

Introduction
67
241. Dreyling, M.H., J.A. Martinez-Climent, M. Zheng, J. Mao, J.D. Rowley, and S.K.
Bohlander. 1996. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the
AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein
family. Proc Natl Acad Sci U S A 93:4804-4809.
242. Asnafi, V., I. Radford-Weiss, N. Dastugue, C. Bayle, D. Leboeuf, C. Charrin, R.
Garand, M. Lafage-Pochitaloff, E. Delabesse, A. Buzyn, X. Troussard, and E.
Macintyre. 2003. CALM-AF10 is a common fusion transcript in T-ALL and is specific
to the TCRgammadelta lineage. Blood 102:1000-1006.
243. Dreyling, M.H., K. Schrader, C. Fonatsch, B. Schlegelberger, D. Haase, C. Schoch, W.
Ludwig, H. Loffler, T. Buchner, B. Wormann, W. Hiddemann, and S.K. Bohlander.
1998. MLL and CALM are fused to AF10 in morphologically distinct subsets of acute
leukemia with translocation t(10;11): both rearrangements are associated with a poor
prognosis. Blood 91:4662-4667.
244. Dik, W.A., W. Brahim, C. Braun, V. Asnafi, N. Dastugue, O.A. Bernard, J.J. van
Dongen, A.W. Langerak, E.A. Macintyre, and E. Delabesse. 2005. CALM-AF10+ T-
ALL expression profiles are characterized by overexpression of HOXA and BMI1
oncogenes. Leukemia 19:1948-1957.
245. Hershfield, M.S., J. Kurtzberg, E. Harden, J.O. Moore, J. Whang-Peng, and B.F.
Haynes. 1984. Conversion of a stem cell leukemia from a T-lymphoid to a myeloid
phenotype induced by the adenosine deaminase inhibitor 2'-deoxycoformycin. Proc
Natl Acad Sci U S A 81:253-257.
246. Aplan, P.D., D.P. Lombardi, G.H. Reaman, H.N. Sather, G.D. Hammond, and I.R.
Kirsch. 1992. Involvement of the putative hematopoietic transcription factor SCL in T-
cell acute lymphoblastic leukemia. Blood 79:1327-1333.
247. Aplan, P.D., S.C. Raimondi, and I.R. Kirsch. 1992. Disruption of the SCL gene by a
t(1;3) translocation in a patient with T cell acute lymphoblastic leukemia. J Exp Med
176:1303-1310.
248. Fitzgerald, T.J., G.A. Neale, S.C. Raimondi, and R.M. Goorha. 1991. c-tal, a helix-
loop-helix protein, is juxtaposed to the T-cell receptor-beta chain gene by a reciprocal
chromosomal translocation: t(1;7)(p32;q35). Blood 78:2686-2695.
249. Francois, S., E. Delabesse, L. Baranger, M. Dautel, C. Foussard, M. Boasson, O.
Blanchet, O. Bernard, E.A. Macintyre, and N. Ifrah. 1998. Deregulated expression of
the TAL1 gene by t(1;5)(p32;31) in patient with T-cell acute lymphoblastic leukemia.
Genes Chromosomes Cancer 23:36-43.
250. Kelliher, M.A., D.C. Seldin, and P. Leder. 1996. Tal-1 induces T cell acute
lymphoblastic leukemia accelerated by casein kinase IIalpha. Embo J 15:5160-5166.

Introduction
68
251. O'Neil, J., M. Billa, S. Oikemus, and M. Kelliher. 2001. The DNA binding activity of
TAL-1 is not required to induce leukemia/lymphoma in mice. Oncogene 20:3897-3905.
252. O'Neil, J., J. Shank, N. Cusson, C. Murre, and M. Kelliher. 2004. TAL1/SCL induces
leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 5:587-596.
253. Aplan, P.D., C.A. Jones, D.S. Chervinsky, X. Zhao, M. Ellsworth, C. Wu, E.A.
McGuire, and K.W. Gross. 1997. An scl gene product lacking the transactivation
domain induces bony abnormalities and cooperates with LMO1 to generate T-cell
malignancies in transgenic mice. Embo J 16:2408-2419.
254. Herblot, S., A.M. Steff, P. Hugo, P.D. Aplan, and T. Hoang. 2000. SCL and LMO1
alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain
expression. Nat Immunol 1:138-144.
255. Leroy-Viard, K., M.A. Vinit, N. Lecointe, H. Jouault, U. Hibner, P.H. Romeo, and D.
Mathieu-Mahul. 1995. Loss of TAL-1 protein activity induces premature apoptosis of
Jurkat leukemic T cells upon medium depletion. Embo J 14:2341-2349.
256. Bernard, M., E. Delabesse, S. Novault, O. Hermine, and E.A. Macintyre. 1998.
Antiapoptotic effect of ectopic TAL1/SCL expression in a human leukemic T-cell line.
Cancer Res 58:2680-2687.
257. Engel, I., and C. Murre. 1999. Ectopic expression of E47 or E12 promotes the death of
E2A-deficient lymphomas. Proc Natl Acad Sci U S A 96:996-1001.
258. Park, S.T., G.P. Nolan, and X.H. Sun. 1999. Growth inhibition and apoptosis due to
restoration of E2A activity in T cell acute lymphoblastic leukemia cells. J Exp Med
189:501-508.
259. Lecuyer, E., and T. Hoang. 2004. SCL: from the origin of hematopoiesis to stem cells
and leukemia. Exp Hematol 32:11-24.
260. Aplan, P.D., C.G. Begley, V. Bertness, M. Nussmeier, A. Ezquerra, J. Coligan, and I.R.
Kirsch. 1990. The SCL gene is formed from a transcriptionally complex locus. Mol Cell
Biol 10:6426-6435.
261. Dhami, P., A.W. Bruce, J.H. Jim, S.C. Dillon, A. Hall, J.L. Cooper, N. Bonhoure, K.
Chiang, P.D. Ellis, C. Langford, R.M. Andrews, and D. Vetrie. 2010. Genomic
approaches uncover increasing complexities in the regulatory landscape at the human
SCL (TAL1) locus. PLoS One 5:e9059.
262. Begley, C.G., L. Robb, S. Rockman, J. Visvader, E.O. Bockamp, Y.S. Chan, and A.R.
Green. 1994. Structure of the gene encoding the murine SCL protein. Gene 138:93-99.
263. Bernard, O., O. Azogui, N. Lecointe, F. Mugneret, R. Berger, C.J. Larsen, and D.
Mathieu-Mahul. 1992. A third tal-1 promoter is specifically used in human T cell
leukemias. J Exp Med 176:919-925.

Introduction
69
264. Courtes, C., N. Lecointe, L. Le Cam, F. Baudoin, C. Sardet, and D. Mathieu-Mahul.
2000. Erythroid-specific inhibition of the tal-1 intragenic promoter is due to binding of
a repressor to a novel silencer. J Biol Chem 275:949-958.
265. Le Clech, M., E. Chalhoub, C. Dohet, V. Roure, S. Fichelson, F. Moreau-Gachelin, and
D. Mathieu. 2006. PU.1/Spi-1 binds to the human TAL-1 silencer to mediate its
activity. J Mol Biol 355:9-19.
266. Sanchez, M., B. Gottgens, A.M. Sinclair, M. Stanley, C.G. Begley, S. Hunter, and A.R.
Green. 1999. An SCL 3' enhancer targets developing endothelium together with
embryonic and adult haematopoietic progenitors. Development 126:3891-3904.
267. Gottgens, B., C. Broccardo, M.J. Sanchez, S. Deveaux, G. Murphy, J.R. Gothert, E.
Kotsopoulou, S. Kinston, L. Delaney, S. Piltz, L.M. Barton, K. Knezevic, W.N. Erber,
C.G. Begley, J. Frampton, and A.R. Green. 2004. The scl +18/19 stem cell enhancer is
not required for hematopoiesis: identification of a 5' bifunctional hematopoietic-
endothelial enhancer bound by Fli-1 and Elf-1. Mol Cell Biol 24:1870-1883.
268. Lecointe, N., O. Bernard, K. Naert, V. Joulin, C.J. Larsen, P.H. Romeo, and D.
Mathieu-Mahul. 1994. GATA-and SP1-binding sites are required for the full activity of
the tissue-specific promoter of the tal-1 gene. Oncogene 9:2623-2632.
269. Bockamp, E.O., F. McLaughlin, A.M. Murrell, B. Gottgens, L. Robb, C.G. Begley, and
A.R. Green. 1995. Lineage-restricted regulation of the murine SCL/TAL-1 promoter.
Blood 86:1502-1514.
270. Bockamp, E.O., J.L. Fordham, B. Gottgens, A.M. Murrell, M.J. Sanchez, and A.R.
Green. 1998. Transcriptional regulation of the stem cell leukemia gene by PU.1 and Elf-
1. J Biol Chem 273:29032-29042.
271. Bockamp, E.O., F. McLaughlin, B. Gottgens, A.M. Murrell, A.G. Elefanty, and A.R.
Green. 1997. Distinct mechanisms direct SCL/tal-1 expression in erythroid cells and
CD34 positive primitive myeloid cells. J Biol Chem 272:8781-8790.
272. Terme, J.M., M. Wencker, A. Favre-Bonvin, F. Bex, L. Gazzolo, M. Duc Dodon, and P.
Jalinot. 2008. Cross talk between expression of the human T-cell leukemia virus type 1
Tax transactivator and the oncogenic bHLH transcription factor TAL1. J Virol 82:7913-
7922.
273. Delabesse, E., S. Ogilvy, M.A. Chapman, S.G. Piltz, B. Gottgens, and A.R. Green.
2005. Transcriptional regulation of the SCL locus: identification of an enhancer that
targets the primitive erythroid lineage in vivo. Mol Cell Biol 25:5215-5225.
274. Gottgens, B., A. Nastos, S. Kinston, S. Piltz, E.C. Delabesse, M. Stanley, M.J. Sanchez,
A. Ciau-Uitz, R. Patient, and A.R. Green. 2002. Establishing the transcriptional
programme for blood: the SCL stem cell enhancer is regulated by a multiprotein
complex containing Ets and GATA factors. Embo J 21:3039-3050.

Introduction
70
275. Goldfarb, A.N., S. Goueli, D. Mickelson, and J.M. Greenberg. 1992. T-cell acute
lymphoblastic leukemia--the associated gene SCL/tal codes for a 42-Kd nuclear
phosphoprotein. Blood 80:2858-2866.
276. Cheng, J.T., M.H. Cobb, and R. Baer. 1993. Phosphorylation of the TAL1 oncoprotein
by the extracellular-signal-regulated protein kinase ERK1. Mol Cell Biol 13:801-808.
277. Hsu, H.L., I. Wadman, and R. Baer. 1994. Formation of in vivo complexes between the
TAL1 and E2A polypeptides of leukemic T cells. Proc Natl Acad Sci U S A 91:3181-
3185.
278. Bernard, M., E. Delabesse, L. Smit, C. Millien, I.R. Kirsch, J.L. Strominger, and E.A.
Macintyre. 1998. Helix-loop-helix (E2-5, HEB, TAL1 and Id1) protein interaction with
the TCRalphadelta enhancers. Int Immunol 10:1539-1549.
279. Hsu, H.L., L. Huang, J.T. Tsan, W. Funk, W.E. Wright, J.S. Hu, R.E. Kingston, and R.
Baer. 1994. Preferred sequences for DNA recognition by the TAL1 helix-loop-helix
proteins. Mol Cell Biol 14:1256-1265.
280. Hsu, H.L., I. Wadman, J.T. Tsan, and R. Baer. 1994. Positive and negative
transcriptional control by the TAL1 helix-loop-helix protein. Proc Natl Acad Sci U S A
91:5947-5951.
281. Aplan, P.D., K. Nakahara, S.H. Orkin, and I.R. Kirsch. 1992. The SCL gene product: a
positive regulator of erythroid differentiation. Embo J 11:4073-4081.
282. Xu, Z., S. Huang, L.S. Chang, A.D. Agulnick, and S.J. Brandt. 2003. Identification of a
TAL1 target gene reveals a positive role for the LIM domain-binding protein Ldb1 in
erythroid gene expression and differentiation. Mol Cell Biol 23:7585-7599.
283. Lahlil, R., E. Lecuyer, S. Herblot, and T. Hoang. 2004. SCL assembles a multifactorial
complex that determines glycophorin A expression. Mol Cell Biol 24:1439-1452.
284. Huang, S., Y. Qiu, R.W. Stein, and S.J. Brandt. 1999. p300 functions as a
transcriptional coactivator for the TAL1/SCL oncoprotein. Oncogene 18:4958-4967.
285. Zhao, X.F., and P.D. Aplan. 1999. The hematopoietic transcription factor SCL binds the
p44 subunit of TFIIH. J Biol Chem 274:1388-1393.
286. Huang, S., and S.J. Brandt. 2000. mSin3A regulates murine erythroleukemia cell
differentiation through association with the TAL1 (or SCL) transcription factor. Mol
Cell Biol 20:2248-2259.
287. Hu, X., X. Li, K. Valverde, X. Fu, C. Noguchi, Y. Qiu, and S. Huang. 2009. LSD1-
mediated epigenetic modification is required for TAL1 function and hematopoiesis.
Proc Natl Acad Sci U S A 106:10141-10146.
288. Wen, J., S. Huang, S.D. Pack, X. Yu, S.J. Brandt, and C.T. Noguchi. 2005. Tal1/SCL
binding to pericentromeric DNA represses transcription. J Biol Chem 280:12956-12966.

Introduction
71
289. Osada, H., G. Grutz, H. Axelson, A. Forster, and T.H. Rabbitts. 1995. Association of
erythroid transcription factors: complexes involving the LIM protein RBTN2 and the
zinc-finger protein GATA1. Proc Natl Acad Sci U S A 92:9585-9589.
290. Wadman, I.A., H. Osada, G.G. Grutz, A.D. Agulnick, H. Westphal, A. Forster, and T.H.
Rabbitts. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an
erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and
Ldb1/NLI proteins. Embo J 16:3145-3157.
291. Lecuyer, E., S. Herblot, M. Saint-Denis, R. Martin, C.G. Begley, C. Porcher, S.H.
Orkin, and T. Hoang. 2002. The SCL complex regulates c-kit expression in
hematopoietic cells through functional interaction with Sp1. Blood 100:2430-2440.
292. Ono, Y., N. Fukuhara, and O. Yoshie. 1998. TAL1 and LIM-only proteins
synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute
lymphoblastic leukemia by acting as cofactors for GATA3. Mol Cell Biol 18:6939-
6950.
293. Barton, L.M., B. Gottgens, and A.R. Green. 1999. The stem cell leukaemia (SCL) gene:
a critical regulator of haemopoietic and vascular development. Int J Biochem Cell Biol
31:1193-1207.
294. Elwood, N.J., H. Zogos, D.S. Pereira, J.E. Dick, and C.G. Begley. 1998. Enhanced
megakaryocyte and erythroid development from normal human CD34(+) cells:
consequence of enforced expression of SCL. Blood 91:3756-3765.
295. Visvader, J.E., Y. Fujiwara, and S.H. Orkin. 1998. Unsuspected role for the T-cell
leukemia protein SCL/tal-1 in vascular development. Genes Dev 12:473-479.
296. Bradley, C.K., E.A. Takano, M.A. Hall, J.R. Gothert, A.R. Harvey, C.G. Begley, and
J.A. van Eekelen. 2006. The essential haematopoietic transcription factor Scl is also
critical for neuronal development. Eur J Neurosci 23:1677-1689.
297. Krosl, G., G. He, M. Lefrancois, F. Charron, P.H. Romeo, P. Jolicoeur, I.R. Kirsch, M.
Nemer, and T. Hoang. 1998. Transcription factor SCL is required for c-kit expression
and c-Kit function in hemopoietic cells. J Exp Med 188:439-450.
298. Landry, J.R., S. Kinston, K. Knezevic, M.F. de Bruijn, N. Wilson, W.T. Nottingham,
M. Peitz, F. Edenhofer, J.E. Pimanda, K. Ottersbach, and B. Gottgens. 2008. Runx
genes are direct targets of Scl/Tal1 in the yolk sac and fetal liver. Blood 111:3005-3014.
299. Chasis, J.A., and N. Mohandas. 1992. Red blood cell glycophorins. Blood 80:1869-
1879.
300. Branton, D., C.M. Cohen, and J. Tyler. 1981. Interaction of cytoskeletal proteins on the
human erythrocyte membrane. Cell 24:24-32.

Introduction
72
301. Lausen, J., O. Pless, F. Leonard, O.N. Kuvardina, B. Koch, and A. Leutz. 2010. Targets
of the Tal1 transcription factor in erythrocytes: E2 ubiquitin conjugase regulation by
Tal1. J Biol Chem 285:5338-5346.
302. Hansson, A., C. Manetopoulos, J.I. Jonsson, and H. Axelson. 2003. The basic helix-
loop-helix transcription factor TAL1/SCL inhibits the expression of the p16INK4A and
pTalpha genes. Biochem Biophys Res Commun 312:1073-1081.
303. Ono, Y., N. Fukuhara, and O. Yoshie. 1997. Transcriptional activity of TAL1 in T cell
acute lymphoblastic leukemia (T-ALL) requires RBTN1 or -2 and induces TALLA1, a
highly specific tumor marker of T-ALL. J Biol Chem 272:4576-4581.
304. Chang, P.Y., K. Draheim, M.A. Kelliher, and S. Miyamoto. 2006. NFKB1 is a direct
target of the TAL1 oncoprotein in human T leukemia cells. Cancer Res 66:6008-6013.
305. Kusy, S., B. Gerby, N. Goardon, N. Gault, F. Ferri, D. Gerard, F. Armstrong, P.
Ballerini, J.M. Cayuela, A. Baruchel, F. Pflumio, and P.H. Romeo. 2010. NKX3.1 is a
direct TAL1 target gene that mediates proliferation of TAL1-expressing human T cell
acute lymphoblastic leukemia. J Exp Med 207:2141-2156.
306. Takagi, S., K. Fujikawa, T. Imai, N. Fukuhara, K. Fukudome, M. Minegishi, S.
Tsuchiya, T. Konno, Y. Hinuma, and O. Yoshie. 1995. Identification of a highly
specific surface marker of T-cell acute lymphoblastic leukemia and neuroblastoma as a
new member of the transmembrane 4 superfamily. Int J Cancer 61:706-715.
307. Zhao, D., P. McCaffery, K.J. Ivins, R.L. Neve, P. Hogan, W.W. Chin, and U.C. Drager.
1996. Molecular identification of a major retinoic-acid-synthesizing enzyme, a
retinaldehyde-specific dehydrogenase. Eur J Biochem 240:15-22.
308. Shen, M.M., and C. Abate-Shen. 2003. Roles of the Nkx3.1 homeobox gene in prostate
organogenesis and carcinogenesis. Dev Dyn 228:767-778.
309. Hayden, M.S., and S. Ghosh. 2004. Signaling to NF-kappaB. Genes Dev 18:2195-2224.
310. Chang, P.Y., and S. Miyamoto. 2006. Nuclear factor-kappaB dimer exchange promotes
a p21(waf1/cip1) superinduction response in human T leukemic cells. Mol Cancer Res
4:101-112.
311. Palomero, T., D.T. Odom, J. O'Neil, A.A. Ferrando, A. Margolin, D.S. Neuberg, S.S.
Winter, R.S. Larson, W. Li, X.S. Liu, R.A. Young, and A.T. Look. 2006.
Transcriptional regulatory networks downstream of TAL1/SCL in t-cell acute
lymphoblastic leukemia. Blood 986.
312. Prasad, K.S., J.E. Jordan, M.J. Koury, M.C. Bondurant, and S.J. Brandt. 1995.
Erythropoietin stimulates transcription of the TAL1/SCL gene and phosphorylation of
its protein products. J Biol Chem 270:11603-11611.
313. Prasad, K.S., and S.J. Brandt. 1997. Target-dependent effect of phosphorylation on the
DNA binding activity of the TAL1/SCL oncoprotein. J Biol Chem 272:11457-11462.

Introduction
73
314. Talora, C., S. Cialfi, C. Oliviero, R. Palermo, M. Pascucci, L. Frati, A. Vacca, A.
Gulino, and I. Screpanti. 2006. Cross talk among Notch3, pre-TCR, and Tal1 in T-cell
development and leukemogenesis. Blood 107:3313-3320.
315. Palamarchuk, A., A. Efanov, V. Maximov, R.I. Aqeilan, C.M. Croce, and Y. Pekarsky.
2005. Akt phosphorylates Tal1 oncoprotein and inhibits its repressor activity. Cancer
Res 65:4515-4519.
316. Tang, T., K.S. Prasad, M.J. Koury, and S.J. Brandt. 1999. Mitogen-activated protein
kinase mediates erythropoietin-induced phosphorylation of the TAL1/SCL transcription
factor in murine proerythroblasts. Biochem J 343 Pt 3:615-620.
317. Tang, T., J.L. Arbiser, and S.J. Brandt. 2002. Phosphorylation by mitogen-activated
protein kinase mediates the hypoxia-induced turnover of the TAL1/SCL transcription
factor in endothelial cells. J Biol Chem 277:18365-18372.
318. Terme, J.M., L. Lhermitte, V. Asnafi, and P. Jalinot. 2009. TGF-beta induces
degradation of TAL1/SCL by the ubiquitin-proteasome pathway through AKT-
mediated phosphorylation. Blood 113:6695-6698.
319. Sloan, S.R., C.P. Shen, R. McCarrick-Walmsley, and T. Kadesch. 1996.
Phosphorylation of E47 as a potential determinant of B-cell-specific activity. Mol Cell
Biol 16:6900-6908.
320. Johnson, S.E., X. Wang, S. Hardy, E.J. Taparowsky, and S.F. Konieczny. 1996. Casein
kinase II increases the transcriptional activities of MRF4 and MyoD independently of
their direct phosphorylation. Mol Cell Biol 16:1604-1613.
321. Lluis, F., E. Ballestar, M. Suelves, M. Esteller, and P. Munoz-Canoves. 2005. E47
phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific
gene transcription. Embo J 24:974-984.
322. Page, J.L., X. Wang, L.M. Sordillo, and S.E. Johnson. 2004. MEKK1 signaling through
p38 leads to transcriptional inactivation of E47 and repression of skeletal myogenesis. J
Biol Chem 279:30966-30972.
323. Hsu, H.L., J.T. Cheng, Q. Chen, and R. Baer. 1991. Enhancer-binding activity of the
tal-1 oncoprotein in association with the E47/E12 helix-loop-helix proteins. Mol Cell
Biol 11:3037-3042.
324. Begley, C.G., and A.R. Green. 1999. The SCL gene: from case report to critical
hematopoietic regulator. Blood 93:2760-2770.
325. Elefanty, A.G., C.G. Begley, D. Metcalf, L. Barnett, F. Kontgen, and L. Robb. 1998.
Characterization of hematopoietic progenitor cells that express the transcription factor
SCL, using a lacZ "knock-in" strategy. Proc Natl Acad Sci U S A 95:11897-11902.

Introduction
74
326. Robb, L., I. Lyons, R. Li, L. Hartley, F. Kontgen, R.P. Harvey, D. Metcalf, and C.G.
Begley. 1995. Absence of yolk sac hematopoiesis from mice with a targeted disruption
of the scl gene. Proc Natl Acad Sci U S A 92:7075-7079.
327. Kallianpur, A.R., J.E. Jordan, and S.J. Brandt. 1994. The SCL/TAL-1 gene is expressed
in progenitors of both the hematopoietic and vascular systems during embryogenesis.
Blood 83:1200-1208.
328. Gering, M., Y. Yamada, T.H. Rabbitts, and R.K. Patient. 2003. Lmo2 and Scl/Tal1
convert non-axial mesoderm into haemangioblasts which differentiate into endothelial
cells in the absence of Gata1. Development 130:6187-6199.
329. Patterson, L.J., M. Gering, C.E. Eckfeldt, A.R. Green, C.M. Verfaillie, S.C. Ekker, and
R. Patient. 2007. The transcription factors Scl and Lmo2 act together during
development of the hemangioblast in zebrafish. Blood 109:2389-2398.
330. Chung, Y.S., W.J. Zhang, E. Arentson, P.D. Kingsley, J. Palis, and K. Choi. 2002.
Lineage analysis of the hemangioblast as defined by FLK1 and SCL expression.
Development 129:5511-5520.
331. Lancrin, C., P. Sroczynska, A.G. Serrano, A. Gandillet, C. Ferreras, V. Kouskoff, and
G. Lacaud. 2010. Blood cell generation from the hemangioblast. J Mol Med 88:167-
172.
332. Lazrak, M., V. Deleuze, D. Noel, D. Haouzi, E. Chalhoub, C. Dohet, I. Robbins, and D.
Mathieu. 2004. The bHLH TAL-1/SCL regulates endothelial cell migration and
morphogenesis. J Cell Sci 117:1161-1171.
333. Robb, L., N.J. Elwood, A.G. Elefanty, F. Kontgen, R. Li, L.D. Barnett, and C.G.
Begley. 1996. The scl gene product is required for the generation of all hematopoietic
lineages in the adult mouse. Embo J 15:4123-4129.
334. Porcher, C., W. Swat, K. Rockwell, Y. Fujiwara, F.W. Alt, and S.H. Orkin. 1996. The T
cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic
lineages. Cell 86:47-57.
335. Zhang, Y., K.J. Payne, Y. Zhu, M.A. Price, Y.K. Parrish, E. Zielinska, L.W. Barsky,
and G.M. Crooks. 2005. SCL expression at critical points in human hematopoietic
lineage commitment. Stem Cells 23:852-860.
336. Hall, M.A., D.J. Curtis, D. Metcalf, A.G. Elefanty, K. Sourris, L. Robb, J.R. Gothert,
S.M. Jane, and C.G. Begley. 2003. The critical regulator of embryonic hematopoiesis,
SCL, is vital in the adult for megakaryopoiesis, erythropoiesis, and lineage choice in
CFU-S12. Proc Natl Acad Sci U S A 100:992-997.
337. Curtis, D.J., M.A. Hall, L.J. Van Stekelenburg, L. Robb, S.M. Jane, and C.G. Begley.
2004. SCL is required for normal function of short-term repopulating hematopoietic
stem cells. Blood 103:3342-3348.

Introduction
75
338. Reynaud, D., E. Ravet, M. Titeux, F. Mazurier, L. Renia, A. Dubart-Kupperschmitt,
P.H. Romeo, and F. Pflumio. 2005. SCL/TAL1 expression level regulates human
hematopoietic stem cell self-renewal and engraftment. Blood 106:2318-2328.
339. Mikkola, H.K., J. Klintman, H. Yang, H. Hock, T.M. Schlaeger, Y. Fujiwara, and S.H.
Orkin. 2003. Haematopoietic stem cells retain long-term repopulating activity and
multipotency in the absence of stem-cell leukaemia SCL/tal-1 gene. Nature 421:547-
551.
340. Lacombe, J., S. Herblot, S. Rojas-Sutterlin, A. Haman, S. Barakat, N.N. Iscove, G.
Sauvageau, and T. Hoang. 2010. Scl regulates the quiescence and the long-term
competence of hematopoietic stem cells. Blood 115:792-803.
341. Brunet de la Grange, P., F. Armstrong, V. Duval, M.C. Rouyez, N. Goardon, P.H.
Romeo, and F. Pflumio. 2006. Low SCL/TAL1 expression reveals its major role in
adult hematopoietic myeloid progenitors and stem cells. Blood
342. Green, A.R., E. Salvaris, and C.G. Begley. 1991. Erythroid expression of the 'helix-
loop-helix' gene, SCL. Oncogene 6:475-479.
343. Green, A.R., T. Lints, J. Visvader, R. Harvey, and C.G. Begley. 1992. SCL is
coexpressed with GATA-1 in hemopoietic cells but is also expressed in developing
brain. Oncogene 7:653-660.
344. van Eekelen, J.A., C.K. Bradley, J.R. Gothert, L. Robb, A.G. Elefanty, C.G. Begley,
and A.R. Harvey. 2003. Expression pattern of the stem cell leukaemia gene in the CNS
of the embryonic and adult mouse. Neuroscience 122:421-436.
345. Muroyama, Y., Y. Fujiwara, S.H. Orkin, and D.H. Rowitch. 2005. Specification of
astrocytes by bHLH protein SCL in a restricted region of the neural tube. Nature
438:360-363.
346. Acharya, M.R., A. Sparreboom, J. Venitz, and W.D. Figg. 2005. Rational development
of histone deacetylase inhibitors as anticancer agents: a review. Mol Pharmacol 68:917-
932.
347. Kornberg, R.D., and Y. Lorch. 1999. Twenty-five years of the nucleosome,
fundamental particle of the eukaryote chromosome. Cell 98:285-294.
348. Hadnagy, A., R. Beaulieu, and D. Balicki. 2008. Histone tail modifications and
noncanonical functions of histones: perspectives in cancer epigenetics. Mol Cancer
Ther 7:740-748.
349. Marks, P., R.A. Rifkind, V.M. Richon, R. Breslow, T. Miller, and W.K. Kelly. 2001.
Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer 1:194-202.
350. Marks, P.A., V.M. Richon, and R.A. Rifkind. 2000. Histone deacetylase inhibitors:
inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst
92:1210-1216.

Introduction
76
351. Dokmanovic, M., C. Clarke, and P.A. Marks. 2007. Histone deacetylase inhibitors:
overview and perspectives. Mol Cancer Res 5:981-989.
352. Strahl, B.D., and C.D. Allis. 2000. The language of covalent histone modifications.
Nature 403:41-45.
353. Pan, L.N., J. Lu, and B. Huang. 2007. HDAC inhibitors: a potential new category of
anti-tumor agents. Cell Mol Immunol 4:337-343.
354. Cress, W.D., and E. Seto. 2000. Histone deacetylases, transcriptional control, and
cancer. J Cell Physiol 184:1-16.
355. Mahlknecht, U., and D. Hoelzer. 2000. Histone acetylation modifiers in the
pathogenesis of malignant disease. Mol Med 6:623-644.
356. Allfrey, V.G. 1966. Structural modifications of histones and their possible role in the
regulation of ribonucleic acid synthesis. Proc Can Cancer Conf 6:313-335.
357. Roth, S.Y., J.M. Denu, and C.D. Allis. 2001. Histone acetyltransferases. Annu Rev
Biochem 70:81-120.
358. Kouzarides, T. 2000. Acetylation: a regulatory modification to rival phosphorylation?
Embo J 19:1176-1179.
359. Cohen, H.Y., S. Lavu, K.J. Bitterman, B. Hekking, T.A. Imahiyerobo, C. Miller, R.
Frye, H. Ploegh, B.M. Kessler, and D.A. Sinclair. 2004. Acetylation of the C terminus
of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell 13:627-638.
360. Blander, G., N. Zalle, Y. Daniely, J. Taplick, M.D. Gray, and M. Oren. 2002. DNA
damage-induced translocation of the Werner helicase is regulated by acetylation. J Biol
Chem 277:50934-50940.
361. Ogiwara, H., A. Ui, A. Otsuka, H. Satoh, I. Yokomi, S. Nakajima, A. Yasui, J. Yokota,
and T. Kohno. 2011. Histone acetylation by CBP and p300 at double-strand break sites
facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end
joining factors. Oncogene 30:2135-2146.
362. Lee, K.K., and J.L. Workman. 2007. Histone acetyltransferase complexes: one size
doesn't fit all. Nat Rev Mol Cell Biol 8:284-295.
363. Huang, S., Y. Qiu, Y. Shi, Z. Xu, and S.J. Brandt. 2000. P/CAF-mediated acetylation
regulates the function of the basic helix-loop-helix transcription factor TAL1/SCL.
Embo J 19:6792-6803.
364. Bradney, C., M. Hjelmeland, Y. Komatsu, M. Yoshida, T.P. Yao, and Y. Zhuang. 2003.
Regulation of E2A activities by histone acetyltransferases in B lymphocyte
development. J Biol Chem 278:2370-2376.
365. Stimson, L., V. Wood, O. Khan, S. Fotheringham, and N.B. La Thangue. 2009. HDAC
inhibitor-based therapies and haematological malignancy. Ann Oncol 20:1293-1302.

Introduction
77
366. Carey, N., and N.B. La Thangue. 2006. Histone deacetylase inhibitors: gathering pace.
Curr Opin Pharmacol 6:369-375.
367. Bolden, J.E., M.J. Peart, and R.W. Johnstone. 2006. Anticancer activities of histone
deacetylase inhibitors. Nat Rev Drug Discov 5:769-784.
368. Lane, A.A., and B.A. Chabner. 2009. Histone deacetylase inhibitors in cancer therapy. J
Clin Oncol 27:5459-5468.
369. Grozinger, C.M., and S.L. Schreiber. 2000. Regulation of histone deacetylase 4 and 5
and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad
Sci U S A 97:7835-7840.
370. Parra, M., H. Kasler, T.A. McKinsey, E.N. Olson, and E. Verdin. 2005. Protein kinase
D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor
activation. J Biol Chem 280:13762-13770.
371. Glozak, M.A., N. Sengupta, X. Zhang, and E. Seto. 2005. Acetylation and deacetylation
of non-histone proteins. Gene 363:15-23.
372. Martinez-Balbas, M.A., U.M. Bauer, S.J. Nielsen, A. Brehm, and T. Kouzarides. 2000.
Regulation of E2F1 activity by acetylation. Embo J 19:662-671.
373. Watamoto, K., M. Towatari, Y. Ozawa, Y. Miyata, M. Okamoto, A. Abe, T. Naoe, and
H. Saito. 2003. Altered interaction of HDAC5 with GATA-1 during MEL cell
differentiation. Oncogene 22:9176-9184.
374. Bali, P., M. Pranpat, J. Bradner, M. Balasis, W. Fiskus, F. Guo, K. Rocha, S.
Kumaraswamy, S. Boyapalle, P. Atadja, E. Seto, and K. Bhalla. 2005. Inhibition of
histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock
protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J
Biol Chem 280:26729-26734.
375. Tran, A.D., T.P. Marmo, A.A. Salam, S. Che, E. Finkelstein, R. Kabarriti, H.S. Xenias,
R. Mazitschek, C. Hubbert, Y. Kawaguchi, M.P. Sheetz, T.P. Yao, and J.C. Bulinski.
2007. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J
Cell Sci 120:1469-1479.
376. Hubbert, C., A. Guardiola, R. Shao, Y. Kawaguchi, A. Ito, A. Nixon, M. Yoshida, X.F.
Wang, and T.P. Yao. 2002. HDAC6 is a microtubule-associated deacetylase. Nature
417:455-458.
377. Halkidou, K., L. Gaughan, S. Cook, H.Y. Leung, D.E. Neal, and C.N. Robson. 2004.
Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer.
Prostate 59:177-189.
378. Choi, J.H., H.J. Kwon, B.I. Yoon, J.H. Kim, S.U. Han, H.J. Joo, and D.Y. Kim. 2001.
Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res
92:1300-1304.

Introduction
78
379. Wilson, A.J., D.S. Byun, S. Nasser, L.B. Murray, K. Ayyanar, D. Arango, M. Figueroa,
A. Melnick, G.D. Kao, L.H. Augenlicht, and J.M. Mariadason. 2008. HDAC4 promotes
growth of colon cancer cells via repression of p21. Mol Biol Cell 19:4062-4075.
380. Zhang, Z., H. Yamashita, T. Toyama, H. Sugiura, Y. Ando, K. Mita, M. Hamaguchi, Y.
Hara, S. Kobayashi, and H. Iwase. 2005. Quantitation of HDAC1 mRNA expression in
invasive carcinoma of the breast*. Breast Cancer Res Treat 94:11-16.
381. Song, J., J.H. Noh, J.H. Lee, J.W. Eun, Y.M. Ahn, S.Y. Kim, S.H. Lee, W.S. Park, N.J.
Yoo, J.Y. Lee, and S.W. Nam. 2005. Increased expression of histone deacetylase 2 is
found in human gastric cancer. Apmis 113:264-268.
382. Duong, V., C. Bret, L. Altucci, A. Mai, C. Duraffourd, J. Loubersac, P.O. Harmand, S.
Bonnet, S. Valente, T. Maudelonde, V. Cavailles, and N. Boulle. 2008. Specific activity
of class II histone deacetylases in human breast cancer cells. Mol Cancer Res 6:1908-
1919.
383. Zhang, Z., H. Yamashita, T. Toyama, H. Sugiura, Y. Omoto, Y. Ando, K. Mita, M.
Hamaguchi, S. Hayashi, and H. Iwase. 2004. HDAC6 expression is correlated with
better survival in breast cancer. Clin Cancer Res 10:6962-6968.
384. Moreno, D.A., C.A. Scrideli, M.A. Cortez, R. de Paula Queiroz, E.T. Valera, V. da
Silva Silveira, J.A. Yunes, S.R. Brandalise, and L.G. Tone. 2010. Differential
expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival
in childhood acute lymphoblastic leukaemia. Br J Haematol 150:665-673.
385. Lin, R.J., T. Sternsdorf, M. Tini, and R.M. Evans. 2001. Transcriptional regulation in
acute promyelocytic leukemia. Oncogene 20:7204-7215.
386. Wang, J., T. Hoshino, R.L. Redner, S. Kajigaya, and J.M. Liu. 1998. ETO, fusion
partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the
human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A 95:10860-10865.
387. Wang, J., Y. Saunthararajah, R.L. Redner, and J.M. Liu. 1999. Inhibitors of histone
deacetylase relieve ETO-mediated repression and induce differentiation of AML1-ETO
leukemia cells. Cancer Res 59:2766-2769.
388. Dhordain, P., R.J. Lin, S. Quief, D. Lantoine, J.P. Kerckaert, R.M. Evans, and O.
Albagli. 1998. The LAZ3(BCL-6) oncoprotein recruits a SMRT/mSIN3A/histone
deacetylase containing complex to mediate transcriptional repression. Nucleic Acids Res
26:4645-4651.
389. Pasqualucci, L., O. Bereschenko, H. Niu, U. Klein, K. Basso, R. Guglielmino, G.
Cattoretti, and R. Dalla-Favera. 2003. Molecular pathogenesis of non-Hodgkin's
lymphoma: the role of Bcl-6. Leuk Lymphoma 44 Suppl 3:S5-12.
390. Ocker, M., and R. Schneider-Stock. 2007. Histone deacetylase inhibitors: signalling
towards p21cip1/waf1. Int J Biochem Cell Biol 39:1367-1374.

Introduction
79
391. Glaser, K.B., M.J. Staver, J.F. Waring, J. Stender, R.G. Ulrich, and S.K. Davidsen.
2003. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors:
defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma
cell lines. Mol Cancer Ther 2:151-163.
392. Peart, M.J., G.K. Smyth, R.K. van Laar, D.D. Bowtell, V.M. Richon, P.A. Marks, A.J.
Holloway, and R.W. Johnstone. 2005. Identification and functional significance of
genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad
Sci U S A 102:3697-3702.
393. Mitsiades, C.S., N.S. Mitsiades, C.J. McMullan, V. Poulaki, R. Shringarpure, T.
Hideshima, M. Akiyama, D. Chauhan, N. Munshi, X. Gu, C. Bailey, M. Joseph, T.A.
Libermann, V.M. Richon, P.A. Marks, and K.C. Anderson. 2004. Transcriptional
signature of histone deacetylase inhibition in multiple myeloma: biological and clinical
implications. Proc Natl Acad Sci U S A 101:540-545.
394. Li, H., and X. Wu. 2004. Histone deacetylase inhibitor, Trichostatin A, activates
p21WAF1/CIP1 expression through downregulation of c-myc and release of the
repression of c-myc from the promoter in human cervical cancer cells. Biochem Biophys
Res Commun 324:860-867.
395. Xu, Y., S. Voelter-Mahlknecht, and U. Mahlknecht. 2005. The histone deacetylase
inhibitor suberoylanilide hydroxamic acid down-regulates expression levels of Bcr-abl,
c-Myc and HDAC3 in chronic myeloid leukemia cell lines. Int J Mol Med 15:169-172.
396. Duan, H., C.A. Heckman, and L.M. Boxer. 2005. Histone deacetylase inhibitors down-
regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol
25:1608-1619.
397. Tong, X., L. Yin, and C. Giardina. 2004. Butyrate suppresses Cox-2 activation in colon
cancer cells through HDAC inhibition. Biochem Biophys Res Commun 317:463-471.
398. Yamaguchi, K., A. Lantowski, A.J. Dannenberg, and K. Subbaramaiah. 2005. Histone
deacetylase inhibitors suppress the induction of c-Jun and its target genes including
COX-2. J Biol Chem 280:32569-32577.
399. You, J.S., J.K. Kang, E.K. Lee, J.C. Lee, S.H. Lee, Y.J. Jeon, D.H. Koh, S.H. Ahn,
D.W. Seo, H.Y. Lee, E.J. Cho, and J.W. Han. 2008. Histone deacetylase inhibitor
apicidin downregulates DNA methyltransferase 1 expression and induces repressive
histone modifications via recruitment of corepressor complex to promoter region in
human cervix cancer cells. Oncogene 27:1376-1386.
400. Januchowski, R., M. Dabrowski, H. Ofori, and P.P. Jagodzinski. 2007. Trichostatin A
down-regulate DNA methyltransferase 1 in Jurkat T cells. Cancer Lett 246:313-317.
401. Krishnan, M., A.B. Singh, J.J. Smith, A. Sharma, X. Chen, S. Eschrich, T.J. Yeatman,
R.D. Beauchamp, and P. Dhawan. 2010. HDAC inhibitors regulate claudin-1

Introduction
80
expression in colon cancer cells through modulation of mRNA stability. Oncogene
29:305-312.
402. Nimmanapalli, R., L. Fuino, P. Bali, M. Gasparetto, M. Glozak, J. Tao, L. Moscinski,
C. Smith, J. Wu, R. Jove, P. Atadja, and K. Bhalla. 2003. Histone deacetylase inhibitor
LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and
induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous
leukemia-blast crisis cells. Cancer Res 63:5126-5135.
403. Wu, J., J.P. Issa, J. Herman, D.E. Bassett, Jr., B.D. Nelkin, and S.B. Baylin. 1993.
Expression of an exogenous eukaryotic DNA methyltransferase gene induces
transformation of NIH 3T3 cells. Proc Natl Acad Sci U S A 90:8891-8895.
404. Xiong, Y., S.C. Dowdy, K.C. Podratz, F. Jin, J.R. Attewell, N.L. Eberhardt, and S.W.
Jiang. 2005. Histone deacetylase inhibitors decrease DNA methyltransferase-3B
messenger RNA stability and down-regulate de novo DNA methyltransferase activity in
human endometrial cells. Cancer Res 65:2684-2689.
405. Wu, P., Y. Tian, G. Chen, B. Wang, L. Gui, L. Xi, X. Ma, Y. Fang, T. Zhu, D. Wang,
L. Meng, G. Xu, S. Wang, D. Ma, and J. Zhou. 2010. Ubiquitin B: an essential mediator
of trichostatin A-induced tumor-selective killing in human cancer cells. Cell Death
Differ 17:109-118.
406. Rahmani, M., C. Yu, E. Reese, W. Ahmed, K. Hirsch, P. Dent, and S. Grant. 2003.
Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase
inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of
p21(CIP1/WAF1) induction rather than AKT inhibition. Oncogene 22:6231-6242.
407. Yu, X., Z.S. Guo, M.G. Marcu, L. Neckers, D.M. Nguyen, G.A. Chen, and D.S.
Schrump. 2002. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer
cells by depsipeptide FR901228. J Natl Cancer Inst 94:504-513.
408. Cha, T.L., M.J. Chuang, S.T. Wu, G.H. Sun, S.Y. Chang, D.S. Yu, S.M. Huang, S.K.
Huan, T.C. Cheng, T.T. Chen, P.L. Fan, and P.W. Hsiao. 2009. Dual degradation of
aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M
arrest and apoptosis of renal cancer cells. Clin Cancer Res 15:840-850.
409. Kawamata, N., J. Chen, and H.P. Koeffler. 2007. Suberoylanilide hydroxamic acid
(SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells.
Blood 110:2667-2673.
410. Kramer, O.H., P. Zhu, H.P. Ostendorff, M. Golebiewski, J. Tiefenbach, M.A. Peters, B.
Brill, B. Groner, I. Bach, T. Heinzel, and M. Gottlicher. 2003. The histone deacetylase
inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. Embo J
22:3411-3420.

Introduction
81
411. Qiu, L., A. Burgess, D.P. Fairlie, H. Leonard, P.G. Parsons, and B.G. Gabrielli. 2000.
Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective
in tumor cells. Mol Biol Cell 11:2069-2083.
412. Ungerstedt, J.S., Y. Sowa, W.S. Xu, Y. Shao, M. Dokmanovic, G. Perez, L. Ngo, A.
Holmgren, X. Jiang, and P.A. Marks. 2005. Role of thioredoxin in the response of
normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S
A 102:673-678.
413. Warrener, R., H. Beamish, A. Burgess, N.J. Waterhouse, N. Giles, D. Fairlie, and B.
Gabrielli. 2003. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints.
Faseb J 17:1550-1552.
414. Ruefli, A.A., M.J. Ausserlechner, D. Bernhard, V.R. Sutton, K.M. Tainton, R. Kofler,
M.J. Smyth, and R.W. Johnstone. 2001. The histone deacetylase inhibitor and
chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death
pathway characterized by cleavage of Bid and production of reactive oxygen species.
Proc Natl Acad Sci U S A 98:10833-10838.
415. Nishiyama, A., M. Matsui, S. Iwata, K. Hirota, H. Masutani, H. Nakamura, Y. Takagi,
H. Sono, Y. Gon, and J. Yodoi. 1999. Identification of thioredoxin-binding protein-
2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function
and expression. J Biol Chem 274:21645-21650.
416. Butler, L.M., X. Zhou, W.S. Xu, H.I. Scher, R.A. Rifkind, P.A. Marks, and V.M.
Richon. 2002. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-
regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl
Acad Sci U S A 99:11700-11705.
417. Tsapis, M., M. Lieb, F. Manzo, P. Shankaranarayanan, R. Herbrecht, P. Lutz, and H.
Gronemeyer. 2007. HDAC inhibitors induce apoptosis in glucocorticoid-resistant acute
lymphatic leukemia cells despite a switch from the extrinsic to the intrinsic death
pathway. Int J Biochem Cell Biol 39:1500-1509.
418. Balasubramanian, S., J. Ramos, W. Luo, M. Sirisawad, E. Verner, and J.J. Buggy. 2008.
A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis
in T-cell lymphomas. Leukemia 22:1026-1034.
419. Bots, M., and R.W. Johnstone. 2009. Rational combinations using HDAC inhibitors.
Clin Cancer Res 15:3970-3977.
420. Cameron, E.E., K.E. Bachman, S. Myohanen, J.G. Herman, and S.B. Baylin. 1999.
Synergy of demethylation and histone deacetylase inhibition in the re-expression of
genes silenced in cancer. Nat Genet 21:103-107.

Introduction
82
421. Kim, M.S., M. Blake, J.H. Baek, G. Kohlhagen, Y. Pommier, and F. Carrier. 2003.
Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting
DNA. Cancer Res 63:7291-7300.
422. Yu, C., G. Dasmahapatra, P. Dent, and S. Grant. 2005. Synergistic interactions between
MEK1/2 and histone deacetylase inhibitors in BCR/ABL+ human leukemia cells.
Leukemia 19:1579-1589.
423. Bali, P., P. George, P. Cohen, J. Tao, F. Guo, C. Sigua, A. Vishvanath, A. Scuto, S.
Annavarapu, W. Fiskus, L. Moscinski, P. Atadja, and K. Bhalla. 2004. Superior activity
of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase
inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3.
Clin Cancer Res 10:4991-4997.
424. George, P., P. Bali, S. Annavarapu, A. Scuto, W. Fiskus, F. Guo, C. Sigua, G.
Sondarva, L. Moscinski, P. Atadja, and K. Bhalla. 2005. Combination of the histone
deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against
human CML-BC cells and AML cells with activating mutation of FLT-3. Blood
105:1768-1776.
425. Ropero, S., M.F. Fraga, E. Ballestar, R. Hamelin, H. Yamamoto, M. Boix-Chornet, R.
Caballero, M. Alaminos, F. Setien, M.F. Paz, M. Herranz, J. Palacios, D. Arango, T.F.
Orntoft, L.A. Aaltonen, S. Schwartz, Jr., and M. Esteller. 2006. A truncating mutation
of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat
Genet 38:566-569.
426. Bandyopadhyay, D., A. Mishra, and E.E. Medrano. 2004. Overexpression of histone
deacetylase 1 confers resistance to sodium butyrate-mediated apoptosis in melanoma
cells through a p53-mediated pathway. Cancer Res 64:7706-7710.
427. Hauswald, S., J. Duque-Afonso, M.M. Wagner, F.M. Schertl, M. Lubbert, C. Peschel,
U. Keller, and T. Licht. 2009. Histone deacetylase inhibitors induce a very broad,
pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by
modulation of multiple ABC transporter genes. Clin Cancer Res 15:3705-3715.
428. Cardoso, B.A., L.R. Martins, C.I. Santos, L.M. Nadler, V.A. Boussiotis, A.A. Cardoso,
and J.T. Barata. 2009. Interleukin-4 stimulates proliferation and growth of T-cell acute
lymphoblastic leukemia cells by activating mTOR signaling. Leukemia 23:206-208.
429. La Motte-Mohs, R.N., E. Herer, and J.C. Zuniga-Pflucker. 2005. Induction of T-cell
development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro.
Blood 105:1431-1439.
430. Littlewood, T.D., D.C. Hancock, P.S. Danielian, M.G. Parker, and G.I. Evan. 1995. A
modified oestrogen receptor ligand-binding domain as an improved switch for the
regulation of heterologous proteins. Nucleic Acids Res 23:1686-1690.

Introduction
83

Interleukin-4 stimulates proliferation and growth of T-ALL cells
84
Chapter 2
Interleukin-4 stimulates proliferation and growth of T-
cell acute lymphoblastic leukemia cells by activating
mTOR signaling
Bruno A. Cardoso*, Leila R. Martins*, Cristina I. Santos*, Lee M. Nadler, Vassiliki A.
Boussiotis, Angelo A. Cardoso and João T. Barata
*these authors contributed equally to this work
Adapted from Leukemia, 2009; 23 (1): 206-8

Interleukin-4 stimulates proliferation and growth of T-ALL cells
85
Abstract
IL-4 is produced within the bone marrow microenvironment and stimulates the
proliferation of T-cell acute lymphoblastic leukemia (T-ALL) cells by as yet unknown
mechanisms. In this study, we showed that IL-4 induced cell cycle progression of
primary T-ALL cells from G0/G1 into S and G2/M phases of the cell cycle, by up-
regulating cyclins D2, E and A, and down-regulating the cyclin-dependent kinase
inhibitor p27kip1
. These events were paralleled by sequential activation of CDK4 and
CDK2 and hyperphosphorylation of Rb. By transfecting T-ALL cells with the chimeric
protein VP22/p27kip1
, which is able to translocate into the cytoplasm and nucleus of
target cells, we showed that down-regulation of p27kip1
is mandatory for IL-4-mediated
proliferation. Furthermore, IL-4 stimulated mTOR activation, as determined by
increased phosphorylation of S6K, S6 and 4E-BP1. To evaluate the functional
contribution of mTOR activation to IL-4 mediated signaling in T-ALL cells, we
specifically inhibited mTOR using rapamycin. Flow cytometry analysis of FSCxSSC
distribution and BrdU+PI staining demonstrated that rapamycin completely prevented
IL-4-dependent T-ALL cell growth and cell cycle progression. Consequently,
proliferation was also abrogated, as assessed by 3H-Thymidine incorporation. In
summary, our data identify mTOR as a critical regulator of IL-4 mediated effects in T-
ALL cells and support the rationale for using mTOR pharmacological inhibitors in T-
ALL therapy.
Introduction
Interleukin-4 (IL-4) is produced within the BM microenvironment either by
nonresident circulating cells, namely T lymphocytes, mast cells, and basophils or by
BM stromal cells. Importantly, IL-4 is able to induce proliferation of T-cell acute
lymphoblastic leukemia (T-ALL) cells (1). Therefore, IL-4 produced in the BM milieu
might influence the progression of T-ALL by stimulating proliferation of tumor cells.
However, the exact mechanisms by which IL-4 induces leukemia expansion remain
unknown. The effects of IL-4 on normal lymphocytes involve at least two signaling
pathways, JAK/STAT and PI3K/PKB (2). In addition, IL-4 activates the PI3K
downstream target mTOR, which regulates cell cycle completion in activated mature T-
cells (3). Constitutive activation of mTOR has been reported in some T-ALL cases (4)

Interleukin-4 stimulates proliferation and growth of T-ALL cells
86
and suggested to regulate viability, cell size and proliferation of tumor cells. However,
leukemia cells depend not only on constitutive, cell-autonomous mechanisms but also
on cues from the microenvironment to fully activate key signaling molecules that are
essential for tumor expansion and decreased sensitivity to chemotherapy (5, 6).
Therefore, we investigated whether mTOR is involved in IL-4-mediated proliferation
and growth of T-ALL cells.
Materials and Methods
T-ALL cells. Primary T-ALL cells were obtained from the peripheral blood and/or the
bone marrow of patients with high leukemia involvement (85–100%). Informed consent
and Institutional Review Board approval was obtained for all sample collections.
Samples were enriched by density centrifugation over Ficoll-Hypaque (GE Healthcare
Life Sciences), washed twice in RPMI 1640 (Invitrogen) supplemented with 10%
(vol/vol) FBS and 2 mM L-glutamine (hereafter referred to as RPMI 10 medium),
subjected to immunophenotypic analysis by flow cytometry and classified according to
their maturation stage using the criteria defined by the European Group for
Immunological Characterization of Leukemias. The TAIL7 cell line was maintained as
described previously (7).
Cell culture. Primary T-ALL cells and TAIL7 cell line were cultured in RPMI-10
medium as 2 x 106 cells/mL at 37ºC with 5% CO2 in the following conditions: Medium
alone (with the appropriate vehicle), or medium with 10ng/mL IL-4 (Endogen) or with
IL-4 plus 100nM Rapamycin (Calbiochem) for the indicated time points. For short-term
stimulation TAIL7 cells were starved overnight in RPMI with 1 % FBS, and in the next
day stimulated in the indicated conditions and time points in PBS 1x. Cells were then
harvested and processed as indicated below for assessment of cell viability, cell cycle,
and proliferation, and protein extraction for Immunobloting.
Assessment of viability and cell size. Cell viability was examined using an apoptosis
detection kit, following the manufacturer‟s protocol (R&D Systems). Briefly, cells were
stained with fluorescein isothiocyanate- conjugated Annexin V and propidium iodide at
room temperature for 15 min in the appropriate binding buffer, and subsequently

Interleukin-4 stimulates proliferation and growth of T-ALL cells
87
analyzed by flow cytometry (FACSCalibur; Becton- Dickinson). Cell size increase was
measured by flow cytometry. Percentage of „„activated‟‟ cells (with increased cell size)
was calculated by defining a threshold gate that excluded most of the bulk, small-sized
fraction of cultured cells.
Proliferation. Cells were cultured (2 x 106 cells/mL) in triplicates in RPMI-10, in flat-
bottom 96-well plates at 37°C with 5% CO2. Cultures were carried out for 72 h in the
following conditions: medium without IL-4 (control condition); with IL-4; with IL-4
plus Rapamycin; with IL-4 plus VP22/VP22 fused with p27Kip1
(8). Cells were
incubated with 3H -thymidine (1µCi/well) for 16 h prior to harvest. Proliferation was
determined by analysis of DNA synthesis, which was assessed by 3H-thymidine
incorporation using a β-scintillation counter.
Cell cycle analysis. Determination of the percentage of cells at each stage of the cell
cycle was performed by assessment of DNA content after staining with propidium
iodide. Briefly, 3-5 x 105 cells per sample were resuspended in 0.5 mL PBS and then
fixed with ice-cold 80% ethanol. Samples were then incubated for 30 min at 37°C in the
dark after addition of propidium iodide (2.5 mg/mL) and ribonuclease A (50 mg/mL).
Flow cytometry acquisition was performed in a FACScanto (Becton Dickinson
Biosciences) and analysis of cell cycle histograms was carried out using ModFit LT
(Verity) or WinCycle DNA Analysis software (Phoenix Flow Systems). In some
experiments, cell cycle profile was determined using BrdU incorporation. The cells
were incubated with 1mM BrdU for 24 h, harvested and ressuspended in PBS, and then
fixed and permeabilized with 1% paraformaldehyde and 0.01% Tween-20. After
permeabilization, the cells were treated with DNase to expose BrdU epitopes and
incubated with anti-BrdU-FITC antibody. Cells were then treated with RNase A
(500µg/mL) and propidium iodide was added to a final concentration of 200µg/mL.
Immunobloting, immunoprecipitation and in vitro kinase reaction. After the
indicated conditions and time intervals of culture, cell lysates were prepared as
described (8) and equal amounts of protein were analyzed by 10% SDS-PAGE,
transferred onto nitrocellulose membranes, and immunoblotted with the following
antibodies: CDK2, CDK4, CDK6, cyclin A, cyclin D2, cyclin E, ZAP70 (Santa-Cruz
Biotechnology), Phospho-4EBP1, Phospho-S6, Phospho-p70S6K
(Cell Signaling

Interleukin-4 stimulates proliferation and growth of T-ALL cells
88
Technology) and p27Kip1
(Becton Dickinson Transduction Laboratories). To examine
the phosphorylation status of Rb, proteins were analyzed by 6% SDS-PAGE, transferred
onto nitrocellulose membrane, and blotted with Rb-specific mAb (Becton Dickinson
Pharmingen). After immunobloting with primary antibodies, immunodetection was
performed using HRP-conjugated anti-mouse IgG (Promega), anti-rabbit IgG (Promega)
or anti-goat IgG (Santa-Cruz Biotecnology) as indicated by the host origin of the
primary antibody and developed by chemiluminescence (Thermo Scientific). In vitro
kinase reactions were performed as described previously (8).
Results and Discussion
IL-4 signaling promotes proliferation of T-ALL cells by inducing cell cycle
progression
We previously showed that IL-4 promotes in vitro proliferation of a significant
proportion of primary T-ALL samples (1). Here, we selected 12 diagnostic patient
samples that proliferated to IL-4 as assessed by 3H-thymidine incorporation, to
investigate the mechanisms of IL-4-driven T-ALL cell expansion. We first evaluated the
effect of IL-4 on cell cycle progression by analyzing the DNA content of primary T-
ALL cells by flow cytometry. IL-4 promoted the transition from G0/G1 to S-phase and
G2/M in all five samples analyzed (Figure 2.1A). IL-4 also induced cell size increase
(cell growth) that paralleled the effect on cell cycle (Figure 2.1B and Table 2.1).
As proliferation may result not only from an effect on cell cycle progression but
also from increased survival, we evaluated the effect of IL-4 on T-ALL cell viability. In
accordance with previous studies (9), we found that IL-4 had heterogeneous effects on
T-ALL cell survival. IL-4 prevented T-ALL in vitro apoptosis in 6 of 12 cases (50%),
promoted cell death in four (33%) and had no significant effects in two cases (17%;
Table 2.1). Nonetheless, IL-4-mediated proliferation occurred irrespectively of the
effect on cell survival, and cell cycle progression was observed both in patients where
IL-4 promoted viability and apoptosis (Table 2.1). These data suggest that IL-4
promotes proliferation of primary T-ALL cells mainly through regulation of the cell
cycle machinery.

Interleukin-4 stimulates proliferation and growth of T-ALL cells
89
Table 2.1. Immunophenotype, classification, and response to IL-4 of primary T-ALL specimens
T-cell maturation stages were defined as described by the European Group for the Immunological Characterization of Leukemias - EGIL.
Effects of IL-4 on viability: death, IL-4 promoted apoptosis; survival, IL-4 prevented spontaneous apoptosis; no effect, IL-4 did not
prevent or increase apoptosis. nd, not determined.

Interleukin-4 stimulates proliferation and growth of T-ALL cells
90

Interleukin-4 stimulates proliferation and growth of T-ALL cells
91
IL4 down-regulates p27kip1
and up-regulates cyclin expression, CDK
activity and Rb hyperphosphorylation
Next we evaluated the mechanisms by which IL-4 mediated cell cycle
progression in T-ALL cells. IL-4 did not affect the expression of cyclin-dependent
kinases CDK6, CDK4 and CDK2 (Figure 2.1C). In contrast, cyclins were up-regulated
by IL-4 in a sequential manner (Figure 2.1D). The early G1 molecule cyclin D2 peaked
around 12–24 h of culture with IL-4, whereas expression of cyclins E and A, which are
associated with late G1 and S-phase, reached a plateau at later time points (48 and 72 h).
These effects were paralleled by hyperphosphorylation of Rb, a critical substrate of
cyclin/CDK activity in the cell (Figure 2.1E), indicating that IL-4 induced cyclin/cdk
activity. To confirm these results, we performed in vitro kinase assays with CDK4 and
CDK2 immunoprecipitated from IL-4-stimulated primary T-ALL cells. IL-4 clearly
augmented CDK activity (Figure 2.1F). In addition, IL-4 induced the down-regulation
of the CDK inhibitor p27kip1
(Figure 2.1G). This event was mandatory for IL-4-
mediated cell cycle progression, because forced expression of p27kip1
completely
abrogated IL-4-mediated proliferation (Figure 2.1H).
Figure 2.1. IL-4 stimulates cell cycle progression of primary T-ALL cells. (A) Primary
T-ALL cells were cultured with or without 10 ng/mL IL-4 for the indicated time points. The
percentage of cells at each phase of the cell cycle was examined within the viable population
by propidium iodide staining. Left: results from one representative patient; right: results from
all patients analyzed (n=5), 0 vs 72 h of culture with IL-4, P=0.0159 (2-tailed Mann–
Whitney). Cells in medium alone did not show significant cell cycle progression (not
shown). (B) Cell size of T-ALL cells cultured with or without 10 ng/mL IL-4 for 48 h was
evaluated by flow cytometry analysis. Representative results from two of twelve patients
analyzed are shown. (C-E) T-ALL cells cultured with IL-4 during the indicated periods were
lysed and analyzed by immunoblot for the expression of CDK6, CDK4 and CDK22 (C),
cyclin D2, cyclin E and cyclin A (D), and phosphorylation of Rb (E). (F) T-ALL cells were
cultured with IL-4 for the indicated time points and in vitro kinase activity of
immunoprecipitated CDK4 and CDK2 was performed using Rb-GST and Histone H1 as
exogenous substrates, respectively. (G) Expression of p27kip1
was evaluated by immunoblot
at the indicated time points. (H) T-ALL cells were cultured with IL-4 alone or with
rapamycin, VP22 control protein or VP22/p27kip1
fusion protein. Proliferation was
determined at 72 h by 3H-thymidine incorporation.

Interleukin-4 stimulates proliferation and growth of T-ALL cells
92
Activation of mTOR pathway is mandatory for IL4-induced proliferation
As mTOR-dependent signaling has been associated with regulation of cell cycle
and size, we next evaluated whether IL-4 activated mTOR in the T-ALL cell line
TAIL7, whose biological features are similar to those from primary leukemia cells (7).
IL-4 induced phosphorylation of mTOR downstream targets p70S6K
, S6 and 4E-BP1 in
TAIL7 cells (Figure 2.2A). As expected, IL-4 mediated phosphorylation of these
molecules was inhibited by treatment with the mTOR-specific antagonist rapamycin
(Figure 2.2B). These data strongly indicate that IL-4 activates mTOR signaling in T-
ALL cells. To evaluate the functional consequences of IL-4-mediated mTOR activation,
we cultured T-ALL cells with IL-4 in the presence of rapamycin. At the molecular level,
inhibition of mTOR prevented IL-4-dependent p27kip1
down-regulation (Figure 2.2C).
Accordingly, rapamycin completely blocked IL-4-mediated proliferation (Figure 2.1H),
cell cycle progression (Figures 2.2D and E) and growth (Figures 2.2F and G) of both
TAIL7 and primary T-ALL cells.
In summary, we demonstrated that IL-4 mediates proliferation of T-ALL cells
through mTOR-dependent regulation of cell cycle progression. In showing that mTOR
is activated by a BM microenvironmental factor that positively stimulates leukemia T-
cells, our observations strengthen the emphasis on mTOR as a key molecular target in
T-ALL, and suggest that the inhibition of IL-4 signaling may also have therapeutic
potential in T-cell leukemia.

Interleukin-4 stimulates proliferation and growth of T-ALL cells
93
Figure 2.2. IL-4-mediated activation of mTOR pathway is critical for cell cycle
progression of T-ALL cells. (A) TAIL7 T-ALL cells were stimulated with 10 ng/mL IL-4
for the indicated time points, lysed and analyzed by immunoblot for phosphorylation of the
indicated mTOR downstream targets. (B and C) TAIL7 cells were cultured with 10 ng/mL
IL-4 in the presence or absence of rapamycin (rapa) and phosphorylation of the indicated
mTOR downstream target proteins (B) or expression of p27kip1
(c) was evaluated at 72 h.
TAIL7 (D and F) or primary (E and G) T-ALL cells were cultured in the presence of IL-4
with or without rapamycin and analyzed for cell cycle distribution (D and E) and cell
growth (F and G) at the indicated time points. (D and E) Percentage of cells in G0/G1
(lower left region), S-phase (upper region) and G2/M (lower right region) were identified
using propidium iodide and BrdU + anti-BrdU-FITC. (F and G) Cell growth was determined
by FSC vs SSC flow cytometry analysis. Percentage of large-sized (FSC high) cells was
estimated relative to the control, small-size bulk population. Results in this figure are
representative of at least two independent experiments.

Interleukin-4 stimulates proliferation and growth of T-ALL cells
94
References
1. Barata, J.T., T.D. Keenan, A. Silva, L.M. Nadler, V.A. Boussiotis, and A.A.
Cardoso. 2004. Common gamma chain-signaling cytokines promote
proliferation of T-cell acute lymphoblastic leukemia. Haematologica 89:1459-
1467.
2. Acacia de Sa Pinheiro, A., A. Morrot, S. Chakravarty, M. Overstreet, J.H.
Bream, P.M. Irusta, and F. Zavala. 2007. IL-4 induces a wide-spectrum
intracellular signaling cascade in CD8+ T cells. J Leukoc Biol 81:1102-1110.
3. Stephenson, L.M., D.S. Park, A.L. Mora, S. Goenka, and M. Boothby. 2005.
Sequence motifs in IL-4R alpha mediating cell-cycle progression of primary
lymphocytes. J Immunol 175:5178-5185.
4. Avellino, R., S. Romano, R. Parasole, R. Bisogni, A. Lamberti, V. Poggi, S.
Venuta, and M.F. Romano. 2005. Rapamycin stimulates apoptosis of childhood
acute lymphoblastic leukemia cells. Blood 106:1400-1406.
5. Brown, V.I., J. Fang, K. Alcorn, R. Barr, J.M. Kim, R. Wasserman, and S.A.
Grupp. 2003. Rapamycin is active against B-precursor leukemia in vitro and in
vivo, an effect that is modulated by IL-7-mediated signaling. Proc Natl Acad Sci
U S A 100:15113-15118.
6. Qiuping, Z., X. Jei, J. Youxin, J. Wei, L. Chun, W. Jin, W. Qun, L. Yan, H.
Chunsong, Y. Mingzhen, G. Qingping, Z. Kejian, S. Zhimin, L. Qun, L. Junyan,
and T. Jinquan. 2004. CC chemokine ligand 25 enhances resistance to apoptosis
in CD4+ T cells from patients with T-cell lineage acute and chronic lymphocytic
leukemia by means of livin activation. Cancer Res 64:7579-7587.
7. Barata, J.T., V.A. Boussiotis, J.A. Yunes, A.A. Ferrando, L.A. Moreau, J.P.
Veiga, S.E. Sallan, A.T. Look, L.M. Nadler, and A.A. Cardoso. 2004. IL-7-
dependent human leukemia T-cell line as a valuable tool for drug discovery in
T-ALL. Blood 103:1891-1900.
8. Barata, J.T., A.A. Cardoso, L.M. Nadler, and V.A. Boussiotis. 2001. Interleukin-
7 promotes survival and cell cycle progression of T-cell acute lymphoblastic
leukemia cells by down-regulating the cyclin-dependent kinase inhibitor
p27(kip1). Blood 98:1524-1531.

Interleukin-4 stimulates proliferation and growth of T-ALL cells
95
9. Karawajew, L., V. Ruppert, C. Wuchter, A. Kosser, M. Schrappe, B. Dorken,
and W.D. Ludwig. 2000. Inhibition of in vitro spontaneous apoptosis by IL-7
correlates with bcl-2 up-regulation, cortical/mature immunophenotype, and
better early cytoreduction of childhood T-cell acute lymphoblastic leukemia.
Blood.96:297-306.

TAL1 and LMO2 expression impair human T-cell development
96
Chapter 3
TAL1 and LMO2 ectopic expression in human T-cell
progenitors impacts T-cell development in vitro
Bruno A. Cardoso, Nádia Correia, Ana Gírio, Patricia Fuentes, María J. García, Miguel
Abecasis, Nuno Clode, Helena Ferreira, João Gonçalves, Maria L. Toribio and João T.
Barata

TAL1 and LMO2 expression impair human T-cell development
97
Abstract
The transcription factor TAL1 is critical in early hematopoiesis and is aberrantly
expressed in T-cell acute lymphoblastic leukemia (T-ALL). TAL1 was shown to have
leukemogenic potential, since transgenic mice develop aggressive leukemias of T-cell
phenotype. However, there are clear discrepancies between mice and humans regarding
the stage of maturation block in T-cell differentiation and the expression of putative
TAL1 downstream targets. Most importantly, it is still unknown whether this gene can
trigger leukemogenesis in humans. Here, we show that transduction of human
hematopoietic progenitors with TAL1 or its partner LMO2 affected human T-cell
differentiation in co-culture with stromal cells. Interestingly, the concomitant expression
of TAL1 and LMO2 led to the accumulation of a minor, discrete subpopulation of
CD3+CD4
+CD8
+ thymocytes with increased cell size that was absent from other
conditions. Furthermore, efficient transduction of human T-cell progenitors with TAL1
and LMO2 strongly abrogated T-cell differentiation, decreasing the expression of all T-
cell markers tested. These preliminary results show that the TAL1 and LMO2 can
disrupt normal human T-cell development. This may predispose thymocytes to
malignant transformation.
Introduction
Several oncogenic transcription factors have been shown to be over-expressed in
T-ALL due to chromosomal lesions and other as yet unknown mechanisms (1-3). One
of these transcription factors, TAL1, is crucial for early hematopoiesis, although it is
rapidly down-regulated upon T-cell lineage commitment. TAL1 ectopic expression
occurs in up to 65% of T-ALL patients, whereas overexpression of LMO genes (LMO1
or LMO2) occurs with lower frequency, commonly in association with TAL1 (4).
TAL1 was initially identified in a translocation that juxtaposes TCRA/D regulatory
sequences to the TAL1 coding region, t(1;14)(p32;q11), driving TAL1 abnormal
expression in developing T-cells (5, 6). However, the main reason for TAL1 ectopic
expression in T-ALL is the interstitial deletion of 90 Kb (TAL1d) which places TAL1
coding region under the control of SIL regulatory elements (7). The TAL1 gene encodes
a class II bHLH transcription factor that heterodimerizes (through the bHLH domain)
with class I proteins to bind the major groove of the DNA to a specific E- box
consensus sequence, CANNTG (8). The usual TAL1 interacting partners are the E

TAL1 and LMO2 expression impair human T-cell development
98
proteins E2A and HEB (9, 10). These proteins regulate T-cell development and act as
tumor suppressors in T-cells (11, 12). Importantly, TAL1 transgenic mice develop fatal
T-cell leukemia, although with a long latency period (13-15), and inhibition of E2A
function by TAL1 was shown to be responsible for the development of T-cell leukemia
in one of the TAL1 transgenic mouse models (15).
LMO2 is also a transcription factor, although there is no evidence of its binding to
DNA. Instead, LMO2 acts as a bridging molecule, establishing important protein-
protein interactions with TAL1, E2A, GATA and LDB1 through its cystein-rich LIM
domains that are zinc binding motifs (16-18). Similar to TAL1, LMO2 is expressed in
T-ALL patients due to abnormal chromosomal rearrangements t(11;14)(p13;q11)] (19)
and del(11)(p12p13) (20), and other unknown reasons (21). LMO2 transgenic mice were
shown to develop T-cell leukemia with shorter latency periods than TAL1 transgenic
mice (22-24). Interestingly, TAL1 and LMO2 expression appears to be synergistic, since
double transgenic mice develop leukemia with shorter latency periods (22). Transgenic
mice for the LMO2 closely related gene LMO1 also develop leukemia and synergize
with the TAL1 gene (22, 25-27). The development of leukemia in these mouse models
appears to be preceded by an impairment in T-cell development, with a decrease in the
double positive population CD4+CD8
+ (13, 22, 26) and an increase in the double
negative (DN) population (CD4-CD8
-). Additionally, TAL1 and LMO1 expression was
reported not only to inhibit T-cell differentiation but to promote the expansion of a
population of DN thymocytes (27).
The evidence for the T-cell oncogenic potential of LMO2 stems not only from
mouse studies but also from unfortunate events in gene therapy trials for human SCID,
in which several patients developed T-cell leukemia-like disease. It was subsequently
observed that, during the retroviral delivery of the γ common chain (IL2RG) gene into
hematopoietic stem cells, the vector integrated near the LMO2 locus, and the strong
promoter in Long Terminal Repeat (LTR) drove LMO2 aberrant expression in the T-
cell precursor population in some of the patients (28-32). In contrast, the tumorigenic
potential of TAL1 has never been formally demonstrated in human cells. Moreover, one
TAL1 transgenic mouse model indicated that TAL1 may be incapable of triggering
leukemia per se (33). In addition, there are significant differences between mice and
humans regarding TAL1 involvement in T-ALL. For instance, T-ALL patients with
TAL1 ectopic expression typically display a phenotype of late cortical thymocytes
(CD4+CD8
+) (4), whereas in TAL1 transgenic mice T-cell development is inhibited at

TAL1 and LMO2 expression impair human T-cell development
99
early stages, when thymocytes lack the expression of CD4 and CD8 (14, 15). Moreover,
TAL1-positive patients show up-regulation of genes that are important for T-cell
development (LCK, TCRA, TCRB, CD2, CD6) (4), whereas in TAL1 transgenic mice the
T-cell differentiation gene expression program appears to be inhibited (15). These
discrepancies raise the possibility that the involmente of TAL1 in human T-ALL may
not be accurately reflected in mouse studies. Further, they raise the question of whether
TAL1 is actually a leukemogenic trigger in human T-ALL or merely a late contributor
to the process in already transformed cells.
To try and answer this question, we forced the expression of TAL1 and/or LMO2
in human hematopoietic stem cells and early thymic progenitors and analyzed their
impact on T-cell development in vitro. Our preliminary data suggest that, in the least,
both TAL1 and LMO2 can significantly deregulate human T-cell differenciation.
Materials and Methods
Cloning procedures. The MAT vectors described in this study were derived from the
#318.pRRE.sin.cPPT.pA.cte.Luci.mCMV.hPGK.GFP.Wpre (#318) vector (34), kindly
provided by Prof. Luigi Naldini. The TAL1 and LMO2 genes were amplified by PCR
from the pcDNA3.1(+) zeo TAL1 and pcDNA3.1 (+) LMO2-HA plasmids respectively,
using primers that span the CMV promoter and the poly-A site in pcDNA3.1 (+) zeo
plasmid (Table 3.1).
These two fragments replaced the luciferase gene in the #318 vector (using the EcoRV
and XbaI restriction enzymes). The IRES sequence from the pMigR1 vector was
amplified by PCR with a forward primer containing AgeI, MluI and NdeI restriction
Designation 5' to 3'
TATA+12 ATCAAGCTTCTCGAGGGTAGGCGTGTACGGTGG
XbaI GATGGCTGGCAACTAGAAGG
AgeI-MluI-NdeI-IRES ATACCGGTACGCGTCATATGAATTCCGCCCCTCTC
IRES-AgeI CGATACCGGTGGTTGTGGCCATATTATC
MluI-5‟ TAL1 ATACGCGTCCCAGGATGACCGAGCGG
NdeI-3‟ TAL1 TCATATGCATTCACCGAGGGCCGGCTCCATC
MluI-5‟ LMO2 ATACGCGTAGGATGTCCTCGGCCATCGAAAGG
NdeI-3‟ LMO2 ATCATATGCGGTCAAGAAGCGTAGTCCGGAACGTCGTACGGGTA
Table 3.1. List of primers used in the cloning of the MAT plasmids

TAL1 and LMO2 expression impair human T-cell development
100
sites upstream of the IRES sequence and a reverse primer containing the AgeI site. This
fragment was cloned upstream to the Green Fluorescent Protein (GFP) AgeI restriction
site. Finally, TAL1 and LMO2 were amplified by PCR from the respective plasmids
with primers containing the MluI and NdeI restriction sites and cloned upstream of the
IRES sequence. The pHR-SIN vectors were described previously (35) and were kindly
provided by Prof. Maria Toribio. We cloned TAL1 and LMO2 in the pSIN-BX-IR/EMW
vector (also provided by Prof. Maria Toribio) in the BamHI and XhoI restriction sites
upstream of the IRES-Emerald sequence. Next we removed the TAL1-IRES-Emerald
and LMO2-IRES-Emerald fragments from the pSIN-BX-IR/EMW vector and replaced
the GFP on the pHR-SIN-CSGW vector.
Production of VSVG-pseudotyped lentiviruses. Vesicular-Stomatitis-Virus-
pseudotyped third-generation lentiviruses were produced by transient four-plasmid
cotransfection into 293T cells. Briefly, a total of 2 x 106 293T cells were seeded in 10
cm-diameter dishes 24 h prior to transfection in 10 mL DMEM (Gibco) with 10% fetal
bovine serum, penicillin (100 IU/mL), and streptomycin (100mg/mL) in a 5% CO2
incubator. 25µM Chloroquine (Sigma) was added to the culture medium 1 h prior to
transfection. A total of 11.6 µg of plasmid DNA was used for the transfection of one
dish: 2 µg of the envelope plasmid pMD2.VSVG, 1.4µg of helper plasmid pRSV-REV
plasmid, 3,1 µg of packaging plasmid pMDLg and 5.1 µg of transfer vector plasmid
where TAL1 and LMO2 were cloned. The precipitate was formed by adding the
plasmids to a final volume of 450 µl of sterile water and 50 µl of 2.5 M CaCl2, mixing
well, then adding dropwise to 500 µl of 2x HBSP buffer (280 mM NaCl, 10mM KCl,
50 mM HEPES, 1.6 mM Na2HPO4, 10mM D(+) Glucose [pH 7.05]) while vortexing
and immediately adding the precipitate to the cultures. The medium (10 mL) was
replaced after 14 to 16 h with medium containing 20mM HEPES (pH=7.9) and 10mM
Sodium Butyrate (6 mL), which was then replaced again after 8 h to 10 h by fresh
medium (6 mL). The conditioned medium (Lentiviral Supernatants) was collected after
another 24 h, filtered through 0.45 mm pore-size cellulose acetate filters, concentrated
using Amicon Columns (Millipore) and then stored at -80°C until use.
Cell lines. The OP9-Dll1 cells were kindly provided by Prof. Zuñiga-Pflücker (36),
maintained in MEM medium supplemented with 20% fetal bovine serum (MEM-20).

TAL1 and LMO2 expression impair human T-cell development
101
293T cells were maintained in DMEM medium supplemented with 10% fetal bovine
serum (DMEM-10).
Transduction of 293T cells. 100 x 103 293T cells were seeded with 2 mL of DMEM-
10 per well in 6-well plates. In the following day, the medium was removed and a 1:1
dilution of lentiviral supernatant in DMEM-10 was added. 72 h post-transduction, the
cells were washed and sorted for high expression of GFP. Sorted 293T cells were
expanded and lysed for protein extracts when indicated.
Isolation and transduction of hematopoietic and thymic progenitors. Institutional
review board approval was obtained for umbilical cord-blood and thymic sample
collections from Comissão de Ética, Faculdade de Medicina da Universidade de Lisboa
(Lisbon, Portugal) and Comissão de Ética of Centro Hospitalar de Lisboa Ocidental
(Carnaxide, Portugal), respectively. Umbilical cord blood (UCB) was collected in sterile
heparinized bags and mononuclear cells were isolated by density gradient centrifugation
(Lympholyte, Cedarlane). CD34+ cells were isolated using the CD34 Microbead kit
(Miltenyi Biotec) and separated in MidiMACS separation columns (Miltenyi Biotec)
according to the manufacturer‟s instructions. The CD34+ cells were further sorted for
CD34+CD38
- cells. Normal thymocytes were isolated from thymic tissue obtained from
children undergoing cardiac surgery. The tissue was gently minced in medium, the
resulting cell suspension was filtered through a cell strainer, and thymocytes were
enriched by density centrifugation to greater than 95% purity. The thymocytes were
enriched in CD34high
using the Human Cord Blood CD34 pre-enrichment cocktail (Stem
Cell Technologies) and depleted of CD1a cells with MidiMACS (Miltenyi biotec).
Purified precursors were transduced in retronectin (Takara) coated plates. Briefly, 105
cells were incubated in 500µl IMDM with 1% BSA plus 10ng/mL of SCF and FLT3L
in a retronectin-coated well (5µg/well) of a 24-well plate with 500µl of the respective
concentrated lentiviral supernatant. The cells were incubated overnight at 37ºC, and in
the next day, more lentiviral supernatant was added.
OP9-Dll1 co-cultures. Co-cultures of OP9-Dll1 stromal cells with hematopoietic cells
were performed as described (37). Briefly, 105 transduced thymic-derived CD34
+CD1a
-
or umbilical-cord blood derived CD34+CD38
- cells were added to a sub-confluent layer
of OP9-Dll1 cells. The co-cultures were maintained in MEM-20 medium supplemented

TAL1 and LMO2 expression impair human T-cell development
102
with 10ng/mL of IL-7 and FLT3L and the medium replaced every two days. At the
indicated time points the cells were harvested, counted (when indicated) and analyzed
by flow cytometry.
Flow Cytometry. Standard procedures were used to stain the cells with fluorochrome-
conjugated antibodies. The antibodies used in this study were CD34-PerCP-Cy5.5 and
TCRαβ-PE (Becton-Dickinson), CD1a-PE, CD3-PerCP-Cy5.5, CD4-PE-Cy7, CD7-PE,
CD8-APC, CD19-PerCP-Cy5.5 and CD38-PE (ebioscience). Samples were acquired in
a FACScalibur (Becton-Dickinson) or FACScanto (Becton-Dickinson) and analyzed
using Flow Jo.
Immunobloting. Transduced or transfected 293T cell lysates were lised as described
(38) and equal amounts of protein were resolved by 12% SDS-PAGE, transferred onto
nitrocellulose membranes, and immunoblotted with the following antibodies or antisera:
HA (Santa-Cruz Biotechnology), Actin (Santa-Cruz Biotechnology), TAL1 (Millipore)
and GFP (Roche). After immunobloting with primary antibodies, immunodetection was
performed using HRP-conjugated anti-mouse IgG (Promega), anti-rabbit IgG (Promega)
or anti-goat IgG (Santa-Cruz Biotecnology), as indicated by the host origin of the
primary antibody, and developed by chemiluminescence (Thermo Scientific).
RNA extraction, RT-PCR and semi-quantitative-PCR. When indicated, RNA was
extracted using High Pure RNA Isolation Kit (Roche) according to the manufacturer‟s
instructions. For the RT-PCR, a total of 65ng of total RNA was reverse transcribed
using SuperScript II (Invitrogen) and random hexamers. Primers used in the semi-
quantitative PCR are described in Table 3.2. The PCR reactions were performed in
50µl, using 3 µl cDNA, 2U of Gotaq DNA polymerase (Promega) and 200nM of each
primer, according to manufacturer‟s instructions. All the amplifications were performed
in a 96-well MyCycler (Bio-Rad). After the initial denaturation of 5 min at 95ºC, PCR
products were amplified by 40 cycles of 30 seg at 95ºC, 1 min at 60ºC and 1 min at
72ºC. 15µl aliquots were removed at cycles 20, 30 and 40. The PCR products were
separated in a 2% agarose gel and visualized under UV light.

TAL1 and LMO2 expression impair human T-cell development
103
Gene Forward primer 5' to 3' Reverse primer 5' to 3'
TAL1 AACAATCGAGTGAAGAGGAG CTTTGGTGTGGGGACCAT
LMO2 TCAGAGGAACCAGTGGATGAG CCGGCCCAGTTTGTAGTAGA
IRES ATACCGGTACGCGTCATATGAATTCCGCCCCTCTC CGATACCGGTGGTTGTGGCCATATTATC
GAPDH GGAGTCAACGGATTTGGTCG GACAAGCTTCCCGTTCTCAG
Results
Establishment of a system to simultaneously transduce target cells with
three genes
In order to simultaneously and stably express the TAL1 gene together with LMO2
and a reporter gene into human Hematopoietic stem cells (HSCs) or early T-cell
progenitors (ETPs) we used a third generation lentiviral vector developed in Naldini‟s
Lab (34), which displays an internal bi-directional promoter that allows the cloning and
efficient expression of two genes in opposite directions. Engineering this vector to
become bi-cistronic in one of its directions, we were able to express both TAL1 and
LMO2 genes, as well as eGFP (enhanced Green Fluorescent Protein), in the same target
cell.
The schematic representation of the five different constructs we generated is
described in Figure 3.1A. The empty vector (MAT-IRES-eGFP) consists of an IRES-
eGFP cassette cloned downstream of the human Phosphoglycerate kinase promoter
(hPGK) and WPRE, which is a post-transcriptional regulatory element from the
woodchuck hepatitis virus. This expression cassette is flanked by the 5‟ and 3‟ LTR.
The TAL1 and LMO2 genes were cloned upstream of the IRES-eGFP cassette. To create
the plasmids minCMV-TAL1/LMO2-MAT-TAL1/LMO2-IRES-eGFP, TAL1 and
LMO2 were cloned downstream of the minimal (TATA box and +1 nucleotide)
Cytomegalovirus (CMV) promoter, and inserted in the opposite direction to the hPGK.
All the constructs were confirmed by sequencing.
These constructs (Figure 3.1A) were then used to produce VSVG-pseudotyped
lentiviruses to transduce 293T fibroblasts. After transduction, we sorted the GFP
positive 293T cells and expanded them in culture.
Table 3.2. List of primers used in semi-quantitative-PCR

TAL1 and LMO2 expression impair human T-cell development
104
Figure 3.1. Transduction of MAT vectors into 293T and CD34+ CD38
- cord blood
cells. (A) Schematic representation of the MAT transfer plasmids. MAT viruses were
produced in 293T cells using four different plasmids: the transfer vector (shown in the
figure), and the packaging vector, the REV expressing vector and the envelop protein
vector Vesicular Stomatitis Virus G Glycoprotein (not shown). (B) 293T cells (not used to
produce the viruses) were transduced with the indicated MAT vectors and 72 h post-
transduction the GFP-positive cells were sorted and expanded for 5 days. GFP expression
of sorted cells was analyzed by flow cytometry and confirmed to be higher than 50% in all
cases. Cells were then lysed and TAL1, LMO2 and GFP expression were analyzed by
immunoblot. Actin was used as loading control. (C) Umbilical cord blood-derived
CD34+CD38
- cells were transduced with the indicated MAT vectors and 48 h post-
transduction the expression of TAL1, LMO2 and IRES was analyzed. GAPDH was used as
loading control.

TAL1 and LMO2 expression impair human T-cell development
105
As shown in Figure 3.1B, we detected LMO2 protein (tagged with HA) in the
lanes 4, 5 and 6 that contain the conditions where 293T cells were transduced with
MAT-LMO2-IRES-eGFP, mTAL1-MAT-LMO2-IRES-eGFP and mLMO2-MAT-
TAL1-IRES-eGFP, respectively. The level of expression in lane 6 is lower than the
previous ones, due to the fact that the LMO2 gene was cloned downstream of the
minCMV promoter that is less efficient than hPGK (34). TAL1 was detected in lanes 3,
5 and 6 that contain the conditions where 293T cells were transduced with MAT-TAL1-
IRES-eGFP, mTAL1-MAT-LMO2-IRES-eGFP and mLMO2-MAT-TAL1-IRES-eGFP,
respectively. The expression was lower in lane 5 for the same reason as for LMO2. As
expected, we detected GFP in all lanes containing transduced cells. These results show
that we were able to successfully clone three genes in the same vector and to detect the
proteins encoded by these genes.
Detection of TAL1 and LMO2 in cord-blood CD34+CD38
- cells
Human hematopoietic stem cells are essentially quiescent, and lentiviral vectors
are considered the most efficient means to deliver genes into these cells (42). We
transduced isolated umbilical cord blood CD34+CD38
- cells, enriched in hematopoetic
stem (HSC) and progenitor cells, with the constructs described in Figure 3.1A. In order
to detect TAL1 and LMO2 gene expression in transduced cells, we extracted the RNA
and performed semi-quantitative PCR. As expected, TAL1 and LMO2 RNA expression
was detected in cells that were not transduced or transduced with the empty vector
(Figure 3.1C). This is in agreement with the know expression and essential function of
TAL1 and LMO2 in hematopoietic stem cells (39, 40). Nonetheless, the basal levels of
TAL1 and LMO2 were clearly upregulated upon transduction with the respective vectors
(Figure 3.1C). High TAL1 expression was detected in cells transduced with MAT-
TAL1-IRES-eGFP, mTAL1-MAT-LMO2-IRES-eGFP and mLMO2-MAT-TAL1.
Similar results were obtained in cells transduced with MAT-LMO2-IRES-eGFP,
mTAL1-MAT-LMO2-IRES-eGFP and mLMO2-MAT-TAL1-IRES-eGFP, which
increased the levels of LMO2 gene expression. To confirm that the cells have been in
fact transduced, and the levels of TAL1 and LMO2 expression were not due to
differential culture conditions, we also measured the expression levels of the IRES
cassette that the vector harbors internally (Figure 3.1C). These results show that we

TAL1 and LMO2 expression impair human T-cell development
106
were able to transduce umbilical cord-blood hematopoietic CD34+CD38
- cells using
bidirectional vectors cloned with both TAL1 and LMO2 genes.
Forced TAL1 and LMO2 expression in CD34+CD38
- cells affect human T-
cell differentiation in vitro
In transgenic mouse models, the role for TAL1 and LMO2 in T-cell leukemia is
well established and associates with an arrest in T-cell development (13-15, 22-25). In
order to investigate whether TAL1 could have an actual role in the development of
human T-cell leukemia, we started by analyzing the function of this gene in co-culture
systems that allow the differentiation of human T-cells. Since TAL1 is often co-express
with LMO2 in T-ALL (4), we also analyzed the possible synergistic effect between
these two genes.
Mouse hematopoietic progenitor cells can differentiate into mature and functional
T-cells after 17 days in culture with a stromal layer of OP9-Dll1 cells (36).
Furthermore, the same co-culture system was used to differentiate human hematopoietic
stem cells into functional T-cells (37). We transduced CD34+CD38
- isolated from
umbilical cord-blood with the vectors described in Figure 3.1A and co-cultured these
cells with OP9-Dll1 cells for 3 weeks. Forced TAL1 expression per se did not affect
cell size. However, when TAL1 was expressed under a strong promoter it clearly
synergized with LMO2 (Figure 3.2A).
T-cell development was also affected by the expression of TAL1 and LMO2. CD7
expression decreased in all conditions when compared to empty transduced cells (Figure
3.2B). Furthermore, there was a tendency for a minor percent increase in CD34+ CD7-
stem/progenitor cells (Figure 3.2B). The co-culture system allowed the development of
a small amount of CD4+CD8
+ T-cells in the control condition (Figure 3.2C) that express
CD3 (Figure 3.2D). The proportion of this population decreased when cells were
transduced with TAL1 or LMO2 (Figure 3.2C), and also in the condition where TAL1 is
less expressed than LMO2 (mTAL1-MAT-LMO2-IRES-eGFP), in agreement with an
earlier developmental block. However, when TAL1 expression is higher than LMO2
(mLMO2-MAT-TAL1-IRES-eGFP), cells differentiated into a distinct population of
CD4+CD8
+ (Figure 3.2C) with high levels of CD3 (Figure 3.2D). Notably, our results
are in agreement with reports indicating that TAL1-expressing T-ALL patients are
associated with a CD3+ CD4
+ CD8
+ phenotype (4).

TAL1 and LMO2 expression impair human T-cell development
107
These results indicate that this co-culture system is a valid model for further
dissection of the mechanisms behind TAL1/LMO2-induced human leukemogenesis.
Figure 3.2. Coordinated expression of TAL1 and LMO2 in human hematopoietic
progenitors promotes cell growth and leads to the differentiation of CD3+CD4
+CD8
+
cells. (A-D) Umbilical cord blood-derived CD34+CD38
- were transduced with the indicated
MAT vectors. At 48 h post-transduction, the cells were co-cultured with OP9-Dll1 cells.
After 3 weeks of co-culture (A) cell size (as determined by FSC/SSC distribution), and
expression of (B) CD34 and CD7, (C) CD4 and CD8, and (D) CD3 within the CD4+CD8
+
population were analyzed by flow cytometry.

TAL1 and LMO2 expression impair human T-cell development
108
Taken together, the preliminary results obtained with this co-culture system
indicate that TAL1 and LMO2 ectopic expression can disrupt normal human T-cell
development.
High TAL1 and LMO2 expression in human thymic progenitors increases
cell proliferation and has a striking effect on T-cell differentiation
The vectors described in Figure 3.1 offer a simple strategy to analyze the possible
synergistic impact of TAL1 and LMO2 gene expression in human hematopoietic
progenitors. In fact, transduction of hematopoietic progenitor cells with these genes
partially disrupted in vitro T-cell development. However, the efficiency of transduction
was very low (data not shown), which could result in an underestimation of the effects
of these two genes. To address this question, we cloned TAL1 and LMO2 into a new
lentiviral vector (pHR-SIN). We detected both TAL1 (middle lane) and LMO2 (right
lane) in transfected 293T cells (Figure 3.3A). However, using this cloning strategy, we
could not transduce the hematopoietic stem cells with a single vector carrying both
genes. Instead, we transduced the cells with both vectors (pHR-SIN-TAL1 and pHR-
SIN-LMO2).
To allow for a more direct assessment of the effect of TAL1 and LMO2 in human
T-cell development, we next isolated CD34+CD1a
- thymic progenitor cells (instead of
cord-blood HSCs), transduced them with the pHR-SIN lentiviral vectors and co-
cultured these cells with OP9-Dll1 cells. The efficiency of transduction of these cells
was high, allowing the distinction between GFP positive and negative cells (data not
shown). As shown in Figure 3.3B, cells proliferated in a time-dependent manner.
Forced expression of LMO2 induced strong proliferation of thymic progenitors after day
17 (Figure 3.3B), in contrast to cells transduced with the empty vector and with TAL1.
In fact, TAL1 expression in these thymic progenitors did not increase the cell number
relative to the empty vector transduced cells. TAL1 and LMO2 co-expression in these
progenitors had a slight increase relative to empty transduced cells but only at the later
days of co-culture (Figure 3.3B).
Ectopic expression of both TAL1 and LMO2 decreased the expression of the CD3
marker (Figure 3.3C) in both days analyzed, when compared to empty transduced cells.
LMO2 expression had higher impact on CD3 expression than TAL1, and the
transduction with both genes did not seem to have a synergistic effect.

TAL1 and LMO2 expression impair human T-cell development
109
Moreover, TAL1 and LMO2 expression also decreased the percentage of double-
positive cells (CD4+CD8
+) at day 17 and to a minor extent at day 26 (Figure 3.3D). This
Figure 3.3. High TAL1 and LMO2 expression in human T-cell progenitors disrupts
normal T-cell development. (A) 293T cells were transfected with the indicated pHR-SIN
vectors and 48 h post-transfection the cells were lysed. TAL1, LMO2 and GFP expression
were analyzed by immunoblot. (B-E) Thymic-derived CD34+CD1a
- progenitors were
transduced with the respective pHR-SIN vectors for 48 h and then co-cultured in a stromal
layer of OP9-Dll1 cells for 26 days. (B) At the indicated time points, viable cells were counted
by trypan-blue exclusion and GFP expression was analyzed by flow cytometry. The bars
indicate the number of cells, normalized to the expression of GFP in the respective time point.
(C-D) At day 17 and 26 of co-culture, expression levels of (C) CD3 and (D) CD4/CD8 were
analyzed by flow cytometry. (E) The emergence of TCRαβ/CD3-expressing cells was analyzed
by flow cytometry at day 26.

TAL1 and LMO2 expression impair human T-cell development
110
effect appears to be associated with an earlier block at the double negative stage and not
to increased differentiation into CD4 or CD8 single-positive cells, as shown by
concomitant analysis of CD3 and TCR expression. A small percentage of cells
differentiated into CD3+TCRαβ
+ T-cells. This differentiation was strongly inhibited by
the expression of either TAL1 or LMO2 genes (Figure 3.3E). Notably, when cells were
transduced with both genes the differentiation of CD3+TCRαβ
+ T-cells was inhibited in
a synergistic fashion.
The results presented here clearly show that TAL1 and LMO2 genes have a
profound impact in human T-cell differentiation, and this is in agreement with the data
collected from TAL1 and LMO2 transgenic mice.
Discussion
TAL1 and LMO2 are fundamental for normal hematopoietic development (41, 42),
and are down-regulated upon T-cell differentiation (39, 40). These genes have been
associated with T-ALL, since they are over-expressed in a high percentage of T-ALL
patients (4). Moreover, TAL1 and LMO2 transgenic mice develop fatal and aggressive
T-cell leukemias (13, 22, 43). In contrast to LMO2 (28, 39), and despite being
commonly associated with human T-ALL, a clear causal role in the origin of the disease
has not been established for the TAL1 gene.
T-ALL is characterized by an accumulation of T-cells arrested at different stages
of development (44), and one of the first steps in malignant transformation may be the
arrest in T-cell differentiation. In fact, in TAL1 and LMO2 transgenic mice pre-
leukemic thymocytes are arrested at the double negative stage (CD4-CD8
-) (13, 22, 43).
Here we show that, TAL1 and LMO2 ectopic expression in hematopoietic progenitors
and early T-cell progenitors has a clear impact in normal T-cell differentiation (Figure
3.2-3.3).
To test whether the ectopic expression of these genes could impact human T-cell
differentitiation we used the OP9-Dll1 co-culture system. As described previously (37),
this co-culture system allows the differentiation of mature and functional human T-cells
in 52 days. We transduced umbilical cord-blood CD34+CD38
- cells and cultured them
in an OP9-Dll1 stromal layer. The transduction of both genes had a moderate effect in
T-cell differentiation and cellular activation, but the coordinated expression of both
genes (mLMO2-MAT-TAL1-IRES-eGFP) allowed the differentiation of a population

TAL1 and LMO2 expression impair human T-cell development
111
with a clear CD3+CD4
+CD8
+ phenotype and with a striking increase in cell size (Figure
3.2). This effect on cell size may have resulted from differences in differentiation
(different T-cell precursor subpopulations can have different sizes), increased
proliferation (dividing cells augment their size before division) and/or a process of
transformation (T-ALL blasts are bigger than normal thymocytes). Apart the
considerations on cell growth, these results are in agreement with previous reports
indicating that T-ALL patients with aberrant TAL1 expression are associated with a late
cortical phenotype (CD3+CD4
+CD8
+) (4). It is interesting to observe that TAL1 and
LMO2 expression per se in hematopoietic progenitors only slightly impaired normal T-
cell development (Figure 3.2). However, when both genes were co-expressed (where
TAL1 is expressed at higher levels in the mLMO2-MAT-TAL1-IRES-eGFP construct),
the blockade in T-cell development was higher (Figure 3.2). These results suggest that
TAL1 ectopic expression is able to reduce T-cell differentiation, but it likely requires
further events leading to malignant transformation, namely LMO2 expression. The
requirement for a second event was also observed in TAL1 transgenic mouse models
(13, 15). TAL1 transgenic mice develop leukemia with long latency periods, but when
combined with another pro-leukemogenic event, such as CK2 (13), LMO1 or LMO2
(22, 26) expression and heterozigoty of E2A/HEB genes (15), the latency period is
reduced. Surprisingly, LMO2 expression per se did not have a great impact in T-cell
development (Figure 3.2), not even when combined with low TAL1 expression
(mTAL1-MAT-LMO2-IRES-eGFP). This is in contrast with reports in transgenic mice
where LMO2 expression completely abrogates normal T-cell differentiation that
precedes the development of leukemia (22, 24, 43).
The initial lentiviral system that we used, allowed the coordinated expression of
both TAL1 and LMO2 genes in the same target cell, but with low efficiency of
transduction. This fact could explain why we observed moderate effects upon
transduction of hematopoietic progenitor cells with the TAL1 and LMO2 genes. To
circumvent this technical problem, we cloned both genes into pHR-SIN based lentiviral
vectors that are more efficient in the transduction of hematopoietic stem cells. The
transduction of thymic T-cell progenitors with the TAL1 and LMO2 genes cloned in
pHR-SIN based-vectors severely impaired T-cell differentiation, decresing the
expression of all the T-cell markers tested (Figure 3.3). Interestingly, in contrast with
the results generated using the other lentiviral system (Figure 3.2), ectopic expression of
LMO2 had a higher impact in T-cell differentiation than TAL1 (Figure 3.3), in

TAL1 and LMO2 expression impair human T-cell development
112
agreement with data collect from transgenic mouse models (22). Our results with this
lentiviral system did not show a synergistic impact of TAL1 and LMO2 expression in T-
cell differentiation. This could be due to a technical limitation of the system, since we
transduced the genes in separate vectors using the same reporter gene. Thus, we could
not assess whether the same cell is transduced simultaneous with TAL1 and LMO2. In
these conditions the GFP-positive population likely consists in a mixture of cells that
express either TAL1 or LMO2 and a fraction that expresses both genes.
Several reports (22, 25-27, 40) indicate that TAL1 cooperates with LMO proteins
(LMO1 and LMO2) to induce leukemia in transgenic mice. In these models, leukemia
development is preceded by an arrest in T-cell differentiation due to the accumulation of
immature CD4-CD8
- cells in the thymus. The reason for this accumulation could be due
to increased apoptosis of CD4+CD8
+ cells (26), but the inability of CD4
-CD8
- cells to
differentiate has also been suggested (40). Recently, it was observed that CD4-CD8
-
immature cells increase proliferation upon expression of TAL1 and LMO1 genes (27). In
our co-culture experiment, LMO2 expression increased cell counts by 15 fold relative to
empty transduced cells (Figure 3.3), which could also reflect increased proliferation of
immature T-cells.
Despite the heterogeneity between the two experimental settings reported here
(which could be due not only to the different vectors used and their specific limitations,
but also to the fact that we transduced different populations in each case), our results
provide the first indication that TAL1 ectopic expression can impair human T-cell
development. The deregulation of normal T-cell development seems to precede the
development of leukemogenesis in TAL1 and LMO2 transgenic mice. Our preliminary
data reported here, supports the notion that TAL1 and also LMO2 ectopic expression
can be leukemogenic triggers in human T-ALL. The analysis of how TAL1 and LMO2
deregulate normal T-cell development may be a critical step in understanding the
leukemogenic signatures of these genes in T-ALL.
References
1. Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias.
Science 278:1059-1064.
2. De Keersmaecker, K., P. Marynen, and J. Coolsi. 2005. Genetic insights in the
pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica 90:1116-1127.

TAL1 and LMO2 expression impair human T-cell development
113
3. Graux, C., J. Cools, L. Michaux, P. Vandenberghe, and A. Hagemeijer. 2006.
Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from
thymocyte to lymphoblast. Leukemia 20:1496-1510.
4. Ferrando, A.A., D.S. Neuberg, J. Staunton, M.L. Loh, C. Huard, S.C. Raimondi, F.G.
Behm, C.H. Pui, J.R. Downing, D.G. Gilliland, E.S. Lander, T.R. Golub, and A.T.
Look. 2002. Gene expression signatures define novel oncogenic pathways in T cell
acute lymphoblastic leukemia. Cancer Cell 1:75-87.
5. Chen, Q., J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A. Carroll, W. Crist, B.
Ozanne, M.J. Siciliano, and R. Baer. 1990. The tal gene undergoes chromosome
translocation in T cell leukemia and potentially encodes a helix-loop-helix protein.
Embo J 9:415-424.
6. Begley, C.G., P.D. Aplan, M.P. Davey, K. Nakahara, K. Tchorz, J. Kurtzberg, M.S.
Hershfield, B.F. Haynes, D.I. Cohen, T.A. Waldmann, and et al. 1989. Chromosomal
translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor
delta-chain diversity region and results in a previously unreported fusion transcript.
Proc Natl Acad Sci U S A 86:2031-2035.
7. Aplan, P.D., D.P. Lombardi, A.M. Ginsberg, J. Cossman, V.L. Bertness, and I.R.
Kirsch. 1990. Disruption of the human SCL locus by "illegitimate" V-(D)-J
recombinase activity. Science 250:1426-1429.
8. Hsu, H.L., L. Huang, J.T. Tsan, W. Funk, W.E. Wright, J.S. Hu, R.E. Kingston, and R.
Baer. 1994. Preferred sequences for DNA recognition by the TAL1 helix-loop-helix
proteins. Mol Cell Biol 14:1256-1265.
9. Bernard, M., E. Delabesse, L. Smit, C. Millien, I.R. Kirsch, J.L. Strominger, and E.A.
Macintyre. 1998. Helix-loop-helix (E2-5, HEB, TAL1 and Id1) protein interaction with
the TCRalphadelta enhancers. Int Immunol 10:1539-1549.
10. Hsu, H.L., I. Wadman, and R. Baer. 1994. Formation of in vivo complexes between the
TAL1 and E2A polypeptides of leukemic T cells. Proc Natl Acad Sci U S A 91:3181-
3185.
11. Bain, G., I. Engel, E.C. Robanus Maandag, H.P. te Riele, J.R. Voland, L.L. Sharp, J.
Chun, B. Huey, D. Pinkel, and C. Murre. 1997. E2A deficiency leads to abnormalities
in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol
Cell Biol 17:4782-4791.
12. Yan, W., A.Z. Young, V.C. Soares, R. Kelley, R. Benezra, and Y. Zhuang. 1997. High
incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Mol
Cell Biol 17:7317-7327.
13. Kelliher, M.A., D.C. Seldin, and P. Leder. 1996. Tal-1 induces T cell acute
lymphoblastic leukemia accelerated by casein kinase IIalpha. Embo J 15:5160-5166.

TAL1 and LMO2 expression impair human T-cell development
114
14. O'Neil, J., M. Billa, S. Oikemus, and M. Kelliher. 2001. The DNA binding activity of
TAL-1 is not required to induce leukemia/lymphoma in mice. Oncogene 20:3897-3905.
15. O'Neil, J., J. Shank, N. Cusson, C. Murre, and M. Kelliher. 2004. TAL1/SCL induces
leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 5:587-596.
16. Wadman, I.A., H. Osada, G.G. Grutz, A.D. Agulnick, H. Westphal, A. Forster, and T.H.
Rabbitts. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an
erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and
Ldb1/NLI proteins. Embo J 16:3145-3157.
17. Ono, Y., N. Fukuhara, and O. Yoshie. 1997. Transcriptional activity of TAL1 in T cell
acute lymphoblastic leukemia (T-ALL) requires RBTN1 or -2 and induces TALLA1, a
highly specific tumor marker of T-ALL. J Biol Chem 272:4576-4581.
18. Ono, Y., N. Fukuhara, and O. Yoshie. 1998. TAL1 and LIM-only proteins
synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute
lymphoblastic leukemia by acting as cofactors for GATA3. Mol Cell Biol 18:6939-
6950.
19. Boehm, T., L. Foroni, Y. Kaneko, M.F. Perutz, and T.H. Rabbitts. 1991. The rhombotin
family of cysteine-rich LIM-domain oncogenes: distinct members are involved in T-cell
translocations to human chromosomes 11p15 and 11p13. Proc Natl Acad Sci U S A
88:4367-4371.
20. Van Vlierberghe, P., M. van Grotel, H.B. Beverloo, C. Lee, T. Helgason, J. Buijs-
Gladdines, M. Passier, E.R. van Wering, A.J. Veerman, W.A. Kamps, J.P. Meijerink,
and R. Pieters. 2006. The cryptic chromosomal deletion del(11)(p12p13) as a new
activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood
108:3520-3529.
21. Ferrando, A.A., S. Herblot, T. Palomero, M. Hansen, T. Hoang, E.A. Fox, and A.T.
Look. 2004. Biallelic transcriptional activation of oncogenic transcription factors in T-
cell acute lymphoblastic leukemia. Blood 103:1909-1911.
22. Larson, R.C., I. Lavenir, T.A. Larson, R. Baer, A.J. Warren, I. Wadman, K. Nottage,
and T.H. Rabbitts. 1996. Protein dimerization between Lmo2 (Rbtn2) and Tal1 alters
thymocyte development and potentiates T cell tumorigenesis in transgenic mice. Embo
J 15:1021-1027.
23. Larson, R.C., P. Fisch, T.A. Larson, I. Lavenir, T. Langford, G. King, and T.H.
Rabbitts. 1994. T cell tumours of disparate phenotype in mice transgenic for Rbtn-2.
Oncogene 9:3675-3681.
24. Larson, R.C., H. Osada, T.A. Larson, I. Lavenir, and T.H. Rabbitts. 1995. The
oncogenic LIM protein Rbtn2 causes thymic developmental aberrations that precede
malignancy in transgenic mice. Oncogene 11:853-862.

TAL1 and LMO2 expression impair human T-cell development
115
25. Aplan, P.D., C.A. Jones, D.S. Chervinsky, X. Zhao, M. Ellsworth, C. Wu, E.A.
McGuire, and K.W. Gross. 1997. An scl gene product lacking the transactivation
domain induces bony abnormalities and cooperates with LMO1 to generate T-cell
malignancies in transgenic mice. Embo J 16:2408-2419.
26. Chervinsky, D.S., X.F. Zhao, D.H. Lam, M. Ellsworth, K.W. Gross, and P.D. Aplan.
1999. Disordered T-cell development and T-cell malignancies in SCL LMO1 double-
transgenic mice: parallels with E2A-deficient mice. Mol Cell Biol 19:5025-5035.
27. Tremblay, M., C.S. Tremblay, S. Herblot, P.D. Aplan, J. Hebert, C. Perreault, and T.
Hoang. Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1
oncogenes. Genes Dev 24:1093-1105.
28. Hacein-Bey-Abina, S., C. Von Kalle, M. Schmidt, M.P. McCormack, N. Wulffraat, P.
Leboulch, A. Lim, C.S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P.
Fraser, J.I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A.
Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E.
Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L.E. Leiva, M. Wissler, C. Prinz, T.H.
Rabbitts, F. Le Deist, A. Fischer, and M. Cavazzana-Calvo. 2003. LMO2-associated
clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science
302:415-419.
29. Schmidt, M., S. Hacein-Bey-Abina, M. Wissler, F. Carlier, A. Lim, C. Prinz, H. Glimm,
I. Andre-Schmutz, C. Hue, A. Garrigue, F. Le Deist, C. Lagresle, A. Fischer, M.
Cavazzana-Calvo, and C. von Kalle. 2005. Clonal evidence for the transduction of
CD34+ cells with lymphomyeloid differentiation potential and self-renewal capacity in
the SCID-X1 gene therapy trial. Blood 105:2699-2706.
30. Hacein-Bey-Abina, S., A. Garrigue, G.P. Wang, J. Soulier, A. Lim, E. Morillon, E.
Clappier, L. Caccavelli, E. Delabesse, K. Beldjord, V. Asnafi, E. MacIntyre, L. Dal
Cortivo, I. Radford, N. Brousse, F. Sigaux, D. Moshous, J. Hauer, A. Borkhardt, B.H.
Belohradsky, U. Wintergerst, M.C. Velez, L. Leiva, R. Sorensen, N. Wulffraat, S.
Blanche, F.D. Bushman, A. Fischer, and M. Cavazzana-Calvo. 2008. Insertional
oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin
Invest 118:3132-3142.
31. Deichmann, A., S. Hacein-Bey-Abina, M. Schmidt, A. Garrigue, M.H. Brugman, J. Hu,
H. Glimm, G. Gyapay, B. Prum, C.C. Fraser, N. Fischer, K. Schwarzwaelder, M.L.
Siegler, D. de Ridder, K. Pike-Overzet, S.J. Howe, A.J. Thrasher, G. Wagemaker, U.
Abel, F.J. Staal, E. Delabesse, J.L. Villeval, B. Aronow, C. Hue, C. Prinz, M. Wissler,
C. Klanke, J. Weissenbach, I. Alexander, A. Fischer, C. von Kalle, and M. Cavazzana-
Calvo. 2007. Vector integration is nonrandom and clustered and influences the fate of
lymphopoiesis in SCID-X1 gene therapy. J Clin Invest 117:2225-2232.

TAL1 and LMO2 expression impair human T-cell development
116
32. Howe, S.J., M.R. Mansour, K. Schwarzwaelder, C. Bartholomae, M. Hubank, H.
Kempski, M.H. Brugman, K. Pike-Overzet, S.J. Chatters, D. de Ridder, K.C. Gilmour,
S. Adams, S.I. Thornhill, K.L. Parsley, F.J. Staal, R.E. Gale, D.C. Linch, J. Bayford, L.
Brown, M. Quaye, C. Kinnon, P. Ancliff, D.K. Webb, M. Schmidt, C. von Kalle, H.B.
Gaspar, and A.J. Thrasher. 2008. Insertional mutagenesis combined with acquired
somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients.
J Clin Invest 118:3143-3150.
33. Robb, L., J.E. Rasko, M.L. Bath, A. Strasser, and C.G. Begley. 1995. scl, a gene
frequently activated in human T cell leukaemia, does not induce lymphomas in
transgenic mice. Oncogene 10:205-209.
34. Amendola, M., M.A. Venneri, A. Biffi, E. Vigna, and L. Naldini. 2005. Coordinate
dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters.
Nat Biotechnol 23:108-116.
35. Caton, D., A. Calabrese, C. Mas, V. Serre-Beinier, A. Charollais, D. Caille, R. Zufferey,
D. Trono, and P. Meda. 2003. Lentivirus-mediated transduction of connexin cDNAs
shows level- and isoform-specific alterations in insulin secretion of primary pancreatic
beta-cells. J Cell Sci 116:2285-2294.
36. Schmitt, T.M., and J.C. Zuniga-Pflucker. 2002. Induction of T cell development from
hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17:749-756.
37. La Motte-Mohs, R.N., E. Herer, and J.C. Zuniga-Pflucker. 2005. Induction of T-cell
development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro.
Blood 105:1431-1439.
38. Barata, J.T., A.A. Cardoso, L.M. Nadler, and V.A. Boussiotis. 2001. Interleukin-7
promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia
cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood
98:1524-1531.
39. Pike-Overzet, K., D. de Ridder, F. Weerkamp, M.R. Baert, M.M. Verstegen, M.H.
Brugman, S.J. Howe, M.J. Reinders, A.J. Thrasher, G. Wagemaker, J.J. van Dongen,
and F.J. Staal. 2007. Ectopic retroviral expression of LMO2, but not IL2Rgamma,
blocks human T-cell development from CD34+ cells: implications for leukemogenesis
in gene therapy. Leukemia 21:754-763.
40. Herblot, S., A.M. Steff, P. Hugo, P.D. Aplan, and T. Hoang. 2000. SCL and LMO1
alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain
expression. Nat Immunol 1:138-144.
41. Robb, L., I. Lyons, R. Li, L. Hartley, F. Kontgen, R.P. Harvey, D. Metcalf, and C.G.
Begley. 1995. Absence of yolk sac hematopoiesis from mice with a targeted disruption
of the scl gene. Proc Natl Acad Sci U S A 92:7075-7079.

TAL1 and LMO2 expression impair human T-cell development
117
42. Yamada, Y., A.J. Warren, C. Dobson, A. Forster, R. Pannell, and T.H. Rabbitts. 1998.
The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis.
Proc Natl Acad Sci U S A 95:3890-3895.
43. Neale, G.A., J.E. Rehg, and R.M. Goorha. 1995. Ectopic expression of rhombotin-2
causes selective expansion of CD4-CD8- lymphocytes in the thymus and T-cell tumors
in transgenic mice. Blood 86:3060-3071.
44. Uckun, F.M., M.G. Sensel, L. Sun, P.G. Steinherz, M.E. Trigg, N.A. Heerema, H.N.
Sather, G.H. Reaman, and P.S. Gaynon. 1998. Biology and treatment of childhood T-
lineage acute lymphoblastic leukemia. Blood 91:735-746.

Identification of novel TAL1 targets in T-ALL
118
Chapter 4
Identification of novel TAL1 target genes with
potential impact on T-cell acute lymphoblastic
leukemia
Ana Gírio, Bruno A. Cardoso, Nádia Correia, Ana R. Grosso and João T. Barata

Identification of novel TAL1 targets in T-ALL
119
Abstract
TAL1, a transcription factor involved in early hematopoiesis, is overexpressed in
a high percentage of T-cell acute lymphoblastic leukemia (T-ALL) cases, and TAL1
transgenic mice develop T-cell malignancies. However, the mechanisms by which
TAL1 possibly contributes to leukemogenesis are very ill defined, particularly in
humans. To identify novel TAL1 effectors we generated an inducible system, in which
TAL1, fused to the hormone binding domain of the estrogen receptor (ER), translocates
to the nucleus on addition of 4-hydroxy-tamoxifen (4-OHT). Microarray analysis of the
gene expression profile in HPB-ALL, a TAL1-negative T-ALL cell line, stably
transduced with ER-TAL1, revealed a total of 26 genes up- or down-regulated upon 4-
OHT treatment, in at least two independent experiments. We selected seven of those
genes on the basis of their function/potential interest in cancer and confirmed the
differential expression of three (CASZ1, DMGDH and OR5M3) by qRT-PCR.
Accordingly, transfection of another TAL1-negative T-ALL cell line, P12, with TAL1,
also led to increased expression of the validated TAL1 target genes. Conversely, knock-
down of TAL1 with siRNA in the TAL1-positive T-ALL cell line Jurkat decreased the
expression of CASZ1, correlating with loss of cell viability. Furthermore, similar to
TAL1, CASZ1 knock-down negatively affected cellular viability and proliferation of T-
ALL cells. The genes identified in this study can provide new clues on how TAL1 may
be involved in T-ALL.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a heterogeneous disease,
characterized by the clonal expansion of malignant hematopoietic progenitors arrested
at different stages of development. Ectopic expression of oncogenic transcription factors
is associated with T-ALL (1-3). TAL1 is over-expressed in up to 65% of T-ALL
patients, and TAL1 expression was reported to associate with poor prognosis (4).
Several chromosomal translocations are responsible for TAL1 ectopic expression in T-
ALL patients (5-9). However, the interstitial deletion [del(1)p32p32 or TAL1d], which
renders TAL1 gene expression under the control of the SIL promoter (10), is the most
common mechanism TAL1 deregulation in T-ALL. Still, a significant number of T-

Identification of novel TAL1 targets in T-ALL
120
ALL cases present TAL1 ectopic expression without evidence of genomic alterations
involving its locus (4, 11).
TAL1 expression in T-ALL is associated with resistance to apoptosis. TAL1
ectopic expression in cell lines protects from apoptosis (12) and loss of TAL1
expression and activity results in increased apoptosis (13, 14). However, the
mechanisms by which TAL1 likely exerts its leukemogenic effect are still far from
being understood. The TAL1 gene codes for a class II bHLH transcription factor (5) that
heterodimerizes with the class I bHLH transcription factors E2A and HEB (15, 16) to
bind the DNA in specific sequences termed E-boxes (17). Several studies demonstrated
that TAL1 inhibits E2A mediated gene transcription (18-20). More importantly, this
inhibition was shown to be the trigger for the development of leukemia in a TAL1
transgenic mouse model (21). In addition to this negative role, TAL1 has also been
shown to promote the transcription of several genes, although validated direct targets
are relatively few. In the context of leukemia, RALDH2 (22); NFKB1 (23) and NKX3.1
(24) were shown to be regulated by TAL1. TAL1 target genes have also been reported
in other cellular contexts, including normal hematopoiesis (25, 26), embryogenesis (27)
and erythropoiesis (28, 29).
Recently, in a genome-wide study, TAL1 was shown to bind the promoter of a
high number of genes in association with E2A and HEB. Importantly, TAL1 modulated
the expression of several genes (TRAF3; RAB40B; EPBH1; PTPRU; TTC3 and RPS3A),
which are involved in important cellular functions (30).Genome-wide studies are
important to understand which genes could be regulated by TAL1. However, they do
not allow to pinpoint which genes contribute to the malignant process elicited by TAL1.
To identify new TAL1 target genes in the context of T-ALL, we analyzed TAL1-
induced gene expression profile. We identified and validated three genes (CASZ1,
DMGDH and OR5M3) whose expression is positively regulated by TAL1. Importantly,
knock-down of CASZ1 expression decreases T-ALL cell viability and proliferation. Full
characterization of the genes identified in this study, particularly CASZ1, might
contribute to the understanding of how TAL1 can participate in T-ALL.
Materials and Methods
Cloning procedures. The #-ER-empty and #-ER-TAL1 vectors were derived from the
#304.pCCL.sin.cPPT.pA.CTE.eGFP.minCMV.hPGK.deltaNGFR.WPRE (31). TAL1

Identification of novel TAL1 targets in T-ALL
121
was amplified by PCR from the pcDNA3.1 (+) zeo TAL1 and cloned into the pcDNA3-
ER plasmid (kindly provided by Prof. P. J. Coffer) that contains the hormone-binding
domain of the estrogen receptor (32). TAL1 was fused in frame in the C-terminus of the
Estrogen-Receptor and the results were confirmed by sequentiation. Both ER and ER-
TAL1 were then amplified by PCR and cloned into the
#304.pCCL.sin.cPPT.pA.CTE.eGFP.minCMV.hPGK.deltaNGFR.WPRE with the
appropriate restriction enzymes (SphI-SalI). TAL1 was amplified by PCR from the
pcDNA3.1 (+) zeo TAL1 and cloned in frame with a HA-tag to create the pMT2-HA-
TAL1 vector. The results were confirmed by sequentiation. The primers used in the
cloning procedures are listed in Table 4.1.
Designation 5' to 3'
ERNTAL1F TAGATATCGGAGGAATGACCGAGCG
ERNTAL1R TAGATATCTCACCGAGGGCCGGCTCCATC
ER F CATGCATGCTGGATGCGAAATGAAATGGGTGC
ER R GCGTCGACTAATCCTCCGATATCGTTGGGGAAG
ER-TAL1 R GCGTCGACTAATCACCGAGGGCCGGCTCCATC
SmaI-TAL1 AGCCCGGGATGACCGAGCGGCCGCCGAGC
TAL1-XhoI ATCTCGAGTCACCGAGGGCCGGCTCCATC
Production of VSVG-pseudotyped lentiviruses. Vesicular-Stomatitis-Virus-
pseudotyped third-generation lentiviruses were produced as described in Material and
Methods section in Chapter 3 (page 99).
Cell lines. 293T cells were maintained in DMEM medium (Invitrogen) supplemented
with 10% fetal bovine serum (DMEM-10) and were splitted every 2 days. The human
T-ALL cell lines HPB-ALL, P12 and Jurkat have already been described and
extensively studied. These three cell lines were maintained in RPMI medium
(Invitrogen) supplemented with 10% FBS (RPMI-10) and splitted every 2-3 days. The
human CML cell line K562 was maintained in RPMI-10 and splitted every 2 days.
HPB-ALL and transfected 293T cells were cultured in the presence of 4-Hidroxy-
Tamoxifen (Sigma) at a concentration of 2.0µM. At the indicated time points, the cells
were harvested and processed as indicated below for RNA extraction and protein lysates
for Immunobloting.
Table 4.1. List of primers used in the cloning procedures

Identification of novel TAL1 targets in T-ALL
122
Establishment of HPB-ALL-ER-empty and HPB-ALL-ER-TAL1 cell lines. HPB-
ALL cells were transduced in retronectin (Takara) coated plates. Briefly, 0.5 x 106 cells
were incubated in 500µl RPMI-10 in a retronectin-coated well (5µg/well) of a 24-well-
plate with 500µl #-ER-empty and #-ER-TAL1 lentiviral supernatant. Cells were
incubated over-night at 37ºC, and in the following day, were washed and fresh media
added. Cells were expanded and sorted for high GFP expression.
Immunofluorescence. 293T cells were grown in coverslips and transfected with 1.0µg
of each plasmid (pcDNA3.1 (+) zeo TAL1 and pcDNA3-ER-TAL1) using Fugene 6
reagent (Roche) according to the manufacturer‟s instructions. After transfection, cells
were washed with PBS, fixed with 1% PFA and permeabilized with PGS (PBS + 0.2%
Gelatin + 0.05% Saponin). TAL1 was detected using a mouse α-TAL1 primary
antibody (Millipore), followed by incubation with Alexa Fluor 488–conjugated goat
anti-mouse antibody (Invitrogen). The images were acquired by confocal microscopy
using a Zeiss LSM 510 Meta microscope, using identical acquisition parameters in one
imaging session.
RNA isolation and Affymetrix GeneChip analysis. RNA labeling, hybridization to
the Affymetrix HuGene 1.0 ST arrays and scanning was performed by the Affymetrix
Core Facility, Instituto Gulbenkian de Ciência, Portugal, as described below. Total
RNA was extracted using the High Pure RNA extraction Kit (Roche). Concentration
and purity were determined by spectrophotometry and integrity was confirmed using an
Agilent 2100 Bioanalyzer with a RNA 6000 Nano Assay (Agilent Technologies). RNA
was processed for use on Affymetrix HuGene 1.0 ST arrays, according to the
manufacturer‟s One-Cycle Target Labeling Assay. Arrays were scanned on an
Affymetrix GeneChip scanner 3000 7G. All the microarray data analysis was performed
using R and several packages available from CRAN (R Development Core Team, 2008)
and Bioconductor. The raw data (CEL files) was normalized and summarized with the
Robust MultiArray Average method from the affy package. The differentially expressed
genes were selected using linear models and empirical Bayes methods as implemented
in limma package, verifying the p-values corresponding to moderated “F statistics”, and
selecting as differentially expressed genes those that had adjusted p-values lower than
0.005.

Identification of novel TAL1 targets in T-ALL
123
RNA extraction, RT-PCR and (semi)-quantitative-PCR. When indicated, RNA was
extracted using the High Pure RNA Isolation Kit (Roche) according to the
manufacturer‟s instructions. For the RT-PCR, up to 1µg of total RNA was reverse
transcribed using SuperScript II (Invitrogen) and random hexamers. Expression of each
gene was normalized to the expression levels of GAPDH. Primers used in the semi and
quantitative -PCR are described in Table 4.2. Semi-quantitative PCR was performed in
20ul reactions, using 2ul of cDNA, 200nM each primer and 2U of Gotaq polymerase
(Promega) according to manufacturer‟s instructions. PCR reactions were performed in
an 96-Well MyCycler (Bio-Rad), after the initial denaturation of 5 min at 95ºC, PCR
products were amplified by 30 cycles of 30 seg at 95ºC, 1 min at 60ºC and 45 seg at
72ºC with a final extension step of 5 min at 72ºC. The PCR products were separated in a
2% agarose gel and visualized under UV light. For quantitative PCR, the transcripts
were amplified in 25µl reactions, using 10 µl cDNA, 12.5 µl Power SYBR Green
(Applied Biosystems) and 200nM of each primer, according to manufacturer‟s
instructions. All the amplifications were performed in an ABI PRISM 7500
thermocycler (Applied Biosystems) for 2 min at 50ºC, 10 min at 95ºC, followed by 45
cycles of 15 seg at 95ºC, and 1 min at 60ºC.
Gene Forward primer 5' to 3' Reverse primer 5' to 3'
TAL1 AACAATCGAGTGAAGAGGAG CTTTGGTGTGGGGACCAT
RALDH2 AGGAGATCTTTGGCCCTGTT TCTGGGCATTTAAGGCATTGTAAC
CASZ1-TV1 CAGGCTAGGTTGCAAGTACA CTCATCTGTCTCAGCATCCA
CASZ1-TV2 AGAAGTGAGTCCCTCGATGA AGCATCTTTGGCTAGAAGGA
DRAM ATTTCCATAACCAAGCTGGA GGGTGACACTCTGGAAATCT
DMGDH GGCCAGGACACTCAGTACA TAAAACCCACCTTTGGAATG
KLRK1 GATGGCAAAAGCAAAGATGT CAGTAACTTTCGGTCAAGGG
OR5M3 GCAATTGGGAATCCTCTGCTTTATGGC GCTGCCAGACTCGTCAGAAAACCA
S100A1 AGACCCTCATCAACGTGTTC CACATCCTTCTGGGCATC
TM4SF1 CAAAGTATGCCTCCGAAAAC TACAGAAGAAAGCATCGCAC
GAPDH GGAGTCAACGGATTTGGTCG GACAAGCTTCCCGTTCTCAG
Electroporation of P12 cells. Electroporation of P12 cells was performed using the
Gene Pulser II (Bio-Rad). P12 cells were washed in RPMI-10 medium and
ressuspended at a concentration of 40 x 106/mL in RPMI-10 medium (without
antibiotics) with 40 μg of pMT2-HA or pMT2-HA-TAL1 plasmid DNA. 300µl samples
Table 4.2. List of primers for quantitative and semi-quantitative-PCR.

Identification of novel TAL1 targets in T-ALL
124
were placed in 4 cm–gap cuvettes (Bio-Rad) and electroporation was performed using
350V and 750µF parameters. After electroporation, cells were washed and cultured for
24 h in RPMI-10 medium.
Nucleofection of Jurkat cells. Nucleofection of Jurkat cells was performed using the
Amaxa Nucleofector II (Lonza) according to the manufacturer‟s instructions. Jurkat
cells were washed in RPMI-10 medium and 2 x 106 cells were ressuspended in 100µl of
solution V with 2µM of the respective small interfering RNA (siRNA) (Dharmacon).
Jurkat cells were nucleofected using the X-001 program and after the nucleofection the
cells were cultured for 24 h in RPMI-10 medium.
Viability assays. 48 h post-nucleofection cellular viability was examined using
Annexin-V and 7-Aminoactinomycin D. The cells were stained with Fluorescein
Isothiocyanate- conjugated Annexin V (eBiosciences) and 7-Aminoactinomycin D
(eBiosciences) at room temperature for 15 min in the appropriate binding buffer, and
subsequently analyzed by flow cytometry (FACSCalibur; Becton- Dickinson,).
Proliferation assays. 24 h post-nucleofection the cells were cultured (2 x 106 cells/mL)
in triplicate in RPMI-10 medium, in flat-bottomed 96-well plates at 37°C with 5% CO2
and incubated with 3H-thymidine (1µCi/well) for 16 h prior to harvest. DNA synthesis
was assessed by 3H-thymidine incorporation at 48 h post-nucleofection.
Immunobloting. At the indicated conditions, cell lysates were prepared and equal
amounts of protein were analyzed by 12% SDS-PAGE. The proteins were transferred
onto nitrocellulose membranes and immunoblotted with an antibody for ER (Santa-Cruz
Biotechnology). After immunobloting with the primary antibody, immunodetection was
performed using HRP-conjugated anti-mouse IgG (Promega) and developed by
chemiluminescence (Thermo Scientific).
Statistical analysis. Differences between populations were calculated using unpaired 2-
tailed Student‟s t test or One-way ANOVA, when appropriate (p<0.05 was considered
significant).

Identification of novel TAL1 targets in T-ALL
125
Results
TAL1 inducible system
To identify possible target genes that could be directly induced upon TAL1
expression, we used the 4OHT inducible system. The modified Hormone-binding
domain (HDB) of the Estrogen Receptor (ER) (32, 33) was fused to the N-terminus of
the TAL1 protein (ER-TAL1). As shown in Figure 4.1A, the ER-TAL1 protein is
sequestered in the cytoplasm due to the interaction of the ER domain with chaperones
(HSPs). When 4OHT is added to the cells, it binds to the ER-TAL1 fusion protein,
allowing its release from the chaperones and translocation into the nucleus. Once in the
nucleus, the ER-TAL1 fusion protein can activate the expression of TAL1 target genes
(Figure 4.1A). This type of induction system was developed to allow the refined
regulation of transcription factor activity (32, 33), although it has also been used to
study the function of signal transducer proteins (34).
TAL1 physiological localization is nuclear (Figure 4.1B, second panel) (35, 36).
We tested the efficacy of the ER-TAL1 fusion protein in 293T cells transfected with the
pcDNA3-ER-TAL1 plasmid. In the control condition, the ER-TAL1 fusion remained in
the cytoplasm of the 293T cells (Figure 4.1B, third panel). Upon upon induction with
2.0µM 4OHT, TAL1 localization shifted to the nucleus (Figure 4.1B, fourth panel).
These results indicate that the ER inducible system works properly.
Next, we subcloned ER and ER-TAL1 into lentiviral vectors that we transduced
into the TAL1-negative T-ALL cell line HPB-ALL (22), in order to identify genes that
are regulated by TAL1 activity in the context of T-ALL. As shown in Figure 4.1C,
HPB-ALL cells properly expressed both ER and ER-TAL1 proteins. To determine
whether the ER-TAL1 fusion effectively worked in HPB-ALL cells, we treated HPB-
ALL-ER and HPB-ALL-ER-TAL1 cells with 2.0µM 4OHT for 24 h and measured the
expression level of the RALDH2 gene, a known TAL1 target gene (22). Induction of the
ER-TAL1 fusion protein increased the expression of RALDH2 by roughly two-fold
when compared to ER alone (Figure 4.1D) in a time-dependent manner (Figure 4.1E).
These results indicate that the ER-TAL1 inducible system worked properly in HPB-
ALL cells.

Identification of novel TAL1 targets in T-ALL
126
Figure 4.1. Overview of the TAL1 inducible system. (A) In the non-induced condition,
Heat-Shock-Proteins (HSPs) bind to ER-TAL1 and arrest the fusion protein in the
cytoplasm. Upon addition of 4OHT, HSPs are released from ER-TAL1 allowing its
translocation to the nucleus and subsequent transcription of TAL1 target genes. (B) 293T
cells were transfected with the pcDNA3-ER-TAL1 plasmid, and 24 h post-transfection
2.0µM 4OHT was added. TAL1 localization was determined by confocal microscopy using
an α-TAL1 antibody. 293T cells transfected with pcDNA3.1(+) zeo TAL1 plasmid were
used as a positive control. (C) Wild type (WT) HPB-ALL cells, or stably expressing empty
ER or ER-TAL1 were lysed and ER and ER-TAL1 expression determined by immunoblot
using an α-ER antibody. Arrows indicate ER and ER-TAL1 expression respectively. (D-E)
HPB-ALL-ER-empty and HPB-ALL-ER-TAL1 cells were treated with 2.0µM 4OHT in the
indicated time points and RALDH2 expression analyzed. The graphs represent the
expression of RALDH2 mRNA measured by quantitative RT-PCR normalized to GAPDH.
The values were normalized relative to the untreated condition and represent mean ±
standard deviation of duplicates ( ** p<0.01).

Identification of novel TAL1 targets in T-ALL
127
TAL1 activity up-regulates genes associated with cancer
In order to identify potential TAL1 target genes that might have relevance in
leukemogenesis and leukemia progression, we analyzed the gene expression profile of
HPB-ALL-ER-TAL1 cells induced with 4OHT. We treated HPB-ALL-ER-TAL1 cells
with 2.0µM 4OHT for 24 h in three independent experiments, and analyzed the gene
expression profile using an Affymetrix microarray platform (Human Gene 1.0 ST). We
selected the genes whose expression changed by at least 1.5 fold upon TAL1 induction.
The Venn-diagram (Figure 4.2) shows the genes whose expression was altered in the
three experiments and the overlap between them. We considered as potential TAL1
targets exclusively those genes whose regulation showed the same trend in at least two
experiments (Table 4.3) and discarded all the rest.
Figure 4.2. Venn diagram of the number of genes differentially expressed in each
of three independent experiments upon TAL1 activity induction. mRNA extracted
from HPB-ALL-ER-TAL1 cells treated with 4OHT or vehicle for 24 h was used for
gene expression analysis by microarrays. A gene was considered positive when its
expression in the presence, relative to the absence, of 4OHT was changed more than 1.5
fold.

Identification of novel TAL1 targets in T-ALL
128
Gene
Up/Down-regulation
References This study
Described in
Cancer
CASZ1 (SRG) Up Up 38, 39
CYP2B6 Up no reports
DMGDH Up Up 37
DRAM Down Down 42
EYS Up no reports
FGF16 Up no reports
FLJ43390 Up no reports
FLJ43763 Up no reports
HIST1H2AJ Down no reports
KCNC1 (NGK2) Up no reports
KLRK1 (NGK2D) Down Down 43
KRT18P49 Up no reports
LOC283588 Down no reports
LOC391742 Up no reports
LOC644714 Up no reports
OR5M3 Up no reports
OR6C76 Up no reports
REG3G Up no reports
RNU13P1 Up no reports
RNU5F Down no reports
S100A1 Up Up 44-45
SNORD56B Down no reports
SNRPN Down no reports
SUMO1P3 Down no reports
TISP43 Up no reports
TM4SF1 Up Up 46
Next, we further narrowed the list of 26 genes to those already associated with
cancer (Table 4.3). Therefore, among the genes whose expression changed similarly in
at least two independent experiments, we selected six to proceed with our study
(CASZ1, DMGDH, DRAM, KLRK1, S100A1 and TM4SF1)(37-46). We also included
OR5M3 (47), which was up-regulated in all the three microarray experiments. Although
OR5M3 has not been described, as far as we know, as being involved in any form of
cancer, it belongs to the family of genes coding for G-coupled receptor proteins that
Table 4.3. List of genes identified in the microarray experiments (similarly regulated in at least
2 of 3 experiments).

Identification of novel TAL1 targets in T-ALL
129
have been associated with cancer progression (48). Notably, none of the genes identified
have been described in T-ALL.
CASZ1, DMGDH and OR5M3 are potential TAL1 target genes
To validate the microarray data, we next performed quantitative RT-PCR
analyses. The transcript levels of DRAM and KLRK1 decreased upon addition of 4OHT,
but the effect was minor (Figure 4.3A) and we could not detect the expression of
S100A1 and TM4SF1 in HPB-ALL cells (data not shown). In contrast, we confirmed
that induction of TAL1 activity in HPB-ALL cells increased the expression levels of
CASZ1 [both transcriptional variant 1 (TV1) and 2 (TV2)] ,DMGDH and OR5M3
(Figure 4.3A).

Identification of novel TAL1 targets in T-ALL
130
To further confirm whether the expression of these three genes is regulated by
TAL1, we electroporated another the TAL1-negative cell line P12 with a plasmid
expressing TAL1 and measured the expression levels of CASZ1 (TV1 and TV2),
DMGDH and OR5M3 at 24 and 48 h post-electroporation. In both time points
measured, the expression of TAL1 increased dramatically, indicating that the
electroporation was successful (Figure 4.3C). TAL1 ectopic expression in P12 cells
significantly up-regulated CASZ1 and OR5M3 and slightly induced DMGDH (Figure
4.3C). Taken together, these results support the hypothesis that TAL1 regulates the
expression of CASZ1, DMGDH and OR5M3.
CASZ1 knock-down decreases T-ALL cell viability and proliferation
The CASZ1 gene is associated with neuronal development (39) and reported to
protect from apoptosis (38). Interestingly, TAL1 expression and activity has also been
associated with protection from apoptosis (12-14). To analyze the impact of CASZ1
expression in T-ALL cells, we knocked down CASZ1 in Jurkat cells using siRNA. As
shown in Figure 4.4A, CASZ1 siRNA decreased the expression of both transcription
variants. We also analyzed TAL1 knockdown in the same cell line, which decreased
resulted in diminished CASZ1 expression, further supporting a role for TAL1 in the
regulation of this gene.
Figure 4.3. CASZ1, DMGDH and OR5M3 are potential TAL1 target genes. (A) HPB-
ALL-ER-TAL1 cells were treated for 24 h with 2.0µM 4OHT and the expression of the
indicated genes was measured. The graphs represent the transcript levels of the indicated
genes measured by quantitative RT-PCR normalized to GAPDH. The values were further
normalized relative to the untreated control condition and represent mean ± standard
deviation of duplicates. (B) HPB-ALL-ER-empty and HPB-ALL-ER-TAL1 cells were
incubated with 2.0µM 4OHT for 24 h and the expression of OR5M3 was analyzed by semi-
quantitative-RT-PCR. GAPDH amplification was used as a loading control. RNA extracted
from regularly cultured K562 cells was used as a positive control. (C) P12 cells were
electroporated with pMT2-HA and pMT2-HA-TAL1 plasmids and the transcript levels of the
indicated genes were measured in the indicated time points. The graphs represent the
transcript levels of the indicated genes measured by quantitative RT-PCR and normalized to
GAPDH. The values were further normalized relative to the pMT2-HA control condition and
represent mean ± standard deviation of duplicates.

Identification of novel TAL1 targets in T-ALL
131
The decrease in TAL1 gene expression had a small, but significant, negative
effect on T-ALL cell proliferation, whereas CASZ1 knockdown had a higher impact on
the reduction of cellular proliferation (Figure 4.4B). Furthermore, TAL1 knockdown
increased apoptosis (Figure 4.4C). This result is agreement with previous reports
indicating that TAL1 expression impacts on apoptosis (12, 13). Importantly, CASZ1
knockdown increased apoptosis similarly to the TAL1 knockdown (Figure 4.4C). Taken
together, our results suggest that CASZ1 might be involved in the biology of T-ALL
Figure 4.4. CASZ1 knockdown decreases T-ALL cell viability and proliferation. Jurkat
cells were nucleofected with siRNA for TAL1 and CASZ1 genes and the functional effects
were analyzed. (A) TAL1 and CASZ1 mRNA levels were analyzed by quantitative-RT-PCR
24 h post-nucleofection. The graphs represent the transcript levels of the indicated genes
and transcription variants measured by quantitative RT-PCR and normalized to 18S. The
values were further normalized relative to the non-targeting siRNA control condition and
represent mean ± standard deviation of duplicates. 48 h post-nucleofection proliferation (B)
and viability (C) were analyzed by 3H-thymidine incorporation and by flow cytometry
analysis of Annexin-V-FITC/7-AAD dot-plots, respectively. (A and C) Values represent
mean ± standard deviation of duplicates (** p<0.01; *** p<0.001).

Identification of novel TAL1 targets in T-ALL
132
and indicate that CASZ1 is positively regulated by TAL1. Analysis of OR5M3 and
DMGDH is warranted.
Discussion
Although TAL1 has been associated with human T-ALL (4, 49), little is known
about the transcriptional program promoted by this transcription factor in the context of
the disease. In our preliminary study, we identified novel putative TAL1 target genes by
showing that TAL1 forced expression up-regulates genes reported to be involved in
other cancers (CASZ1 and DMGDH) (37-41) and a member of the olfactory receptor
family (OR5M3) (47).
To identify these genes, we made use of the 4OHT inducible system (33), fusing
TAL1 with the hormone-binding-domain (HDB) of the estrogen receptor (ER), which
allowed the translocation of TAL1 to the nucleus upon 4OHT induction (Figure
4.1).Surprisingly, the translocation of TAL1 into the nucleus only clearly occurred after
24 h of induction with 4OHT. This result was in contrast with other reported ER-
fusions, which are induced much faster upon 4OHT induction (32). Since the TAL1
nuclear-localization-sequence (NLS) is N-terminal (36) it could be affected by the
fusion with the ER. Importantly, the ER-TAL1 fusion protein was able to induce the
expression of RALDH2 (a described TAL1-target gene) (Figure 4.1) even in the absence
of LMO proteins (22).
Our gene expression analyses showed that most of the genes were up-regulated
upon TAL1 induction by 4OHT (Table 4.3), suggesting that the main action of TAL1 is
to activate the expression of its target genes. From the list of genes described in Table
4.3, we chose to validate only those already reported as being involved in cancer
(CASZ1; DMGDH; DRAM; KLRK1; S100A1 and TM4SF1) and the only gene up-
regulated in all the three independent experiments (OR5M3). Of course, this strategy of
simplification implicates that other potentially relevant genes may have been
overlooked. We validated CASZ1, DMGDH and OR5M3 as new potential TAL1 targets
(Figure 4.3). Ectopic expression of TAL1 in another T-ALL cell line increased the
expression of these genes (Figure 4.3). Moreover, knockdown of TAL1 expression in T-
ALL cells decreased the expression of the CASZ1 gene. Despite the fact that our results
suggest these genes might be TAL1 targets in T-ALL, more experiments are required to
establish a direct link between TAL1 and their expression. Interestingly, the promoter

Identification of novel TAL1 targets in T-ALL
133
region of all the three genes contains consensus sequences (E-boxes) for TAL1 binding
(data not shown), supporting the possibility that these are TAL1 direct target genes.
The CASZ1 gene encodes for a transcription factor up-regulated in neuronal cell
differentiation and transcribes two different mRNAs (39). Interestingly, TAL1 was
shown to also play a role in neuronal differentiation (50, 51) and CASZ1 could be one of
its targets. Moreover, CASZ1 was also shown to be expressed in a variety of human
tumors and implicated in resistance to apoptosis (38). Importantly, TAL1 expression is
also directly associated with resistance to apoptosis in T-ALL cell lines (12-14). Here,
we demonstrated that CASZ1 expression regulates T-ALL cell viability and
proliferation (Figure 4.4). These results suggest that CASZ1 may act downstream of the
TAL1 leukemogenic pathway in the maintenance of T-ALL viability and proliferation.
DMGDH, which encodes for dimethylglycine dehydrogenase, was also up-
regulated upon induction of TAL1. The protein is a mitochondrial matrix enzyme that
generates sarcosine from dimethylglycine (40, 41). Its association with cancer comes
from the fact that high levels of sarcosine are associated with the aggressiveness of
prostate cancer cells (37).
OR5M3 was the only gene systematically up-regulated by TAL1 in all the three
independent experiments (Figure 4.2). OR5M3 is a member of the large family of G-
protein-coupled olfactory receptors (47). These receptors are expressed in sensory
neurons of mammals and other tissues (52) and can be activated by a variety of
chemical compounds (47). Recently, it was described that steroid hormones can bind to
OR51E2, an olfactory receptor expressed in prostate cancer, and inhibit cellular
proliferation by activation of p38 MAPK pathway (53). The ligands and activity of
OR5M3 are unknown. Nonetheless, it is conceivable that up-regulation of OR5M3
might contribute to TAL1-mediated leukemia.
Overall, our study identified three novel downstream targets of TAL1 with the
aim to characterize the possible mechanisms by which TAL1 might exert its
leukemogenic function. The fact that T-ALL patients with TAL1 ectopic expression
apparently have a less favorable prognosis (4), prompts for a better understanding of the
actual participation of TAL1 in T-ALL progression, including the identification of
functionally relevant effectors. Determining the exact participation of CASZ1, DMGDH
and OR5M3, in the network regulated by TAL1 in the context of leukemia and whether
they may represent novel therapeutic targets in the treatment of TAL1+ T-ALL requires
considerable investigation.

Identification of novel TAL1 targets in T-ALL
134
References
1. Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias.
Science 278:1059-1064.
2. Graux, C., J. Cools, L. Michaux, P. Vandenberghe, and A. Hagemeijer. 2006.
Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from
thymocyte to lymphoblast. Leukemia 20:1496-1510.
3. De Keersmaecker, K., P. Marynen, and J. Coolsi. 2005. Genetic insights in the
pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica 90:1116-1127.
4. Ferrando, A.A., D.S. Neuberg, J. Staunton, M.L. Loh, C. Huard, S.C. Raimondi, F.G.
Behm, C.H. Pui, J.R. Downing, D.G. Gilliland, E.S. Lander, T.R. Golub, and A.T.
Look. 2002. Gene expression signatures define novel oncogenic pathways in T cell
acute lymphoblastic leukemia. Cancer Cell 1:75-87.
5. Chen, Q., J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A. Carroll, W. Crist, B.
Ozanne, M.J. Siciliano, and R. Baer. 1990. The tal gene undergoes chromosome
translocation in T cell leukemia and potentially encodes a helix-loop-helix protein.
Embo J 9:415-424.
6. Begley, C.G., P.D. Aplan, M.P. Davey, K. Nakahara, K. Tchorz, J. Kurtzberg, M.S.
Hershfield, B.F. Haynes, D.I. Cohen, T.A. Waldmann, and et al. 1989. Chromosomal
translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor
delta-chain diversity region and results in a previously unreported fusion transcript.
Proc Natl Acad Sci U S A 86:2031-2035.
7. Fitzgerald, T.J., G.A. Neale, S.C. Raimondi, and R.M. Goorha. 1991. c-tal, a helix-
loop-helix protein, is juxtaposed to the T-cell receptor-beta chain gene by a reciprocal
chromosomal translocation: t(1;7)(p32;q35). Blood 78:2686-2695.
8. Aplan, P.D., S.C. Raimondi, and I.R. Kirsch. 1992. Disruption of the SCL gene by a
t(1;3) translocation in a patient with T cell acute lymphoblastic leukemia. J Exp Med
176:1303-1310.
9. Francois, S., E. Delabesse, L. Baranger, M. Dautel, C. Foussard, M. Boasson, O.
Blanchet, O. Bernard, E.A. Macintyre, and N. Ifrah. 1998. Deregulated expression of
the TAL1 gene by t(1;5)(p32;31) in patient with T-cell acute lymphoblastic leukemia.
Genes Chromosomes Cancer 23:36-43.
10. Aplan, P.D., D.P. Lombardi, G.H. Reaman, H.N. Sather, G.D. Hammond, and I.R.
Kirsch. 1992. Involvement of the putative hematopoietic transcription factor SCL in T-
cell acute lymphoblastic leukemia. Blood 79:1327-1333.

Identification of novel TAL1 targets in T-ALL
135
11. Ferrando, A.A., S. Herblot, T. Palomero, M. Hansen, T. Hoang, E.A. Fox, and A.T.
Look. 2004. Biallelic transcriptional activation of oncogenic transcription factors in T-
cell acute lymphoblastic leukemia. Blood 103:1909-1911.
12. Bernard, M., E. Delabesse, S. Novault, O. Hermine, and E.A. Macintyre. 1998.
Antiapoptotic effect of ectopic TAL1/SCL expression in a human leukemic T-cell line.
Cancer Res 58:2680-2687.
13. Leroy-Viard, K., M.A. Vinit, N. Lecointe, H. Jouault, U. Hibner, P.H. Romeo, and D.
Mathieu-Mahul. 1995. Loss of TAL-1 protein activity induces premature apoptosis of
Jurkat leukemic T cells upon medium depletion. Embo J 14:2341-2349.
14. Park, S.T., G.P. Nolan, and X.H. Sun. 1999. Growth inhibition and apoptosis due to
restoration of E2A activity in T cell acute lymphoblastic leukemia cells. J Exp Med
189:501-508.
15. Hsu, H.L., I. Wadman, and R. Baer. 1994. Formation of in vivo complexes between the
TAL1 and E2A polypeptides of leukemic T cells. Proc Natl Acad Sci U S A 91:3181-
3185.
16. Bernard, M., E. Delabesse, L. Smit, C. Millien, I.R. Kirsch, J.L. Strominger, and E.A.
Macintyre. 1998. Helix-loop-helix (E2-5, HEB, TAL1 and Id1) protein interaction with
the TCRalphadelta enhancers. Int Immunol 10:1539-1549.
17. Hsu, H.L., L. Huang, J.T. Tsan, W. Funk, W.E. Wright, J.S. Hu, R.E. Kingston, and R.
Baer. 1994. Preferred sequences for DNA recognition by the TAL1 helix-loop-helix
proteins. Mol Cell Biol 14:1256-1265.
18. Hansson, A., C. Manetopoulos, J.I. Jonsson, and H. Axelson. 2003. The basic helix-
loop-helix transcription factor TAL1/SCL inhibits the expression of the p16INK4A and
pTalpha genes. Biochem Biophys Res Commun 312:1073-1081.
19. Tremblay, M., S. Herblot, E. Lecuyer, and T. Hoang. 2003. Regulation of pT alpha gene
expression by a dosage of E2A, HEB, and SCL. J Biol Chem 278:12680-12687.
20. Herblot, S., A.M. Steff, P. Hugo, P.D. Aplan, and T. Hoang. 2000. SCL and LMO1
alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain
expression. Nat Immunol 1:138-144.
21. O'Neil, J., J. Shank, N. Cusson, C. Murre, and M. Kelliher. 2004. TAL1/SCL induces
leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 5:587-596.
22. Ono, Y., N. Fukuhara, and O. Yoshie. 1998. TAL1 and LIM-only proteins
synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute
lymphoblastic leukemia by acting as cofactors for GATA3. Mol Cell Biol 18:6939-
6950.
23. Chang, P.Y., K. Draheim, M.A. Kelliher, and S. Miyamoto. 2006. NFKB1 is a direct
target of the TAL1 oncoprotein in human T leukemia cells. Cancer Res 66:6008-6013.

Identification of novel TAL1 targets in T-ALL
136
24. Kusy, S., B. Gerby, N. Goardon, N. Gault, F. Ferri, D. Gerard, F. Armstrong, P.
Ballerini, J.M. Cayuela, A. Baruchel, F. Pflumio, and P.H. Romeo. 2010. NKX3.1 is a
direct TAL1 target gene that mediates proliferation of TAL1-expressing human T cell
acute lymphoblastic leukemia. J Exp Med 207:2141-2156.
25. Lecuyer, E., S. Herblot, M. Saint-Denis, R. Martin, C.G. Begley, C. Porcher, S.H.
Orkin, and T. Hoang. 2002. The SCL complex regulates c-kit expression in
hematopoietic cells through functional interaction with Sp1. Blood 100:2430-2440.
26. Krosl, G., G. He, M. Lefrancois, F. Charron, P.H. Romeo, P. Jolicoeur, I.R. Kirsch, M.
Nemer, and T. Hoang. 1998. Transcription factor SCL is required for c-kit expression
and c-Kit function in hemopoietic cells. J Exp Med 188:439-450.
27. Landry, J.R., S. Kinston, K. Knezevic, M.F. de Bruijn, N. Wilson, W.T. Nottingham,
M. Peitz, F. Edenhofer, J.E. Pimanda, K. Ottersbach, and B. Gottgens. 2008. Runx
genes are direct targets of Scl/Tal1 in the yolk sac and fetal liver. Blood 111:3005-3014.
28. Lahlil, R., E. Lecuyer, S. Herblot, and T. Hoang. 2004. SCL assembles a multifactorial
complex that determines glycophorin A expression. Mol Cell Biol 24:1439-1452.
29. Xu, Z., S. Huang, L.S. Chang, A.D. Agulnick, and S.J. Brandt. 2003. Identification of a
TAL1 target gene reveals a positive role for the LIM domain-binding protein Ldb1 in
erythroid gene expression and differentiation. Mol Cell Biol 23:7585-7599.
30. Palomero, T., D.T. Odom, J. O'Neil, A.A. Ferrando, A. Margolin, D.S. Neuberg, S.S.
Winter, R.S. Larson, W. Li, X.S. Liu, R.A. Young, and A.T. Look. 2006.
Transcriptional regulatory networks downstream of TAL1/SCL in t-cell acute
lymphoblastic leukemia. Blood 986.
31. Amendola, M., M.A. Venneri, A. Biffi, E. Vigna, and L. Naldini. 2005. Coordinate
dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters.
Nat Biotechnol 23:108-116.
32. Dijkers, P.F., R.H. Medema, C. Pals, L. Banerji, N.S. Thomas, E.W. Lam, B.M.
Burgering, J.A. Raaijmakers, J.W. Lammers, L. Koenderman, and P.J. Coffer. 2000.
Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional
regulation of p27(KIP1). Mol Cell Biol 20:9138-9148.
33. Littlewood, T.D., D.C. Hancock, P.S. Danielian, M.G. Parker, and G.I. Evan. 1995. A
modified oestrogen receptor ligand-binding domain as an improved switch for the
regulation of heterologous proteins. Nucleic Acids Res 23:1686-1690.
34. van Gorp, A.G., K.M. Pomeranz, K.U. Birkenkamp, R.C. Hui, E.W. Lam, and P.J.
Coffer. 2006. Chronic protein kinase B (PKB/c-akt) activation leads to apoptosis
induced by oxidative stress-mediated Foxo3a transcriptional up-regulation. Cancer Res
66:10760-10769.

Identification of novel TAL1 targets in T-ALL
137
35. Goldfarb, A.N., S. Goueli, D. Mickelson, and J.M. Greenberg. 1992. T-cell acute
lymphoblastic leukemia--the associated gene SCL/tal codes for a 42-Kd nuclear
phosphoprotein. Blood 80:2858-2866.
36. Bernard, M., L. Smit, E. Macintyre, D. Matthieu-Mahul, and K. Pulford. 1995. Nuclear
localization of the SCL/TAL1 basic helix-loop-helix protein is not dependent on the
presence of the basic domain. Blood 85:3356-3357.
37. Sreekumar, A., L.M. Poisson, T.M. Rajendiran, A.P. Khan, Q. Cao, J. Yu, B. Laxman,
R. Mehra, R.J. Lonigro, Y. Li, M.K. Nyati, A. Ahsan, S. Kalyana-Sundaram, B. Han, X.
Cao, J. Byun, G.S. Omenn, D. Ghosh, S. Pennathur, D.C. Alexander, A. Berger, J.R.
Shuster, J.T. Wei, S. Varambally, C. Beecher, and A.M. Chinnaiyan. 2009.
Metabolomic profiles delineate potential role for sarcosine in prostate cancer
progression. Nature 457:910-914.
38. Yuan, Z.R., R. Wang, J. Solomon, X. Luo, H. Sun, L. Zhang, and Y. Shi. 2005.
Identification and characterization of survival-related gene, a novel cell survival gene
controlling apoptosis and tumorigenesis. Cancer Res 65:10716-10724.
39. Liu, Z., X. Yang, F. Tan, K. Cullion, and C.J. Thiele. 2006. Molecular cloning and
characterization of human Castor, a novel human gene upregulated during cell
differentiation. Biochem Biophys Res Commun 344:834-844.
40. Binzak, B.A., J.G. Vockley, R.B. Jenkins, and J. Vockley. 2000. Structure and analysis
of the human dimethylglycine dehydrogenase gene. Mol Genet Metab 69:181-187.
41. Binzak, B.A., R.A. Wevers, S.H. Moolenaar, Y.M. Lee, W.L. Hwu, J. Poggi-Bach, U.F.
Engelke, H.M. Hoard, J.G. Vockley, and J. Vockley. 2001. Cloning of dimethylglycine
dehydrogenase and a new human inborn error of metabolism, dimethylglycine
dehydrogenase deficiency. Am J Hum Genet 68:839-847.
42. Crighton, D., S. Wilkinson, J. O'Prey, N. Syed, P. Smith, P.R. Harrison, M. Gasco, O.
Garrone, T. Crook, and K.M. Ryan. 2006. DRAM, a p53-induced modulator of
autophagy, is critical for apoptosis. Cell 126:121-134.
43. Osaki, T., H. Saito, T. Yoshikawa, S. Matsumoto, S. Tatebe, S. Tsujitani, and M.
Ikeguchi. 2007. Decreased NKG2D expression on CD8+ T cell is involved in immune
evasion in patients with gastric cancer. Clin Cancer Res 13:382-387.
44. Teratani, T., T. Watanabe, F. Kuwahara, H. Kumagai, S. Kobayashi, U. Aoki, A.
Ishikawa, K. Arai, and R. Nozawa. 2002. Induced transcriptional expression of calcium-
binding protein S100A1 and S100A10 genes in human renal cell carcinoma. Cancer
Lett 175:71-77.
45. Kanamori, T., K. Takakura, M. Mandai, M. Kariya, K. Fukuhara, M. Sakaguchi, N.H.
Huh, K. Saito, T. Sakurai, J. Fujita, and S. Fujii. 2004. Increased expression of calcium-

Identification of novel TAL1 targets in T-ALL
138
binding protein S100 in human uterine smooth muscle tumours. Mol Hum Reprod
10:735-742.
46. Lekishvili, T., E. Fromm, M. Mujoomdar, and F. Berditchevski. 2008. The tumour-
associated antigen L6 (L6-Ag) is recruited to the tetraspanin-enriched microdomains:
implication for tumour cell motility. J Cell Sci 121:685-694.
47. Fleischer, J., H. Breer, and J. Strotmann. 2009. Mammalian olfactory receptors. Front
Cell Neurosci 3:9.
48. Dorsam, R.T., and J.S. Gutkind. 2007. G-protein-coupled receptors and cancer. Nat Rev
Cancer 7:79-94.
49. Bash, R.O., S. Hall, C.F. Timmons, W.M. Crist, M. Amylon, R.G. Smith, and R. Baer.
1995. Does activation of the TAL1 gene occur in a majority of patients with T-cell
acute lymphoblastic leukemia? A pediatric oncology group study. Blood 86:666-676.
50. Muroyama, Y., Y. Fujiwara, S.H. Orkin, and D.H. Rowitch. 2005. Specification of
astrocytes by bHLH protein SCL in a restricted region of the neural tube. Nature
438:360-363.
51. Bradley, C.K., E.A. Takano, M.A. Hall, J.R. Gothert, A.R. Harvey, C.G. Begley, and
J.A. van Eekelen. 2006. The essential haematopoietic transcription factor Scl is also
critical for neuronal development. Eur J Neurosci 23:1677-1689.
52. Feldmesser, E., T. Olender, M. Khen, I. Yanai, R. Ophir, and D. Lancet. 2006.
Widespread ectopic expression of olfactory receptor genes. BMC Genomics 7:121.
53. Neuhaus, E.M., W. Zhang, L. Gelis, Y. Deng, J. Noldus, and H. Hatt. 2009. Activation
of an olfactory receptor inhibits proliferation of prostate cancer cells. J Biol Chem
284:16218-16225
.

HDACis down-regulate TAL1 expression in T-ALL
139
Chapter 5
TAL1/SCL is down-regulated upon histone deacetylase
inhibition in T-cell acute lymphoblastic leukemia cells
Bruno A. Cardoso, Sérgio F. de Almeida, Angelo B. A. Laranjeira, Maria Carmo-
Fonseca, J. Andrés Yunes, Paul J. Coffer and João T. Barata
Adapted from Leukemia, 2011; 25(10): 1578-86.

HDACis down-regulate TAL1 expression in T-ALL
140
Abstract
The transcription factor TAL1 is a major T-cell oncogene associated with poor
prognosis in T-cell Acute Lymphoblastic Leukemia (T-ALL). TAL1 binds histone
deacetylase 1 (HDAC1) and incubation with histone deacetylase inhibitors (HDACis)
promotes apoptosis of leukemia cells derived from TAL1 transgenic mice. Here, we
show for the first time that TAL1 protein expression is strikingly down-regulated upon
HDAC inhibition in T-ALL cells. This is due to decreased TAL1 gene transcription in
cells with intact TAL1 locus, and to impaired TAL1 mRNA translation in cells that
harbor the TAL1d microdeletion and consequently express TAL1 under the control of the
SIL promoter. Notably, HDACi-triggered apoptosis of T-ALL cells is significantly
reversed by TAL1 forced overexpression. Our results indicate that the HDACi-mediated
apoptotic program in T-ALL cells is partially dependent on their capacity to down-
regulate TAL1, and provide support for the therapeutic use of HDACis in T-ALL.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is characterized by the clonal
expansion of T-cell progenitors arrested at different stages of development. The T-cell
acute lymphocytic leukemia 1 (TAL1) transcription factor is essential for normal
hematopoeisis. However, TAL1 expression is rapidly down-regulated upon commitment
to the T-cell lineage (1, 2). Importantly, TAL1 is ectopically expressed in 65% of T-
ALL patients (3, 4). Aberrant expression of TAL1 in T-ALL results from relatively rare
non-random translocations that include t(1;14)(p32;q11), t(1;7)(p32;q34),
t(1;3)(p32;q21) and t(1;5)(p32;32) (5-9), and more frequently from a small interstitial
deletion [del(1)p32 or TAL1d] that renders the TAL1 gene expression dependent on the
upstream SIL promoter (10). Bi-allelic transcriptional activation, possibly resulting
from deregulation of the machinery normally involved in TAL1 gene repression during
normal T-cell development, can also account for TAL1 over-expression in some T-ALL
cases, although the precise mechanism is still unknown (11). The relevance of TAL1 in
T-ALL is supported by studies reporting the development of fatal T-cell leukemia in
TAL1 transgenic mice (12-15) and showing that TAL1 expression in T-ALL cell lines is
associated with protection from apoptosis (16-18). Furthermore, TAL1 expression has
been associated with poor prognosis in T-ALL (4).

HDACis down-regulate TAL1 expression in T-ALL
141
Histone acetylation, which plays a pivotal role in chromatin organization and gene
expression, is mainly regulated by two complexes of enzymes. Histone acetyl
transferases (HAT) add acetyl groups to the N-terminus of histones, and histone
deacetylases (HDAC) catalyze the opposite reaction (19). Increased histone acetylation
induces an open chromatin conformation that allows the transcriptional machinery to
access promoters and, in general, drives transcription. HDAC over-expression is
commonly observed in cancer (20), and HDAC inhibitors (HDACis) induce apoptosis
and growth arrest in cancer cell lines (19). Interestingly, gene expression profiling has
shown that cancer cells treated with HDACis not only up-regulate pro-apoptotic and
growth arrest genes but also commonly down-regulate oncogenic and proliferative
genes (21, 22).
TAL1 is a class II helix-loop-helix transcription factor that heterodimerizes with
the class I transcription factors E2A, E2-2 and HEB (23-25) and binds to specific DNA
sequences termed E-boxes (26). In addition, TAL1 was shown to interact with HDAC1
(27), as well as with chromatin remodeling complexes such as mSin3a (27) and HP1
(28), which influence TAL1 transcriptional activity. Moreover, TAL1 interaction with
mSin3a has been observed in pre-leukemic thymocytes from TAL1 transgenic mice, and
HDACi treatment was shown to selectively induce apoptosis of TAL1 transgenic
leukemia cells without affecting TAL1-negative cells (15).
Given that HDACis were previously shown to decrease the expression of different
oncogenes (29-31), we speculated that TAL1 could be regulated by HDACs. We treated
T-ALL cell lines and primary cells with Sodium Butyrate (SB) and Suberoylanilide
Hydroxamic Acid (SAHA) and found that TAL1 protein levels were strikingly reduced
upon treatment with these HDACis. This was due to inhibition of TAL1 gene
transcription in leukemia cells with an intact TAL1 locus, and to decreased protein
translation in cells bearing the TAL1d allele. Moreover, over-expression of TAL1
rescued HDACi-induced T-ALL cell apoptosis. Overall, these results indicate that
HDACis target TAL1 expression in T-ALL, and suggest that the apoptotic effect of
HDAC inhibition in T-ALL cells is partially dependent on TAL1 down-regulation.
Materials and Methods
T-ALL Primary cells and cell lines. T-ALL patient samples were obtained from
peripheral blood and/or bone marrow of patients with high leukemia involvement (85–

HDACis down-regulate TAL1 expression in T-ALL
142
100%). Informed consent and Institutional Review Board approval was obtained for all
sample collections in accordance with the Declaration of Helsinki. Samples were
enriched by density centrifugation over Lympholyte (Cedarlane Laboratories), washed
twice in RPMI 1640 (Invitrogen) supplemented with 10% (vol/vol) FBS and 2 mM l-
glutamine (hereafter referred to as RPMI10 medium), subjected to immunophenotypic
analysis by flow cytometry as described previously (32). The T-ALL cell lines CEM,
Jurkat, PF382 and SupT1 were maintained in RPMI10 medium and split every 2-3 days.
T-ALL primary cells and cell lines were cultured at 37ºC with 5% CO2 in RPMI10
alone (with the appropriate vehicle when necessary), or in RPMI10 plus: Sodium
Butyrate (SB) in the indicated concentrations (Sigma-Aldrich), 5mM Sodium Phenyl
Butyrate (SPB; Biomol International), Suberoylanilide Hydroxamic Acid (SAHA) in
the indicated concentrations (Cayman Chemicals), 150nM Trichostatin A (TSA; Sigma-
Aldrich), 2.5µg/mL Actinomycin D (Act.D; Sigma-Aldrich), 500µM Cyclohexamide
(CHX; Sigma-Aldrich) and 20µM QVD-OPH (Biovision). At the indicated time points,
the cells were harvested and processed as indicated below for assessment of cell
viability, and RNA and protein extraction.
Cloning procedures. The #-empty and #-TAL1 vectors were derived from the
pCCL.sin.cPPT.PGK.GFP.WPRE (33). In the empty vector, ΔLNGFR was removed
and the vector re-ligated. TAL1 gene was subcloned from the pcDNA 3.1 (+) zeo TAL1
vector using the appropriate restriction enzymes.
Production of VSVG-pseudotyped lentiviruses. Vesicular-Stomatitis-Virus-
pseudotyped third-generation lentiviruses were produced as described in Material and
Methods section in Chapter 3 (page 99).
Transduction of T-ALL cells. PF382 cells were transduced with VSVG-pseudotyped
TAL1-expressing lentiviruses. Briefly, 0.5 x 106 cells were incubated in 500µl RPMI10
plus 500µl of the corresponding lentiviral supernatant. The cells were then centrifuged
for 2 h at 2000 rpm at 33ºC, incubated overnight at 37ºC, washed and cultured in fresh
medium. After expansion, cells were sorted for high GFP expression. Viral supernatants
were produced by transient transfection of 293T cells, as described (33).

HDACis down-regulate TAL1 expression in T-ALL
143
Assessment of cell viability. Determination of cell viability was performed by flow
cytometry analysis of Forward Scatter versus Side Scatter (FSC/SSC) distribution using
a FACScalibur (Becton-Dickinson). We have previously confirmed that this strategy
measure lymphocyte viability as accurately as using Annexin V and propidium iodide
staining (32, 34). In experiments using primary T-ALL patient samples viable cells
were counted by trypan-blue exclusion.
Immunobloting. Cell lysates were prepared as described (35) and equal amounts of
protein were analyzed by 12% SDS-PAGE, transferred onto nitrocellulose membranes,
and immunoblotted with the following antibodies or antiserum: Actin (Santa-Cruz
Biotechnology); PARP (Novus Biologicals); phospho-S6 (S235/S236) (Cell signaling
technology); P-Akt/PKB (S473) (Cell signaling technology) and TAL1 (Millipore).
After immunobloting with primary antibodies, immunodetection was performed using
HRP-conjugated anti-mouse IgG (Promega), anti-rabbit IgG (Promega) or anti-goat IgG
(Santa-Cruz Biotecnology) as indicated by the host origin of the primary antibody and
developed by chemiluminescence (Thermo Scientific). Where indicated, densitometry
analysis was performed using Adobe Photoshop CS3 software (version 10.0). Each
band was analyzed with a constant frame and normalized to the respective loading
control.
RNA extraction, RT-PCR and quantitative-PCR. Where indicated, RNA was
extracted using High Pure Isolation Kit (Roche) according to the manufacturer‟s
instructions. For the RT-PCR, up to 1µg of total RNA was reverse transcribed using
SuperScript II (Invitrogen) and random hexamers. Expression of each gene was
normalized to the expression levels of GAPDH, 18S or ABL where indicated. Primers
used for the quantitative-PCR are indicated in Table 5.1. The transcripts were amplified
in 25µl reactions, using 10 µl cDNA, 12.5 µl Power SYBR Green (Applied Biosystems)
and 200nM of each primer, according to manufacturer‟s instructions. All the
amplifications were performed in an ABI PRISM 7500 thermocycler (Applied
Biosystems) for 2 min at 50ºC, 10 min at 95ºC, followed by 45 cycles of 15 seg at 95ºC,
and 1 min at 60ºC.

HDACis down-regulate TAL1 expression in T-ALL
144
Gene Forward primer 5' to 3' Reverse primer 5' to 3'
TAL1 AACAATCGAGTGAAGAGGAG CTTTGGTGTGGGGACCAT
TAL1 E5 TTGGGGAGCCGGATGCCTTC GTAATCTCCATCTCATAGGGGG
TAL1d AAGGGGAGCTAGTGGGAGAAA AGAGCCTGTCGCCAAGAA
p21 CAGCAGAGGAAGACCATGTG GGCGTTTGGAGTGGTAGAAA
GAPDH GGAGTCAACGGATTTGGTCG GACAAGCTTCCCGTTCTCAG
18S GGAGAGGGAGCCTGAGAAACG CGCGGCTGCTGGCACCAGACTT
ABL TGGAGATAACACTCTAAGCATAACTAAAGGT GATGTAGTTGCTTGGGACCCA
Chromatin Immunoprecipitation. For Chromatin Immunoprecipitation (ChIP), 2x108
Jurkat cells were used for each condition. Cells were washed twice in 1x Phosphate
Buffered Saline (PBS) and cross-linked with 1% formaldehyde at room temperature.
Cells were then lysed in SDS lysis buffer and sonicated. Chromatin was pre-cleared for
1 h at 4ºC using protein A-sepharose beads and then incubated overnight with 5µg
rabbit polyclonal antibody against RNA polymerase II (Santa Cruz Biotechnology) and
5µg rabbit polyclonal antibody against H3K9ac (Abcam). DNA-protein complexes were
pulled down with protein A-sepharose beads (Sigma-Aldrich) for 4 h at 4ºC, washed
and eluted in 1% SDS, 0.1 M NaHCO3 elution buffer. Cross-link was reversed
overnight at 65ºC in 0.2M NaCl (the input samples were also reverse cross-linked).
DNA was purified using phenol:chloroform extraction and ethanol precipitation, eluted
in DNase/RNase free water and used for quantitative-RT-PCR analysis. Primers were
designed to amplify specific regions of the TAL1 gene and are indicated in Table 5.2.
Region Forward primer 5'to 3' Reverse primer 5' to 3'
Promoter 1a GGATAGGGAGACTGCCCATTG CACCTCCCAGGGCTTCTTTC
Exon 4 TGAACGGCGTCGCCAAGGAG CGCGTCGCGGCCCTTTAAGT
Exon 6 TCGGCCTTTTGGGGGTGGGT GGGCCCGCCCACAGAAACAA
Statistical analysis. Differences between populations were calculated using unpaired 2-
tailed Student‟s t test or One-way ANOVA, when appropriate (p<0.05 was considered
significant).
Table 5.1. List of primers used in quantitative-PCR.
Table 5.2. List of primers used in Chromatin Immunoprecipitation experiments.

HDACis down-regulate TAL1 expression in T-ALL
145
Results
HDAC inhibition down-regulates TAL1 protein levels in T-ALL cells
HDACis are potent inducers of apoptosis in T-cell tumors derived from TAL1
transgenic mice (15). Since TAL1 has been implicated in prevention of T-ALL cell
apoptosis (16-18), we hypothesized that TAL1 expression and/or activity could be
altered by treatment with HDACi. To answer this question, we treated several T-ALL
cell lines with the HDACi SB. TAL1 protein expression significantly decreased in all
the cell lines tested (Figure 5.1A,B). Similar results were obtained with other HDACis,
including, SPB, TSA and SAHA (Figure 5.1B). Moreover, the negative effect of
HDACis on TAL1 protein expression was time- (Figure 5.1C,D) and dose-dependent
(Figure 5.1E,F).

HDACis down-regulate TAL1 expression in T-ALL
146
HDACi-mediated TAL1 protein down-regulation in T-ALL cells is not due
to increased apoptosis or protein degradation.
HDACis were shown to induce apoptosis of acute lymphoblastic leukemia cell
lines, including T-ALL (36). Analysis of PARP cleavage indicated that both SB (Figure
5.2A) and SAHA (Figure 5.2B) promoted apoptosis of Jurkat T-ALL cells. The
temporal down-regulation of TAL1 (Figure 5.1E,F) coincided with the induction of
apoptosis (Figure 5.2A,B). To exclude the possibility that TAL1 down-regulation
resulted from increased apoptosis, we treated Jurkat and PF382 cells with the pan-
caspase inhibitor QVD-OPH, and analyzed TAL1 protein expression upon incubation
with HDACis. Although QVD-OPH significantly rescued apoptosis in both cell lines, as
shown by analysis of PARP cleavage (Figure 5.2C) and percent cell viability (Figure
5.2D), it did not restore TAL1 protein levels (Figure 5.2C).
Several reports indicate that HDACis affect protein stability, inducing degradation
of oncogenes and cellular proteins (29, 37). To test whether protein degradation could
play a role in the TAL1 protein down-regulation induced by HDACis, we treated Jurkat
cells (Figure 5.2E) with the protein translation inhibitor Cycloheximide (CHX). The
half-life of TAL1 protein, which is roughly 6 h in Jurkat cells, is not significantly
altered by treatment with SB (Figure 5.2F), indicating that HDACis do not have a major
effect on TAL1 protein stability. Similar results were obtained in PF382 cells upon
treatment with CHX and SB, although TAL1 protein half-life was significantly longer
in these cells (around 12-16 h; data not shown).
Figure 5.1. HDACis down-regulate TAL1 protein in T-ALL cells. T-ALL cell lines
treated with HDACi, lysed and analyzed by immunoblot for the expression of TAL1. Actin
was used as loading control. (A) Jurkat, SupT1 and PF382 cells were treated for 24 h with the
indicated concentrations of SB. (B) Jurkat, SupT1, PF382 and CEM cells were treated for 24
h with 5mM SB, 5mM SPB, 150nM or 10µM SAHA. (C-F) Jurkat cells were treated with
5mM SB (C) or 2µM SAHA (D) for the indicated time points or treated for 24 h with
increasing concentrations of SB (E) or SAHA (F). Data are representative of at least three
independent experiments.

HDACis down-regulate TAL1 expression in T-ALL
147
Figure 5.2. HDACi-mediated TAL1 protein down-regulation is not due to increased
apoptosis or increased protein degradation. (A, B) Jurkat cells were treated for 24 h with
5mM SB (A) or 2µM SAHA (B). At the indicated time points the cells were lysed and PARP
cleavage was analyzed by immunoblot. Actin was used as loading control. (C, D) Jurkat and
PF382 were incubated for 24 h with the pan-caspase inhibitor QVD-OPH (20µM), SB (5mM)
or both. (C) The cells were lysed and TAL1 expression and PARP cleavage analyzed by
immunoblot. Actin was used as loading control. (D) Cell viability was determined by flow
cytometry analyzis of FSC/SSC distribution. Viability index was calculated as described in
“Materials and Methods”. Mean values ± standard deviation of duplicates of each condition are
represented ( ** p<0.01; *** p<0.001). (E, F) Jurkat cells were treated with 500µM CHX or
the combination of CHX and 5mM SB. At the indicated time points the cells were lysed and
TAL1 expression analyzed by immunoblot. Actin was used as a loading control (E). TAL1
protein levels were determined by densitometry analysis and are normalized to Actin (F). The
data are representative of two independent experiments.

HDACis down-regulate TAL1 expression in T-ALL
148
HDAC inhibition down-regulates TAL1 transcript levels without affecting
TAL1 splicing or mRNA stability
HDACis have been reported to diminish the transcript levels of specific genes by
inhibiting gene transcription (31) and by decreasing mRNA stability (30, 38). Hence,
we next evaluated whether HDACis decreased TAL1 protein expression in T-ALL cells
through down-regulation of TAL1 mRNA levels. Treatment of Jurkat and SupT1 cells
with SB and SAHA for 24 h induced a dramatic down-regulation of TAL1 mRNA levels
(Figure 5.3A). Similar results were obtained using three primary leukemia samples
collected from T-ALL patients at diagnosis (Figure 5.3B). Moreover, down-regulation
of TAL1 mRNA levels was rapid (Figure 5.3C,D) and dose-dependent (Figure 5.3E,F).
Notably, the decrease in TAL1 mRNA was not due to increased apoptosis
(Supplementary Figure 5.1) or to a generalized negative effect on gene expression, since
HDACi treatment clearly upregulated the transcript levels of CDKN1A, the gene coding
for the cell cycle inhibitor p21cip1
(Supplementary Figure 5.2), in accordance with
previous reports (39, 40).
Given the rapid down-regulation of TAL1 transcript levels and the fact that the
TAL1 gene is composed of several exons (Supplementary Figure 5.3A), we
hypothesized that TAL1 splicing was impaired upon HDAC inhibition, leading to an
accumulation of unspliced RNA that would result in decreased detection of the
processed TAL1 mRNA. To address this possibility, we treated Jurkat cells for up to 6 h
with 5mM SB. No difference between the levels of total and processed TAL1 transcripts
was observed in any of the time points analyzed (Figure 5.3G), suggesting that HDACi
treatment did not affect TAL1 mRNA splicing.
Several lines of evidence indicate that HDACis may decrease mRNA stability (30,
38). To test whether this could be the cause for decreased TAL1 mRNA expression, we
treated Jurkat cells with Actinomycin D (Act D), a known inhibitor of gene
transcription.
We treated Jurkat cells with 5mM SB, 2.5µg/mL Act D or the combination of
both inhibitors for 6 h and analyzed TAL1 mRNA levels. Treatment with Act D
decreased TAL1 mRNA similarly to SB, and the combination of both did not further
down-regulate TAL1 transcript levels (Figure 5.3H). These data suggest that HDACi
treatment did not affect TAL1 mRNA stability.

HDACis down-regulate TAL1 expression in T-ALL
149

HDACis down-regulate TAL1 expression in T-ALL
150
HDAC inhibition abrogates TAL1 transcription in TAL1-positive T-ALL
cells with an intact TAL1 locus
Since neither mRNA splicing nor stability were significantly affected by HDAC
inhibition, we next asked whether HDACis could directly affect TAL1 gene
transcription. We treated Jurkat cells with 5mM SB for 4 h and performed Chromatin
Immunoprecipition (ChIP) to analyze the binding of RNA polymerase II and the levels
of Histone H3 Lysine-9 acetylation (H3K9Ac), which is a marker of actively
transcribed euchromatin (41). SB incubation abrogated the binding of RNA polymerase
II to the TAL1 promoter 1a and to exon 4 (Figure 5.3I). It is interesting to note that
TAL1 exon 4 had a higher binding of RNA polymerase II than the promoter 1a in the
control condition (Figure 5.3I), suggesting that the TAL1 promoter IV (42) located
within this exon is active. Notably, SB incubation substantially decreased H3K9Ac,
both at TAL1 promoter 1a and at exon 4 (Figure 5.3J). Altogether, these results clearly
indicate that HDACis actively reduce binding of RNA polymerase II to TAL1 promoters
and inhibit TAL1 gene transcription.
Figure 5.3. HDACi down-regulate TAL1 through inhibition of TAL1 gene transcription
in TAL1wt
T-ALL cells lines. TAL1 mRNA levels from T-ALL cell lines and primary cells
were analyzed by quantitative-PCR, normalized to GAPDH (A, C-G), ABL (B) or 18S (H)
housekeeping genes and depicted as relative values to each control condition. (A) Jurkat and
SupT1 cells were treated with 5mM SB or 10µM SAHA for 24 h. (B) Primary T-ALL cells
collected from 3 different patients at diagnosis were treated with 5mM SB and 5µM SAHA
for 6 h. (C, D) Jurkat cells were treated with 5mM SB (C) and 2µM SAHA (D) for the
indicated time. (E, F) Jurkat cells were treated for 6 h with increasing concentrations of SB
(E) and SAHA (F). (G) Jurkat cells were incubated with 5mM SB, for the indicated time,
and TAL1 mRNA was amplified with primers that allow the detection of both processed and
total TAL1 transcripts (Supplementary figure 2B). (H) Jurkat cells were incubated with 5mM
SB, 2.5µg/mL Act.D or both, for the indicated time. (I, J) Jurkat cells were treated with
5mM SB for 4 h, cells were lysed and ChIP was performed to evaluate the binding of RNA
polymerase II (I) and H3K9Ac (J) to the indicated regions of the TAL1 locus. Fold
enrichment relative to the input is indicated. (A-J) Values represent mean ± standard
deviation of duplicates (* p<0.05; ** p<0.01; *** p<0.001).

HDACis down-regulate TAL1 expression in T-ALL
151
HDAC inhibition up-regulates TAL1 transcripts in T-ALL cells with TAL1d
As shown above, cell lines with an intact TAL1 locus (hereafter referred to as
TAL1wt
), such as Jurkat and SupT1, down-regulated TAL1 protein levels after HDACi
treatment, due to decreased TAL1 mRNA levels that resulted from diminished gene
transcription. However, we found that other T-ALL cell lines, such as PF382 and CEM,
paradoxically up-regulated TAL1 transcript levels upon incubation with HDACis
(Figure 5.4A), despite the fact that TAL1 protein expression was clearly decreased by
all the HDACis tested (Figure 5.1A,B).
We speculated that this could be due to the fact that TAL1 gene expression is not
driven by its native promoters in these cell lines, in such way that HDACi treatment
would not negatively affect TAL1 transcription. Similar to primary patient samples,
some TAL1+ T-ALL cell lines display a small interstitial deletion in the TAL1 promoter
(TAL1d) that renders TAL1 gene transcription dependent on regulatory elements of the
upstream gene SIL (10) (Supplementary Figure 5.4A).

HDACis down-regulate TAL1 expression in T-ALL
152
By using primers that specifically detect SIL-TAL1 fusion transcripts (43), we
confirmed that PF382 cells (Supplementary Figure 5.4B, right panel) and CEM (data
not shown) displayed the TAL1d microdelection and up-regulated TAL1 mRNA levels
in response to HDACi treatment (Supplementary Figure 5.4B, right panel, and data not
shown). In contrast, we did not detect SIL-TAL1 fusion transcripts in Jurkat cells
(Supplementary Figure 5.3B left panel) or SupT1 cells (data not shown).
HDAC inhibition down-regulates TAL1 protein levels by decreasing
translation in T-ALL cells with TAL1d
Next, we sought to determine the mechanism by which HDACis decreased TAL1
protein expression in TAL1d cell lines while augmenting TAL1 transcript levels. Our
results indicated that HDACi treatment did not affect TAL1 protein stability in PF382
cells (data not shown), excluding increased protein degradation as a compensatory
mechanism for increased TAL1 mRNA expression. Comparison of the kinetics of TAL1
protein down-regulation induced by HDACis in Jurkat (TAL1wt
) and PF382 (TAL1d)
cells showed that SB induced faster down-regulation of TAL1 protein in Jurkat cells
than in PF382 cells (Figure 5.4B). A recent report demonstrated that SAHA inhibits
Figure 5.4. HDACis down-regulate TAL1 by affecting TAL1 protein translation in
TAL1d T-ALL cell lines. (A) PF382 and CEM cells were treated with 5mM SB or 10µM
SAHA for 24 h. TAL1 mRNA levels were analyzed by quantitative-PCR, normalized to the
levels of GAPDH and depicted as relative values to the medium control condition. Values
indicate mean ± standard deviation of duplicates. (B) Jurkat and PF382 cells were treated
with 5mM SB. At the indicated time points, the cells were lysed and analyzed by
immunoblot for the expression of TAL1. Actin was used as loading control. TAL1 protein
levels were quantified by densitometry analysis, normalized to Actin and depicted as
relative values to the medium control condition at 6 h of treatment. (C) PF382 cells were
incubated for 6 h with 500µM CHX, washed, and then treated for further 24 h with 5mM
SB. Cells were lysed in the indicated time points that represent the time point where SB
was added to the cells. TAL1 expression was analyzed by immunoblot and Actin used as a
loading control. TAL1 protein levels were quantified by densitometry analysis, normalized
to Actin and depicted as relative values to the medium control condition at 6 h of
treatment. The data are representative of at least three independent experiments.

HDACis down-regulate TAL1 expression in T-ALL
153
cyclin D1 translation (44). Therefore, we speculated that HDACi impaired TAL1
protein translation in TAL1d cells and thereby negatively regulated TAL1 protein levels.
To test this hypothesis, we pretreated PF382 cells for 6 h with CHX, and then removed
the translation inhibitor to allow de novo protein synthesis. SB was incubated for
additional 24 h to analyze its effect on de novo translation of TAL1. Incubation with SB
inhibited TAL1 protein translation (Figure 5.4C), suggesting that HDAC inhibition
impaired TAL1 protein translation in TAL1d cell lines.
Forced TAL1 expression partially rescues T-ALL cell death induced by
HDAC inhibition
HDACis were demonstrated to induce apoptosis in human leukemia cell lines (36,
45) and primary cells (46). In accordance, we observed that primary T-ALL samples
(Supplementary Figure 5.5A) and cell lines (Supplementary Figure 5.5B) entered
apoptosis when treated with different HDACis. TAL1 expression was shown to prevent
apoptosis of T-ALL cells (16, 17), and HDACis were reported to promote cell death of
TAL1-expressing mouse leukemia cells (15). Thus, we next compared the efficacy of
HDAC inhibition upon forced expression of TAL1 in T-ALL cells. We transduced
PF382 (Figure 5.5A) and Jurkat (Supplementary Figure 5.6) cells with a lentiviral
vector driving the expression of TAL1. Forced expression of TAL1 significantly
reversed T-ALL cell death induced by treatment with as shown by the viability index
(Figure 5.5A and Supplementary Figure S6) and PARP cleavage (Figure 5.5B). These
results suggest that HDACis promote apoptosis of T-ALL cells in part via down-
regulation of TAL1.
Discussion
HDACis affect global gene expression. In cancer cells, HDACis have been shown
to induce the transcription of genes associated with apoptosis and cell cycle arrest, while
decreasing the expression of oncogenes and genes that promote proliferation (21, 22,
29-31). Here, we showed for the first time that TAL1, a major T-cell oncogene, was
strikingly down-regulated in T-ALL cells upon treatment with HDACi and that TAL1
forced expression partially reversed the pro-apoptotic effect of HDACis on the leukemic
cells.

HDACis down-regulate TAL1 expression in T-ALL
154
TAL1 ectopic expression in T-ALL cells can result from different non-random
chromosomal translocations (5-9) that, for example, juxtapose the promoter region of
TCR genes to the TAL1 locus. More frequently, TAL1 is expressed due to a small
interstitial deletion (TAL1d) that places TAL1 under the control of the regulatory
elements of the SIL locus (10). In both instances, TAL1 over-expression results from the
presence of a non-native „TAL1 promoter‟ that aberrantly drives TAL1 transcription. In
Figure 5.5. Enforced TAL1 expression partially rescues HDACi-mediated T-ALL cell
death. (A, B) PF382 cells were transduced with empty or TAL1 expressing lentiviruses and
incubated for 24 h with the indicated concentrations of SB. (A) Viability was determined by
flow cytometry analysis of FSC/SSC distribution. (B) Cells were lysed and TAL1 expression
and PARP cleavage analyzed by immunoblot. Actin was used as loading control. The data are
representative of at least two independent experiments.

HDACis down-regulate TAL1 expression in T-ALL
155
most cases, however, TAL1 ectopic expression results from other, as yet unknown,
mechanisms that apparently do not involve genomic alterations in the TAL1 locus
(TAL1wt
). In these cases TAL1 expression is aberrantly promoted by native regulatory
elements, perhaps due to epigenetic alterations affecting the chromatin structure. Our
present studies indicate that HDACis negatively affected TAL1 gene transcription in
TAL1wt
T-ALL cells by suppressing the binding of RNA polymerase II to the TAL1
promoter(s). In contrast, HDACis up-regulated TAL1 mRNA levels in TAL1d cells
(Supplementary Figure 5.7). This discrepancy likely reflects differences in the
respective promoters and their regulation. Normal T-cell precursors do not express
TAL1, which suggests that TAL1 ectopic expression in TAL1wt
cells results from an
aberrantly accessible TAL1 locus and/or from the inhibition of negative regulators of
TAL1 transcription. It is possible that HDACs have been selected by T-ALL cells to
partake in this process. In this regard, it is noteworthy that the transcripts of HDAC1 and
HDAC4 were recently shown to be highly expressed in primary T-ALL patient samples
(47). Whether HDAC1 and HDAC4 positively regulate TAL1 gene transcription in T-
ALL cells warrants investigation. HDACis can also induce the acetylation of proteins
other than histones (48). Thus, one can picture at least two mechanisms by which
HDACis can down-regulate TAL1 gene transcription: histone acetylation nearby the
promoter of a negative regulator of TAL1 gene expression (with consequent increase in
its transcription) and/or direct acetylation and functional activation of the TAL1
repressor. Whatever the mechanism, HDACis down-regulate TAL1 mRNA expression
in TAL1wt
similarly to what has been frequently reported for other oncogenes (30, 31,
49). In TAL1d T-ALL cells, TAL1 is under the control of the SIL promoter that is
normally active in hematopoietic cells (50). In this scenario, it is not surprising that
HDACis act by inducing an open-chromatin conformation that further activates TAL1
transcription.
TAL1 protein levels decreased upon treatment with HDACis in both TAL1wt
and
TAL1d cells, indicating the existence of an additional mechanism of regulation that
counterbalances increased TAL1 mRNA expression in the latter. Indeed, we found that
HDACis impair TAL1 translation, leading to a slow but significant decrease in TAL1
protein levels (Supplementary Figure 5.7). It is probable that HDACis have the potential
to inhibit TAL1 translation also in TAL1wt
T-ALL cells, although the rapid shutdown of
TAL1 gene transcription in these cells renders this mechanism irrelevant or vestigial. It
has been shown that treatment of mantle cell lymphoma cells with SAHA abrogates

HDACis down-regulate TAL1 expression in T-ALL
156
cyclin D1 translation, by direct inhibition of PI3K activity (44). However, inhibition of
PI3K or its downstream target mTOR does not mimic the effect of HDACis on TAL1
protein expression (Supplementary Figure 5.8), suggesting that a different pathway
controls TAL1 translation in T-ALL.
In summary, we showed that HDACis promote human T-ALL cell death at least
in part via a previously unrecognized mechanism involving down-regulation of TAL1
expression. Since TAL1 is one of the most commonly deregulated oncogenes in T-ALL,
defining a major cytogenetic subgroup with bad prognosis, our data provide further
rationale for the inclusion of HDACis into T-ALL therapeutic regimens.
References
1. Herblot, S., A.M. Steff, P. Hugo, P.D. Aplan, and T. Hoang. 2000. SCL and
LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-
T alpha chain expression. Nat Immunol 1:138-144.
2. Pike-Overzet, K., D. de Ridder, F. Weerkamp, M.R. Baert, M.M. Verstegen,
M.H. Brugman, S.J. Howe, M.J. Reinders, A.J. Thrasher, G. Wagemaker, J.J.
van Dongen, and F.J. Staal. 2007. Ectopic retroviral expression of LMO2, but
not IL2Rgamma, blocks human T-cell development from CD34+ cells:
implications for leukemogenesis in gene therapy. Leukemia 21:754-763.
3. Bash, R.O., S. Hall, C.F. Timmons, W.M. Crist, M. Amylon, R.G. Smith, and R.
Baer. 1995. Does activation of the TAL1 gene occur in a majority of patients
with T-cell acute lymphoblastic leukemia? A pediatric oncology group study.
Blood 86:666-676.
4. Ferrando, A.A., D.S. Neuberg, J. Staunton, M.L. Loh, C. Huard, S.C. Raimondi,
F.G. Behm, C.H. Pui, J.R. Downing, D.G. Gilliland, E.S. Lander, T.R. Golub,
and A.T. Look. 2002. Gene expression signatures define novel oncogenic
pathways in T cell acute lymphoblastic leukemia. Cancer Cell 1:75-87.
5. Chen, Q., J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A. Carroll, W.
Crist, B. Ozanne, M.J. Siciliano, and R. Baer. 1990. The tal gene undergoes
chromosome translocation in T cell leukemia and potentially encodes a helix-
loop-helix protein. Embo J 9:415-424.
6. Begley, C.G., P.D. Aplan, M.P. Davey, K. Nakahara, K. Tchorz, J. Kurtzberg,
M.S. Hershfield, B.F. Haynes, D.I. Cohen, T.A. Waldmann, and et al. 1989.

HDACis down-regulate TAL1 expression in T-ALL
157
Chromosomal translocation in a human leukemic stem-cell line disrupts the T-
cell antigen receptor delta-chain diversity region and results in a previously
unreported fusion transcript. Proc Natl Acad Sci U S A 86:2031-2035.
7. Fitzgerald, T.J., G.A. Neale, S.C. Raimondi, and R.M. Goorha. 1991. c-tal, a
helix-loop-helix protein, is juxtaposed to the T-cell receptor-beta chain gene by a
reciprocal chromosomal translocation: t(1;7)(p32;q35). Blood 78:2686-2695.
8. Aplan, P.D., S.C. Raimondi, and I.R. Kirsch. 1992. Disruption of the SCL gene
by a t(1;3) translocation in a patient with T cell acute lymphoblastic leukemia. J
Exp Med 176:1303-1310.
9. Francois, S., E. Delabesse, L. Baranger, M. Dautel, C. Foussard, M. Boasson, O.
Blanchet, O. Bernard, E.A. Macintyre, and N. Ifrah. 1998. Deregulated
expression of the TAL1 gene by t(1;5)(p32;31) in patient with T-cell acute
lymphoblastic leukemia. Genes Chromosomes Cancer 23:36-43.
10. Aplan, P.D., D.P. Lombardi, G.H. Reaman, H.N. Sather, G.D. Hammond, and
I.R. Kirsch. 1992. Involvement of the putative hematopoietic transcription factor
SCL in T-cell acute lymphoblastic leukemia. Blood 79:1327-1333.
11. Ferrando, A.A., S. Herblot, T. Palomero, M. Hansen, T. Hoang, E.A. Fox, and
A.T. Look. 2004. Biallelic transcriptional activation of oncogenic transcription
factors in T-cell acute lymphoblastic leukemia. Blood 103:1909-1911.
12. Kelliher, M.A., D.C. Seldin, and P. Leder. 1996. Tal-1 induces T cell acute
lymphoblastic leukemia accelerated by casein kinase IIalpha. Embo J 15:5160-
5166.
13. Aplan, P.D., C.A. Jones, D.S. Chervinsky, X. Zhao, M. Ellsworth, C. Wu, E.A.
McGuire, and K.W. Gross. 1997. An scl gene product lacking the transactivation
domain induces bony abnormalities and cooperates with LMO1 to generate T-
cell malignancies in transgenic mice. Embo J 16:2408-2419.
14. O'Neil, J., M. Billa, S. Oikemus, and M. Kelliher. 2001. The DNA binding
activity of TAL-1 is not required to induce leukemia/lymphoma in mice.
Oncogene 20:3897-3905.
15. O'Neil, J., J. Shank, N. Cusson, C. Murre, and M. Kelliher. 2004. TAL1/SCL
induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer
Cell 5:587-596.
16. Leroy-Viard, K., M.A. Vinit, N. Lecointe, H. Jouault, U. Hibner, P.H. Romeo,
and D. Mathieu-Mahul. 1995. Loss of TAL-1 protein activity induces premature

HDACis down-regulate TAL1 expression in T-ALL
158
apoptosis of Jurkat leukemic T cells upon medium depletion. Embo J 14:2341-
2349.
17. Bernard, M., E. Delabesse, S. Novault, O. Hermine, and E.A. Macintyre. 1998.
Antiapoptotic effect of ectopic TAL1/SCL expression in a human leukemic T-
cell line. Cancer Res 58:2680-2687.
18. Park, S.T., G.P. Nolan, and X.H. Sun. 1999. Growth inhibition and apoptosis
due to restoration of E2A activity in T cell acute lymphoblastic leukemia cells. J
Exp Med 189:501-508.
19. Schrump, D.S. 2009. Cytotoxicity mediated by histone deacetylase inhibitors in
cancer cells: mechanisms and potential clinical implications. Clin Cancer Res
15:3947-3957.
20. Nakagawa, M., Y. Oda, T. Eguchi, S. Aishima, T. Yao, F. Hosoi, Y. Basaki, M.
Ono, M. Kuwano, M. Tanaka, and M. Tsuneyoshi. 2007. Expression profile of
class I histone deacetylases in human cancer tissues. Oncol Rep 18:769-774.
21. Glaser, K.B., M.J. Staver, J.F. Waring, J. Stender, R.G. Ulrich, and S.K.
Davidsen. 2003. Gene expression profiling of multiple histone deacetylase
(HDAC) inhibitors: defining a common gene set produced by HDAC inhibition
in T24 and MDA carcinoma cell lines. Mol Cancer Ther 2:151-163.
22. Peart, M.J., G.K. Smyth, R.K. van Laar, D.D. Bowtell, V.M. Richon, P.A.
Marks, A.J. Holloway, and R.W. Johnstone. 2005. Identification and functional
significance of genes regulated by structurally different histone deacetylase
inhibitors. Proc Natl Acad Sci U S A 102:3697-3702.
23. Hsu, H.L., I. Wadman, and R. Baer. 1994. Formation of in vivo complexes
between the TAL1 and E2A polypeptides of leukemic T cells. Proc Natl Acad
Sci U S A 91:3181-3185.
24. Bernard, M., E. Delabesse, L. Smit, C. Millien, I.R. Kirsch, J.L. Strominger, and
E.A. Macintyre. 1998. Helix-loop-helix (E2-5, HEB, TAL1 and Id1) protein
interaction with the TCRalphadelta enhancers. Int Immunol 10:1539-1549.
25. Tanaka, A., F. Itoh, S. Itoh, and M. Kato. 2009. TAL1/SCL relieves the E2-2-
mediated repression of VEGFR2 promoter activity. J Biochem 145:129-135.
26. Hsu, H.L., L. Huang, J.T. Tsan, W. Funk, W.E. Wright, J.S. Hu, R.E. Kingston,
and R. Baer. 1994. Preferred sequences for DNA recognition by the TAL1 helix-
loop-helix proteins. Mol Cell Biol 14:1256-1265.

HDACis down-regulate TAL1 expression in T-ALL
159
27. Huang, S., and S.J. Brandt. 2000. mSin3A regulates murine erythroleukemia cell
differentiation through association with the TAL1 (or SCL) transcription factor.
Mol Cell Biol 20:2248-2259.
28. Wen, J., S. Huang, S.D. Pack, X. Yu, S.J. Brandt, and C.T. Noguchi. 2005.
Tal1/SCL binding to pericentromeric DNA represses transcription. J Biol Chem
280:12956-12966.
29. Nimmanapalli, R., L. Fuino, P. Bali, M. Gasparetto, M. Glozak, J. Tao, L.
Moscinski, C. Smith, J. Wu, R. Jove, P. Atadja, and K. Bhalla. 2003. Histone
deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal
degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -
refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res 63:5126-
5135.
30. Krishnan, M., A.B. Singh, J.J. Smith, A. Sharma, X. Chen, S. Eschrich, T.J.
Yeatman, R.D. Beauchamp, and P. Dhawan. 2010. HDAC inhibitors regulate
claudin-1 expression in colon cancer cells through modulation of mRNA
stability. Oncogene 29:305-312.
31. Yamaguchi, K., A. Lantowski, A.J. Dannenberg, and K. Subbaramaiah. 2005.
Histone deacetylase inhibitors suppress the induction of c-Jun and its target
genes including COX-2. J Biol Chem 280:32569-32577.
32. Silva, A., J.A. Yunes, B.A. Cardoso, L.R. Martins, P.Y. Jotta, M. Abecasis, A.E.
Nowill, N.R. Leslie, A.A. Cardoso, and J.T. Barata. 2008. PTEN
posttranslational inactivation and hyperactivation of the PI3K/Akt pathway
sustain primary T cell leukemia viability. J Clin Invest 118:3762-3774.
33. Amendola, M., M.A. Venneri, A. Biffi, E. Vigna, and L. Naldini. 2005.
Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic
bidirectional promoters. Nat Biotechnol 23:108-116.
34. Barata, J.T., A. Silva, J.G. Brandao, L.M. Nadler, A.A. Cardoso, and V.A.
Boussiotis. 2004. Activation of PI3K is indispensable for interleukin 7-mediated
viability, proliferation, glucose use, and growth of T cell acute lymphoblastic
leukemia cells. J Exp Med 200:659-669.
35. Silva, A., P.Y. Jotta, A.B. Silveira, D. Ribeiro, S.R. Brandalise, J.A. Yunes, and
J.T. Barata. 2010. Regulation of PTEN by CK2 and Notch1 in primary T-cell
acute lymphoblastic leukemia: rationale for combined use of CK2- and gamma-
secretase inhibitors. Haematologica 95:674-678.

HDACis down-regulate TAL1 expression in T-ALL
160
36. Tsapis, M., M. Lieb, F. Manzo, P. Shankaranarayanan, R. Herbrecht, P. Lutz,
and H. Gronemeyer. 2007. HDAC inhibitors induce apoptosis in glucocorticoid-
resistant acute lymphatic leukemia cells despite a switch from the extrinsic to
the intrinsic death pathway. Int J Biochem Cell Biol 39:1500-1509.
37. Cha, T.L., M.J. Chuang, S.T. Wu, G.H. Sun, S.Y. Chang, D.S. Yu, S.M. Huang,
S.K. Huan, T.C. Cheng, T.T. Chen, P.L. Fan, and P.W. Hsiao. 2009. Dual
degradation of aurora A and B kinases by the histone deacetylase inhibitor
LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer
Res 15:840-850.
38. Januchowski, R., M. Dabrowski, H. Ofori, and P.P. Jagodzinski. 2007.
Trichostatin A down-regulate DNA methyltransferase 1 in Jurkat T cells.
Cancer Lett 246:313-317.
39. Fandy, T.E., S. Shankar, D.D. Ross, E. Sausville, and R.K. Srivastava. 2005.
Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated
with changes in mitochondrial functions and expressions of cell cycle regulatory
genes in multiple myeloma. Neoplasia 7:646-657.
40. Siavoshian, S., J.P. Segain, M. Kornprobst, C. Bonnet, C. Cherbut, J.P.
Galmiche, and H.M. Blottiere. 2000. Butyrate and trichostatin A effects on the
proliferation/differentiation of human intestinal epithelial cells: induction of
cyclin D3 and p21 expression. Gut 46:507-514.
41. Pokholok, D.K., C.T. Harbison, S. Levine, M. Cole, N.M. Hannett, T.I. Lee,
G.W. Bell, K. Walker, P.A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Lewitter,
D.K. Gifford, and R.A. Young. 2005. Genome-wide map of nucleosome
acetylation and methylation in yeast. Cell 122:517-527.
42. Bernard, O., O. Azogui, N. Lecointe, F. Mugneret, R. Berger, C.J. Larsen, and
D. Mathieu-Mahul. 1992. A third tal-1 promoter is specifically used in human T
cell leukemias. J Exp Med 176:919-925.
43. Delabesse, E., M. Bernard, J. Landman-Parker, F. Davi, D. Leboeuf, B. Varet, F.
Valensi, and E.A. Macintyre. 1997. Simultaneous SIL-TAL1 RT-PCR detection
of all tal(d) deletions and identification of novel tal(d) variants. Br J Haematol
99:901-907.
44. Kawamata, N., J. Chen, and H.P. Koeffler. 2007. Suberoylanilide hydroxamic
acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell
lymphoma cells. Blood 110:2667-2673.

HDACis down-regulate TAL1 expression in T-ALL
161
45. Bernhard, D., M.J. Ausserlechner, M. Tonko, M. Loffler, B.L. Hartmann, A.
Csordas, and R. Kofler. 1999. Apoptosis induced by the histone deacetylase
inhibitor sodium butyrate in human leukemic lymphoblasts. Faseb J 13:1991-
2001.
46. Batova, A., L.E. Shao, M.B. Diccianni, A.L. Yu, T. Tanaka, A. Rephaeli, A.
Nudelman, and J. Yu. 2002. The histone deacetylase inhibitor AN-9 has
selective toxicity to acute leukemia and drug-resistant primary leukemia and
cancer cell lines. Blood 100:3319-3324.
47. Moreno, D.A., C.A. Scrideli, M.A. Cortez, R. de Paula Queiroz, E.T. Valera, V.
da Silva Silveira, J.A. Yunes, S.R. Brandalise, and L.G. Tone. 2010. Differential
expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and
survival in childhood acute lymphoblastic leukaemia. Br J Haematol 150:665-
673.
48. Dokmanovic, M., C. Clarke, and P.A. Marks. 2007. Histone deacetylase
inhibitors: overview and perspectives. Mol Cancer Res 5:981-989.
49. You, J.S., J.K. Kang, E.K. Lee, J.C. Lee, S.H. Lee, Y.J. Jeon, D.H. Koh, S.H.
Ahn, D.W. Seo, H.Y. Lee, E.J. Cho, and J.W. Han. 2008. Histone deacetylase
inhibitor apicidin downregulates DNA methyltransferase 1 expression and
induces repressive histone modifications via recruitment of corepressor complex
to promoter region in human cervix cancer cells. Oncogene 27:1376-1386.
50. Aplan, P.D., D.P. Lombardi, and I.R. Kirsch. 1991. Structural characterization
of SIL, a gene frequently disrupted in T-cell acute lymphoblastic leukemia. Mol
Cell Biol 11:5462-5469.
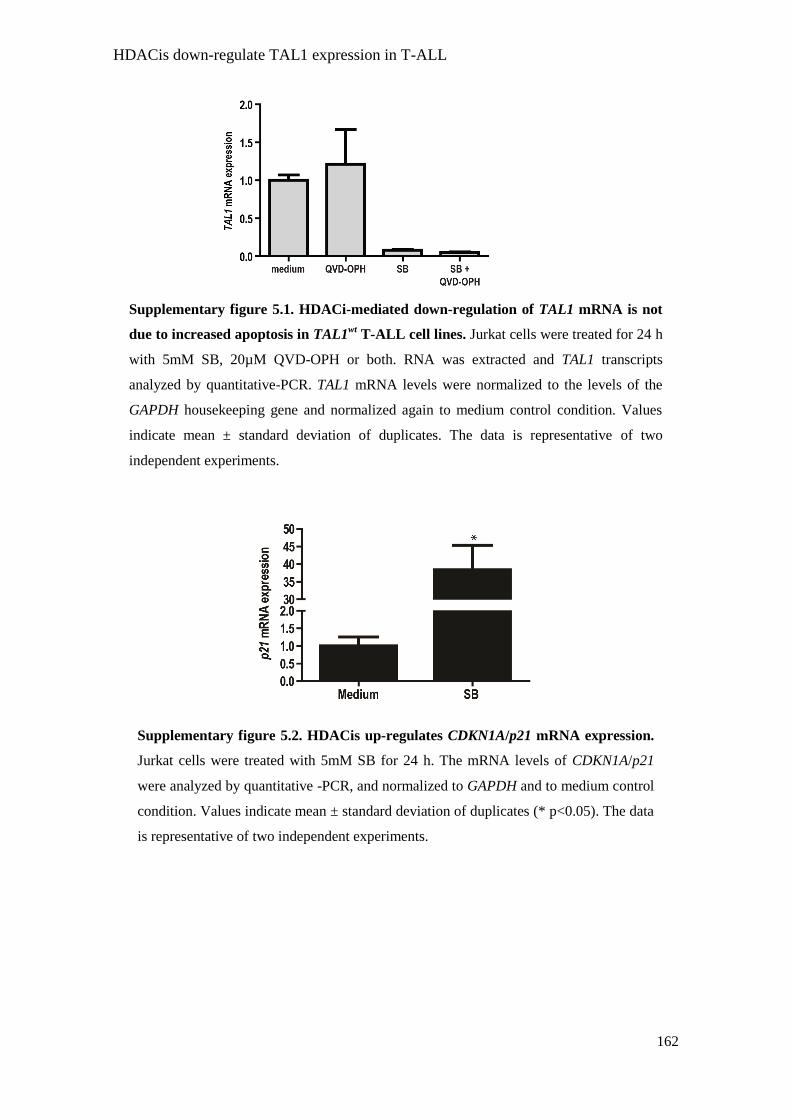
HDACis down-regulate TAL1 expression in T-ALL
162
Supplementary figure 5.1. HDACi-mediated down-regulation of TAL1 mRNA is not
due to increased apoptosis in TAL1wt
T-ALL cell lines. Jurkat cells were treated for 24 h
with 5mM SB, 20µM QVD-OPH or both. RNA was extracted and TAL1 transcripts
analyzed by quantitative-PCR. TAL1 mRNA levels were normalized to the levels of the
GAPDH housekeeping gene and normalized again to medium control condition. Values
indicate mean ± standard deviation of duplicates. The data is representative of two
independent experiments.
Supplementary figure 5.2. HDACis up-regulates CDKN1A/p21 mRNA expression.
Jurkat cells were treated with 5mM SB for 24 h. The mRNA levels of CDKN1A/p21
were analyzed by quantitative -PCR, and normalized to GAPDH and to medium control
condition. Values indicate mean ± standard deviation of duplicates (* p<0.05). The data
is representative of two independent experiments.

HDACis down-regulate TAL1 expression in T-ALL
163
Supplementary figure 5.3. Schematic representation of TAL1 locus and the primers
used to detect total and processed TAL1 mRNA. (A) TAL1 locus is schematic summarized.
TAL1 exons are represented as boxes. White boxes depict untranslated exons or regions,
while the black boxes show the translated exons and regions [adapted from (10)]. The SIL
locus is located at the 5‟ and the MAP17 locus is located at the 3‟ end of the TAL1 locus. (B)
Primers that detect total TAL1 mRNA were designed to bind exclusively within exon 5 in
order to detect both the processed and unprocessed transcripts. Primers that detect processed
TAL1 mRNA bind within exon 5 and in the exon-exon boundary between exons 5 and 6,
allowing the detection of TAL1 mRNA only after splicing when exon 5 and 6 are juxtaposed.

HDACis down-regulate TAL1 expression in T-ALL
164
Supplementary figure 5.4. TAL1d-expressing T-ALL cells up-regulated TAL1 mRNA
upon HDACi treatment. (A) Structural comparison of TAL1wt
(left panel) and TAL1d
(right panel) locus [Adapted from (10)]. The SIL locus is located at the 5‟ and the MAP17
locus is located at the 3‟ end of the TAL1 locus. Exons are represented as boxes. The white
boxes represent exons or regions that are not translated, while the black boxes show exons
or regions that are translated. The grey box indicates the SIL exon that is placed in the
TAL1 locus upon the TAL1d deletion. The arrows represent the TAL1 promoters (Pr.) (B)
Jurkat and PF382 cells were treated with 5mM SB or 10µM SAHA, RNA was extracted
and TAL1 and SIL-TAL1 transcripts analyzed by quantitative-PCR. TAL1 and SIL-TAL1
mRNA levels were normalized to the levels of the GAPDH housekeeping gene and
depicted as relative values to the medium control condition. Values represent mean ±
standard deviation of duplicates (** p<0.01). The data is representative of two independent
experiments.

HDACis down-regulate TAL1 expression in T-ALL
165
Supplementary figure 5.5. HDACis induce T-ALL cell death. T-ALL cells were treated
with HDACi and cellular viability was determined as described in “Materials and Methods.
Viability index represents the viability values normalized to the viability in medium control
condition. (A) Primary T-ALL cells (n=3) were treated for 24 h in the indicated
concentrations of SB and SAHA. (B) Jurkat, PF382 and SupT1 cells were treated for 24 h
with the indicated concentrations of SB or TSA. Values indicate the mean ± standard
deviation of duplicates (* p<0.05; ** p<0.01; *** p<0.001). The data is representative of at
least two independent experiments.

HDACis down-regulate TAL1 expression in T-ALL
166
Supplementary figure 5.6. Enforced TAL1 expression partially rescues HDACi-
mediated apoptosis of Jurkat cells. Jurkat cells were transduced with empty or TAL1
expressing lentiviruses and incubated for 24 h with the indicated concentrations of SB.
Viability was determined by flow cytometry analysis of FSC/SSC distribution. Viability
index represents the viability values normalized to the viability in medium control
condition. Values indicate the mean ± standard deviation of triplicates (*** p<0.001).

HDACis down-regulate TAL1 expression in T-ALL
167
Supplementary figure 5.7. Model for HDACi-mediated TAL1 down-regulation in T-
ALL cells. In T-ALL cells that contain an intact TAL1 locus (TAL1wt
), treatment with
HDACi impairs TAL1 gene transcription by displacing RNA polymerase II from TAL1
native promoters. In T-ALL cells that contain the TAL1d allele and consequently express
TAL1 under the control of the SIL promoter, HDACi do not negatively affect TAL1 gene
transcription, (on the contrary, they appear to up-regulate it) but shut down TAL1 protein
translation. Both mechanisms lead to the same outcome: down-regulation of TAL1 protein
expression in HDACi-treated T-ALL cells.

HDACis down-regulate TAL1 expression in T-ALL
168
Supplementary figure 5.8. TAL1 expression is not affect by inhibition of PI3K and
mTOR. PF382 cells were cultured for 24 h with 5mM SB, 50µM LY294002 (LY), 200nM
Rapamycin (Rap.) or the indicated combinations. Cells were lysed and the phosphorylation
levels of S6 and Akt/PKB, as well as TAL1 protein expression were analyzed by
immunoblot. Actin was used as loading control.

Discussion
169
Chapter 6
DISCUSSION

Discussion
170
Cytokine signaling in T-ALL: the role of IL-4
Cytokines that bind γ-common-chain receptors have a clear impact on the
proliferation of T-ALL cells. We previously showed that incubation of primary T-ALL
cells with γ-common chain cytokines increases their proliferation. In fact, from the
panel of cytokines tested, IL-7 and IL-4 showed the highest proliferative responses.
While IL-7 induced proliferation in T-ALL cells in all the maturation stages, IL-4
induced-proliferation is largely restricted to cortical and mature T-ALL (1). Regarding
the role of IL-7 in T-ALL, it was demonstrated that the incubation of T-ALL cells with
this cytokine led to increased proliferation and viability through the activation of the
PI3K pathway (2).
IL-4 is a cytokine produced by several types of cells (stromal cells, lymphocytes)
(3, 4) and is involved in Th2-type immune responses (4). Importantly, IL-4 is also
produced within the bone marrow (5, 6), which is a major niche where T-ALL cells
proliferate. In chapter 2 we showed that IL-4 promotes T-ALL cell proliferation through
the activation of mTOR signaling pathway and inhibition of the cyclin-dependent kinase
inhibitor p27Kip1
(Figure 6.1). It is possible that IL-4 could also activate other signaling
pathways in T-ALL cells, since studies indicate that IL4 can activate other signaling
pathways in lymphoid cells (7, 8). This notwithsatnding, when we treat T-ALL cells
with the mTOR pathway inhibitor Rapamycin, IL-4-induced proliferation is completely
abrogated. These results clearly show that the mTOR signaling pathway is the main
driver of IL-4 induced proliferation (chapter 2). Increasing lines of evidence suggest
that IL-4 can have a role in cancer development. Recently, it has been described that IL-
4 can stimulate the proliferation of pancreatic cancer cell lines (9). Furthermore, human
colon cancer cells express the IL-4 receptor and its signaling promotes proliferation of
these cell lines (10). Despite the increasing evidence that IL-4 promotes tumor cell
growth, IL-4 is not classified as an oncogene. The IL-4 transgenic mouse was described
in 1991 and did not show any signs of tumorigenesis. However, it is interesting that IL-
4 transgenic mice display enlarged spleens and B cells with increased size and number
(11).
It was also demonstrated that IL-4 can modulate the immune response against
tumors. It has been postulated that IL-4 can impair tumor growth by inhibiting
angiogenesis and activating several innate immune effectors, such as granulocytes and

Discussion
171
eosinophils. In contrast, other studies suggest that IL-4 impairs tumor control by the
immune system by depleting the pool of primary and memory CD8+ cells (12).
Figure 6.1. IL-4 signaling promotes the proliferation of T-ALL cells through the
activation of the mTOR pathway. In T-ALL cells, binding of IL-4 to its cognate
receptor activates the mTOR pathway. The activation is measured by the increased
phosphorylation in mTOR and its downstream targets p70S6K
and 4EBP1, which in turn
activate protein synthesis and therefore contribute to increase the cell size. Activation of
mTOR also down-regulates the expression of the cyclin-dependent kinase inhibitor
p27Kip1
, promoting cell cycle entry. Importantly, blockade of the mTOR pathway with
Rapamycin completely abrogates IL-4-induced proliferation of T-ALL cells.

Discussion
172
Our results implicate the mTOR pathway as a main driver of IL-4-induced T-ALL
proliferation. Treatment of T-ALL cells with IL-4 increased the phosphorylation levels
of mTOR and its downstream targets S6, 4EBP1 and p70S6K
(Figure 2.1). The mTOR
kinase associates in two multimeric complexes, mTORC1 and mTORC2 (13, 14). The
mTORC1 complex is directly activated by PKB (13, 15) and it is involved in the
regulation of protein synthesis and cell cycle progression (16). The PI3K pathway has
been shown to be constitutively activated in several human cancers (17-19). Recently,
we described that T-ALL cells also display constitutive activation of PI3K-PKB
pathway (19). Importantly, blockade of PI3K and mTOR pathways with
pharmacological inhibitors (LY294002 and Rapamycin) severely impairs the viability
of these cells (19, 20). Due to its efficacy in promoting apoptosis in human tumors,
several mTOR pathway inhibitors, including CCI-779 and RAD001, were successfully
tested in clinical trials (21). Our results reinforce the idea that the PI3K-mTOR axis,
activated by both microenvironmental stimuli, such as IL-4, and by cell-autonomous
alterations, has a pivotal and non-redundant role in T-ALL.
Is TAL1 a human T-cell oncogene?
While the importance of microenvironmental cues as contributors to T-ALL
disease progression is increasingly recognized, it remains inquestionable that the
trigger(s) for leukemogenesis lie within the developing T-cell itself. A common feature
in T-ALL is the occurrence of non-random chromosomal translocations that lead to
increased expression of several oncogenes (22, 23). In fact, the TAL1 and LMO2 genes
were initially identified due to the occurrence of these non-random chromosomal
translocations (24-26). The TAL1 gene, essential for hematopoietic development (27,
28), is down-regulated upon commitment to the lymphoid lineage (29-31). TAL1
ectopic expression is arguably the most common oncogenic alteration in T-ALL, being
ectopically expressed in up to 65% of T-ALL patients (32, 33). Despite the fact that this
gene is commonly expressed in a high percentage of T-ALL patients, it is still unknown
whether it could be at the origin of human leukemia or it is merely a by-product of
already transformed and genetically unstable cells.
The LMO2 gene is also frequently over-expressed in T-ALL and is commonly
associated with TAL1 ectopic expression (33). In contrast to TAL1, ectopic expression
of LMO2 in human hematopoietic progenitors was already shown to have an impact in

Discussion
173
T-cell development (29). In a recent gene therapy trial for correction of human SCID,
two patients developed leukemia due to the integration of a retrovirus carrying the
IL2RG gene near the LMO2 locus leading to its expression in developing T-cells (34-
38). These observations indicate that LMO2 actually acts as a human T-cell oncogene,
by impairing normal T-cell differentiation (29) and promoting clonal T-cell expansion
that originated leukemia (34). However, it is possible that the synergism between the
LMO2 and the IL2RG gene, rather than LMO2 per se, was the driving force of this T-
cell proliferation.
In transgenic mouse models, the role for the TAL1 gene in the etiology of T-cell
leukemia is well documented and its expression results in the development of T-cell
tumors (39-41). Importantly, the combined expression of both TAL1 and LMO genes in
transgenic mouse models also led to the development of T-cell tumors with a decreased
latency period suggestive of a synergistic effect (30, 42, 43). The development of T-cell
leukemia in TAL1 transgenic mouse models is preceded by an arrest in normal T-cell
differentiation by the accumulation of immature T-cells arrested at the DN stage (40,
41). In these mouse models, the abrogation of normal T-cell differentiation seems to be
a requirement for the development of T-cell leukemia. Our results described in chapter 3
are in agreement with these findings and provide the first evidence that TAL1 ectopic
gene expression can negatively affect normal human T-cell development in vitro. The
data presented on LMO2 confirms previous observations from other groups (29).
Moreover, our results support, in part, the possibility that the effect of TAL1 and LMO2
is synergistic and recapitulate what happens in transgenic mouse models for the TAL1
and LMO2 genes, where the T-cell differentiation program is strongly inhibited (42, 43).
Since in these transgenic mice T-cell developmental arrest precedes leukemia, it is
tempting to speculate that thymic progenitor cells upon ectopic expression of these
genes would also became leukemic, provide they would have enough time. Further
studies are required to clarify this question.
Previous reports show that TAL1 gene expression and activity in T-ALL cells
protects from apoptosis (44-46), observations that we recapitulated in chapter 4.
Notably however, the results in chapter 3 demonstrate that ectopic expression of the
TAL1 gene in association with LMO2 in normal human hematopoietic progenitors can
increase the cell size, which is normally associated with increase in cellular metabolism
and proliferation. This may indicate that metabolism, rather than viability, is the
primary effect of TAL1/LMO2 aberrant expression in normal T cell precursors, and that

Discussion
174
possibly the impact on viability in T-ALL cells a late effect resulting from oncogene
addiction.
Taken together, as stated above, our results indicate that TAL1 ectopic gene
expression can inhibit normal human T-cell development in synergism with the LMO2
gene. However, further studies are still necessary to formally test whether the TAL1
gene is responsible for human T-ALL. The ectopic expression of TAL1 in hematopoietic
and thymic progenitors and further transplantation into immunocompromised mice will
confirm whether this gene is a human oncogene capable of triggering T-ALL or its
expression is merely a secondary event in already transformed T-ALL cells, in which
case its importance could be limited to advanced-stage disease.
Novel TAL1 target genes in T-ALL and beyond
Despite the multiple processes, from regulation of early hematopoiesis to neural
development, in which TAL1 is involved, the actual number of validated and well
characterized TAL1 target genes described so far is surprisingly short. TAL1 targets are
associated with different cellular mechanisms. NKX3.1 and NFKB1 (47, 48) regulate
cellular proliferation; pTα (30, 49), is involved in T-cell differentiation; RALDH2 (50),
in metabolism; c-Kit (51, 52), in hematopoietic development; Runx1 and Runx3 (53), in
embryogenesis; and GPA and P4.2 (54, 55), in red blood cell differentiation.
Even fewer TAL1 target genes were identified in the context of T-ALL. In chapter
4, we identified three novel putative TAL1 target genes. CASZ1, DMGDH and OR5M3
were identified by gene expression profiling of a T-ALL cell line upon ectopic
expression of TAL1. Among the genes identified in the microarray screen, we only
validated those that had been reported to associate with cancer in the literature.
Evidently, this option implicates that there may be genes that we have not yet explored
which may nonetheless play a role in TAL1-mediated leukemia.
The genes identified in our screen are associated with very different cellular
processes such as apoptosis (CASZ1) (56), cellular metabolism (DMGDH) (57-59) and
extracellular signaling (OR5M3) (60). These results indicate that TAL1 can activate a
broad transcriptional program, involved in the regulation of an array of different cellular
functions. In addition, we demonstrated that CASZ1 gene is involved in T-ALL cell
maintenance by promoting not only viability but also proliferation (Figure 4.4). The
mechanisms by which CASZ1 mediates these effects require further investigation.

Discussion
175
We identified these genes in a leukemia context, by ectopic expression of TAL1
in a T-ALL cell line (61). However, the genes that we identified may also be associated
with the physiological transcriptional program of TAL1. Given the known role of TAL1
in neuronal differentiation (62), it is noteworthy that the TAL1 target genes that we
have identified are normally expressed in neuronal tissues. CASZ1 encodes a
transcription factor up-regulated during neuronal differentiation (63). DMDGH encodes
for Dimethylglycine Dehydrogenase which is a mitochondrial enzyme that produces
sarcosine (57, 58), which in turn can also play a role as a neuronal transmitter (64, 65).
Finally, OR5M3 is a member of the olfactory family of receptors (60), which is
expressed in the sensory neurons of mammals (66). Determining whether any of these
genes plays a role in TAL1-mediated functions in other contexts besides T-ALL is an
interesting challenge ahead, which may arise from the studies described in this thesis.
The TAL1 protein heterodimerizes with E2A and HEB proteins to bind the DNA
(49, 67-69) and it has been suggested that TAL1 induces leukemia by inhibiting the
transcriptional activity of E2A and HEB (41). Under this light, it is conceivable to
consider at least three possible scenarios to explain how TAL1 may regulate the
expression of the genes we have now identified (Figure 6.2). In the simplest scenario
(Figure 6.2A), E2A/HEB transcription factors repress the transcription of CASZ1,
DMGDH and OR5M3 genes. Upon expression of TAL1, E2A/HEB homodimers are
inhibited and the transcription of CASZ1, DMGDH and OR5M3 genes occurs. Another
possibility is that upon expression, TAL1 heterodimerizes with E2A/HEB and these
transcription factors leave the promoters of their target genes and occupy the promoters
of CASZ1, DMGDH and OR5M3 genes activating their transcription (Figure 6.2B). It is
also conceivable that TAL1 activates the expression of a still unknown gene which in
turn would activate the transcription of these genes (Figure 6.2C). However, the
promoters of the three genes harbor consensus binding sites for TAL1, reinforcing the
idea that the role of TAL1 in the expression of CASZ1, DMGDH and OR5M3 genes
could be direct (Figure 6.2A and 6.2B).

Discussion
176
HDAC inhibitors: a novel therapeutic approach for T-ALL?
T-ALL is characterized by the clonal expansion of T-cell progenitors arrested at
different stages of development, and associates with poor prognosis even though
intensive and risk adjusted chemotherapy have led to significantly improved outcome
(70, 71). Despite the therapeutic successes, T-ALL patients remain in need of novel
approaches that diminish the aggressiveness of the current treatment protocols, in order
to minimize side-effects and augment efficacy.
Figure 6.2. Several hypothetical mechanisms could explain TAL1-mediated up-
regulation of CASZ1, DMGDH and OR5M3. (A) E2A/HEB homodimers repress the
transcription of the CASZ1, DMGDH and OR5M3 genes. Upon ectopic expression, TAL1
heterodimerizes with E2A/HEB and the transcription of CASZ1, DMGDH and OR5M3
genes is activated. (B) E2A/HEB homodimers are bound to the promoters of their target
genes (gene X) promoting their transcription. However, when TAL1 is expressed it
heterodimerizes with E2A/HEB promoting the transcription of its target-genes (CASZ1,
DMGDH and OR5M3). (C) Upon TAL1 expression, it binds to E2A/HEB and activates the
expression of a yet unknown gene (the Y gene) which in turns activates the expression of
CASZ1, DMGDH and OR5M3.

Discussion
177
In chapter 5, we demonstrate that TAL1 expression is rapidly and strikingly
down-regulated upon treatment with HDACis. Interestingly, we found that HDACis
inhibit not only TAL1 gene transcription but also TAL1 protein translation. T-ALL
patients with TAL1 expression are associated with a poorer prognosis when compared
with patients that express other T-cell oncogenes (33). This can be related with the fact
that TAL1 expression and activity in T-ALL protects from apoptosis (44-46). Why this
is the case remains to be determined, but it could be the result of the fact that TAL1 is
involved in a broad transcriptional program that may be required for proper cell
function. Interestingly, ectopic expression of TAL1 in a T-ALL cell line protected it
from apoptosis induced by standard chemotherapeutic drugs (45). We propose that the
combined use of HDACis and standard chemotherapeutic drugs could be useful in the
treatment of T-ALL cases with TAL1 ectopic expression and poorer prognosis. It is
tempting to speculate that the use of HDACis to decrease TAL1 expression followed by
the combined use of standard chemotherapeutic drugs (72) would be efficient in the
treatment of TAL1-expressing T-ALL cases.
Acetylation, a new clue on TAL1 regulation?
We showed in chapter 5 that TAL1 expression is down-regulated by HDACi
incubation via two independent mechanisms. In T-ALL cells that contain the TAL1
native promoter HDACi treatment displaces the RNA polymerase II from the promoter
impairing TAL1 gene transcription (Figure 5.3). In contrast, in T-ALL cells that harbor
the TAL1d deletion, HDACi treatment down-regulates TAL1 expression by inhibiting
TAL1 translation (Figure 5.4).
The fact that HDACi treatment impairs TAL1 gene transcription could help
uncover how this gene is transcriptionally regulated. The TAL1 gene is controlled by
several elements that include promoters (73, 74), enhancers (75, 76) and also a silencer
(77, 78). HDACi treatment not only displaces the RNA polymerase II from the
promoter Ia and IV, but also decreases the levels of H3K9Ac, a chromatin marker
highly associated with active transcription (79). The TAL1 promoters Ia and Ib are
regulated by several transcription factors that include SP1, SP3 and GATA1 (80-83).
Importantly, the transcriptional and DNA binding activity of these proteins was shown
to be regulated by acetylation (84-86). In fact, acetylation of these proteins can increase
or decrease their transcriptional activity depending on the cellular and genomic

Discussion
178
environment (87, 88). Thus, it is tempting to speculate that HDACis increase the
acetylation levels of the transcription factors that regulate the TAL1 Ia promoter,
particularly the GATA1 transcription factor. However, additional experiments are
required to test whether incubation with HDACis increase the level of GATA1
acetylation in T-ALL cells and whether this increased acetylation results in diminished
activity at the TAL1 Ia promoter. It is also possible that incubation with HDACis in T-
ALL also impairs TAL1 gene transcription at the Ib promoter that is regulated by SP1
and SP3 (82), but we only analyzed the genomic region of the TAL1 promoter Ia.
HDACis incubation in T-ALL cells that display a TAL1d allele also down-
regulates TAL1 expression (Figure 5.1 and 5.4). As described previously, the TAL1d
mutation juxtaposes the SIL regulatory sequences to the coding region of the TAL1
gene, rendering TAL1 gene expression under the control of the former (89). In chapter 5,
we describe that HDACis up-regulate TAL1 mRNA levels in TAL1d T-ALL cells but
impair the translation of these transcripts (Figure 5.4). This is not the first report of
impaired mRNA translation by HDACis. Previously, Kawamata and colleagues
described that HDACi treatment of Mantle Cell Lymphoma cells impaired cyclin D1
translation, through the inhibition of the PI3K pathway (90). Treatment of T-ALL cells
with HDACis also impairs the PI3K-mTOR axis. However the inhibition of these
pathways fails to decrease TAL1 expression in T-ALL cells (Supplementary Figure 5.7
and 5.8). It is possible that the incubation of HDACis in T-ALL cells can alter the
acetylation levels of proteins involved in translation regulation. In fact, Fenton and
colleagues described that p300 and P/CAF proteins interact and acetylate in vitro and in
vivo S6 kinases, and importantly, HDACis enhance the acetylation levels of the S6
kinases. However, the authors did not report any alterations in the activity or
localization of the S6 kinases upon increased acetylation (91). Still, is possible that
HDACi treatment in TAL1d T-ALL cells could alter the activity of proteins involved in
translation control by increasing their protein acetylation levels. However further
experiments are required to clarify whether this is the mechanism responsible for the
decrease in TAL1 mRNA translation levels.
Concluding remarks
Increasing evidence demonstrates that both cell-autonomous (24, 33, 39, 41, 92-
97) and extra-cellular cues regulate and contribute to leukemia progression (1, 98, 99).

Discussion
179
We previously demonstrated that primary T-ALL cells display constitutive activation of
the PI3K pathway (19), but also that IL-7 signaling can increase viability and
proliferation of T-ALL cells through the activation of this pathway (2, 99). In other
words, cell-intrinsic and external cues converge on the activation of PI3K pathway, to
promote leukemia expansion.
In this thesis, we describe in chapter 2, that IL-4, yet another cytokine produced in
the leukemic milieu, can contribute to T-ALL progression by increasing proliferation
through the activation of the mTOR pathway, which lies downstream of PI3K.
On the other hand, we also looked at the cell-autonomous mechanisms that may
promote leukemia progression. We showed that TAL1 and LMO2 (two frequently
ectopically expressed genes in T-ALL) could contribute to T-ALL through the
inhibition of normal T-cell development (chapter 3). Moreover, we identified new
TAL1 target genes up-regulated in the context of T-ALL (chapter 4). Finally, the
demonstration that incubation of HDACis down-regulated TAL1 expression in T-ALL
cells opens a novel therapeutic window in the treatment of the cohort of T-ALL patients
associated with poor prognosis (chapter 5).
The results that we describe in this PhD thesis are summarized in Figure 6.3.

Discussion
180
Figure 6.3. The role of extra-cellular cues and cell-autonomous mechanisms in the
progression of T-ALL. An environmental factor like IL-4 increases the proliferation of T-
ALL cells through the activation of key signaling components such as those belonging to the
the mTOR pathway (chapter 2). Extracellular cues complement the effect of cell-
autonomous aberrations, such as the ectopic expression of TAL1 and/or LMO2 in T-cell
progenitors. Abnormal TAL1/LMO2 levels impair normal thymocyte differentiation,
possibly predisposing cells to leukemogenesis (chapter 3). The activation of genes
potentially reported to be involved in cancer (chapter 4) may be a mechanism by which
TAL1 contributes to T-ALL. Importantly, HDACi incubation of T-ALL cells decreases
TAL1 expression (chapter 5). This result opens the possibility of using HDACis to treat T-
ALL patients, especially those that display TAL1 expression. Overall, the results described
in the present thesis support the notion that both microenvironmental cues and cell-
autonomous mechanisms contribute to T-ALL progression, through the complementary
activation of signaling pathways and up-regulation of genes involved in the maintenance of
viability and proliferation of T-ALL cells.

Discussion
181
References
1. Barata, J.T., T.D. Keenan, A. Silva, L.M. Nadler, V.A. Boussiotis, and A.A. Cardoso.
2004. Common gamma chain-signaling cytokines promote proliferation of T-cell acute
lymphoblastic leukemia. Haematologica 89:1459-1467.
2. Barata, J.T., A. Silva, J.G. Brandao, L.M. Nadler, A.A. Cardoso, and V.A. Boussiotis.
2004. Activation of PI3K is indispensable for interleukin 7-mediated viability,
proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J
Exp Med 200:659-669.
3. Gessner, A., K. Mohrs, and M. Mohrs. 2005. Mast cells, basophils, and eosinophils
acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are
sufficient for rapid cytokine production. J Immunol 174:1063-1072.
4. Min, B., M. Prout, J. Hu-Li, J. Zhu, D. Jankovic, E.S. Morgan, J.F. Urban, Jr., A.M.
Dvorak, F.D. Finkelman, G. LeGros, and W.E. Paul. 2004. Basophils produce IL-4 and
accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med 200:507-
517.
5. Yoshimoto, T., H. Tsutsui, K. Tominaga, K. Hoshino, H. Okamura, S. Akira, W.E.
Paul, and K. Nakanishi. 1999. IL-18, although antiallergic when administered with IL-
12, stimulates IL-4 and histamine release by basophils. Proc Natl Acad Sci U S A
96:13962-13966.
6. Gutierrez-Ramos, J.C., C. Olsson, and R. Palacios. 1992. Interleukin (IL1 to IL7) gene
expression in fetal liver and bone marrow stromal clones: cytokine-mediated positive
and negative regulation. Exp Hematol 20:986-990.
7. Stephenson, L.M., D.S. Park, A.L. Mora, S. Goenka, and M. Boothby. 2005. Sequence
motifs in IL-4R alpha mediating cell-cycle progression of primary lymphocytes. J
Immunol 175:5178-5185.
8. Acacia de Sa Pinheiro, A., A. Morrot, S. Chakravarty, M. Overstreet, J.H. Bream, P.M.
Irusta, and F. Zavala. 2007. IL-4 induces a wide-spectrum intracellular signaling
cascade in CD8+ T cells. J Leukoc Biol 81:1102-1110.
9. Prokopchuk, O., Y. Liu, D. Henne-Bruns, and M. Kornmann. 2005. Interleukin-4
enhances proliferation of human pancreatic cancer cells: evidence for autocrine and
paracrine actions. Br J Cancer 92:921-928.
10. Koller, F.L., D.G. Hwang, E.A. Dozier, and B. Fingleton. Epithelial interleukin-4
receptor expression promotes colon tumor growth. Carcinogenesis 31:1010-1017.
11. Burstein, H.J., R.I. Tepper, P. Leder, and A.K. Abbas. 1991. Humoral immune
functions in IL-4 transgenic mice. J Immunol 147:2950-2956.

Discussion
182
12. Olver, S., S. Apte, A. Baz, and N. Kienzle. 2007. The duplicitous effects of interleukin
4 on tumour immunity: how can the same cytokine improve or impair control of tumour
growth? Tissue Antigens 69:293-298.
13. Steelman, L.S., S.L. Abrams, J. Whelan, F.E. Bertrand, D.E. Ludwig, J. Basecke, M.
Libra, F. Stivala, M. Milella, A. Tafuri, P. Lunghi, A. Bonati, A.M. Martelli, and J.A.
McCubrey. 2008. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and
Jak/STAT pathways to leukemia. Leukemia 22:686-707.
14. Guertin, D.A., and D.M. Sabatini. 2007. Defining the role of mTOR in cancer. Cancer
Cell 12:9-22.
15. Inoki, K., Y. Li, T. Zhu, J. Wu, and K.L. Guan. 2002. TSC2 is phosphorylated and
inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648-657.
16. Panwalkar, A., S. Verstovsek, and F.J. Giles. 2004. Mammalian target of rapamycin
inhibition as therapy for hematologic malignancies. Cancer 100:657-666.
17. Xu, Q., S.E. Simpson, T.J. Scialla, A. Bagg, and M. Carroll. 2003. Survival of acute
myeloid leukemia cells requires PI3 kinase activation. Blood 102:972-980.
18. Hsu, J., Y. Shi, S. Krajewski, S. Renner, M. Fisher, J.C. Reed, T.F. Franke, and A.
Lichtenstein. 2001. The AKT kinase is activated in multiple myeloma tumor cells.
Blood 98:2853-2855.
19. Silva, A., J.A. Yunes, B.A. Cardoso, L.R. Martins, P.Y. Jotta, M. Abecasis, A.E.
Nowill, N.R. Leslie, A.A. Cardoso, and J.T. Barata. 2008. PTEN posttranslational
inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell
leukemia viability. J Clin Invest 118:3762-3774.
20. Avellino, R., S. Romano, R. Parasole, R. Bisogni, A. Lamberti, V. Poggi, S. Venuta,
and M.F. Romano. 2005. Rapamycin stimulates apoptosis of childhood acute
lymphoblastic leukemia cells. Blood 106:1400-1406.
21. Faivre, S., G. Kroemer, and E. Raymond. 2006. Current development of mTOR
inhibitors as anticancer agents. Nat Rev Drug Discov 5:671-688.
22. Graux, C., J. Cools, L. Michaux, P. Vandenberghe, and A. Hagemeijer. 2006.
Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from
thymocyte to lymphoblast. Leukemia 20:1496-1510.
23. De Keersmaecker, K., P. Marynen, and J. Coolsi. 2005. Genetic insights in the
pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica 90:1116-1127.
24. Begley, C.G., P.D. Aplan, M.P. Davey, K. Nakahara, K. Tchorz, J. Kurtzberg, M.S.
Hershfield, B.F. Haynes, D.I. Cohen, T.A. Waldmann, and et al. 1989. Chromosomal
translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor
delta-chain diversity region and results in a previously unreported fusion transcript.
Proc Natl Acad Sci U S A 86:2031-2035.

Discussion
183
25. Chen, Q., J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A. Carroll, W. Crist, B.
Ozanne, M.J. Siciliano, and R. Baer. 1990. The tal gene undergoes chromosome
translocation in T cell leukemia and potentially encodes a helix-loop-helix protein.
Embo J 9:415-424.
26. Boehm, T., L. Foroni, Y. Kaneko, M.F. Perutz, and T.H. Rabbitts. 1991. The rhombotin
family of cysteine-rich LIM-domain oncogenes: distinct members are involved in T-cell
translocations to human chromosomes 11p15 and 11p13. Proc Natl Acad Sci U S A
88:4367-4371.
27. Robb, L., I. Lyons, R. Li, L. Hartley, F. Kontgen, R.P. Harvey, D. Metcalf, and C.G.
Begley. 1995. Absence of yolk sac hematopoiesis from mice with a targeted disruption
of the scl gene. Proc Natl Acad Sci U S A 92:7075-7079.
28. Porcher, C., W. Swat, K. Rockwell, Y. Fujiwara, F.W. Alt, and S.H. Orkin. 1996. The T
cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic
lineages. Cell 86:47-57.
29. Pike-Overzet, K., D. de Ridder, F. Weerkamp, M.R. Baert, M.M. Verstegen, M.H.
Brugman, S.J. Howe, M.J. Reinders, A.J. Thrasher, G. Wagemaker, J.J. van Dongen,
and F.J. Staal. 2007. Ectopic retroviral expression of LMO2, but not IL2Rgamma,
blocks human T-cell development from CD34+ cells: implications for leukemogenesis
in gene therapy. Leukemia 21:754-763.
30. Herblot, S., A.M. Steff, P. Hugo, P.D. Aplan, and T. Hoang. 2000. SCL and LMO1
alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain
expression. Nat Immunol 1:138-144.
31. Ferrando, A.A., S. Herblot, T. Palomero, M. Hansen, T. Hoang, E.A. Fox, and A.T.
Look. 2004. Biallelic transcriptional activation of oncogenic transcription factors in T-
cell acute lymphoblastic leukemia. Blood 103:1909-1911.
32. Bash, R.O., S. Hall, C.F. Timmons, W.M. Crist, M. Amylon, R.G. Smith, and R. Baer.
1995. Does activation of the TAL1 gene occur in a majority of patients with T-cell
acute lymphoblastic leukemia? A pediatric oncology group study. Blood 86:666-676.
33. Ferrando, A.A., D.S. Neuberg, J. Staunton, M.L. Loh, C. Huard, S.C. Raimondi, F.G.
Behm, C.H. Pui, J.R. Downing, D.G. Gilliland, E.S. Lander, T.R. Golub, and A.T.
Look. 2002. Gene expression signatures define novel oncogenic pathways in T cell
acute lymphoblastic leukemia. Cancer Cell 1:75-87.
34. Hacein-Bey-Abina, S., C. Von Kalle, M. Schmidt, M.P. McCormack, N. Wulffraat, P.
Leboulch, A. Lim, C.S. Osborne, R. Pawliuk, E. Morillon, R. Sorensen, A. Forster, P.
Fraser, J.I. Cohen, G. de Saint Basile, I. Alexander, U. Wintergerst, T. Frebourg, A.
Aurias, D. Stoppa-Lyonnet, S. Romana, I. Radford-Weiss, F. Gross, F. Valensi, E.
Delabesse, E. Macintyre, F. Sigaux, J. Soulier, L.E. Leiva, M. Wissler, C. Prinz, T.H.

Discussion
184
Rabbitts, F. Le Deist, A. Fischer, and M. Cavazzana-Calvo. 2003. LMO2-associated
clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science
302:415-419.
35. Hacein-Bey-Abina, S., A. Garrigue, G.P. Wang, J. Soulier, A. Lim, E. Morillon, E.
Clappier, L. Caccavelli, E. Delabesse, K. Beldjord, V. Asnafi, E. MacIntyre, L. Dal
Cortivo, I. Radford, N. Brousse, F. Sigaux, D. Moshous, J. Hauer, A. Borkhardt, B.H.
Belohradsky, U. Wintergerst, M.C. Velez, L. Leiva, R. Sorensen, N. Wulffraat, S.
Blanche, F.D. Bushman, A. Fischer, and M. Cavazzana-Calvo. 2008. Insertional
oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin
Invest 118:3132-3142.
36. Schmidt, M., S. Hacein-Bey-Abina, M. Wissler, F. Carlier, A. Lim, C. Prinz, H. Glimm,
I. Andre-Schmutz, C. Hue, A. Garrigue, F. Le Deist, C. Lagresle, A. Fischer, M.
Cavazzana-Calvo, and C. von Kalle. 2005. Clonal evidence for the transduction of
CD34+ cells with lymphomyeloid differentiation potential and self-renewal capacity in
the SCID-X1 gene therapy trial. Blood 105:2699-2706.
37. Deichmann, A., S. Hacein-Bey-Abina, M. Schmidt, A. Garrigue, M.H. Brugman, J. Hu,
H. Glimm, G. Gyapay, B. Prum, C.C. Fraser, N. Fischer, K. Schwarzwaelder, M.L.
Siegler, D. de Ridder, K. Pike-Overzet, S.J. Howe, A.J. Thrasher, G. Wagemaker, U.
Abel, F.J. Staal, E. Delabesse, J.L. Villeval, B. Aronow, C. Hue, C. Prinz, M. Wissler,
C. Klanke, J. Weissenbach, I. Alexander, A. Fischer, C. von Kalle, and M. Cavazzana-
Calvo. 2007. Vector integration is nonrandom and clustered and influences the fate of
lymphopoiesis in SCID-X1 gene therapy. J Clin Invest 117:2225-2232.
38. Howe, S.J., M.R. Mansour, K. Schwarzwaelder, C. Bartholomae, M. Hubank, H.
Kempski, M.H. Brugman, K. Pike-Overzet, S.J. Chatters, D. de Ridder, K.C. Gilmour,
S. Adams, S.I. Thornhill, K.L. Parsley, F.J. Staal, R.E. Gale, D.C. Linch, J. Bayford, L.
Brown, M. Quaye, C. Kinnon, P. Ancliff, D.K. Webb, M. Schmidt, C. von Kalle, H.B.
Gaspar, and A.J. Thrasher. 2008. Insertional mutagenesis combined with acquired
somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients.
J Clin Invest 118:3143-3150.
39. Kelliher, M.A., D.C. Seldin, and P. Leder. 1996. Tal-1 induces T cell acute
lymphoblastic leukemia accelerated by casein kinase IIalpha. Embo J 15:5160-5166.
40. O'Neil, J., M. Billa, S. Oikemus, and M. Kelliher. 2001. The DNA binding activity of
TAL-1 is not required to induce leukemia/lymphoma in mice. Oncogene 20:3897-3905.
41. O'Neil, J., J. Shank, N. Cusson, C. Murre, and M. Kelliher. 2004. TAL1/SCL induces
leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell 5:587-596.
42. Larson, R.C., I. Lavenir, T.A. Larson, R. Baer, A.J. Warren, I. Wadman, K. Nottage,
and T.H. Rabbitts. 1996. Protein dimerization between Lmo2 (Rbtn2) and Tal1 alters

Discussion
185
thymocyte development and potentiates T cell tumorigenesis in transgenic mice. Embo
J 15:1021-1027.
43. Chervinsky, D.S., X.F. Zhao, D.H. Lam, M. Ellsworth, K.W. Gross, and P.D. Aplan.
1999. Disordered T-cell development and T-cell malignancies in SCL LMO1 double-
transgenic mice: parallels with E2A-deficient mice. Mol Cell Biol 19:5025-5035.
44. Leroy-Viard, K., M.A. Vinit, N. Lecointe, H. Jouault, U. Hibner, P.H. Romeo, and D.
Mathieu-Mahul. 1995. Loss of TAL-1 protein activity induces premature apoptosis of
Jurkat leukemic T cells upon medium depletion. Embo J 14:2341-2349.
45. Bernard, M., E. Delabesse, S. Novault, O. Hermine, and E.A. Macintyre. 1998.
Antiapoptotic effect of ectopic TAL1/SCL expression in a human leukemic T-cell line.
Cancer Res 58:2680-2687.
46. Park, S.T., G.P. Nolan, and X.H. Sun. 1999. Growth inhibition and apoptosis due to
restoration of E2A activity in T cell acute lymphoblastic leukemia cells. J Exp Med
189:501-508.
47. Kusy, S., B. Gerby, N. Goardon, N. Gault, F. Ferri, D. Gerard, F. Armstrong, P.
Ballerini, J.M. Cayuela, A. Baruchel, F. Pflumio, and P.H. Romeo. NKX3.1 is a direct
TAL1 target gene that mediates proliferation of TAL1-expressing human T cell acute
lymphoblastic leukemia. J Exp Med 207:2141-2156.
48. Chang, P.Y., K. Draheim, M.A. Kelliher, and S. Miyamoto. 2006. NFKB1 is a direct
target of the TAL1 oncoprotein in human T leukemia cells. Cancer Res 66:6008-6013.
49. Tremblay, M., S. Herblot, E. Lecuyer, and T. Hoang. 2003. Regulation of pT alpha gene
expression by a dosage of E2A, HEB, and SCL. J Biol Chem 278:12680-12687.
50. Ono, Y., N. Fukuhara, and O. Yoshie. 1998. TAL1 and LIM-only proteins
synergistically induce retinaldehyde dehydrogenase 2 expression in T-cell acute
lymphoblastic leukemia by acting as cofactors for GATA3. Mol Cell Biol 18:6939-
6950.
51. Krosl, G., G. He, M. Lefrancois, F. Charron, P.H. Romeo, P. Jolicoeur, I.R. Kirsch, M.
Nemer, and T. Hoang. 1998. Transcription factor SCL is required for c-kit expression
and c-Kit function in hemopoietic cells. J Exp Med 188:439-450.
52. Lecuyer, E., S. Herblot, M. Saint-Denis, R. Martin, C.G. Begley, C. Porcher, S.H.
Orkin, and T. Hoang. 2002. The SCL complex regulates c-kit expression in
hematopoietic cells through functional interaction with Sp1. Blood 100:2430-2440.
53. Landry, J.R., S. Kinston, K. Knezevic, M.F. de Bruijn, N. Wilson, W.T. Nottingham,
M. Peitz, F. Edenhofer, J.E. Pimanda, K. Ottersbach, and B. Gottgens. 2008. Runx
genes are direct targets of Scl/Tal1 in the yolk sac and fetal liver. Blood 111:3005-3014.
54. Lahlil, R., E. Lecuyer, S. Herblot, and T. Hoang. 2004. SCL assembles a multifactorial
complex that determines glycophorin A expression. Mol Cell Biol 24:1439-1452.

Discussion
186
55. Xu, Z., S. Huang, L.S. Chang, A.D. Agulnick, and S.J. Brandt. 2003. Identification of a
TAL1 target gene reveals a positive role for the LIM domain-binding protein Ldb1 in
erythroid gene expression and differentiation. Mol Cell Biol 23:7585-7599.
56. Yuan, Z.R., R. Wang, J. Solomon, X. Luo, H. Sun, L. Zhang, and Y. Shi. 2005.
Identification and characterization of survival-related gene, a novel cell survival gene
controlling apoptosis and tumorigenesis. Cancer Res 65:10716-10724.
57. Binzak, B.A., J.G. Vockley, R.B. Jenkins, and J. Vockley. 2000. Structure and analysis
of the human dimethylglycine dehydrogenase gene. Mol Genet Metab 69:181-187.
58. Binzak, B.A., R.A. Wevers, S.H. Moolenaar, Y.M. Lee, W.L. Hwu, J. Poggi-Bach, U.F.
Engelke, H.M. Hoard, J.G. Vockley, and J. Vockley. 2001. Cloning of dimethylglycine
dehydrogenase and a new human inborn error of metabolism, dimethylglycine
dehydrogenase deficiency. Am J Hum Genet 68:839-847.
59. Sreekumar, A., L.M. Poisson, T.M. Rajendiran, A.P. Khan, Q. Cao, J. Yu, B. Laxman,
R. Mehra, R.J. Lonigro, Y. Li, M.K. Nyati, A. Ahsan, S. Kalyana-Sundaram, B. Han, X.
Cao, J. Byun, G.S. Omenn, D. Ghosh, S. Pennathur, D.C. Alexander, A. Berger, J.R.
Shuster, J.T. Wei, S. Varambally, C. Beecher, and A.M. Chinnaiyan. 2009.
Metabolomic profiles delineate potential role for sarcosine in prostate cancer
progression. Nature 457:910-914.
60. Fleischer, J., H. Breer, and J. Strotmann. 2009. Mammalian olfactory receptors. Front
Cell Neurosci 3:9.
61. Ono, Y., N. Fukuhara, and O. Yoshie. 1997. Transcriptional activity of TAL1 in T cell
acute lymphoblastic leukemia (T-ALL) requires RBTN1 or -2 and induces TALLA1, a
highly specific tumor marker of T-ALL. J Biol Chem 272:4576-4581.
62. Bradley, C.K., E.A. Takano, M.A. Hall, J.R. Gothert, A.R. Harvey, C.G. Begley, and
J.A. van Eekelen. 2006. The essential haematopoietic transcription factor Scl is also
critical for neuronal development. Eur J Neurosci 23:1677-1689.
63. Liu, Z., X. Yang, F. Tan, K. Cullion, and C.J. Thiele. 2006. Molecular cloning and
characterization of human Castor, a novel human gene upregulated during cell
differentiation. Biochem Biophys Res Commun 344:834-844.
64. Zhang, H.X., K. Hyrc, and L.L. Thio. 2009. The glycine transport inhibitor sarcosine is
an NMDA receptor co-agonist that differs from glycine. J Physiol 587:3207-3220.
65. Zhang, H.X., A. Lyons-Warren, and L.L. Thio. 2009. The glycine transport inhibitor
sarcosine is an inhibitory glycine receptor agonist. Neuropharmacology 57:551-555.
66. Feldmesser, E., T. Olender, M. Khen, I. Yanai, R. Ophir, and D. Lancet. 2006.
Widespread ectopic expression of olfactory receptor genes. BMC Genomics 7:121.

Discussion
187
67. Hsu, H.L., I. Wadman, and R. Baer. 1994. Formation of in vivo complexes between the
TAL1 and E2A polypeptides of leukemic T cells. Proc Natl Acad Sci U S A 91:3181-
3185.
68. Bernard, M., E. Delabesse, L. Smit, C. Millien, I.R. Kirsch, J.L. Strominger, and E.A.
Macintyre. 1998. Helix-loop-helix (E2-5, HEB, TAL1 and Id1) protein interaction with
the TCRalphadelta enhancers. Int Immunol 10:1539-1549.
69. Park, S.T., and X.H. Sun. 1998. The Tal1 oncoprotein inhibits E47-mediated
transcription. Mechanism of inhibition. J Biol Chem 273:7030-7037.
70. Schrappe, M., A. Reiter, W.D. Ludwig, J. Harbott, M. Zimmermann, W. Hiddemann, C.
Niemeyer, G. Henze, A. Feldges, F. Zintl, B. Kornhuber, J. Ritter, K. Welte, H. Gadner,
and H. Riehm. 2000. Improved outcome in childhood acute lymphoblastic leukemia
despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-
BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 95:3310-3322.
71. Silverman, L.B., R.D. Gelber, V.K. Dalton, B.L. Asselin, R.D. Barr, L.A. Clavell, C.A.
Hurwitz, A. Moghrabi, Y. Samson, M.A. Schorin, S. Arkin, L. Declerck, H.J. Cohen,
and S.E. Sallan. 2001. Improved outcome for children with acute lymphoblastic
leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 97:1211-1218.
72. Pui, C.H., and W.E. Evans. 2006. Treatment of acute lymphoblastic leukemia. N Engl J
Med 354:166-178.
73. Begley, C.G., L. Robb, S. Rockman, J. Visvader, E.O. Bockamp, Y.S. Chan, and A.R.
Green. 1994. Structure of the gene encoding the murine SCL protein. Gene 138:93-99.
74. Bernard, O., O. Azogui, N. Lecointe, F. Mugneret, R. Berger, C.J. Larsen, and D.
Mathieu-Mahul. 1992. A third tal-1 promoter is specifically used in human T cell
leukemias. J Exp Med 176:919-925.
75. Sanchez, M., B. Gottgens, A.M. Sinclair, M. Stanley, C.G. Begley, S. Hunter, and A.R.
Green. 1999. An SCL 3' enhancer targets developing endothelium together with
embryonic and adult haematopoietic progenitors. Development 126:3891-3904.
76. Gottgens, B., C. Broccardo, M.J. Sanchez, S. Deveaux, G. Murphy, J.R. Gothert, E.
Kotsopoulou, S. Kinston, L. Delaney, S. Piltz, L.M. Barton, K. Knezevic, W.N. Erber,
C.G. Begley, J. Frampton, and A.R. Green. 2004. The scl +18/19 stem cell enhancer is
not required for hematopoiesis: identification of a 5' bifunctional hematopoietic-
endothelial enhancer bound by Fli-1 and Elf-1. Mol Cell Biol 24:1870-1883.
77. Courtes, C., N. Lecointe, L. Le Cam, F. Baudoin, C. Sardet, and D. Mathieu-Mahul.
2000. Erythroid-specific inhibition of the tal-1 intragenic promoter is due to binding of
a repressor to a novel silencer. J Biol Chem 275:949-958.

Discussion
188
78. Le Clech, M., E. Chalhoub, C. Dohet, V. Roure, S. Fichelson, F. Moreau-Gachelin, and
D. Mathieu. 2006. PU.1/Spi-1 binds to the human TAL-1 silencer to mediate its
activity. J Mol Biol 355:9-19.
79. Pokholok, D.K., C.T. Harbison, S. Levine, M. Cole, N.M. Hannett, T.I. Lee, G.W. Bell,
K. Walker, P.A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Lewitter, D.K. Gifford, and
R.A. Young. 2005. Genome-wide map of nucleosome acetylation and methylation in
yeast. Cell 122:517-527.
80. Lecointe, N., O. Bernard, K. Naert, V. Joulin, C.J. Larsen, P.H. Romeo, and D.
Mathieu-Mahul. 1994. GATA-and SP1-binding sites are required for the full activity of
the tissue-specific promoter of the tal-1 gene. Oncogene 9:2623-2632.
81. Bockamp, E.O., F. McLaughlin, A.M. Murrell, B. Gottgens, L. Robb, C.G. Begley, and
A.R. Green. 1995. Lineage-restricted regulation of the murine SCL/TAL-1 promoter.
Blood 86:1502-1514.
82. Bockamp, E.O., J.L. Fordham, B. Gottgens, A.M. Murrell, M.J. Sanchez, and A.R.
Green. 1998. Transcriptional regulation of the stem cell leukemia gene by PU.1 and Elf-
1. J Biol Chem 273:29032-29042.
83. Bockamp, E.O., F. McLaughlin, B. Gottgens, A.M. Murrell, A.G. Elefanty, and A.R.
Green. 1997. Distinct mechanisms direct SCL/tal-1 expression in erythroid cells and
CD34 positive primitive myeloid cells. J Biol Chem 272:8781-8790.
84. Braun, H., R. Koop, A. Ertmer, S. Nacht, and G. Suske. 2001. Transcription factor Sp3
is regulated by acetylation. Nucleic Acids Res 29:4994-5000.
85. Boyes, J., P. Byfield, Y. Nakatani, and V. Ogryzko. 1998. Regulation of activity of the
transcription factor GATA-1 by acetylation. Nature 396:594-598.
86. Waby, J.S., H. Chirakkal, C. Yu, G.J. Griffiths, R.S. Benson, C.D. Bingle, and B.M.
Corfe. Sp1 acetylation is associated with loss of DNA binding at promoters associated
with cell cycle arrest and cell death in a colon cell line. Mol Cancer 9:275.
87. Ammanamanchi, S., J.W. Freeman, and M.G. Brattain. 2003. Acetylated sp3 is a
transcriptional activator. J Biol Chem 278:35775-35780.
88. White, N.R., P. Mulligan, P.J. King, and I.R. Sanderson. 2006. Sodium butyrate-
mediated Sp3 acetylation represses human insulin-like growth factor binding protein-3
expression in intestinal epithelial cells. J Pediatr Gastroenterol Nutr 42:134-141.
89. Aplan, P.D., D.P. Lombardi, A.M. Ginsberg, J. Cossman, V.L. Bertness, and I.R.
Kirsch. 1990. Disruption of the human SCL locus by "illegitimate" V-(D)-J
recombinase activity. Science 250:1426-1429.
90. Kawamata, N., J. Chen, and H.P. Koeffler. 2007. Suberoylanilide hydroxamic acid
(SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells.
Blood 110:2667-2673.

Discussion
189
91. Fenton, T.R., J. Gwalter, J. Ericsson, and I.T. Gout. Histone acetyltransferases interact
with and acetylate p70 ribosomal S6 kinases in vitro and in vivo. Int J Biochem Cell
Biol 42:359-366.
92. Pear, W.S., J.C. Aster, M.L. Scott, R.P. Hasserjian, B. Soffer, J. Sklar, and D.
Baltimore. 1996. Exclusive development of T cell neoplasms in mice transplanted with
bone marrow expressing activated Notch alleles. J Exp Med 183:2283-2291.
93. Weng, A.P., A.A. Ferrando, W. Lee, J.P.t. Morris, L.B. Silverman, C. Sanchez-Irizarry,
S.C. Blacklow, A.T. Look, and J.C. Aster. 2004. Activating mutations of NOTCH1 in
human T cell acute lymphoblastic leukemia. Science 306:269-271.
94. Tycko, B., S.D. Smith, and J. Sklar. 1991. Chromosomal translocations joining LCK
and TCRB loci in human T cell leukemia. J Exp Med 174:867-873.
95. Abraham, K.M., S.D. Levin, J.D. Marth, K.A. Forbush, and R.M. Perlmutter. 1991.
Thymic tumorigenesis induced by overexpression of p56lck. Proc Natl Acad Sci U S A
88:3977-3981.
96. Lacronique, V., A. Boureux, V.D. Valle, H. Poirel, C.T. Quang, M. Mauchauffe, C.
Berthou, M. Lessard, R. Berger, J. Ghysdael, and O.A. Bernard. 1997. A TEL-JAK2
fusion protein with constitutive kinase activity in human leukemia. Science 278:1309-
1312.
97. Carron, C., F. Cormier, A. Janin, V. Lacronique, M. Giovannini, M.T. Daniel, O.
Bernard, and J. Ghysdael. 2000. TEL-JAK2 transgenic mice develop T-cell leukemia.
Blood 95:3891-3899.
98. Irish, J.M., R. Hovland, P.O. Krutzik, O.D. Perez, O. Bruserud, B.T. Gjertsen, and G.P.
Nolan. 2004. Single cell profiling of potentiated phospho-protein networks in cancer
cells. Cell 118:217-228.
99. Barata, J.T., A.A. Cardoso, L.M. Nadler, and V.A. Boussiotis. 2001. Interleukin-7
promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia
cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood
98:1524-1531.
Related Documents











