IGF-I Treatment Reduces Hyperphagia, Obesity, and Hypertension in Metabolic Disorders Induced by Fetal Programming MARK H. VICKERS, BETTINA A. IKENASIO, AND BERNHARD H. BREIER Liggins Institute for Medical Research, Faculty of Medical and Health Sciences, University of Auckland, Auckland, New Zealand 92019 The discovery of a link between in utero experience and later metabolic and cardiovascular disease is one of the most im- portant advances in epidemiology research of recent years. There is increasing evidence that alterations in the fetal en- vironment may have long-term consequences on cardiovas- cular, metabolic, and endocrine pathophysiology in adult life. This process has been termed programming, and we have shown that undernutrition of the mother during gestation leads to programming of hyperphagia, obesity, hypertension, hyperinsulinemia, and hyperleptinemia in the offspring. Us- ing this model of maternal undernutrition throughout preg- nancy combined with postnatal hypercaloric nutrition of the offspring, we examined the effects of IGF-I therapy. Virgin Wistar rats (age 75 5 d, n 20 per group) were time mated and randomly assigned to receive food either ad libitum or 30% of ad libitum intake (UN) throughout pregnancy. At wean- ing, female offspring were assigned to one of two diets (control or hypercaloric [30% fat]). Systolic blood pressure was mea- sured at day 175 and following infusion with 3 g/g per day recombinant human IGF-1 (rh-IGF-I) by minipump for 14 d. Before treatment, UN offspring were hyperinsulinemic, hy- perleptinemic, hyperphagic, obese, and hypertensive on both diets, compared with ad libitum offspring and this was exacerbated by hypercaloric nutrition. IGF-I treatment in- creased body weight in all treated animals. However, systolic blood pressure, food intake, retroperitoneal and gonadal fat pad weights, and plasma leptin and insulin concentrations were markedly reduced with IGF-I treatment. IGF-I treat- ment resulted in a 3- to 5-fold increase in 38 – 44 kDa and 28 –30 kDa IGF binding proteins, although in UN animals, there was an impaired and differential up-regulation of these insulin- like growth factor binding proteins following IGF-I treatment. The 24-kDa IGF binding protein representing IGF binding protein-4 was down-regulated in all IGF-I-treated animals, but the decrease was more marked in UN animals. Our data suggest that IGF-I treatment alleviates hyperphagia, obesity, hyperinsulinemia, hyperleptinemia, and hypertension in rats programmed to develop the metabolic syndrome X. (Endocrinology 142: 3964 –3973, 2001) T HERE IS INCREASING evidence that metabolic disor- ders that manifest in adult life have their roots before birth. This concept of fetal programming is based on epide- miological and experimental observations of close associa- tions between an adverse intrauterine environment and the later onset of adult metabolic and cardiovascular disorders (1–3). We have defined fetal programming as an adaptive process to an adverse intrauterine environment that alters the fetal metabolic and hormonal milieu, resulting in reset- ting of developmental processes to ensure fetal survival. The persistence of these adaptive responses, designed for sur- vival in a fetal environment, into postnatal life, leads to metabolic and cardiovascular disorders (4). We have developed an animal model of fetal program- ming in which we apply maternal undernutrition through- out gestation, generating a nutrient-deprived intrauterine environment that results in fetal growth retardation and postnatal growth failure and leads to changes in allometric growth patterns and endocrine parameters of the somatotro- phic axis (5, 6). We have recently shown that programmed offspring develop profound hyperphagia, obesity, hyperten- sion, hyperinsulinemia, and hyperleptinemia during adult life and that postnatal hypercaloric nutrition amplifies the metabolic and cardiovascular abnormalities induced by fetal programming (4). Thus, our animal model closely resembles the clinical and metabolic abnormalities seen in humans born of low birth weight and, furthermore, displays the pheno- type of syndrome X (7, 8). Epidemiological studies have shown that babies born of low birth weight develop in- creased rates of obesity in adult life (9). This was most clearly shown in a recent report from the Dutch Famine Study in which poor nutrition in the first trimester of pregnancy re- sulted in increased rates of obesity during adult life (10). Animal studies have also shown that maternal malnutrition during pregnancy results in the development of adult-onset obesity in offspring (9, 11, 12). IGF-I is one of the most important regulators of growth, and IGF-I deficiency is associated with prenatal and post- natal growth failure (13, 14). Under conditions of adequate nutrition, IGF-I has been shown to promote postnatal catch-up growth in rats with intrauterine growth retardation caused by gestational protein deficiency (15). IGF-I therapy is associated with increased insulin sensitivity in normal subjects as well as in patients with GH deficiency, type 2 diabetes mellitus, and type A insulin resistance (16). IGF-I can reduce hyperglycemia in patients with severe insulin resistance by direct effects mediated via the IGF-I receptor Abbreviations: AD, Ad libitum; IGFBP, IGF-binding protein; IRS, in- sulin receptor substrate; RAS, renin-angiotensin system; rh-IGF-1, recombinant human IGF-1; SBP, systolic blood pressure; UN offspring, offspring from mothers that were undernourished throughout pregnancy. 0013-7227/01/$03.00/0 Endocrinology 142(9):3964 –3973 Printed in U.S.A. Copyright © 2001 by The Endocrine Society 3964

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

IGF-I Treatment Reduces Hyperphagia, Obesity, andHypertension in Metabolic Disorders Induced byFetal Programming
MARK H. VICKERS, BETTINA A. IKENASIO, AND BERNHARD H. BREIER
Liggins Institute for Medical Research, Faculty of Medical and Health Sciences, University ofAuckland, Auckland, New Zealand 92019
The discovery of a link between in utero experience and latermetabolic and cardiovascular disease is one of the most im-portant advances in epidemiology research of recent years.There is increasing evidence that alterations in the fetal en-vironment may have long-term consequences on cardiovas-cular, metabolic, and endocrine pathophysiology in adult life.This process has been termed programming, and we haveshown that undernutrition of the mother during gestationleads to programming of hyperphagia, obesity, hypertension,hyperinsulinemia, and hyperleptinemia in the offspring. Us-ing this model of maternal undernutrition throughout preg-nancy combined with postnatal hypercaloric nutrition of theoffspring, we examined the effects of IGF-I therapy. VirginWistar rats (age 75 � 5 d, n � 20 per group) were time matedand randomly assigned to receive food either ad libitum or30% of ad libitum intake (UN) throughout pregnancy. At wean-ing, female offspring were assigned to one of two diets (controlor hypercaloric [30% fat]). Systolic blood pressure was mea-sured at day 175 and following infusion with 3 �g/g per dayrecombinant human IGF-1 (rh-IGF-I) by minipump for 14 d.
Before treatment, UN offspring were hyperinsulinemic, hy-perleptinemic, hyperphagic, obese, and hypertensive on bothdiets, compared with ad libitum offspring and this wasexacerbated by hypercaloric nutrition. IGF-I treatment in-creased body weight in all treated animals. However, systolicblood pressure, food intake, retroperitoneal and gonadal fatpad weights, and plasma leptin and insulin concentrationswere markedly reduced with IGF-I treatment. IGF-I treat-ment resulted in a 3- to 5-fold increase in 38–44 kDa and 28–30kDa IGF binding proteins, although in UN animals, there wasan impaired and differential up-regulation of these insulin-like growth factor binding proteins following IGF-I treatment.The 24-kDa IGF binding protein representing IGF bindingprotein-4 was down-regulated in all IGF-I-treated animals,but the decrease was more marked in UN animals. Our datasuggest that IGF-I treatment alleviates hyperphagia, obesity,hyperinsulinemia, hyperleptinemia, and hypertension inrats programmed to develop the metabolic syndrome X.(Endocrinology 142: 3964–3973, 2001)
THERE IS INCREASING evidence that metabolic disor-ders that manifest in adult life have their roots before
birth. This concept of fetal programming is based on epide-miological and experimental observations of close associa-tions between an adverse intrauterine environment and thelater onset of adult metabolic and cardiovascular disorders(1–3). We have defined fetal programming as an adaptiveprocess to an adverse intrauterine environment that altersthe fetal metabolic and hormonal milieu, resulting in reset-ting of developmental processes to ensure fetal survival. Thepersistence of these adaptive responses, designed for sur-vival in a fetal environment, into postnatal life, leads tometabolic and cardiovascular disorders (4).
We have developed an animal model of fetal program-ming in which we apply maternal undernutrition through-out gestation, generating a nutrient-deprived intrauterineenvironment that results in fetal growth retardation andpostnatal growth failure and leads to changes in allometricgrowth patterns and endocrine parameters of the somatotro-phic axis (5, 6). We have recently shown that programmedoffspring develop profound hyperphagia, obesity, hyperten-
sion, hyperinsulinemia, and hyperleptinemia during adultlife and that postnatal hypercaloric nutrition amplifies themetabolic and cardiovascular abnormalities induced by fetalprogramming (4). Thus, our animal model closely resemblesthe clinical and metabolic abnormalities seen in humans bornof low birth weight and, furthermore, displays the pheno-type of syndrome X (7, 8). Epidemiological studies haveshown that babies born of low birth weight develop in-creased rates of obesity in adult life (9). This was most clearlyshown in a recent report from the Dutch Famine Study inwhich poor nutrition in the first trimester of pregnancy re-sulted in increased rates of obesity during adult life (10).Animal studies have also shown that maternal malnutritionduring pregnancy results in the development of adult-onsetobesity in offspring (9, 11, 12).
IGF-I is one of the most important regulators of growth,and IGF-I deficiency is associated with prenatal and post-natal growth failure (13, 14). Under conditions of adequatenutrition, IGF-I has been shown to promote postnatalcatch-up growth in rats with intrauterine growth retardationcaused by gestational protein deficiency (15). IGF-I therapyis associated with increased insulin sensitivity in normalsubjects as well as in patients with GH deficiency, type 2diabetes mellitus, and type A insulin resistance (16). IGF-Ican reduce hyperglycemia in patients with severe insulinresistance by direct effects mediated via the IGF-I receptor
Abbreviations: AD, Ad libitum; IGFBP, IGF-binding protein; IRS, in-sulin receptor substrate; RAS, renin-angiotensin system; rh-IGF-1,recombinant human IGF-1; SBP, systolic blood pressure; UN offspring,offspring from mothers that were undernourished throughoutpregnancy.
0013-7227/01/$03.00/0 Endocrinology 142(9):3964–3973Printed in U.S.A. Copyright © 2001 by The Endocrine Society
3964

(17). IGF-I infusion lowers insulin and lipid levels in healthyhumans and reduces plasma leptin concentrations in rats(18), suggesting that IGF-I may reduce the degree of insulinresistance in type 2 diabetes, obesity, and hyperlipidemia(19). Clinical studies of IGF-I in hypertension are limited, butIGF-I has previously been shown to have vasodilatory effectsand to improve cardiac function in healthy volunteers (20).In animal studies, IGF-I treatment has been shown to causepartial reversion of hypertension-induced changes in cardiacfunction and to increase cardiac output and stroke volume(21). Furthermore, recent evidence suggests that IGF-I caninteract with the renin-angiotensin system (RAS) and mayalter angiotensin II expression via angiotensin type 1 receptorregulation (22, 23). The reported effects of IGF-I on cardio-vascular and metabolic homeostasis may be mediated by theIGF-binding proteins (IGFBPs). IGFBP-1 and -2 levels closelyreflect changes related to nutrition, insulin secretion, anddisease states such as obesity and type 2 diabetes. IGFBP-3correlates with IGF-I and is a chronic indicator of GH-dependent growth status (24). Previous work by our group(5) and others (25, 26) has shown differential expression ofIGFBPs following fetal growth retardation.
The present study investigates the morphometric, meta-bolic, and endocrine responses to IGF-I treatment in post-natal life following fetal programming alone or in combina-tion with hypercaloric nutrition. The aim of the present studywas to establish whether IGF-I treatment can alleviate hy-perinsulinemia, hyperleptinemia, hyperphagia, obesity, andhypertension caused by fetal programming and postnatalhypercaloric nutrition.
Materials and Methods
Virgin Wistar rats (age 100 � 5 d, n � 15 per group) were time matedusing a rat oestrus cycle monitor to assess the stage of oestrus of the animalsbefore introducing the male. After confirmation of mating, rats were housedindividually in standard rat cages containing wood shavings as beddingand free access to water. All rats were kept in the same room with a constanttemperature maintained at 25 C and a 12-h light:12-h darkness cycle. An-imals were assigned to one of two nutritional groups: group 1 consisted ofundernutrition ([UN], 30% of ad libitum) of a standard diet throughoutgestation; group 2 consisted of a standard diet fed ad libitum (AD) through-out pregnancy. Food intake and maternal weights were recorded daily untilbirth. After birth, pups were weighed and litter size recorded. Pups fromundernourished mothers were cross-fostered onto dams that received ADfeeding throughout pregnancy. Litter size was adjusted to eight pups perlitter to assure adequate and standardized nutrition until weaning. Afterweaning, female offspring from the two groups of dams a) AD offspringand b) offspring from UN mothers were divided into two balanced post-natal nutritional groups to be fed either a standard diet (total digestibleenergy 2959 kcal/kg, protein 19.4%, fat 5%, fat/energy ratio 15.21%, proteinenergy ratio 26.23) or a hypercaloric diet (total digestible energy 4846kcal/kg, protein 31.8%, fat 30%, fat/energy ratio 55.72%, protein/energyratio 26.25%). The mineral and vitamin content in the two diets wereidentical and in accordance with the requirements for standard rat diets.The fat content of the hypercaloric diet is typical of that seen in manyWestern diets. Weights and food intake of all offspring were measureddaily for the first 2 wk and then every second day. At day 175, systolic bloodpressure measurements were recorded using tail cuff plethysmography.Rats were then weight matched and received either rh-IGF-I (3 �g/g perday) or saline by osmotic minipump (model 2002, Alzet Corp., Palo Alto,CA) for 14 d. On the day before the rats were killed, a repeated systolic bloodpressure was recorded. Rats were then fasted overnight and killed byhalothane anesthesia followed by decapitation. Blood was collected intoheparinized vacutainers and stored on ice until centrifugation and removal
of supernatant for analysis. All animal work was approved by the AnimalEthics Committee of the University of Auckland.
Blood pressure measurements
Systolic blood pressure (SBP) was recorded by tail cuff plethysmog-raphy according to the manufacturer’s instructions (blood pressure anal-yser IITC, Life Science, Woodland Hills, CA). Rats were restrained in aclear plastic tube in a prewarmed room (25–28 C). After the rats hadacclimatized (10–15 min), the cuff was placed on the tail and inflated to240 mm Hg. Pulses were recorded during deflation at a rate of 3 mmHg/sec, and reappearance of a pulse was used to determine SBP. Aminimum of three clear SBP recordings per animal was taken, and thecoefficient of variation for repeated measurements was �5%.
IGF-I infusion
At day 175, rats were weight matched (n � 6 per group) and receivedeither rh-IGF-I (Genentech, Inc., San Francisco, CA, code no. G117AZ,batch c9831AY) or saline by osmotic minipump (model 2002, AlzetCorp.). The dose was 3 �g/g per day for 14 days with a pump deliveryrate of 5 �l/h. The mean pump rate for the batch (lot no. 167258) ofpumps used was 5.23 � 0.2 �l/h. Pumps containing the IGF-I or salinesolution were incubated in sterile saline for 4 h at 37 C before implan-tation. The osmotic pumps were implanted sc, under halothane anes-thesia, using a small incision made in the skin between the scapulae.Using a hemostat, a small pocket was formed by spreading apart the scconnective tissues. The pump was inserted into the pocket with the flowmoderator pointing away from the incision. The skin incision was thenclosed with sutures. All animals (n � 48) were housed individually forthe duration of the study.
Endocrine analyses
IGF-I in rat blood plasma was measured using an IGFBP-blocked RIAdescribed previously (27). The ED50 was 0.1 ng/tube, and the intra- andinterassay coefficients of variation were �5% and �10% respectively.
Rat insulin was measured by RIA as described previously (4). Bloodplasma was diluted 1:4 in assay buffer (0.01 m PBS containing 0.37% NaEDTA and 0.5% BSA, pH 6.2). In brief, the primary antibody (guinea pigantiovine insulin) was diluted in assay buffer to an initial workingdilution of 1:80,000. After 0.1 ml diluted sample, control, or standard (ratinsulin, 0.01–10 ng/ml, Crystal Chem, Chicago, IL) was incubated with0.2 ml primary antibody for 24 h at room temperature, 0.2 ml 125I-rh-Insulin (lot no. 615–707-208, Eli Lilly, Indianapolis, IN) was added at15–20,000 counts per tube. Equilibrium conditions were established after24-h incubation at 4 C. A second antibody was used to separate boundfrom free ligand as outlined previously (28) and the pellet counted by� counter. Rat plasma samples showed parallel displacement to thestandard curve, and recovery of unlabeled rat insulin was 96.5 � 4.4%(mean � sem, n � 11). The ED50 was 0.5 ng/ml.
A double-antibody RIA was developed for measurement of leptin in ratplasma. An antibody was raised in rabbits against a fragment (aa 30–45)of bovine leptin. Standard preparation was rm-leptin (Crystal Chem,#CR-6781) used in concentrations ranging from 0.5 to 20 ng/ml. Sampleswere assayed neat or diluted 1:2–1:4 in assay buffer (0.05 m PBS, pH 7.4containing 0.1 m NaCl, 0.5% BSA, 10 mm EDTA, 0.05% NaN3). In brief,100-�l primary antibody (1:25,000) was added to tubes containing 100-�lsample or standard. Following incubation for 24 h at 4 C, 100 �l tracer(125I-rm-leptin, 20 000 cpm per tube) was added to all tubes followed by afurther incubation for 24 h at 4 C. A second antibody technique to separatebound from free ligand was used as outlined previously (28). Rat plasmasamples showed parallel displacement to the standard curve, and recoveryof unlabeled rm-leptin was 101.4 � 2.7% (mean � sem, n � 26). The ED50was 0.37 ng/ml, and the intra-assay coefficient of variation was � 5% (allsamples measured within a single assay).
Fasting plasma glucose concentrations from samples taken at the timeof sacrifice were measured using a glucose analyzer (model 2300, YellowSprings Instrument Co., Yellow Springs, OH). Blood plasma FFA weremeasured by diagnostic kit (no. 1383175, Roche Molecular Biochemicals,Indianapolis, IN). All other plasma analytes were measured by a BM 737analyzer (Hitachi, Roche Diagnostics, Indianapolis, IN) by AucklandHealthcare Laboratory Services.
Vickers et al. • IGF-I Treatment and Fetal Programming Endocrinology, September 2001, 142(9):3964–3973 3965
IGFBPs in rat plasma (2-�l sample, n � 6 per treatment group) wereanalyzed by ligand blotting (29) as described in detail elsewhere (30). Rat125I-IGF-II was used as radiolabel. Nitrocellulose blots were air dried andexposed to X-Omat AR diagnostic film (Eastman Kodak Co., Rochester,NY) in hyperscreen cassettes with intensifier screens (Amersham Phar-macia Biotech, Piscataway, NJ). For quantification, nitrocellulose blotswere exposed overnight to phospor imaging screens and analyzed on aStorm PhosporImager system using ImageQuant software (MolecularDynamics, Inc., Sky Valley, CA). All values were expressed relative toa normal rat plasma pool and standardized to 100% for control group.The IGFBPs were identified on the basis of their molecular size usingnomenclature previously described (31).
Statistical analyses were carried out using SigmaStat (Jandel Sci-entific, San Rafael, CA) and StatView (SAS Institute, Inc., Cary, NC)statistical packages. Differences between groups were determined bytwo-way (pre-IGF-I treatment) or three-way ANOVA (post-IGF-Itreatment) followed by Bonferroni post hoc analysis, and data areshown as mean � sem.
Results
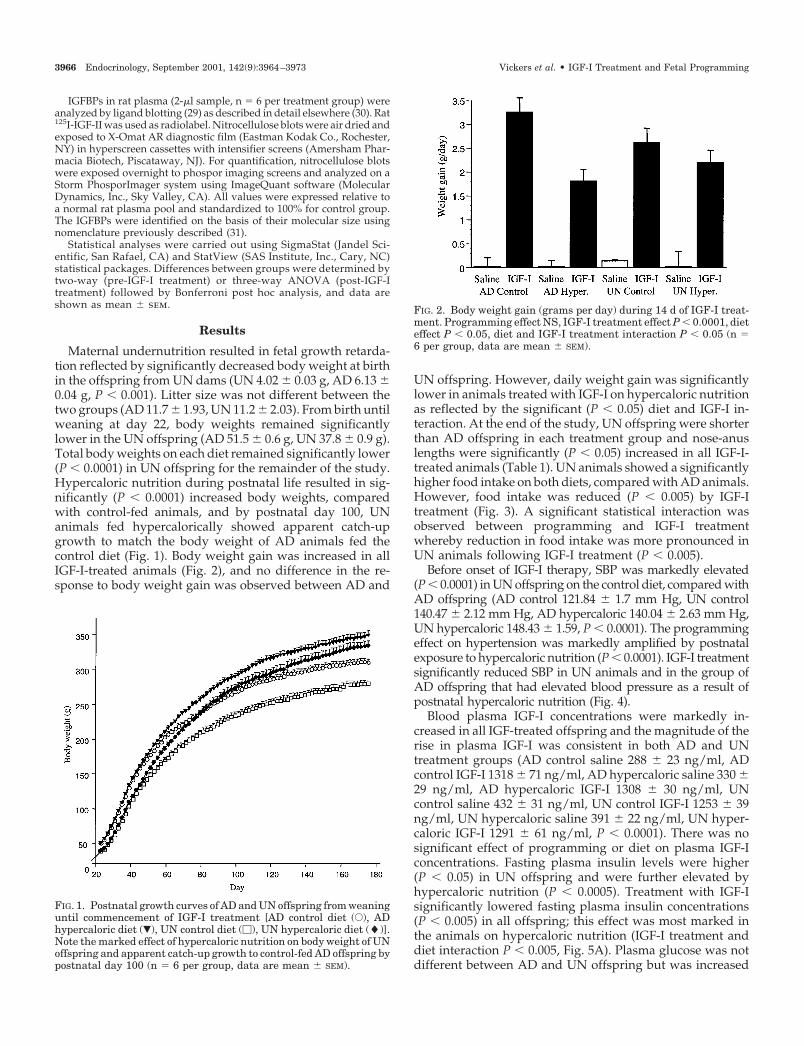
Maternal undernutrition resulted in fetal growth retarda-tion reflected by significantly decreased body weight at birthin the offspring from UN dams (UN 4.02 � 0.03 g, AD 6.13 �0.04 g, P � 0.001). Litter size was not different between thetwo groups (AD 11.7 � 1.93, UN 11.2 � 2.03). From birth untilweaning at day 22, body weights remained significantlylower in the UN offspring (AD 51.5 � 0.6 g, UN 37.8 � 0.9 g).Total body weights on each diet remained significantly lower(P � 0.0001) in UN offspring for the remainder of the study.Hypercaloric nutrition during postnatal life resulted in sig-nificantly (P � 0.0001) increased body weights, comparedwith control-fed animals, and by postnatal day 100, UNanimals fed hypercalorically showed apparent catch-upgrowth to match the body weight of AD animals fed thecontrol diet (Fig. 1). Body weight gain was increased in allIGF-I-treated animals (Fig. 2), and no difference in the re-sponse to body weight gain was observed between AD and
UN offspring. However, daily weight gain was significantlylower in animals treated with IGF-I on hypercaloric nutritionas reflected by the significant (P � 0.05) diet and IGF-I in-teraction. At the end of the study, UN offspring were shorterthan AD offspring in each treatment group and nose-anuslengths were significantly (P � 0.05) increased in all IGF-I-treated animals (Table 1). UN animals showed a significantlyhigher food intake on both diets, compared with AD animals.However, food intake was reduced (P � 0.005) by IGF-Itreatment (Fig. 3). A significant statistical interaction wasobserved between programming and IGF-I treatmentwhereby reduction in food intake was more pronounced inUN animals following IGF-I treatment (P � 0.005).
Before onset of IGF-I therapy, SBP was markedly elevated(P � 0.0001) in UN offspring on the control diet, compared withAD offspring (AD control 121.84 � 1.7 mm Hg, UN control140.47 � 2.12 mm Hg, AD hypercaloric 140.04 � 2.63 mm Hg,UN hypercaloric 148.43 � 1.59, P � 0.0001). The programmingeffect on hypertension was markedly amplified by postnatalexposure to hypercaloric nutrition (P � 0.0001). IGF-I treatmentsignificantly reduced SBP in UN animals and in the group ofAD offspring that had elevated blood pressure as a result ofpostnatal hypercaloric nutrition (Fig. 4).
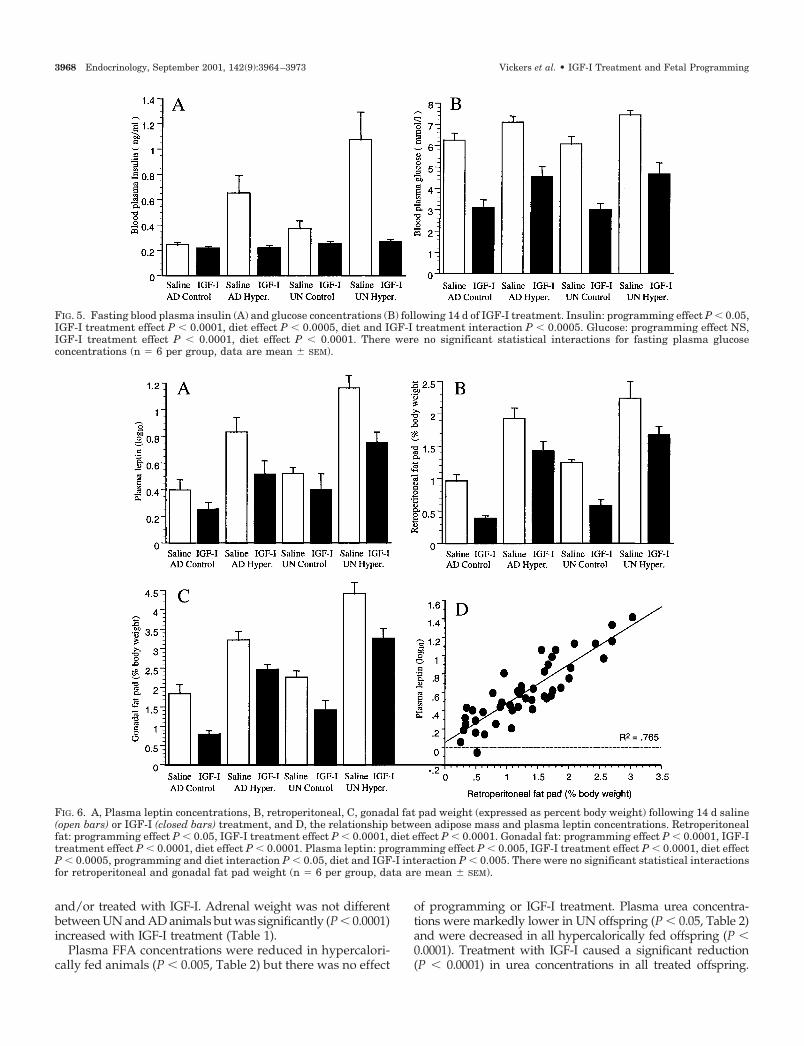
Blood plasma IGF-I concentrations were markedly in-creased in all IGF-treated offspring and the magnitude of therise in plasma IGF-I was consistent in both AD and UNtreatment groups (AD control saline 288 � 23 ng/ml, ADcontrol IGF-I 1318 � 71 ng/ml, AD hypercaloric saline 330 �29 ng/ml, AD hypercaloric IGF-I 1308 � 30 ng/ml, UNcontrol saline 432 � 31 ng/ml, UN control IGF-I 1253 � 39ng/ml, UN hypercaloric saline 391 � 22 ng/ml, UN hyper-caloric IGF-I 1291 � 61 ng/ml, P � 0.0001). There was nosignificant effect of programming or diet on plasma IGF-Iconcentrations. Fasting plasma insulin levels were higher(P � 0.05) in UN offspring and were further elevated byhypercaloric nutrition (P � 0.0005). Treatment with IGF-Isignificantly lowered fasting plasma insulin concentrations(P � 0.005) in all offspring; this effect was most marked inthe animals on hypercaloric nutrition (IGF-I treatment anddiet interaction P � 0.005, Fig. 5A). Plasma glucose was notdifferent between AD and UN offspring but was increased
FIG. 1. Postnatal growth curves of AD and UN offspring from weaninguntil commencement of IGF-I treatment [AD control diet (E), ADhypercaloric diet (�), UN control diet (�), UN hypercaloric diet (�)].Note the marked effect of hypercaloric nutrition on body weight of UNoffspring and apparent catch-up growth to control-fed AD offspring bypostnatal day 100 (n � 6 per group, data are mean � SEM).
FIG. 2. Body weight gain (grams per day) during 14 d of IGF-I treat-ment. Programming effect NS, IGF-I treatment effect P � 0.0001, dieteffect P � 0.05, diet and IGF-I treatment interaction P � 0.05 (n �6 per group, data are mean � SEM).
3966 Endocrinology, September 2001, 142(9):3964–3973 Vickers et al. • IGF-I Treatment and Fetal Programming
(P � 0.0001) by hypercaloric nutrition. IGF-I-treated animalsshowed markedly reduced plasma glucose concentrations(P � 0.0001) (Fig. 5B). Plasma leptin concentrations werehigher (P � 0.005) in UN offspring and were increased (P �0.0001) by hypercaloric diet. IGF-I treatment significantlylowered plasma leptin concentrations (P � 0.0005). As ob-served with insulin, there was a strong diet and IGF-I treat-ment interaction (P � 0.005, Fig. 6A) with plasma leptinlevels being most markedly reduced by IGF-I treatment inoffspring fed hypercalorically. Regression analysis revealeda strong positive relationship between plasma leptin andfasting insulin concentrations (r2 � 0.75, P � 0.0001). Ret-roperitoneal and gonadal fat pads were significantly largerin UN offspring (P � 0.05) and were further increased byhypercaloric nutrition in both AD and UN offspring (P �0.0001). Treatment with IGF-I significantly reduced fat padmass in all treated animals (P � 0.0001, Fig. 6, B and C).Regression analysis showed a highly significant positive re-
lationship between fat mass and fasting plasma leptin (r2 �0.765, P � 0.001, Fig. 6D).
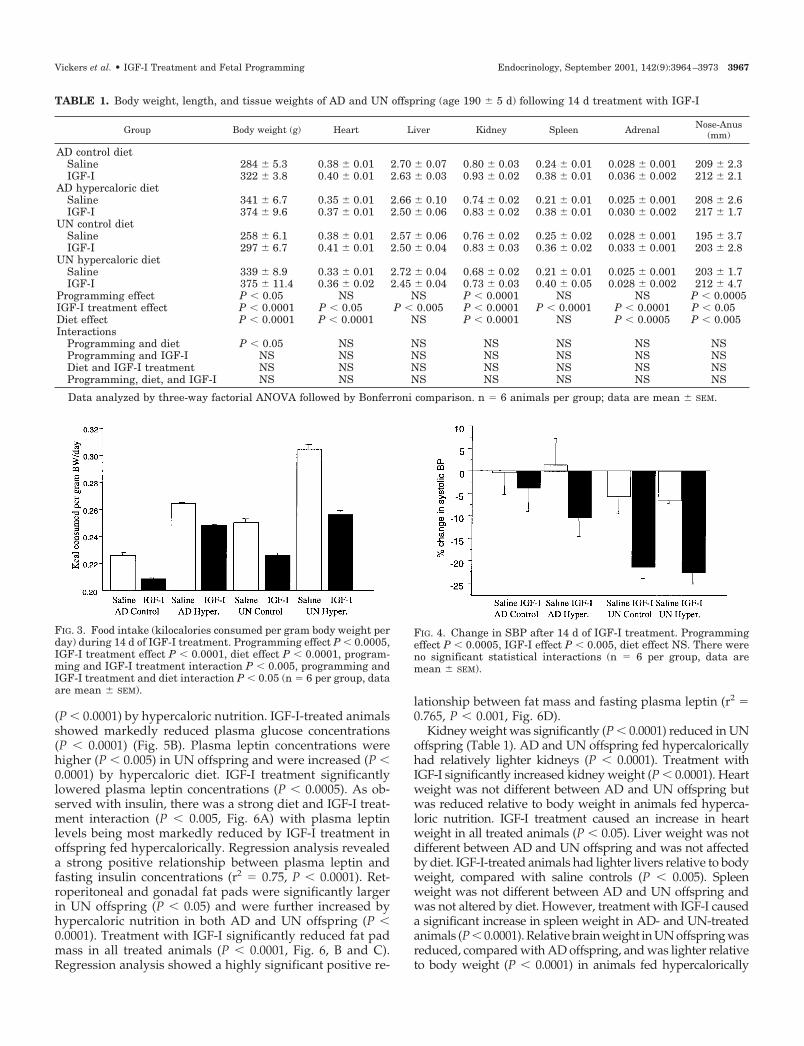
Kidney weight was significantly (P � 0.0001) reduced in UNoffspring (Table 1). AD and UN offspring fed hypercaloricallyhad relatively lighter kidneys (P � 0.0001). Treatment withIGF-I significantly increased kidney weight (P � 0.0001). Heartweight was not different between AD and UN offspring butwas reduced relative to body weight in animals fed hyperca-loric nutrition. IGF-I treatment caused an increase in heartweight in all treated animals (P � 0.05). Liver weight was notdifferent between AD and UN offspring and was not affectedby diet. IGF-I-treated animals had lighter livers relative to bodyweight, compared with saline controls (P � 0.005). Spleenweight was not different between AD and UN offspring andwas not altered by diet. However, treatment with IGF-I causeda significant increase in spleen weight in AD- and UN-treatedanimals (P � 0.0001). Relative brain weight in UN offspring wasreduced, compared with AD offspring, and was lighter relativeto body weight (P � 0.0001) in animals fed hypercalorically
FIG. 3. Food intake (kilocalories consumed per gram body weight perday) during 14 d of IGF-I treatment. Programming effect P � 0.0005,IGF-I treatment effect P � 0.0001, diet effect P � 0.0001, program-ming and IGF-I treatment interaction P � 0.005, programming andIGF-I treatment and diet interaction P � 0.05 (n � 6 per group, dataare mean � SEM).
FIG. 4. Change in SBP after 14 d of IGF-I treatment. Programmingeffect P � 0.0005, IGF-I effect P � 0.005, diet effect NS. There wereno significant statistical interactions (n � 6 per group, data aremean � SEM).
TABLE 1. Body weight, length, and tissue weights of AD and UN offspring (age 190 � 5 d) following 14 d treatment with IGF-I
Group Body weight (g) Heart Liver Kidney Spleen Adrenal Nose-Anus(mm)
AD control dietSaline 284 � 5.3 0.38 � 0.01 2.70 � 0.07 0.80 � 0.03 0.24 � 0.01 0.028 � 0.001 209 � 2.3IGF-I 322 � 3.8 0.40 � 0.01 2.63 � 0.03 0.93 � 0.02 0.38 � 0.01 0.036 � 0.002 212 � 2.1
AD hypercaloric dietSaline 341 � 6.7 0.35 � 0.01 2.66 � 0.10 0.74 � 0.02 0.21 � 0.01 0.025 � 0.001 208 � 2.6IGF-I 374 � 9.6 0.37 � 0.01 2.50 � 0.06 0.83 � 0.02 0.38 � 0.01 0.030 � 0.002 217 � 1.7
UN control dietSaline 258 � 6.1 0.38 � 0.01 2.57 � 0.06 0.76 � 0.02 0.25 � 0.02 0.028 � 0.001 195 � 3.7IGF-I 297 � 6.7 0.41 � 0.01 2.50 � 0.04 0.83 � 0.03 0.36 � 0.02 0.033 � 0.001 203 � 2.8
UN hypercaloric dietSaline 339 � 8.9 0.33 � 0.01 2.72 � 0.04 0.68 � 0.02 0.21 � 0.01 0.025 � 0.001 203 � 1.7IGF-I 375 � 11.4 0.36 � 0.02 2.45 � 0.04 0.73 � 0.03 0.40 � 0.05 0.028 � 0.002 212 � 4.7
Programming effect P � 0.05 NS NS P � 0.0001 NS NS P � 0.0005IGF-I treatment effect P � 0.0001 P � 0.05 P � 0.005 P � 0.0001 P � 0.0001 P � 0.0001 P � 0.05Diet effect P � 0.0001 P � 0.0001 NS P � 0.0001 NS P � 0.0005 P � 0.005Interactions
Programming and diet P � 0.05 NS NS NS NS NS NSProgramming and IGF-I NS NS NS NS NS NS NSDiet and IGF-I treatment NS NS NS NS NS NS NSProgramming, diet, and IGF-I NS NS NS NS NS NS NS
Data analyzed by three-way factorial ANOVA followed by Bonferroni comparison. n � 6 animals per group; data are mean � SEM.
Vickers et al. • IGF-I Treatment and Fetal Programming Endocrinology, September 2001, 142(9):3964–3973 3967
and/or treated with IGF-I. Adrenal weight was not differentbetween UN and AD animals but was significantly (P � 0.0001)increased with IGF-I treatment (Table 1).
Plasma FFA concentrations were reduced in hypercalori-cally fed animals (P � 0.005, Table 2) but there was no effect
of programming or IGF-I treatment. Plasma urea concentra-tions were markedly lower in UN offspring (P � 0.05, Table 2)and were decreased in all hypercalorically fed offspring (P �0.0001). Treatment with IGF-I caused a significant reduction(P � 0.0001) in urea concentrations in all treated offspring.
FIG. 5. Fasting blood plasma insulin (A) and glucose concentrations (B) following 14 d of IGF-I treatment. Insulin: programming effect P � 0.05,IGF-I treatment effect P � 0.0001, diet effect P � 0.0005, diet and IGF-I treatment interaction P � 0.0005. Glucose: programming effect NS,IGF-I treatment effect P � 0.0001, diet effect P � 0.0001. There were no significant statistical interactions for fasting plasma glucoseconcentrations (n � 6 per group, data are mean � SEM).
FIG. 6. A, Plasma leptin concentrations, B, retroperitoneal, C, gonadal fat pad weight (expressed as percent body weight) following 14 d saline(open bars) or IGF-I (closed bars) treatment, and D, the relationship between adipose mass and plasma leptin concentrations. Retroperitonealfat: programming effect P � 0.05, IGF-I treatment effect P � 0.0001, diet effect P � 0.0001. Gonadal fat: programming effect P � 0.0001, IGF-Itreatment effect P � 0.0001, diet effect P � 0.0001. Plasma leptin: programming effect P � 0.005, IGF-I treatment effect P � 0.0001, diet effectP � 0.0005, programming and diet interaction P � 0.05, diet and IGF-I interaction P � 0.005. There were no significant statistical interactionsfor retroperitoneal and gonadal fat pad weight (n � 6 per group, data are mean � SEM).
3968 Endocrinology, September 2001, 142(9):3964–3973 Vickers et al. • IGF-I Treatment and Fetal Programming
Plasma creatinine levels were not different between AD andUN offspring and were unaffected by diet. Treatment withIGF-I lowered (P � 0.0001) creatinine concentrations in alltreated animals (Table 2).
Alanine aminotransferase concentrations were signifi-cantly increased (P � 0.0001) in IGF-I-treated offspring butwere not different between AD or UN offspring and wereunaltered by hypercaloric nutrition (Table 2). Albumin con-centrations were significantly (P � 0.05) lower in UN off-spring, but there was no effect on diet or treatment. Calciumlevels were higher (P � 0.05) in UN offspring, but there wasno effect on diet or treatment. Plasma magnesium concen-trations were markedly increased (P � 0.0001) with IGF-Itreatment but were unaffected by diet and were not differentbetween AD and UN offspring (Table 2).
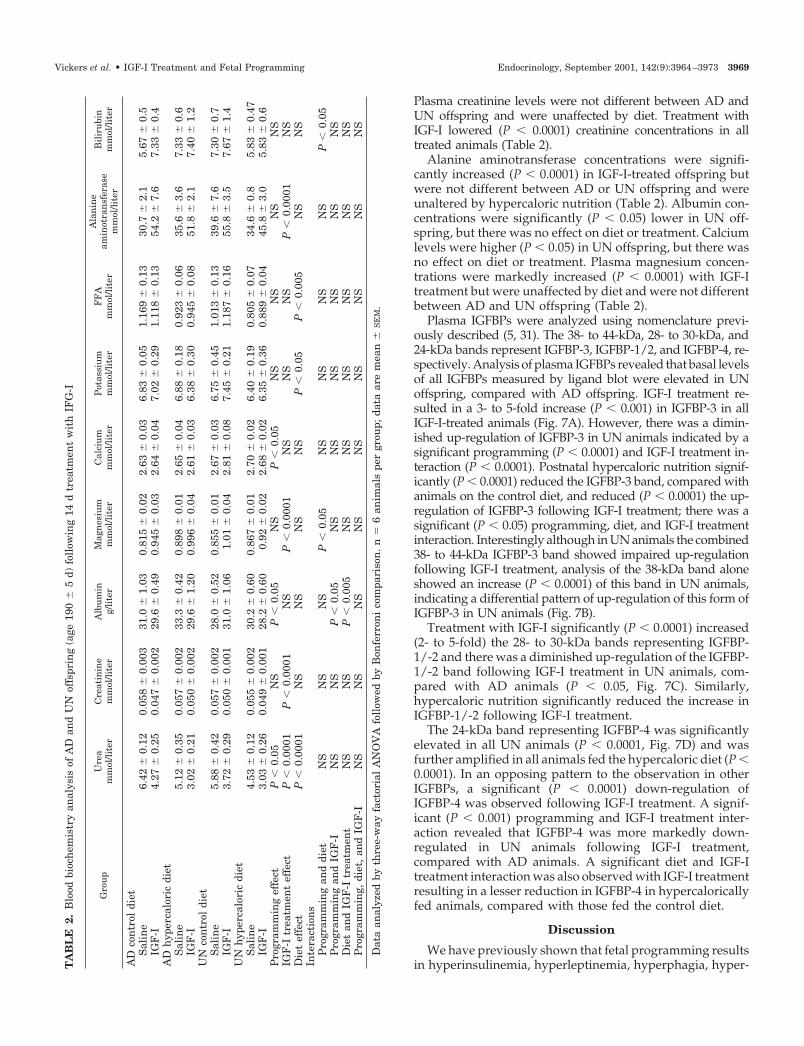
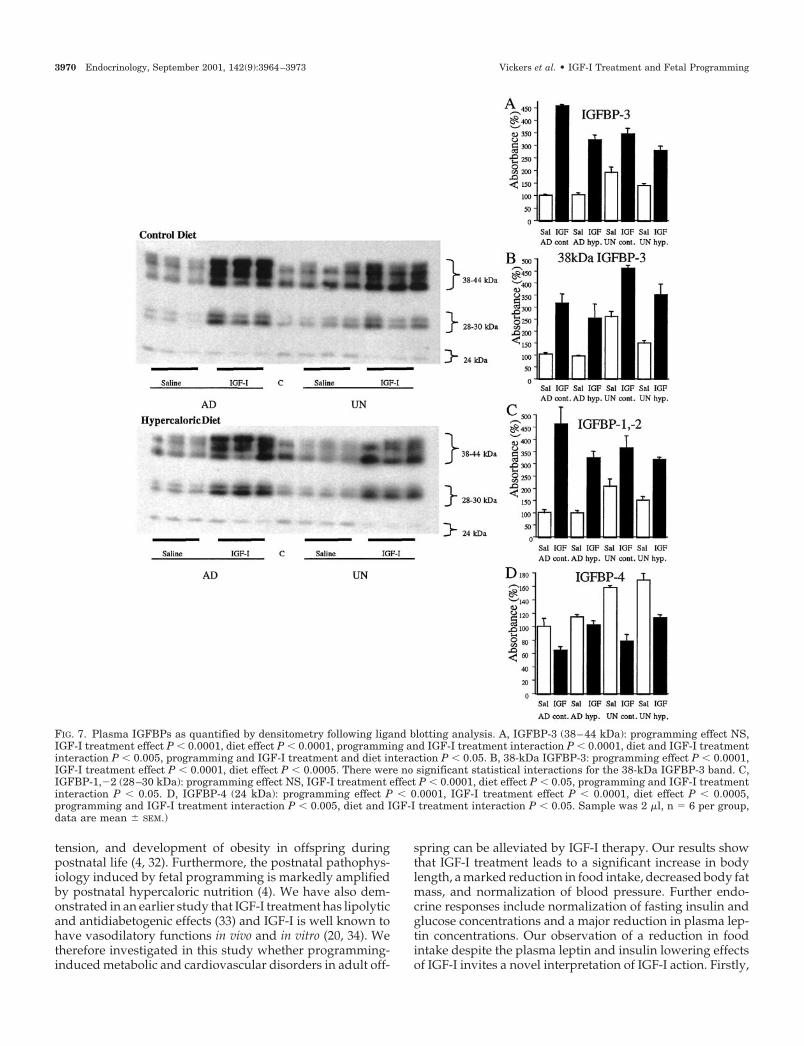
Plasma IGFBPs were analyzed using nomenclature previ-ously described (5, 31). The 38- to 44-kDa, 28- to 30-kDa, and24-kDa bands represent IGFBP-3, IGFBP-1/2, and IGFBP-4, re-spectively. Analysis of plasma IGFBPs revealed that basal levelsof all IGFBPs measured by ligand blot were elevated in UNoffspring, compared with AD offspring. IGF-I treatment re-sulted in a 3- to 5-fold increase (P � 0.001) in IGFBP-3 in allIGF-I-treated animals (Fig. 7A). However, there was a dimin-ished up-regulation of IGFBP-3 in UN animals indicated by asignificant programming (P � 0.0001) and IGF-I treatment in-teraction (P � 0.0001). Postnatal hypercaloric nutrition signif-icantly (P � 0.0001) reduced the IGFBP-3 band, compared withanimals on the control diet, and reduced (P � 0.0001) the up-regulation of IGFBP-3 following IGF-I treatment; there was asignificant (P � 0.05) programming, diet, and IGF-I treatmentinteraction. Interestingly although in UN animals the combined38- to 44-kDa IGFBP-3 band showed impaired up-regulationfollowing IGF-I treatment, analysis of the 38-kDa band aloneshowed an increase (P � 0.0001) of this band in UN animals,indicating a differential pattern of up-regulation of this form ofIGFBP-3 in UN animals (Fig. 7B).
Treatment with IGF-I significantly (P � 0.0001) increased(2- to 5-fold) the 28- to 30-kDa bands representing IGFBP-1/-2 and there was a diminished up-regulation of the IGFBP-1/-2 band following IGF-I treatment in UN animals, com-pared with AD animals (P � 0.05, Fig. 7C). Similarly,hypercaloric nutrition significantly reduced the increase inIGFBP-1/-2 following IGF-I treatment.
The 24-kDa band representing IGFBP-4 was significantlyelevated in all UN animals (P � 0.0001, Fig. 7D) and wasfurther amplified in all animals fed the hypercaloric diet (P �0.0001). In an opposing pattern to the observation in otherIGFBPs, a significant (P � 0.0001) down-regulation ofIGFBP-4 was observed following IGF-I treatment. A signif-icant (P � 0.001) programming and IGF-I treatment inter-action revealed that IGFBP-4 was more markedly down-regulated in UN animals following IGF-I treatment,compared with AD animals. A significant diet and IGF-Itreatment interaction was also observed with IGF-I treatmentresulting in a lesser reduction in IGFBP-4 in hypercaloricallyfed animals, compared with those fed the control diet.
Discussion
We have previously shown that fetal programming resultsin hyperinsulinemia, hyperleptinemia, hyperphagia, hyper-T
AB
LE
2.B
lood
bioc
hem
istr
yan
alys
isof
AD
and
UN
offs
prin
g(a
ge19
0�
5d)
foll
owin
g14
dtr
eatm
ent
wit
hIF
G-I
Gro
up
Ure
am
mol
/lite
rC
reat
inin
em
mol
/lite
rA
lbu
min
g/li
ter
Mag
nes
ium
mm
ol/li
ter
Cal
ciu
mm
mol
/lite
rP
otas
siu
mm
mol
/lite
rF
FA
mm
ol/li
ter
Ala
nin
eam
inot
ran
sfer
ase
mm
ol/li
ter
Bil
iru
bin
mm
ol/li
ter
AD
con
trol
diet
Sal
ine
6.42
�0.
120.
058
�0.
003
31.0
�1.
030.
815
�0.
022.
63�
0.03
6.83
�0.
051.
169
�0.
1330
.7�
2.1
5.67
�0.
5IG
F-I
4.27
�0.
250.
047
�0.
002
29.6
�0.
490.
945
�0.
032.
64�
0.04
7.02
�0.
291.
118
�0.
1354
.2�
7.6
7.33
�0.
4A
Dh
yper
calo
ric
diet
Sal
ine
5.12
�0.
350.
057
�0.
002
33.3
�0.
420.
898
�0.
012.
65�
0.04
6.88
�0.
180.
923
�0.
0635
.6�
3.6
7.33
�0.
6IG
F-I
3.02
�0.
210.
050
�0.
002
29.6
�1.
200.
996
�0.
042.
61�
0.03
6.38
�0.
300.
945
�0.
0851
.8�
2.1
7.40
�1.
2U
Nco
ntr
oldi
etS
alin
e5.
88�
0.42
0.05
7�
0.00
228
.0�
0.52
0.85
5�
0.01
2.67
�0.
036.
75�
0.45
1.01
3�
0.13
39.6
�7.
67.
30�
0.7
IGF
-I3.
72�
0.29
0.05
0�
0.00
131
.0�
1.06
1.01
�0.
042.
81�
0.08
7.45
�0.
211.
187
�0.
1655
.8�
3.5
7.67
�1.
4U
Nh
yper
calo
ric
diet
Sal
ine
4.53
�0.
120.
055
�0.
002
30.2
�0.
600.
867
�0.
012.
70�
0.02
6.40
�0.
190.
805
�0.
0734
.6�
0.8
5.83
�0.
47IG
F-I
3.03
�0.
260.
049
�0.
001
28.2
�0.
600.
92�
0.02
2.68
�0.
026.
35�
0.36
0.88
9�
0.04
45.8
�3.
05.
83�
0.6
Pro
gram
min
gef
fect
P�
0.05
NS
P�
0.05
NS
P�
0.05
NS
NS
NS
NS
IGF
-Itr
eatm
ent
effe
ctP
�0.
0001
P�
0.00
01N
SP
�0.
0001
NS
NS
NS
P�
0.00
01N
SD
iet
effe
ctP
�0.
0001
NS
NS
NS
NS
P�
0.05
P�
0.00
5N
SN
SIn
tera
ctio
ns
Pro
gram
min
gan
ddi
etN
SN
SN
SP
�0.
05N
SN
SN
SN
SP
�0.
05P
rogr
amm
ing
and
IGF
-IN
SN
SP
�0.
05N
SN
SN
SN
SN
SN
SD
iet
and
IGF
-Itr
eatm
ent
NS
NS
P�
0.00
5N
SN
SN
SN
SN
SN
SP
rogr
amm
ing,
diet
,an
dIG
F-I
NS
NS
NS
NS
NS
NS
NS
NS
NS
Dat
aan
alyz
edby
thre
e-w
ayfa
ctor
ial
AN
OV
Afo
llow
edby
Bon
ferr
oni
com
pari
son
.n
�6
anim
als
per
grou
p;da
taar
em
ean
�S
EM
.
Vickers et al. • IGF-I Treatment and Fetal Programming Endocrinology, September 2001, 142(9):3964–3973 3969
tension, and development of obesity in offspring duringpostnatal life (4, 32). Furthermore, the postnatal pathophys-iology induced by fetal programming is markedly amplifiedby postnatal hypercaloric nutrition (4). We have also dem-onstrated in an earlier study that IGF-I treatment has lipolyticand antidiabetogenic effects (33) and IGF-I is well known tohave vasodilatory functions in vivo and in vitro (20, 34). Wetherefore investigated in this study whether programming-induced metabolic and cardiovascular disorders in adult off-
spring can be alleviated by IGF-I therapy. Our results showthat IGF-I treatment leads to a significant increase in bodylength, a marked reduction in food intake, decreased body fatmass, and normalization of blood pressure. Further endo-crine responses include normalization of fasting insulin andglucose concentrations and a major reduction in plasma lep-tin concentrations. Our observation of a reduction in foodintake despite the plasma leptin and insulin lowering effectsof IGF-I invites a novel interpretation of IGF-I action. Firstly,
FIG. 7. Plasma IGFBPs as quantified by densitometry following ligand blotting analysis. A, IGFBP-3 (38–44 kDa): programming effect NS,IGF-I treatment effect P � 0.0001, diet effect P � 0.0001, programming and IGF-I treatment interaction P � 0.0001, diet and IGF-I treatmentinteraction P � 0.005, programming and IGF-I treatment and diet interaction P � 0.05. B, 38-kDa IGFBP-3: programming effect P � 0.0001,IGF-I treatment effect P � 0.0001, diet effect P � 0.0005. There were no significant statistical interactions for the 38-kDa IGFBP-3 band. C,IGFBP-1,�2 (28–30 kDa): programming effect NS, IGF-I treatment effect P � 0.0001, diet effect P � 0.05, programming and IGF-I treatmentinteraction P � 0.05. D, IGFBP-4 (24 kDa): programming effect P � 0.0001, IGF-I treatment effect P � 0.0001, diet effect P � 0.0005,programming and IGF-I treatment interaction P � 0.005, diet and IGF-I treatment interaction P � 0.05. Sample was 2 �l, n � 6 per group,data are mean � SEM.)
3970 Endocrinology, September 2001, 142(9):3964–3973 Vickers et al. • IGF-I Treatment and Fetal Programming

IGF-I treatment may abolish the programming-induced lep-tin resistance at the leptin-hypothalamic circuitry and at thepancreatic adipoinsular feedback system. Secondly, IGF-Itreatment may also ameliorate insulin resistance, both cen-trally and peripherally.
During treatment with IGF-I, we observed no significantdifference in body weight response between AD and UNoffspring, although a lower body weight gain was observedin all hypercalorically fed animals treated with IGF-I, com-pared with control-fed animals. As shown previously, treat-ment with IGF-I significantly reduced fasting plasma insulinand glucose concentrations in all treated animals (20, 35). Amore pronounced decrease in plasma insulin concentrationswas observed with IGF-I treatment in all animals fed a hy-percaloric diet. However, in UN offspring animals (whichwere profoundly hyperinsulinemic) fed a hypercaloric diet,IGF-I treatment resulted in a return to basal fasting plasmainsulin concentrations. In the present study, UN offspringwere hyperphagic on both postnatal diets, compared withAD animals, confirming our previous observations. How-ever, the significant decrease in plasma leptin concentrationsfollowing IGF-I treatment was associated with a decrease infood intake. More importantly however, the decrease in foodintake following IGF-I treatment was more pronounced inoffspring that were programmed to become obese and hy-perphagic in adult life, which may explain the lower bodyweight gain observed in IGF-I-treated offspring fed hyper-caloric nutrition. Although IGF-I treatment caused a signif-icant reduction in food intake, weight gain was significantlyincreased in all IGF-I-treated animals. This may be a resultof increased food conversion efficiency and significant in-creases in nitrogen balance and carcass nitrogen content fol-lowing IGF-I treatment as reported previously (36). Thiswould suggest that increased body weight as observed in thepresent study may be a result of altered partitioning of nu-trients from fat to lean body tissue mass.
Data for a role of IGF-I in appetite regulation are limited,although early work by Tannenbaum et al. (37) showed thatintracerebroventricular administration of IGFs resulted in areduction in food intake. More recent work has also showna reduction in food intake in tumor-bearing rats followinginfusion with either IGF-I or LR3-IGF-I (38). It is thereforetempting to speculate that our observation of reduced foodintake following IGF-I treatment may be the result of theanorectic effect of IGF-I via its insulin-sensitizing effects andreduction of chronic hyperinsulinemia. Food intake wasmost markedly reduced in programmed animals fed hyper-caloric nutrition; the same animals that showed the mostmarked decrease and normalization of fasting insulin andglucose concentrations following IGF-I treatment.
Our data on the lipolytic effect of IGF-I support resultspublished previously (33, 39–41) and suggest that the effectsof prolonged IGF-I treatment on adipose tissue are notinsulin-like as reflected by increased lipolysis and decreasedbody fat mass. We propose that IGF-I treatment may reducebody fat mass via an inhibition of the lipogenic capacity ofadipocytes and reduction of lipogenesis in adipose tissue viainhibition of insulin secretion. The lipolytic effects of IGF-Itreatment were also concomitant with a significant decreasein fasting plasma leptin concentrations. This agrees with
recent work in normal rats showing decreased plasma leptinand fat mass following constant infusion with rh-IGF-I for 6 d(18). It is unlikely that IGF-I acts via cross-reactivity with theinsulin receptor. An insulin-like action would rather stimu-late lipogenesis and thus increase fat pad weight and en-hance leptin expression. It is also unlikely that IGF-I actsdirectly on adipose tissue via the IGF-I type 1 receptor. Ratadipose tissue lacks functional type 1 IGF receptors (41, 42)and IGF-I has been shown to have no effect on leptin secre-tion by mature adipocytes in vitro (43). Reduction of adiposetissue mass and suppression of leptin by IGF-I may be dueto a reduction in circulating insulin leading to enhanced fatmobilization and nonesterified fatty acid oxidation as well asto increased gluconeogenesis from glycerol (18). However, inour study, although IGF-I infusion reduced adipose tissueweight, we observed no significant effect on plasma FFAconcentrations. This may be due to increased triglyceridedeposition and utilization in muscle tissue (author’s unpub-lished observations).
A further explanation for the metabolic effects of IGF-Iobserved in the present study may relate to the interactionsbetween the leptin and insulin signaling networks (44),which may be disrupted as a result of fetal programming andfurther exacerbated by postnatal hypercaloric nutrition (32).Such dysregulation of the adipoinsular axis may contributeto the progression to insulin resistance and adipogenic di-abetes (45, 46). Insulin receptor substrates 1 and 2 (IRS-1 andIRS-2) co-ordinate essential effects of insulin/IGF on periph-eral metabolism and � cell function. Recent evidence sug-gests that impaired IRS-1 expression and downstream sig-naling events in adipocytes in response to insulin areassociated with insulin resistance and the pentad of hyper-tension, hyperinsulinemia, dyslipidemia, obesity, and car-diovascular disease, known as syndrome X (7). IGF-I hasbeen shown to inhibit insulin secretion from � cells throughan IGF-I receptor-mediated pathway (47, 48) and the IGF-I-IRS-2 signaling pathway has been proposed to be critical forpostnatal � cell function (49). It is tempting therefore tospeculate that treatment with IGF-I may restore some of thefunctional feedback between the insulin signaling systemand leptin action via modification of IRS-1 and IRS-2 anddownstream signaling events.
Insulin resistance is often accompanied by hypertension,and obesity-induced hyperinsulinemia may induce alter-ations in sympathetic nervous system activity to increaseblood pressure via vascular constriction. Because insulin-sensitizing agents have been shown to reduce blood pressurein obese, hypertensive subjects (50), it is possible that ourobservation of decreased SBP following IGF-I treatment maybe a result of improved insulin sensitivity and glycemiccontrol in conjunction with the known vasodilatory effects ofIGF-I treatment (34). Further support for the antihyperten-sive effects of IGF-I as a result of improved insulin sensitivitystems from the observation that calcium and magnesiumconcentrations in circulation may regulate cellular respon-siveness to insulin (51). In human hypertension, basal cal-cium levels are significantly elevated while basal magnesiumconcentrations are significantly decreased. Furthermore, el-evated calcium or reduced magnesium concentrations arealso observed in clinical states linked to hypertension, such
Vickers et al. • IGF-I Treatment and Fetal Programming Endocrinology, September 2001, 142(9):3964–3973 3971

as obesity and type 2 diabetes (52). Our observations ofelevated calcium in hypertensive UN animals and a signif-icant increase in plasma magnesium after IGF-I treatmentagrees with these findings and suggests that IGF-I treatmentmay alleviate insulin resistance and reduce hypertension inpart by changing the calcium/magnesium ratio in plasma.
The highly significant increase in kidney weight with IGF-Itreatment may also be an important factor in reduction ofSBP via changes in renal plasma flow and glomerular filtra-tion rate. Given recent in vitro observations (22, 53), it istempting to speculate that IGF-I treatment may also reduceblood pressure by down-regulating the local RAS and lim-iting angiotensin II formation through mediation of the an-giotensin type 1 receptor (23). Heart weight in all IGF-I-treated animals was significantly increased, which mayreflect myocyte growth and improved contractility. Othershave shown cardiac hypertrophy and increased left ventric-ular mass following IGF-I treatment (20, 54, 55). Importantly,IGF-I treatment reduced SBP only in animals that were hy-pertensive as a result of fetal programming or postnatalhypercaloric nutrition, and systolic blood pressure in nor-motensive animals remained unaltered.
Some effects of IGF-I treatment on improving insulin sen-sitivity and ameliorating the postnatal pathophysiology fol-lowing fetal programming may be mediated by changes incirculating IGFBPs. Previous work by our group (5) hasshown that circulating IGFBPs are differentially regulated asa result of fetal programming. However, data on the effect ofIGF-I therapy on IGFBPs in postnatal life following fetalprogramming are limited. In the present study, fetal pro-gramming led to an increase in circulating levels of IGFBP-1/-2 and -4 concentrations as shown by Western ligand blot-ting. Similar results have been observed before in the serumof growth-retarded fetuses, compared with control fetuses(25, 26). The IGFBP-1/-2 doublet has also been previouslyshown to be increased in growth-retarded fetuses using amodel of uterine artery ligation in the rat; because serumimmunoreactive IGFBP-2 was unchanged among the groups,it was suggested that IGFBP-1 accounted for the increase indoublet intensity (56). Elevated IGFBP-1 is normally associ-ated with poor glycemic control and implicated in the patho-genesis of type 2 diabetes because a rise in IGFBP-1 has beenrelated to inadequate portal delivery of insulin (57–59). Pre-vious work by our group (60) has shown that plasma levelsof IGFBP-3 are increased 2-fold following treatment withh-IGF-I in the GH-deficient dwarf rat. Work by others (61)has shown a highly significant increase in IGFBP-3 followingIGF-I therapy in the normal rat. The mechanism underlyingthe preferential up-regulation of the 38-kDa IGFBP-3 band inUN animals following IGF-I treatment is still to be elucidatedbut may relate to changes in phosphorylation or glycosyla-tion of IGFBP-3 (62).
Fetal programming resulted in a significant elevation in cir-culating IGFBP-4 levels, which were amplified by postnatalhypercaloric nutrition. Treatment with IGF-I decreased circu-lating IGFBP-4 in all treated animals; moreover, IGF-I treatmentwas more effective in reducing IGFBP-4 concentrations in thoseanimals that had become obese as a result of fetal programmingand hypercaloric nutrition. This observation is not surprisingbecause IGFBP-4 appears to inhibit IGF-I action under most, if
not all, experimental conditions (63). It is tempting to speculatethat IGF-I treatment causes activation of IGFBP-4 proteases andmay result in the degradation and inactivation of IGFBP-4 asreported by others (64–66). The increase in circulating IGFBP-1,-2, and -3 and the decrease in IGFBP-4 with IGF-I treatment mayrepresent a mechanism of increasing IGF-I activity at the dif-ferent target tissues discussed above.
In conclusion, our animal model of fetal programming bymaternal undernutrition during pregnancy results in pro-found hyperphagia, obesity, hypertension, hyperinsulin-emia, and hyperleptinemia during adult life. Postnatalhypercaloric nutrition amplifies the metabolic and cardio-vascular pathophysiology consistent with the clinical settingof syndrome X (7). Treatment with IGF-1 at an adult ageshowed a significant increase in body length, a marked re-duction in food intake and body fat mass, and normalizationof blood pressure. Further intriguing findings include re-duction of fasting plasma insulin and leptin concentrations.Thus, IGF-1 treatment may alleviate insulin and leptin re-sistance and improve obesity, hyperphagia, and hyperten-sion by differential effects on IGF-I receptor-signaling path-ways or downstream signaling networks including the IRSand RAS. IGF-I treatment may also restore functional inter-actions between insulin and leptin following perturbations ofthe hypothalamic circuitry that controls food intake and ofthe adipoinsular axis as a result of fetal programming.
Acknowledgments
We thank Christine Keven, Andrzej Surus, and Janine Street for theirexpert technical assistance.
Received December 15, 2000. Accepted May 24, 2001.Address all correspondence and requests for reprints to: Associate
Professor Bernhard H. Breier, Liggins Institute for Medical Research,Faculty of Medical and Health Sciences, University of Auckland, PrivateBag 92019, Auckland, New Zealand. E-mail: [email protected].
This work was supported by the Health Research Council of NewZealand and the National Child Health Research Foundation.
References
1. Godfrey KM, Barker DJP 2000 Fetal nutrition and adult disease. Am J ClinNutr 1344S–1352S
2. Reynolds RM, Phillips DI 1998 Long-term consequences of intrauterinegrowth retardation. Horm Res 49(Suppl 2):28–31
3. Barker DJ 1995 The fetal and infant origins of disease. Eur J Clin Invest25:457–463
4. Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD 2000 Fetalorigins of hyperphagia, obesity and hypertension and its postnatal amplifi-cation by hypercaloric nutrition. Am J Physiol 279:E83–E87
5. Woodall SM, Breier BH, Johnston BM, Gluckman PD 1996 A model ofintrauterine growth retardation caused by chronic maternal undernutrition inthe rat: effects on the somatotropic axis and postnatal growth. J Endocrinol150:231–242
6. Woodall SM, Johnston BM, Breier BH, Gluckman PD 1996 Chronic maternalundernutrition in the rat leads to delayed postnatal growth and elevated bloodpressure of offspring. Pediatr Res 40:438–443
7. Reaven GM 1993 Role of insulin resistance in human disease (syndrome X):an expanded definition. Ann Rev Med 44:121–131
8. Smith U, Axelsen M, Carvalho E, Eliasson B, Jansson PA, Wesslau C 1999Insulin signaling and action in fat cells: associations with insulin resistance andtype 2 diabetes. Ann NY Acad Sci 892:119–126
9. Jackson AA, Langley-Evans SC, McCarthy HD 1996 Nutritional influences inearly life upon obesity and body proportions. Ciba Found Symp 201:118–129
10. Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP 1999Obesity at the age of 50 y in men and women exposed to famine prenatally.Am J Clin Nutr 70:811–816
11. Anguita RM, Sigulem DM, Sawaya AL 1993 Intrauterine food restriction isassociated with obesity in young rats. J Nutr 123:1421–1428
3972 Endocrinology, September 2001, 142(9):3964–3973 Vickers et al. • IGF-I Treatment and Fetal Programming

12. Jones AP, Pothos EN, Rada P, Olster DH, Hoebel BG 1995 Maternal hormonalmanipulations in rats cause obesity and increase medial hypothalamic nor-epinephrine release in male offspring. Brain Res Dev Brain Res 88:127–131
13. Baker J, Liu JP, Robertson EJ, Efstratiadis A 1993 Role of insulin-like growthfactors in embryonic and postnatal growth. Cell 75:73–82
14. Powell-Braxton L, Hollingshead P, Warburton C, et al. 1993 IGF-I is requiredfor normal embryonic growth in mice. Genes Dev 7:2609–2617
15. Muaku SM, Thissen JP, Gerard G, Ketelslegers JM, Maiter D 1997 Postnatalcatch-up growth induced by growth hormone and insulin-like growth factor-Iin rats with intrauterine growth retardation caused by maternal protein mal-nutrition. Pediatr Res 42:370–377
16. Froesch ER, Bianda T, Hussain MA 1996 Insulin-like growth factor-I in thetherapy of non-insulin-dependent diabetes mellitus and insulin resistance.Diabetes Metab 22:261–267
17. Dunger DB, Acerini CL 1997 Does recombinant human insulin-like growthfactor-1 have a role in the treatment of diabetes? Diabet Med 14:723–731
18. Boni-Schnetzler M, Hauri C, Zapf J 1999 Leptin is suppressed during infusionof recombinant human insulin-like growth factor I (rhIGF I) in normal rats.Diabetologia 42:160–166
19. Zenobi PD, Jaeggi Groisman SE, Riesen WF, Roder ME, Froesch ER 1992Insulin-like growth factor-I improves glucose and lipid metabolism in type 2diabetes mellitus. J Clin Invest 90:2234–2241
20. Donath MY, Sutsch G, Yan XW, et al. 1998 Acute cardiovascular effects ofinsulin-like growth factor I in patients with chronic heart failure. J Clin En-docrinol Metab 83:3177–3183
21. Ambler GR, Johnston BM, Maxwell L, Gavin JB, Gluckman PD 1993 Im-provement of doxorubicin induced cardiomyopathy in rats treated withinsulin-like growth factor I. Cardiovasc Res 27:1368–1373
22. Leri A, Liu Y, Wang X, Kajstura J, Malhotra A, Anversa P 1999 Overexpressionof insulin-like growth factor-1 attenuates the myocyte renin-angiotensin sys-tem in transgenic mice. Circ Res 84:752–762
23. Nilsson ABM, Nitescu N, Chen Y, et al. 2000 IGF-I treatment attenuates renalabnormalities induced by neonatal ACE inhibition. Am J Physiol 279:R1050–R1060
24. Blum WF, Ranke MB 1990 Use of insulin-like growth factor-binding protein3 for the evaluation of growth disorders. Horm Res 34(Suppl 1):31–37
25. Unterman TG, Lascon R, Gotway MB, et al. 1990 Circulating levels of insulin-likegrowth factor binding protein-1 (IGFBP-1) and hepatic mRNA are increased in thesmall for gestational age (SGA) fetal rat. Endocrinology 127:2035–2037
26. Tapanainen PJ, Bang P, Wilson K, Unterman TG, Vreman HJ, Rosenfeld RG1994 Maternal hypoxia as a model for intrauterine growth retardation: effectson insulin-like growth factors and their binding proteins. Pediatr Res 36:152–158
27. Blum WF, Breier BH 1994 Radioimmunoassays for IGFs and IGFBPs. GrowthRegul 4:11–19
28. Breier BH, Vickers MH, Gravance CG, Casey PJ 1996 Growth hormone (GH)therapy markedly increases the motility of spermatozoa and the concentrationof insulin-like growth factor-I in seminal vesicle fluid in the male GH-deficientdwarf rat. Endocrinology 137:4061–4064
29. Hossenlop P, Seurin D, Segovia-Quinson B, Hardouin S, Binoux M 1986Analysis of serum insulin-like growth factor binding proteins using Western-ligand blotting: use of the method for titration of the binding proteins andcompetitive binding studies. Ann Biochem 154:138–143
30. Gallaher BW, Breier BH, Oliver MH, Harding JE, Gluckman PD 1992 On-togenic differences in the nutritional regulation of circulating IGF bindingproteins in sheep plasma. Acta Endocrinol (Copenh) 126:49–54
31. Gargosky SE, Tapainen P, Rosenfeld RG 1994 Administration of growthhormone (GH), but not insulin-like growth factor-I (IGF-I), by continuousinfusion can induce the formation of the 150-kilodalton IGF-binding protein-3complex in GH-deficient rats. Endocrinology 134:2267–2276
32. Vickers MH, Ikenasio BA, Reddy S, Breier BH 2000 Dysregulation of theadipoinsular axis may be an important determinant for the development ofhyperphagia, obesity and type 2 diabetes during adult life. Proceedings of the82nd Meeting of The Endocrine Society, Toronto, Canada, 2000 (Abstract)
33. Min SH, MacKenzie DD, Breier BH, McCutcheon SN 1996 Responses ofyoung energy-restricted sheep to chronically administered insulin-like growthfactor I (IGF-I): evidence that IGF-I suppresses the hepatic growth hormonereceptor. Endocrinology 137:1129–1137
34. Izhar U, Hasdai D, Richardson DM, Cohen P, Lerman A 2000 Insulin andinsulin-like growth factor-I cause vasorelaxation in human vessels in vitro.Coron Artery Dis 11:69–76
35. Vestergaard H, Rossen M, Urhammer SA, Muller J, Pedersen O 1997 Short-and long-term metabolic effects of recombinant human IGF-I treatment inpatients with severe insulin resistance and diabetes mellitus. Eur J Endocrinol136:475–482
36. Tomas FM, Knowles SE, Chandler CS, Francis GL, Owens PC, Ballard FJ1993 Anabolic effects of insulin-like growth factor-I (IGF-I) and an IGF-I variantin normal female rats. J Endocrinol 137:413–421
37. Tannenbaum GS, Guyda HJ, Posner BI 1983 Insulin-like growth factors: a rolein growth hormone negative feedback and body weight regulation via brain.Science 220:77–79
38. Tomas FM, Chandler CS, Coyle P, Bourgeois CS, Burgoyne JL, Rofe AM 1994
Effects of insulin and insulin-like growth factors on protein and energy me-tabolism in tumour-bearing rats. Biochem J 301:769–775
39. Guler H-P, Zapf J, Scheiwiller E, Froesch ER 1988 Recombinant humaninsulin-like growth factor I stimulates growth and has distinct effects on organsize in hypophysectomised rats. Proc Natl Acad Sci USA 85:4889–4893
40. Tomas FM, Knowles SE, Owens PC, Chandler CS, Francis GL, Ballard FJ1993 Insulin-like growth factor-I and more potent variants restore growth ofdiabetic rats without inducing all characteristic insulin effects. Biochem J291:781–786
41. Frick F, Oscarsson J, Vikman-Adolfsson K, Ottosson M, Yoshida N, Eden S2000 Different effects of IGF-I on insulin-stimulated glucose uptake in adiposetissue and skeletal muscle. Am J Physiol Endocrinol Metab 278:E729–E737
42. Hussain MA, Schmitz O, Christiansen JS, Zapf J, Froesch ER 1995 Metaboliceffects of insulin-like growth factor-I: a focus on insulin sensitivity. Metabolism44:108–112
43. Hardie LJ, Guilhot N, Trayhurn P 1996 Regulation of leptin production incultured mature white adipocytes. Horm Metab Res 28:685–689
44. Szanto I, Kahn CR 2000 Selective interaction between leptin and insulinsignaling pathways in a hepatic cell line. Proc Natl Acad Sci USA 97:2355–2360
45. Seufert J, Kieffer TJ, Leech CA, et al. 1999 Leptin suppression of insulin secretionand gene expression in human pancreatic islets: implications for the developmentof adipogenic diabetes mellitus. J Clin Endocrinol Metab 84:670–676
46. Kieffer TJ, Habener JF 2000 The adipoinsular axis: effects of leptin on pan-creatic �-cells. Am J Physiol 278:E1–E14
47. Zhao AZ, Zhao H, Teague J, Fujimoto W, Beavo JA 1997 Attenuation of insulinsecretion by insulin-like growth factor 1 is mediated through activation ofphosphodiesterase 3B. Proc Natl Acad Sci USA 94:3223–3228
48. Van Schravendijk CF, Heylen L, Van den Brande JL, Pipeleers DG 1990Direct effect of insulin and insulin-like growth factor-I on the secretory activityof rat pancreatic � cells. Diabetologia 33:649–653
49. Withers DJ, White MF 2000 Perspective: the insulin signaling system—a commonlink in the pathogenesis of type 2 diabetes. Endocrinology 141:1917–1921
50. Corry DB, Tuck ML 1996 Glucose and insulin metabolism in hypertension.Am J Nephrol 16:223–236
51. Barbagallo M, Gupta RK, Bardicef O, Bardicef M 1997 Altered ionic effectsof insulin in hypertension: role of basal ion levels in determining cellularresponsiveness. J Clin Endocrinol Metab 82:1761–1765
52. Resnick L 1992 Cellular calcium and magnesium metabolism in the patho-physiology and treatment of hypertension and related metabolic disorders.Am J Med 93:2A-11S–2A-21S
53. Leri A, Liu Y, Claudio PP, et al. 1999 Insulin-like growth factor-1 inducesMdm2 and down-regulates p53, attenuating the myocyte renin-angiotensinsystem and stretch-mediated apoptosis. Am J Pathol 154:567–580
54. Isgaard J, Tivesten A, Friberg P, Bengtsson BA 1999 The role of the GH/IGF-Iaxis for cardiac function and structure. Horm Metab Res 31:50–54
55. Burren CP, Wilson EM, Hwa V, Oh Y, Rosenfeld RG 1999 Binding propertiesand distribution of insulin-like growth factor binding protein-related protein3 (IGFBP-rP3/NovH), an additional member of the IGFBP superfamily. J ClinEndocrinol Metab 84:1096–1103
56. Price WA, Rong L, Stiles AD, D’Ercole AJ 1992 Changes in IGF-I and -II, IGFbinding protein, and IGF receptor transcript abundance after uterine arteryligation. Pediatr Res 32:291–295
57. Janssen JA, Lamberts SW 2000 Circulating IGF-I and its protective role in thepathogenesis of diabetic angiopathy. Clin Endocrinol (Oxf) 52:1–9
58. Brismar K, Fernqvistforbes E, Wahren J, Hall K 1994 Effect of insulin on thehepatic production of insulin- like growth factor-binding protein-1 (IGFBP-1),IGFBP-3, and IGF-I in insulin-dependent diabetes. J Clin Endocrinol Metab79:872–878
59. Bereket A, Lang CH, Wilson TA 1999 Alterations in the growth hormone-insulin-like growth factor axis in insulin dependent diabetes mellitus. HormMetab Res 31:172–181
60. Butler AA, Gallaher BW, Ambler GR, Gluckman PD, Breier BH 1996 Insulin-like growth factor-I (IGF-I) and IGF-binding protein-3 in plasma of growth-hormone-deficient rats. J Endocrinol 150:67–76
61. Villafuerte BC, Zhang WN, Phillips LS 1996 Insulin and insulin-like growthfactor-1 regulate hepatic insulin-like growth factor binding protein-3 by dif-ferent mechanisms. Mol Endocrinol 10:622–630
62. Baxter RC 1988 The insulin-like growth factors and their binding proteins.Comp Biochem Physiol 91B:229–235
63. Jones JI, Clemmons DR 1995 Insulin-like growth factors and their bindingproteins: biological actions. Endocr Rev 16:3–34
64. Fowlkes J, Schultz HD 1992 Evidence for a novel insulin-like growth factor(IGF)-dependent protease regulating IGF-binding protein-4 in dermal fibro-blasts. Endocrinology 131:2071–2076
65. Neely EK, Rosenfeld RG 1992 Insulin-like growth factors (IGFs) reduce IGF-binding protein-4 (IGFBP-4) concentration and stimulate IGFBP-3 indepen-dently of IGF receptors in human fibroblasts and epidermal cells. Endocri-nology 130:985–993
66. Kuemmerle JF, Teng B 2000 Regulation of IGFBP-4 levels in human intestinalmuscle by an IGF-I-activated, confluence-dependent protease. Am J Physiol279:G975–G982
Vickers et al. • IGF-I Treatment and Fetal Programming Endocrinology, September 2001, 142(9):3964–3973 3973
Related Documents











