Mol Divers (2012) 16:193–202 DOI 10.1007/s11030-011-9338-x FULL-LENGTH PAPER Identification of novel potential HIF-prolyl hydroxylase inhibitors by in silico screening Mahesh Kumar Teli · G. K. Rajanikant Received: 30 June 2011 / Accepted: 7 October 2011 / Published online: 1 November 2011 © Springer Science+Business Media B.V. 2011 Abstract Suppression of HIF-prolyl hydroxylase (PHD) activity by small-molecule inhibitors leads to the stabilization of hypoxia inducible factor and has been recognized as prom- ising drug target for the treatment of ischemic diseases. In this study, pharmacophore-based virtual screening and molecu- lar docking approaches were concurrently used with suit- able modifications to identify target-specific PHD inhibitors with better absorption, distribution, metabolism, and excre- tion properties and to readily minimize false positives and false negatives. A customized method based on the active site information of the enzyme was used to generate a phar- macophore hypothesis (AAANR). The hypothesis was val- idated and utilized for chemical database screening and the retrieved hit compounds were subjected to molecular docking for further refinement. AAANR hypothesis comprised three H-bond acceptor, one negative ionizable group and one aro- matic ring feature. The hypothesis was validated using decoy set with a goodness of fit score of 2 and was used as a 3D query for database screening. After manual selection, molec- ular docking and further refinement based on the molecular interactions of inhibitors with the essential amino acid resi- dues, 18 hits with good absorption, distribution, metabolism, and excretion (ADME) properties were selected as excel- lent PHD inhibitors. The best pharmacophore hypothesis, AAANR, contains chemical features required for the effec- tive inhibition of PHD. Using AAANR, we have identified 18 potential and diverse virtual leads, which can be readily Electronic supplementary material The online version of this article (doi:10.1007/s11030-011-9338-x) contains supplementary material, which is available to authorized users. M. K. Teli · G. K. Rajanikant (B ) School of Biotechnology, Bioinformatics Infrastructure Facility, National Institute of Technology Calicut, Calicut 673601, Kerala, India e-mail: [email protected] evaluated for their potency as novel inhibitors of PHD. It can be concluded that the combination of pharmacophore, molec- ular docking, and manual interpretation approaches can be more successful than the traditional approach alone for discovering more effective inhibitors. Keywords HIF-prolyl hydroxylase inhibitor · Virtual screening · Ischemia · Pharmacophore model · Docking Introduction Mounting evidence indicates that hypoxia inducible factor (HIF) is a key molecule regulating the cellular response to tis- sue hypoxia [1, 2]. HIF is a heterodimeric transcription factor that regulates cellular stress responses [3]. It has been increas- ingly appreciated for its key role in transcriptional control of a large array of target genes that influence a wide-spectrum of cellular functions, including angiogenesis, vasomotor con- trol, erythropoiesis, stem cell homing, energy metabolism, radical production/scavenging, iron homeostasis, pH regula- tion, cell proliferation, and viability [2–4]. HIF has earned the epithet of being the ‘master regulator’ of a hypoxically reg- ulated transcriptional system. Taken together, these observa- tions have suggested that pharmacological activation of HIF may be beneficial in stroke and other ischemic conditions [5, 6]. A family of dioxygenases called HIF-prolyl hydroxylases (PHDs) has been demonstrated to regulate the stability of the HIF in an oxygen-dependent manner [7]. Suppression of PHD activity by small-molecule inhibitors leads to sta- bilization of HIF and offers a potential therapeutic option for various ischemic disorders such as the peripheral artery occlusive disease, myocardial infarction, renal ischemia and 123

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Mol Divers (2012) 16:193–202DOI 10.1007/s11030-011-9338-x
FULL-LENGTH PAPER
Identification of novel potential HIF-prolyl hydroxylase inhibitorsby in silico screening
Mahesh Kumar Teli · G. K. Rajanikant
Received: 30 June 2011 / Accepted: 7 October 2011 / Published online: 1 November 2011© Springer Science+Business Media B.V. 2011
Abstract Suppression of HIF-prolyl hydroxylase (PHD)activity by small-molecule inhibitors leads to the stabilizationof hypoxia inducible factor and has been recognized as prom-ising drug target for the treatment of ischemic diseases. In thisstudy, pharmacophore-based virtual screening and molecu-lar docking approaches were concurrently used with suit-able modifications to identify target-specific PHD inhibitorswith better absorption, distribution, metabolism, and excre-tion properties and to readily minimize false positives andfalse negatives. A customized method based on the activesite information of the enzyme was used to generate a phar-macophore hypothesis (AAANR). The hypothesis was val-idated and utilized for chemical database screening and theretrieved hit compounds were subjected to molecular dockingfor further refinement. AAANR hypothesis comprised threeH-bond acceptor, one negative ionizable group and one aro-matic ring feature. The hypothesis was validated using decoyset with a goodness of fit score of 2 and was used as a 3Dquery for database screening. After manual selection, molec-ular docking and further refinement based on the molecularinteractions of inhibitors with the essential amino acid resi-dues, 18 hits with good absorption, distribution, metabolism,and excretion (ADME) properties were selected as excel-lent PHD inhibitors. The best pharmacophore hypothesis,AAANR, contains chemical features required for the effec-tive inhibition of PHD. Using AAANR, we have identified18 potential and diverse virtual leads, which can be readily
Electronic supplementary material The online version of thisarticle (doi:10.1007/s11030-011-9338-x) contains supplementarymaterial, which is available to authorized users.
M. K. Teli · G. K. Rajanikant (B)School of Biotechnology, Bioinformatics Infrastructure Facility,National Institute of Technology Calicut, Calicut 673601, Kerala, Indiae-mail: [email protected]
evaluated for their potency as novel inhibitors of PHD. It canbe concluded that the combination of pharmacophore, molec-ular docking, and manual interpretation approaches canbe more successful than the traditional approach alone fordiscovering more effective inhibitors.
Keywords HIF-prolyl hydroxylase inhibitor ·Virtual screening · Ischemia · Pharmacophore model ·Docking
Introduction
Mounting evidence indicates that hypoxia inducible factor(HIF) is a key molecule regulating the cellular response to tis-sue hypoxia [1,2]. HIF is a heterodimeric transcription factorthat regulates cellular stress responses [3]. It has been increas-ingly appreciated for its key role in transcriptional control of alarge array of target genes that influence a wide-spectrum ofcellular functions, including angiogenesis, vasomotor con-trol, erythropoiesis, stem cell homing, energy metabolism,radical production/scavenging, iron homeostasis, pH regula-tion, cell proliferation, and viability [2–4]. HIF has earned theepithet of being the ‘master regulator’ of a hypoxically reg-ulated transcriptional system. Taken together, these observa-tions have suggested that pharmacological activation of HIFmay be beneficial in stroke and other ischemic conditions[5,6].
A family of dioxygenases called HIF-prolyl hydroxylases(PHDs) has been demonstrated to regulate the stability ofthe HIF in an oxygen-dependent manner [7]. Suppressionof PHD activity by small-molecule inhibitors leads to sta-bilization of HIF and offers a potential therapeutic optionfor various ischemic disorders such as the peripheral arteryocclusive disease, myocardial infarction, renal ischemia and
123

194 Mol Divers (2012) 16:193–202
stroke [7,8]. Despite growing interest in PHD inhibition, onlya few inhibitor structural classes have been published [9–11].However, some of these compounds lack sufficient propertiesto be useful PHD inhibitors because of low selectivity dueto the presence of iron chelating groups or poor blood–brainbarrier (BBB) permeability [12,13].
This study was carried out to identify novel target-spe-cific PHD inhibitors with better ADME properties, usingin silico approaches [14–19]. In this study, pharmacophore-based virtual screening and molecular docking approacheswere concurrently used with some modifications to mini-mize false positive and false negative results, encounteredby either ligand-based or structure based virtual screeningalone [20–22].
Methods
Receptor X-ray structure
The 3D coordinates of X-ray crystallographic structure ofhuman PHD complexed with respective inhibitors or activa-tor (PDB code: 2HBT, 2G19, 3OUH, 3OUI, and 3OUJ) wasselected to generate a pharmacophore model. Before retriev-ing ligands from proteins, all structures were prepared by pro-tein preparation wizard of Schrödinger Suite (SchrödingerLLC., Portland, USA) and all hetero atoms (except inhibi-tor and metal ion) were removed from protein files [23–25].All water molecules (beyond 3 Å from the inhibitor) and therest of the chains (except A) were removed from the com-plex and the protein was minimized using OPLS-2005 forcefield. Further, H-atoms were added to the protein to correctionization and tautomeric states of amino acid residues.
Active site analysis
The active site was analyzed by selecting neighbors within5 Å around the ligand. The active site contains highly con-served amino acid residues (Arg383, Tyr329, Tyr310, andTyr303) and Fe+2, essential for binding of ligand to the activesite. Arg383 and Tyr329 are responsible for making saltbridge interaction and H-bonding with the inhibitor, respec-tively. Tyr310 and Tyr303 are responsible for π–π stackingand H-bonding with the inhibitor, respectively (Fig. 1a). Thepresence of Trp389, Trp258, Met299, and Ilu256 at the open-ing of the active site renders hydrophobicity [23].
Pharmacophore modeling
The Phase module was used to develop a pharmacophoremodel of these inhibitors derived from their respective pro-teins [26]. The chemical features of ligands that may facilitatenon-covalent bonding with its target receptor were defined by
Fig. 1 Active site analysis of PHD. a Docking pose showing majorinteractions of the inhibitor (UN9) with the amino acid residues ofPHD (PDB ID: 2HBT). b Narrow cylindrical active site occupied bythe inhibitor (UN9)
using built-in six pharmacophoric features: H-bond accep-tor (A), H-bond donor (D), hydrophobic group (H), nega-tively charged group (N), positively charged group (P), andaromatic ring (R). The following conserved features in allaligned inhibitors responsible for interaction with the aminoacid residues were considered: (1) carboxyl group of theinhibitor to make salt bridge and H-bonding interactions withArg383 and Tyr329, respectively, (2) presence of two elec-tron negative groups in the inhibitor for interaction with Fe+2,(3) the inhibitor to make H-bonding with Tyr303, and (4) theinhibitor bearing aromatic hydrophobic group required for itsstabilization was preferred, owing to the hydrophobic natureof the opening of active site due to the presence of Trp389,Trp258, Met299, and Ilu256. Further, aromatic moiety is alsoresponsible for π–π stacking with Tyr310 [27].
123

Mol Divers (2012) 16:193–202 195
Virtual-screening (VS) workflow validation
The ability of VS workflow to identify PHD inhibitors in adatabase was tested by applying the AAARN pharmacophoremodel on a group of ten known inhibitors (other than thoseused to generate the pharmacophore) along with ten inactivecompounds [28]. The model was considered best if the VSretrieves back all those inhibitors with good fitness value. Fit-ness value is the contribution from align, vector, and volumescores, and carries maximum value three. It is a measure ofhow well the pharmacophoric sites of a ligand are matchingwith the hypothesis and how well the conformation superim-poses with the vector features (acceptors, donors, and aro-matic rings). If any compound scores ≈2 (or above), it maybe considered as a good inhibitor and if it scores below one,then it is considered as inactive [27].
Ligand preparation
All the ligands used were processed in the similar man-ner. The Asinex compound database was downloaded and3D structures were incorporated into the LigPrep module[26]. The cleaning process was carried out with the follow-ing parameters: (a) the force field used was OPLS-2005; (b)all possible ionization states at pH 7.0 ± 2.0 were generatedwith ionizer; (c) the desalt option was activated; (d) tautom-ers were generated for all ionization states at pH 7.0±2.0; (e)chiralities, when present, were determined from the 3D struc-ture; and (f) one low-energy ring conformation per ligandwas generated [24]. Conformations and sites for the resultingligand structures were determined during the generation ofthe corresponding Phase databases with the Generate PhaseDatabase graphic front-end. The parameter values used dur-ing this conformer generation were by default with the max-imum number of conformers (100) per structure. The con-former sites were generated with definitions made by thedefault built-in Phase [27].
Database screening
The validated pharmacophore hypothesis (AAANR) wasused as a 3D query for screening Asinex database. The pur-pose of this screening was to retrieve novel and potential leadssuitable for further development. Conformers were generatedfor each molecule in the database using Phase. The data-base screening was carried out using Phase virtual screen-ing protocol implemented in Schrödinger with Best/FlexibleSearch option. The retrieved compounds were filtered by fit-ness value and the obtained compounds were further refinedusing molecular docking study [27].
The Asinex database contains 3D structure of more than500,000 commercially available derivatives and was used asthe source of molecules to which our VS scheme was applied.
Initially, these molecules were submitted to an ADME filter(QikProp). QikProp module efficiently evaluates pharmaceu-tically relevant properties (ADME) of the ligand, which is aprerequisite to eliminate unnecessary testing on compoundsthat will ultimately fail. Thus, the drug-like properties of acompound were evaluated using the Lipinski rule, and onlyone violation of this rule was allowed [29,30].
Molecules with clinically acceptable ADME propertieswere then set up with LigPrep and were used to generate aPhase database. Next, this database was filtered through thecommon pharmacophore (AAANR) using the following run-ning conditions: (a) search for potential hits in the conform-ers database generated by Phase, (b) do not score in placethe conformers into the structure-based common pharmaco-phore and refine (i.e., allow reorientation of the conformersto determine if they match the pharmacophore or not), (c)match at least four out of the five sites of the structure-basedcommon pharmacophore, (d) do not consider any site as man-datory, and (e) do not prefer partial matches involving moresites. The rest of the options and parameters used during thatsearch were the default values [27].
Molecular docking
Pharmacophore modeling is normally associated with dock-ing procedure, in which the first step flexibly aligns the lig-and into a flexible macromolecule environment and thenestimates the tightness of the interaction by assigningscores.
Glide was used to perform automated docking with fullacyclic ligand flexibility, partial cyclic ligand flexibility, andpartial protein flexibility in the neighborhood of the pro-tein active site [31]. The crystal structure of PHD (PDB ID:2HBT) complexed with an inhibitor, UN9, was taken basedon its high resolution (1.60 Å). The inhibitor was extractedfrom the active site and redocked. The redocking confor-mation was aligned with the original inhibitor conformationto check Root Mean Square Deviation (RMSD) in order toconfirm the accuracy of docking program [32].
Extra precision docking with receptor flexibility was usedfor all Glide docking runs with default settings for someparameters and no constraints of similarity scoring wasapplied. The protein was prepared using the protein prep-aration and refinement tool. For the active site, a grid boxcentered at the ligand (UN9) was used to accommodate amaximum ligand length of 15 Å. The positional constraintand constraint for Fe+2, Arg383 and H-bond with Tyr329were applied while grid preparation. Default values wereaccepted for van der Waals scaling and input partial chargeswere used. We docked the ligands flexibly, allowing for theflip of 5- and 6-membered rings, writing out a maximumof one poses per ligand and also enabling the post-dockingminimization of the ligands [31,32].
123

196 Mol Divers (2012) 16:193–202
The retrieved database hits from VS were docked into theactive site of the PHD. Water molecules were not removednear the ligand prior to docking since they were found to playan important role in PHD–ligand interactions.
Results and discussion
In general, a pharmacophore model can be generated byeither custom based selection of sites or by QSAR method.In custom-based selection, desired important pharmacophoresites are selected manually, while in QSAR-based approach,already known inhibitors are aligned computationally to findcommon sites among the given input structures to providepharmacophore hypotheses. Then, the suitable hypothesis isconsidered for virtual screening and docking.
In this study, we have used a combination of both themethods with the following modifications to improve theefficiency of virtual screening protocol: (1) five structurallydiverse ligands for PHD were extracted from Protein DataBank (PDB) and were aligned with their original bindingactive confirmations to get common features responsible fortheir interaction with the protein, (2) the interactions of allfive ligands with their respective PDB proteins were checked,analyzed and all the common sites that are interacting withthe same amino acid residues of their respective proteins wereselected for screening the chemical database, and (3) Aftervirtual screening with the pharmacophore model, hits wereselected manually based on the structural information of theactive site to reduce false positives and false negatives.
Pharmacophore description
All five ligands obtained from respective PHD crystal struc-tures (PDB code: 2HBT, 2G19, 3OUH, 3OUI, and 3OUJ)with original conformation were aligned and the RMSDvalue (<0.5) was obtained. This indicated that all the activecompounds are binding in the same orientation and confor-mation (Fig. 2a). Further, it demonstrated that all interac-tive groups in the ligand are interacting with the receptors,which are aligned at the same place and orientation (OnlineResource 1). The active site information and aligned ligandposes that have common groups to most of them were used tobuild the pharmacophore model. Then, custom based phar-macophore model was generated by the Phase module bykeeping all the information in mind. The resulting commonstructure and ligand-based pharmacophore comprised threeH-bond acceptor (A), one negatively charged group (N), andone aromatic site (R) that were common to most of the alignedligands (Fig. 2b).
It is always worthy to take into account of the active siteinformation followed by the alignment of a ligand (if bind-ing to the same place) and find out the common as well as
Fig. 2 Generation of pharmacophore hypothesis for PHD inhibitors. aBiologically active conformations [retrieved from their respective pro-teins (PDB ID: 2HBT, 2G19, 3OUH, 3OUI, and 3OUJ)] of ligandsaligned together. b Three dimensional (3D) spatial arrangement of thefive feature pharmacophore model (AAANR) indicating the distance(Å) between pharmacophore features. The features, A1, A2 and A3are for H-bond acceptor, N7 for negative ionizable group and R8 foraromatic ring.
newer interactions to keep maximum interaction sites. Fur-ther, it is advantageous to preserve substrate interactive site inthe pharmacophore with some extra sites (gained by aligningvariety of ligands collected from different PDB structures butbinding to the same active site). Since the structures attainedby proteins are not rigid but are very dynamic, these substratefeatures need to be preserved to get a good activity in labo-ratory conditions. The particular combination of 3D sites ofsubstrates is expected to guide the ligands to their respectiveactive site.
VS workflow validation
Virtual-screening workflow was validated by applying it toa set consisting of ten known PHD inhibitors (different fromthose used for the structure-based pharmacophore genera-tion) and ten decoys obtained from the random selection ofinactives. VS recovered all active molecules with a fitnessvalue of ≈2, while it was <1.5 for inactive compounds, sug-gesting that our VS protocol was effectively selective to dis-criminate between actives and inactives (Online Resource 2).
Database screening
One efficient approach to drug discovery is virtual screen-ing of molecule libraries. The validated pharmacophore
123
Mol Divers (2012) 16:193–202 197
Table 1 ADME properties of selected hits with their respective docking scores
IDa %Human oral absorptionb QPlogBBc QPlogPo/wd QPlogSe QPlogHERGf Docking score
202127 39.324 −0.969 −0.599 −2.4 −3.375 −7.58034
107270 38.736 −2.584 0.128 −2.32 −2.947 −7.50601
74095 26.917 −2.343 −0.94 −1.657 −1.988 −7.48511
68307 56.307 −1.931 1.271 −3.55 −3.154 −7.29168
73446 65.159 −1.036 1.292 −2.161 −2.076 −7.24972
205455 67.931 −1.121 1.614 −2.976 −2.663 −7.10446
93330 66.087 −1.087 1.301 −2.64 −2.806 −7.06487
283081 61.021 −1.213 0.727 −2.214 −2.472 −7.0204
273206 68.538 −1.16 1.768 −3.087 −2.339 −6.98423
430008 55.473 −1.408 0.332 −1.705 −1.927 −6.97439
412853 65.522 −1.088 1.004 −2.062 −2.81 −6.90713
263858 53.092 −1.491 0.309 −1.902 −2.073 −6.90345
262508 69.993 −1.041 1.483 −2.182 −2.486 −6.84076
387620 61.55 −1.071 0.925 −2.113 −2.531 −6.82064
342088 59.431 −1.091 0.779 −1.37 −1.417 −6.79534
96064 66.546 −0.953 0.959 −2.207 −2.122 −6.77151
387628 65.727 −0.96 1.18 −2.331 −2.278 −6.69167
283051 54.252 −1.556 0.257 −1.874 −2.338 −6.66043
a Ligand IDs are of the Asinex database.b Percentage of human oral absorption (25% is poor and 80% is high).c Predicted BBB permeability (acceptable range: −3 to 1.2)d Predicted octanol/water partition co-efficient logP (acceptable range: −2.0 to 6.5).e Predicted aqueous solubility; S in mol/L (acceptable range: −6.5 to 0.5).f Predicted IC50 value for blockage of HERG K+ channels (acceptable range: below −5.0).
hypothesis, AAANR, was used as a 3D structural query forretrieving compounds from the Asinex chemical database. Asa result of first screening, 10,000 compounds were retrievedwith fitness value range of 0.5–2.3. Since the active site ofPHD resembles narrow cylindrical cavity, the experimen-tally known most active inhibitors are also small and linear(Fig. 1b). The top 5,000 hits with highest fit values were sub-sequently analyzed manually to find out the best inhibitors,keeping in mind the overall structural information of activesite, fitness value of ligand to pharmacophore (more than1.5) and physiochemical properties of ligands (Table 1). Themanual selection was done to exclude false positives, sincethey may also show affinity towards the receptor by bindingto other places. Hits were selected based on their good fitnessvalue to the pharmacophoric site (greater than 1.5), presenceof one negatively charged group (for salt bridge interaction)and two acceptors (to interact with Fe+2) (Online Resource3). After considering all these features, 189 molecules wereselected for docking analysis.
Molecular docking
Docking was carried out during the second step of VSworkflow to increase the power of pharmacophore-based
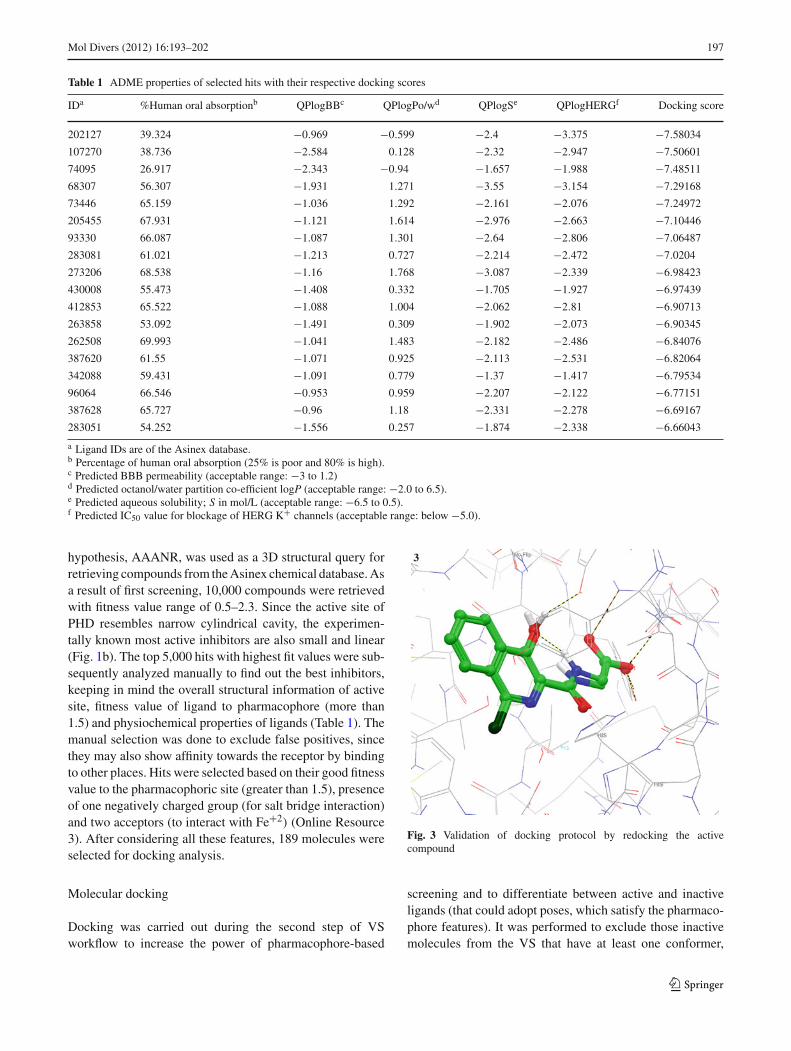
Fig. 3 Validation of docking protocol by redocking the activecompound
screening and to differentiate between active and inactiveligands (that could adopt poses, which satisfy the pharmaco-phore features). It was performed to exclude those inactivemolecules from the VS that have at least one conformer,
123
198 Mol Divers (2012) 16:193–202
which matches the sites in the pharmacophore but was steri-cally hindered by other residues in the active site.
The RMSD value of superimposition after redockingwas substantially low (0.24), confirming the accuracy ofour docking program (Fig. 3). Subsequently, 189 hits weredocked into the active site of PHD to refine the retrievedhits. The binding modes of all 189 compounds were rankedbased on the docking score and their ability to fulfillthe pre-defined constraints. The 50 highest scoring com-pounds were selected for further evaluation. After visualinspection, the most favorable compounds with the bestbinding mode (exact matching of π–π overlap with Tyr310,
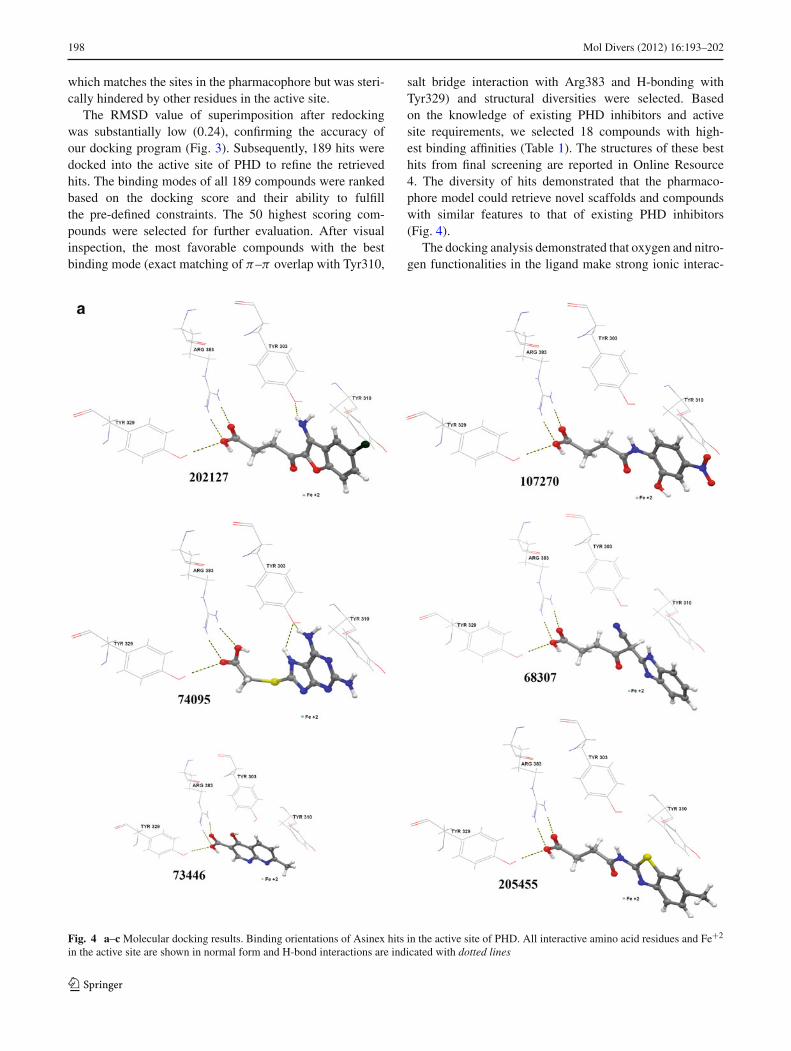
salt bridge interaction with Arg383 and H-bonding withTyr329) and structural diversities were selected. Basedon the knowledge of existing PHD inhibitors and activesite requirements, we selected 18 compounds with high-est binding affinities (Table 1). The structures of these besthits from final screening are reported in Online Resource4. The diversity of hits demonstrated that the pharmaco-phore model could retrieve novel scaffolds and compoundswith similar features to that of existing PHD inhibitors(Fig. 4).
The docking analysis demonstrated that oxygen and nitro-gen functionalities in the ligand make strong ionic interac-
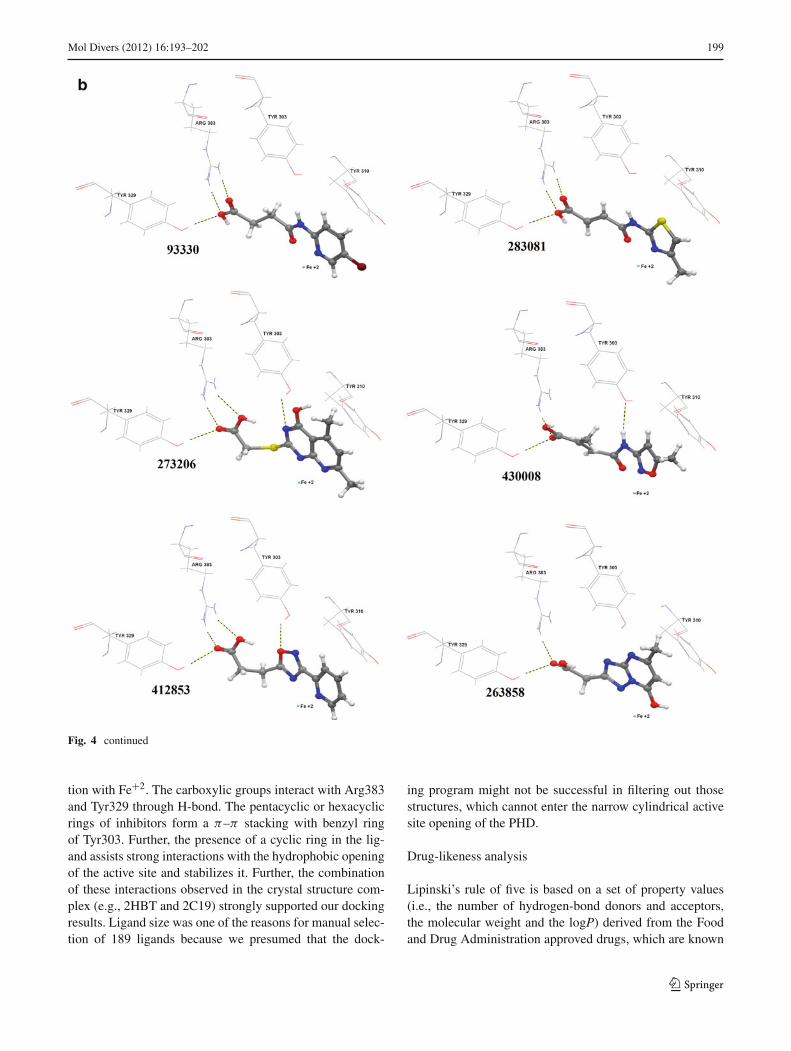
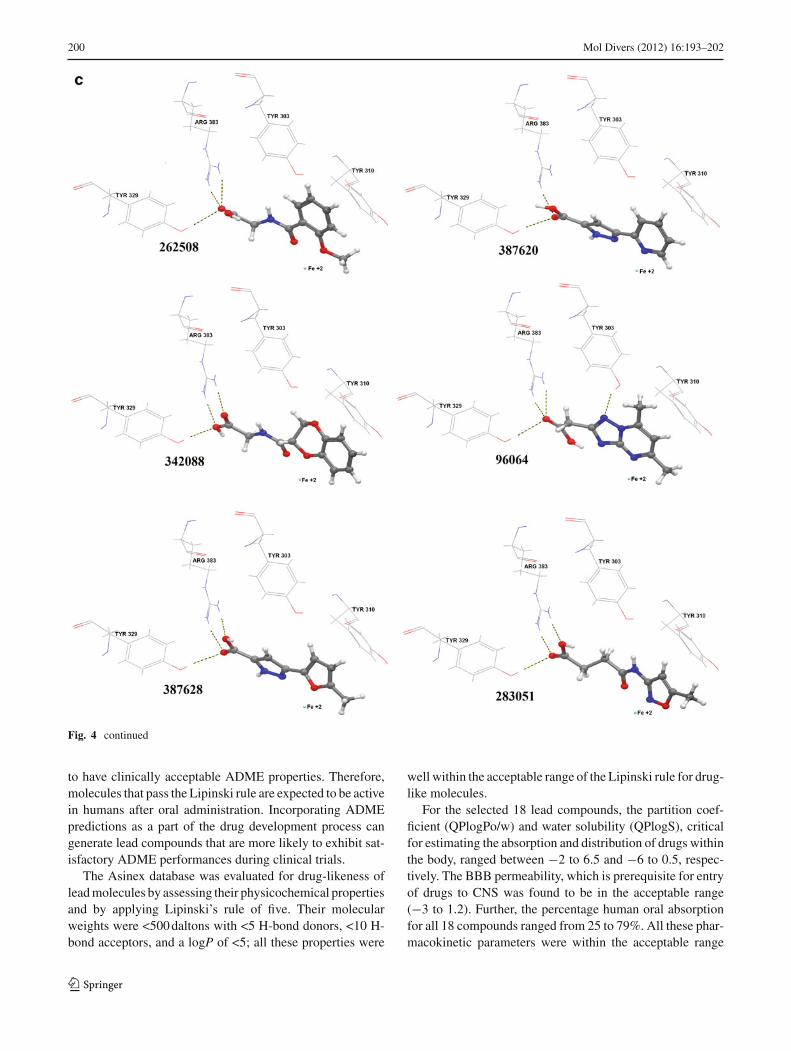
Fig. 4 a–c Molecular docking results. Binding orientations of Asinex hits in the active site of PHD. All interactive amino acid residues and Fe+2
in the active site are shown in normal form and H-bond interactions are indicated with dotted lines
123
Mol Divers (2012) 16:193–202 199
Fig. 4 continued
tion with Fe+2. The carboxylic groups interact with Arg383and Tyr329 through H-bond. The pentacyclic or hexacyclicrings of inhibitors form a π–π stacking with benzyl ringof Tyr303. Further, the presence of a cyclic ring in the lig-and assists strong interactions with the hydrophobic openingof the active site and stabilizes it. Further, the combinationof these interactions observed in the crystal structure com-plex (e.g., 2HBT and 2C19) strongly supported our dockingresults. Ligand size was one of the reasons for manual selec-tion of 189 ligands because we presumed that the dock-
ing program might not be successful in filtering out thosestructures, which cannot enter the narrow cylindrical activesite opening of the PHD.
Drug-likeness analysis
Lipinski’s rule of five is based on a set of property values(i.e., the number of hydrogen-bond donors and acceptors,the molecular weight and the logP) derived from the Foodand Drug Administration approved drugs, which are known
123
200 Mol Divers (2012) 16:193–202
Fig. 4 continued
to have clinically acceptable ADME properties. Therefore,molecules that pass the Lipinski rule are expected to be activein humans after oral administration. Incorporating ADMEpredictions as a part of the drug development process cangenerate lead compounds that are more likely to exhibit sat-isfactory ADME performances during clinical trials.
The Asinex database was evaluated for drug-likeness oflead molecules by assessing their physicochemical propertiesand by applying Lipinski’s rule of five. Their molecularweights were <500 daltons with <5 H-bond donors, <10 H-bond acceptors, and a logP of <5; all these properties were
well within the acceptable range of the Lipinski rule for drug-like molecules.
For the selected 18 lead compounds, the partition coef-ficient (QPlogPo/w) and water solubility (QPlogS), criticalfor estimating the absorption and distribution of drugs withinthe body, ranged between −2 to 6.5 and −6 to 0.5, respec-tively. The BBB permeability, which is prerequisite for entryof drugs to CNS was found to be in the acceptable range(−3 to 1.2). Further, the percentage human oral absorptionfor all 18 compounds ranged from 25 to 79%. All these phar-macokinetic parameters were within the acceptable range
123

Mol Divers (2012) 16:193–202 201
defined for human use (Table 1), thereby indicating theirpotential to act like drug-like molecules.
Structural analysis of the inhibition
The docking analysis of 18 compounds showed that all mol-ecules share common intermolecular interactions with theenzyme. They formed a salt bridge and H-bond with theArg383 and Tyr329 residues, respectively. All 18 compoundshave shown interaction with the Fe+2 and formed π−π over-lap with Tyr310. Hydrophobic interactions were also noticedfor all hits. Moreover, several ligands (Asinex ID # 74095,96064, 202127, 273206, 430008, and 412853) have formedH-bond with Tyr303 as well and showed strong interactions(Fig. 4).
Conclusions
By combining the pharmacophore model, docking, and man-ual interpretation approach altogether, we could perform vir-tual screening on a data set of compounds to identify noveltarget-specific PHD inhibitors and to delineate importantinteractions responsible for binding of ligands to the activesite of PHD more effectively. A 3D pharmacophore hypoth-esis (AAANR) of PHD inhibitors was developed usingcustom-based pharmacophore generation protocol, whichcomprised three H-bond acceptor, one negative ionizablegroup and one ring aromatic feature. AAANR was furthervalidated by decoy set method and was used as a 3D query indatabase screening. The database of hit compounds were sub-sequently subjected to filtering by checking desired features.To further refine the retrieved hits, all 189 compounds wereselected for molecular docking study and the result was ana-lyzed based on the docking score, binding modes, and molec-ular interactions with essential active site residues (Tyr310,Tyr303, Arg383, and Tyr329). Finally, 18 hits of diverse scaf-folds with good docking score (−7.58 to −6.88) and ADMEproperties were chosen. These compounds as such or on fur-ther optimization can be used as potential leads in designingnew PHD inhibitors.
In light of the pharmacophore model developed in thisstudy and the knowledge gained from the observations ofinteractions between PHD and potential inhibitors, it canbe deduced that combination of pharmacophore, moleculardocking, and manual interpretation efforts can be more effi-cient than the traditional approaches alone for discoveringeffective enzyme inhibitors that may have a great impact forfuture experimental studies. It is believed that appropriate useof these methods in a drug design process should improvethe ability to identify and optimize hits and confirm their
potentials to serve as scaffolds for the development of newtherapeutic agents.
Conflict of interest The authors report no declarations ofinterest.
References
1. Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DMet al (2000) Role of hypoxia-inducible factor-1 in hypoxia-inducedischemic tolerance in neonatal rat brain. Ann Neurol 48: 285–296. doi:10.1002/1531-8249(200009)48:3<285::AID-ANA2>3.3.CO;2-#
2. Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat RevCancer 3: 721–732. doi:10.1038/nrc1187
3. Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J et al(2006) Concordant regulation of gene expression by hypoxia and2-oxoglutarate dependent dioxygenase inhibition; the role of HIF-1α, HIF-2α and other pathways. J Biol Chem 281: 15215–15226.doi:10.1074/jbc.M511408200
4. Wenger RH, Stiehl DP, Camenisch G (2005) Integration of oxygensignaling at the consensus HRE. Sci Signal 306:re12. doi:10.1126/stke.3062005re12
5. Gidday JM, Fitzgibbons JC, Shah AR, Park TS (1994) Neuro-protection from ischemic brain injury by hypoxic precondition-ing in the neonatal rat. Neurosci Lett 168: 221–224. doi:10.1016/0304-3940(94)90455-3
6. Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS et al(2001) Cerebral protection by hypoxic preconditioning in a murinemodel of focal ischemia-reperfusion. Neuroreport 12: 1663–1669.doi:10.1097/00001756-200106130-00030
7. Harten SK, Ashcroft M, Maxwell PH (2010) Prolyl hydroxylasedomain inhibitors: a route to HIF activation and neuroprotection.Antioxid Redox Signal 12: 459–480. doi:10.1089/ars.2009.2870
8. Tian YM, Yeoh KK, Lee MK, Eriksson T, Kessler BM et al(2011) Differential sensitivity of hypoxia inducible factor hydrox-ylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem286: 13041–13051. doi:10.1074/jbc.M110.211110
9. Murray JK, Balan C, Allgeier AM, Kasparian A, ViswanadhanV et al (2010) Dipeptidyl-quinolone derivatives inhibit hypoxiainducible factor-1α prolyl hydroxylases-1, -2, and -3 with alteredselectivity. J Comb Chem 12: 676–686. doi:10.1021/cc100073a
10. Mecinovic J, Loenarz C, Chowdhury R, Schofield CJ (2009) 2-Oxoglutarate analogue inhibitors of prolyl hydroxylase domain 2.Bioorg Med Chem Lett 19: 6192–6195. doi:10.1016/j.bmcl.2009.09.005
11. Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S et al(2005) Hypoxia-inducible factor prolyl 4-hydroxylase inhibition.A target for neuroprotection in the central nervous system. J BiolChem 280: 41732–41743. doi:10.1074/jbc.M504963200
12. Semenza GL (2009) Regulation of vascularization by hypoxia-inducible factor 1. Ann N Y Acad Sci 1177: 2–8. doi:10.1111/j.1749-6632.2009.05032.x
13. Takeda K, Ichiki T, Narabayashi E, Inanaga K, Miyazaki Ret al (2009) Inhibition of prolyl hydroxylase domain-contain-ing protein suppressed lipopolysaccharide-induced TNF-α expres-sion. Arterioscler Thromb Vasc Biol 29: 2132–2137. doi:10.1161/ATVBAHA.109.196071
14. Pala N, Dallocchio R, Dessì A, Brancale A, Carta F et al(2011) Virtual screening-driven identification of human car-bonic anhydrase inhibitors incorporating an original, newpharmacophore. Bioorg Med Chem Lett 21: 2515–2520. doi:10.1016/j.bmcl.2011.02.059
123

202 Mol Divers (2012) 16:193–202
15. Gupta AK, Bhunia SS, Balaramnavar VM, Saxena AK (2011)Pharmacophore modelling, molecular docking and virtual screen-ing for EGFR (HER 1) tyrosine kinase inhibitors. SAR QSAREnviron Res 22: 239–263. doi:10.1080/1062936X.2010.548830
16. Bagga V, Silakari O, Ghorela VS, Bahia MS, Rambabu G et al(2011) A three-dimensional pharmacophore modelling of ITKinhibitors and virtual screening for novel inhibitors. SAR QSAREnviron Res 22: 171–190. doi:10.1080/1062936X.2010.510480
17. Sala E, Guasch L, Iwaszkiewicz J, Mulero M, Salvadó MJ et al(2011) Identification of human IKK-2 inhibitors of natural origin(part I): modeling of the IKK-2 kinase domain, virtual screen-ing and activity assays. PLoS One 6:e16903. doi:10.1371/journal.pone.0016903
18. John S, Thangapandian S, Sakkiah S, Lee KW (2011) PotentBACE-1 inhibitor design using pharmacophore modeling, in sil-ico screening and molecular docking studies. BMC Bioinformatics12(suppl 1):S28. doi:10.1186/1471-2105-12-S1-S28
19. Lu SH, Wu JW, Liu HL, Zhao JH, Liu KT et al (2011) The discov-ery of potential acetylcholinesterase inhibitors: a combination ofpharmacophore modeling, virtual screening, and molecular dock-ing studies. J Biomed Sci 18:8. doi:10.1186/1423-0127-18-8
20. Noha SM, Atanasov AG, Schuster D, Markt P, Fakhrudin N et al(2011) Discovery of a novel IKK-β inhibitor by ligand-based vir-tual screening techniques. Bioorg Med Chem Lett 21: 577–583.doi:10.1016/j.bmcl.2010.10.051
21. Chen Z, Tian G, Wang Z, Jiang H, Shen J et al (2010) Multiplepharmacophore models combined with molecular docking: a reli-able way for efficiently identifying novel PDE4 inhibitors with highstructural diversity. J Chem Inf Model 50: 615–625. doi:10.1021/ci9004173
22. Peach ML, Nicklaus MC (2009) Combining docking with phar-macophore filtering for improved virtual screening. J Cheminform1:6. doi:10.1186/1758-2946-1-6
23. Maestro, version 9.1 (2010) Schrödinger, LLC, New York24. LigPrep, version 2.4 (2010) Schrödinger, LLC, New York25. Schrödinger Suite 2009 (2009) Protein Preparation Wizard; Epik
version 2.0; Impact version 5.5; Prime version 2.1, Schrödinger,LLC, New York
26. Phase, version 3.2 (2010) Schrödinger, LLC, New York, NY27. Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Fries-
ner RA (2006) PHASE: a new engine for pharmacophore percep-tion, 3D QSAR model development, and 3D database screening:1. Methodology and preliminary results. J Comput Aided Mol Des20: 647–671. doi:10.1007/s10822-006-9087-6
28. Warshakoon NC, Wu S, Boyer A, Kawamoto R, Sheville J, BhattRT, Renock S, Xu K, Pokross M, Zhou S, Walter R, Mekel M,Evdokimov AG, East S (2006) Design and synthesis of substitutedpyridine derivatives as HIF-1alpha prolyl hydroxylase inhibitors.Bioorg Med Chem Lett 16: 5616–5620. doi:10.1016/j.bmcl.2006.08.026
29. QikProp, version 3.3 (2010) Schrödinger, LLC, New York, NY30. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Exper-
imental and computational approaches to estimate solubility andpermeability in drug discovery and development settings. AdvDrug Deliv Rev 46: 3–26. doi:10.1016/S0169-409X(00)00129-0
31. Schrödinger Suite 2010 (2010) Induced fit docking protocol; Glideversion 5.6; Prime version 2.2, Schrödinger, LLC, New York
32. Glide, version 5.5 (2009) Schrödinger, LLC, New York, NY
123
Related Documents











