International Pharmaceutical Excipients Council Collaborative solutions for excipient industry stakeholders Multiple stakeholders; one objective. ICH Q3D Elemental Impurities - Implementation Considerations ExcipientFest 2017 Phyllis Walsh: Vice Chair for Harmonization and Compendial Monographs Dave Schoneker: Vice Chair for Science & Regulatory Policy Copyright 2017, All Rights Reserved, IPEC-Americas 2 Elemental Impurities Workshop Agenda Back Ground Information on Elemental Impurities Scope of ICH Q3D Guideline Compendial Requirements Specific Metals Tests in Monograph EMA Guideline Draft FDA Guideline ICH Q3D Developments for Topicals Pb As Hg Cd Co V Ni ???

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

International Pharmaceutical Excipients Council
Collaborative solutions for excipient industry stakeholders
Multiple
stakeholders;
one objective.
ICH Q3D Elemental
Impurities -
Implementation
Considerations
ExcipientFest 2017
Phyllis Walsh: Vice Chair for
Harmonization and Compendial
Monographs
Dave Schoneker: Vice Chair for
Science & Regulatory Policy
Copyright 2017, All Rights Reserved, IPEC-Americas 2
Elemental Impurities Workshop Agenda
Back Ground Information on Elemental
Impurities
Scope of ICH Q3D Guideline
Compendial Requirements
Specific Metals Tests in Monograph
EMA Guideline
Draft FDA Guideline
ICH Q3D Developments for Topicals
Pb As Hg
Cd Co V
Ni ???

Copyright 2017, All Rights Reserved, IPEC-Americas 3
Elemental Impurities Workshop Agenda
Implementation by Region
Implementation Timelines & ICH regions - Regulatory Filings
Developments in other countries
Life Cycle Management
Copyright 2017, All Rights Reserved, IPEC-Americas 4
Elemental Impurities Workshop Agenda
Analytical and Risk Assessment Challenges
Methodologies concerns
Collaborative study performed
PQRI Phase 2 testing initiated
Lhasa database
Supplier information (API and Excipient)
Risk Assessment Approaches
Q&A

Copyright 2017, All Rights Reserved, IPEC-Americas 5
Elemental Impurities Workshop Agenda
Case studies – ICH Modules
Solid Oral Dosage
Working Example
Copyright 2017, All Rights Reserved, IPEC-Americas
Back Ground Information on Elemental Impurities
6
Scope of ICH Q3D Guideline
Compendial Requirements
Specific Metals Tests in Monograph
EMA Guideline
Draft FDA Guideline
ICH Q3D Developments for Topicals

Copyright 2017, All Rights Reserved, IPEC-Americas 7
ICH Q3D Approach to Risk Assessment
ICH Q3D defines a science and risk based assessment process to identify, evaluate, and define controls to limit elemental impurities in drug products.
Identify known and potential sources of elemental
impurities that may find their way into the drug product.
Evaluate the presence of a particular elemental impurity in the drug product by determining the observed or predicted level of the impurity and comparing with the established PDE.
Summarize and document the risk assessment. Identify if controls built into the process are sufficient or identify additional controls to be considered to limit elemental impurities in the drug product.
Scope of ICH Q3D Guideline
Copyright 2017, All Rights Reserved, IPEC-Americas 8
Risk Process – General Principles
ICH Q3D advocates a 3 step process:
Identify
Evaluate
Summarize Control
Different approaches to each stage are examined through a series of actual risk assessments.

Copyright 2017, All Rights Reserved, IPEC-Americas 9
Risk Assessments
ICH allows you to use the Component Approach or Finish Product Analysis.
The Component Approach is ICH Q3D option 1,
2A or 2B.
The Finish Product Analysis is option 3.
Copyright 2017, All Rights Reserved, IPEC-Americas 10
Risk Assessments
Finish Product Analysis
The Finish Product Analysis is option 3.
Assessment of potential elemental impurities from
each component of the drug product (API,
excipients, container closure system).
Need to know what potential elemental impurities
are present.
May need to test finished product.

Copyright 2017, All Rights Reserved, IPEC-Americas 11
Risk Assessments
Component Approach
The component approach is ICH Q3D option 1, 2A or 2B.
Assessment of potential elemental impurities from each component of the drug product (API, excipients,
container closure system).
Assess each component for potential sources of elemental impurities.
Identify known or likely elemental impurities.
Determine the contribution of each component or source of elemental impurity to the levels in the final drug product.
Copyright 2017, All Rights Reserved, IPEC-Americas 12
Potential Sources of Elemental Impurities

Copyright 2017, All Rights Reserved, IPEC-Americas 13
Compendial Requirements
USP
General Chapters <232> & <233> are
official but not yet implemented.
<232> revisions proposed in PF to align with ICH Q3D
General Notices 5.60.30 Elemental Impurities official
01-Jan-2018.
General Notices 5.60.30 Elemental Impurities official on
January 1, 2018 is the date on which General Chapter
<232> will become broadly applicable to drug products.
Heavy Metals General Chapter <231>
Chapter will be deleted from USP-NF on 01-Jan-2018.
Copyright 2017, All Rights Reserved, IPEC-Americas 14
Compendial Requirements
European Pharmacopoeia (Ph. Eur.)
Prior to 2008 No guideline for safety limits
only Heavy Metals General Chapter.
2008 EMA guideline on specification limits for
residues of metal catalysts or metal reagents
(known metals).
General Chapter 5.20
2014 ICH Q3D development and implementation
(Official 01-Jan-2018 Ph. Eur. 9.3)
Replace General Chapter 5.20 - Only parts of the
introduction and the scope of ICH Q3D together with
information specific to Q3D in the Ph. Eur.
Will not republish entire ICH Q3D into Ph. Eur.

Copyright 2017, All Rights Reserved, IPEC-Americas 15
Compendial Requirements
European Pharmacopoeia (Ph. Eur.) (cont.)
General method 2.4.20 Determination of elemental
impurities.
Editorial revision to align the wording with the ICH Q3D
guideline will be published in Ph. Eur. 9.3.
“metal catalyst and metal reagent residues” to “elemental
impurities.
General Method 2.4.8 Heavy Metals
Will not be deleted since needed for Veterinary use.
All references in monographs for Human products is
deleted.
Copyright 2017, All Rights Reserved, IPEC-Americas 16
Compendial Requirements
European Pharmacopoeia (Ph. Eur.)
(cont.)
General monograph on Substances for
Pharmaceutical use (2034):
Introduce requirements for the control of elemental
impurities intentionally added during production and
explains the absence of a test for elemental impurities from
individual monographs except for special cases.
The identity of the elemental impurities derived from
intentionally added catalysts and reagents is known and
strategies for controlling them should be established by
using the principles of risk management.

Copyright 2017, All Rights Reserved, IPEC-Americas 17
Compendial Requirements
European Pharmacopoeia (Ph. Eur.) (cont.)
General monograph on Pharmaceutical Preparations (2619)
Refers to chapter 5.20, rendering it and by extension the ICH Q3D guideline legally binding.
Clarification for medicinal products outside of the scope of ICH Q3D guideline manufacturers of these products remain responsible for controlling the levels of elemental impurities using the principles of risk management.
■ If appropriate, testing is performed using suitable analytical procedures according to general chapter 2.4.20 Determination of elemental impurities.
■ Elemental Impurities should at least be considered in risk management strategy
Copyright 2017, All Rights Reserved, IPEC-Americas 18
Specific Metals Tests in Monographs - USP
USP published a Stimuli Article Future of
Element - Specific Chapters in the USP–NF
with questions on specific metal tests in monographs.
What will be the future of USP chapters that provide specific
information regarding the analysis of individual elements, such as arsenic (As) Arsenic ⟨211⟩, lead (Pb) Lead ⟨251⟩, selenium
(Se) Selenium ⟨291⟩, mercury (Hg) Mercury ⟨261⟩, and others?
What about USP monographs that may have limit tests for
specific elements and refer to their respective element-
specific chapters for methodology?
What about USP monographs that include limits for specific elements that differ from the limits established in ⟨232⟩?
Limit tests and references to element specific chapters are
included in about 1000 monographs

Copyright 2017, All Rights Reserved, IPEC-Americas 19
Specific Metals Tests in Monographs Industry Concerns
Industry response to USP Pharmacopeias should not change an existing monograph
specific element requirement (methods/limits) unless evaluated as part of an individual monograph modernization activity designed to include specific metal requirement.
Existing element limit requirements and test methods should stay in the monographs and not be removed – to allow for comparisons with historical methods/data.
History supports limits/test methods which can be used in risk assessments as worst case examples AND provide useful information to users since actual detailed information is limited.
No changes should be made to the limits and no new elements should be added based on a limited amount of batch testing, since excursions won’t show up except over long-term history.
Copyright 2017, All Rights Reserved, IPEC-Americas 20
Specific Metals Tests in Monographs Industry Concerns
Industry response to USP (cont.) Current monograph limits and test methods are linked,
USP should not change the monographs to use the approaches for methodology and analysis in <233> or the existing limits unless validation work conducted demonstrates that the current methods in the monograph and any alternative methods give equivalent results
API suppliers: Elemental Impurities is for the finished drug product not the Drug Substances.
Removing specific metal tests from USP would impact API supplier, FDA would not allow deletion without justification.

Copyright 2017, All Rights Reserved, IPEC-Americas 21
Specific Metals Tests in Monographs Industry Assistance
IPEC-Americas is assisting USP in developing a priority list of monographs containing specific metals.
IPEC-Americas is requesting Industry to supply historical data to USP directly on specific metals in the monographs.
USP will evaluate the historical data and determine if any changes are needed in the monographs.
If changes are needed they will proceed through the normal PF process.
Copyright 2017, All Rights Reserved, IPEC-Americas 22
Specific Metals Tests in Monographs Ph. Eur.
In Ph. Eur. approx. 300 monographs describe more than 450 specific metal tests
Concerns - Specific Elemental Impurities in
individual monographs
Historical reason for the presence of the test in the
monograph
No change control for updating production pathways
(intentionally added metals)
Substances of natural origin (e.g. mined excipients) =>
Elemental Impurities potentially present but not intentionally
added

Copyright 2017, All Rights Reserved, IPEC-Americas 23
Specific Metals Tests in Monographs Ph. Eur.
EDQM announced actions:
Test for heavy metals (method 2.4.8) will be deleted from all individual monographs except from monographs of substances for veterinary use only (9th edition).
Other tests for specific Elemental Impurities in individual monographs will be reviewed by groups of experts on a case by case basis. Secretariat provided lists of monographs concerned to the groups.
Specific tests in individual monographs for elements not covered by ICH Q3D will remain untouched but may be considered upon discussion of a monograph in the group.
Copyright 2017, All Rights Reserved, IPEC-Americas 24
Specific Metals Tests in Monographs Ph. Eur.
EDQM decided to keep the published specific
elemental impurities tests in monographs on
substances of Natural Origin only.
Given the intrinsic nature of elemental impurities in Natural Origin
substances, they are amongst the major potential sources of elemental
contamination in medicinal products.
Recommended keeping the different tests for elements for which no
Permitted Daily Exposure limits have been established, i.e. those
identified as “other elements” in the ICH Q3D guideline (such as
aluminum and iron), in individual monographs.
Specific elemental impurities tests will be deleted from monographs on
other substances (i.e. not from natural origin), unless otherwise justified.
Specific tests for elemental contaminants originating from the
production process will be deleted.

Copyright 2017, All Rights Reserved, IPEC-Americas 25
Specific Metals Tests in Monographs Ph. Eur.
EDQM Focus: Substances of Natural
Origin (mainly mined excipients)
Obtain batch data and revise tests / or add new
ones, if necessary based on batch data
Need for more expertise and support (especially
from manufacturers) to revise / maintain the tests
Copyright 2017, All Rights Reserved, IPEC-Americas 26
EMA - Implementation Strategy of ICH Q3D Guideline
EMA Implementation strategy of ICH Q3D guideline published as final on 08-Mar-2017
Same Risk Assessment Approaches as ICH Q3D for Drug Product or Component
Particulars for Intentionally Added Element(s) Any element intentionally added during manufacturing
must be included in the description of the drug substance manufacturing process in the marketing authorization dossier, an ASMF or a CEP application, as well as its fate and the need for any controls (for instance the use of a metal catalyst in the last step of the synthesis).

Copyright 2017, All Rights Reserved, IPEC-Americas 27
FDA Draft Guidance FDA draft Guidance for Industry published June 2016
Scope
How applicants submitting new drug applications (NDAs) or abbreviated new drug applications (ANDAs) for non-compendial drug products should control elemental impurities as described in ICH Q3D. ICH Q3D contains recommendations on applying a risk-based approach to control elemental impurities and permitted daily exposure (PDE).
How manufacturers of compendial drug products that are not marketed under an approved NDA or ANDA can comply with USP General Chapters Elemental Impurities—Limits and Elemental Impurities—Procedures and the Federal Food, Drug, and Cosmetic (FD&C) Act.
How holders of NDAs or ANDAs for compendial drug products should report changes in chemistry, manufacturing, and controls specifications to FDA to comply with General Chapters and 21 CFR 314.70.
How manufacturers of non-compendial drug products that are marketed without an approved NDA or ANDA should control elemental impurities.
Copyright 2017, All Rights Reserved, IPEC-Americas 28
FDA Draft Guidance
FDA Final Guidance is expected soon
Expected clarity on Biologics.
Expected clarity on OTC products documentation.
Expected filings by Annual Reports.

Copyright 2017, All Rights Reserved, IPEC-Americas 29
FDA Advice to Industry
Risk Assessment: Potential considerations during review
Intentionally added elements.
Contributions from raw materials derived from plant or marine origins.
Contributions from raw materials that are mined, e.g., Inorganic drug substances and excipients.
Contributions from manufacturing, e.g., high shear micronization using metal discs.
Leachable elemental impurities from container/closure.
Extractables information from container/closure components typically included in a supplier Type III DMF.
Copyright 2017, All Rights Reserved, IPEC-Americas 30
ICH Q3D Developments for Topicals
Currently topicals fall under ICH
“Other Routes of Administration”.
ICH Q3D announced the start of Q3D(R1) - Revised PDEs for the cutaneous and transdermal Route of Administration on September 15, 2016.
Develop PDEs for EIs for products administered on skin and its appendages (e.g., hair, nails); these products remain the largest area where PDEs for EIs have not been established.
Cutaneous and transdermal PDEs were not developed at that time due to the late request for inclusion (post Step 2). These products include both prescription and over-the-counter products.
Since the intact skin serves as a barrier to absorption, it is possible that not all EIs in Q3D will require cutaneous and transdermal PDEs, streamlining the risk assessment process for these products.

Copyright 2017, All Rights Reserved, IPEC-Americas
Implementation by Region
31
Implementation Timelines & ICH regions -
Regulatory Filings.
Developments in other Countries.
Life cycle management.
Copyright 2017, All Rights Reserved, IPEC-Americas 32
Implementation Timelines for US
FDA Draft Guidance Recommendations and Timelines for risk assessment and documentation of risk assessment:
New NDA and ANDA applications submitted after June 1, 2016 should follow the recommendations of Q3D.
Consistent with the EMA implementation timeline
For existing marketed products, manufactures should follow the recommendations of Q3D and/or comply with USP <232> by January 1, 2018. Consistent with USP implementation timeline for <232>
and <233>.

Copyright 2017, All Rights Reserved, IPEC-Americas 33
ICH Regions - Regulatory Filings - FDA
Anticipates most approved drug products marketed in the United States do not contain any elemental impurities that exceed the Q3D/ <232>PDEs.
Products that meet PDE recommendations of Q3D or comply with <232>PDEs.
Perform risk assessment to determine if additional controls (e.g. upstream controls, specifications) are needed by 1 January 2018.
Document changes in the next Annual Report.
Copyright 2017, All Rights Reserved, IPEC-Americas 34
ICH Regions - Regulatory Filings - FDA
New NDAs or ANDAs. Include a summary of the risk assessment application. Cite
supporting material (e.g., controls) as warranted.
The P.2 section (Pharmaceutical Development) is an appropriate location for the risk assessment summary.
Approved NDAs or ANDAs. Include a summary in the next Annual Report following the
completion of the risk assessment. Document changes to controls.
See FDA Draft Guidance for details if drug products exceed PDEs and changes are implemented to reduce EI levels
For drug products not approved under an NDA or ANDA. Include risk assessment in the documentation maintained at
the manufacturing site for Agency review during an inspection.

Copyright 2017, All Rights Reserved, IPEC-Americas 35
Implementation Timelines for EU
CHMP recommendations and timelines:
New MA for new product (new active substance).
Implementation date June 2016
New MA for product with existing active substance.
Implementation date June 2016
Marketed products including new MR applications of already approved products.
Implementation date December 2017
Copyright 2017, All Rights Reserved, IPEC-Americas 36
ICH Regions - Regulatory Filings - EU
New Marketing Authorizations:
Compliance with the Q3D PDEs.
Should document the risk assessment and the
control approaches.
The documentation of the risk assessment should be
kept available for inspection on site.
A summary of the Risk Assessment and any
measures taken to ascertain compliance is needed
in the regulatory filing.
The overall Control Strategy for elemental impurities
including any specifications included in the
regulatory filing.

Copyright 2017, All Rights Reserved, IPEC-Americas 37
ICH Regions - Regulatory Filings - EU
Existing Marketed Products
Risk Assessment should be performed, documented
and be kept available.
No variation is necessary if the Risk Assessment shows
compliance: No further controls on elemental impurities to materials such as
the designated active substance starting material, synthesis
intermediates, active substance, excipients or the finished
product are needed.
No replacement or change of quality of materials such as the
designated active substance starting material, synthesis
intermediates, active substance, excipients or of the
manufacturing equipment is needed.
No change of the manufacturing process is needed.
Copyright 2017, All Rights Reserved, IPEC-Americas 38
ICH Regions - Regulatory Filings - EU
Existing marketed products
In other cases a variation is needed:
Categorized according the Variation Guidelines
(Official Journal 2013/C 223/01)
Accompanied with the documentation required in
the Variation Guideline.
In addition contain a summary of the Risk
Assessment and the conclusions drawn.

Copyright 2017, All Rights Reserved, IPEC-Americas 39
ICH Regions - Regulatory Filings - EU
Submission expectation
A Summary of the Risk Assessment to be submitted.
Full documentation of Risk Assessment available at site.
What should the Summary look like?
Should follow the principles lined out in ICH Q3D
Contain what is needed to evaluate the appropriateness and
completeness of the elemental impurities Risk Assessment.
Tell a story to the assessor on what has been considered,
done and concluded.
Raw data not expected, but summary of findings may be
necessary.
The justification for the Control Strategy (what to control and
not to control).
Copyright 2017, All Rights Reserved, IPEC-Americas 40
ICH Regions - Regulatory Filings - EU
Where to be put in the dossier?
It is suggested: Summary of the Risk Assessment
3.2.P.5.5 Characterization of impurities (DP) (rather
than 3.2.P.2 Pharmaceutical development)
Depending on the outcome, data may also go into e.g.:
3.2.S.3.2 Impurities (DS)
3.2.S.4.5 Justification of specification (DS)
3.2.P.4 Control of Excipients
3.2.P.5.6 Justification of specification

Copyright 2017, All Rights Reserved, IPEC-Americas 41
Implementation Timelines for Japan
Japan MHLW recommendations and timelines:
For New Drugs – Implementation date: April 1, 2017.
For Marketed Products – Application of Q3D to
existing products approved before April 1, 2017 will
not be expected to 36 months (January 1, 2018) after
publication of the ICH guideline.
Official date on application to existing Marketed
products have not been announced.
The sponsor should study the feasibility of Q3D.
MHLW will evaluate applicability of Q3D to existing
products.
Copyright 2017, All Rights Reserved, IPEC-Americas 42
Developments in other Countries Australia TGA Q3D applies to registration
applications for prescription medicines.
The date for coming into effect aligns with implementation in the EU. New products containing new drug substance/s:
from June 2016.
New products containing existing drug substance/s: from December 2017.
No official statement for current marketed product.

Copyright 2017, All Rights Reserved, IPEC-Americas 43
Developments in other Countries Health Canada
Document for new
application/supplement Implementation date
Submission of a new abbreviated new
drug submission (ANDS) or drug
identification number (DIN) application
for a drug product should include the
content requirements as per Q3D.
Submissions received after
December 31, 2016.
Submission of a new Supplemental
(A)NDS or Post-DIN Change for a major
change to an existing Drug Product as a
result of the risk assessment per Q3D.
Submissions received after
December 31, 2016.
Copyright 2017, All Rights Reserved, IPEC-Americas 44
Developments in other Countries Health Canada
Document for marketed drugs Implementation date
Completion of the risk assessment for
elemental impurities. January 1, 2018.
Implementation of any manufacturing
changes to control the levels of elemental
impurities.
January 1, 2018.
Updated drug product specifications with a
statement confirming compliance with ICH
Q3D.
January 1, 2018.

Copyright 2017, All Rights Reserved, IPEC-Americas 45
Developments in other Countries Health Canada
OTC should comply with Q3D; natural health products
excluded
The risk assessment should be documented and
available for inspection
The locations where the elemental impurities-related
information can be placed for new submissions
Summarized in Module 2.3.P.5: Control of Drug Product of the
Quality Overall Summary, QOS.
Module 3.2.P.5.6 Justification of Specifications.
Risk assessment for the container closure system may be cross-
referenced to a master file.
Copyright 2017, All Rights Reserved, IPEC-Americas 46
Developments in other Countries Health Canada
The locations where the elemental impurities-related information can be placed for currently marketed products.
Quality Overall Summary Module 2.3.P.5: Control of Drug Product: a summary of the Module 3 locations where the elemental impurities-related information can be found.
Module 3.2.P.5.6 Justification of Specifications includes the overall risk assessment summary for elemental impurities.
Appropriate data to support any changes made to comply with Q3D or Canadian changes guidance.

Copyright 2017, All Rights Reserved, IPEC-Americas 47
Developments in other Countries Swissmedic
For New Drugs or Existing drugs.
Both new drug substances and existing drug
substances apply Implementation date: July 1, 2016.
For Marketed products
Implementation date: January 1, 2018.
Complied with Ph. Eur. Supplement 9.3.
During the transition period companies should
perform a risk assessment covering all potential
sources of elemental impurities.
Copyright 2017, All Rights Reserved, IPEC-Americas 48
Developments in other Countries Taiwan FDA
For New Drugs
Both new drug substances and existing drug
substances apply.
Implementation date: July 1, 2016.
For Marketed Products
Implementation date is not published.
Guiding principle would be:
Perform risk-based assessment according to Q3D Guideline.
Reduce regulatory reviewing burden without oversight.

Copyright 2017, All Rights Reserved, IPEC-Americas 49
Developments in other Countries
China, India, or South American Countries have not indicated the implementation of
ICH Q3D or any timelines for implementation at this time.
China may start discussions on ICH Q3D in 2020.
Copyright 2017, All Rights Reserved, IPEC-Americas 50
Summary on Implementation of ICH Q3D
New drugs (new drug substances and existing
drug substances).
The Health Authorities in US, EU, Japan, Canada,
Switzerland, Australia and Taiwan announced the
Q3D implementation schedule.
Currently marketed products.
The Health Authorities in US, EU, Canada and
Switzerland announced the Q3D implementation
schedule.
Risk assessment is a must
Australia, Japan and Taiwan are still in the planning
stage.

Copyright 2017, All Rights Reserved, IPEC-Americas 51
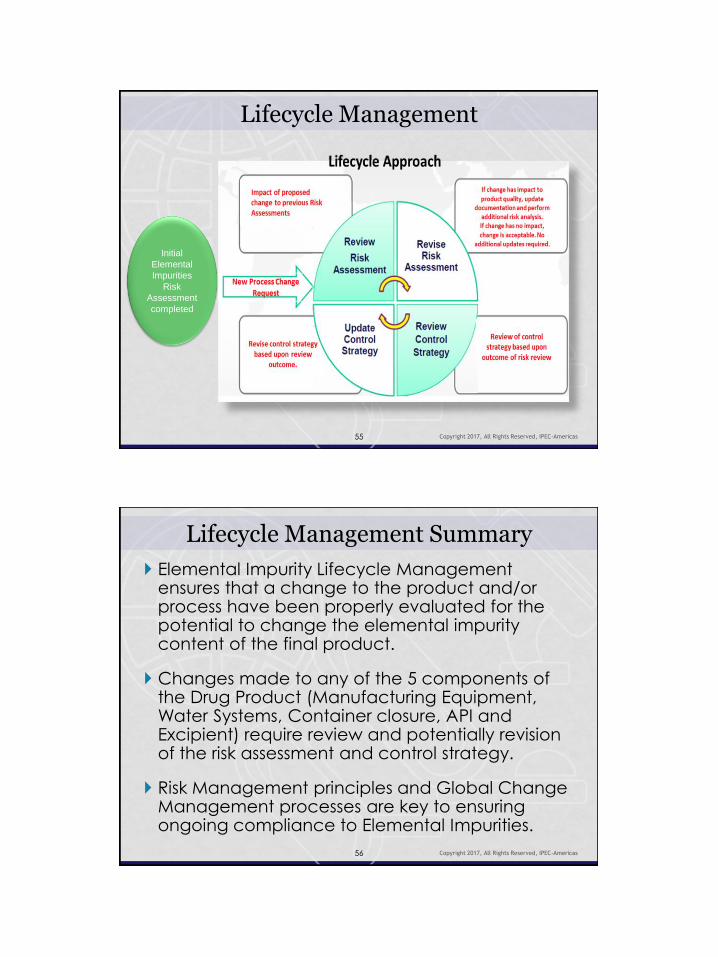
Lifecycle Management
The Lifecycle approach is to ensure ongoing compliance to Elemental Impurity requirements.
It is defined as an understanding of the product and/or process changes which have the potential to change the Elemental Impurity content of the final product and therefore may require a re-evaluation of the overall risk assessment and control strategy to ensure ongoing compliance to Elemental Impurity requirements.
Copyright 2017, All Rights Reserved, IPEC-Americas 52
Lifecycle Management Essential to Lifecycle management is an
understanding of how changes in the product/process and/or components may impact the Elemental Impurity risk assessment and the need for a control strategy on the drug product.
Changes Potentially Impacting Elemental Impurities:
Risk Assessment Fishbone Diagram
Elemental
Impurities in
Drug Product
Drug
Substance Excipients
Manufacturing
Equipment
Utilities (e.g.,
Water)
Container
Closure
System

Copyright 2017, All Rights Reserved, IPEC-Americas 53
Lifecycle Management
Lifecycle Approach
Copyright 2017, All Rights Reserved, IPEC-Americas 54
Lifecycle Management
Possible Changes to a Drug Product
Examples of changes which may lead to reviews of
the original risk assessment and control strategy are:
Synthetic routes
Excipient suppliers / changes to the source or
manufacturing process of the excipient
API suppliers
Materials
Processing equipment
Container closure
Water supply
Copyright 2017, All Rights Reserved, IPEC-Americas
Lifecycle Management
55
Initial
Elemental
Impurities
Risk
Assessment
completed
Copyright 2017, All Rights Reserved, IPEC-Americas 56
Lifecycle Management Summary
Elemental Impurity Lifecycle Management ensures that a change to the product and/or process have been properly evaluated for the potential to change the elemental impurity content of the final product.
Changes made to any of the 5 components of the Drug Product (Manufacturing Equipment, Water Systems, Container closure, API and Excipient) require review and potentially revision of the risk assessment and control strategy.
Risk Management principles and Global Change Management processes are key to ensuring ongoing compliance to Elemental Impurities.

Copyright 2017, All Rights Reserved, IPEC-Americas
Analytical and Risk Assessment Challenges
57
Methodology Concerns
Collaborative study performed
PQRI Phase 2 testing initiated
Lhasa database
Supplier information (API and Excipient)
Risk assessment approaches
Copyright 2017, All Rights Reserved, IPEC-Americas 58
Methodology Concerns
Analytical Testing Considerations:
Difficult sample matrices— implications for sample preparation and subsequent analysis.
Fundamentals of ICP-MS with respect to solving problems for pharmaceutical samples.
Contract lab perspective on common misconceptions and practical aspects of sample analysis and validation.
API and excipient supplier perspectives demonstrating risk assessment principles.
Alternative methods—WD-XRF as a complimentary technique to ICP-MS.

Copyright 2017, All Rights Reserved, IPEC-Americas 59
Methodology Concerns
Risk assessment requires some basis in data.
Key question for industry and the regulatory community. How reliably can we measure elemental impurities in drug
products, APIs and excipients at the levels outlined in ICH Q3D and USP <232>/<233>?
Variety and complexity of pharmaceutical samples.
Many labs expanding capabilities. Pharmaceutical labs adapting to ICP-MS analysis
Existing spectroscopy labs adapting to the requirements of <233>
Technical/Analytical Challenges Project Team formed in 2013 as a sub-team of the Coalition for Rational Implementation – now working with PQRI.
TECHNICAL/ANALYTICAL CHALLENGES PROJECT TEAM
Team Chartered June 2013
Membership (42+ colleagues)
• Comprised of scientists from – Coalition companies
• 5 pharmaceutical companies
• 7 raw material suppliers (API/excipient)
– 8 Contract laboratories
– 1 Government laboratory
– 1 University laboratory

Examples of Key Challenges
• Sample Preparation
– Ensure appropriate and effective solution preparation
– Total metal extraction implies clear solutions
• Instrumental Analysis
– System suitability/data integrity
– Options for sample introduction and interference reduction
• Sample introduction accessories, reaction gasses & collision cells, correction equations, etc.
– Calibration & LOQs
• LOQ can be a concern for large dose products and for raw material analysis
• Data review & interpretation
– Recognition of issues
• Drift, carryover/memory, interferences or element-specific pitfalls, non-ideal recoveries
– Reportable data
• Multiple modes of analysis possible
• No pharmaceutically relevant reference materials available
61
Inter-laboratory Study Objectives
• Address some of the key technical challenges faced by industry in preparation for compliance to ICH Q3D and USP <232>/<233>
• Provide a data-driven way to discuss technical aspects and expected variation of ICP-MS analysis of elemental impurities
• More specific objectives:
– Inter-laboratory data comparison for standardized samples
– Inter-laboratory evaluation of effectiveness of microwave digestion
– Comparison of acid leach/extraction techniques to total metal extraction
– Examination of the correlation (good or bad) between the analysis of individual components (summation) vs. the formulated tablet analysis
– Comparison of ICP-MS and alternative techniques (ICP-OES and XRF)
• First round study reported preliminary results at PQRI 2015, final results at AAPS 2015, and XRF arm at PQRI 2016
• Second round benefits from PQRI Sponsorship—allows wider participation
& Study Administrator—RTI International
62

Second Round Design Improvements & Best Practices
General
• Consistency among alternative techniques and digestion methods to ensure adequate data for comparison
• ICP-OES (14) and XRF (6) analysis considered proactively
• Raw materials distributed widely for summation approach comparison
Uniform Sample Preparation
• Specify parameters such as sample size, sampling technique, acid mixtures, and digestion temperature/pressure
• Document type of digestion vessels and microwave model used
Uniform Analysis
• Define procedures around LOQs, calibration, and data reporting
• Document interference management (reaction/collision gases, correction equations, etc.) and internal standards
63
Evaluation Samples and Analysis
64
Second Round Improvements— Evaluation
Samples
• Liquid sample to assess instrumental variation
• Evaluation samples with higher EI levels overall
• Multiple powdered or tableted evaluation samples targeting different levels
• EI source from pharma materials wherever possible
Powdered or tableted material at three concentrations and liquid
sample are aliquoted and shipped to each participating laboratory.
Samples extracted in triplicate using the
uniform method. Alternate extraction
methods may be used if available in
addition to the uniform method.
Uniform method
sample extracts/liquid
sample must be
analyzed by ICP-MS.
Optional alternate
method sample
extracts are analyzed
by ICP-MS.
Replicate results and average/SD reported by extraction method and
analysis method. Total samples for ICP-MS analysis are 12, alternative
digestion and analysis techniques will be in addition to basic results.
Minimum samples: 12, maximum: 36.
Uniform method
sample extracts, liquid
may be analyzed by
ICP-OES or other
instrument.
Samples extracted in
triplicate using the uniform
acid leaching method.
Samples extracted by
acid leaching method
must be analyzed by
ICP-MS.
Results for replicates and
average/SD are reported. Total
samples for acid leach analysis
are 12.

Recruiting
• Distributed participant questionnaire for analytical laboratories in early August 2016.
• 29 laboratories enrolled
• Pharma manufacturers: 16 laboratories
• Contract/CRO: 9 laboratories
• Instrument manufacturers: 2 laboratories
• Government: 2 laboratories
65
1 lab
11 labs
17
labs
Next Steps
• Tablet and liquid sample production – Partnering with Liverpool John Moore’s University
• Uniform method and acid leach method development—critical step – To be developed with finalized tablets prior to distribution
• Package assembly & shipment
• Sample analysis during summer months
• Results readout at PQRI in November
66

Copyright 2017, All Rights Reserved, IPEC-Americas 67
Lhasa Database
Database Development: Database created by EI Pharma Consortium to gather a
critical mass of data for excipients.
Aid in risk assessment and overall understanding of excipient risks.
Intent to publish key findings to de-risk common excipients.
Current membership mainly from big pharma in partnership with Lhasa.
Contact: Crina Heghes ([email protected])
Appropriate Use of Published Data – Data will be blinded so it will be important to establish a scientifically sound bridge from this data to the grades and suppliers actually used in the drug product – cannot simply use the data in your risk assessment!
Copyright 2017, All Rights Reserved, IPEC-Americas 68
Supplier Information (API and Excipient)
There is NO regulatory requirement for API or Excipient suppliers to perform or supply elemental impurity information to pharmaceutical users.
Some suppliers will want to provide some useful information to support their customers but this will depend on the amount of business done in the pharmaceutical market.
The level of information provided will typically vary quite a bit from supplier to supplier.
Users should not try to pressure suppliers for EI data that may not exist, otherwise supplier may leave the market and availability will then be a problem.
Requests should be for whatever information the supplier is willing to share about their products and processes but not focus on the development of specifications since many suppliers will not agree to specifications on EI.
IPEC-Americas developed an information sharing mechanism which is well used throughout the industry.

Sharing Information between Makers & Users IPEC Template Information Exchange Request
IDEAL WORLD….
Pro-actively completed by suppliers and sent to
users
download THE TEMPLATE
URGENT industry need for BASE-LINE DATA!
REAL WORLD…
Few suppliers have data or will complete and
return the form to users
download THE LETTER
PDE Calculator also available on IPEC-Americas website
to assist in Risk Assessment
Copyright 2017, All Rights Reserved, IPEC-Americas 70
Risk Assessment Approaches
ICH Q3D is a global policy for limiting elemental impurities in drug products.
Harmonised, safety-based limits for elemental impurities, especially those of highest toxicological concern.
Selection of elements to control.
Methodology for establishing safety-based limits.
Permitted daily exposure for specific elements.
Appropriate risk-based approach to ensure control for elements likely to be present in drug products and ingredients.

Copyright 2017, All Rights Reserved, IPEC-Americas 71
Risk Assessment Approaches
ICH Q3D guideline has different options (1, 2 and 3).
Option 1, & 2 are Component Approach
Option 3 Finished Product Approach
Regulatory authorities accept any of these options.
It may be easier for Industry to use Component
option to set control strategies per component.
Copyright 2017, All Rights Reserved, IPEC-Americas 72
Risk Assessment Approaches
Potential considerations during review of Risk
Assessments:
Intentionally added elements
Contributions from raw materials derived from plant or marine origins.
Contributions from raw materials that are mined, e.g.,
Inorganic drug substances and excipients.
Contributions from manufacturing, e.g., high shear micronization using metal discs.
Leachable elemental impurities from container/closure.
Extractables information from container/closure components typically included in a supplier Type III DMF.

Copyright 2017, All Rights Reserved, IPEC-Americas 73
Risk Assessment Reports
The Risk Assessment Reports tell the story of the risk assessment thinking / process.
Risk Assessment Reports are kept at the company manufacturing sites.
Risk Assessment Reports may be requested by regulatory agency inspectors.
Complete Risk assessment reports are not sent to
the FDA or EMA.
FDA updated through Annual Reports process
containing a summary of the risk assessment.
Copyright 2017, All Rights Reserved, IPEC-Americas 74
Risk Assessment Reports
Title page
Generic Name
Introduction
State purpose of report - ICH Q3D risk assessment.
Formulation
The manufacturing formulation for the finished product.
If report will cover different strengths of the same product then each formulation should be included.
Dosing information from label
List the range of doses. Identify the maximum possible dosing per day (This is needed to determine the PDEs).

Copyright 2017, All Rights Reserved, IPEC-Americas 75
Risk Assessment Reports
Risk Assessment section (Major portion of the report is based on the fish bone diagram in the ICH guidance).
Include the fish bone diagram with the elements
/sections.
Manufacturing Equipment Brief Summary of the
manufacturing steps
Process flow diagram
Overview of summary of worksheets
Conclusion
If necessary, what testing or other control measures will be
put in place
Elemental
Impurities in
Drug Product
Drug
Substance Excipients
Manufacturing
Equipment
Utilities (e.g.,
Water)
Container
Closure
System
Copyright 2017, All Rights Reserved, IPEC-Americas 76
Risk Assessment Reports
Water system Statement of grade(s) of water used.
Source of water
Overview of summary of worksheets
Conclusion
If necessary, what testing or other control measures will be
put in place
Container closure Summary description of the
container closure system including
materials of construction
Overview of summary of worksheets
Conclusion
If necessary, what testing or other control measures will be
put in place
Elemental
Impurities in
Drug Product
Drug
Substance Excipients
Manufacturing
Equipment
Utilities
(e.g., Water)
Container
Closure
System
Elemental
Impurities in
Drug Product
Drug
Substance Excipients
Manufacturing
Equipment
Utilities
(e.g., Water)
Container
Closure
System

Copyright 2017, All Rights Reserved, IPEC-Americas 77
Risk Assessment Reports API
Brief Summary of the manufacturing
steps
Overview of summary of worksheets
Conclusion
If necessary, what testing or other control measures will be
put in place
Excipient
Summary of the excipients used in
the formulation including suppliers
Overview of summary of worksheets
Conclusion
If necessary, what testing or other control measures will be
put in place.
Elemental
Impurities in
Drug Product
Drug
Substance Excipients
Manufacturing
Equipment
Utilities
(e.g., Water)
Container
Closure
System
Elemental
Impurities in
Drug Product
Drug
Substance Excipients
Manufacturing
Equipment
Utilities
(e.g., Water)
Container
Closure
System
Copyright 2017, All Rights Reserved, IPEC-Americas 78
Risk Assessment Reports
Excipient and API - ICP/MS Procedure –
include only if data generated Method (Sample preparation and analysis)
Statement on validation including description of the
validation.
Summary of Patient exposure risk compared to PDEs.
Identify any areas of risk and the sources.
Summary/ Conclusion.
Discussion if additional controls are needed.

Copyright 2017, All Rights Reserved, IPEC-Americas
QUESTIONS
79
Copyright 2017, All Rights Reserved, IPEC-Americas
CASE STUDIES
80

Copyright 2017, All Rights Reserved, IPEC-Americas 81
Case Studies – ICH Modules
Solid Oral Dosage – Initial Information
Greatstuff Tablets is a new drug product for the treatment of hypertension.
It is film coated and available in 2 strengths 50 & 100 mg.
Intended to be dosed once per day with a maximum daily dose of 100 mg.
The drug product is packaged in individual blister packages (PVC-91 Aclar) and HDPE bottles.
Samples tested were from Manufacturing and Development sites:
Drug substance development: Anywhere, Kansas
Drug product development: Anywhere, Kansas
Drug substance and drug product manufacturing: Humacao, Puerto Rico
Copyright 2017, All Rights Reserved, IPEC-Americas 82
Case Studies – ICH Modules
See ICH Q3D Case Study 1A for discussion
Handout of Module ICH Q3D Case Study 1A

Copyright 2017, All Rights Reserved, IPEC-Americas 83
ICH Q3D Elemental Impurities
Thank You
Pb As Hg
Cd Co V
Ni ???
Related Documents











