Hzf, a key modulator of p53 mediated transcription, functions as a critical determinant of cell survival and death upon genotoxic stress Sanjeev Das 1 , Lakshmi Raj 1 , Bo Zhao 2 , Alan Bernstein 3 , Stuart A. Aaronson 2 , and Sam W. Lee 1,* 1 Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, USA 2 Department of Oncological Sciences, Mount Sinai School of Medicine, New York, New York 10029, USA 3 Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, ON, Canada M5G IX5 Summary A critical unresolved issue about the DNA damage/genotoxic stress response is how the resulting activation of the p53 tumor suppressor can lead either to cell cycle arrest and DNA repair, or to apoptosis. We show here that Hematopoietic Zinc Finger (Hzf), a zinc finger containing p53 target gene, modulates p53 transactivation functions in an autoregulatory feedback loop. Hzf is induced by p53 and binds to its DNA binding domain, resulting in preferential transactivation of pro-arrest p53 target genes over its pro-apoptotic target genes. Thus p53 activation results in cell cycle arrest in Hzf wt-MEFs while in Hzf −/− MEFs apoptosis is induced. Additionally, prolonged exposure to stress results in Hzf degradation concomitant with induction of apoptosis. Exposure of Hzf-null mice to IR resulted in enhanced apoptosis in several organs including skin and prostate, as compared to that of wt-mice. These findings provide novel insights into the regulation of p53 transactivation function that plays an important role in cell fate decisions in response to genotoxic stress. Introduction p53 is an important component of pathways mediating cellular response to genotoxic stress by inducing the transcription of a variety of genes that regulate diverse cellular processes including cell cycle progression, apoptosis and genomic stability (Harris and Levine, 2005; Vogelstein et al., 2000; Vousden and Lu, 2002). However little is known about the mechanism(s) that determines which sets of target genes i.e. cell cycle arrest genes like p21 (El-Deiry et al., 1993), 14-3-3σ (Hermeking et al., 1997) or pro-apoptotic genes such as Bax (Miyashita and Reed, 1995), Noxa (Oda et al., 2000; Villunger et al., 2003), Pidd (Lin et al., 2000), Puma (Nakano and Vousden, 2001; Villunger et al., 2003), Perp (Attardi et al., 2000) etc. are transactivated by p53 under a specific condition. p53 is a transcription factor that plays a central role in cellular responses to genotoxic stress like DNA damage, hypoxia, oncogene activation etc (Harris and Levine, 2005; Laptenko and Prives, 2006). In order to perform its cellular *Correspondence should be addressed to S.L. ([email protected]). Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. NIH Public Access Author Manuscript Cell. Author manuscript; available in PMC 2009 November 19. Published in final edited form as: Cell. 2007 August 24; 130(4): 624–637. doi:10.1016/j.cell.2007.06.013. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Hzf, a key modulator of p53 mediated transcription, functions asa critical determinant of cell survival and death upon genotoxicstress
Sanjeev Das1, Lakshmi Raj1, Bo Zhao2, Alan Bernstein3, Stuart A. Aaronson2, and Sam W.Lee1,*1 Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard MedicalSchool, Charlestown, Massachusetts 02129, USA2 Department of Oncological Sciences, Mount Sinai School of Medicine, New York, New York 10029,USA3 Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, ON, Canada M5G IX5
SummaryA critical unresolved issue about the DNA damage/genotoxic stress response is how the resultingactivation of the p53 tumor suppressor can lead either to cell cycle arrest and DNA repair, or toapoptosis. We show here that Hematopoietic Zinc Finger (Hzf), a zinc finger containing p53 targetgene, modulates p53 transactivation functions in an autoregulatory feedback loop. Hzf is induced byp53 and binds to its DNA binding domain, resulting in preferential transactivation of pro-arrest p53target genes over its pro-apoptotic target genes. Thus p53 activation results in cell cycle arrest in Hzfwt-MEFs while in Hzf−/− MEFs apoptosis is induced. Additionally, prolonged exposure to stressresults in Hzf degradation concomitant with induction of apoptosis. Exposure of Hzf-null mice to IRresulted in enhanced apoptosis in several organs including skin and prostate, as compared to that ofwt-mice. These findings provide novel insights into the regulation of p53 transactivation functionthat plays an important role in cell fate decisions in response to genotoxic stress.
Introductionp53 is an important component of pathways mediating cellular response to genotoxic stress byinducing the transcription of a variety of genes that regulate diverse cellular processes includingcell cycle progression, apoptosis and genomic stability (Harris and Levine, 2005; Vogelsteinet al., 2000; Vousden and Lu, 2002). However little is known about the mechanism(s) thatdetermines which sets of target genes i.e. cell cycle arrest genes like p21 (El-Deiry et al.,1993), 14-3-3σ (Hermeking et al., 1997) or pro-apoptotic genes such as Bax (Miyashita andReed, 1995), Noxa (Oda et al., 2000; Villunger et al., 2003), Pidd (Lin et al., 2000), Puma(Nakano and Vousden, 2001; Villunger et al., 2003), Perp (Attardi et al., 2000) etc. aretransactivated by p53 under a specific condition. p53 is a transcription factor that plays a centralrole in cellular responses to genotoxic stress like DNA damage, hypoxia, oncogene activationetc (Harris and Levine, 2005; Laptenko and Prives, 2006). In order to perform its cellular
*Correspondence should be addressed to S.L. ([email protected]).Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customerswe are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resultingproof before it is published in its final citable form. Please note that during the production process errors may be discovered which couldaffect the content, and all legal disclaimers that apply to the journal pertain.
NIH Public AccessAuthor ManuscriptCell. Author manuscript; available in PMC 2009 November 19.
Published in final edited form as:Cell. 2007 August 24; 130(4): 624–637. doi:10.1016/j.cell.2007.06.013.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

functions p53 must rapidly accumulate in response to these stressful conditions, as its basallevel is very low. Activation of p53 has two major outcomes: cell cycle arrest or apoptosis.Cell cycle arrest allows DNA repair to take place before replication occurs thereby maintaininggenomic integrity. On the other hand apoptosis results in elimination of irreparably damagedcells.
The regulation of p53 is usually achieved by post-translational modifications and through itsinteractions with various other proteins (Lavin and Gueven, 2006). p53 undergoesphosphorylations on numerous serine residues both in N-and C-terminal regions (Lavin andGueven, 2006). The N-terminal phosphorylations inhibit its interactions with its negativeregulator MDM2 (Canman et al., 1998; Chehab et al., 2000; Khosravi et al., 1999) while theC-terminal phosphorylations are thought to enhance the sequence specific DNA binding abilityof p53 by inducing a conformational change (Hupp et al., 1992; Wang and Prives, 1995).Similarly, other modifications like ubiquitination, acetylation, and sumolation also affect itsproteolytic turnover and sequence specific DNA binding ability (Brooks and Gu, 2006;Rodriguez et al., 1999). This can also be achieved by its interaction with cellular proteins suchas Pin-1, ASPP family etc (Braithwaite et al., 2006). When Pin-1 binds to p53, it undergoesconformational change which enhances its transactivation ability (Zacchi et al., 2002; Zhenget al., 2002). Recently, a new family of proteins, known as ASPPs, were found to be potentactivators of p53, providing an important insight into how p53 responds to apoptotic signals(Trigiante and Lu, 2006). The ASPP family consists of three members –ASPP1, ASPP2, andiASPP. ASPP1 and ASPP2 interact with p53 and specifically enhance p53-induced apoptosisbut not cell cycle arrest while iASPP binds and inhibits p53-mediated apoptosis (Bergamaschiet al., 2006; Samuels-Lev et al., 2001).
While studying the genome-wide transcriptional response to p53 induction we found that oneof the genes upregulated was the hematopoietic zinc finger gene (Hzf). Hzf was originallyidentified as a gene induced in hematopoietic progenitor cells derived from differentiatingembryonic stem cells (Hidaka et al., 2000). It encodes a zinc finger protein of 366 amino acids.It has three C2H2-type zinc finger domains. The zinc finger domains in Hzf are widely spacedwith long linker regions connecting the fingers as a consequence of which it cannot form anystable nucleic acid-protein complex (Sharma et al., 2004). Recently, it was reported that Hzfis a direct transcriptional target of p53, which plays a role in p53-mediated cell cycle arrest inresponse to DNA damage in NIH 3T3 cells (Sugimoto et al., 2006). We found that Hzf is uniqueamong the different p53 transcriptional targets in that upon induction by p53 or DNA damage,it binds to the p53 DNA binding domain and modulates its transactivation function in anautoregulatory feedback loop. We further investigated the role of Hzf in the p53-mediatedDNA damage response. Here, we show that when Hzf binds to p53, it is preferentially recruitedto the promoters of its pro-cell cycle arrest target genes rather than its pro-apoptotic targetgenes. Thus, in presence of Hzf, p53-mediated cell cycle arrest is promoted while apoptosis isrepressed in response to genotoxic stress.
ResultsInhibition of Hzf induction in response to DNA damage represses p21 expression butenhances Bax levels
Through microarray analysis of cDNA expression, we identified Hzf as one of the upregulatedgenes whose transcript levels were elevated in response to p53 induction (Han et al., 2002).To confirm the array result, we carried out several Northern and Western blot analyses toexamine Hzf expression in response to different types of stresses in several cell lines of diversetissue origin and different p53 background. Under various stress conditions including DNAdamage and oxidative stress, Hzf was induced in a p53-dependent manner (Figure 1A).
Das et al. Page 2
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Moreover, promoter analysis including the reporter gene assay and ChIP analysis supportedthat Hzf is a direct p53 transcriptional target (Figure S1).
To investigate the role of Hzf in p53-mediated stress response, we examined whether Hzfinduction contributes directly to the ability of p53 to transactivate p21 and Bax. To do so, weused shRNA against Hzf to knockdown the induction of endogenous Hzf in response to p53-dependent DNA damage response or ectopic p53 expression. We transiently transfected U2OS(wt p53) and EJ (nonfunctional p53) cell lines with Hzf shRNA or scrambled shRNA constructsfollowed by treatment with chemotherapeutic agent etoposide or infection with Ad-p53 toinduce p53 expression (Figure 1B). Transfection of Hzf shRNA resulted in the suppression ofendogenous Hzf induction by etoposide treatment or Ad-p53 infection, while control scrambledshRNA had no effect on induced Hzf levels. In U2OS cells transfected with the scrambledshRNA construct, upon etoposide treatment or Ad-p53 infection, concomitant with p53induction, Hzf and p21 induction was seen while Bax levels was minimally increased.However, depletion of Hzf expression resulted in enhanced Bax induction while p21 inductionwas significantly reduced in response to DNA damage (Figure 1B, left panel). In EJ cellscontaining nonfunctional p53, similar results were obtained with Ad-p53 infection (Figure 1B,right panel). To exclude off-target effects of shRNA, two other Hzf shRNAs were used, andsimilar results were obtained (data not shown).
We next examined the levels of p21 and Bax transcripts in response to DNA damage (etoposidetreatment) in Hzf+/+ and Hzf−/− MEFs. As shown Figure 1C, Hzf−/− MEFs showed drasticallylower the induction levels of p21 mRNA than Hzf+/+ MEFs, while Bax induction wassignificantly enhanced in Hzf−/− MEFs. To further investigate the role of Hzf in p53-mediatedtranscription of these target genes, we used reporter assays with either the p21 or Bax promoterfused to a luciferase reporter (pGL3-basic, p21-Luc, and Bax-Luc). U2OS and EJ cells wereco-transfected with Hzf shRNA or scrambled shRNA constructs together with pGL3 basic,p21-Luc or Bax-Luc constructs followed by treatment with a DNA damaging agent etoposideor infection with Ad-p53 (Figure 1D). We found that the ability of p53 to transactivate the p21promoter was severely compromised upon the knockdown of Hzf while its ability totransactivate the Bax promoter was significantly enhanced in response to etoposide treatmentor Ad-p53 infection in U2OS cells containing wt-p53 (Figure 1D, upper panel). In the EJ cells,this effect was also observed upon ectopic p53 expression (Figure 1D, lower panel). All ofthese results suggested that Hzf affected p53-mediated transcription in a positive way for p21transcription and in a negative way for Bax transcription.
Hzf is induced by p53 and binds directly to its DNA binding domainSince Hzf was identified as a novel zinc finger protein (Hidaka et al., 2000), is induced by p53and modulates p53 mediated transactivation, we next tested whether Hzf physically interactswith p53. For this purpose, we used extracts made from U2OS cells treated with etoposide orEJ cells infected with Ad-p53 for immunoprecipitation using the p53-specific monoclonalantibody (PAb1801). In both cell lines we found that Hzf co-immunoprecipitated with p53(Figure 2A, top panels). We also performed the reverse co-IP experiment whereimmunoprecipitation was carried out using anti-Hzf polyclonal antibody (Figure 2A, bottompanels). As shown in Figure 2A, p53 also co-immunoprecipitated with Hzf. As a control an IPwas carried out in etoposide treated Hzf−/− MEF using a Hzf antibody. As shown in Figure S2,p53 was not in the precipitated complex, and no Hzf was detected, confirming the specificityof the Hzf antibody used.
To determine whether the interaction between Hzf and p53 was a direct one, a yeast two hybridassay was performed. Expression constructs in which p53 was fused to the Gal4 DNA bindingdomain (pGBKT7-p53), and Hzf was fused to Gal4 transactivation domain to generatepGADT7-Hzf, and then transformed into yeast strains AH109 and Y187, respectively. Blue
Das et al. Page 3
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

colored Ade+/His+ colonies were obtained when yeast transformants containing the pGBKT7-p53 and pGADT7-Hzf were mated, as observed in a positive control mating experimentbetween p53 (pGBKT7-p53) and SV40 T antigen (pGADT7-T Ag) (Figure 2B) (Iwabuchi etal., 1993; Li and Fields, 1993).
To map the domain of p53 to which Hzf bound, we carried out GST-pull-down experimentswhere constructs expressing different domains of p53 fused to GST were transfected into EJcells followed by infection with control Ad-GFP or Ad-Hzf. As shown in Figure 2C, Hzf boundto GST-p53 (full length) but not to GST alone supporting our immunoprecipitation and yeasttwo hybrid assay results. The GST-p53 segment containing aa 100–300 specifically bound tothe Hzf protein, which represents the DNA binding domain of p53 (Figure 2C). To study theHzf-p53 interaction more closely, we determined whether Hzf could bind to tumor-derivedp53 DNA binding domain mutants including p53/175Pro, p53/175His and p53/143Ala. It wasreported that p53/175His and p53/143Ala had no DNA binding ability while p53/175Pro wascapable of some DNA binding activity but could trigger only arrest but not apoptosis (Kern etal., 1991; Rowan et al., 1996; Zhang et al., 1994). As shown in Figure 2D, Hzf bound to wt-p53 robustly but reduced binding was observed with p53/175Pro and p53/175His. The bindingof Hzf with p53/143Ala was severely repressed. Nevertheless, none of these p53 mutationswas able to abolish the Hzf-p53 interaction completely. This suggests that p53-Hzf interactioninvolves several key residues in DNA binding domain of p53 and mutations in any one of themweaken the binding but cannot fully abolish it. Together, these results indicate that Hzf isinduced by p53 and directly binds to the DNA binding domain of p53.
Hzf influences the specificity of p53-mediated transactivationSince our results demonstrated that Hzf binds to p53 and differentially modulates expressionlevels of p53 targets p21 and Bax, we next determined whether this regulation of p53transactivation function by Hzf is a wide-ranging phenomenon encompassing other p53 pro-arrest and pro-apoptotic targets. Thus we infected Hzf+/+ and Hzf−/− MEFs with adenovirusexpressing p53 (Ad-p53) or treated the cells with etoposide, and analyzed the expression ofp53 target genes including both pro-arrest and pro-apoptotic targets. In the presence of Hzf,ectopic p53 or etoposide treatment preferentially induced expression of pro-arrest p53 targetsp21 and 14-3-3σ, whereas in the absence of Hzf, the expression of pro-apoptotic targets Bax,Perp, Puma and Noxa was selectively induced (Figure 3A and 3B). Mdm2 is a p53 targetinvolved in p53 protein regulation but does not specifically influence neither arrest norapoptosis, Mdm2 levels were not affected by Hzf inactivation (Figure 3A and 3B). In addition,exogenous expression of Hzf in Hzf−/− MEFs restored preferential induction of cell cycle arresttargets p21 and 14-3-3σ and lowered the levels of Perp, Puma, Noxa and Bax expression, butcaused no change seen in p53 induction of Mdm2 (Figure 3C).
To further evaluate the effect of Hzf on the transactivation function of p53, we measured DNAbinding activity of p53 in response to DNA damage on the promoters of p53 targets includingp21, 14-3-3σ, Bax, Noxa, Perp and Mdm2 in Hzf+/+ and Hzf−/−MEFs by using chromatinimmunoprecipitation assay (ChIP). Hzf+/+ and Hzf−/− MEFs were exposed to a DNA damagingagent, etoposide, for 24 hours, and ChIP assays were performed using control IgG or p53antibody for several of p53 target genes. Upon DNA damage treatment of Hzf+/+ MEFs p53predominantly bound to the promoter of pro-arrest targets such as p21 and 14-3-3σ but DNAbinding activity of p53 on the promoters of pro-apoptotic target genes such as Bax, Perp andNoxa was significantly lower (Figure 3D). In contrast, in Hzf-null MEFs upon DNA damageDNA binding activity of p53 was significantly enhanced for the promoters of its pro-apoptotictarget genes such as Bax, Perp and Noxa, as compared to that of p21 and 14-3-3σ (Figure 3D).The binding ability of p53 to the Mdm2 and Hzf promoter remained unchanged irrespectiveof Hzf status. Quantitative ChIP assays by real-time PCR also confirmed the above results with
Das et al. Page 4
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

wt- and Hzf−/− MEFs (Figure S3). These results strongly suggest that upon Hzf binding top53, p53 is preferentially recruited to the promoters of its cell cycle arrest mediating targetgenes. To further verify this, we performed “ReChIP” experiments using Hzf antibody. Asshown in Figure 3E, Hzf was detected only at the promoters of p21, 14-3-3σ Hzf and Mdm2but not at the promoters of Bax, Noxa and Perp. Moreover, quantitative ChIP assays by real-time PCR were carried out with etoposide treated p53+/+ and p53−/− MEFs using IgG controlantibody and Hzf antibody (Figure S4). Our results indicate that Hzf is at the cell cycle arrestpromoters only in presence of p53. To further strengthen the differential effects of Hzf bindingon the DNA binding activities of p53 to its targets, EMSA was carried out in etoposide treatedHzf+/+ and Hzf−/− MEFs using oligonucleotides specific for p53 binding site in p21 promoter(El-Deiry et al., 1993), mdm2 promoter (Wu et al., 1993) and Bax first intron (Thornborrowet al., 2002). As shown in Figure 3F, the DNA binding activity of p53 on p21 promoter wassignificantly increased in etoposide-treated wt-MEF, whereas there was no strong DNAbinding activity of p53 on Bax promoter. However, in the absence of Hzf no significant bindingactivity of p53 was observed on the p21 promoter but significantly higher DNA bindingactivities seen on the Bax first intron (Figure 3G). The binding of p53 to Mdm2 promoterremained unchanged irrespective of Hzf status. Taken together, these data demonstrate thatHzf preferentially enhances the DNA binding and transactivation functions of p53 on cell cyclearrest targets such as p21 and 14-3-3σ.
Hzf promotes the cell cycle arrest function of p53We next examined the effects of Hzf on p53-mediated cell cycle arrest or apoptosis inHzf+/+ and Hzf−/− MEFs. In Hzf+/+ MEFs, the ectopic expression of p53 by Ad-p53 infectioninduced prominent G1 arrest but little apoptosis (Figure 4). In Hzf−/− MEFs, the apoptoticfunction of p53 was significantly enhanced, as measured by TUNEL assay, sub-G1 populationand DNA fragmentation (Figures 4A and 4B). To further verify if indeed Hzf deficiencymodulates p53 functions under physiological conditions, Hzf+/+ and Hzf−/− MEFs wereexposed to etoposide, and then TUNEL positive apoptotic cells were measured. The loss ofHzf led to enhanced apoptosis sensitivity to etoposide (Figure 4C, left panel). Re-expressionof Hzf in Hzf −/− MEFs through Ad-Hzf infection reversed this effect of Hzf deficiency andresulted in a decrease in apoptosis induced by etoposide treatment or overexpression of p53similar to that seen in etoposide-treated or Ad-p53 infected Hzf+/+ MEF (Figure 4C) bymodulating p53-mediated transactivation of its target genes (Figure 3C) that was due to alteredDNA binding activity of p53 (Figure S5). These results indicate that Hzf modulates the p53mediated stress response favoring cell cycle arrest over apoptosis.
Prolonged p53 expression/activation or extended exposure to stress results in Hzf proteindownregulation/degradation leading to apoptosis
To examine whether levels of Hzf expression are correlated with levels of genotoxic stress, weinvestigated the kinetics of Hzf induction at extended time points during stress response inU2OS cells. The results shown in Figure 5A suggest that Hzf protein expression is rapidlyinduced in response to etoposide treatment or ectopic p53 up to 36 hours, but beyond 36 hoursHzf protein levels start declining and are markedly decreased at the 72 hours. p21 expressionalso followed a similar pattern to that observed with Hzf. In contrast, Bax protein levels rosevery slowly up to 36 hours but beyond that time exhibited a sharp increase (Figure 5A). Wealso immunoprecipitated p53 over the time courses of etoposide treatment to examine theamounts of Hzf bound to p53. We found that there was a sharp decline in the amounts of p53-bound Hzf beyond the 36-hour time point to the undetectable levels at 72 hours (Figure 5B).However, Hzf mRNA levels remained steady throughout the time course while mRNA levelsof p21 and Bax were similar to their protein levels (Figure 5C), suggesting that Hzf down-regulation is due to the degradation of the Hzf protein. To determine the implication for thesedifferential responses in Hzf protein levels in response to genotoxic stress or p53, we next
Das et al. Page 5
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

tested the cell cycle profile under these conditions by flow cytometry. Ad-p53 overexpressioninduced a pronounced arrest in G1 phase of the cell cycle at 24–36 hours (Figure 5D), afterwhich there was a marked increase in the apoptotic population. TUNEL staining confirmedthese results (Figure 5E). These results indicate that extended exposure to stress and theinduction of a p53-mediated apoptotic response correlated with a reduction of Hzf, suggestingthat Hzf may play a role in controlling the switch in the p53-dependent stress response.
Since ubiquitin-mediated proteasome degradation pathway is a frequently engaged mechanismfor protein downregulation, we investigated the potential role of the proteasome in stress-induced Hzf protein degradation in cells undergoing p53-dependent apoptotic response. Wetreated U2OS cells with etoposide for 0, 36 and 72 hours. At 10 hours prior to the end of the72-hour time point, the cells were treated with the proteasome inhibitor, MG132. Loss of Hzftriggered by genotoxic stress at 72 hours time point was significantly attenuated by theproteasome inhibitor MG132 resulting in elevated p21 levels (Figure 5F). However, Bax levelswere diminished similar to those at 36 hours in MG132 treated U2OS cells (Figure 5F).Furthermore, northern blot analysis showed that the proteosome inhibitor treatment elevatedthe levels of p21 mRNA, but decreased Bax mRNA (Figure 5G), supporting the hypothesisthat the preferential DNA binding activities of p53 to p21 promoter results from stabilizationof Hzf protein. This was confirmed by chromatin IP as well as quantitative ChIP assays byreal-time PCR (Figure 5H; Figure S6A). We next determined whether prolonged stress in cellstriggered the polyubiquitination of Hzf prior to Hzf degradation. Treatment of U2OS cells withetoposide in the presence of MG132 revealed that endogenous Hzf undergoes extensiveubiquitination after prolonged exposure (72 hours treatment) to genotoxic stress, while no suchubiquitinated forms were observed at 36 hours treatment (Figure 5I). Moreover, proteosomeinhibitor MG132-mediated stabilization of Hzf could indeed prevent the p53-mediatedapoptosis upon prolonged exposure to genotoxic stress (Figures S6B and S6C). Thus, theappearance of ubiquitinated forms of Hzf in cells treated with MG132 supports the idea thatthe Hzf protein is targeted for degradation during prolonged exposure to genotoxic stress dueto activation of an ubiquitination-proteasome pathway resulting in initiation of the apoptoticprocess.
Hzf−/− mice show increased sensitivity to γ-irradiationHzf-null mice have been generated and while mice lacking Hzf seemed to be relatively normal,initial characterization suggested a role for Hzf in megakaryopoiesis and hemostasis (Kimuraet al., 2002). To further study the role of Hzf in genotoxic stress response in vivo, weinvestigated the functional consequence of Hzf deficiency in Hzf-null mice exposed to γ-irradiation. Hzf−/− and wt-mice littermates (14 weeks old) were exposed to 5 Gy total body γ-irradiation. 6.5 hours after treatment, 3 mice from each group were evaluated by examiningsensitivity to cell death using TUNEL staining and histopathological analysis of target organsincluding skin, spleen, small intestine, and prostate. As shown in Figure 6A and B, skin andprostate from irradiated Hzf−/− mice showed increased sensitivity to DNA damage induced byγ-irradiation as measured by TUNEL staining. However, the extent of apoptosis in wt-prostatewas less than that in skin which was due to increased expression of Hzf in prostate as comparedto skin (Figure 6C). TUNEL staining and immunostaining for Hzf was also carried out in IR-sensitive tissues like spleen and small intestine epithelium, where we found extensive apoptosistaking place due to meager levels of Hzf expression upon γ-irradiation (Figure S7).Consequently, in spleen no difference could be seen in the level of apoptosis between Hzf+/+
and Hzf−/− samples in response to IR but in small intestine due to marginally increased levelsof Hzf expression there were slightly higher levels of apoptotis detected in Hzf−/− samples.Thus our results show that sensitivity to γ-irradiation is inversely correlated with the levels ofHzf expression in those tissues. We also performed immunostaining for p53 targets, p21 andBax, in the same sections of skin and prostate from wt- and Hzf−−-mice. In response to γ-
Das et al. Page 6
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

irradiation, the level of Bax expression was increased in Hzf−/− skin compared to that of wt-mice while p21 levels were reduced in γ-irradiated Hzf−/− skin samples (Figure 7A). Similarresults were obtained in prostate tissue from irradiated Hzf−/− mice (Figure 7B). In concurrencewith TUNEL staining, Bax staining in Hzf−/− prostate was also less pronounced as comparedto skin while the pattern of p21 staining was reversed. These results support the role of Hzffunctioning downstream of p53 in the p53-dependent DNA damage response acting as a criticalswitch that prevents apoptosis by repressing transactivation of pro-apoptotic p53 targets bothin vitro and in vivo. Thus, the results from the mouse model suggest that under in vivoconditions, Hzf modulates the p53 mediated stress response but there is an element of tissuespecificity in this regulation due to varying degree of Hzf expression in different tissues uponγ-irradiation.
DiscussionIt is widely accepted that the p53 tumor suppressor restricts the proliferation of cells exposedto genotoxic stress by induction of growth arrest or by triggering cell death (Vousden and Lu,2002). This process depends mainly on the expression of genes that regulate cell-cycle arrestor apoptosis. A critical unresolved issue about the DNA damage response is how the resultingup-regulation of the p53 tumor suppressor can lead either to cell cycle arrest and DNA repair,or to apoptosis. Here we present evidence that Hzf plays a critical role in p53-mediatedtranscription and functions as a key player in controlling a cellular regulatory switch thatdictates the cellular decision toward cell cycle arrest in response to stress. An autoregulatoryfeedback loop involving p53 and its target proteins such as Mdm2, Pirh2, and Cop1 is wellestablished (Dornan et al., 2004; Leng et al., 2003; Wu et al., 1993). These genes are knownto have ubiquitin ligase activity, and can target p53 for ubiquitination and degradation tomaintain low basal levels of the p53 protein. Unlike these p53 target proteins, Hzf encodes azinc-finger domain protein, and its binding to p53 does not seem to regulate p53 stability inresponse to stress. Instead, induction of Hzf and its binding to p53 selectively influences thespecificity of the p53-mediated transcription, resulting in preferential transactivation of its cellcycle arrest mediating target genes such as p21 and 14-3-3σ.
It has been previously shown that Hzf is a direct p53 transcriptional target in NIH3T3 cells(Sugimoto et al., 2006). Promoter analyses revealed that p53 could bind directly to the p53responsive element in the first intron of the Hzf gene. Moreover, Hzf was found to have a rolein cell cycle checkpoint control, specifically at the G2/M checkpoint. Strikingly, knockdownof Hzf expression promoted p21 ubiquitination and degradation, the mechanism of which wasnot elucidated. We independently identified Hzf as a p53 target gene in human cells, and ourpromoter analysis strongly suggest that unlike mouse Hzf, the p53-responsive element inhuman Hzf is at the −1044 bp of the promoter region (Figure S1). Our results show further thatHzf binds to p53, which results in preferential transactivation for pro-arrest p53 target genes.Thus, upon genotoxic stress, abrogation of Hzf expression results in preferential transactivationof pro-apoptotic p53 target genes such as Bax, Perp, Puma and Noxa over its pro-arrest targetssuch as p21 and 14-3-3σ. Hence, our results argue that down regulation of p21 and other pro-arrest p53 target genes such as 14-3-3σ in the absence of Hzf is transcriptional in nature.
Our results also indicate that the level of Hzf protein is inversely correlated with the extent ofgenotoxic stress. Prolonged p53 activation/expression or extended exposure to stress inducedHzf protein degradation through activation of an ubiquitination-proteasome pathway whoseconsequences were induction of pro-apoptotic p53 targets such as Bax, Puma, Noxa and Perpand apoptosis. Thus, we propose a model (Figure 7C) wherein p53 activates Hzf, which thenbinds to p53 in an autoregulatory feedback loop. Hzf-bound p53 is specifically recruited to cellcycle arrest gene promoters resulting in growth arrest. Prolonged p53 expression is associatedwith Hzf degradation, which then allows p53 activation of pro-apoptotic targets, resulting in
Das et al. Page 7
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

apoptosis. However, if the cells lack the mechanism for Hzf degradation, prolonged p53expression leads to senescence and shRNA mediated inhibition of Hzf expression makes thecells highly apoptotic (Figure S8). Therefore, our results provide a novel mechanism formodulation of p53-dependent DNA damage response. It will be of interest to determine theexpression pattern of p53-regulated genes by the microarray analysis in the absence or presenceof Hzf as well as the specific interacting partner(s) that mediates Hzf ubiquitination/degradation. Recently, it was shown that Tip60-dependent acetylation of p53 at lysine120modulates the decision between cell-cycle arrest and apoptosis, leading to enhanced p53-dependent apoptotic response (Sykes et al., 2006; Tang et al., 2006). Thus it will be interestingto know whether the acetylation of K120 within the DNA-binding domain of p53 affects Hzf-mediated preferential transactivation of pro-arrest p53 target genes. While the role of Mdm2in p53 regulation is well-established, we found that there is no role of Mdm2 in Hzf degradation(Figure S9).
Another question remains as to whether cells that have deficiency of p53 pro-arrest or pro-apoptotic target(s) can affect Hzf-mediated cell fate decision upon genotoxic stress. In HCT116and HCT116 p21−/− cells genotoxic stress results in cell cycle arrest in both cells lines withp21−/− cells showing a higher extent of G2/M arrest. If under such conditions Hzf is alsoknocked down, the cells show much increased apoptosis levels with marginal differencesbetween them (Figure S10). Thus, Hzf is likely to play a key role in sustaining p53 mediatedcell cycle arrest through other pro-arrest p53 target genes such as 14-3-3σ in addition to p21.It is well established that HCT116 Bax−/− cells are resistant to apoptosis upon DNA damage(Zhang et al., 2000). When Hzf was depleted in parental HCT116 and HCT116 Bax−/− cellsand then exposed to DNA damage, there was a significant induction of apoptosis in both thecell types, but the extent of apoptosis was higher in the wild type cells (data not shown). Thus,Hzf can act through modulation of p53-mediated transactivation of multiple pro-arrest and pro-apoptotic genes.
Targeted Hzf disruption in the mouse germ line has no reported effects on cell cycle dynamics,but general growth retardation and internal hemorrhage in brain of Hzf −/− mice were observed(Kimura et al., 2002). While mice lacking Hzf seemed to be otherwise normal, the initialcharacterization of Hzf-null mice suggested a role for Hzf in megakaryopoiesis and hemostasis.Based on our in vitro characterization of the role of Hzf in p53-dependent stress response, wecarried out an analysis of Hzf function in response to irradiation under in vivo conditions usingHzf knock out mice. We observed that exposure to ionizing radiation results in increased celldeath in Hzf−/− mice as compared to their wild type littermates, especially in the epidermis andprostate epithelium. However, comparison of the extent of apoptosis between the differenttissues of γ-irradiated Hzf−/− mice revealed significant differences indicative of some tissuespecificity with respect to Hzf function due to varying degree of Hzf expression in differenttissues upon γ-irradiation. While our preliminary investigations reveal some evidence forepithelial hyperplasia in organs like lung, mammary gland, etc. of Hzf−/−mice, further studieswill be needed to elucidate the role of Hzf in tumor susceptibility of mice and its implicationsfor human cancer. Nevertheless, our present findings establish that Hzf functions downstreamof p53 in DNA damage response and acts as a critical switch that favors cell cycle arrest overapoptosis by promoting preferential transactivation of pro-arrest targets of p53 both in vitroand in vivo. Thus, Hzf is both a p53 target and an important modulator of p53 functions, addinga new layer of complexity to the mechanisms by which p53 determines cell fate decisions inresponse to cellular stresses.
Das et al. Page 8
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Experimental ProceduresCell lines and culture conditions
U2OS, LNCaP, Saos2, HCT116, HCT116 p53−/− (Bunz et al., 1998) HCT116p21−/−(Waldman et al., 1996) and HCT116 bax−/− (Zhang et al., 2000) were cultured in DMEMcontaining fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), 100 U/ml penicillin and100μg/ml streptomycin at 37°C. Hzf+/+ and Hzf−/− early passage MEFs (Kimura et al., 2002)were also maintained in DMEM containing 10% FBS. For drug treatment, cells were grownto ~50% confluency prior to exposure to the DNA-damaging agents for the indicated time.Adenoviruses expressing Hzf or GFP were generated, amplified and titrated as previouslyreported (Ongusaha et al., 2003). Cells were grown to ~50–70% confluency and infected withrecombinant adenovirus at multiplicity of infection (MOI) of 10–20 for the indicated time.GFP expressing adenovirus (Ad-GFP) was used as control.
GST-pull down assay and yeast two hybrid assayCells were lysed in lysis buffer (20 mM Tris-HCl at pH 7.4, 5 mM EDTA, 10 mM Na4P2O7,100 mM NaF, 2 mM Na3VO4, 1% NP-40, 1 mM phenyl methylsulphonyl fluoride (PMSF),1X Protease inhibitor cocktail (Roche). 500μg cell extracts pre-treated with MNase asdescribed (Groisman et al., 2003) was subjected to GST-pull down following themanufacturer’s protocol (Amersham). Yeast two hybrid assay was performed following themanufacturer’s protocol (Clontech). Briefly, Hzf cloned in the pGADT7 vector wastransformed into yeast strain Y187, mated with yeast strain AH109 carrying the pGBKT7-p53plasmid and then plated on SD/-Trp/-Leu and SD/-Trp/-Leu/-Ade/-His plates. Representativecolonies for the positive control, negative controls and the test interaction were then streakedon to a SD/-Trp/-Leu/-Ade/-His/X-α-Gal plate.
Apoptosis and flow cytometryApoptosis was detected using the Cell Death Detection ELISA (Roche) and TUNEL assay(Roche). For the Cell Death Detection ELISA cells were lysed and the amount of nucleosomesin the cytoplasmic fraction of the cell lysates was measured using anti-histone antibody andanti-DNA antibody linked to peroxidase in a sandwich ELISA based protocol. For calculatingrelative nucleosome content the absorbance of all the samples was normalized with respect tothat of untreated/untransfected cells. Error bars are means ± SD of three independentexperiments with triplicate samples. For TUNEL assay the cells were fixed withparaformaldehyde and stained by TUNEL reaction using TMR red conjugated nucleotides andcounterstained with DAPI. For each sample 5 random fields were counted and error bars aremeans ± SD of three such independent experiments. Cell cycle analysis using flow cytometrywas carried out as described previously (Han et al., 2002).
shRNAVectors expressing shRNA against Hzf (5′-ATCCGCTTCAATTCTCAGA-3′), and thescrambled sequence (5′-GAGCCCTATTTCACAACTT-3′) were generated using the pBabe-U6-shRNA plasmid. Cells were transfected with Lipofectamine 2000 (Invitrogen) accordingto the manufacturer’s protocol.
Total body irradiation and immunohisochemistry14 weeks old Hzf−/− mice and age- and sex-matched wild-type littermates were subject to 5Gy of total body irradiation with a 137Cs gamma source at a rate of 0.6 Gy/min. All animalprotocols were approved by an Institutional Animal Care and Use Committee. 6.5 hours laterthe animals were euthanised. Tissues were harvested, fixed in formalin, washed with PBS andfinally embedded in paraffin for sectioning. Immunohistochemical analyses of the tissues were
Das et al. Page 9
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

carried out by fluorescence microscopy. Briefly, 5μm paraffin sections were dewaxed andimmunostained with Bax (Santa-Cruz) or p21 (Calbiochem) antibody followed bycounterstaining with TO-PRO-3 (Invitrogen). TUNEL staining of dewaxed sections werecarried out as described above.
Supplementary MaterialRefer to Web version on PubMed Central for supplementary material.
AcknowledgmentsWe would like to thank the Cutaneous Biology Research Center for supporting the work, S. Boswell, A. Mandinovaand L. Brown for reading the manuscript, and Y. Minamishima for initiation of the project. We also thank J. Manfredi,Mount Sinai School of Medicine for providing the mutant p53 constructs. This work was supported by NIH grants(2RO1 CA085681, 1RO1 CA097216 and 2RO1 CA078356 to SWL, and 2RO1 CA085214 and PO1 CA80058 toSAA), and Shiseido Research Core funding.
ReferencesAttardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T. PERP, an
apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev2000;14:704–718. [PubMed: 10733530]
Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, SmithP, Gasco M, et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic functionof codon 72-polymorphic p53. Nat Genet 2006;38:1133–1141. [PubMed: 16964264]
Braithwaite AW, Del Sal G, Lu X. Some p53-binding proteins that can function as arbiters of life anddeath. Cell Death Differ 2006;13:984–993. [PubMed: 16575404]
Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006;21:307–315. [PubMed:16455486]
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, VogelsteinB. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282:1497–1501. [PubMed: 9822382]
Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, SilicianoJD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science1998;281:1677–1679. [PubMed: 9733515]
Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpointin G(1) by stabilizing p53. Genes Dev 2000;14:278–288. [PubMed: 10673500]
Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O’Rourke K, Koeppen H, Dixit VM. Theubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004;429:86–92. [PubMed:15103385]
El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, KinzlerKW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75:817–825.[PubMed: 8242752]
Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, NakataniY. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by theCOP9 signalosome in response to DNA damage. Cell 2003;113:357–367. [PubMed: 12732143]
Han JA, Kim JI, Ongusaha PP, Hwang DH, Ballou LR, Mahale A, Aaronson SA, Lee SW. P53-mediatedinduction of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis. Embo J 2002;21:5635–5644. [PubMed: 12411481]
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene 2005;24:2899–2908. [PubMed: 15838523]
Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B.14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1997;1:3–11. [PubMed:9659898]
Das et al. Page 10
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Hidaka M, Caruana G, Stanford WL, Sam M, Correll PH, Bernstein A. Gene trapping of two novel genes,Hzf and Hhl, expressed in hematopoietic cells. Mech Dev 2000;90:3–15. [PubMed: 10585558]
Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53.Cell 1992;71:875–886. [PubMed: 1423635]
Iwabuchi K, Li B, Bartel P, Fields S. Use of the two-hybrid system to identify the domain of p53 involvedin oligomerization. Oncogene 1993;8:1693–1696. [PubMed: 8502489]
Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53as a sequence-specific DNA-binding protein. Science 1991;252:1708–1711. [PubMed: 2047879]
Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylationof MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci USA1999;96:14973–14977. [PubMed: 10611322]
Kimura Y, Hart A, Hirashima M, Wang C, Holmyard D, Pittman J, Pang XL, Jackson CW, Bernstein A.Zinc finger protein, Hzf, is required for megakaryocyte development and hemostasis. J Exp Med2002;195:941–952. [PubMed: 11927637]
Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell DeathDiffer 2006;13:951–961. [PubMed: 16575405]
Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006;13:941–950. [PubMed: 16601750]
Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S.Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003;112:779–791.[PubMed: 12654245]
Li B, Fields S. Identification of mutations in p53 that affect its binding to SV40 large T antigen by usingthe yeast two-hybrid system. Faseb J 1993;7:957–963. [PubMed: 8344494]
Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotesapoptosis. Nat Genet 2000;26:122–127. [PubMed: 10973264]
Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene.Cell 1995;80:293–299. [PubMed: 7834749]
Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001;7:683–694. [PubMed: 11463392]
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N.Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis.Science 2000;288:1053–1058. [PubMed: 10807576]
Ongusaha PP, Kim JI, Fang L, Wong TW, Yancopoulos GD, Aaronson SA, Lee SW. p53 induction andactivation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation througha positive feedback loop. Embo J 2003;22:1289–1301. [PubMed: 12628922]
Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activatesthe transcriptional response of p53. Embo J 1999;18:6455–6461. [PubMed: 10562557]
Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, Oren M, Vousden KH. Specific loss of apoptotic but notcell-cycle arrest function in a human tumor derived p53 mutant. Embo J 1996;15:827–838. [PubMed:8631304]
Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I,Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate the apoptotic function of p53.Mol Cell 2001;8:781–794. [PubMed: 11684014]
Sharma S, Dimasi D, Higginson K, Della NG. RZF, a zinc-finger protein in the photoreceptors of humanretina. Gene 2004;342:219–229. [PubMed: 15527981]
Sugimoto M, Gromley A, Sherr CJ. Hzf, a p53-responsive gene, regulates maintenance of the G2 phasecheckpoint induced by DNA damage. Mol Cell Biol 2006;26:502–512. [PubMed: 16382142]
Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of thep53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006;24:841–851. [PubMed:17189187]
Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision betweencell-cycle arrest and apoptosis. Mol Cell 2006;24:827–839. [PubMed: 17189186]
Das et al. Page 11
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ. A conserved intronicresponse element mediates direct p53-dependent transcriptional activation of both the human andmurine bax genes. Oncogene 2002;21:990–999. [PubMed: 11850816]
Trigiante G, Lu X. ASPP and cancer. Nat Rev Cancer 2006;6:217–226. [PubMed: 16498444]Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A.
p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science2003;302:1036–1038. [PubMed: 14500851]
Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000;408:307–310. [PubMed:11099028]
Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer 2002;2:594–604. [PubMed:12154352]
Waldman T, Lengauer C, Kinzler KW, Vogelstein B. Uncoupling of S phase and mitosis induced byanticancer agents in cells lacking p21. Nature 1996;381:713–716. [PubMed: 8649519]
Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature 1995;376:88–91. [PubMed: 7596441]
Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev1993;7:1126–1132. [PubMed: 8319905]
Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C,Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxicinsults. Nature 2002;419:853–857. [PubMed: 12397362]
Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticanceragents. Science 2000;290:989–992. [PubMed: 11062132]
Zhang W, Guo XY, Hu GY, Liu WB, Shay JW, Deisseroth AB. A temperature-sensitive mutant of humanp53. Embo J 1994;13:2535–2544. [PubMed: 8013454]
Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX. The prolylisomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002;419:849–853. [PubMed:12397361]
Das et al. Page 12
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
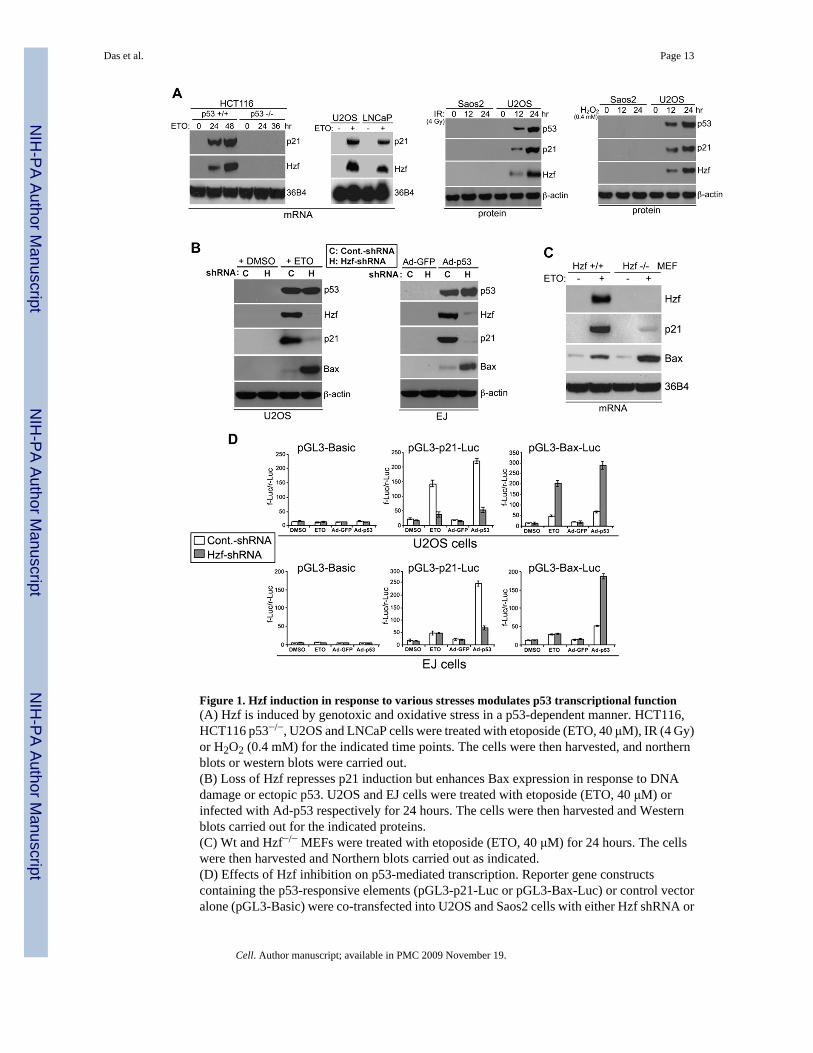
Figure 1. Hzf induction in response to various stresses modulates p53 transcriptional function(A) Hzf is induced by genotoxic and oxidative stress in a p53-dependent manner. HCT116,HCT116 p53−/−, U2OS and LNCaP cells were treated with etoposide (ETO, 40 μM), IR (4 Gy)or H2O2 (0.4 mM) for the indicated time points. The cells were then harvested, and northernblots or western blots were carried out.(B) Loss of Hzf represses p21 induction but enhances Bax expression in response to DNAdamage or ectopic p53. U2OS and EJ cells were treated with etoposide (ETO, 40 μM) orinfected with Ad-p53 respectively for 24 hours. The cells were then harvested and Westernblots carried out for the indicated proteins.(C) Wt and Hzf−/− MEFs were treated with etoposide (ETO, 40 μM) for 24 hours. The cellswere then harvested and Northern blots carried out as indicated.(D) Effects of Hzf inhibition on p53-mediated transcription. Reporter gene constructscontaining the p53-responsive elements (pGL3-p21-Luc or pGL3-Bax-Luc) or control vectoralone (pGL3-Basic) were co-transfected into U2OS and Saos2 cells with either Hzf shRNA or
Das et al. Page 13
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

control scrambled shRNA, and 24 hours later cells were treated with etoposide or infected withAd-p53 for another 24 hours. At the end of the time point Luciferase assay was carried out.Error bars are means ± SD of three independent experiments with duplicate samples.
Das et al. Page 14
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
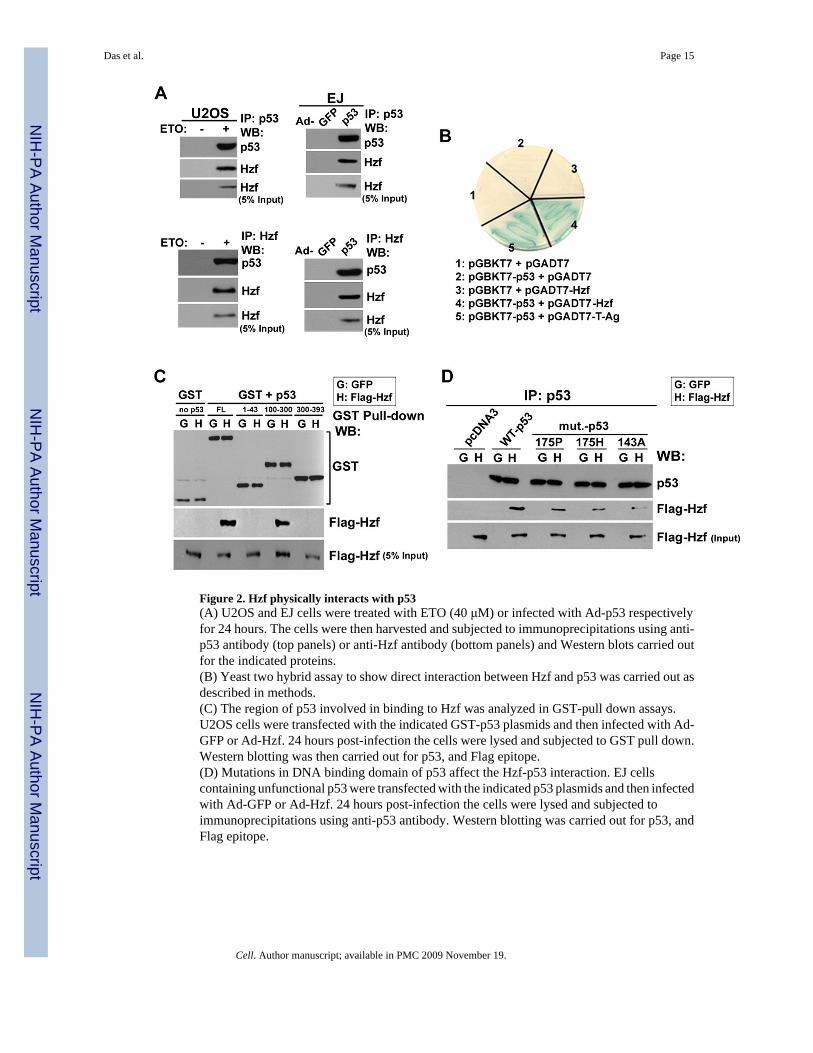
Figure 2. Hzf physically interacts with p53(A) U2OS and EJ cells were treated with ETO (40 μM) or infected with Ad-p53 respectivelyfor 24 hours. The cells were then harvested and subjected to immunoprecipitations using anti-p53 antibody (top panels) or anti-Hzf antibody (bottom panels) and Western blots carried outfor the indicated proteins.(B) Yeast two hybrid assay to show direct interaction between Hzf and p53 was carried out asdescribed in methods.(C) The region of p53 involved in binding to Hzf was analyzed in GST-pull down assays.U2OS cells were transfected with the indicated GST-p53 plasmids and then infected with Ad-GFP or Ad-Hzf. 24 hours post-infection the cells were lysed and subjected to GST pull down.Western blotting was then carried out for p53, and Flag epitope.(D) Mutations in DNA binding domain of p53 affect the Hzf-p53 interaction. EJ cellscontaining unfunctional p53 were transfected with the indicated p53 plasmids and then infectedwith Ad-GFP or Ad-Hzf. 24 hours post-infection the cells were lysed and subjected toimmunoprecipitations using anti-p53 antibody. Western blotting was carried out for p53, andFlag epitope.
Das et al. Page 15
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
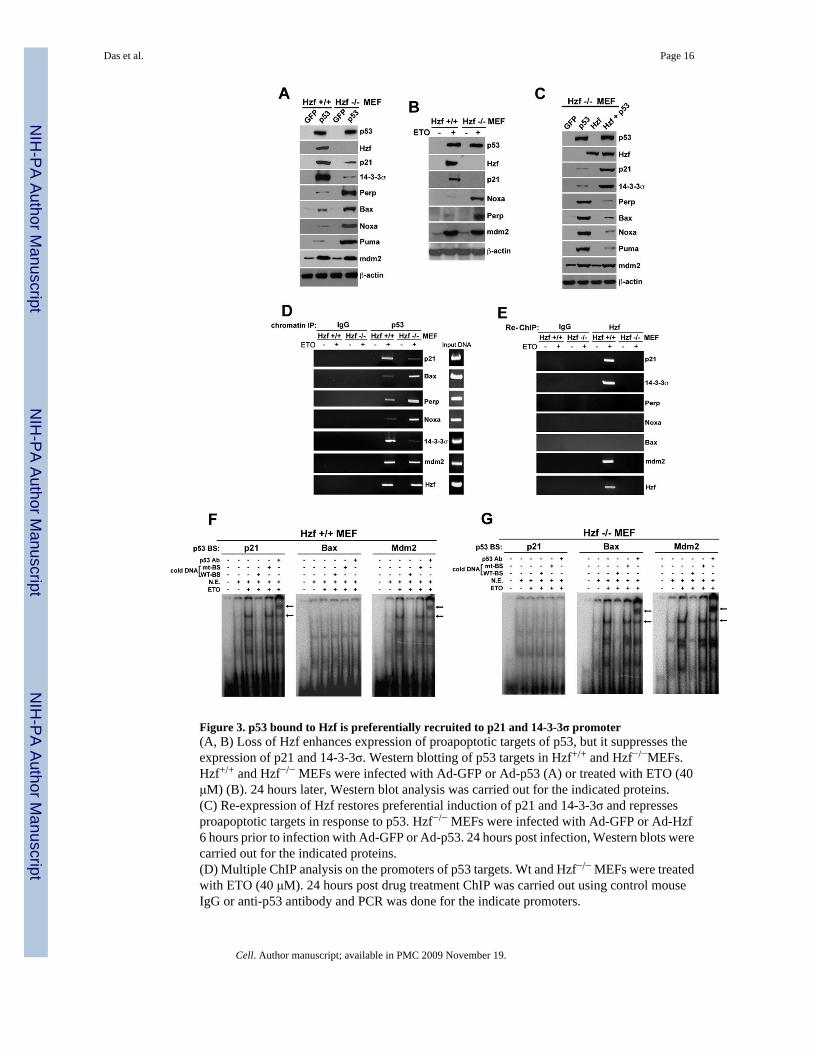
Figure 3. p53 bound to Hzf is preferentially recruited to p21 and 14-3-3σ promoter(A, B) Loss of Hzf enhances expression of proapoptotic targets of p53, but it suppresses theexpression of p21 and 14-3-3σ. Western blotting of p53 targets in Hzf+/+ and Hzf−/−MEFs.Hzf+/+ and Hzf−/− MEFs were infected with Ad-GFP or Ad-p53 (A) or treated with ETO (40μM) (B). 24 hours later, Western blot analysis was carried out for the indicated proteins.(C) Re-expression of Hzf restores preferential induction of p21 and 14-3-3σ and repressesproapoptotic targets in response to p53. Hzf−/− MEFs were infected with Ad-GFP or Ad-Hzf6 hours prior to infection with Ad-GFP or Ad-p53. 24 hours post infection, Western blots werecarried out for the indicated proteins.(D) Multiple ChIP analysis on the promoters of p53 targets. Wt and Hzf−/− MEFs were treatedwith ETO (40 μM). 24 hours post drug treatment ChIP was carried out using control mouseIgG or anti-p53 antibody and PCR was done for the indicate promoters.
Das et al. Page 16
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

(E) Part of the chromatin immunoprecipitated with p53 antibody was again subjected to ChIPusing control rabbit IgG or Hzf antibody. Input represents 2% of total chromatin from untreatedHzf+/+ MEFs used for immunoprecipitation.(F, G) EMSA showing the DNA binding activity of p53 to oligonucleotides containing the p53binding site in p21 promoter, Bax first intron, and Mdm2 promoter in wt-MEF (F) and Hzf-null MEF (G). Wt and Hzf−/− MEFs were treated with ETO (40 μM). 24 hours post drugtreatment, nuclear extracts were prepared, and EMSA carried out as described in supplementarymethods. The arrows indicate the position of the p53-DNA complex (lower) and supershiftedcomplex containing anti-p53 antibody (pAb421).
Das et al. Page 17
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
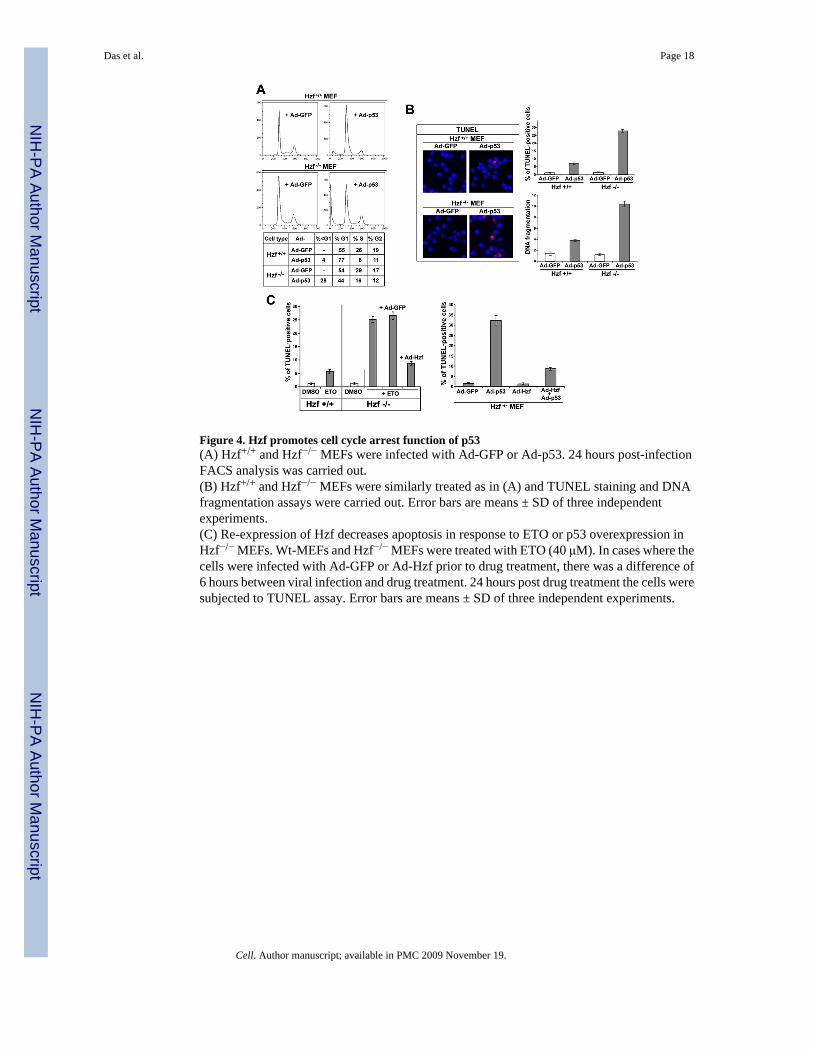
Figure 4. Hzf promotes cell cycle arrest function of p53(A) Hzf+/+ and Hzf−/− MEFs were infected with Ad-GFP or Ad-p53. 24 hours post-infectionFACS analysis was carried out.(B) Hzf+/+ and Hzf−/− MEFs were similarly treated as in (A) and TUNEL staining and DNAfragmentation assays were carried out. Error bars are means ± SD of three independentexperiments.(C) Re-expression of Hzf decreases apoptosis in response to ETO or p53 overexpression inHzf−/− MEFs. Wt-MEFs and Hzf−/− MEFs were treated with ETO (40 μM). In cases where thecells were infected with Ad-GFP or Ad-Hzf prior to drug treatment, there was a difference of6 hours between viral infection and drug treatment. 24 hours post drug treatment the cells weresubjected to TUNEL assay. Error bars are means ± SD of three independent experiments.
Das et al. Page 18
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Figure 5. Sustained p53 activation promotes ubiquitination and degradation of Hzf(A) Prolonged p53 activation induces Hzf protein down-regulation. U2OS cells were exposedto ETO (40 μM) or were infected with Ad-GFP or Ad-p53 for the indicated time points. Thecells were lysed and Western blots were carried out for the indicated proteins.(B) U2OS cells exposed to ETO (40 μM) for the indicated time points were immunoprecipitatedwith p53 Ab, and the precipitates were immunoblotted with Hzf Ab.(C) Hzf mRNA levels in response to ETO treatment remain steady. U2OS cells were exposedto ETO (40 μM) for the indicated time points and Northern blots were carried out as indicated.(D) U2OS cells were infected with Ad-GFP or Ad-p53 for the indicated time points and FACSanalysis was carried out.(E) U2OS cells were similarly treated as in (A) and TUNEL assays were carried out for theindicated time points. Error bars are means ± SD of three independent experiments.(F, G, H) Proteasome-dependent Hzf degradation. U2OS cells were treated for 36 and 72 hourswith ETO (40 μM), and MG132 (10 μM) was added during the last 10 hours prior to the endof the 72 hours time point. The cells were then harvested, and Western blotting, Northernblotting or ChIP was carried out with the indicated antibodies. Input represents 2% of totalchromatin from untreated U2OS cells used for immunoprecipitation.(I) Genotoxic stress-induced ubiquitination of Hzf. U2OS cells were treated with ETO (40μM) for 36 and 72 hours, and with MG132 for the last 10 hours. Cell extracts wereimmunoprecipitated with anti-Hzf Ab, and immunoblotted with anti-ubiquitin Ab.
Das et al. Page 19
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
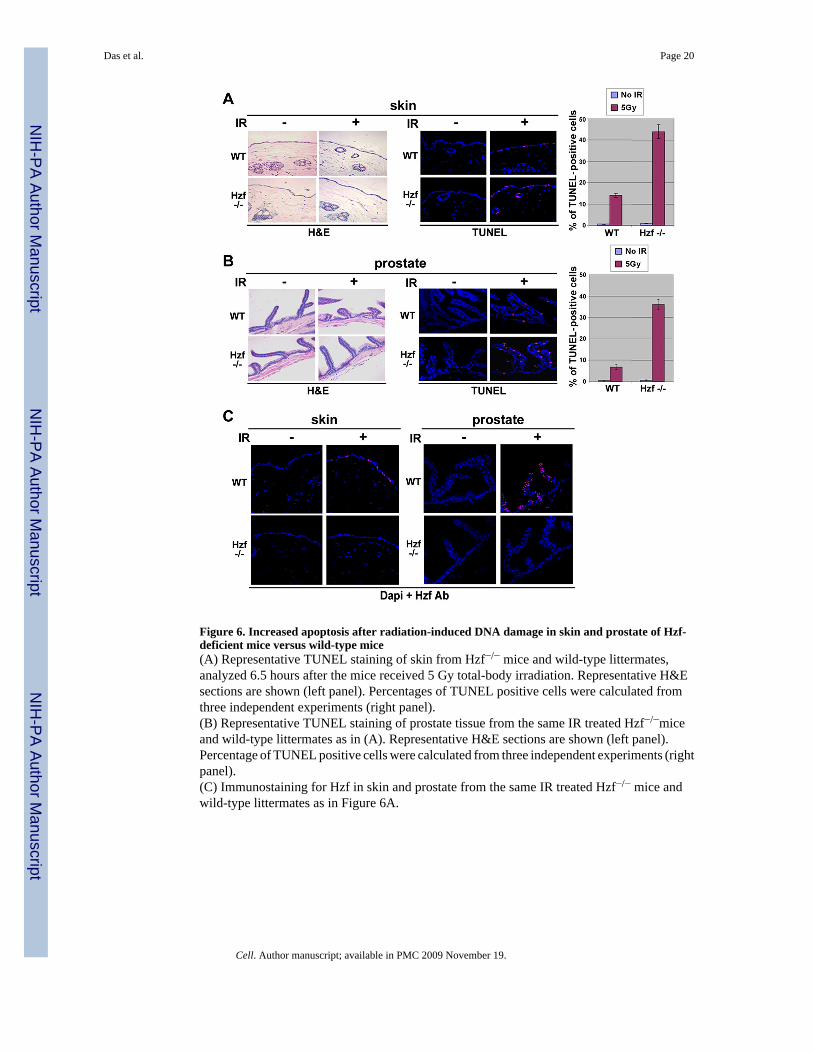
Figure 6. Increased apoptosis after radiation-induced DNA damage in skin and prostate of Hzf-deficient mice versus wild-type mice(A) Representative TUNEL staining of skin from Hzf−/− mice and wild-type littermates,analyzed 6.5 hours after the mice received 5 Gy total-body irradiation. Representative H&Esections are shown (left panel). Percentages of TUNEL positive cells were calculated fromthree independent experiments (right panel).(B) Representative TUNEL staining of prostate tissue from the same IR treated Hzf−/−miceand wild-type littermates as in (A). Representative H&E sections are shown (left panel).Percentage of TUNEL positive cells were calculated from three independent experiments (rightpanel).(C) Immunostaining for Hzf in skin and prostate from the same IR treated Hzf−/− mice andwild-type littermates as in Figure 6A.
Das et al. Page 20
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
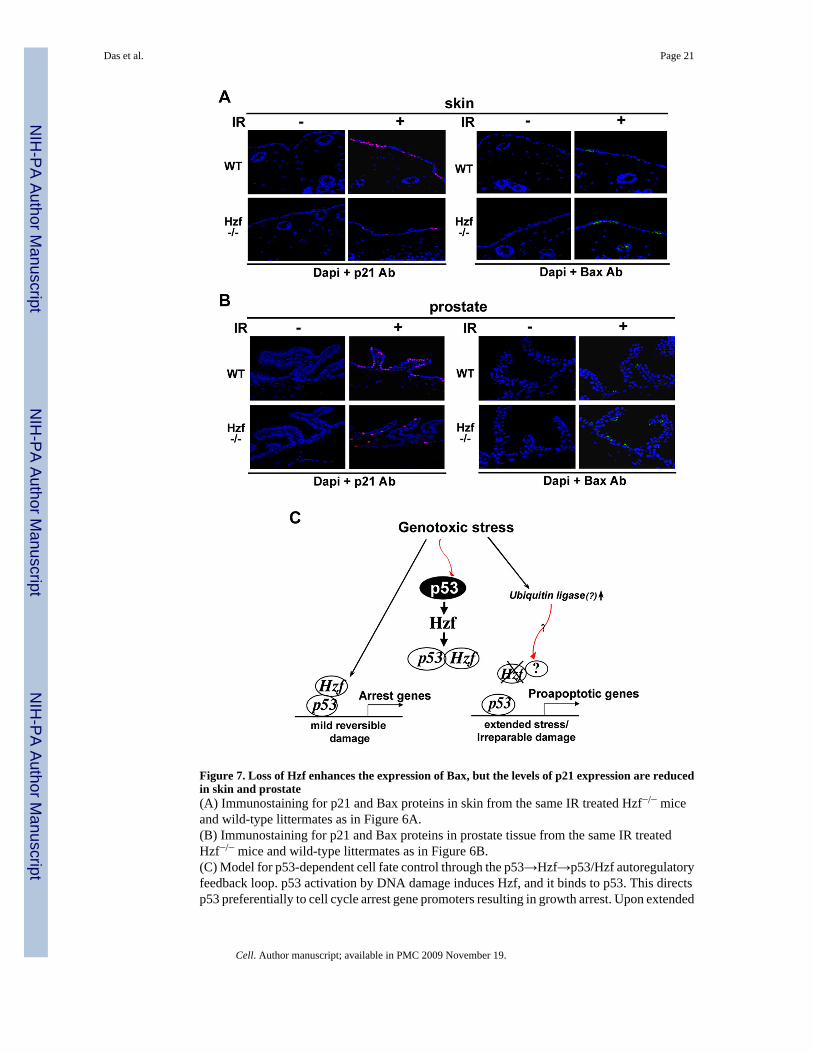
Figure 7. Loss of Hzf enhances the expression of Bax, but the levels of p21 expression are reducedin skin and prostate(A) Immunostaining for p21 and Bax proteins in skin from the same IR treated Hzf−/− miceand wild-type littermates as in Figure 6A.(B) Immunostaining for p21 and Bax proteins in prostate tissue from the same IR treatedHzf−/− mice and wild-type littermates as in Figure 6B.(C) Model for p53-dependent cell fate control through the p53→Hzf→p53/Hzf autoregulatoryfeedback loop. p53 activation by DNA damage induces Hzf, and it binds to p53. This directsp53 preferentially to cell cycle arrest gene promoters resulting in growth arrest. Upon extended
Das et al. Page 21
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

or irreparable stress, Hzf ubiquitination/degradation prevents this from occurring, thusallowing p53 to activate pro-apoptotic targets, and resulting in cell to trigger apoptosis.
Das et al. Page 22
Cell. Author manuscript; available in PMC 2009 November 19.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Related Documents











