Hyposialylation of neprilysin possibly affects its expression and enzymatic activity in hereditary inclusion-body myopathy muscle Aldobrando Broccolini,* Teresa Gidaro,* Raimondo De Cristofaro, Roberta Morosetti,* , à Carla Gliubizzi,* , à Enzo Ricci,* , à Pietro A. Tonali* , à and Massimiliano Mirabella* , à *Department of Neuroscience, Catholic University, School of Medicine, Rome, Italy Department of Internal Medicine, Catholic University, School of Medicine, Rome, Italy àFondazione Don Carlo Gnocchi ONLUS, Rome, Italy Hereditary inclusion-body myopathy (h-IBM, MIM No. 600737) is an autosomal recessive disorder characterized by progressive weakness of lower and upper limb muscles with relative sparing of the quadriceps (Argov and Yarom 1984). h-IBM muscle shares many similarities with that of sporadic IBM, including vacuolated muscle fibers and the abnormal accumulation of proteins commonly observed in Alzheimer disease (AD) brain such as amyloid-b (Ab) (Askanas and Engel 1998, 2002). h-IBM is caused by mutations in the UDP-N-acetylglucosamine 2-epimerase/ N-acetylmannosamine kinase (GNE; MIM No. 603824) gene on chromosome 9p12–13, that codes for a bifunctional enzyme that catalyzes the first two steps in sialic acid biosynthesis (Hinderlich et al. 1997; Eisenberg et al. 2001). Mutations of the GNE gene responsible of h-IBM result in reduced activity of both UDP-N-acetylglucosamine 2-epim- erase and N-acetylmannosamine kinase and in abnormal sialylation of muscle glycoproteins (Noguchi et al. 2004; Saito et al. 2004; Broccolini et al. 2005; Tajima et al. 2005; Received November 6, 2007; revised manuscript received December 7, 2007; accepted December 12, 2007. Address correspondence and reprint requests to Aldobrando Brocco- lini, MD PhD, and Massimiliano Mirabella, MD PhD, Department of Neuroscience, Catholic University, L.go A, Gemelli 8, Rome 00168, Italy. E-mail: [email protected] and [email protected]. Abbreviations used: AD, Alzheimer disease; Ab, amyloid-b;AbPP, amyloid-b precursor protein; CEX, cycloheximide; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GNE, UDP-N-acetylglucosamine 2-epimerase/N-acetyl mannosamine kinase; h-IBM, hereditary inclusion-body myopathy; IGF-I, insulin-like growth factor-I; MAL, Maackia amurensis lectin; NEP, neprilysin; VCN, Vibrio Cholerae neuraminidase. Abstract Autosomal recessive hereditary inclusion-body myopathy (h-IBM) is caused by mutations of the UDP-N-acetylglucos- amine 2-epimerase/N-acetylmannosamine kinase gene, a rate-limiting enzyme in the sialic acid metabolic pathway. Previous studies have demonstrated an abnormal sialylation of glycoproteins in h-IBM. h-IBM muscle shows the abnormal accumulation of proteins including amyloid-b (Ab). Neprilysin (NEP), a metallopeptidase that cleaves Ab, is characterized by the presence of several N-glycosylation sites, and changes in these sugar moieties affect its stability and enzymatic activity. In the present study, we found that NEP is hyposial- ylated and its expression and enzymatic activity reduced in all h-IBM muscles analyzed. In vitro, the experimental removal of sialic acid by Vibrio Cholerae neuraminidase in cultured myotubes resulted in reduced expression of NEP. This was most likely because of a post-translational modification con- sisting in an abnormal sialylation of the protein that leads to its reduced stability. Moreover, treatment with Vibrio Cholerae neuraminidase was associated with an increased immunore- activity for Ab mainly in the form of distinct cytoplasmic foci within myotubes. We hypothesize that, in h-IBM muscle, hyposialylated NEP has a role in hampering the cellular Ab clearing system, thus contributing to its abnormal accumula- tion within vulnerable fibers and possibly promoting muscle degeneration. Keywords: hereditary inclusion-body myopathy, neprilysin, UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J. Neurochem. (2008) 105, 971–981. d JOURNAL OF NEUROCHEMISTRY | 2008 | 105 | 971–981 doi: 10.1111/j.1471-4159.2007.05208.x ȑ 2008 The Authors Journal Compilation ȑ 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981 971

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Hyposialylation of neprilysin possibly affects its expression andenzymatic activity in hereditary inclusion-body myopathymuscle
Aldobrando Broccolini,* Teresa Gidaro,* Raimondo De Cristofaro,� Roberta Morosetti,*,�Carla Gliubizzi,*,� Enzo Ricci,*,� Pietro A. Tonali*,� and Massimiliano Mirabella*,�
*Department of Neuroscience, Catholic University, School of Medicine, Rome, Italy
�Department of Internal Medicine, Catholic University, School of Medicine, Rome, Italy
�Fondazione Don Carlo Gnocchi ONLUS, Rome, Italy
Hereditary inclusion-body myopathy (h-IBM, MIM No.600737) is an autosomal recessive disorder characterizedby progressive weakness of lower and upper limb muscleswith relative sparing of the quadriceps (Argov and Yarom1984). h-IBM muscle shares many similarities with that ofsporadic IBM, including vacuolated muscle fibers and theabnormal accumulation of proteins commonly observed inAlzheimer disease (AD) brain such as amyloid-b (Ab)(Askanas and Engel 1998, 2002). h-IBM is caused bymutations in the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE; MIM No. 603824) geneon chromosome 9p12–13, that codes for a bifunctionalenzyme that catalyzes the first two steps in sialic acidbiosynthesis (Hinderlich et al. 1997; Eisenberg et al. 2001).Mutations of the GNE gene responsible of h-IBM result inreduced activity of both UDP-N-acetylglucosamine 2-epim-
erase and N-acetylmannosamine kinase and in abnormalsialylation of muscle glycoproteins (Noguchi et al. 2004;Saito et al. 2004; Broccolini et al. 2005; Tajima et al. 2005;
Received November 6, 2007; revised manuscript received December 7,2007; accepted December 12, 2007.Address correspondence and reprint requests to Aldobrando Brocco-
lini, MD PhD, and Massimiliano Mirabella, MD PhD, Department ofNeuroscience, Catholic University, L.go A, Gemelli 8, Rome 00168,Italy. E-mail: [email protected] and [email protected] used: AD, Alzheimer disease; Ab, amyloid-b; AbPP,
amyloid-b precursor protein; CEX, cycloheximide; FBS, fetal bovineserum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GNE,UDP-N-acetylglucosamine 2-epimerase/N-acetyl mannosamine kinase;h-IBM, hereditary inclusion-body myopathy; IGF-I, insulin-like growthfactor-I; MAL, Maackia amurensis lectin; NEP, neprilysin; VCN, VibrioCholerae neuraminidase.
Abstract
Autosomal recessive hereditary inclusion-body myopathy
(h-IBM) is caused by mutations of the UDP-N-acetylglucos-
amine 2-epimerase/N-acetylmannosamine kinase gene, a
rate-limiting enzyme in the sialic acid metabolic pathway.
Previous studies have demonstrated an abnormal sialylation
of glycoproteins in h-IBM. h-IBM muscle shows the abnormal
accumulation of proteins including amyloid-b (Ab). Neprilysin
(NEP), a metallopeptidase that cleaves Ab, is characterized
by the presence of several N-glycosylation sites, and changes
in these sugar moieties affect its stability and enzymatic
activity. In the present study, we found that NEP is hyposial-
ylated and its expression and enzymatic activity reduced in all
h-IBM muscles analyzed. In vitro, the experimental removal of
sialic acid by Vibrio Cholerae neuraminidase in cultured
myotubes resulted in reduced expression of NEP. This was
most likely because of a post-translational modification con-
sisting in an abnormal sialylation of the protein that leads to its
reduced stability. Moreover, treatment with Vibrio Cholerae
neuraminidase was associated with an increased immunore-
activity for Ab mainly in the form of distinct cytoplasmic foci
within myotubes. We hypothesize that, in h-IBM muscle,
hyposialylated NEP has a role in hampering the cellular Ab
clearing system, thus contributing to its abnormal accumula-
tion within vulnerable fibers and possibly promoting muscle
degeneration.
Keywords: hereditary inclusion-body myopathy, neprilysin,
UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine
kinase.
J. Neurochem. (2008) 105, 971–981.
d JOURNAL OF NEUROCHEMISTRY | 2008 | 105 | 971–981 doi: 10.1111/j.1471-4159.2007.05208.x
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981 971

Ricci et al. 2006). Very recently, it has been also shown thata mouse model of h-IBM exhibits marked hyposialylation ofserum, muscle and other organs (Malicdan et al. 2007).
The zinc metallopeptidase neprilysin (NEP, EP24.11, alsoknown as CD10, and enkephalinase) is a neutral endopep-tidase that was originally described in the brush-bordermembranes of rabbit kidney (Kerr and Kenny 1974). NEP isable to hydrolyze small peptides in the immediacy of theplasma membrane, and in the CNS it regulates the level ofneuropeptides close to the peptidergic synapses (Turner et al.2001). In addition, NEP has been shown to cleave Ab atmultiple sites (Howell et al. 1995) and may play a role in thepathogenesis of AD through the regulation of Ab levels inthe brain (Akiyama et al. 2001; Iwata et al. 2001; Leissringet al. 2003). In skeletal muscle, NEP appears to play a roleduring development and regeneration and is abnormallyaccumulated in Ab-bearing abnormal muscle fibers ofsporadic IBM (Broccolini et al. 2006).
Neprilysin protein is characterized by the presence ofseveral N-glycosylation sites (Devault et al. 1987) andcontains large amounts of sialic acid (Johnson et al. 1984),and previous studies have shown that changes in these sugarmoieties of the protein affect its stability and enzymaticactivity (Lafrance et al. 1994).
These observations provide the rationale to investigatewhether NEP participates to the pathogenic scenario under-lying muscle degeneration in h-IBM.
Materials and methods
PatientsFive h-IBM patients from four unrelated families were diagnosed as
having h-IBM based on clinical findings, muscle pathology, and
genetic study (Askanas and Engel 1998; Eisenberg et al. 2001). Allmuscle biopsies showed typical h-IBM abnormalities including
absence of inflammation and necrosis, vacuolated muscle fibers, and
variable amount of angulated atrophic fibers (Askanas and Engel
1998). Clinical and genetic data of our h-IBM patients are
summarized in Table 1. Controls were polymyositis (n = 4),
dermatomyositis (n = 4), Duchenne muscular dystrophy (n = 3),
Becker muscular dystrophy (n = 3). The muscle specimens of 8
subjects, proven to be free from any neuromuscular disorder after all
diagnostic tests were performed, were used as normal controls.
All muscle samples used in this study were obtained for
diagnostic purposes with informed consent. This research was
approved by the Ethical Committee of our Institution.
Primary muscle culturesHuman primary muscle cultures were obtained from the biopsies of
five h-IBM and five normal control patients using the explantation
re-explantation method, as previously described (Askanas and Engel
1992). Myoblasts were kept in a replicative state up to the third-
fourth passage using a medium containing 15% (v/v) fetal bovine
serum (FBS; Cambrex Bioscience, Baltimore, MD, USA) and a
cocktail of growth factors. The fusion of subconfluent mononucle-
ated myoblasts into multinucleated myotubes was obtained using a
culturing medium with 5% (v/v) FBS and without growth factors
(differentiation medium) for 5 days. Fusion index was expressed as
number of myonuclei/number of total nuclei, visualized by Hoechst
33258 staining (Molecular Probes Inc., Eugene, OR, USA), and
varied between 0.8 and 0.9 for all the culture sets used in this study.
Before any experimental procedure, myotubes from h-IBM and
control patients were maintained for 2 days in culturing medium
containing only serum replacement factors (Ultraculture; Cambrex
Bioscience) to avoid the uptake of exogenous sialic acid and
glycoproteins. Reagents used in specific experiments included
Vibrio Cholerae neuraminidase (VCN; Sigma, St Louis, MO,
USA) suspended in aqueous solution, and cycloheximide (CEX;
Sigma) dissolved in dimethyl sulfoxide at an initial concentration of
100 mg/mL and kept at 4�C until used.
Immunochemistry on muscle biopsies and primary muscle culturesThis was performed on 8-lm thick unfixed cryostat sections and on
myotubes grown in optical grade bottom culture dishes (Ibidi,
Martinsried, Germany), as previously described (Broccolini et al.2006). The following well-characterized primary antibodies were
used: (i) monoclonal anti-NEP (NCL-CD10–270 clone 56C6;
Novocastra, Newcaste Upon Tyne, UK) diluted 1/25; (ii) polyclonal
anti-b-amyloid40, recognizing Ab1–40 and not cross-reacting with
Table 1 Clinical and genetic data of h-IBM
patients
Patient/gender
Age at
onset
(years)
Clinical
involvement
GNE mutation
Protein
change Protein Domain
1/F 25 Severe p.N519S+
p.N519S
Kinase
2/M 37 Moderate p.N519S+
p.N519S
Kinase
3/F 35 Moderate p.P27S+
p.P27S
Epimerase
4/F 21 Moderate p.R246Q+
p.Q355_C357del/p.I377fsX16
Epimerase kinase
5/F 23 Moderate p.M171V+
p.M712T
Epimerase kinase
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981� 2008 The Authors
972 | A. Broccolini et al.

amyloid-b precursor protein (AbPP), Ab1–42 and Ab1–43 (Ab40,
Calbiochem, San Diego, CA, USA) diluted 1/100; and (iii)
polyclonal anti-skeletal muscle myosin (Sigma) diluted 1/100. The
reaction with the primary antibodies was performed for 16–18 h at
4�C, followed by the incubation with the appropriate Alexa Fluor�-
conjugated secondary antisera (Invitrogen, Carlsbad, CA, USA)
diluted 1/100. In control experiments for staining specificity the
primary antibody was omitted or replaced by a non-immune serum.
Sections were then analyzed using a Leica TCS SP5 laser scanning
confocal microscope (Leica Microsystems, Wetzlar, Germany).
Glycoprotein enrichmentThis was performed on diagnostic muscle biopsies and primary
muscle cultures by lectin affinity chromatography using the
Qproteome Sialic Glycoprotein kit (Qiagen, Valencia, CA, USA)
according to the manufacturer’s instructions. In brief, muscle
samples were homogenized in binding buffer provided by the
supplier containing protease inhibitors and Nonidet P40 as detergent
and then centrifuged at 10 000 g at 4�C for 20 min. Supernatants
were then incubated with Maackia amurensis lectin (MAL). The
eluted glycoproteins were then used for western blot analysis.
Western blotDiagnostic muscle biopsies and primary muscle cultures were
homogenized in Tris-buffered saline containing Triton X-100 and
protease inhibitors and stored at )80�C until used. Glycoprotein
eluted from chromatography columns were concentrated using
Ultrafree-0.5 columns (Millipore, Billerica, MA, USA) and quan-
tified by the Bradford colorimetric assay (Biorad, Hercules, CA,
USA). Proteins were separated by sodium dodecyl sulfate–poly-
acrylamide gel electrophoresis and blotted onto a nitrocellulose
membrane (Schleicher & Schuell, Relliehausen, Germany). Blots
were incubated for 16–18 h at 4�C with the monoclonal anti-NEP
antibody (Novocastra) diluted 1 : 80. In experiments where total
homogenates from either muscle biopsies or muscle cultures were
used, equal loading of samples was confirmed by probing the
membranes with the monoclonal anti-muscle specific actin antibody
(Novocastra) diluted 1/2000. On the contrary, in experiments using
glycoprotein extracts, the equal loading of samples was confirmed
by Coomassie brilliant blue staining of the gel. After incubation with
the appropriate peroxidase-conjugated secondary antibody (GE
Healthcare, Piscataway, NJ, USA), blots were developed using the
ECL Western Blotting Analysis System (GE Healthcare). Densi-
tometry on autoradiographic films was carried out using the
TOTALLAB 2.01 software (Nonlinear Dynamics Ltd, Newcastle upon
Tyne, UK).
NEP enzyme assayThis was performed according to a previously published protocol
(Li and Hersh 1995). Muscle biopsies and cultured myotubes were
homogenized in 10 mM HEPES buffer and protein concentration
was determined by the Bradford colorimetric assay (Biorad). Forty
micrograms of proteins from each experimental sample were loaded
in the reaction mixture containing 0.025 U/mL of aminopeptidase M
(EC 3.4.11.2; Sigma) and 50 lM of N-benzyloxycarbonyl-L-alanyl-L-alanyl-L-leucyl-p-nitroanilide (Z-Ala-Ala-Leu-pNA; Bachem,
Bubendorf, Switzerland) as substrate. For each experimental sample
the assay was conducted in the presence and absence of 10 lM
phosphoramidon (Sigma), a highly specific inhibitor of NEP (Iwata
et al. 2001; Shirotani et al. 2001; Marr et al. 2004). Samples were
then incubated for 1 h at 37�C and the amount of chromogenic
substrate released was measured in a microplate reader at 405 nM
(Biorad). The amount of absorbance generated by NEP was
calculated by subtracting the arbitrary absorbance units produced
by samples containing phosphoramidon from the arbitrary absor-
bance units produced by the corresponding samples without
phosphoramidon.
Northern blottingTotal RNA was extracted from experimental cultures using RNA-
Trizol reagent, according to the manufacturer’s instructions (Invi-
trogen). Ten micrograms of total RNA of each sample were
electrophoretically separated on a 1% agarose/0.6 M formaldehyde
gel, blotted onto a Hybond-N+ membrane (GE Healthcare) and
cross-linked by UV exposure. NEP and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) probes were generated by PCR from total
human cDNA using the following primers: NEP forward 5¢-ATC-AGCCTCTCGGTCCTTG-3¢, NEP reverse 5¢-TGTAGGTTC-GGCTGAGGCT-3¢ (GenBank Accession No. NM_007289);
GAPDH forward 5¢-ATGGGGAAGGTGAAGGTCG-3¢, GAPDHreverse 5¢-GTGGGTGTCGCTGTTGAAGTC-3¢ (GenBank Acces-
sion No. AY340484). PCR products were radiolabeled with
[a-32P]dCTP (3000 lCi/mM; ICN Biomedicals, Asse, Belgium)
using a random primers labeling kit (Invitrogen), and then
hybridized on blots in standard hybridization buffer for 16–18 h at
68�C. After high stringency rinsing, the membranes were scanned in
a Molecular Imager Typhoon 8006 (GE Healthcare). Band densi-
tometry was assessed as above.
Statistical analysisAll data were expressed as mean ± SE. Analysis of repeated
measures was performed by ANOVA and comparisons between
groups was assessed by Student’s t-test. A value of p £ 0.05 was
considered significant.
Results
NEP expression and enzymatic activity is reduced in h-IBMmuscle biopsiesBy immunohistochemistry, in h-IBM muscle biopsies NEPimmunoreactivity was increased in rare vacuolated fibers,mainly in the form of cytoplasmic foci surrounding thevacuoles, and co-localized with the Ab40 immunosignal(Fig. 1). NEP immunoreactivity was also increased inregenerating muscle fibers in all the myopathies thatcontained them (not shown), as previously demonstrated(Broccolini et al. 2006).
Neprilysin expression was then analyzed by western blotanalysis in total protein extracts from muscles of h-IBMpatients and normal and disease-controls. The mean densi-tometric value of NEP signal in normal control musclebiopsies was arbitrarily set as 1 and the measured values ofall samples were expressed as a percentage of it. Despite the
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981
Neprilysin in h-IBM | 973
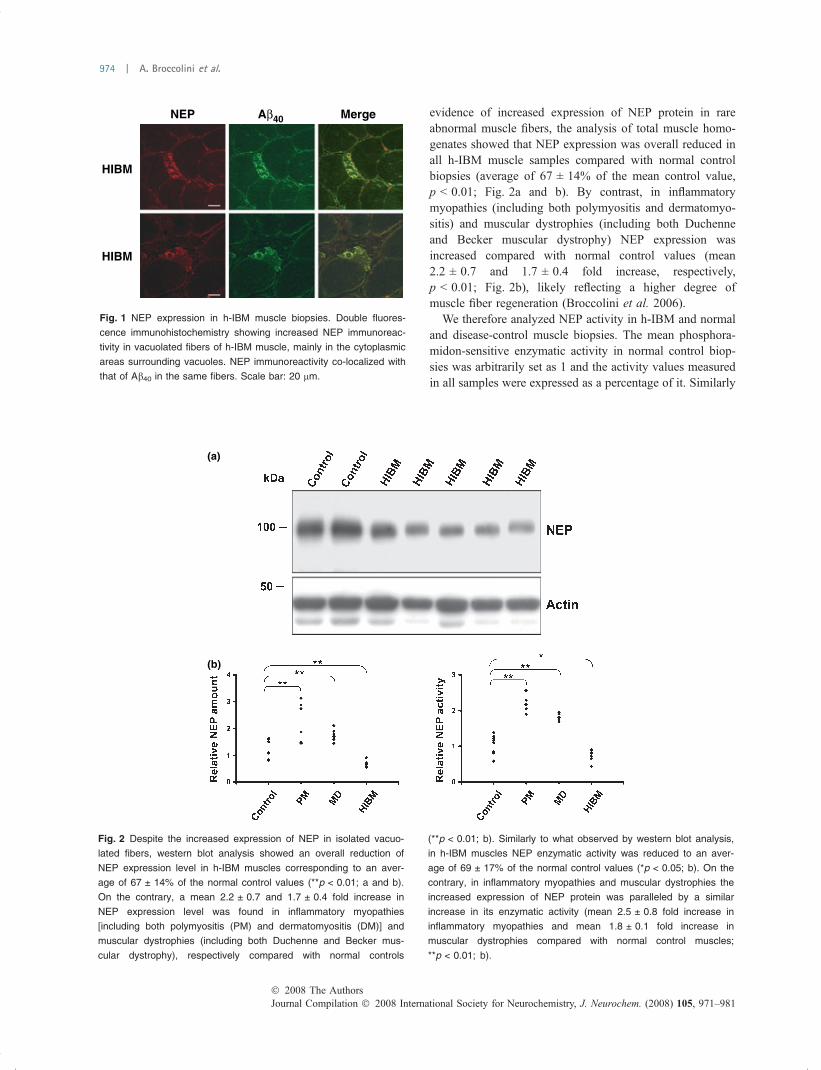
evidence of increased expression of NEP protein in rareabnormal muscle fibers, the analysis of total muscle homo-genates showed that NEP expression was overall reduced inall h-IBM muscle samples compared with normal controlbiopsies (average of 67 ± 14% of the mean control value,p < 0.01; Fig. 2a and b). By contrast, in inflammatorymyopathies (including both polymyositis and dermatomyo-sitis) and muscular dystrophies (including both Duchenneand Becker muscular dystrophy) NEP expression wasincreased compared with normal control values (mean2.2 ± 0.7 and 1.7 ± 0.4 fold increase, respectively,p < 0.01; Fig. 2b), likely reflecting a higher degree ofmuscle fiber regeneration (Broccolini et al. 2006).
We therefore analyzed NEP activity in h-IBM and normaland disease-control muscle biopsies. The mean phosphora-midon-sensitive enzymatic activity in normal control biop-sies was arbitrarily set as 1 and the activity values measuredin all samples were expressed as a percentage of it. Similarly
NEP
HIBM
HIBM
Aβ40 Merge
Fig. 1 NEP expression in h-IBM muscle biopsies. Double fluores-
cence immunohistochemistry showing increased NEP immunoreac-
tivity in vacuolated fibers of h-IBM muscle, mainly in the cytoplasmic
areas surrounding vacuoles. NEP immunoreactivity co-localized with
that of Ab40 in the same fibers. Scale bar: 20 lm.
(a)
(b)
Fig. 2 Despite the increased expression of NEP in isolated vacuo-
lated fibers, western blot analysis showed an overall reduction of
NEP expression level in h-IBM muscles corresponding to an aver-
age of 67 ± 14% of the normal control values (**p < 0.01; a and b).
On the contrary, a mean 2.2 ± 0.7 and 1.7 ± 0.4 fold increase in
NEP expression level was found in inflammatory myopathies
[including both polymyositis (PM) and dermatomyositis (DM)] and
muscular dystrophies (including both Duchenne and Becker mus-
cular dystrophy), respectively compared with normal controls
(**p < 0.01; b). Similarly to what observed by western blot analysis,
in h-IBM muscles NEP enzymatic activity was reduced to an aver-
age of 69 ± 17% of the normal control values (*p < 0.05; b). On the
contrary, in inflammatory myopathies and muscular dystrophies the
increased expression of NEP protein was paralleled by a similar
increase in its enzymatic activity (mean 2.5 ± 0.8 fold increase in
inflammatory myopathies and mean 1.8 ± 0.1 fold increase in
muscular dystrophies compared with normal control muscles;
**p < 0.01; b).
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981� 2008 The Authors
974 | A. Broccolini et al.

to what observed by western blot analysis, in h-IBM musclesthe average NEP enzymatic activity was reduced to69 ± 17% of the mean normal control value (p < 0.05;Fig. 2b). On the contrary in inflammatory myopathies and inmuscular dystrophies the increased expression of NEPprotein was paralleled by a similar increase of its enzymaticactivity (mean 2.5 ± 0.8 fold increase in inflammatorymyopathies and mean 1.8 ± 0.1 fold increase in musculardystrophies compared with normal control muscles;p < 0.01; Fig. 2b).
NEP is hyposialylated in h-IBM muscleNeprilysin protein is characterized by the presence of severalN-glycosylation sites (Devault et al. 1987) and contains largeamounts of sialic acid (Johnson et al. 1984). Previous studieshave shown that changes in the sugar moieties of the proteinaffect its stability and enzymatic activity (Lafrance et al.1994). As several lines of evidence suggest that hyposialy-lation of glycoproteins occurs in h-IBM muscle (Noguchiet al. 2004; Saito et al. 2004; Broccolini et al. 2005; Tajimaet al. 2005; Ricci et al. 2006), we therefore asked whether anabnormal sialylation of NEP could be responsible for itsreduced expression and enzymatic activity. First, we lookedfor an abnormal sialylation of NEP in h-IBM muscle bywestern blot analysis of glycoprotein enriched samples. Totalmuscle homogenates were purified by lectin affinity chro-matography using MAL that is capable of binding toglycoproteins via their sialic acid residues. Western blotanalysis carried on glycoprotein samples showed a reductionof NEP more pronounced than what observed in total musclehomogenates. Specifically, NEP level was reduced to39 ± 7% of the normal control mean value (p < 0.05;Fig. 3). The further reduction of NEP signal on western blot
of h-IBM samples after glycoprotein enrichment indicates areduced binding of NEP to MAL and, therefore, that NEP ishyposialylated. All the experiments were run in duplicate andalways gave the same results.
The experimental removal of sialic acid from muscleglycoproteins results in reduced NEP stability andenzymatic activity in vitroTo possibly explain the reduced expression of NEP in h-IBMmuscle, we looked whether the experimental removal ofsialic acid from NEP was sufficient to reduce its stability andenzymatic activity in vitro. Primary subconfluent myoblasts,obtained from both normal control and h-IBM musclebiopsies (five different culture sets from each group), wereinduced to differentiate into myotubes by keeping them indifferentiation medium for 5 days. Myotubes were thenmaintained in a medium devoid of FBS (to avoid the uptakeof exogenous sialic acid and glycoproteins) for additional2 days and then de-sialylated using VCN at a final concen-tration of 0.03 U/mL for 24 h. By western blot analysis oftotal cellular homogenates, in cultures treated with VCN,NEP was reduced to a mean value of 58.4 ± 23.9%compared with untreated controls (p < 0.01, n = 5; Fig. 4aand b). The efficacy of the enzyme treatment was verified bythe fact that NEP protein appeared as a weaker band in MAL-purified glycoproteins from VCN-treated cultures comparedwith what observed in total homogenates (Fig. 4a). Thisreduced binding of NEP to MAL confirms that NEP wasindeed hyposialylated in VCN-treated cultures. The reducedNEP protein level in VCN-treated cultures was associatedwith a reduction of the phosphoramidon-sensitive endo-peptidase activity corresponding to a mean value of40.8 ± 25.3% of the control values (p < 0.01, n = 5;Fig. 4b).
To verify whether the reduction of NEP protein expres-sion, following experimental de-sialylation, reflected similarchanges of NEP mRNA levels, we performed northern blotanalysis on total RNA samples extracted from culturesexposed to VCN for 24 h. This showed that NEP mRNAlevel did not change following treatment with VCN duringthe entire time course, thus suggesting that the reducedexpression of NEP protein might be rather because of areduced stability of the de-sialylated peptide (Fig. 5a). To testthis hypothesis, cultured myotubes were exposed to 35 mMCEX (an antibiotic known to inhibit protein synthesis) eitheralone or in combination with VCN 0.03 U/mL for 24 h.Myotubes kept in no-serum medium containing vehicle only(dimethyl sulfoxide) for the same time span were used ascontrols. In myotubes exposed to CEX, NEP protein wasreduced to 43% of control values after 24 h (Fig. 5b and c).Based on this evidence, the predicted half-life of NEP in ourcellular system is 22 h, less than what previously describedby other investigators (Lorkowski et al. 1987), possibly inconnection with our different culturing conditions. However,
Fig. 3 Western blot analysis of glycoproteins enriched by lectin affinity
chromatography using MAL. Following glycoprotein extraction, the
reduction of NEP signal appeared more pronounced than what
observed in total muscle homogenates (39 ± 7% of the mean normal
control value; p < 0.05). This further reduction of NEP signal indicates
a reduced binding of NEP to MAL, thus suggesting abnormal sialyla-
tion of the protein. The Coomassie brilliant blue staining confirms an
even loading of glycoprotein extracts from different samples. Analo-
gous results were obtained with all h-IBM samples.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981
Neprilysin in h-IBM | 975

the combined exposure of cultured myotubes to both CEXand VCN resulted in a further reduction of NEP protein levelat 24 h corresponding to 20% of the value measured inuntreated muscle cultures (Fig. 5b and c). This evidenceargue in favor of a reduced stability of NEP followingexperimental de-sialylation of cultured myotubes. It isinteresting to note that no difference, regarding the expres-sion and molecular characteristics of NEP, could be observedbetween normal control and h-IBM muscle cultures, either inbasal conditions or after VCN treatment. This suggests thath-IBM muscle cultures are still able to autonomouslyproduce sialic acid despite the presence of mutations in theGNE gene.
The experimental removal of sialic acid from muscleglycoproteins is associated with an increasedimmunoreactivity for Ab40 in cultured myotubesNEP has been shown to be an Ab cleaving enzyme (Howellet al. 1995) and possibly plays a role in the pathogenicmechanism underlying AD. Provided that the enzymaticremoval of sialic acid from muscle glycoproteins led to areduced expression and activity of NEP, we then verifiedwhether such an experimental setting was also associatedwith the accumulation of Ab within cultured myotubes.Primary myotubes obtained from both normal controls and
h-IBM patients were therefore analyzed for the expression ofAb40 and NEP after the addition of VCN in the culturingmedium. By immunocytochemistry, in normal myotubes keptfor 24 h in medium containing only serum replacementfactors without the addiction of VCN, Ab40 was detected as afaint cytoplasmic signal. An identical result was obtainedalso in h-IBM myotubes kept in the same culturingconditions for 24 h (Fig. 6a and c). On the contrary, theexperimental de-sialylation of cultured cells from bothnormal control and h-IBM muscles resulted in the presenceof distinct foci strongly immunoreactive for Ab40 within thecytoplasm of myosin-positive myotubes (Fig. 6d–f). Simi-larly to what observed in abnormal fibers of h-IBM muscle,Ab40 immunoreactivity co-localized with that of NEP(Fig. 6g–l).
Discussion
Despite the identification of the genetic defect responsible forh-IBM, the pathogenic mechanisms leading to progressivemuscle degeneration and weakness remain elusive. Severallines of evidence suggest that mutations of the GNE generesult in hyposialylation of glycoproteins in h-IBM muscle(Noguchi et al. 2004; Saito et al. 2004; Broccolini et al.2005; Tajima et al. 2005; Ricci et al. 2006), but it has not
(a)
(b)
Fig. 4 Western blot analysis of NEP expression after the experi-
mental removal of sialic acid in culture. Cultured human primary
myotubes were exposed to VCN at a final concentration of 0.03 U/
mL for 24 h. In total homogenates of VCN-treated cultures NEP
expression was reduced to a mean value of 58.4 ± 23.9% com-
pared with untreated controls (**p < 0.01, n = 5; a and b). The
efficacy of the enzyme treatment was verified by the fact that NEP
protein appeared as a weaker band in MAL-purified glycoproteins
from VCN-treated cultures compared with what observed in total
homogenates. The Coomassie brilliant blue staining confirms an
even loading of glycoprotein extracts from different samples (a). The
reduced NEP protein level in VCN-treated cultures was associated
with a reduction of the phosphoramidon-sensitive endopeptidase
activity corresponding to a mean value of 40.8 ± 25.3% of the
control values (**p < 0.01, n = 5; b). Of note, the illustrated data
refer to cultured myotubes from normal control individuals. Identical
results were obtained when muscle cultures derived from h-IBM
patients were used.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981� 2008 The Authors
976 | A. Broccolini et al.

been definitely determined whether this abnormality plays acrucial role in the activation of pathogenic cellular mecha-nisms. h-IBM muscle is characterized by the abnormalaccumulation of proteins commonly observed in AD brainincluding Ab (Askanas and Engel 1998, 2002). Theseabnormalities have also been recently replicated in a mousemodel of the disease carrying the p.D176V mutation of theGNE gene (Malicdan et al. 2007).
In the present study, we investigated a possible role ofNEP, a glycoprotein known to be capable of cleaving Ab atmultiple sites (Howell et al. 1995), in the pathogenicmechanism taking place in h-IBM muscle.
In all h-IBM muscles analyzed the overall expression ofNEP was reduced, although in rare vacuolated muscle fibersimmunohistochemistry analysis showed the presence of focalaccumulation of the protein. This apparent discrepancy maybe simply related to the fact that the cellular NEP becomesaggregated within distinct foci, together with Ab40, in themore advanced stages of muscle fiber degeneration, thusresulting in the presence of cytoplasmic areas of increasedimmunoreactivity.
A lower level of NEP protein expression was associatedwith a parallel significant reduction of its enzymatic activity.Of note, in our h-IBM patients the observed NEP abnormal-ities were not connected with the impairment of a specificdomain of the enzyme, as they were detected in patientsharboring a homozygous mutation in either the epimerasedomain or the kinase domain, and in 2 patients who werecompound heterozygous for GNE mutations affecting differ-ent domains.
Previous studies have proven that the experimentalremoval of some or all the glucidic chains of NEP resultsin the impairment of protein stability and, although to a lesserextent, enzymatic activity (Lafrance et al. 1994). Such aremoval of the glucidic chains produces a noticeablereduction of NEP molecular weight (Lafrance et al. 1994)that was not observed in our h-IBM samples, thus ruling outmajor glycosylation abnormalities of the protein. However,reduced sialylation of NEP appears to be present in h-IBMmuscle as suggested by its reduced binding to MAL. In h-IBM muscle, we found a direct correlation between NEPamount and its specific enzymatic activity, similarly to whatobserved in all the other normal and diseased musclesanalyzed. This suggests that the overall reduction of NEPenzymatic activity is likely caused by a reduced expressionof the protein rather than an impairment of its catalyticproperties. As a matter of fact, we found that the experi-mental removal of sialic acid from glycoproteins of culturedmyotubes results in a reduced expression, and thereforeenzymatic activity, of NEP. This was not because of areduced transcription of the gene, as the analysis of NEPmRNA did not reveal any significant change of its expressionthroughout the entire time course of VCN treatment, thussuggesting that such change in NEP protein expression was
(a)
(b)
(c)
Fig. 5 NEP expression level is down-regulated at the post-tran-
scriptional level in VCN-treated cultures. Exposure of cultured myo-
tubes to VCN 0.03 U/mL for 24 h did not result in a change of NEP
mRNA level as evidenced by northern blot analysis. (a) To test
whether the down-regulation of NEP expression was due to a re-
duced stability of the protein in VCN-treated cultures, myotubes were
exposed to 35 mM cycloheximide (CEX) either alone or in combi-
nation with VCN 0.03 U/mL for 24 h. In myotubes exposed to CEX
alone, NEP protein was reduced to 43% of control values after 24 h
(b and c), whereas the combined exposure of cultures to both CEX
and VCN resulted in a further reduction of NEP protein level at 24 h
corresponding to 20% of the value measured in untreated control
muscle cultures (b and c).
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981
Neprilysin in h-IBM | 977

mainly as a result of post-transcriptional mechanism/s.Indeed, it appears that de-sialylation of NEP results in asignificant reduction of protein stability as suggested by ourin vitro analysis after inhibition of protein synthesis by CEX.It is worth mentioning that overlapping results were obtainedusing either normal control or h-IBM muscle cultures. Thisindicates that, differently from what observed in the corre-sponding mature muscle samples, h-IBM myotubes do notshow substantial abnormalities in the sialylation level ofglycoproteins, and is in agreement with a previous reportshowing no significant difference in sialic acid contentbetween h-IBM and normal control muscle cells in culture(Salama et al. 2005). It has to be considered that our resultswere obtained in rather ‘young’ h-IBM cultured myotubesand it cannot be excluded that the metabolic set-up maychange with their increasing age in culture. Nonetheless, ourongoing experiments have so far shown that the sialic acid
biosynthetic pathway is functionally maintained in theimmature h-IBM muscle despite the presence of mutationsin the GNE gene (A. Broccolini and M. Mirabella, unpub-lished observations). However, it is not known whether, inthe immature h-IBM fiber (i) the GNE defect is rescued bythe up-regulation of other enzyme/s of the sialic acidbiosynthetic pathway or (ii) sialic acid can be synthesizedby other yet unknown metabolic pathway/s, that are specif-ically activated in the course of muscle fiber developmentand then progressively shut down as differentiation pro-gresses.
Previous studies have demonstrated that increased sialy-lation of specific glycoproteins results in their increasedstability and polysialylation of pharmacologically activepeptides has been proposed as a potentially promising routeto enhance their therapeutic value (Fernandes and Gregori-adis 2001; Jain et al. 2003; Bork et al. 2007). Our results
(a) (b) (c) (d) (e) (f)
(g) (h) (i) (j) (k) (l)
Fig. 6 Immunocytochemistry for NEP and
Ab40 in cultured myotubes treated with
VCN. In myotubes kept for 24 h in medium
containing only serum replacement factors
without the addiction of VCN, Ab40 was
detected as a faint cytoplasmic signal.
Myosin staining was used to identify myo-
tubes (a–c). On the contrary, the experi-
mental de-sialylation of cultured cells
derived from both normal control and h-IBM
muscles resulted in the presence of distinct
foci strongly immunoreactive for Ab40 within
the cytoplasm of myosin-positive myotubes
(d–f). Similarly to what observed in some
fibers of h-IBM muscle, Ab40 immunoreac-
tivity co-localized with that of NEP within
myotubes (g–l). Scale bar: 10 lm.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981� 2008 The Authors
978 | A. Broccolini et al.

suggest that, in h-IBM muscle, the reduced expression ofNEP might be primarily because of its reduced sialylation.Although a reduced stability of NEP has been so fardemonstrated only after removal of the entire glucidic chains(either complete or partial) (Lafrance et al. 1994), we cannotexclude that also a reduction of the sialic acid content of NEPmay per se be sufficient to impair protein stability. We canalso speculate that, in h-IBM muscle, hyposialylation of NEPmay perturb its proper folding, thus impairing the traffickingthrough the endoplasmic reticulum and the Golgi networkand the translocation to the plasma membrane, as previouslydemonstrated for NEP and for other glycoproteins indifferent cell systems (Lafrance et al. 1994; Hebert et al.2005). This would probably activate a mechanism ofendoplasmic reticulum stress ultimately leading to increaseddegradation of the protein by the proteasome (Zhang andKaufman 2006). Whether such mechanism is responsible forthe reduced expression of NEP in h-IBM muscle remains tobe elucidated.
To date numerous studies have demonstrated an inversecorrelation between NEP level and Ab aggregation, thusconfirming a crucial role of NEP in Ab clearance. In fact, ithas been shown that up-regulation of NEP in the brain oftransgenic mice over-expressing AbPP results in reducedamyloid plaque formation (Leissring et al. 2003; Marr et al.2004). On the contrary, the complete or partial disruption ofthe NEP gene in mouse models expressing relative lowlevels of human AbPP consistently leads to a variableincrease in Ab40 and Ab42 levels and amyloid plaqueburden in the brain (Farris et al. 2007). Moreover, thetypical Ab deposition in the brain of AD patients, as wellas in that of aged normal subjects, is associated with areduced expression of NEP in vulnerable areas (Akiyamaet al. 2001; Yasojima et al. 2001; Hellstrom-Lindahlet al. 2006). In our cellular system, the experimentalde-sialylation was associated with increased immunoreac-tivity for Ab40, mainly in the form of rounded or linearinclusions within the cytoplasm of cultured myotubes,similarly to what observed in h-IBM abnormal musclefibers. Provided that the experimental de-sialylation resultsin lower expression, and therefore overall reduced enzy-matic activity, of NEP, we can hypothesize that thiscontributes, at least in part, to the abnormal accumulationof Ab40.
Of course, we cannot rule out the possibility that theexperimental de-sialylation of cultured myotubes mayinterfere with other metabolic pathway/s involved in Abclearance. We do not know whether the functional defectof NEP, as found in h-IBM muscle, is per se sufficient totrigger Ab accumulation. In fact, it has to be consideredthat h-IBM muscle is also characterized by an increasedexpression of AbPP mRNA and protein (Askanas andEngel 1998, 2002), promoted by abnormal cellular mech-anisms possibly connected with mutations in the GNE
gene. However, in the complex and yet unexplainedmolecular pathogenic scenario of h-IBM muscle, thepossibility exists that hyposialylated and dysfunctionalNEP has a role in hampering the cellular Ab clearingsystem, thus contributing to its abnormal accumulationwithin vulnerable fibers.
We have previously shown that NEP participates tomuscle fiber development/regeneration and suggested thatNEP is capable of cleaving the insulin-like growth factor-I(IGF-I) binding protein 5, thus modulating the activationof the IGF-I/Akt pathway within muscle fibers (Broccoliniet al. 2006). In fact, down-regulation of IGF-I bindingprotein 5 results in a higher level of free and metabolicallyactive IGF-I that is therefore available for interaction withother molecular partners and activation of the IGF-I/Aktpathway, thus promoting mechanisms of cell survival(Datta et al. 1999). It is worth mentioning that veryrecently a possible derangement of apoptotic mechanismsinvolving the IGF-I/Akt pathway has been proposed to beinvolved in the pathophysiology of h-IBM (Amsili et al.2007). Taking into account these considerations, thepossibility exists that a dysfunctional NEP may be alsoinstrumental in hindering the IGF-I/Akt cellular survivalpathway, thus favoring the progressive degenerative pro-cess of h-IBM muscle.
We are aware of the fact that only a partial reduction ofNEP activity (correspondent to an average of 31% of thenormal values), as detected in h-IBM muscle, can raisedoubts regarding its efficacy in mediating pathology, as theimpact of this isolated defect on the muscle fiber homeostasismay be negligible at a given time point. However, it ispossible that the relentless effects of this abnormality over alonger period of time, in conjunction with other yet unknownpathogenic mechanisms, may contribute to muscle fiberdegeneration.
In conclusion, we have demonstrated that hyposialylationof NEP may have a detrimental effect in its stability andenzymatic function and possibly plays a role in thepathogenic mechanism activated by mutations of the GNEgene in h-IBM muscle. More in general, we have providedevidence that hyposialylation of muscle glycoproteins mayindeed represent an important aspect in h-IBM patho-physiology, as already suggested by other authors (Noguchiet al. 2004; Saito et al. 2004; Tajima et al. 2005; Malicdanet al. 2007). If this will be proven true also by future studies,then to better understand and possibly modulate the molec-ular mechanism/s underlying the biosynthesis of sialic acid inh-IBM muscle fibers can potentially offer a therapeuticavenue for this crippling disorder.
Acknowledgements
This study was supported by Grants from the Catholic University to
AB and MM.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981
Neprilysin in h-IBM | 979

References
Akiyama H., Kondo H., Ikeda K., Kato M. and McGeer P. L. (2001)Immunohistochemical localization of neprilysin in the humancerebral cortex: inverse association with vulnerability to amyloidbeta-protein (Ab) deposition. Brain Res. 902, 277–281.
Amsili S., Shlomai Z., Levitzki R., Krause S., Lochmuller H., Ben-Bassat H. and Mitrani-Rosenbaum S. (2007) Characterization ofhereditary inclusion body myopathy myoblasts: possible primaryimpairment of apoptotic events. Cell Death Differ. 14, 1916–1924.
Argov Z. and Yarom R. (1984) ‘Rimmed vacuole myopathy’ sparing thequadriceps. A unique disorder in Iranian Jews. J. Neurol. Sci. 64,33–43.
Askanas V. and Engel W. K. (1992) Cultured normal and geneticallyabnormal human muscle, in Handbook of Clinical Neurology(Rowland L. P. and Di Mauro S., eds), pp. 85–116. ElsevierScience Publisher, Amsterdam, the Netherlands.
Askanas V. and Engel W. K. (1998) Sporadic inclusion-body myositisand hereditary inclusion-body myopathies: current concepts ofdiagnosis and pathogenesis. Curr. Opin. Rheumatol. 10, 530–542.
Askanas V. and Engel W. K. (2002) Inclusion-body myositis andmyopathies: different etiologies, possibly similar pathogenicmechanisms. Curr. Opin. Neurol. 15, 525–531.
Bork K., Reutter W., Weidemann W. and Horstkorte R. (2007) Enhancedsialylation of EPO by overexpression of UDP-GlcNAc 2-epimer-ase/ManAc kinase containing a sialuria mutation in CHO cells.FEBS Lett. 581, 4195–4198.
Broccolini A., Gliubizzi C., Pavoni E. et al. (2005) a-Dystroglycandoes not play a major pathogenic role in autosomal recessivehereditary inclusion-body myopathy. Neuromuscul. Disord. 15,177–184.
Broccolini A., Gidaro T., Morosetti R., Gliubizzi C., Servidei T., Pesc-atori M., Tonali P. A., Ricci E. and Mirabella M. (2006) Neprilysinparticipates in skeletal muscle regeneration and is accumulated inabnormal muscle fibres of inclusion body myositis. J. Neurochem.96, 777–789.
Datta S. R., Brunet A. and Greenberg M. E. (1999) Cellular survival: aplay in three Akts. Genes Dev. 13, 2905–2927.
Devault A., Lazure C., Nault C. et al. (1987) Amino acid sequence ofrabbit kidney neutral endopeptidase 24.11 (enkephalinase) deducedfrom a complementary DNA. EMBO J. 6, 1317–1322.
Eisenberg I., Avidan N., Potikha T. et al. (2001) The UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase gene ismutated in recessive hereditary inclusion body myopathy. Nat.Genet. 29, 83–87.
Farris W., Schutz S. G., Cirrito J. R. et al. (2007) Loss of neprilysinfunction promotes amyloid plaque formation and causes cerebralamyloid angiopathy. Am. J. Pathol. 171, 241–251.
Fernandes A. I. and Gregoriadis G. (2001) The effect of polysialylationon the immunogenicity and antigenicity of asparaginase: implica-tion in its pharmacokinetics. Int. J. Pharm. 217, 215–224.
Hebert D. N., Garman S. C. and Molinari M. (2005) The glycan code ofthe endoplasmic reticulum: asparagine-linked carbohydrates asprotein maturation and quality-control tags. Trends Cell Biol. 15,364–370.
Hellstrom-Lindahl E., Ravid R. and Nordberg A.(2008) Age-dependentdecline of neprilysin in Alzheimer’s disease and normal brain:inverse correlation with Ab levels. Neurobiol. Aging 29, 210–221.
Hinderlich S., Stasche R., Zeitler R. and Reutter W. (1997) A bifunc-tional enzyme catalyzes the first two steps in N-acetylneuraminicacid biosynthesis of rat liver. Purification and characterization
of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosaminekinase. J. Biol. Chem. 272, 24313–24318.
Howell S., Nalbantoglu J. and Crine P. (1995) Neutral endopeptidase canhydrolyze beta-amyloid(1-40) but shows no effect on beta-amyloidprecursor protein metabolism. Peptides 16, 647–652.
Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N. P.,Gerard C., Hama E., Lee H. J. and Saido T. C. (2001) Meta-bolic regulation of brain Ab by neprilysin. Science 292, 1550–1552.
Jain S., Hreczuk-Hirst D. H., McCormack B., Mital M., Epenetos A.,Laing P. and Gregoriadis G. (2003) Polysialylated insulin: syn-thesis, characterization and biological activity in vivo. Biochim.Biophys. Acta 1622, 42–49.
Johnson A. R., Skidgel R. A., Gafford J. T. and Erdos E. G. (1984)Enzymes in placental microvilli: angiotensin I converting enzyme,angiotensinase A, carboxypeptidase, and neutral endopeptidase(‘enkephalinase’). Peptides 5, 789–796.
Kerr M. A. and Kenny A. J. (1974) The purification and specificity of aneutral endopeptidase from rabbit kidney brush border. Biochem.J. 137, 477–488.
Lafrance M. H., Vezina C., Wang Q., Boileau G., Crine P. and LemayG.(1994) Role of glycosylation in transport and enzymic activityof neutral endopeptidase-24.11. Biochem. J. 302 (Pt. 2), 451–454.
Leissring M. A., Farris W., Chang A. Y., Walsh D. M., Wu X., Sun X.,Frosch M. P. and Selkoe D. J. (2003) Enhanced proteolysis ofbeta-amyloid in APP transgenic mice prevents plaque formation,secondary pathology, and premature death. Neuron 40, 1087–1093.
Li C. and Hersh L. B. (1995) Neprilysin: assay methods, purification,and characterization. Methods Enzymol. 248, 253–263.
Lorkowski G., Zijderhand-Bleekemolen J. E., Erdos E. G., von Figura K.and Hasilik A. (1987) Neutral endopeptidase-24.11 (enkephalin-ase). Biosynthesis and localization in human fibroblasts. Biochem.J. 248, 345–350.
Malicdan M. C., Noguchi S., Nonaka I., Hayashi Y. K. and Nishino I.(2007) A Gne knockout mouse expressing human GNE D176Vmutation develops features similar to distal myopathy with rimmedvacuoles or hereditary inclusion body myopathy. Hum. Mol. Genet.16, 2669–2682.
Marr R. A., Guan H., Rockenstein E., Kindy M., Gage F. H., Verma I.,Masliah E. and Hersh L. B. (2004) Neprilysin regulates amyloidbeta peptide levels. J. Mol. Neurosci. 22, 5–11.
Noguchi S., Keira Y., Murayama K. et al. (2004) Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinaseactivity and sialylation in distal myopathy with rimmed vacuoles.J. Biol. Chem. 279, 11402–11407.
Ricci E., Broccolini A., Gidaro T., Morosetti R., Gliubizzi C., FruscianteR., Di Lella G. M., Tonali P. A. and Mirabella M. (2006) NCAM ishyposialylated in hereditary inclusion body myopathy due to GNEmutations. Neurology 66, 755–758.
Saito F., Tomimitsu H., Arai K., Nakai S., Kanda T., Shimizu T.,Mizusawa H. and Matsumura K. (2004) A Japanese patient withdistal myopathy with rimmed vacuoles: missense mutations in theepimerase domain of the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene accompanied byhyposialylation of skeletal muscle glycoproteins. Neuromuscul.Disord. 14, 158–161.
Salama I., Hinderlich S., Shlomai Z. et al. (2005) No overall hyposial-ylation in hereditary inclusion body myopathy myoblasts carryingthe homozygous M712T GNE mutation. Biochem. Biophys. Res.Commun. 328, 221–226.
Shirotani K., Tsubuki S., Iwata N. et al. (2001) Neprilysin degrades bothamyloid beta peptides 1-40 and 1-42 most rapidly and efficiently
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981� 2008 The Authors
980 | A. Broccolini et al.

among thiorphan- and phosphoramidon-sensitive endopeptidases.J. Biol. Chem. 276, 21895–21901.
Tajima Y., Uyama E., Go S. et al. (2005) Distal myopathy with rimmedvacuoles: impaired O-glycan formation in muscular glycoproteins.Am. J. Pathol. 166, 1121–1130.
Turner A. J., Isaac R. E. and Coates D. (2001) The neprilysin (NEP)family of zinc metalloendopeptidases: genomics and function.Bioessays 23, 261–269.
Yasojima K., Akiyama H., McGeer E. G. and McGeer P. L. (2001)Reduced neprilysin in high plaque areas of Alzheimer brain: apossible relationship to deficient degradation of beta-amyloidpeptide. Neurosci. Lett. 297, 97–100.
Zhang K. and Kaufman R. J. (2006) The unfolded protein response: astress signaling pathway critical for health and disease. Neurology66, S102–S109.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 971–981
Neprilysin in h-IBM | 981
Related Documents











