FULL PAPER DOI: 10.1002/ejoc.201100846 Special Section Hyaluronan–Carbon Nanotube Derivatives: Synthesis, Conjugation with Model Drugs, and DOSY NMR Characterization Riccardo Marega, [a] Massimo Bergamin, [b] Vincent Aroulmoji, [b] Francesca Dinon, [c] Maurizio Prato,* [d] and Erminio Murano* [b] Dedicated to Professor Gianfranco Scorrano on the occasion of his 72nd birthday Keywords: Nanotubes / NMR spectroscopy / Drug delivery / Conjugation Carbon nanotube (CNTs) derivatives are nowadays under thorough investigation as biomedically interesting materials. In this paper we describe a method for the preparation of water-soluble CNTs by condensation of the carboxylic groups introduced onto the carbon framework and the pri- mary amine moieties inserted in the naturally occurring bio- polymer hyaluronan (HA). The covalent conjugation be- tween CNTs and HA should merge the biocompatibility and further processability of the HA chains with the well-known Introduction The emerging applications of carbon nanotubes (CNTs) in the biomedical field are nowadays under thorough inves- tigation. [1] In fact, after appropriate derivatization tech- niques [2] it is possible to improve CNT solubility in water [3,4] and to covalently attach known drugs. [5] As an example, after 1,3-dipolar cycloaddition of azomethine ylides, [3,6] the anticancer drug methotrexate (MTX), [7] the antifungal amphotericin B (AmB), [8] and an indium-based radiotracer [9] were efficiently conjugated to the CNT struc- ture, yielding derivatives that show interesting biological properties. [10] After noncovalent functionalization of CNTs, some interesting derivatives bearing anticancer drugs, for example, cisplatinum complexes [11] and doxorubicin, [12] were also reported. In the present study, we report on the covalent function- alization of oxidized single-walled CNTs (ox-SWNTs) with [a] Department of Chemistry, University of Namur, Rue Bruxelles 61, Namur, 5000, Belgium [b] Protos Research Institute, c/o BIC Incubatori FVG, Via Flavia 23/1, Trieste, 34148, Italy E-mail: [email protected] [c] Cimteclab Spa, c/o Area Science Park, Padriciano 99, Trieste, 34149, Italy [d] Department of Chemical and Pharmaceutical Sciences, University of Trieste, Piazzale Europa 1, Trieste, 34127, Italy E-mail: [email protected] Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/ejoc.201100846. Eur. J. Org. Chem. 2011, 5617–5625 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 5617 cellular penetration properties of the CNT derivatives to pro- duce novel drug delivery platforms. In fact, thanks to the pri- mary amino groups introduced in the HA chains, HA–CNT derivatives can be further covalently modified with model drugs like ibuprofen and methotrexate. We describe also the monitoring of all the CNT derivatization steps by diffusion- ordered NMR spectroscopy (DOSY), a technique that allows fast and reliable characterization of these novel derivatives. a hyaluronan (HA) derivative to impart significant water solubility to ox-SWNTs and to provide functional groups for further attachment of model drugs. HA is a naturally occurring biopolymer consisting of the linear sequence of β-(13)-sodium glucuronate-β-(14)-N-acetyl-d-glucos- amine, [13] which can be easily functionalized with amino groups at the primary hydroxy groups of the N-acetyl-d- glucosamine moiety, as recently reported. [14] Such amino groups were exploited for both the condensation with car- boxylic groups along the ox-SWNT structure and for fur- ther attachment of model drugs, like the anti-inflammatory drug ibuprofen (Ibu) and the anticancer drug methotrexate (MTX). By this way, thebiological properties, the water sol- ubility, and the further chemical processability of the HA chains can be merged with the well-known cellular penetra- tion and multivalent properties of the CNTs. [10] Usually, characterization of CNT derivatives is carried out by a number of analytical techniques, such as microscopy-based analysis (transmission-electron microscopy and atomic- force microscopy), spectroscopy-based analysis (Raman, UV/Vis, fluorescence, X-ray photoelectron spectroscopy, and infrared analysis), thermogravimetric analysis (TGA), [15] and more recently 1 H NMR spectroscopy and diffusion-ordered spectroscopy (DOSY). [16,17] We report here the DOSY results obtained by measuring the reduced diffusion in solution of the organic moieties attached to the CNT framework. Owing to the water solubility of ox- SWNTs, given by the HA derivatives moiety, it was possible

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

FULL PAPER
DOI: 10.1002/ejoc.201100846
Sp
ecia
lS
ecti
on
Hyaluronan–Carbon Nanotube Derivatives: Synthesis, Conjugation with ModelDrugs, and DOSY NMR Characterization
Riccardo Marega,[a] Massimo Bergamin,[b] Vincent Aroulmoji,[b] Francesca Dinon,[c]
Maurizio Prato,*[d] and Erminio Murano*[b]
Dedicated to Professor Gianfranco Scorrano on the occasion of his 72nd birthday
Keywords: Nanotubes / NMR spectroscopy / Drug delivery / Conjugation
Carbon nanotube (CNTs) derivatives are nowadays underthorough investigation as biomedically interesting materials.In this paper we describe a method for the preparation ofwater-soluble CNTs by condensation of the carboxylicgroups introduced onto the carbon framework and the pri-mary amine moieties inserted in the naturally occurring bio-polymer hyaluronan (HA). The covalent conjugation be-tween CNTs and HA should merge the biocompatibility andfurther processability of the HA chains with the well-known
IntroductionThe emerging applications of carbon nanotubes (CNTs)
in the biomedical field are nowadays under thorough inves-tigation.[1] In fact, after appropriate derivatization tech-niques[2] it is possible to improve CNT solubility inwater[3,4] and to covalently attach known drugs.[5] As anexample, after 1,3-dipolar cycloaddition of azomethineylides,[3,6] the anticancer drug methotrexate (MTX),[7] theantifungal amphotericin B (AmB),[8] and an indium-basedradiotracer[9] were efficiently conjugated to the CNT struc-ture, yielding derivatives that show interesting biologicalproperties.[10] After noncovalent functionalization of CNTs,some interesting derivatives bearing anticancer drugs, forexample, cisplatinum complexes[11] and doxorubicin,[12]
were also reported.In the present study, we report on the covalent function-
alization of oxidized single-walled CNTs (ox-SWNTs) with
[a] Department of Chemistry, University of Namur,Rue Bruxelles 61, Namur, 5000, Belgium
[b] Protos Research Institute, c/o BIC Incubatori FVG,Via Flavia 23/1, Trieste, 34148, ItalyE-mail: [email protected]
[c] Cimteclab Spa, c/o Area Science Park,Padriciano 99, Trieste, 34149, Italy
[d] Department of Chemical and Pharmaceutical Sciences,University of Trieste,Piazzale Europa 1, Trieste, 34127, ItalyE-mail: [email protected] information for this article is available on theWWW under http://dx.doi.org/10.1002/ejoc.201100846.
Eur. J. Org. Chem. 2011, 5617–5625 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 5617
cellular penetration properties of the CNT derivatives to pro-duce novel drug delivery platforms. In fact, thanks to the pri-mary amino groups introduced in the HA chains, HA–CNTderivatives can be further covalently modified with modeldrugs like ibuprofen and methotrexate. We describe also themonitoring of all the CNT derivatization steps by diffusion-ordered NMR spectroscopy (DOSY), a technique that allowsfast and reliable characterization of these novel derivatives.
a hyaluronan (HA) derivative to impart significant watersolubility to ox-SWNTs and to provide functional groupsfor further attachment of model drugs. HA is a naturallyoccurring biopolymer consisting of the linear sequence ofβ-(1�3)-sodium glucuronate-β-(1�4)-N-acetyl-d-glucos-amine,[13] which can be easily functionalized with aminogroups at the primary hydroxy groups of the N-acetyl-d-glucosamine moiety, as recently reported.[14] Such aminogroups were exploited for both the condensation with car-boxylic groups along the ox-SWNT structure and for fur-ther attachment of model drugs, like the anti-inflammatorydrug ibuprofen (Ibu) and the anticancer drug methotrexate(MTX). By this way, the biological properties, the water sol-ubility, and the further chemical processability of the HAchains can be merged with the well-known cellular penetra-tion and multivalent properties of the CNTs.[10] Usually,characterization of CNT derivatives is carried out by anumber of analytical techniques, such as microscopy-basedanalysis (transmission-electron microscopy and atomic-force microscopy), spectroscopy-based analysis (Raman,UV/Vis, fluorescence, X-ray photoelectron spectroscopy,and infrared analysis), thermogravimetric analysis(TGA),[15] and more recently 1H NMR spectroscopy anddiffusion-ordered spectroscopy (DOSY).[16,17] We reporthere the DOSY results obtained by measuring the reduceddiffusion in solution of the organic moieties attached to theCNT framework. Owing to the water solubility of ox-SWNTs, given by the HA derivatives moiety, it was possible
R. Marega, M. Bergamin, V. Aroulmoji, F. Dinon, M. Prato, E. MuranoFULL PAPER
Sp
ecia
lS
ecti
on
to determine, in a fast and reliable way, the conjugationeffects by using one dimensional[18] and two dimensionaldiffusion-ordered NMR spectroscopy[19] (1D DOSY and2D DOSY).
Results and Discussion
Production of Hyaluronan Oligomers
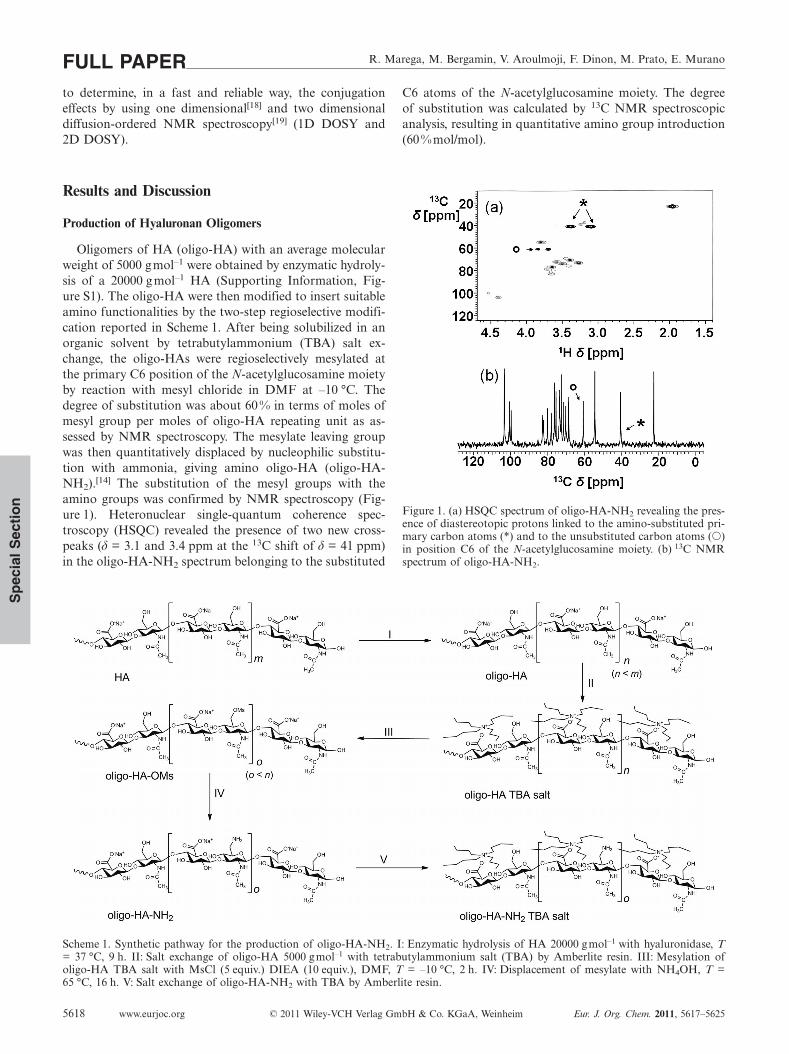
Oligomers of HA (oligo-HA) with an average molecularweight of 5000 gmol–1 were obtained by enzymatic hydroly-sis of a 20000 g mol–1 HA (Supporting Information, Fig-ure S1). The oligo-HA were then modified to insert suitableamino functionalities by the two-step regioselective modifi-cation reported in Scheme 1. After being solubilized in anorganic solvent by tetrabutylammonium (TBA) salt ex-change, the oligo-HAs were regioselectively mesylated atthe primary C6 position of the N-acetylglucosamine moietyby reaction with mesyl chloride in DMF at –10 °C. Thedegree of substitution was about 60 % in terms of moles ofmesyl group per moles of oligo-HA repeating unit as as-sessed by NMR spectroscopy. The mesylate leaving groupwas then quantitatively displaced by nucleophilic substitu-tion with ammonia, giving amino oligo-HA (oligo-HA-NH2).[14] The substitution of the mesyl groups with theamino groups was confirmed by NMR spectroscopy (Fig-ure 1). Heteronuclear single-quantum coherence spec-troscopy (HSQC) revealed the presence of two new cross-peaks (δ = 3.1 and 3.4 ppm at the 13C shift of δ = 41 ppm)in the oligo-HA-NH2 spectrum belonging to the substituted
Scheme 1. Synthetic pathway for the production of oligo-HA-NH2. I: Enzymatic hydrolysis of HA 20000 gmol–1 with hyaluronidase, T= 37 °C, 9 h. II: Salt exchange of oligo-HA 5000 gmol–1 with tetrabutylammonium salt (TBA) by Amberlite resin. III: Mesylation ofoligo-HA TBA salt with MsCl (5 equiv.) DIEA (10 equiv.), DMF, T = –10 °C, 2 h. IV: Displacement of mesylate with NH4OH, T =65 °C, 16 h. V: Salt exchange of oligo-HA-NH2 with TBA by Amberlite resin.
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 5617–56255618
C6 atoms of the N-acetylglucosamine moiety. The degreeof substitution was calculated by 13C NMR spectroscopicanalysis, resulting in quantitative amino group introduction(60 %mol/mol).
Figure 1. (a) HSQC spectrum of oligo-HA-NH2 revealing the pres-ence of diastereotopic protons linked to the amino-substituted pri-mary carbon atoms (*) and to the unsubstituted carbon atoms (�)in position C6 of the N-acetylglucosamine moiety. (b) 13C NMRspectrum of oligo-HA-NH2.

Hyaluronan–Carbon Nanotube Derivatives
Sp
ecia
lS
ecti
on
Coupling of Oligo-HA to SWNTs
The oxidation of SWNTs to generate carboxyl groups wascarried out stepwise by using the mineral-acid-based wetprocess previously reported.[16,20] The carboxylic groupswere then converted into N-hydroxysuccinimidyl estersthrough carbodiimide chemistry to give a ox-SWNT-OSucompound, which was isolated from the reaction mixtureto avoid intra- and intermolecular cross-linking side reac-tions of the oligo-HA-NH2 chains in the subsequent amid-ation step. Isolated ox-SWNT-OSu was allowed to reactwith oligo-HA-NH2 as TBA salt salt to form an oligo-HA-NHCO-ox-SWNT conjugate (Scheme 2). The reactionswere performed in DMF at room temperature over 48 h,and the conjugates were recovered from the reaction mix-ture by solubilization in water (see the Experimental Sec-tion). The water-soluble conjugate was then treated with abasic solution of NaOH at pH 11 to remove the ester link-ages that could have possibly formed during the condensa-tion. The conjugate oligo-HA-NHCO-ox-SWNT was thenpurified by discontinuous diafiltration, a membrane-basedpurification technique able to selectively separate soluteswith different molecular weights.[17] The excess amounts ofthe reagents were found in the filtered portion of the dia-filtration (Supporting Information, Figure S2). The purifiedconjugate was then isolated as a freeze-dried material. Themoisture content, determined by TGA analysis under an
Scheme 2. Synthetic routes for the production of oligo-HA-NHCO-ox-SWNT. I: 2.6 m HNO3, reflux 48 h. II: H2SO4/H2O2 (4:1), 1 h,35 °C. III: N-Hydroxysuccinimide (NHS), N-ethyl-N�-(dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), and 4-dimethylami-nopyridine (DMAP) in DMF, r.t., 220 min. IV: DMF, r.t. 48 h, then NaOH, pH 11 overnight.
Eur. J. Org. Chem. 2011, 5617–5625 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5619
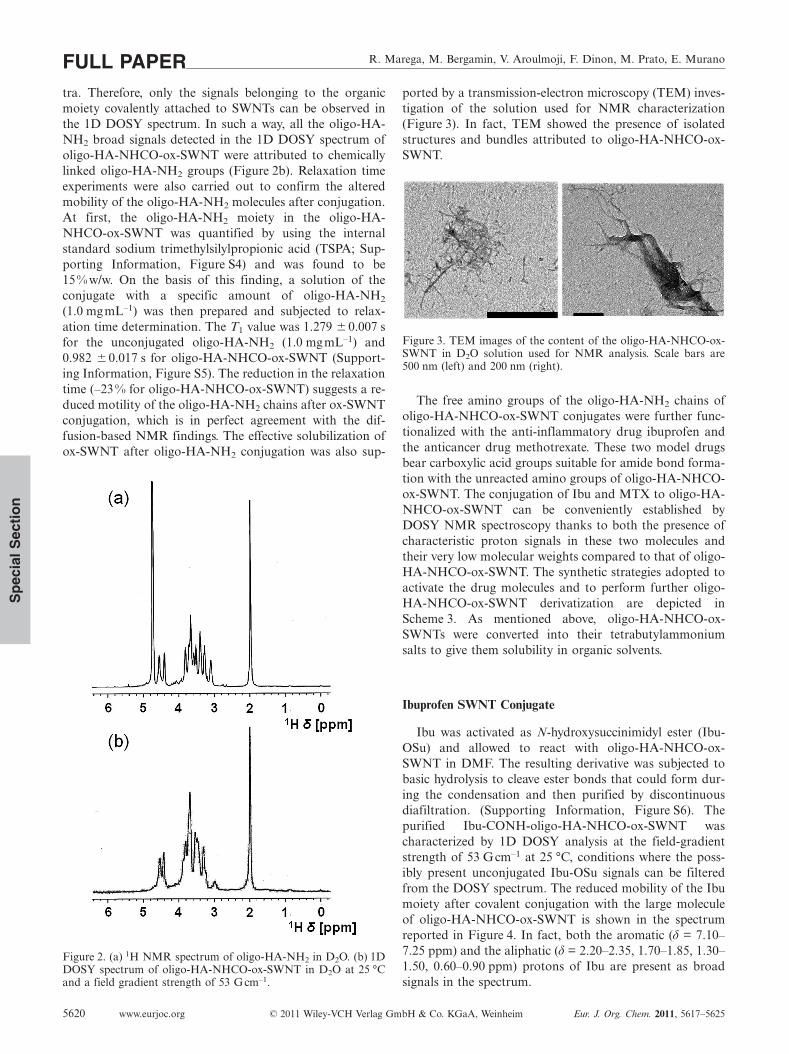
inert atmosphere in the range 50–250 °C, was in the orderof ≈4 %w/w (Supporting Information, Figure S3). The pres-ence of the oligo-HA-NH2 moieties on the SWNT back-bone was confirmed by NMR spectroscopy (Figure 2). The1H NMR spectrum of oligo-HA-NH2 reported in Figure 2ashows the typical signal pattern of the HA disaccharide re-peating unit, with the methyl protons of the N-acetylglu-cosamine moiety (δ = 1.96 ppm), the protons of the HAbackbone (δ = 3.19–3.71 ppm), and the anomeric ones (δ =4.42, 4.52 ppm). On the contrary, CNTs do not possess anyprotons, so that 1H NMR spectroscopic analysis does notdiscriminate between the oligo-HA-NHCO-ox-SWNT con-jugates and a physical mixture of ox-SWNTs-OSu andoligo-HA-NH2. For this reason, 1D DOSY was used tocharacterize the products after covalent conjugation. We re-cently reported, for the first time, the use of DOSY NMRto detect the covalent derivatization of ox-SWNTs.[16,17] Infact, 1D DOSY NMR detects the reduction of the diffusionof an organic moiety after its conjugation to the ox-SWNTdue to the formation of bigger macromolecules with slowerdiffusional behavior. Indeed, CNT derivatives show molec-ular weights in the order of millions of gmol–1 and diffusevery slowly if compared to the free, unconjugated organicmoieties. 1D DOSY analysis of oligo-HA-NHCO-ox-SWNT (Figure 2b) was performed at strong field-gradientstrengths to avoid the detection of lower molecular weightunconjugated oligo-HA-NH2 signals in the 1D DOSY spec-
R. Marega, M. Bergamin, V. Aroulmoji, F. Dinon, M. Prato, E. MuranoFULL PAPER
Sp
ecia
lS
ecti
on
tra. Therefore, only the signals belonging to the organicmoiety covalently attached to SWNTs can be observed inthe 1D DOSY spectrum. In such a way, all the oligo-HA-NH2 broad signals detected in the 1D DOSY spectrum ofoligo-HA-NHCO-ox-SWNT were attributed to chemicallylinked oligo-HA-NH2 groups (Figure 2b). Relaxation timeexperiments were also carried out to confirm the alteredmobility of the oligo-HA-NH2 molecules after conjugation.At first, the oligo-HA-NH2 moiety in the oligo-HA-NHCO-ox-SWNT was quantified by using the internalstandard sodium trimethylsilylpropionic acid (TSPA; Sup-porting Information, Figure S4) and was found to be15% w/w. On the basis of this finding, a solution of theconjugate with a specific amount of oligo-HA-NH2
(1.0 mgmL–1) was then prepared and subjected to relax-ation time determination. The T1 value was 1.279 � 0.007 sfor the unconjugated oligo-HA-NH2 (1.0 mgmL–1) and0.982 �0.017 s for oligo-HA-NHCO-ox-SWNT (Support-ing Information, Figure S5). The reduction in the relaxationtime (–23 % for oligo-HA-NHCO-ox-SWNT) suggests a re-duced motility of the oligo-HA-NH2 chains after ox-SWNTconjugation, which is in perfect agreement with the dif-fusion-based NMR findings. The effective solubilization ofox-SWNT after oligo-HA-NH2 conjugation was also sup-
Figure 2. (a) 1H NMR spectrum of oligo-HA-NH2 in D2O. (b) 1DDOSY spectrum of oligo-HA-NHCO-ox-SWNT in D2O at 25 °Cand a field gradient strength of 53 Gcm–1.
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 5617–56255620
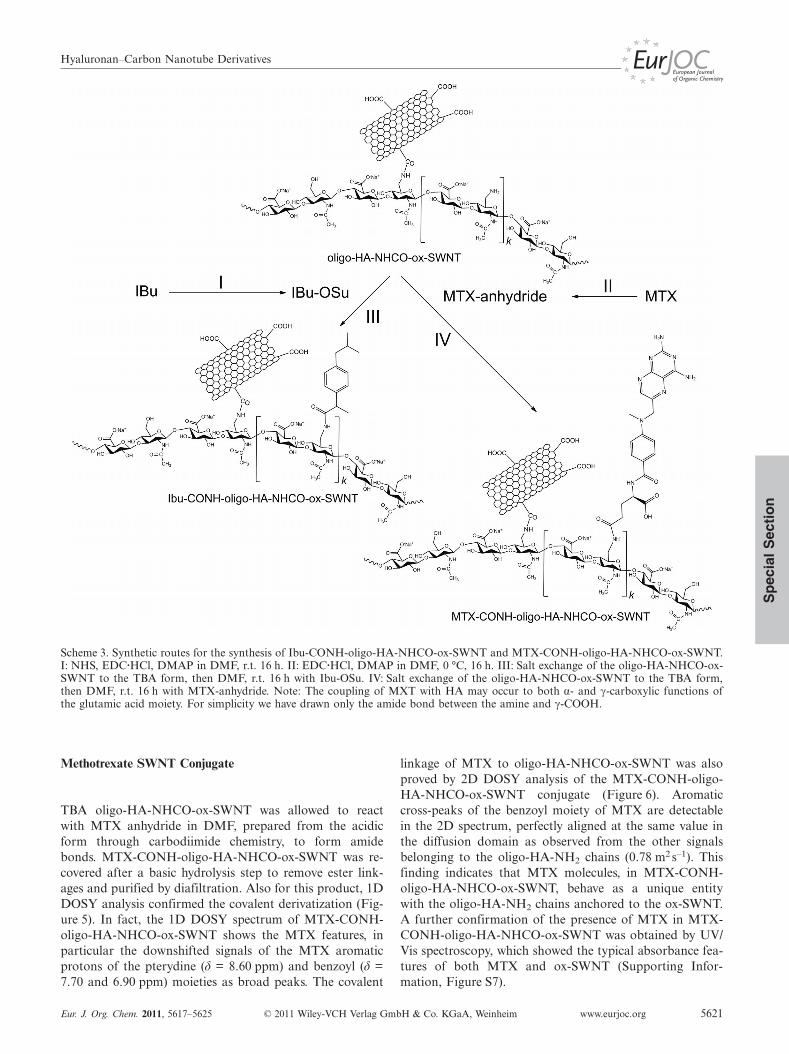
ported by a transmission-electron microscopy (TEM) inves-tigation of the solution used for NMR characterization(Figure 3). In fact, TEM showed the presence of isolatedstructures and bundles attributed to oligo-HA-NHCO-ox-SWNT.
Figure 3. TEM images of the content of the oligo-HA-NHCO-ox-SWNT in D2O solution used for NMR analysis. Scale bars are500 nm (left) and 200 nm (right).
The free amino groups of the oligo-HA-NH2 chains ofoligo-HA-NHCO-ox-SWNT conjugates were further func-tionalized with the anti-inflammatory drug ibuprofen andthe anticancer drug methotrexate. These two model drugsbear carboxylic acid groups suitable for amide bond forma-tion with the unreacted amino groups of oligo-HA-NHCO-ox-SWNT. The conjugation of Ibu and MTX to oligo-HA-NHCO-ox-SWNT can be conveniently established byDOSY NMR spectroscopy thanks to both the presence ofcharacteristic proton signals in these two molecules andtheir very low molecular weights compared to that of oligo-HA-NHCO-ox-SWNT. The synthetic strategies adopted toactivate the drug molecules and to perform further oligo-HA-NHCO-ox-SWNT derivatization are depicted inScheme 3. As mentioned above, oligo-HA-NHCO-ox-SWNTs were converted into their tetrabutylammoniumsalts to give them solubility in organic solvents.
Ibuprofen SWNT Conjugate
Ibu was activated as N-hydroxysuccinimidyl ester (Ibu-OSu) and allowed to react with oligo-HA-NHCO-ox-SWNT in DMF. The resulting derivative was subjected tobasic hydrolysis to cleave ester bonds that could form dur-ing the condensation and then purified by discontinuousdiafiltration. (Supporting Information, Figure S6). Thepurified Ibu-CONH-oligo-HA-NHCO-ox-SWNT wascharacterized by 1D DOSY analysis at the field-gradientstrength of 53 Gcm–1 at 25 °C, conditions where the poss-ibly present unconjugated Ibu-OSu signals can be filteredfrom the DOSY spectrum. The reduced mobility of the Ibumoiety after covalent conjugation with the large moleculeof oligo-HA-NHCO-ox-SWNT is shown in the spectrumreported in Figure 4. In fact, both the aromatic (δ = 7.10–7.25 ppm) and the aliphatic (δ = 2.20–2.35, 1.70–1.85, 1.30–1.50, 0.60–0.90 ppm) protons of Ibu are present as broadsignals in the spectrum.
Hyaluronan–Carbon Nanotube Derivatives
Sp
ecia
lS
ecti
on
Scheme 3. Synthetic routes for the synthesis of Ibu-CONH-oligo-HA-NHCO-ox-SWNT and MTX-CONH-oligo-HA-NHCO-ox-SWNT.I: NHS, EDC·HCl, DMAP in DMF, r.t. 16 h. II: EDC·HCl, DMAP in DMF, 0 °C, 16 h. III: Salt exchange of the oligo-HA-NHCO-ox-SWNT to the TBA form, then DMF, r.t. 16 h with Ibu-OSu. IV: Salt exchange of the oligo-HA-NHCO-ox-SWNT to the TBA form,then DMF, r.t. 16 h with MTX-anhydride. Note: The coupling of MXT with HA may occur to both α- and γ-carboxylic functions ofthe glutamic acid moiety. For simplicity we have drawn only the amide bond between the amine and γ-COOH.
Methotrexate SWNT Conjugate
TBA oligo-HA-NHCO-ox-SWNT was allowed to reactwith MTX anhydride in DMF, prepared from the acidicform through carbodiimide chemistry, to form amidebonds. MTX-CONH-oligo-HA-NHCO-ox-SWNT was re-covered after a basic hydrolysis step to remove ester link-ages and purified by diafiltration. Also for this product, 1DDOSY analysis confirmed the covalent derivatization (Fig-ure 5). In fact, the 1D DOSY spectrum of MTX-CONH-oligo-HA-NHCO-ox-SWNT shows the MTX features, inparticular the downshifted signals of the MTX aromaticprotons of the pterydine (δ = 8.60 ppm) and benzoyl (δ =7.70 and 6.90 ppm) moieties as broad peaks. The covalent
Eur. J. Org. Chem. 2011, 5617–5625 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5621
linkage of MTX to oligo-HA-NHCO-ox-SWNT was alsoproved by 2D DOSY analysis of the MTX-CONH-oligo-HA-NHCO-ox-SWNT conjugate (Figure 6). Aromaticcross-peaks of the benzoyl moiety of MTX are detectablein the 2D spectrum, perfectly aligned at the same value inthe diffusion domain as observed from the other signalsbelonging to the oligo-HA-NH2 chains (0.78 m2 s–1). Thisfinding indicates that MTX molecules, in MTX-CONH-oligo-HA-NHCO-ox-SWNT, behave as a unique entitywith the oligo-HA-NH2 chains anchored to the ox-SWNT.A further confirmation of the presence of MTX in MTX-CONH-oligo-HA-NHCO-ox-SWNT was obtained by UV/Vis spectroscopy, which showed the typical absorbance fea-tures of both MTX and ox-SWNT (Supporting Infor-mation, Figure S7).

R. Marega, M. Bergamin, V. Aroulmoji, F. Dinon, M. Prato, E. MuranoFULL PAPER
Sp
ecia
lS
ecti
on
Figure 4. (a) Presaturated (δ =4.7 ppm) 1H NMR spectrum of Ibuas sodium salt. (b) 1D DOSY spectrum of Ibu-CONH-oligo-HA-NHCO-ox-SWNT at 25 °C in D2O.
Figure 5. (a) 1H NMR spectrum of MTX as sodium salt. (b) 1DDOSY spectrum of MTX-CONH-oligo-HA-NHCO-ox-SWNT at53 Gcm–1 at 25 °C in D2O.
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 5617–56255622
Figure 6. 2D DOSY contour mode plot of MTX-CONH-oligo-HA-NHCO-ox-SWNT at 25 °C in D2O.
Conclusions
In conclusion, we have demonstrated the feasibility ofthe synthesis of novel hyaluronan–CNTs covalent adductsby exploiting amide bond formations and their furtherfunctionalization with model drugs like ibuprofen and me-thotrexate. The NMR results obtained by classical 1HNMR analysis and relaxation time measurements were sup-ported by 1D and 2D DOSY results, which suggest a re-duced mobility of the organic moieties due to covalent con-jugation with the CNT structure. The covalent conjugationof the modified hyaluronan to the CNTs can allow furtherfunctionalization with molecules bearing bearing one ormore carboxylic acid groups. The obtained CNT derivativeswill be evaluated for their biological activity.
Experimental SectionPreparation of Oligo-HA: To a stirred solution of sodium hyaluron-ate (MW = 18210, Mn = 10808, 10.0 g, 24.9 mmol) in acetate buffer(10 mm, pH 5.0 NaCl = 0.1 m, 500 mL) at 37 °C was addedhyaluronidase (420 mg, 189.42 KUnits). The reaction mixture wasstirred at 37 °C for 9 h, and then it was heated to 100 °C over10 min and kept at 100 °C for an additional 10 min while stirring.The reaction mixture was cooled to 25 °C, filtered through a 7 μmglass microfiber filter and then through a 1.2 μm polycarbonatefilter. The resulting clear, yellowish solution was purified by tangen-tial flow filtration (polyether sulfone membrane, cut-off1000 gmol–1) against water and then freeze-dried to give 7.35 g ofa white powder (73.5%w/w). 1H NMR (500 MHz, D2O): δ = 1.92(br. s, 3 H, NHCOCH3), 3.24–3.81 (m, HA backbone), 4.35 (m,anomeric), 4.45 (m, anomeric), 5.05 (d, anomeric) ppm. 13C NMR(125 MHz, D2O): δ = 22.0 (NHCOCH3), 53.8, 60.0 (CH2OH), 68.0,72.0, 73.1, 74.9, 75.8, 79.5, 82.1, 100.0, 102.8, 173.7, 174.5 ppm.HPSEC-MALS-RI: MW = 4923 gmol–1, Mn = 2668 gmol–1.
Preparation of Oligo-HA TBA Salt: Amberlite IRA-120 resin wastreated with an excess amount of 20% tetrabutylammonium hy-droxide solution for 24 h and then washed with H2O. A solutionof oligo-HA in H2O (5%) was then gently mixed with the resin for4 h. Filtration through a 7 μm glass microfiber filter, concentrationand freeze-drying gave the oligo-HA TBA salt with stoichiometricTBA content, as confirmed by 1H NMR spectroscopy.

Hyaluronan–Carbon Nanotube Derivatives
Sp
ecia
lS
ecti
on
Preparation of Oligo-HA-OMs: Oligo-HA TBA salt (1.50 g,2.42 mmol) and diisopropylethylamine (DIEA, 4.756 mL,28.20 mmol) were solubilized in DMF (30 mL) and stirred at–10 °C. MsCl (1.125 mL, 14.36 mmol) was then added dropwise,and the resulting mixture was stirred at –10 °C for 2 h. The reactionmixture was quenched by adding H2O (300 mL, to pH 12); the pHof the solution was adjusted to 11 with 10% HCl and stirring wasmaintained overnight. The resulting solution was filtered througha 1.2 μm polycarbonate filter and then purified by tangential flowfiltration (polyethersulfone membrane, cut-off 1000 gmol–1) againstH2O, before being concentrated in vacuo. The solution was freeze-dried to give 0.820 g of a white solid (78.6% mol/mol). 1H NMR(500 MHz, D2O): δ = 1.96 (s, 3 H, NHCOCH3), 3.23 (s, OMs),3.14–3.86 (m, HA backbone), 4.4–4.55 (m, anomeric + CH2OMs)ppm. 13C NMR (125 MHz, D2O): δ = 23 (NHCOCH3), 37 (OMs),55, 61 (CH2OH), 68, 69.5 (CH2OMs), 72, 74, 75, 76, 80, 83, 101,103, 174, 175 ppm. Loading of mesylate by 1H NMR was 60% molOMs/mol oligo-HA dimer.
Preparation of Oligo-HA-NH2. A solution of oligo-HA-OMs(700 mg, 1.42 mmol) in 25 % aqueous NH4OH (40 mL) was stirredat 65 °C for 16 h in a pressure-tight reactor. The excess amount ofammonia was removed under reduced pressure, and the reactionmixture was diluted to a final volume of 500 mL with H2O andfiltered through a 7 μm glass microfiber filter and then through apolycarbonate 1.2 μm filter before purification by tangential-flowfiltration (polyetheresulfone membrane, cut-off 1000 gmol–1)against water. Freeze-drying yielded 0.480 g of oligo-HA-NH2 as awhite powder (84.1 % mol/mol). 1H NMR (500 MHz, D2O): δ =1.96 (br. s, 3 H, NHCOCH3), 2.9 (m, CH2NH2), 3.19–3.71 (m, HAbackbone), 3.10 (m, CH2NH2), 3.70 (m, CH2OH), 3.77 (m,CHOH), 3.84 (m, CH2OH), 4.42 (m, anomeric), 4.52 (m, anomeric)ppm. 13C NMR (125 MHz, D2O): δ = 22.7 (NHCOCH3), 40.6(CH2NH2), 54.5, 60.7 (CH2OH), 68.6, 70.3, 71.6, 72.7, 73.5, 73.8,75.6, 76.4, 78.1, 80.1, 82.3, 82.7, 99.7, 100.8, 103.4, 174.2, 174.7,175.1 ppm. Loading of amino groups by 13C NMR was 60% molNH2/mol of oligo-HA dimer. HPSEC-MALS-RI: MW =4796 gmol–1, Mn = 2503 gmol–1.
Preparation of Oligo-HA-NH2 TBA Salt: Amberlite IRA-120 resinwas treated with an excess amount of 20% tetrabutylammoniumhydroxide solution for 24 h and then washed with H2O. A solutionof oligo-HA-NH2 in H2O (5%) was then gently mixed with theresin for 4 h. Filtration through a 7 μm glass microfiber filter, con-centration, and freeze-drying gave oligo-HA-NH2 TBA salt withstoichiometric TBA content, as confirmed by 1H NMR spec-troscopy.
Preparation of Ox-SWNTs: Pristine single-walled carbon nanotu-bes were first purified by treatment with nitric acid and then furtheroxidized by treatment with sulfuric acid/hydrogen peroxide.
Purification: A suspension of 1.0 g of SWNTs in 2.6 m HNO3 (1 L)was stirred at 125 °C for 48 h. Filtration under reduced pressurethrough 0.2 μm polycarbonate filters led to the isolation of a blackcake, which was subsequently washed with 1 mm NaOH (20 mL),H2O (1000 mL) and MeOH (10 mL) and dried under vacuum for30 min to give a black powder. Yield = 0.700 g, 70% w/w.
Shortening: A suspension of purified SWNTs (100 mg) in H2SO4/H2O2 (4:1, 100 mL) was ultrasonicated for 1 h at 35 °C. The mix-ture was diluted with H2O to a final volume of 2 L and filteredthrough 0.2 μm polycarbonate filter. The resulting black cake wasthen washed with H2O (600 mL), Na2CO3 (100 mL), and H2O(200 mL), and dried under vacuum for 30 min to give 0.0750 g of
Eur. J. Org. Chem. 2011, 5617–5625 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5623
a black powder (75% w/w). IR (KBr): ν̃ = 1389 (b; OCOH), 1727(s; RCO), 3470 (s; OCOH) cm–1. Loading of carboxylic groups(TGA) = 3.4 μmolmg–1.
Synthesis of Ox-SWNT-OSu: Ox-SWNTs (50.0 mg, 170.0 μmol ofCOOH) were sonicated for 20 min in DMF (15 mL) to give a blackhomogeneous suspension; after that DMAP (21.0 mg, 173.3 μmol)was added and sonication was continued for 10 min. NHS(61.2 mg, 521.1 μmol) was then added to ox-SWNTs dispersion andsonication continued for 10 min. Subsequently, a solution ofEDC·HCl (83.7 mg, 425.0 μmol) and DMAP (51.5 mg,425.0 μmol) in DMF (5 mL) was slowly added to the ox-SWNTsdispersion. The reaction mixture was sonicated for 1 h and thenstirred at room temperature for 2 h. Ox-SWNT-OSu were then iso-lated by precipitation with Et2O (100 mL) and filtration under re-duced pressure through 0.2 μm polytetrafluoroethylene filter. Theblack precipitate was then washed with Et2O (100 mL), dispersedin DMF (10 mL), sonicated for 5 min, and then precipitated againin phosphate buffer (50 mm, pH 7.0). Filtration under reducedpressure through 0.2 μm polycarbonate filter allowed a black pre-cipitate to be isolated, which was washed with H2O (200 mL) andacetone (10 mL), before being dried under reduced pressure. Theox-SWNT-OSu (0.0420 g) were recovered as a black powder.
Preparation of Oligo-HA-NHCO-ox-SWNT: Ox-SWNTs-OSu(40 mg) was dispersed in DMF (10 mL) and sonicated for 20 min.A solution of oligo-HA-NH2 TBA salt (430 mg, 280 μmol) andDMAP (34 mg, 280 μmol) in DMF (10 mL) was added to the blackdispersion of ox-SWNT-OSu and the sonication continued for fur-ther 5 min. The resulting black dispersion was stirred at room tem-perature for 48 h. The reaction mixture was then precipitated inH2O (100 mL), and then the pH of the black solution was adjustedto 11 with 1 n NaOH. After stirring at room temperature over-night, the resulting saturated black solution was centrifuged at4500 g for 20 min to remove the unreacted and unsolubilized ox-SWNTs. The supernatant was collected, while the precipitate wasdiluted with H2O (10 mL), filtered through 0.2 μm polycarbonatefilter, and washed with H2O (50 mL). The filtered was combinedwith the supernatant and the resulting black solution was concen-trated to 20 mL in centrifugal concentrators (30000 gmol–1 cut-off)and purified by using the same device by subsequent additions ofbrine to ensure complete TBA removal and H2O to desalt the mix-ture until the conductivity of the filtered matched that of the deion-ized water. The resulting black solution was freeze-dried, giving0.0460 g of a black powder. 1H NMR (500 MHz, D2O): δ = 1.95(br. s, 3 H, NHCOCH3), 2.94 (m, CH2NH2), 3.19–3.69 (br. m, HAbackbone), 3.21 (m, CH2NH2), 3.69 (m, CH2OH), 3.77 (m,CHOH), 3.84 (m, CH2OH), 4.38 (m, anomeric), 4.49 (m, anomeric)ppm. 13C NMR (125 MHz, D2O): δ = 22.4 (NHCOCH3), 40.8(CH2NH2), 54.3, 58.1, 60.5 (CH2OH), 68.4, 70.0, 72.5, 73.3, 73.5,75.3, 76.2, 78.5, 79.9, 82.6, 99.9, 100.5, 103.2, 174.1, 174.4,174.9 ppm. Composition (w/w) as evaluated by 1H NMR andTGA: 14.4 % oligo-HA-NH2, 81.6% ox-SWNTs, 4.0% H2O.
Preparation of Oligo-HA-NHCO-ox-SWNT TBA Salt: To a sus-pension of tetrabutylammonium-activated Amberlite (IR-120,5.0 g) in H2O (15 mL) was added oligo-HA-NHCO-ox-SWNT(25.6 mg), and the resulting solution was gently stirred at roomtemperature for 4 h. The oligo-HA-NHCO-ox-SWNT solution wasfiltered through a 7 μm glass microfiber filter and concentrated to10 mL under reduced pressure; freeze-drying of the solution af-forded 0.0302 g of oligo-HA-NHCO-ox-SWNT-TBA as a blackpowder, which showed stoichiometric TBA content, as confirmedby 1H NMR spectroscopy.

R. Marega, M. Bergamin, V. Aroulmoji, F. Dinon, M. Prato, E. MuranoFULL PAPER
Sp
ecia
lS
ecti
on
Preparation of Ibu-CONH-oligo-HA-NHCO-ox-SWNT
Synthesis of IBu-OSu: To a solution of ibuprofen (373 mg,1.81 mmol) in THF (10 mL) was sequentially added NHS(535.3 mg, 456 mmol) and a solution of EDC·HCl (780.1 mg,3.96 mmol) and DMAP (486.0 mg, 3.96 mmol) in THF (5 mL).The resulting reaction mixture was stirred at room temperature for16 h, and then the solvent was removed under reduced pressure.The precipitate was solubilized in EtOAc (30 mL) and washed with0.1 n HCl (3 �50 mL), 0.5 m NaHCO3 (pH 8.2, 3�50 mL), andbrine (3 � 50 mL). The organic phase was dried with anhydrousNa2SO4, filtered, and concentrated under reduced pressure, yield-ing Ibu-OSu as a white powder (0.3922 g, 71.3%). 1H NMR(500 MHz, CDCl3): δ = 0.91 (s, 6 H), 1.62 (d, 3 H), 1.8–1.92 (m, 1H), 2.46 (d, 2 H), 2.84 (s, 4 H, OSu), 4.2 (d, 1 H), 7.0–7.3 (m, 4HAr) ppm.
Condensation of Ibu-OSu with Oligo-HA-NHCO-ox-SWNT TBASalt: Ibu-OSu (5.8 mg, 19.1 μmol) and oligo-HA-NHCO-ox-SWNT TBA salt (12.5 mg, 4.0 μmol of NH2) were dissolved inDMF (5 mL). The resulting black solution was sonicated for 5 minand then stirred at room temperature overnight. The reaction mix-ture was poured into 0.5 m NaHCO3 (pH 12.5, 25 mL) and stirredfor 4 h. The resulting solution was concentrated to 20 mL in cen-trifugal concentrators (30000 gmol–1 cut-off) and purified using thesame device by subsequent additions of 0.5 m NaHCO3 to ensurecomplete removal of unreacted Ibu-OSu and H2O to desalt themixture until the conductivity of the filtered matched that of thedeionized water. Ibu-CONH-oligo-HA-NHCO-ox-SWNT was iso-lated by precipitation with acetone (30 mL) followed by filtrationthrough a 0.2 μm polytetrafluoroethylene filter to yield 0.0112 g ofa dark-grey powder. 1H NMR (500 MHz, D2O): δ = 0.81 (s, 2 CH3
Ibu), 1.62 (d, CH3 Ibu), 2.03 (br. s, 3H NHCOCH3), 2.46 (d, CH2
Ibu), 2.80–4.0 (br. m, HA backbone + Ibu), 4.47 (m, anomeric),4.57 (m, anomeric), 7.1–7.3 (br. m, Ar Ibu) ppm.
Preparation of MTX-CONH-oligo-HA-NHCO-ox-SWNT: To astirred solution of methotrexate (161.5 mg, 355.4 μmol) in DMF(10 mL) was added DCC (73.5 mg, 352.7 μmol), and the mixturewas stirred continuously overnight at 4 °C. The resulting yellowsuspension was filtered through a 0.2 μm polytetrafluoroethylenefilter. Oligo-HA-NHCO-ox-SWNT TBA salt (43.0 mg, approxi-mately 13.9 μmol NH2) was added to the filtered solution alongwith DMAP (2.70 mg, 22.2 μmol) dissolved in DMF (3 mL). Theresulting dark-yellowish mixture was sonicated for 5 min and thenstirred at room temperature overnight. The reaction mixture waspoured in 0.5 m NaHCO3 (pH 12.5, 50 mL) and stirred for 4 h.The resulting solution was concentrated to 20 mL in centrifugalconcentrators (30000 gmol–1 cut-off) and purified using the samedevice by subsequent additions of 0.5 m NaHCO3 to ensure com-plete removal of unreacted MTX and H2O to desalt the mixtureuntil the conductivity of the filtered matched that of the deionizedwater. MTX-CONH-oligo-HA-NHCO-ox-SWNT was isolated byprecipitation with acetone (30 mL), followed by filtration througha 0.2 μm polytetrafluoroethylene filter to yield 0.0420 g of grey-yellowish powder. 1H NMR (500 MHz, D2O): δ = 2.03 (br. s, 3HNHCOCH3), 2.0–2.4 (br. m, 1.25 H, CH2 MTX), 2.80–4.0 (br. m,HA backbone + MTX), 4.47 (m, anomeric), 4.57 (m, anomeric),6.4–7.0 (br. m, MTX), 7.4–7.85 (br. m, MTX), 8.22–8.65 (br. m,MTX) ppm. UV/Vis (H2O): λmax = 370.7, 306.5, 258.7 nm.
Supporting Information (see footnote on the first page of this arti-cle): Detailed experimental procedures, size exclusion and ionic ex-change chromatographic characterizations of oligo-HA. 1H NMR
www.eurjoc.org © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2011, 5617–56255624
spectrum of the permeate after diafiltration of the oligo-HA-NHCO-ox-SWNT reaction mixture; evaluation of the moisturelevel of oligo-HA-NHCO-ox-SWNT by thermal analysis; quantifi-cation of the oligo-HA-NH2 moiety in oligo-HA-NHCO-ox-SWNT by 1H NMR analysis in the presence of standard 3-(tri-methylsilyl)propionic acid 2,2,3,3-d4 sodium salt (TSP-d4). 1HNMR inversion recovery experiments of oligo-HA-NH2 and oligo-HA-NHCO-ox-SWNT. 1H NMR spectrum of the permeate afterdiafiltration of the oligo-HA-NHCO-ox-SWNT-Ibu reaction mix-ture. UV/Vis spectroscopy analysis of ox-SWNT, MTX and MTX-CONH-oligo-HA-NHCO-ox-SWNT.
Acknowledgments
The authors wish to thank the University of Trieste, the ConsorzioInteruniversitario Nazionale per la Scienza e Tecnologia dei Mate-riali (INSTM), Eurand spa, Ministero dell’Università e dellaRicerca (MIUR) (Cofin Prot. 20085M27SS), and Regione FriuliVenezia-Giulia for financial support.
[1] a) F. Lu, l. Gu, M. J. Meziani, X. Wang, P. G. Luo, L. M. Veca,L. Cao, Y. P. Sun, Adv. Mater. 2009, 21, 139–152; b) K. Kostar-elos, A. Bianco, M. Prato, Nat. Nanotechnol. 2009, 4, 627–633.
[2] a) P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco,M. Prato, Chem. Soc. Rev. 2009, 38, 2214–2230; b) X. Peng,S. S. Wong, Adv. Mater. 2008, 20, 625–642.
[3] V. Georgakilas, N. Tagmatarchis, D. Pantarotto, A. Bianco,J. P. Briand, M. Prato, Chem. Commun. 2002, 3050–3051.
[4] a) F. Pompeo, D. E. Resasco, Nano Lett. 2002, 2, 369–373; b)B. Zhao, H. Hu, R. C. Haddon, Adv. Funct. Mater. 2004, 14,71–76; c) J. L. Hudson, M. J. Casavant, J. M. Tour, J. Am.Chem. Soc. 2004, 126, 11158–11159.
[5] a) M. Prato, K. Kostarelos, A. Bianco, Acc. Chem. Res. 2008,41, 60–68; b) A. Bianco, K. Kostarelos, M. Prato, Expert Opin.Drug Deliv. 2008, 5, 331–342.
[6] V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holz-inger, A. Hirsch, J. Am. Chem. Soc. 2002, 124, 760–761.
[7] C. Samori, H. Ali-Boucetta, R. Sainz, C. Guo, F. M. Toma, C.Fabbro, T. da Ros, M. Prato, K. Kostarelos, A. Bianco, Chem.Commun. 2010, 46, 1494–1496.
[8] W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp,J. P. Briand, R. Gennaro, M. Prato, A. Bianco, Angew. Chem.Int. Ed. 2005, 44, 6358–6362.
[9] R. Singh, D. Pantarotto, L. Lacerda, G. Pastorin, C. Klumpp,M. Prato, A. Bianco, K. Kostarelos, Proc. Natl. Acad. Sci.USA 2006, 103, 3357–3362.
[10] K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski,J. Luangsivilay, S. Godefroy, D. Pantarotto, J. P. Briand, S.Muller, M. Prato, A. Bianco, Nat. Nanotechnol. 2007, 2, 108–113.
[11] a) R. P. Feazell, N. Nakayama-Ratchford, H. Dai, S. J. Lip-pard, J. Am. Chem. Soc. 2007, 129, 8438–8439; b) M. Adeli, F.Hakimpoor, M. Ashiri, R. Kabiri, M. Bavadi, Soft Matter2011, 7, 4062–4070.
[12] a) A. Di Crescenzo, D. Velluto, J. A. Hubbell, A. Fontana,Nanoscale 2011, 3, 925–928; b) H. Ali-Boucetta, K. T. Al-Ja-mal, D. McCarthy, M. Prato, A. Bianco, K. Kostarelos, Chem.Commun. 2008, 459–461.
[13] B. Weissman, K. Meyer, J. Am. Chem. Soc. 1954, 76, 1753–1757.
[14] S. Norbedo, F. Dinon, M. Bergamin, S. Bosi, V. Aroulmoji, R.Khan, E. Murano, Carbohydr. Res. 2009, 344, 98–104.
[15] D. Tasis, N. Tagmatarchis, A. Bianco, M. Prato, Chem. Rev.2006, 106, 1105–1136.

Hyaluronan–Carbon Nanotube Derivatives
Sp
ecia
lS
ecti
on
[16] R. Marega, V. Aroulmoji, F. Dinon, L. Vaccari, S. Giordani,A. Bianco, E. Murano, M. Prato, J. Am. Chem. Soc. 2009, 131,9086–9093.
[17] R. Marega, V. Aroulmoji, M. Bergamin, L. Feruglio, F. Dinon,A. Bianco, E. Murano, M. Prato, ACS Nano 2010, 4, 2051–2058.
[18] M. N. Loening, J. Keeler, G. A. Morris, J. Magn. Reson. 2001,153, 103–112.
Eur. J. Org. Chem. 2011, 5617–5625 © 2011 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5625
[19] K. F. Morris, C. S. Johnson, J. Am. Chem. Soc. 1992, 112,3139–3141.
[20] D. Bonifazi, C. Nacci, R. Marega, S. Campidelli, G. Ceballos,S. Modesti, M. Meneghetti, M. Prato, Nano Lett. 2006, 6,1408–1414.
Received: June 12, 2011Published Online: August 31, 2011

Eur. J. Org. Chem. 2011 · © WILEY-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2011 · ISSN 1434–193X
SUPPORTING INFORMATION
DOI: 10.1002/ejoc.201100846 Title: Hyaluronan–Carbon Nanotube Derivatives: Synthesis, Conjugation with Model Drugs, and DOSY NMR Characterization Author(s): Riccardo Marega, Massimo Bergamin, Vincent Aroulmoji, Francesca Dinon, Maurizio Prato,* Erminio Murano*

Detailed experimental procedure:
Materials
Materials. All the solvents were obtained from Merck, (Acetone, CH2Cl2, CHCl3, DMF, EtOAc, EtOH, MeOH, THF),
Fluka (HNO3 and H2SO4) and from Riedel de Haen (H2O2 30%), All the reagents were purchased as commercial
grade from Sigma-Aldrich (hyaluronidase from bovine testes type I-S, EDC·HCl, NHS, Ibuprofen) or Fluka (DMAP,
DCC) and used without further purification. Hyaluronan sodium salt was purchased from Bioiberica as cosmetic
grade. Methotrexate was obtained from Fermion (Orion). Single walled carbon nanotubes produced through
high pressure carbon monoxide disproportion (HiPco SWNTs) were obtained from Carbon Nanotechnologies Inc.
(lot # R0510c). D2O (99.9%) and 3-(trimethylsilyl)propionic acid 2,2,3,3-d4 sodium salt (TSPA-d4) were purchased
from Cambridge Isotope Laboratories. Polyetheresulfone membranes (Centramate Omega) where purchased
from Pall inc., while centrifugal concentrators (Vivaspin R20) where obtained by Sartorius Stedim biotech.
Methods
NMR characterizations
High resolution NMR spectra were performed on a Varian INOVA (500 MHz) NMR spectrometer
equipped with Performa II-Z gradient coils (Varian, Palo Alto, CA, USA). The longitudinal relaxation
time (T1) determinations were performed at 25 °C by employing an inversion recovery pulse sequence.
The gradient strength was calibrated with pure water (D= 2.229 x 10-9
m2 s
-1 at 298.2 K) and the
maximum gradient strength was about 54 G cm-1
. The Doneshot pulse sequence (Dbppste) was used for
the measurements of diffusion coefficient at 25 °C. This sequence has an elaborate PFG arrangement
compensating for pulse imperfections without the need of an extensive phase-cycle, hence making
single-transient DOSY experiments possible.
Experiments were performed by keeping the z-gradient pulse length constant and fixing the gradient
strength, in the 1D DOSY experiments, or gradually increasing the gradient from 0.5 to 53.6 G cm-1
in
22 steps, in the 2 D-DOSY experiments. The diffusion coefficients (D) were obtained from the slope of
the following equation:
ln(Ig/I0) = − [γ2δ
2G
2(Δ−/3)]D
Where Ig and I0 are intensities of the NMR signal in the presence and absence of field gradient pulses; γ
is the gyromagnetic constant for 1H; δ is the duration of the z-gradient pulse; G is the gradient strength;
and Δ is the time interval between the gradient pulses (diffusion time, 200 msec).
Thermogravimetric analysis
Thermogravimetric measurements were performed using a Perkin Elmer TGA 7 thermal analyzer connected to a
Perkin-Elmer TGA 7/DX controlling console, under a constant N2 flow of 20.0 ml min-1 with a temperature

increase of 10 °C min-1. For the moisture evaluation, the samples were brought to 50 °C and immediately
subjected to a temperature scan from 50 to 250 °C with a temperature increase of 10 °C·min-1.
IR spectroscopy characterization
IR spectrum of ox-SWNT was performed on a KBr dispersion of with 1% w/w of the compound, by
using a Perkin Elmer 2000 spectrometer.
TEM characterization
All the images were obtained from the deposition onto Nickel grids (with carbon layer coating, 200
mesh) of the D2O solution of oligo-HA-NHCO-ox-SWNT used in the NMR characterizations; these
grids were analyzed with a Philips 208 electron microscope at a 100 kV voltage, and the resulting
images were collected with an AMT high-resolution digital imaging camera.

Figure S1. (a) HPSEC-MALLS-RI of oligo-HA: Multi Angle Laser Light Scattering (MALLS, ······), UV@220 nm (——)
and refractive index (RI, ——) traces. (b) molecular weight distribution profiles calculated from the traces reported in (a):
MW 4,923 g mol-1
, Mn 2,668 g mol-1
, polydispersity index = 1.85. (c) high performance ionic exchange (HPIEC)
chromatogram of the oligo-HA fraction, showing the abundance of the single chains of oligomers after the hydrolysis step,
which resulted (retention times of the repeating disaccharides units, mass percentage): 2-mer (5.67 min, 4.86%), 3-mer (7.25
min, 4.61%), 4-mer (8.87 min, 6.94%), 5-mer (10.37 min, 6.62%), 6-mer (11.85 min, 7.14%), 7-mer (13.26 min, 6.72%), 8-
mer (14.6 min, 6.11%), 9-mer (15.68 min, 5.38%), 10-mer (16.66 min, 4.90%), 11-mer (17.57 min, 4.46%), 12-mer (18.44
min, 3.98%), 13-mer (19.27 min, 3.52%), 14-mer (20.11 min, 2.97%), 15-mer (21.48 min, 2.83%) and traces of 16-mer to 24-
mer for a total amount of 9.86%.
Figure S2. 1H NMR spectrum of the permeate after diafiltration of the oligo-HA-NHCO-ox-SWNT reaction mixture. The
signals found correspond to dimethylaminopyridine (8.35, 7.8 and 6.6 ppm), dimethylformammide (7.96, 3.05 and 2.95 ppm),
excess oligo-HA-NH2 (4.50 4.35, 3.8 3 and 1.96 ppm) and tetrabutylammonium salt (2.7, 1.5, 1.2 and 0.8 ppm),
respectively.

Figure S3. Evaluation of the moisture level of oligo-HA-NHCO-ox-SWNT, estimated by TGA (temperature scan from 50 to
250 °C under N2 atmosphere). Weight loss at 160 °C was in the order of 4% w/w.
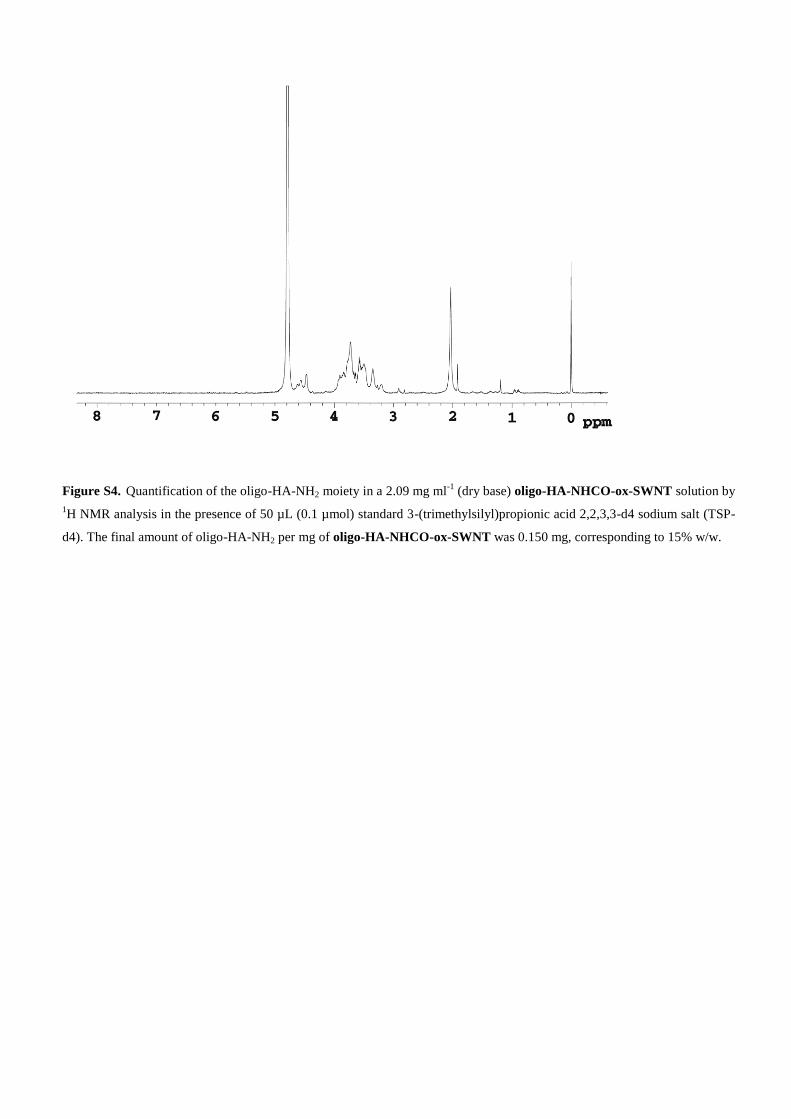
Figure S4. Quantification of the oligo-HA-NH2 moiety in a 2.09 mg ml-1
(dry base) oligo-HA-NHCO-ox-SWNT solution by
1H NMR analysis in the presence of 50 µL (0.1 µmol) standard 3-(trimethylsilyl)propionic acid 2,2,3,3-d4 sodium salt (TSP-
d4). The final amount of oligo-HA-NH2 per mg of oligo-HA-NHCO-ox-SWNT was 0.150 mg, corresponding to 15% w/w.
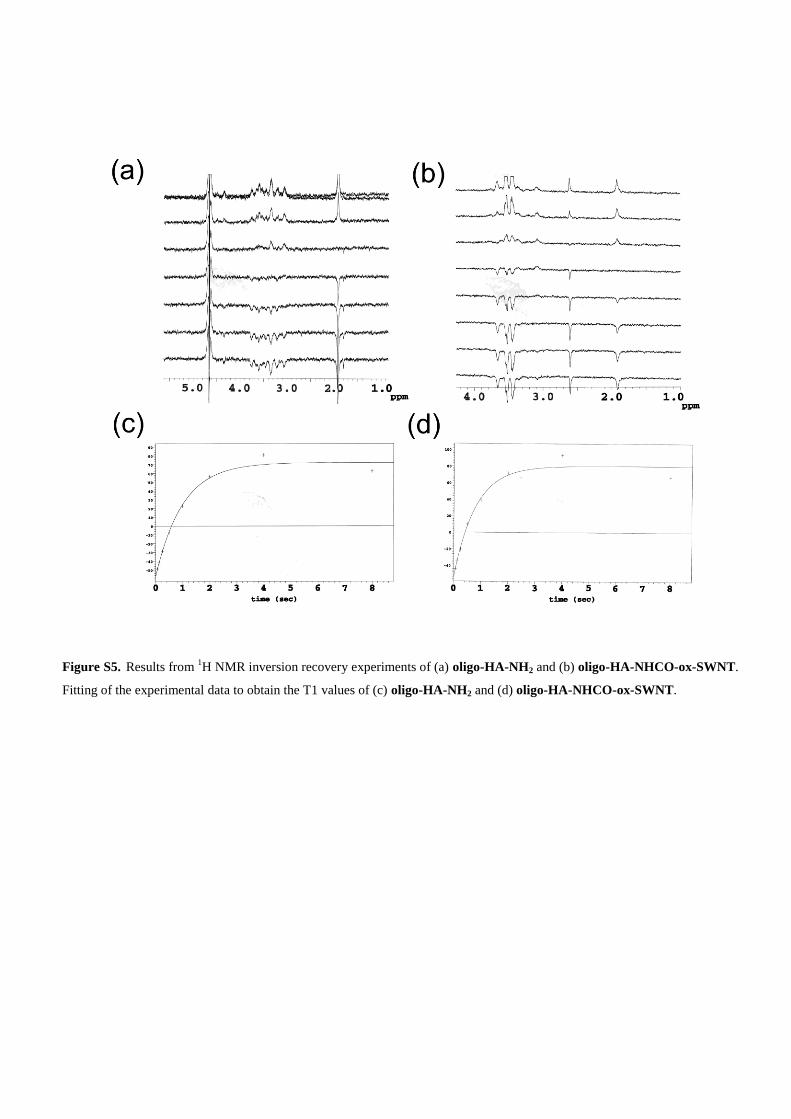
Figure S5. Results from 1H NMR inversion recovery experiments of (a) oligo-HA-NH2 and (b) oligo-HA-NHCO-ox-SWNT.
Fitting of the experimental data to obtain the T1 values of (c) oligo-HA-NH2 and (d) oligo-HA-NHCO-ox-SWNT.
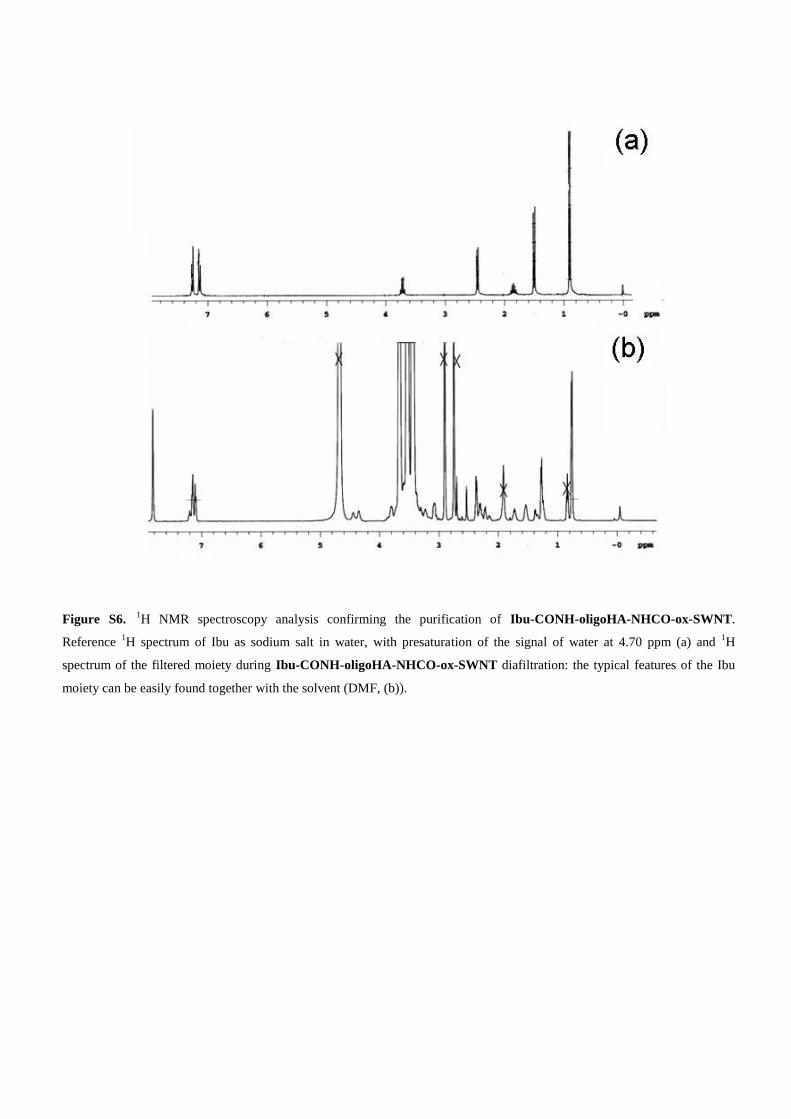
Figure S6. 1H NMR spectroscopy analysis confirming the purification of Ibu-CONH-oligoHA-NHCO-ox-SWNT.
Reference 1H spectrum of Ibu as sodium salt in water, with presaturation of the signal of water at 4.70 ppm (a) and
1H
spectrum of the filtered moiety during Ibu-CONH-oligoHA-NHCO-ox-SWNT diafiltration: the typical features of the Ibu
moiety can be easily found together with the solvent (DMF, (b)).

Figure S7. UV-vis spectroscopy analysis of ox-SWNT (black), MTX (red) and MTX-CONH-oligoHA-
NHCO-ox-SWNT (blue) in H2O.
Related Documents











