Human Sirt-1: Molecular Modeling and Structure- Function Relationships of an Unordered Protein Ida Autiero 1. , Susan Costantini 1,2,3. *, Giovanni Colonna 1,3 1 CRISCEB (Interdepartmental Research Center for Computational and Biotechnological Sciences) Second University of Naples, Naples, Italy, 2 CROM (Oncology Research Centre of Mercogliano) ‘‘Fiorentino Lo Vuolo’’, Mercogliano, Italy, 3 Department of Biochemistry and Biophysics, Second University of Naples, Naples, Italy Abstract Background: Sirt-1 is a NAD+-dependent nuclear deacetylase of 747 residues that in mammals is involved in various important metabolic pathways, such as glucose metabolism and insulin secretion, and often works on many different metabolic substrates as a multifunctional protein. Sirt-1 down-regulates p53 activity, rising lifespan, and cell survival; it also deacetylases peroxisome proliferator-activated receptor-gamma (PPAR-c) and its coactivator 1 alpha (PGC-1a), promoting lipid mobilization, positively regulating insulin secretion, and increasing mitochondrial dimension and number. Therefore, it has been implicated in diseases such as diabetes and the metabolic syndrome and, also, in the mechanisms of longevity induced by calorie restriction. Its whole structure is not yet experimentally determined and the structural features of its allosteric site are unknown, and no information is known about the structural changes determined by the binding of its allosteric effectors. Methodology: In this study, we modelled the whole three-dimensional structure of Sirt-1 and that of its endogenous activator, the nuclear protein AROS. Moreover, we modelled the Sirt-1/AROS complex in order to study the structural basis of its activation and regulation. Conclusions: Amazingly, the structural data show that Sirt-1 is an unordered protein with a globular core and two large unordered structural regions at both termini, which play an important role in the protein-protein interaction. Moreover, we have found on Sirt-1 a conserved pharmacophore pocket of which we have discussed the implication. Citation: Autiero I, Costantini S, Colonna G (2009) Human Sirt-1: Molecular Modeling and Structure-Function Relationships of an Unordered Protein. PLoS ONE 4(10): e7350. doi:10.1371/journal.pone.0007350 Editor: Dafydd Jones, Cardiff University, United Kingdom Received August 2, 2009; Accepted September 14, 2009; Published October 8, 2009 Copyright: ß 2009 Autiero et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Funding: The funding is from the Institute in which this project was carried out. Funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. IA is supported by Doctorate in Computational Biology, CRISCEB, Second University of Naples, Naples, Italy. Competing Interests: The authors have declared that no competing interests exist. * E-mail: [email protected] . These authors contributed equally to this work. Introduction The sirtuin family is widely distributed from archaea and eubacteria to eukaryotes and seven different homologous proteins are found in the humans that show a central highly conserved region, defined as the catalytic core [1]. In comparison to other proteins of the family, Sirt-1 presents two large regions, i.e. the amino and carboxyl terminals, that are missing in all the other sirtuins. Sirt-1 is a NAD+-dependent deacetylase closely related to yeast Sir2, the first gene discovered in sirtuin family, which has NAD+ dependent class III histone deacetylase activity [2]. Sites of phosphorylation and SUMOylation consensus were recently found also in the amino- and in carboxyl-terminal regions, and these were proposed having regulation and localization functions [3]. Experimental data support the Sirt-1 implication in processes including chromatin remodelling, transcriptional silencing, chro- mosomal stability, cell cycle progression, apoptosis, autophagy, metabolism, growth suppression, inflammation, and stress re- sponse [4]. In fact, the Sirt-1 regulation activity occurs through the deacetylation reaction of various and different substrates such as p53 [5], forkhead box class O (FOXO) transcription factors [6], peroxisome proliferator activated receptor (PPAR)g co-activator 1a (PGC-1a) [7], nuclear factor (NF)-kB and others, which are closely linked to some age-related diseases [5]. Also Sirt-1 stimulates eNOS activity and increases the endothelial NO. Its inhibition in the endothelium of arteries inhibits endothelium dependent vasodilation and decreases bioavailable NO [8]. Moreover, it was seen that Sirt-1 is a negative modulator of adipogenesis by docking with the nuclear receptor co-repressor (NcoR) [9] and induces a decrease of pro-inflammatory cytokine release [10] and a promotion of carcinogenesis [11] by the negative control of Nuclear factor-kB (NF-kB). Experimental data demonstrated that the inhibition of NF-kB in hepatocytes in vivo promotes hepatocarcinogenesis. Kaeberlein et al. demonstrated firstly the anti-aging effects of Sir2 showing that in Saccharomyces cerevisiae the integration of extra copies of Sir2 extended lifespan up to 30% [12]. Similar effects of Sir2 were subsequently observed in C. elegans and Drosophila melanogaster [13–14]. In fact, the overexpression of Sir-2 [15] increased lifespan up to 50% in C. elegans but in Drosophila, an extra copy of the Sir2 gene extended lifespan in female and male by 29 and 18%, respectively. PLoS ONE | www.plosone.org 1 October 2009 | Volume 4 | Issue 10 | e7350

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Human Sirt-1: Molecular Modeling and Structure-Function Relationships of an Unordered ProteinIda Autiero1., Susan Costantini1,2,3.*, Giovanni Colonna1,3
1 CRISCEB (Interdepartmental Research Center for Computational and Biotechnological Sciences) Second University of Naples, Naples, Italy, 2 CROM (Oncology Research
Centre of Mercogliano) ‘‘Fiorentino Lo Vuolo’’, Mercogliano, Italy, 3 Department of Biochemistry and Biophysics, Second University of Naples, Naples, Italy
Abstract
Background: Sirt-1 is a NAD+-dependent nuclear deacetylase of 747 residues that in mammals is involved in variousimportant metabolic pathways, such as glucose metabolism and insulin secretion, and often works on many differentmetabolic substrates as a multifunctional protein. Sirt-1 down-regulates p53 activity, rising lifespan, and cell survival; it alsodeacetylases peroxisome proliferator-activated receptor-gamma (PPAR-c) and its coactivator 1 alpha (PGC-1a), promotinglipid mobilization, positively regulating insulin secretion, and increasing mitochondrial dimension and number. Therefore, ithas been implicated in diseases such as diabetes and the metabolic syndrome and, also, in the mechanisms of longevityinduced by calorie restriction. Its whole structure is not yet experimentally determined and the structural features of itsallosteric site are unknown, and no information is known about the structural changes determined by the binding of itsallosteric effectors.
Methodology: In this study, we modelled the whole three-dimensional structure of Sirt-1 and that of its endogenousactivator, the nuclear protein AROS. Moreover, we modelled the Sirt-1/AROS complex in order to study the structural basisof its activation and regulation.
Conclusions: Amazingly, the structural data show that Sirt-1 is an unordered protein with a globular core and two largeunordered structural regions at both termini, which play an important role in the protein-protein interaction. Moreover, wehave found on Sirt-1 a conserved pharmacophore pocket of which we have discussed the implication.
Citation: Autiero I, Costantini S, Colonna G (2009) Human Sirt-1: Molecular Modeling and Structure-Function Relationships of an Unordered Protein. PLoSONE 4(10): e7350. doi:10.1371/journal.pone.0007350
Editor: Dafydd Jones, Cardiff University, United Kingdom
Received August 2, 2009; Accepted September 14, 2009; Published October 8, 2009
Copyright: � 2009 Autiero et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: The funding is from the Institute in which this project was carried out. Funders had no role in study design, data collection and analysis, decision topublish, or preparation of the manuscript. IA is supported by Doctorate in Computational Biology, CRISCEB, Second University of Naples, Naples, Italy.
Competing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected]
. These authors contributed equally to this work.
Introduction
The sirtuin family is widely distributed from archaea and
eubacteria to eukaryotes and seven different homologous proteins
are found in the humans that show a central highly conserved
region, defined as the catalytic core [1]. In comparison to other
proteins of the family, Sirt-1 presents two large regions, i.e. the
amino and carboxyl terminals, that are missing in all the other
sirtuins. Sirt-1 is a NAD+-dependent deacetylase closely related to
yeast Sir2, the first gene discovered in sirtuin family, which has
NAD+ dependent class III histone deacetylase activity [2]. Sites of
phosphorylation and SUMOylation consensus were recently found
also in the amino- and in carboxyl-terminal regions, and these
were proposed having regulation and localization functions [3].
Experimental data support the Sirt-1 implication in processes
including chromatin remodelling, transcriptional silencing, chro-
mosomal stability, cell cycle progression, apoptosis, autophagy,
metabolism, growth suppression, inflammation, and stress re-
sponse [4]. In fact, the Sirt-1 regulation activity occurs through the
deacetylation reaction of various and different substrates such as
p53 [5], forkhead box class O (FOXO) transcription factors [6],
peroxisome proliferator activated receptor (PPAR)g co-activator
1a (PGC-1a) [7], nuclear factor (NF)-kB and others, which are
closely linked to some age-related diseases [5]. Also Sirt-1
stimulates eNOS activity and increases the endothelial NO. Its
inhibition in the endothelium of arteries inhibits endothelium
dependent vasodilation and decreases bioavailable NO [8].
Moreover, it was seen that Sirt-1 is a negative modulator of
adipogenesis by docking with the nuclear receptor co-repressor
(NcoR) [9] and induces a decrease of pro-inflammatory cytokine
release [10] and a promotion of carcinogenesis [11] by the
negative control of Nuclear factor-kB (NF-kB). Experimental data
demonstrated that the inhibition of NF-kB in hepatocytes in vivo
promotes hepatocarcinogenesis.
Kaeberlein et al. demonstrated firstly the anti-aging effects of
Sir2 showing that in Saccharomyces cerevisiae the integration of extra
copies of Sir2 extended lifespan up to 30% [12]. Similar effects of
Sir2 were subsequently observed in C. elegans and Drosophila
melanogaster [13–14]. In fact, the overexpression of Sir-2 [15]
increased lifespan up to 50% in C. elegans but in Drosophila, an extra
copy of the Sir2 gene extended lifespan in female and male by 29
and 18%, respectively.
PLoS ONE | www.plosone.org 1 October 2009 | Volume 4 | Issue 10 | e7350

Because Sirt-1 deacetylates non histone proteins, including
various transcription factors, it is involved in the control of
important biological mechanisms. Through its catalytic activity, it
exhibits diversified functions in cell type-specific manner, which
have pathophysiological implications in cancer, obesity, inflamma-
tion and neurodegenerative diseases [4,16–17]. Its modulation leads
an increase in mitochondrial biogenesis, and an improvement of
glucose metabolism in mitochondria but also in skeletal muscle and
adipose tissues [18]. These evidences suggest that Sirt-1 could be a
novel target to treat metabolic disorders such as type 2 diabetes.
Numerous experimental data [2,19–20] have shown the
modulation of the catalytic activity of Sirt-1 exerted through its
allosteric effectors. Kim et al (2007) demonstrated that the nuclear
protein AROS is an endogenous activator of Sirt-1 which
increases its activity interacting with the allosteric site. Accordingly
with the functional relevance of Sirt-1 activation, recent works
focused on its interaction with small allosteric effectors [19–21].
In particular, the phenolic compound, named resveratrol, is the
first found allosteric activator, that has been reported to extend
lifespan in yeast [21], Caenorhabditis elegans, Drosophila [22] and
rodents [20]. It improves the metabolism and glucose tolerance, as
well as the overall physical performance in various stress tests [7].
However recent efforts have identified new compounds signifi-
cantly more potent than resveratrol [19–20].
Even if the substrates and the effects of Sirt-1 activation have
been well characterized, no structural information about its
activation and regulation is known.
On the basis of the biological importance of this protein, we
focused our attention to create a model of the whole structure of
human Sirt-1 in order to understand its interaction with the
allosteric effectors and to propose the molecular basis of its
activation and regulation.
Results
Modelling of the catalytic core and allosteric site of Sirt-1The best model has been obtained for the catalytic core of
human Sirt-1 using as template the structures of human Sirt-2
[23], Hst2 from Saccharomyces cerevisae [21] and Sir2-Af1 from
Archaeoglobus flugidus [22] is shown in Figure 1. This model has
94,9% residues in the most favoured regions and a Prosa Z-score
of 8,7. These values, compared also with those of the template
structures, indicate that a good quality model has been created.
The catalytic core appears well structured in agreement with its
low propensity to the disorder, as shown in Figure 2. Secondary
structure predictions made by JPRED program agree with the
structure of catalytic core obtained by comparative modeling
(Figure 3). The core (from residue 244 to 498) is well structured. The
model (Figure 1) shows the two structural domains typical of the
sirtuin family of which the first one is a Rossman fold with the
characteristic b-a-b motives of NAD proteins, and the second one is
a smaller sub-domain where the zinc is located. At the interface
between domains there is the binding site for NAD. The NAD
molecule fits in a specific pocket constituted of a hydrophobic patch
Figure 1. Model of human Sirt-1. The different structural/functional regions are shown in different colors: in white the N-terminal region, in cyanthe allosteric site, in green the catalytic core and in magenta the C-terminal region. The region of the zinc binding (down on the right) shows the fourtetrahedrically coordinated cysteines.doi:10.1371/journal.pone.0007350.g001
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 2 October 2009 | Volume 4 | Issue 10 | e7350

on the small sub-domain, and a hydrophilic patch on the large
domain. In particular, the NAD molecule presents the adenine and
ribose moieties inside the pocket, as described for other sirtuins
[22–25], but the nicotinamide group is near to the residues 269–295
of human Sirt-1. These residues (indicated with a white arrow in
Figure 1) represent another conserved region that forms a flexible
loop near the pocket. This loop was proposed as a ‘‘frontwall’’ [25]
of the C-site, or a ‘‘ceiling’’[25] on the pocket. Its flexible
organization allows a structural rearrangement of the catalytic
domain during the NAD binding, which seems essential for the
catalysis. This central role is demonstrated by the orientation of this
region in the crystallized sirtuins. In fact, the crystallized complexes
with the NAD+ molecule show a same order orientation, but
without NAD, this loop region shows a more poorly defined or
disordered orientation [21,23]. This suggests that the NAD+binding influences the orientation of this flexible loop by triggering
the assembly and disassembly of the C pocket [25] as well as the
loop’s orientation is a measure of the accessibility into the pocket for
the nicotinamide or other small inhibitors [1].
The zinc ion is tetrahedrally coordinated by the thiol groups of four
Cys residues (at positions 371, 374, 395, 398) which are folded into a
single structural unit. This tetrad of cysteine residues are that is broadly
conserved in the small sub-domain of all the sirtuin family [23]. and this
small zinc-binding domain (see Figure 1) is thought to play a role in
substrate-specific binding by the sirtuin proteins [26]. In the classic zinc
finger, one zinc atom is bound to two cysteines and two histidines while
in this case we have a Cys4-Zn. This structure is composed by about 30
residues included in a flexible structural environment that sterically
favours the tetrahedrally coordination of the cysteines.
The allosteric site (from residue 181 to 243) is straddling
between the N-terminal and the compact globular core of the
protein and shows an all-alpha structure composed by four alpha
helices in agreement with secondary structure and disorder
predictions (Figure 2b and 3).
Both the catalytic core and the allosteric site have compact and
globular shapes according to the definition of globularity recently
published [27] with a score of 5.1 and 4.3, respectively.
The two models were linked, as reported in Methods, by a
flexible loop and subjected to molecular dynamics simulation. The
state of equilibrium was reached after 8 ns simulation. The
structure remained very stable during the whole simulation time,
as confirmed by all the indicators commonly used to analyse MD
simulations (Fig. 4A). In particular, the Rossman fold, as well as
the smaller domain and the four helices of the allosteric site were
well conserved during the simulation. Only two flexible loops, i.e.
the final loop of the allosteric site and the ‘‘frontwall’’ in the
catalytic domain, presented some fluctuation. An evaluation of the
distance between the allosteric site and the catalytic core was
obtained in terms of center of mass distances along the trajectories.
This distance decreased of 17 A after 10 ns of simulation and was
maintained constant during the last 5 ns of simulation.
Modeling of the N-terminal and C-terminal regionsVarious programs for predicting the secondary structure of
globular proteins were unable to have a good consensus for the N-
terminal and C-terminal segments of Sirt-1. They have differently
predicted in amount and in sequence strings the scattered presence
of non alfa and non beta regions in both terminal zones. This
observation raised the suspect that the Sirt-1 could be an
unordered protein and more appropriate algorithms were used.
The N-terminal and C-terminal regions were found largely
unordered by two specific structural tests. DISOPRED, a software
devoted to the search of unordered regions predicted long
segments in N-terminal region (1–150 and 160–182) as well as
in C-terminal region (510–580, 585–640 and 680–740). Moreover,
the Anchor program, in addition, was also able to predict the
presence of definite protein binding regions [28] in the unordered
segments (Figure 2 and Table S1). The N-terminal and C-terminal
regions were modelled as reported in Methods and resulted made
of six very short helices inserted with seven short b-strands and of
five short helices inserted with five short b-strands, respectively.
The remaining large part of residues are in irregular conforma-
tions according to the predictions.
The two modelled regions were independently subjected to
molecular dynamics simulations with the same protocol used for
the allosteric and catalytic sites. The N-terminus reached a stable
equilibrated state after 8 ns and the C-terminal after 4 ns of
simulation, respectively. Both models looked very stable (Fig. 4B
and C) and the secondary structures present in both models were
well kept during the simulation time.
Complete model of Sirt-1We created a whole model of the human Sirt-1 by comparing
modelling using as templates the previously obtained models of N-
terminal, allosteric site, catalytic site and C-terminal site. The
model was minimized according to our recent papers [29].
The resulting model has 87.8% residues in the most favoured
regions and a Prosa Z-score of -6.57.
The various structural parts that compose the global architec-
ture of the protein are shown in Figure 1. The N-terminal and C-
terminal regions are positioned behind the catalytic core, but the
groove site of Sirt-1 is exposed to the solvent to receive its
substrates. Distances among the centers of mass of catalytic core,
N-terminal and C-terminal regions were evaluated. In details, the
catalytic core-N-terminal distance resulted to be 38 A, and that
between the catalytic core and C-terminal of 48 A. The catalytic
core exchanges 2 hydrogen bonds and 8 salt bridges with the N-
terminal site, and 5 hydrogen bonds and 13 salt bridges with the
C-terminal site.
These values confirm that the three regions have distinct
structural roles even if one can easily hypothesize that their very
Figure 2. Predictions of disorder regions. Top - The disorderpredictions (blue) made by DISOPRED for the N-terminal region (a), theallosteric site (b), the catalytic core (c) and the C-terminal region (d).Bottom - The prediction of disordered binding regions (black) made byAnchor.doi:10.1371/journal.pone.0007350.g002
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 3 October 2009 | Volume 4 | Issue 10 | e7350

Figure 3. Primary and secondary structures of Sirt-1 and AROS. a) catalitic core (from residue 244 to 498), b) allosteric site (from residue 181to 243) and c) AROS. Helix and beta strands regions are shown in red and cyan, respectively. Moreover, JPred predictions are reported.doi:10.1371/journal.pone.0007350.g003
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 4 October 2009 | Volume 4 | Issue 10 | e7350

close relationships suggests an involvement in the functional
activities of the Sirt-1. In fact, the allosteric site is positioned
between the N- terminus and the catalytic core in the best position
to regulate the enzymatic activity of Sirt-1. Moreover, the N-
terminal and C-terminal sites are positioned in a region that does
not prevent the catalytic core activity, and this confirms that they
have a role of regulation and localization of Sirt-1.
AROS modelAROS is the endogenous protein that is able to increase the
Sirt-1 activity [2], interacting with its allosteric site. Its model was
obtained by fold recognition strategy and presents an all-alpha fold
composed from 4 alpha helices in agreement with the secondary
structure predictions made by JPred [30] (Figure 3C) and with the
structural class prediction made by PRECLASSPRO server [31].
The AROS model was subjected to molecular dynamics
simulations by using the protocol reported in the Methods. A
stable state was reached after 7 ns of simulation time and was kept
constant up to the end of the dynamics (Figure 4D).
Sirt-1/AROS complexAROS is the endogenous protein that is able to increase the
Sirt-1 activity [2], interacting with its allosteric site. To investigate
the best binding-groove of Sirt-1 for allosteric effectors we used the
principal docking servers (i.e. PATCHDOCK, GRAMMX,
CLUSPRO) to obtain a set of possible complexes. We firstly
Figure 4. Molecular dynamics results. RMSD evolution and analysis of the secondary structures during the molecular dynamics.doi:10.1371/journal.pone.0007350.g004
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 5 October 2009 | Volume 4 | Issue 10 | e7350

selected the complexes having AROS near to the allosteric site
because experimental truncation data have suggested that some
amino acids of the allosteric site are important for the binding of
allosteric activators [19]. It is worthy of note that in the most part
of simulated complexes, AROS is placed in the region between the
N-terminal site and the allosteric site (Figure 5). For a more
detailed analysis, we selected two of the best complexes for each
set, in terms of energy binding, interface ASA, number of atoms
and residues at the interface, number of hydrogen bonds and salt
bridges (see Table 1). Moreover, we compared, in terms of
physical-chemical and geometric properties, the exposed residues
of Sirt-1 to the binding pocket in each complex to identify
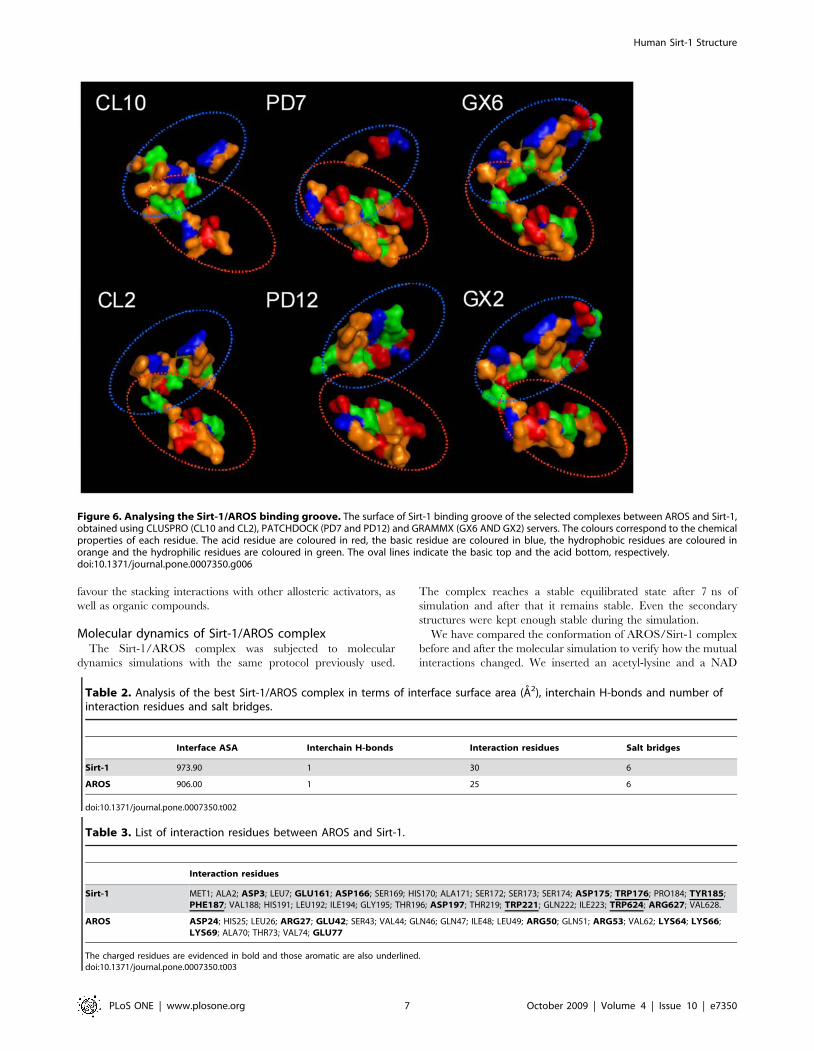
pharmacophore features. As shown in Figure 6, the binding
groove presents basic residues at the top of the cavity, acidic
residues at its bottom and hydrophobic residues as edges.
The best Sirt-1/AROS complex was obtained using the
CLUSPRO web server [32] that uses one of the best docking
algorithm tested in CAPRI experiments with a success rate of about
71% [32]. The good quality of this complex is also suggested by the
very low value of its binding free energy (210.19 Kcal/mol),
calculated by the DCOMPLEX server [33]. For this complex, we
have evaluated the interacting residues, the number of interchain
H-bonds and salt bridges and the interface surface area (Tables 2
and 3). The AROS-Sirt-1 complex shows that AROS is located
behind to the catalytic groove. This can be considered one of the
most favourable structural site to increase the enzyme activity by
conformational changes without penalizing the correct interaction
between Sirt-1 and its substrates. In particular, AROS and Sirt-1
chains should form one H-bond and 6 salt bridges at their
interaction surface. At the interface Sirt-1 also exposed four
aromatic residues, one positively charged residue and five negatively
charged residues. The interface region of AROS is composed by six
and three positively and negatively charged residues, respectively.
These data suggest that the predominant interaction between Sirt-1
and AROS is on electrostatic basis and that the four aromatic
residues structurally closely in Sirt-1 might play an important role to
Figure 5. Model of human Sirt-1/AROS complex.doi:10.1371/journal.pone.0007350.g005
Table 1. The selected complex obtained using CLUSPRO, PATCHDOCK and GRAMMX server.
Complexes Binding free energy Interface ASA Interaction Residues H-bonds Salt Bridges
CL2 29.96 1023.49 34 4 24
CL10 210.19 973.9 30 1 29
PD7 210.51 1231.39 34 7 39
PD12 28.92 1230.1 39 4 29
GX2 210.73 1471.8 48 5 11
GX6 212.65 1332.27 40 4 12
For each one are reported the value of: energy binding (Kcal/mol), interface ASA (A2), number of atoms and residues at the interface, number of hydrogen bonds andsalt bridge are reported.doi:10.1371/journal.pone.0007350.t001
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 6 October 2009 | Volume 4 | Issue 10 | e7350
favour the stacking interactions with other allosteric activators, as
well as organic compounds.
Molecular dynamics of Sirt-1/AROS complexThe Sirt-1/AROS complex was subjected to molecular
dynamics simulations with the same protocol previously used.
The complex reaches a stable equilibrated state after 7 ns of
simulation and after that it remains stable. Even the secondary
structures were kept enough stable during the simulation.
We have compared the conformation of AROS/Sirt-1 complex
before and after the molecular simulation to verify how the mutual
interactions changed. We inserted an acetyl-lysine and a NAD
Figure 6. Analysing the Sirt-1/AROS binding groove. The surface of Sirt-1 binding groove of the selected complexes between AROS and Sirt-1,obtained using CLUSPRO (CL10 and CL2), PATCHDOCK (PD7 and PD12) and GRAMMX (GX6 AND GX2) servers. The colours correspond to the chemicalproperties of each residue. The acid residue are coloured in red, the basic residue are coloured in blue, the hydrophobic residues are coloured inorange and the hydrophilic residues are coloured in green. The oval lines indicate the basic top and the acid bottom, respectively.doi:10.1371/journal.pone.0007350.g006
Table 2. Analysis of the best Sirt-1/AROS complex in terms of interface surface area (A2), interchain H-bonds and number ofinteraction residues and salt bridges.
Interface ASA Interchain H-bonds Interaction residues Salt bridges
Sirt-1 973.90 1 30 6
AROS 906.00 1 25 6
doi:10.1371/journal.pone.0007350.t002
Table 3. List of interaction residues between AROS and Sirt-1.
Interaction residues
Sirt-1 MET1; ALA2; ASP3; LEU7; GLU161; ASP166; SER169; HIS170; ALA171; SER172; SER173; SER174; ASP175; TRP176; PRO184; TYR185;PHE187; VAL188; HIS191; LEU192; ILE194; GLY195; THR196; ASP197; THR219; TRP221; GLN222; ILE223; TRP624; ARG627; VAL628.
AROS ASP24; HIS25; LEU26; ARG27; GLU42; SER43; VAL44; GLN46; GLN47; ILE48; LEU49; ARG50; GLN51; ARG53; VAL62; LYS64; LYS66;LYS69; ALA70; THR73; VAL74; GLU77
The charged residues are evidenced in bold and those aromatic are also underlined.doi:10.1371/journal.pone.0007350.t003
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 7 October 2009 | Volume 4 | Issue 10 | e7350

molecule in the AROS/Sirt-1 complex using as template the
crystallographic structure of Saccharomyces cerevisae Sirt-1. Then, we
focused our attention on the catalytic groove. We noted that,
during the molecular dynamics, a conformational change of the
protein occurs mainly in active site region. In details, the residues
His 363, Asn 346, Ser 265, Gly 261 and Pro 447, indicated as
important for its catalytic role [34], are sterically changed (see
Figure S1). Also Pro 447, that makes Van der Waals contacts with
the aliphatic portion of the acetyl-lysine side chain and may affect
the positioning of residues that contact the acetyl group, is more
close to the Acetyllisine while four other residues (His 363, Asn
346, Ser 265, Gly 261) that affect the NAD orientation,
approaches to the NAD.
Therefore these results could indicate that the molecular
dynamics improves the interaction between Sirt-1 and AROS by
some structural parameters (see Table S2) even if longer time scale
will be used for studying this type of long range coupling.
Discussion
Human sirtuin-1, an oxidative stress-response and chromatin-
silencing factor, is an unordered protein. About half of its sequence
appears to be in a disordered state with long flexible poorly
structured regions observed at N- and C-terminus. The modeling
has also evidenced a large central globular part very well structured
made by two close regions, the catalytic core and the allosteric site.
Therefore, the protein can be considered composed by four
different regions: N-terminal domain, allosteric site, catalytic core
and C-terminal domain. The catalytic core is a central highly
conserved structured region common to all the sirtuin family [1]
which has the role of catalyzing the NAD+-dependent deacetylation
reaction, involved in various nuclear events such as transcription,
DNA replication, and DNA repair. The core is made by a well
organized Rossman fold typical for NAD-dependent proteins. In
details, the acetyl lysine substrate has been proposed to bind in a
narrow channel that terminates near to the nicotinamide ribose.
Moreover, the binding of acetyl-substrate is believed to mediate
bending of the nicotinamide ring of NAD+ in a strained
conformation also referred to productive NAD+ binding, promoting
the cleavage of the ribosyl–nicotinamide bond [21,24].
In this domain we find an atom of Zinc tetrahedrically
coordinated with four cysteine residues also present in the other
members of the sirtuin family. The allosteric site is a small
structural domain made of four helices and interacting with the
core. It is sited in a structural location where it can easily exert a
control of the catalytic activity by conformational changes. Several
articles report the presence of activators modulating the functional
activity of the human sirtuin. One of the activators, the resveratrol,
decreases the Michaelis constant of SIRT1 for both the acetylated
substrate and NAD+, and increases cell survival by stimulating
SIRT1-dependent deacetylation of p53 [35]. Moreover, a small
nuclear protein, AROS (Active Regulator Of Sirtuin), has been
found to be the first known endogenous active modulator of
SIRT1 which directly regulates SIRT1 function. AROS enhances
SIRT1-mediated deacetylation of p53 both in vitro and in vivo,
and it inhibits p53-mediated transcriptional activity. It is
interesting to observe that AROS was unable to inactivate p53
when was used an AROS-binding-defective SIRT1 mutant. This
clearly indicates a direct interaction of AROS with a specific site of
Sirt-1. Our best model of AROS presents an all-alpha fold
composed from 4 alpha helices. We found that AROS binds the
same allosteric site of some small synthetic compounds (not shown;
manuscript in preparation) proposed as putative therapeutics for the
treatment of type 2 diabetes [2,19].
The catalytic core and the allosteric site make a central
structured domain (about from residues 181 to 498), compact and
globular as assessed by various globularity indices.
Moreover Sirt-1 presents two long disordered structural
segments at the terminal regions, missing in the other six
homologous proteins belonging to the same family. The N-
terminal region is 180 residues long and that C-terminal 249.
These regions were predicted largely disordered by DISOPRED
program that resulted one of the best algorithm for the accuracy of
disorder prediction in CASP7 [36–37]. Figure S2 shows the
secondary structure of the best model of Sirt-1 calculated by DSSP
algorithm that evaluates the W and C dihedral angles of each
residue and the backbone Hydrogen bonds. From this table as one
can see both termini are characterized by numerous and large
unstructured regions even if in the C-terminal more structured
segments are presents.
Many proteins or protein domains show an intrinsic inability to
form a well defined tertiary structure. This property is encoded in
their sequence owing to the local depletion of typically buried
amino acid residues as well as enrichment of typically exposed
amino acid residues (about 40%) (see Table S3). Amino acid
residues located in highly mobile regions of protein also possess the
smallest volumes and molecular weights as well as the lowest
hydrophobicities and the highest flexibility (see Figures S3, S4, S5
and S6). Only in this way the protein can be so dynamically
flexible to minimize unfavorable pairwise contacts.
In the last years it has become evident that there is a large
number of proteins that do not require a stable structure even
under physiological conditions in order to fulfil their biological role
[38–40]. The importance of protein disorder is underlined by the
abundance of partially or fully disordered proteins that are
encoded in higher eukaryotic genomes [36] and are involved in
many important biological functions [38], which complement the
functional repertoire of globular proteins [41]. Disordered
segments often act as flexible linkers between folded domains in
multidomain proteins [38] and their function is often that of
binding specifically other proteins, DNA or RNA through a
process, termed coupled folding and binding, that involves a
transition disordered-ordered with stable secondary and tertiary
structural elements [38]. This ‘‘coupled binding and folding’’
confers several functional advantages in certain types of molecular
interactions that often are essential for signaling processes. The
high propensity of their residues to stay disordered makes these
regions predominantly not structured and this can favour their
putative functionality.
In fact, recent papers have shown the importance in Sirt-1 of
various phosphorilations sites, nuclear localization signals (NLS)
and nuclear export signals (NESs, amino acids ) that were mainly
found in the terminal regions [3,42,43]. These authors suggested
that these regions seem to be involved in regulating enzyme
activity and have the peculiarity of being present only in Sirt-1.
Moreover, this fact also suggests that C- and N-terminal regions of
the human Sirt-1 might be involved in a more fine regulating role
in order to exploit its biological mechanisms. In the disordered
regions specific protein binding sites were identified by using
Anchor program [28]. The prediction of these binding sites is
based on estimating the energy content in free and in the bound
states, and identifying segments that are potentially sensitive to
these changes. In particular, Anchor program has predicted in
human Sirt-1 fourtheen disordered binding regions of which four
located in the N-terminal domain, two in the allosteric site and
other eight in the C-terminal domain (Figure 2-down and Table
S1). The two disordered binding regions predicted in the allosteric
site agree with those evidenced in Sirt-1/AROS complex (see
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 8 October 2009 | Volume 4 | Issue 10 | e7350

Table 3). All these data indicate that human Sirt-1 is an unordered
protein and that its terminal domains can play different roles. The
inherent flexibility of the two termini suggests that the protein has
a malleable interface that can allow the binding of several partners
or adopt different conformations, as manifested by its high binding
capability.
Moreover the flexibility and the open structure of Sirt-1 termini
could also favour the binding of phosphorylating proteins in order
to activate regulation processes mediated by phoshorylation [3]. In
fact, very recently the presence of phosphorylation sites located in
the amino- and in carboxyl-terminal regions were found [3].
Therefore, in order to assess the ability of human Sirt-1 to be
regulated by phorphorylation [43] and if there are the structural
features inducing the interaction with phosphorylating proteins,
we have evaluated the solvent accessibility of each residue,
suggested as a putative site of phosphorylation [3]. Our data
indicate that Ser14, Ser173, Ser538, Ser 539, Ser540, Thr 544
and Ser747 are largely exposed to the solvent (Figure 7)
confirming that these residues have a high capability to be
phosphorylated. If one considers the sequence position of Ser and
Thr residues they are almost located in the N- and C- termini:
Ser14, Ser26 and Ser27 are inserted in the segment 1–33
predicted by Anchor as a protein binding site. The residues
Ser47 and Ser 159 are also included in the predicted segments 43–
49 and 133–161, respectively, while the remaining Ser169,
Ser173, Ser174, Thr196 and Thr 219 are very close to the above
predicted segments. This suggests that these residues are actually
involved in some way in the control of protein-protein binding
and, as a consequence, in the control of the Sirt-1 function. The
Sirt-1/AROS complex explains well this view by suggesting that
the interaction between the two proteins occurs primarily through
the involvement of the N-terminal segment with the achievement
of greater compactness of the complex and the presence of
changes that propagate to the active site in order to modulate the
function. Work is in progress to simulate the presence of phosphate
at various sites in the unordered regions in order to assess their
structural or functional effects during the formation of the
complex. The numerous and different structural features of
unordered proteins such as conformational heterogeneity, second-
ary structural propensities, and tertiary contacts within disordered
protein states can in principal generate many different interaction
modes in proteins. Frequently these proteins mediate in signaling
networks dynamic protein interactions that exhibit unusual
binding characteristics, such as multisite dependence and ultra-
sensitivity. Such interactions are frequently modulated by
phosphorylation, which requires disorder in the target protein
both for optimal kinase accessibility and for subsequent accessi-
bility of the binding motif [44,45]. Therefore this binding appears
to require multiple sites to be phosphorylated, suggesting a binding
mode in which multiple phosphoepitopes engage a single receptor
site in dynamic equilibrium. These observations allows to
hypothesize a binding mode in which the multiple sites found in
Sirt-1 might engage the putative receptor sites of AROS (5 Ser and
4 Thr residues) in a dynamic equilibrium. Moreover, Sirt-1
contains numerous additional phosphorylation sites remote from
the targeting regions, making their participation in the complex
unlikely. It is possible, however, that they may serve as decoy sites
that compete with the key binding sites for phosphorylation by the
targeting kinase [46].
Finally, a detailed study of the binding patches between AROS
and Sirt-1 has shown that in Sirt-1 there is a conserved
pharmacophore pocket composed by basic residues at the top of
the cavity, acidic residues at its bottom and hydrophobic residues
as edges. Moreover we have focused our attention on the presence
of five aromatic residues (i.e. Trp176, Tyr185, Phe187, Trp221,
Trp624) in the pocket that could be involved in putative stacking-
interactions. These data could explain the high affinity of the
allosteric site for synthetic activators containing aromatic rings and
hydrophobic anchor points. Investigations are in progress to study
the Sirt-1 interaction also with small allosteric effectors that have
been recently identified [19].
Methods
Modelling of catalytic and allosteric sites of human Sirt-1The three-dimensional model of catalytic site of human Sirt-1
(UniProt code: Q96EB6, region 244–498) was performed by
comparative modelling strategy using the template structures of
human Sirt-2 (PDB code: 1J8F chain A) [23], Hst2 from
Saccharomyces cerevisae (PDB code: 1Q1A chain A) [21] and
Sir2-Af1 from Archaeoglobus flugidus (PDB code: 1M2G chain A)
[22] because the percentage of sequence identity between these
proteins and human Sirt-1 was equal to 44%, 38% and 30%,
respectively. Protein sequences were aligned with CLUSTALW
[47]. The MODELLER9v5 program [48] was used to build 10
full-atom models of catalytic site, we used the ProsaII program to
check the fitness of the sequences relative to the obtained
structures and to assign a scoring function, and the PROCHECK
program [49] to evaluate their stereochemical and structural
packing quality. Secondary structures were assigned by the DSSP
program [50]. Secondary structure predictions were performed
with Jpred [29] server but structural class predictions were made
by using PRECLASSPRO server [31]. The globularity of the best
model was evaluated according to our recent work [27].
Moreover, the three-dimensional model of allosteric site of
human Sirt-1 region 181–243 was performed using the template
structure of a hexokinase from Sulfolobus Tokodaii by compar-
ative modelling (PDB code: 2E2N) [51]. As the sequence identity
between this Sirt-1 region and the homologous template model
was lower than 30% (i.e. 26%), we used a procedure strategy in
agreement with the rules recently reviewed to improve the quality
of the modelling results at low target-template sequence similarity
[52]. Allosteric and catalytic sites have been connected by using
the Builder module of Insight II and the related model was
subjected to molecular dynamics simulations.
Figure 7. Analysis of phosphorilated sites. The Solvent accessi-bility profile for the residues belonging to putative phosphorilation site,valuated using Surface program (red line) and ASAview server (blueline). The number indicates the sequence position and the one lettercode identifies the amino acid. The figure also reports the segmentalposition of residues in the protein.doi:10.1371/journal.pone.0007350.g007
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 9 October 2009 | Volume 4 | Issue 10 | e7350

Modelling of N-terminal and C-terminal regions of humanSirt-1
The BLAST research [53] has found in databases only protein
structures related to small sequences similar to the N-terminal and
C-terminal regions of human Sirt-1 [region 1–180 and 499–747,
respectively]. The prediction of unordered regions in the protein
was made by Disopred server [36]. Moreover, a prediction of
protein binding sites in the unordered regions was made by
Anchor program that identifies specific binding regions undergo-
ing disorder-to-order transition using a general disorder prediction
method IUPred based on the assumption that disordered proteins
have a specific amino acid composition that does not allow the
formation of a stable well-defined structure [28]. The prediction of
binding sites is based on estimating the energy content in free and
in the bound states, and identifying segments that are potentially
sensitive to these changes.
Therefore these regions were modelled by using a new
approach combining fold recognition and comparative modelling.
The preliminary models for N-terminal and C-terminal regions of
Sirt-1 were obtained by fold recognition strategy using the Fugue
and SAM-T06 servers [54–55]. Then, these models with the
crystallographic structures suggested by BLAST were used as
template for applying the comparative modelling strategy in order
to obtain the complete models for both regions. In details, we used
as template the following structures deposited in PDB: 1W36
(577–625), 1QHZ (1–34) for the N-terminal region and 1TYC
(76–101) for C-terminal region.
In the obtained models the loop regions were refined using the
LOOPY module of Jackal package [56]. LOOPY appeared to
yield the best results for loop modelling, with models that are on
average of 2–8% better than those generated by other programs
[52]. The models obtained for N-terminal and C-terminal regions
of Sirt-1 were subjected to molecular dynamics simulations.
Molecular dynamics simulationsMD simulations were performed with GROMACS software
package (v3.3.1) [57]. Models of different Sirt-1 regions were put
in cubic boxes filled with SPC216 water molecules and
GROMOS43a1 was selected as force-field. In order to optimize
the system, the models were previously subjected to energy
minimization and position restraints cycles. The simulations were
carried out with periodic boundary conditions by adding sodium
ions in order to the net electrostatic charge of the system is zero.
The bond lengths were constrained by the all atoms LINCS
algorithm. Particle Mesh Ewald (PME) algorithm was used for the
electrostatic interactions with a cut-off of 0.9 nm, according to
recent papers [29]. Simulations were conducted at neutral pH
where the tritable groups of His, Glu and Asp residues are
unprotonated. All simulations were run for 15 ns at room
temperature (300 K) coupling the system to an external bath.
GROMACS routines were utilized to check the trajectories and
the quality of the simulations.
Simulation of complete model of Sirt-1The whole model of human Sirt-1 was obtained by comparing
modelling using as template the models obtained for N-terminal
region, allosteric site, catalytic site, and C-terminal region and the
same procedure and programs described above. This model was
minimized by using 500 steps of energy minimization under
conjugate gradient algorithm to optimize side chain conformations
and avoid sterical clashes according to the commonly used
procedure [29,58–59].
Modelling and Simulation of Sirt-1/AROS complexThe three-dimensional model of AROS (UniProt code:
Q86WX3, region 47–123) was performed by fold recognition
strategy using the Fugue server [54]. The model obtained for AROS
was subjected for 15 ns to molecular dynamics simulations using the
protocol reported above to assess its conformational stability.
To simulate the Sirt-1/AROS complex we used the docking
web server CLUSPRO [32] that resulted the best docking
program in CAPRI experiments with a success rate of about
71% [32]. The models were selected by evaluating some features
and parameters. The ‘‘Protein–Protein Interaction Server’’ [60]
were used to identify the amino acids at the interface and to
evaluate their solvent accessibility.
Moreover, the binding free energy between the different chains
was calculated by using the DCOMPLEX program [33].
Supporting Information
Table S1 Regions predicted as protein binding sites by Anchor
program
Found at: doi:10.1371/journal.pone.0007350.s001 (0.03 MB
DOC)
Table S2 Analysis of interaction between AROS and Sirt-1
before and after MD
Found at: doi:10.1371/journal.pone.0007350.s002 (0.03 MB
DOC)
Table S3 Amino acid composition of Sirt-1
Found at: doi:10.1371/journal.pone.0007350.s003 (0.18 MB
DOC)
Figure S1 Details of catalytic groove before and after molecular
dynamics are shown. We reported in pink and green the carbon
atoms related to Sirt-1 before and after dynamics but N, O and H
atoms always in blue, red and white, respectively. The Acetil-
lysine, NAD and Sirt-1 residues are evidenced with labels.
Found at: doi:10.1371/journal.pone.0007350.s004 (0.19 MB
DOC)
Figure S2 Secondary structure of the whole Sirt-1 structure
assigned by DSSP program. The N-terminal and C-terminal region
sequences are reported in green and blue, respectively. The helices
and beta-strands are indicated in red and cyan, respectively.
Found at: doi:10.1371/journal.pone.0007350.s005 (0.07 MB
DOC)
Figure S3 Flexibility plot for Sirt-1 sequence. Ordinate reports
the value of Hydrophobicity x Volume obtained with a shifting
window of 5 according to Ragone et al. Protein Eng. 1989
2(7):497–504. Abscissa reports the residue position.
Found at: doi:10.1371/journal.pone.0007350.s006 (0.09 MB
DOC)
Figure S4 Average area buried. Lower values indicates higher
exposures of residues. The graph shows that the residues in the
globular part of the protein are in average more buried than the N
and C termini. In particular, residues in the N-terminus are in
average more exposed.
Found at: doi:10.1371/journal.pone.0007350.s007 (0.03 MB
DOC)
Figure S5 The average molecular weight of residues with a
shifting window of 5. The graph shows that the compact globular
core is made in average of high molecular weight residues while the
N- and C- termini are made of low molecular weight residues and
thus smaller residues are located in the more fluctuating or flexible
structural regions. It is interesting to note the highly fluctuating
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 10 October 2009 | Volume 4 | Issue 10 | e7350

values in the C-terminal region in agreement with the presence of
more structured segments in respect to the N-terminal region.
Found at: doi:10.1371/journal.pone.0007350.s008 (0.03 MB
DOC)
Figure S6 Ramachandran Plot
Found at: doi:10.1371/journal.pone.0007350.s009 (0.46 MB
DOC)
Author Contributions
Conceived and designed the experiments: SC GC. Performed the
experiments: IA. Analyzed the data: IA SC GC. Wrote the paper: IA
SC GC.
References
1. Huhtiniemi T, Wittekindt C, Laitinen T, Leppanen J, Salminen A, et al. (2006)
Comparative and pharmacophore model for deacetylase SIRT1. J ComputAided Mol Des 20: 589–99.
2. Kim EJ, Kho JH, Kang MR, Um SJ (2007) Active regulator of SIRT1
cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell 28:
277–90.
3. Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, et al. (2008)Phosphorylation regulates SIRT1 function. PLoS ONE 3: e4020.
4. Saunders LR, Verdin E (2007) Sirtuins: critical regulators at the crossroads
between cancer and aging. Oncogene 26: 5489–5504.
5. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, et al. (2001)
hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107:149–159.
6. Yang Y, Hou H, Haller EM, Nicosia SV, Bai W (2005) Suppression of FOXO1activity by FHL2 through SIRT1-mediated deacetylation. EMBO J 24:
1021–1032.
7. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, et al. (2005) Nutrientcontrol of glucose homeostasis through a complex of PGC-1alpha and SIRT1.
Nature 434: 113–118.
8. Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, et al. (2007)
SIRT1 promotes endothelium-dependent vascular relaxation by activatingendothelial nitric oxide synthase. Proc Natl Acad Sci USA 104: 14855–14860.
9. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, et al. (2004)Sirt1 promotes fat mobilization in white adipocytes by repressing PPARgamma.
Nature 429: 771–776.
10. Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I (2008) SIRT1, anantiinflammatory and antiaging protein, is decreased in lungs of patients with
chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177:
861–870.
11. Ghosh HS, Spencer JV, Ng B, McBurney MW, Robbins PD (2007) Sirt1interacts with transducin-like enhancer of split-1 to inhibit nuclear factor
kappaB-mediated transcription. Biochem J 408: 105–111.
12. Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2
alone promote longevity in Saccharomyces cerevisiae by two differentmechanisms. Genes Dev 13: 2570–80.
13. Wang Y, Oh SW, Deplancke B, Luo J, Walhout AJ, et al. (2006) C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO.
Mech Ageing Dev 127: 741–747.
14. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, et al. (2004) Sirtuinactivators mimic caloric restriction and delay ageing in metazoans. Nature 430:
686–689.
15. Yamamoto H, Schoonjans K, Auwerx J (2007) Sirtuin functions in health and
disease. Mol Endocrinol 21: 1745–1755.
16. Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, et al. (2007)
Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med39: 335–345.
17. Haigis MC, Guarente LP (2006) Mammalian sirtuins–emerging roles in
physiology, aging, and calorie restriction. Genes Dev 20: 2913–2921.
18. Hollander P (2007) Diabetes Spectrum 20: 159.
19. Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, et al. (2007) Small
molecule activators of SIRT1 as therapeutics for the treatment of type 2diabetes. Nature 450: 712–6.
20. Bemis JE, Vu CB, Xie R, Nunes JJ, Ng PY, et al. (2009) Discovery ofoxazolo4,5-b.pyridines and related heterocyclic analogs as novel SIRT1
activators. Bioorg Med Chem Lett 19: 2350–3.
21. Zhao K, Harshaw R, Chai X, Marmorstein R (2004) Structural basis fornicotinamide cleavage and ADP-ribose transfer by NAD(+)-dependent Sir2
histone/protein deacetylases. Proc Natl Acad Sci USA 101: 8563.
22. Chang JH, Kim HC, Hwang KY, Lee JW, Jackson SP, et al. (2002) Structural
basis for the NAD-dependent deacetylase mechanism of Sir2. J Biol Chem 277:34489–34498.
23. Finnin MS, Donigian JR, Pavletich NP (2001) Structure of the histonedeacetylase SIRT2. Nat Struct Biol 8: 621–5.
24. Min J, Landry J, Sternglanz R, Xu RM (2001) Crystal structure of a SIR2
homolog-NAD complex. Cell 105: 269.
25. Avalos JL, Bever KM, Wolberger C (2005) Mechanism of sirtuin inhibition by
nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. MolCell 17: 855.
26. Zhao K, Chai X, Marmorstein R (2004) Structure and Substrate Binding
Properties of cobB, a Sir2 Homolog Protein Deacetylase from Escherichia coli.
Journal of Molecular Biology 337: 731.
27. Costantini S, Facchiano AM, Colonna G (2007) Evaluation of the structuralquality of modeled proteins by using globularity criteria. BMC Structural
Biology 7: 9.
28. Meszaros B, Simon I, Dosztanyi Z (2009) Prediction of protein binding regions
in disordered proteins. Plos Computational Biology 5: e1000376.
29. Paladino A, Costantini S, Colonna G, Facchiano AM (2008) Molecular
modelling of miraculin: structural analyses and functional hypotheses.Biochemical and Biophysical Research Communications 367: 26–32.
30. Cuff JA, Barton GJ (2000) Application of enhanced multiple sequence alignmentprofiles to improve protein secondary structure prediction. Proteins: Structure,
Function and Genetics 40: 502–511.
31. Costantini S, Facchiano AM (2009) Prediction of the protein structural class by
specific peptide frequencies. Biochimie 91: 226–9.
32. Comeau SR, Kozakov D, Brenke R, Shen Y, Beglov D, et al. (2007) ClusPro:
performance in CAPRI rounds 6-11 and the new server. Proteins 69: 781–5.
33. Liu S, Zhang C, Zhou H, Zhou Y (2004) A physical reference state unifies the
structure-derived potential of mean force for protein folding and binding.
Proteins 56: 93–101.
34. Khan AN, Lewis PN (2006) Use of substrate analogs and mutagenesis to study
substrate binding and catalysis in the Sir2 family of NAD-dependent proteindeacetylases. J Biol Chem 281: 11702–11.
35. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, et al. (2003)Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan.
Nature 425: 191–196.
36. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (2004) Prediction and
functional analysis of native disorder in proteins from the three kingdoms of life.Journal of Molecular Biology 337: 635–645.
37. Bordoli L, Kiefer F, Schwede T (2007) Assessment of disorder predictions inCASP7. Proteins 69S: 129–136.
38. Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and theirfunctions. Nat Rev Mol Cell Biol 6: 197–208.
39. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, et al. (2001)Intrinsically disordered protein. J Mol Graph Model 19: 26–59.
40. Tompa P (2002) Intrinsically unstructured proteins. Trends Biochem Sci 27:527–533.
41. Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, et al. (2007)Functional anthology of intrinsic disorder. 1. Biological processes and functions
of proteins with long disordered regions. J Proteome Res 6: 1882–1898.
42. Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y (2007) Nucleocyto-
plasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J BiolChem 282: 6823–32.
43. Ford J, Ahmed S, Allison S, Jiang M, Milner J (2008) JNK2-dependentregulation of SIRT1 protein stability Cell Cycle 19: 3091–3097.
44. Lamming DW, Wood JG, Sinclair DA (2004) Small molecules that regulatelifespan: evidence for xenohormesis. Mol Microbiol 53: 1003–9.
45. Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, et al. (2004)The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids
Res 32: 1037.
46. Kim SY, Ferrell JE (2007) Substrate competition as a source of ultrasensitivity in
the inactivation of Wee1. Cell 128: 1133.
47. Thompson JD, Higgins DG, Gibson T (1994) CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence
weighting, position-specific gap penalties and weight matrix choice. NucleicAcids Res 22: 4673–4680.
48. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of
spatial restraints. J Mol Biol 234: 779–815.
49. Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK:
a program to check the stereochemical quality of protein structures. J Appl Cryst
26: 283–291.
50. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern
recognition of hydrogen-bonded and geometrical features. Biopolymers 2:2577–2637.
51. Nishimasu H, Fushinobu S, Shoun H, Wakagi T (2007) Crystal structures of anATP-dependent hexokinase with broad substrate specificity from the hyperther-
mophilic archaeon Sulfolobus tokodaii. J Biol Chem 282: 9923–9931.
52. Dalton JAR, Jackson RM (2007) An evaluation of automated homology
modelling methods at low target-template sequence similarity. Bioinformatics23: 1901–1908.
53. Altschul SF, Gish W, Miller W, Myers E, Lipman DJ (1990) Best local alignmentsearch tool. J Mol Biol 215: 403–410.
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 11 October 2009 | Volume 4 | Issue 10 | e7350

54. Shi J, Blundell TL, Mizuguchi K (2001) FUGUE: sequence-structure homology
recognition using environment-specific substitution tables and structure-
dependent gap penalties. J Mol Biol 310: 243–257.
55. Karplus K, Karchin R, Draper J, Casper J, Mandel-Gutfreund Y, et al. (2003)
Combining local-structure, fold-recognition, and new fold methods for protein
structure prediction. Proteins 6: 491–6.
56. Xiang Z, Soto CS, Honig B (2002) Evaluating conformational free energies: the
colony energy and its application to the problem of loop prediction. PNAS 99:
7432–7437.
57. Van Der Spoel D Lindahl E, Hess B, Groenhof G, Mark AE, et al. (2005)
GROMACS: fast, flexible, and free. J Comput Chem 26: 1701–1718.58. Costantini S, Colonna G, Facchiano AM (2007) Simulation of conformational
changes occurring when a protein interacts with its receptor. Computational
Biology and Chemistry 31: 196–206.59. Costantini S, Buonocore F, Facchiano AM (2008) Molecular modelling of co-
receptor CD8aa and its complex with MHC class I and T-cell receptor in seabream (Sparus aurata). Fish Shellfish Immunology 25: 782–90.
60. Jones S, Thornton JM (1996) Principals of protein-protein interactions derived
from structural studies. PNAS 93: 13–20.
Human Sirt-1 Structure
PLoS ONE | www.plosone.org 12 October 2009 | Volume 4 | Issue 10 | e7350
Related Documents











