Human Leukocyte Antigen–Associated Sequence Polymorphisms in Hepatitis C Virus Reveal Reproducible Immune Responses and Constraints on Viral Evolution Joerg Timm, 1 Bin Li, 1 Marcus G. Daniels, 2 Tanmoy Bhattacharya, 2 Laura L. Reyor, 1 Rachel Allgaier, 1 Thomas Kuntzen, 1 Will Fischer, 2 Brian E. Nolan, 1 Jared Duncan, 1 Julian Schulze zur Wiesch, 1 Arthur Y. Kim, 1 Nicole Frahm, 1 Christian Brander, 1 Raymond T. Chung, 1,3 Georg M. Lauer, 1 Bette T. Korber, 2 and Todd M. Allen 1 CD8 T cell responses play a key role in governing the outcome of hepatitis C virus (HCV) infection, and viral evolution enabling escape from these responses may contribute to the inabil- ity to resolve infection. To more comprehensively examine the extent of CD8 escape and adap- tation of HCV to human leukocyte antigen (HLA) class I restricted immune pressures on a population level, we sequenced all non-structural proteins in a cohort of 70 chronic HCV genotype 1a-infected subjects (28 subjects with HCV monoinfection and 42 with HCV/human immunodeficiency virus [HIV] coinfection). Linking of sequence polymorphisms with HLA allele expression revealed numerous HLA-associated polymorphisms across the HCV proteome. Multiple associations resided within relatively conserved regions, highlighting attractive targets for vaccination. Additional mutations provided evidence of HLA-driven fixation of sequence polymorphisms, suggesting potential loss of some CD8 targets from the population. In a sub- group analysis of mono- and co-infected subjects some associations lost significance partly due to reduced power of the utilized statistics. A phylogenetic analysis of the data revealed the substan- tial influence of founder effects upon viral evolution and HLA associations, cautioning against simple statistical approaches to examine the influence of host genetics upon sequence evolution of highly variable pathogens. Conclusion: These data provide insight into the frequency and reproducibility of viral escape from CD8 T cell responses in human HCV infection, and clarify the combined influence of multiple forces shaping the sequence diversity of HCV and other highly variable pathogens. (HEPATOLOGY 2007;46:339-349.) R ecent studies suggest that immune control of hep- atitis C virus (HCV) is possible 1-3 and the role of CD8 T cells is supported by studies linking par- ticular human leukocyte antigen (HLA) class I alleles with control of HCV. 4 How viral infection persists in the face of an activated host immune response is poorly under- stood. Several mechanisms have been suggested that may contribute to the failure to contain HCV: these include impairment of cellular effector functions, 3,5 suppression of antigen-specific cells by regulatory T cells, 6-8 dendritic cell dysfunction, 9 T cell exhaustion, 10,11 or deletion of antigen-specific T cells in the liver. 12 However, persis- tence of HCV may also be facilitated by viral evolution that enables evasion of host immune responses occurring over the course of an individual infection. In the chim- panzee model, a strong association has been demonstrated between viral persistence and the development of CD8 escape mutations. 13,14 Moreover, recent studies have be- gun to clarify the propensity for viral escape from CD8 Abbreviations: aa, amino acid; CTL, cytotoxic T lymphocyte; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HLA, human leukocyte antigen; nt, nucleotide; SIV, Simian immunodeficiency virus. From the 1 Partners AIDS Research Center, Infectious Disease Division, Massachu- setts General Hospital, Harvard Medical School, Boston, MA; 2 Los Alamos National Laboratory, Los Alamos, NM, and Santa Fe Institute, Santa Fe, NM; and 3 Gastrointestinal Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA. Received November 2, 2006; accepted February 16, 2007. Supported by the National Institutes of Health grants AI067926 (TMA) and AI066345 (TMA and GL). B.K., M.D., W.F., and T.B. were supported by an internal Los Alamos Research Development grant, and through NIH HIV-RAD grant # P01 AI061734-02. J.T. was supported by the Deutsche Forschungsgemeinschaft (DFG 323/ 1-1). G.M.L. is a Liver Scholar (the American Liver Foundation). Dr. Timm is currently affiliated with the University of Essen, Institute for Virol- ogy, Essen, Germany. Address reprint requests to: Todd M. Allen, Ph.D., 149 13th Street, Charles- town, MA 02129. E-mail: [email protected]; fax: 617-724-8586. Copyright © 2007 by the American Association for the Study of Liver Diseases. Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hep.21702 Potential conflict of interest: Nothing to report Supplementary material for this article can be found on the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html). 339

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Human Leukocyte Antigen–Associated SequencePolymorphisms in Hepatitis C Virus Reveal
Reproducible Immune Responses and Constraints onViral Evolution
Joerg Timm,1 Bin Li,1 Marcus G. Daniels,2 Tanmoy Bhattacharya,2 Laura L. Reyor,1 Rachel Allgaier,1 Thomas Kuntzen,1
Will Fischer,2 Brian E. Nolan,1 Jared Duncan,1 Julian Schulze zur Wiesch,1 Arthur Y. Kim,1 Nicole Frahm,1
Christian Brander,1 Raymond T. Chung,1,3 Georg M. Lauer,1 Bette T. Korber,2 and Todd M. Allen1
CD8� T cell responses play a key role in governing the outcome of hepatitis C virus (HCV)infection, and viral evolution enabling escape from these responses may contribute to the inabil-ity to resolve infection. To more comprehensively examine the extent of CD8 escape and adap-tation of HCV to human leukocyte antigen (HLA) class I restricted immune pressures on apopulation level, we sequenced all non-structural proteins in a cohort of 70 chronic HCVgenotype 1a-infected subjects (28 subjects with HCV monoinfection and 42 with HCV/humanimmunodeficiency virus [HIV] coinfection). Linking of sequence polymorphisms with HLAallele expression revealed numerous HLA-associated polymorphisms across the HCV proteome.Multiple associations resided within relatively conserved regions, highlighting attractive targetsfor vaccination. Additional mutations provided evidence of HLA-driven fixation of sequencepolymorphisms, suggesting potential loss of some CD8 targets from the population. In a sub-group analysis of mono- and co-infected subjects some associations lost significance partly due toreduced power of the utilized statistics. A phylogenetic analysis of the data revealed the substan-tial influence of founder effects upon viral evolution and HLA associations, cautioning againstsimple statistical approaches to examine the influence of host genetics upon sequence evolutionof highly variable pathogens. Conclusion: These data provide insight into the frequency andreproducibility of viral escape from CD8� T cell responses in human HCV infection, and clarifythe combined influence of multiple forces shaping the sequence diversity of HCV and otherhighly variable pathogens. (HEPATOLOGY 2007;46:339-349.)
Recent studies suggest that immune control of hep-atitis C virus (HCV) is possible1-3 and the role ofCD8 T cells is supported by studies linking par-
ticular human leukocyte antigen (HLA) class I alleles withcontrol of HCV.4 How viral infection persists in the faceof an activated host immune response is poorly under-stood. Several mechanisms have been suggested that maycontribute to the failure to contain HCV: these includeimpairment of cellular effector functions,3,5 suppressionof antigen-specific cells by regulatory T cells,6-8 dendriticcell dysfunction,9 T cell exhaustion,10,11 or deletion ofantigen-specific T cells in the liver.12 However, persis-tence of HCV may also be facilitated by viral evolutionthat enables evasion of host immune responses occurringover the course of an individual infection. In the chim-panzee model, a strong association has been demonstratedbetween viral persistence and the development of CD8escape mutations.13,14 Moreover, recent studies have be-gun to clarify the propensity for viral escape from CD8�
Abbreviations: aa, amino acid; CTL, cytotoxic T lymphocyte; HCV, hepatitis Cvirus; HIV, human immunodeficiency virus; HLA, human leukocyte antigen; nt,nucleotide; SIV, Simian immunodeficiency virus.
From the 1Partners AIDS Research Center, Infectious Disease Division, Massachu-setts General Hospital, Harvard Medical School, Boston, MA; 2Los Alamos NationalLaboratory, Los Alamos, NM, and Santa Fe Institute, Santa Fe, NM; and 3GastrointestinalUnit, Massachusetts General Hospital and Harvard Medical School, Boston, MA.
Received November 2, 2006; accepted February 16, 2007.Supported by the National Institutes of Health grants AI067926 (TMA) and
AI066345 (TMA and GL). B.K., M.D., W.F., and T.B. were supported by an internalLos Alamos Research Development grant, and through NIH HIV-RAD grant # P01AI061734-02. J.T. was supported by the Deutsche Forschungsgemeinschaft (DFG 323/1-1). G.M.L. is a Liver Scholar (the American Liver Foundation).
Dr. Timm is currently affiliated with the University of Essen, Institute for Virol-ogy, Essen, Germany.
Address reprint requests to: Todd M. Allen, Ph.D., 149 13th Street, Charles-town, MA 02129. E-mail: [email protected]; fax: 617-724-8586.
Copyright © 2007 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.21702Potential conflict of interest: Nothing to reportSupplementary material for this article can be found on the HEPATOLOGY website
(http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
339

T cell responses in human HCV infections15-21 and accu-mulation of escape mutations in a region of non-struc-tural protein 3 (NS3) has been reported.15,22 In HIV-1the majority of mutations arising outside of the envelopegene are in fact driven by host CD8� T cell responses.23
Therefore, viral escape in targeted epitopes can be wide-spread, and such studies are beginning to solidify ourunderstanding of the role of host immune pressures inshaping the diversity of these pathogens. Unfortunately,the limited size of previous CD8 escape studies in humanHCV infection,15-21 combined with the difficulty of de-tecting ex vivo CD8� T cell responses in the peripheralblood,24-26 have precluded a broad assessment of the ex-tent and frequency of CD8 escape mutations in HCVinfection.
We sought, therefore, to determine the extent to whichCD8 escape mutations were arising in a cohort of chronicHCV-infected subjects by identifying HLA class I-associ-ated sequence polymorphisms across the HCV proteome.The relationship between HLA allele expression and viralsequence diversity in HCV will elucidate the extent towhich CD8 escape occurs in human HCV infection, andprovide insight into possible mechanisms by which vari-ous HLA alleles are associated with resolution of HCVinfection.4 We sequenced all non-structural proteins from70 chronic genotype 1a-infected subjects, including aportion of E2, and related viral sequence polymorphismsto the HLA class I alleles expressed by each subject. Uti-lizing both a previously published statistical approach,27
and a novel phylogenetic approach, multiple HLA-asso-ciated sequence polymorphisms were identified bothwithin and outside of previously described CD8 epitopes,revealing the commonality by which viral escape fromCD8� T cell responses is occurring in human HCV in-fection.
Patients and MethodsSubjects
Seventy subjects with chronic HCV infection were re-cruited from the Hepatology outpatient clinic of the Mas-sachusetts General Hospital in Boston, the Lemuel-Shattuck Hospital and the Fenway Community HealthCenter. Subjects were included if they presented with apositive HCV-RNA test in serum and no clinical evidencefor acute infection. Only subjects infected with genotype1a were chosen. Twenty-four of the 70 subjects were non-Caucasian, reflecting individuals of mostly African-Amer-ican and Hispanic descent, and 42 of the 70 subjects wereco-infected with HIV-1. The study was approved by thelocal Institutional Review Board, and all subjects gavewritten informed consent.
Detection of HLA Class I-Associated SequencePolymorphisms
Two methods for the detection of HLA-class I-associ-ated sequence polymorphisms were used, a pure statisticalanalysis algorithm based on a previously publishedmethod by Moore et al.27 and a novel phylogenetic anal-ysis algorithm that controls for a potential sample bias dueto a shared phylogeny of the analyzed sequences. A de-tailed description of both methods is provided as Supple-mentary Material (Available at the Hepatology website:http://interscience.wiley.com/jpages/0270-9139/supp-mat/index.html) together with detailed informationabout the amplification and sequencing procedures.
ResultsIdentification of Positive HLA-Associated SequencePolymorphisms in HCV
Seventy subjects chronically infected with HCV geno-type 1a were identified from our Hepatology outpatientclinics in Boston. We population sequenced a 7208 nu-cleotide region of the HCV genome (nt1944-9152 ofH77) spanning amino acid 535 of E2 to amino acid 2937of NS5B, and representing 80% of the expressed openreading frame. HLA typing revealed that the frequency ofthe most common HLA-A, -B, and -C alleles in our co-hort were largely reflective of North American Caucasianpopulations [http://www.ashi-hla.org] (Fig. 1). To iden-tify HLA-associated sequence polymorphisms we modi-fied an algorithm previously utilized for the detection ofHLA-allele associated polymorphisms in HIV-1 reversetranscriptase by Moore et al.,27 and included an analysisfor associations within a 9 amino acid sliding window (seemethods in Supplementary Material).
A total of 15 HLA-associated sequence polymorphisms(adjusted P � 0.05) were identified across the HCV pro-teome (Table 1A), some at single residues and othersthrough a 9 amino acid sliding window (w). Associationswere found for both HLA-A, B, and C alleles and withinall proteins except for NS4A/B. Sequence alignments forthree examples (#2, #5, and #12) that illustrate an in-creased frequency of mutations in subjects expressing thecorresponding HLA allele are shown in Fig. 2. Three ofthe identified associations were within, or overlappedwith, described CD8� T cell epitopes for which HCVescape has been documented during acute or chronicHCV infection. Notably, mutations had occurred withinthe recently described B27-ARMILMTH2841-2849 epitopein 100% of the B27 subjects (Fig. 2A), similar to the highfrequency of escape previously observed by Neumann-Haefelin et al.28 Because of such associations within de-scribed epitopes, we included a subsequent analysis ofassociations within a panel of 123 mapped and HLA-
340 TIMM ET AL. HEPATOLOGY, August 2007

defined human HCV CD8� T cell epitopes previouslypublished [http://hcv.lanl.gov] or identified in our labo-ratory (data not shown). Unadjusted P values were deter-mined for this more focused query to identify significantassociations below P � 0.05. Here a total of 11 additionalHLA associations were identified (Table 1B), includingthe three denoted above in Table 1A. Some of these ad-ditional associations were borderline significant when ad-justed for multiple comparisons, suggesting thatpotentially larger datasets would detect these in an unbi-ased screen as in Table 1A.
Therefore, a total of 23 positive HLA-associated se-quence polymorphisms were detected in this cohort ofgenotype 1a infected subjects, suggesting the potential for
numerous CD8� T cell responses to be exerting immunepressure and driving sequence variation in human HCVinfection. 15 out of 23 associations (65.2%) were re-stricted by HLA-B alleles, a striking result given the ob-served dominant role for HLA-B alleles in mediating theevolution of HIV-1.29
Co-infection with HIV may lead to decreased cellularimmunity against HCV.30 To determine whether HIV co-infection status has an impact on the observed HLA-associ-ations we reevaluated five of the strongest positiveassociations based on HIV status and CD4 count (Supple-mentary Fig. 1). In 2/5 cases the association holds up with asignificant P value in both subgroups (HIV� and HIV�),in two cases the association was lost in the HIV negativegroup and in one other case the association was lost in bothgroups. No significant associations were detected when sub-jects were stratified by CD4 counts (�300/�l; data notshown). However, due to the decreased sample size the sta-tistical power was substantially decreased in these subgroupanalyses and a much larger sample size is needed to addressthis question adequately. We also tested if sequence poly-morphisms are located in the flanking region of describedepitopes potentially blocking proteasomal processing. NoHLA-class I associated sequence polymorphisms were de-tected in the flanking five residues of previously describedCD8 epitopes that passed the threshold of significance afteradjustment for multiple comparisons.
Negative HLA Associations or “Negatopes”A few studies have begun to identify negative HLA asso-
ciations, or ‘negatopes’ in HIV-1.27,31,32 Negatopes representassociations between expression of an HLA allele and aminoacid substitutions that already represent highly prevalent orconsensus residues (�50%) in the population.31 An analysissimilar to those of Table 1 identified a total of seven negativeHLA associations across the HCV proteome, including twolocated in previously defined CD8 epitopes (Table 2A andB). Sequence alignments for two examples (#1 and #6) thatillustrate preferential selection of consensus residues in sub-jects expressing the corresponding HLA allele are shown inFig. 3. Notably, three of these negatopes were restricted byHLA-A02, the most frequent HLA allele in our cohort (Fig.1). These data raised the possibility that escape mutationswithin HCV CD8 epitopes restricted by common HLAclass I alleles may be more prone to accumulate in the pop-ulation due to continuous exposure to these selection pres-sures.32,33
Phylogenetic Analysis of HLA AssociatedPolymorphisms
The above statistical approach to identify HLA-associ-ated sequence polymorphisms examines at face value the
Fig. 1. HLA class I allele distribution of cohort. The frequency of themost common HLA-A, -B, and -C alleles in the study cohort werereflective of the allele frequencies commonly observed in North AmericanCaucasian populations.
HEPATOLOGY, Vol. 46, No. 2, 2007 TIMM ET AL. 341
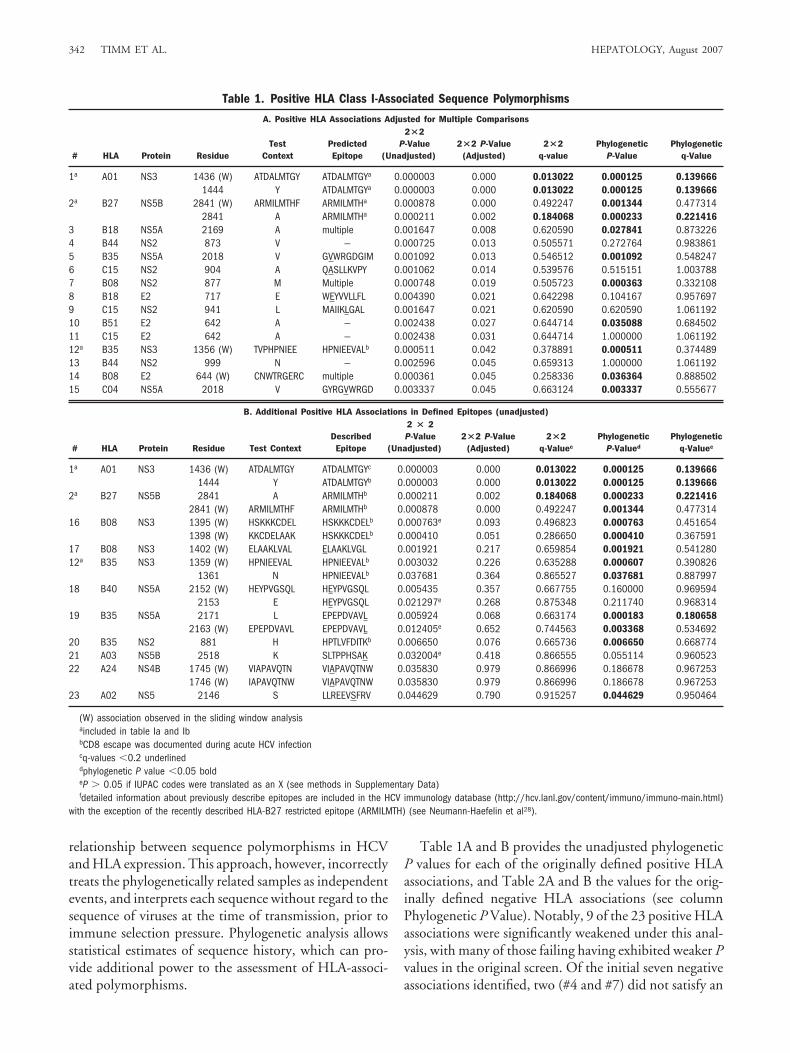
relationship between sequence polymorphisms in HCVand HLA expression. This approach, however, incorrectlytreats the phylogenetically related samples as independentevents, and interprets each sequence without regard to thesequence of viruses at the time of transmission, prior toimmune selection pressure. Phylogenetic analysis allowsstatistical estimates of sequence history, which can pro-vide additional power to the assessment of HLA-associ-ated polymorphisms.
Table 1A and B provides the unadjusted phylogeneticP values for each of the originally defined positive HLAassociations, and Table 2A and B the values for the orig-inally defined negative HLA associations (see columnPhylogenetic P Value). Notably, 9 of the 23 positive HLAassociations were significantly weakened under this anal-ysis, with many of those failing having exhibited weaker Pvalues in the original screen. Of the initial seven negativeassociations identified, two (#4 and #7) did not satisfy an
Table 1. Positive HLA Class I-Associated Sequence Polymorphisms
A. Positive HLA Associations Adjusted for Multiple Comparisons
# HLA Protein ResidueTest
ContextPredictedEpitope
2�2P-Value
(Unadjusted)2�2 P-Value
(Adjusted)2�2
q-valuePhylogenetic
P-ValuePhylogenetic
q-Value
1a A01 NS3 1436 (W) ATDALMTGY ATDALMTGYa 0.000003 0.000 0.013022 0.000125 0.1396661444 Y ATDALMTGYa 0.000003 0.000 0.013022 0.000125 0.139666
2a B27 NS5B 2841 (W) ARMILMTHF ARMILMTHa 0.000878 0.000 0.492247 0.001344 0.4773142841 A ARMILMTHa 0.000211 0.002 0.184068 0.000233 0.221416
3 B18 NS5A 2169 A multiple 0.001647 0.008 0.620590 0.027841 0.8732264 B44 NS2 873 V — 0.000725 0.013 0.505571 0.272764 0.9838615 B35 NS5A 2018 V GVWRGDGIM 0.001092 0.013 0.546512 0.001092 0.5482476 C15 NS2 904 A QASLLKVPY 0.001062 0.014 0.539576 0.515151 1.0037887 B08 NS2 877 M Multiple 0.000748 0.019 0.505723 0.000363 0.3321088 B18 E2 717 E WEYVVLLFL 0.004390 0.021 0.642298 0.104167 0.9576979 C15 NS2 941 L MAIIKLGAL 0.001647 0.021 0.620590 0.620590 1.06119210 B51 E2 642 A — 0.002438 0.027 0.644714 0.035088 0.68450211 C15 E2 642 A — 0.002438 0.031 0.644714 1.000000 1.06119212a B35 NS3 1356 (W) TVPHPNIEE HPNIEEVALb 0.000511 0.042 0.378891 0.000511 0.37448913 B44 NS2 999 N — 0.002596 0.045 0.659313 1.000000 1.06119214 B08 E2 644 (W) CNWTRGERC multiple 0.000361 0.045 0.258336 0.036364 0.88850215 C04 NS5A 2018 V GYRGVWRGD 0.003337 0.045 0.663124 0.003337 0.555677
B. Additional Positive HLA Associations in Defined Epitopes (unadjusted)
# HLA Protein Residue Test ContextDescribedEpitope
2 � 2P-Value
(Unadjusted)2�2 P-Value
(Adjusted)2�2
q-Valuec
PhylogeneticP-Valued
Phylogeneticq-Valuec
1a A01 NS3 1436 (W) ATDALMTGY ATDALMTGYc 0.000003 0.000 0.013022 0.000125 0.1396661444 Y ATDALMTGYb 0.000003 0.000 0.013022 0.000125 0.139666
2a B27 NS5B 2841 A ARMILMTHb 0.000211 0.002 0.184068 0.000233 0.2214162841 (W) ARMILMTHF ARMILMTHb 0.000878 0.000 0.492247 0.001344 0.477314
16 B08 NS3 1395 (W) HSKKKCDEL HSKKKCDELb 0.000763e 0.093 0.496823 0.000763 0.4516541398 (W) KKCDELAAK HSKKKCDELb 0.000410 0.051 0.286650 0.000410 0.367591
17 B08 NS3 1402 (W) ELAAKLVAL ELAAKLVGL 0.001921 0.217 0.659854 0.001921 0.54128012a B35 NS3 1359 (W) HPNIEEVAL HPNIEEVALb 0.003032 0.226 0.635288 0.000607 0.390826
1361 N HPNIEEVALb 0.037681 0.364 0.865527 0.037681 0.88799718 B40 NS5A 2152 (W) HEYPVGSQL HEYPVGSQL 0.005435 0.357 0.667755 0.160000 0.969594
2153 E HEYPVGSQL 0.021297e 0.268 0.875348 0.211740 0.96831419 B35 NS5A 2171 L EPEPDVAVL 0.005924 0.068 0.663174 0.000183 0.180658
2163 (W) EPEPDVAVL EPEPDVAVL 0.012405e 0.652 0.744563 0.003368 0.53469220 B35 NS2 881 H HPTLVFDITKb 0.006650 0.076 0.665736 0.006650 0.66877421 A03 NS5B 2518 K SLTPPHSAK 0.032004e 0.418 0.866555 0.055114 0.96052322 A24 NS4B 1745 (W) VIAPAVQTN VIAPAVQTNW 0.035830 0.979 0.866996 0.186678 0.967253
1746 (W) IAPAVQTNW VIAPAVQTNW 0.035830 0.979 0.866996 0.186678 0.96725323 A02 NS5 2146 S LLREEVSFRV 0.044629 0.790 0.915257 0.044629 0.950464
(W) association observed in the sliding window analysisaincluded in table Ia and IbbCD8 escape was documented during acute HCV infectioncq-values �0.2 underlineddphylogenetic P value �0.05 boldeP � 0.05 if IUPAC codes were translated as an X (see methods in Supplementary Data)fdetailed information about previously describe epitopes are included in the HCV immunology database (http://hcv.lanl.gov/content/immuno/immuno-main.html)
with the exception of the recently described HLA-B27 restricted epitope (ARMILMTH) (see Neumann-Haefelin et al28).
342 TIMM ET AL. HEPATOLOGY, August 2007

unadjusted P value of 0.05 upon reanalysis using the phy-logenetic approach (Table 2A and B; see Phylogenetic PValue). We will discuss three examples at length.
One negative association, an HLA-A02 associationwithin a defined CD8 epitope (#7; A02-ALSTGLIHL684-
692), was decidedly refuted by the phylogenetic analysis.Fig. 4A illustrates the HLA sorted alignment of sequenceswithin this epitope, while Fig. 5A illustrates the phyloge-netic relationship of these sequences and the pattern ofamino acid substitutions within this epitope. Although an
association was found between the expression of HLA-A02 and expression of the consensus serine (S) residue(P � 0.039; Fig. 4A), no significance was found (P �1.000 when assessing whether sequences likely to haveoriginally contained a threonine residue [T] preferentiallymutated to consensus serine in the presence of HLA-A02(measurement of an A02 negatope; Fig. 5A; “escape” ta-ble). Rather, the tree reveals that all sequences containingthe non-consensus threonine (T) residue are in fact phy-logenetically related to one another, regardless of the ex-
Fig. 2. Amino acid alignments of positiveHLA-associations. Sequence alignments forHLA-associated polymorphisms #2, #5, and#12 from Table 1A are shown. Sequences de-rived from subjects expressing the correspond-ing HLA allele are shown above the line, whilethose from subjects not expressing the alleleare shown below the line. The residue/windowwith the strongest association is highlighted ingrey, regions for which CD8 epitopes thatmatch the associated HLA-allele are describedor predicted are boxed. (A) Alignment of anassociation in the previously described B27-ARMILMTH epitope. (B) Alignment of an asso-ciation overlapping with the previouslydescribed B35-HPNIEEVAL epitope. C) Align-ment of an association inside a predicted HLA-B35 epitope.
Table 2. Negative HLA Class-I Associated Sequence Polymorphisms
A. Negative HLA Associations Adjusted for Multiple Comparisons
# HLA Protein Residue Test ContextPredictedEpitope
2 � 2 P-Value(Unadjusted)
2�2 P-Value(Adjusted) 2�2 q-Value
PhylogeneticP-Value
Phylogeneticq-Value
1 A02 NS3 1019 V multiple 0.000485 0.008 0.380565 0.002215 0.5813302 A33 NS3 1444 Y — 0.005560 0.008 0.650952 0.070985 0.9606633 B27 E2 610 H PRCLVHYPY 0.019565 0.025 0.878834 0.012351 0.9071934 C06 NSSA 2320 Q PPPQSPPV 0.010981 0.043 0.696846 0.100000 0.9039545 C02 NS3 1200 N KAVDFIPVENL 0.044751 0.046 0.917556 0.014286 1.048138
B. Additional Negative HLA Associations in Defined Epitopes (unadjusted)
# HLA Protein Residue Test ContextDescribedEpitope
2 � 2 P-Value(Unadjusted)
2�2 P-Value(Adjusted) 2�2 q-value
PhylogeneticP-Value
Phylogeneticq-Value
6 A02 E2 723 (W) FLLLADARV FLLLADARV 0.002483 0.211 0.624181 0.007091 0.6594257 A02 E2 686 S ALSTGLIHL 0.038958 0.472 0.871650 1.000000 1.061192
(W) association observed in the sliding window analysis.aPhylogenetic P value �0.05 boldbDetailed information about previously described epitopes are included in the HCV immunology database (http://hcv.lanl.gov/content/immuno/immuno-main.html)
HEPATOLOGY, Vol. 46, No. 2, 2007 TIMM ET AL. 343

pression of HLA-A02. That is, the sequences with a non-consensus residue were all related by common descent;the non-consensus residue was not associated with expres-sion of HLA-A02. Together these data reveal a profoundinfluence of founder effects within this epitope.
In contrast, the phylogenetic method (tree-based Fish-er’s exact tests) supported HLA-associated immune pres-sure for association #1, epitope A01-ATDALMTGY1436-
1444 (Figs. 4B and 5B). Here, 10/11 sequences were likelyto have mutated from the consensus Y to the variant F inthe presence of the selecting HLA-A01 allele, versus only11/43 in the absence of the HLA allele (Fig. 5B; “escape”table, (P � 0.0001). The phylogenetic approach also en-ables identifying reversions, cases where mutations areselectively being driven back toward consensus residues in
the absence of a given HLA allele. Here the “reversion”table indicates whether sequences have mutated towardsthe consensus Y in the absence of the HLA-A01 allele.There was no significant P value for “reversion” (Fig. 5B).However, there was a trend indicating that potentially alarger dataset would have revealed reversion at this site inthe absence of HLA-A01 mediated selection pressure, po-tentially indicating a fitness cost for this substitution ingenotype 1a.
Finally, the HLA-B35 restricted epitope in NS5A(B35-EPEPDVAVL2163-2171), association #19, is illus-trated in which the tree strengthens an HLA associationfor the single residue in position 2171 (Figs. 4C and 5C).In this case, there is no indication of an elevated rate ofreversion in the absence of the presenting HLA.
Application of q-ValuesDue to the potential of any screening approach to iden-
tify false positives we also included an analysis of q-values,which are based on the concept of the false discovery rateand designed specifically for the analysis of genome-widedata sets.34 Here we utilized a q-value criteria of 0.2,which corresponds roughly to a P value of 0.003, andindicates that only 20% of the associations significant atthis threshold are likely to represent false positives. Withthis criteria we identified three particularly strong positiveHLA associations within our dataset (Table 1A; see col-umn Phylogenetic q-Value for Associations #1, #2, and#19), all in previously described epitopes. Note, also in-cluded for comparison purposes is the q-value for theoriginal corresponding 2�2 P value (Table 1A; see col-umn 2�2 q-Value). In examining the 2�2 q-value andthe phylogenetic q-value analyses two positive associa-tions were shared (#1, #2), while the phylogenetic analysisstrengthened one (#19). Notably, however, none of thenegative HLA associations remained significant with q-values �0.2 by either approach (Table 2, see columnPhylogenetic q-Value and 27times;2 q-Value).
Thus, from this analysis of a modest-sized data set ofHCV sequences a total of 23 positive and 7 negativeHLA-associations were identified using a previously pub-lished statistical approach. Upon implementation of anovel phylogenetic approach, and the use of q-values tomore appropriately deal with false discovery rates, threeparticularly strong positive associations exhibiting q-val-ues �0.2 were identified. It is notable that three of theassociations in particular that failed the stricter q � 0.2criteria, HLA-B08 HSKKKCDEL1395-1403, HLA-B35HPNIEEVAL1359-1367 and HLA-B35 HPTLVFDITK881-
890, reside within CD8 epitopes in which viral escape dur-ing acute HCV infection has previously been welldescribed.15,17 Therefore, these data suggest that larger
Fig. 3. Amino acid alignments of negative HLA-associations. Sequencealignments for two examples of negative HLA-associated polymorphismsare shown. Sequences derived from subjects expressing the correspond-ing HLA allele are shown above the line, while those from subjects notexpressing the allele are shown below the line. The residue/window withthe strongest association is highlighted in grey, regions for which CD8epitopes that match the associated HLA-allele are described or predictedare boxed. A) Alignment of an association inside a predicted HLA-A2epitope. B) Alignment of an association in the previously describedA2-FLLLADARV epitope.
344 TIMM ET AL. HEPATOLOGY, August 2007

datasets are needed to sufficiently power the identification ofHLA associations in highly variable pathogens such as HCV.
Correlation of HLA-Associated Polymorphisms withDetection of CD8 Epitopes
To evaluate the data from the sequencing approach wecompared the HLA-associated sequence polymorphismswith Elispot data from 10 patients utilizing a comprehen-sive method with overlapping peptides.30,35 In these 10patients a total of six CD8 responses were detected (Table3). Notably the autologous viral sequence in five of thesetargeted epitopes showed sequence polymorphisms con-sistent with escape (bolded). We further analyzed all re-gions with HLA-associated polymorphisms in describedepitopes restricted by each subject’s HLA-alleles. Despitelack of detection of CD8 T cell responses with standardtechniques ex vivo, 11 of 21 (52.4%) previously describedepitopes also showed the corresponding polymorphism.Interestingly, after bulk-stimulation of PBMC the A1-1436 epitope (ATDALMTGY) was detectable in an ad-ditional two of seven HLA-A1 positive subjects withoutan ex vivo response suggesting that utilizing standard tech-
niques may underestimate the CD8 immune response(data not shown).
DiscussionRecent reports have clarified the role of viral escape in
leading to the loss of CD8� T cell responses in humanHCV infection,15-21 contributing to our understanding ofthe multitude of mechanisms impacting viral persistencein the face of an active immune response. However, littleis known regarding the rate at which HCV actually es-capes from CD8� T cell responses at the population level,nor the extent to which HLA class I-associated immunepressures are driving the sequence diversity of HCV. Arecent study reports adaptation of HCV to HLA-classI-associated selection pressure in a region of NS3.22 Herewe demonstrate that numerous positive HLA class I asso-ciations can be detected throughout the HCV proteomeat the population level, some occurring with surprisingreproducibility, and supporting viral escape from numer-ous CD8� T cell responses in human HCV infection.Furthermore, only a few potential negative associations(negatopes) were identified. This suggests that, although
Fig. 4. Examples of HLA-associations differ-entially impacted by the phylogenetic analysis.(A) The A02-ALSTGLIHL epitope for which anegative association was detected. (B) TheA01-ATDALMTGY epitope for which a positiveassociation was detected. (C) The B35-EPEP-DVAVL epitope for which a positive associationwas detected.
HEPATOLOGY, Vol. 46, No. 2, 2007 TIMM ET AL. 345

CD8 immune pressures are likely to affect the frequenciesof viral epitopes, their proposed role in driving extinctionof particular CD8 epitopes at the population level mustbe interpreted cautiously. Together these data provideclearer insight into the extent to which viral escape from
CD8� T cell responses is occurring in human HCV in-fection.
The detection of multiple HLA-associated sequencepolymorphisms across the HCV proteome reveals thatsome CD8� T cell responses are predictably mounted
346 TIMM ET AL. HEPATOLOGY, August 2007

against specific CD8 epitopes, and that CD8 escape canreproducibly occur. For example, in the HLA-A01epitope ATDALMTGY1436-1444 only 16/52 (31%) ofsubjects lacking HLA-A01 exhibited a tyrosine to phenyl-alanine (Y3F) substitution, but in subjects expressingHLA-A01 the frequency of this substitution was highlyelevated (17/18; 94%), suggesting that HLA-A01 sub-jects must routinely target this epitope. In line with thishypothesis, 11/28 (39%) HLA-A01 positive subjects inour Boston cohort have a detectable ex vivo responseagainst this epitope (unpublished results). Another exam-ple is the B27-ARMILMTHF2841-2849 epitope, where 6/6(100%) HLA-B27 subjects exhibited sequence polymor-
phisms as compared to only 9/64 (14%) non-B27-pre-senting subjects. Neumann-Haefelin et al. havepreviously observed that 5 of 6 HLA-B27 subjects thatspontaneously resolved infection mounted detectableCD8 responses against this epitope, but only 3 of 8 sub-jects with chronic HCV infection did so.28 This HLA-B27 association was one of the strongest associations andis notable because it identified a potential novel B27epitope in this region prior to its recent publication. Rayet al. recently observed many amino acid substitutionsassociated with the expression of particular HLA alleleswithin another cohort of women accidentally infectedwith HCV in a common-source outbreak.16 While thisstudy was limited to only 22 subjects, and only withindefined CD8 epitopes, knowledge of the infecting strainpermitted a similar analysis. Notably, many of the sameHLA-associated polymorphisms identified by Ray et al.were evident in our dataset, clearly illustrating the repro-ducibility of viral escape within HCV, even across differ-ent cohorts. Thus, the commonality of some HLA-associated polymorphisms, which reflect CD8� T cellimmune pressures, indicates that indeed some CD8responses are highly reproducible and consistentlydriving viral escape in HCV. In previous studies noclear immunodominance of specific CD8 epitopescould be detected during chronic infection.25,35 How-ever, the current sequencing approach suggests thatsome escaped epitopes were previously targeted andimmune responses are no longer detectable with stan-dard techniques. Indeed, this loss of the associatedCD8 T cell response following CTL escape is alsohighly typical in both HIV and SIV.36 Examining viralsequence evolution may therefore provide a powerfulsurrogate marker for the detection of CD8� T cellresponses against HCV and a better understanding ofthe breadth and specificities of these responses.
Table 3. Sequence Polymorphisms in Targeted and Untargeted Previously Described CD8 Epitopes
Patient A A B B Cw Cw Targeted CD8 Epitopesa untargeted HLA-matched CD8 epitopes
1 03 30 13 51 06 15 negative A3-25182 01 02 08 55 03 07 A1-1426, B55-2898b, B55-2568b A2-2146, B8-1395, B8-14023 01 01 15 15 03 03 negative A1-14364 02 26 35 38 04 12 B35-881 B35-1359, B35-2171, A2-21465 02 36 52 65 08 15 negative A2-21466 01 02 08 44 01 01 ND-2197 A1-1436, B8-1395, B8-1402, A2-21467 23 30 07 52 07 16 negative none8 02 02 40 44 03 05 negative A2-2146, B40-21529 02 24 08 41 07 17 negative B8-1395, B8-1402, A24-1745, A2-214610 01 74 08 18 02 07 A1-1436 B8-1395, B8-1402
Epitopes that are consistent with escape are in boldfaceND epitope restriction not determinedaThese CD8 responses have been previously published.25,30,35
bConsistent with escape - not included in manuscript because of minimum HLA criteria.
4™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™Fig. 5. Phylogenetic tree illustrating Tree-based Fisher’s exact calcu-
lations. A phylogenetic tree based on sequences derived from all 70subjects was constructed. At the end of each branch of the tree isindicated the inferred amino acid present in the ancestral sequence‘ancestral’ and the amino acid at the residue under investigation in theparticular strain ‘current’. Also indicated at each branch and internalnode of the tree is an additional indicator, a number from 0-9 in tenthsor X for 1.0, provided to indicate the probability of each sequence toexhibit the consensus form. Sequences derived from subjects expressingthe particular HLA allele of interest are distinguished with a purple line,while sequences derived from other subjects in the study exhibit a boldgrey line. Additional untyped genotype 1a control sequences shown inlight grey are added to strengthen the tree. At the top of each figure isindicated the specific consensus residue under consideration shown inred, with flanking residues in black. Presented at the bottom of eachfigure three tables are provided. The first of these, a standard 2x2 Fisher’sexact test, considers all HLA typed sequences in the tree indicatingwhether there is significant selection for residues in subjects expressingthe given HLA allele. Two additional tree-based Fisher’s exact tests arethen shown. The first, “Reversion”, tests whether at the test residuesequences are reverting towards the consensus residue versus remainingstable. The second, “Escape” tests whether at the test residue sequencesare mutating away from the consensus residue versus remaining stable.(A) The A02-ALSTGLIHL epitope for which a negative association wasdetected. (B) The A01-ATDALMTGY epitope for which a positive associ-ation was detected. (C) The B35-EPEPDVAVL epitope for which a positiveassociation was detected.
HEPATOLOGY, Vol. 46, No. 2, 2007 TIMM ET AL. 347

Studies in both HIV-1 and SIV are now revealing thatthere are limitations to the ability of these highly variablepathogens to support sequence polymorphisms. Viral es-cape in HIV-1 is often limited to a single residue withinthe CD8 epitope, and often further limited to substitu-tion by only a single alternative residue.23 In addition,reversion of escape mutations has now been commonlyobserved upon transmission of viruses to a subsequenthost.15,16,37,38 Here we described various HLA-associatedsequence polymorphisms in HCV CD8 epitopes thatexhibit varying degrees of conservation. Again the A01-ATDALMTGY1436-1444 and B27-ARMILMTH2841-2849
epitopes provide illustrative examples (Figs. 2A, 4B, 5B).In the case of the A01 epitope, these data indicate thatthere may be fewer functional constraints at work tomaintain one particular residue at this position. In a sim-ilar analysis in a different cohort a negative association hasbeen described for this epitope indicating potential dele-tion from this population.22 However, phylogenetic eval-uation of the same association in our cohort reveals atrend towards reversion in the absence of selection pres-sure. Alternatively, the B27 epitope region appears muchmore conserved and therefore may exact higher fitnesscosts to the virus upon escape. Recent data in both HIV-1and SIV now illustrate the specific impact that particularCD8 escape mutations can have on viral replication ca-pacity,39,40 and suggest some finite space within whichthese pathogens can functionally exist. Indeed, it is nota-ble that some of the HLA associations we report weredriven by a single polymorphic site within the epitope,suggesting strict constraints on sequence variation atmany residues across the HCV proteome.
Recent studies suggest that continuous exposure ofhighly polymorphic viruses to focused immune pressuresmay result in the accumulation of CD8 escape mutations,and thus the eventual loss of some CD8 epitopes within apopulation.31,32 A follow-up evaluation of these data us-ing the novel phylogenetic approach described herein re-vealed that most of the those negative HLA associationswere due to founder effects rather than immune pres-sure.42 Applying this phylogenetic approach to the cur-rent HCV dataset revealed that some of the negative HLAassociations in our study were similarly influenced byfounder effects. Overall, comparison of the Fisher’s exacttest and the phylogenetic approach in this modestly sizeddata set also revealed that while some associations wereweakened by the phylogenetic approach, still others werestrengthened. Larger datasets are needed, therefore, to de-termine to what degree both positive and negative HLAassociations are present in both HIV-1 and HCV at thepopulation level, and to what degree these two approachesto identifying HLA associations are complementary.
Taken together, these data reveal the combined influ-ence of multiple forces shaping the sequence diversity ofHCV in the human population. The vast sequence diver-sity of viruses such as HCV, HIV-1, and SIV, and thehighly polymorphic nature of the MHC class I loci, rep-resent critical evolutionary characteristics governing thesurvival of both pathogen and host. Examination of thesecomplex interactions in larger cohorts reveals patterns ofhost/pathogen co-evolution and a clearer picture of fac-tors governing immune control.27,29,31,41 Our data pro-vide an important step towards elucidating the role ofCD8 escape mutations in contributing to viral persistenceand control of HCV.
Acknowledgment: We thank David Heckerman forpointing us to the q-test.
References1. Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohren-
wend P, et al. Analysis of successful immune responses in persons infectedwith hepatitis C virus. J Exp Med 2000;191:1499-1512.
2. Folgori A, Capone S, Ruggeri L, Meola A, Sporeno E, Ercole BB, et al. AT-cell HCV vaccine eliciting effective immunity against heterologous viruschallenge in chimpanzees. Nat Med 2006;12:190-197.
3. Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. De-terminants of viral clearance and persistence during acute hepatitis C virusinfection. J Exp Med 2001;194:1395-1406.
4. McKiernan SM, Hagan R, Curry M, McDonald GS, Kelly A, Nolan N, etal. Distinct MHC class I and II alleles are associated with hepatitis C viralclearance, originating from a single source. HEPATOLOGY 2004;40:108-114.
5. Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB,Hoofnagle JH, et al. Impaired effector function of hepatitis C virus-specificCD8� T cells in chronic hepatitis C virus infection. J Immunol 2002;169:3447-3458.
6. Rushbrook SM, Ward SM, Unitt E, Vowler SL, Lucas M, Klenerman P, etal. Regulatory T cells suppress in vitro proliferation of virus-specific CD8�T cells during persistent hepatitis C virus infection. J Virol 2005;79:7852-7859.
7. Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM.Suppression of HCV-specific T cells without differential hierarchy dem-onstrated ex vivo in persistent HCV infection. HEPATOLOGY 2003;38:1437-1448.
8. Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S,Ferrari C, et al. T cells with a CD4�CD25� regulatory phenotype sup-press in vitro proliferation of virus-specific CD8� T cells during chronichepatitis C virus infection. J Virol 2005;79:7860-7867.
9. Bain C, Fatmi A, Zoulim F, Zarski JP, Trepo C, Inchauspe G. Impairedallostimulatory function of dendritic cells in chronic hepatitis C infection.Gastroenterology 2001;120:512-524.
10. Urbani S, Amadei B, Tola D, Massari M, Schivazzappa S, Missale G, et al.PD-1 expression in acute hepatitis C is associated with HCV-specific CD8exhaustion. J Virol 2006;80:11398-11403.
11. Kantzanou M, Lucas M, Barnes E, Komatsu H, Dusheiko G, Ward S, et al.Viral escape and T cell exhaustion in hepatitis C virus infection analysedusing Class I peptide tetramers. Immunol Lett 2003;85:165-171.
12. Iken K, Huang L, Bekele H, Schmidt EV, Koziel MJ. Apoptosis of acti-vated CD4� and CD8� T cells is enhanced by co-culture with hepato-cytes expressing hepatitis C virus (HCV) structural proteins through FasLinduction. Virology 2006;346:363-372.
13. Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J,et al. The outcome of hepatitis C virus infection is predicted by escape
348 TIMM ET AL. HEPATOLOGY, August 2007

mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity2001;15:883-895.
14. Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J,et al. HCV persistence and immune evasion in the absence of memory Tcell help. Science 2003;302:659-662.
15. Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, et al.CD8 epitope escape and reversion in acute HCV infection. J Exp Med2004;200:1593-1604.
16. Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL.Divergent and convergent evolution after a common-source outbreak ofhepatitis C virus. J Exp Med 2005;201:1753-1759.
17. Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, et al. Cellularimmune selection with hepatitis C virus persistence in humans. J Exp Med2005;201:1741-1752.
18. Seifert U, Liermann H, Racanelli V, Halenius A, Wiese M, Wedemeyer H,et al. Hepatitis C virus mutation affects proteasomal epitope processing.J Clin Invest 2004;114:250-259.
19. Tester I, Smyk-Pearson S, Wang P, Wertheimer A, Yao E, Lewinsohn DM,et al. Immune evasion versus recovery after acute hepatitis C virus infectionfrom a shared source. J Exp Med 2005;201:1725-1731.
20. Urbani S, Amadei B, Cariani E, Fisicaro P, Orlandini A, Missale G, et al.The impairment of CD8 responses limits the selection of escape mutationsin acute hepatitis C virus infection. J Immunol 2005;175:7519-7529.
21. Guglietta S, Garbuglia AR, Pacciani V, Scotta C, Perrone MP, Laurenti L,et al. Positive selection of cytotoxic T lymphocyte escape variants duringacute hepatitis C virus infection. Eur J Immunol 2005;35:2627-2637.
22. Gaudieri S, Rauch A, Park LP, Freitas E, Herrmann S, Jeffrey G, et al.Evidence of Viral Adaptation to HLA Class I-Restricted Immune Pressurein Chronic Hepatitis C Virus Infection. J Virol 2006;80:11094-11104.
23. Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, O’Sullivan K M, et al.Selective escape from CD8� T-cell responses represents a major drivingforce of human immunodeficiency virus type 1 (HIV-1) sequence diversityand reveals constraints on HIV-1 evolution. J Virol 2005;79:13239-13249.
24. Rehermann B, Chang KM, McHutchison JG, Kokka R, Houghton M,Chisari FV. Quantitative analysis of the peripheral blood cytotoxic T lym-phocyte response in patients with chronic hepatitis C virus infection. J ClinInvest 1996;98:1432-1440.
25. Lauer GM, Ouchi K, Chung RT, Nguyen TN, Day CL, Purkis DR, et al.Comprehensive analysis of CD8(�)-T-cell responses against hepatitis Cvirus reveals multiple unpredicted specificities. J Virol 2002;76:6104-6113.
26. Koziel MJ, Walker BD. Characteristics of the intrahepatic cytotoxic Tlymphocyte response in chronic hepatitis C virus infection. Springer SeminImmunopathol 1997;19:69-83.
27. Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA.Evidence of HIV-1 adaptation to HLA-restricted immune responses at apopulation level. Science 2002;296:1439-1443.
28. Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg HC,Killinger T, et al. Dominant influence of an HLA-B27 restricted CD8� T
cell response in mediating HCV clearance and evolution. HEPATOLOGY
2006;43:563-572.29. Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S,
et al. Dominant influence of HLA-B in mediating the potential co-evolu-tion of HIV and HLA. Nature 2004;432:769-775.
30. Kim AY, Lauer GM, Ouchi K, Addo MM, Lucas M, Wiesch JS, et al. Themagnitude and breadth of hepatitis C virus-specific CD8� T cells dependon absolute CD4� T-cell count in individuals coinfected with HIV-1.Blood 2005;105:1170-1178.
31. Leslie A, Kavanagh D, Honeyborne I, Pfafferott K, Edwards C, Pillay T, etal. Transmission and accumulation of CTL escape variants drive negativeassociations between HIV polymorphisms and HLA. J Exp Med 2005;201:891-902.
32. Altfeld M, Allen TM, Kalife ET, Frahm N, Addo MM, Mothe BR, et al.The majority of currently circulating human immunodeficiency virus type1 clade B viruses fail to prime cytotoxic T-lymphocyte responses against anotherwise immunodominant HLA-A2-restricted epitope: implications forvaccine design. J Virol 2005;79:5000-5005.
33. Trachtenberg E, Korber B, Sollars C, Kepler TB, Hraber PT, Hayes E, etal. Advantage of rare HLA supertype in HIV disease progression. Nat Med2003;9:928-935.
34. Storey JD, Tibshirani R. Statistical significance for genomewide studies.Proc Natl Acad Sci U S A 2003;100:9440-9445.
35. Lauer GM, Barnes E, Lucas M, Timm J, Ouchi K, Kim AY, et al. Highresolution analysis of cellular immune responses in resolved and persistenthepatitis C virus infection. Gastroenterology 2004;127:924-936.
36. Allen TM, O’Connor DH, Jing P, Dzuris JL, Mothe BR, Vogel TU, et al.Tat-specific cytotoxic T lymphocytes select for SIV escape variants duringresolution of primary viraemia. Nature 2000;407:386-390.
37. Allen TM, Altfeld M, Yu XG, O’Sullivan K, Lichterfeld M, Le Gall S, et al.Selection, Transmission, and Reversion of an Antigen Processing CTLEscape Mutation in HIV-1 Infection. J Virol 2007;8:193-201.
38. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, et al.HIV evolution: CTL escape mutation and reversion after transmission.Nat Med 2004;10:282-289.
39. Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, et al.Fitness cost of escape mutations in p24 Gag in association with control ofhuman immunodeficiency virus type 1. J Virol 2006;80:3617-3623.
40. Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, et al.Reversion of CTL escape-variant immunodeficiency viruses in vivo. NatMed 2004;10:275-281.
41. Yusim K, Kesmir C, Gaschen B, Addo MM, Altfeld M, Brunak S, et al.Clustering patterns of cytotoxic T-lymphocyte epitopes in human immu-nodeficiency virus type 1 (HIV-1) proteins reveal imprints of immuneevasion on HIV-1 global variation. J Virol 2002;76:8757-8768.
42. Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, etal. Founder effects in the assessment of HIV polymorphisms and HLAallele associations. Science 2007;315:1583-1586.
HEPATOLOGY, Vol. 46, No. 2, 2007 TIMM ET AL. 349
Related Documents











