2000;60:5803-5814. Cancer Res Kasirajan Ayyanathan, William J. Fredericks, Carola Berking, et al. Oncogene Transcriptional Repressor Directed at the PAX3-FKHR by an Inducible in Vivo Hormone-dependent Tumor Regression Updated version http://cancerres.aacrjournals.org/content/60/20/5803 Access the most recent version of this article at: Cited Articles http://cancerres.aacrjournals.org/content/60/20/5803.full.html#ref-list-1 This article cites by 39 articles, 24 of which you can access for free at: Citing articles http://cancerres.aacrjournals.org/content/60/20/5803.full.html#related-urls This article has been cited by 12 HighWire-hosted articles. Access the articles at: E-mail alerts related to this article or journal. Sign up to receive free email-alerts Subscriptions Reprints and . [email protected] Department at To order reprints of this article or to subscribe to the journal, contact the AACR Publications Permissions . [email protected] Department at To request permission to re-use all or part of this article, contact the AACR Publications Research. on February 6, 2014. © 2000 American Association for Cancer cancerres.aacrjournals.org Downloaded from Research. on February 6, 2014. © 2000 American Association for Cancer cancerres.aacrjournals.org Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

2000;60:5803-5814. Cancer Res Kasirajan Ayyanathan, William J. Fredericks, Carola Berking, et al. Oncogene Transcriptional Repressor Directed at the PAX3-FKHR
by an Induciblein VivoHormone-dependent Tumor Regression
Updated version
http://cancerres.aacrjournals.org/content/60/20/5803
Access the most recent version of this article at:
Cited Articles
http://cancerres.aacrjournals.org/content/60/20/5803.full.html#ref-list-1
This article cites by 39 articles, 24 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/60/20/5803.full.html#related-urls
This article has been cited by 12 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

[CANCER RESEARCH 60, 5803–5814, October 15, 2000]
Hormone-dependent Tumor Regressionin Vivo by an Inducible TranscriptionalRepressor Directed at the PAX3-FKHR Oncogene1
Kasirajan Ayyanathan, William J. Fredericks, Carola Berking, Meenhard Herlyn, Christopher Balakrishnan,Edward Gunther, and Frank J. Rauscher, III 2
The Wistar Institute, Philadelphia, Pennsylvania 19104
ABSTRACT
In alveolar rhabdomyosarcomas (ARMSs), a specific chromosomaltranslocation creates a fusion transcription factor, PAX3-FKHR, that isoncogenic due to transcriptional activation. As a strategy for down-regulation of PAX3-FKHR target genes, we created conditional PAX3repressors by fusing the PAX3 DNA-binding motifs to the hormonebinding domain (HBD) of the estrogen receptor and to the KRAB repres-sion domain. We validated proper expression, specific DNA binding,corepressor interaction, and nuclear localization for the KRAB-PAX3-HBD protein and showed it to be a 4-hydroxytamoxifen-dependent tran-scriptional repressor of transiently transfected and integrated PAX3 re-porters in ARMS cells. We established ARMS cell lines that exhibitedstable expression of the conditional PAX3 repressor proteins and usedthem to down-regulate the malignant growth under low serum or anchor-age-independent conditions in a hormone-dependent manner. Terminaldeoxynucleotidyl transferase-mediated nick end labeling assays revealedthat hormonal activation of the PAX3 repressors induced extensive apo-ptosis that correlated with down-regulation of BCL-XL expression. SCIDmice that were engrafted with the KRAB-PAX3-HBD ARMS cell linesand were implanted with 4-hydroxytamoxifen timed-release pellets exhib-ited suppression of tumor growth and an altered vascularity that was notobserved in the control mice. These observations strongly suggest that wehave directly repressed the PAX3 target genes that are deregulated by thePAX3-FKHR oncogene in ARMS.
INTRODUCTION
Chromosomal translocations that result in the creation of chimerictranscription factors that deregulate specific target genes are a type ofgenetic alteration frequently associated with oncogenesis (1). Differ-ent modes of deregulation include gain or loss of transcriptionalactivation or repression function. The model system, ARMS3, chosenin this study provides a clear example for this phenomenon for humanpediatric solid tumors (2).
ARMSs occur due to a highly specific chromosomal translocationevent [t(2;13) (q35;q14)] that juxtaposes the DNA-binding domains ofPAX3 with the transcriptional activation domain of FKHR (3). Intransfection assays,PAX3-FKHR, the causative oncogene in ARMS,functions as a more potent activator of transcription than the wild-typePAX3 (4). Less frequently, another translocation [t(1;13) (p36;q14)]fuses the PAX7 DNA-binding domain to the same FKHR activation
domain (5). These studies suggest that these PAX proteins havesustained a gain of function that leads to ARMS tumorigenesis (2).However, the PAX3-FKHR-activated target genes responsible forARMS have not been defined.
ThePAXgene family consists of nine members that are unified bythe presence of the paired box DNA-binding domain and are subclas-sified based on their genomic organization. PAX proteins play regu-latory roles in pattern formation during organogenesis (6). Ectopicexpression of severalPAX genes in NIH/3T3 cells induces cellulartransformation and tumor formation in nude mice, suggesting thatderegulated expression of PAX proteins could play a role in humantumorigenesis (6, 7). Furthermore, suppression of apoptosis by PAXproteins is crucial for their complex developmental role and couldaccount for their tumorigenic potential (8, 9). In accordance with this,antisense inhibition ofPAX genes results in growth arrest and apo-ptosis in tumor cell lines (10–12). Although evidence supports a rolefor PAX3 in protection of cells from apoptosis during development,the mechanism has not been determined (11).
The developmental abnormalities and alterations in gene expressionobserved in thesplotchmouse model, which contains mutations in thePAX3 DNA-binding domains, has suggested several downstreamtarget genes regulated by PAX3 (13, 14). Candidate targets includemi(15), myoD and myogenin(16), pax7 (11) and others. Similarly,transfection of the PAX3-FKHR fusion protein present in ARMS intoheterologous cells has been shown to up-regulate the expression ofpdgfr-a and c-met, the receptor for hepatocyte scatter factor (17).Whether any of these candidate genes play a role in ARMS tumori-genesis remains to be clarified.
It is generally hypothesized that the enhanced transcriptional acti-vation potential of PAX3-FKHR is responsible for ARMS. We havepreviously engineered synthetic PAX3 repressors using the KRABrepression domain and demonstrated that expression of a PAX3-KRAB repressor in the ARMS Rh30 cell line could inhibit malignantgrowth (18). The KRAB domain functions as a potent DNA binding-dependent transcriptional repression module by recruiting the KAP-1corepressor (19, 20). Other repression domains such as the SNAGdomain from theGFI-1 proto-oncogene (21) and the WT-1 repressiondomain derived from the Wilms’ tumor gene (22) do not use theKAP-1 corepressor mechanism. The KRAB and the SNAG domainsare well suited for the creation of engineered repressors due to theirsmall size and strong repression potentials when fused to heterologousDNA-binding domains.
We were interested in developing conditional PAX3 repressors toexamine the immediate consequences of repressingPAX3target genesin ARMS cell clones. The use of conditional repressors avoids sec-ondary changes in cells that might be selected by constitutive expres-sion of transcriptional repressors. Several conditional eukaryotic ex-pression systems based on either inducible transcription or conditionalactivity due to fusion to the HBD of steroid receptors have beendeveloped (23). Of these, the HBD fusion confers rapid temporalregulation to the functionality of heterologous proteins. Furthermore,specific mutations in the HBDs make these receptors very selective tosynthetic ligands without being influenced by endogenous hormones(24). HBD fusions with transcription factors including PAX5 (25–27)
Received 5/5/00; accepted 8/17/00.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby markedadvertisementin accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1 K. A. was supported by the American Cancer Society NP-954 Research TrainingGrant. W. J. F. was supported by Wistar Basic Cancer Research Training Grant CA09171.F. J. R. is supported in part by NIH Grants CA52009, Core Grant CA10815, DK49210,GM54220, DAMD17-96-1-6141, ACS NP-954, the Irving A. Hansen Memorial Founda-tion, the Mary A. Rumsey Memorial Foundation, and the Pew Scholars Program in theBiomedical Sciences.
2 To whom requests for reprints should be addressed, at The Wistar Institute, 3601Spruce Street, Philadelphia, PA 19104. Phone: (215) 898-0995; Fax: (215) 898-3929;E-mail: [email protected].
3 The abbreviations used are: ARMS, alveolar rhabdomyosarcoma; 4-OHT, 4-hydroxytamoxifen; EMSA, electrophoretic mobility shift assay; RT-PCR, reversetranscription-PCR; DD-PCR, differential display RT-PCR; TUNEL, terminal deoxynucle-otidyl transferase-mediated nick end labeling; HBD, hormone-binding domain; FBS, fetalbovine serum; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
5803
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
and enzymes such as STAT6 (28) have been successfully made togenerate hormone-dependent conditional alleles.
In this study, we generated hormone-inducible, conditional allelesof a KRAB-PAX3 protein by fusing it to the HBD of the murineestrogen receptor ERTM, which exhibits selectivity to 4-OHT (24).Hormone-dependent changes in biological properties such as growthin low-serum medium, apoptosis, anchorage-independent growth, and
growth as tumors in SCID mice, were studied using ARMS Rh30 cellclones expressing the KRAB-PAX3-HBD repressors. The results ofthese experiments suggest that we have successfully used the induc-ible repressor strategy to down-regulate the set of PAX3 target genesthat are activated by the PAX3-FKHR oncoprotein. Furthermore, wehave used the conditional PAX3 repressors in ARMS cells to explorewhether the cellular survival factor BCL-XL might be a PAX3 target
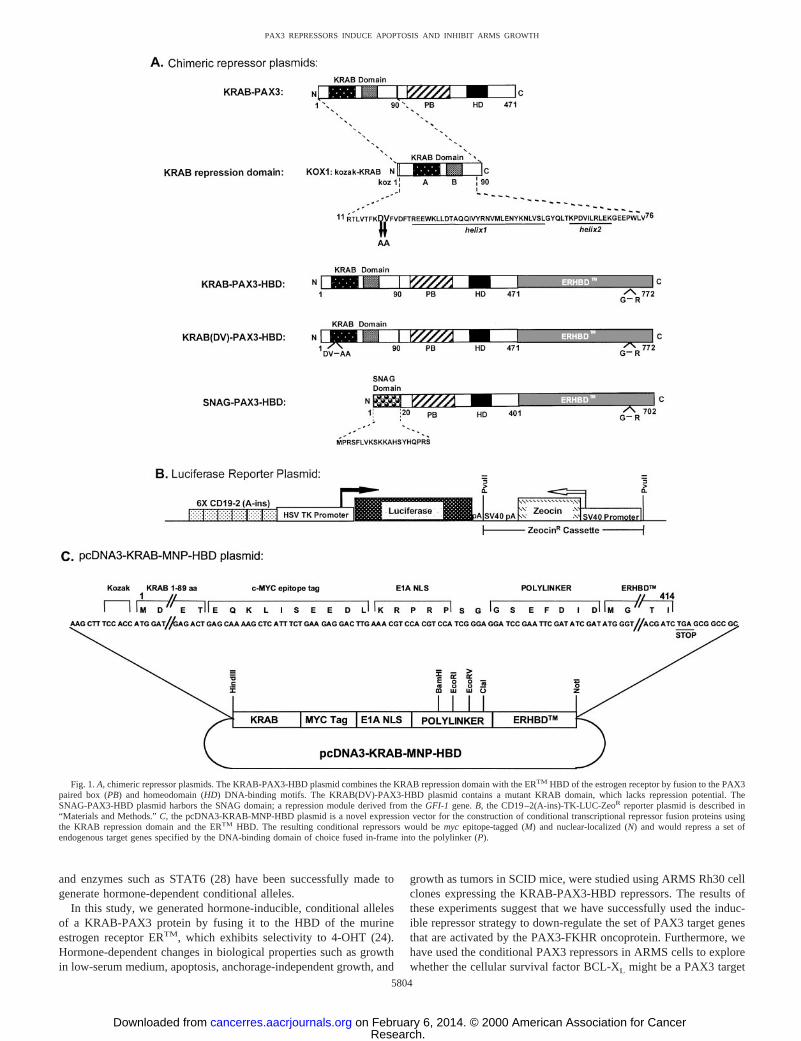
Fig. 1.A, chimeric repressor plasmids. The KRAB-PAX3-HBD plasmid combines the KRAB repression domain with the ERTM HBD of the estrogen receptor by fusion to the PAX3paired box (PB) and homeodomain (HD) DNA-binding motifs. The KRAB(DV)-PAX3-HBD plasmid contains a mutant KRAB domain, which lacks repression potential. TheSNAG-PAX3-HBD plasmid harbors the SNAG domain; a repression module derived from theGFI-1 gene.B, the CD19–2(A-ins)-TK-LUC-ZeoR reporter plasmid is described in“Materials and Methods.”C, the pcDNA3-KRAB-MNP-HBD plasmid is a novel expression vector for the construction of conditional transcriptional repressor fusion proteins usingthe KRAB repression domain and the ERTM HBD. The resulting conditional repressors would bemyc epitope-tagged (M) and nuclear-localized (N) and would repress a set ofendogenous target genes specified by the DNA-binding domain of choice fused in-frame into the polylinker (P).
5804
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

gene and be involved in ARMS tumorigenesis. These studies repre-sent a first step in the identification of important oncogenic targets inARMS by creation of a biological system well suited for analysisusing differential gene expression array technologies.
MATERIALS AND METHODS
Cell Lines. The NIH/3T3 cell line was maintained in DMEM supple-mented with 10% calf serum, 2 mM glutamine, 100 IU/ml penicillin, and 100mg/ml streptomycin at 37°C in 5% CO2 under sterile conditions. The COS-1cells were grown in Iscove’s modified Dulbecco’s medium containing 10%FBS and other components, as above. The parental Rh30 cell line and its clonalderivatives were maintained in RPMI 1640 containing 10% calf serum sup-plemented as above.
Construction of Expression Plasmids.The previously describedpcDNA3-PAX3-STOP plasmid, which expresses a hybrid mouse-humanPAX3 protein, was used as a base to construct the KRAB-PAX3 wild-type andmutant fusion genes (4, 18). This plasmid was digested withHindIII andBamHI and ligated to the wild-type or the mutant (D18V19 changed to A18A19)KRAB repression domains (19). The KRAB domain-encoding fragments weregenerated by PCR amplification from the pM1-KOX-1 template (29) and were
derived asHindIII and BamHI fragments encoding amino acids 1–90 of theKOX1 cDNA. The 59oligonucleotide primer incorporated aHindIII site and aKozak consensus immediately before the KOX-1 initiator methionine(59 primer, 59-TTTTAAGCTTCCACCATGGATGCTAAGTCAC-39). The 39oligonucleotide primer incorporated aBamHI site after amino acid 90 ofKOX-1 (39 primer, 59-TTTTGGATCCAGTCTCTGAATCAGGATG-39). Theresulting pcDNA3-KRAB-PAX3-STOP plasmid contains the KRAB domain,followed by a small linker encoding amino acids GSGVP, followed by aminoacids 11–381 of the PAX3 DNA-binding domain. After amino acid 381, thePAX3-STOP protein is terminated by a vector-derived stop codon (18). ThepcKRAB-PAX3-HBD plasmid was constructed by fusing the HBD, ERTM ofthe murine estrogen receptor, to pcKRAB-PAX3-STOP. The ERTM DNA wasgenerated by PCR amplification from the pBS1ERTM plasmid (24) templateusing a pair of oligonucleotide primers designed to incorporate flankingEcoRIsites (59 primer, 59-GCATGAATTCTATGGGTGCTTCAGGAG-39; 39 T3promoter primer, 59-AATTAACCCTCACTAAAGGG-39). The PCR productwas digested withEcoRI and ligated to the uniqueEcoRI site just 59of thevector-derived stop codon in the pcKRAB-PAX3-STOP plasmid to create anin-frame fusion. The pcSNAG-PAX3-HBD plasmid was constructed by fusingthe HBD to the pcDNA3-SNAG-PAX3 plasmid, as described above. Thefragment encoding the SNAG domain was generated by overlapping PCR
Fig. 2.A, characterization of KRAB-PAX3 andKRAB-PAX3-HBD proteins expressed in COS-1cells that were transiently transfected with thepcDNA3, pcKRAB-PAX3, and pcKRAB-PAX3-HBD expression plasmids and metabolically la-beled with35S-methionine. Whole cell lysates pre-pared in radioimmunoprecipitation assay bufferwere immunoprecipitated using preimmune-, ora-PAX3-, and a-KRAB-IgGs and analyzed bySDS-PAGE and fluorography. KRAB-PAX3 (Mr55,000) and KRAB-PAX3-HBD (Mr 88,000) pro-teins are indicated by thehalf-arrows. B, DNA-binding properties of KRAB-PAX3 and KRAB-PAX3-HBD proteins. Recombinant KRAB-PAX3and KRAB-PAX3-HBD proteins were synthesizedin vitro and used in gel shift assays using32P-labeled e5 probe. For KRAB: PAX3: KAP-1 com-plex analysis, the binding reactions contained 1ml(;3 mg) of COS-1 nuclear extract as a source ofthe KAP-1 corepressor. Binary complexes (KRAB-PAX3:e5DNA and KRAB-PAX3-HBD:e5DNA)and ternary complexes (KRAB-PAX3: KAP-1:e5DNA and KRAB-PAX3-HBD:KAP-1:e5DNA)are indicated on the autoradiogram.FP, free probe.C, expression of the KRAB-PAX3-HBD transcriptin Rh30-KPHBD-cl22 cells. Northern blot analysisof 20 mg of total RNA from un-induced and4-OHT-induced Rh30-KPHBD-cl22 cells. The po-sitions of the 28S and 18S rRNA species are indi-cated (top). The ethidium bromide-stained gel isshown to control for loading (bottom).
5805
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
amplification. The 59primer was designed to incorporate anEcoRI and aBamHI site, a Kozak consensus sequence, and the SNAG domain sequences(encoding amino acids 1–15; 59primer, 59-GAATTCGGATCC ACCATGC-CACGTTCTTTCCTGGTTAAATCTAAAAAAGCGCACTCTTACC-39).The39 primer contained the remaining portion of the SNAG domain (aminoacids 16–20 in an antisense orientation), followed byBglII and SalI sites(39-primer, 59-GTCGACAGATCTGGAGTAGTCCGGACCCGGAG-AACGCGGCTGGTGGTAAGAGTGCGCTTTTTTAG-39). These two oligo-nucleotides were annealed and amplified to yield a 105-bp fragment that wasused as a template in the PCR reaction with a pair of flanking primers:(59-GTCAGAATTCGGATCCACC-39; and 39primer, 59-CCAAGTCGACA-GATCTGGAG-39). The resulting PCR product was digested withBamHI andBglII and cloned into theBamHI site in the pcDNA3-PAX3-STOP plasmid.The general-purpose vector pcDNA3-KRAB-MNP-HBD was constructed intwo steps. First, the KRAB domain was amplified using the 59HindIII primerdescribed above and a 39primer that incorporated amyc-epitope tag (EQKLI-SEEDL) and a nuclear localization signal of theE1A gene (KRPRP) imme-diately after amino acid 89 of the KRAB domain, followed by aBamHI site.Next, the ERTM DNA was generated by PCR amplification using a 59primerdesigned to incorporate a polylinker composed ofBamHI,EcoRI,EcoRV, andClaI sites, and the 39T3 primer. This fragment was cleaved withBamHI andNotI, and then these two fragments were cloned into theHindIII and NotI sitesof the pcDNA3 vector to generate the final construct. The nucleotide sequencesof all PCR-derived constructs were confirmed by sequencing both strands. Thepreviously described PAX reporter plasmid, CD19–2(A-ins)-TK-LUC (30),was modified by incorporation of a ZeocinR cassette derived as aPvuIIfragment from the pcDNA3.1Zeo plasmid (Invitrogen) to create theCD19 –2(A-ins)-TK-LUC-ZeoR plasmid.
COS-1 Transfection, Extract Preparation, and Immunoprecipitation.The expression plasmids depicted in Fig. 1Awere transfected into COS-1 cellsfor 6 h with a mixture of DNA:lipofectAMINE in the ratio 1:6 in optiMEM,followed by growth for 48 h in DMEM containing 10% FBS. Transfected cellswere metabolically labeled with35S-methionine, and the cell extracts preparedin radioimmunoprecipitation assay buffer were subjected to immunoprecipita-tion analysis witha-KRAB, a-PAX3, anda-HBD antibodies, as describedpreviously (4, 18).
EMSA. The DNA-binding potentials of KRAB-PAX3 and KRAB-PAX3-HBD proteins were assessed by EMSA performed using32P-labeled e5 DNAprobe, as described previously (4, 31). The KRAB-PAX3 and KRAB-PAX3-
HBD proteins used for EMSA were synthesized using the Promega TnT T7transcription and translation system. In parallel reactions,35S-labeled proteinswere prepared and analyzed by 10% SDS-PAGE and fluorography to confirmand normalize specificin vitro synthesis. For the corepressor supershift stud-ies, COS-1 nuclear extract was used as a source of the KAP-1 corepressor (;3mg/ml, prepared as described previously; Refs. 4 and 31). For the antibodysupershift studies,a-KRAB or a-PAX3 IgG (2 mg/ml) was included in theEMSA-binding reactions.
Northern Analysis for KRAB-PAX3-HBD Transcript. Total RNA wasisolated from Rh30-KPHBD-cl22 cells using the TRIzol reagent (Life Tech-nologies, Inc., Rockville, MD) and electrophoresed on 1% formaldehyde-agarose gels. Prior to RNA isolation, Rh30-KPHBD-cl22 cells were eitherun-induced (treated with 0.1% ethanol as a solvent control) or induced with500 nM 4-OHT. The gel was stained with ethidium bromide to assess theintegrity and equal loading of the samples and then transferred to a nylonmembrane (Hybond). The membrane was prehybridized, hybridized withradiolabeled PAX3-KRAB probe, and washed to a final stringency of 0.23SSC, 0.2% SDS, at 65°C, before autoradiography.
Indirect Immunofluorescence. Subcellular localization of KRAB-PAX3and KRAB-PAX3-HBD proteins in Rh30, Rh30-pcDNA-cl5, and Rh30-KPHBD-cl22 cells grown on cover glasses was conducted using previouslydescribed procedures for immunofluorescence (31). After fixation with 1%paraformaldehyde and permeabilization with 0.2% Triton X-100 (SigmaChemical Co.), the antigens were localized usinga-PAX3 or a-HBD primaryrabbit antibodies, followed by detection with a secondary biotinylateda-rabbitIgG and avidin-FITC (Vector Laboratories, Inc.). The nuclei were counter-stained for DNA with 0.5mg/ml Hoechst 33258 (Sigma Chemical Co.) and thecells were visualized using a Leica confocal laser-scanning microscope. TheHC-20 antibody to the HBD of the murine estrogen receptor was obtained fromSanta Cruz Biotechnology Inc.
Transient Transfections and Reporter Assays.The transcription assayswere performed on NIH/3T3 cells that were transiently transfected with aLipofectAMINE mixture containing 1 or 2.5mg of the expression plasmids(pcDNA3, pcKRAB-PAX3, and pcKRAB-PAX3-HBD), 0.5mg of CD19–2(A-ins)-TK-LUC, and 0.25mg of CMV-b-D-galactosidase plasmids, as de-scribed previously (18). Transfected cells were treated with 0.1% ethanol as asolvent control for un-induced dishes or were induced with 500 nM 4-OHT(Research Biochemicals International, Natick, MA). After 24 h, the cells werewashed twice with Tris-buffered saline, and the cell extracts were prepared inreporter lysis buffer and assayed for luciferase andb-galactosidase activities asdescribed (31).
Generation of Stable Rh30 Cell Clones.Rh30 cell transfectants contain-ing the conditional PAX3 repressor plasmids depicted in Fig. 1Aor thepcDNA3 vector were isolated as individual colonies using cloning rings andwere expanded into cell lines after selection for stable resistance to 500mg/mlG418 (Mediatech, Inc., Herndon, VA). Twenty-four independent cell lineswere tested for expression of the PAX3-HBD repressor proteins by immuno-precipitation witha-PAX3 IgG. Dual-stable inducible PAX3 repressor/PAX3reporter cell clones were generated in the Rh30-KPHBD-cl22 cell line aftertransfection with the CD19–2(A-ins)-TK-LUC-ZeoR plasmid and selectionwith 500 mg/ml G418 and 100mg/ml Zeocin. The PAX3 repressor/PAX3reporter cell lines, designated as HBDLUC clones, were screened for repres-sion of luciferase after induction with 4-OHT. The luciferase activities of theHBDLUC clones were normalized to a protein content of 1.0 A595 unit in theBioRad protein assay.
DD-PCR. Gene expression profiles of un-induced and 4-OHT-inducedRh30-KPHBD-cl22 cells were analyzed using DD-PCR. Total RNA wasisolated using the TRIzol reagent and poly(A)1 mRNA was purified using theoligo-(dT)25 Dynabeads (Dynal, Inc., Lake Success, NY). First-strand cDNAwas synthesized using the Life Technologies, Inc. cDNA synthesis system andquantitated by spectrophotometry. Differential subtraction display PCR wasconducted as described (32), and the samples were electrophoresed on asequencing gel and autoradiographed.
Low-Serum and Poly-HEMA-MTT Assays. The growth assays of Rh30,Rh30-KPHBD-cl22, Rh30-SPHBD-cl8, and Rh30-K(DV)PHBD-cl24 celllines in low-serum (0.1% FBS) medium was conducted in 24-well tissueculture plates and was repeated twice with at least 10 replicates. The anchor-age-independent growth of parental Rh30 and Rh30-KPHBD-cl22 cells wasevaluated using poly-HEMA-coated plates. In both assays, the proportion of
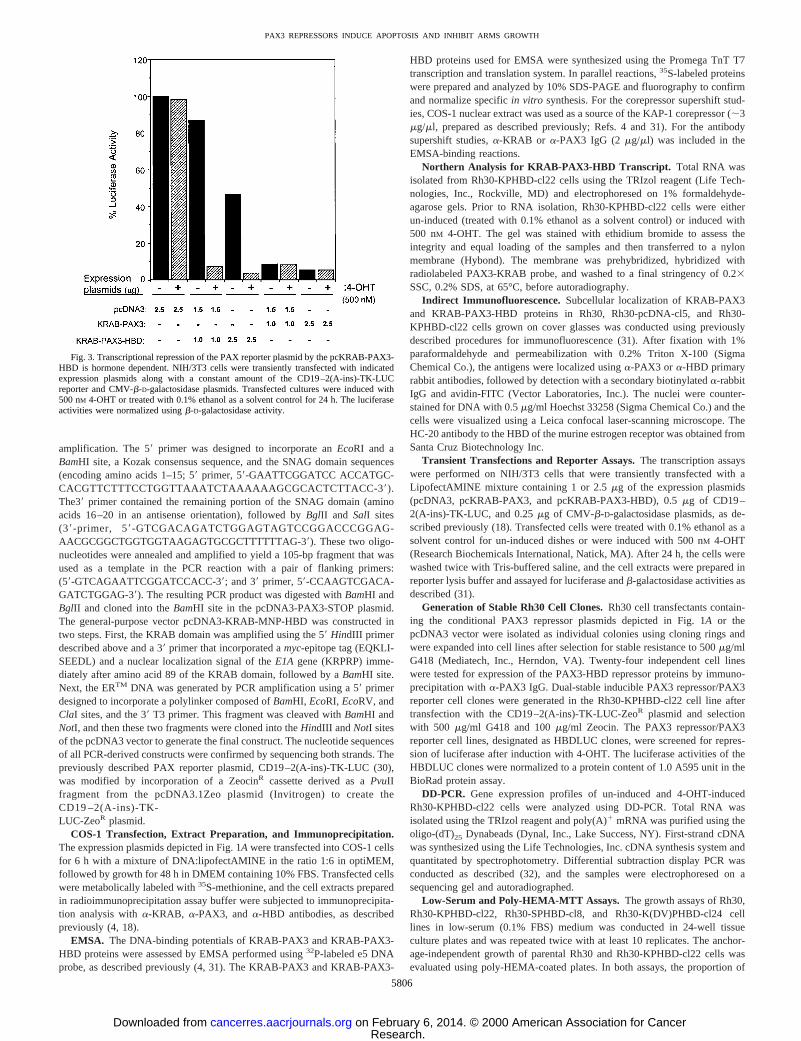
Fig. 3. Transcriptional repression of the PAX reporter plasmid by the pcKRAB-PAX3-HBD is hormone dependent. NIH/3T3 cells were transiently transfected with indicatedexpression plasmids along with a constant amount of the CD19–2(A-ins)-TK-LUCreporter and CMV-b-D-galactosidase plasmids. Transfected cultures were induced with500 nM 4-OHT or treated with 0.1% ethanol as a solvent control for 24 h. The luciferaseactivities were normalized usingb-D-galactosidase activity.
5806
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
viable cells at each time point was determined by MTT assay, as describedpreviously (33).
Apoptosis Assays.Apoptosis assays were performed using the ApoAlertDNA Fragmentation and the ApoAlert Annexin V Assay kits according to themanufacturer’s instructions (Clontech Laboratories, Inc.). Assays were con-ducted on SCID mouse tumor sections or on Rh30-KPHBD-cl22 cells thatwere grown on coverslips in low-serum medium under either un-induced(0.1% ethanol) or 4-OHT-induced conditions (500 nM, 48 h). The polyclonalrabbit antibody for immunoblot detection of BCL-XL (bcl-x, Ab-1) and theantibody for detection of humana-tubulin as a loading control were obtainedfrom Oncogene Research Products (Cambridge, MA).
Semiquantitative RT-PCR Analysis. For RT-PCR analysis, RNA wasisolated from the un-induced or the 4-OHT-induced Rh30-KPHBD-cl22 andRh30-SPHBD-cl8 cells grown in low-serum medium using the TRIzol reagent.The reverse transcription reactions were performed on 5mg of total RNA usingoligo-dT primers with the Ready-To-Go You-Prime First-Strand SynthesisBeads (Pharmacia Biotech). The PCR reactions were carried out with 2.5ml ofreverse transcription reactions as templates. A pair of primers specific for thehuman BCL-XL transcript (59primer, 59-CAGCAGCAGTTTGGATGC-39;39-primer, 59-CCACAGTCATGC CCG TC-39) was used to amplify the448-bp product. A specific primer pair (59-primer, 59-TCAGCGCAGGGG-CGCCCGGTTCTT T-39; and 39-primer, 59-ATCGACAAGACCGGCTTC-CATCCGA-39) was used to amplify the 345-bp product from theNeoR gene.A pair of primers specific for the HBD of the murine estrogen receptor
(59-primer, 59-GCGACGGGCCCATGGGTGCTTCAG G-39; and 39-primer,59GGTGGGCCCCTGATATCACAAGTCCTCTTCAGAAATGAGCTTTTG-CTCGATCGTGTTGGGGAAGCC-39) was used to amplify a 1010-bp productfrom the SCID mouse tumor samples that were derived from Rh30-KPHBD-cl22 cells.
Tumor Growth Inhibition Assays in SCID Mice. The tumorigenic po-tentials of Rh30-pcDNA-cl5 and Rh30-KPHBD-cl22 cell lines were evaluatedafter s.c. injection into female CB17-SCID mice, 6 weeks of age. Evidence oftumor growth became apparent after 10 days, at which time the mice weredivided into two groups of five mice each (un-induced and 4-OHT induced).The mice of the 4-OHT-induced group were implanted with 35-mg timed-release pellets specified to maintain a 200-nM circulating concentration of4-OHT for 21 days (Innovative Research of America, Sarasota, FL). Improvedimplant success was ensured by application of DERMABOND topical skinadhesive (Ethicon, Inc., Somerville, NJ) over the wounds. Alternate daymeasurements of tumor volumes were made using a tumorimeter (CancerTechnologies, Inc., Tucson, AZ). After 3 weeks, the mice were sacrificed andthe wet weights of the tumors were recorded. A portion of each tumor wasfixed in formalin for H&E staining and histopathological evaluation.
RESULTS
Conditional PAX3 Repressors and Integrated Reporter.Wehave previously demonstrated that we could engineer a PAX3 tran-
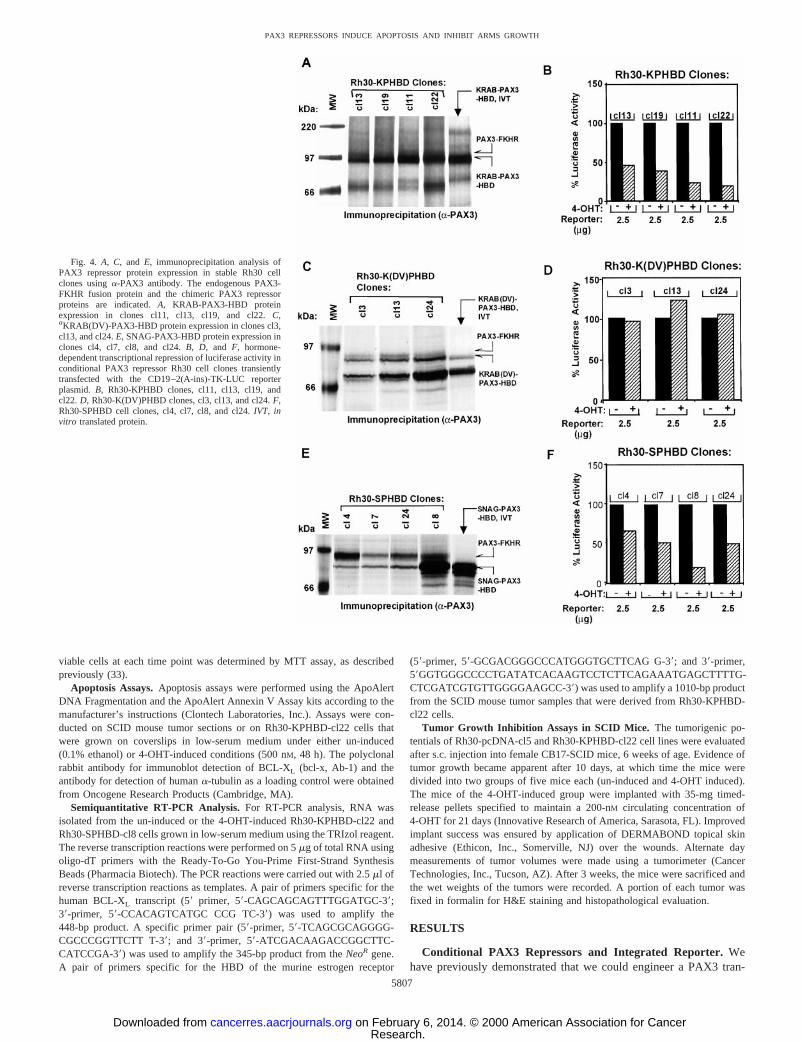
Fig. 4. A, C, and E, immunoprecipitation analysis ofPAX3 repressor protein expression in stable Rh30 cellclones usinga-PAX3 antibody. The endogenous PAX3-FKHR fusion protein and the chimeric PAX3 repressorproteins are indicated.A, KRAB-PAX3-HBD proteinexpression in clones cl11, cl13, cl19, and cl22.C,aKRAB(DV)-PAX3-HBD protein expression in clones cl3,cl13, and cl24.E, SNAG-PAX3-HBD protein expression inclones cl4, cl7, cl8, and cl24.B, D, and F, hormone-dependent transcriptional repression of luciferase activity inconditional PAX3 repressor Rh30 cell clones transientlytransfected with the CD19–2(A-ins)-TK-LUC reporterplasmid. B, Rh30-KPHBD clones, cl11, cl13, cl19, andcl22. D, Rh30-K(DV)PHBD clones, cl3, cl13, and cl24.F,Rh30-SPHBD cell clones, cl4, cl7, cl8, and cl24.IVT, invitro translated protein.
5807
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
scriptional repressor by fusing the KRAB domain to the PAX3 DNA-binding domains. We found that constitutive expression of a PAX3-KRAB protein could inhibit the malignant growth of ARMS cells(18). In the present study, a KRAB-PAX3 plasmid was converted toa conditional repressor, KRAB-PAX3-HBD, by fusion of the ERTM
domain in-frame to the COOH terminus (Fig. 1A). The ERTM domainis a well-established, tamoxifen-selective, mutant version of the HBDof the murine estrogen receptor that confers hormone-dependent func-tionality to heterologous fusion proteins (24). The KRAB(DV)-PAX3-HBD protein features a mutant KRAB repression domain thatlacks transcriptional repression potential and is a useful control toconfirm that the elicited biological responses depend on a functionalrepression module (19). Furthermore, by stable integration of the 63CD19–2(A-ins)-TK-LUC luciferase reporter plasmid (Fig. 1B), wehave created inducible PAX3 repressor/reporter (HBDLUC) cell linesthat permit convenient monitoring of the repression function in achromatin-mediated state. The modular nature of the engineered re-pressor is shown by the use of another conditional repressor plasmid,SNAG-PAX3-HBD, which was created using the SNAG repressionmodule of theGFI-1 gene (21).4 Clearly, the use of engineered
repressors is not limited to the set of target genes selected by thePAX3 DNA-binding domains as in this study. The general purposeKRAB-MNP-HBD vector (Fig. 1C) was designed for use with otherDNA-binding domains of interest that could be inserted into thepoly-linker between the KRAB repression domain and the ERTM
domain. This plasmid features incorporation of a nuclear localizationsignal and a c-mycepitope tag to aid in detection. The resultingconditional chimeric repressors can be applied for repression of the setof endogenous target genes of choice determined by the inserted DNAbinding motif.
Characteristics of Engineered Repressor Proteins.Immunopre-cipitation analysis of transfected COS-1 cell extracts using botha-KRAB anda-PAX3 IgGs (Fig. 2A) demonstrates that the pcKRAB-PAX3 (55 kDa) and pcKRAB-PAX3-HBD (88 kDa) proteins areexpressedin vivo at the size predicted by their cDNAs and arerecognized by the appropriate antibodies. The EMSA analysis (Fig.2B) indicated the presence of clear binary complexes of both KRAB-PAX3:e5DNA and KRAB-PAX3-HBD:e5DNA, which confirmedthat both KRAB-PAX3 and KRAB-PAX3-HBD proteins exhibitedthe anticipated DNA-binding properties. We have previously shown,
4 Unpublished results.
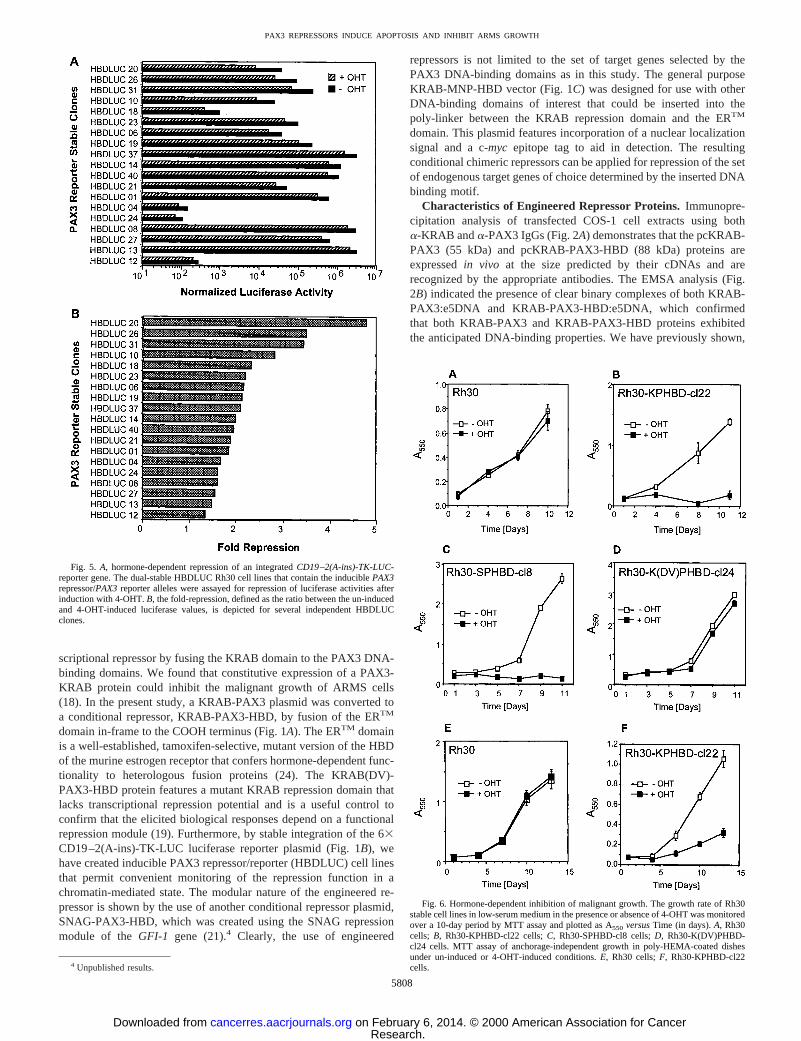
Fig. 5. A, hormone-dependent repression of an integratedCD19–2(A-ins)-TK-LUC-reporter gene. The dual-stable HBDLUC Rh30 cell lines that contain the induciblePAX3repressor/PAX3reporter alleles were assayed for repression of luciferase activities afterinduction with 4-OHT.B, the fold-repression, defined as the ratio between the un-inducedand 4-OHT-induced luciferase values, is depicted for several independent HBDLUCclones.
Fig. 6. Hormone-dependent inhibition of malignant growth. The growth rate of Rh30stable cell lines in low-serum medium in the presence or absence of 4-OHT was monitoredover a 10-day period by MTT assay and plotted as A550 versusTime (in days).A, Rh30cells; B, Rh30-KPHBD-cl22 cells;C, Rh30-SPHBD-cl8 cells;D, Rh30-K(DV)PHBD-cl24 cells. MTT assay of anchorage-independent growth in poly-HEMA-coated dishesunder un-induced or 4-OHT-induced conditions.E, Rh30 cells;F, Rh30-KPHBD-cl22cells.
5808
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

by competition with unlabeled homologous oligonucleotide, thatbinding of PAX3 protein to the e5 binding site probe is specific (4).Furthermore, these binary complexes were confirmed to contain thepredicted proteins because they were efficiently super-shifted whena-KRAB or a-PAX3 IgG was included during the DNA-bindingreactions (data not shown). It was also evident that the KRAB domainpresent in the binary complex was amenable for protein-protein in-teractions because ternary complexes of KRAB-PAX3:KAP-1:e5DNA or KRAB-PAX3-HBD:KAP-1:e5DNA were observed whenCOS-1 nuclear extract was included in the gel-shift reaction as asource of the KAP-1 corepressor (20). We have also found that theSNAG-PAX3-HBD protein efficiently binds the e5 DNA probe butdoes not form a ternary complex with KAP-1 (data not shown). Aswould be expected with the ERTM posttranslational regulation system,we have shown that similar levels of KRAB-PAX3-HBD transcriptare expressed in Rh30-KPHBD-cl22 cells grown under both un-induced and 4-OHT-induced conditions (Fig. 2C). Immunofluores-cence microscopy indicated that in the absence of hormone theKRAB-PAX3-HBD protein was predominantly cytoplasmic (Fig.10A,top), but after induction with 4-OHT it was located in the nucleus(Fig. 10A,bottom).
Hormone-dependent Luciferase Repression.Transcriptional re-pression assays that were carried out in NIH/3T3 cells transientlytransfected with the KRAB-PAX3 plasmid showed constitutive re-pression that depended on the dose of the input expression plasmid butnot on the presence of 4-OHT. In contrast, cells transfected with 1mgof the KRAB-PAX3-HBD plasmid exhibited a significant dependenceon the presence of 4-OHT to repress the luciferase reporter andtypically manifested a 10-fold repression (Fig. 3). A hormone-inde-pendent repression was also observed at a high input dose (2.5mg) ofKRAB-PAX3-HBD plasmid. As expected, the vector-transfected cellsshowed no repression (Fig. 3). Overall, our characterization of the
KRAB-PAX3-HBD repressor indicated proper expression, DNA-binding activity, and a significant degree of hormone-dependent tran-scriptional repression activity in NIH/3T3 cells. NIH/3T3 cells do notexpress PAX3, hence, lack any endogenous PAX3-dependent tran-scriptional activation function (data not shown; Ref. 34).
Conditional PAX3 Repressor in ARMS Stable Cell Lines.Ourprimary interest, however, was to evaluate the conditional PAX3repressor in Rh30 ARMS cell lines that express the endogenousPAX3-FKHR oncogenic transcriptional activator. Therefore, we gen-erated stable Rh30 cell clones, which expressed the conditional PAX3repressors and then evaluated their repression potentials after transienttransfection of the CD19–2(A-ins)-TK-LUC reporter plasmid. In con-trast to the 10-fold repression observed in transiently transfectedNIH/3T3 cells (Fig. 3), in Rh30 cells the repression achieved rangedfrom 2–4-fold, consistent with the presence of a competing endoge-nous activation due to PAX3-FKHR. Hormone-dependent repressionof the reporter plasmid was observed in all of the Rh30-KPHBDclones (e.g.,2cl11, cl13, cl19, and cl22; Fig. 4B) that correlated wellwith the expression levels of the KRAB-PAX3-HBD fusion protein(Fig. 4A). The observed conditional transcriptional repression ap-peared to be dominant over transcriptional activation elicited by theendogenousPAX3-FKHR oncogene. Similarly, all of the SNAG-PAX3-HBD-expressing Rh30 cell clones (e.g., 2cl4, cl7, cl8, andcl24; Fig. 4F) showed a good correlation between hormone-dependentrepression of luciferase activities and expression levels of the SNAG-PAX3-HBD protein (Fig. 4E). As expected, the Rh30 cell clones (e.g.,2cl3, cl13, and cl24; Fig. 4C) that expressed the mutant KRAB(DV)-PAX3-HBD repressor protein (Fig. 4C) showed no repression (Fig.4D). Thus, repression was found to completely depend on the pres-ence of a functional repression domain (KRAB or SNAG) but was notrestricted to a particular type of repression mechanism.
Fig. 7.A, BCL-XL expression in Rh30-SPHBD-cl8 (Lanes 1and 2) and Rh30-KPHBD-cl22 celllysates(Lanes 3and 4) obtained after growth inlow-serum medium (0.1%) under un-induced(Lanes 1and3) and 4-OHT-induced(Lanes 2and4) conditions. Depicted are immunoblot analyses of100 mg of whole cell lysates usinga-BCL-XL-specific antibody (top). Expression ofa-tubulin inthe same lysates was assessed as a control for equalloading (bottom). Semiquantitative RT-PCR analy-sis for BCL-XL (top) and NeoR (bottom) transcriptsunder un-induced and 4-OHT-induced conditionsin the absence of cycloheximide (B) and in thepresence of 50mg/ml cycloheximide (C).
5809
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
Repression of the Integrated PAX3 Reporter and EndogenousGenes.Our intended goal was to apply the PAX3 conditional repres-sor strategy to specifically down-regulate endogenous target genesthat are activated by thePAX3-FKHRoncogene. Hence, we producedstable cell clones containing both the KRAB-PAX3-HBD plasmid(NeoR) and the CD19–2(A-ins)-TK-LUC reporter plasmid (ZeoR),designated as HBDLUC cell lines. This allowed us to correlate re-pression of an integrated artificial reporter gene target with reversionof ARMS malignant growth characteristics and changes in expressionof endogenousPAX3 target genes. Several independent clonal celllines were tested for hormone-dependent repression of the chromatin-integrated luciferase gene, and the results obtained for a selectednumber of clones are depicted in Fig. 5A, which shows that thenormalized luciferase activities varied from 103–106 luciferase units.The fold repression ranged from.1.5–5 for different clonal cell lines(Fig. 5B). The variation observed between clones most likely reflectsdifferences in reporter copy number and integration site. The range ofrepression potentials obtained with the chromatin-integrated reporterclones are consistent with those observed in transient assays in Rh30cells (Fig. 4), further supporting the contention that the PAX3 repres-sors could dominantly repress endogenous genes despite the presenceof the PAX3-FKHR protein.
Our studies have indicated that repression of PAX3-FKHR targetgenes is a strategy for reversion of malignant growth of ARMS cells.Thus, we have established a relevant system to model the conditionalrepression of endogenous PAX3-FKHR target genes. To demonstratethat hormone-dependent repression of endogenous genes was occur-ring in this ARMS cell system, DD-PCR analysis was performedusing RNA made from un-induced and 4-OHT-induced Rh30-KPHBD-cl22 cells. When different combinations of anchor and arbi-trary primers were used, the DD-PCR products displayed significantexpression pattern differences (data not shown). Isolation and char-
acterization of these differentially expressed mRNA species is cur-rently under investigation, and candidate genes are being evaluated forfunctional relevance to the malignant phenotype of ARMS.
Growth Properties of Conditional PAX3 Repressor ARMS CellLines. To further substantiate the inducible PAX3 repressor Rh30cell lines as pertinent model systems for the study of PAX-3-FKHRtarget genes in ARMS, we used the Rh30-KPHBD-cl22 cell line toexamine whether KRAB-PAX3-HBD would function as a conditionalrepressor of ARMS malignant growth. Growth properties of Rh30 andRh30-KPHBD-cl22 cells were studied in the presence or absence of4-OHT under full-serum (10%) and reduced-serum (0.1%) conditions.The growth in full serum was not significantly changed relative togrowth of a parallel un-induced culture on activation of KRAB-PAX3-HBD protein by 4-OHT (data not shown). However, underlow-serum conditions, a significant reduction in the number of viableRh30-KPHBD-cl22 cells over a 10-day period was observed when theKRAB-PAX3-HBD repressor was activated by 4-OHT and growthwas evaluated by MTT assay (Fig. 6B). Rh30-SPHBD-cl8 cells ex-pressing the SNAG-PAX3-HBD protein manifested similar hormone-dependent inhibition of cell growth (Fig. 6C). As expected, Rh30-K(DV)PHBD-cl24 cells did not show any growth retardation undereither conditions (Fig. 6D), similar to parental Rh30 cells (Fig. 6A).Growth of un-induced Rh30-KPHBD-cl22 cells (Fig. 6B) was alsosimilar to that of Rh30 cell line (Fig. 6A). These results clearlyindicate that conversion of the inactive repressor protein to an activeform by 4-OHT is responsible for this growth inhibition in Rh30-KPHBD-cl22 and Rh30-SPHBD-cl8 cells. The malignant phenotypeof the Rh30-KPHBD-cl22 cells was further examined in poly-HEMA-coated tissue culture plates to evaluate anchorage-independent growthpotential. As expected, the parental Rh30 cell line exhibited abundantgrowth under the conditions of the poly-HEMA assay, and the growthwas equivalent under un-induced and 4-OHT-induced conditions (Fig.
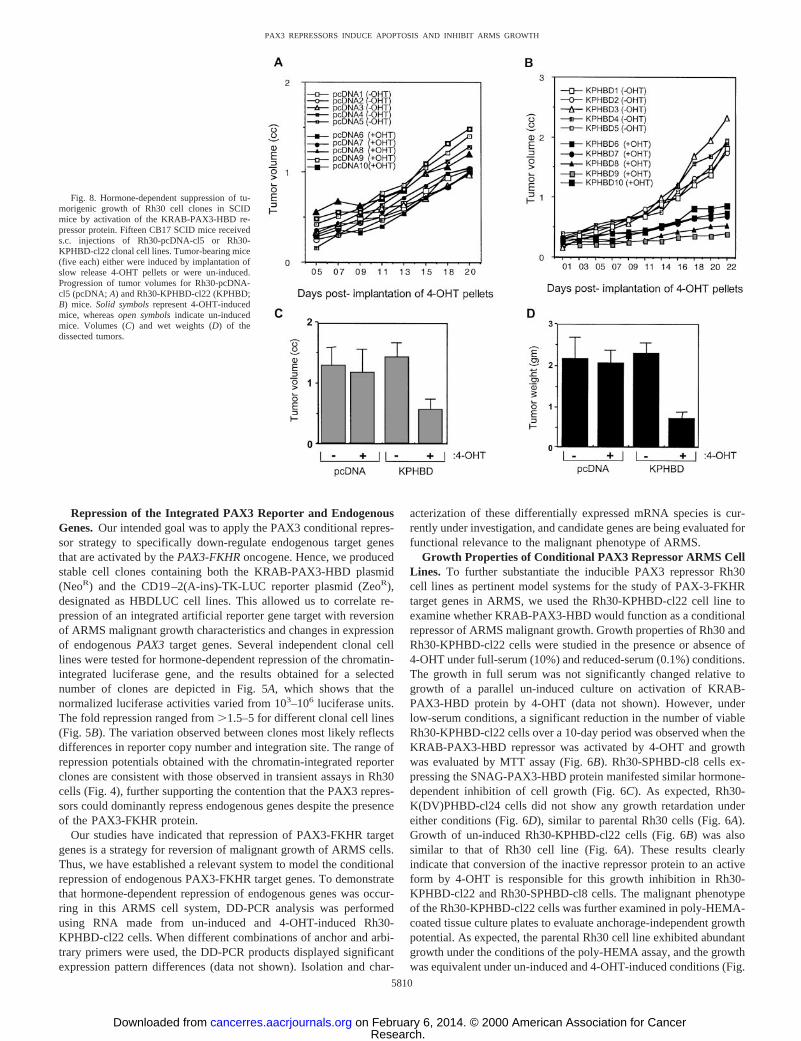
Fig. 8. Hormone-dependent suppression of tu-morigenic growth of Rh30 cell clones in SCIDmice by activation of the KRAB-PAX3-HBD re-pressor protein. Fifteen CB17 SCID mice receiveds.c. injections of Rh30-pcDNA-cl5 or Rh30-KPHBD-cl22 clonal cell lines. Tumor-bearing mice(five each) either were induced by implantation ofslow release 4-OHT pellets or were un-induced.Progression of tumor volumes for Rh30-pcDNA-cl5 (pcDNA; A) and Rh30-KPHBD-cl22 (KPHBD;B) mice. Solid symbolsrepresent 4-OHT-inducedmice, whereasopen symbolsindicate un-inducedmice. Volumes (C) and wet weights (D) of thedissected tumors.
5810
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

6E). In marked contrast, the Rh30-KPHBD-cl22 cell line showed adramatic suppression of growth potential only on 4-OHT treatment(Fig. 6F).
Our data indicate that activation of the KRAB-PAX3-HBD repres-sor renders the Rh30-KPHBD-cl22 cells very sensitive to inhibition ofgrowth in low-serum medium as evidenced by a loss of cell viabilityby day 8 (Fig. 6B). The PAX3 repressor also inhibited growth underanchorage-independent conditions as seen above, however, there wasno evidence of drastic cell loss. We were interested to know whetherthe difference in growth kinetics observed between the two differentassays could be due to apoptosis. Hence, we carried out two inde-pendent apoptosis assays (TUNEL and Annexin V) using Rh30-KPHBD-cl22 cells that were grown in low serum under un-induced or4-OHT-induced conditions. On 4-OHT induction, the TUNEL assaysshowed a significant increase in the proportion of cells stained withstreptavidin-FITC (Fig. 10B). This enhanced staining reflects incor-poration of biotin-16-dUTP at DNA strand breaks and is a sensitiveindication of apoptotic cell death. The level of nuclear staining bypropidium iodide was the same in the presence or absence of 4-OHTinduction, ruling out nonspecific cytotoxicity (data not shown). En-hanced immunodetection of Annexin V was also observed after4-OHT induction (data not shown), supporting the correlation be-tween KRAB-PAX3-HBD activity and apoptosis in low-serum cul-ture.
These findings suggested that the KRAB-PAX3-HBD proteinmight have repressed a PAX3 target gene linked to protection fromapoptosis, consistent with the role described for PAX proteins duringdevelopment (10, 11). It has previously been reported that PAX8 canpromote cell survival by activating antiapoptotic factors of the BCLfamily (8). We evaluated expression of the BCL-XL survival factor inRh30-KPHBD-cl22 cells that were grown in low serum under un-induced or 4-OHT-induced conditions. Immunoblot analysis of wholecell lysates indicated that the BCL-XL protein levels from 4-OHT-induced cells were decreased compared with un-induced cells (Fig.7A). RT-PCR analysis using BCL-XL-specific primers indicated areduced amplification of the BCL-XL product from reactions from4-OHT-induced samples compared with the un-induced samples.These data indicate that activation of KRAB-PAX3-HBD proteinresults in repression of BCL-XL transcription (Fig. 7B).
To test whether the KRAB-PAX3-HBD protein is a direct repressorof BCL-XL, Rh30-KPHBD-cl22 cells were induced with 4-OHT for10 h in the presence of cycloheximide. The RT-PCR analyses (Fig.7C) indicated that amplification of the BCL-XL product continued tobe diminished compared with the un-induced controls. However, theBCL-XL product generated from the cycloheximide-treated samplewas not repressed as fully as the 4-OHT-induced, noncycloheximide-treated sample. This diminution may be attributed to a reduced levelof the KRAB-PAX3-HBD protein after the cycloheximide treatment(data not shown). Overall, these results suggest that repression of theBCL-XL product does not requirede novoprotein synthesis andBCL-XL is a probable candidate for a direct target gene of PAX3-KRAB-HBD.
Hormone-dependent Suppression of Tumorigenesis in SCIDMice. Our previous studies had demonstrated that PAX3-KRAB ex-pression could suppress Rh30 cell tumorigenesis in SCID mice (18).Thus, we were interested in whether conditional suppression of tu-morigenesisin vivo could be demonstrated using the newly estab-lished conditional PAX3 repressor cell line Rh30-KPHBD-cl22. Wegenerated tumors by injecting SCID mice with Rh30-pcDNA-cl5 orRh30-KPHBD-cl22 cells. After a 10-day period to establish tumorgrowth, half of the mice having comparably sized tumors were im-planted with slow-release pellets containing the 4-OHT inducer. Sub-sequently, tumor measurements were made at regular intervals for 3weeks, and these results over time are shown in Fig. 8A for Rh30-pcDNA-cl5 mice and in Fig. 8Bfor Rh30-KPHBD-cl22 mice. Datafor the measured tumor volumes are presented in Fig. 8C. Aftersacrifice, tumors were isolated by dissection, and their wet weights areplotted in Fig. 8D. These results agree with the tumor volume esti-mates and clearly indicate that in the presence of 4-OHT only theRh30-KPHBD-cl22 mice showed a reduction in tumor growth,whereas the Rh30-pcDNA-cl5 mice did not. The mice bearing theRh30-pcDNA-cl5 and the Rh30-KPHBD-cl22 tumors were photo-graphed before sacrifice (Fig. 9A). RT-PCR analysis of tumor RNAusing the HBD 59and 39primers indicated that only Rh30-KPHBD-cl22 (-/1 4-OHT)-injected mice expressed the KRAB-PAX3-HBDtranscript (Fig. 9B). Furthermore, the tumors from the un-induced or4-OHT-induced pcDNA-cl5 mice and the un-induced KPHBD-cl22mice contained many blood vessels in the H&E-stained tumor tissue
Fig. 9. A, un-induced and 4-OHT-induced tumors in SCID micebearing Rh30-pcDNA-cl5 or Rh30-KPHBD-cl22 cell tumors. The site ofimplantation of 4-OHT pellets is indicated by thearrows. B, RT-PCRanalysis of tumors derived from Rh30-KPHBD-cl22 SCID mice (un-induced:Lanes 1,2, and3; 4-OHT-induced:Lanes 4,5, and6) and oftumors derived from Rh30-pcDNA-cl5 control mice (un-induced:Lanes7, 8, and9; 4-OHT-induced:Lanes 10,11, and12). Thearrow indicatesthe specific product from the pcKRAB-PAX3-HBD plasmid (KPHBD).
5811
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
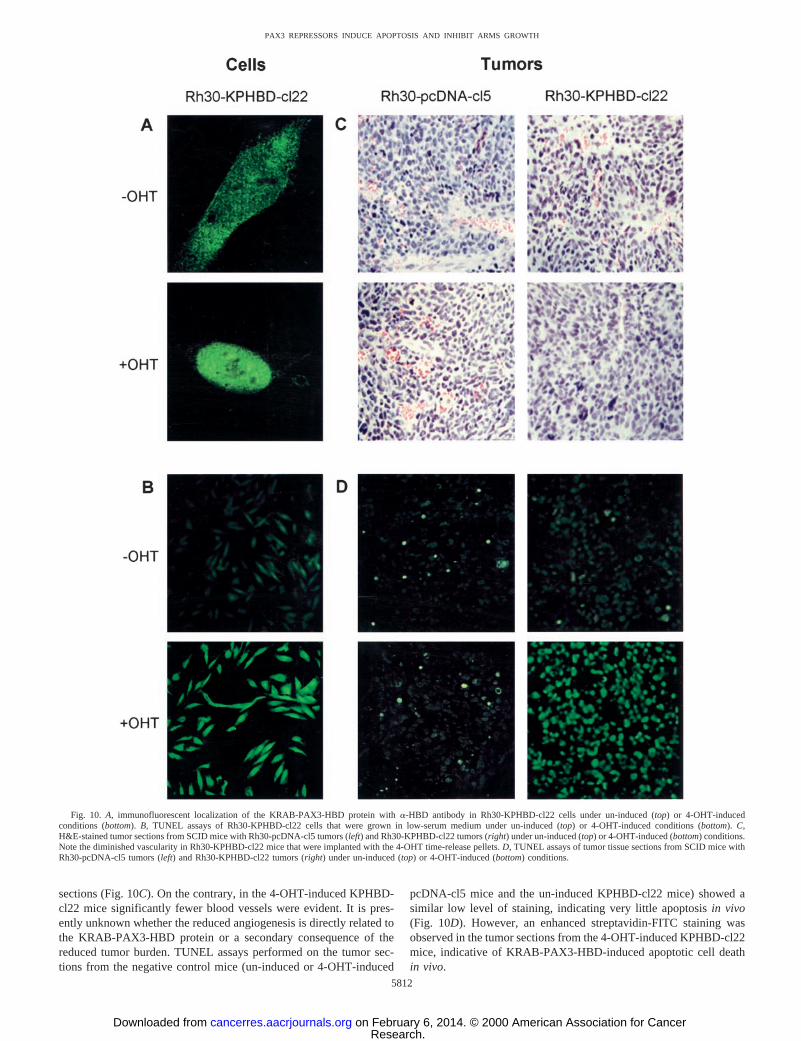
sections (Fig. 10C). On the contrary, in the 4-OHT-induced KPHBD-cl22 mice significantly fewer blood vessels were evident. It is pres-ently unknown whether the reduced angiogenesis is directly related tothe KRAB-PAX3-HBD protein or a secondary consequence of thereduced tumor burden. TUNEL assays performed on the tumor sec-tions from the negative control mice (un-induced or 4-OHT-induced
pcDNA-cl5 mice and the un-induced KPHBD-cl22 mice) showed asimilar low level of staining, indicating very little apoptosisin vivo(Fig. 10D). However, an enhanced streptavidin-FITC staining wasobserved in the tumor sections from the 4-OHT-induced KPHBD-cl22mice, indicative of KRAB-PAX3-HBD-induced apoptotic cell deathin vivo.
Fig. 10. A, immunofluorescent localization of the KRAB-PAX3-HBD protein witha-HBD antibody in Rh30-KPHBD-cl22 cells under un-induced (top) or 4-OHT-inducedconditions (bottom).B, TUNEL assays of Rh30-KPHBD-cl22 cells that were grown in low-serum medium under un-induced (top) or 4-OHT-induced conditions (bottom).C,H&E-stained tumor sections from SCID mice with Rh30-pcDNA-cl5 tumors (left) and Rh30-KPHBD-cl22 tumors (right) under un-induced (top) or 4-OHT-induced (bottom) conditions.Note the diminished vascularity in Rh30-KPHBD-cl22 mice that were implanted with the 4-OHT time-release pellets.D, TUNEL assays of tumor tissue sections from SCID mice withRh30-pcDNA-cl5 tumors (left) and Rh30-KPHBD-cl22 tumors (right) under un-induced (top) or 4-OHT-induced (bottom) conditions.
5812
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

DISCUSSION
In this study, we have generated ARMS cell lines with conditionalPAX3 repressors to establish a system to understand the biologicalrole and identify endogenous target genes of the PAX3-FKHR onco-protein. The conditional PAX3 repressors were generated by fusingthe KRAB or SNAG repression domains and PAX3 DNA-bindingmotifs to the tamoxifen-selective ERTM HBD of the estrogen receptor.We believe that application of our conditional repressors to the ARMSmodel system is especially relevant for evaluating the cellular path-ways aberrantly regulated by the PAX3-FKHR protein. One importantadvantage of using the ERTM system is that rapid activation of thetranscription factor-ER fusion protein can be achieved by a simpleligand-mediated on/off mechanism (24, 35). Thus, the immediateconsequences of the KRAB-PAX3-HBD repressor on biological prop-erties contributing to malignant growth and tumorigenesis can bestudied without selection for secondary mechanisms.
Our confidence in the conditional repressors applied in this study issupported by the following principal results that support their effec-tive dominant negative function. First, the KRAB-PAX3-HBD andSNAG-PAX3-HBD proteins were fully competent in binding to the e5target DNA in EMSA analysis. The super-shift experiments showedthat the KRAB-PAX3-HBD protein was also fully capable of bindingthe KAP-1 corepressor, which is a prerequisite for KRAB-mediatedtranscriptional repression (20). Conditional repressors using theSNAG domain do not rely on KAP-1 association, thus, may have anextended applicability in biological systems in which KAP-1 is notpresent, such as in insect cells, or in certain differentiated cell types(31). Second, the ability of the KRAB-PAX3-HBD and SNAG-PAX3-HBD conditional repressors to induce transcriptional repres-sion was tightly regulated by the ligand, 4-OHT. Moreover, the degreeof transcriptional repression achieved by the conditional KRAB-PAX3-HBD repressor was as potent as the repression observed withthe constitutive KRAB-PAX3 repressor, implying that fusion of theER HBD did not interfere with the expected function. Third, in Rh30stable clones, the KRAB-PAX3-HBD and the SNAG-PAX3-HBDprotein expression levels were higher than that of the endogenousPAX3-FKHR protein; hence, the repressors would be expected tohave a competitive advantage for PAX3-FKHR target genes. Fourth,the conditional PAX3 repressor proteins exhibited hormone-depen-dent nuclear localization, which may account for the hormone-depen-dent biological effects in Rh30 cells. Fifth, the KRAB-PAX3-HBDand SNAG-PAX3-HBD proteins exhibited potent repression of eithertransiently transfected or integrated reporter genes in Rh30 ARMScells. The conditional repressors were as effective in repression ofintegrated reporter plasmids as transiently transfected reporter plas-mid. Furthermore, the repression of endogenous genes by the condi-tional repressors was confirmed by the DD-PCR analysis. In all cases,the conditional repression was dominant over any activation mediatedby the endogenous PAX3-FKHR. In addition, there was a strikingcorrelation between the expression levels of the PAX3 repressors andthe degree of transcriptional repression. Sixth, the conditional PAX3repressors exhibited hormone-dependent suppression of characteristicmalignant properties such as: (a) growth in low serum; (b) anchorage-independent growth; and (c) tumorigenicity in SCID mice. Further-more, both the KRAB-based as well as the SNAG-based conditionalPAX3 repressors exhibited similar inhibitory properties, demonstrat-ing that the growth inhibition did not depend on a particular type ofrepression mechanism. Finally, the temporal control afforded by theconditional PAX3 repressor allowed observation of immediatechanges in growth under low-serum conditions that led us to demon-strate that the KRAB-PAX3-HBD protein induces apoptosis.
The hormone-dependent apoptosis exhibited in the conditional
PAX3 repressor Rh30 cell lines under low-serum conditions is en-tirely consistent with previous studies in which an antisense approachwas used to down-regulate PAX3-FKHR in Rh30 ARMS cells (10).Furthermore, it has been clearly demonstrated that down-regulation ofPAX3 function, either by mutation insplotchmice (14), or by reducedexpression in diabetic mice (36), leads to extensive apoptosis. It iswell established that many of the PAX family proteins function duringorganogenesis to protect cells from apoptosis (9, 11, 14). Previousstudies have linked the antiapoptotic function of PAX8 to activationof the BCL family protein, BCL-2 (8). Our studies have demonstratedthat the KRAB-PAX3-HBD protein can down-regulate expression ofthe BCL-XL cellular survival factor and induce apoptosis in Rh30 celllines in vitro and in SCID micein vivo. These observations suggestthat PAX3-FKHR may contribute to oncogenesis in ARMS by acti-vating the antiapoptotic BCL-XL survival factor. One advantage of theER system for studying transcription factors like KRAB-PAX3-HBDis that activation of the ER-fusion protein can occur in the absence ofde novoprotein synthesis. Thus, in the presence of protein synthesisinhibitors such as cycloheximide, a distinction can be made betweendirectly regulated target genes and those secondary targets whoseregulation depends on transcription and translation of a primary targetgene. Our data supports the hypothesis thatBCL-XL is a direct PAX3target gene, because the repression by KRAB-PAX3-HBD waslargely insensitive to inhibition by cycloheximide. Recent studieshave confirmed that PAX3 and PAX3-FKHR can directly regulate theBCL-XL via direct binding to the promoter (37).
It is likely that regulation of apoptosis by PAX3 may involveseveral other mechanisms in addition to BCL-XL. Recent expressionprofiling has shown that PAX3-FKHR can activate expression of theSLUG protein (34). Other studies have shown that SLUG plays anantiapoptotic role in E2A-HLF-induced tumorigenesis (38). In addi-tion, PAX3 is known to regulate c-MET, the receptor for the hepato-cyte growth factor/scatter factor, which has also been implicated inapoptosis (39, 40). It is interesting that apoptosis was only observedwhen the KRAB-PAX3-HBD protein was activated under low-serumconditions, implying that the Rh30 cells may only depend on PAX3-FKHR for protection against apoptosis when deprived of certainfactors present in serum. Substantial evidence supports a role forgrowth factor signaling in ARMS. PAX3-FKHR has been shown toactivate expression of insulin-like growth factor II, which has awell-established role in ARMS malignant growth (34, 41). Otherstudies have defined a role for the wild-type FKHR protein in linkinginsulin signaling to a cellular proapoptotic response (41, 42). It is notknown whether PAX3-FKHR exerts any dominant-negative effect onthe wild-type FKHR protein derived from the nontranslocated allele inARMS. Protection from apoptosis by PAX3-FKHR is likely to be akey oncogenic mechanism in ARMS that will require further identi-fication and validation of the essential target genes.
Our demonstration that KRAB-PAX3-HBD Rh30 cell tumors showpronounced hormone-dependent inhibition of growth confirms ourprevious findings and emphasizes that in ARMS the tumorigenicpotential is dependent on PAX3-FKHR. In SCID mice experiments,use of the timed-release pellets allowed maintenance of the bloodlevels of 4-OHT and activation of the conditional repressor through-out the course of the experiment. The tumors that grew from controlcells without the repressor or from Rh30-KPHBD-cl22 cells withoutthe 4-OHT inducer were heavily vascularized. It is interesting thatonly the Rh30-KPHBD-cl22 tumors derived from the 4-OHT-treatedmice showed very little evidence of vascularization in the H&E-stained tumor sections. We are currently investigating whether theKRAB-PAX3-HBD protein may have repressed angiogenic factors.
We believe that our conditional PAX3 repressor strategies havetargeted a major oncogenic mechanism in ARMS, protection from
5813
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from

apoptosis by PAX3-FKHR. Characterization of repressed target genesthat comprise the PAX3-FKHR “oncogenic transcriptome” in ARMSidentified by differential display RT-PCR analysis is our current focus.
ACKNOWLEDGMENTS
We are grateful to Dr. Satish Parimoo (Skin Biology, Johnson & Johnson,Skillman, NJ) for providing reagents and guidance with DD-PCR protocols.We thank Dr. Beat Schafer for the 6xCD19–2 (A-ins)-TK-LUC reporterplasmid. We thank Trevor Littlewood (ICRF, London) for the HBD-ER™
plasmid. Richelle Takemoto and Rachel Beurmann of Dr. Meenhard Herlyn’slaboratory at the Wistar Institute provided assistance with tumorigenicitystudies in SCID mice. We are grateful to members of Dr. Frank Rauscher’slaboratory for helpful suggestions and comments.
REFERENCES
1. Rauscher, F. J., III., and Vogt, P. K. Chromosomal translocations and oncogenictranscription factors. Current Topics in Microbiology and Immunology, vol. 220,pp.1–166. Berlin, Heidelberg: Springer-Verlag, 1997.
2. Barr, F. G. The role of chimeric paired box transcription factors in the pathogenesisof pediatric rhabdomysarcoma. Cancer Res.,59: 1711s–1715s, 1999.
3. Galili, N., Davis, R. J., Fredericks, W. J., Mukhopadhyay, S., Rauscher, F. J., III.,Emanuel, B. S., Rovera, G., and Barr, F. G. Fusion of a fork head domain gene toPAX3 in the solid tumour alveolar rhabdomyosarcoma [published erratum appears inNat. Genet.,6: 214, 1994]. Nat Genet.,5: 230–235, 1993.
4. Fredericks, W. J., Galili, N., Mukhopadhyay, S., Rovera, G., Bennicelli, J., Barr,F. G., and Rauscher, F. J., III. The PAX3-FKHR fusion protein created by the t(2;13)translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activa-tor than PAX3. Mol. Cell. Biol.,15: 1522–1535, 1995.
5. Davis, R. J., and Barr, F. G. Fusion genes resulting from alternative chromosomaltranslocations are overexpressed by gene-specific mechanisms in alveolar rhabdomy-osarcoma. Proc. Natl. Acad. Sci. USA,94: 8047–8051, 1997.
6. Mansouri, A. The role of Pax3 and Pax7 in development and cancer. Crit. Rev.Oncog.,9: 141–149, 1998.
7. Maulbecker, C. C., and Gruss, P. The oncogenic potential of Pax genes. EMBO J.,12:2361–2367, 1993.
8. Hewitt, S. M., Hamada, S., Monarres, A., Kottical, L. V., Saunders, G. F., andMcDonnell, T. J. Transcriptional activation of the bcl-2 apoptosis suppressor gene bythe paired box transcription factor PAX8. Anticancer Res.,17: 3211–3215, 1997.
9. Ostrom, L., Tang, M. J., Gruss, P., and Dressler, G. R. Reduced Pax2 gene dosageincreases apoptosis and slows the progression of renal cystic disease. Dev. Biol.,219:250–258, 2000.
10. Bernasconi, M., Remppis, A., Fredericks, W. J., Rauscher, F. J., III., and Schafer,B. W. Induction of apoptosis in rhabdomyosarcoma cells through down- regulation ofPAX proteins. Proc. Natl. Acad. Sci. USA,93: 13164–9, 1996.
11. Borycki, A. G., Li, J., Jin, F., Emerson, C. P., and Epstein, J. A. Pax3 functions in cellsurvival and in pax7 regulation. Development,126: 1665–1674, 1999.
12. Reeves, F. C., Burdge, G. C., Fredericks, W. J., Rauscher, F. J., III., and Lillycrop,K. A. Induction of antisense Pax-3 expression leads to the rapid morphologicaldifferentiation of neuronal cells and an altered response to the mitogenic growthfactor bFGF. J. Cell Sci.,112: 253–261, 1999.
13. Goulding, M., Lumsden, A., and Paquette, A. J. Regulation of Pax-3 expression in thedermomyotome and its role in muscle development. Development,120: 957–971,1994.
14. Dickman, E. D., Rogers, R., and Conway, S. J. Abnormal skeletogenesis occurscoincident with increased apoptosis in the Splotch (Sp2H) mutant: putative roles forPax3 and PDGFRa in rib patterning. Anat. Rec.,255: 353–361, 1999.
15. Watanabe, A., Takeda, K., Ploplis, B., and Tachibana, M. Epistatic relationshipbetween Waardenburg syndrome genes MITF and PAX3. Nat. Genet.,18: 283–286,1998.
16. Arnold, H. H., and Winter, B. Muscle differentiation: more complexity to the networkof myogenic regulators. Curr. Opin. Genet. Dev.,8: 539–544, 1998.
17. Epstein, J. A., Song, B., Lakkis, M., and Wang, C. Tumor-specific PAX3-FKHRtranscription factor, but not PAX3, activates the platelet-derived growth factorareceptor. Mol. Cell. Biol.,18: 4118–4130, 1998.
18. Fredericks, W. J., Ayyanathan, K., Friedman, J. R., and Rauscher, F. J., III. Anengineered PAX3-KRAB transcriptional repressor inhibits the malignant phenotypeof alveolar rhabdomyosarcoma cells harboring the PAX3-FKHR oncogene. Mol.Cell. Biol., 20: 5019–5031, 2000.
19. Margolin, J. F., Friedman, J. R., Meyer, W. K., Vissing, H., Thiesen, H. J., andRauscher, F. J., III. Kruppel-associated boxes are potent transcriptional repressiondomains. Proc. Natl. Acad. Sci. USA,91: 4509–4513, 1994.
20. Friedman, J. R., Fredericks, W. J., Jensen, D. E., Speicher, D. W., Huang, X. P.,Neilson, E. G., and Rauscher, F. J., III. KAP-1, a novel corepressor for the highlyconserved KRAB repression domain. Genes Dev.,10: 2067–2078, 1996.
21. Grimes, H. L., Chan, T. O., Zweidler-McKay, P. A., Tong, B., and Tsichlis, P. N. TheGfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG,and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol. Cell. Biol.,16:6263–6272, 1996.
22. Madden, S. L., Cook, D. M., Morris, J. F., Gashler, A., Sukhatme, V. P., andRauscher, F. J., III. Transcriptional repression mediated by the WT1 Wilms tumorgene product. Science (Washington DC),253: 1550–1553, 1991.
23. Wang, Y., Xu, J., Pierson, T., O’Malley, B. W., and Tsai, S. Y. Positive and negativeregulation of gene expression in eukaryotic cells with an inducible transcriptionalregulator. Gene Ther.,4: 432–441, 1997.
24. Littlewood, T. D., Hancock, D. C., Danielian, P. S., Parker, M. G., and Evan, G. I. Amodified oestrogen receptor ligand-binding domain as an improved switch for theregulation of heterologous proteins. Nucleic Acids Res.,23: 1686–1690, 1995.
25. Nutt, S. L., Morrison, A. M., Dorfler, P., Rolink, A., and Busslinger, M. Identificationof BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. EMBO J.,17: 2319–2333, 1998.
26. Kruse, U., Iacovoni, J. S., Goller, M. E., and Vogt, P. K. Hormone-regulatableneoplastic transformation induced by a Jun-estrogen receptor chimera. Proc. Natl.Acad. Sci. USA,94: 12396–12400, 1997.
27. Alarcon, R. M., Rupnow, B. A., Graeber, T. G., Knox, S. J., and Giaccia, A. J.Modulation of c-Mycactivity and apoptosisin vivo. Cancer Res.,56: 4315–4319,1996.
28. Milocco, L. H., Haslam, J. A., Rosen, J., and Seidel, H. M. Design of conditionallyactive STATs: insights into STAT activation and gene regulatory function. Mol. Cell.Biol., 19: 2913–2920, 1999.
29. Thiesen, H. J., and Meyer, W. Krab domains analyzed in human Cys/His-typezinc-finger proteins KOX 1, KOX 8, and KOX 19. Ann. NY Acad. Sci.,684:243–245, 1993.
30. Schafer, B. W., Czerny, T., Bernasconi, M., Genini, M., and Busslinger, M. Molec-ular cloning and characterization of a humanPAX-7cDNA expressed in normal andneoplastic myocytes. Nucleic Acids Res.,22: 4574–4582, 1994.
31. Ryan, R. F., Schultz, D. C., Ayyanathan, K., Singh, P. B., Friedman, J. R., Fredericks,W. J., and Rauscher, F. J., III. KAP-1 corepressor protein interacts and colocalizeswith heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol.Cell. Biol., 19: 4366–4378, 1999.
32. Pardinas, J. R., Combates, N. J., Prouty, S. M., Stenn, K. S., and Parimoo, S.Differential subtraction display: a unified approach for isolation of cDNAs fromdifferentially expressed genes. Anal. Biochem.,257: 161–168, 1998.
33. Fukazawa, H., Mizuno, S., and Uehara, Y. A microplate assay for quantitation ofanchorage-independent growth of transformed cells. Anal. Biochem.,228: 83–90,1995.
34. Khan, J., Bittner, M. L., Saal, L. H., Teichmann, U., Azorsa, D. O., Gooden, G. C.,Pavan, W. J., Trent, J. M., and Meltzer, P. S. cDNA microarrays detect activation ofa myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc. Natl.Acad. Sci. USA,96: 13264–13269, 1999.
35. Chambraud, B., Berry, M., Redeuilh, G., Chambon, P., and Baulieu, E. E. Severalregions of human estrogen receptor are involved in the formation of receptor-heatshock protein 90 complexes. J. Biol. Chem.,265: 20686–20691, 1990.
36. Fine, E. L., Horal, M., Chang, T. I., Fortin, G., and Leoken, M. R. Evidence thatelevated glucose causes altered gene expression, apoptosis, and neural tube defects ina mouse model of diabetic pregnancy. Diabetes,48: 2454–2462, 1999.
37. Margue, C. M., Bernasconi, M., Barr, F. G., and Schafer, B. W. Transcriptionalmodulation of the anti-apoptotic protein BCL-XL by the paired box transcriptionfactors PAX3 and PAX3/FKHR, Oncogene,19: 2921–2929, 2000.
38. Inukai, T., Inoue, A., Kurosawa, H., Goi, K., Shinjyo, T., Ozawa, K., Mao, M., Inaba,T., and Look, A. T. SLUG, a ces-1-related zinc finger transcription factor gene withantiapoptotic activity, is a downstream target of the E2A-HLF oncoprotein. MolecularCell., 4: 343–352, 1999.
39. Bardelli, A., Longati, P., Albero, D., Goruppi, S., Schneider, C., Ponzetto, C., andComoglio, P. M. HGF receptor associates with the anti-apoptotic protein BAG-1 andprevents cell death. EMBO J.,15: 6205–6212, 1996.
40. Fan, S., Wang, J. A., Yuan, R. Q., Rockwell, S., Andres, J., Zlatapolskiy, A.,Goldberg, I. D., and Rosen, E. M. Scatter factor protects epithelial and carcinomacells against apoptosis induced by DNA-damaging agents. Oncogene,17: 131–141,1998.
41. Wang, W., Kumar, P., Epstein, J., Helman, L., Moore, J. V., and Kumar, S. Insulin-like growth factor II and PAX3-FKHR cooperate in the oncogenesis of rhabdomyo-sarcoma. Cancer Res.,58: 4426–4433, 1998.
42. Tang, E. D., Nunez, G., Barr, F. G., and Guan, K. L. Negative regulation of theforkhead transcription factor FKHR by Akt. J. Biol. Chem.,274: 16741–6, 1999.
5814
PAX3 REPRESSORS INDUCE APOPTOSIS AND INHIBIT ARMS GROWTH
Research. on February 6, 2014. © 2000 American Association for Cancercancerres.aacrjournals.org Downloaded from
Related Documents











