of August 25, 2012. This information is current as Suppression of HIV-Specific CD8 T Cells Death-1 Ligand Upregulation and Activation in APCs Leads to Programmed Threonine Kinase - 3-Kinase/Serine HIV-Mediated Phosphatidylinositol David B. Weiner Pablo Tebas, Michael A. Chattergoon, Andrew Y. Choo and Chaoran B. Bian, Aarti A. Ramanathan, Parikh Atman, Paolo Fagone, Omkar U. Kawalekar, Jonathan Goodman, Karuppiah Muthumani, Devon J. Shedlock, Daniel K. Choo, http://www.jimmunol.org/content/187/6/2932 doi: 10.4049/jimmunol.1100594 August 2011; 2011; 187:2932-2943; Prepublished online 19 J Immunol References http://www.jimmunol.org/content/187/6/2932.full#ref-list-1 , 21 of which you can access for free at: cites 50 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2011 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology at Univ of Pennsylvania Library on August 25, 2012 http://jimmunol.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

of August 25, 2012.This information is current as Suppression of HIV-Specific CD8 T Cells
Death-1 Ligand Upregulation andActivation in APCs Leads to Programmed
Threonine Kinase−3-Kinase/SerineHIV-Mediated Phosphatidylinositol
David B. WeinerPablo Tebas, Michael A. Chattergoon, Andrew Y. Choo and Chaoran B. Bian, Aarti A. Ramanathan, Parikh Atman,Paolo Fagone, Omkar U. Kawalekar, Jonathan Goodman, Karuppiah Muthumani, Devon J. Shedlock, Daniel K. Choo,
http://www.jimmunol.org/content/187/6/2932doi: 10.4049/jimmunol.1100594August 2011;
2011; 187:2932-2943; Prepublished online 19J Immunol
Referenceshttp://www.jimmunol.org/content/187/6/2932.full#ref-list-1
, 21 of which you can access for free at: cites 50 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

The Journal of Immunology
HIV-Mediated Phosphatidylinositol 3-Kinase/Serine–Threonine Kinase Activation in APCs Leads to ProgrammedDeath-1 Ligand Upregulation and Suppression ofHIV-Specific CD8 T Cells
Karuppiah Muthumani,* Devon J. Shedlock,* Daniel K. Choo,* Paolo Fagone,*
Omkar U. Kawalekar,* Jonathan Goodman,* Chaoran B. Bian,* Aarti A. Ramanathan,*
Parikh Atman,* Pablo Tebas,† Michael A. Chattergoon,‡ Andrew Y. Choo,x and
David B. Weiner*
Recent evidence demonstrates that HIV-1 infection leads to the attenuation of cellular immune responses, which has been correlated
with the increased expression of programmed death (PD)-1 on virus-specific CD8+ T cells. PD-1 is induced upon T cell activation,
and its prolonged expression facilitates CD8+ T cell inhibitory signals when bound to its B7 family ligands, PD-ligand (L)1/2,
which are expressed on APCs. Importantly, early reports demonstrated that blockade of the PD-1/PD-L interaction by Abs may
help to counter the development of immune exhaustion driven by HIV viral persistence. To better understand the regulation of the
PD-1 pathway during HIV infection, we examined the ability of the virus to induce PD-L expression on macrophages and dendritic
cells. We found a direct relationship between the infection of APCs and the expression of PD-L1 in which virus-mediated
upregulation induced a state of nonresponsiveness in uninfected HIV-specific T cells. Furthermore, this exhaustion phenotype
was revitalized by the blockade of PD-L1, after which T cells regained their capacity for proliferation and the secretion of
proinflammatory cytokines IFN-g, IL-2, and IL-12 upon restimulation. In addition, we identify a critical role for the PI3K/
serine–threonine kinase signaling pathway in PD-L1 upregulation of APCs by HIV, because inhibition of these intracellular signal
transducer enzymes significantly reduced PD-L1 induction by infection. These data identify a novel mechanism by which HIV
exploits the immunosuppressive PD-1 pathway and suggest a new role for virus-infected cells in the local corruption of immune
responses required for viral suppression. The Journal of Immunology, 2011, 187: 2932–2943.
TheHIV-1 epidemic continues to be amajor issueworldwidewith ∼41.3 million adults and 2.1 million children cur-rently living with the virus and ∼16,000 new infections/y.
This retrovirus preferentially infects and kills CD4+ T cells andmacrophages (1–4) resulting in the progressive dysfunction of thehost immune system and increased susceptibility to various op-portunistic infections and neoplasms. Although slowing diseaseprogression is typically achieved with highly active antiretroviraltherapy (HAART), there is growing evidence that the immunesystem, on its own, can limit HIV replication in some cases. Forinstance, a subset of HIV-infected patients termed “elite con-trollers” have viral loads maintained below the detectable limit(,50 copies HIV RNA/ml) without HAART (5–7). It has beensuggested that this enhanced viral control directly correlated with
CD8+ T cell activation and function as HIV-specific CD8+ T cellsfrom human controllers (long-term progressors) exhibit greater
degree of activation and function (8) than those from non-
controllers. Furthermore, in SIV infection models, CD8+ T cells arenecessary for control of viremia, and vaccines that induce the most
potent CD8+ T cell responses have proven to be themost effective in
controlling disease progression (3, 9, 10).Recent reports from several groups suggested that HIV persis-
tence may be caused in certain instances by the inability of host
HIV-specific CD8+ T cells to mount effective immune responses
(11, 12). This deficit correlated with increased expression of pro-
grammed death (PD)-1 (also CD279), a receptor that inhibits T cell
activation on HIV-specific CD8+ T cells (9, 13–19), and a decrease
of CD4 T cell help (20). Moreover, PD-1 upregulation correlatedwith impaired immune function and disease progression (12, 15,
21). Induced expression of PD-1 on CD4+, CD8+, and NK T cells,
engagement of its ligands, and subsequent signaling attenuates
T cell function via the inhibition of cellular kinases that signal
through the TCR and CD28 to promote cytokine production and
cell proliferation (21). Through recruitment of phosphatases suchas protein–tyrosine phosphatase Src homology region 2 domain-
containing phosphatase 2, PD-1 decreases the phosphorylation
and activation of kinases such as Spleen-tyrosine kinase (22),
PI3K, and serine–threonine kinase (Akt) (21). The essential role of
PD-1 in suppressing T cell activation and promoting immune
homeostasis is underscored by theobservation that pcdc2/2 mice
develop spontaneous, late-onset lupus-like disease and a dilatedcardiomyopathy characterized by autoantibodies to troponin (23).
*Department of Pathology and Laboratory Medicine, University of PennsylvaniaSchool of Medicine, Philadelphia, PA 19104; †Division of Infectious Diseases, Uni-versity of Pennsylvania School of Medicine, Philadelphia, PA 19104; ‡Division ofInfectious Diseases, The Johns Hopkins Hospital, Baltimore, MD 21205; and xDe-partment of Cell Biology, Harvard Medical School, Boston, MA 02115
Received for publication March 1, 2011. Accepted for publication July 14, 2011.
Address correspondence and reprint requests to Dr. Karuppiah Muthumani, De-partment of Pathology and Laboratory Medicine, University of PennsylvaniaSchool of Medicine, 422 Curie Boulevard, Philadelphia, PA 19104. E-mail address:[email protected]
Abbreviations used in this article: Akt, serine–threonine kinase; DC, dendritic cell;HAART, highly active antiretroviral therapy; IRF, IFN regulatory factor; L, ligand;mDC, myeloid dendritic cell; MFI, mean fluorescence intensity; PD-1, programmeddeath-1; pDC, plasmacytoid dendritic cell; siRNA, small interfering RNA.
Copyright� 2011 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100594
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

The effect of PD-1 in T cell regulation and its role in the main-tenance of a chronic viral infection was shown in the lymphocyticchoriomeningitis virus murine infection model where PD-1 Abblockade restored the function of Ag-specific T cells and led toclearance of the chronic infection (17–19, 24). A role for PD-1 inretroviral infection has also been suggested by several studies (13,15, 17, 18, 25). During HIV infection, PD-1 expression on HIV-specific CD8+ T cells correlates with disease progression as mea-sured by viral load and CD4+ T cell counts (9, 17), and in chroni-cally infected individuals PD-1 expression is high on HIV-specificCD8+ T cells. Furthermore, administration of Abs that interferewith PD-1/PD-ligand (L)1 (also CD274) binding leads to an in-crease in the activity of HIV-specific, PD-1highCD8+ T cells (18).Finally, in an SIV infection model, direct in vivo interruption ofPD-1 signaling using a specific mAb increased the number of SIV-specific CD8+ T cells, decreased the viral load, and prolonged thesurvival of chronically infected animals (19). Taken together, thesedata suggest that interruption of PD-1/PD-L1 binding and pre-sumably signaling can augment the host’s immune system to achronic viral infection and possibly help mediate control.Although the expression of PD-1 onHIV-specificCD4+ andCD8+
T cells has been well documented, the natural source of PD-L1 iscurrently unknown. Furthermore, the signals regulating PD-L1 ex-pression are also not well studied, and, most importantly, it is notknownwhether HIVinfection can directly influence these. Althoughfew studies have directly investigated the role of PD-L1 in HIV-induced immune suppression, several have shown a clear corre-lation between HIV infection and PD-L1 upregulation (26–28).Baosso et al. (26) demonstrated that direct HIV-1 infection of mono-cytes in patient-derived samples showed increased levels of PD-L1expression. Trabattoni et al. (29) reported that expression of PD-L1in HIV-1–infected typical progressors and AIDS patients correlateswith viral load and is inversely correlated with CD4+ T cell count.We hypothesized that HIV infection directly disrupts the gen-
eration and/or activation of HIV-specific T cells by attenuating/disrupting Ag presentation via upregulation of PD-L1/2 expressionon APCs and hence signaling through PD-1 on T cells. In an effort tounderstand the role of PD-L1 in mediating HIV-induced immunesystem suppression, we explored the effect onHIV-specific T cells ofdisrupting the PD-1/PD-L1 interaction between HIV-specific T cellsand APCs derived from the T cell donor using PD-L1 small in-terfering RNA (siRNA) to modulate PD-L1 expression. In addition,we demonstrated that APCs including monocytes and dendritic cells(DCs) from HIV-positive humans have higher PD-L1 expressioncompared with normal donors. Infection of APCs was sufficient todirectly induce PD-L1 expression, and interruption of PD-L1 invitroby siRNA resulted in significant enhancement of proliferation andantiviral cytokine secretion by HIV-specific T cells. Understandingthemechanism of HIV-induced PD-L1may provide new therapeutictargets for enhancing the host’s immune response, and the resultsreported in this paper have significant implications for the de-velopment of novel immunotherapeutic approaches (e.g., by block-ing negative T cell regulators) that will restore T cell function inthis widespread disease.
Materials and MethodsCell isolation and culture
Leukopacks fromindividualhealthydonorswereobtained fromtheUniversityof Pennsylvania School ofMedicine Immunology Clinical Core, and PBMCswere isolated by Ficoll-Hypaque (Pharmacia, Piscataway, NJ) density cen-trifugation. PBMCs, serum, and plasma from HIV-infected individuals wereobtained from the University of Pennsylvania-Center for AIDS Research andthe University of Pennsylvania School Immunology Core. Samples wereobtained in accordance with protocols approved by the Institutional ReviewBoard of the Hospital of the University of Pennsylvania. Leukophoresis
procedures were conducted in accordance with protocols approved by theInstitutional Review Board of the Hospital of the University of Pennsylvania.Leukophoresis procedures were conducted in accordance with protocolsapproved by the Institutional Review Board of the Hospital of the Universityof Pennsylvania. PBMCs were separated and cryopreserved in liquid N2.HIV-1 RNA level was determined from plasma using the Roche Amplicor 1.5Kit (Roche Diagnostic Systems, Somerville, NJ), as per the manufacturer’srecommendations (Table II). All primary cells were maintained in LPS-freeRPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine,20 mM HEPES, 100 U/ml penicillin, 100 mg/ml streptomycin, and in thepresence of recombinant human IL-2 (50 U/ml). CD3MicroBeads were usedfor the positive selection of T cells from PBMCs using MACS Technologyisolation kit reagents and protocol (Miltenyi Biotec, Auburn, CA). The re-sulting preparation was stained with anti–CD3-FITC (BD Biosciences, SanJose, CA) to confirm ,95% purity of T cells. Human monocytic leukemiacell lines THP-1 and U937 were purchased from the American Type CultureCollection (Manassas, VA) and maintained in their appropriate growth me-dium (30).
Monocytes were isolated from PBMCs using the human monocyteisolation kit (Automacs; Miltenyi Biotec), according to the manufacturer’sspecifications. The isolated monocytes were washed four times with ice-cold PBS containing 0.3% (w/v) BSA and 0.6% (w/v) Na3 citrate·3H2O,and the preparations were .90% pure as determined by flow cytometryusing anti-human CD14 Abs. Elutriated monocytes were cultured inRPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine,100 U/ml penicillin, 100 mg/ml streptomycin, 20 mg/ml gentamicin (LifeTechnologies/Invitrogen), 50 ng/ml GM-CSF (R&D Systems, Minneap-olis, MN), and 25 ng/ml IL-4 (PeproTech, Rocky Hill, NJ) at 37˚C for2 d and then for an additional 5 d in medium lacking M-CSF before use intransfection, infection, ELISPOT, and other assays (26, 30–33).
Cloning of PD-L1 promoter and luciferase assay
To assess modulation of PD-L1 transcription by viral infection, luciferasereporter plasmids expressing PD-L1 were assembled from synthetic oli-gonucleotides (GeneArt; Invitrogen, Darmstadt, Germany) and were clonedby inserting PD-L1 promoter sequences into specific restriction cloningsites (XhoI and BamHI) of the pMet-Luciferase reporter vector (BDClontech, Mountain View, CA) (34). For the PD-L1 reporter assay, THP-1cells were seeded onto a 6-well culture plate at a density of 500,000 cells/ml medium and transiently cotransfected as previously described (34, 35)with a constant amount of the luciferase reporter PD-L1 promoter plasmidand various plasmids expressing viral gene–GFP fusion proteins. Fur-thermore, the total amount of plasmid DNA was kept constant by addingempty vectors. Cells were harvested 48 h posttransfection, lysed, and thentested for luciferase activity with the luciferase assay kit (Promega,Madison, WI) using LUMAT-LB9501 (Berthold, Bad Wildbad, Germany).Transfection efficiency was normalized by cotransfection with pLacZ andassayed for b-galactosidase expression (34).
PD-L1/2 siRNA synthesis and transfection
Pools of three to five target-specific 19–25 nt siRNAs designed to knockdown gene expression were used. Three different PD-L1 and PD-L2 StealthsiRNA duplexes and one recommended negative control siRNA duplexeswere synthesized (Invitrogen, Carlsbad, CA) and displayed in Table II.These target sequences were submitted to a basic local alignment search toolsearch to ensure that only the PD-L1 or PD-L2 genes were targeted (36, 37).Similarly, human Akt1 siRNA and nonspecific control pool (sc-29195)were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). AllsiRNAs were dissolved or diluted to a concentration of 25 mM in diethylpyrocarbonate-treated water and stored at220˚C. siRNAs were transfectedinto 5 3 106 experimental cells, using the Amaxa Nucleofector device(Amaxa, Walkersville, MD) with cell-specific Nucleofector kit V, accordingto the manufacturer’s recommendations. After electroporation, cells weretransferred to a complete RPMI 1640 medium for 48 h before analysis forprotein knockdown.
RT-PCR analysis of PD-L1 expression
HIV-infected patient samples were added to Lysing Matrix D tubes (BIO101, Carlsbad, CA) containing TRIzol reagent and stored at 270˚C. TotalRNAwas later extracted per the manufacturer’s protocol. To eliminate anyresidual genomic DNA, the RNA samples were incubated with DNase I for15 min at room temperature. The DNase I was later inactivated by adding 1ml 25 mM EDTA to the reaction at 65˚C for 10 min. The yield and purityof the total RNA were determined from the A260/280 absorbance value.
PCR primers for the genes of interest and b-actin (used as an internalcontrol) were designed using Primer Express Software version 2.0 anda software program specially provided with the 7700 SDS (Applied
The Journal of Immunology 2933
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

Biosystems). The following gene-specific primers and TaqMan probes wereused: PD-L1 (XM-219775), 59-TGTACCACGTCT CCCACATAACAG-39(forward primer) and 59-ACCCCACGATG AGGAACAAA-39 (reverseprimer); PD-L2 (XM-219777), 59-TGACCCTCTGAGTT GGATGGA-39(forward primer) and 59-GCCGGGATGAAAGCATGA-39 (reverse primer)(38); and b-actin (NM-031144), 59-TGCTGACAGGATGCAGAAGGA-39(forward primer) and 59-CGCTCAGGAGGAGCAATGAT-39 (reverse primer).
RT-PCR assays using SYBR Green were performed with the ABIPRISMER 7700 Sequence Detection System (PerkinElmer-Applied Bio-systems, Foster City, CA). For each primer pair, the dissociation curveanalysis was conducted to ensure the specificity of the amplification at theend of PCR. Different concentrations of primers (50–100 nM) were testedto determine the optimal PCR conditions. Briefly, 90 ml Master Mix,containing 200 nM primers, 45 ml 23 Quantitative PCR MasterMix Plusfor Sybr Green I, and 2 ml cDNAwere mixed before aliquoting in triplicateto a 96-well microtitre plate. The cDNAwas amplified under the followinguniversal conditions: one cycle at 50˚C for 2 min and 95˚C for 10 min,followed by 40 cycles at 95˚C for 15 s and 60˚C for 1 min. To correct forvariations in input RNA amounts and the efficiency of reverse transcrip-tion, an endogenous “housekeeping” gene, b-actin, was also quantified andused to normalize the results (39–41).
Virus production and infection
The full-lengthproviralDNAconstructs used for productionofHIV-1wereasfollows: HIV-1 NL4-3Wt, a chimeric VSV-G envelope (NL43ΔEnv/VSV-GEnv), macrophage-tropic envelope AD8, 89.6, and Bal-mac (33, 34). Theseproviral vectors were obtained from the National Institutes of Health AIDSResearch and Reference Reagent Program (Germantown, MD). HIV-1 env,vif, vpu, vpr, and nef were individually cloned using PCR and characterizedas described previously (34). For production of viruses, 293T cells weretransfected in 10-cm-diameter dishes with 10 mg each of proviral vectors asdescribed previously. Briefly, we used 10 mg defective HIV-1 genomicvector, under control of the LTR promoter. HIV-1 stocks were produced in293T cells (American Type Culture Collection) were cultured at 37˚C in 5%CO2 and DMEM supplemented with FBS (10%), penicillin (50 IU/ml), andstreptomycin (50 mg/ml) and pseudotyped using 5 mg VSV-G or AD8 toreplace Env (29, 32) with FuGENE 6 transfection reagent (Roche AppliedScience, Nutley, NJ) (30, 33). Twelve hours after transfection, the cells werewashed with 5 ml PBS, and 4 ml complete medium was added. Con-trol viruses were produced in the presence of equivalent concentrationsof control vector. Three days posttransfection, virus stocks were preparedby collecting culture supernatants and passing them through 0.45-mm-pore-size cellulose acetate syringe filters (Millex 0.45-mmfilters; Millipore,Bedford, MA) and after adding FBS (10%) stored at 280˚C.
Viral titers were determined by infection of the human Jurkat T or U937cells, and p24gag Ag was measured by capture ELISA at the Viral andMolecular Core Services, Center for AIDS Research, University of Penn-sylvania School of Medicine. For infection studies, human PBMCs wereisolated from normal, HIV-1–negative donors as described above. PBMCs(2 3 105 cells/well) were mock infected (with media from the cell culturesused to grow cells) or infected with cell-free HIV-1 virus at a concentrationof 100 tissue culture-infective dose50/10
6 cells/ml. After 4–6 h of incubationat 37˚C, cells were gently washed, resuspended with complete medium, andmaintained for the indicated time periods. Culture supernatants and cellswere then harvested for p24 Gag by ELISA and by flow cytometry as de-scribed below.
IFN-g ELISPOT assay
ELISPOT was performed as described previously (42, 43). Briefly, 96-wellELISPOT plates (Millipore) were coated with anti-human IFN-g capture Ab(R&D Systems) and incubated for 24 h at 4˚C. The following day, plateswere washed with PBS and blocked for 2 h with 1% BSA. Two hundredthousand PBMCs from HIV-1–positive patient were plated in triplicate andstimulated with overlappingHIV-1 peptide consisting of 15-mer overlappingby nine amino acids and spanning the length of the appropriate protein (Gagor Env). Autologous monocytes with and without PD-L1 depletion usingspecific siRNA were then added to the corresponding wells and incubatedovernight at 37˚C in 5% CO2 in the presence of media alone (negativecontrol), media with Con A (positive control), or media with peptide pools(Gag/Env) (10mg/ml). As an additional control, anti–PD-L1was added at 10mg/ml. After 24 h, the cells werewashed and then incubated for an additional24 h at 4˚C with biotinylated anti-human IFN-g Ab (R&D Systems).Streptavidin–alkaline phosphatase (R&D Systems) was added to each wellafter washing, and then, the plate was incubated for 2 h at room temperature.The plate was then washed, and 5-bromo-4-chloro-39-indolylphosphate p-toluidine salt and NBT chloride (chromogen color reagent; R&D Systems)were added. Last, the plate was rinsed with distilled water, dried at room
temperature, and spot-forming units were quantified by an automated ELI-SPOT reader (CTL Limited, Bowling Green, OH), and the raw values werenormalized to spot-forming units per million cells.
Flow cytometry and CFSE labeling
Anti-human Abs were used in cell surface staining and intracellularstaining: CD3 (clone UCHT1; BD Pharmingen, San Diego, CA), CD4(clone RPA-T4; BD Pharmingen), CD8 (clone RPA-T8; BD Pharmingen),CD14 (clone M5E2; BD Pharmingen), CD80 (L307.4; BD Pharmingen),CD86 (clone IT2.2; Pharmingen), PD-L1 (clone MIH1; eBioscience, SanDiego, CA), and PD-L2 (clone MIH18; eBioscience). The PD-L1–blockingmAb (10F.9G2) was purchased from BioLegend (San Diego, CA). Forstaining, the experimental cells were incubated with combinations offluorochrome-labeled Abs for 30 min on ice. Cell surface staining wasperformed at 4˚C in FACS buffer (1% FCS, PBS, and 2 mM EDTA;Invitrogen). For intracellular staining, cells were permeabilized using BDFixPerm (42) following staining. The percentages of cells expressingintracytoplasmic HIV-1 Gag-related products were evaluated using KC57-RD1/PE– or KC57/FITC-conjugated anti–HIV-1 Gag mAb (BeckmanCoulter, Miami, FL). Electronic compensation was conducted with Abcapture beads (BD Biosciences) stained separately with individual mAbs.Forward light scatter area versus forward light scatter height was used togate out the cell aggregates. In addition, ViViD dye staining was used toexclude dead and dying cells (44). Cells were analyzed with a modifiedLSRII flow cytometry (BD Immunocytometry Systems, San Jose, CA) orCoulter EPICS Flow Cytometer (Beckman Coulter, Miami, FL) usingFlowJo software (Tree Star, Ashland, OR).
For CFSE labeling, T cells (0.5–2 3 106) were labeled with 0.2–1 mMCFSE (Invitrogen) just before stimulation (44). Cell division accompaniedby CFSE dilution was analyzed by flow cytometry. Cell-free culturesupernatants were stored at 280˚C, thawed, and subsequently analyzed forthe presence of IFN-g, IL-2, IL-12, and IL-10 by ELISA (R&D Systems),according to the manufacturer’s instructions. ELISA for phospho- andtotal Akt were performed using PathScan kits (7160 and 7170) from CellSignaling Technology (Danvers, MA); the phospho-Akt–specific ELISAdetects Akt, which is phosphorylated on Ser473. Although equal samplevolumes were added to the wells, a correction for differing protein con-centrations was made before the calculation of relative Akt.
Western blotting
Infected and mock-infected cells were lysed in 60-mm dishes for 5 min at4˚C by using 250 ml Nonidet P-40 lysis buffer (Invitrogen) supplementedwith a phosphatase inhibitor mixture (PhosSTOP) and a protease inhibitormixture (Complete) as directed by the manufacturer (Roche AppliedScience). Lysates were collected and spun at 10,000 rpm for 5 min at 4˚C,and then, 100 ml supernatant was added to 20 ml 63 sample buffer(Invitrogen) for 12% SDS-PAGE using equal volumes of lysate. Afterward,proteins were transferred to nitrocellulose using the iBlot Dry BlottingSystem (Invitrogen), immunodetection was performed with the SNAP i.d.Protein Detection System (Millipore) using specific mouse antiserum, andthe expressed proteins were visualized with HRP-conjugated goat anti-mouse IgG using an ECL detection system (Amersham Biosciences, Pis-cataway, NJ). Primary Abs were diluted in 5% (w/v) BSA (fraction V)-TBST as recommended by the Ab manufacturer. PI3K and Akt i.v.inhibitors were used and were purchased from Invitrogen and Calbiochem(San Diego, CA), respectively. Detection Abs for Akt and phospho-AktSer473 were purchased from Cell Signaling Technology, and the Ab againstb-actin (1:5000) was purchased from Santa Cruz Biotechnology.
Statistical analysis
Data was collected from cellular assays and presented as the mean 6 SD,which was calculated from triplicate wells of pooled samples from eachexperimental group. Prior to all statistical analysis, the normality of thedata was confirmed with Levine’s test. An analysis between groups wasperformed using an independent samples t test. Comparisons among threegroups were performed with ANOVA with a post hoc Fisher’s Least Sig-nificant Difference test to correct for multiple comparisons betweengroups. In each case, p # 0.05 was considered to be significant. All sta-tistical analysis was carried out using SPSS.
ResultsHIV infection increases PD-L1/2 on APCs
It is has been reported that different viruses induce unique com-binations of costimulatory ligands that determine the fate of antiviralactivity (45). In this manner, viruses that specifically induce ex-pression of PD-L1/2 (PD-Ls) on APCs might downregulate the
2934 HIV INFECTION REGULATES PD-1 LIGANDS ON APCs
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from
adaptive immune response and favor viral persistence. Althoughthe expression of PD-1 on HIV-specific CD4+ and CD8+ T cells hasbeen extensively investigated, neither the expression nor the sourceof PD-Ls in activating PD-1 on HIV-specific T cells is known. Forexample, we still do not know whether HIV infection can directlyinfluence the expression of not only PD-1 but also the PD-Ls. If thisis the case, then the source of the PD-Ls, which activate PD-1,remains to be characterized. Although very few studies have di-rectly investigated the importance of PD-L1 in HIV-induced im-mune suppression, some recent work suggests a clear association(26, 27). For example, Trabattoni et al. (29) reported that the ex-
pression of the PD-Ls in HIV-positive patients is directly correlatedwith viral load and inversely correlated with CD4+ T cell counts,andMeier et al. (41) illustrated that uridine-richHIV-derived siRNAis sufficient to stimulate PD-Ls expression through a MyD88-dependent mechanism. Unfortunately, the latter experiment failedto show whether HIV-derived siRNA or MyD88 is required for thePD-Ls’ increase in an appropriate infection setting. To determinewhether HIV-1 infection modulates the expression of PD-Ls, wecompared the expression of PD-L1 and PD-L2 on infected (CD4+
CD14+p24gag+) and uninfected (CD4+CD14+p24gag2) monocytesfromHIV-1–infected patients (Fig. 1, Table I). Both PD-L1 and PD-
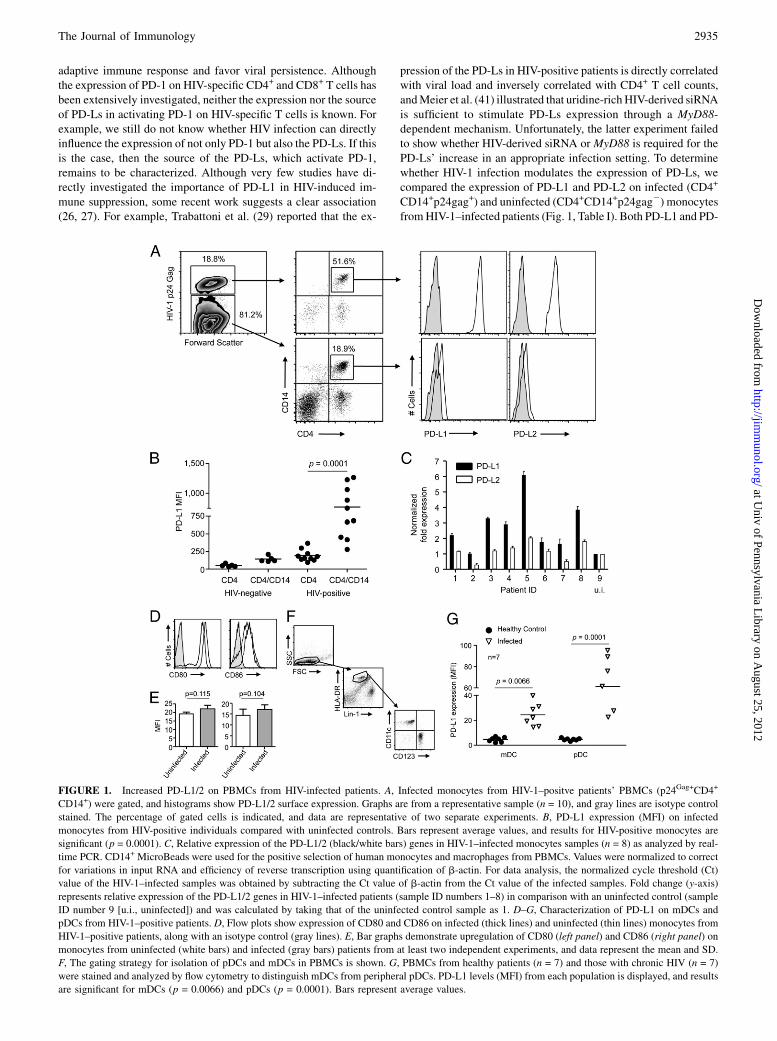
FIGURE 1. Increased PD-L1/2 on PBMCs from HIV-infected patients. A, Infected monocytes from HIV-1–positve patients’ PBMCs (p24Gag+CD4+
CD14+) were gated, and histograms show PD-L1/2 surface expression. Graphs are from a representative sample (n = 10), and gray lines are isotype control
stained. The percentage of gated cells is indicated, and data are representative of two separate experiments. B, PD-L1 expression (MFI) on infected
monocytes from HIV-positive individuals compared with uninfected controls. Bars represent average values, and results for HIV-positive monocytes are
significant (p = 0.0001). C, Relative expression of the PD-L1/2 (black/white bars) genes in HIV-1–infected monocytes samples (n = 8) as analyzed by real-
time PCR. CD14+ MicroBeads were used for the positive selection of human monocytes and macrophages from PBMCs. Values were normalized to correct
for variations in input RNA and efficiency of reverse transcription using quantification of b-actin. For data analysis, the normalized cycle threshold (Ct)
value of the HIV-1–infected samples was obtained by subtracting the Ct value of b-actin from the Ct value of the infected samples. Fold change (y-axis)
represents relative expression of the PD-L1/2 genes in HIV-1–infected patients (sample ID numbers 1–8) in comparison with an uninfected control (sample
ID number 9 [u.i., uninfected]) and was calculated by taking that of the uninfected control sample as 1. D–G, Characterization of PD-L1 on mDCs and
pDCs from HIV-1–positive patients. D, Flow plots show expression of CD80 and CD86 on infected (thick lines) and uninfected (thin lines) monocytes from
HIV-1–positive patients, along with an isotype control (gray lines). E, Bar graphs demonstrate upregulation of CD80 (left panel) and CD86 (right panel) on
monocytes from uninfected (white bars) and infected (gray bars) patients from at least two independent experiments, and data represent the mean and SD.
F, The gating strategy for isolation of pDCs and mDCs in PBMCs is shown. G, PBMCs from healthy patients (n = 7) and those with chronic HIV (n = 7)
were stained and analyzed by flow cytometry to distinguish mDCs from peripheral pDCs. PD-L1 levels (MFI) from each population is displayed, and results
are significant for mDCs (p = 0.0066) and pDCs (p = 0.0001). Bars represent average values.
The Journal of Immunology 2935
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from
L2 were expressed at much higher levels on infected cells (Fig. 1A)when compared with uninfected monocytes. This was further vali-datedwhenwe comparedwith totalmonocytes fromhealthy donors tothose fromHIV-infected patients (Fig. 1B). To determinewhether thisincreased expression of PD-L1 and L2 on infected monocytes is aposttranslation effect, RT-PCR analysis of total RNA extracted fromthe HIV-1–infected purified CD14+ monocytes was performed. Asshown in Fig. 1C, HIV infection also increased the amount of PD-L1and L2 mRNA transcript when compared with levels from uninfectedcontrol cells (patient ID number 9; uninfected). Taken together, thesedata demonstrate that HIV infection significantly increased both thetranscription and surface expression of PD-L1/2 on monocytes fromHIV-infected individuals.It is known that numbers of circulating myeloid DCs (mDCs) and
plasmacytoid DCs (pDCs), the two main subsets of blood DCs, aremodified during different viral infections (46). The capacity ofDCs to stimulate T cells depends on the net costimulatory signaldelivered by DCs to T cells. Increased costimulatory marker ex-pression (such as CD83, CD86, and CD40) can facilitate T cellactivation (47). Thus, in addition to CD14+ cells, we also analyzedPD-L1 expression on DCs. Monocytic CD80 and CD86 expressionin PBMCs was determined by dual-color staining analysis both inHIV-1 patients and uninfected controls. Most CD14+ monocytesexpressed CD80 and CD86 (Fig. 1D), and there was no statisticallysignificant difference in these levels (Fig. 1E), as determinedby mean fluorescence intensity (MFI) values. We next examinedwhether this could also be the case during HIV infection by ana-lyzing the frequency of mDCs and pDCs in seven HIV-positivepatients. PBMCs were stained, acquired, and gated during analy-sis to exclude debris, and most polymorphonuclear cells and DCsare identified as negative for the lineage markers CD3, CD14,CD16, CD19, CD20, and CD56 and positive for HLA-DR. Fur-thermore, high-lineage low DCs were then phenotyped as mye-loid (CD11chigh,CD123low) or plasmacytoid (CD11clow,CD123high)(Fig. 1F). As shown in Fig. 1G, MFI of PD-L1 on peripheral bloodpDCs and mDCs from HIV-infected patients was significantlygreater than that from HIV-uninfected patients. Taken together,these data show that HIV-1 infection increases the expression ofPD-L1 on blood DCs from HIV-infected patients.Although it appears that upregulation of PD-1 correlates with dis-
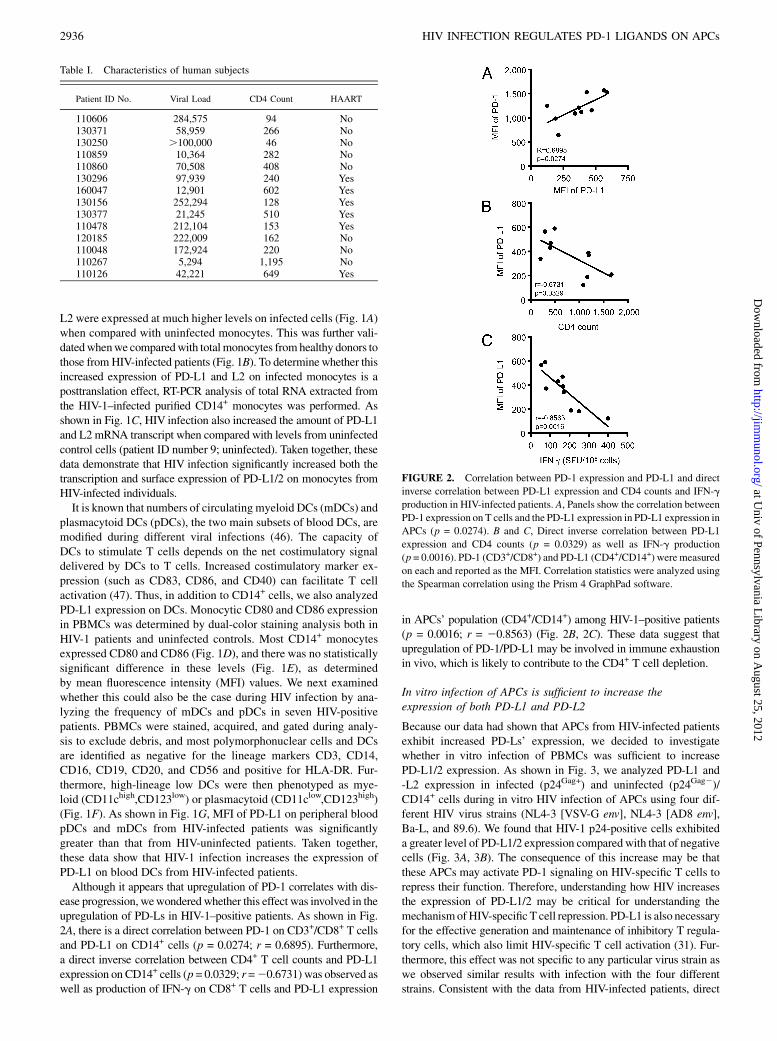
ease progression, wewonderedwhether this effect was involved in theupregulation of PD-Ls in HIV-1–positive patients. As shown in Fig.2A, there is a direct correlation between PD-1 on CD3+/CD8+ T cellsand PD-L1 on CD14+ cells (p = 0.0274; r = 0.6895). Furthermore,a direct inverse correlation between CD4+ T cell counts and PD-L1expression on CD14+ cells (p = 0.0329; r =20.6731) was observed aswell as production of IFN-g on CD8+ T cells and PD-L1 expression
in APCs’ population (CD4+/CD14+) among HIV-1–positive patients(p = 0.0016; r = 20.8563) (Fig. 2B, 2C). These data suggest thatupregulation of PD-1/PD-L1 may be involved in immune exhaustionin vivo, which is likely to contribute to the CD4+ T cell depletion.
In vitro infection of APCs is sufficient to increase theexpression of both PD-L1 and PD-L2
Because our data had shown that APCs from HIV-infected patientsexhibit increased PD-Ls’ expression, we decided to investigatewhether in vitro infection of PBMCs was sufficient to increasePD-L1/2 expression. As shown in Fig. 3, we analyzed PD-L1 and-L2 expression in infected (p24Gag+) and uninfected (p24Gag2)/CD14+ cells during in vitro HIV infection of APCs using four dif-ferent HIV virus strains (NL4-3 [VSV-G env], NL4-3 [AD8 env],Ba-L, and 89.6). We found that HIV-1 p24-positive cells exhibiteda greater level of PD-L1/2 expression compared with that of negativecells (Fig. 3A, 3B). The consequence of this increase may be thatthese APCs may activate PD-1 signaling on HIV-specific T cells torepress their function. Therefore, understanding how HIV increasesthe expression of PD-L1/2 may be critical for understanding themechanismofHIV-specific T cell repression. PD-L1 is also necessaryfor the effective generation and maintenance of inhibitory T regula-tory cells, which also limit HIV-specific T cell activation (31). Fur-thermore, this effect was not specific to any particular virus strain aswe observed similar results with infection with the four differentstrains. Consistent with the data from HIV-infected patients, direct
FIGURE 2. Correlation between PD-1 expression and PD-L1 and direct
inverse correlation between PD-L1 expression and CD4 counts and IFN-g
production in HIV-infected patients. A, Panels show the correlation between
PD-1 expression on T cells and the PD-L1 expression in PD-L1 expression in
APCs (p = 0.0274). B and C, Direct inverse correlation between PD-L1
expression and CD4 counts (p = 0.0329) as well as IFN-g production
(p = 0.0016). PD-1 (CD3+/CD8+) and PD-L1 (CD4+/CD14+) weremeasured
on each and reported as the MFI. Correlation statistics were analyzed using
the Spearman correlation using the Prism 4 GraphPad software.
Table I. Characteristics of human subjects
Patient ID No. Viral Load CD4 Count HAART
110606 284,575 94 No130371 58,959 266 No130250 .100,000 46 No110859 10,364 282 No110860 70,508 408 No130296 97,939 240 Yes160047 12,901 602 Yes130156 252,294 128 Yes130377 21,245 510 Yes110478 212,104 153 Yes120185 222,009 162 No110048 172,924 220 No110267 5,294 1,195 No110126 42,221 649 Yes
2936 HIV INFECTION REGULATES PD-1 LIGANDS ON APCs
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from
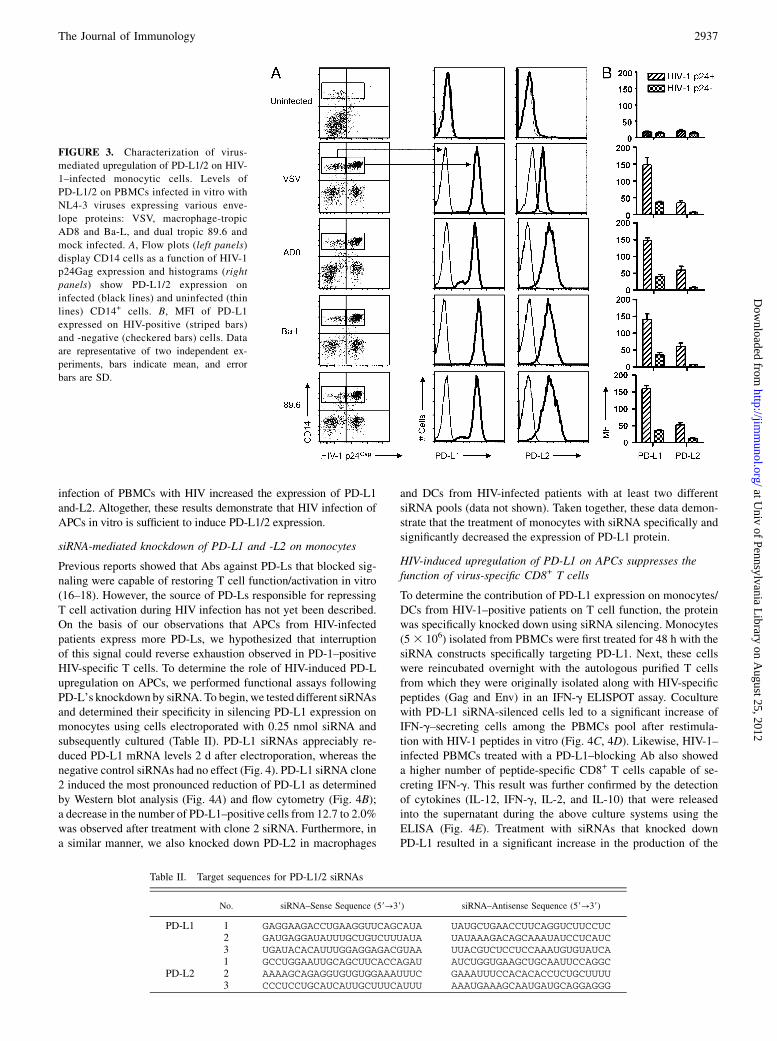
infection of PBMCs with HIV increased the expression of PD-L1and-L2. Altogether, these results demonstrate that HIV infection ofAPCs in vitro is sufficient to induce PD-L1/2 expression.
siRNA-mediated knockdown of PD-L1 and -L2 on monocytes
Previous reports showed that Abs against PD-Ls that blocked sig-naling were capable of restoring T cell function/activation in vitro(16–18). However, the source of PD-Ls responsible for repressingT cell activation during HIV infection has not yet been described.On the basis of our observations that APCs from HIV-infectedpatients express more PD-Ls, we hypothesized that interruptionof this signal could reverse exhaustion observed in PD-1–positiveHIV-specific T cells. To determine the role of HIV-induced PD-Lupregulation on APCs, we performed functional assays followingPD-L’s knockdown by siRNA. To begin, we tested different siRNAsand determined their specificity in silencing PD-L1 expression onmonocytes using cells electroporated with 0.25 nmol siRNA andsubsequently cultured (Table II). PD-L1 siRNAs appreciably re-duced PD-L1 mRNA levels 2 d after electroporation, whereas thenegative control siRNAs had no effect (Fig. 4). PD-L1 siRNA clone2 induced the most pronounced reduction of PD-L1 as determinedby Western blot analysis (Fig. 4A) and flow cytometry (Fig. 4B);a decrease in the number of PD-L1–positive cells from 12.7 to 2.0%was observed after treatment with clone 2 siRNA. Furthermore, ina similar manner, we also knocked down PD-L2 in macrophages
and DCs from HIV-infected patients with at least two differentsiRNA pools (data not shown). Taken together, these data demon-strate that the treatment of monocytes with siRNA specifically andsignificantly decreased the expression of PD-L1 protein.
HIV-induced upregulation of PD-L1 on APCs suppresses thefunction of virus-specific CD8+ T cells
To determine the contribution of PD-L1 expression on monocytes/DCs from HIV-1–positive patients on T cell function, the proteinwas specifically knocked down using siRNA silencing. Monocytes(53 106) isolated from PBMCs were first treated for 48 h with thesiRNA constructs specifically targeting PD-L1. Next, these cellswere reincubated overnight with the autologous purified T cellsfrom which they were originally isolated along with HIV-specificpeptides (Gag and Env) in an IFN-g ELISPOT assay. Coculturewith PD-L1 siRNA-silenced cells led to a significant increase ofIFN-g–secreting cells among the PBMCs pool after restimula-tion with HIV-1 peptides in vitro (Fig. 4C, 4D). Likewise, HIV-1–infected PBMCs treated with a PD-L1–blocking Ab also showeda higher number of peptide-specific CD8+ T cells capable of se-creting IFN-g. This result was further confirmed by the detectionof cytokines (IL-12, IFN-g, IL-2, and IL-10) that were releasedinto the supernatant during the above culture systems using theELISA (Fig. 4E). Treatment with siRNAs that knocked downPD-L1 resulted in a significant increase in the production of the
FIGURE 3. Characterization of virus-
mediated upregulation of PD-L1/2 on HIV-
1–infected monocytic cells. Levels of
PD-L1/2 on PBMCs infected in vitro with
NL4-3 viruses expressing various enve-
lope proteins: VSV, macrophage-tropic
AD8 and Ba-L, and dual tropic 89.6 and
mock infected. A, Flow plots (left panels)
display CD14 cells as a function of HIV-1
p24Gag expression and histograms (right
panels) show PD-L1/2 expression on
infected (black lines) and uninfected (thin
lines) CD14+ cells. B, MFI of PD-L1
expressed on HIV-positive (striped bars)
and -negative (checkered bars) cells. Data
are representative of two independent ex-
periments, bars indicate mean, and error
bars are SD.
Table II. Target sequences for PD-L1/2 siRNAs
No. siRNA–Sense Sequence (59→39) siRNA–Antisense Sequence (59→39)
PD-L1 1 GAGGAAGACCUGAAGGUUCAGCAUA UAUGCUGAACCUUCAGGUCUUCCUC2 GAUGAGGAUAUUUGCUGUCUUUAUA UAUAAAGACAGCAAAUAUCCUCAUC3 UGAUACACAUUUGGAGGAGACGUAA UUACGUCUCCUCCAAAUGUGUAUCA1 GCCUGGAAUUGCAGCUUCACCAGAU AUCUGGUGAAGCUGCAAUUCCAGGC
PD-L2 2 AAAAGCAGAGGUGUGUGGAAAUUUC GAAAUUUCCACACACCUCUGCUUUU3 CCCUCCUGCAUCAUUGCUUUCAUUU AAAUGAAAGCAAUGAUGCAGGAGGG
The Journal of Immunology 2937
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

proinflammatory cytokines IFN-g, IL-2, and IL-12, whereas levelsof the anti-inflammatory IL-10 were significantly decreased. Togain more insight into the levels of various cytokines produced bythe different cell subsets upon stimulation with PD-L knockdownmonocytes, coculture cells were collected and analyzed by in-tracellular flow cytometry for delineation of cell-specific contri-butions of the observed cytokines. PD-L knockdown evidentlyelevated IL-2 and IFN-g production by CD4+ as well as CD8+
T cells, whereas CD11c+ and CD14+ produced IL-12. Furthermore,production of IL-10 by CD14+ cells was decreased (data notshown). Altogether, these data confirm previous results showingthat increased PD-1 signaling results in T cell suppression anddemonstrate that siRNA-mediated silencing of PD-L1 onAPCswassufficient for significant improvement in T cell immune function.
Another hallmark of T cell function is the capacity to mount Ag-specific proliferative responses. Because proinflammatory cytokineresponses were markedly improved in T cells from HIV-infectedpatients after PD-L1 silencing in peptide-presenting APCs, wenext analyzed the proliferative ability of HIV-specific CD8+ T cellsfrom HIV-infected PBMCs upon siRNA treatment (Fig. 5). PurifiedT cells from infected cells were labeled with CFSE and mixed withautologous monocytes (at a ratio of 1:10) that were untreated orelectroporated with PD-L1 or control siRNA. Mixed cells wereincubated in medium alone or stimulated with Gag or Env pep-tides, and 5 d later, cells were stained and analyzed by flowcytometry. Although only modest peptide-specific proliferativeCD4+ and CD8+ T cell responses were observed in the absence ofsiRNA treatment, knockdown of PD-L1 on APCs resulted in
FIGURE 4. PD-L1–mediated functional suppression of HIV-specific CD8+ T cells. PD-L1–specific siRNA-silencing (A) Western blot analysis for PD-L1
content of whole-cell lysates from PBMCs 72 h postelectroporation using the Amaxa nucleofector system with the indicated siRNAs at a concentration of
25nM. The lower panel shows b-actin levels as a control. B, Representative FACS plots showing PD-L1 expression on CD14+ cells 5 d following siRNA
treatment. Percentages are from gates, and data are representative of two independent experiments. Monocytes and T cells were isolated from the PBMCs of
an HIV-infected donor. Monocytes (5 3 106) were electroporated with 50 nmol PD-L1 or scrambled (control) siRNA. Two days later, the transfected
monocytes were recombined with the purified T cells (at a ratio of 1:10), cocultured in the presence of Gag and Env peptides, and IFN-g secretion was
measured 3 d later by ELISPOT using T cell Gag-specific (C) and Env-specific (D) secretion of IFN-g in vitro when cocultured with PD-L1 siRNA-silenced
monocytes or in the presence of anti–PD-L1–blocking Abs. E, Blockade of PD-L1 can restore immune cytokine production following PD-L1 silencing.
Env-stimulated samples were tested for cytokine responses by ICS of CD3+/ CD8+ or CD3+CD4+ T cells for IFN-g or IL-2 and CD14+ cells for the IL-12
and IL-10. The percentage for the specific cytokines is shown. These data are representative of at least two independent experiments.
2938 HIV INFECTION REGULATES PD-1 LIGANDS ON APCs
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from
a marked increase in the number of responding T cells (Fig. 5A,5B); CD4+ and CD8+ T cell responses were 3- and 4-fold higherfor Gag stimulation and 4.4- and 2.7-fold for Env (Fig. 5C). Thesedata show a clear increase in the proliferation of HIV-specificCD8+ T cells in the presence of PD-L1 knockdown on APCs ascompared with expansion following stimulation with peptides inuntreated cells. Altogether, our findings confirm previous reportsthat PD-1/PD-L1 signaling inhibits HIV-specific CD8+ T cell pro-liferation and that the blockade of the PD-L signal pathway canrescue T cell function.
HIV infection of APCs induces PD-L1/2 through the expressionof its accessory genes
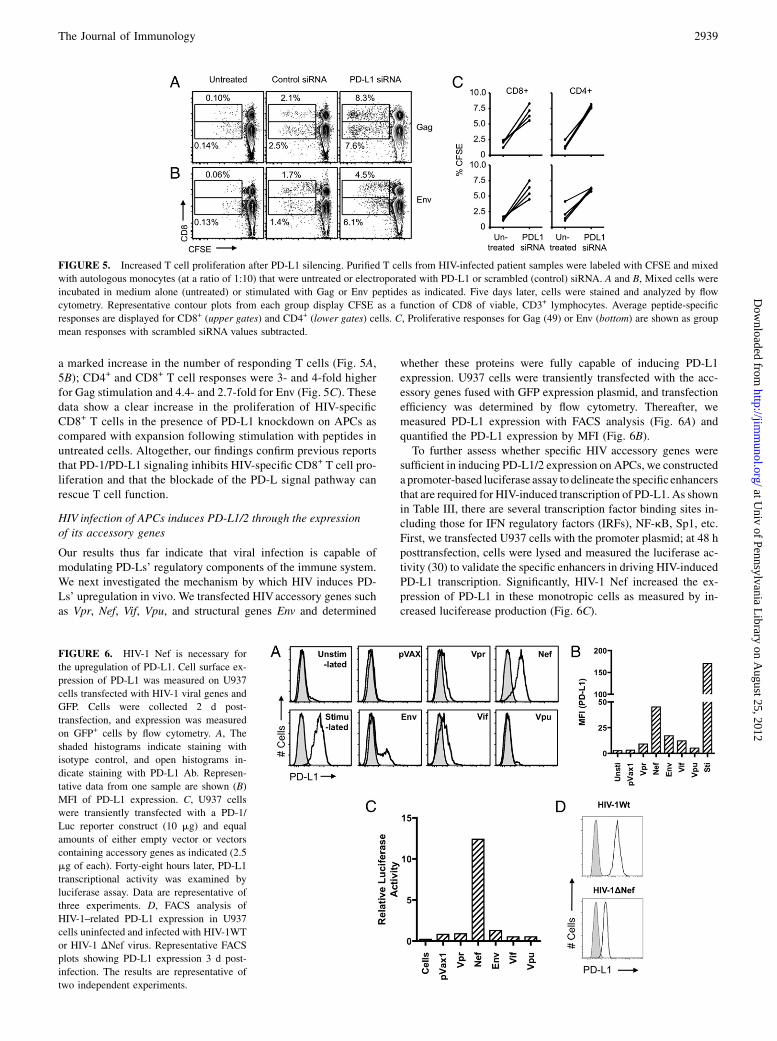
Our results thus far indicate that viral infection is capable ofmodulating PD-Ls’ regulatory components of the immune system.We next investigated the mechanism by which HIV induces PD-Ls’ upregulation in vivo. We transfected HIVaccessory genes suchas Vpr, Nef, Vif, Vpu, and structural genes Env and determined
whether these proteins were fully capable of inducing PD-L1expression. U937 cells were transiently transfected with the acc-essory genes fused with GFP expression plasmid, and transfectionefficiency was determined by flow cytometry. Thereafter, wemeasured PD-L1 expression with FACS analysis (Fig. 6A) andquantified the PD-L1 expression by MFI (Fig. 6B).To further assess whether specific HIV accessory genes were
sufficient in inducing PD-L1/2 expression on APCs, we constructeda promoter-based luciferase assay to delineate the specificenhancersthat are required for HIV-induced transcription of PD-L1. As shownin Table III, there are several transcription factor binding sites in-cluding those for IFN regulatory factors (IRFs), NF-kB, Sp1, etc.First, we transfected U937 cells with the promoter plasmid; at 48 hposttransfection, cells were lysed and measured the luciferase ac-tivity (30) to validate the specific enhancers in driving HIV-inducedPD-L1 transcription. Significantly, HIV-1 Nef increased the ex-pression of PD-L1 in these monotropic cells as measured by in-creased luciferease production (Fig. 6C).
FIGURE 6. HIV-1 Nef is necessary for
the upregulation of PD-L1. Cell surface ex-
pression of PD-L1 was measured on U937
cells transfected with HIV-1 viral genes and
GFP. Cells were collected 2 d post-
transfection, and expression was measured
on GFP+ cells by flow cytometry. A, The
shaded histograms indicate staining with
isotype control, and open histograms in-
dicate staining with PD-L1 Ab. Represen-
tative data from one sample are shown (B)
MFI of PD-L1 expression. C, U937 cells
were transiently transfected with a PD-1/
Luc reporter construct (10 mg) and equal
amounts of either empty vector or vectors
containing accessory genes as indicated (2.5
mg of each). Forty-eight hours later, PD-L1
transcriptional activity was examined by
luciferase assay. Data are representative of
three experiments. D, FACS analysis of
HIV-1–related PD-L1 expression in U937
cells uninfected and infected with HIV-1WT
or HIV-1 DNef virus. Representative FACS
plots showing PD-L1 expression 3 d post-
infection. The results are representative of
two independent experiments.
FIGURE 5. Increased T cell proliferation after PD-L1 silencing. Purified T cells from HIV-infected patient samples were labeled with CFSE and mixed
with autologous monocytes (at a ratio of 1:10) that were untreated or electroporated with PD-L1 or scrambled (control) siRNA. A and B, Mixed cells were
incubated in medium alone (untreated) or stimulated with Gag or Env peptides as indicated. Five days later, cells were stained and analyzed by flow
cytometry. Representative contour plots from each group display CFSE as a function of CD8 of viable, CD3+ lymphocytes. Average peptide-specific
responses are displayed for CD8+ (upper gates) and CD4+ (lower gates) cells. C, Proliferative responses for Gag (49) or Env (bottom) are shown as group
mean responses with scrambled siRNA values subtracted.
The Journal of Immunology 2939
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from
Furthermore, we investigated whether Nef is necessary to inducethe expression of PD-L1. To address this question, we deleted theNef gene and developed a pseudoviral system as a tool to in-vestigate the PD-L1 expression. We infected purified monocyteswith this HIV-1 pseudovirus and measured the expression of PD-L1/2 at 96 h postinfection. Specifically, cells were infected withVSV-G pseudotyped HIV-1WT or HIV-1DNef, with 50 ng CAp24equivalent of virus/105 cells. Loss of Nef led to an attenuation ofHIV-mediated PD-L1 upregulation upon infection (Fig. 6D). Thiseffect did not appear to be a consequence of efficiency of infectionbecause the mutant viruses all exhibited similar levels of p24Gag
production (data not shown). Therefore, in the context of HIVinfection of target cells, Nef activity may be a primary cause ofthe surface expression of PD-L1 on APCs. Taken together, theseexperiments strongly suggest that HIV-1 Nef genes are necessaryfor HIV to induce PD-Ls upon infection.
PI3K/Akt activity is important for PD-L1 upregulation
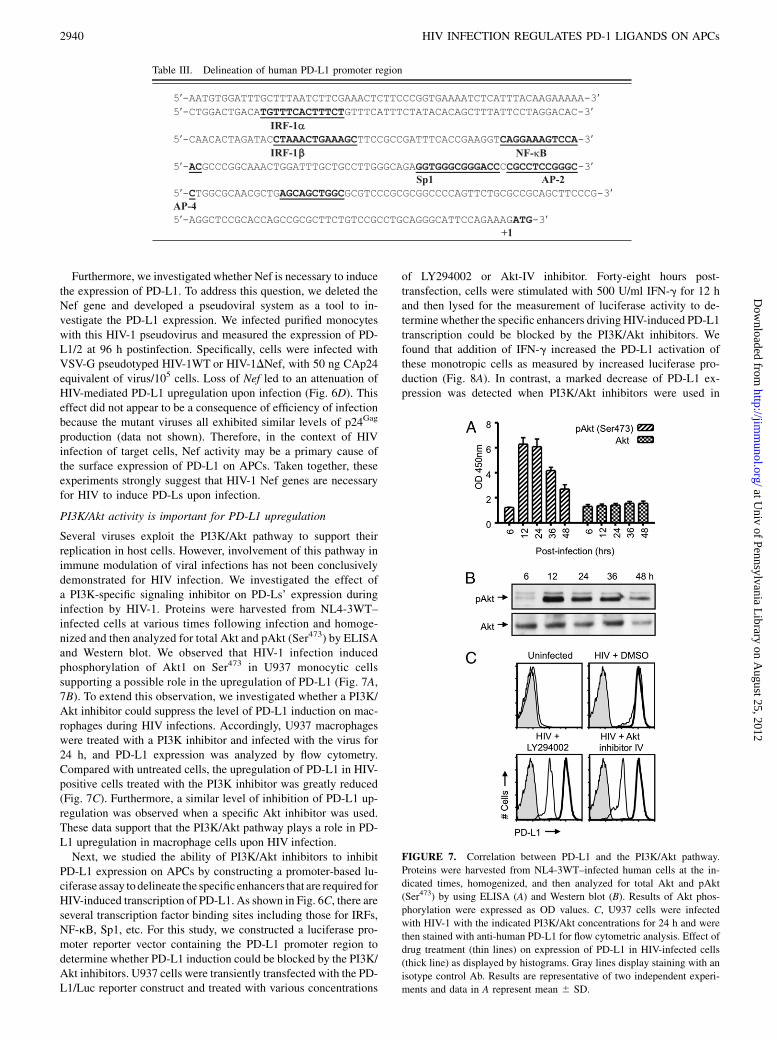
Several viruses exploit the PI3K/Akt pathway to support theirreplication in host cells. However, involvement of this pathway inimmune modulation of viral infections has not been conclusivelydemonstrated for HIV infection. We investigated the effect ofa PI3K-specific signaling inhibitor on PD-Ls’ expression duringinfection by HIV-1. Proteins were harvested from NL4-3WT–infected cells at various times following infection and homoge-nized and then analyzed for total Akt and pAkt (Ser473) by ELISAand Western blot. We observed that HIV-1 infection inducedphosphorylation of Akt1 on Ser473 in U937 monocytic cellssupporting a possible role in the upregulation of PD-L1 (Fig. 7A,7B). To extend this observation, we investigated whether a PI3K/Akt inhibitor could suppress the level of PD-L1 induction on mac-rophages during HIV infections. Accordingly, U937 macrophageswere treated with a PI3K inhibitor and infected with the virus for24 h, and PD-L1 expression was analyzed by flow cytometry.Compared with untreated cells, the upregulation of PD-L1 in HIV-positive cells treated with the PI3K inhibitor was greatly reduced(Fig. 7C). Furthermore, a similar level of inhibition of PD-L1 up-regulation was observed when a specific Akt inhibitor was used.These data support that the PI3K/Akt pathway plays a role in PD-L1 upregulation in macrophage cells upon HIV infection.Next, we studied the ability of PI3K/Akt inhibitors to inhibit
PD-L1 expression on APCs by constructing a promoter-based lu-ciferase assay to delineate the specificenhancers that are required forHIV-induced transcription of PD-L1. As shown in Fig. 6C, there areseveral transcription factor binding sites including those for IRFs,NF-kB, Sp1, etc. For this study, we constructed a luciferase pro-moter reporter vector containing the PD-L1 promoter region todetermine whether PD-L1 induction could be blocked by the PI3K/Akt inhibitors. U937 cells were transiently transfected with the PD-L1/Luc reporter construct and treated with various concentrations
of LY294002 or Akt-IV inhibitor. Forty-eight hours post-transfection, cells were stimulated with 500 U/ml IFN-g for 12 hand then lysed for the measurement of luciferase activity to de-terminewhether the specific enhancers driving HIV-induced PD-L1transcription could be blocked by the PI3K/Akt inhibitors. Wefound that addition of IFN-g increased the PD-L1 activation ofthese monotropic cells as measured by increased luciferase pro-duction (Fig. 8A). In contrast, a marked decrease of PD-L1 ex-pression was detected when PI3K/Akt inhibitors were used in
FIGURE 7. Correlation between PD-L1 and the PI3K/Akt pathway.
Proteins were harvested from NL4-3WT–infected human cells at the in-
dicated times, homogenized, and then analyzed for total Akt and pAkt
(Ser473) by using ELISA (A) and Western blot (B). Results of Akt phos-
phorylation were expressed as OD values. C, U937 cells were infected
with HIV-1 with the indicated PI3K/Akt concentrations for 24 h and were
then stained with anti-human PD-L1 for flow cytometric analysis. Effect of
drug treatment (thin lines) on expression of PD-L1 in HIV-infected cells
(thick line) as displayed by histograms. Gray lines display staining with an
isotype control Ab. Results are representative of two independent experi-
ments and data in A represent mean 6 SD.
Table III. Delineation of human PD-L1 promoter region
2940 HIV INFECTION REGULATES PD-1 LIGANDS ON APCs
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

combination with IFN-g stimulation. Taken together, these ex-periments demonstrated a role for PI3K/Akt signaling pathways inthe regulation of the PD-L1 promoter and their necessity for HIVinfection to induce PD-L1 in APCs.Because phospho-Akt was involved in PD-L1 expression during
HIV infection, the presence of this protein may contribute to theexpression of PD-L1 in these cells. To further validate our resultsfor the inhibitor experiments, we tested adapted Akt1 siRNA con-structs targeting Akt into macrophages. U937 cells were transfectedwith Akt1 siRNA or control siRNA, and 72 h after transfection,whole-cell lysates were prepared for Western blot analysis. Thistreatment resulted in a 50–60% reduction in Akt1 protein ex-pression levels; however, Akt2 levels were not affected by Akt1siRNA (Fig. 8B), further demonstrating the specificity of this ef-
fect. To better explore the role of Akt in regulating PD-L1 ex-pression, we used U937 cells that are silenced for active Akt andhave been infected with HIV. When Akt expression was sup-pressed by siRNA, PD-L1 levels were blocked significantly uponHIV infection (Fig. 8C). To our knowledge, these data are the firstto demonstrate that downregulation of Akt leads to decreasedexpression of PD-L1. Collectively, the data summarized in thispaper indicate that PI3K/Akt signaling regulates PD-L1 expres-sion and that Akt is a regulator of PD-L1 expression that is tar-geted by HIV during infection.
DiscussionA hallmark of chronic viral infections, including HIV infection ofhumans, is the inability of the host to sustain effector immune
FIGURE 9. Schematic of the sig-
naling pathway depicting possible
sites of action by PD-1/PD-L1.
FIGURE 8. PI3K/Akt pathway plays a critical role in PD-L1 expression. A and B, U937 cells were transiently transfected with a PD-L1/Luc reporter
construct and treated with the indicated concentrations of LY294002 or Akt-IV inhibitor. Forty-eight hours posttransfection, cells were stimulated with
500 U/ml IFN-g for 6 h, and PD-L1 transcriptional activity was examined by luciferase assay. Data were normalized to luciferase activity in DMSO-treated
cells. C, Akt1 siRNA transfection of U937 cells resulted in decreased Akt1 but not Akt2 protein levels. U937 cells were transfected with Akt1 siRNA or
control siRNA, and 72 h after transfection, whole-cell lysates were prepared for Western blot analysis using the indicated Abs. D, Representative histograms
illustrating IFN-g–induced expression of PD-L1 on nonspecific siRNA (control) and Akt-1 siRNA in HIV-infected subjects. Thin lines represent no siRNA,
thick lines represent treatment with the indicated siRNA, and gray lines are staining with an isotype control.
The Journal of Immunology 2941
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

responses sufficient to clear infection (12). Recent reports suggestthat this may be explained in part by PD-1 upregulation andPD-1 signaling to Ag-specific CD8+ T cells continually exposed totheir Ag (9, 13, 18, 48). Indeed, steric inhibition of PD-1 ability toengage its ligands can increase the activity of Ag-specific CD8+
T cells (12, 18).A poorly understood aspect of the relationship between PD-1 and
HIV infection is the role of the natural ligands of PD-1. In par-ticular, it is currently unknown whether HIV-induced immunesuppression via PD-1 signaling is a direct or indirect effect me-diated by APCs. Two naturally occurring ligands for PD-1 havebeen described thus far (23), and little is known about the tissuedistribution, expression, and distribution of these in people withchronic viral infections.Several studies have demonstrated APC dysfunction during
ineffective HIV-specific T cell immune responses and viral per-sistence (13, 18, 48). Our studies demonstrate a connection be-tween HIV and PD-1Ls on APCs including monocytes and bothmajor subsets of DCs. Monocytes and DCs infected with HIVexpress high levels of the PD-1Ls (Fig. 1). Furthermore, the up-regulation of PD-L1/2 was a direct effect of HIV infection inprimary monocytes (Fig. 3). The expression of PD-1Ls by thesecells changes the outcome of the interaction between HIV-specificT cells and APCs, rendering the T cells nonfunctional.We hypothesized that the upregulation of PD-L1 in pDCs during
HIV infection is due to the molecule’s pivotal role in Ag pre-sentation and elicitation of T cell effectors. To examine this, wesilenced PD-L’s expression on monocytes using siRNA duplexes,which are highly specific for PD-L1 mRNA and stably limit up-regulation of PD-L1, allowing us to study the role PD-1/PD-Lsin the APC–T cell interaction. Prevention of this interaction byknockdown of PD-L1 and PD-L2 expression with siRNA re-covered the functionality of HIV-specific T cells presented withHIV peptides (Fig. 4). We observed that PD-L1 silencing in APCsfrom patients with long-standing HIV infection resulted in theenhanced production of proinflammatory antiviral cytokines (IL-2,IL-12, and IFN-g) by uninfected HIV-specific T cells (Fig. 5).Furthermore, this recovery was closely associated with a de-creased secretion of the anti-inflammatory IL IL-10. This cytokineprofile has been shown to play a critical role in the regulation ofvirus-specific immune responses, and elevated proinflammatorytiters measured in this study following siRNA treatment mighthelp to bolster T cell function in the midst of a chronic infection.The effect of interrupting the PD-1/PD-L’s pathway is anotherfunctional property of virus-specific T cells, and their proliferativecapacity upon restimulation was also found to be greatly enhancedfollowing siRNA treatment. Furthermore, we tested whether Nefis necessary for the upregulation of PD-L1. Our study suggestedthat HIV’s accessory protein Nef is both necessary and sufficientto regulate PD-Ls’ upregulation in infected cells in vitro (Fig. 6).Taken together, these data showed that inhibiting PD-L’s ex-pression on monocytic cells significantly influences HIV-specificCD8+ T cell responses and further supports the growing datademonstrating that PD-1/PD-L’s interactions play an importantrole in attenuating immune responses and that interruption ofthese interactions can reverse T cell dysfunction. What remainsunclear and requires further investigation is whether PD-Ls ex-pressed on APCs in vivo are the major tolergenic mechanism forHIV-specific T cells in vivo.Finally, we investigated the PI3K/Akt signaling pathway in HIV-
induced PD-L expression, a pathway prominent for the activationof many cell types and one that has long been recognized for itssignificance during virus infection. Previously, Parsa et al. reportedthat the loss of PTEN, which negatively controls PI3K activation,
increased the expression of PD-L1 (50), suggesting that PI3Kactivity correlates with PD-L1 expression. On the basis of ourprevious observations, we anticipated that the mechanism bywhich HIV induces PD-Ls’ expression used the PI3K/Akt pathwayand that PD-Ls’ dysregulation can be blocked by inhibiting thissignaling pathway. Indeed, we observed that inhibitors of PI3Kand Akt caused similar changes in PD-L1 expression character-ized by reduced expression (Figs. 7, 8). Early activation of thePI3K/Akt pathway by HIV may also have implications in thepathogenesis of macrophages, which are among the primary tar-gets of HIV and respond to infection by providing a number ofimportant immune signals.In conclusion, data in this article indicate that in patients with
long-standing chronic HIV infection, blocking the PD-1 pathwaymay help to rescue the capacity of peripheral HIV-specific T cellsand profoundly exhausted T cells to mount effective antiviralimmune responses (Fig. 9) without CD4+ T cell help. The possi-ble benefit of understanding the mechanism of HIV-induced PD-Lregulation would be the identification of new therapeutic targetsfor reducing CTL suppression during chronic viral infection.Results from this investigation may help lead to new and morespecific immune therapeutic interventions for HIV infection andmay also provide information that is relevant for other chronicinfections such as HCV, an elusive goal that current drug therapyhas not yet attained.
AcknowledgmentsWe thank the National Institutes of Health AIDS Reagent Program for pro-
viding the reagents for this study.
DisclosuresD.B.W. would like to note several commercial relationships for purposes
of disclosure. These relations include service on a scientific advisory board
or advisory committee service, speaking, project consultations, stock own-
ership, sponsored research, or license royalties with commercial entities
including Novartis, Inovio, VGXi, BMS, Medarex, Pfizer, Virxsys, Ichor,
Merck, and Althea.
References1. Hewson, T., N. Lone, M. Moore, and S. Howie. 1999. Interactions of HIV-1 with
antigen-presenting cells. Immunol. Cell Biol. 77: 289–303.2. Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho.
1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, andviral generation time. Science 271: 1582–1586.
3. Barouch, D. H., S. Santra, J. E. Schmitz, M. J. Kuroda, T. M. Fu, W. Wagner,M. Bilska, A. Craiu, X. X. Zheng, G. R. Krivulka, et al. 2000. Control of viremiaand prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNAvaccination. Science 290: 486–492.
4. Shedlock, D. J., D. Hwang, A. Y. Choo, C. W. Chung, K. Muthumani, andD. B. Weiner. 2008. HIV-1 viral genes and mitochondrial apoptosis. Apoptosis13: 1088–1099.
5. Butler, S. L., M. S. Hansen, and F. D. Bushman. 2001. A quantitative assay forHIV DNA integration in vivo. Nat. Med. 7: 631–634.
6. Kiepiela, P., K. Ngumbela, C. Thobakgale, D. Ramduth, I. Honeyborne,E. Moodley, S. Reddy, C. de Pierres, Z. Mncube, N. Mkhwanazi, et al. 2007.CD8+ T-cell responses to different HIV proteins have discordant associationswith viral load. Nat. Med. 13: 46–53.
7. Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, andM. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes inHIV-1 infection. Nature 373: 123–126.
8. Migueles, S. A., C. M. Osborne, C. Royce, A. A. Compton, R. P. Joshi,K. A. Weeks, J. E. Rood, A. M. Berkley, J. B. Sacha, N. A. Cogliano-Shutta,et al. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infectedcell elimination associated with immune control. Immunity 29: 1009–1021.
9. Barber, D. L., E. J. Wherry, D. Masopust, B. Zhu, J. P. Allison, A. H. Sharpe,G. J. Freeman, and R. Ahmed. 2006. Restoring function in exhausted CD8T cells during chronic viral infection. Nature 439: 682–687.
10. Betts, M. R., B. Exley, D. A. Price, A. Bansal, Z. T. Camacho, V. Teaberry,S. M. West, D. R. Ambrozak, G. Tomaras, M. Roederer, et al. 2005. Charac-terization of functional and phenotypic changes in anti-Gag vaccine-inducedT cell responses and their role in protection after HIV-1 infection. Proc. Natl.Acad. Sci. USA 102: 4512–4517.
11. Stevenson, M. 2008. Can HIV be cured? Sci. Am. 299: 78–83.
2942 HIV INFECTION REGULATES PD-1 LIGANDS ON APCs
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from

12. Walker, B. D., and D. R. Burton. 2008. Toward an AIDS vaccine. Science 320:760–764.
13. Day, C. L., D. E. Kaufmann, P. Kiepiela, J. A. Brown, E. S. Moodley, S. Reddy,E. W. Mackey, J. D. Miller, A. J. Leslie, C. DePierres, et al. 2006. PD-1 ex-pression on HIV-specific T cells is associated with T-cell exhaustion and diseaseprogression. Nature 443: 350–354.
14. D’Souza, M., A. P. Fontenot, D. G. Mack, C. Lozupone, S. Dillon, A. Meditz,C. C. Wilson, E. Connick, and B. E. Palmer. 2007. Programmed death 1 ex-pression on HIV-specific CD4+ T cells is driven by viral replication and asso-ciated with T cell dysfunction. J. Immunol. 179: 1979–1987.
15. Freeman, G. J., E. J. Wherry, R. Ahmed, and A. H. Sharpe. 2006. Reinvigoratingexhausted HIV-specific T cells via PD-1–PD-1 ligand blockade. J. Exp. Med.203: 2223–2227.
16. Kaufmann, D. E., D. G. Kavanagh, F. Pereyra, J. J. Zaunders, E. W. Mackey,T. Miura, S. Palmer, M. Brockman, A. Rathod, A. Piechocka-Trocha, et al. 2007.Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with diseaseprogression and defines a reversible immune dysfunction. Nat. Immunol. 8:1246–1254.
17. Petrovas, C., J. P. Casazza, J. M. Brenchley, D. A. Price, E. Gostick,W. C. Adams, M. L. Precopio, T. Schacker, M. Roederer, D. C. Douek, andR. A. Koup. 2006. PD-1 is a regulator of virus-specific CD8+ T cell survival inHIV infection. J. Exp. Med. 203: 2281–2292.
18. Trautmann, L., L. Janbazian, N. Chomont, E. A. Said, S. Gimmig, B. Bessette,M. R. Boulassel, E. Delwart, H. Sepulveda, R. S. Balderas, et al. 2006. Up-regulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversibleimmune dysfunction. Nat. Med. 12: 1198–1202.
19. Velu, V., S. Kannanganat, C. Ibegbu, L. Chennareddi, F. Villinger, G. J. Freeman,R. Ahmed, and R. R. Amara. 2007. Elevated expression levels of inhibitoryreceptor programmed death 1 on simian immunodeficiency virus-specificCD8 T cells during chronic infection but not after vaccination. J. Virol. 81:5819–5828.
20. Shedlock, D. J., and H. Shen. 2003. Requirement for CD4 T cell help in gen-erating functional CD8 T cell memory. Science 300: 337–339.
21. Keir, M. E., M. J. Butte, G. J. Freeman, and A. H. Sharpe. 2008. PD-1 and itsligands in tolerance and immunity. Annu. Rev. Immunol. 26: 677–704.
22. Deacon, N. J., A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting,D. J. Hooker, D. A. McPhee, A. L. Greenway, A. Ellett, C. Chatfield, et al. 1995.Genomic structure of an attenuated quasi species of HIV-1 from a blood trans-fusion donor and recipients. Science 270: 988–991.
23. Sharpe, A. H., E. J. Wherry, R. Ahmed, and G. J. Freeman. 2007. The function ofprogrammed cell death 1 and its ligands in regulating autoimmunity and in-fection. Nat. Immunol. 8: 239–245.
24. Urbani, S., B. Amadei, D. Tola, M. Massari, S. Schivazappa, G. Missale, andC. Ferrari. 2006. PD-1 expression in acute hepatitis C virus (HCV) infection isassociated with HCV-specific CD8 exhaustion. J. Virol. 80: 11398–11403.
25. Zhang, J. Y., Z. Zhang, X. Wang, J. L. Fu, J. Yao, Y. Jiao, L. Chen, H. Zhang,J. Wei, L. Jin, et al. 2007. PD-1 up-regulation is correlated with HIV-specificmemory CD8+ T-cell exhaustion in typical progressors but not in long-termnonprogressors. Blood 109: 4671–4678.
26. Boasso, A., A. W. Hardy, A. L. Landay, J. L. Martinson, S. A. Anderson,M. J. Dolan, M. Clerici, and G. M. Shearer. 2008. PDL-1 upregulationon monocytes and T cells by HIV via type I interferon: restricted expression oftype I interferon receptor by CCR5-expressing leukocytes. Clin. Immunol. 129:132–144.
27. Rodrıguez-Garcıa, M., F. Porichis, O. G. de Jong, K. Levi, T. J. Diefenbach,J. D. Lifson, G. J. Freeman, B. D. Walker, D. E. Kaufmann, andD. G. Kavanagh. 2011. Expression of PD-L1 and PD-L2 on human macrophagesis up-regulated by HIV-1 and differentially modulated by IL-10. J. Leukoc. Biol.89: 507–515.
28. Trabattoni, D., M. Saresella, M. Pacei, I. Marventano, L. Mendozzi, M. Rovaris,D. Caputo, M. Borelli, and M. Clerici. 2009. Costimulatory pathways in multiplesclerosis: distinctive expression of PD-1 and PD-L1 in patients with differentpatterns of disease. J. Immunol. 183: 4984–4993.
29. Trabattoni, D., M. Saresella, M. Biasin, A. Boasso, L. Piacentini, P. Ferrante,H. Dong, R. Maserati, G. M. Shearer, L. Chen, and M. Clerici. 2003. B7-H1 isup-regulated in HIV infection and is a novel surrogate marker of disease pro-gression. Blood 101: 2514–2520.
30. Muthumani, K., A. Y. Choo, D. S. Hwang, A. Premkumar, N. S. Dayes,C. Harris, D. R. Green, S. A. Wadsworth, J. J. Siekierka, and D. B. Weiner. 2005.HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPKactivation. Blood 106: 2059–2068.
31. Francisco, L. M., V. H. Salinas, K. E. Brown, V. K. Vanguri, G. J. Freeman,V. K. Kuchroo, and A. H. Sharpe. 2009. PD-L1 regulates the development,
maintenance, and function of induced regulatory T cells. J. Exp. Med. 206:3015–3029.
32. Breton, G., B. Yassine-Diab, L. Cohn, M. R. Boulassel, J. P. Routy, R. P. Sekaly,and R. M. Steinman. 2009. siRNA knockdown of PD-L1 and PD-L2 inmonocyte-derived dendritic cells only modestly improves proliferative responsesto Gag by CD8+ T cells from HIV-1–infected individuals. J. Clin. Immunol. 29:637–645.
33. Swingler, S., B. Brichacek, J. M. Jacque, C. Ulich, J. Zhou, and M. Stevenson.2003. HIV-1 Nef intersects the macrophage CD40L signalling pathway to pro-mote resting-cell infection. Nature 424: 213–219.
34. Muthumani, K., A. Y. Choo, D. J. Shedlock, D. J. Laddy, S. G. Sundaram,L. Hirao, L. Wu, K. P. Thieu, C. W. Chung, K. M. Lankaraman, et al. 2008.Human immunodeficiency virus type 1 Nef induces programmed death 1 ex-pression through a p38 mitogen-activated protein kinase-dependent mechanism.J. Virol. 82: 11536–11544.
35. Muthumani, K., A. Y. Choo, W. X. Zong, M. Madesh, D. S. Hwang,A. Premkumar, K. P. Thieu, J. Emmanuel, S. Kumar, C. B. Thompson, andD. B. Weiner. 2006. The HIV-1 Vpr and glucocorticoid receptor complex isa gain-of-function interaction that prevents the nuclear localization of PARP-1.Nat. Cell Biol. 8: 170–179.
36. Latchman, Y., C. R. Wood, T. Chernova, D. Chaudhary, M. Borde, I. Chernova,Y. Iwai, A. J. Long, J. A. Brown, R. Nunes, et al. 2001. PD-L2 is a second ligandfor PD-1 and inhibits T cell activation. Nat. Immunol. 2: 261–268.
37. Freeman, G. J., A. J. Long, Y. Iwai, K. Bourque, T. Chernova, H. Nishimura,L. J. Fitz, N. Malenkovich, T. Okazaki, M. C. Byrne, et al. 2000. Engagement ofthe PD-1 immunoinhibitory receptor by a novel B7 family member leads tonegative regulation of lymphocyte activation. J. Exp. Med. 192: 1027–1034.
38. Franceschini, D., M. Paroli, V. Francavilla, M. Videtta, S. Morrone, G. Labbadia,A. Cerino, M. U. Mondelli, and V. Barnaba. 2009. PD-L1 negatively regulatesCD4+CD25+Foxp3+ Tregs by limiting STAT-5 phosphorylation in patientschronically infected with HCV. J. Clin. Invest. 119: 551–564.
39. Lubelchek, R. J., B. Max, C. J. Sandusky, B. Hota, and D. E. Barker. 2009.Reliability at the lower limits of HIV-1 RNA quantification in clinical samples:a comparison of RT-PCR versus bDNA assays. PLoS ONE 4: e6008.
40. Liang, S. C., Y. E. Latchman, J. E. Buhlmann, M. F. Tomczak, B. H. Horwitz,G. J. Freeman, and A. H. Sharpe. 2003. Regulation of PD-1, PD-L1, and PD-L2expression during normal and autoimmune responses. Eur. J. Immunol. 33:2706–2716.
41. Meier, A., A. Bagchi, H. K. Sidhu, G. Alter, T. J. Suscovich, D. G. Kavanagh,H. Streeck, M. A. Brockman, S. LeGall, J. Hellman, and M. Altfeld. 2008.Upregulation of PD-L1 on monocytes and dendritic cells by HIV-1 derived TLRligands. AIDS 22: 655–658.
42. Kutzler, M. A., K. A. Kraynyak, S. J. Nagle, R. M. Parkinson, D. Zharikova,M. Chattergoon, H. Maguire, K. Muthumani, K. Ugen, and D. B. Weiner. 2010.Plasmids encoding the mucosal chemokines CCL27 and CCL28 are effectiveadjuvants in eliciting antigen-specific immunity in vivo. Gene Ther. 17: 72–82.
43. Muthumani, K., K. M. Lankaraman, D. J. Laddy, S. G. Sundaram, C. W. Chung,E. Sako, L. Wu, A. Khan, N. Sardesai, J. J. Kim, et al. 2008. Immunogenicity ofnovel consensus-based DNA vaccines against Chikungunya virus. Vaccine 26:5128–5134.
44. Shedlock, D. J., K. T. Talbott, M. P. Morrow, B. Ferraro, D. A. Hokey,K. Muthumani, and D. B. Weiner. 2010. Ki-67 staining for determinationof rhesus macaque T cell proliferative responses ex vivo. Cytometry A 77:275–284.
45. Golden-Mason, L., B. Palmer, J. Klarquist, J. A. Mengshol, N. Castelblanco, andH. R. Rosen. 2007. Upregulation of PD-1 expression on circulating and intra-hepatic hepatitis C virus-specific CD8+ T cells associated with reversible im-mune dysfunction. J. Virol. 81: 9249–9258.
46. Oestreich, K. J., H. Yoon, R. Ahmed, and J. M. Boss. 2008. NFATc1 regulatesPD-1 expression upon T cell activation. J. Immunol. 181: 4832–4839.
47. Steinman, R. M., D. Hawiger, and M. C. Nussenzweig. 2003. Tolerogenicdendritic cells. Annu. Rev. Immunol. 21: 685–711.
48. Wherry, E. J., S. J. Ha, S. M. Kaech, W. N. Haining, S. Sarkar, V. Kalia,S. Subramaniam, J. N. Blattman, D. L. Barber, and R. Ahmed. 2007. Molecularsignature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684.
49. Morrow, M. P., J. Yan, P. Pankhong, D. J. Shedlock, M. G. Lewis, K. Talbott,R. Toporovski, A. S. Khan, N. Y. Sardesai, and D. B. Weiner. 2010. IL-28B/IFN-l3 drives granzyme B loading and significantly increases CTL killing activity inmacaques. Mol. Ther. 18: 1714–1723.
50. Parsa, A. T., J. S. Waldron, A. Panner, C. A. Crane, I. F. Parney, J. J. Barry,K. E. Cachola, J. C. Murray, T. Tihan, M. C. Jensen, et al. 2007. Loss of tumorsuppressor PTEN function increases B7-H1 expression and immunoresistance inglioma. Nat Med. 13: 84–88.
The Journal of Immunology 2943
at Univ of Pennsylvania L
ibrary on August 25, 2012
http://jimm
unol.org/D
ownloaded from
Related Documents











