High Throughput Sequencing Xiaole Shirley Liu STAT115, STAT215, BIO298, BIST520

High Throughput Sequencing Xiaole Shirley Liu STAT115, STAT215, BIO298, BIST520.
Dec 18, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

High Throughput Sequencing
Xiaole Shirley Liu
STAT115, STAT215, BIO298, BIST520
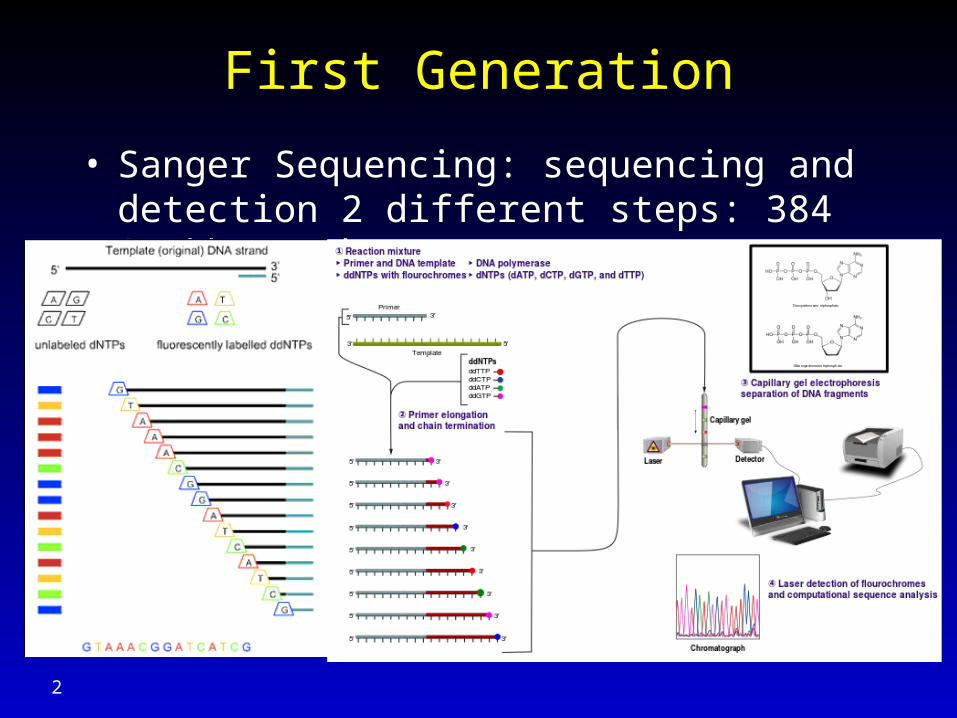
First Generation
• Sanger Sequencing: sequencing and detection 2 different steps: 384 * 1kb / 3 hours
2

Second Generation
• Massively parallel sequencing by synthesis
• Many different technologies: Illumina, 454, SOLiD, Helicos, etc
• Illumina: HiSeq, MiSeq, NextSeq• 1-16 samples • 25M-4B reads• 30-300bp • 1-8 days• 15GB-1TB output• Moving targets
3
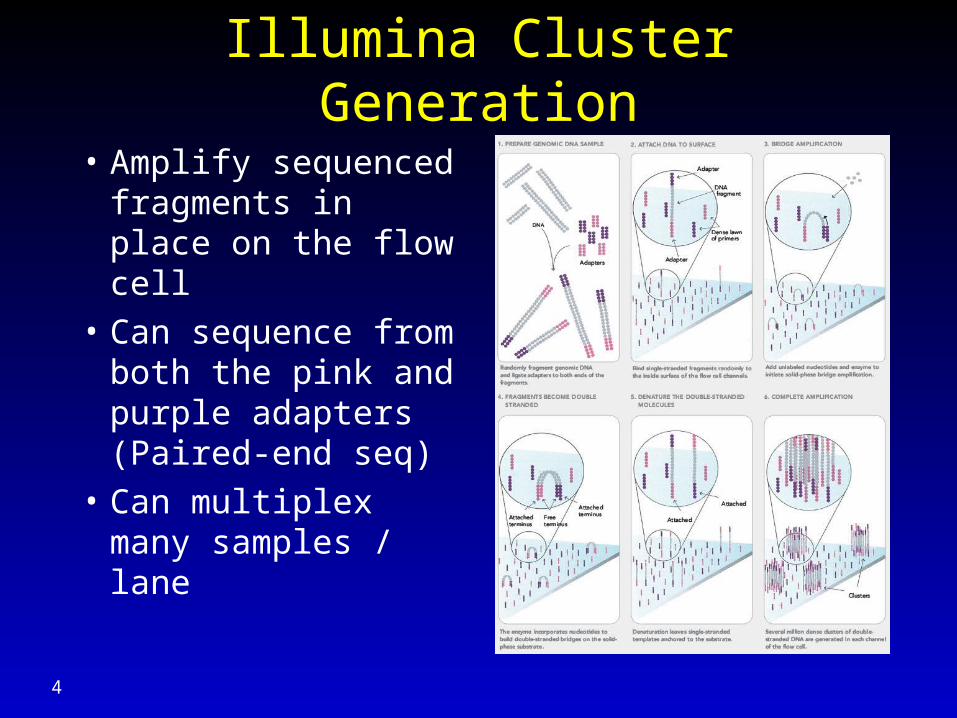
Illumina Cluster Generation
• Amplify sequenced fragments in place on the flow cell
• Can sequence from both the pink and purple adapters (Paired-end seq)
• Can multiplex many samples / lane
4
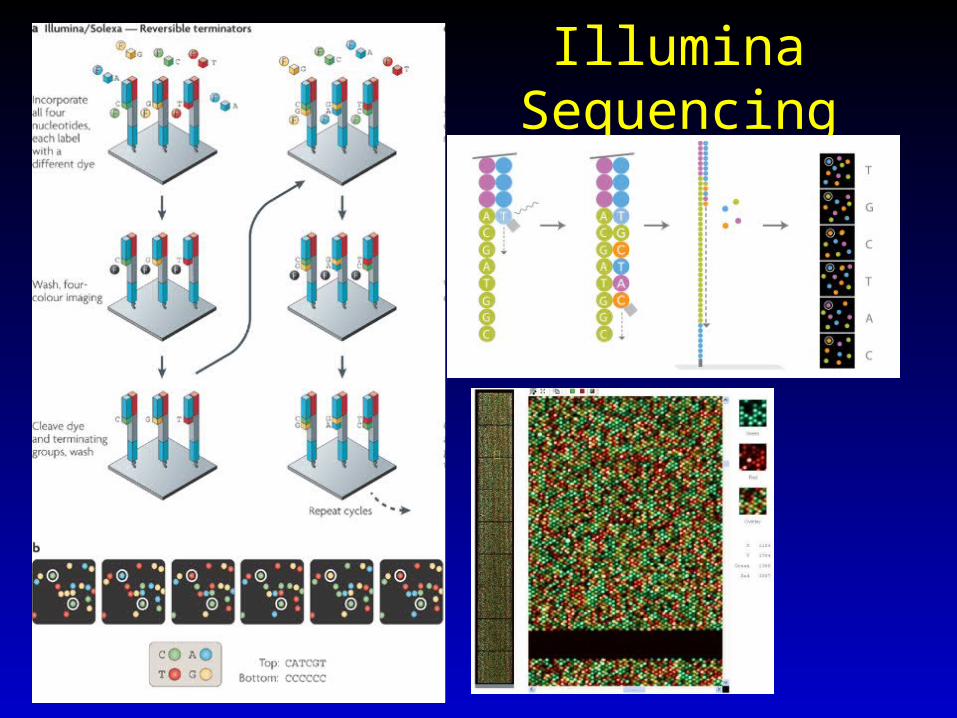
Illumina Sequencing
5

Third Generation• Single molecule sequencing: no amp• Fewer but much longer reads• Good for sequencing long reads, but not for read count
applications, technology still in developmenthttp://www.youtube.com/watch?v=v8p4ph2MAvI
• https://www.nanoporetech.com/news/movies#movie-28-minion
6
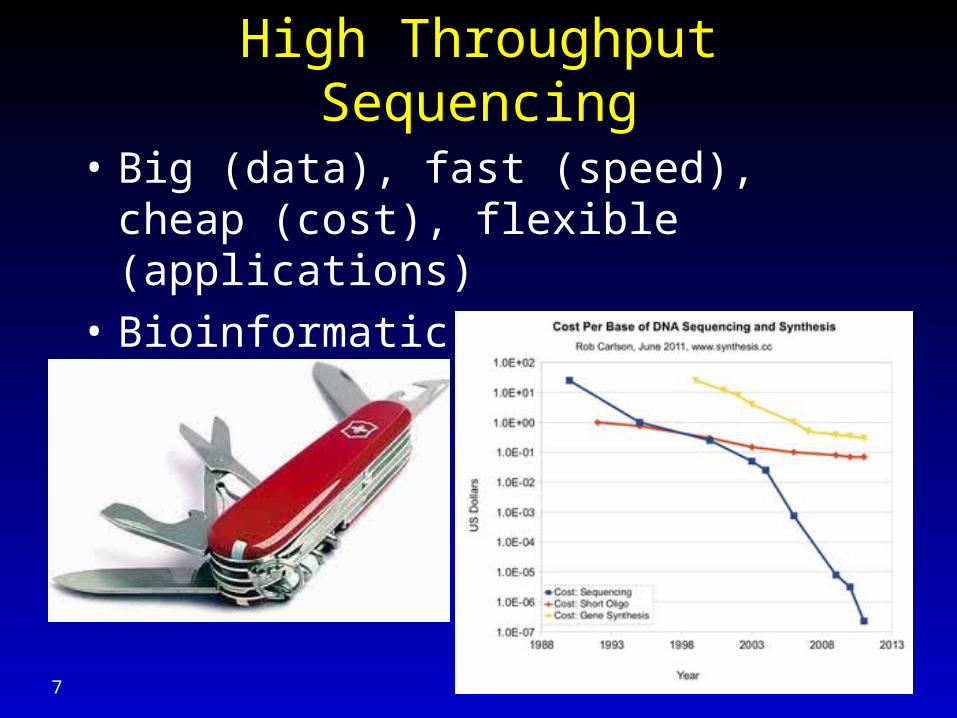
High Throughput Sequencing
• Big (data), fast (speed), cheap (cost), flexible (applications)
• Bioinformatic analyses become bottleneck
7

High Throughput Sequencing Data Analysis
8

FASTQ File
• Format– Sequence ID, sequence
– Quality ID, quality score
• Quality score using ASCII (higher -> better)
9
@HWI-EAS305:1:1:1:991#0/1
GCTGGAGGTTCAGGCTGGCCGGATTTAAACGTAT
+HWI-EAS305:1:1:1:991#0/1
MVXUWVRKTWWULRQQMMWWBBBBBBBBBBBBBB
@HWI-EAS305:1:1:1:201#0/1
AAGACAAAGATGTGCTTTCTAAATCTGCACTAAT
+HWI-EAS305:1:1:1:201#0/1
PXX[[[[XTXYXTTWYYY[XXWWW[TMTVXWBBB
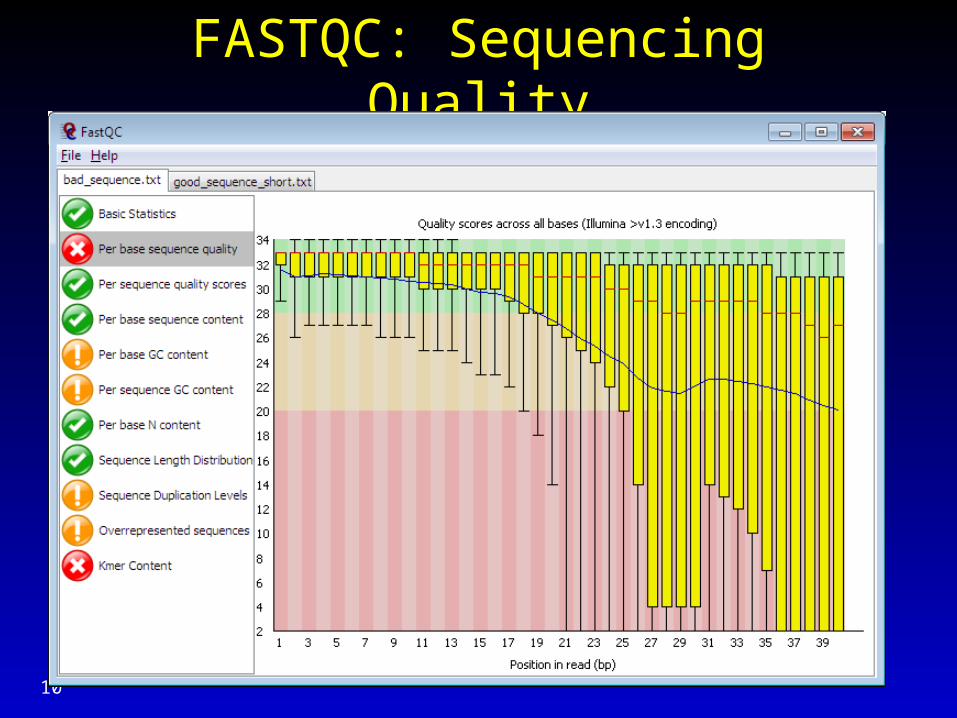
FASTQC: Sequencing Quality
10

Read Mapping
• Mapping hundreds of millions of reads back to the reference genome is CPU and RAM intensive and slow
• Read quality decreases with length (small single nucleotide mismatches or indels)
• Most mappers allow ~2 mismatches within first 30bp (4 ^ 28 could still uniquely identify most 30bp sequences in a 3GB genome), slower when allowing indels
• Mapping output: SAM (BAM) or BED11
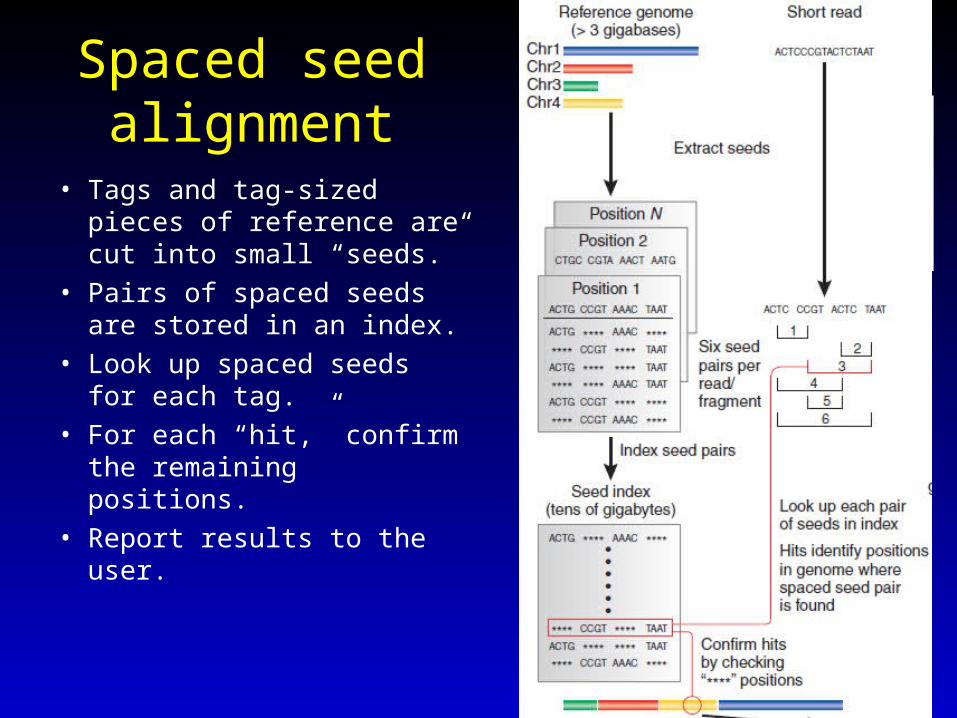
Spaced seed alignment
• Tags and tag-sized pieces of reference are cut into small “seeds.”
• Pairs of spaced seeds are stored in an index.
• Look up spaced seeds for each tag.
• For each “hit,” confirm the remaining positions.
• Report results to the user.
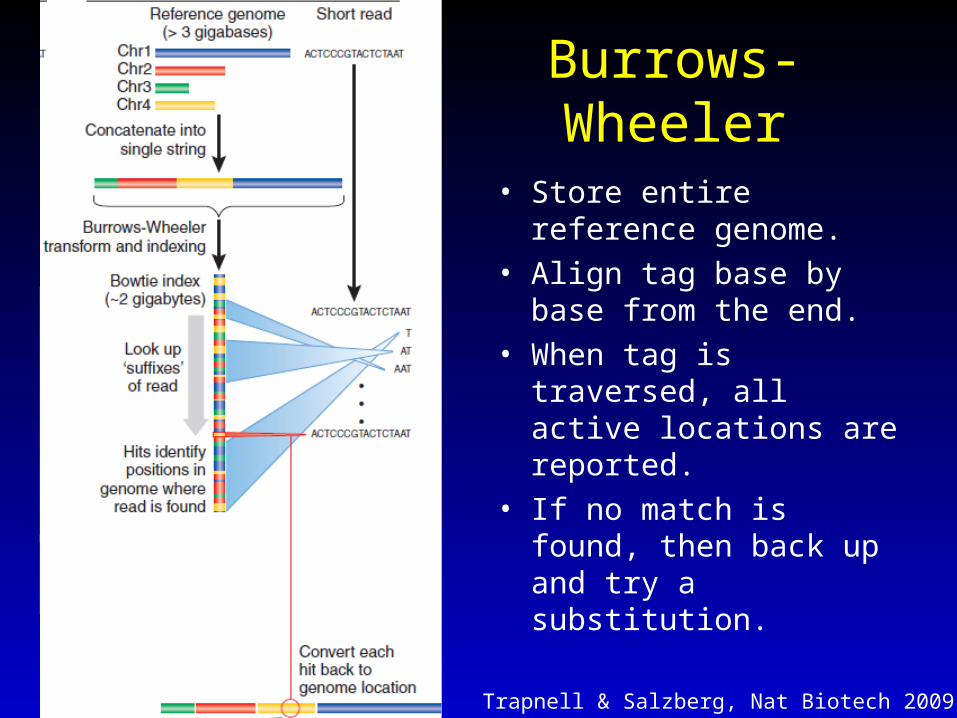
Burrows-Wheeler
• Store entire reference genome.
• Align tag base by base from the end.
• When tag is traversed, all active locations are reported.
• If no match is found, then back up and try a substitution.
Trapnell & Salzberg, Nat Biotech 2009
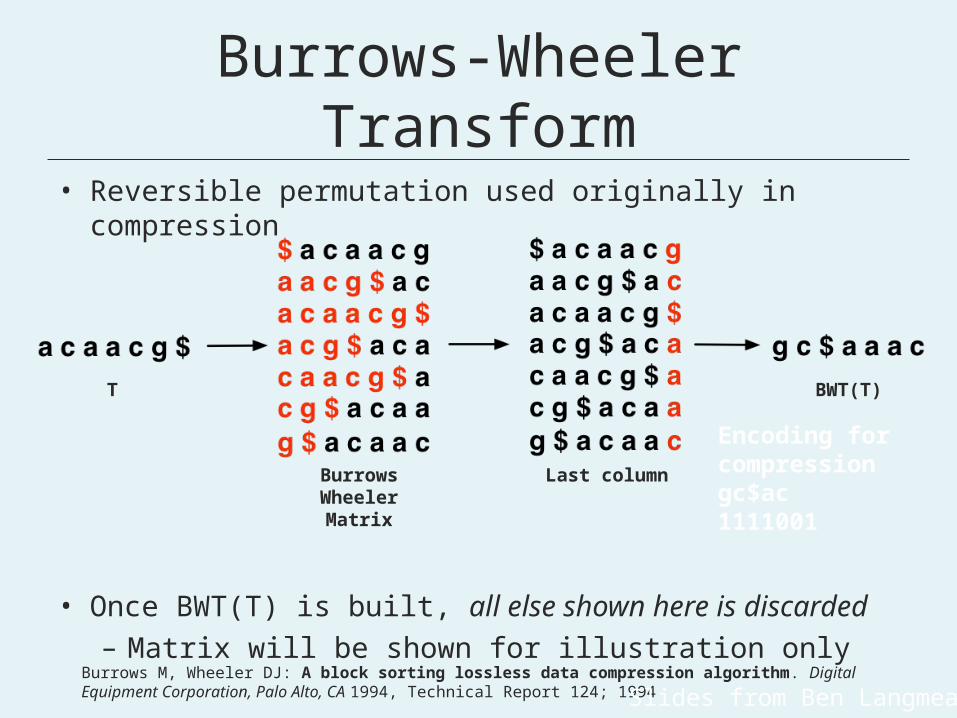
Burrows-Wheeler Transform
• Reversible permutation used originally in compression
• Once BWT(T) is built, all else shown here is discarded– Matrix will be shown for illustration only
BurrowsWheelerMatrix
Last column
BWT(T)T
Burrows M, Wheeler DJ: A block sorting lossless data compression algorithm. Digital Equipment Corporation, Palo Alto, CA 1994, Technical Report 124; 1994Slides from Ben Langmead
Encoding for compressiongc$ac1111001

Burrows-Wheeler Transform
• Property that makes BWT(T) reversible is “LF Mapping”– ith occurrence of a character in Last column is
same text occurrence as the ith occurrence in First column
T
BWT(T)
Burrows WheelerMatrix
Rank: 2
Rank: 2
Slides from Ben Langmead
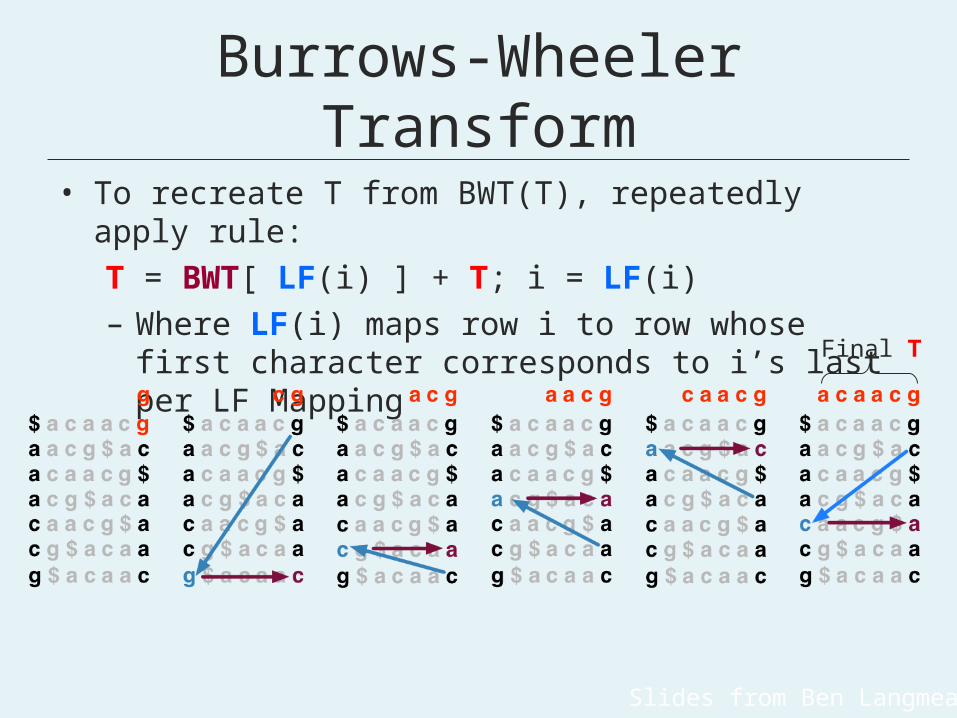
Burrows-Wheeler Transform
• To recreate T from BWT(T), repeatedly apply rule:T = BWT[ LF(i) ] + T; i = LF(i)– Where LF(i) maps row i to row whose first
character corresponds to i’s last per LF Mapping Final T
Slides from Ben Langmead

Exact Matching with FM Index
• To match Q in T using BWT(T), repeatedly apply rule:top = LF(top, qc); bot = LF(bot, qc)– Where qc is the next character in Q (right-to-
left) and LF(i, qc) maps row i to the row whose first character corresponds to i’s last character as if it were qc
Slides from Ben Langmead

Exact Matching with FM Index
• In progressive rounds, top & bot delimit the range of rows beginning with progressively longer suffixes of Q (from right to left)
• If range becomes empty the query suffix (and therefore the query) does not occur in the text
• If no match, instead of giving up, try to “backtrack” to a previous position and try a different base (mismatch, much slower)
Slides from Ben Langmead

STAR Alignment
• Suffix Tree
• Very fast and accuracy for mapping PE-seq and high read counts
• O(n) time to build
• O(mlogn) time to
search
19

Suffix tree (Example)
Let s=abab, a suffix tree of s is a compressed trie of all suffixes of s=abab$
{ $ b$ ab$ bab$ abab$ }
ab
ab
$
ab$
b
$
$
$
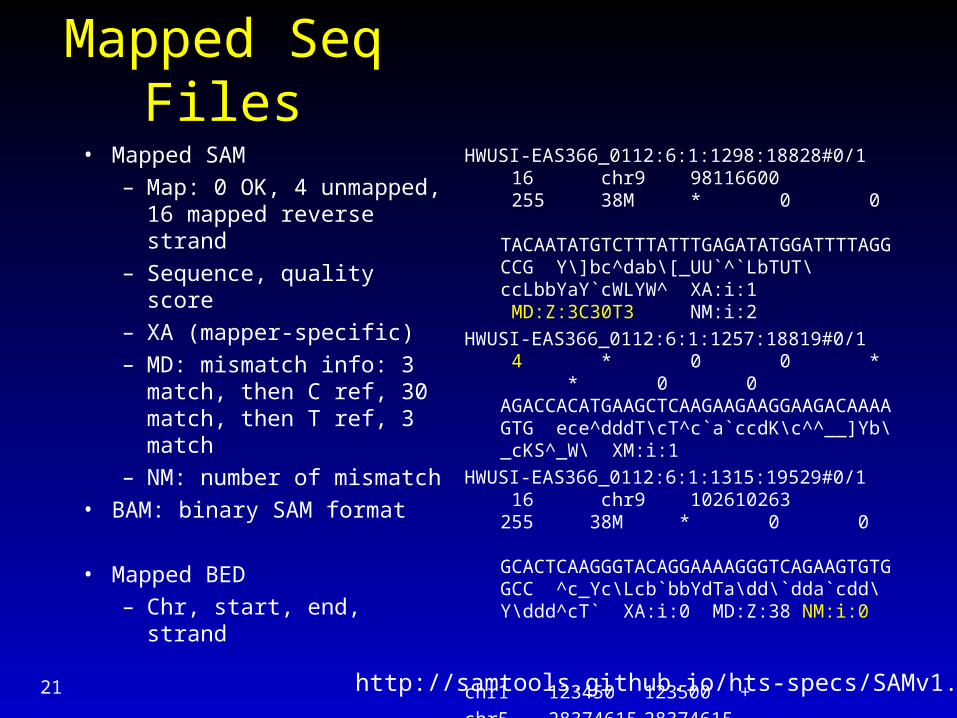
Mapped Seq Files
• Mapped SAM
– Map: 0 OK, 4 unmapped, 16 mapped reverse strand
– Sequence, quality score
– XA (mapper-specific)
– MD: mismatch info: 3 match, then C ref, 30 match, then T ref, 3 match
– NM: number of mismatch
• BAM: binary SAM format
• Mapped BED
– Chr, start, end, strand
21
HWUSI-EAS366_0112:6:1:1298:18828#0/1 16 chr9 98116600 255 38M * 0 0 TACAATATGTCTTTATTTGAGATATGGATTTTAGGCCG Y\]bc^dab\[_UU`^`LbTUT\ccLbbYaY`cWLYW^ XA:i:1 MD:Z:3C30T3 NM:i:2
HWUSI-EAS366_0112:6:1:1257:18819#0/1 4 * 0 0 * * 0 0 AGACCACATGAAGCTCAAGAAGAAGGAAGACAAAAGTG ece^dddT\cT^c`a`ccdK\c^^__]Yb\_cKS^_W\ XM:i:1
HWUSI-EAS366_0112:6:1:1315:19529#0/1 16 chr9 102610263 255 38M * 0 0 GCACTCAAGGGTACAGGAAAAGGGTCAGAAGTGTGGCC ^c_Yc\Lcb`bbYdTa\dd\`dda`cdd\Y\ddd^cT` XA:i:0 MD:Z:38 NM:i:0
chr1 123450 123500 +chr5 2837461528374615-http://samtools.github.io/hts-specs/SAMv1.pdf

Mapping Statistics Terms
• Mappable locations: reads that can find match to A location in the genome
• Uniquely mapped reads: reads that can find match to A SINGLE location in the genome– Repeat sequences in the genome, length-
dependent
• Uniquely mapped locations: number of unique locations hit by uniquely mapped reads– Redundancy: potential PCR amplification bias
22

Summary
• Sequencing technologies– 1st, 2nd, 3rd generation
• Sequence quality assessment– FASTQC
• Read mapping– Spaced seed
– BWA: Borrows Wheeler transformation, LF mapping
– STAR: Suffix Tree, fast
• SAM / BAM format
23
Related Documents











