Big Data Analytics in Bioinformatics and Healthcare Baoying Wang Waynesburg University, USA Ruowang Li Pennsylvania State University, USA William Perrizo North Dakota State University, USA A volume in the Advances in Bioinformatics and Biomedical Engineering (ABBE) Book Series

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Big Data Analytics in Bioinformatics and HealthcareBaoying WangWaynesburg University, USA
Ruowang LiPennsylvania State University, USA
William PerrizoNorth Dakota State University, USA
A volume in the Advances in Bioinformatics and Biomedical Engineering (ABBE) Book Series

Published in the United States of America by Medical Information Science Reference (an imprint of IGI Global)701 E. Chocolate AvenueHershey PA, USA 17033Tel: 717-533-8845Fax: 717-533-8661 E-mail: [email protected] site: http://www.igi-global.com
Copyright © 2015 by IGI Global. All rights reserved. No part of this publication may be reproduced, stored or distributed in any form or by any means, electronic or mechanical, including photocopying, without written permission from the publisher.Product or company names used in this set are for identification purposes only. Inclusion of the names of the products or companies does not indicate a claim of ownership by IGI Global of the trademark or registered trademark. Library of Congress Cataloging-in-Publication Data
British Cataloguing in Publication DataA Cataloguing in Publication record for this book is available from the British Library.
All work contributed to this book is new, previously-unpublished material. The views expressed in this book are those of the authors, but not necessarily of the publisher.
For electronic access to this publication, please contact: [email protected].
Big data analytics in bioinformatics and healthcare / Baoying Wang, Ruowang Li, and William Perrizo, editors. p. ; cm. Includes bibliographical references and index. Summary: “This book merges the fields of biology, technology, and medicine in order to present a comprehensive study on the emerging information processing applications necessary in the field of electronic medical record management”--Provid-ed by publisher. ISBN 978-1-4666-6611-5 (hardcover) -- ISBN 978-1-4666-6612-2 (ebook) -- ISBN 978-1-4666-6614-6 (print & perpetual access) I. Wang, Baoying, 1964- , editor. II. Li, Ruowang, 1988- , editor. III. Perrizo, W. (William), editor. [DNLM: 1. Computational Biology--methods. 2. Biomedical Research--methods. 3. Data Mining--methods. 4. Elec-tronic Health Records. QU 26.5] R858 610.285--dc23 2014032346 This book is published in the IGI Global book series Advances in Bioinformatics and Biomedical Engineering (ABBE) (ISSN: 2327-7033; eISSN: 2327-7041)
Managing Director: Managing Editor: Director of Intellectual Property & Contracts: Acquisitions Editor: Production Editor: Development Editor: Typesetter: Cover Design:
Lindsay Johnston Austin DeMarco Jan Travers Kayla Wolfe Christina Henning Erin O’Dea Cody Page Jason Mull

138
Copyright © 2015, IGI Global. Copying or distributing in print or electronic forms without written permission of IGI Global is prohibited.
Chapter 7
Heuristic Principal Component Analysis-Based Unsupervised
Feature Extraction and Its Application to Bioinformatics
ABSTRACT
Feature Extraction (FE) is a difficult task when the number of features is much larger than the number of samples, although that is a typical situation when biological (big) data is analyzed. This is espe-cially true when FE is stable, independent of the samples considered (stable FE), and is often required. However, the stability of FE has not been considered seriously. In this chapter, the authors demonstrate that Principal Component Analysis (PCA)-based unsupervised FE functions as stable FE. Three bioin-formatics applications of PCA-based unsupervised FE–detection of aberrant DNA methylation associated with diseases, biomarker identification using circulating microRNA, and proteomic analysis of bacterial culturing processes–are discussed.
INTRODUCTION
Feature extraction (FE) is a task that reduces the number of features (independent variables) for predicting/estimating (a dependent variable). For example, when performing face recognition as a computational task, the specific parts of facial
photos, e.g., eye lines, colors of irises, or shapes of jaws, should be considered. Alternatively, to predict tomorrow’s stock prices computationally, certain factors, e.g., today’s prices, economical indices, or weather, should be taken into account. The problem is that increased numbers of fea-tures considered does not always result in better
Y-H. TaguchiChuo University, Japan
Mitsuo IwadateChuo University, Japan
Hideaki UmeyamaChuo University, Japan
Yoshiki MurakamiOsaka City University, Japan
Akira OkamotoAichi University of Education, Japan
DOI: 10.4018/978-1-4666-6611-5.ch007

139
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
performance. To perform better face recognition, considering hairstyle as a key feature is not useful, since it can vary. For stock price prediction, what your wife or husband cooks this morning is not useful as an important factor. However, neglect-ing the shape of the nose in face recognition or ignorance of this week’s unemployment rate for stock price prediction may reduce performance of the task. Thus, clearly, there should be mini-mum number of critical features to achieve the best performance. The current problem is how to determine the specific set of such features.
The era of big data has added additional dif-ficulty to this problem: a small number of samples (cases) versus too many features observed. For example, in facial recognition, it is not difficult to obtain many features from a facial photo that often consists of several millions of pixels, each of which has more than a million color grades. However, it is not a realistic requirement to collect a million facial photos, especially from a cost point of view, if payment is required for research use of individual photos. For stock market prediction, the situation appears better, because stock prices, even measured per second over months, can be collected and recorded. However, the use of many sample (case) numbers does not always resolve the “many features vs. small cases” difficulty, since these records are not always independent of each other. Stock price varies periodically over time, e.g., daily, weekly, or even seasonally. Thus, huge amounts of data are often simply replicates. What is required are samples taken under different economic situations. For example, if the market is lively, data should be obtained from when the market is bad. However, this kind of data is only available after the economic crash, thus stock price prediction based only on measurements taken under good economic situations naturally fails to predict price reduction (and money is therefore lost). Thus, “many features vs small samples” problems must be resolved to compete with mas-sive data flowing into “prediction” systems.
In this chapter, we would like to propose an alternative solution to the difficulty of FE when the number of samples is much lower than the number of features. There have been many pro-posals to overcome this problem, e.g., stepwise feature extraction (Prasad, 2008), regression with regularization (Girosi, 1995) and Bayesian work frame (Chu, 2006). Despite these efforts, FE prob-lems with small sample size and large number of features have not been solved completely.
Recently, unsupervised FE (De Backer, 1998) has gained the interest of many researchers. Un-supervised FE is robust and thus, it is expected to provide more stable FEs, i.e., unsupervised FEs have weaker sample dependence than other meth-ods with optimization procedures in all senses.
Although many kinds of implementations of unsupervised FE are possible, we employed one of the simplest, principal component analysis (PCA) based FE (Murakami, 2012; Taguchi, 2013) in this chapter. Since PCA is a linear method, it is ex-pected to have greater robustness than other more complicated methods such as those with kernel tricks (Scholkopf, 2001) or Bayesian statistics. Linear methods, such as PCA, are also expected to be less computationally challenging. Finally, it is easier to interpret the obtained results, since it is a linear combination of the original features. In contrast to these advantages, linear methods including PCA usually have poorer performances than other more complicated methods (Cao, 2003). In this chapter, we also discuss how these difficul-ties can be overcome when applying PCA to FEs.
The applications considered in this chapter are the detection of aberrant DNA methylation associ-ated with diseases (Ishida, 2014; Kinoshita 2014), biomarker identification of circulating microRNA (miRNA) (Murakami, 2012; Taguchi, 2013) and proteomic analysis of bacterial culture (Taguchi, 2012). In the background section, we introduce FE methods and compare them to our methods. Then, we illustrate details of our proposed method and an illustrative (artificial) example. Each ap-

140
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
plication of PCA based FE is described separately, and then feature research directions, additional readings section, and conclusion are discussed.
BACKGROUND
As mentioned in the introduction, the purpose of this chapter is to propose a new methodology to resolve difficulties within FE when many features are available, but a small number of samples (cases) are provided (“many features vs small cases” difficulty). These difficulties are two-fold: combinatorial divergence of many features and bias of small sample numbers. Combinatorial divergence of many features occurs because the possible number of feature combinations becomes divergent exponentially as the number of consid-ered features increase. For example, if ten features are selected from one hundred available features, the total number of possible combinations is over one billion. If the number of features considered or available features increase, this already huge number further increases exponentially. Thus, small numbers of samples cannot distinctly evaluate or rank these combinations. Therefore, there is no effective way to judge which combination is best. In addition, small numbers of samples inevitably result in bias that prevents selecting the best com-binations within innumerous possibilities. Finally, FE is highly sample dependent, i.e., distinct sample sets indicates which combinations are best.
Considering these two difficulties, it is appar-ent that the stability of FE resolves this difficulty to some extent. If FE can select features stably, not all possible combinations can be evaluated every time new sample data is provided and bias caused by a small number of samples is reduced. As can be seen later, our proposed methodology can resolve these problems by maintaining the stability of FE.
In the following section, we briefly discuss a few conventional FEs. Further information can be obtained in another well-written review
article by Gyon (2003). There have been several efforts to overcome difficulties of using FE when small numbers of samples with many features are available (Table 1). The standard and simplest method is stepwise FE (Prasad, 2008) (Figure 1(a)), where FE usually starts from a small num-ber of pre-selected features. Then, using some criterion, either the addition of a new feature or elimination of a selected feature is employed. If neither of them passes the criterion, then itera-tion is terminated. There are no unique definite criterion that exist for stepwise FE. However, variables such as residuals of linear regression cannot be used for this criterion since residuals are monotonic and decrease the function of the number of features. Optimal numbers of employed features are always diverse. Thus, variables used for the criterion must include a penalty term for the addition of features. One of the most frequently used criterion is Akaike Information Criterion (AIC) (Bozdogan, 1987). AIC is the sum between the number of features and negative likelihood, and decreases as the number of features increases. Likelihood generally describes the “goodness” of prediction, i.e., “smallness” of the difference between prediction and true answer. Larger like-lihood means better prediction. Thus, AIC can balance between the “goodness” of prediction and the number of features. When too many features are considered, the AIC becomes larger. Since AIC is not a monotonic decreasing function of the number of features, optimal (minimum) AIC can determine the optimal number of features. The most critical drawback of stepwise FE is conver-gence. If the number of possible features is huge, the occasional selection of optimal combination of features is difficult. Stepwise FE is even often captured at local minimum, which is far from the true optimal combination of features. Because of the local optimization nature of stepwise FE, once it is captured at local minimum, there is no way to escape it. This forces the use of stepwise FE from a huge number of initial conditions (a trial

141
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
of starting combinations of features) that result in a heavy time-consuming process.
Another useful concept often used for optimal FE is regulation (Girosi, 1995). As denoted above, since residuals and likelihood are monotonic decreasing functions of the number of features, an additional penalty term is required. Regula-tion (Figure 1(b)) limits the number of features by the addition of “total amount” of coefficients of employed features. The most frequently used regulatory procedure is lasso (Tibshirani, 1996)
that is a shrinkage and selection method for linear regression. Lasso tries to minimize the sum of residuals by restricting the sum of absolute values of coefficients of features to less than the constant. Because of the singular property of absolute function at zero, this forces some coefficients to be zero, causing a limited number of non-zero coefficients, which automatically results in FE. As the upper limit of sum of absolute values of coefficients increases (decreases), the number of features with non-zero coefficients increases
Table 1. Summary of three conventional FEs
Method Description Advantages Disadvantages
Stepwise Adding and removing features iteratively until no improvements are achieved
Simple, thus easy to understand and to implement
Time consuming, thus hard to converge
Regulation Try to minimize the number of features by restricting the sum of coefficients attributed to features
Rapid to converge. Greater opportunity to reach solution
Results are strongly dependent upon how to restrict sum of coefficients. Optimal restriction must be evaluated by other criterion (e.g., cross validation)
Bayesian Optimizing model parameters such that likelihood is maximized
Once the model is assigned, results can be obtained automatically. Additional evaluation (e.g., confidence interval) can be easily driven from the obtained probabilities
Requires massive computation because of huge size of parameter search. The results are completely model dependent
Figure 1. Schematic diagrams that illustrate conventional FE (a) STEPWISE FE (b) FE with regulation (c) Bayesian

142
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
(decreases). Although regression with regulation terms often works very well, one drawback of this method is the decision of upper limit of sum of absolute values of coefficients, since there are no universal criteria for the optimal decision of this value.
Recent and more popular criterion have been obtained by Bayesian statistics (Chu, 2006). The Bayesian method (Figure 1(c)) tries to maximize conditional probability P(y|x), where y and x are dependent and independent variables, respec-tively. If some x are causally independent of y, elimination of x simply increases conditional probability. This means no additional criterion are required to obtain the optimal combination of features, although some arbitrariness on the decision of the functional form of P(y|x) still ex-ists. An obvious drawback of the Bayesian work frame is the divergence of search space. Ideally, we must search among all combinations of y and x, and the number of combinations between y and x will increase exponentially. Since situations with a huge number of features (independent variables) are considered, the Bayesian work frame inevita-bly results in heavy computationally challenging problems. Thus, although the Bayesian work frame is free from the difficulties of FE, it is not useful for real problems.
In contrast to these optimization-based meth-ods, unsupervised FE (De Backer, 1998) does not use labeling information for FE. Instead of minimizing the discrepancy between predictions and true answers, unsupervised FE attempts to find a “simpler pattern” hidden in the data set. Although there are no unique definitions of simpler patterns, this often means a smaller number of clusters or smaller number of dimensions to which objects are embedded. Elimination of useless (non-informative) features often provides a simpler pattern than when considering all features. If the obtained simpler pattern reflects the true answer, it is employed and FE is regarded to be success-ful. A clear drawback of unsupervised FE is that there are no guarantees to obtaining meaningful
simpler patterns coincident with true answers, in contrast to the optimal based methods that always provide answers independent of their usefulness. If unsupervised FE fails to provide simpler patterns, the unsupervised FE should be discarded. Thus, the important question is whether unsupervised FE works for a specific problem or not.
Classically, “unsupervised FE” often means “dimensionality reduction” (Jimenez-Rodriguez, 2007). In dimensionality reduction, new feature vectors are constructed from given features directly (linear transformation) or indirectly (non-linear transformation). Although many studies have fol-lowed this policy, there is a disadvantage, as every time new features are constructed, all features must be measured in advance. Thus, although dimensionality reduction technique can provide a smaller number (low dimensional) of features, the effort to measure huge numbers of features is not reduced. Unsupervised FE in our context differs from the conventional procedure. Similar to the supervised FE mentioned above, unsupervised FE in our context reduces the number of features that must be measured. Thus, we believe, in this context, this is the real meaning of unsupervised FE. To our knowledge, no previous studies have investigated this context of unsupervised FE.
In the following, we demonstrate that PCA based unsupervised FE works in at least three examples.
INTRODUCTION OF PCA BASED UNSUPERVISED FE AND ITS APPLICATIONS
Illustrative Examples of PCA Based Unsupervised FE
Before describing the application of PCA based unsupervised FE to biological examples, we would like to illustrate briefly how PCA works as an unsupervised FE. For simplicity, assume the following situation. There are M samples, each of

143
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
which has N features attributed to it. Then sup-pose the ith feature associated with the jth sample takes the value of xij. M samples can be classified as follows. M/2 samples (j=1,...,M/2) belong to the first class and the other M/2 samples (j=M/2 +1,...,M) belong to the second class. In addition, only N’ < N features have distinct values between the two classes and the other N-N’ features do not have any distinct values between the two classes. To demonstrate this situation, xijs are assumed to have the following values.
N(1,1/2) (i ≤N’ <N, 1 ≤ j ≤ Μ/2)
xij ∈ N(0,1/2) (i ≤N’ <N, M/2 < j ≤ Μ)
N(εij,1/2) (N’<i ≤N),
where N(μ,σ) is normal distribution with mean μ and standard deviation σ, and εij is a random number that obeys probability
P(εij =0)=P(εij=1)=1/2.
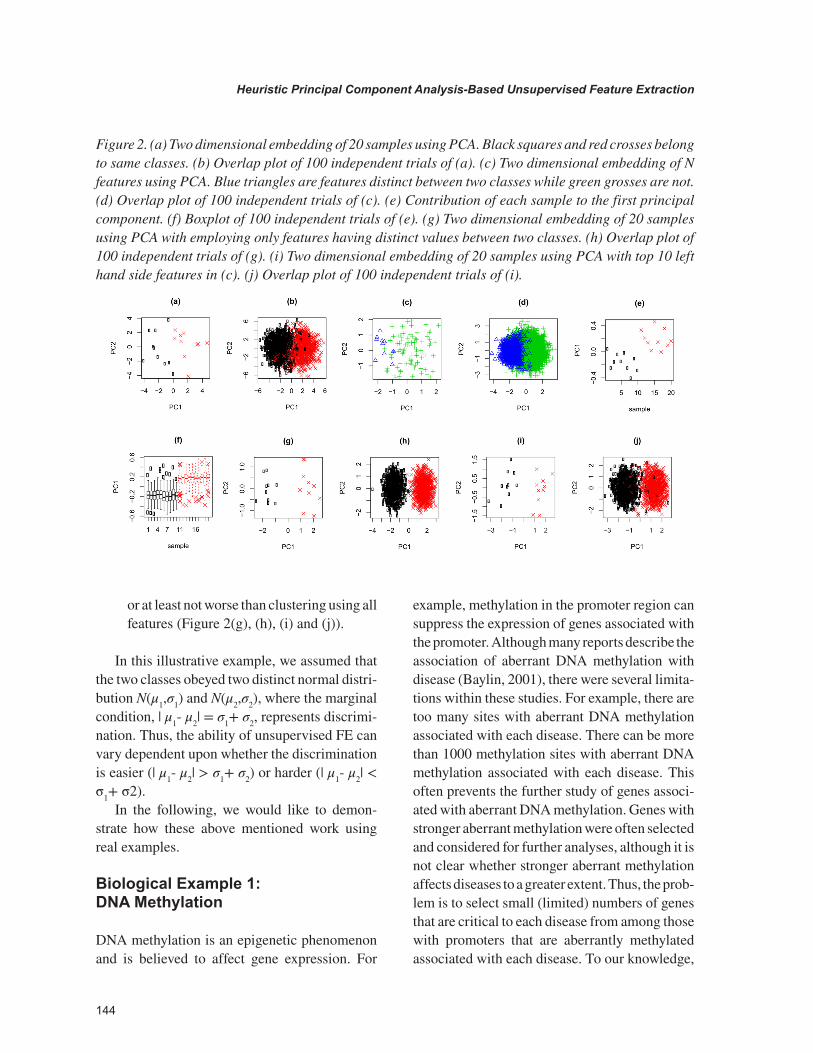
Figure 2(a) shows two dimensional embedding using PCA, of M(=20) samples with N(=100) features within which only the first N’(=10) features have distinct values between the two classes. Figure 2(b) is the overlap plot of 100 independent trials. It is obvious that M samples are divided into two classes. The problem is how to select only features that have distinct values between the two classes, i.e., i ≤N’, using PCA based unsupervised FE. Figure 2(c) shows the two dimensional embedding of N features and Figure 2(d) is the overlap plot of 100 independent trials. Ten features having distinct values between two classes (i ≤N’) are clearly located at the left hand side. The mean probability that 10 features are included in the top most 10 left hand side features in Figure 2 (d) is 0.7. Although we did not use label information to obtain Figures 2(c) and (d), PCA-based unsupervised FE clearly has ability to select features having distinct values
between two classes (i ≤N’). The reason why PCA based unsupervised FE can select features having distinct values between two features (i ≤N’) can be understood from Figure 2(e) that shows the contribution of each sample to the first principal component (PC1). Since PC1 clearly has distinct values between two classes, features having distinct values between two classes (i ≤N’) have more projections to PC1, thus are located at the left hand side (Figure 2(f) is the overlap plots of 100 independent trials).
Using features with distinct values between two classes can provide clearer clustering. Figure 2(g) shows the two dimensional embedding of M samples using only features with distinct values between two classes (i ≤N’) and Figure 2(h) shows the overlap plot of 100 independent trials. Even using the top 10 left hand side features in Figure 2(c), better clustering can be obtained than in Figure 2(a) (Figure 2 (i), Figure 2(j) shows the overlap plot of 100 independent trials), although only 70% of selected features are taken from features i ≤N’.
All of these demonstrations show how well PCA based unsupervised FE can work and the great potential PCA based unsupervised FE has for selecting features with distinct values between two classes, without using labeling information. That is,
1. Even if features having distinct values be-tween two classes are a minority (N’ << N), PCA can identify distinctions between two classes (Figure 2 (a) and (b)).
2. PCA based unsupervised FE can extract features with distinct values between two classes (Figure 1(c) and (d)).
3. Principal component that extracts features with distinct values between two classes can be selected by investigating contributions of each sample to the PC (Figure 1(e) and (f)).
4. Using only features having distinct values between two classes, clustering can be better
144
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
or at least not worse than clustering using all features (Figure 2(g), (h), (i) and (j)).
In this illustrative example, we assumed that the two classes obeyed two distinct normal distri-bution N(μ1,σ1) and N(μ2,σ2), where the marginal condition, | μ1- μ2| = σ1+ σ2, represents discrimi-nation. Thus, the ability of unsupervised FE can vary dependent upon whether the discrimination is easier (| μ1- μ2| > σ1+ σ2) or harder (| μ1- μ2| < σ1+ σ2).
In the following, we would like to demon-strate how these above mentioned work using real examples.
Biological Example 1: DNA Methylation
DNA methylation is an epigenetic phenomenon and is believed to affect gene expression. For
example, methylation in the promoter region can suppress the expression of genes associated with the promoter. Although many reports describe the association of aberrant DNA methylation with disease (Baylin, 2001), there were several limita-tions within these studies. For example, there are too many sites with aberrant DNA methylation associated with each disease. There can be more than 1000 methylation sites with aberrant DNA methylation associated with each disease. This often prevents the further study of genes associ-ated with aberrant DNA methylation. Genes with stronger aberrant methylation were often selected and considered for further analyses, although it is not clear whether stronger aberrant methylation affects diseases to a greater extent. Thus, the prob-lem is to select small (limited) numbers of genes that are critical to each disease from among those with promoters that are aberrantly methylated associated with each disease. To our knowledge,
Figure 2. (a) Two dimensional embedding of 20 samples using PCA. Black squares and red crosses belong to same classes. (b) Overlap plot of 100 independent trials of (a). (c) Two dimensional embedding of N features using PCA. Blue triangles are features distinct between two classes while green grosses are not. (d) Overlap plot of 100 independent trials of (c). (e) Contribution of each sample to the first principal component. (f) Boxplot of 100 independent trials of (e). (g) Two dimensional embedding of 20 samples using PCA with employing only features having distinct values between two classes. (h) Overlap plot of 100 independent trials of (g). (i) Two dimensional embedding of 20 samples using PCA with top 10 left hand side features in (c). (j) Overlap plot of 100 independent trials of (i).

145
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
there is no effective method to perform this task currently.
In contrast to the selection of aberrant DNA methylation sites based on pre-defined criterion of thresholds, e.g., significance or the amount of fold change, PCA based unsupervised FE is based on more data oriented criterion, because only PC used for FE need be specified. A priori criteria were not set up for the evaluation of features that represent aberrant DNA methylation. Thus, our PCA based unsupervised FE can potentially select limited numbers of critical genes among those with promoters that are aberrantly methylated and associated with each disease.
The first example of applying PCA based unsupervised FE to aberrant DNA methylation is to specify genes whose promoters are aber-rantly methylated associated with autoimmune diseases (Ballestar, 2011). Autoimmune diseases are complex diseases whose causes are rarely well understood, although internal abnormality of the immune systems may play critical roles in disease pathogenesis. Aberrant DNA methylation is also a potential candidate mechanism involved in the pathogenesis of autoimmune disease, because with monozygotic (MZ) twins, one might be healthy and the other develop autoimmune disease despite sharing the same genome. Javierre (2010) tried to identify genes whose promoters were commonly and aberrantly methylated in three autoimmune diseases: systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and dermatomyositis (DM). However, aberrant DNA methylation was only observed in SLE but not DM or RA. The data set they used was as follows. First, they found MZ twins that consisted of pairs of healthy controls and patients. They added two healthy age-gender matched controls to the twins. These four consisted of a unit and they collected five units for each of the three diseases. Thus, four individuals (patient or healthy control) × 5 units × 3 diseases = 60 samples. Promoter methylation of each sample was measured using microarray that had 1350 probes corresponding to promoters of potential
cancer-related genes. Thus, this demonstrates the problem of FE where 1350 features are analyzed in 60 samples. Thus, can PCA-based unsupervised FE identify undiscovered genes whose promoters are commonly and aberrantly methylated in three autoimmune diseases from this data set (Ishida, 2014)?
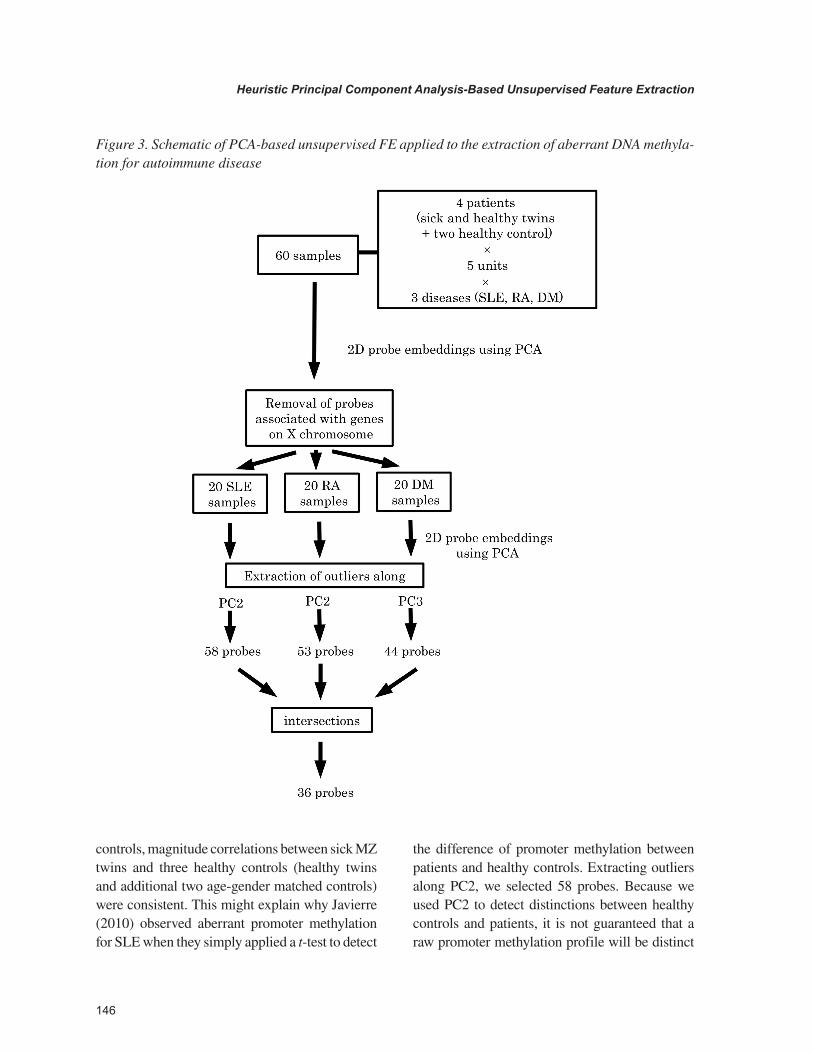
Figure 3 illustrates a schematic of PCA-based unsupervised FE applied to the extraction of aber-rant DNA methylation for autoimmune disease. First, PCA was applied to all 60 samples and probes were embedded to a 2D space. The first principal component (PC1) has almost equal contribution from each sample although the proportion of PC1 contribution is greater than 90%. Thus, we ignored PC1 while PC2 had distinct contributions from male and female samples, but contributions were not distinct between healthy controls and patients. Thus, PC2 was not interesting and we tried to suppress PC2. After the investigation, we found that the gender-dependent component PC2 expressed the differential promoter methylation of genes on chromosome X. Since females and males have different numbers of X chromosomes, it is well known that promoter methylation on chromosome X is gender dependent such that two genders have the same gene expression of genes on chromosome X. By excluding genes on X chromosome from the analysis, we successfully suppressed gender dependent PC2.
Next, we applied PCA to 20 samples in each disease separately. Again, for all three diseases, PC1 did not have a disease specific contribution from samples despite its large contribution. In contrast to PC1, PC2 (or PC3) exhibited a distinct contribution for all three diseases between patients and healthy controls, but its manner differed from disease to disease. Thus, we will discuss the dis-tinct contributions from healthy controls and each individual disease to PC2.
For SLE, the distinct contribution from healthy controls and patients of each disease to PC2 is relatively simple. In 20 samples, for all 5 units that consist of sick and healthy MZ twins and two healthy
146
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
controls, magnitude correlations between sick MZ twins and three healthy controls (healthy twins and additional two age-gender matched controls) were consistent. This might explain why Javierre (2010) observed aberrant promoter methylation for SLE when they simply applied a t-test to detect
the difference of promoter methylation between patients and healthy controls. Extracting outliers along PC2, we selected 58 probes. Because we used PC2 to detect distinctions between healthy controls and patients, it is not guaranteed that a raw promoter methylation profile will be distinct
Figure 3. Schematic of PCA-based unsupervised FE applied to the extraction of aberrant DNA methyla-tion for autoimmune disease

147
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
between healthy controls and patients. This point is checked by t-test between 58 selected probes and the remaining probes. For all five units, 58 probes were significantly more hypomethylated in sick MZ twins than in healthy twins (P < 10-11). Thus, PCA-based unsupervised FE successfully extracted aberrant methylated probes associated with SLE.
The case of RA was more complicated. PC1 did not have a distinct contribution between healthy controls and patients as described above while PC2 did. Although this point was similar to SLE, the magnitude of correlations between contributions of three healthy controls and that of patients to PC2 were not consistent among the units. Two additional healthy controls had larger or smaller contributions than sick twins and were dependent upon units. In addition, among five pairs of MZ twins, the contributions of the third pair to PC2 were not distinct between healthy and RA twins. Despite these difficulties, 53 probes selected by PCA-based unsupervised FE using PC2 exhibited significant differences (hypomethylation in patients) between healthy and sick twins excluding the third pair (P< 10-12, by t-test). This also demonstrated the difficulties in finding aberrantly methylated promoter regions us-ing simple statistical tests. Comparisons between sick twins and healthy controls, including healthy twins, may provide a different set of aberrantly methylated promoter regions because PC2 does not reflect consistent differences between sick twins and healthy controls. This might also explain why Javierre (2010) failed to identify aberrantly methylated promoter regions for RA.
Finally, a DM case was considered. This was even harder than RA to analyze. While PC1 did not have any distinct contributions between patients and healthy controls as mentioned above, PC2 exhibited a distinction between genders but not between healthy controls and patients. Although PC3 represented a distinction between healthy and sick twins, it did not exhibit a consistent magni-tude correlation between sick twins and healthy controls including healthy twins. In addition, the
magnitude correlation between healthy and sick twins was also consistent but opposite between males and females; hyper (hypo) methylated in male (female) DM twins. Despite these difficulties, 44 probes selected by PCA-based unsupervised FE using PC3 exhibited significant differences between healthy and sick twins (P< 10-10, by t-test).
Although we successfully identified promot-ers with aberrant methylation associated with autoimmune diseases, the feasibility of selected probes was not sufficient. There may be more suitable methods that identify a different selec-tion of probes. To confirm feasibility, we counted the number of probes in the intersection among the 58, 53 and 44 probes selected for SLE, RA and DM, respectively. Then 36 probes were com-monly selected. Because there are 1350 probes in a microarray, the P-value for this accidentally occurring was less than 10-96. As all three diseases are autoimmune diseases, it was plausible that probes with aberrant methylation associated with diseases were coincident. Thus, the feasibility of our analysis was high.
We also compared our results with those ob-tained by an independent data set. For SLE, we used the study by Jeffries (2011), where female genome methylation of T-cells from 11 SLE and 12 normal controls were measured. Because no single PCs exhibited distinct contributions be-tween normal controls and patients, combination of PC2 and PC3 was used for probe selection. This combination exhibited distinct methylation between patients and normal controls and the selected probes significantly overlapped with probes selected in the present study. Thus, the methodology used in this study is also valid for independent data sets. For RA, we used results from the study by Liu (2013) that measured pe-ripheral blood lymphocyte genome methylation from 335 patients and 354 normal controls. The probes selected in this study significantly over-lapped with probes selected by PCA-based FE with PC2 that exhibited significant differences between patients and normal controls. Thus, the
148
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
methodology used in this study was again valid for independent data sets.
To further confirm the feasibility of selected probes in this study, we checked the disease as-sociation of genes associated with the selected probes using the Gendoo server (Nakazato, 2009) (Table 2). As can be seen, most selected genes were associated with autoimmune diseases or immune related compounds, although not all of the associated P-values were significant. This
also confirmed the feasibility and superiority of our methodology.
Another example of the application of PCA based unsupervised FE to problems related to DNA methylation was the detection of genotype specific DNA methylation in esophageal squamous cell carcinoma (ESCC) (Kinoshita, 2014). ESCC is a lethal cancer disease whose 5-year survival rate is less than 20%. Yang (2010) investigated DNA methylation and genotypes using single nucleotide
Table 2. Disease association of genes selected by PCA based unsupervised FE
Gene Name Disease P-Value Compounds P-Value
AIM2 ---- Interferon-gamma 4.00E-10
CARD15 Autoimmune Diseases 0.02
Immune System Diseases 1.00E-04
Arthritis, Juvenile Rheumatoid 8.00E-04
CD82 Arthritis, Rheumatoid 0.13
CSF1R Arthritis, Rheumatoid 0.14
CSF3 Autoimmune Diseases 0.08
CSF3R Multiple Sclerosis 0.05
DHCR24 ----
ERCC3 ----
GRB7 ---- Immunoglobulins 0.03
HGF Autoimmune Diseases 0.28
Immune System Diseases 0.19
Arthritis, Rheumatoid 0.69
HOXB2 ---- Immune Sera 9.00E-03
IFNGR2 SLE 0.09
Multiple Sclerosis 0.09
LCN2 SLE 0.31
Multiple Sclerosis 0.32
LMO2 Severe Combined Immunodeficiency
1.00E-73
LTB4R Arthritis, Rheumatoid 2.00E-04
MMP14 Arthritis, Rheumatoid 0.08
MMP8 Arthritis, Rheumatoid 3.00E-05
Multiple Sclerosis 0.09
MPL SLE 0.02
PADI4 Arthritis, Rheumatoid 4.00E-89
continued on following page
149
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
polymorphism (SNP) arrays that made use of two restriction enzymes, Nsp and Sty. Although each array included only about 200,000 probes that were much lower than the estimated total number of potential methylation sites (>500,000), they could detect SNP abundance and SNP specific DNA methylation simultaneously when combined
with bisulfite treatment. They collected blood (non-mutated genome), tumor (mutated genome) and normal tissue adjusting to tumor (in between) samples from 30 patients.
Figure 4 illustrates how genes were selected using PCA-based FE. First, PCA was applied to genotype and DNA methylation separately for Sty
Table 2. Continued
Gene Name Disease P-Value Compounds P-Value
Multiple Sclerosis 7.00E-09
SLE 0.01
Autoimmune Diseases 0.01
PECAM1 Multiple Sclerosis 0.12
Autoimmune Diseases 0.13
PI3 Arthritis, Rheumatoid 0.11
RARA ---- Immunoglobulin M 0.03
Interferon-gamma 0.2
S100A2 ---- Receptors, Immunologic 3.00E-05
Interferon-gamma 0.1
SEPT9 ---- ----
SLC22A18 ---- ----
SPI1 Immunologic Deficiency Syndromes
5.00E-03
Immune System Diseases 0.02
Autoimmune Diseases 0.31
SPP1 SLE 4.00E-05
Arthritis, Rheumatoid 5.00E-03
Multiple Sclerosis, Chronic Progressive
0.02
STAT5A Autoimmune Diseases 0.21
SYK Arthus Reaction 5.00E-07
Autoimmune Diseases 8.00E-03
Arthritis, Rheumatoid 0.02
Immunologic Deficiency Syndromes
0.04
SLE 0.12
TIE1 Arthritis, Rheumatoid 0.03
TM7SF3 ---- ----
TRIP6 ---- ----
VAMP8 Multiple Sclerosis 0.01
SLE, systemic lupus erythematosus.
150
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
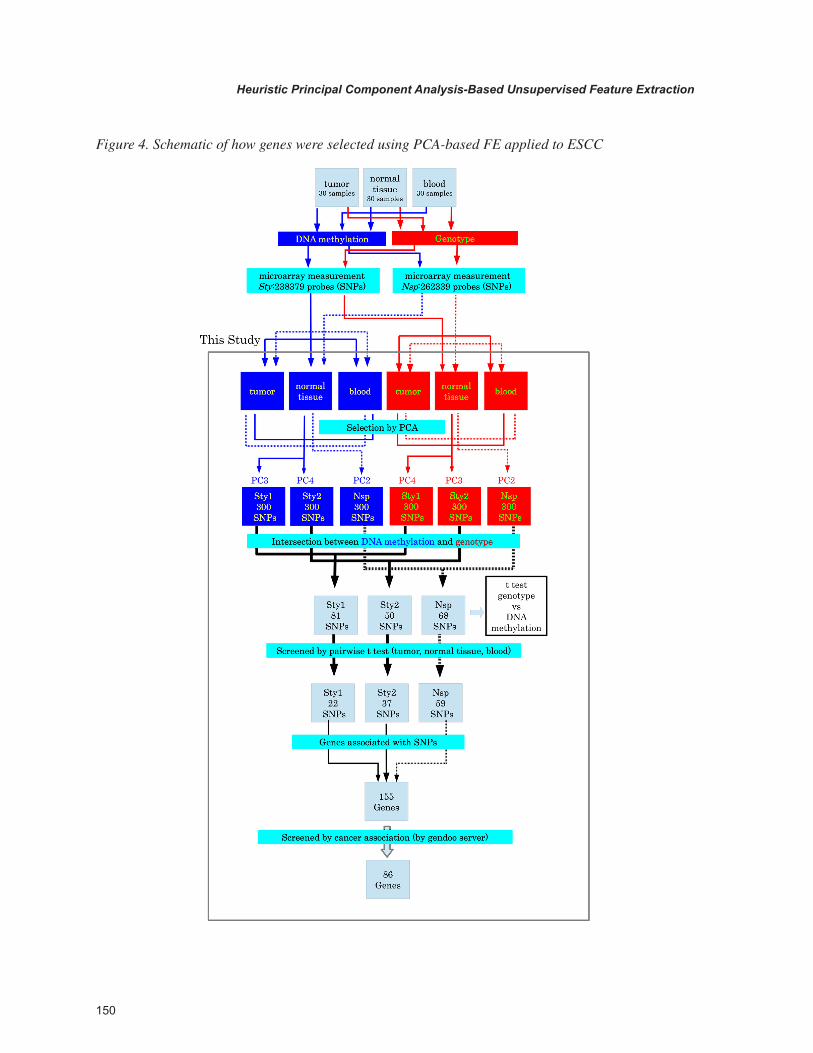
Figure 4. Schematic of how genes were selected using PCA-based FE applied to ESCC

151
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
and Nsp. Then, PC3 and PC4 (PC2) reflected dis-tinctions between blood, normal tissues and tumors for Nsp (Sty) array. Then, 300 outliers (probes or SNPs) along PC3 and PC4 (PC2) were extracted for Sty (Nsp) arrays of genotype/DNA methyla-tion measurements. Next, intersection between outliers from genotype and DNA methylation was selected. For Sty, PC3 (PC4) from DNA meth-ylation measurements was compared with PC4 (PC3) from genotype measurements. This set of SNPs was named Sty1(Sty2). For Nsp, PC2 from DNA methylation measurements was compared with PC2 from genotype measurements. This set of SNPs was named Nsp. The number of SNPs included in Sty1, Sty2 and Nsp SNP sets was 81, 50 and 68. Comparison between genotype and DNA methylation, demonstrated these selected SNPs were hypomethylated. Applying three pairwise t-tests (normal tissue vs tumor, blood vs tumor, and blood vs normal tissue), 22, 37 and 59 SNPs were significantly abundant in tumors compared with blood and normal tissues. Thus, we could successfully identify SNPs that were abundant and hypomethylated in tumors. Because this was a detection of SNP specific methylated SNPs, we called this “genotype-specific DNA methylation.”
To determine whether the selection of these SNPs was biologically meaningful, 155 genes associated with the SNPs were tested by Gendoo server (Nakazato, 2009) that reports disease asso-ciation of genes based on a text mining database. Interestingly, 86 of 155 genes were reported to be associated with cancers by the Gendoo server. Es-pecially, nine genes (CCND1, CKAP4, CRABP1, FGF3, MYEOV, PKP4, RPL14, SMAD3, and ZNF639) were associated with “Esophageal Neo-plasms” and “Carcinoma, Squamous cell.” This is coincident with the fact that the cancer considered in this study was ESCC. This demonstrated the biological significance of SNPs selected by PCA-based unsupervised FE.
Finally, we compared performance of PCA based unsupervised FE with other methods. Since this problem is a three classes problem, no other
methods were proposed since any other methods require ranking information between three classes (blood, tumor and normal tissue), while ranking was automatically decided when PCs used for selecting outliers were selected for PCA-based unsupervised FE. However, owing to PCA-based unsupervised FE, we identified that an abundance of SNPs associated with genes with genotype-specific DNA hypomethylation were ordered from blood, through normal tissue to tumor. Thus, we assigned these ranks when applying the other three classes of FE to the present problem. The methods employed were Pearson and Spearman correlation based FE, partial least square (PLS) based FE, stepwise FE and lasso. In correlation based FE, 300 SNPs whose abundance correlated with the ranking of three classes (tumor, normal tissue and blood) were selected. For PLS based FE, 300 SNPs with more contributions to PLS were selected. In stepwise FE, 3 class linear discriminant analysis (LDA) was applied by adding and removing SNPs until 300 SNPs were selected. In lasso, multivariate liner regression was performed with regularization terms and regularization constants were chosen such that 300 SNPs remained. Among these, stepwise FE did not converge and lasso could not be performed because of too many (>200,000) SNPs. Although PLS and two correlation based FE worked, the number of SNPs selected for Sty and SNPs were 14 and 49 (Pearson), 18 and 39 (Spearman), and 13 and 7 (PLS), which is lower than for PCA-based unsupervised FE (81, 50 and 69 for Sty1, Sty2 and Nsp 300 SNPs sets, respectively). Thus, we concluded that PCA-based unsupervised FE outperformed other FEs despite the need for three class ranking that was not required by PCA-based unsupervised FE.
In summary, we have applied PCA based unsu-pervised FE to two distinct examples where genes with aberrant promoter methylation associated with disease are sought. In the first example, we identified genes with promoters that are commonly and aberrantly methylated, for three autoimmune

152
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
diseases, SLE, RA, and DM. In the second ex-ample, we identified genotype-specific promoter aberrant methylation associated with ESCC. To our knowledge, these two examples are the first demonstration of successful application of PCA based unsupervised FE.
Biological Example 2: Biomarkers
The next example of applying PCA-based unsuper-vised FE to biological problems was its use in two studies of disease biomarker identification, using circulating microRNAs (miRNAs) (Murakami, 2012; Taguchi, 2013). Identification of disease biomarkers is always an important problem for diagnosis since effective biomarkers enables us to free medical doctors from diagnosis. By simply measuring the amount of biomarkers present, some diseases can easily be identified. One problem of biomarkers is their measurement. If it is not easy to measure the particular biomarker, such as by sur-gery, the usefulness of the biomarker is drastically decreased since doctors must be recruited for both diagnosis and surgery. Thus, biomarkers are more useful if they can be measured non-invasively.
From this point of view, circulating biomark-ers have attracted much interest (Almufti, 2014). Circulating biomarkers can be measured from various body fluids, such as blood, urine, and saliva. The advantages of circulating biomarkers are twofold; since fluid circulates through the body, it can reflect the various stages of the whole body and since it is easily obtained, it is non-invasive. However, one important disadvantage is that it might be too non-specific, in terms of disease and tissue involved. Reduced specificity makes it difficult to identify disease-circulating biomarkers. Circulating miRNAs might be prom-ising circulating biomarker candidates (Kosaka, 2013) although miRNA alone can suppress gene expression at the post-transcriptional level. In this section, we apply PCA-based unsupervised FE to identify circulating miRNA disease biomarkers.
Thus, the problem is to identify circulating miRNAs that reflect disease status. Since it is not expected that each miRNA reflect a specific disease status, combinatorial selections must be performed. Thus, this task is a typical FE since it selects a set of miRNAs. Because disease specific-ity of each miRNA is not expected to be strong, an effective set of miRNAs with slight specificity to disease must be identified. This is a difficult task because to be a suitable target by our PCA based unsupervised FE it does not require label-ing information.
The first example is hepatitis, an inflamma-tory disorder of the liver (Murakami, 2012). Although acute hepatitis is undoubtedly severe, chronic hepatitis often results in hepatic cancer through liver cirrhosis. Currently, there is no effective therapy for progressive chronic hepa-titis excluding exceptional antiviral therapies. Hepatocellular carcinoma is a severe hepatic cancer that often redevelops, although the 5-year survival rate is as high as 50–60%. Thus, frequent diagnosis using non-invasive diagnosis is very important. We recently developed a non-invasive biomarker for multiple chronic hepatitis, chronic hepatitis C (CHC), chronic hepatitis B (CHB), and non-alcoholic steatohepatitis (NASH), us-ing circulating miRNAs isolated from exosome purified blood (Murakami, 2012). Many reports studied miRNAs in progressing/suppressing liver diseases and identified multiple miRNAs as biomarkers. PCA-based unsupervised FE was applied to this problem. We collected total RNA from exosomes in blood from 64 CHC, 4 CHB, 12 NASH patients and 24 healthy controls. The amount of miRNA in each sample was measured by microarray. PCA-based unsupervised FE was applied to obtain miRNA profiles. First, 64 CHC samples and 24 normal controls were analyzed together. Nine miRNAs were selected and suc-cessfully discriminated between normal controls and CHC, with an accuracy of 96.59% using PCA-based LDA evaluated by leaving out one cross validation. Since microarray includes more

153
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
than 800 miRNAs, successful discrimination us-ing only nine selected miRNAs was remarkable. PCA-based LDA is the LDA using PCs up to some numbers. However, when all samples (64 CHC, 4 CHB, 12 NASH and 24 healthy controls) were analyzed together using twelve selected miRNAs, four classes (CHC, CHB, NASH and normal controls) were discriminated (87.50% samples were discriminated successfully). Furthermore, other pairwise discriminations between distinct diseases, i.e., CHC vs CHB, CHC vs NASH, and CHB vs NASH, achieved accuracies of 98.35, 97.37 and 87.50%, respectively, using less than 10 miRNAs. Finally, these were validated using an independent set that consisted of 31 CHC, 16 CHB, and 8 NASH patients. Using the same miRNAs selected for the original samples, CHC
vs CHB, CHC vs NASH, and CHB vs NASH, achieved accuracies of 74.47, 87.18 and 79.19%, respectively. Although performances in the inde-pendent sets were poorer than in the original set, they were still informative. This demonstrated the usefulness and robustness (stability) of PCA-based unsupervised FE when applied to the identification of circulating biomarkers. Figure 5(a) illustrates these processes.
The performance of PCA-based unsupervised FE was used not only for discrimination between diseases but also for the diagnosis of disease progression. There are two clinical measures of chronic hepatitis progression: inflammation and fibrosis. Inflammation is caused by cell injury as a result of immune system responses to virus infec-tion (CHB and CHC) or by accumulation of fat
Figure 5. (a) Diagrams that illustrate FE applied to liver diseases; (b) Diagrams that illustrate FE ap-plied to 14 diseases

154
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
(NASH). Fibrosis is caused by cell replacement by cell regeneration that gives rise to cirrhosis. Thus, inflammation and fibrosis are important measures of chronic hepatitis progression. However, it is not easy to diagnose these two diseases, because direct observation of liver tissue is required. If a non-invasive biomarker could differentially diagnose these two disorders, it would be useful. PCA-based unsupervised FE was applied to the discrimina-tion of inflammation (A0, A1, A2 and A3 stages) and fibrosis grades (F0, F1, F2 and F3 stages) in CHC. Three pairwise discriminations of inflam-mation (A1 vs A0+A2+A3, A2 vs. A0+A1+A3, A3 vs A0+A1+A2) achieved accuracies of 71.88, 75.00, and 82.81%, respectively. Similarly, three pairwise discriminations of inflammation (F1 vs. F0+F2+F3, F2 vs. F0+F1+F3, F3 vs. F0+F1+F2) achieved accuracies of 64.62, 70.31, and 73.44%, respectively. This also demonstrated the usefulness of PCA-based unsupervised FE.
Although the previous examples were limited to chronic hepatitis, many other diseases can be discriminated using circulating miRNAs in the blood. Keller (2011) tried to discriminate 14 diseases (lung cancer, pancreatitis, other pancre-atic tumors and diseases, ovarian cancer, chronic obstructive pulmonary disease, ductal pancreatic cancer, gastric cancer, sarcoidosis, prostate can-cer, acute myocardial infarction, periodontitis, multiple sclerosis, melanoma and Wilms’ tumor) from healthy controls using circulating miRNAs in serum. Although Keller (2011) successfully discriminated 14 diseases from normal controls, they failed to list specific miRNAs used for the discrimination. From the point of clinical applica-tion, definitive and limited numbers of miRNAs used for discrimination is better. Thus, we tried to discriminate 14 diseases from normal controls using ten specific miRNAs based on their data set. First, we applied PCA-based unsupervised FE for 14 pairs that consisted of one disease and normal controls. Achieved accuracies ranged from 0.713 to 0.892. Thus, we could successfully identify 10 disease-specific circulating miRNAs
that discriminated patients of each of the 14 diseases from normal controls. To test stability of the selected biomarkers, we applied a cross validation study of FE. We randomly selected 90% of samples and selected 10 miRNAs using PCA-based unsupervised FE. Repeating this procedure 100 times, we estimated the stability of selected biomarkers. Since there are 14 diseases, in total 140 miRNAs were selected for each trial. Among 140 miRNAs, 129 miRNAs were selected for all 100 trials. Thus, miRNAs selected by PCA-based unsupervised FE are highly stable. To compare stability of PCA-based unsupervised FE with other methods, we tested the stability of multiple FE methods. FE methods tested were t-test based FE (Keller, 2011), significance analysis of microar-rays (SAM) (Tusher, 2001), gene selection based on a mixture of marginal distributions (gsMMD) (Qiu, 2008), ensemble recursive feature elimina-tion (RFE) (Abeel, 2005), and unsupervised feature filtering (UFF) (Varshavsky, 2007). Among them, the only method that was competitive with PCA-based unsupervised FE was UFF, where 111 of 140 miRNAs were selected for all 100 trials. The performance of the other methods were 30 (SAM), 5 (upregulation by gsMMD), 1 (downregulation by gsMMD), 1 (RFE) and 0 (ensemble RFE), re-spectively. Since UFF was the only unsupervised method other than PCA-based unsupervised FE, the use of unsupervised procedures was essential to achieve good stability of FE.
We followed the study of Keller (2011) to discriminate between each disease and healthy controls. We have found that PCA-based unsuper-vised FE combined with PCA-based LDA using 10 selected miRNAs discriminated between diseases with accuracies typically of 0.8.
Finally, we validated the biological feasibility of our selected set of miRNAs using DIANA-mirPath (Papadopoulos, 2009) that evaluates a set of miRNAs based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of miRNA target genes. For all 14 diseases, reported KEGG pathways by DIANA-mirPath were also

155
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
related to corresponding diseases. Thus, from a biological point of view, PCA-based unsupervised FE successfully selected plausible sets of miRNAs as circulating biomarkers.
Although these analyses demonstrated the usefulness of PCA-based unsupervised FE for the selection of circulating biomarkers, there was an additional interesting finding: the possibility of a universal disease biomarker. As mentioned above, 140 miRNAs were potentially selected to discriminate between 14 diseases and normal controls. However, these 140 miRNAs were taken from only 12 miRNAs: hsa-miR-425, hsa-miR-15b, hsa-miR-185, hsa-miR-92a, hsa-miR-140-3p, hsa-miR-320a, hsa-miR-486-5p, hsa-miR-16, hsa-miR-191, hsa-miR-106b, hsa-miR-19b, and hsa-miR-30d. Even after extending to the discrimi-nation between diseases, i.e., compared with the possible number of selected miRNAs, 140 × 14 = 1960, the method selected the above 12 miRNAs plus three additional miRNAs, miR-130, miR-22, miR-720. Figure 5(b) illustrates these processes. This suggests that miRNAs selected by PCA-based unsupervised FE can serve as universal disease biomarkers independent of the disease analyzed. Since the miRNAs selected from the study by Keller et al (2011) and normal controls were shared, we also evaluated the performance of the 12 miRNAs for seven other diseases (Alzheimer disease (AD) (Leidinger, 2013), carcinoma (Ma-clellan, 2012), coronary artery disease (CAD) (GEO ID: GSE49823), nasopharyngeal carcinoma (NPC) (GEO ID: GSE43329), hepatocellular carcinoma (HCC) (Shen, 2013), breast cancer (BC) (Chan, 2013) and acute myeloid leukemia (AML) (Rommer, 2013), using an independent data set. Combining 12 miRNAs with support vector machine (SVM) resulted in accuracies ranging from 0.72 to 0.94. These performances were also competitive with the fully supervised method (lasso). Considering the 12 miRNAs were selected from a distinct disease in an independent set, this performance was impressive. These results demonstrated that PCA-based unsupervised FE
can achieve very high performance not possible by other methods (i.e., the proposal of universal disease biomarker) (Taguchi, 2014).
In summary, we applied PCA based unsuper-vised FE for the identification of two distinct cir-culating miRNA biomarkers. In the first example, we successfully discriminated between multiple liver diseases. This is a difficult task because both diseases are classified as hepatitis. In the second disease model, we also successfully identified specific (stable) circulating miRNA biomarkers that could discriminate between 14 diseases and normal controls. To our knowledge, this is the first study to demonstrate disease discrimination by the stable identification of circulating miRNAs.
Biological Example 3: Proteome
The last biological application to be tested by PCA-based unsupervised FE is the proteome of bacteria under different culture conditions (Taguchi, 2012). After the development of antibiotics, bacterial infectious diseases were eradicated. Recently, antibiotic-resistance has become a new problem to be solved. Because of the frequent appearance of antibiotic-resistant bacteria, alternative and new antibiotics have been developed. However, the frequency of antibiotic-resistance has outper-formed the frequency of the development of new antibiotics. It is now believed that most antibiotics will become ineffective in the near future because of the appearance of antibiotic-resistant bacteria. Why antibiotic-resistance can appear so frequently and quickly might be because of the unplanned overuse of antimicrobial agents and horizontal gene transfer among bacterial species, including harmful bacterial strains. In health-care or animal husbandry facilities, the overuse of antimicrobial agents allows bacterial flora drug-sensitive to become drug-resistant by selection of resistant bacteria in the guts of humans or animals. More-over, bacteria possess horizontal gene transfer systems such as gene transformation, conjugation of plasmids, bacterial phages, and transposons.

156
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
Drug-resistant genes often transfer to other bac-teria, even beyond the barrier of bacterial species, and function as drug-resistant phenotypes. Thus, if antibiotics can be developed that target only specific virulent bacteria, drug-resistance will become less frequent. However, to develop such a drug, detailed information regarding the bacterial metabolic systems is required.
To understand bacterial metabolic systems in detail, we employed proteomic analysis of bacte-rial growth conditions. Culturing processes are also very important for the infectious process, since bacteria must effectively increase their population to cause infectious diseases. How culturing processes differ dependent upon external culture conditions will enable us to understand the metabolic status during bacterial growth. For this process, we applied PCA-based unsupervised feature extraction.
Thus, the problem is to identify proteins that characterize the bacterial culturing process with-out pre-defined criterion. Proteins may monotoni-cally decrease/increase as culturing time increases or may be expressed/suppressed only at a specific time point. Some proteins may be expressed ei-ther intra- or extracellularly or both. We aimed to collect all proteins with characteristic behavior regardless of their class. To our knowledge, no methods other than unsupervised FE can fulfill this kind of requirement.
The samples were prepared as follows. A bacte-rial strain, Streptococcus pyogenes, was cultured under two distinct incubation conditions: stable and shaking. S. pyogenes occasionally causes life-threatening disease, streptococcal toxic shock syndrome, although it is sometimes part of the typical normal bacteria flora. The estimated annual number of S. pyogenes infection cases is greater than 700 million, although not all infections are life threatening. The total proteome was measured at four time points from initial to final growth stages. Incubation was repeated 3 times. Thus, in total, two incubation conditions × four time points × three biological replicates = 24 samples
available. Prior to proteome measurement, each sample was divided into two parts, the complete cellular fraction (wc) and the supernatant fraction (snt) using centrifugation. Thus, in total, there were 48 proteome profiles.
PCA was applied to the 48 proteome profiles and they were embedded into a two dimensional space. Three clear clusters were observed, i.e., wc cluster, early snt cluster and late snt cluster. In addition, while snt under shaking incubation conditions were equally divided into the early and late snt clusters, four snts under stable incubation were divided into an early snt cluster and three that belonged to the late cluster. This suggests that incubation processes under stable incubation conditions progress faster than that under shak-ing incubation conditions. This observation was reasonable since oxidizing stress under shaking incubation conditions is more severe than under stable incubation conditions, which may result in a delayed growth process.
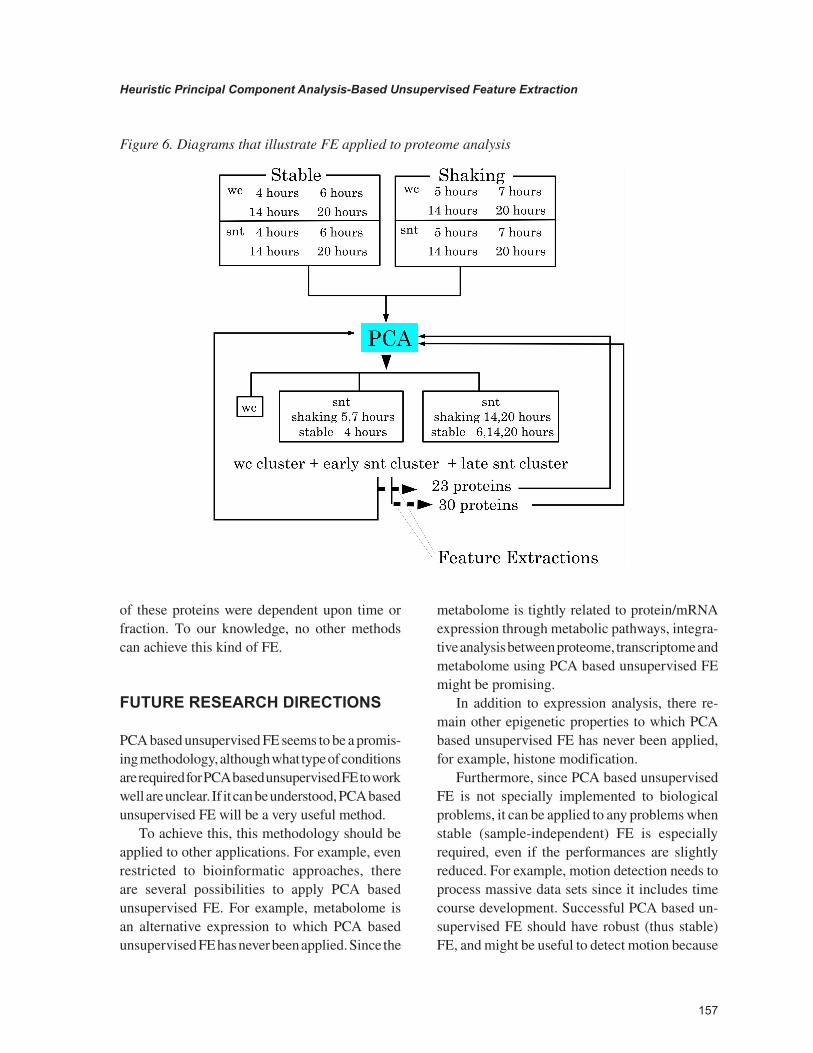
Because more than 400 proteins were ob-served by proteomic analyses, we tried to select a smaller number of proteins that contributed to the formation of the three cluster formation ob-served. PCA-based unsupervised FE was applied and 23 proteins were selected. Only using the 23 selected proteins, we obtained almost identical two dimensional embeddings of 48 proteome profiles, i.e., three clusters formation. Thus, 23 proteins were essential to form the three clusters observed and PCA-based unsupervised FE suc-cessfully extracted the important proteins. After removing 23 proteins, the 48 proteome profiles were again clustered into three clusters with a different shape by PCA, and PCA-based unsuper-vised FE was applied again. Then, we obtained 30 proteins where the same three clusters in two dimensional embeddings were formed by PCA. Thus, PCA-based unsupervised FE was successful. Figure 6 illustrates this process.
In summary, we identified critical and limited numbers of proteins that contribute to forming three clusters, regardless of how the expression
157
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
of these proteins were dependent upon time or fraction. To our knowledge, no other methods can achieve this kind of FE.
FUTURE RESEARCH DIRECTIONS
PCA based unsupervised FE seems to be a promis-ing methodology, although what type of conditions are required for PCA based unsupervised FE to work well are unclear. If it can be understood, PCA based unsupervised FE will be a very useful method.
To achieve this, this methodology should be applied to other applications. For example, even restricted to bioinformatic approaches, there are several possibilities to apply PCA based unsupervised FE. For example, metabolome is an alternative expression to which PCA based unsupervised FE has never been applied. Since the
metabolome is tightly related to protein/mRNA expression through metabolic pathways, integra-tive analysis between proteome, transcriptome and metabolome using PCA based unsupervised FE might be promising.
In addition to expression analysis, there re-main other epigenetic properties to which PCA based unsupervised FE has never been applied, for example, histone modification.
Furthermore, since PCA based unsupervised FE is not specially implemented to biological problems, it can be applied to any problems when stable (sample-independent) FE is especially required, even if the performances are slightly reduced. For example, motion detection needs to process massive data sets since it includes time course development. Successful PCA based un-supervised FE should have robust (thus stable) FE, and might be useful to detect motion because
Figure 6. Diagrams that illustrate FE applied to proteome analysis

158
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
it reduces the effort required to record movies. Finally, all previous examples to which FE has been applied are potential targets of PCA based unsupervised FE.
CONCLUSION
In this chapter, we introduced PCA based un-supervised FE and applied it to three biological examples: detections of aberrant DNA methylation associated with diseases, biomarker identifica-tion using circulating microRNA and bacterial proteome analysis.
In the first example, the main finding is that minor PCs can have potential roles for FE. This is because we have to select a small number of features. Naturally, major PCs cannot represent these minority features. One may wonder whether minor PCs simply reflect noise. To avoid this situation, multiple information sources should be considered. When aberrant promoter methylation associated with autoimmune disease is considered, the multiple information sources are multiple autoimmune diseases. When genotype-specific DNA aberrant methylation associated with ESCC is considered, genotype and DNA methylation are considered together. Since it is not likely that simple noises are identically associated with dis-tinct observations, this kind of integrated analysis prevents us from selecting the wrong PCs for FE.
In the second example, we found that even if each feature (circulating miRNA) does not exhibit disease specific expression, combinatorial selec-tions of features can exhibit disease specificity. In this case, unsupervised (labeling free) FE was very useful, since we could not select features based on disease specificity.
In the third example, we selected limited numbers of features (proteins) that contributed to cluster formation. Again, no labeling informa-tion was used for FE because we did not know what kind of difference contributed to the cluster
formation. This is why we successfully selected proteins that exhibited three clusters.
For all three examples, PCA based unsuper-vised FE worked very well. Further applications of PCA-based unsupervised FE in biological examples should be tested.
REFERENCES
Abeel, T., Helleputte, T., Van de Peer, Y., Du-pont, P., & Saeys, Y. (2010). Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics (Oxford, England), 26(3), 392–398. doi:10.1093/bioinfor-matics/btp630 PMID:19942583
Almufti, R., Wilbaux, M., Oza, A., Henin, E., Freyer, G., & Tod, M. et al. (2014). A critical review of the analytical approaches for circulat-ing tumor biomarker kinetics during treatment. Annals of Oncology, 25(1), 41–56. doi:10.1093/annonc/mdt382 PMID:24356619
Ballestar, E. (Ed.). (2011). Epigenetic contribu-tions in autoimmune disease. Heidelberg, Ger-many: Springer. doi:10.1007/978-1-4419-8216-2
Baylin, S. B., Esteller, M., Rountree, M. R., Bach-man, K. E., Schuebel, K., & Herman, J. G. (2001). Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Human Molecular Genetics, 10(7), 687–692. doi:10.1093/hmg/10.7.687 PMID:11257100
Bozdogan, H. (1987). Model selection and Akaike’s Information Criterion (AIC): The general theory and its analytical extensions. Psychometri-ka, 52(3), 345–370. doi:10.1007/BF02294361
Cao, L. J., Chua, K. S., Chong, W. K., Lee, H. P., & Gu, Q. M. (2003). A comparison of PCA, KPCA and ICA for dimensionality reduction in support vector machine. Neurocomputing, 55(1&2), 321–336.

159
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
Chan, M., Liaw, C. S., Ji, S. M., Tan, H. H., Wong, C. Y., & Thike, A. A. et al. (2013). Identification of circulating microRNA signatures for breast cancer detection. Clinical Cancer Research, 19(16), 4477–4487. doi:10.1158/1078-0432.CCR-12-3401 PMID:23797906
Chu, S., Keerthi, S., Ong, C. J., & Ghahramani, Z. (2006). Bayesian Support Vector Machines for Feature Ranking and Selection. In I. Guyon & A. Elisseeff (Eds.), Feature Extraction: Studies in Fuzziness and Soft Computing (Vol. 207, pp. 403–418). Berlin: Springer. doi:10.1007/978-3-540-35488-8_19
De Backer, S., Naud, A., & Scheunders, P. (1998). Non-linear dimensionality reduction techniques for unsupervised feature extraction. Pattern Rec-ognition Letters, 19(8), 711–720. doi:10.1016/S0167-8655(98)00049-X
Diaz-Uriarte, R., & Alvarez de Andres, S. (2006). Gene selection and classification of microarray data using random forest. BMC Bio-informatics, 7(1), 3. doi:10.1186/1471-2105-7-3 PMID:16398926
Girosi, F., Jones, M., & Poggio, T. (1995). Regu-larization Theory and Neural Networks Archi-tectures. Neural Computation, 7(2), 219–269. doi:10.1162/neco.1995.7.2.219
Gyon, I., & Elisseeff, A. (2003). An Introduc-tion to Variable and Feature Selection. Journal of Machine Learning Research, 3, 1157–1182.
Ishida, S., Umeyama, H., Iwadate, M., & Ta-guchi, Y.-h. (2014). Bioinformatic Screening of Autoimmune Disease Genes and Protein Structure Prediction with FAMS for Drug Discovery. Protein Pept Lett. 21(8), 828-39. doi: 10.2174/09298665113209990052 PMID: 23855671
Javierre, B. M., Fernandez, A. F., Richter, J., Al-Shahrour, F., Martin-Subero, J. I., & Rodri-guez-Ubreva, J. et al. (2010). Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Research, 20(2), 170–179. doi:10.1101/gr.100289.109 PMID:20028698
Jeffries, M. A., Dozmorov, M., Tang, Y., Mer-rill, J. T., Wren, J. D., & Sawalha, A. H. (2011). Genomewide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythe-matosus. Epigenetics, 6(5), 593–601. doi:10.4161/epi.6.5.15374 PMID:21436623
Jimenez-Rodriguez, L. O., Arzuaga-Cruz, E., & Velez-Reyes, M. (2007). Unsupervised Linear Feature-Extraction Methods and Their Effects in the Classification of High-Dimensional Data. Geoscience and Remote Sensing, 45(2), 469–483. doi:10.1109/TGRS.2006.885412
Keller, A., Leidinger, P., Bauer, A., Elsharawy, A., Haas, J., & Backes, C. et al. (2011). Toward the blood-borne miRNome of human diseases. Nature Methods, 8(10), 841–843. doi:10.1038/nmeth.1682 PMID:21892151
Kinoshita, R., Iwadate, M., Umeyama, H., & Taguchi, Y.h. (2014). Genes associated with genotype-specific DNA methylation in squamous cell carcinoma as candidate drug targets. BMC Systems Biology, 8(S1), S4. doi:10.1186/1752-0509-8-S1-S4 PMID:24565165
Kosaka, N. (Ed.). (2013). Circulating microRNAs. Heidelberg, Germany: Springer. doi:10.1007/978-1-62703-453-1
Leidinger, P., Backes, C., Deutscher, S., Schmitt, K., Mueller, S. C., & Frese, K. et al. (2013). A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biology, 14(7), R78. doi:10.1186/gb-2013-14-7-r78 PMID:23895045

160
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
Liu, Y., Aryee, M. J., Padyukov, L., Fallin, M. D., Hesselberg, E., & Runarsson, A. et al. (2013). Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nature Biotechnology, 31(2), 142–147. doi:10.1038/nbt.2487 PMID:23334450
Maclellan, S. A., Lawson, J., Baik, J., Guillaud, M., Poh, C. F., & Garnis, C. (2012). Differential expression of miRNAs in the serum of patients with high-risk oral lesions. Cancer Med, 1(2), 268–274. doi:10.1002/cam4.17 PMID:23342275
Murakami, Y., Toyoda, H., Tanahashi, T., Tanaka, J., Kumada, T., & Yoshioka, Y. et al. (2012). Comprehensive miRNA expression analysis in peripheral blood can diagnose liver disease. PLoS ONE, 7(10), e48366. doi:10.1371/journal.pone.0048366 PMID:23152743
Nakazato, T., Bono, H., Matsuda, H., & Takagi, T. (2009). Gendoo: functional profiling of gene and disease features using MeSH vocabulary. Nucleic Acids Res, 37(Web Server), W166–169.
Papadopoulos, G. L., Alexiou, P., Maragkakis, M., Reczko, M., & Hatzigeorgiou, A. G. (2009). DIANAmirPath: Integrating human and mouse microRNAs in pathways. Bioinformatics (Oxford, England), 25(15), 1991–1993. doi:10.1093/bio-informatics/btp299 PMID:19435746
Prasad, S., & Bruce, L. M. (2008). Overcoming the Small Sample Size Problem in Hyperspectral Classification and Detection Tasks. In Geoscience and Remote Sensing Symposium: Vol.5. IGARSS 2008 (pp. V381-V384) IEEE International. doi:10.1109/IGARSS.2008.4780108
Qiu, W., He, W., Wang, X., & Lazarus, R. (2008). A marginal mixture model for selecting differentially expressed genes across two types of tissue samples. The International Journal of Biostatistics, 4(1), 20. doi:10.2202/1557-4679.1093 PMID:20231912
Rommer, A., Steinleitner, K., Hackl, H., Sch-neckenleithner, C., Engelmann, M., & Scheideler, M. et al. (2013). Overexpression of primary microRNA 221/222 in acute myeloid leukemia. BMC Cancer, 13(1), 364. doi:10.1186/1471-2407-13-364 PMID:23895238
Scholkopf, B. (2001). In T. K. Leen, T. G. Diet-terich, & V. Tresp (Eds.), The kernel trick for distances (pp. 301–307). Advances in neural information processing systems. MIT Press.
Shen, J., Wang, A., Wang, Q., Gurvich, I., Siegel, A. B., Remotti, H., & Santella, R. M. (2013). Exploration of Genome-wide Circulating Mi-croRNA in Hepatocellular Carcinoma (HCC): MiR-483-5p as a Potential Biomarker. Cancer Epidemiology, Biomarkers & Prevention, 22(12), 2364–2373. doi:10.1158/1055-9965.EPI-13-0237 PMID:24127413
Taguchi, Y.-h., & Murakami, Y. (2014). Univer-sal disease biomarker: Can a fixed set of blood microRNAs diagnose multiple diseases? BMC Research Notes, 7, 581. doi:10.1186/1756-0500-7-581
Taguchi, Y.-, & Murakami, Y. (2013). Principal component analysis based feature extraction approach to identify circulating microRNA bio-markers. PLoS ONE, 8(6), e66714. doi:10.1371/journal.pone.0066714 PMID:23874370
Taguchi, Y.-h., & Okamoto, A. (2012). Principal Component Analysis for Bacterial Proteomic Analysis. In T. Shibata, H. Kashima, J. Sese, & S. Ahmad. (Eds.), Pattern Recognition in Bioin-formatics: 7th IAPR International Conference, PRIB 2012 (pp. 441–452). Berlin: Springer. doi:10.1007/978-3-642-34123-6_13
Tibshirani, R. (1996). Regression shrinkage and selection via the lasso. Journal of the Royal Sta-tistical Society. Series B. Methodological, 58(1), 267–288.

161
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
Tusher, V. G., Tibshirani, R., & Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America, 98(9), 5116–5121. doi:10.1073/pnas.091062498 PMID:11309499
Varshavsky, R., Gottlieb, A., Horn, D., & Linial, M. (2007). Unsupervised feature selection under perturbations: Meeting the challenges of biological data. Bioinformatics (Oxford, England), 23(24), 3343–3349. doi:10.1093/bioinformatics/btm528 PMID:17989091
Wang, Y., Tetko, I. V., Hall, M. A., Frank, E., Facius, A., Mayer, K. F., & Mewes, H. W. (2005). Gene selection from microarray data for cancer classification–a machine learning approach. Computational Biology and Chemistry, 29(1), 37–46. doi:10.1016/j.compbiolchem.2004.11.001 PMID:15680584
Yang, H. H., Hu, N., Wang, C., Ding, T., Dunn, B. K., & Goldstein, A. M. et al. (2010). Influ-ence of genetic background and tissue types on global DNA methylation patterns. PLoS ONE, 5(2), e9355. doi:10.1371/journal.pone.0009355 PMID:20186319
ADDITIONAL READING
Bishop, C. M. (2006). Pattern Recognition and Machine Learning. New York: Springer.
Broemeling, L. D. (2011). Advanced Bayesian Methods for Medical test Accuracy. Boca Raton, FL: CRC Press. doi:10.1201/b11055
Chen, M., & Hofestädt, R. (2014). Approaches in Integrative Bioinformatics. Heidelberg: Springer. doi:10.1007/978-3-642-41281-3
Cho, W. C. S. (Ed.). (2011). MicroRNAs in Cancer Translational Research. Heidelberg: Springer. doi:10.1007/978-94-007-0298-1
Etheridge, A., Lee, I., Hood, L., Galas, D., & Wang, K. (2011). Extracellular microRNA: A new source of biomarkers. Mutation Research, 717(1-2), 85–90. doi:10.1016/j.mrfmmm.2011.03.004 PMID:21402084
Hamelryck, T., Mardia, K., & Ferkinghoff-Borg, J. (Eds.). (2012). Bayesian Methods in Structural Bioinformatics. Heidelberg: Springer. doi:10.1007/978-3-642-27225-7
Hastie, T., Tibshrani, R., & Friedman, J. (2009). The elements of Statistical Learning: Data Mining, Inference and Prediction. New York: Springer. doi:10.1007/978-0-387-84858-7
Hollander, M., Wolfe, D. A., & Chicken, E. (2013). Nonparametric statistical methods. New Jersey: Willy.
Karpf, A. R. (Ed.). (2013). Epigenetic Alter-nations in Oncogenesis. New York: Springer. doi:10.1007/978-1-4419-9967-2
Kim, D., Shin, H., Sohn, K. A., Verma, A., Ritchie, M. D., & Kim, J. H. (2014). Incorporat-ing inter-relationships between different levels of genomic data into cancer clinical outcome prediction. Methods (San Diego, Calif.), 67(3), 344–353. doi:10.1016/j.ymeth.2014.02.003 PMID:24561168
Kosaka, N., Iguchi, H., & Ochiya, T. (2010). Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Science, 101(10), 2087–2092. doi:10.1111/j.1349-7006.2010.01650.x PMID:20624164
Lopez-Serra, P., & Esteller, M. (2012). DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene, 31(13), 1609–1622. doi:10.1038/onc.2011.354 PMID:21860412
Mallick, B., & Ghosh, Z. (Eds.). (2012). Regulato-ry RNAs. Heidelberg: Springer. doi:10.1007/978-3-642-22517-8

162
Heuristic Principal Component Analysis-Based Unsupervised Feature Extraction
Mamitsuka, H., DeLisi, C., & Kanehisa, M. (Eds.). (2013). Data Mining for Systems Biology: Meth-ods and Protocols. New York: Humana Press. doi:10.1007/978-1-62703-107-3
Mathivanan, S., Ji, H., & Simpson, R. J. (2010). Exosomes: Extracellular organelles important in intercellular communication. Journal of Proteomics, 3(10), 1907–1920. doi:10.1016/j.jprot.2010.06.006 PMID:20601276
Monticelli, S. (Ed.). (2010). MicroRNAs and the Immune System. New York: Humana Press. doi:10.1007/978-1-60761-811-9
Novianti, P. W., Roes, K. C., & Eijkemans, M. J. (2014). Evaluation of gene expression classification studies: Factors associated with classification performance. PLoS ONE, 9(4), e96063. doi:10.1371/journal.pone.0096063 PMID:24770439
Rangwala, H., & Karypis, G. (2010). In-troduction to Protein Structure Prediction: Methods and Algorithms. New Jersey: Willy. doi:10.1002/9780470882207
Rodriguez-Ezpeleta, N., Hackenberg, M., & Aransay, A. M. (Eds.). (2012). Bioinformatics for High Throughput Sequencing. Heidelberg: Springer. doi:10.1007/978-1-4614-0782-9
Shen, B. (Ed.). (2013). Bioinformatics for Di-agnosis, Prognosis and Treatment of Complex Diseases. Heidelberg: Springer. doi:10.1007/978-94-007-7975-4
Wan, J. (Ed.). (2013). Introduction to Genetics: DNA Methylation, Histone Modification and Gene Regulation. Hong Kong: iConcept press.
Witten, I. H., & Frank, E. (2005). Data Mining. San Francisco: Elsevier.
Wu, W. (Ed.). (2011). MicroRNA and Cancer. New York: Humana Press. doi:10.1007/978-1-60761-863-8
Yousef, M., & Allmer, J. (Eds.). (2014). miRNom-ics. New York: Humana Press.
Zen, K., & Zhang, C. Y. (2012). Circulating mi-croRNAs: A novel class of biomarkers to diagnose and monitor human cancers. Medicinal Research Reviews, 32(2), 326–348. doi:10.1002/med.20215 PMID:22383180
KEY TERMS AND DEFINITIONS
Circulating Biomarker: Biomarker that discriminates diseases and are circulating within body. Typical example is blood.
Feature Extraction (FE): FE is the method to select limited number of (useful) variables among given many variables.
Linear Discriminant Analysis (LDA): LDA is linear method that constructs combinatorial features that discriminate something, e.g., patient vs healthy control.
Principal Component Analysis (PCA): PCA is the linear method that embeds high dimensional structure onto the lower dimensional space.
Promoter or DNA Methylation: Methylation takes place along genome sequences. It is generally known to be the most significant epigenetic effects that regulate gene expression. Promoter methyla-tion is supposed to suppress gene expression.
Proteome: Omics of proteins, i.e., a set of proteins expressed in whole bodies, cells, or environments.
Supervised and Unsupervised Learning: These are terminologies of machine or statisti-cal learning theory. Supervised learning is to learn something, (e.g., pattern recognition, with regard to “teacher” signals). On the other hand, unsupervised learning is performed without any “correct” answers.
Related Documents











