Cliriical Genetics 1977: 11: 137-146 Heterozygote detection in phenylketonuria FLEMMING G+ER AND GERT HANSEN John F. Kennedy Institute, Glostrup, Denmark Phenylalanine loading was carried out on 105 parents of children with phenylalanine hydroxylase deficiency and 33 apparently normal individuals with no family history of phenylketonuria. The best discriminant was found to be the logarithmic transformation of the slope of the rise in serum tyrosine multiplied by the maximum serum tyrosine con- centration over the maximum serum phenylalanine concentration obtained after an oral load with a pure solution of L-phenylalanine. The overlap between heterozygotes for phenylketonuria and normal homozygotes was 2.4 %. The distribution of the discriminant values suggested three heterozygous phenotypes for phenylalanine hydroxylase deficiency, and the phenotypic combination of parents could be correlated to the phenotype of their affected offspring, i.e. classical phenylketonuria, mild phenylketonuria or hyperphenyl- alaninemia. The probability of heterozygosity for phenylketonuria was determined by means of the distribution of the discriminant values of the heterozygotes and that of normal homozygotes. The likelihood of being a heterozygote was corrected for the genetic background of the person requiring genetic counseling, and was finally expressed as the percentage probability of being a heterozygote for phenylketonuria. Received I0 September, accepted for pirblicntiori 30 September I976 The enzyme deficient in phenylketonuria, phenylalanine hydroxylase, occurs in the liver (Kaufman 1969), but not in phytohe- magglutinin-stimulated human lymphocytes (Horn & Giittler 1976). Heterozygous car- riers for this deficiency are therefore prefer- ably detected by indirect methods, e.g. by the administration of a loading dose of phe- nylalanine (Hsia et al. 1956. Rampini et al. 1969, Westwood 8: Raine 1973). In the last decade, a growing number of observations have shown heterogeneity in phenylketonuria. Instead of a single clinical and biochemical entity, a spectrum of dis- orders in the hydroxylation of phenylala- nine to tyrosine has been recognized (Ram- pini et al. 1969, Menkes & Holtzman 1970, Blaskovics et al. 1974). The aim of the present investigation was to develop a test of heterozygosity for phenylketonuria with high discriminatory ability, in order to examine whether the heterogeneity among affected homozygotes, mentioned above, is reflected by a similar heterogeneity of the heterozygous phenotypes. Material and Methods The criteria for distinguishing classical ver- Part of this material was presented at The Fourth International Congress for the Inter- national Association for the Scientific Study of Mental Deficiency, Washington, D.C., August 1976.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Cliriical Genetics 1977: 11: 137-146
Heterozygote detection in phenylketonuria
FLEMMING G+ER AND GERT HANSEN John F. Kennedy Institute, Glostrup, Denmark
Phenylalanine loading was carried out on 105 parents of children with phenylalanine hydroxylase deficiency and 33 apparently normal individuals with no family history of phenylketonuria. The best discriminant was found to be the logarithmic transformation of the slope of the rise in serum tyrosine multiplied by the maximum serum tyrosine con- centration over the maximum serum phenylalanine concentration obtained after an oral load with a pure solution of L-phenylalanine. The overlap between heterozygotes for phenylketonuria and normal homozygotes was 2.4 %. The distribution of the discriminant values suggested three heterozygous phenotypes for phenylalanine hydroxylase deficiency, and the phenotypic combination of parents could be correlated to the phenotype of their affected offspring, i.e. classical phenylketonuria, mild phenylketonuria or hyperphenyl- alaninemia. The probability of heterozygosity for phenylketonuria was determined by means of the distribution of the discriminant values of the heterozygotes and that of normal homozygotes. The likelihood of being a heterozygote was corrected for the genetic background of the person requiring genetic counseling, and was finally expressed as the percentage probability of being a heterozygote for phenylketonuria.
Received I 0 September, accepted for pirblicntiori 30 September I976
The enzyme deficient in phenylketonuria, phenylalanine hydroxylase, occurs in the liver (Kaufman 1969), but not in phytohe- magglutinin-stimulated human lymphocytes (Horn & Giittler 1976). Heterozygous car- riers for this deficiency are therefore prefer- ably detected by indirect methods, e.g. by the administration of a loading dose of phe- nylalanine (Hsia et al. 1956. Rampini et al. 1969, Westwood 8: Raine 1973).
In the last decade, a growing number of observations have shown heterogeneity in phenylketonuria. Instead of a single clinical and biochemical entity, a spectrum of dis- orders in the hydroxylation of phenylala-
nine to tyrosine has been recognized (Ram- pini e t al. 1969, Menkes & Holtzman 1970, Blaskovics et al. 1974). The aim of the present investigation was t o develop a test of heterozygosity for phenylketonuria with high discriminatory ability, in order to examine whether the heterogeneity among affected homozygotes, mentioned above, is reflected by a similar heterogeneity of the heterozygous phenotypes.
Material and Methods
The criteria for distinguishing classical ver-
Part of this material was presented at The Fourth International Congress for the Inter- national Association for the Scientific Study of Mental Deficiency, Washington, D.C., August 1976.
138 G U T T L E R A N D H A N S E N
Fasling serum phenylalanine correlaled lo the eeuvalent ra l io of phenylalanins to tyrosine
82 Helerozygoles lor P K U A 33 Controls
i 3.0
. . :
'.I . i
c
Obi I I
1 I I I I I I 0 90 100 110 120 130 140 150 160 170
Serum phnylalanine I i m o l / I I
sus mild phenylketonuria, and these forms versus persistent hyperphenylalaninemia, have been described in previous papers (Giittler & Wamberg 1972, Giittler & Han- sen 1976). Briefly, to keep serum phenyl- alanine levels within 180-425 prnolil (3-7 mg/100 ml), children with classical phenyl- ketonuria tolerate 9-18 % and children with the mild form of phenylketonuria 22-36 % of a normal daily intake of phenyl- alanine. This distinction cannot be made until the child is more than 2% years of age (c.f. Giittler & Hansen 1976). Children with persistent hyperphenylalaninemia show serum phenylalanine values within
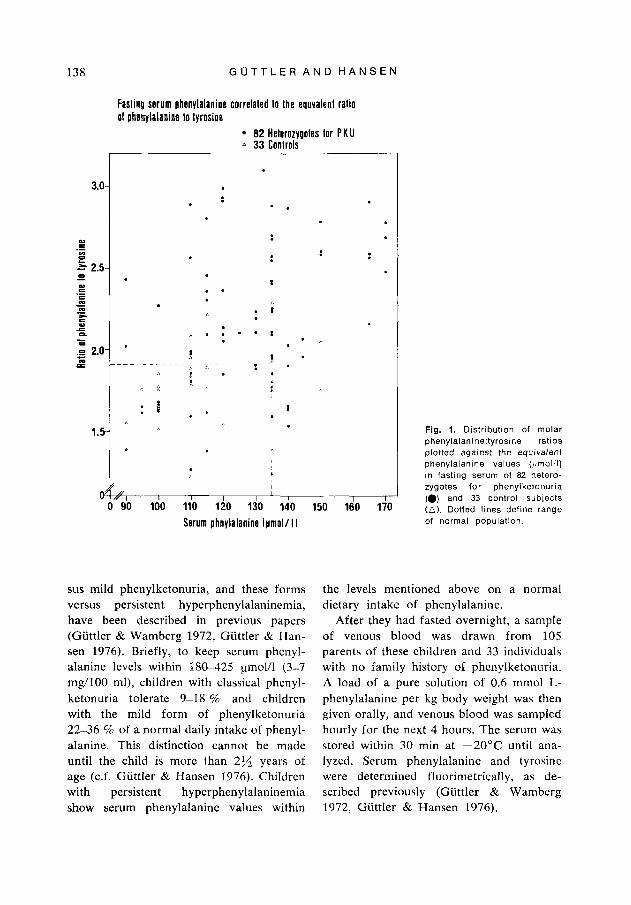
Fig. 1. Distribution of molar phenyla1anine:tyrosine ratios plotted against the equivalenl phenylalanine values ((,mol/l) in fasting serum of 82 hetero- zygotes for phenylketonuria (0) and 33 control subjects (A). Dotted lines define range of normal population.
the levels mentioned above on a normal dietary intake of phenylalanine.
After they had fasted overnight, a sample of venous blood was drawn from 105 parents of these children and 33 individuals with no family history of phenylketonuria. A load of a pure solution of 0.6 mmol L- phenylalanine per kg body weight was then given orally, and venous bIood was sampled hourly for the next 4 hours. The serum was stored within 30 min at -20°C until ana- lyzed. Serum phenylalanine and tyrosine were determined fluorimetrically, as de- scribed previously (Giittler & Wamberg 1972, Giittler & Hansen 1976).
H E T E R O Z Y G O T E D E T E C T I O N I N P H E N Y L K E T O N U R I A 139
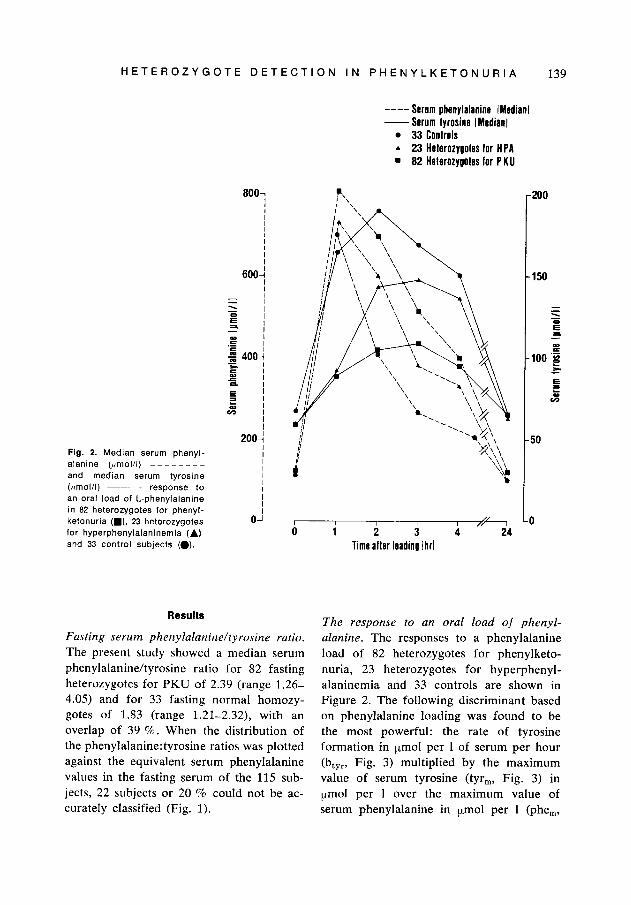
Flg. 2. Median serum phenyl- alanine (!rnol/l) - - - - - - - - and median serum tyrosine (pmolll) __- response to an oral load of L-phenylalanine in 82 heterozygotes for phenyl- ketonuria (W), 23 heterozygotes for hyperphenylalaninemia (A) and 33 control subjects (0).
---- Serum phenylalanine Median1 -Serum tyrosine IMedianl
33 Controls A 23 Helerozygoles for H PA . 82 Helerozygoles lor P K U
Results
Fasting serum phenylalanine/tyrosine ratio. The present study showed a median serum phenylalanine/tyrosine ratio for 82 fasting heterozygotes for PKU of 2.39 (range 1.26- 4.05) and for 33 fasting normal homozy- gotes of 1.83 (range 1.21-2.32), with an overlap of 39 %. When the distribution of the phenyla1anine:tyrosine ratios was plotted against the equivalent serum phenylalanine values in the fasting serum of the 115 sub- jects, 22 subjects or 20 % could not be ac- curately classified (Fig. 1).
I I I I I /h 0 1 2 3 4 24
l ime alter loading I hrl
The response to an oral load of phenyl- alanine. The responses to a phenylalanine load of 82 heterozygotes for phenylketo- nuria, 23 heterozygotes for hyperphenyl- alaninemia and 33 controls are shown in Figure 2. The following discriminant based on phenylalanine loading was found to be the most powerful: the rate of tyrosine formation in pmol per 1 of serum per hour (btyr, Fig. 3) multiplied by the maximum value of serum tyrosine (tyr,, Fig. 3) in ymol per 1 over the maximum value of serum phenylalanine in pmol per 1 (phe,,
G O T T L E R A N D H A N S E N
I I 1 I I I /h Lo 0 I 2 3 4 24 Time alter load in i lhrl
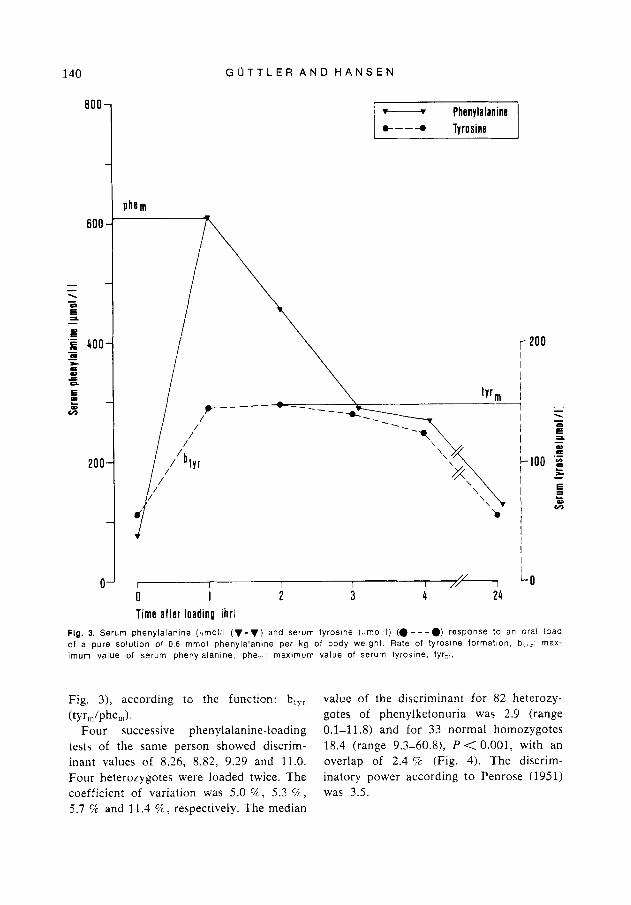
Fig. 3. Serum phenylalanine (umoi;l ( v - v ) and serum tyrosine ((tmol I) (0 ---a) response to an oral load of a pure solution of 0 6 mmol phenylalanine per kg of body weight. Rate of tyrosine formation, b , > r ; max- imum value of serum phenylalanine, phe,,,; maximum value of serum tyrosine, tyr,,,.
Fig. 3), according to the function: btyr value of the discriminant for 82 heterozy- (tY r,,/phe,,) ' gotes of phenylketonuria was 2.9 (range
Four successive phenylalanine-loading 0.1-1 1.8) and for 33 normal homozygotes tests of the same person showed discrim- 18.4 (range 9.3-60.8), P < 0.001, with an inant values of 8.26, 8.82, 9.29 and 11.0. overlap of 2.4 '2 (Fig. 4). The discrim- Four heterozygotes were loaded twice. The inatory power according to Penrose (1951) coefficient of variation was 5.0 %, 5.3 52, was 3.5. 5.7 % and 11.4 %, respectively. The median

H E T E R O Z Y G O T E D E T E C T I O N I N P H E N Y L K E T O N U R I A 141
NO I I N
I I
0.5 1.0 1.5 2.0 2.5 3.0 I O U 10 X h t y r [ l ~ r m / ~ h e m ]
Fig. 4. Distribution of individual values of the logarithm to the discriminant, b,,-,(tyr,,’phe,,,) (for explanation see Fig. 3) of 33 control subjects (N) and 82 heterozygotes for phenylketonuria (H).
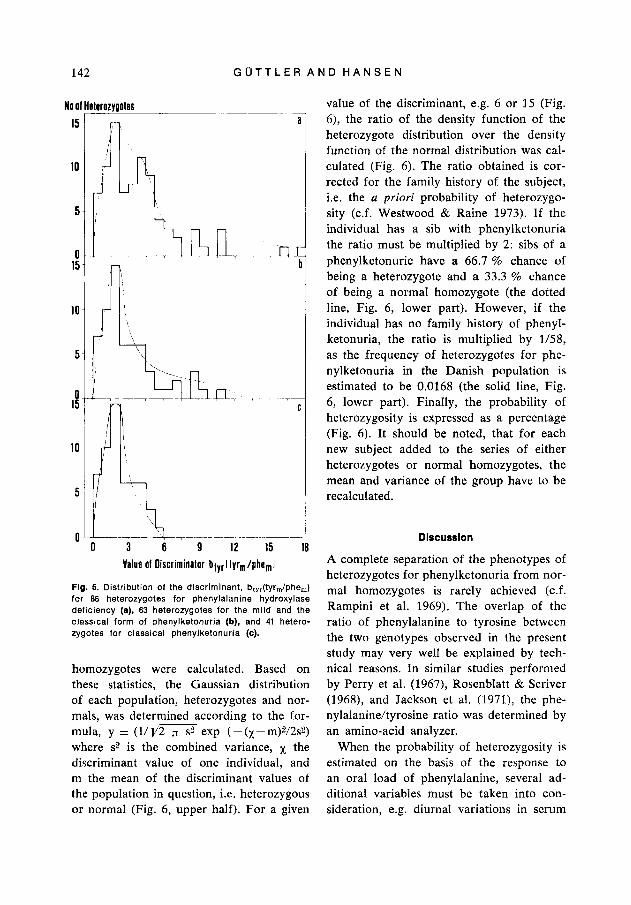
Phenotypic combination o f parents related to the phenotype o f their affected offspring. The distribution of the discriminant describ- ed above revealed a heterogenous popula- tion of parents of children in whom the type of phenylalanine deficiency was veri- fied, i.e. classical phenylketonuria, mild phenylketonuria or hyperphenylalaninemia (see Material and Methods) (Fig. 5a). In order to examine whether subpopulations of heterozygotes could be distinguished, parents of children with hyperphenylala- ninemia were excluded. This operation eliminated one top and much of the tailing of the distribution (Fig. 5b). Parents of children in whom the mild form of phenyl- ketonuria could be established were sub- sequently excluded. A fairly symmetrical distribution of the discriminant was then obtained (Fig. 5c). T h e remaining popula- tion of parents had children with classical phenylketonuria (Fig. 5c). The observation that the successive exclusion of parents of children with hyperphenylalaninemia and parents of children with the mild form of
phenylketonuria resulted in a symmetrical distribution of the discriminant might be explained by assuming three heterozygous phenotypes for phenylalanine hydroxylase deficiency. According to their discriminant value the heterozygous parents were divided into three groups, and the phenotypic com- bination of the parents was compared with the phenotype of their affected offspring. All children with the classical form of phe- nylketonuria had parents with a discrimi- nant value of 6 or lower, whereas most of the parents of children with the mild form of phenylketonuria had discriminant values above 6 ( P < 0.002, Table I ) . All children with hyperphenylalaninemia had parents with discriminant values above 2 (Table 2). In five out of 11 families with the mild form of phenylketonuria, one parcnt showed a discriminant value below 2 (Table 2 ) .
The percentage probability o f heterozygo- sity. T h e means and variance\ of the di\- criminant value5 of 82 hetcrozygotes for phenylketonuria and of 33 presumed normal
142 G U T T L E R A N D H A N S E N
No o 15
10
5
0 15
10
5
0 15
10
5
0
n I l.,- -ar b
i L I T . , ---
C
0 3 6 9 12 15 18 Value of Discriminator btyr l lyrm/phemI
Fig. 5. Distribution of the discriminant, btyr(tyr,/phe,) for 86 heterozygotes for phenylalanine hydroxylase deficiency (a), 63 heterozygotes for the mild and the classical form of phenylketonuria (b), and 41 hetero- zygotes for classical phenylketonuria (c).
homozygotes were calculated. Based on these statistics, the Gaussian distribution of each population, heterozygotes and nor- mals, was determined according to the for- mula, y = (UV2 n se exp (-(~-m)V2s2) where s2 is the combined variance, x the discriminant value of one individual, and m the mean of the discriminant values of the population in question, i.e. heterozygous or normal (Fig. 6, upper half). For a given
value of the discriminant, e.g. 6 or 15 (Fig. 6), the ratio of the density function of the heterozygote distribution over the density function of the normal distribution was cal- culated (Fig. 6). The ratio obtained is cor- rected for the family history of the subject, i.e. the a priori probability of heterozygo- sity (c.f. Westwood & Raine 1973). If the individual has a sib with phenylketonuria the ratio must be multiplied by 2: sibs of a phenylketonuric have a 66.7 % chance of being a heterozygote and a 33.3 % chance of being a normal homozygote (the dotted line, Fig. 6, lower part). However, if the individual has no family history of phenyl- ketonuria, the ratio is multiplied by 1/58, as the frequency of heterozygotes for phe- nylketonuria in the Danish population is estimated to be 0.0168 (the solid line, Fig. 6, lower part). Finally, the probability of heterozygosity is expressed as a percentage (Fig. 6). It should be noted, that for each new subject added to the series of either heterozygotes or normal homozygotes, the mean and variance of the group have to be recalculated.
Discussion
A complete separation of the phenotypes of heterozygotes for phenylketonuria from nor- mal homozygotes i s rarely achieved (c.f. Rampini et al. 1969). The overlap of the ratio of phenylalanine to tyrosine between the two genotypes observed in the present study may very well be explained by tech- nical reasons. In similar studies performed by Perry et al. (1967), Rosenblatt & Scriver (1968), and Jackson et al. (1971), the phe- nylalanine/tyrosine ratio was determined by an amino-acid analyzer.
When the probability of heterozygosity is estimated on the basis of the response to an oral load of phenylalanine, several ad- ditional variables must be taken into con- sideration, e.g. diurnal variations in serum

H E T E R O Z Y G O T E D E T E C T I O N I N P H E N Y L K E T O N U R I A 143
Table 1
The parental phenotypes in 16 families with classical phenylketonuria (classical PKU) and in 11 families with mild phenylketonuria (mild PKU), related to t h e phenotype of their af-
fected offspring
Table 2
The parental phenotypes in 12 families with hyperphenylalaninemia (HPA) and in 11 fami- lies with mild phenylketonuria (mild PKU), related to the phenotype of their affected off-
spring
Phenotype of Classical Mild both parents' PKU PKU
Phenotype of Mild both parents' H PA PKU
>6 0 12 <6 32 10
x z 22.4 P < 0.002
* As reflected by the discriminant
For explanation, see Figure 3. bt, dtyr&he,,).
phenylalanine (Guttler et al. 1969), rate of phenylalanine absorption and stimulation of tyrosine transamination (Rose & Cramp 1970). Accordingly, in the present study phenylalanine was fully dissolved in 0.01 N HCI and the pure solution was given orally after an overnight fast. Subjects to be loaded stayed overnight before the test and no meals or medicine were allowed from 6 p.m. before the Ioading and during the first 4 hours of the loading test. Females were tested on days when they were not taking oral contraceptives. The subject was asked to sit down for 15 min before blood sampling.
Among the variables that theoretically might account for the shape of the phenyl- alanine and tyrosine curves following a load of phenylalanine is the absorption of L- phenylalanine (c.f. Goodwin 1964). Pre- liminary investigations showed that the maximum serum phenylalanine concentra- tion is obtained within 60 min after an oral load of fully dissolved L-phenylalanine (c.f. Fig. 2). The test was repeated in individuals showing a maximum serum phenylalanine concentration later than 60 min after the load. As observed in a number of investiga- tions, the phenylalanine values within the first 4 hours following oral phenylalanine
<2 >2
~
0 5 24 17
x 2 6.12 P < 0.05 * As reflected by the discriminant
bt,,(tYr,/phe,,). For explanation, see Figure 3.
loading were higher in heterozygotes for phenylketonuria than in normal homozy- gotes (c.f. Giittler & Wamberg 1972, West- wood & Raine 1973). The observation was made that the slope of the rectilinear part of the tyrosine curve within 30 min after oral phenylalanine loading was significantly smaller in heterozygotes for phenylketonuria than in normal homozygotes (to be pub- lished), and this may explain the differences in the 1st or 2nd hour serum phenylalanine values in loaded heterozygotes as compared with normal homozygotes. Thus, the 1st hour serum phenylalanine value may reflect the rate of conversion of phenylalanine to tyrosine (c.f. Fig. 2). This may explain the present observation that the combination of the slope of the rise in serum tyrosine multiplied by the maximum serum tyrosine concentration over the maximum serum phenylalanine concentration improved the discriminatory ability of the phenylalanine loading test.
The distribution of the discriminant de- scribed above revealed a heterogeneous population of heterozygotes for phenylala- nine hydroxylase deficiency (c.f. Fig. 5a). Similar observations have been published by Wang et al. (1961), and Kaariainen & Karlsson (1973). The present study sup-
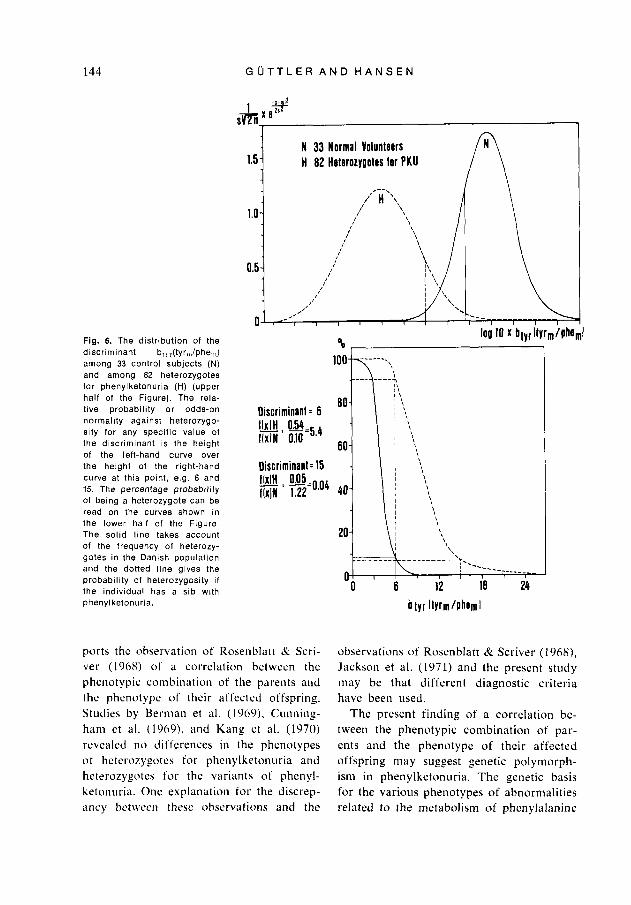
144 G U T T L E R A N D H A N S E N
Fig. 6. The distribution of the discriminant b,,,(tyr,,,/phe,,,) among 33 control subjects (N) and among 82 heterozygotes for phenylketonuria (H) (upper half of the Figure). The rela- tive probability or odds-on normality against heterozygo- sity for any specific value of the discriminant is the height of the left-hand curve over the height of the right-hand curve at this point, e.g. 6 and 15. The percentage probability of being a heterozygote can be read on the curves shown in the lower half of the Figure The solid l ine takes account of the frequency of heterozy- gotes in the Danish population and the dotted l ine gives the probabil ity of heterozygosity i f the individual has a sib with phenyl ketonuria.
Discriminant = 6 IMH 054 rlxln: G=5.4 Oiscriminanl= 15
ports the observation of Rosenblatt & Scri- ver (1968) of a correlation between the phenotypic combination of the parents and the phenotype o f their affected offspring. Studies by Berman et al. (19691, Cunning- hani et al. (19691, and Kang et al. (1970) revealed no differences in the phenotypes of heterozygotes f o r phenylketonuria and heterozygotes for the variants of phenyl- ketonuria. One explanation for the discrep- ancy between these observations and the
observations of Rosenblatt & Scriver (1968). Jackson et al. (1971) and the present study may be that different diagnostic criteria have been used.
The present finding of a correlation be- tween the phenotypic combination of par- ents and the phenotype of their affected offspring may suggest genetic polymorph- ism in phenylketonuria. The genetic basis for the various phenotypes of abnormalities related to the metabolism of phenylalanine

H E T E R O Z Y G O T E D E T E C T I O N I N P H E N Y L K E T O N U R I A 145
to tyrosine has been outlined by Hsia (1970). Multiple alleles a t the phenylalanine hydroxylase locus or a major modifier are the most convincing possibilities.
The probability of heterozygosity a t a given discriminant value was expressed as the percentage probability of being a hetero- zygote (c.f. Westwood & Raine 1975). Ac- cording to our experiences this interpre- tation has proved to be the most meaningful way of explaining the loading test result of the person requiring genetic counseling.
Acknowledgments
We are indebted to Dr. Erik Wamberg for encouragement, helpful discussions and advice. We thank the staff of the John F. Kennedy Institute for their considerable help and advice. Financial support was received from the Danish Health Insurance Founda- tion (grant no. H6/87.74), the Danish Med- ical Research Council (grant no. 512-5313 and 512-6840), the Research Commitee of the Danish Mental Retardation Service (project no. 93), and P. Carl Petersens Fund.
References
Berman, J . L., G. C. Cunningham, R. W. Day, R. Ford & D. Y. Y . Hsia (1969). Causes for high phenylalanine with normal tyrosine in newborn screening programs. Anier. J . Dis. Child. 117, 51-65.
Blaskovics, M. E., G. E. Schaeffler & S. Hack (1971). Phenylalaninaemia. Differential diag- nosis. Arch. Dis. Child/,. 49, 835-843.
Cunningham, G. C., R. W. Day, J. L. Berman & D. Y . Y. Hsia (1969). Phenylalanine tol- erance tests in families with phenylketonuria and hyperphenylalaninemia. Amer. J . Dis. Child. 117, 626-635.
Goodwin, B. L. (1964). The metabolism of aromatic amino acids in health and disease. D. Phil. Thesis. University of Oxford.
Guttler, F., E. S. Olesen & E. Wamberg (1969). Inverse diurnal variations of serum phenyl- alanine and tyrosine in phenylketonuric
children on low-phenylalanine diet. Enzymo- penic Anaemias, Lysosomes and Other Pa- pers, Proceedings of the 6th Symposium of The Society for the Study of Inborn Er- rors of Metabolism, ed. J. D. Allan, K. A. Holt, J. T. Ireland & R. J. Pollitt. Edinburgh and London, E. & S. Livingstone, pp. 149- 158.
Guttler, F. & E. Wamberg (1972). Persistent hyperphenylalaninemia. Actn padiat. scnnd.
Guttler, F. & G. Hansen (1976). Different phenotypes for phenylalanine hydroxylase deficiency. Ann. clin. Biochem. In press.
Horn, N. & F. Guttler (1976). Presence of di- hydropteridine reductase and absence of phenylalanine hydroxylase in cultured foetal fibroblasts and amniotic fluid cells. To be published.
Hsia, D. Y. Y . (1970). Phenylketonuria and its variants. Progress in Medical Genetics, Vol. VII, ed. A. G. Steinberg & A. G. Beam New York and London, Grune & Stratton,
Hsia, D. Y. Y., K. W. Driscoll, W. Troll & W. E. Knox (1956). Detection by phenyl- alanine tolerance tests of heterozygous car- riers of phenylketonuria. Nature (Lond.) 178, 1239-1240.
Jackson, S. H., W. B. Hanley, T. Gero & G. D. Gosse (1971). Detection of phenylketonuric heterozygotes. Clin. Chem. 17, 538-543.
Kang, E. S., S. Kaufman & P. S. Gerald (1970). Clinical and biochemical observations of patients with atypical phenylketonuria. Pediatrics 45, 83-92.
Kaufman, S. (1969). Phenylalanine hydro- xylase of human liver: Assay and some prop- erties. Arch. Biochem. Biophys. 134, 249- 252.
Kaariainen, R. & R. Karlsson (1973). Hetero- zygous carriers in the relatives of a case of phenylketonuria. Hereditas (Lcrnd) 75, 109- 118.
Menkes, J . H. & N. A. Holtzman (1970). Neo- natal hyperphenylalaninemia: a differential diagnosis. Neuropiidiatrie 1, 434-446.
Penrose, L. S. (1951). Measurement of pleio- tropic effects in phenylketonuria. Ann.
Perry, Th. L., €3. Tischler, S. Hansen & L. MacDougal (1967). A simple test for hetero- zygosity for phenylketonuria. Clin. Chin].
Rampini, S., P. W. Anders, H. Ch. Curtius & Th. Marthaler (1969). Detection of hetero-
61, 321-328.
pp. 29-68.
EtrgetI. (Lortd.) 16, 134-141.
Acts 15, 47-55.

146 G U T T L E R A N D H A N S E N
zygotes for phenylketonuria by column chromatography and discriminatory anal- ysis. Pediat. Res. 3, 287-291.
Rose, D. P. & D. G. Cramp (1970). Reduction of plasma tyrosine by oral contraceptives and oestrogens: A possible consequence of tyrosine aminotransferase induction. Clin. Chim. Acta 29, 49-53.
Rosenblatt, D. BC C. R. Scriver (1968). Hetero- geneity in genetic control of phenylalanine metabolism in man. Nature (Lond.) 218,
Wang, H. L., N. E. Morton & H. A. Waisman (1961). Increased reliability for the deter- mination of the carrier state in phenyl- ketonuria. Amer. J . hum. Genet. 13, 255- 261.
Westwood, A. & D. N. Raine (1913). Some
617-618.
problems of heterozygote recognition in inherited metabolic disease with special ref- erence to phenylketonuria. Treatment of Inborn Errors of Metabolism, ed. J. W. T. Seakins, R. A. Saunders .& C. Toothill. Edinburgh and London, Churchill Living- stone, pp. 63-76,
Westwood, A. & D. N. Raine (1975). Hetero- zygote detection in phenylketonunia. J . med. Genet. 12, 327-333.
Address: FIemrning Giittler, M.D. The John F. Kennedy Institute DK-2600 Glostricp Denmark
Related Documents











