Published Ahead of Print 8 June 2011. 2011, 85(16):8093. DOI: 10.1128/JVI.02689-10. J. Virol. Yuan Xueqiao Liu, David M. Knipe, Peter Cresswell and Weiming Ping Rao, Hong Thanh Pham, Arpita Kulkarni, Yang Yang, Presentation and NKT Cell Function US3 Collaborate To Inhibit CD1d Antigen Herpes Simplex Virus 1 Glycoprotein B and http://jvi.asm.org/content/85/16/8093 Updated information and services can be found at: These include: REFERENCES http://jvi.asm.org/content/85/16/8093#ref-list-1 at: This article cites 75 articles, 40 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on February 1, 2013 by KANSAS STATE UNIV http://jvi.asm.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Published Ahead of Print 8 June 2011. 2011, 85(16):8093. DOI: 10.1128/JVI.02689-10. J. Virol.
YuanXueqiao Liu, David M. Knipe, Peter Cresswell and Weiming Ping Rao, Hong Thanh Pham, Arpita Kulkarni, Yang Yang, Presentation and NKT Cell FunctionUS3 Collaborate To Inhibit CD1d Antigen Herpes Simplex Virus 1 Glycoprotein B and
http://jvi.asm.org/content/85/16/8093Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/85/16/8093#ref-list-1at:
This article cites 75 articles, 40 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from

JOURNAL OF VIROLOGY, Aug. 2011, p. 8093–8104 Vol. 85, No. 160022-538X/11/$12.00 doi:10.1128/JVI.02689-10Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Herpes Simplex Virus 1 Glycoprotein B and US3 Collaborate To InhibitCD1d Antigen Presentation and NKT Cell Function�
Ping Rao,1 Hong Thanh Pham,1 Arpita Kulkarni,1 Yang Yang,1 Xueqiao Liu,2 David M. Knipe,2Peter Cresswell,3* and Weiming Yuan1*
Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Los Angeles,California1; Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, Massachussattes2; and
Howard Hughes Medical Institute, Department of Immunobiology, Yale University School of Medicine, New Haven, Connecticut3
Received 29 December 2010/Accepted 25 May 2011
Herpes simplex viruses (HSVs) are prevalent human pathogens that establish latency in human neuronalcells and efficiently evade the immune system. It has been a major medical challenge to eradicate them and,despite intensive efforts, an effective vaccine is not available. We previously showed that upon infection ofantigen-presenting cells, HSV type 1 (HSV-1) rapidly and efficiently downregulates the major histocompati-bility complex class I-like antigen-presenting molecule, CD1d, and potently inhibits its recognition by CD1d-restricted natural killer T (NKT) cells. It suppresses CD1d expression primarily by inhibiting its recycling tothe cell surface after endocytosis. We identify here the viral glycoprotein B (gB) as the predominant CD1d-interacting protein. gB initiates the interaction with CD1d in the endoplasmic reticulum and stably associateswith it throughout CD1d trafficking. However, an additional HSV-1 component, the serine-threonine kinaseUS3, is required for optimal CD1d downregulation. US3 expression in infected cells leads to gB enrichment inthe trans-Golgi network (TGN) and enhances the relocalization of both gB and CD1d to this compartment,suggesting that following internalization CD1d is translocated from the endocytic pathway to the TGN by itsassociation with gB. Importantly, both US3 and gB are required for efficient inhibition of CD1d antigenpresentation and NKT cell activation. In summary, our results suggest that HSV-1 uses gB and US3 to rapidlyinhibit NKT cell function in the initial antiviral response.
During a viral infection the host uses both innate and adap-tive arms of the immune system to prevent, suppress, or clearthe pathogens, while many viruses encode proteins that allowthem to evade the host response. Conventional CD8� andCD4� T cells are critical in antiviral immune responses, andviral evasion of antigen presentation by major histocompati-bility complex (MHC) class I and II molecules have beenstudied extensively (reviewed in references 24 and 33). Naturalkiller T (NKT) cells are an unusual subset of T cells thatcoexpress T-cell receptors (TCRs) and surface receptors typi-cal of NK cells (5). NKT cells influence diverse immune re-sponses, including immunity to tumors and infectious diseases,as well as autoimmune diseases and allergies (5). Most NKTcells express identical or similar TCRs, with predominantV�24J�18 in human and V�14J�18 in mouse, and are oftencalled invariant NKT (iNKT) cells. Distinct from conventionalCD4� and CD8� T cells, iNKT cells can be activated by bothexogenous or endogenous lipid ligands. NKT cells are amongthe first peripheral responders during an immune response andare typically activated within hours, rapidly producing both
Th1 and Th2 cytokines, and exerting critical immunomodula-tory functions in the ensuing adaptive immune response (18).
NKT cells are restricted by the nonpolymorphic MHC classI-like surface glycoprotein, CD1d, a member of CD1 genefamily (3). In humans, there are five CD1 gene members:CD1a, CD1b, CD1c, CD1d, and CD1e. Based on sequencehomology, they are classified into two groups. Group I includesCD1a, CD1b, CD1c, and CD1e, while CD1d is the sole mem-ber of group II. There are no group I CD1 members in mice;however, there are two CD1d genes (CD1d1 and CD1d2) (5).Structurally, CD1 is similar to MHC class I molecules in that itis a heterodimer formed by a 43- to 49-kDa heavy chain and �2
microglobulin (�2m). The heavy chain consists of three extra-cytoplasmic domains (�1, �2, and �3), a transmembrane do-main, and a short cytoplasmic tail. CD1d molecules presentantigenic lipids or glycolipids to NKT cells. The extracytoplas-mic �1 and �2 domains form a binding cleft similar to thepeptide binding site of MHC class I molecules, except that itcontains extremely hydrophobic pockets which can accommo-date a variety of lipid antigens with hydrocarbon chains (17,75). As a transmembrane protein, CD1d is synthesized in theendoplasmic reticulum (ER) and traffics through trans-Golginetwork (TGN) to the cell surface. Mediated by its cytoplasmictail, cell surface CD1d can be endocytosed to endosomes andthen recycle back to the cell surface. It is believed that matureCD1d molecules constantly recycle between cell surface andintracellular endosomal compartment, presumably to surveylipid antigens (reviewed in references 5 and 22).
CD1d-restricted NKT cells play critical roles in immunity todifferent groups of viruses. Although the exact function of
* Corresponding author. Mailing address for W. Yuan: Departmentof Molecular Microbiology and Immunology, University of SouthernCalifornia, 1450 Biggy St., NRT 5516, Los Angeles, CA 90033. Phone:(323) 442-7938. Fax: (323) 442-1721. E-mail: [email protected] address for P. Cresswell: Howard Hughes Medical Institute,Department of Immunobiology, Yale University School of Medicine, 1Gilbert Street, TAC S670, New Haven, CT 06720. Phone: (203) 785-5176.Fax: (203) 785-4461. E-mail: [email protected].
� Published ahead of print on 8 June 2011.
8093
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from

NKT cells in antiviral immunity remains unclear, it has beenshown that innate-like NKT cells are rapidly activated afterviral infection (14). Patients with deficiencies in NKT cell de-velopment are hypersensitive to herpesvirus infection (59).There is a selective loss of circulating NKT cells during HIV-1infection (68), and current treatments of HIV-1 patients byinterleukin-2 (IL-2) or highly active antiretroviral therapy rap-idly restore iNKT cell populations (49, 67). In animal models,NKT-cell-deficient mice are much more susceptible to infec-tion by herpes simplex virus type 1 (HSV-1) (21), influenzaviruses (13), respiratory syncytial virus (34), and encephalo-myocarditis virus (EMCV) (30). On the other hand, the acti-vation of NKT cells, mostly by the prototypical NKT cell li-gand, �-galactosylceramide (�-GalCer), greatly reduces thereplication of mouse cytomegalovirus (69), hepatitis B virus(35), influenza virus (29), respiratory syncytial virus (34), lym-phocytic choriomeningitis virus (LCMV), and EMCV (71).This critical role for NKT cells is also substantiated by thediverse mechanisms used by different groups of viruses toevade NKT cell function and the CD1d antigen presentationpathway (9, 62, 71, 73).
A common tactic adopted by viruses to evade T cell functionis to downregulate antigen-presenting molecules, includingMHC class I and II glycoproteins, on the surface of antigen-presenting cells (24, 33). To antagonize the potent antiviralfunction of NKT cells, many viruses, including HIV (8, 9, 23,48), HSV-1 (56, 73), Kaposi’s sarcoma herpesvirus (KSHV)(62), and LCMV (43) have evolved different mechanisms toinhibit CD1d expression and block the function of NKT cells.HIV Nef protein interacts with the cytoplasmic tail of CD1d,accelerates its endocytosis and relocalizes it to the TGN (63).In addition, HIV-1 also uses Vpu protein, to relocalize CD1dfrom the late endosome/lysosome to the early endosomal com-partment (48). KSHV uses the protein MIR-2 (for modulatorof immune recognition 2) to ubiquitinate the cytoplasmic tailof human CD1d and enhance CD1d endocytosis, apparentlywithout accelerating degradation (62). LCMV also downregu-lates CD1d from the cell surface, but the mechanism is un-known (43). Other viruses, including vaccinia and vesicularstomatitis viruses, inhibit NKT cell function not by inhibitingCD1d surface expression but rather by modulating the signaltransduction pathways and the quality of CD1d lipid presen-tation (57, 58, 71).
Herpesvirus infection is a major public health concern, caus-ing both acute and life-long recurring infections that pose aconstant threat to both immunocompetent and immunocom-promised patients. Herpesviruses are categorized into threemajor groups, alpha-, beta-, and gammaherpesviruses. HSV isa member of the alphaherpesvirus family and can cause oral,ocular, and genital infections. Despite intensive studies ourunderstanding of HSV pathogenesis is still limited. Currently,there are no vaccines available. Understanding the viral inter-action with the immune system, particularly the innate immunesystem, may help to improve the vaccine efficiency and provideclues for novel antivirals. We have previously shown thatHSV-1, upon infection, rapidly and substantially downregu-lates CD1d expression and inhibits NKT cell function. Unlikeother viruses, HSV-1 does so primarily through suppressingCD1d recycling (73). In this report we identify glycoprotein B
(gB) and US3 as two viral factors functioning in suppressingCD1d antigen presentation.
MATERIALS AND METHODS
Viruses, cells, antibodies, and DNA constructs. Wild-type HSV-1 F strain wasa gift from David Johnson (Oregon Health Science University, Portland, OR).gB-deficient virus and adenovirus expressing gB protein were generous gifts fromKonstantin Kousoulas (Louisiana State University, New Orleans, LA). The gB-deficient virus was amplified and titered in gB-complementing Vero (D6) cells,also a gift from K. Kousoulas. US3-deficient (R7041) and vhs-deficient (R7621)viruses were gifts from Bernard Roizman (University of Chicago, Chicago, IL).The mouse monoclonal antibodies (MAbs) against human CD1d, CD1d51, andD5 were from Steven Porcelli (Albert Einstein College of Medicine, Bronx, NY)and Steven Balk (Harvard Medical School, Boston, MA), respectively. Theanti-viperin MAb MaP.VIP was generated against the C-terminal domain ofmouse viperin. Phycoerythrin (PE)-conjugated anti-CD1d monoclonal antibody(42.1) was purchased from BD Pharmingen. The anti-gB MAbs, H126 and 10B7,are from Virusys Co. Rabbit polyclonal anti-HSV1 antibody is from Dako Co.The rabbit polyclonal anti-gH/gL antibody was from Gary Cohen and RoselynEisenberg (University of Pennsylvania, Philadelphia, PA). Rabbit polyclonalanti-US3 antibody was from Bernard Roizman. MAbs, anti-mouse MHC class I,Y3, and anti-HLA DM, Map.DM1, have been described previously (36, 37).MAbs against TGN46, Grp94, and LAMP1 were from Abcam, Stressgen, andBD Pharmingen, respectively. Alexa 488-, 568-, and 647-conjugated goat anti-mouse secondary antibodies were from Invitrogen/Molecular Probes. Alexa 647-conjugated anti-CD1d (CD1d51) antibody was generated by using purifiedCD1d51 antibody and a labeling kit from Invitrogen/Molecular Probes accordingto the manufacturer’s instructions. The HSV-1 gB gene was subcloned by PCRfrom pSR175 construct (Gary Cohen and Roselyn Eisenberg, University ofPennsylvania, Philadelphia, PA) into pcDNA3.1.myc.hisA vector (Invitrogen).The HSV-1 strain 17 US3 gene was subcloned by PCR into pLPCX at a BglII/NotI site from cosmid 48 (kindly provided by Andrew Davidson, MRC Virology,Glasgow, United Kingdom) (12). US3 K220A (US3KA) mutant was generatedby PCR mutating lysine 220 (codon AAG) to alanine (codon GCG) as reportedby Wisner et al. (72). The US3 gene from the HSV-1 KOS strain was cloned byPCR into pcDNA3.1 vector as reported previously (45).
Pulse-chase labeling and immunoprecipitation. Radiolabeling of infectedHeLa.CD1d cells and immunoprecipitation and reimmunoprecipitation wereperformed as described previously (73). Briefly, HeLa.CD1d cells were infectedwith vhs-null HSV-1 virus for 6 h, starved in methionine-cysteine-free mediumfor 1 h, pulse-labeled using [35S]methionine-cysteine for 15 min, and chased withnonradioactive methionine-cysteine-containing medium for the indicated times.Radiolabeled cells were lysed in 1% Brij98-containing Tris-buffered saline(TBS), immunoprecipitated with MAbs against CD1d, eluted, and reprecipitatedusing anti-gB antibodies as described above. Endoglycosidase H (Endo H; NewEngland Biolabs) digestion of immunoprecipitated proteins was performed ac-cording to the manufacturer’s instructions.
Coimmunoprecipitation. Coimmunoprecipitation with anti-CD1d monoclo-nal antibody of HSV-1-infected HeLa.CD1d cells were performed essentiallyas described previously (37). Briefly, HeLa or HeLa.CD1d was infected byvhs-deficient virus at a multiplicity of infection (MOI) of 10 for 6 h, starvedin methionine-cysteine-free medium for 1 h, and labeled using [35S]methio-nine-cysteine for 4 h before being lysed in TBS (pH 7.4) containing 1% Brij98(Sigma). Postnuclear lysates were immunoprecipitated with the anti-CD1dMAb CD1d51. Precipitated protein was either directly loaded onto an SDS-PAGE gel or eluted in 1% SDS and reimmunoprecipitated by polyclonalanti-HSV-1 antibody, anti-gB MAb 10B7, or anti-gH/gL polyclonal antibody.To determine whether the coimmunoprecipitated protein was an N-linkedglycoprotein, immunoprecipitated proteins were treated with peptide N-gly-canase (PNGase; New England Biolabs) overnight before loading them ontoSDS-PAGE gel. For direct cell surface coimmunoprecipitation of gB byanti-CD1d antibodies, HeLa.CD1d cells were infected by vhs-null HSV-1 for6 h and pulse-labeled for 4 h before being incubated with anti-CD1d anti-bodies (CD1d51) or two isotype-matched control MAbs, eBioscience mIgG2bisotype control (eBMG2b) and anti-mouse MHC class I MAb (Y3), for 30min on ice. After lysis and removal of the nuclei, CD1d-gB complexes wereprecipitated directly by using protein G-Sepharose beads. After severalwashes, the immunoprecipitated proteins were resolved by SDS-PAGE andautoradiography was performed.
Virus infection and flow cytometry. HeLa or HeLa.CD1d cells were infectedby HSV-1 at an MOI of 10 for 6 or 12 h and then harvested in phosphate-buffered saline containing 1 mM EDTA. A half million cells were stained in 100
8094 RAO ET AL. J. VIROL.
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from

�l with anti-CD1d, anti-HSV-1, or anti-gB antibodies and analyzed on a BDFACS Canto II flow cytometer as described previously (73). Adenovirus-express-ing gB was amplified in HEK293 cells and used to infect HeLa.CD1d cells at anMOI of 10.
Measurement of CD1d recycling by flow cytometry. HeLa.CD1d cells wereeither uninfected or infected by wild-type HSV-1 F strain or US3-deficient virusfor 12 h or else cotransfected with pLPCX.US3 and peGFP-N1 for 40 h andtreated with cycloheximide (10 �g/ml) for 1 h before being used for recyclingassays as described previously (73). The cells were incubated with a purifiedanti-CD1d MAb, CD1d51, on ice for 30 min to block the surface CD1d beforebeing incubated at 37°C for 20 min. Newly surface expressed, recycled, CD1dmolecules, not associated with anti-CD1d antibodies, were then stained by a PE-conjugated anti-CD1d MAb, 42.1, and fixed with 3.7% formaldehyde before beinganalyzed by flow cytometry. This approach separates the recycled CD1d from theCD1d molecules previously existing on cell surface. The percentage of recycledmean fluorescence intensity (MFI) was calculated as follows: [(postrecycling MFI �postblocking MFI)/(unblocked sample MFI � isotype control MFI)] � 100%.
NKT cell stimulation assay. Human NKT cell stimulations using uninfected orinfected HeLa.CD1d cells were performed as described previously (73). Briefly,HeLa.CD1d cells were uninfected or infected with HSV-1 virus at an MOI of 10for 12 h and then incubated with �-GalCer (a gift from Gurdyal S. Besra,University of Birmingham, Birmingham, United Kingdom) at a concentration of100 ng/ml. Aliquots of 2 � 104 loaded cells were incubated with 105 V�24-positive DN2.D6 invariant NKT cells (a gift of Steven Porcelli, Albert EinsteinCollege of Medicine, Bronx, NY) for 48 h. The culture supernatant was assayedby enzyme-linked immunosorbent assay for gamma interferon secretion usingantibodies from BD Pharmingen.
Transient transfections, immunofluorescence, and Western blotting. Theseprocedures were performed essentially as described previously (73, 74). Trans-fection was performed using Lipofectamine 2000 (Invitrogen) or Bio-T reagent(Bioland). For immunofluorescence, HeLa.CD1d cells were seeded on a 10-mmcoverslip (Fisher) and infected by HSV-1 for 6 h, fixed with 3.7% formaldehyde,and permeabilized with 0.1% saponin. The cells were then stained with anti-gB(H126), anti-CD1d (CD1d51), anti-Lamp1 (H4A3), or anti-TGN46 MAbs atconcentrations of 5, 1, 2, and 2 �g/ml, respectively. The cells were then stainedwith Hoechst 33258 (Invitrogen/Molecular Probes) at 2 �g/ml. Alexa 488-, 568-,and 647-conjugated goat anti-mouse secondary antibodies were used at a con-centration of 2 �g/ml. Images were acquired and processed using a Nikon EclipseC1 laser scanning microscope (Nikon Instruments, Inc., Melville, NY) fitted witha �60 Nikon objective and Nikon Element image software. Immunoblotting forUS3 expression was performed with rabbit polyclonal anti-US3 primary anti-body, followed by horseradish peroxidase-conjugated secondary goat anti-rabbitantibody (Jackson Immunoresearch) and developed by ECL (Pierce) chemilu-minescence.
RESULTS
HSV-1 gB stably interacts with CD1d in infected cells. As afirst step to elucidate the molecular mechanism of HSV-1downregulation of CD1d in infected cells, we performed co-immunoprecipitation experiments to identify proteins that in-teract with CD1d. HeLa or HeLa.CD1d cells were infected byvhs-null HSV-1 virus as previously reported (73) for 6 h, andcell lysates were immunoprecipitated using a specific MAb(CD1d51) against CD1d or a control MAb and subjected toSDS-PAGE analysis. As shown in Fig. 1A, anti-CD1d antibodyspecifically precipitates CD1d from uninfected and infectedHeLa.CD1d cells but only coprecipitates a protein of �120kDa (p120) in the infected sample. Mature CD1d acquiresheavily sialylated complex N-linked glycans at three of its fourglycosylation sites (52), resulting in a smeared appearance.Reimmunoprecipitation by anti-CD1d antibody has confirmedthat the smear band is CD1d (data not shown). The coprecipi-tated p120 protein was absent from the control immunopre-cipitations. To determine whether the p120 protein was a viralor cellular protein, we used a polyclonal anti-HSV-1 antibody,which was generated in rabbits to a lysate of rabbit cornealcells infected with HSV-1 and should recognize HSV-1-en-
coded proteins rather than cellular proteins. The proteins co-precipitated with CD1d were SDS-eluted and reimmunopre-cipitated using the anti-HSV-1 antibody (Fig. 1B). The p120protein can be precipitated by the anti-HSV-1 antibody (lane8), but not by normal rabbit serum (lane 5), suggesting it is anHSV-1 protein (6).
The HSV-1 genome encodes approximately 80 proteins and12 glycoproteins (53). To determine whether p120 was a gly-coprotein, we performed a PNGase digestion. A shift of �15kDa suggested that the p120 protein was a heavily N-linkedglycosylated protein (Fig. 1C). In the HSV-1 genome, there aretwo glycoproteins with molecular masses close to 120 kDa,glycoproteins B and H (gB and gH). We therefore used anti-bodies against gB or gH for reimmunoprecipitation. Theanti-gB MAb reacted with the p120 protein (Fig. 1D), whileanti-gH/gL did not (data not shown). gB has six potentialN-linked glycosylation sites (6), a finding consistent with thesubstantial shift after PNGase digestion (Fig. 1C). Thus, theHSV-1 envelope glycoprotein gB was the predominant proteinthat interacts with CD1d.
To estimate the percentages of total CD1d and gB mole-cules present in the form of CD1d/gB complexes, we per-formed reciprocal immunoprecipitation and Western blotexperiments. About 3 to 5% of CD1d and gB proteins can bedetected as CD1d/gB complexes when either anti-CD1d(Fig. 1E, compare lanes 5 and 8) or anti-gB (Fig. 1F, com-pare lanes 6 and 9) antibodies were used for coimmunopre-cipitation. The presence of two CD1d bands coprecipitatedby anti-gB antibody suggest that both the two forms ofCD1d, the immature unfolded CD1d heavy chain andfolded, �2m-associated CD1d (44) can be associated withgB. The percentage of CD1d associated with gB appears tobe low. However, we reason that CD1d-gB interaction, sim-ilar to other protein-protein interactions, is a dynamic andtransient process. On the other hand, both CD1d and gBproteins are membrane proteins, detergents have to be usedto solubilize the proteins and even mild detergents, such asBrij98, used in our assays may disrupt the CD1d/gB com-plex. Therefore, the actual percentage of CD1d and gB inthe complexes is likely to be higher, and this interaction maysubstantially affect CD1d expression and function.
At least some of the gB-CD1d interaction initiates in the ER,and the interaction is maintained during CD1d intracellulartransport. To examine the kinetics of gB-CD1d interaction, weperformed a pulse-chase analysis. HeLa.CD1d cells were in-fected with HSV-1 for 6 h and pulse-labeled for 15 min with[35S]methionine-cysteine. At various time points cells werelysed and immunoprecipitations were performed with anti-CD1d MAbs. Immunoprecipitated proteins were eluted inSDS and reimmunoprecipitated with an anti-gB MAb (10B7)or anti-CD1d antibody again to detect the associated gB pro-tein and CD1d protein itself. Two anti-CD1d antibodies wereused, D5 and CD1d51, which recognize unfolded CD1d heavychain and folded, �2m-associated CD1d heavy chain, respec-tively (36, 73). Coimmunoprecipitation with both anti-CD1dantibodies can isolate the gB protein, which is consistent withcoimmunoprecipitation results achieved with the anti-gB anti-body (Fig. 1F). Immunoprecipitation by D5 detected nascentCD1d heavy chain immediately after synthesis in the ER, asindicated by Endo H sensitivity, and most of these heavy chains
VOL. 85, 2011 HSV-1 gB AND US3 COLLABORATE TO DOWNREGULATE CD1d 8095
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
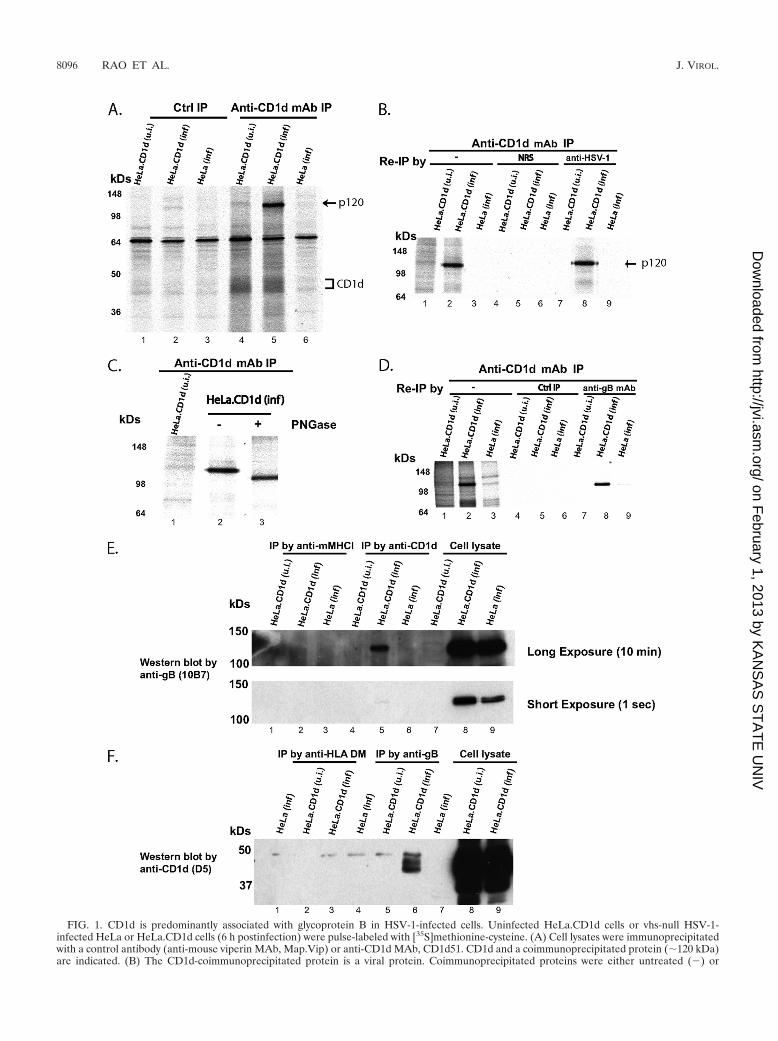
FIG. 1. CD1d is predominantly associated with glycoprotein B in HSV-1-infected cells. Uninfected HeLa.CD1d cells or vhs-null HSV-1-infected HeLa or HeLa.CD1d cells (6 h postinfection) were pulse-labeled with [35S]methionine-cysteine. (A) Cell lysates were immunoprecipitatedwith a control antibody (anti-mouse viperin MAb, Map.Vip) or anti-CD1d MAb, CD1d51. CD1d and a coimmunoprecipitated protein (�120 kDa)are indicated. (B) The CD1d-coimmunoprecipitated protein is a viral protein. Coimmunoprecipitated proteins were either untreated (�) or
8096 RAO ET AL. J. VIROL.
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
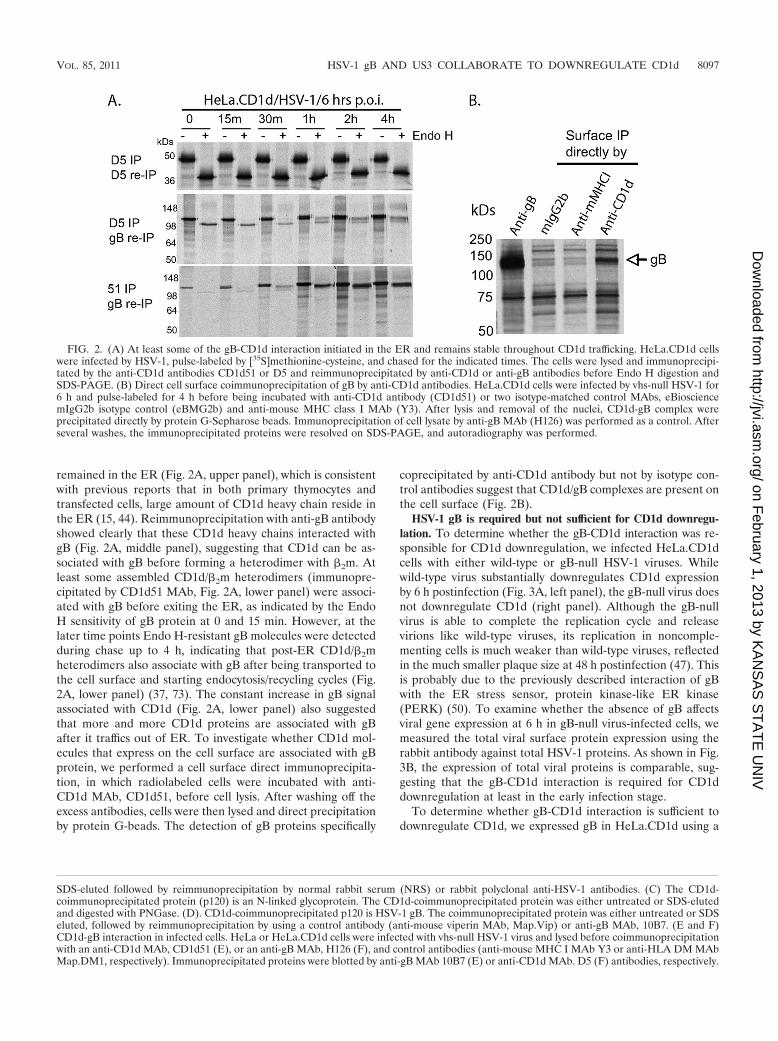
remained in the ER (Fig. 2A, upper panel), which is consistentwith previous reports that in both primary thymocytes andtransfected cells, large amount of CD1d heavy chain reside inthe ER (15, 44). Reimmunoprecipitation with anti-gB antibodyshowed clearly that these CD1d heavy chains interacted withgB (Fig. 2A, middle panel), suggesting that CD1d can be as-sociated with gB before forming a heterodimer with �2m. Atleast some assembled CD1d/�2m heterodimers (immunopre-cipitated by CD1d51 MAb, Fig. 2A, lower panel) were associ-ated with gB before exiting the ER, as indicated by the EndoH sensitivity of gB protein at 0 and 15 min. However, at thelater time points Endo H-resistant gB molecules were detectedduring chase up to 4 h, indicating that post-ER CD1d/�2mheterodimers also associate with gB after being transported tothe cell surface and starting endocytosis/recycling cycles (Fig.2A, lower panel) (37, 73). The constant increase in gB signalassociated with CD1d (Fig. 2A, lower panel) also suggestedthat more and more CD1d proteins are associated with gBafter it traffics out of ER. To investigate whether CD1d mol-ecules that express on the cell surface are associated with gBprotein, we performed a cell surface direct immunoprecipita-tion, in which radiolabeled cells were incubated with anti-CD1d MAb, CD1d51, before cell lysis. After washing off theexcess antibodies, cells were then lysed and direct precipitationby protein G-beads. The detection of gB proteins specifically
coprecipitated by anti-CD1d antibody but not by isotype con-trol antibodies suggest that CD1d/gB complexes are present onthe cell surface (Fig. 2B).
HSV-1 gB is required but not sufficient for CD1d downregu-lation. To determine whether the gB-CD1d interaction was re-sponsible for CD1d downregulation, we infected HeLa.CD1dcells with either wild-type or gB-null HSV-1 viruses. Whilewild-type virus substantially downregulates CD1d expressionby 6 h postinfection (Fig. 3A, left panel), the gB-null virus doesnot downregulate CD1d (right panel). Although the gB-nullvirus is able to complete the replication cycle and releasevirions like wild-type viruses, its replication in noncomple-menting cells is much weaker than wild-type viruses, reflectedin the much smaller plaque size at 48 h postinfection (47). Thisis probably due to the previously described interaction of gBwith the ER stress sensor, protein kinase-like ER kinase(PERK) (50). To examine whether the absence of gB affectsviral gene expression at 6 h in gB-null virus-infected cells, wemeasured the total viral surface protein expression using therabbit antibody against total HSV-1 proteins. As shown in Fig.3B, the expression of total viral proteins is comparable, sug-gesting that the gB-CD1d interaction is required for CD1ddownregulation at least in the early infection stage.
To determine whether gB-CD1d interaction is sufficient todownregulate CD1d, we expressed gB in HeLa.CD1d using a
SDS-eluted followed by reimmunoprecipitation by normal rabbit serum (NRS) or rabbit polyclonal anti-HSV-1 antibodies. (C) The CD1d-coimmunoprecipitated protein (p120) is an N-linked glycoprotein. The CD1d-coimmunoprecipitated protein was either untreated or SDS-elutedand digested with PNGase. (D). CD1d-coimmunoprecipitated p120 is HSV-1 gB. The coimmunoprecipitated protein was either untreated or SDSeluted, followed by reimmunoprecipitation by using a control antibody (anti-mouse viperin MAb, Map.Vip) or anti-gB MAb, 10B7. (E and F)CD1d-gB interaction in infected cells. HeLa or HeLa.CD1d cells were infected with vhs-null HSV-1 virus and lysed before coimmunoprecipitationwith an anti-CD1d MAb, CD1d51 (E), or an anti-gB MAb, H126 (F), and control antibodies (anti-mouse MHC I MAb Y3 or anti-HLA DM MAbMap.DM1, respectively). Immunoprecipitated proteins were blotted by anti-gB MAb 10B7 (E) or anti-CD1d MAb. D5 (F) antibodies, respectively.
FIG. 2. (A) At least some of the gB-CD1d interaction initiated in the ER and remains stable throughout CD1d trafficking. HeLa.CD1d cellswere infected by HSV-1, pulse-labeled by [35S]methionine-cysteine, and chased for the indicated times. The cells were lysed and immunoprecipi-tated by the anti-CD1d antibodies CD1d51 or D5 and reimmunoprecipitated by anti-CD1d or anti-gB antibodies before Endo H digestion andSDS-PAGE. (B) Direct cell surface coimmunoprecipitation of gB by anti-CD1d antibodies. HeLa.CD1d cells were infected by vhs-null HSV-1 for6 h and pulse-labeled for 4 h before being incubated with anti-CD1d antibody (CD1d51) or two isotype-matched control MAbs, eBiosciencemIgG2b isotype control (eBMG2b) and anti-mouse MHC class I MAb (Y3). After lysis and removal of the nuclei, CD1d-gB complex wereprecipitated directly by protein G-Sepharose beads. Immunoprecipitation of cell lysate by anti-gB MAb (H126) was performed as a control. Afterseveral washes, the immunoprecipitated proteins were resolved on SDS-PAGE, and autoradiography was performed.
VOL. 85, 2011 HSV-1 gB AND US3 COLLABORATE TO DOWNREGULATE CD1d 8097
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
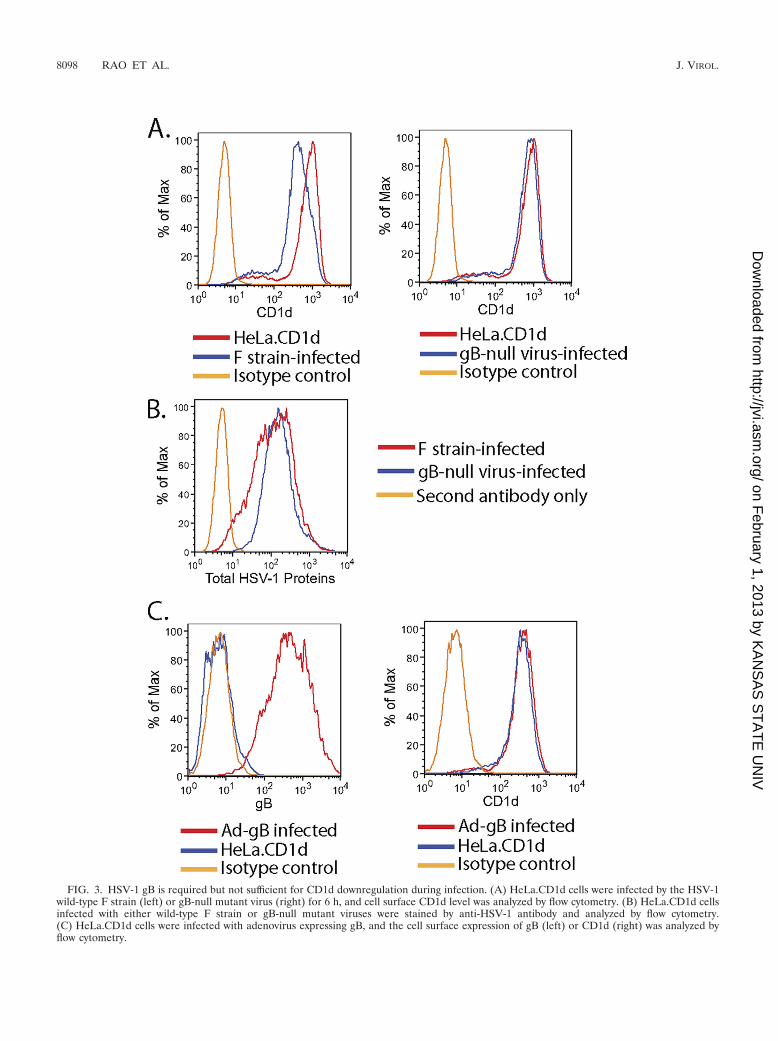
FIG. 3. HSV-1 gB is required but not sufficient for CD1d downregulation during infection. (A) HeLa.CD1d cells were infected by the HSV-1wild-type F strain (left) or gB-null mutant virus (right) for 6 h, and cell surface CD1d level was analyzed by flow cytometry. (B) HeLa.CD1d cellsinfected with either wild-type F strain or gB-null mutant viruses were stained by anti-HSV-1 antibody and analyzed by flow cytometry.(C) HeLa.CD1d cells were infected with adenovirus expressing gB, and the cell surface expression of gB (left) or CD1d (right) was analyzed byflow cytometry.
8098 RAO ET AL. J. VIROL.
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
recombinant adenovirus expressing gB under the control of thecytomegalovirus promoter (Fig. 3C, left panel). No CD1ddownregulation was observed (right panel), suggesting that gBalone cannot reduce the cell surface expression of CD1d.
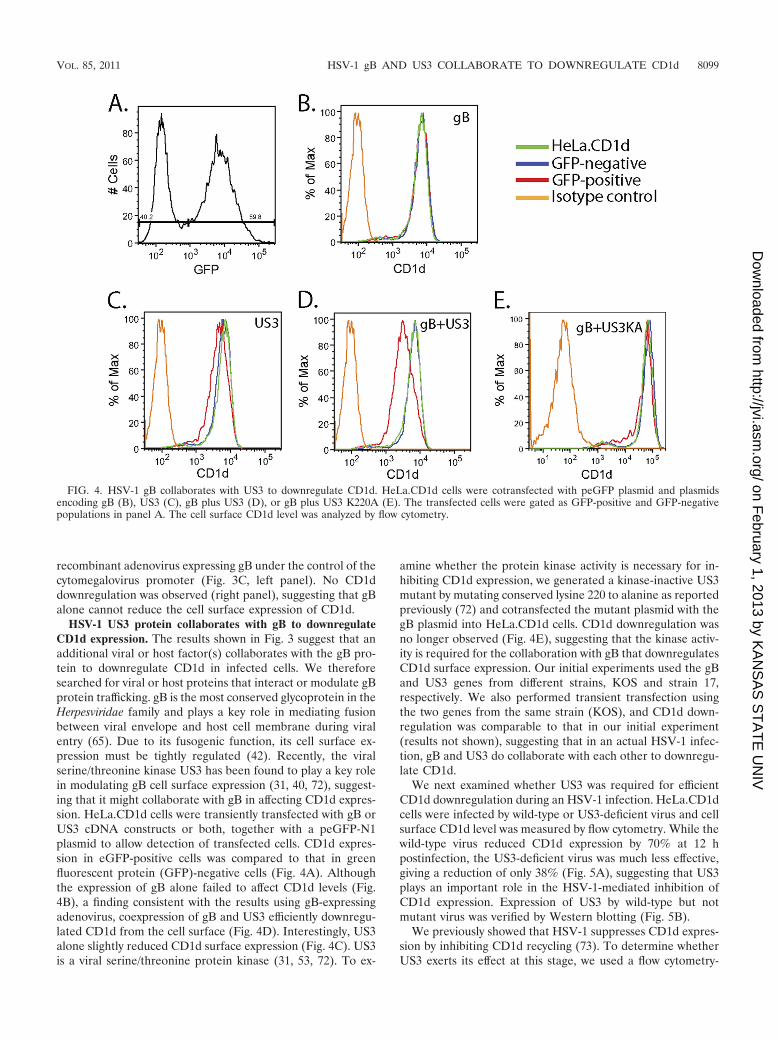
HSV-1 US3 protein collaborates with gB to downregulateCD1d expression. The results shown in Fig. 3 suggest that anadditional viral or host factor(s) collaborates with the gB pro-tein to downregulate CD1d in infected cells. We thereforesearched for viral or host proteins that interact or modulate gBprotein trafficking. gB is the most conserved glycoprotein in theHerpesviridae family and plays a key role in mediating fusionbetween viral envelope and host cell membrane during viralentry (65). Due to its fusogenic function, its cell surface ex-pression must be tightly regulated (42). Recently, the viralserine/threonine kinase US3 has been found to play a key rolein modulating gB cell surface expression (31, 40, 72), suggest-ing that it might collaborate with gB in affecting CD1d expres-sion. HeLa.CD1d cells were transiently transfected with gB orUS3 cDNA constructs or both, together with a peGFP-N1plasmid to allow detection of transfected cells. CD1d expres-sion in eGFP-positive cells was compared to that in greenfluorescent protein (GFP)-negative cells (Fig. 4A). Althoughthe expression of gB alone failed to affect CD1d levels (Fig.4B), a finding consistent with the results using gB-expressingadenovirus, coexpression of gB and US3 efficiently downregu-lated CD1d from the cell surface (Fig. 4D). Interestingly, US3alone slightly reduced CD1d surface expression (Fig. 4C). US3is a viral serine/threonine protein kinase (31, 53, 72). To ex-
amine whether the protein kinase activity is necessary for in-hibiting CD1d expression, we generated a kinase-inactive US3mutant by mutating conserved lysine 220 to alanine as reportedpreviously (72) and cotransfected the mutant plasmid with thegB plasmid into HeLa.CD1d cells. CD1d downregulation wasno longer observed (Fig. 4E), suggesting that the kinase activ-ity is required for the collaboration with gB that downregulatesCD1d surface expression. Our initial experiments used the gBand US3 genes from different strains, KOS and strain 17,respectively. We also performed transient transfection usingthe two genes from the same strain (KOS), and CD1d down-regulation was comparable to that in our initial experiment(results not shown), suggesting that in an actual HSV-1 infec-tion, gB and US3 do collaborate with each other to downregu-late CD1d.
We next examined whether US3 was required for efficientCD1d downregulation during an HSV-1 infection. HeLa.CD1dcells were infected by wild-type or US3-deficient virus and cellsurface CD1d level was measured by flow cytometry. While thewild-type virus reduced CD1d expression by 70% at 12 hpostinfection, the US3-deficient virus was much less effective,giving a reduction of only 38% (Fig. 5A), suggesting that US3plays an important role in the HSV-1-mediated inhibition ofCD1d expression. Expression of US3 by wild-type but notmutant virus was verified by Western blotting (Fig. 5B).
We previously showed that HSV-1 suppresses CD1d expres-sion by inhibiting CD1d recycling (73). To determine whetherUS3 exerts its effect at this stage, we used a flow cytometry-
FIG. 4. HSV-1 gB collaborates with US3 to downregulate CD1d. HeLa.CD1d cells were cotransfected with peGFP plasmid and plasmidsencoding gB (B), US3 (C), gB plus US3 (D), or gB plus US3 K220A (E). The transfected cells were gated as GFP-positive and GFP-negativepopulations in panel A. The cell surface CD1d level was analyzed by flow cytometry.
VOL. 85, 2011 HSV-1 gB AND US3 COLLABORATE TO DOWNREGULATE CD1d 8099
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
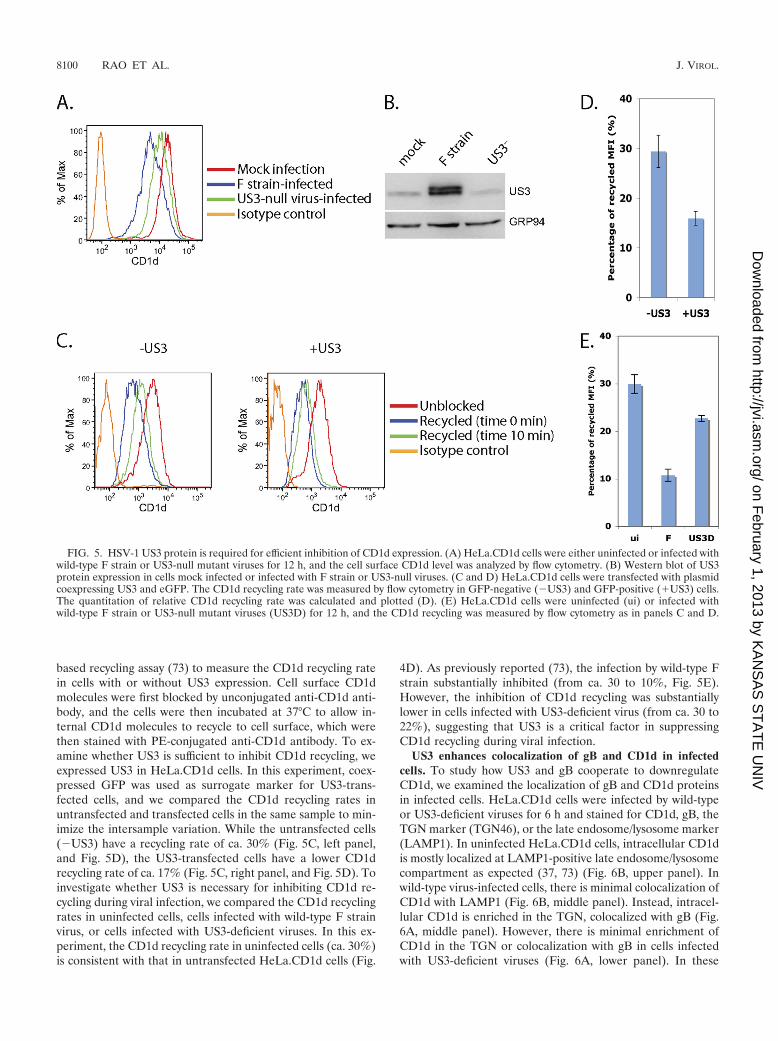
based recycling assay (73) to measure the CD1d recycling ratein cells with or without US3 expression. Cell surface CD1dmolecules were first blocked by unconjugated anti-CD1d anti-body, and the cells were then incubated at 37°C to allow in-ternal CD1d molecules to recycle to cell surface, which werethen stained with PE-conjugated anti-CD1d antibody. To ex-amine whether US3 is sufficient to inhibit CD1d recycling, weexpressed US3 in HeLa.CD1d cells. In this experiment, coex-pressed GFP was used as surrogate marker for US3-trans-fected cells, and we compared the CD1d recycling rates inuntransfected and transfected cells in the same sample to min-imize the intersample variation. While the untransfected cells(�US3) have a recycling rate of ca. 30% (Fig. 5C, left panel,and Fig. 5D), the US3-transfected cells have a lower CD1drecycling rate of ca. 17% (Fig. 5C, right panel, and Fig. 5D). Toinvestigate whether US3 is necessary for inhibiting CD1d re-cycling during viral infection, we compared the CD1d recyclingrates in uninfected cells, cells infected with wild-type F strainvirus, or cells infected with US3-deficient viruses. In this ex-periment, the CD1d recycling rate in uninfected cells (ca. 30%)is consistent with that in untransfected HeLa.CD1d cells (Fig.
4D). As previously reported (73), the infection by wild-type Fstrain substantially inhibited (from ca. 30 to 10%, Fig. 5E).However, the inhibition of CD1d recycling was substantiallylower in cells infected with US3-deficient virus (from ca. 30 to22%), suggesting that US3 is a critical factor in suppressingCD1d recycling during viral infection.
US3 enhances colocalization of gB and CD1d in infectedcells. To study how US3 and gB cooperate to downregulateCD1d, we examined the localization of gB and CD1d proteinsin infected cells. HeLa.CD1d cells were infected by wild-typeor US3-deficient viruses for 6 h and stained for CD1d, gB, theTGN marker (TGN46), or the late endosome/lysosome marker(LAMP1). In uninfected HeLa.CD1d cells, intracellular CD1dis mostly localized at LAMP1-positive late endosome/lysosomecompartment as expected (37, 73) (Fig. 6B, upper panel). Inwild-type virus-infected cells, there is minimal colocalization ofCD1d with LAMP1 (Fig. 6B, middle panel). Instead, intracel-lular CD1d is enriched in the TGN, colocalized with gB (Fig.6A, middle panel). However, there is minimal enrichment ofCD1d in the TGN or colocalization with gB in cells infectedwith US3-deficient viruses (Fig. 6A, lower panel). In these
FIG. 5. HSV-1 US3 protein is required for efficient inhibition of CD1d expression. (A) HeLa.CD1d cells were either uninfected or infected withwild-type F strain or US3-null mutant viruses for 12 h, and the cell surface CD1d level was analyzed by flow cytometry. (B) Western blot of US3protein expression in cells mock infected or infected with F strain or US3-null viruses. (C and D) HeLa.CD1d cells were transfected with plasmidcoexpressing US3 and eGFP. The CD1d recycling rate was measured by flow cytometry in GFP-negative (�US3) and GFP-positive (�US3) cells.The quantitation of relative CD1d recycling rate was calculated and plotted (D). (E) HeLa.CD1d cells were uninfected (ui) or infected withwild-type F strain or US3-null mutant viruses (US3D) for 12 h, and the CD1d recycling was measured by flow cytometry as in panels C and D.
8100 RAO ET AL. J. VIROL.
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
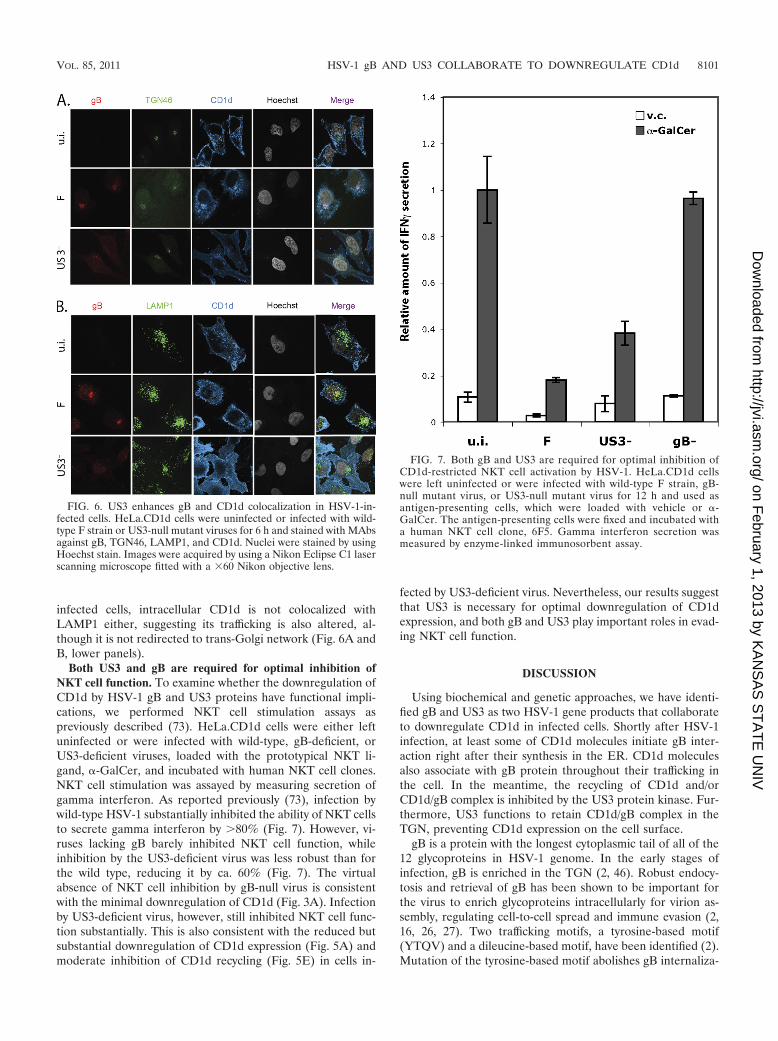
infected cells, intracellular CD1d is not colocalized withLAMP1 either, suggesting its trafficking is also altered, al-though it is not redirected to trans-Golgi network (Fig. 6A andB, lower panels).
Both US3 and gB are required for optimal inhibition ofNKT cell function. To examine whether the downregulation ofCD1d by HSV-1 gB and US3 proteins have functional impli-cations, we performed NKT cell stimulation assays aspreviously described (73). HeLa.CD1d cells were either leftuninfected or were infected with wild-type, gB-deficient, orUS3-deficient viruses, loaded with the prototypical NKT li-gand, �-GalCer, and incubated with human NKT cell clones.NKT cell stimulation was assayed by measuring secretion ofgamma interferon. As reported previously (73), infection bywild-type HSV-1 substantially inhibited the ability of NKT cellsto secrete gamma interferon by �80% (Fig. 7). However, vi-ruses lacking gB barely inhibited NKT cell function, whileinhibition by the US3-deficient virus was less robust than forthe wild type, reducing it by ca. 60% (Fig. 7). The virtualabsence of NKT cell inhibition by gB-null virus is consistentwith the minimal downregulation of CD1d (Fig. 3A). Infectionby US3-deficient virus, however, still inhibited NKT cell func-tion substantially. This is also consistent with the reduced butsubstantial downregulation of CD1d expression (Fig. 5A) andmoderate inhibition of CD1d recycling (Fig. 5E) in cells in-
fected by US3-deficient virus. Nevertheless, our results suggestthat US3 is necessary for optimal downregulation of CD1dexpression, and both gB and US3 play important roles in evad-ing NKT cell function.
DISCUSSION
Using biochemical and genetic approaches, we have identi-fied gB and US3 as two HSV-1 gene products that collaborateto downregulate CD1d in infected cells. Shortly after HSV-1infection, at least some of CD1d molecules initiate gB inter-action right after their synthesis in the ER. CD1d moleculesalso associate with gB protein throughout their trafficking inthe cell. In the meantime, the recycling of CD1d and/orCD1d/gB complex is inhibited by the US3 protein kinase. Fur-thermore, US3 functions to retain CD1d/gB complex in theTGN, preventing CD1d expression on the cell surface.
gB is a protein with the longest cytoplasmic tail of all of the12 glycoproteins in HSV-1 genome. In the early stages ofinfection, gB is enriched in the TGN (2, 46). Robust endocy-tosis and retrieval of gB has been shown to be important forthe virus to enrich glycoproteins intracellularly for virion as-sembly, regulating cell-to-cell spread and immune evasion (2,16, 26, 27). Two trafficking motifs, a tyrosine-based motif(YTQV) and a dileucine-based motif, have been identified (2).Mutation of the tyrosine-based motif abolishes gB internaliza-
FIG. 6. US3 enhances gB and CD1d colocalization in HSV-1-in-fected cells. HeLa.CD1d cells were uninfected or infected with wild-type F strain or US3-null mutant viruses for 6 h and stained with MAbsagainst gB, TGN46, LAMP1, and CD1d. Nuclei were stained by usingHoechst stain. Images were acquired by using a Nikon Eclipse C1 laserscanning microscope fitted with a �60 Nikon objective lens.
FIG. 7. Both gB and US3 are required for optimal inhibition ofCD1d-restricted NKT cell activation by HSV-1. HeLa.CD1d cellswere left uninfected or were infected with wild-type F strain, gB-null mutant virus, or US3-null mutant virus for 12 h and used asantigen-presenting cells, which were loaded with vehicle or �-GalCer. The antigen-presenting cells were fixed and incubated witha human NKT cell clone, 6F5. Gamma interferon secretion wasmeasured by enzyme-linked immunosorbent assay.
VOL. 85, 2011 HSV-1 gB AND US3 COLLABORATE TO DOWNREGULATE CD1d 8101
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from

tion, whereas disruption of the dileucine motif impairs thetrafficking of gB from early endosomes to the TGN (2).How this dileucine motif contributes to the retrieval of gB tothe TGN is not clear. Our immunofluorescence results sug-gests that US3 protein kinase may play a critical role in thisstep, since gB fails to relocalize to the TGN in most of the cellsinfected by US3-deficient virus (Fig. 6). Some cell surfacereceptors, such as Memapsin 2, are phosphorylated at theirdi-leucine motif, and this phosphorylated motif acquires theability to interact with Golgi-localized -ear-containing ARF-binding (GGA) proteins, redirecting them from the recyclingpathway to the TGN (25). gB can be directly phosphorylated byUS3 protein kinase and, in fact, multiple phosphorylation siteshave been discovered near the dileucine motif (40, 72). It willbe interesting to determine whether US3 phosphorylation ofgB at these sites leads to TGN retrieval.
US3 can also inhibit CD1d recycling directly (Fig. 5C andD), likely because of its kinase activity (Fig. 4E). The targets ofthis phosphorylation and how they block CD1d recycling areunknown, and this is an area we are currently investigating.Phosphorylation of G-protein-coupled receptors (GPCRs),cell surface receptors by protein kinase A (PKA), and GRKsleads to retention of these receptors intracellularly (7, 55).Tyrosine phosphorylation of CD1d has been reported andshown to be important for autocrine secretion of IL-10 inintestinal epithelial cells (10). However, serine/threonine phos-phorylation of CD1d has not been described. It has been pro-posed that US3 has an overlapping substrate specificity tocellular PKA (4). Using phosphorylation site prediction soft-ware for serine/threonine kinases, we identified the two puta-tive noncanonical phosphorylation sites for PKA-like kinase(T329 and S330), in the sequence RFKRQT329S330, within theCD1d cytoplasmic tail. However, overexpression of US3 inCD1d-expressing cells did not result in CD1d phosphorylation(P. Rao and W. Yuan, unpublished results), suggesting US3 isunlikely to suppress recycling by direct phosphorylation ofCD1d, but rather by phosphorylating an associated protein ora factor(s) in the CD1d recycling pathway.
Little is known about the CD1d recycling pathway, but re-cycling is required to present both endogenous and exogenousantigens to iNKT cells (37, 60). At steady state, CD1d is lo-calized in the late endosomal/lysosomal compartment (22, 37,66). The recycling routes of CD1d are not clear, namely,whether endocytosed CD1d is recycled through early/recyclingendosomes (rab4-mediated short route) or endocytic recyclingcompartment (rab11-mediated long route) (19). Regulation ofeither of these two recycling pathways by phosphorylation hasbeen reported. For the short route, it has been found that inmitotic cells or insulin-stimulated cells, cdc2/cyclin B kinasephosphorylates rab4 and relocalizes it from early endosomalmembranes to the cytosol and shuts off this vesicular recyclingpathway (1). For the long route, two phosphorylation-depen-dent regulatory events have been described. In Drosophila, ithas been shown that myosin V, the major motor protein for therab11-mediated recycling pathway in the cellular periphery, isphosphorylated by calcium/calmodulin-dependent protein ki-nase II (CaMKII), which efficiently shuts down endosomalrecycling during mitosis (38). In mammalian cells, the associ-ation of rab11-interacting protein, RIP11/FIP5, with rab11 isregulated by a phosphorylation/dephosphorylation cycle (54).
The Rip11/FIP5 protein mediates the interaction of rab11-asso-ciated recycling endosomes and microtubules for long-rangetransport of these endosomes (28). Since CD1d is predominantlylocalized in late endosomal/lysosomal compartment, it is likelythat at least a portion of CD1d recycles to cell surface through therab11-mediated long route. Recently, the motor protein associ-ated with Rip11/FIP5 has been identified as KIF3a (64), and wehave found that purified US3 protein can phosphorylate KIF3a invitro (Rao and Yuan, unpublished). We are currently investigat-ing whether this phosphorylation efficiently shuts off CD1d recy-cling in vivo.
Our studies have demonstrated that HSV-1 uses two geneproducts, gB and US3, to efficiently downregulate CD1d fromthe cell surface. Nevertheless, as shown in Fig. 5A, infectionusing US3-deficient HSV-1 still leads to CD1d downregulationto a certain extent, suggesting the existence of additional viralfactors downregulating CD1d expression. Due to the potentantiviral function of NKT cells, it is plausible that HSV-1dedicates multiple genes to inhibit the expression of NKTcell-stimulating CD1d molecules. In fact, it has been shownthat, for a single virus, multiple herpesvirus genes are dedi-cated to inhibiting MHC class I antigen presentation. For ex-ample, HCMV uses US2, US3, US6, and US11 to downregu-late MHC class I expression (24). We are currently identifyingthese putative additional HSV-1 factors downregulating CD1dexpression.
NKT cells, as a group of potent immunoregulatory T cells,play critical roles in various immune responses (5). Our dis-covery that HSV-1 efficiently downregulates CD1d expression(73) and similar findings from another group (56) support anantiviral function of NKT cells in anti-HSV-1 immunity. How-ever, the exact role of NKT cells in HSV-1 pathogenesis is notclear. In addition, whether the NKT cells play a critical role inanti-HSV-1 immunity has been under debate (11, 20, 21, 39). Itis possible that different results from different studies are dueto differences in the virus and mouse strains used. Differencesin viral virulence and host resistance can both contribute to theoutcome of viral infection.
We have identified gB and US3 as two viral factors thattogether downregulate CD1d surface expression. However,gB is an essential gene for viral replication, so only US3-deficient virus can be used for in vivo pathogenesis studies.US3 is a critical in vivo virulence factor despite the fact thatthe growth of US3-deficient virus is comparable to that ofwild-type virus in tissue culture cells (31, 41). In vivo, US3-deficient HSV-1 and HSV-2 are almost avirulent whenintroduced by conventional routes, such as ocular or peri-toneal inoculation (32, 41, 51, 61). However, when US3-deficient HSV is inoculated directly into the brain, an im-munoprivileged location, replication of US3-deficient virusis similar to that of wild-type virus (41), suggesting thatimmune-mediated clearance at least partially contributes tothe growth defect of US3-deficient virus in vivo. Recently, amutant virus with an attenuated US3 (with the autophos-phorylation site S217 mutated) showed a weakened US3function and yet still causes infection in vivo (61). It will beintriguing to compare the function of NKT cells in miceinfected by this US3-attenuated virus versus wild-type virus.
8102 RAO ET AL. J. VIROL.
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from

ACKNOWLEDGMENTS
We thank Jae U. Jung (University of Southern California) for sup-port, advice, and helpful suggestions; Konstantin Kousoulas and Ber-nard Roizman for generously providing reagents; and Ralf Leonhardt(Yale University, New Haven, CT) for valuable discussions.
This study is supported by the University of Southern CaliforniaStartup Fund, the American Cancer Society (IRG-58-007-48), the Uni-versity of Southern California Research Center for Liver Disease (NIHP30 DK048522 Satellite Project), a Robert T. Poe Faculty Develop-ment Grant and the Margaret E. Early Medical Research Trust(W.Y.), by NIH grant R01 AI059167 and the Howard Hughes MedicalInstitute (P.C.), and by NIH grant P01AI083215 to D.M.K.
REFERENCES
1. Ayad, N., M. Hull, and I. Mellman. 1997. Mitotic phosphorylation of rab4prevents binding to a specific receptor on endosome membranes. EMBO J.16:4497–4507.
2. Beitia Ortiz de Zarate, I., L. Cantero-Aguilar, M. Longo, C. Berlioz-Torrent,and F. Rozenberg. 2007. Contribution of endocytic motifs in the cytoplasmictail of herpes simplex virus type 1 glycoprotein B to virus replication andcell-cell fusion. J. Virol. 81:13889–13903.
3. Bendelac, A., et al. 1995. CD1 recognition by mouse NK1� T lymphocytes.Science 268:863–865.
4. Benetti, L., and B. Roizman. 2004. Herpes simplex virus protein kinase US3activates and functionally overlaps protein kinase A to block apoptosis. Proc.Natl. Acad. Sci. U. S. A. 101:9411–9416.
5. Brigl, M., and M. B. Brenner. 2004. CD1: antigen presentation and T cellfunction. Annu. Rev. Immunol. 22:817–890.
6. Calistri, A., et al. 2007. Intracellular trafficking and maturation of herpessimplex virus type 1 gB and virus egress require functional biogenesis ofmultivesicular bodies. J. Virol. 81:11468–11478.
7. Cao, T. T., H. W. Deacon, D. Reczek, A. Bretscher, and M. von Zastrow.1999. A kinase-regulated PDZ-domain interaction controls endocytic sortingof the �2-adrenergic receptor. Nature 401:286–290.
8. Chen, N., et al. 2006. HIV-1 downregulates the expression of CD1d via Nef.Eur. J. Immunol. 36:278–286.
9. Cho, S., et al. 2005. Impaired cell surface expression of human CD1d by theformation of an HIV-1 Nef/CD1d complex. Virology 337:242–252.
10. Colgan, S. P., R. M. Hershberg, G. T. Furuta, and R. S. Blumberg. 1999.Ligation of intestinal epithelial CD1d induces bioactive IL-10: critical role ofthe cytoplasmic tail in autocrine signaling. Proc. Natl. Acad. Sci. U. S. A.96:13938–13943.
11. Cornish, A. L., et al. 2006. NKT cells are not critical for HSV-1 diseaseresolution. Immunol. Cell Biol. 84:13–19.
12. Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for con-structing mutants of herpes simplex virus type 1. Virology 197:116–124.
13. De Santo, C., et al. 2008. Invariant NKT cells reduce the immunosuppressiveactivity of influenza A virus-induced myeloid-derived suppressor cells inmice and humans. J. Clin. Invest. 118:4036–4048.
14. Diana, J., et al. 2009. NKT cell-plasmacytoid dendritic cell cooperation viaOX40 controls viral infection in a tissue-specific manner. Immunity 30:289–299.
15. Exley, M., et al. 2000. CD1d structure and regulation on human thymocytes,peripheral blood T cells, B cells and monocytes. Immunology 100:37–47.
16. Favoreel, H. W., G. Van Minnebruggen, H. J. Nauwynck, L. W. Enquist, andM. B. Pensaert. 2002. A tyrosine-based motif in the cytoplasmic tail ofpseudorabies virus glycoprotein B is important for both antibody-inducedinternalization of viral glycoproteins and efficient cell-to-cell spread. J. Virol.76:6845–6851.
17. Gadola, S. D., et al. 2002. Structure of human CD1b with bound ligands at2.3 Å, a maze for alkyl chains. Nat. Immunol. 3:721–726.
18. Godfrey, D. I., and M. Kronenberg. 2004. Going both ways: immune regu-lation via CD1d-dependent NKT cells. J. Clin. Invest. 114:1379–1388.
19. Grant, B. D., and J. G. Donaldson. 2009. Pathways and mechanisms ofendocytic recycling. Nat. Rev. Mol. Cell. Biol. 10:597–608.
20. Grubor-Bauk, B., J. L. Arthur, and G. Mayrhofer. 2008. Importance of NKTcells in resistance to herpes simplex virus, fate of virus-infected neurons, andlevel of latency in mice. J. Virol. 82:11073–11083.
21. Grubor-Bauk, B., A. Simmons, G. Mayrhofer, and P. G. Speck. 2003. Im-paired clearance of herpes simplex virus type 1 from mice lacking CD1d orNKT cells expressing the semivariant V alpha 14-J. alpha 281 TCR. J. Im-munol. 170:1430–1434.
22. Gumperz, J. E. 2006. The ins and outs of CD1 molecules: bringing lipidsunder immunological surveillance. Traffic 7:2–13.
23. Hage, C. A., et al. 2005. Human immunodeficiency virus gp120 downregu-lates CD1d cell surface expression. Immunol. Lett. 98:131–135.
24. Hansen, T. H., and M. Bouvier. 2009. MHC class I antigen presentation:learning from viral evasion strategies. Nat. Rev. Immunol. 9:503–513.
25. He, X., F. Li, W. P. Chang, and J. Tang. 2005. GGA proteins mediate therecycling pathway of memapsin 2 (BACE). J. Biol. Chem. 280:11696–11703.
26. Heineman, T. C., P. Connolly, S. L. Hall, and D. Assefa. 2004. Conservedcytoplasmic domain sequences mediate the ER export of VZV, HSV-1, andHCMV gB. Virology 328:131–141.
27. Heineman, T. C., and S. L. Hall. 2001. VZV gB endocytosis and Golgilocalization are mediated by YXX motifs in its cytoplasmic domain. Virol-ogy 285:42–49.
28. Hirokawa, N., Y. Noda, Y. Tanaka, and S. Niwa. 2009. Kinesin superfamilymotor proteins and intracellular transport. Nat. Rev. Mol. Cell. Biol. 10:682–696.
29. Ho, L. P., et al. 2008. Activation of invariant NKT cells enhances the innateimmune response and improves the disease course in influenza A virusinfection. Eur. J. Immunol. 38:1913–1922.
30. Ilyinskii, P. O., R. Wang, S. P. Balk, and M. A. Exley. 2006. CD1d mediatesT-cell-dependent resistance to secondary infection with encephalomyocar-ditis virus (EMCV) in vitro and immune response to EMCV infection invivo. J. Virol. 80:7146–7158.
31. Imai, T., K. Sagou, J. Arii, and Y. Kawaguchi. 2010. Effects of phosphory-lation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase invivo and in vitro. J. Virol. 84:153–162.
32. Inagaki-Ohara, K., T. Iwasaki, D. Watanabe, T. Kurata, and Y. Nishiyama.2001. Effect of the deletion of US2 and US3 from herpes simplex virus type2 on immune responses in the murine vagina following intravaginal infection.Vaccine 20:98–104.
33. Johnson, D. C., and N. R. Hegde. 2002. Inhibition of the MHC class IIantigen presentation pathway by human cytomegalovirus. Curr. Top. Micro-biol. Immunol. 269:101–115.
34. Johnson, T. R., S. Hong, L. Van Kaer, Y. Koezuka, and B. S. Graham. 2002.NK T cells contribute to expansion of CD8� T cells and amplification ofantiviral immune responses to respiratory syncytial virus. J. Virol. 76:4294–4303.
35. Kakimi, K., L. G. Guidotti, Y. Koezuka, and F. V. Chisari. 2000. Naturalkiller T cell activation inhibits hepatitis B virus replication in vivo. J. Exp.Med. 192:921–930.
36. Kang, S. J., and P. Cresswell. 2002. Calnexin, calreticulin, and ERp57 co-operate in disulfide bond formation in human CD1d heavy chain. J. Biol.Chem. 277:44838–44844.
37. Kang, S. J., and P. Cresswell. 2002. Regulation of intracellular trafficking ofhuman CD1d by association with MHC class II molecules. EMBO J. 21:1650–1660.
38. Karcher, R. L., et al. 2001. Cell cycle regulation of myosin-V by calcium/calmodulin-dependent protein kinase II. Science 293:1317–1320.
39. Kastrukoff, L. F., et al. 2010. Redundancy in the immune system restricts thespread of HSV-1 in the central nervous system (CNS) of C57BL/6 mice.Virology 400:248–258.
40. Kato, A., et al. 2009. Herpes simplex virus 1 protein kinase Us3 phosphor-ylates viral envelope glycoprotein B and regulates its expression on the cellsurface. J. Virol. 83:250–261.
41. Kurachi, R., et al. 1993. The pathogenicity of a US3 protein kinase-deficientmutant of herpes simplex virus type 2 in mice. Arch. Virol. 133:259–273.
42. Lin, E., and P. G. Spear. 2007. Random linker-insertion mutagenesis toidentify functional domains of herpes simplex virus type 1 glycoprotein B.Proc. Natl. Acad. Sci. U. S. A. 104:13140–13145.
43. Lin, Y., T. J. Roberts, P. M. Spence, and R. R. Brutkiewicz. 2005. Reductionin CD1d expression on dendritic cells and macrophages by an acute virusinfection. J. Leukoc. Biol. 77:151–158.
44. Liu, J., et al. 2010. A threonine-based targeting signal in the human CD1dcytoplasmic tail controls its functional expression. J. Immunol. 184:4973–4981.
45. Liu, X., K. Fitzgerald, E. Kurt-Jones, R. Finberg, and D. M. Knipe. 2008.Herpesvirus tegument protein activates NF-�B signaling through theTRAF6 adaptor protein. Proc. Natl. Acad. Sci. U. S. A. 105:11335–11339.
46. Maresova, L., T. J. Pasieka, E. Homan, E. Gerday, and C. Grose. 2005.Incorporation of three endocytosed varicella-zoster virus glycoproteins, gE,gH, and gB, into the virion envelope. J. Virol. 79:997–1007.
47. Melancon, J. M., R. E. Luna, T. P. Foster, and K. G. Kousoulas. 2005.Herpes simplex virus type 1 gK is required for gB-mediated virus-inducedcell fusion, while neither gB and gK nor gB and UL20p function redundantlyin virion de-envelopment. J. Virol. 79:299–313.
48. Moll, M., S. K. Andersson, A. Smed-Sorensen, and J. K. Sandberg. 2010.Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpuinterference with CD1d recycling from endosomal compartments. Blood116:1876–1884.
49. Moll, M., et al. 2006. Expansion of CD1d-restricted NKT cells in patientswith primary HIV-1 infection treated with interleukin-2. Blood 107:3081–3083.
50. Mulvey, M., C. Arias, and I. Mohr. 2007. Maintenance of endoplasmicreticulum (ER) homeostasis in herpes simplex virus type 1-infected cellsthrough the association of a viral glycoprotein with PERK, a cellular ERstress sensor. J. Virol. 81:3377–3390.
51. Nishiyama, Y., Y. Yamada, R. Kurachi, and T. Daikoku. 1992. Constructionof a US3 lacZ insertion mutant of herpes simplex virus type 2 and charac-terization of its phenotype in vitro and in vivo. Virology 190:256–268.
VOL. 85, 2011 HSV-1 gB AND US3 COLLABORATE TO DOWNREGULATE CD1d 8103
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from

52. Paduraru, C., et al. 2006. An N-linked glycan modulates the interactionbetween the CD1d heavy chain and �2-microglobulin. J. Biol. Chem. 281:40369–40378.
53. Poon, A. P., and B. Roizman. 2007. Mapping of key functions of the herpessimplex virus 1 US3 protein kinase: the US3 protein can form functionalheteromultimeric structures derived from overlapping truncated polypep-tides. J. Virol. 81:1980–1989.
54. Prekeris, R., J. Klumperman, and R. H. Scheller. 2000. A Rab11/Rip11protein complex regulates apical membrane trafficking via recycling endo-somes. Mol. Cell 6:1437–1448.
55. Prossnitz, E. R. 2004. Novel roles for arrestins in the post-endocytic traf-ficking of G protein-coupled receptors. Life Sci. 75:893–899.
56. Raftery, M. J., F. Winau, S. H. Kaufmann, U. E. Schaible, and G. Schonrich.2006. CD1 antigen presentation by human dendritic cells as a target forherpes simplex virus immune evasion. J. Immunol. 177:6207–6214.
57. Renukaradhya, G. J., M. A. Khan, D. Shaji, and R. R. Brutkiewicz. 2008.Vesicular stomatitis virus matrix protein impairs CD1d-mediated antigenpresentation through activation of the p38 MAPK pathway. J. Virol. 82:12535–12542.
58. Renukaradhya, G. J., et al. 2005. Virus-induced inhibition of CD1d1-medi-ated antigen presentation: reciprocal regulation by p38 and ERK. J. Immu-nol. 175:4301–4308.
59. Rigaud, S., et al. 2006. XIAP deficiency in humans causes an X-linkedlymphoproliferative syndrome. Nature 444:110–114.
60. Roberts, T. J., et al. 2002. Recycling CD1d1 molecules present endogenousantigens processed in an endocytic compartment to NKT cells. J. Immunol.168:5409–5414.
61. Sagou, K., T. Imai, H. Sagara, M. Uema, and Y. Kawaguchi. 2009. Regula-tion of the catalytic activity of herpes simplex virus 1 protein kinase Us3 byautophosphorylation and its role in pathogenesis. J. Virol. 83:5773–5783.
62. Sanchez, D. J., J. E. Gumperz, and D. Ganem. 2005. Regulation of CD1dexpression and function by a herpesvirus infection. J. Clin. Invest. 115:1369–1378.
63. Schaefer, M. R., E. R. Wonderlich, J. F. Roeth, J. A. Leonard, and K. L.
Collins. 2008. HIV-1 Nef targets MHC-I and CD4 for degradation via a finalcommon beta-COP-dependent pathway in T cells. PLoS Pathog. 4:e1000131.
64. Schonteich, E., et al. 2008. The Rip11/Rab11-FIP5 and kinesin II complexregulates endocytic protein recycling. J. Cell Sci. 121:3824–3833.
65. Spear, P. G. 2004. Herpes simplex virus: receptors and ligands for cell entry.Cell Microbiol. 6:401–410.
66. Sugita, M., et al. 2002. Failure of trafficking and antigen presentation by CD1in AP-3-deficient cells. Immunity 16:697–706.
67. van der Vliet, H. J., et al. 2006. Cutting edge: rapid recovery of NKT cellsupon institution of highly active antiretroviral therapy for HIV-1 infection.J. Immunol. 177:5775–5778.
68. van der Vliet, H. J., et al. 2002. Selective decrease in circulating V alpha24�V beta 11� NKT cells during HIV type 1 infection. J. Immunol. 168:1490–1495.
69. van Dommelen, S. L., H. A. Tabarias, M. J. Smyth, and M. A. Degli-Esposti.2003. Activation of natural killer (NK) T cells during murine cytomegalovi-rus infection enhances the antiviral response mediated by NK cells. J. Virol.77:1877–1884.
70. Wang, B., et al. 2001. Human CD1d functions as a transplantation antigenand a restriction element in mice. J. Immunol. 166:3829–3836.
71. Webb, T. J., et al. 2006. Inhibition of CD1d1-mediated antigen presentationby the vaccinia virus B1R and H5R molecules. Eur. J. Immunol. 36:2595–2600.
72. Wisner, T. W., et al. 2009. Herpesvirus gB-induced fusion between the virionenvelope and outer nuclear membrane during virus egress is regulated by theviral US3 kinase. J. Virol. 83:3115–3126.
73. Yuan, W., A. Dasgupta, and P. Cresswell. 2006. Herpes simplex virus evadesnatural killer T cell recognition by suppressing CD1d recycling. Nat. Immu-nol. 7:835–842.
74. Yuan, W., S. J. Kang, J. E. Evans, and P. Cresswell. 2009. Natural lipidligands associated with human CD1d targeted to different subcellular com-partments. J. Immunol. 182:4784–4791.
75. Zeng, Z., et al. 1997. Crystal structure of mouse CD1: an MHC-like fold witha large hydrophobic binding groove. Science 277:339–345.
8104 RAO ET AL. J. VIROL.
on February 1, 2013 by K
AN
SA
S S
TA
TE
UN
IVhttp://jvi.asm
.org/D
ownloaded from
Related Documents











