JOURNAL OF VIROLOGY, 0022-538X/98/$04.0010 July 1998, p. 6119–6130 Vol. 72, No. 7 Copyright © 1998, American Society for Microbiology. All Rights Reserved. Heparan Sulfate Proteoglycan Binding by Herpes Simplex Virus Type 1 Glycoproteins B and C, Which Differ in Their Contributions to Virus Attachment, Penetration, and Cell-to-Cell Spread SYLVIE LAQUERRE, 1 RAFAELA ARGNANI, 2 DINA B. ANDERSON, 1 SILVIA ZUCCHINI, 2 ROBERTO MANSERVIGI, 2 AND JOSEPH C. GLORIOSO 1 * Department of Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15261, 1 and Biotechnology Center, University of Ferrara, Ferrara 1-44100, Italy 2 Received 14 January 1998/Accepted 21 April 1998 Herpes simplex virus type 1 (HSV-1) mutants defective for envelope glycoprotein C (gC) and gB are highly impaired in the ability to attach to cell surface heparan sulfate (HS) moieties of proteoglycans, the initial virus receptor. Here we report studies aimed at defining the HS binding element of HSV-1 (strain KOS) gB and determining whether this structure is functionally independent of gB’s role in extracellular virus penetration or intercellular virus spread. A mutant form of gB deleted for a putative HS binding lysine-rich (pK) sequence (residues 68 to 76) was transiently expressed in Vero cells and shown to be processed normally, leading to exposure on the cell surface. Solubilized gBpK 2 also had substantially lower affinity for heparin-acrylic beads than did wild-type gB, confirming that the HS binding domain had been inactivated. The gBpK 2 gene was used to rescue a KOS gB null mutant virus to produce the replication-competent mutant KgBpK 2 . Compared with wild-type virus, KgBpK 2 showed reduced binding to mouse L cells (ca. 20%), while a gC null mutant virus in which the gC coding sequence was replaced by the lacZ gene (KCZ) was substantially more impaired (ca. 65%-reduced binding), indicating that the contribution of gC to HS binding was greater than that of gB. The effect of combining both mutations into a single virus (KgBpK 2 gC 2 ) was additive (ca. 80%-reduced binding to HS) and displayed a binding activity similar to that observed for KOS virus attachment to sog9 cells, a glycosaminoglycan-deficient L-cell line. Cell-adsorbed individual and double HS mutant viruses exhibited a lower rate of virus entry following attachment, suggesting that HS binding plays a role in the process of virus penetration. Moreover, the KgBpK 2 mutant virus produced small plaques on Vero cells in the presence of neutralizing antibody where plaque formation depended on cell-to-cell virus spread. These studies permitted the following conclusions: (i) the pK sequence is not essential for gB processing or function in virus infection, (ii) the lysine-rich sequence of gB is responsible for HS binding, and (iii) binding to HS is cooperatively linked to the process of efficient virus entry and lateral spread but is not absolutely required for virus infectivity. Herpes simplex virus type 1 (HSV-1) is a neurotropic human pathogen capable of infection and spread in a variety of cells. Infection is mediated by the viral envelope glycoproteins, which have been assigned specific and often redundant func- tional roles. Of the 10 virus envelope glycoproteins, only gB, gD, gH, and gL are essential to the process of infection in cell culture, while the other six contribute to virus infectivity and spread in the host (2, 4, 5, 10, 14, 27, 29, 42, 43, 54). An additional glycoprotein, gK, has been shown to be absent from the virus envelope; however, it is required for the production of infectious virions (30, 31). Infection involves virus attachment to the cell surface mem- brane followed by virus penetration and entry of the nucleo- capsid into the cytoplasm (53, 57). Current evidence indicates that virus attachment is a two-step process (48) involving dif- ferent glycoproteins and several receptors. Glycoprotein B (gB) and gC have been shown to be involved in the initial attachment phase through the interaction of positively charged glycoprotein structures with negatively charged heparan sul- fate (HS) moieties located on cell surface proteoglycans (44, 56). This HS-dependent attachment may facilitate a second attachment in which gD binds to a cellular receptor, one of them recently reported to be a member of the tumor necrosis factor-nerve growth factor receptor family (50). Following at- tachment, the virus penetrates the cell by fusion of the virus envelope with the cell plasma membrane (57). Genetic studies have shown that gB, gD, and gH are required to carry out the fusion-penetration process (4, 10, 32, 42) and that gL is essen- tial for proper processing and insertion of gH into the virus envelope (29). These studies have demonstrated that virus penetration is a highly complex process involving the cooper- ative activities of multiple viral glycoproteins. Different lines of evidence have identified HS as an initial receptor for HSV infection. First, HS proteoglycans are com- monly found on the surface of most vertebrate cell types (15), including those susceptible to HSV infection (16, 21, 44, 58, 64). Second, removal of HS from the cell surface, either by enzymatic treatment or by selection of cell lines defective in the pathway of HS (3, 17, 41, 56), renders the cells at least partially resistant to HSV infection by reducing virus attach- ment to the cell surface. Third, heparin, a molecule chemically similar to HS (35), has been shown to inhibit viral infection by masking the HS binding domain on the virus envelope (21, 22, 55), and immobilized heparin columns bind to the principal * Corresponding author. Mailing address: Department of Molecular Genetics and Biochemistry, University of Pittsburgh School of Medi- cine, E1240 Biomedical Science Tower, Pittsburgh, PA 15261. Phone: (412) 648-8106. Fax: (412) 624-8997. E-mail: [email protected] .edu. 6119 on May 19, 2018 by guest http://jvi.asm.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF VIROLOGY,0022-538X/98/$04.0010
July 1998, p. 6119–6130 Vol. 72, No. 7
Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Heparan Sulfate Proteoglycan Binding by Herpes Simplex VirusType 1 Glycoproteins B and C, Which Differ in Their
Contributions to Virus Attachment, Penetration,and Cell-to-Cell Spread
SYLVIE LAQUERRE,1 RAFAELA ARGNANI,2 DINA B. ANDERSON,1 SILVIA ZUCCHINI,2
ROBERTO MANSERVIGI,2 AND JOSEPH C. GLORIOSO1*
Department of Molecular Genetics and Biochemistry, University of Pittsburgh School of Medicine,Pittsburgh, Pennsylvania 15261,1 and Biotechnology Center, University of Ferrara, Ferrara 1-44100, Italy2
Received 14 January 1998/Accepted 21 April 1998
Herpes simplex virus type 1 (HSV-1) mutants defective for envelope glycoprotein C (gC) and gB are highlyimpaired in the ability to attach to cell surface heparan sulfate (HS) moieties of proteoglycans, the initial virusreceptor. Here we report studies aimed at defining the HS binding element of HSV-1 (strain KOS) gB anddetermining whether this structure is functionally independent of gB’s role in extracellular virus penetrationor intercellular virus spread. A mutant form of gB deleted for a putative HS binding lysine-rich (pK) sequence(residues 68 to 76) was transiently expressed in Vero cells and shown to be processed normally, leading toexposure on the cell surface. Solubilized gBpK2 also had substantially lower affinity for heparin-acrylic beadsthan did wild-type gB, confirming that the HS binding domain had been inactivated. The gBpK2 gene was usedto rescue a KOS gB null mutant virus to produce the replication-competent mutant KgBpK2. Compared withwild-type virus, KgBpK2 showed reduced binding to mouse L cells (ca. 20%), while a gC null mutant virus inwhich the gC coding sequence was replaced by the lacZ gene (KCZ) was substantially more impaired (ca.65%-reduced binding), indicating that the contribution of gC to HS binding was greater than that of gB. Theeffect of combining both mutations into a single virus (KgBpK2gC2) was additive (ca. 80%-reduced binding toHS) and displayed a binding activity similar to that observed for KOS virus attachment to sog9 cells, aglycosaminoglycan-deficient L-cell line. Cell-adsorbed individual and double HS mutant viruses exhibited alower rate of virus entry following attachment, suggesting that HS binding plays a role in the process of viruspenetration. Moreover, the KgBpK2 mutant virus produced small plaques on Vero cells in the presence ofneutralizing antibody where plaque formation depended on cell-to-cell virus spread. These studies permittedthe following conclusions: (i) the pK sequence is not essential for gB processing or function in virus infection,(ii) the lysine-rich sequence of gB is responsible for HS binding, and (iii) binding to HS is cooperatively linkedto the process of efficient virus entry and lateral spread but is not absolutely required for virus infectivity.
Herpes simplex virus type 1 (HSV-1) is a neurotropic humanpathogen capable of infection and spread in a variety of cells.Infection is mediated by the viral envelope glycoproteins,which have been assigned specific and often redundant func-tional roles. Of the 10 virus envelope glycoproteins, only gB,gD, gH, and gL are essential to the process of infection in cellculture, while the other six contribute to virus infectivity andspread in the host (2, 4, 5, 10, 14, 27, 29, 42, 43, 54). Anadditional glycoprotein, gK, has been shown to be absent fromthe virus envelope; however, it is required for the productionof infectious virions (30, 31).
Infection involves virus attachment to the cell surface mem-brane followed by virus penetration and entry of the nucleo-capsid into the cytoplasm (53, 57). Current evidence indicatesthat virus attachment is a two-step process (48) involving dif-ferent glycoproteins and several receptors. Glycoprotein B(gB) and gC have been shown to be involved in the initialattachment phase through the interaction of positively chargedglycoprotein structures with negatively charged heparan sul-
fate (HS) moieties located on cell surface proteoglycans (44,56). This HS-dependent attachment may facilitate a secondattachment in which gD binds to a cellular receptor, one ofthem recently reported to be a member of the tumor necrosisfactor-nerve growth factor receptor family (50). Following at-tachment, the virus penetrates the cell by fusion of the virusenvelope with the cell plasma membrane (57). Genetic studieshave shown that gB, gD, and gH are required to carry out thefusion-penetration process (4, 10, 32, 42) and that gL is essen-tial for proper processing and insertion of gH into the virusenvelope (29). These studies have demonstrated that viruspenetration is a highly complex process involving the cooper-ative activities of multiple viral glycoproteins.
Different lines of evidence have identified HS as an initialreceptor for HSV infection. First, HS proteoglycans are com-monly found on the surface of most vertebrate cell types (15),including those susceptible to HSV infection (16, 21, 44, 58,64). Second, removal of HS from the cell surface, either byenzymatic treatment or by selection of cell lines defective inthe pathway of HS (3, 17, 41, 56), renders the cells at leastpartially resistant to HSV infection by reducing virus attach-ment to the cell surface. Third, heparin, a molecule chemicallysimilar to HS (35), has been shown to inhibit viral infection bymasking the HS binding domain on the virus envelope (21, 22,55), and immobilized heparin columns bind to the principal
* Corresponding author. Mailing address: Department of MolecularGenetics and Biochemistry, University of Pittsburgh School of Medi-cine, E1240 Biomedical Science Tower, Pittsburgh, PA 15261. Phone:(412) 648-8106. Fax: (412) 624-8997. E-mail: [email protected].
6119
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

mediators of virus attachment, gB and gC, either derived fromHSV-1-infected cells or produced in a baculovirus expressionsystem (24, 59). Fourth, construction of deletion mutants forthe glycoproteins involved in HS binding, namely gC and gB,impairs virus binding to the cell surface (23).
Glycoprotein C interactions with HS have been extensivelystudied (6, 12, 16, 23, 24, 48), and HS binding domains havebeen identified (62). HSV-1 gC-negative mutants (23, 24, 28,59) are impaired in binding and slightly impaired in penetra-tion but remain highly infectious, presumably because the HSbinding function of gB is sufficient to mediate virus attachmentto HS (24). A deletion mutant lacking both gC and gB codingsequences was demonstrated to be substantially impaired inbinding compared with a gC null mutant virus, suggesting thatgB also contributes to HS binding (23). The residual attach-ment ability of this mutant may indicate either that other viralglycoproteins possess limited HS binding activity or that addi-tional receptors are recognized in the absence of HS binding.Mutants deleted for gB cannot be used to determine whethergB binding to HS is essential to the process of penetration andcell-to-cell spread, since gB is required for both functions.
In this report, experiments were performed to (i) identify theHS binding domain of gB and quantify its contribution to HSbinding, (ii) determine whether the HS binding domain of gBis required for gB’s essential role in virus penetration andlateral spread, and (iii) compare the contributions of both gB-and gC-mediated HS binding to the efficiency of virus attach-ment, rate of penetration, and lateral spread in plaque forma-tion. To achieve these goals, a lysine-rich (pK) sequence (ami-no acids 68 to 76) was deleted from the N terminus of gB(gBpK2) and analyzed for its ability to bind heparin-acrylicbeads. The reduction in gBpK2 binding to heparin confirmedthat an HS binding domain had been removed. A mutant viruscarrying this altered gB molecule showed reduced adsorptionto the cell membrane and reduced capacity to spread from cellto cell but remained infectious, demonstrating that gB bindingto HS is not essential for gB function. A double mutant virus(KgBpK2gC2) deleted for gC in addition to the pK domain ofgB, was constructed and shown to be highly impaired in bind-ing and to have a reduced rate of penetration. Taken together,these data demonstrate that HS binding is required for efficientpenetration by both extra- and intercellular virus.
MATERIALS AND METHODS
Cells and viruses. Vero cells were used to propagate the KOS strain of HSV-1,from which all recombinant viruses were derived. The Vero cell line A1 (60)(kindly provided by Fred Homa, Kalamazoo, Mich.), stably transfected with theHSV-1 genes encoding gB and ICP 18.5, was used to propagate a double-mutantvirus, KD4BX (9), deleted for both genes. The Vero cell line C1 (60) (kindlyprovided by Fred Homa), stably transfected with the HSV-1 gene encoding ICP18.5, was used to passage the double-mutant virus (KgBpK2gC2) to generate avirus particle deleted for gB and gC only. Mouse L cells and their glycosamino-glycan (GAG)-negative derivatives (sog9 [3]) were kindly provided by FrankTufaro, Vancouver, British Columbia, Canada. All cell lines were maintained at37°C in Dulbecco’s modified minimum essential medium (DMEM) (Gibco BRL,Grand Island, N.Y.) supplemented with 10% fetal bovine serum.
Construction of gB mutant and gC-deleted plasmids. The HSV-1 gB codingsequence (UL27) was excised by enzymatically digesting the BamG fragment ofthe viral genome with KpnI and SalI endonucleases and subcloned with theKpnI-XhoI restriction sites of the pTZ18U vector (pTZ18UgB1). Site-specificmutagenesis was performed to delete 27 nucleotides encoding the putative HSbinding domain of gB (amino acids 68 to 76 [KPKKNKKPK]), and a BamHIrecognition site was inserted in frame at the site of the mutation. The resultinggBpK2 mutation was subsequently inserted into the pKBXX vector (4, 25) tocreate pgBpK2, and both plasmids (pKBXX and pgBpK2) were further modi-fied by inserting the human cytomegalovirus immediate early promoter (HCMV-IEp) at their 59 ends to create HCMV-BXX and HCMV-gBpK2.
The HSV-1 gC coding sequence (UL44) contained in the pgC1 plasmid (27)was deleted by digestion with the restriction endonucleases XhoI and EcoRV,and the 1,622-bp fragment was replaced with the SalI/BamHI fragment encoding
the HCMV-lacZ cassette of the pIEP-lacZ plasmid (12) in order to createpDgC:lacZ.
Construction and isolation of recombinant viruses. Mutant HSV-1 viruseswere constructed by standard methods for transfer by using LipofectAmine(Gibco BRL) for cotransfection, and mutant or recombinant virus plaques werethrice plaque purified by limiting dilution prior to characterization. To constructthe KgBpK2 recombinant virus, plasmid pgBpK2 DNA was cotransfected withviral DNA from KD4BX on the complementing A1 cell line. The KgBpK2
recombinant virus was selected for growth on Vero cells and screened by South-ern hybridization with a gB probe that hybridized to the gB coding sequence (seeFig. 3A). The pDgC:lacZ plasmid was used to cotransfect Vero cells with viralDNA from KOS. A virus recombinant (KCZ) deleted for 1,622 bp of the gCcoding sequence was selected by complement-dependent neutralization with apool of gC-specific monoclonal antibodies (MAbs) (47). The viral plaquesformed by the neutralization escape mutants were further screened for the“blue-plaque” phenotype in presence of X-Gal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside) substrate. The same pDgC:lacZ plasmid was used to co-transfect A1 cells with KD4BX viral DNA in order to create the KD4BXCZ virus,which was deleted for genes encoding ICP 18.5, gB, and gC. The recombinantvirus was selected on A1-complementing cells, as described above for KCZ onVero cells. Southern blot hybridization with a gC probe was used to confirm theabsence of the gC coding sequence in the KCZ (see Fig. 3B) and KD4BXCZviruses. The double mutant KgBpKgB2gC2 was produced by cotransfection ofpgBpK2 plasmid DNA with viral DNA from KD4BXCZ on A1 cells. Virusesfrom the cotransfection were plated on Vero cells, and the recombinant virusdeleted for both the pK region of gB and the gC coding sequence (KgBpK2gC2)was selected for the presence of the gBpK2 sequence, as described above for theKgBpK2 recombinant virus, and for the absence of the gC coding sequence, asdescribed above for KCZ. KgBpK2gC2 recombinant virus was rescued for thefull-length gB and gC coding sequences in order to produce KgBpKRgC2 andKgBpK2gCR, respectively. Viral DNA from KgBpK2gC2 was cotransfectedwith wild-type KOS gB DNA (pKBXX [4]) or wild-type KOS gC DNA (pgC1[27]). Vero cell plaques from both cotransfections were isolated and tested forthe rescue of each respective gene by Southern blot hybridization, as shown inFig. 3. The rescue of gC was also confirmed by complement-dependent neutral-ization of the recombinant virus with gC-specific MAbs and by a “clear-plaque”phenotype following X-Gal staining.
Immunofluorescence. Thirty hours posttransfection with HCMV-BXX orHCMV-gBpK2, the Vero cell monolayers were incubated for 1 h at 4°C with apool of gB MAbs (46), rinsed with cold Tris-buffered saline (TBS), pH 7.4, andincubated for an additional hour with a cy3-conjugated anti-mouse antibody(Jackson Immunoresearch Laboratories, West Grove, Pa.); the monolayer wasthen fixed with 2% paraformaldehyde. Immunofluorescence-positive cells werephotographed with a Nikon TMS microscope-camera (model 211910).
Binding of gB and gBpK2 to heparin-acrylic beads. Plasmids pKBXX (4) andpgBpK2 were used individually to transfect Vero cells. Twenty-four hours post-transfection, the cell monolayers were infected at a multiplicity of infection(MOI) of 3 with KD4BX virus followed by overlay with DMEM containing[35S]methionine/cysteine. Ten hours postinfection (p.i.), the cell monolayerswere scraped, harvested, and resuspended in 200 ml of lysis buffer (0.5 M Tris-HCl, 150 mM NaCl, 1% Triton X-100, 10 mM phenylmethylsulfonyl fluoride,and 1 mM TLCK [Na-p-tosyl-L-lysine chloromethyl ketone]). The Triton-solubleextracts were incubated for 2 h at 4°C in the presence of heparin-acrylic beads(Sigma, St. Louis, Mo.). The beads were rinsed three times with lysis buffer orlysis buffer supplemented with 10 mg of heparin (Sigma) per ml. The Triton-soluble extract and lysis buffer washes or lysis buffer with heparin washes wereimmunoprecipitated with a pool of gB-specific MAbs (46) and analyzed bysodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Quan-tification of precipitated radiolabeled proteins was determined with the 1-D Scanand Report program (Biomed Instruments, Fullerton, Calif.).
Purification of radiolabeled virus. Virions used for binding and immunopre-cipitation assays were labeled and purified as follows. Confluent Vero cell mono-layers in T150s flasks (Falcon; Becton Dickinson, Franklin Lakes, N.J.) wereinfected with viruses at an MOI of 10. Four hours p.i., 16 ml of DMEM withoutmethionine and cysteine (Gibco BRL) and supplemented with 1% fetal calfserum was added to the infected cell monolayers. [35S]methionine/cysteine (Ex-preSS; Dupont, NEN, Boston, Mass.) having a specific activity of 50 mCi/ml wasadded after 4 h. Twenty-four hours p.i., medium containing radiolabeled viruswas harvested and virions were purified by centrifugation (SWTi-40 Beckmanrotor) through sucrose gradients (30 to 65% sucrose). The fractions containingthe radiolabeled virus were pooled, diluted in sterile TBS, and centrifuged at20,000 3 g for 1 h at 4°C in a SWTi-40 rotor. The virus pellet was resuspendedin DMEM containing 10% fetal bovine serum, and radioactivity was determinedwith a beta counter (Beckman, Fullerton, Calif.).
Immunoprecipitation analysis of surface glycoproteins. Aliquots of radiola-beled virus were immunoprecipitated with a pool of gB-specific (46), gC-specific(47), or gD-specific (26) MAbs. Each virus aliquot was diluted in 200 ml of lysisbuffer containing 2 ml of antibody and incubated at 4°C for a minimum of 4 h.The quantity of antibody used in this assay was in 10-fold excess of the amountneeded for complete immunoprecipitation of gB from wild-type virus-infectedcells. The immune complexes were incubated with protein A-Sepharose (Sigma)for 1 h, centrifuged at 500 3 g, and washed five times with 600 ml of lysis buffer.
6120 LAQUERRE ET AL. J. VIROL.
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

The protein A-Sepharose complexes were resuspended in Laemmli loadingbuffer (36), boiled for 2 min, and subjected to SDS-PAGE. After electrophoresis,the gels were fixed, treated with En3Hance solution (Dupont, NEN), vacuumdried, and exposed to X-Omat-XAR film (Kodak, Rochester, N.Y.). Quantifi-cation of radiolabeled proteins precipitated was determined with the 1-D Scanand Report program (Biomed Instruments).
Binding of radiolabeled virus to cells. Monolayers of confluent Vero, murineL, or sog9 cells in 24-well plates were incubated at 4°C with radiolabeled purifiedvirions. The viruses were allowed to bind to the cell surface for 10 to 320 min,after which the unbound viruses were removed and the cell monolayer waswashed three times with cold TBS. Cell monolayers with bound virions wereresuspended in 100 ml of lysis buffer and transferred to vials for liquid scintilla-tion counting. Quantification of cell-associated labeled virions was determinedwith a beta counter.
Elution of bound virus with heparin. Confluent monolayers of Vero cells in6-well plates were infected with 200 PFU of the KOS, KCZ, KgBpK2,KgBpK2gC2, KgBpKRgC2, or KgBpK2gCR virus. Following a 4-h incubation at4°C, the cells were washed three times with complete medium supplemented with500 mg of heparin per ml (or without heparin in control wells). The cell mono-layers were then overlaid with medium containing 0.5% methyl-cellulose at 37°Cto allow virus plaques to form. Cells were then fixed and stained with crystalviolet for plaque quantification.
Virus titration in the presence of heparin. Confluent monolayers of Vero cellswere infected for 2 h at different dilutions with all mutant viruses or withvesicular stomatitis virus (VSV) (kindly provided by Patricia A. Dowling, Uni-versity of Pittsburgh) in the absence or presence of 50 or 500 and 10,000 mg ofheparin per ml. Virus inoculates were removed, and the cell monolayer wasrinsed twice with medium or heparin-supplemented medium. Two additionalwashes were performed with medium only in all samples to remove the presenceof heparin that could reduce the efficiency of plaque formation. The cell mono-layers were then overlaid with medium containing 0.5% methyl-cellulose at 37°Cto allow virus plaques to form. Cells were fixed and stained with crystal violet toquantify plaque numbers.
Virus penetration assay. The rate of virus penetration was assessed by deter-mining the rate at which adsorbed virus became resistant to inactivation by alow-pH glycine buffer (0.1 M glycine [pH 3.0] [8]). Confluent Vero, murine L,and sog9 cells in 6-well plates were incubated at 4°C for 2 h with 300 PFU ofwild-type KOS, HS binding mutants, or rescued viruses. Following virus adsorp-tion the cells were rinsed three times, overlaid with complete medium, andshifted to 37°C to allow virus penetration. At selected times after temperatureshift, wells were treated with 2 ml of glycine buffer for 1 min while control wellswere treated with 2 ml of TBS for 1 min. The monolayers were then washed threetimes with complete medium, overlaid with DMEM containing 0.5% methyl-cellulose, and incubated at 37°C, allowing virus plaques to form. Cells were thenfixed and stained with crystal violet to visualize and count viral plaques.
In vitro cell-to-cell spread of virus. Vero and sog9 cell monolayers wereinfected with KOS, KCZ, KgBpK2, and KgBpK2gC2 viruses for 2 h, the me-dium was aspirated, and the cells were washed twice with TBS and overlaid withfresh medium supplemented with 0.2% human gamma globulin (HGG) (BayerCorporation, Elkhart, Ind.) containing anti-HSV neutralizing antibody. Twenty-four, 36, and 48 h p.i., cell monolayers were fixed with methanol, washed threetimes with TBS, and processed for immunofluorescence as described above byusing rabbit anti-HSV-1 antibodies combined with secondary cy3-conjugatedantibody (Jackson Immunoresearch Laboratories). Plaque sizes were determinedby a Zeiss Axiophot microscope linked to a Xillix digital camera with personalcomputer-based image analysis (MCID) from Imaging Resources Incorporated(Brock, Ontario, Canada).
RESULTS
Construction of a gB mutant molecule impaired in heparinbinding. The role of gB in HS proteoglycan binding may be
distinguished from its essential role in penetration if an HSbinding domain can be identified and removed from the mol-ecule without inactivating its ability to function in membranefusion. If so, then a double mutant virus also lacking gC couldbe constructed since gC is nonessential for virus infectivity(27). Polylysine inhibits HSV infection (38) presumably byinteracting with HS, a property shared with other cationicsubstances such as polyarginine and neomycin (37, 38). Ac-cordingly, a lysine-rich (pK) sequence (residues 68 to 76) wasdeleted from the N-terminal region of gB (gBpK2) for studiesof its ability to be processed and bind heparin, an HS-likemolecule.
Deletion or substitution of peptide sequences from HSV-1glycoproteins can result in altered glycoprotein processing andprevent analysis of the effect of the mutation on the glycopro-tein’s functional role in virus infection. For example, modifiedglycoproteins can be retained within the endoplasmic reticu-lum and fail to be transported to the cell surface membraneand incorporated into virus particles (9). In order to evaluatethe posttranslational processing of the mutated gB molecule,Vero cells were transfected with expression vectors encodingwild-type (HCMV-BXX) (Fig. 1A) or mutant (HCMV-gBpK2) (Fig. 1B) gB; mock-transfected Vero cells were usedas a negative control (Fig. 1C). Twenty-four hours posttrans-fection, the cells were analyzed by immunofluorescence for thepresence and localization of gB molecules. Untreated cellmonolayers were incubated at 4°C with a pool of gB-specificMAbs (46) to determine whether the mutant gB molecule wasprocessed and transported to the cell surface membrane. Asdemonstrated in Fig. 1, both wild-type (panel A) and mutant(panel B) gB (gBpK2) were recognized by a pool of specificMAbs, and both proteins were incorporated into the cell sur-face membrane. No fluorescence was detected on mock-trans-fected cells (panel C). These data demonstrate that mutant gB(gBpK2) was transported to the cell membrane of transfectedcells, showing that deletion of the pK domain of gB permitsendoplasmic reticulum and Golgi processing of the mutantmolecule. Moreover, the molecular size of the fully processedmutant molecule was similar to the wild-type gB molecule,revealing no obvious differences in the addition of complexcarbohydrates (see Fig. 4).
Since the mutant form of gB was apparently normally pro-cessed it could be compared with the wild-type molecule for itsreactivity with heparin, an HS-like molecule, to determinewhether the pK deletion had removed the HS binding activityattributed to the gB molecule. Expression plasmids encodingeither wild-type gB (pKBXX) or gBpK2 (pgBpK2) were usedto transfect Vero cells in order to produce wild-type and mu-tant proteins, respectively, after induction of gB expression byinfection with a gB–ICP 18.5-deleted virus (KD4BX). The
FIG. 1. Cell surface detection of gBpK2 mutant glycoprotein. Vero cells were transfected with expression plasmids encoding the wild-type gB protein (HCMV-BXX) (A) and the pK-deleted gB protein (HCMV-gBpK2) (B) or were mock transfected (C). After 30 h, unfixed transfected monolayers were incubated with a poolof monoclonal anti-gB antibody for 1 h at 4°C, washed with cold TBS, and incubated for 1 h at 4°C with a cy3 anti-mouse antibody. Monolayers were visualized witha Nikon microscope (model 211910) and photographed.
VOL. 72, 1998 HS BINDING IN HSV-1 ATTACHMENT, ENTRY, AND SPREAD 6121
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

transfected/infected cells were radiolabeled with [35S]methi-onine/cysteine, and the cell monolayers were harvested andsolubilized prior to analysis of binding to heparin-acrylic beads.As shown in Fig. 2, approximately the same quantity of gB orgBpK2 molecules was incubated with the heparin beads (lanes1). However, as demonstrated by the quantity of unboundglycoprotein (lanes 2), gBpK2 displayed a lower affinity forheparin-acrylic beads than did wild-type gB; more than 40% ofthe gBpK2 molecules were recovered in the unbound fractioncompared to less than 3% of the gB wild-type molecules. Fig-ure 2, lanes 3, demonstrates that similar amounts of gB mol-ecules were bound nonspecifically to the heparin beads andcould be eluted with lysis buffer. In order to show that wild-type gB molecules were bound specifically to heparin-coatedbeads, the bound glycoprotein was eluted by washing the beadswith lysis buffer supplemented with heparin. The results re-vealed that bound wild-type gB was efficiently eluted by hep-arin washes (Fig. 2A, lane 4), while a small amount of gBpK2
was released by heparin wash due to the small quantity ofgBpK2 nonspecifically associated with the heparin beads (Fig.2B, lane 4). Together, these studies confirmed that the gBpK2
mutant molecule was fully glycosylated, incorporated into thecell surface membrane, and that a domain responsible forheparin binding had been inactivated by deletion of the pKregion of gB.
Construction of HS attachment-defective mutant viruses.An HSV-1 viral mutant carrying the gBpK2 coding sequence(KgBpK2) was constructed to evaluate the effect of the pKdeletion on virus infection. The gBpK2 gene was insertedtogether with the UL28 gene (ICP 18.5) by marker rescue ofthe gB and UL28 double-deletion virus (KD4BX) (9). Therecombinant viruses (KgBpK2) were selected by growth onnoncomplementing Vero cells. The DNA sequence encodingthe major HSV-1 HS binding protein, gC, was deleted fromwild-type KOS virus and replaced with the human cytomega-lovirus immediate early gene promoter (HCMV-IEp) lacZgene expression cassette in order to create KCZ. The virusrecombinants were selected by complement-dependent neu-tralization escape with gC-reactive MAbs and identified bytheir blue-plaque phenotype following X-Gal staining. A dou-ble HS attachment-defective virus encoding gBpK2 and de-leted for the gC coding sequence (KgBpK2gC2) was createdto determine the role of virus binding to HS in virus infection.The gBpK2 gene, together with the UL28 gene (ICP 18.5), wasused to marker rescue KD4BXCZ, a virus deleted for gC, gB,and UL28. The recombinant KD4BXCZ mutant virus was cre-
ated by replacing the gC coding sequence of KD4BX with theHCMV-IEp-lacZ cassette, as described above for the genera-tion of KCZ. Both gB and gC wild-type genes were usedsubsequently to rescue either the gB or gC mutation of theKgBpK2gC2 mutant virus to produce the KgBpKRgC2 andKgBpK2gCR viruses, respectively.
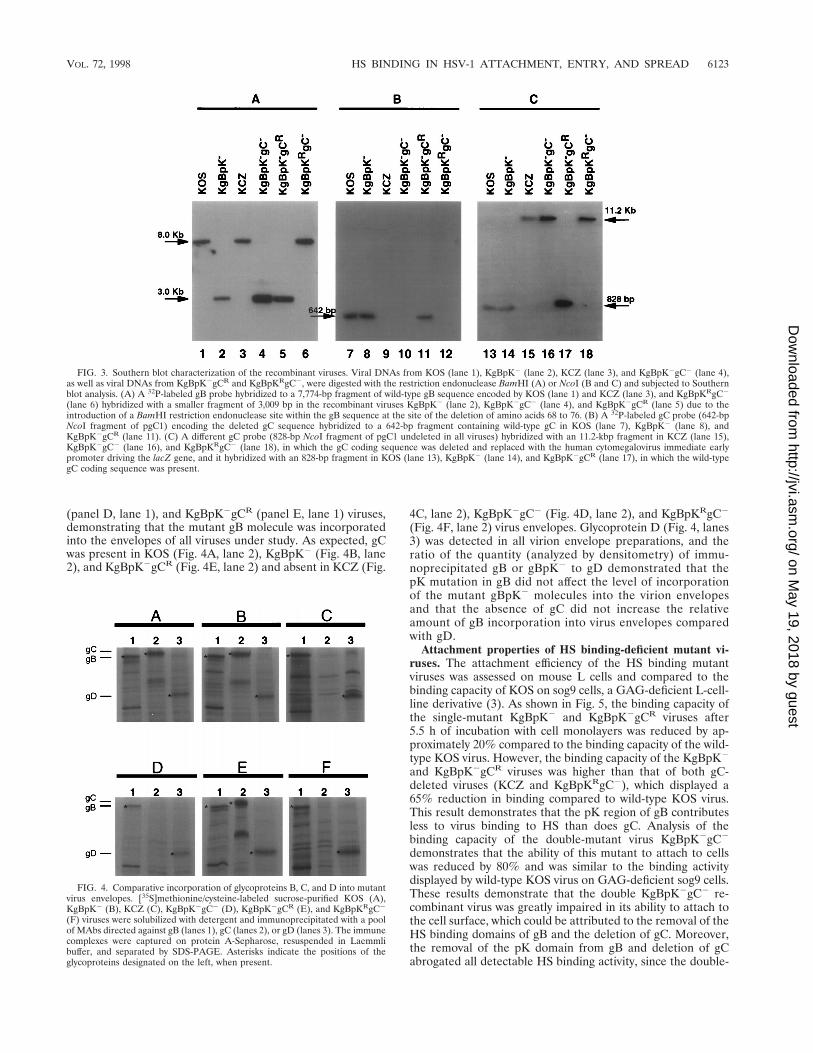
The mutant and recombinant virus genotypes were analyzedby Southern blotting to confirm their genotypes. Viral DNAswere extracted from purified virions, digested with BamHI(Fig. 3A) or NcoI (Fig. 3B and C) endonuclease, and Southernblotted with a 32P-labeled gB (Fig. 3A) or gC (Fig. 3B and C)probe. As shown in Fig. 3A, the 32P-labeled gB probe hybrid-ized to the BamG fragment (7,774 bp) of HSV-1 viral DNAcontaining the wild-type gB coding sequence encoded by theKOS, KCZ, and KgBpKRgC2 viruses (lanes 1, 3, and 6, re-spectively). The same probe hybridized to a 3,009-bp fragmentin the recombinant KgBpK2, KgBpK2gC2, and KgBpK2gCR
viruses (lanes 2, 4, and 5, respectively). This confirmed thepresence of the mutant gB gene because a BamHI recognitionsequence was introduced at the site of the pK mutation in themutant viruses, resulting in the production of two subfrag-ments (3,009 and 4,738 bp) after digestion. Replacement of thegC coding sequence by the HCMV-IEp-lacZ cassette was con-firmed by Southern blot hybridization of NcoI-digested viralDNA using a 32P-labeled gC probe (642-bp NcoI fragmentof pgC1) that hybridized to a gC sequence deleted in thegC-negative recombinant viruses. As shown in Fig. 3B, the 32P-labeled gC probe hybridized to a 642-bp fragment in KOS,KgBpK2, and KgBpK2gCR digested viral DNA (lanes 7, 8,and 11, respectively) containing the wild-type gC coding se-quence. The same probe, however, failed to hybridize withKCZ, KgBpK2gC2, and KgBpKRgC2 viral DNA (lanes 9,10, and 12, respectively), demonstrating that the gC codingsequence was deleted. The presence of the HCMV-IEp-lacZcassette was identified with an 828-bp NcoI 32P-labeled gCprobe that hybridized to the wild-type gC gene (KOS, KgBpK2,and KgBpK2gCR viruses [Fig. 3C, lanes 13, 14, and 17, respec-tively]) but with an 11.2-kb fragment derived from viral DNAcontaining the HCMV-IEp-lacZ cassette (KCZ, KgBpK2gC2,and KgBpKRgC2 viruses [Fig. 3C, lanes 15, 16, and 18, respec-tively]). This hybridization pattern confirmed the absence ofthe NcoI endonuclease restriction site within the HCMV-IEp-lacZ cassette and its presence within the wild-type gC se-quence. Together, these data confirm the isolation of single-and double-mutant viruses deleted for one or both HS bindingelements (pK of gB and gC), as well as both mutant virusrescuants.
Evaluation of the envelope glycoprotein composition of themutant viruses. Since the quantity of viral glycoproteins in thevirus envelope could influence the ability of individual glyco-proteins to carry out virus attachment, penetration, and lateralspread, the mutant virus envelopes were analyzed for glyco-protein content to determine whether the amounts of the in-dividual HSV-1 glycoproteins were similar to that of wild-typeKOS virus. The level of incorporation of these glycoproteinsinto mutant virions was determined by specific immunoprecipi-tation of solubilized radiolabeled envelopes from wild-typevirus (KOS), the mutants KCZ, KgBpK2, and KgBpK2gC2,and the rescued KgBpK2gCR and KgBpKRgC2 viruses byusing pools of monoclonal antibodies specific for gB, gC, andgD. The protein A-captured immune complexes were thenanalyzed by SDS-PAGE and autoradiography. As shown inFig. 4, wild-type gB was immunoprecipitated from the KOS(panel A, lane 1), KCZ (panel C, lane 1), and KgBpKRgC2
(panel F, lane 1) viruses and the mutant gBpK2 was immuno-precipitated from the KgBpK2 (panel B, lane 1), KgBpK2gC2
FIG. 2. Heparin binding capacity of gBpK2 mutant glycoprotein. Vero cellswere transfected with plasmids encoding wild-type gB (A) and gBpK2 (B).Twenty-four hours posttransfection, the cell monolayers were infected withKD4BX virus in the presence of [35S]methionine/cysteine, as described in Ma-terials and Methods. Ten hours p.i., the monolayers were harvested and solubi-lized with 0.1% Triton X-100-containing buffer and equivalent amounts of de-tergent-extracted protein (lanes 1) and were incubated for 2 h at 4°C withheparin-acrylic beads. The unbound proteins (lanes 2) and proteins eluted fromthe heparin-acrylic beads with detergent buffer (lanes 3) or detergent buffersupplemented with 10 mg of heparin per ml (lanes 4) were immunoprecipitatedwith a pool of gB-specific MAbs. Each sample was analyzed by SDS-PAGE andautoradiography. The arrows indicate the positions of gB wild-type or gBpK2
mutant glycoproteins.
6122 LAQUERRE ET AL. J. VIROL.
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
(panel D, lane 1), and KgBpK2gCR (panel E, lane 1) viruses,demonstrating that the mutant gB molecule was incorporatedinto the envelopes of all viruses under study. As expected, gCwas present in KOS (Fig. 4A, lane 2), KgBpK2 (Fig. 4B, lane2), and KgBpK2gCR (Fig. 4E, lane 2) and absent in KCZ (Fig.
4C, lane 2), KgBpK2gC2 (Fig. 4D, lane 2), and KgBpKRgC2
(Fig. 4F, lane 2) virus envelopes. Glycoprotein D (Fig. 4, lanes3) was detected in all virion envelope preparations, and theratio of the quantity (analyzed by densitometry) of immu-noprecipitated gB or gBpK2 to gD demonstrated that thepK mutation in gB did not affect the level of incorporationof the mutant gBpK2 molecules into the virion envelopesand that the absence of gC did not increase the relativeamount of gB incorporation into virus envelopes comparedwith gD.
Attachment properties of HS binding-deficient mutant vi-ruses. The attachment efficiency of the HS binding mutantviruses was assessed on mouse L cells and compared to thebinding capacity of KOS on sog9 cells, a GAG-deficient L-cell-line derivative (3). As shown in Fig. 5, the binding capacity ofthe single-mutant KgBpK2 and KgBpK2gCR viruses after5.5 h of incubation with cell monolayers was reduced by ap-proximately 20% compared to the binding capacity of the wild-type KOS virus. However, the binding capacity of the KgBpK2
and KgBpK2gCR viruses was higher than that of both gC-deleted viruses (KCZ and KgBpKRgC2), which displayed a65% reduction in binding compared to wild-type KOS virus.This result demonstrates that the pK region of gB contributesless to virus binding to HS than does gC. Analysis of thebinding capacity of the double-mutant virus KgBpK2gC2
demonstrates that the ability of this mutant to attach to cellswas reduced by 80% and was similar to the binding activitydisplayed by wild-type KOS virus on GAG-deficient sog9 cells.These results demonstrate that the double KgBpK2gC2 re-combinant virus was greatly impaired in its ability to attach tothe cell surface, which could be attributed to the removal of theHS binding domains of gB and the deletion of gC. Moreover,the removal of the pK domain from gB and deletion of gCabrogated all detectable HS binding activity, since the double-
FIG. 3. Southern blot characterization of the recombinant viruses. Viral DNAs from KOS (lane 1), KgBpK2 (lane 2), KCZ (lane 3), and KgBpK2gC2 (lane 4),as well as viral DNAs from KgBpK2gCR and KgBpKRgC2, were digested with the restriction endonuclease BamHI (A) or NcoI (B and C) and subjected to Southernblot analysis. (A) A 32P-labeled gB probe hybridized to a 7,774-bp fragment of wild-type gB sequence encoded by KOS (lane 1) and KCZ (lane 3), and KgBpKRgC2
(lane 6) hybridized with a smaller fragment of 3,009 bp in the recombinant viruses KgBpK2 (lane 2), KgBpK2gC2 (lane 4), and KgBpK2gCR (lane 5) due to theintroduction of a BamHI restriction endonuclease site within the gB sequence at the site of the deletion of amino acids 68 to 76. (B) A 32P-labeled gC probe (642-bpNcoI fragment of pgC1) encoding the deleted gC sequence hybridized to a 642-bp fragment containing wild-type gC in KOS (lane 7), KgBpK2 (lane 8), andKgBpK2gCR (lane 11). (C) A different gC probe (828-bp NcoI fragment of pgC1 undeleted in all viruses) hybridized with an 11.2-kbp fragment in KCZ (lane 15),KgBpK2gC2 (lane 16), and KgBpKRgC2 (lane 18), in which the gC coding sequence was deleted and replaced with the human cytomegalovirus immediate earlypromoter driving the lacZ gene, and it hybridized with an 828-bp fragment in KOS (lane 13), KgBpK2 (lane 14), and KgBpK2gCR (lane 17), in which the wild-typegC coding sequence was present.
FIG. 4. Comparative incorporation of glycoproteins B, C, and D into mutantvirus envelopes. [35S]methionine/cysteine-labeled sucrose-purified KOS (A),KgBpK2 (B), KCZ (C), KgBpK2gC2 (D), KgBpK2gCR (E), and KgBpKRgC2
(F) viruses were solubilized with detergent and immunoprecipitated with a poolof MAbs directed against gB (lanes 1), gC (lanes 2), or gD (lanes 3). The immunecomplexes were captured on protein A-Sepharose, resuspended in Laemmlibuffer, and separated by SDS-PAGE. Asterisks indicate the positions of theglycoproteins designated on the left, when present.
VOL. 72, 1998 HS BINDING IN HSV-1 ATTACHMENT, ENTRY, AND SPREAD 6123
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

mutant virus KgBpK2gC2 bound to mouse L cells to an extentsimilar to wild-type KOS virus binding to the GAG-deficientsog9 cells. The binding capacity of a double mutant deleted forgB and gC (KD4BXCZ) showed a binding activity on L cellscomparable to that of the KgBpK2gC2 virus, demonstratingthat removal of the pK domain of gB eliminated all of itsdetectable HS binding activity and confirmed studies de-scribed above (Fig. 2) which determined that the pK se-quence of gB was solely responsible for the HS bindingfunction of gB. In similar studies using Vero cells, the ratiosof bound virus to input were higher for all viruses on Verothan on L cells; however, a proportional reduction in bind-ing for all recombinant viruses compared to wild-type KOSvirus was observed, confirming studies using L cells (datanot shown).
Although removal of the pK domain of gB and deletion ofgC greatly impaired the adsorption of the recombinant virusKgBpK2gC2 to L cells (Fig. 5), there remained a 20% residualbinding activity. This residual binding capacity was also ob-served for wild-type KOS virus on sog9 cells (Fig. 5), confirm-ing that it was HS independent. In order to determine whetherthis residual binding was HS independent on cell types otherthan L cells, the HS binding-deficient recombinant viruseswere titrated on Vero cells in the presence or absence ofheparin (50 mg/ml), an HS-like molecule. The data presentedin Table 1 demonstrate that the wild-type KOS and mutantKgBpK2 and KCZ viruses infected Vero cells more efficientlyin the absence than in the presence of heparin, confirming thatthese viruses contained HS binding activity. However, the ratioof the titers of the double-mutant KgBpK2gC2 virus was sim-ilar on Vero cells in the presence or absence of heparin. Thesmall observed difference (relative ratio of 2.1) could be ac-counted for by the nonspecific inhibitory effect of heparin on
virus binding, since this effect also was observed when an HSbinding independent virus, VSV, was titrated in the presenceof heparin (data not shown). Similar results were also obtainedat different concentrations (500 mg/ml and 10 mg/ml) of hep-arin, and the rescued viruses KgBpKRgC2 and KgBpK2gCR
showed results similar to the KCZ and KgBpK2 mutant vi-ruses, respectively (data not shown). In experiments in whichthe viruses were bound at 4°C in the presence or absence ofheparin rather than titrated in heparin-supplemented media(Table 1), similar results were observed (data not shown),demonstrating that the inhibitory effect of heparin was at theadsorption level. Moreover, similar titers for the double-mu-tant KgBpK2gC2 virus were obtained on L and sog9 cells(relative ratio of 1.1), demonstrating that this virus was defi-cient for HS binding (data not shown).
To confirm that the impaired binding of KgBpK2gC2 wasdue to the loss in virus-specific HS recognition sites, virusesbound to the cell surface were washed with heparin and thequantity of released virus particles was taken as a measure ofvirus specifically bound to HS. Vero cells were incubated for4 h at 4°C with the KOS, KCZ, KgBpK2, and KgBpK2gC2
FIG. 5. Cell surface binding capacity of viruses altered in HS proteoglycan binding domains. Vero cells infected with KOS, KCZ, KgBpK2, KgBpK2gC2,KgBpKRgC2, or KgBpK2gCR viruses and C1 cells infected with KD4BXCZ virus were labeled with [35S]methionine/cysteine, and the virions from cell supernatantswere subsequently purified on sucrose gradients. The binding capacity of each purified virion was determined on mouse L cells and compared to the binding of wild-typeKOS virus on sog9 cells (GAG-deficient L-cell derivatives). Aliquots from the different virus preparations were incubated on the two cell lines at 4°C for up to 320 minand washed with cold TBS, and the cells were scraped, harvested, and counted for virus-associated radioactivity. The percentage of bound virus was determined asradioactive counts representing the bound fraction divided by the total counts per minute (input). The binding capacity of KOS virus on mouse L cells after 320 minwas designated 100% binding. Error bars indicate results determined for triplicate wells.
TABLE 1. Effects of heparin on infection of Vero cells by HSV
Virus
Titer (PFU/ml)
RatioWithoutheparin
Withheparin
KOS 4.8 3 109 1.1 3 108 44.6KgBpK2 1.9 3 109 1.0 3 108 19.0KCZ 2.2 3 108 2.0 3 107 11.0KgBpK2gC2 8.8 3 108 4.2 3 108 2.1
6124 LAQUERRE ET AL. J. VIROL.
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

viruses, and washes were performed in the presence or absenceof heparin to elute the fraction of virus bound to HS. As shownin Fig. 6, the binding of the KgBpK2 virus was less sensitive(12%) to heparin washes than that of KOS, consistent withother evidence showing that the HS binding region of gB hadbeen deleted. Moreover, the binding of the double-mutantKgBpK2gC2 virus was highly resistant to heparin washes butonly to an extent slightly higher (2.4%) than the resistancedemonstrated by KCZ, again showing that the majority of HSbinding activity was attributable to gC. The residual boundvirus that was resistant to heparin washes depended on bindingto a non-HS receptor, most likely due to the binding of gD toits specific receptor (50).
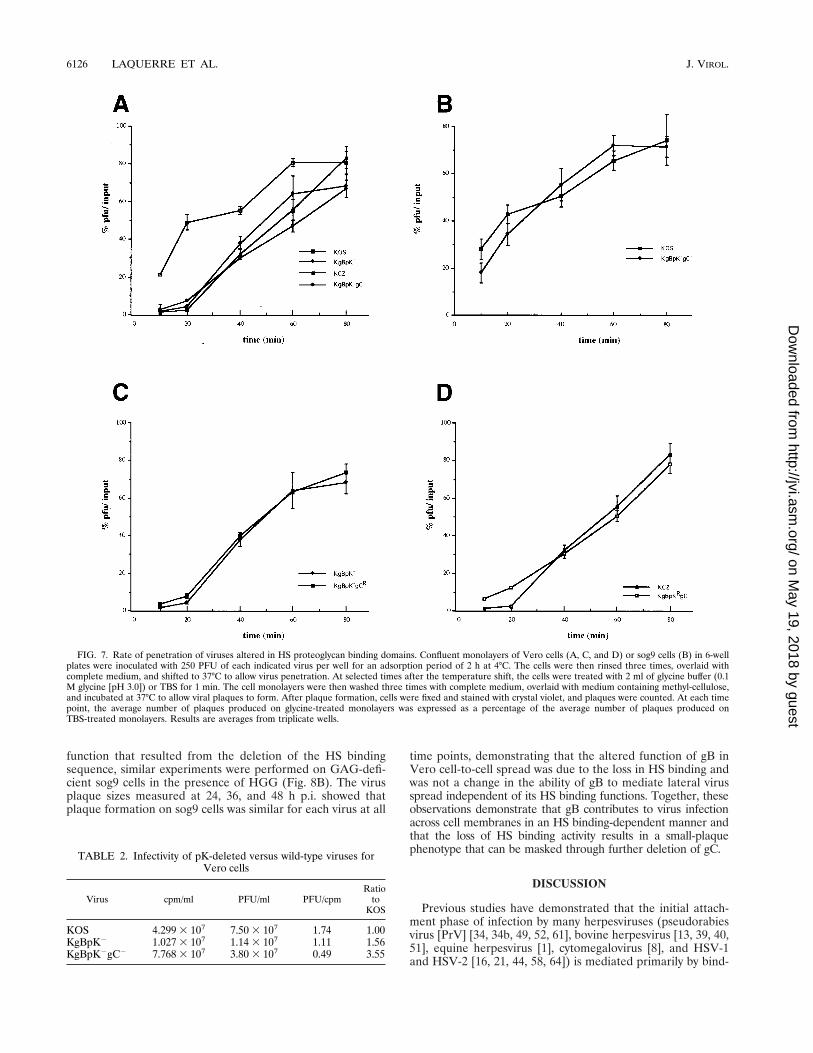
Virus mutants altered in HS binding domains also showeda reduced capacity for viral entry and infectivity. The entry ofHSV-1 capsids into the host cell is believed to involve a two-step process requiring the binding of the virus to the cellsurface prior to fusion of the viral envelope with the cellularplasma membrane. Whether the binding and fusion events areindependent is unknown. In particular, does binding to HScontribute to the efficiency of entry of bound virus or does HSbinding-defective virus enter cells with the same kinetics asbound wild-type virus? Assays of the rate of virus entry wereperformed to answer this question. The rate of virus entry intohost cells was determined as the rate at which a virus bound tothe cell surface becomes resistant to acid treatment as a con-sequence of penetration of the virus into the host cell, com-pared with untreated virus controls. The results presented inFig. 7A show that the single-mutant KgBpK2 and KCZ virusesdisplayed reduced kinetics of penetration compared to wild-type KOS virus. The rate of entry of the double-mutantKgBpK2gC2 virus was slightly lower than each individual sin-gle mutant virus but equivalent to the additive effect of eachindividual mutation. Similar data were also obtained with Lcells (data not shown). These data suggest either that stableattachment to the cell surface HS receptor is required forefficient virus entry or that deletion of the pK domain of gBreduces its fusion capacity in addition to its contribution tovirus adsorption to the cell surface. In order to discriminatebetween these two possibilities, the rates of penetration of thewild-type and double-mutant KgBpK2gC2 viruses were deter-mined on GAG-deficient sog9 cells (Fig. 7B). We reasoned
that if the fusion function of gB was altered by deletion of thepK domain, the rates of penetration of both viruses should bedifferent on HS-deficient sog9 cells, while if the differences inthe rates of penetration were due to the absence of binding toHS then both viruses should have the same rate of penetrationon these cells. The data presented in Fig. 7B show that the rateof penetration of the double-mutant virus was similar to that ofthe wild-type virus on sog9 cells, confirming that the fusionfunction of gB was not altered by deletion of the pK domain ofgB. Taken together, these data demonstrate that stable attach-ment to cell surface HS is required for efficient virus entry.The rate of entry of KCZ virus was compared to that ofKgBpKRgC2 (Fig. 7C), and the rate of entry of KgBpK2 viruswas compared to that of KgBpK2gCR (Fig. 7D). Both combi-nations gave similar results, confirming that the double-mutantviruses rescued for gB or gC showed rates of penetration sim-ilar to the corresponding single-mutant viruses.
The infectivity of the pK-deleted viruses (KgBpK2 andKgBpK2gC2) compared to the wild-type KOS virus was mea-sured on Vero cells by calculating the capacity of the 35S-labeled sucrose-purified recombinant viruses to form plaqueson Vero cells and expressed as a ratio of PFU per counts perminute, where counts per minute was used to normalize thenumber of input particles. As demonstrated in Table 2, thesingle-mutant virus KgBpK2 demonstrated 1.56-fold-less in-fectivity than wild-type KOS virus, while the double-mutantKgBpK2gC2 virus demonstrated 3.55-fold-less infectivity thanKOS virus. These data confirm results reported previously forgC deletion mutants (23) where a decrease in infectivitycorresponded with a reduction in virus binding to the cellsurface.
Virus mutants altered in gB-dependent HS binding alsoshowed reduced cell-to-cell virus spread. Virus penetrationand cell-to-cell spread of viruses have been uncoupled throughisolation of virus mutants deleted for glycoprotein gE and gI(11) that are defective in cell-to-cell spread but competent forvirus entry. These mutants retained the ability to initiate in-fection of cells but could not form plaques in the presence ofvirus-neutralizing antibody. Glycoprotein B is required forboth penetration and cell-to-cell spread; however, the role ofits HS binding domain in lateral virus transmission has notbeen examined. To determine whether gB binding to HS in-fluences lateral virus spread, Vero cells were infected withKOS, KgBpK2, KCZ, and KgBpK2gC2 in the presence ofHGG, which neutralized extracellular virus particles. As dem-onstrated in Fig. 8A, at 24, 36, and 48 h p.i., the average plaquesize of mutant KgBpK2 virus was significantly smaller thanthat of wild-type KOS virus at all time points, showing thatdeletion of the HS binding function of gB reduced lateral virusspread. These data demonstrate that the HS binding activity ofgB is important to this mode of virus infection, an activitywhich could not be compensated for by the HS binding activityof gC. However, deletion of gC from wild-type KOS virusincreased the efficiency of the mutant virus (KCZ) in spreadingfrom cell to cell, as reported by Manservigi et al. (45), produc-ing significantly larger plaques at 36 and 48 h p.i. These find-ings demonstrate that unlike virus penetration (Fig. 7A), thepresence of gC is not required for efficient virus transmissionto neighboring cells. However, similar experiments carried outwith the double-mutant virus (KgBpK2gC2) that was deletedfor the HS binding activity of gB and gC did not show signif-icant reduction in plaque size compared with wild-type virus,suggesting that the opposing effects of each mutation onplaque development mask their individual phenotypes. In or-der to determine if the reduction in plaque size observed withthe KgBpK2 virus was related to an additional alteration in gB
FIG. 6. Contribution of a viral glycoprotein(s) other than gB and gC to HSbinding. Confluent monolayers of Vero cells in 12-well plates were inoculatedwith 100 PFU of each test virus per well for an adsorption period of 2 h at 4°C.The cells were then rinsed three times with complete medium in the absence orpresence of 500 mg of heparin per ml, overlaid with medium containing methyl-cellulose, and shifted to 37°C to allow viral plaque formation. The cells were thenfixed and stained with crystal violet, and plaques were counted. At each timepoint, the average number of plaques produced on heparin-washed monolayerswas expressed as a percentage of the average number of plaques produced oncomplete medium-washed monolayers. Error bars indicate results determinedfor triplicate wells.
VOL. 72, 1998 HS BINDING IN HSV-1 ATTACHMENT, ENTRY, AND SPREAD 6125
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
function that resulted from the deletion of the HS bindingsequence, similar experiments were performed on GAG-defi-cient sog9 cells in the presence of HGG (Fig. 8B). The virusplaque sizes measured at 24, 36, and 48 h p.i. showed thatplaque formation on sog9 cells was similar for each virus at all
time points, demonstrating that the altered function of gB inVero cell-to-cell spread was due to the loss in HS binding andwas not a change in the ability of gB to mediate lateral virusspread independent of its HS binding functions. Together, theseobservations demonstrate that gB contributes to virus infectionacross cell membranes in an HS binding-dependent manner andthat the loss of HS binding activity results in a small-plaquephenotype that can be masked through further deletion of gC.
DISCUSSION
Previous studies have demonstrated that the initial attach-ment phase of infection by many herpesviruses (pseudorabiesvirus [PrV] [34, 34b, 49, 52, 61], bovine herpesvirus [13, 39, 40,51], equine herpesvirus [1], cytomegalovirus [8], and HSV-1and HSV-2 [16, 21, 44, 58, 64]) is mediated primarily by bind-
FIG. 7. Rate of penetration of viruses altered in HS proteoglycan binding domains. Confluent monolayers of Vero cells (A, C, and D) or sog9 cells (B) in 6-wellplates were inoculated with 250 PFU of each indicated virus per well for an adsorption period of 2 h at 4°C. The cells were then rinsed three times, overlaid withcomplete medium, and shifted to 37°C to allow virus penetration. At selected times after the temperature shift, the cells were treated with 2 ml of glycine buffer (0.1M glycine [pH 3.0]) or TBS for 1 min. The cell monolayers were then washed three times with complete medium, overlaid with medium containing methyl-cellulose,and incubated at 37°C to allow viral plaques to form. After plaque formation, cells were fixed and stained with crystal violet, and plaques were counted. At each timepoint, the average number of plaques produced on glycine-treated monolayers was expressed as a percentage of the average number of plaques produced onTBS-treated monolayers. Results are averages from triplicate wells.
TABLE 2. Infectivity of pK-deleted versus wild-type viruses forVero cells
Virus cpm/ml PFU/ml PFU/cpmRatio
toKOS
KOS 4.299 3 107 7.50 3 107 1.74 1.00KgBpK2 1.027 3 107 1.14 3 107 1.11 1.56KgBpK2gC2 7.768 3 107 3.80 3 107 0.49 3.55
6126 LAQUERRE ET AL. J. VIROL.
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

ing of one or more envelope glycoproteins to HS proteoglycanson the surface of susceptible cells. The HS binding activity ofHSV-1 is attributable primarily to gC and gB (23), an obser-vation confirmed in the present study. Mutants deleted forboth gB and gC are severely impaired for binding to HS, to anextent similar to the highly impaired adsorption of wild-typevirus to mutant cells defective for HS expression (17, 23, 55).Glycoprotein C accounts for the most avid binding, since mu-tations that result in gC-deficient enveloped particles show atleast a 60% reduction in the efficiency of virus attachmentcompared to gB null virion particles (4, 24). Moreover, mutantviruses deleted for gC penetrate cells at a reduced rate (24),suggesting that attachment to HS may be required either toincrease binding to a second receptor potentially recognized bygD or to initiate changes in the bound particle envelope thattrigger the process of envelope fusion with the cell surfacemembrane (33). The interdependence of attachment to HSwith virus entry and spread previously could not be studied in
HSV with either gB or gB and gC null viruses, since gB isessential for both processes and may itself initiate fusion.
Initial experiments were aimed at determining whether anHS binding domain could be identified within gB based onanalysis of the predicted amino acid sequence of the gB geneproduct. Studies of other HS binding molecules indicate thatthe negatively charged proteoglycan molecules would be rec-ognized by peptide domains rich in positively charged residues,particularly lysine (38). Adenovirus vectors modified to containa polylysine sequence within the fiber protein, for example,bind to HS and increase virus infection of cells low in adeno-virus receptor density (63). Glycoprotein B contains a polyly-sine region (pK) that lies within amino acid residues 68 to 76and has the sequence KPKKNKKPK. This sequence is similarto the consensus sequences predicted for the HS binding do-main of proteins (7, 62) and was deleted from gB to produce amutant form designated gBpK2. SDS-PAGE analysis of thetransiently expressed mutant gene revealed a molecular sizeconsistent with the predicted sequence. The processed glyco-protein was transported to the cell surface membrane inamounts similar to wild-type gB, and the gBpK2 molecule wasreactive with gB-specific monoclonal antibodies, indicatingthat these conformational epitopes were preserved. ThegBpK2 transiently expressed molecule was highly impaired inits ability to bind to heparin-conjugated acrylic beads, confirm-ing that at least the major HS binding domain of gB waslocalized to this lysine-rich sequence. These results showedthat although the HS binding domain of gB was deleted, itsremoval did not appear to significantly alter its conformationalstructure, glycosylation, or intracellular trafficking. The mutantgB molecule also proved to be functional for virus penetrationby rescue of gB null mutant virus. Biochemical analysis ofpurified mutant virus particles indicated that normal levels ofthe mutant molecules were incorporated into virions, rulingout the possibility that any phenotypic change in mutant virusbehavior was due to a reduction of gB in virus particles.
The KgBpK2 mutant virus showed a decreased capacity forattachment to susceptible cells in culture. However, previousstudies showed that gB-negative mutant viruses demonstratedthe same binding capacity as wild-type virus to susceptible cells(4, 23). The discrepancy between our data and these reportscould be explained on the basis of altered capacity for incor-poration of the remaining glycoproteins into the virus envelope(9, 18, 19). For example, deletion of gB from the virus envelopemight provide added space for increasing the amount of otherglycoproteins such as gC in the envelope of mature particles,which in turn compensates for and thereby masks the HSbinding activity of gB. Removal of both the HS binding func-tion of gB and gC by construction of a double-mutant virus(KgBpK2gC2) had a profound effect on HS binding; however,the mutant virus still retained the ability to attach to murine Lcells to an extent similar to the binding of wild-type virus tosog9 cells, a GAG-deficient mutant derivative (3). Moreover,the binding of the double mutant virus (KgBpK2gC2) wassimilar to the binding of the double deletion mutant gB/gCvirus (K4BXCZ), demonstrating that notwithstanding the sen-sitivity of our binding assay gB and gC are the only HS bindingproteins encoded by HSV-1 and that the pK domain of gB isthe only binding domain of gB. The ratios of the titer of thedouble-mutant (KgBpK2gC2) virus on Vero cells in the ab-sence or presence of heparin on L cells compared to sog9 cellswere close to unity, demonstrating that all detectable HS-dependent binding activity had been deleted from the double-mutant virus. This conclusion was also supported by resultsdemonstrating that the double-mutant virus was resistant toheparin washes, indicating that attachment had occurred
FIG. 8. Effect of HS binding mutations on lateral mutant virus spread. Con-fluent monolayers of Vero and sog9 cells were infected with 300 PFU of KOS,KCZ, KgBpK2, and KgBpK2gC2 viruses per well. Twenty-four, 36, and 48 h p.i.,Vero cell (A) and sog9 cell (B) monolayers were fixed with ice-cold methanoland processed for immunofluorescence with an anti-rabbit HSV-1 polyclonalantibody for 1 h at 37°C, washed with TBS, and incubated for 1 h at 37°C witha cy3 anti-rabbit antibody. Plaque sizes were determined with a Zeiss Axiophotmicroscope linked to a Xillix digital camera. For Vero cells, statistically signifi-cant differences (P , 0.05) in plaque sizes between viruses are marked by likesymbols above the measured values; for sog9 cells, the plaque sizes were notstatistically different (P . 0.39).
VOL. 72, 1998 HS BINDING IN HSV-1 ATTACHMENT, ENTRY, AND SPREAD 6127
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

through recognition of a non-HS receptor, presumably gDattachment to its specific receptor (50). We cannot rule out thepossibility, however, that binding to the non-HS receptor ismore avid in the absence of HS binding, making bound virusresistant to heparin washes and thus masking additional HSbinding activity associated with gB and/or other glycoproteins.The ability of heparin to release bound virus to the HS recep-tor where, presumably, the non-HS receptor is also recognizedmight be explained by the order of receptor recognition. Thatis, HS receptor binding may preceed binding to the non-HSreceptor; thus, heparin may release those particles in transi-tion. Comparison of the infectivity of the double mutant withthe wild-type virus demonstrated that in the absence of HSbinding more input mutant virus was required to obtain infec-tivity comparable to the wild-type virus, indicating that thenon-HS receptor was less abundant or less accessible. Similarstudies have been performed with a gC-deleted PrV mutantwhere gC is solely responsible for the virus HS binding func-tion. The PrV HS binding-deficient mutant showed reducedattachment to HS-bearing cells but not to HS-deficient mutantcells, a finding consistent with our studies of HSV mutantsdefective for HS binding. Both the HSV and PrV mutantsremain active, demonstrating that HS binding is not essentialfor virus infectivity (34a).
The reduction in double-mutant virus attachment to the cellsurface might also reflect a change in the ability of the virusparticle to “neutralize” a repulsive charge on the cell surface,resulting in a change in the kinetics of recognition of thenon-HS virus receptor. Such repulsive forces may not bepresent on HS-deficient sog9 cells; thus, the kinetics of virusattachment would appear similar to the wild-type virus. How-ever, studies of the specificity of HSV-1 and HSV-2 for differ-ent locations of virus transmission may be related to the abilityof the two serotypes to recognize different HS moieties on cellsurface proteoglycans, and the HS specificity and recognitionsequence of gC of PrV is apparently different from that ofHSV-1 (24a, 61). In the present study, we show that the HSrecognition sequence of gB differs from earlier reports for theHS binding element of gC (12a, 62). Thus, although HS rec-ognition involves a charge interaction, the specific HS recep-tors recognized by different viruses may vary, suggesting thatthe interaction between the virus envelope and cell surface HSspecies are specific (3a, 12a).
Because gBpK2 was functional in virus infection, it could bedirectly determined whether HS binding was also essential tothe process of virus penetration. The rate of cell-boundKgBpK2 mutant virus penetration was reduced compared withthe wild-type virus, indicating that gB recognition of HS wasrequired in this process. Moreover, the gC null virus (KCZ)also entered cells more slowly, in agreement with an earlierreport (24), whereas the double-mutant (KgBpK2gC2) virusdid not differ significantly from either single-mutant virus, sug-gesting that HS recognition by both glycoproteins is requiredfor efficient virus entry and that they may cooperate in thisprocess. This conclusion was supported by the finding that theKgBpK2gC2 mutant entered cells with kinetics similar to thewild-type virus on sog9 cells. Moreover, this result suggests thatdeletion of the HS binding function of gB does not alter thefusion function of gB. Since the double-mutant KgBpK2gC2
virus was capable of binding to the cell surface through anon-HS receptor but demonstrated a lower rate of penetrationthan wild-type virus, we conclude that virus binding to theinherent non-HS receptor can substitute for HS binding inpromoting virus entry but with less efficiency. The biochemicalmechanism which underlies these cooperative functions is un-known but may relate to the possibility that HS binding brings
the virus in closer proximity to the cell membrane, therebyenhancing the ability of gB to contribute to virus envelope cellmembrane fusion. The binding to HS, for example, may alterthe conformation of gB or other viral fusogens in a mannerthat allows invasion of the fusogenic peptide into the cellmembrane (20).
The role of HS receptor binding glycoproteins in entry ofextracellular virus may be different when virus infection pro-ceeds from cell to cell. For example, mutants deficient in gCform larger plaques in the presence of neutralizing antibodiesthan does the wild-type virus, suggesting that gC reduces theefficiency of virus spread. In our studies we have made similarobservations, indicating that the HS binding activity of gC maynot be required for intercellular virus entry into neighboringcells. Shieh and Spear (55) reported that binding to HS isinvolved in virus spread from cell to cell since wild-type HSV-1cannot spread laterally in CHO cells deficient for HS but canbe enhanced in such cells if soluble heparin is provided. Incontrast, Gruenheid et al. (17) showed that HS may be dis-pensable for lateral virus spread, since virus plaques were ofsimilar size on murine L cells or their HS-deficient derivatives,gro2C cells. These mutant cells retained condroitin sulfatemoieties that were subsequently demonstrated to be recog-nized by HSV in virus attachment to murine L cells (3). In thepresent study, we compared the ability of both single anddouble HS binding mutant viruses to form plaques on Verocells in the presence of virus-neutralizing antiserum whereplaque formation depended exclusively on cell-to-cell virusspread. The results showed that the KgBpK2 mutant virusproduced significantly smaller plaques than wild-type virus onVero cells, while the gC null mutant produced larger-than-wild-type virus plaques in the presence of antiserum. However,deletion of both the pK domain of gB and the gC gene createda mutant virus (KgBpK2gC2) that formed plaques of similarsize to the wild-type virus, suggesting that only gB binding tothe HS receptor is required for efficient intercellular infectionand indicating that the roles of HS binding are different inextra- and intercellular infection. These conclusions werestrengthened by comparisons of the virus plaque sizes onGAG-deficient cells in which all mutants tested formedplaques of comparable size to wild-type KOS virus. Becausethe presence of gC appeared to inhibit lateral virus spread,deletion of both the gB and gC HS binding activity resulted ina normal-size plaque phenotype in which the plaque-size-re-ducing effect of mutant gB appeared to be masked by theplaque-size-enhancing phenotype associated with the deletionof gC. The reason(s) for these opposing effects is unknown butmight be explained by gB and gC recognition of different HSstructures such as, for instance, those that might be associatedwith cell junctions where virus transmission occurs. Alterna-tively, these differences could be due to a change in the stoi-chiometry of gC-deleted virus envelope components, some ofwhich may also contribute to lateral virus spread. While theseobservations distinguished between the roles of gB and gC inplaque size development, deletion of either HS binding activityreduced the binding of virus to cell surfaces and slowed therate of extracellular virus entry. Further studies are in progressto determine whether HS binding is important to the patho-genesis of HSV where virus spread occurs from cell to cell.These studies should determine whether the HS binding func-tion of gB is essential to the process of virus infection in vivo.
ACKNOWLEDGMENTS
We thank Frank Tufaro for providing murine L cells and derivativesog9 cells, Patricia Dowling for VSV, and David Fink for assistance in
6128 LAQUERRE ET AL. J. VIROL.
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

measuring plaque size areas. We also thank Thomas Holland for crit-ical reading of the manuscript.
This work was supported by Public Health Service grant R01CA66141-07 from the National Institutes of Health (J.C.G.) and bygrants from Telethon A100 and AIRC (R.M.). Rafaela Argnani wassupported by an AIRC fellowship, and Sylvie Laquerre was supportedby L’Association Francaise contre les Myopathies (AFM).
REFERENCES
1. Allen, G. P., and L. D. Coogle. 1988. Characterization of an equine herpes-virus type 1 gene encoding a glycoprotein (gp13) with homology to herpessimplex virus glycoprotein C. J. Virol. 62:2850–2858.
2. Baines, J. D., and B. Roizman. 1993. The UL10 gene of herpes simplex virus1 encodes a novel glycoprotein, gM, which is present in the virion and in theplasma membrane of infected cells. J. Virol. 67:1441–1452.
3. Banfield, B. W., Y. Leduc, L. Esford, K. Schubert, and F. Tufaro. 1995.Sequential isolation of proteoglycan synthesis mutants by using herpes sim-plex virus as a selective agent: evidence for a proteoglycan-independent virusentry pathway. J. Virol. 69:3290–3298.
3a.Bergstrom, T., E. Trybala, and D. Spillmann. 1997. Heparan sulfate andviral tropism. Nat. Med. 3:1177.
4. Cai, W., B. Gu, and S. Person. 1988. Role of glycoprotein B of herpessimplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596–2604.
5. Cai, W., S. Person, S. C. Warner, J. Zhou, and N. A. DeLuca. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycop-rotein gene of herpes simplex virus type 1. J. Virol. 61:714–721.
6. Campadelli-Fiume, G., D. Stirpe, A. Boscaro, E. Avitabile, L. Foa-Tomasi, D.Barker, and B. Roizman. 1990. Glycoprotein C-dependent attachment ofherpes simplex virus to susceptible cells leading to productive infection.Virology 178:213–222.
7. Cardin, A. D., and H. J. R. Weintraub. 1989. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9:21–32.
8. Compton, T., D. M. Nowlin, and N. R. Cooper. 1993. Initiation of humancytomegalovirus infection requires initial interaction with cell surface hepa-ran sulfate. Virology 193:834–841.
9. Desai, P., F. L. Homa, S. Person, and J. C. Glorioso. 1994. A geneticselection method for the transfer of HSV-1 glycoprotein B mutations fromplasmid to the viral genome: preliminary characterization of transdominanceand entry kinetics of mutant viruses. Virology 204:312–322.
10. Desai, P. J., P. A. Schaffer, and A. C. Minson. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitivemutant of herpes simplex virus type 1: evidence that gH is essential for virioninfectivity. J. Gen. Virol. 69:1147–1156.
11. Dingwell, K. S., C. R. Brunetti, R. L. Hendricks, Q. Tang, M. Tang, A. J.Rainbow, and D. C. Johnson. 1994. Herpes simplex virus glycoproteins E andI facilitate cell-to-cell spread in vivo and across junctions of cultured cells.J. Virol. 68:834–845.
12. Dolter, K. E., W. F. Goins, M. Levine, and J. C. Glorioso. 1992. Geneticanalysis of type-specific antigenic determinants of herpes simplex virus gly-coprotein C. J. Virol. 66:4864–4873.
12a.Feyzi, E., E. Trybala, T. Bergstrom, U. Lindahi, and D. Spillmann. 1997.Structural requirement of heparan sulfate for interaction with herpes sim-plex virus type 1 glycoprotein C. J. Biol. Chem. 272:24850–24857.
13. Fitzpatrick, D. R., L. A. Babiuk, and T. J. Zamb. 1989. Nucleotide sequenceof bovine herpesvirus type 1 glycoprotein gIII, a structural model of gIII asa new member of the immunoglobulin superfamily, and implications for thehomologous glycoproteins of other herpesviruses. Virology 173:46–57.
14. Fuller, A. O., and W.-C. Lee. 1992. Herpes simplex virus type 1 entry througha cascade of virus-cell interactions requires different roles of gD and gH inpenetration. J. Virol. 66:5002–5012.
15. Gallagher, J. T., M. Lyon, and W. P. Steward. 1986. Structure and functionof heparan sulfate proteoglycans. Biochem. J. 236:313–325.
16. Gerber, S. I., B. J. Belval, and B. C. Herold. 1995. Differences in the role ofglycoprotein C of HSV-1 and HSV-2 in viral binding may contribute toserotype differences in cell tropism. Virology 214:29–39.
17. Gruenheid, S., L. Gatzke, H. Meadows, and F. Tufaro. 1993. Herpes simplexvirus infection and propagation in a mouse L-cell mutant lacking heparansulfate proteoglycans. J. Virol. 67:93–100.
18. Handler, G. C., R. J. Eisenberg, and G. H. Cohen. 1996. Oligomeric structureof glycoproteins in herpes simplex virus type 1. J. Virol. 70:6067–6075.
19. Handler, G. C., G. H. Cohen, and R. J. Eisenberg. 1996. Oligomeric structureof glycoproteins in herpes simplex virus type 1. J. Virol. 70:6076–6082.
20. Haywood, A. M. 1994. Virus receptors: binding, adhesion strengthening, andchanges in viral structure. J. Virol. 68:1–5.
21. Herold, B. C., S. I. Gerber, B. J. Belval, A. M. Siston, and N. Shulman. 1996.Differences in the susceptibility of herpes simplex virus types 1 and 2 tomodified heparin compounds suggest serotype differences in viral entry.J. Virol. 70:3461–3469.
22. Herold, B. C., S. I. Gerber, T. Polonsky, B. J. Belval, P. N. Shaklee, and K.Holme. 1995. Identification of structural features of heparin required forinhibition of herpes simplex virus type 1 binding. Virology 206:1108–1116.
23. Herold, B. C., R. J. Visalli, N. Sumarski, C. R. Brandt, and P. G. Spear. 1994.Glycoprotein C-independent binding of herpes simplex virus to cells requirescell surface heparan sulfate and glycoprotein B. J. Gen. Virol. 75:1211–1222.
24. Herold, B. C., D. WuDunn, N. Soltys, and P. G. Spear. 1991. Glycoprotein Cof herpes simplex virus type 1 plays a principal role in the adsorption of virusto cells and in infectivity. J. Virol. 65:1090–1098.
24a.Herold, B. C., S. I. Gerber, B. J. Belval, A. M. Siston, and N. Scrulman. 1996.Differences in the susceptibility of herpes simplex virus types 1 and 2 tomodified heparin compounds suggest serotype differences in viral entry.J. Virol. 70:3461–3469.
25. Highlander, S. L., W. F. Goins, S. Person, T. C. Holland, M. Levine, and J. C.Glorioso. 1991. Oligomer formation of the gB glycoprotein of herpes simplexvirus type 1. J. Virol. 50:805–812.
26. Highlander, S. L., S. L. Sutherland, P. J. Gage, D. C. Johnson, M. Levine,and J. C. Glorioso. 1987. Neutralizing monoclonal antibodies specific forherpes simplex virus glycoprotein D inhibit virus penetration. J. Virol. 61:3356–3364.
27. Holland, T. C., F. L. Homa, S. D. Marlin, M. Levine, and J. C. Glorioso.1984. Herpes simplex virus type 1 glycoprotein C-negative mutants exhibitmultiple phenotypes, including secretion of truncated glycoproteins. J. Virol.52:566–574.
28. Homa, F. L., D. J. M. Purifoy, J. C. Glorioso, and M. Levine. 1986. Molecularbasis of the glycoprotein C-negative phenotypes of herpes simplex virus type1 mutants selected with a virus-neutralizing monoclonal antibody. J. Virol.58:281–289.
29. Hutchinson, L., H. Browne, V. Wargent, N. Davis-Poynter, S. Primorac, K.Goldsmith, A. C. Minson, and D. C. Johnson. 1992. A novel herpes simplexglycoprotein, gL, forms a complex with glycoprotein H (gH) and affectsnormal folding and surface expression of gH. J. Virol. 66:2240–2250.
30. Hutchinson, L., K. Goldsmith, D. Snoddy, H. Ghosh, F. L. Graham, andD. C. Johnson. 1992. Identification and characterization of a novel herpessimplex virus glycoprotein, gK, involved in cell fusion. J. Virol. 66:5603–5609.
31. Hutchinson, L., and D. C. Johnson. 1995. Herpes simplex virus glycoproteinK promotes egress of virus particles. J. Virol. 69:5401–5413.
32. Johnson, D. C., and M. W. Ligas. 1988. Herpes simplex viruses lackingglycoprotein D are unable to inhibit viral penetration: quantitative evidencefor virus-specific cell surface receptors. J. Virol. 62:4605–4612.
33. Johnson, D. C., M. R. McDermott, C. Chrisp, and J. C. Glorioso. 1986.Pathogenicity in mice of herpes simplex virus type 2 mutants unable toexpress glycoprotein C. J. Virol. 58:36–42.
34. Karger, A., and T. C. Mettenleiter. 1993. Glycoproteins gIII and gp50 playdominant roles in the biphasic attachment of pseudorabies virus. Virology194:654–664.
34a.Karger, A., A. Saalmuller, F. Tufaro, B. W. Banfield, and T. C. Mettenleiter.1995. Cell surface proteoglycans are not essential for infection by pseudor-abies virus. J. Virol. 69:3482–3489.
35. Kjellen, L., and U. Lindahl. 1991. Proteoglycans: structures and interactions.Annu. Rev. Biochem. 60:443–475.
36. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature (London) 227:680–685.
37. Langeland, N., H. Holmsen, J. R. Lillehaug, and L. Haarr. 1987. Evidencethat neomycin inhibits binding of herpes simplex virus type 1 to the cellularreceptor. J. Virol. 61:3388–3393.
38. Langeland, N., L. J. Moore, H. Holmsen, and L. Haarr. 1988. Interaction ofpolylysine with the cellular receptor for herpes simplex virus type 1. J. Gen.Virol. 69:1137–1145.
39. Liang, X., L. A. Babiuk, S. van Drunen Littel-van den Hurk, D. R. Fitz-patrick, and T. J. Zamb. 1991. Bovine herpesvirus 1 attachment to permis-sive cells is mediated by its major glycoproteins gI, gIII, and gIV. J. Virol.65:1124–1132.
40. Liang, X., L. A. Babiuk, and T. J. Zamb. 1993. Mapping of heparin-bindingstructures of bovine herpesvirus 1 and pseudorabies virus gIII glycoproteins.Virology 194:233–243.
41. Lidholt, K., J. L. Weinke, C. S. Kiser, F. N. Lugemwa, K. J. Bame, S.Cheifetz, J. Massague, U. Lindahl, and J. D. Esko. 1992. A single mutationaffects both N-acetylglucosaminyltransferase and glucoronosyltransferase ac-tivities in a Chinese hamster ovary cell mutant defective in heparan sulfatebiosynthesis. Proc. Natl. Acad. Sci. USA 89:2267–2271.
42. Ligas, M. W., and D. C. Johnson. 1988. A herpes simplex virus mutant inwhich glycoprotein D sequences are replaced by b-galactosidase sequencesbinds to but is unable to penetrate into cells. J. Virol. 62:1486–1494.
43. Little, S. P., J. T. Jofre, R. J. Courtney, and P. A. Schaffer. 1981. A virion-associated glycoprotein essential for infectivity of herpes simplex virus type1. Virology 115:149–160.
44. Lycke, E., M. Johansson, B. Svennerholm, and U. Lindahl. 1991. Binding ofherpes simplex virus to cellular heparan sulfate, an initial step in the adsorp-tion process. J. Gen. Virol. 72:1131–1137.
45. Manservigi, R., P. G. Spear, and A. Buchan. 1977. Cell fusion induced byherpes simplex virus is promoted and suppressed by different viral glycop-roteins. Proc. Natl. Acad. Sci. USA 74:3913–3917.
46. Marlin, S. D., S. L. Highlander, T. C. Holland, M. Levine, and J. C. Glorioso.1986. Antigenic variation (mar mutations) in herpes simplex virus glycopro-
VOL. 72, 1998 HS BINDING IN HSV-1 ATTACHMENT, ENTRY, AND SPREAD 6129
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from

tein B can induce temperature-dependent alterations in gB processing andvirus production. J. Virol. 59:142–153.
47. Marlin, S. D., T. C. Holland, M. Levine, and J. C. Glorioso. 1985. Epitopesof herpes simplex virus type 1 glycoprotein gC are clustered in two distinctantigenic sites. J. Virol. 53:128–136.
48. McClain, D. S., and A. O. Fuller. 1994. Cell-specific kinetics and efficiency ofherpes simplex virus type 1 are determined by two distinct phases of attach-ment. Virology 198:690–702.
49. Mettenleiter, T. C., L. Zsak, F. Zuckermann, N. Sugg, H. Kern, and T.Ben-Porat. 1990. Interaction of glycoprotein gIII with a cellular heparinlikesubstance mediates adsorption of pseudorabies virus. J. Virol. 64:278–286.
50. Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpessimplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436.
51. Okazaki, K., T. Matsuzaki, Y. Sugahara, J. Okada, M. Hasebe, Y. Iwamura,M. Ohnishi, T. Kanno, M. Shimizu, E. Honda, and Y. Kono. 1991. BHV-1adsorption is mediated by the interaction of glycoprotein gIII with heparin-like moiety on the cell surface. Virology 181:666–670.
52. Robbins, A. K., R. J. Watson, M. E. Whealy, W. W. Hays, and L. W. Enquist.1986. Characterization of a pseudorabies virus glycoprotein gene with ho-mology to herpes simplex virus type 1 and type 2 glycoprotein C. J. Virol.58:339–347.
53. Roizman, B., and A. E. Sears. 1996. Herpes simplex viruses and their repli-cation, p. 2231–2295. In B. N. Fields, D. M. Knipe, P. M. Howley, et al. (ed.),Fields virology, 3rd ed. Lippincot-Raven Publishers, Philadelphia, Pa.
54. Roop, C., L. Hutchinson, and D. C. Johnson. 1993. A mutant herpes simplexvirus type 1 unable to express glycoprotein L cannot enter cells, and itsparticles lack glycoprotein H. J. Virol. 67:2285–2297.
55. Shieh, M.-T., and P. G. Spear. 1994. Herpesvirus-induced cell fusion that is
dependent on cell surface heparan sulfate or soluble heparin. J. Virol. 68:1224–1228.
56. Shieh, M.-T., D. WuDunn, R. I. Montgomery, J. D. Esko, and P. G. Spear.1992. Cell surface receptors for herpes simplex virus are heparan sulfateproteoglycans. J. Cell Biol. 116:1273–1281.
57. Spear, P. G. 1993. Entry of alphaherpesviruses into cells. Semin. Virol.4:167–180.
58. Subramanian, G., D. S. McClain, A. Perez, and A. O. Fuller. 1994. Swinetestis cells contain functional heparan sulfate but are defective in entry ofherpes simplex virus. J. Virol. 68:5667–5676.
59. Tal-Singer, R., C. Peng, M. Ponce de Leon, W. R. Abrams, B. W. Banfield, F.Tufaro, G. H. Cohen, and R. J. Eisenberg. 1995. Interaction of herpessimplex virus glycoprotein gC with mammalian cell surface molecules. J. Vi-rol. 69:4471–4483.
60. Tengelsein, L. A., N. E. Pederson, P. R. Shaver, M. W. Wathen, and F. L.Homa. 1993. Herpes simplex virus type 1 DNA cleavage and encapsidationrequire the product of the UL28 gene: isolation and characterization of twoUL28 deletion mutants. J. Virol. 67:3470–3480.
61. Trybala, E., T. Bergstrom, D. Spillmann, B. Svennerholm, S. Olofsson, S. J.Flynn, and P. Ryan. 1996. Mode of interaction between pseudorabies virusand heparan sulfate/heparin. Virology 218:35–42.
62. Trybala, E., T. Bergstrom, B. Svennerholm, S. Jeansson, J. C. Glorioso, andS. Olofsson. 1994. Localization of the functional site on herpes simplex virustype 1 glycoprotein C involved in binding to cell surface heparan sulfate.J. Gen. Virol. 75:743–752.
63. Wickham, T. J., P. W. Roelvink, D. E. Brough, and I. Kovesdi. 1996. Ade-novirus targeted to heparan containing receptors increases its gene deliveryefficiency to multiple cell types. Nat. Biotechnol. 14:1570–1573.
64. WuDunn, D., and P. G. Spear. 1989. Initial interaction of herpes simplexvirus with cells is binding to heparan sulfate. J. Virol. 63:52–58.
6130 LAQUERRE ET AL. J. VIROL.
on May 19, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Related Documents











