Hemoglobin and hemoglobinpathies Srbová M., Průša R.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Hemoglobin and hemoglobinpathies
Srbová M., Průša R.

Hemoproteins
Consist of hem
– cyclic tetrapyrrole
– 1 iron cation Fe2+ bound in the middle of tetrapyrrole scelet by coordination covalent bonds
– conjugated system of double bonds
methine bridge
pyrrole ring







Types of hemoglobin Adult HbA: 2α and 2β subunits (98%HbA)
Adult HbA2: 2α and 2δ subunits (2% HbA) Fetal HbF: 2α and 2γ have higher O2 affinity than HbA – take up oxygen
from the maternal circulation Embryoinic: 2and 2 2 and 2 2 and 2 have higher O2 affinity than HbA

Hemoglobin switching
Alteration of globin gene expresion during development


Hemoproteins
Hemoglobin (transports O2 to the tissues)
Myoglobin (stores O2 in the muscles)
Cytochromes (e- carriers in ETC)
Catalase + peroxidases (decomposition of peroxides)
Cytochrome P-450 (hydroxylation)
Desaturasases FA (desaturation FA)
Redox state Fe 2+ Fe 3+
Redox state Fe 2+

Structure of Hemoglobin
• 4 polypetide subunits (globins)
• Hb A (adults) heterotetramer 2α a 2β
• Each subunit contains 1 hem group
• 8 helices (A-H) β subunit
• 7 helices α subunit
• Hydrofobic pocket
- protect hem against oxidation

• Hem binding to globin – Fe 2+ is coordinated by N atom from proximal histidin F8
• Binding of O2 – distal histidin E7 hydrogen bonds to the O2
Structure of Hemoglobin

• Quaternary structure
Interactions between subunits 1) hydrofobic ( between α-β)
2) electrostatic (between α-α; β-β, α-β)
– O2 binding – loss of these interactions
Structure of Hemoglobin
α1
α2
β1 β2

• 1 polypeptide chain (153 AA)
• 1 heme
• Tertiary structures of the α and β subunits are remarkably similar, both to each other and to that of Mb
• Skeletal and heart muscles
Structure of Myoglobin

Binding of O2 (oxygenation)
• Oxygenation changes the electronic state of the Fe2+ - heme
• Color change of blood from dark purplish (venous) to the brilliant scarlet color (arterial)

• The binding of the first O2 to Hb enhances the binding futher O2 molecules
• O2 affinity of Hb increases with increasing pO2
• Sigmoidal saturation curve
• Hyperbolic curve for Mb - no cooperative behavior
Mechanism of oxygen-binding cooperativity

Satu
rati
on
O2
•Hb loads O2 to about 90% saturation under the arterial partial pressure • Hb travels to the tissue where the O2 partial pressure is 20 torr, most of Hb´s bound O2 is released

Sa
tura
tio
n O
2
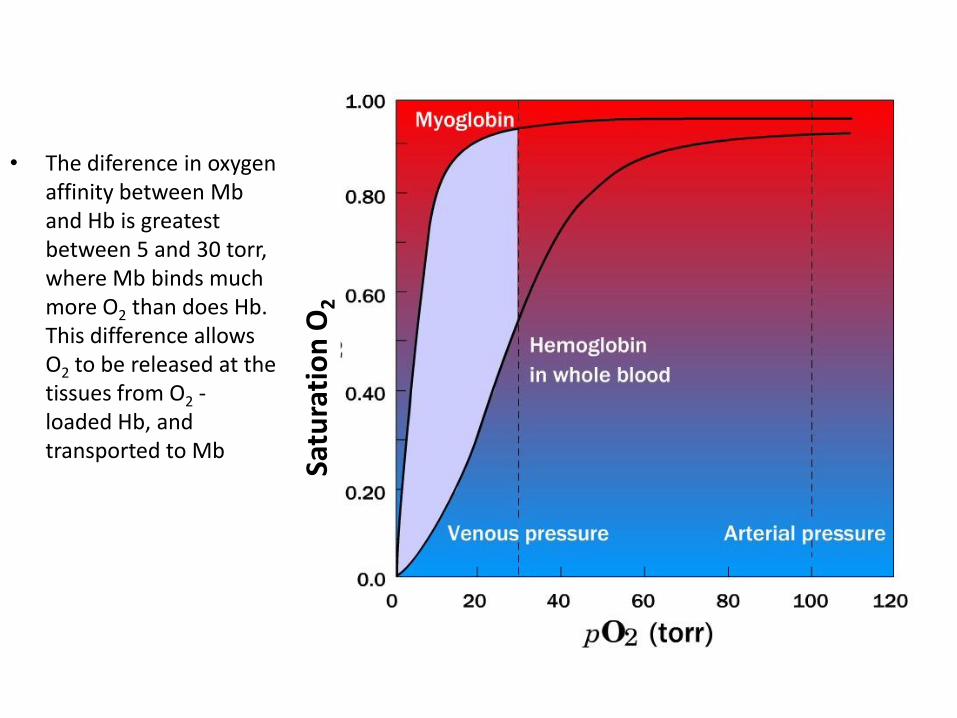
• The diference in oxygen affinity between Mb and Hb is greatest between 5 and 30 torr, where Mb binds much more O2 than does Hb. This difference allows O2 to be released at the tissues from O2 - loaded Hb, and transported to Mb
Satu
rati
on
O2

Oxygen binding to Hb

• The movement of Fe 2+ into the heme plane triggers the T→R conformational shift
•The loss of electrostatic interactions induce conformational changes in all other subunits


Conversion of T form→R form
T form (tense)
R form (relaxed)
The binding of the first O2 molecule to subunit of the T-form leads to a local conformational change that weakens association between the subunits R-form

Allosteric effectors
• CO2
• H+
• 2,3-bisphosphoglycerate
Decrease O2 affinity of Hb
Influence the equilibrium between T and R forms

Oxygen transport regulation

2,3 - bisphosphoglycerate
•binds selectively to deoxy-Hb •stabilizes T form •lowers the affinity of Hb for oxygen •oxygen is more readily released in tissues

2,3 - bisphosphoglycerate Clinical aspects:
In people with high-altitude adaptation or smokers the concentration of 2,3-BPG in the blood is increased increases the amount of oxygen that Hb unloads in the capilaries
Fetal hemoglobin (HbF α2γ2), has low BPG affinity – the higher O2 affinity – facilitates the transfer of O2 to the fetus via the placenta

Bohr effect
• The binding of protons H+ by Hb lowers its affinity for O2
• Increasing pH, that is, removing protons,stimulates Hb to bind O2
• pH of the blood decreases as it enters
tissues because CO2 produced by
metabolism is converted to H2CO3
• Dissociation of H2CO3 produces protons
• Promote the release of oxygen
In the tissues

Bohr effect
In the lungs
Oxygen binds to Hb, causing a release protons, which combine with bicarbonate to form H2CO3
Carbonic anhydrase cleaves H2CO3 to H2O and CO2 CO2 is exhaled

Hemoglobin and transport CO2

Hemoglobin determination
2. Direct spectrophotometry of plasma 415 – 460 nm
1.

Total Hb and Free Hb
• Reference values of total Hb – age and sex dependent, about 150 g/l
• Free Hb: 125 – 300 mg/l

Derivatives of hemoglobin
Deoxyhemoglobin – Hb without O2
Oxyhemoglobin – Hb with O2
Carbaminohemoglobin – Hb with CO2
– CO2 is bound to globin chain
– about 15% of CO2 is transported in blood bound to Hb
Carbonylhemoglobin – Hb with CO
– CO binds to Fe2+ 200x higher affinity to Fe2+ than O2 – poisoning, smoking

Methemoglobin – (metHb) contains Fe3+ instead of Fe2+
Autooxidation of hemoglobin
3% of hemoglobin undergoes oxidation every day
Hem – Fe2+- O2 Hem - Fe3+ + O2•-
Methemoglobin reductase
reduces methemoglobin
FAD, cytochrom b5 a NADH
Methemoglobinemia
1. Hereditary deficit of methemoglobin reductase
2. Abnormal hemoglobin HbM (Hb mutation)
3. Exposure to exogenous oxidizing drugs (sulfonamides, aniline)
Clinical aspects: cyanosis (10% Hb forms metHb) treatment: administration of methylene blue or ascorbic acid

Glycohemoglobin (HbA1c)
Formed by Hb‘s exposure to high levels of glucose
Nonenzymatic glycation of terminal NH2 group (Val) β-chain
Normally about 4 % of Hb is glycated (proportional to blood Glc concentration)
People with DM have more HbA1c than normal ( 5%)
Measurement of blood HbA1c is useful to get information about long-term control of glycemia


β1
β2
α1
α2
α1
α2
α1
α2
γ1
γ2
δ1
δ2
Hb A > 96,5% Hb F < 1% Hb A2 < 3,5%
HbA1c: What are we looking for?

1st Step: Unstable, reversible reaction between Glucose and the N-terminal valine of the β-chain (Schiff base)
2nd Step: During red blood cell circulation, some of the labile A1C is
converted to form a stable HbA1c (Amadori rearrangement)

HbA1c is currently defined as:
Hemoglobin A which is irreversibly glycated at one or both N-terminal Valines of the chains in the tetramer.
Glycation elsewhere on the or chains is irrelevant.
G
G
G
G
G
G
G
G
G
N
N
N
N
N
All of these are HbA1c
The nature of the problem – what is HbA1c?

Glycohemoglobin, or GHb, or Total GHb, is defined as:
Hb having one or more sugars irreversibly attached at any point in any
of the globin chains.
(This also includes all forms of HbA1c).
G
G
N
G
G
G
N
G
N
G
G
G
G N
All of these are GHb (but not HbA1c)
The nature of the problem – what is HbA1c?

Hb A0 93-95%
Hb A1 = GHb
Glycated Hbs
5-7%
Hb A
+ + +
Hb A1a 0,5%
Fructose-1,6-diphosphate Glucose-6-phosphate
Hb A1b 0,5%
pyruvate
Hb A1c 4-6%
glucose
Hb F Hb A2
HbA1c: What are we looking for?
Glycation at the N-terminal Valin
of the β-globin chain

The Pros and the Cons of using HbA1c for Diabetes Diagnosis David B.Sacks; AACC Webinar April 10th 2012


Glycohemoglobin Assay

HPLC – TOSOH G7





Fructosamine 3- kinase
• AGE (advanced glycation endproducts)
• Deglycation protective enzyme
• Brain, erytrocytes, lens – high activity
• D.M. and fasting – no influence on activity
• Polymorphisms of gene – lower or higher activity

Hemoglobinopathies
mutation → abnormal structure of the hemoglobin
Large number of haemoglobin mutations, a fraction has deleterious effects: sickling, change in O2 affinity, heme loss or dissociation of tetramer
hemoglobin M and S, thalassemias
1. Hemoglobin M
• Replacement of His E7α by Tyr (Hb Boston) or
• Replacement of Val E11β by Glu (Hb Milwaukee)
• the iron in the heme group is in the Fe3+ state (methemoglobin) stabilized by the tyrosine or by glutamate
• Methemoglobin reductase cannot reduce Fe3+
• methemoglobin can not bind oxygen

2. Thalassemias
• Mutation that results in decreased synthesis of α or β-chains
• thalassemia mutations provide resistence to malaria in the heterozygous state
α- thalassemias – complete gene deletion
4 α globin genes per cell:
1 copy of gen is deleted: without symptoms
2 copies are deleted: RBC are of decreased size (microcytic) and reduced Hb concentration (hypochromic), individual is usually not anemic
3 copies are deleted: moderately severe microcytic hypochromic anemia with splenomegaly
4 copies are deleted: hydrops fetalis: fatal in utero
Excess β chains form homotetramer HbH which is useless for delivering oxygen to the tissues (high oxygen affinity)






• β+ – some globin chain synthesis
• β0 – no globin chain synthesis
Heterozygotes: microcytic hypochromic RBC, mild anemia
Homozygotes β0 β0 : severe anemia
Excess α chains precipitate in erythroid precursor – their destruction-ineffective erythropoiesis
β- thalassemias



3. Hemoglobin S (sickle-cell)
• Causes a sickle-cell anemia
• Replacing Glu A3β with the less polar amino acid Val - forming „an adhesive region“ of the β chain
• HbS proteins aggregate into a long rodlike helical fiber




67
G7 x G8



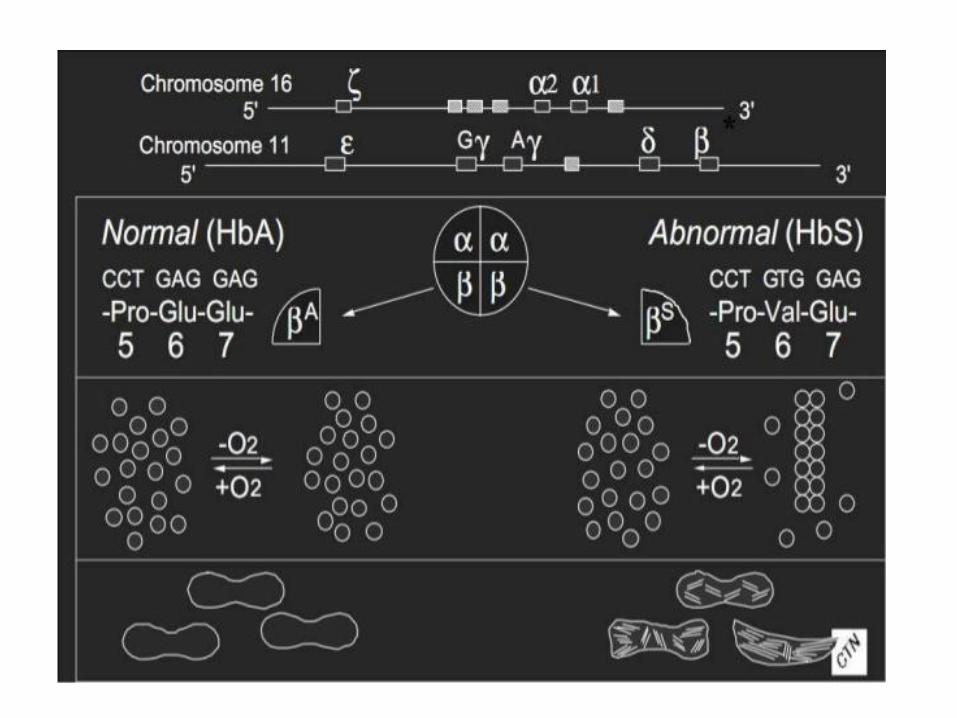


Sickle-cell anemia Red blood cells adopt a sickle shape in a consequence of the forming haemoglobin S fibers The high incidence of sickle-cell disease coincides with a high incidence of malaria Individuals heterozygous in HbS have a higher resistance to malaria; the malarial parasite spends a portion of its life cycle in red cells, and the increased fragility of the sickled cells tends to interrupt this cycle


Pictures used in the presentation:
• Marks´ Basic Medical Biochemistry, A Clinical Approach, third edition, 2009 (M. Lieberman, A.D. Marks)
• Principles of Biochemistry, 2008, (Voet D, Voet J.G., and Pratt C.W)
• Color Atlas of Biochemistry, second edition, 2005 (J. Koolman and K.H. Roehm)
Related Documents