ch1.1 1 1 Introduction 1.1 Principles of Heterogeneous Catalysis 1 James A. Dumesic ∗ , George W. Huber and Michel Boudart 1.1.1 Introduction Heterogeneous catalysis is of vital importance to the world’s economy, allowing us to convert raw materi- als into valuable chemicals and fuels in an economical, efficient, and environmentally benign manner. For ex- ample, heterogeneous catalysts have numerous industrial applications in the chemical, food, pharmaceutical, au- tomobile and petrochemical industries [1–5], and it has been estimated that 90% of all chemical processes use het- erogeneous catalysts [6]. Heterogeneous catalysis is also finding new applications in emerging areas such as fuel cells [7 – 9], green chemistry [10 – 12], nanotechnology [13], and biorefining/biotechnology [14 – 18]. Indeed, contin- ued research into heterogeneous catalysis is required to allow us to address increasingly complex environmental and energy issues facing our industrialized society. Discussing the principles of heterogeneous catalysis is difficult, because catalysts are used for a wide range of applications, involving a rich range of surface chemistries. Moreover, the field of heterogeneous catalysis is highly interdisciplinary in nature, requiring the cooperation between chemists and physicists, between surface scientists and reaction engineers, between theorists and experimentalists, between spectroscopists and kineticists, and between materials scientists involved with catalyst synthesis and characterization. Furthermore, industrial catalysts are complex materials, with highly optimized chemical compositions, structures, morphologies, and pellet shapes; moreover, the physical and chemical characteristics of these materials may depend on hidden or unknown variables. Accordingly, principles of heterogeneous catalysis are typically formulated from studies of model catalysts in ideal reactors with simplified reactants under mild pressure conditions (e.g., 1 bar), rather than from catalytic performance data obtained with commercial catalysts in complex reactors using mixed feed streams under industrial reaction conditions. The principles derived from these more simplified studies advance the science of heterogeneous catalysis, and they guide the researcher, inventor, and innovator of new catalysts and catalytic processes. 1.1.2 Definitions of Catalysis and Turnover The definition of a catalyst has been discussed many times [19]. For example, a catalyst is a material that converts reactants into products, through a series of elementary steps, in which the catalyst participates while being regenerated to its original form at the end of each cycle during its lifetime. A catalyst changes the kinetics of the reaction, but does not change the thermodynamics. Another definition is that a catalyst is a substance that transforms reactants into products, through an uninterrupted and repeated cycle of elementary steps in which the catalyst participates while being regenerated to its original form at the end of each cycle during its lifetime [20]. The main advantage of using a heterogeneous catalyst is that, being a solid material, it is easy to separate from the gas and/or liquid reactants and products of the overall catalytic reaction. The heart of a heterogeneous catalyst involves the active sites (or active centers) at the surface of the solid. The catalyst is typically a high-surface area material (e.g., 10 – 1000 m 2 g −1 ), and it is usually desirable to maximize the number of active sites per reactor volume. Identifying the reaction intermediates – and hence the 1 A list of abbreviations/acronyms used in the text is provided at the end of the chapter. ∗ Corresponding author. References see page 14 HANDBOOK OF HETEROGENEOUS CATALYSIS Second, Completely Revised and Enlarged Edition Volume 1 Gerhard Ertl, Helmuth Knözinger, Ferdi Schüth, Jens Weitkamp (Editors) Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim, Germany, ISBN: 978-3-527-31241-2, 2008

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ch1.1
1
1Introduction
1.1Principles of Heterogeneous Catalysis1
James A. Dumesic∗, George W. Huber and Michel Boudart
1.1.1Introduction
Heterogeneous catalysis is of vital importance to theworld’s economy, allowing us to convert raw materi-als into valuable chemicals and fuels in an economical,efficient, and environmentally benign manner. For ex-ample, heterogeneous catalysts have numerous industrialapplications in the chemical, food, pharmaceutical, au-tomobile and petrochemical industries [1–5], and it hasbeen estimated that 90% of all chemical processes use het-erogeneous catalysts [6]. Heterogeneous catalysis is alsofinding new applications in emerging areas such as fuelcells [7–9], green chemistry [10–12], nanotechnology [13],and biorefining/biotechnology [14–18]. Indeed, contin-ued research into heterogeneous catalysis is required toallow us to address increasingly complex environmentaland energy issues facing our industrialized society.
Discussing the principles of heterogeneous catalysis isdifficult, because catalysts are used for a wide range ofapplications, involving a rich range of surface chemistries.Moreover, the field of heterogeneous catalysis is highlyinterdisciplinary in nature, requiring the cooperationbetween chemists and physicists, between surfacescientists and reaction engineers, between theorists andexperimentalists, between spectroscopists and kineticists,and between materials scientists involved with catalystsynthesis and characterization. Furthermore, industrialcatalysts are complex materials, with highly optimizedchemical compositions, structures, morphologies, andpellet shapes; moreover, the physical and chemicalcharacteristics of these materials may depend on
hidden or unknown variables. Accordingly, principlesof heterogeneous catalysis are typically formulated fromstudies of model catalysts in ideal reactors with simplifiedreactants under mild pressure conditions (e.g., 1 bar),rather than from catalytic performance data obtained withcommercial catalysts in complex reactors using mixedfeed streams under industrial reaction conditions. Theprinciples derived from these more simplified studiesadvance the science of heterogeneous catalysis, and theyguide the researcher, inventor, and innovator of newcatalysts and catalytic processes.
1.1.2Definitions of Catalysis and Turnover
The definition of a catalyst has been discussed manytimes [19]. For example, a catalyst is a material thatconverts reactants into products, through a series ofelementary steps, in which the catalyst participates whilebeing regenerated to its original form at the end of eachcycle during its lifetime. A catalyst changes the kineticsof the reaction, but does not change the thermodynamics.Another definition is that a catalyst is a substancethat transforms reactants into products, through anuninterrupted and repeated cycle of elementary stepsin which the catalyst participates while being regeneratedto its original form at the end of each cycle during itslifetime [20].
The main advantage of using a heterogeneous catalystis that, being a solid material, it is easy to separate fromthe gas and/or liquid reactants and products of the overallcatalytic reaction. The heart of a heterogeneous catalystinvolves the active sites (or active centers) at the surfaceof the solid. The catalyst is typically a high-surface areamaterial (e.g., 10–1000 m2 g−1), and it is usually desirableto maximize the number of active sites per reactor volume.Identifying the reaction intermediates – and hence the
1 A list of abbreviations/acronyms used in the text is provided at theend of the chapter.
∗ Corresponding author. References see page 14
HANDBOOK OF HETEROGENEOUS CATALYSISSecond, Completely Revised and Enlarged Edition
Volume 1Gerhard Ertl, Helmuth Knözinger, Ferdi Schüth, Jens Weitkamp (Editors)
Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim, Germany,ISBN: 978-3-527-31241-2, 2008

ch1.1
2 1.1 Principles of Heterogeneous Catalysis
mechanism – for a heterogeneous catalytic reaction isoften difficult, because many of these intermediates aredifficult to detect using conventional methods (e.g., gaschromatography or mass spectrometry) because they donot desorb at significant rates from the surface of thecatalyst (especially for gas-phase reactions).
Heterogeneous catalysts typically contain differenttypes of surface sites, because crystalline solids exhibitcrystalline anisotropy. Equilibrated single crystals exposedifferent faces with different atomic structures soas to minimize total surface energy. It would besurprising, in fact, if different crystallographic planesexposing sites with different coordination environmentspossessed identical properties for chemisorption andcatalytic reactions. Moreover, most catalytic solids arepolycrystalline. Furthermore, in order to achieve highsurface areas, most catalysts contain particles with sizesin the nanometer length scale. The surfaces of thesenanoscopic particles contain sites associated with terraces,edges, kinks, and vacancies [21]. If the catalyst containsmore than one component (as is generally the case), thesurface composition may be different from that of the bulkand differently so for each exposed crystallographic plane.Solids normally contain defects of electronic or atomicnature; in addition, they contain impurities which areeither known or unknown in the bulk, but are mostlyunknown at the surface. Finally, the surface atomicstructure and composition may change with time-on-stream as the catalytic reaction proceeds. In short, itis normal to expect that a catalytic surface exposes avariety of surface sites, in contrast to displaying a singletype of active site. Indeed, it is so normal today toexpect such complexity that it seems surprising that,in 1925, when Taylor formulated his principle of activesites or active centers, the report created so muchattention and remains one of the most often cited inheterogeneous catalysis [22]. The relative importance ofsurface structure – as influenced by crystalline anisotropy,surface defects, and surface composition – underlines thedifficulty of identifying the active sites, either simpleor complex, that are responsible for turning over thecatalytic cycle. The identification and counting of activesites in heterogeneous catalysis became the ‘‘Holy Grail’’of heterogeneous catalysis in 1925, and the situationremains the same today.
The activity of a catalyst is defined by the number ofrevolutions of the catalytic cycle per unit time, given inunits of turnover rate (TOR) or turnover frequency (TOF).In cases where the rate is not uniform within the catalyticreactor or within the catalyst pellets, it is useful to reportthe rate as a site time yield (STY), defined as the overall rateof the catalytic reaction within the reactor normalized bythe total number of active sites within the reactor, againin units of reciprocal time. Catalysis by solid materials
has been observed quantitatively at temperatures as lowas 78 K and as high as 1500 K; at pressures between10−9 and 103 bar; with reactants in the gas phase or inpolar or non-polar solvents; with or without assistanceof photons, radiation or electron transfer at electrodes;with pure metals as unreactive as gold and as reactive assodium; with multicomponent and multiphase inorganiccompounds and acidic organic polymers; and at STYsas low as 10−5 s−1 (one turnover per day) and as highas 109 s−1 (gas kinetic collision rate at 10 bar). TOFs ofcommonly used heterogeneous catalysts are commonlyon the order of one per second. The life of the catalyst canbe defined as the number of turnovers observed beforethe catalyst ceases to operate at an acceptable rate. Clearly,this number must be larger than unity, otherwise thesubstance used is not a catalyst but a reagent. Catalystlife can either be short, as in catalytic cracking of oil, orvery long, corresponding to as many as 109 turnovers inammonia synthesis.
1.1.3Steps in a Heterogeneous Catalytic Reaction
During an overall catalytic reaction, the reactantsand products undergo a series of steps over thecatalyst, including:
1. Diffusion of the reactants through a boundary layersurrounding the catalyst particle.
2. Intraparticle diffusion of the reactants into the catalystpores to the active sites.
3. Adsorption of the reactants onto active sites.4. Surface reactions involving formation or conversion
of various adsorbed intermediates, possibly includingsurface diffusion steps.
5. Desorption of products from catalyst sites.6. Intraparticle diffusion of the products through the
catalyst pores.7. Diffusion of the products across the boundary layer
surrounding the catalyst particle.
Accordingly, different regimes of catalytic rate controlcan exist, including: (i) film diffusion control (Steps 1and 7); (ii) pore diffusion control (Steps 2 and 6); and(iii) intrinsic reaction kinetics control (Steps 3 to 5) ofcatalyst performance. In addition to mass transfer effects,heat transfer effects can also occur in heterogeneouscatalysis for highly exothermic or endothermic reactions(especially in combustion or steam reforming).
Figure 1 shows a general effect of temperature onthe reaction rate for a heterogeneous catalyst. At lowtemperatures, diffusion through the film and pores isfast compared to rates of surface reactions, and theoverall reaction rate is controlled by the intrinsic reaction

ch1.1
1.1.4 Desired Characteristics of a Catalyst 3
1/Temperature
ln (
rate
)
IncreasingTemperature
Pore diffusioncontrolled regime
Intrinsicregime
Film diffusioncontrolled regime
Slope = Ea/R
Slope = Ea/2R
Slope = 3-5 kJ/mol
Fig. 1 General effects of temperature on catalytic activity. Theintrinsic activation energy is equal to Ea, and R is the gas constant.
kinetics. As the temperature is increased, the rates ofsurface reactions typically increase more rapidly than therates of diffusion, and the overall rate of the catalyticprocess becomes controlled by intraparticle diffusion.The apparent activation energy in this regime is equalto the intrinsic activation energy divided by two. As thetemperature is increased further, mass transfer throughthe external boundary layer becomes the controllingstep. The onset of diffusion limited regimes can bealtered by changing the reactor design, the catalyst porestructure, the catalyst particle size, and the distributionof the active sites in the catalyst particles. Valuesof various dimensionless groups can be calculated toestimate the extents to which transport phenomenamay control catalytic performance for specific operatingconditions [23–31]; however, these calculations are mostreliable for cases where the intrinsic reaction kinetics areknown. In these cases, it is possible to make catalystswith structures designed to provide adequate rates ofdiffusion and yet offering high surfaces areas, leadingto high rates of reaction per reactor volume, such asthe design of specific pore size distributions (e.g., bi-modal distributions containing large pores leading tohigh accessibility of the active sites within the interior ofthe catalytic pellet, and small pores that branch from thelarger pores leading to high surface areas), the formulationof unique pellet shapes (that lead to high accessibility ofthe active sites but do not cause large pressure dropsthrough the catalytic reactor), and the synthesis of catalystpellets containing a spatial distribution of the activematerial within the catalyst pellet [32]. In some cases,transport effects can be used to improve the selectivity ofa catalyst, such as in the case of shape-selective catalysisin zeolites [33–36]. In the following sections, we focus onvarious factors controlling the intrinsic reaction kinetics
of catalysts, and we refer the reader to other articles forfurther discussion on transport effects in heterogeneouscatalysis [23–31].
1.1.4Desired Characteristics of a Catalyst
The following list provides several of the key attributes ofa good catalyst:
• The catalyst should exhibit good selectivity for produc-tion of the desired products and minimal production ofundesirable byproducts.
• The catalyst should achieve adequate rates of reactionat the desired reaction conditions of the process(remembering that achieving good selectivity is usuallymore important than achieving high catalytic activity).
• The catalyst should show stable performance at reactionconditions for long periods of time, or it should bepossible to regenerate good catalyst performance byappropriate treatment of the deactivated catalyst aftershort periods.
• The catalyst should have good accessibility of reactantsand products to the active sites such that high rates canbe achieved per reactor volume.
The first three key attributes of a good catalyst areinfluenced primarily by the interactions of the catalystsurface with the reactants, products, and intermediatesof the catalytic process. In addition, other species mayform on the catalyst surface (e.g., hydrogen-deficientcarbonaceous deposits denoted as coke) that are notdirectly part of the reaction scheme (or mechanism) forthe overall catalytic process.
The principle of Sabatier states that a good heteroge-neous catalyst is a material that exhibits an intermediatestrength of interaction with the reactants, products, andintermediates of the catalytic process [37, 38]. Interactionsof the catalyst surface with the various adsorbed speciesof the reaction mechanism that are too weak lead tohigh activation energies for surface reactions and thuslow catalytic activity, whereas interactions of the catalystwith adsorbed species that are too strong lead to excessiveblocking of surface sites by these adsorbed species, againleading to low catalytic activity.
The principle of Sabatier is elegant in its simplicityand generality, but it is deceptively difficult to use inpractice. In particular, this principle applies to a catalystin its working state, and the nature of the catalyst surfacecan be expected to be dependent on the nature of thecatalytic reaction conditions. For example, one may beginthe catalytic reaction with the heterogeneous catalyst
References see page 14

ch1.1
4 1.1 Principles of Heterogeneous Catalysis
in a given oxidation state (e.g., containing zero-valentmetal particles following treatment of the catalyst inH2 at elevated temperature); however, the nature of thesurface can be changed dramatically upon interactionwith strongly adsorbed species, such as the formation ofcarbonaceous deposits (coke), and formation of oxides,carbides, nitrides, or sulfides upon interaction with O,C, N, or S species, respectively [39–44]. In this case,the interactions of these oxide, carbide, nitride, orsulfide surfaces with the adsorbed species enter intothe reaction mechanism. Of even greater complexityis the fact that a variety of different types of sites aretypically present on a catalyst surface (e.g., sites havingdifferent coordination and/or chemical composition), anda majority of the observed catalytic activity may becaused by the contributions from a small fraction ofthe sites present on the catalyst surface. In this case,the adsorbed species interact with these special surfacesites (e.g., steps and defect sites on a metal nanoparticle,or sites present at the metal–support interface of asupported metal catalyst). Another factor that complicatescatalyst design is that the strengths of interaction ofthe surface with adsorbed species typically depend onthe surface coverages by adsorbed species. For example,the interaction of a transition metal surface with adsorbedCO may be very strong at low surface coverages (e.g.,binding energy of nearly 200 kJ mol−1), suggesting thatthese surfaces would be completely covered and thuspoisoned by adsorbed CO at moderate pressures andtemperatures; however, these surfaces may carry outcatalytic reactions in the presence of gaseous CO at thesepressures and temperatures because the differential heatof CO adsorption decreases significantly (e.g., to bindingenergies near 100 kJ mol−1) as the surface coverage byadsorbed CO increases [45, 46]. Accordingly, there is arelationship between activity and the interaction of thesurface with adsorbed species at the surface coverageregime appropriate for the catalytic reaction conditions.
The aforementioned complications caused by thepresence of different types of sites on the surface, andthe effects on the surface binding energies caused bychanges in surface coverages, clearly make it difficult tointerpret the performance of a heterogeneous catalystin quantitative detail. Tools are certainly available toaddress these complications, such as kinetic MonteCarlo simulations combined with results from densityfunctional theory (DFT) calculations [47–50]. Yet, froma different point of view, the presence of differenttypes of sites and the effects of surface coverage maywell contribute to the robustness of the heterogeneouscatalyst for operation over a wide range of reactionconditions. In general, the presence of different typesof sites and the effects of surface coverage bothcontribute to surface non-uniformity (different types
of sites producing a prior non-uniformity, and effectsof surface coverage causing induced non-uniformity).At a selected set of reaction conditions, an optimalset of surface binding energies exists that satisfy theprinciple of Sabatier (as discussed below). Accordingly,the performance of a heterogeneous catalyst with anon-uniform surface will be dominated by the subsetof the sites having surface binding energies closest tothe optimal values. At higher temperatures, other siteshaving stronger binding energies with adsorbed specieswill become the dominant contributors to the observedcatalytic activity, whereas sites having weaker bindingenergies with adsorbed species will control catalyticactivity at lower temperatures. Thus, while the effects ofsurface non-uniformity make it more difficult to predictthe performance of a heterogeneous catalyst from amolecular-level understanding, these effects may serveto broaden the range of reaction conditions over whichthe catalyst can operate effectively. In this respect, ourdesire to design catalysts having very high selectivityis guided by the synthesis of uniform catalysts, whereeach site has the optimal properties for production ofthe desired reaction product. This strategy leads to theidea of highly selective, single-site catalysis as discussedby Thomas et al. [51]. In contrast, the design of catalyststhat operate over a wide range of reaction conditions isguided by the synthesis of non-uniform catalysts, suchthat different subsets of sites control catalyst performanceat different reaction conditions. The disadvantage of usingnon-uniform catalysts, however, is that different sites maydisplay different selectivities for the production of variousproducts, and control over catalytic selectivity may thusbe limited [22].
1.1.5Reaction Schemes and Adsorbed Species
We now explore further the principle of Sabatier usinga specific example: water-gas shift over a metal catalyst(e.g., Cu). This reaction (CO + H2O −−−→←−−− CO2 + H2) isof importance for the production of H2 from steam-reforming of fossil fuels, and for controlling the CO : H2ratio in synthesis gas mixtures used in methanol andFischer–Tropsch synthesis processes. For this example,we consider the reaction scheme shown in Fig. 2, where* represents a surface site. The stoichiometric numbers,σi,1 and σi,2, indicate the number of times that step i
occurs to give the overall reaction for reaction schemes 1and 2, respectively.
In this sequence of steps, the water-gas shift reactioncan take place via the formation of carboxyl species(COOH) or through the formation of formate species(HCOO) [52]. In the absence of a catalyst, the rate of water-gas shift via this mechanism is negligible, because the

ch1.1
1.1.5 Reaction Schemes and Adsorbed Species 5
1. 1
2. 1
3. 2
4. 1
5. 1
6. 1
7. 1
8. 0
9. 0 1
overall
1
1
1
0
0
1
1
1
si,1 si,2
CO +∗ CO∗
H2O +∗ H2O∗
H2O∗ +∗ OH ∗+H∗
CO∗ + OH ∗ COOH ∗+∗
COOH ∗ + OH ∗ CO2∗ + H2O ∗
CO2∗ CO2 + ∗
2H ∗ H2 + 2∗
CO ∗ + OH ∗ HCOO ∗∗
HCOO∗∗ CO2∗ + H ∗
CO + H2O CO2 + H2
Fig. 2 Assumed reaction mechanism for water-gas shift reactionover Cu. Adapted from Ref. [52].
reaction intermediates (e.g., OH∗, H∗, COOH∗, HCOO∗)are at very low concentrations in the gas phase. Forexample, the enthalpy change for step 3 in the gas phaseis approximately 500 kJ mol−1. However, adsorption ofthe reaction intermediates onto the catalyst surface allowsthese steps to take place with small enthalpy changes. Inthe case of copper, the binding energies of H and OHare approximately 250 and 280 kJ mol−1 on Cu(111), suchthat the enthalpy change for step 3 on the catalyst surfaceis now slightly exothermic. According to the principle ofSabatier, a good catalyst is a material that adsorbs reactionintermediates with intermediate strength. However, wenow must distinguish between reactive intermediates andspectator species on the catalyst surface.
In the above reaction scheme, we see that the water-gas shift reaction can take place through adsorbedcarboxyl species or formate species. Results from DFTcalculations indicate that adsorbed formate species havelower energy compared to adsorbed carboxyl species oncopper surfaces, suggesting that path 2 for water-gasshift (σi,2) would be favored versus path 1 (σi,1) basedon thermodynamic arguments. However, the activationenergy for step 4 is considerably lower than that forstep 8, and the primary path for water-gas shift overcopper involves the formation and subsequent reaction ofadsorbed carboxyl species. Accordingly, the most stablespecies on the catalyst surface are not necessarily themost reactive species. This idea leads us to distinguishbetween a most abundant surface intermediate (MASI) anda most abundant reactive intermediate (MARI). In certaincases, the MASI and the MARI may be the same species,but in other cases (such as in this case of water-gasshift on copper), the MASI is a spectator species that
does not participate in the overall reaction. In this lattercase, the spectator species inhibits the overall reactionby blocking surface sites, and it serves no useful role inthe overall reaction scheme. For purposes of elucidatingcatalytic reaction schemes it is essential to distinguishbetween reactive intermediates and spectator species. Thisdistinction is of paramount importance in spectroscopicstudies of adsorbed species on catalyst surfaces, wherethe detection of a specific adsorbed species using aspectroscopic method (e.g., the detection by infra-red(IR) studies of adsorbed ethylidyne species on platinumsurfaces during ethylene hydrogenation [53]) does notguarantee that this species is a reactive intermediate.Instead, these spectroscopic studies must be conductedunder dynamic conditions (e.g., so-called operandomeasurements, where spectroscopic and reaction kineticsdata are collected simultaneously) to determine thatthe time constant for the formation or disappearanceof the surface species is the same as the time constant forthe overall catalytic reaction [54, 55].
The overall catalytic reaction is given by a linearcombination of elementary steps, and the enthalpy changefor the overall reaction, �H , is given by:
�H =∑
i
σi�Hi (1)
where �Hi are the enthalpy changes for elementarysteps i. From the principle of Sabatier, it is now clearthat the overall value of �H should be composed ofapproximately equal contributions from each of thevalues of �Hi , giving rise to a relatively flat potentialenergy diagram of energy versus reaction coordinate inmoving from reactants, through adsorbed intermediates,to products. Specifically, any value of �Hj that is verynegative must be balanced by a value of �Hk that isvery positive, such that the surface will become highlycovered (and poisoned) by the adsorbed species producedin step j , and the activation energy for step k will be high.Both of these effects lead to low catalytic activity. We notethat the reaction mechanism can certainly contain stepswith positive values of �Hi , because the intermediatesproduced in such a step can be consumed by followingsteps having negative values of �Hi . This situation istermed ‘‘kinetic coupling’’, where the conversion of anunfavorable step is increased by its combination with afavorable step that consumes the unstable products of thefirst step. The highest value of �Hi for a surface reactionthat can be tolerated can be estimated from transitionstate theory. The value of �Hi,max depends on the overallrate of the reaction (TOF), the surface coverage by thesurface species that reacts in this step (θA), a frequency
References see page 14

ch1.1
6 1.1 Principles of Heterogeneous Catalysis
factor ν (of the order of 1013 s−1), and the temperature T ,as given by:
TOF = ν exp
(−�Hi,max
RT
)θA (2)
For a reaction operating at 500 K with a TOF of 1 s−1,the maximum value of �Hi that can be tolerated for aspecies with high surface coverage (θA approaching unity)corresponds to 125 kJ mol−1, which is still a rather highvalue. In practice, the highest value of �Hi that could betolerated would be lower than this value of 125 kJ mol−1,because the surface coverage by the reactive intermediatewould typically be lower than unity and the above analysisassumes that the activation energy for the reverse ofstep i (i.e., the exothermic direction for this step) is equalto zero. This situation where the overall enthalpy changeis shared fairly equally between the various steps of thereaction scheme is a necessary condition for high catalyticactivity, but it is not a sufficient condition, because wehave not yet considered the transition states for the variouselementary steps.
The aforementioned reaction scheme for water-gas shiftinvolving the formation of carboxyl species contains sevensteps, thereby requiring the determination (or estimation)of 13 rate constants to describe the reaction kineticscompletely; that is, a forward and reverse rate constant foreach step (kfor,i and krev,i ) constrained by the relationshipthat these rate constants must give the proper equilibriumconstant for the overall reaction, Keq, as given below:
∏i
(kfor,i
krev,i
)σi
= Keq (3)
However, it is a rare case that all of these rateconstants are kinetically significant. Thus, while wegenerally have the desire to know the values for asmany rate constants as possible, we typically need toknow only the values of a limited number of these rateconstants to describe the performance of the catalyst
for the reaction conditions of interest. Unfortunately,at the outset of research on a given catalyst process,we usually do not know which rate constants will bekinetically significant. Accordingly, an important objectiveof research into a given catalytic process is to identifywhich steps are kinetically significant, such that furtherresearch can focus on altering the nature of the catalystand the reaction conditions to enhance the rates of thesekinetically controlling steps. This situation is illustratedin Fig. 3 for the above case of water-gas shift involvingcarboxyl species, according to which the rate is controlledby steps 3 and 4, whereas steps 1, 2, 5, 6, and 7 arequasi-equilibrated.
The net rate of step 3 in Fig. 3, is twofold faster thanthe net rates of all other steps, because the stoichiometricnumber of step 3 is equal to 2 whereas all other stoichio-metric numbers are equal to 1. Importantly, the net rate ofeach step divided by its stoichiometric number is equal tothe net rate of the overall catalytic reaction. This equalityis due to the principle of kinetic steady state, as stated byBodenstein (see Ref. [38]), according to which determin-ing the rate of one single reaction (typically the overallreaction) allows one to calculate the net rates of all theother individual reactions. The Bodenstein principle is animportant foundation of our thinking about how catalyticcycles turn over. This principle also shows that the notionof a ‘‘slow’’ step in a catalytic cycle at the steady state is amisnomer, because all steps proceed at the same net rate.
1.1.6Conditions for Catalyst Optimality
It can be shown that the net rate of the overall catalyticreaction is controlled by kinetic parameters which dependonly on the properties of the transition states for thekinetically significant steps relative to the reactants (andpossibly the products) of the overall reaction [56]. Theoverall rate is also controlled by an additional kineticparameter for each surface species that is abundant on
1.
2.
3.
4.
5.
6.
7.
CO +∗ CO∗
H2O +∗ H2O∗
H2O∗+∗ OH ∗ + H∗
CO∗+OH ∗ COOH ∗+∗
COOH ∗+OH ∗ CO2∗+H2O ∗
CO2∗ CO2 + ∗
2H ∗ H2 + 2∗
Fig. 3 Rates of forward and reverse steps in the water-gas shift reaction on Cu.

ch1.1
1.1.6 Conditions for Catalyst Optimality 7
the catalyst surface. Specifically, the net rate of the overallreaction is determined by the kinetic parameters as wellas by the fraction of the surface sites, θ∗, that is availablefor the formation of the transition states; the value of θ∗is determined by the extent of site blocking by abundantsurface species.
To illustrate how to determine the optimal activityof a catalyst, we consider an example in which thereaction scheme contains a single rate-controlling stepand a single abundant surface species. According toresults obtained using DeDonder relations (discussedin Section 5.2.1.10) [56], we may write this reactionscheme in terms of a quasi-equilibrated step involvingthe transition state for the rate-controlling step, TSi , anda second equilibrated step involving the formation of themost abundant surface species, A∗, as given below and:
1. Reactants + 2∗ −−−→←−−− T Si (4)
2. A + ∗ −−−→←−−− A∗ (5)
The overall rate of the reaction, rnet, as will be discussedin Section 5.2.1.12, is now given by:
rnet = ν‡
σ1exp
(�S
o‡1
R− �H
o‡1
RT
)F(ai)θ
2∗ (1 − z1/σ1tot )
(6)
θ∗ = 1
1 + exp(
�So2
R− �Ho
2
RT
)aA
(7)
where F(ai) is a function of the activities (ai) of the reac-tants and/or products of the overall reaction. Neglectingentropy effects, as we change the nature of the catalystfor constant reaction conditions, the primary items in theabove equations that change are �H
o‡1 and �Ho
2 . (Note,we implicitly assume that the reaction mechanism doesnot change.) Accordingly, the overall rate of the reactionfor different catalysts is given by:
rnet =C1 exp
(−�H
o‡1
RT
)(
1 + C2 exp(
−�Ho2
RT
))2 (8)
C1 = ν‡
σ1exp
(�S
o‡1
R
)F(ai)
(1 − z
1/σ1tot
)(9)
C2 = exp(
�So2
R
)aA (10)
We next consider that the surface properties of thecatalyst are described in terms of some fundamentalcatalyst property, x. This property x could be a heat
of adsorption of one of the reactants [57], the heat offormation of a bulk compound that can be correlatedwith a heat of adsorption [58], the position of the catalyticelement along a horizontal series in the Periodic Table,an electronic property of the catalyst such as Pauling’sd-band character of the metal [59], or the d-band center ofthe metal [60]. The optimal catalyst can thus characterizedby the following relationship:
drnet
dx= 0 =
−C1 exp
(−�H
o‡1
RT
)d�H
o‡1
RT dx(1 + C2 exp
(−�Ho
2
RT
))2
+2C1 exp
(−�H
o‡1
RT
)C2 exp
(−�Ho
2
RT
)d�Ho
2
RT dx(1 + C2 exp
(−�Ho
2
RT
))3 (11)
This relationship may be simplified to give:
d�Ho‡1
dx=
2C2 exp(
−�Ho2
RT
)d�Ho
2
dx(1 + C2 exp
(−�Ho
2
RT
)) = 2θAd�Ho
2
dx
(12)
Thus, for the optimal catalyst, the surface coverage bythe most abundant surface species is equal to:
θA =d�H
o‡1
dx
2d�Ho
2
dx
= ω‡1
2ω2(13)
where the values of ωi are defined as:
ω‡1 = d�H
o‡1
dx(14)
ω2 = d�Ho2
dx(15)
In the above derivation, we assume that ω‡1 and ω2
have the same sign, such that variations in x changethe enthalpy of the transition state and the MASI inthe same direction. We also assume that (d2�H
o‡1 )/dx2
and (d2�Ho‡2 )/dx2 are small or zero. This assumption
is valid if we are searching for improved catalysts overa small range of x, which typically occurs when testingcatalysts. In fact, when we vary x over a large range, thenthe mechanism of the catalytic reaction would probablychange.
References see page 14

ch1.1
8 1.1 Principles of Heterogeneous Catalysis
x (Fundamental catalyst parameter)
Inte
rmed
iate
ene
rgie
s
Rat
e
∆HMASI
∆H‡tran
Ratenetis maximum as∆H‡
tran → 0
Case 1: w‡1 >> w2
x (Fundamental catalyst parameter)
Inte
rmed
iate
ene
rgie
s
Rat
e
∆HMASI
∆H‡tran
Ratenet is maximum at w‡1 = 2qMASIw2
Case 2: w‡1 ≈ w2
x (Fundamental catalyst parameter)
Inte
rmed
iate
ene
rgie
s
Rat
e
∆HMASI
∆H‡tran
Ratenet is maximum at w‡1 = 2qMASIw2
Case 3: w‡1 << w2
Fig. 4 Reaction rates and energies of transition state and most abundant surface intermediate (MASI) as functions of fundamentalcatalyst parameter ‘‘x’’.
First, we will consider Case 1 shown in Fig. 4, whereω
‡1 � ω2. In this case, a maximum rate does not exist
and the parameter x should be adjusted to its lowestpossible value (i.e., the strongest bonding to the surface)which would decrease the enthalpy of the transitionstate as much as possible. In this case the optimalcatalyst operates at high surface coverage. We nowconsider Case 2, where ω
‡1 ≈ ω2; that is, x changes
the enthalpies of the transition state and the MASIby similar amounts. This situation is probably morephysically realistic, and if the MASI is in fact a reactiveintermediate (i.e., if it is a MARI), then the family ofcatalysts described by the variation of x follows theBrønsted–Evans–Polanyi–Semenov relationship, whichrelates the thermodynamics and kinetics of the system.Here, there is a clear maximum in the rate versus x,as shown in Fig. 4. The plot of rates versus x appearsas a volcano-type curve which decreases symmetricallyon both sides. In the case where ω
‡1 is equal to ω2, the
surface coverage by the MASI on the optimal catalyst isequal to 0.5. For Case 3, ω
‡1 � ω2, corresponding to the
situation where x changes the enthalpy of the MASI moresignificantly than the transition state. The optimal catalystin this case has a low surface coverage by the MASI. Amaximum rate occurs in this case, as shown in Fig. 4,provided that
∣∣ω‡1
∣∣ > 0; however, the plot of rate versus x
is not symmetric with respect to x. We note that as x
increases for these three cases, the number of vacant siteson the catalyst increases.
Cases 2 and 3 clearly illustrate the principle of Sabatier,in which a maximum rate occurs at some moderate levelof interaction of the catalyst surface with the intermediatesand adsorbed species. While Case 1 appears to contradictSabatier’s principle, the situation where ω
‡1 � ω2 is highly
unlikely. In particular, this situation corresponds to thecase where the catalyst interacts more strongly with thetransition state than with any of the reactive intermediates.However, if the activation energies for the elementarysteps of the mechanism are positive, then the reactantsand/or products of the elementary step involving the rate-controlling transition state are more strongly adsorbedon the surface than is the transition state, leading to thesituation described by Case 2 or 3.
We may generalize the above expression for catalystoptimality to the case where the surface contains severalabundant surfaces species, A∗, B∗, C∗, and D∗, leading tothe following expression:
ω‡1 = 2(ωAθA + ωBθB + ωCθC + ωDθD) (16)
In this case, the nature of the optimal catalyst iscontrolled by the change in the binding energy of thetransition state with respect to x (ω‡
1 ) compared to thechanges in the bindings energies of species A, B, C, and

ch1.1
1.1.6 Conditions for Catalyst Optimality 9
D (ωA, ωB, ωC, ωD) weighed by their respective surfacecoverages at the steady state.
A bridge between the thermodynamics and kineticsof a reaction is provided by the Brønsted–Evans–Pol-anyi–Semenov relationship, which states that there is alinear relationship between the activation energy Eact ofan elementary step and the heat of reaction if entropyeffects are neglected [38]:
Eact = E0 + α�H (17)
where �H is the enthalpy of reaction, α is the transfercoefficient that varies between zero and one, and E0is a constant. In other words, if we neglect entropiceffects, the activation energy of an elementary step inthe exothermic direction is lower when the heat ofreaction becomes more favorable (i.e., �H becomesmore negative). DFT calculations have recently shownthat Brønsted–Evans–Polanyi–Semenov relationshipsare generally upheld in chemical reactions on catalystsurfaces [61–64].
We now consider the following catalytic reaction:
A∗ −−−→ B∗ + C∗ (18)
If we use the gas phase A species as the zero energylevel (as shown in Fig. 5) we can define the activationenergy as:
Eact = E0 + α(HBgas + B.E.B + HCgas + B.E.C
− HAgas − B.E.A) = E0 + α(B.E.B
+ B.E.C − B.E.A) + α�Hgas (19)
where the B.E. terms are the binding energies of thevarious species on the surface.
Agas
Agas → Bgas + Cgas
0
B.E.A
A*
Bgas + Cgas
B* + C*
Eact
∆H‡
A*
B* + C*
a = 0
a = 1
Assume:dB.E.CdB.E.A dB.E.B
dx dx dx= =
Fig. 5 Schematic potential energy diagram of energy versusreaction coordinate, showing the relationship between the energyof transition state, �H �=, and changes in the energies of adsorbedspecies.
We now define the standard enthalpy for the formationof the transition state from gaseous species A as:
�Ho‡1 =Eact + B.E.A = E0 + α(�Hgas)
+ α(B.E.B + B.E.C) + (1 − α)B.E.A (20)
and we differentiate the enthalpy of the transition statewith respect to x, leading to the following result:
d�Ho‡1
dx= α
(dB.E.B
dx+ dB.E.C
dx
)+ (1 − α)
dB.E.A
dx(21)
This relationship shows how the change of thetransition state enthalpy with respect to x(ω
‡1 ) is related to
the changes in the binding energies B.E. of the adsorbedspecies (ω2). If α is equal to zero, then the change of thetransition state enthalpy depends only on the change ofthe binding energy of A, and ω
‡1 = ω2. In this case, the
transition state is an early transition state that chemicallylooks similar to A [65]. If α is equal to 1, then the transitionstate is a late transition state that resembles the productsof the reaction, and the change of the transition stateenthalpy is related to changes of the binding energies of Band C. If (dB.E.A)/dx = (dB.E.B)/dx = (dB.E.C)/dx,then the late transition state gives rise to the situationwhere ω
‡1 = 2ω2.
The above examples show how it is possible to maximizethe activity of the catalyst. However, it is often moreimportant to optimize the selectivity of the catalyst.Similar types of analyses can be carried out for these cases.In general, these situations are classified as being seriesselectivity challenges, such as A → B → C, where B isthe desired product, and/or parallel selectivity challenges,such as A → B coupled with A → C. In these cases, if wewant to optimize the selectivity of the catalyst, we searchfor some catalyst property, x, that decreases the enthalpyof the transition state for the desired reaction more thanit decreases the enthalpies of the transition states for theundesired reactions.
An undesired reaction that leads to progressiveblocking of surface sites leads to deactivation of thecatalyst with respect to time-on-stream. More generally,various mechanisms exist by which a catalyst canundergo deactivation, such as: (i) poisoning; (ii) thermaldegradation (sintering); (iii) leaching of the active site;and (iv) attrition [66]. The first three of these mechanismsare chemical in nature, whereas the last mechanismis physical (e.g., the catalyst pellet breaks apart). Somecatalysts do not show any measurable deactivation overperiods of years, such as in ammonia synthesis. However,other catalysts lose an important fraction of their activity
References see page 14

ch1.1
10 1.1 Principles of Heterogeneous Catalysis
after less than a minute of contact with feed, as in catalyticcracking. In the latter case, if deactivation is caused bycoking, the catalyst must be regenerated by continuousregeneration in an oxidizing atmosphere.
1.1.7Catalyst Design
Given that the performance of a catalyst is controlledby a limited number of kinetic parameters, it is unclearwhy it is so difficult to design a catalyst from molecular-level concepts. As noted above, during the early stagesof research into a catalytic process, first we do not knowwhich steps in the reaction mechanism are kineticallysignificant, and which species are most abundant on thecatalyst surface under reaction conditions. Second, wedo not often know the structure of the active site andits dependence on the nature of the reaction conditions.Third, we do not usually know how the activity andselectivity for the catalytic reaction depend on the structureof the active sites. Fourth, we do not typically know duringthese early stages the rates of various modes of catalystdeactivation (e.g., sintering, phase changes, depositionof carbonaceous deposits on the surface, etc.), and wedo not know whether the catalyst can be regeneratedfollowing deactivation. Finally, we must ensure that thetexture of the catalyst and the geometry of the reactor aredesigned in such a way that mass transport of reactantsand products to and from the active sites is sufficientlyrapid that high rates of reaction per unit volume of reactorcan be achieved.
Because of these difficulties, the field of heteroge-neous catalysis is highly interdisciplinary in nature, and
involves close collaboration between experts in such areasas catalyst synthesis, catalyst characterization, surfacespectroscopy, chemical kinetics, chemical reaction engi-neering and, most recently, in theoretical calculations ofcatalyst structure and performance using density func-tion theory. These broad studies can be grouped intothree levels, as shown in Fig. 6.
All studies of heterogeneous catalysis begin at theMaterials Level. High-surface area catalytic materialsmust be synthesized with specific structures and textures,the latter referring to such features as the sizes ofthe various phase domains and the details of the porestructure. Clearly, the synthesis of catalytic materialsmust be guided by detailed characterization studies todetermine the structures, compositions, and texturesof the materials that have been prepared. Thesecharacterization studies should be conducted after thecatalyst has been subjected to various treatment steps(such as those treatments employed during activation ofthe catalytic material), and it is most desirable to carryout characterization studies of the catalyst under theactual reaction conditions of the catalytic process. Indeed,the properties of a heterogeneous catalyst are inherentlydynamic in nature, and these properties often changedramatically with changes in the reaction conditions(e.g., phase changes, surface reconstructions, changesin surface versus bulk composition, etc.) [67].
The central level of research and development of het-erogeneous catalysts involves the quantification of catalystperformance (this is known as the Catalyst PerformanceLevel). These studies can be carried out in a prelimi-nary fashion over a wide range of catalytic materials (e.g.,high-throughput studies) to identify promising catalysts
Experimental studies:interactions of probe
molecules withwell-defined sites
Theoretical studies:stability & reactivity
of species onwell-defined sites
Exploratory studies:promising leads for newcatalytic materials & new
catalytic reactions
Materials synthesis:catalytic materials
with specific structures& textures
Characterization studies:catalyst structure, composition,
& texture, (ideally underreaction conditions)
Reaction kinetics studies:activity, selectivity &stability for variousreaction conditions
Surface studies:surface composition and natureof surface sites, (ideally under
reaction conditions)
Materials level
Catalyst performance level
Elucidation level
Fig. 6 Levels of study in heterogeneous catalysis research.

ch1.1
1.1.8 Catalyst Development 11
and reaction conditions for further studies. The perfor-mance of the catalyst is then documented in greater detailby determining catalytic activity, selectivity, and stabilitywith respect to time-on-stream for various reaction con-ditions. These measurements must be made at variousconversions when multiple reaction pathways exist, be-cause catalytic selectivities in these cases are different,depending on whether the desired products are formedin primary versus secondary reactions, or in series versusparallel pathways. We note here that various definitionsof catalytic activity are used, depending on the nature ofthe study. For practical studies, catalytic activities can bereported as rates per gram of catalyst or per unit surfacearea. However, for more detailed studies or for researchpurposes, it is often desirable to report catalytic activitiesas rates per surface site (i.e., TOFs), with the number ofsurface sites measured most often by selective adsorptionmeasurements (e.g., adsorption of H2 or CO to titratemetal sites, adsorption of ammonia or pyridine to titrateacid sites). In some cases it is possible to report catalyticactivity as rate per active site (also called TOF), when it ispossible to distinguish active sites from the larger num-ber of surface sites using special probe molecules (e.g.,dissociative adsorption of N2 to titrate sites for ammo-nia synthesis [68]; selective poisoning by adsorbates thatcompete with the reactants of the catalytic reaction [69]);or by transient isotopic tracing [70].
For the purposes of catalyst development, it is prob-ably sufficient to work at the Materials Level and theCatalyst Performance Level. However, research into het-erogeneous catalysis is dominated by studies conductedat a third level – the Elucidation Level – where the aimis to identify the fundamental building blocks of knowl-edge which can be assembled to build a molecular-levelunderstanding of catalyst performance in order to guidefurther investigations to improve catalyst performance.At the Elucidation Level the studies are designed to deter-mine the surface composition and nature of the surfacesites on the catalyst [71–74]. Clearly, these investigationsmust be conducted with the catalyst under controlled con-ditions (e.g., under ultra-high vacuum, after treatmentwith H2, after calcination, etc.) and, where possible, suchmeasurements should be made with the catalyst underreaction conditions. Moreover, the studies may be carriedout on real catalytic materials and on more well-definedsurfaces (e.g., single crystals, or model samples formedby depositing known amounts of materials onto well-defined supports) [73, 75, 76]. Most measurements at theElucidation Level involve studies of the interactions ofspecific probe molecules with the catalyst surface. Theseprobe molecules may be the reactants, intermediates, orproducts of the catalytic reaction, or they may be moresimple species chosen to monitor a specific functionalityof the surface. Alternatively, a molecule may be used as
a probe because it has an advantageous feature for spec-troscopic identification (e.g., CO for infrared studies, a13C-containing molecule for NMR studies). These studiesof the interaction of probe molecules with surfaces aredesigned to determine the surface concentrations of dif-ferent types of surface site, to determine the nature of theadsorbed species formed on the surface sites, and to de-termine the reactivities of the surface sites by monitoringthe adsorbed species on the surface versus time, versustemperature or, most commonly, during a temperatureramp (e.g., temperature-programmed desorption).
The third pillar of studies at the Elucidation Levelinvolves the use of DFT calculations to assess thestructures, stabilities, and reactivity of species adsorbedonto the surface sites (with the sites being composed ofclusters of atoms or as periodic slabs of atoms) [77–81].These studies are used to help interpret the resultsobtained from spectroscopic studies of catalyst surfaces(e.g., to predict the vibrational spectra of species adsorbedin different orientations on different sites), to calculateheats of adsorption for various intermediates in a reactionmechanism (e.g., to predict which species are expected tobe abundant on the catalyst under reaction conditions),to estimate the energy changes for possible steps in areaction mechanism (thereby eliminating from furtherconsideration steps with very positive energy changes),and to determine activation energy barriers for stepsthat are suspected as being kinetically significant in thereaction scheme. Indeed, a key feature of these theoreticalstudies is the ability to predict how the surface propertiesare expected to change as the nature of the surface isaltered (e.g., by changing the surface structure, or byadding possible promoters). This in turn will providefeedback to the Materials Level with regards to newmaterials that should be synthesized and which are likelyto lead to an improved catalyst performance. In addition,these theoretically based studies provide informationabout highly reactive intermediates which might bedifficult to obtain by direct experimental measurements.Most importantly, studies conducted at the ElucidationLevel provide a scientific basis about the working catalysisthat may, in future, be used to design different reactionpathways.
1.1.8Catalyst Development
Catalyst development typically involves testing a largenumber of catalysts with a feedback loop, as it is currentlydifficult to design catalysts a priori. In this respect, catalystdevelopment studies involve examining a large numberof catalysts, for which recent advances in high-throughput
References see page 14

ch1.1
12 1.1 Principles of Heterogeneous Catalysis
testing have attracted considerable attention [82–87].Catalyst development through the testing of a wide rangeof materials was first practiced in 1909 by Mittasch atBASF who, according to Timm [88], issued the followingdirective to his team who at the time were developing thesynthesis of ammonia:
• The search for a suitable catalyst necessitates carryingout experiments with a number of elements, togetherwith numerous additives.
• The catalytic substances must be tested at highpressures and temperatures, just as in the case ofHaber’s experiments.
• A very large number of tests will be required.
Ten years later, the number of tests conducted hadexceeded 10 000, and more than 4000 catalysts had beenstudied. This extraordinary effort was also extraordinarilysuccessful. What has changed since then, however, isthe way in which the systematic search is assisted.Today, armed with an arsenal of principles, concepts,instrumentation and computers, it is possible to identifyand to improve new catalytic materials in a muchshorter time and with a smaller number of trial samples,especially with the possibility of advanced characterizationmethods (especially in-situ techniques) and insights fromtheoretical calculations (e.g., DFT calculations). Thepractical merit of this ‘‘assisted catalyst design’’ is clear,while its scientific dividend is the possibility of learningas the design proceeds, with the building of a data bankof rate constants and the formulation of more precisemodels of active sites. With new theoretical insightsor principles, quantitative bases of catalyst preparationand reproducibility of catalyst behavior, the future ofheterogeneous catalysis still looks very bright.
The path to the design of an optimal catalytic processwould be clear if the activity, selectivity and stabilityof the catalyst were to move in the same directionupon an increase in a single process variable, suchas temperature. However, this simple behavior is nottypically observed, and choices must be made in everyinstance. For example, while the activity of a catalystmay increase with temperature, its stability usuallydecreases with temperature. In addition, the relationshipbetween catalytic activity and selectivity is typically verycomplex, and is not understood in detail until the surfacechemistry of the catalytic process has been elucidated.Accordingly, selectivity, stability and activity must beconsidered together, and trade-offs may have to benegotiated, perhaps by using multi-functional reactorswith catalytic distillation or catalytic membranes. Successin heterogeneous catalysis begins with chemistry, butends with catalytic reaction engineering.
1.1.9Bridging Gaps in Heterogeneous Catalysis
The above description of research and development intoheterogeneous catalysis as being interdisciplinary in na-ture, involving studies at the levels of materials, catalystperformance and elucidation, can also be cast in the formof building bridges between various types of studies anddifferent types of material. As depicted in Fig. 7, we oftentalk about bringing together the field of surface science(which traditionally is focused on studies of single crystalsurfaces at low pressures) with the field of heterogeneouscatalysis (which traditionally is focused on studies ofhigh-surface area catalytic materials surfaces under high-pressure reaction conditions). More recently, we havetalked about ‘‘bridging the materials gap’’, as we haveattempted to use experimental results from studies of well-defined model materials to interpret the performance ofmore complex, high-surface area catalytic materials. Tra-ditionally, these model materials have been single crystals,cut at various angles to expose surfaces containing differ-ent types of sites, such as surfaces with different symme-tries and atoms present at terraces, steps, and kinks [89].More recently, however, these model materials have be-come highly sophisticated, such as the deposition ofnanoparticles with specific sizes and geometries on well-defined support surfaces (e.g., metal nanoparticles sup-ported on thin films of oxides deposited on single crystalmetal surfaces, or non-metallic nanoparticles supporteddirectly on single crystal metal surfaces) [73, 75, 76]. Wealso talk about ‘‘bridging the pressure gap’’, as we at-tempt to use experimental results from studies conductedat low pressures (less than 10−6 Torr) to interpret theperformance of catalysts under high-pressure reaction
Surfacescience
Heterogeneouscatalysis
Mat
eria
ls g
ap
Pressure gap High PLow P
Low m2
High m2
Fig. 7 Bridging the gap between surface science and heteroge-neous catalysis.

ch1.1
1.1.10 A Philosophical Note 13
conditions. The origin for this pressure gap comes fromthe fact that, whereas some spectroscopic techniques canbe employed to study the surface and bulk properties ofcatalysts under high-pressure reaction conditions (e.g.,FTIR, Raman, XRD, EXAFS, Mossbauer spectroscopy),other spectroscopic and characterization techniques (e.g.,XPS, TEM) are most easily conducted with the sampleat low pressures (e.g., <10−6 Torr) [90]. These latter tech-niques are typically associated with use of electrons toprobe the sample, with the electrons interacting stronglywith molecules in the gas phase. This pressure gap can bebridged directly by designing advanced instrumentation,such that the distance traversed by the electrons in thegas phase is minimized [91, 92]. In addition, the pressuregap can be bridged indirectly by using molecular-basedmodels (e.g., kinetic Monte Carlo calculations, micro-kinetic models), first to describe the experimental resultsobtained at low pressures, and then to extrapolate thisinformation to high-pressure reaction conditions.
The past few years have witnessed an explosion inthe area of nanotechnology, in which researchers havelearned – and are continuing to learn – how to engineermaterials at the nanometer length scale. The field of het-erogeneous catalysis has been involved in the synthesisof nanomaterials for many years (e.g., the synthesis ofzeolites). Indeed, the scheme depicted in Fig. 6 showsthat essentially all studies of heterogeneous catalysisbegin at the Materials Level. Recent advances in nan-otechnology offer new routes for catalyst synthesis (e.g.,atomic layer deposition, self-assembly methods) [93–96]and, importantly, also for catalyst characterization (e.g.,
techniques such as scanning tunneling microscopy thatallow atomic-scale imaging of materials at elevated tem-peratures and pressures) [73, 97]. However, as an asincreasing number of research groups become involvedin nanotechnology, it is possible that an ‘‘applicationsgap’’ will be created in heterogeneous catalysis, wherenew materials are formed without clear applications forcatalytic processes. Clearly, this gap can be bridged by re-alizing that research and development into heterogeneouscatalysis involves the combination of studies at the levelsof materials, catalyst performance, and elucidation. As ad-vances in nanotechnology allow us to create new materials(the Materials Level) and to characterize these materialsin greater detail (Materials and Elucidation Levels), weare positioned to take full advantage of these advances byconducting studies at the Catalyst Performance Level.
1.1.10A Philosophical Note
Today, we live in an era in which it is possible to employa vast range of advanced experimental techniques andtheoretical methods to elucidate in detail the surfacechemistry of catalytic processes. This situation, withrespect to the hierarchy of theoretical methods thatcan be employed to describe the reaction kinetics fora catalytic process, is illustrated schematically in Fig. 8. Atthe lowest level, we use empirical rate expressions to fitreaction kinetics data over a range of process conditions;
References see page 14
Empirical rate expressions
Simplified rate expressions basedon assumed surface chemistry
Micro-kinetic rate expressions basedon results from DFT calculations
Kinetic monte-carlo analyses basedon results from DFT calculationsns
Quantum molecular dynamics calculationsfor surface reactions
Future methods …..
Desire to know more about surface chemistry
Suf
ficie
nt in
sigh
t gai
ned
to g
uide
the
sear
ch fo
rne
w c
atal
ytic
pro
cess
es
Elucidation well
Discovery
Elu
cida
tion
Fig. 8 The catalyst ‘‘elucidation well’’ and catalyst discovery.

ch1.1
14 1.1 Principles of Heterogeneous Catalysis
however, these models typically have questionable successin predicting catalyst performance outside the range ofexperimental conditions used to fit the model.
We then move to rate expressions based on assumedsurface chemistry. These models should have a betterpredictive value, although it is often difficult to determinewhich types of assumption should be made. Accordingly,we turn to results from DFT calculations to build micro-kinetic models that describe catalyst performance withoutthe need to make prior assumptions about which steps arekinetically significant and which species are abundant onthe catalyst surface. These micro-kinetic models, however,are typically based on the mean-field approximation, andthey thus make simplified assumptions about (or neglect)the effects of surface coverage and lateral interactionsbetween adsorbed species. Accordingly, these restrictiveassumptions are relaxed when using kinetic Monte Carlomethods to describe reaction kinetics based on bindingenergies and lateral interaction terms determined fromDFT calculations. Indeed, today’s research groups arebeginning to combine quantum mechanics and moleculardynamics calculations to describe a variety of surfaceprocesses. Who knows what new computational methodsare on the horizon?
The sequential use of the aforementioned methods todescribe reaction kinetics in greater detail is depictedin Fig. 8, as the digging of an ‘‘elucidation well’’. Aswe dig deeper by using more sophisticated methods, welearn more about the details of the surface chemistry.Indeed, we are driven to dig deeper by our desire to learnas much as possible about the fundamental principlesthat control catalyst performance. However, this desireto know as much as possible must be balanced by ourneed to discover new catalysts and new catalytic processes.Clearly, as we learn more about the fundamentals of thecatalytic process (i.e., as we dig deeper), we should havebetter insight to guide our search for better catalysts.Luckily, we need not dig to the deepest levels to begin thesearch for better catalysts. As noted at the beginningof this chapter, our industrialized society is facingincreasingly complex environmental and energy issuesfor sustained growth. Thus, while our scientific curiosityfor fundamental knowledge drives us to dig deeper towarddetailed elucidation of catalytic phenomena, we must alsocontinue to look horizontally as we use our newfoundinsight to develop new catalysts and catalytic processesfor the benefit of society.
List of Abbreviations
DFT density functional theoryEXAFS extended X-ray absorption fine structureFTIR Fourier transform infrared
STY site time yieldTEM transmission electron microscopyTOF turnover frequencyTOR turnover rateXPS X-ray photoelectron spectroscopyXRD X-ray diffraction
References
1. J. M. Thomas, W. J. Thomas, Principles and Practice of Hetero-geneous Catalysis, VCH, Weinheim, 1997, 669 pp.
2. J. N. Armor, Appl. Catal., A 2001, 222, 407.3. I. Chorkendorff, J. W. Niemantsverdriet, Concepts of Mod-
ern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2003,452 pp.
4. R. J. Farrauto, C. H. Bartholomew, Fundamentals of Indus-trial Catalytic Processes, Chapman & Hall, London, 1997,754 pp.
5. R. A. van Santen, P. W. N. M. v. Leeuwen, J. A. Mooulijn,B. A. Averill, Catalysis: An Integrated Approach, ElsevierScience B.V., Amsterdam, 1999, 574 pp.
6. National Research Council Panel on New Directions inCatalytic Sciences and Technology, Catalysis Looks to the Future,National Academy Press, Washington D.C., 1992, p. 1.
7. W. Vielstich, A. Lamm, H. Gasteiger, Handbook of Fuel Cells:Fundamentals, Technology, Applications, Wiley, Chichester,2003, 2690 pp.
8. S. Park, J. M. Vohs, R. J. Gorte, Nature 2000, 404, 265.9. S. Ha, R. Larsen, R. I. Masel, J. Power Sources 2005, 144, 28.
10. A. Corma, H. Garcia, Chem. Rev. 2003, 103, 4307.11. G. Centi, Catal. Today 2003, 77, 287.12. R. A. Sheldon, Green Chem. 2005, 7, 267.13. A. Borgna, L. Balzano, J. E. Herrera, W. E. Alvarez, D. E.
Resasco, J. Catal. 2001, 204, 131.14. H. van Bekkum, P. Gallezot, Top. Catal. 2004, 27, 1.15. G. W. Huber, J. W. Shabaker, J. A. Dumesic, Science 2003,
300, 2075.16. G. W. Huber, J. N. Chheda, C. J. Barrett, J. A. Dumesic, Sci-
ence 2005, 308, 1446.17. I. K. Mbaraka, B. H. Shanks, J. Catal. 2005, 229, 365.18. S. Varadarajan, D. J. Miller, Biotechnol. Progr. 1999, 15, 845.19. E. K. Rideal, H. S. Taylor, Catalysis in Theory and Practice,
Macmillan, London, 1919, Chapter 2.20. M. Boudart, in Perspectives in Catalysis, J. M. Thomas,
K. I. Zamaraev (Eds.), Blackwell, Oxford, 1992, p. 183.21. G. A. Somorjai, Introduction to Surface Chemistry and Catalysis,
John Wiley, New York, 1994, 667 pp.22. H. S. Taylor, Proc. Roy. Soc. (London) 1925, A108, 105.23. J. B. Anderson, Kagaku Kogaku (Chem. Eng. Jpn.) 1962, 147,
191.24. J. J. Carberry, AICHE 1961, 7, 350.25. G. F. Froment, K. B. Bischoff, Chemical Reactor Analysis and
Design, Wiley, New York, 1990, 664 pp.26. C. N. Satterfield, Mass Transfer in Heterogeneous Catalysis, MIT
Press, Cambridge, MA, 1970, 267 pp.27. D. E. Mears, Ind. Eng. Chem. Proc. Des. Dev. 1971, 10, 541.28. D. E. Mears, J. Catal. 1971, 20, 127.29. J. M. Smith, J. Chem. Eng. Japan 1973, 6, 191.30. P. B. Weisz, Z. Phys. Chem. 1954, 11, 1.31. P. B. Weisz, C. D. Prater, Adv. Catal. 1957, 6, 143.

ch1.1
References 15
32. R. Aris, in Catalyst Design: Progress and Perspectives,L. L. Hegedus (Ed.), Wiley Interscience, New York, 1987,Chapter 7.
33. P. A. Jacobs, J. A. Martens, J. Weitkamp, H. K. Beyer, FaradayDiscuss. Chem. Soc. 1981, 72, 353.
34. W. O. Haag, R. M. Lego, P. B. Weisz, Faraday Discuss. Chem.Soc. 1981, 72, 317.
35. E. G. Derouane, P. Dejaifve, Z. Gabelica, Faraday Discuss.Chem. Soc. 1981, 72, 331.
36. J. M. Thomas, G. R. Millward, S. Ramdas, L. A. Busil,M. Audier, Faraday Discuss. Chem. Soc. 1981, 72, 345.
37. P. Sabatier, La catalyse en chimie organique, Berange, Paris,1920, 388 pp.
38. M. Boudart, Kinetics of Chemical Processes, Blackwell, Oxford,Stoneham, MA, 1991, 246 pp.
39. J. V. Lauritsen, M. Nyberg, J. K. Norskøv, J. Catal. 2004, 224,94.
40. B. Hinnemann, J. K. Norskøv, H. Topsøe, J. Phys. Chem. B2005, 109, 2245.
41. H. H. Hwu, J. G. Chen, Chem. Rev. 2005, 105, 185.42. R. B. Levy, M. Boudart, Science 1973, 181, 547.43. M. K. Neylon, S. Choi, H. Kwon, K. E. Curry, L. T. Thompson,
Appl. Catal., A 1999, 183, 253.44. S. T. Oyama, Catal. Today 1992, 15, 179.45. S. G. Podkolzin, J. Shen, J. J. D. Pablo, J. A. Dumesic, J. Phys.
Chem. B 2000, 104, 4169.46. R. M. Watwe, B. E. Spiewak, R. D. Cortright, J. A. Dumesic,
Catal. Lett. 1998, 51, 139.47. C. G. M. Hermse, A. P. van Bave, A. P. J. Jansen, L. A. M. M.
Barbosa, P. Sautet, R. A. van Santen, J. Phys. Chem. B 2004,108, 11035.
48. M. Neurock, S. A. Wasileski, D. Mei, Chem. Eng. Sci. 2004, 59,4703.
49. S. Raimondeau, P. Aghalayam, A. B. Mhadeshwar, D. G.Vlachos, Ind. Eng. Chem. Res. 2003, 42, 1174.
50. F. J. Garcia, E. E. Wolf, Chem. Eng. Sci. 2004, 59, 4723.51. J. M. Thomas, C. R. A. Catlow, G. Sankar, Chem. Commun.
2002, 2921.52. A. A. Gokhale, Water–Gas Shift Reaction and Fischer–Tropsch
Synthesis on Transition Metal Surfaces, Phd thesis, Universityof Wisconsin, 2005.
53. P. S. Cremer, X. Su, Y. R. Shen, G. A. Somorjai, J. Am. Chem.Soc. 1996, 118, 2942.
54. H. Topsøe, J. Catal. 2003, 216, 155.55. I. E. Wachs, Catal. Today 1996, 27, 437.56. J. A. Dumesic, J. Catal. 1999, 185, 496.57. O. Beeck, Disc. Faraday Soc. 1950, 8, 118.58. W. J. M. Rootsaert, W. M. H. Sachtler, Z. Physik. Chem. 1960,
26, 16.59. J. H. Sinfelt, Bimetallic Catalysts, Wiley, New York, 1983 p. 14.60. M. Boudart, J. Am. Chem. Soc. 1950, 72, 1050.61. T. Bligaard, J. K. Norskøv, S. Dahl, J. Matthiesen, C. H.
Christensen, J. Sehested, J. Catal. 2004, 224, 206.62. J. K. Norskøv, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen,
M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M.Mavrikakis, Y. Xu, S. Dahl, C. J. H. Jacobsen, J. Catal. 2002,209, 275.
63. V. Pallassana, M. Neurock, J. Catal. 2000, 191, 301.64. Z. P. Liu, P. J. Hu, J. Chem. Phys. 2001, 114, 8244.65. R. A. van Santen, J. W. Niemantsverdriet, Chemical Kinetics
and Catalysis, Plenum Press, New York, 1995, p. 233.66. C. H. Bartholomew, Appl. Catal., A 2001, 212, 17.67. G. A. Somorjai, Annu. Rev. Phys. Chem. 1994, 45, 721.
68. H. Topsøe, J. A. Dumesic, N. Topsøe, H. Bohlbro, inProceedings of the Seventh International Congress in Catalysis,T. Seiyama, K. Tanabe (Eds.), Elsevier, Amsterdam, 1981,p. 247.
69. H. Knozinger, Adv. Catal. 1976, 25, 183.70. J. G. Goodwin Jr., S. Kim, W. D. Rhodes, Catal. 2004, 17, 320.71. D. W. Goodman, J. Catal. 2003, 216, 213.72. G. A. Somorjai, K. R. McCrea, J. Zhu, Top. Catal. 2002, 18,
157.73. H.-J. Freund, M. Baumer, J. Libuda, T. Risse, G. Rupprechter,
S. Shaikhutdinov, J. Catal. 2003, 216, 223.74. G. Ertl, J. Mol. Catal. A: Chem. 2002, 182–183, 5.75. J. V. Lauritsen, M. Nyberg, J. K. Norskov, B. S. Clausen,
H. Topsoe, E. Laegsgaard, F. Besenbacher, J. Catal. 2004,224, 94.
76. M. S. Chen, D. W. Goodman, Science 2004, 306, 252.77. J. Greeley, J. K. Norskøv, M. Mavrikakis, Annu. Rev. Phys.
Chem. 2002, 53, 319.78. M. Neurock, J. Catal. 2003, 216, 73.79. B. Hammer, J. K. Norskøv, Adv. Catal. 2000, 45, 71.80. S. Linic, H. Piao, K. Adib, M. A. Barteau, Angew. Chem. Int.
Ed. 2004, 43, 2918.81. M. T. M. Koper, R. A. van Santen, M. Neurock, in Catalysis
and Electrocatalysis at Nanoparticle Surfaces, A. Wieckowski,E. R. Savinova, C. G. Vayenas (Eds.), Marcel Dekker, Inc., NewYork, 2003, p. 1.
82. A. Hagemeyer, P. Strasser, J. Anthony, F. Volpe, High-Throughput Screening in Chemical Catalysis: Technologies,Strategies and Applications, Wiley-VCH Verlag, Weinheim,Germany, 2004, 339 pp.
83. A. Hagemeyer, R. Borade, P. Desrosiers, S. Guan, D. M. Lowe,D. M. Poojary, H. Turner, H. Weinberg, X. Zhou, R. Armbrust,G. Fengler, U. Notheis, Appl. Catal., A 2002, 227, 43.
84. R. J. Hendershot, C. M. Snively, J. Lauterbach, Chem. Eur. J.2005, 11, 806.
85. J. M. Serra, E. Guillon, A. Corma, J. Catal. 2005, 232, 342.86. S. Senkan, Angew. Chem. Int. Ed. 2001, 40, 312.87. J. W. Saalfrank, W. F. Maier, Angew. Chem. Int. Ed. 2004, 43,
2028.88. B. Timm, Proceedings of the 8th International Congress on
Catalysis Berlin 1984, Verlag Chemie, Frankfurt, 1984, Vol. I,p. 7.
89. P. L. J. Gunter, J. W. Niemantsverdriet, F. H. Ribeiro, G. A.Somorjai, Cat. Rev. - Sci. Eng. 1997, 39, 77.
90. J. W. Niemantsverdriet, Spectroscopy in Catalysis, Wiley, Wein-heim, Germany, 1993, 288 pp.
91. P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen,B. S. Clausen, H. Topsøe, Science 2002, 295, 2053.
92. D. Teschner, A. Pestryakov, E. Kleimenov, M. Haevecker,H. Bluhm, H. Sauer, A. Knop-Gericke, R. Schloegl, J. Catal.2005, 230, 186.
93. A. Corma, F. Rey, J. Rius, M. J. Sabater, S. Valencia, Nature2004, 431, 287.
94. Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A.Somorjai, A. P. Alivisatos, Science 2004, 304, 711.
95. M. J. Pellin, P. C. Stair, G. Xiong, J. W. Elam, J. Birrell,L. Curtiss, S. M. George, C. Y. Han, L. Iton, H. Kung, M.Kung, H. H. Wang, Catal. Lett. 2005, 102, 127.
96. A. Gervasini, P. Carniti, J. Keranen, L. Niinisto, A. Auroux,Catal. Today 2004, 96, 187.
97. F. Besenbacher, E. Laegsgaard, I. Stensgaard, Mater. Today2005, 8, 26.

16 1.2 Development of the Science of Catalysis
1.2Development of the Science of Catalysis
Burtron H. Davis∗
1.2.1Early Concepts: Berzelius, Liebig, and Faraday
Prior to the introduction of the concept by Berzeliusduring the period of 1835–1836, catalysis was anexperimental fact and a subject of much debate. Thefirst application of catalysis, to produce ethanol byfermentation, is lost to antiquity. However, by the middleages the use of sulfuric acid to catalyze the production ofdiethyl ether was widespread and indeed, written recordsof this synthesis date back to 1552 [1].
The reception by the scientific community of theconcept of catalysis is well illustrated by the studiesof Mitscherlich and Berzelius. In 1834, Mitscherlichreported that when alcohol was run into dilute sulfuricacid at 140 ◦C, ether and water could be distilled fromthe mixture [2]. He extended his observations by statingthat decompositions and combinations of this kind werevery frequent. Mitscherlich introduced the term ‘‘contact’’to describe these actions, and summarized a number ofreactions that were caused by contact – the formationof ether, the oxidation of ethanol to acetic acid, thefermentation of sugar, the production of sugar fromstarch by boiling sulfuric acid, the hydrolysis of ethylacetate by alkali, and the formation of ethene from ethanolby heating with acid.
Berzelius, from 1821 on, summarized and reviewedcritically the scientific investigations conducted world-wide in his ‘‘Annual Report’’ (Jahresberichte) [3]. Thegeneralizations in Berzelius’ reviews added as much ormore to his reputation as his own discoveries, and thesewere many and important. In his annual review of 1835,Berzelius covered a number of reactions which take placein the presence of a substance which remains unaffected.Some roots of catalysis are provided in Fig. 1, whichemphasizes those considered by Berzelius; a number ofadditional examples of reactions that predate Berzelius’definition can be found elsewhere [4]. Trofast [3] presentsan English language version of Berzelius’ conclusions:
‘‘This is a new power to produce chemical activity belonging toboth inorganic and organic nature, which is surely morewidespread than we have hitherto believed and the nature of whichis still concealed from us. When I call it a new power, I do notmean to imply that it is a capacity independent of theelectrochemical properties of the substance. On the contrary, I amunable to suppose that this is anything other than a kind of special
∗ Corresponding author.
manifestation of these, but as long as we are unable to discovertheir mutual relationship, it will simplify our researches to regardit as a separate power for the time being. It will also make it easierfor us to refer to it if it possesses a name of its own. I shall therefore,using a derivation well-known in chemistry, call it the catalyticpower of the substances, and the decomposition by means of thispower catalysis, just as we use the word analysis to denote theseparation of the component parts of bodies by means of ordinarychemical forces. Catalytic power actually means that substancesare able to awaken affinities which are asleep at this temperatureby their mere presence and not by their own affinity.’’
Thus, although the same concept was put forth fromtwo perspectives within a two-year period, one perspectivehad a much more significant and lasting impact. Theconcepts of Berzelius attracted much criticism thatwas directed, for the most part, not at catalysis, butrather at the concept of catalytic force. Berzelius hastherefore been credited with introducing the concept ofcatalysis, even though one could easily conclude thatMitscherlich predated him by two years. Mitscherlich wasnot completely ignored, however, since ‘‘contact catalysis’’was utilized into the middle of the 20th century.
The theories which were advanced during the 19th cen-tury to explain the mechanism of catalysis may be groupedinto three classes: the chemical, vibrational, and physical.Proponents of the chemical theory view the catalyst tooperate through the continuous formation and decompo-sition of unstable intermediate products. The vibrationtheory was applied especially to fermentation, and was atits zenith under the leadership of Liebig. The ferment (en-zyme) was then viewed to possess a particular internal mo-tion that could be transmitted to neighboring molecules,and thereby induce reactions of them. The physical theoryexplains the phenomena as being due to the condensa-tion, and the increase in concentration, of the reactingsubstances at the surface of the catalyst, such increase inconcentration being brought about by capillary forces.
It has long been recognized that a catalyst has thesame chemical composition at the end of the reactionthat it effected; however, it has been equally recognizedthat its physical state may have undergone a profoundalteration [5]. Thus, when ammonia is decomposed bycontact with heated metals there is nearly always analteration of the physical state of the metal, and thishas been attributed to the formation and decompositionof intermediate compounds, metallic nitrides in thisparticular case. While these views are obvious today,it would be a demanding task to trace their history ‘‘to theoriginal statement of the concept’’.
Whereas Berzelius defined ‘‘catalysis’’, ‘‘catalyticforce’’, and ‘‘catalytic action’’, it remained for Arm-strong [6] to define catalyst:
‘‘. . . so little has been done to ascertain the nature of the influenceof the contact-substance, or catalyst, as I would term it, the main

1.2.1 Early Concepts: Berzelius, Liebig, and Faraday 17
The roots
CATALYSIS
Payen & persoz (1833)isolate ferment
(enzyme)
Dobereiner (1820's)
Dobereiner (1808) Dobereiner (1823)
EtOH
EtOH
HAc
Starch
Starch
Starch
Kirchoff (1811)
Sugar
Porcelain
Fe, Pt, etc.
Sugar
H+
H+
H+Parmentier (1781)
Thenard (1813)
2 NH3 EtOET + H2O
H. Davy (∼1800)CH4 + Air (Pt)
MeOH + AirEtOH + EtOHH2 + AirPt Catalyst
H2 + Air Pt
DuLong & Thenard (1824)
2 EtOH EtOET + H2O
Mitscherlich (1835)
Berzelius (1835/36)
Fig. 1 Schematic representation of some of the studies that serve as a background for the formulation of the concept of catalysis byBerzelius.
object in view being the study of the product of the reaction, thatthe importance of the catalyst is not duly appreciated.’’
This is an outstanding example of an accurate historicalfact, and a presentation, on the one hand, of an obviousdefinition, and on the other hand, an insightful statementof needs for future research.
Adsorption is at the heart of any heterogeneously cat-alyzed reaction. In 1800, adsorption was absorption, andscientists were aware of it. The concepts of adsorption areillustrated in an admirably lucid account of its status at thattime by de Saussure in a presentation to the Geneva Soci-ety on April 16, 1812 [7]. The questions that de Saussureasked in the introduction of his paper are equally valid fora manuscript that is being written today. He begins witha statement that no accurate experimental data were avail-able on the question of whether a gas, when it penetratesinto the pores of a solid body, undergoes any diminutionof bulk (volume) when no chemical bonding takes placebetween the gas and the solid. de Saussure, assuming thatsuch contraction of volume does take place, is led to a num-ber of insightful questions: What influence has the sizeof the pores on this condensation? Are all gases equallycondensed by the same bodies? What influence has thedensity of the gas on this condensation? When equal quan-tities of two gases are in contact with a solid, do they adsorbin equal quantities? Do the mixed gases, when condensed
in the solid, enter into combinations which they wouldnot form in the free state? He understood the need forcareful experimental results. Also, he clearly outlined anddefined many scientific aspects of adsorption.
de Saussure confirmed that adsorption in all cases,except for oxygen, was complete at the end of 24 to36 hours. He also demonstrated that the adsorptionof a gas is exothermic, liberating a quantity of heatduring the condensation that is often sensible to thefeel, and sufficient to raise by several degrees thetemperature indicated by a thermometer in contact withthe solid (charcoal). Aware that condensation of a gaswas exothermic and evaporation was endothermic, deSaussure speculated that the solid should become coolerduring evaporation, and demonstrated experimentallythat this was indeed the case.
de Saussure speculated that charcoal should be acti-vated by evacuation as well as by heating, and showedthis to be the case. He compared the amount adsorbed onthe whole and ground charcoal. The pulverized sampleadsorbed less gas, and de Saussure attributed this to theamount of preadsorbed water. The rate of adsorption ofdifferent gases was reported to be the same in all solidsof similar chemical properties, even though the amountsof gases condensed in the various solids may vary greatly.
References see page 35

18 1.2 Development of the Science of Catalysis
de Saussure observed that exposure of a solid saturatedwith one gas to a second gas would cause adsorption of thesecond gas, resulting in the desorption of some fractionof the first gas.
He stated that the affinity of the gas and the solid, aswell as the condensibility of the gas, must be consideredin addition to the porosity of the solid. Furthermore, theforces of affinity and of condensibility act to oppose eachother. de Saussure considered the condensation of gasesin solid bodies to be analogous with the rise of liquids incapillary tubes. Both are in the inverse ratio of the size ofthe interior diameters of the tubes or pores.
Mellor [5] devotes considerable space to Faraday’s ‘‘con-densation theory’’ of catalysis. Taylor wrote [8]: ‘‘Faradaywas the first to indicate the zone of adsorbed material asthe reaction space of a heterogeneous catalytic action.’’
Robertson [9] and Schwab [10], among others whosurvey the concepts of catalysis, attribute the adsorptiontheory to Faraday. However, Faraday’s view of catalysiswas a limited extension of de Saussure’s views that aredescribed above.
Faraday, during about a three-month period, inves-tigated the recombination of oxygen and hydrogen asthey remained in contact with platinum following theirliberation during electrolytic studies [11]. He concludedthat this property belonged to platinum ‘‘. . . at all times,and was always effective when the surface was perfectlyclean.’’ The platinum could be activated by heating in acidor by employing mechanical cleaning; other approaches,such as treating or heating with alkali, were effective foronly part of the time. Faraday was convinced that thephenomenon which he observed had a satisfactory expla-nation based on known principles, and did not requirethe assumption of any new state or new property. Fara-day clearly showed that the presence of hydrogen sulfide,phosphine or ethene, among others, would prevent theplatinum from exhibiting its properties of causing thecombination of hydrogen and oxygen.
Faraday wrote that Dobereiner [12] considered the effectto be an electric action in which hydrogen, being veryhighly positive, represents the zinc of the usual electriccell arrangement, and like it, attracts oxygen and combineswith it. Faraday stressed the need for a perfectly clean andmetallic surface. He considered the effect to be producedby most, if not all, solid bodies, weakly perhaps in manyof them, but rising to a high degree in platinum. He wrotethat he was prepared to admit that the sphere of actionof particles extends beyond those other particles withwhich they are immediately and evidently in union, andin many cases produces effects rising into considerableimportance, and he thought that this kind of attractionwas a determining cause of Dobereiner’s effect. Faradaycompared the spongy platinum to a hygroscopic bodywhich becomes moist due to the condensation of the water
vapor with which it is in contact. He then considers thata gas, even when compressed at high pressure, still hasa low density and possesses sufficient vacant volume thatanother gas can be added in the void space. He wrote thatthe two gases, hydrogen and oxygen, will not react evenwhen compressed unless the platinum surface is presentto suppress, or remove, their elasticity and/or the actionof the metal in condensing them against its surface by anattractive force. He compares the hydrogen/oxygen case tothat where Hall found CO2 and lime to remain combinedunder pressure at temperatures at which they wouldnot have remained combined if the pressure had beenremoved. The course of events may be stated, accordingto Faraday by these principles:
• the deficiency of elastic power and the attraction of themetal for the gases are such that the gases are so farcondensed as to be brought within the action of theirmutual affinities at the existing temperature
• the deficiency of elastic power, not merely subjectingthem more closely to the attractive influence of themetal, but also bringing them into a more favorablestate for union, by abstracting a part of that power(upon which depends their elasticity) which elsewherein the mass of gases is opposing their combination
The consequence of their combination is the productionof the vapor of water and an elevation of temperature.The attraction of Pt for water is not as great as for thegases, and so the water quickly diffuses through theremaining gases. Fresh portions of the gases come intojuxtaposition with the metal, combine, and the vaporformed also diffuses, allowing new portions of gas tobe acted upon. In this way the process advances and isaccelerated by the evolution of heat. The platinum is notconsidered to cause the combination of any particles withitself, but only associating them closely around it; thecompressed particles are free to move from the platinum,being replaced by other particles, as a portion of denseair upon the surface of the globe, or at the bottom of adeep mine, is free to move by the slightest impulse intothe upper and rarer parts of the atmosphere. Thus, apartfrom introducing the need for a clean surface, and whatmay be, in the most optimistic view, a small step in thedirection of stating the concept of chemisorption, Faradayheld views that were essentially the same as those of deSaussure. This is not meant to degrade Faraday’s work;in fact, he applied extraordinary concepts and visionarythought in an experimental program that extended overonly three months.
During the period 1840–1880, Liebig’s view of catalysisprevailed. Liebig matched Berzelius in scientific stature,published his own journal, and had the advantage thatmost chemists had been trained by him, either directly

1.2.2 Wilhelm Ostwald 19
or indirectly by his students. During the period between1836 and 1846, Liebig was engaged in research intoputrefaction, and in developing agriculture science whereorganic ferments were a subject for active investigation.The isolation of an enzyme from barley malt in 1832 [13]ignited these intense research efforts.
Liebig was a proponent of the vibration theory offermentation and catalysis. Pasteur [14] proved thatalcoholic fermentation and putrefaction are caused bythe presence of certain low forms of life, namely bacteria.Liebig attacked this view, and with vigor:
‘‘To suppose that putrefaction or fermentation is caused by thephysiological action of such creatures can only be compared withthe idea entertained by a child who would explain the rapidcurrent of a river through a mill-wheel by supposing that themill-wheel, by its force, drives the water down the stream.’’
However, Liebig’s criticism did push Pasteur to conductmore experiments, and to firmly establish his views. Whenconsidering the decomposition of hydrogen peroxide,Liebig wrote [15]:
‘‘Yet it is singular that the cause of the sudden separation of thecomponent parts of peroxide of hydrogen has been viewed asdifferent from those of common decomposition, and has beenascribed to a new power termed the catalytic force. Now, it has notbeen considered, that the presence of the platinum and silver serveshere only to accelerate the decomposition; for without the contact ofthese metals, the peroxide of hydrogen decomposes spontaneously,although very slowly.’’
Thus, Liebig presented a major concept that was to be apart of the theory of catalysis advanced later by Ostwald,although it did not have the framework of kinetics thatdeveloped during the 40-year period separating the two.
After considering several examples of such decomposi-tions, Liebig [16] concludes that:
‘‘No other explanation of these phenomena can be given, than thata body in the act of combination or decomposition enables anotherbody, with which it is in contact, to enter into the same state. It isevident that the active state of the atoms of one body has aninfluence upon the atoms of a body in contact with it; and if theseatoms are capable of the same change as the former, they likewiseundergo that change; and combinations and decompositions arethe consequence. But when the atoms of the second body are notcapable of such an action, any further disposition to change ceasesfrom the moment at which the atoms of the first body assume thestate of rest, that is when the changes of transformations of thisbody are quite completed.’’‘‘In that state in which they exist within the pores or upon thesurface of solid bodies, their repulsion ceases, and their wholechemical action is exerted. Thus, combinations which oxygencannot enter into, decompositions which it cannot effect while inthe state of gas, take place with the greatest facility in the pores ofplatinum containing condensed oxygen. When a jet of hydrogengas, for instance, is thrown upon spongy platinum, it combineswith the oxygen condensed in the interior of the mass; at their point
of contact water is formed, and as the immediate consequence heatis evolved; the platinum becomes red hot and the gas is inflamed. . .
In finely pulverized platinum, and even in spongy platinum, wetherefore possess a perpetuum mobile – a mechanism like a watchwhich runs out and winds itself up – a force which is neverexhausted – competent to produce effects of the most powerful kind,and self-renewed ad infinitum. Most phenomena, formerlyinexplicable, are satisfactorily explained by these recentlydiscovered properties of porous bodies. The metamorphosis ofalcohol into acetic acid, by the process known as the quick vinegarmanufacture, depends upon principles, at a knowledge of which wehave arrived by a careful study of these properties.’’
1.2.2Wilhelm Ostwald
During the 50 years following the definition of catalysisby Berzelius, very little progress was made in itsunderstanding. Berzelius had organized enzyme catalysis,as well as organic and inorganic catalysis, into asingle discipline, and it remained that way for thenext half-century. During this period, as in the periodpreceding its definition, catalysis was a science basedon trial and error – Edisonian research – and reportsof the observations. However, during this period greatadvances in the understanding of chemistry weretaking place, and these provided the framework foran explosion of soundly based concepts of catalysis.Organic chemistry was gaining a structure through manydevelopments that included van’t Hoff–Le Bel’s three-dimensional molecules, the recognition of four-valentcarbon, the development of reliable chemical formulasbased upon Liebig’s method of elemental analysis oforganic compounds, the recognition of stereoisomers,and the isolation of enantiomers. Physical chemistrywas introduced and developed, primarily by Arrhenius,Ostwald and van’t Hoff; its acceptance was documentedwith the appearance of Ostwald’s two-volume book andthe Zeitschrift fur Physikalische Chemie, edited by van’tHoff and Ostwald. Kinetics became a subject for studyfollowing the measurements by Wilhelmy in 1850 of theacid-catalyzed inversion of cane sugar [17], and attainedits first mathematical maturity in the studies of Harcourtand Esson [18]. Mayer introduced the concept of theconservation of energy in 1842, and others placed theconcept on a firm scientific basis [19]. Thermodynamicswas established, and Gibbs provided advanced conceptsof statistical thermodynamics and the phase rules.Boltzmann provided his theories of the distribution ofenergy, the theory of approach to equilibrium, and entropyand probability, among others.
Ostwald first came to catalysis through his studieson the acceleration of homogeneous reactions by acids.These findings were widely accepted at the time although,
References see page 35

20 1.2 Development of the Science of Catalysis
ultimately, they were shown to be incorrect becauseOstwald believed that the acid, in acting as a catalyst, didnot enter into the chemical change which it influencesbut rather acted by its mere presence (contact catalysis).
Ostwald, when reviewing a paper in which Stohmann[20] utilized the vibrational theory of catalysis, wrote:
‘‘The abstractor has several objections to make to this definition.First, the assumption of a ‘condition of movement of the atoms ina molecule’ is hypothetical and therefore not suitable for purposesof definition. . . . If the abstractor were to formulate for himself theproblem of characterizing the phenomena of catalysis in a generalway, he would consider the following expression as probably mostsuitable: Catalysis is the acceleration of a chemical reaction, whichproceeds slowly, by the presence of a foreign substance. It wouldthen be necessary to give the following explanations. . . Thisacceleration occurs without alteration of the general energyrelations, since after the end of the reaction the foreign body canagain be separated from the field of the reaction, so that the energyused by the addition can once more be obtained by the separation,or the reverse. However, these processes, like all natural ones, mustalways occur in such a direction that the free energy of the entiresystem is decreased. . . . The existence of catalytic processes is to metherefore a positive proof that chemical processes cannot have akinetic nature.’’
With this review, the dominant concept of catalysispassed from Liebig to Ostwald. In these comments,Ostwald showed that a catalyst could not change theequilibrium, and it was he, more than anyone, whobrought down the vibrational theory as not beingamenable to experimental verification. At the same time,his definition emphasized an equally unverifiable concept:a catalyst cannot start a reaction which is not taking placewithout it. In 1901, Ostwald [21] included as catalyticphenomena:
• the release of supersaturation• catalysis in homogeneous mixtures• catalysis in heterogeneous systems• enzyme action.
Ostwald had not accepted the kinetic theory at the time ofhis definitions of catalysis, and incorrectly saw the phe-nomena disproving the atomic hypothesis championedby, among others, Boltzmann. Like Liebig before him,Ostwald saw catalysis theory by analogy: a catalyst acts likeoil on a machine, or as with a whip on a tired horse [22].His investigations into catalysis were recognized with theaward of Nobel prize for chemistry in 1909.
In Ostwald we have an example which demonstratesthat concepts that are shown, ultimately, to have littlevalidity – or even to be incorrect – may have great im-pact in developing science. Indeed, in many instancesit is the drive and forceful dominance by the individualthat establishes the concept, and not the novelty of theconcept. Thus, most authors associate the concept of a
catalyst speeding up an existing reaction with Ostwald,not realizing that Liebig advanced the same suggestionsome 50 years earlier. With the acceptance of physicalchemistry, and the training of a sufficient number of sci-entists to make use of the advances in the field, catalysiswas set to make many advances beginning at about 1900,and the Ostwald school was in the driver’s seat.
1.2.3The Concepts of Kinetics and Intermediate Compounds
Dobereiner believed that platinum black carried oxygenover to hydrogen, while de la Rive suspected that a layerof platinum oxide formed on the surface of platinum.Fusinieri [23] went so far as to propose that waves ofoxidation and reduction were responsible for the reactionof hydrogen and oxygen; he even reported the visualobservation of such wave action. Although Faraday wasunable to form a distinct idea of the power of Fusinieri’stheory, the latter defended his proposals, citing evidenceto show that those with good eyesight could see thesewaves. Berthollet suggested that platinum hydrides wereformed in the presence of hydrogen, and that these reactedwith oxygen to produce hydrogen.
Brodie [24] explained the decomposition of peroxideson the basis of chemical affinity and of catalysis interms of coupled chemical reactions. For the reaction(his uncorrected equations are used in the following)
I2 + Ba2O2 = 2BaI + O2
Brodie considered the reaction to take place by thedecomposition and reformation of water, according tothe two equations:
I2 + BaO2 + H2O = 2HI + Ba2O + O2
2HI + Ba2O = 2BaI + H2O
Viewing water as a catalyst provides the cyclic concept ofcatalytic action.
Brodie [24] also noted that:
‘‘Views as to the polarization of oxygen, and the cause of thedecompositions effected by the alkaline peroxides, which to a greatextent are identical with the preceding, and in which the samelanguage and the same notation are employed, have recently beenput forward, with considerable pretension, as new and originatingwith himself, by Schonbein, Professor of Chemistry at Basle. Thischemist can scarcely be aware of the memoir referred to, as in hisnumerous publications he makes no allusion to it. A reclamationof priority of ten years ought not to be required, but I am compelledto call the attention of chemists to these circumstances in order thatI myself may not be considered to appropriate withoutacknowledgment the ideas of discoveries of another.’’

1.2.3 The Concepts of Kinetics and Intermediate Compounds 21
The intermediate compound theory has guided thethoughts of many individuals whose contributions movedcatalysis to the forefront. For example, Sabatier [25] wrote:
‘‘Having arrived at the end of my career as a chemist, I havethought that it might be of interest, and at the same time of someutility, to relate how I was led to develop the direct hydrogenationmethod by finely divided metals, which won for me the Nobel Prizein 1912. . . It is the faith in the theory of temporary compoundsfurnished by the catalyst which has constantly guided me in thesevarious labors; it is these inductions to which I owe all my newresults.’’
The concept of the intermediate compound remainsdominant today in the many ‘‘volcano plots’’, and is fre-quently associated with the names of Sabatier and/orBalandin. Thus, the rate increases with the heat of ad-sorption (or formation) until a sufficiently high value isobtained so that the species is adsorbed so strongly thatthe rate decreases; this produces the well-known volcanoplot [26] (Fig. 2). As with any principle or correlation, itis frequently utilized in situations where its use is ques-tionable. Thus, without the data point for Au, the volcanocurve in Fig. 3 would be a straight line.
Polanyi [27] utilized this concept in the development ofhis ‘‘transition state’’ theory of catalysis, assuming thatfree homopolar valencies protrude from the surface of asolid, and that a reaction AB + CD = AC + BD would becatalyzed on the surface of such a solid (see Fig. 4). Forthe reaction of H + H2, Polanyi provided the curve shownin Fig. 5, and concluded that the catalytic effect would be
Au
Ag
Ag (110)
Pt
Ru
Cu
NiCu (110)
Cu/Ni (110)
Fe(100)
Ni (110)Ru (1010)
Pt (110)
60060 70 80 90 100 120
500
400
300
200
600
500
400
300
200
∆Hf (HCOOM), (kcal mol−1)
For
mat
e de
com
posi
tion
tem
pera
ture
s (K
)fr
om lo
w p
ress
ure,
sin
gle
crys
tal e
xper
imen
ts
Isok
inet
ic te
mpe
ratu
re (
K)
from
hig
hpr
essu
re c
atal
ytic
rat
e da
ta [1
]
Fig. 2 Example of a typical volcano plot correlating the formatedecomposition rate for both pressure and single crystal studies.(Reproduced from Ref. [26].)
PdO
CuO
BaO CaO
CeO2
Co3O4
NiOAg
Au
Ti2O
Fe2O3
V2O5
Cr2O3Al2O3
ZnO
Pt373
573
773
9730 200 400 600
−∆H°f (kJ mol O−1)T
empe
ratu
re/(
K)
Fig. 3 Example of a volcano plot with little data to support oneside of the mountain. (Reproduced from an anonymous reference.)
K KKK
AA
A
BB
B
C
C
C
D
D
D
Transition state
Initial state
Final state
Final state
Initial state
Fig. 4 Polanyi’s representation of his ‘‘transition state’’ theory.(Redrawn from Ref. [27].)
strongest when there is adsorption of moderate overallenergy. Surface valencies forming links of 50 000–51 000gcal, corresponding to QY = 0–2000, will be the mostefficient.
The highlights in chemical kinetics have been sum-marized by Laidler in his outstanding history of physicalchemistry (Table 1) [28]. The kinetic ideas introduced byWilhelmy, and also by Harcourt and Esson, were put forthby van’t Hoff in a book, Etudes de dynamique chimique,so that a compilation of the concepts of both chemicalthermodynamics and kinetics was available. van’t Hoffintroduced what is now considered to be the order ofthe reaction. Arrhenius also introduced the concept of anintermediate, Z, in the reaction scheme:
A + Ck1←−−−−−−→k3
Z
References see page 35
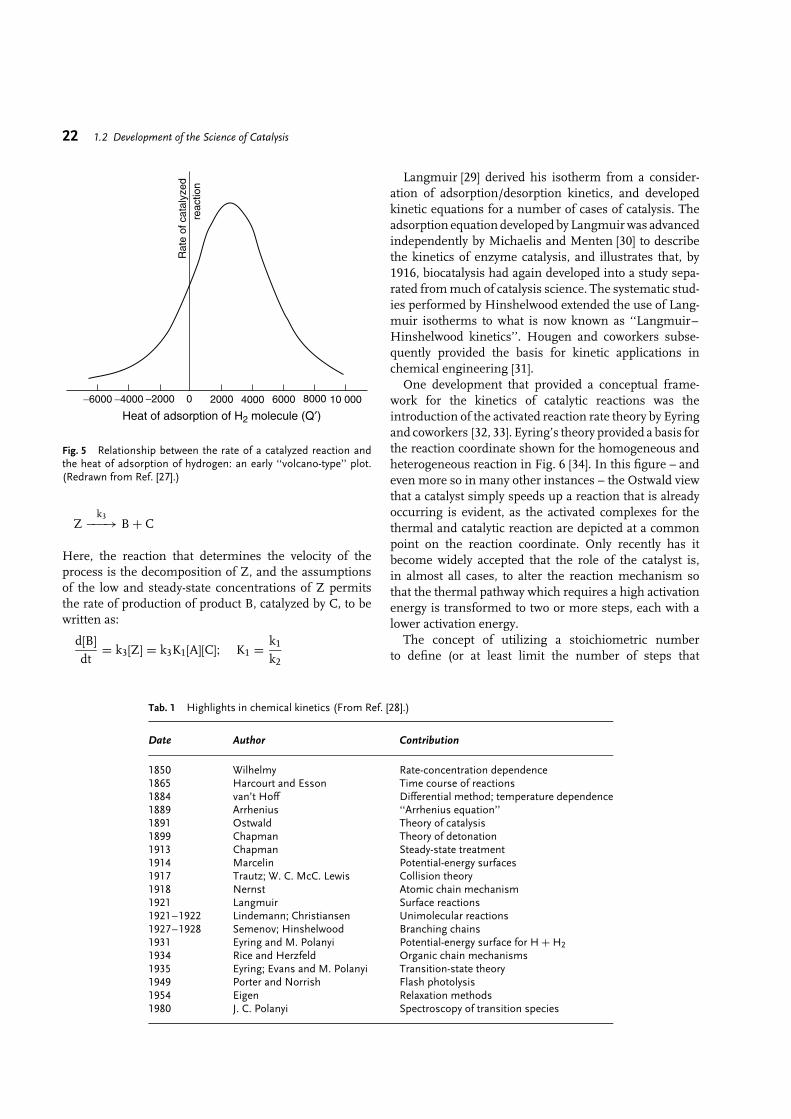
22 1.2 Development of the Science of Catalysis
−6000 −4000 −2000 0 2000 4000 6000 8000 10 000
Heat of adsorption of H2 molecule (Q′)
Rat
e of
cat
alyz
ed re
actio
n
Fig. 5 Relationship between the rate of a catalyzed reaction andthe heat of adsorption of hydrogen: an early ‘‘volcano-type’’ plot.(Redrawn from Ref. [27].)
Zk3−−−→ B + C
Here, the reaction that determines the velocity of theprocess is the decomposition of Z, and the assumptionsof the low and steady-state concentrations of Z permitsthe rate of production of product B, catalyzed by C, to bewritten as:
d[B]
dt= k3[Z] = k3K1[A][C]; K1 = k1
k2
Langmuir [29] derived his isotherm from a consider-ation of adsorption/desorption kinetics, and developedkinetic equations for a number of cases of catalysis. Theadsorption equation developed by Langmuir was advancedindependently by Michaelis and Menten [30] to describethe kinetics of enzyme catalysis, and illustrates that, by1916, biocatalysis had again developed into a study sepa-rated from much of catalysis science. The systematic stud-ies performed by Hinshelwood extended the use of Lang-muir isotherms to what is now known as ‘‘Langmuir–Hinshelwood kinetics’’. Hougen and coworkers subse-quently provided the basis for kinetic applications inchemical engineering [31].
One development that provided a conceptual frame-work for the kinetics of catalytic reactions was theintroduction of the activated reaction rate theory by Eyringand coworkers [32, 33]. Eyring’s theory provided a basis forthe reaction coordinate shown for the homogeneous andheterogeneous reaction in Fig. 6 [34]. In this figure – andeven more so in many other instances – the Ostwald viewthat a catalyst simply speeds up a reaction that is alreadyoccurring is evident, as the activated complexes for thethermal and catalytic reaction are depicted at a commonpoint on the reaction coordinate. Only recently has itbecome widely accepted that the role of the catalyst is,in almost all cases, to alter the reaction mechanism sothat the thermal pathway which requires a high activationenergy is transformed to two or more steps, each with alower activation energy.
The concept of utilizing a stoichiometric numberto define (or at least limit the number of steps that
Tab. 1 Highlights in chemical kinetics (From Ref. [28].)
Date Author Contribution
1850 Wilhelmy Rate-concentration dependence1865 Harcourt and Esson Time course of reactions1884 van’t Hoff Differential method; temperature dependence1889 Arrhenius ‘‘Arrhenius equation’’1891 Ostwald Theory of catalysis1899 Chapman Theory of detonation1913 Chapman Steady-state treatment1914 Marcelin Potential-energy surfaces1917 Trautz; W. C. McC. Lewis Collision theory1918 Nernst Atomic chain mechanism1921 Langmuir Surface reactions1921–1922 Lindemann; Christiansen Unimolecular reactions1927–1928 Semenov; Hinshelwood Branching chains1931 Eyring and M. Polanyi Potential-energy surface for H + H21934 Rice and Herzfeld Organic chain mechanisms1935 Eyring; Evans and M. Polanyi Transition-state theory1949 Porter and Norrish Flash photolysis1954 Eigen Relaxation methods1980 J. C. Polanyi Spectroscopy of transition species

1.2.3 The Concepts of Kinetics and Intermediate Compounds 23
Pot
entia
l ene
rgy
Activatedstate
for gas reaction
Activatedstate for
surface reaction
Gaseousreactants
Gaseousproducts
Adsorbedreactants
Adsorbedproducts
Reaction coordinate
Ehom
Ehet
Fig. 6 Potential-energy curves for a reaction proceeding homo-geneously (solid curve) and on a surface (dotted curve). Thesecurves illustrate retention of the Ostwald concept, that a catalystonly speeds up a homogeneous reaction, but does not change themechanism. (Reproduced from Ref. [34].)
define) the rate was introduced by Horiuti [35], andmany of the mathematical derivations relating to thismethod were developed by his group. The stoichiometricnumber is defined as the number of times that a stepof the reaction mechanism occurs. The first effortsto utilize the technique to define the mechanism forammonia synthesis led to contradictory results; Enomotoand Horiuti [36] reported a stoichiometric number of 2,whereas the results of a combined kinetic and isotopicstudy indicated that it was 1 [37]. Similar problemshave been encountered in applying the stoichiometricnumber to other catalytic systems; thus, while it is anattractive concept and provides assistance in eliminatingsome potential mechanisms from consideration, theexperimental data available today usually limit itsusefulness.
The concept of turnover – the number of catalyticreactions per catalytic site per unit time – came intocommon usage in enzyme catalysis. Only recently hasthis concept been adapted to heterogeneous catalysis,particularly by Boudart and his students. Where thenumber of active sites can be defined precisely (e.g.,H+ in a sulfuric acid solution), the concept has greatutility in a definition of catalysis; however, it hasbeen applied too frequently in instances where themeasure of the number of catalytic sites is uncertain,or worse.
Jost [38] attributes:
‘‘. . . the discovery of chain reactions to Bodenstein in 1913, andthe coining of the word to Christiansen. . . . E. Cremer, in her thesiswith Bodenstein in 1927 on the H2 –Cl2 reaction, first noted theoccurrence of chain branching and instability caused by branching.The implications of this concept, however, were not fully recognizedeither by her or by Bodenstein until Semenoff’s later publications.’’
Kemball, however, wrote that the idea of an atomic chainbased on the repeated reactions was proposed by Nernstin 1916 [39]. In any event, catalysis and chain reactionshave features of a common concept.
One area of kinetics that has attracted much attentionis the compensation effect; here, a high activation energyis compensated for by an increase in the pre-exponentialterm, the effect being derived from a one-page note byCremer and Schwab [40]. The initial experimental datademonstrating the compensation effect was provided byConstable [41]. The debate continues as to the cause ofthe linear relationship between ln A and E; these includeviews that it is based upon one of several theoreticalexplanations, is merely an empirical observation, or evento question its existence [42, 43].
Today, the progress of a heterogeneous catalyticreactions can be resolved into at least five distinct steps:
(i) diffusion of the reactants to the catalyst(ii) formation of the adsorption complex (reactant-
surface)(iii) the chemical change on the surface(iv) decomposition of the adsorption complex (product-
surface)(v) diffusion of the reaction products from the catalyst
Unless all five steps have the same activation energy, theone step with the highest activation energy determinesthe rate.
Steps (i) and (v) received prominent attention duringthe early application of kinetics to catalysis. Nernst,among others, considered the rate-controlling step ofthe dissolution of a solid to be due to the diffusionof the molecule or ions through the nearly saturatedboundary layer that surrounds each particle. By analogy,during the late 1800s and early 1900s, Nernst, Bodenstein,and others proposed that the rate of a catalytic reactionwas determined by the diffusion of reactants and/orproducts through the layer(s) of adsorbed molecules.While this particular view of catalysis gradually fellfrom favor, diffusion remains an important concept incatalysis.
Thiele, while investigating the theory of catalysisat home in his spare time, developed a connection
References see page 35

24 1.2 Development of the Science of Catalysis
between the rate of reaction and the diffusionallimitations imposed by the porosity of a catalyst [44]. Thistheory was amplified by many, particularly Wheeler [45],whose studies proved to be highly original and theresults monumental. Wheeler’s most significant resultsincluded:
• emphasizing the apparent decrease in the activationenergy, pressure and temperature gradients expectedin catalyst pellets
• the influence of catalyst poisons on activity andselectivity
• the role of diffusion in altering the observed kineticsfrom, for example, series to parallel reactions
• an estimate of the percentage of internal catalyst surfaceutilized for many reactions (Fig. 7) [46]
1.2.4Negative Catalysis: Autocatalysis
Negative catalysis was one of the concepts that added tothe confusion about catalysis. The findings of Turner [47]and Faraday [11] clearly showed that some substancesretarded the effectiveness of a catalyst. Mellor [48]noted that:
‘‘The interesting feature is that these gases may be regarded ascatalytic agents, which inhibit the action of another catalyticagent. This phenomenon will be called negative catalysis.’’
Subsequently, Mellor explained that negative catalysisin ester hydrolysis has been explained, with more or lesssuccess, by assuming that:
NH3 → N2 + H2 (10 mesh)
0
20
40
60
80
100
1 2 3 4 5 6
% In
tern
al s
urfa
ce a
vaila
ble
H2 + C2H4 on Ni film
Cat. cracking (1/8" pills)
NH3 synthesis at high pressure (5 mm granules)Butane dehydrogenation (1/8" pills)
Cumene cracking(3.5 mm pills)
kexp./kD
Fig. 7 Wheeler’s estimate for the percent of internal catalystsurface available for effecting a number of important industrialreactions. (Reproduced from Ref. [46].)
(i) The degree of ionization of the ester or catalyzeris diminished, or else the catalyzer combines withthe ‘‘foreign substance’’ so that the quantity of theavailable catalytic agent is diminished.
(ii) The combination of the ester with the retarding saltby which the active mass of the ester is diminished.
However, it was gradually recognized that negativecatalysis was merely the result of catalyst poisoning.Likewise, the chemical concepts slowly developed to showthat a strong adsorptive bond between the poison andthe catalyst caused the effect to be highly specific. Duein a large part to the extensive studies of Maxted [49],it was understood that the effect depended on definitetypes of electronic configuration of both the catalystand the poison. A material would only be regarded asa ‘‘poison’’ if it exerted an appreciable effect on catalysiseven when present in very small concentrations. Maxtedwas careful to distinguish between catalyst activity lossdue to poisoning and that of ‘‘. . . mechanically coveringup of a catalyst surface by less specifically held coatings,such as the cloaking of a catalyst by a layer of gums orwaxes or by a deposit of carbon in organic reactions athigh temperatures.’’ Today, the study of these topics hasbecome so extensive that they are important componentsof International Symposia on Catalyst Deactivation; thebooks based on these symposia contain many reports thatprovide finer details and more complex examples of thesetwo basic concepts.
The term ‘‘autocatalysis’’ was coined by Ostwald toidentify a class of catalytic reactions which are slow duringan initial induction period, after which the rate acceleratesrapidly. This effect will occur when one of the products canact as a catalyst for the reaction. Thus, the acid formedas a product of the hydrolysis of an ester can act as ahydrolysis catalyst. Another instance where autocatalysiswill be observed is during the reduction of a metal oxidewith hydrogen, provided that the metal formed catalyzesthe reduction. Another example occurs in the methanol-to-gasoline conversion, where a product (in this case theC3+ alkenes) combines with methanol more rapidly thanthe hydrocarbons can be formed from only methanol.
1.2.5Adsorption
During the early 1900s, it was gradually recognizedthat there are two types of adsorption. Langmuir’sstudies [29] emphasized the type which has becomeknown as chemisorption, whereas the value of theother type – commonly known as physisorption – wasemphasized by the introduction of the Brunauer, Emmettand Teller (BET) method for measuring surface area.Taylor was primarily responsible for the concept of

1.2.5 Adsorption 25
activated adsorption, and the need for an activation energyfor the transformation from the physisorbed state to thechemisorbed state [50]. This proposal was made by Taylorin an effort to more closely correlate the specific activityof the catalysts with their adsorption characteristics.
Wheeler [45] advanced the use of a physisorbedstate by introducing the concept that, by taking intoaccount the formation of multilayers of physicallyadsorbed gas and capillary condensation in capillaries,one could calculate the size distribution of porespresent in a catalyst. Barrett et al. [51] provided adetailed procedure for quantitative calculations. Wheelersubsequently pioneered the development of the theoryof interpreting the influence of pore size on manycharacteristics of catalytic activity and selectivity. Forexample, the activation energy for a reaction completelycontrolled by pore diffusion will have only one-half thetrue activation energy; likewise, a series reaction A →B → C may be made to appear as parallel reactions by porediffusion control. Moreover, Wheeler pointed out that thecatalyst preparation conditions can be utilized to obtain amaterial with the optimum surface area and porosity forthe production of a desired product. The isomerization ofxylenes to produce greater than 90% of the para-isomer,in contrast to the 20–25% present at equilibrium, simplyby controlling the pore opening of the HZSM-5 catalyst,is one example of the application of the concept selectivitycontrol by diffusion. The concept of diffusion in catalysishas now advanced to become a scientific discipline thatrepresents an independent field of study.
Washburn [52] developed a theory based upon mercurypenetration to provide a method for measuring porosity(especially for larger pores), and this concept was con-verted into an experimental reality by the investigationsof Ritter and Drake [53].
The classification into five types of physical adsorptionisotherms was accomplished by Brunauer and colleagues(see [54]). The desorption hysteresis associated with eachadsorption isotherm came to be related to the type of poresthat the material possessed, and while a single sourcecannot be cited for this concept, de Boer and coworkershad much to do with publicizing the importance of theshapes of the hysteresis loops.
During 1914–15, Langmuir put forth three conceptsimportant to chemisorption and its role in catalysis:kinetics, his checkerboard model of the surface, andchemical forces and bonding [55]. In developing hisconcepts, Langmuir described heterogeneous catalysisas a chemical drama that occurred in a single layer and bythe same chemical forces that held molecules and solidstogether. Langmuir’s concepts dominated the field fordecades, and remain important today. The kinetic natureof the adsorption and desorption steps at equilibrium for
a given pressure led to the Langmuir Isotherm:
θ = kP
1 + kP
where θ is the surface coverage, k is the ratio of theadsorption and desorption rate constants, and P is thepressure of the adsorbent. Langmuir’s notebook containhis ‘‘Fundamental Laws of Heterogeneous Reactions’’:
• The surface of a metal contains atoms spaced accordingto a surface lattice (number per cm2).
• Adsorption films consist of atoms or molecules heldto the atoms forming the surface lattice by chemicalforces [55].
Langmuir’s model implies that the amount of gas (e.g.,CO) adsorbed onto a metal (e.g., Pt) permits a calculationof the surface of the metal exposed to the gas; however,Langmuir did not emphasize this concept. He did,however, introduce the ‘‘checkerboard’’ model for thesurface of the catalyst.
During the 1930s, Brunauer and Emmett utilized thecombination of physical and chemical adsorption to obtaina measure of the fraction of the surface of an iron syntheticammonia catalyst that consists of metallic iron, alkalioxide promoter, and a structural promoter such as silicaor alumina. Thus, low-temperature nitrogen adsorptionprovided a measure of the total surface, chemisorbed COa measure of free metallic iron, and CO2 adsorptionthe surface concentration of alkali oxide promoter,and – by difference – the fraction covered by the structuralpromoter. Significant improvements in the techniquehave occurred since these initial investigations, but littlehas been altered concerning the concepts involved.
In 1935, Lennard–Jones [56] provided a pictorialscheme for the adsorption of, in this example, hydrogen(Fig. 8). This scheme illustrates that the transition fromvan der Waals adsorption to that of the chemisorbed staterequires an activation energy. This concept allows for theactivated adsorption championed by Taylor; at the sametime, in some special cases the activation energy may be solow that adsorption may occur with little or no activation.deBoer [57] considered adsorption from the viewpoint ofan adsorption time, whereby the average stay of a moleculeon the surface should depend upon the interactionenergy. Thus, as the interaction energy increases, theaverage residence time increases; hence, as the energyincreases the residence time increases from essentiallyno adsorption to physical adsorption to chemisorption.
Surface mobility became an important concern follow-ing Langmuir’s studies on chemisorption. Mobility, as itapplies to physical adsorption, means that the adsorbate
References see page 35

26 1.2 Development of the Science of Catalysis
(2) (1) M + A + B
M + AB
D
L
Q P
K W
W0
Ene
rgy
Dist. from metal
Fig. 8 Schematic representation of the Leonard-Jones potential,illustrating the activation energy needed to transfer from thephysically adsorbed to the chemisorbed state. (Redrawn fromRef. [56].)
can move so freely on the surface that its state is that ofa two-dimensional gas. For chemisorption, at least threedefinitions have been advanced [58]. Beeck [59] suggestedthat adsorption be classified as mobile or immobile ac-cording to whether the heat of adsorption fell with increas-ing coverage or remained constant. Thus, if the adsorbateis mobile it should adsorb on high-energy sites first, result-ing with a decrease in the heat of adsorption with increas-ing surface coverage. The field emission microscope [60]provided the first direct method of studying surface mobil-ity; indeed, it is surprising that the developer of this tech-nique, E. Muller, did not receive greater recognition [61].
1.2.6Active Site: Geometric or Electronic?
Langmuir’s checkerboard model provided an initialimpetus to relating physical/chemical properties of thesolid and the catalytic characteristics. Taylor’s nameis usually the one associated with the concept of anactive site [62], although he was not the first to proposea special site of unusually high activity. However, hissimple model and his stature focused much attention tothe active site. Thus, Taylor attributed special activity tothose atoms which, because of the uneven geometry ofthe surface (Fig. 9), have many atoms whose coordinationto other catalyst atoms is very low (unsaturation), andthese are the atoms that were attributed to provide theseat of most catalytic conversions. This view focusedattention on the heterogeneity of the surface of almostall catalysts, and the fact that the total surface wouldnot be equally active in effecting chemical reactions.Constable assumed that these active sites followed anexponential distribution [41]. The application of modern
Ni
Ni
Ni Ni
Ni Ni
Ni Ni Ni Ni Ni Ni Ni Ni Ni Ni Ni
NiNiNiNiNiNiNiNiNi
Ni Ni
Ni
Ni
NiNi
Ni
Ni
Ni
NiNi
Ni Ni
Granule proper
Gas phase
Fig. 9 Taylor’s representation of heterogeneity and the presenceof various types of active site on a nickel catalyst. (Reproducedfrom Ref. [62].)
surface techniques have provided the data to place theconcept of active sites on a firm foundation. At about thesame time that he advanced the active site, Taylor [63]made an observation that is equally insightful, but hasreceived little attention:
‘‘. . . an oxide catalyst surface is to be regarded as composed, not ofa single catalyst, but of two catalysts, metal ions and oxide ionsand the nature of the changes induced in the adsorbed reactant isdetermined by the charge of the ion on which the reactant moleculeis adsorbed.’’
This statement contains the rudiments of an acid–basetheory of metal oxide catalysis.
A liberal historian could trace the concept of active sitesback at least to Loew [64], who suggested that when amolecule of the reactant contacts the catalyst, the ‘‘sharpcorners’’ of the catalyst break up the molecule into atoms,and these are more reactive. Hence, the finer the particle,the greater the number of corners, and the greater theactivity of the catalyst.
Shortly following Taylor’s active-site hypothesis, Ba-landin proposed what has become known as the multiplethypothesis. Balandin proposed that binding between twoatoms of an adsorbed molecule can be broken if theyare attracted by two different catalyst atoms; a bondmay be formed between atoms of an adsorbed moleculebound to the same catalyst atom [65]. This hypothesiswas applied by Balandin to many reactions, includingthe dehydrogenation of cyclohexane; here, the spatial ar-rangement of the catalyst surface atoms must be suchthat metal–hydrogen bonding can result (Fig. 10) [66].Those metals with little mismatch between the spacingof the reactant hydrogen atoms and the catalyst surfaceatoms were considered to be active catalysts, whereasthose with a large mismatch would be inactive. Balandinexpanded his scope to include the activation enthalpy ofthe formation/decomposition of the multiplet complex,and conducted extensive studies to obtain a measure ofthe sum of energies of disrupted bonds in reactants and

1.2.6 Active Site: Geometric or Electronic? 27
6
e
f 1
23
4
5
a
b
d c
Fig. 10 Schematic showing the location of surface atoms of thecatalyst (intersections of light lines) and the adsorbed cyclohexane(heavy lines); the multiplet theory indicated that the spacings hadto match in order to form the appropriate bonding. (Reproducedfrom Ref. [66].)
newly formed ones in products. This view attracted muchattention and, because of Balandin’s position within theUSSR scientific community, probably survived in its orig-inal form much longer than could have been anticipated.
In 1934, Gwathmey began a series of studies toshow, at least qualitatively, that different metal crystalfaces possess different catalytic activities. Gwathmey’sexperiments, which were based on his captivation bythe diffraction experiments of Davisson and Germer in1927 [67], allowed him to demonstrate the anisotropy ofdifferent crystal faces in chemical reactions, as well as theview that anisotropy of surface behavior was widespread,if not universal.
Subsequently, in 1938, Kobozev [68] extended Ba-landin’s concept and introduced the hypothesis of activeensembles. As originally proposed, the theory lackeddetail and was not readily susceptible to experimentalverification. Later, during the 1960s, Van Hardeveld andHartog [69] placed the Kobozev concept upon a morequantitative basis when they made calculations for thedependence of the number of metal atoms of a specifiedcoordination number upon the crystal size. The studiesconducted by Van Hardeveld and Hartog resulted fromtheir initial failure to reproduce the report by Eischens andJacknow [70] of infrared (IR) bands due to the adsorptionof nitrogen on nickel. Only when they reduced a freshcatalyst in situ could they obtain the sought-after IR band,and they then proceeded to combine quantitative IR dataand models of the surface of various sized metal catalystparticles to develop their quantitative model. The situation
became even more complex with the introduction of theconcept of facile and demanding reactions [71]; further-more, it was considered that a single atom should beadequate for a facile reaction, whereas two or many moreatoms would be required for a catalytic site adequate fordemanding reactions. Thus, while some reactions shouldbe dependent on surface geometry, others may not.
The pioneering studies of Beeck, using evaporatedmetal films, led to the concept of catalytic activity beingrelated to the number of holes in the d-band and/or thegeometry (Figs. 11 and 12) (e.g., [72, 73]). The conceptof relating catalytic activity to the electronic theories ofsolid-state properties was given impetus by the theoreticalreport by Dowden and the accompanying experimentaldata which showed that the catalytic activity of nickeldecreased linearly as the nickel was alloyed with copperto cause a parallel decrease in the number of d-bandholes. Furthermore, the activity approached zero for thealloy composition where the number of d-band holesapproached zero [74]. Although these findings providedimpetus for many studies during the following years, itwas subsequently shown that surface enrichment causedthe composition of the outermost layer of the NiCu alloyused by Dowden and Reynolds to differ dramatically fromthat of the bulk.
Schwab [75], using Hume–Rothery alloys, showedthat the addition of a second element to increase theoccupation ratios of the first Brillouin zone (conductionband) provided a corresponding increase in the activation
0
1
2
3
4
38 44 52
% of d-character
Log
(rat
e of
hyd
roge
natio
n)
Cr
W
Fe
Ni
Ta
Ni (110)
Pt
Pd
Rh
Fig. 11 Correlation of the catalytic activity for ethene hydrogena-tion with the percentage d-character of the catalyst. (Reproducedfrom Ref. [72].)
References see page 35

28 1.2 Development of the Science of Catalysis
Rh
Pd
Pt
Fe
Cr
Ni
Ni (110)
W
W
TaTa
0
1
2
3
4
.30 .35 .40 .45
Interatomic distance nm
Log
(rat
e of
hyd
roge
natio
n)
Fig. 12 Correlation of the catalytic activity for ethene hydrogena-tion with the surface atom interatomic distance. (Reproduced fromRef. [73].)
300
200
100
35
30
25
0.6
0.4
0.2
A B C a g e h
1.5 2 2.5EC
Fig. 13 Alteration energy for the conversion of formic acid aselectron levels are filled by varying the composition of binarymetal catalysts [A – , hardness (kg mm−2); B–o–, activationenergy (kcal mol−1); C •••, resistance (�cm·104)]. (Redrawn fromRef. [75].)
energies of donor reactions (Fig. 13). Schwab thereforeconcluded that electron holes must be considered to beinvolved in the catalysis.
The pioneering studies of Farnsworth [76] (whostruggled from his first publication in 1929 until the1950s to develop adequate instrumentation to obtain datashowing the likelihood of differences in atomic spacingwithin the surface layer and the bulk, as well as therelaxation of the surface layer inwards) and Germer [77]showed that the surface layer in many instancesundergoes reorganization to a more stable configurationthat differs dramatically from the structure of the bulk.Furthermore, the gas molecules may adsorb and arrangethemselves into a regular two-dimensional lattice of theirown, and this may be dependent upon surface coverage.
In addition, chemisorption may cause reconstruction ofthe surface arrangement [78]. The pioneering IR studiesof Eischens and Pliskin [79] provided definitive evidencethat CO adsorbed onto a Pt surface in more than oneform, contrary to the checkerboard model of Langmuir,and that, by analogy with carbonyl cluster molecules,these corresponded to the bridged and linear forms ofadsorbed CO.
The staggering number of developments in thetheoretical physics of both metals and semiconductorsduring the mid-1900s led their application in catalysis.For example, most of Volume 7 of Advances in Catalysiswas devoted to some aspect of this issue. Wagner [80]showed that zinc oxide could accommodate excess zincatoms that were associated with interstitial positions ofthe lattice, and that the conductivity of the solid dependedupon the pressure of the oxygen in contact with the solid,as predicted by the theory. Doping ZnO with a metalof valence greater than Zn2+ requires an increase infree electrons, whereas doping with a univalent cationwill decrease the electron concentration. Parravano andBoudart [81] pointed out that, while the literature dataand the activity for hydrogen/deuterium exchange couldbe shown to be compatible with the predictions of thesemiconductor theory, the limitations of the verificationof the concept were numerous. These included deviationsfrom the Arrhenius plots for calculating the activationenergy, and evidence for a compensation effect in thelow-temperature range where activation energies couldbe calculated. Likewise, the data for the changes inactivity for CO oxidation with doping of NiO led tocontradictory conclusions [81]. Even so, it was concludedthat ‘‘. . . catalytic behavior of semiconducting oxides hasbeen modified in opposite directions by the addition ofimpurities which modify their electrical characteristics inopposite directions’’ [81].
During the late 1940s, Garner et al. [82] conductedextensive studies of the catalytic oxidation of CO oncuprous oxide and, at the same time, followed theconductivity of the solid, which resembled that of the solidfollowing saturation with CO. These authors suggestedthat oxygen and CO react on the surface to produce acarbonate complex which then reacts with adsorbed COto produce CO2:
2O−ads + COads −−−→ CO3
2−ads
and CO32−
ads + COads −−−→ 2CO2
Vol’kenshtein [83] attempted to formulate adsorptionon semiconductors in a manner that took into accountnot only the Fermi level but also the surface statesand the gas phase and adsorbed electronic structureof the adsorbate. This concept led Vol’kenshtein to theconclusion that the act of adsorption could create new

1.2.7 Selected Systems 29
sites for adsorption. Thus, there may be different types oflevel even for the same molecules adsorbed on the same(from its chemical nature) surface. A ‘‘homogeneous’’surface could therefore become ‘‘heterogeneous’’ by theact of adsorption.
Studies to elucidate the electronic aspects of catalysisdominated the 1950s, and continue even today. Subse-quently, two models have emerged: (i) the atomistic model(a surface molecule of one or more atoms); and (ii) theband model. The atomistic model essentially ignores thesolid and concentrates attention on the ensemble of one ora few surface atoms. In contrast, the band model describesthe surface in terms of surface states and localized energylevels available at the surface. Vol’kenshtein serves as anexample of an early proponent of the valence band model,while Knor [84] summarizes the other approach. Morri-son [85] surveyed the application of solid state theories tocatalysis in 1977 and in 1989.
Today, the geometric effect is one of two dominant themesof catalysis by metals, the other being the electronic effect.The geometric and electronic effects are usually discussedas separate topics, although it appears that – just as withacid–base catalysis – the two effects are interrelated sothat, at best, one can only document that one of the effectsplays a dominant role in the catalysis. In fact, it appearsthat it is necessary to accept the concept that the initialcatalytic material, and especially with metallic catalysts,may interact with the reactant(s) to redesign itself. Thisconcept can be illustrated by the observation of Beeckinitially [86], by the retention of 14C-labeled acetyleneduring hydrogenation of ethylene [87], as well as by thesurface science of, for example, Somorjai and Ertl, thatdifferent metal catalysts will strongly – and essentiallyirreversibly – bond different amounts of a reactant. Thus,one can view the metallic catalyst as redesigning itself bydoping its surface, and in some cases the bulk, withsufficient reactant so as to attain an electronic statewhere the bonding is sufficiently weakened that chemicalreactions may occur.
1.2.7Selected Systems
1.2.7.1 Ammonia SynthesisLe Chatelier’s principle indicates that ammonia formationis favored at high pressure. Convinced of his principle,Le Chatelier had his assistant compress a mixture ofhydrogen and nitrogen to a very high pressure, andthen apply an electrical spark. An explosive reactionoccurred which killed the assistant, and was the resultof a faulty experimental procedure whereby air waspermitted to mix with the nitrogen–hydrogen mixture.Hence, it was the oxygen–hydrogen reaction, rather than
ammonia formation, that caused the explosion. This wasjust one of many hundreds of unsuccessful early attemptsto synthesize ammonia.
Larson [88] credits Perman with the initial synthesisof ammonia using an iron catalyst. However, it wasthe interactions of Nernst and Haber that led to thesuccessful demonstration of the catalytic synthesis ofammonia and the results that were to generate theexcitement needed for rapid progress. Haber determinedexperimentally the amount of ammonia present atequilibrium. Nernst computed, by using the heats ofreaction and other chemical constants, the equilibriumamounts for many reactions. All values calculated byNernst were in agreement with the experimental values,except for the experimental value for ammonia reportedby Haber. In diplomatic terms, a very spirited debate aroseover the correct value for ammonia [89]. It is sufficienthere to state that Haber’s additional measurements athigh pressure, spurned by Nernst’s comments, resulted inshowing that the equilibrium concentration of ammoniawas sufficiently high to indicate industrial significance.Haber’s most active catalyst, osmium, was not ofindustrial significance. One of Haber’s novel concepts wasto use a process in which the unconverted hydrogen andnitrogen, after removal by condensation of the ammoniaproduced, were recycled to the reactor. In this connection,Haber introduced the concept of ‘‘space-time-yield’’ [90].Even so, Haber’s studies would only have resulted in avictory for ego and academics if he had not been able tointerest BASF in the process.
Mittasch, a student of Ostwald, guided the research atBASF that led to the iron synthetic ammonia catalyst thatis still used today, albeit in an improved form. Mittaschwas led, just as Sabatier, to a successful catalyst followingthe intermediate compound concept; in this case, thecatalyst was viewed briefly to form a nitride intermediatewith subsequent reduction by hydrogen. While conceptswere utilized by Mittasch, the investigations weresystematic and extensive. During about a two-year period,6500 experiments with about 2500 different catalystswere conducted [90], truly an example of Edisonianresearch. A magnetite sample from Sweden proved tobe a surprisingly active catalyst and, because of its relativehigh density, the concept of ‘‘compact while porous’’developed, but was subsequently proved to be wrong.Haber considered, temporarily, that the high density ofosmium and similar metals was the reason for theireffectiveness as an ammonia synthesis catalyst. Duringextensive studies by Mittasch and coworkers, the conceptsof promoter action of catalysis were developed and,building on the meager earlier results, defined the optionsfor the role of promoters during the 1920s (Fig. 14) [91].
References see page 35

30 1.2 Development of the Science of Catalysis
I
I. Simple activationIb. Support effectII. Poisoning
III. and V. Additive effectIV. Mutual activationVI. Deactivation
IIIII
IV
VVI Ib
50 A50 B
0 A100 B
100 A0 B
Composition of the catalyst
Rel
ativ
e ca
taly
tic e
ffici
ency
Fig. 14 The five ways in which a promoter may impact catalyticactivity as the concentration of the promoter is varied. (Reproducedfrom Ref. [91].)
The development of the United States synthetic am-monia industry provides an outstanding example ofexceptional accomplishments in the development ofthe technology first, and then in scientific under-standing, whilst simultaneously showing exceptionallydisastrous political actions. Within six years of commer-cial operations in Germany, Larson and coworkers atthe Fixed Nitrogen Laboratory (FNL) had developed asuitable catalyst. Sir Hugh Taylor [92], in consideringthe scientific studies conducted by Emmett, Brunauerand coworkers at the FNL following the developmentscheme, wrote:
‘‘These authors have given us the most detailed kinetic study evermade by a single reaction, with all aspects of the reaction studied,adsorptions, kinetics, influence of reactant concentrations as afunction of composition and mode of preparations. The treatmentis so comprehensive that it is possible to present an almost completeaccount of the phenomenon of surface catalysis by reference to thisone example alone.’’
The concept of the dissociation of nitrogen as the slowstep was demonstrated using both kinetic and isotopictechniques. The catalysts were carefully characterized.Kinetic concepts were developed and these, together withthe concept of ‘‘virtual pressure’’, which was introducedby Temkin and Pyzhev [93], provides a kinetic modelthat is still valid today. However, the more detailedmodel that has resulted from many surface sciencestudies provides a much sounder basis, with greaterdetail for the concepts of how the catalyst operates(Fig. 15) [94].
~41
N + 3H
NH2 + H
NH + 2H
1129
1400~960
~21
543460
NH3
+ 3/2 H2NH3ad
NH2ad + HadNHad + 2HadNad + 3Had
= 46
389
314
∆H
259
17
1/2 N2 1/2 N2ad~33
50
Fig. 15 Energy profile of the progress of the ammonia synthesison Fe (units of energy are kJ mol−1). (Reproduced from Ref. [94].)
1.2.7.2 Acid CatalysisAcids have been a dominant theme in the study ofreactions, and acid catalysis predates its definition.The dissociation theories advanced by Arrhenius, andapplied by Ostwald, did much to aid in developing theunderstanding of acid catalysis. These authors dominatedinvestigations to show that the catalytic power of asolution of an acid is directly proportional to its electricalconductivity, and independent of the nature of the anion.This indicated that the catalyst is the hydrogen-ion, andthat its effect is directly proportional to its concentrationin the solution. However, by 1900 acid catalysis was ina state of confusion, primarily because of the failure torecognize the role of the undissociated acid and the salteffects. The concepts of acid–base catalysis have beenpresented during the past 50 years in a series of books byBell [95]. Unfortunately, all too frequently scientists forgetthat acid–base catalysis is the title of Bell’s first book, andtreat acid catalysis in isolation from the conjugate base.
Hydrocarbon conversions have been dominant in thestudy of acid catalysis. Early studies on this topic in-volved acids such as the one formed by an aluminumchloride/hydrogen chloride mixture; however, the com-bination of the corrosion and the difficulty of catalystrecovery has limited its use to special situations, andthese two factors led to the failure of the cracking processintroduced by Gulf Oil that was based on this catalyst.The applications of acid catalysis were first realized inpetroleum processing by Eugene Houdry, with his intro-duction of catalytic cracking. Actually, the Houdry processprovided much greater advances in catalyst regenerationand process control than it did in introducing new catalytic
Related Documents











