Guidelines for the validation and application of typing methods for use in bacterial epidemiology A. van Belkum 1 , P. T. Tassios 2 , L. Dijkshoorn 3 , S. Haeggman 4 , B. Cookson 5 , N. K. Fry 6 , V. Fussing 7 , J. Green 8 , E. Feil 9 , P. Gerner-Smidt 10 , S. Brisse 11 and M. Struelens 12 for the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) Study Group on Epidemiological Markers (ESGEM) 1 Erasmus MC, Department of Medical Microbiology and Infectious Diseases, Rotterdam, The Netherlands, 2 National and Kapodistrian University of Athens, Department of Microbiology, Athens, Greece, 3 Leiden University Medical Center, Department of Infectious Diseases, Leiden, The Nether- lands, 4 Swedish Institute for Infectious Disease Control, Department of Bacteriology, Solna, Sweden, 5 Laboratory of Health Care Associated Infections, 6 Respiratory and Systemic Infection Laboratory, Health Protection Agency, Centre for Infections, London, UK, 7 Novo Nordisk, QC Microbiology SDK, Novo Alle, Bagsvaerd, Denmark, 8 Statistics, Modelling and Bioinformatics Department, Health Protection Agency, Centre for Infections,London, 9 University of Bath, Department of Biology, Bath, UK, 10 Centers for Disease Control and Prevention, Foodborne and Diarrheal Diseases Branch, Divison of Bacterial and Mycotic Diseases, Atlanta, GA, USA, 11 Institut Pasteur, Unit BBPE28, Paris, France and 12 Universite ´ Libre de Bruxelles, Ho ˆ pital Erasme, Bacteriologie,Brussels, Belgium ABSTRACT For bacterial typing to be useful, the development, validation and appropriate application of typing methods must follow unified criteria. Over a decade ago, ESGEM, the ESCMID (Europen Society for Clinical Microbiology and Infectious Diseases) Study Group on Epidemiological Markers, produced guidelines for optimal use and quality assessment of the then most frequently used typing procedures. We present here an update of these guidelines, taking into account the spectacular increase in the number and quality of typing methods made available over the past decade. Newer and older, phenotypic and genotypic methods for typing of all clinically relevant bacterial species are described according to their principles, advantages and disadvantages. Criteria for their evaluation and application and the interpretation of their results are proposed. Finally, the issues of reporting, standardisation, quality assessment and international networks are discussed. It must be emphasised that typing results can never stand alone and need to be interpreted in the context of all available epidemiological, clinical and demographical data relating to the infectious disease under investigation. A strategic effort on the part of all workers in the field is thus mandatory to combat emerging infectious diseases, as is financial support from national and international granting bodies and health authorities. CENTRAL THEME Bacterial typing methods generate isolate- specific molecular fingerprints for assessment of epidemiological relatedness INTRODUCTION The ability to quickly and reliably differentiate among related bacterial isolates is essential for epidemiological surveillance, and is an endeav- our as old as the discipline of bacteriology itself. Long-standing ‘conventional’ typing methods, such as bacteriophage typing of Staphylococcus aureus and Listeria monocytogenes [1,2], serotyping of Salmonella spp. and Escherichia coli [3,4], or biochemical typing of Enterobacteriaceae [5], have historically been important contributors to our understanding of the natural history and epidemiology of infections caused by strains of these clinically relevant bacterial species. Corresponding author and reprint requests: A. van Belkum, Erasmus MC, Department of Medical Microbiology and Infec- tious Diseases, Dr. Molewaterplein 40, 3015 GD Rotterdam, The Netherlands. E-mail: [email protected] ȑ 2007 The Authors Journal compilation ȑ 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Guidelines for the validation and application of typing methods for use inbacterial epidemiologyA. van Belkum1, P. T. Tassios2, L. Dijkshoorn3, S. Haeggman4, B. Cookson5, N. K. Fry6, V. Fussing7,J. Green8, E. Feil9, P. Gerner-Smidt10, S. Brisse11 and M. Struelens12 for the European Society of ClinicalMicrobiology and Infectious Diseases (ESCMID) Study Group on Epidemiological Markers (ESGEM)
1Erasmus MC, Department of Medical Microbiology and Infectious Diseases, Rotterdam, TheNetherlands, 2National and Kapodistrian University of Athens, Department of Microbiology, Athens,Greece, 3Leiden University Medical Center, Department of Infectious Diseases, Leiden, The Nether-lands, 4Swedish Institute for Infectious Disease Control, Department of Bacteriology, Solna, Sweden,5Laboratory of Health Care Associated Infections, 6Respiratory and Systemic Infection Laboratory,Health Protection Agency, Centre for Infections, London, UK, 7Novo Nordisk, QC Microbiology SDK,Novo Alle, Bagsvaerd, Denmark, 8Statistics, Modelling and Bioinformatics Department, HealthProtection Agency, Centre for Infections,London, 9University of Bath, Department of Biology, Bath,UK, 10Centers for Disease Control and Prevention, Foodborne and Diarrheal Diseases Branch, Divisonof Bacterial and Mycotic Diseases, Atlanta, GA, USA, 11Institut Pasteur, Unit BBPE28, Paris, France and12Universite Libre de Bruxelles, Hopital Erasme, Bacteriologie,Brussels, Belgium
A B S T R A C T
For bacterial typing to be useful, the development, validation and appropriate application of typingmethods must follow unified criteria. Over a decade ago, ESGEM, the ESCMID (Europen Society forClinical Microbiology and Infectious Diseases) Study Group on Epidemiological Markers, producedguidelines for optimal use and quality assessment of the then most frequently used typing procedures.We present here an update of these guidelines, taking into account the spectacular increase in thenumber and quality of typing methods made available over the past decade. Newer and older,phenotypic and genotypic methods for typing of all clinically relevant bacterial species are describedaccording to their principles, advantages and disadvantages. Criteria for their evaluation andapplication and the interpretation of their results are proposed. Finally, the issues of reporting,standardisation, quality assessment and international networks are discussed. It must be emphasisedthat typing results can never stand alone and need to be interpreted in the context of all availableepidemiological, clinical and demographical data relating to the infectious disease under investigation.A strategic effort on the part of all workers in the field is thus mandatory to combat emerging infectiousdiseases, as is financial support from national and international granting bodies and health authorities.
CENTRAL THEMEBacterial typing methods generate isolate-
specific molecular fingerprints forassessment of epidemiological relatedness
I N T R O D U C T I O N
The ability to quickly and reliably differentiateamong related bacterial isolates is essential forepidemiological surveillance, and is an endeav-our as old as the discipline of bacteriology itself.Long-standing ‘conventional’ typing methods,such as bacteriophage typing of Staphylococcusaureus and Listeria monocytogenes [1,2], serotypingof Salmonella spp. and Escherichia coli [3,4], orbiochemical typing of Enterobacteriaceae [5],have historically been important contributors toour understanding of the natural history andepidemiology of infections caused by strainsof these clinically relevant bacterial species.
Corresponding author and reprint requests: A. van Belkum,Erasmus MC, Department of Medical Microbiology and Infec-tious Diseases, Dr. Molewaterplein 40, 3015 GD Rotterdam, TheNetherlands. E-mail: [email protected]
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Similarly, antibiogram typing has for many yearsbeen and, as a matter of fact, still is, in the field ofclinical microbiology, a first-line method to iden-tify possible cases of bacterial cross-transmissionin healthcare institutions. These methods forbacterial phenotyping have a clear purpose inthe confirmation and elucidation of local andnational healthcare-associated outbreaks due tobacterial strains [1]. However, although stilluseful for specific purposes, they have a numberof practical limitations which render them unsuit-able for comprehensive studies of bacterial popu-lation structure and dynamics, and also for thescientifically less ambitious, but very critical,endeavours of infection control and surveillance[6,7]. Furthermore, most phenotypic methodshave been developed for specific bacterial speciesand are not generally applicable. However,although it is generally accepted that phenotyp-ing cannot usually stand alone, in some cases(e.g., serotyping of salmonellae), it is a veryuseful prerequisite. Nevertheless, the develop-ment, application and quality control of phagetyping and serotyping are labour-intensive andrequire skills and methodologies that are difficultto maintain at levels of quality sufficient to satisfythe standards of today’s accreditation bodies formicrobiology laboratories. More importantly, anygiven phenotype does not always accuratelyreflect the genotype of a microorganism, andtherefore may not provide a reliable and stableepidemiological marker. The rate of geneticexchange within many bacterial species meansthat a given phenotype may not always reflect
evolutionary history. For example, two isolatesthat are identical according to phage typingmight in fact be quite unrelated, and conversely,two isolates that show quite different phenotypesfor a single marker might in fact be closelyrelated. For these reasons, phenotyping has beenlargely replaced by genotypic or ‘molecular’typing over the past two decades [8–13]. Inprinciple, at least, asexual (clonal) reproductionby binary fission implies that genotypic markersshould reflect evolutionary history and wouldtherefore be useful in delineating a naturaltaxonomy. In practice, the ease with which genescan be transferred among different lineagesmeans that the data from multiple markers arerequired, and even then there is no guaranteethat a natural taxonomy will present itself[14]. Polyphasic taxonomy currently uses com-binations of different phenotypic or genotypicdatasets to define genera, species and eventaxonomically relevant subspecies [15–18]. Atthe same time, however, there are inherentlypolymorphic loci present in the genomes of allbacterial species that enable further subspeciesdifferentiation. Thus, DNA typing, which essen-tially comprises the direct or indirect assessment ofsubspecies nucleotide sequence motifs and theirvariation in both primary structure and number ofcopies per chromosome (see Fig. 1 for a generalisedscheme), can reproducibly reveal conserved as wellas variable characteristics, both at different taxo-nomic levels and at levels below species/subspe-cies, the lowest taxonomic rank with officialstanding in nomenclature.
Figure 1. The general features of molecular typing methods. The four boxes show the various molecular conceptsassociated with genetic variability. Below these boxes, the typing techniques most suited for the detection of such nucleicacid changes are indicated. More technical detail can be retrieved from various sections in the text.
2
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Unfortunately, new molecular typing methodsare often proposed for general use without suffi-cient prior critical evaluation. For example, theymay not have been standardised, a minimalnumber of isolates may have been used forvalidation, their agreement with epidemiologicaldata may not have been assessed, or the suitabil-ity of a specific method–microbe combination fora specific bacterial taxon may not have beenaddressed [19–28]. Finally, basic terminol-ogy—including fundamental terms such as ‘iso-late’, ‘strain’, ‘type’ or ‘clone’—is often useddifferently by different workers in the field ofbacterial epidemiology.
Here, we present an update of the previousESGEM guidelines for the correct applicationof methods and interpretation of the resultingdata [29]. We endeavour to define the terminol-ogy used in microbial typing, distinguish themajor means and purposes of bacterial typing,provide criteria for evaluation, and outline theadvantages, limitations and unresolved issuesrelated to the methods currently used. Weintend to increase awareness of the importanceof methodological evaluations and optimisa-tions, and the appropriate use of control andreference strains, as well as prudent data inter-pretation. In short, we aim to define the pur-pose and choice of methods, in combinationwith interpretation of the results, thereby facil-itating the development of practical decisiontrees. We suggest useful ways for the commu-nication of typing data in general, and morespecifically, communication from the laboratoryto the clinic. We include discussions on differ-ent typing applications and their globalisation,and, importantly, on quality control. Finally, thelinks between practical baterial typing andphylogeny, population biology and taxonomyare considered. This position paper has beendeveloped through interactions with microbiol-ogists active in the field, and aims to proposegenuine and applicable general typing guide-lines. These guidelines, however, should alwaysbe applied carefully and their consequencesinterpreted critically in all instances. Theintended audience includes, among many others,general and clinical microbiologists, infectiousdisease specialists, infection control managers,higher degree students, research technologistsinterested in the molecular epidemiology ofbacteria, decision-makers in the context of
public health, and workers in reference labora-tories.
D E F I N I T I O N S R E G A R D I N G I S O L A T ER E L A T I O N S H I P S
Bacterial typing has acquired its own vocabulary,in part borrowed from that of other scientificdisciplines, including population biology, molec-ular biology, taxonomy and ecology. Use of thisterminology is not always consistent and can beconfusing. Prior to presentation of a glossary, wewould like to discuss the terms ‘isolate’, ‘strain’and ‘clone’ in detail, in order to highlight some ofthe debatable issues concerning definitions, andthereby suggest a more standardised and uniformterminology.
The terms ‘isolate’ and ‘strain’ are often usedinterchangeably, but not always appropriately. Abacterial isolate can be defined simply as a singleisolation in pure culture from a clinical speci-men. Depending on the state of characterisation,an isolate may be referred to as, for example,‘urine isolate X’ (if only the sample type isknown) or ‘MRSA isolate Y’ (if the species andsome antimicrobial resistance properties areknown). Ultimately, isolates can be characterisedas descendants of the same strain. However,there is no agreement concerning the minimalsets of characters required to define any kind ofstrain. A reference strain is a well-characterisedstrain that is maintained in pure culture forfurther study, while a type strain is a specialkind of reference strain, i.e., the strain withwhich the name of the species is permanentlyassociated. An isolate can be assigned to adefined type according to the results of theapplication of a particular typing method, e.g.,pulsed-field gel electrophoresis (PFGE) type X,spa type Y. It must be noted that isolates withidentical typing results need not necessarilybelong to the same strain, since different strainsmay be indistinguishable with respect to atyping method. The opposite can also be true;isolates with different types may be part of thesame (pandemic) strain. This can be observedwhen the intrinsic evolutionary clockspeed of agiven species is higher than average. At present,different nomenclatures for bacterial strains,isolates and types exist and these must beconsidered with care and used appropriately.To ensure the consistent use of the terms ‘isolate’
3
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

and ‘strain’, we suggest the following example:two isolates (1 and 2) can be representatives ofone strain (A), but two strains (A and B) cannever be the same isolate (1).
The terms ‘strain’ and ‘clone’ are also usedinterchangeably. The ‘clone’ concept, which isfrequently used in the context of bacterial epi-demiology and population genetics, also illus-trates the importance of correct usage ofdefinitions and nomenclature. ‘Clone’ is a termcoined in the early 20th century in the field ofbotany and used to denote a group of isolatesdescended from a common ancestor as part of ausually direct chain of replication [30,31]. Theclonal relatedness of isolates is manifested bytheir display of a significantly higher level ofsimilarity in their genotype and/or phenotypethan can be expected for randomly occurringand epidemiologically unrelated isolates of thesame species. This epidemiological working def-inition is less stringent than the definitions of aclone used by microbial geneticists [31–35]. Theinterest in clones has increased over the pastdecades, due to the emergence of multiresistantor highly virulent clones of pathogenic bacteriathat have become widespread and seem toremain stable for prolonged periods [24–26,33–38]. Ørskov and Ørskov [31] proposed thefollowing formulation: ‘The word clone will beused to denote bacterial cultures isolated inde-pendently from different sources, in differentlocations, and perhaps at different times, but stillshowing so many identical phenotypic andgenotypic traits that the most likely explanationof this identity is a common origin.’ The oppositeof clonality is called panmixis, reflecting freeDNA recombination among isolates [35,39,40].Examples of panmictic bacterial species areHelicobacter pylori [41] and Neisseria meningitidis[42]. Isolates of panmictic bacterial species tendto display extensive genetic variability, and themolecular fingerprints of a single strain mayvary within a limited number of generations.
Since the terms ‘isolate’, ‘strain’, ‘type’ and‘clone’ have not always been used according tothe definitions given above, we propose defini-tions of a range of terms that are often used bybacterial typists. We hope that these definitionswill contribute to consistent usage amongtypists and scientists from affiliated fields suchas taxonomy and population genetics anddynamics.
G L O S S A R Y O F T E R M S
Some of the general terms defined below havebeen previously described in the literature[29,43,44]. The internet was scanned via theGoogle search engine, using the terms as keywords (search period November 2006). Thesedefinitions may have been adapted slightly tomake them consistent with technological andphilosophical approaches.
Alert organisms: Bacterial species, strains,types or clones of special epidemiologicalsignificance because of their predictable trans-missibility and potential for causing difficult-to-treat infections. Identification of such anorganism should alert healthcare providersand trigger additional control measures suchas barrier isolation of colonised or infectedpatients. Alert organisms are usually impor-tant nosocomial pathogens or organisms withan unusual antibiotic susceptibility profile.
Bacterial epidemiology: The study of thedissemination of human bacterial pathogens,including their transmission patterns, risk-factors for and control of infectious disease inhuman populations.
Clonal complex: A group of bacterial isolatesshowing a high degree of similarity, ideallybased on near-identity of multilocus enzymeprofiles and multilocus sequence types. Clonalcomplexes are identical to clonal groups.
Clonal reproduction: Mode of, usually, asex-ual reproduction in which the offspring areessentially identical to the parent. In bacteria,clonal reproduction proceeds by binary fission.
Clone: Bacterial isolates that, although theymay have been cultured independently fromdifferent sources in different locations andperhaps at different times, still have so manyidentical phenotypic and genotypic traits thatthe most likely explanation for this identity is acommon origin within a relevant time span.
Cluster analysis: Comparative analysis oftyping data collected for a variety of bacterialisolates in order to group the organismsaccording to their similarity in these data.Clusters can be identified by manual (visual)or computerised methods. The partitioning ofa dataset into subsets (clusters) reveals groupsthat share common traits.
4
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Comparative typing: A typing strategy aim-ed at assessing relatedness within a set ofisolates without reference to other isolates.
Convergence: Independent evolution alongparallel paths in unrelated lineages that ren-ders the lineages similar for some trait.
Definitive (library) typing: Type allocationof organisms according to an existing typingscheme aimed at the development of(exchangeable) databases for long-term retro-spective and prospective multicentre studiesas well as epidemiological surveillance studies.
Dendrogram: Binary tree illustrating a clus-ter analysis performed on a number of isolatesfor any chosen number of typing data. Eachtree, depending on the cluster algorithm used,depicts possible relationships between theisolates included in the analysis. The basis forthe tree is all the pairwise comparisons amongthe included isolates.
Endemicity: Constant presence in a com-munity at a significant frequency, typicallyrestricted to, or peculiar to, a locality or region.This usually presents as persistent occurrenceof disease in a population with a stable long-term pattern of incidence around short-termstochastic fluctuations.
Endemic: Strain present in a given settingover a longer period than if it were epidemic,although possibly at a relatively low frequency.
Epidemic: The occurrence of an organismabove the usual endemic level as evidenced by alarger than expected number of infections. Usedas an adjective, the rapid and extensive spreadby infection and/or colonisation that are widelyprevalent, i.e., affecting many individuals in anarea or a population at the same time.
Epidemic strain: A strain that is suddenlypresent in a given setting with an unexpect-edly high incidence. (However, it is sometimesdifficult to determine whether increased inci-dence is due to strain traits, since there maywell be other explanations, e.g., poor hygienicconditions.)
Evolutionary or phylogenetic tree: A dia-gram that depicts the hypothetical phylogeny(evolutionary history) of the taxa under con-sideration. The points at which lineages splitrepresent ancestor taxa to the descendant taxaappearing at the terminal points of the tree.
Fingerprint: A specific pattern (e.g., DNAbanding pattern) or set of marker scores (e.g.,absorbance values) displayed by an isolate onapplication of one or more typing methods.These fingerprints may be used for assessmentof epidemiological relatedness among bacterialisolates.
Fitness: The performance of a bacterialisolate/strain in a particular environment interms of survival and reproductive rates.
Genetic drift: The process of random sam-pling of alleles for each generation, which isrelatively important in small populations, andis an alternative evolutionary force for naturalselection, causing allele frequencies to change.Genetic drift determines the distribution ofalleles in different generations.
Genome: The complete genetic informationof an organism as encoded in its DNA and/orRNA.
Genotype: Genetic constitution of an organ-ism as assessed by a molecular method.
Hierarchical clustering: A method thatemphasises how adjacent spatial units withhigh or low disease rates might cluster byranking the units by disease rate, and thenexamining how probable cluster adjacencieswould be compared to random conditions, andmarking off successive clusters wherever low-probability values occur.
Isolate: A population of bacterial cells inpure culture derived from a single colony. Inclinical microbiology, isolates are usuallyderived from the primary culture of a clinicalspecimen obtained from an individual patient.
Lineage: Group of isolates sharing essentialcharacteristics due to common descent.
Linkage disequilibrium: Non-random re-assortment of alleles occurring at different locidue to physical linkage, usually due to lack orinhibition of recombination; strong in clonalorganisms and absent in freely recombiningpopulations.
Mutation: The simplest mutation (change) in aDNA or RNA sequence is a point mutation (aone-nucleotide change); other mutations includedeletion or insertion of one or more nucleotides.
Niche: A unique environment or set ofecological conditions in which a specific(micro)organism occurs and thrives.
5
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Outbreak: Local, initially small-scale, clusterof disease generally caused by increased fre-quency of infection in a distinct population(may be caused by single epidemic strains orcombinations of different strains).
Panmixis: Situation in which gene exchangeoccurs randomly in the population at a highrate. Isolates of panmictic bacterial species(e.g., H. pylori and N. gonorrhoeae) tend todisplay extensive genetic variability, and abso-lute fingerprint identity may vary even withinlimited numbers of generations.
Pathogenicity: Biological ability to causedisease.
Pattern analysis: The process of comparingdata patterns generated by one or more typingmethods.
Phenotype: The observable characteristics ofa bacterial isolate/strain. Primary phenotypemarkers are the distribution of proteins andother cell components and the morphologyand behaviour of cells.
Phylogeny: Evolutionary relationshipsamong members of the same taxon (species,strains, etc.).
Population: A group of organisms of thesame species inhabiting a given environment.
Population dynamics: The study of factorsaffecting the variability of populations ofmicroorganisms over time and space, includ-ing the interactions of these factors.
Population genetics: The study of variationin genes among a group of individual bacterialstrains, including the genetic evolution ofpopulations.
Selection: A natural process resulting in theevolution of an organism that is best adaptedto a (selective) environment.
Species: The basic taxonomic category ofbacteria; a named group below the genus levelwhose members show a high degree of overallsimilarity as compared with other, more distantlyrelated, strains. There is currently no universallyaccepted species definition in the context ofbacteriology, despite many attempts.
Sporadic: Rare, occurring at unpatternedirregular moments and localities, disconnectedin space and time; the opposite of epidemicand endemic.
Strain: The descendants of a single isolationin pure culture, usually derived from a singleinitial colony on a solid growth medium.A strain may be considered an isolate or groupof isolates that can be distinguished from otherisolates of the same genus and species byphenotypic and genotypic characteristics. Cul-tures of a particular microorganism, isolated atthe same time from multiple body sites of apatient and indistinguishable by typing, alsorepresent a single strain.
Taxonomy: Theoretical study of organismclassification, which involves the sequential,interrelated activities of allocation of organ-isms to taxa, their nomenclature and identifi-cation.
Type: A bacterial isolate may be allocated toa named type according to an existing typingscheme. Type designations aim at facilitatingthe handling and communication of typingresults, and the development of (exchangeable)databases for long-term retrospective and pro-spective multicentre studies, as well as epide-miological surveillance studies.
Type strain: A strain, maintained in pureculture, with which the name of the species ispermanently associated. The type strain of aspecies is marked by a superscript T at the endof its identification number. The type strain issimply one of the first specimens of adescribed species. Unfortunately, many so-called type strains are in fact atypical speciesrepresentatives.
Typing: Phenotypic and/or genetic analysisof bacterial isolates, below the species/subspe-cies level, performed in order to generatestrain/clone-specific fingerprints or datasetsthat can be used, for example, to detect or ruleout cross-infections, elucidate bacterial trans-mission patterns and find reservoirs or sourcesof infection in humans. ‘Subtyping’, a termcommonly seen in American literature, is oftenused as a synonym for typing.
Virulence: The property of an infectiousagent that determines the extent to which anovert disease is produced in an infected pop-ulation.
6
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

W H A T I S T Y P I N G A N D W H A T A R ET Y P I N G M E T H O D S ?
Pathogenic bacteria replicate and persevere inecological niches called reservoirs. Reservoirsmay be humans, including (fellow) patients andhealthcare personnel, animals, plants, water,food and various niches in the environment.Transmission of bacteria from any of thesesources may generate clusters of colonisation orinfection among humans. Such clusters arerecognised mostly as outbreaks of infectiousdiseases. When these outbreaks are not con-trolled, major epidemics (due to unrestrictedfurther transmission) may arise. Bacterial epide-miological typing generates isolate-specific geno-typic or phenotypic characters that can be usedto elucidate the sources and routes of spread ofbacteria [46,47]. The scope of typing studies mayvary from purely ‘clinical’ (dissemination ofinfections from patients, animals or other sourcesto non-colonised and uninfected individuals) to‘environmental’ (the presence or spread oforganisms in inanimate surroundings) or even‘industrial’ (identification of organisms that areeither valuable or a menace to bio-industry).Typing may also be used to identify emergingpathogenic strains or clones within a species,including potential agents of bioterrorism, inforensic biology and as evidence in medico-legalcases. A variety of methods have been developedto generate isolate-specific fingerprints, for epi-demiological typing. These methods should facil-itate the determination of the relatedness amongisolates derived from outbreak situations orobvious and recent chains of transmission, inorder to support or reject the hypothesis that theisolates come from a single source.
Typing data should always be consideredwithin the time-frame and current epidemiolog-ical context that are being evaluated and fromwhich bacterial isolates have been obtained. Forexample, more variability can be expectedbetween related isolates when longer time peri-ods are studied. The main focus of data inter-pretation in the clinical setting would be toidentify sources, as opposed to reservoirs ofinfection or colonisation [48–50]. Thus, typingdata can distinguish between cases linked to anoutbreak of infections and those unrelated casesdue to more complex scenarios. In addition,markers of biological diversity can also be
relevant to taxonomy, ecology and the study ofpathogenesis.
To put it simply, typing applies distinct labels tobacterial isolates. These labels facilitate identifica-tion of transmission routes and sources. However,they can also contribute to in-depth investigationsof infectious disease pathogenesis, bacterial pop-ulation structures and baterial genetics.
Typing can be considered as either comparativeor definitive (library) typing. In comparativetyping, outbreak-related and unrelated isolatesare compared, since comparison of outbreak-related isolates with isolates from the past or thefuture is not relevant. This is sometimes consid-ered sufficient for outbreak investigation [20].However, in many outbreak settings, be theynosocomial or community-based, it is often usefulto compare strains from a current outbreak withprevious strains, in which case a definitive(library) typing method should be used. There-fore, it is important to set up and maintaincollections of alert organisms in any typinglaboratory. Library systems are those that can beused in different laboratories, by different inves-tigators at various time intervals, with the aim ofgenerating high-quality data to be aggregated in asingle database for comparative assessment, ingreat detail at any time [51]. It is thus importantthat the typing methods are robust and suffi-ciently standardised to monitor the organisms ofinterest. While various multicentre studies aimedat standardising potential library typing methodshave been undertaken with varying success, therealready exist a number of international networksincorporating databases compiled on the basis ofmolecular typing data.
Typing can be undertaken at different levels,depending on the situation: locally, at a hospitalor other primary laboratory, for small investiga-tions; regionally or nationally, in a referencelaboratory, to bear upon wider issues of publichealth and surveillance; or internationallythrough collaborative networks, to define orsurvey the worldwide dissemination of majorbacterial clones. At each of these levels, differentmethods may be applied.
S E T T I N G U P S T R A I N C O L L E C T I O N SF O R T Y P I N G L A B O R A T O R I E S
The initiation and maintenance of strain collectionsare prerequisites for an epidemiological typing
7
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

study. The collection should comprise strains of thespecies of interest: epidemiologically unrelatedstrains, sets of strains from outbreaks, and pro-spective clinical isolates with well-defined inclu-sion criteria. The number of strains and thecomplexity of the collection are dependent uponthe objective(s) of the research. The organismsshould be stored preferably in glycerol broth at)80�C or freeze-dried according to accepted guide-lines for strain preservation. Such collections are ofmuch less value in the absence of a(n) (electronic)database of relevant clinical, epidemiological anddemographical data concerning the strains at-tached. Combining typing data with clinical anddemographical data is deemed to be extremelyimportant in deriving useful conclusions frominfectious diseases surveillance data. The com-bined data should comprise: strain designation,eventual other designations, species name, theoriginal specimen and its origin, date of isolation,hospital, department, patient code, city, country,and—for external strains—identity of provider.Other relevant (optional) data are: antibiogram,species identification method, and possible associ-ation with an outbreak or otherwise. For strategicpurposes, it is worthwhile to set up integrateddatabases linking the hospital information system,strain collection database and typing result data-base, using appropriate software, either commer-cially acquired or developed in-house.
R E A S O N S F O R T Y P I N G
Typing methods are used to study the spreadand population dynamics of bacteria and othermicroorganisms in clinical and environmentalsettings, at levels ranging from a single host to aglobal ecosystem. To date, these methods aremost easily and conveniently applied to haploidorganisms [40], but interest in the use of meth-ods for typing of diploid organisms, includingparasites, yeasts, fungi and plants, is growingrapidly [52,53]. Finally, space (flight) microbiol-ogy and the prevention of bioterrorism are newfields in which microbial typing is useful. Inforensic biology, nucleic acid technology isapplied to human materials [54,55]. Interestingly,human forensics and microbial typing meetwhere bacteria can be used to collect criminalevidence or to scan crime scenes [56]. Finally,genotypic methods can also be used in microbialtaxonomy.
Surveillance of infectious diseases
Typing methods contribute useful information toepidemiological surveillance of infectious dis-eases, defined as a systematic, ongoing processof data collection, analysis, interpretation, dis-semination of results, and action taken, aimed atrecording disease trends and designing ways inwhich to curb them [48,57–59]. Detection ofclusters of defined pathogens (alert organisms)with a similar type may constitute an ‘earlywarning’ of a potential outbreak. Library typing,such as serotyping, phage typing, PFGE or mul-tilocus sequence typing (MLST), is mandatory foradequate surveillance of infectious diseases (forexamples, see Pitt [20]).
Outbreak investigation
An outbreak can be defined as a temporalincrease in the incidence of infection (or coloni-sation) by a certain bacterial species, caused byenhanced transmission of a specific strain. It hasto be noted that outbreaks can also be caused bymultiple strains. The increased occurrence of asingle strain, therefore, needs to be distinguishedfrom the fortuitous accumulation of sporadiccases. Nevertheless, while this holds true forhealthcare-associated infections, it should be keptin mind that in the case of foodborne infections,for example, multi-strain outbreaks can alsooccur. This situation is one of the many instanceswhen accurate epidemiological and clinicaldescriptions are needed to prepare the designand corroborate the results of typing.
In this context, typing methods are applied togenerate and test hypotheses. Typing determinesthe number of strains causing the increasedincidence and, ultimately, should help identifythe source(s) of contamination and the route oftransmission. Correct application of bacterial typ-ing will increase the efficacy of control measuresaimed at containing or interrupting the outbreak[60,61]. Unfortunately, the relevance of typing ininfection control strategies is still under-appreci-ated. Didactic instructions should, therefore, beprovided to those using typing in relation toinfection control [62–64]. This should lead to animproved understanding of methodology and abetter overall appreciation of the added value ofepidemiological typing in the clinical setting. Costsavings can be derived from curbing unnecessary
8
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

investigations or control measures when a sus-pected outbreak is dismissed as an accumulationof sporadic cases derived from a single source.
Study of pathogenesis and the course ofinfection
We have already briefly mentioned the two majoruses of typing in studying infections affectingmore than one patient. However, typing can alsobe used to elucidate the progress of infection in asingle patient, e.g., by differentiating between aninfection from endogenous microflora and thatfrom an exogenous source [65]. When typing isused to compare groups of strains that are eithervirulent or non-virulent, pathogenesis-relatedmarkers can be identified. Such markers canultimately be translated into clinically relevantdiagnostic targets.
Study of bacterial population genetics
Last but not least, some molecular typing systemsmay be applied to large numbers of isolates fromvarious origins in order to determine the intra-species population structure, and derive phylo-genetic hypotheses from this structure [33–35,66].For example, PFGE analysis of the Pseudomonasaeruginosa genome indicates that the averagegenomic pattern similarity of unrelated strainsranges between 20% and 60% with an average of35%, whereas clonally derived strains from asingle host cluster at similarity levels above 80%[66,67]. Similarly, high-resolution genomic finger-printing of Acinetobacter has revealed that strainsof the same species cluster at 50% similarity ormore, while the clone and strain delineation levelsare approximately 80% and 90%, respectively[68–70].
The current typing method of choice forperforming bacterial population genetics studies,and the one with the soundest biological basis, isMLST [71]. This sequence-based technique hasbeen applied to many important pathogens andhas provided valuable information concerning theevolution and diversification of these species. Inparticular, these data have provided the means toestimate how commonly bacterial genomes un-dergo horizontal gene transfer and the impor-tance that this process may have for theemergence of clinically relevant strains withheightened virulence or drug resistance [72–75].
Technological aspects of the MLST method will bediscussed in more detail in later sections of theseguidelines.
C R I T E R I A F O R T H E E V A L U A T I O NA N D V A L I D A T I O N O FT Y P I N G M E T H O D S
Before a typing method may be used in a givensituation, its appropriateness must have beenclearly demonstrated. Every typing method there-fore needs to be evaluated and validated withrespect to a number of criteria [76–78]. These canbe divided into performance and conveniencecriteria. Because different investigations maydepend on different means and have differentrequirements, there is no ideal, universally appli-cable bacterial typing method [8]. Nevertheless,the increasing need to communicate among labo-ratories and to exchange outbreak investigation orsurveillance data requires some degree of agree-ment on common methods. Such standardisationis, of course, a lengthy and difficult process, but isgradually being undertaken for the most popularand dependable typing methods.
Performance criteria
A good typing method should assess a markerthat remains stable during the study period, anddoes not vary to a degree that confuses theepidemiological picture. This marker should betestable in every isolate, i.e., it should provideuniversal typeability of all isolates. It should alsousefully discriminate among isolates, and thisdiscrimination should be concordant with theepidemiological picture. Finally, the results of agood typing method should be reproducible, inde-pendently of the operator, place and time [79–81].A high degree of reproducibility will in turn makethe results of the method amenable to inclusion indatabases and analysis by dedicated computersoftware.
StabilityThis refers to the stability of the markers assessedby the typing method: a strain’s marker scoreshould not change rapidly and should correspondwith the strain’s position in the epidemiologicalcontext. For example, the characteristics tested by atyping method should remain stable for eachisolate after its primary isolation and during
9
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

laboratory storage and subculture. Preferably, theassessment of stability should also be performed inan in-vivo system. Although this may not alwaysbe possible, successful examples have beenreported in the literature [92]. Because mutationsand recombination occur at frequencies dependentupon species, strain and environmental condi-tions, the stability of the marker(s) tested by eachmethod should be evaluated for each bacterialspecies studied [93,94]. Stability and reproducibil-ity (see below) are concepts that are sometimesconfused. To test stability, multiple subcultures ofthe same isolate, stored over different periods andunder different conditions, have to be processed inthe same run to minimise laboratory-introducedvariations [95]. A marker can also be considered tobe stable if multiple isolates of an epidemic strainobtained from different patients at differentmoments are indistinguishable by typing basedon that particular marker.
TypeabilityThis refers to a method’s ability to assign a type toall isolates tested by it. It can be expressed as thepercentage of typeable isolates over the totalnumber of typed (typeable and non-typeable)isolates [82–84]. Whereas most of the genotypingmethods can characterise all of the isolates withina population (100% typeability), typeability canbe low with classic phenotypic methods such asserotyping, due to the fact that the existingserotyping schemes do not cover genetic variationin full.
Discriminatory powerThis refers to a method’s ability to assign adifferent type to two unrelated strains sampledrandomly from the population of a given species.It can be expressed as a probability using Simp-son’s index of diversity [85,86]. Hunter andGaston’s modification of Simpson’s index ofdiversity and fixed confidence intervals areimportant parameters used for making a decisionon strain identity or diversity [86]. The formulaused to define the diversity index or, better,Simpson’s index of diversity D is:
D ¼ 1� 1
NðN� 1ÞXS
J¼1
njðnj � 1Þ;
where N is the total number of strains in thesample population, S is the total number of types
described, and nj is the number of strainsbelonging to the jth type. The index should ide-ally be 1.00 but, in practice, it should be at least inthe order of 0.95 for a typing system to be con-sidered more or less ‘ideal’. A 5% probability oferror is accepted by most professionals in thefield. Calculations of the diversity index shouldbe accompanied by critical assessment of theconfidence interval, although this is very rarelydone [87]. Typing methods exploring polymor-phisms at multiple sites of the whole genome aremore likely to be more discriminatory than aremethods exploring variation at a single locus. Forthe purpose of calculation, non-typeable strainscan be either excluded or grouped together,although the latter does not imply that they are ofthe same type. In order to avoid overestimatingthe discriminatory power of a system, it is bestthat all untypeables be assembled into a singlegroup.
Epidemiological concordanceThe results of a typing method should reflect,agree with, and possibly further illuminate theavailable epidemiological information about thecases of colonisation or infection under study.For example, epidemiologically related isolatesderived from presumably single-strain or single-clone outbreaks should be assigned to identical orrelated types [22,23]. When validating a method,it is desirable that several sets, e.g., five or more,of outbreak-related strains (n = five to ten isolatesper set) are included in the test population (seebelow). Phenotypic methods are usually lesslikely to be concordant with epidemiology when,for example, distinct strains display similar phe-notypes (due to evolutionary convergence) [96].
ReproducibilityThis refers to the ability of a typing method toassign the same type to an isolate tested onindependent occasions, separated in time and/orplace [88]. The reproducibility of a marker pattern(or data generation in general) and that of typeassignment (data interpretation) may be different,and both need to be evaluated. Reproducibilitymay be influenced by many steps in a procedure,as a result of either the protocol used or thestringency of its application. Factors to considerinclude: the preparation of materials (e.g., varia-tion in growth conditions, and methods of DNAextraction), different batches or reagents, or
10
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

reagent variation as a result of local preparation,different types of equipment, bias in observingand recording the results, and, finally, analysisand interpretation of results. Reproducibility hasboth intra-laboratory and inter-laboratory dimen-sions. Both require standardised protocols andadequate personnel training to ensure a reliablemethod that produces results that are ‘fit forpurpose’ for different organisms in differentsettings [89–91].
Test populationAn appropriate and well-defined test populationis a prerequisite for evaluating the typeability,discriminatory power and epidemiological con-cordance of typing methods. Note that thenature of such a population is, of course, definedby the epidemiological context, the species oforganism involved, whether the studies are local,regional or global, and whether long-term sur-veillance is required. A large test population ofisolates correctly identified to the species level(preferably n > 100) should be assembled toreflect as much as possible the diversity expectedin the species as a whole, or at least in the sub-population to which the typing method will beapplied [20–23]. It is recommended to cover asmany ecological niches as may be included infuture investigations, such as particular patientpopulations (including age category, immunestatus, type of hospital and ward, geographicalorigin) and relevant environmental reservoirs(e.g., for zoonoses or foodborne and waterborneinfections). The test population should includestrains that are presumably unrelated epidemio-logically, on the basis of detailed clinical andepidemiological data, as well as outbreak-relatedisolates. For these reasons, it is important thathospital epidemiologists invest in prospectivecollections of organisms that have given rise toimportant healthcare-associated outbreaks. Thetest population is distinct from the panels ofcontrol isolates that should be used in manystudies. For example, in outbreak investigations,the appropriate level of discrimination of thetyping method(s) should be confirmed by com-paring the outbreak-related strains to a set ofcontrol strains (n = 10–30) from a similar timeperiod, locality and patient population, butwhich are, a priori, not epidemiologically related.We feel compelled to emphasise that, althoughthe earlier version of the current guidelines was
published more than 10 years ago, it has notbeen adopted very widely. Publications in whichappropriate test populations are analysed indetail are rare, and the mathematics required tosupport the corresponding conclusions arehardly ever applied.
Convenience criteria
Once the intrinsic value of a method, as well as itsappropriateness for the typing of a specific spe-cies, has been established on the basis of theperformance criteria discussed above, another setof criteria, those related to feasibility or conve-nience, need to be considered. These are impor-tant for the selection of an appropriate typingmethod, depending on a number of factors, suchas the scale of the investigation, the timelinessrequired of the results, and the financial andtechnical resources available. The following crite-ria of convenience, therefore, need to be consid-ered: flexibility, rapidity, accessibility, ease of use,costs, and suitability for computerised analysisand storage of results [97]. The portability ofresults is being improved continuously, and thislatter criterion is becoming increasingly impor-tant.
Flexibility (or spectrum)This reflects the range of species that are typeablewith minimal modifications of the method [98].The broader the range of bacterial species that canbe studied, the more central the position of themethod in the general typing laboratory will be.Modern DNA sequence-based methods showoptimal flexibility in the sense that the principle,as well as the skills and equipment required, arethe same for different species. Nevertheless, thesemethods still need to be optimised and validatedfor each species of interest; e.g., amplificationprimers developed for one species are usually notuseful for another.
RapidityThis refers to the total time required to get fromthe bacterial isolates to the final typing results.The highest degree of typing rapidity can beattained with methods that are applied directly toclinical materials, the so-called culture-indepen-dent procedures [99,100]. Ideally, typing shouldbe performed in ‘real time’; having results avail-able within a single working day would strongly
11
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

enhance the clinical impact of epidemiologicaltyping in general medicine.
AccessibilityThis depends upon the availability of reagentsand equipment, as well as the skills required for agiven method in a given laboratory.
Ease of useThis encompasses technical simplicity, workload,suitability for processing large numbers of iso-lates, and ease of scoring and interpreting theresults.
CostThis depends on numerous factors. For example,there is the amount of the initial capital outlay forthe equipment, its depreciation, which willdepend on whether it is out-of-date comparedwith newer versions or totally new platforms, thefrequency and care with which it is used, andfinally, the costs of any modifications to the room.The latter could include the additional options ofextra air-conditioning and floor reinforcement.The costs of servicing, the price, need for andready availability of replacement parts, and thecost of consumable reagents should also beconsidered. Then there are staffing costs, whichwill depend on the time required to performprocedures, the number and grade of personnelrequired, their training and requirements fordemonstration of competencies for accreditationor other purposes. These costs can be offset, forexample, by income generation, which willdepend on the ability to provide typing servicesfor others or income-generating training coursesfor others to learn the typing method.
Amenability to computerised analysis andincorporation of typing results in electronic databasesThese two factors are most important for longi-tudinal comparison of large numbers of isolates.At the local (hospital) level, data obtained byrobust typing methods can be analysed elec-tronically or assessed visually. Visual interpre-tation, even when only small numbers ofisolates are studied, requires normalisation ofthe data prior to inspection [101]. Nevertheless,since clones are spreading among hospitals or inthe community, both regionally and globally, itis important that electronic databases be created,enabling microbiologists and public health insti-
tutes to monitor the spread of such strains orclones beyond the hospital level. Of course,computerised analysis is optimal in combinationwith library methods of typing, with MLST asthe current key example.
V A L I D A T I O N O F N E W M E T H O D –M I C R O B E C O M B I N A T I O N S
Application of any typing method requires care-ful assessment of its suitability for a species notyet analysed by it. New methods or variants ofexisting ones are published on a regular basis[102], but they vary widely in terms of how wellvalidated they are. It cannot be emphasisedenough that testing limited numbers of bacterialisolates without adequate follow-up, using non-validated technology in merely local applications,should be discouraged. In the current era, whencomplete genome sequences are available formultiple strains of most, if not all, clinicallyrelevant microorganisms, such sequence deposi-tories can generate important clues for the selec-tion of appropriate molecular typing targets.Protocols for frequently used typing methodsshould be validated according to the recommen-dations given in this article by networks of expertlaboratories. Subsequently, certified ‘end-user’laboratories should attentively adhere to theseprotocols. Admittedly, the latter simple statementis often difficult to translate into practice; thepersonal preferences of many scientists canseverely compromise the objective of workingaccording to a standardised protocol. In conclu-sion, inter-method validation is important andnecessary, both from a theoretical point of viewand from a practical perspective [103].
P R I N C I P L E S A N D O V E R V I E W O FC U R R E N T T Y P I N G M E T H O D S
Over the past two decades, a plethora of noveland often innovative typing methods has beendeveloped. These range from methods that assesssimple phenotypic traits to DNA sequencing.Previously, the comparison of phenotypic char-acters, which involves the comparison of appar-ent biological features of isolates, was oftenabandoned because of the problems with perfor-mance criteria already mentioned. Instead, meth-ods involving the comparison of genomic DNAfragments were adopted. DNA molecules (or
12
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

restriction fragments or amplified sections there-of) can be separated on the basis of their molec-ular size by gel electrophoresis. Such sizecomparisons assess differences in the length ofDNA fragments obtained from DNA from differ-ent bacterial strains. Whether the fragments ofDNA are natural (e.g., plasmids) or generated atrandom, by restriction enzymes or after amplifi-cation of the DNA using enzymatic DNA repli-cation (PCR), does not matter; size differences,provided that they are accurately determined, canbe excellent markers of strain differences.
By definition, the genome of every bacterialisolate is unique. The mere fact that DNApolymerases make copying mistakes during rep-lication suggests that no genome has a 100%identical counterpart [104]. However, such muta-tions must be compatible with nature; they mustbe neutral or at least in line with existingstructure–function relationships among the corre-sponding gene products. Hence, bacterial strainsdiffer with respect to their complete genomesequence, and DNA sequencing methodologiescan therefore be used to assess similarity ofstrains. A challenge for the near future is toassess which DNA sequences are useful epidemi-ological markers, a task that is greatly assisted bywhole genome sequencing [105–107].
Since far more detailed reviews exist concern-ing the technical aspects of typing methods[50,108], we will restrict ourselves to definingbriefly the common aspects and quality charac-teristics of the methods, without any claim tocompleteness. The diversity and plethora ofmethods available to the scientific communityare such that it is impossible to be comprehensivein the subsequent sections. Strategic literaturereferences will be included to facilitate andstimulate further reading. Important overviewsof typing methods can also be found in severalgeneral textbooks on the practical and theoreticalaspects of bacterial typing.
Phenotypic typing methods
Phenotyping may involve colony morphology,colour, odour and other macroscopic features,but most typing methods rely on traits thatrequire specialised technology in order to bedocumented. For example, they may assess,qualitatively and quantitatively, the ability ofisolates to grow in the presence of specific
substances (be they metabolites, drugs, bacterialtoxins or bacteriophages) and their expression ofspecific molecules (be they surface antigens orallelic variants of housekeeping enzymes). Allmethods require strict standardisation of experi-mental conditions, since phenotypes are generallyquite susceptible to changes in environmentalconditions. In a simple statement: phenotypingresults in the grouping of organisms according totheir similarity in characters resulting from theexpression of their genotypes.
Biotyping assesses biochemical characteristicsthat are known to vary within a given species.Typeability is usually excellent. Discriminatorypower is variable and, to optimise it, a largenumber of well-selected characteristics, e.g., meta-bolic reactions, needs to be included in the testscheme. Stability is dependent on the species andcharacteristic under consideration. The methodsare usually technically easy and inexpensive, thedata generated are simple to score and interpret,and all tests can be performed, even in thesmallest of laboratories, on large numbers ofisolates. If reproducibility is demonstrated, it canbe used as a library typing method [109,110]. Forinstance, commercial systems facilitating the mea-surement of large panels of ‘biotype characteris-tics’ have been developed. These systems useversatile redox technologies, enabling the quanti-fication of various biochemical reactions by colourreadings [111–114]. The main power of the systemlies in its ability to distinguish among strainswithin a species [115,116]. Phenotype reactionarrays are available and are useful tools inaddition to DNA and proteomic technologies.The reproducibility of biotyping is organism- andcharacter-dependent. It is rarely 100%.
Antimicrobial susceptibility testing (antibio-gram-based typing) can be performed either bydrug diffusion in solid growth media or drugdilution in liquid media using a variety ofmeasurement systems. Most clinical microbiologylaboratories perform some sort of antibiogramtyping, since its results are commonly used toguide chemotherapy. Therefore, this method hasimmediate clinical consequences also. Antibio-gram-based typing can, with appropriate selec-tion of drugs, be applied to most species.Discrimination is dependent on the diversity,stability and relative prevalence of the detect-able acquired resistance mechanisms in studyisolates. It is also dependent on the number of
13
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

antimicrobials (including antibiotics no longer inuse, such as neomycin, which are adequate forrevealing specific resistance mechanisms). Testingfor resistance to heavy metals (resistotyping), aswell as to disinfectants and antiseptics, canprovide useful typing information. The utility ofthis method can vary according to the stability ofresistance patterns, which can be insufficient foruse as a clonal marker. Some resistance determi-nants are plasmid-borne and can be readily lost inthe absence of selective conditions; in addition,resistance expression can be under the control ofcomplex regulatory systems [23]. Susceptibilityprofiles expressed as diameters of inhibitionzones combined with cluster analysis can provideuseful typing data as an adjunct to data generatedby other methods [117,118]. There exist large,international databases built around antibio-grams, including data on the geographical originand clinical nature of the isolates. Although theseare primarily used to estimate incidences ofresistance, they may, of course, also be consultedfor epidemiological queries concerning the spreadof specific resistance markers [119,120]. It is ofnote that similar resistance patterns may be due toconvergent evolution (as is the case with manyextended-spectrum b-lactamase-producing micro-organisms, for instance), which is a stronglyconfounding phenomenon.
Serotyping is traditionally the most importantphenotypic method that has been developed fromthe early days of microbiology. It has led tocomprehensive systems for typing of, for exam-ple, Salmonella and E. coli isolates. Most typingsera react with surface antigens. These systemsare still widely used in healthcare-associated orfood-associated microbiology laboratories. High-throughput procedures using defined sets ofpolyclonal or monoclonal antibodies have beenmade available [121]. Typeability and discrimina-tion, complicated by cross-reactions, are variable[8,21,22]. With adequate quality control of bothreagent and method, serotyping can be a repro-ducible, library typing method of wide applica-bility. Standardisation of preparation and testingconditions is important. Discrimination can some-times be improved by combining serotyping withSDS-PAGE, resulting in ‘western’ (immuno)blot-ting [8,23,122]. Some serotyping schemes (e.g., theone for E. coli [4] or M-protein typing of Strepto-coccus pyogenes [123]) are now being replaced bytheir genotypic equivalents, where variability is
assessed at the level of genes encoding for theantigens [124,125]. Similarly, restriction analysisof the amplified O-antigen gene cluster (‘molec-ular serotyping’) has proven to be an interestingalternative for classic serotyping of E. coli andShigella isolates [126,127]. Genetic instabilityper se, horizontal gene transfer and convergencedue to natural or vaccine-driven herd immunityintrinsically limit the power of serotypingmethods.
Phage and bacteriocin typing assess the lyticpatterns of test isolates that have been exposed toa defined set of bacteriophages, or bactericidaltoxins (bacteriocins). These traditional typingmethods are restricted to a limited number ofspecies for which such agents have been identi-fied in numbers large enough to provide a usefuldegree of discrimination. In addition, when newbacterial clones are discovered, additional phagesmay need to be included in the typing scheme.Types can change over the longer term, and thisin itself can be a useful characteristic in endemicsituations. Discrimination is therefore variable,typeability often partial, and reproducibility poor.The production and continuous quality control ofphages is important, requiring extensive expertiseand time-consuming efforts. However, largenumbers of isolates can be processed readily,which is not the case with most current DNAfragment-based typing methods. Interpretation ofresults is not easy and requires training andexperience [128,129]. Nowadays, acquisition orloss of phages, which may play a role in virulence,can be traced by molecular typing, providing amodern extension of the role of phage typing[130].
Phage typing has long been an important toolwith which to study the epidemiology of S. aureusfor example, but today it has lost its position as areference typing method.
SDS-PAGE of cellular and extracellular com-ponents can give rise to highly discriminatorytyping methods, with applications in taxonomyalso [16,131–134]. In the 1980s, these methodswere applied to a variety of organisms, but sincethe 1990s they have been largely superseded byDNA-based methods. Interestingly, the need forcomparative analysis of the complex bandingpatterns obtained by protein SDS-PAGE was thetrigger for the development of dedicated com-puter software that is now successfully applied toDNA fragment analysis. By protein SDS-PAGE,
14
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

cell envelope fractions obtained by sonication andstepwise centrifugation, or whole cells, are solu-bilised in buffer with the denaturing agent SDSand separated under denaturing conditions byPAGE. After staining, the gels are digitised andthe images subjected to cluster analysis. If growthconditions, sample preparation and electrophore-sis are rigorously standardised, the profiles arereproducible and suited for databases for longi-tudinal analysis. Protein SDS-PAGE is ratherlaborious and requires experience; the advantageis that reagents and equipment are relativelyinexpensive.
The step from protein SDS-PAGE to lipopoly-saccharide (LPS) gel electrophoresis is relativelysmall, since the samples prepared for proteinanalysis can be treated with proteinase K, afterwhich they can be used for electrophoretic sepa-ration of LPS molecules, followed by silver stain-ing to visualise them. ‘Ladder-type’ LPS gelelectrophoresis can be strain-specific and hasbeen used for comparative typing, but the methodis not widely used because it is laborious [135–137].
Multilocus enzyme electrophoresis (MLEE)identifies electrophoretic variants of a set ofhousekeeping enzymes, encoded by differentalleles of the same gene, thus giving rise to smallbut detectable variations in protein size andcharge [138]. MLEE has been used as a referencemethod for defining the phylogenetic structure ofclonal lineages in bacterial populations [33,34].Although it is neither a rapid nor a widelyapplied system, it has been very important inshaping the bacterial population biology land-scape. Its molecular progeny, MLST (see below),is much more practical and, hence, more widelyused nowadays.
Mass spectrometry (MS) is a technique origi-nally developed for the identification of (primar-ily organic) molecules of a low molecular weightin complex mixtures [139]. Nowadays, the tech-nology can also be used to characterise mixturesof complex biological macromolecules, throughtheir specific degradation products. Matrix-as-sisted laser desorption ionisation time-of-flight(MALDI-TOF) MS facilitates the generation ofmolecular fingerprints for entire organisms [140–142]. The method uses intense laser light toevaporate the biological material, which is subse-quently subjected to a strong electrical field. Smallions move at high speed and reach a detector
before the larger ones. The signals generated arerecorded and give rise to complex spectra, char-acteristic for the molecular content of a bacterialcell. When these spectra are compared usingappropriate computer software, bacterial typescan be distinguished [143,144]. MALDI-TOF MS isalso suited for the analysis of less complexmixtures of, for instance, DNA molecules [145–148] (Fig. 2). Other spectroscopic methods, basedon alternative biophysical strategies, can be usedas well. Infrared (IR) or Raman spectroscopy aretwo such methods that can be used for isolatecomparison [149–151]. Both use focused illumina-tion of bacterial biomass and record the emissionspectra generated. The complexity of the spec-trum reflects molecular complexity and, althoughnot every peak in the spectrum can be assigned toa submolecular particle, the composite patternscan allow comparisons to be performed and typesto be assigned. Other spectroscopic and chro-matographic methods have been commercialisedsuccessfully and can provide useful platforms forcertain formats of bacterial typing. Gas–liquidchromatography (GLC; the widely used MIDIsystem) and Fourier-transform (FT)-IR spectro-metry/FT-IR microscopy are merely two exam-ples [145,152].
Other methods based on physics approacheswill certainly be developed over the coming
Figure 2. An example of the use of mass spectrometry forthe detection of sequence variation in PCR products. Afteramplification of the DNA stretch under investigation, RNAis transcribed. This is degraded in a sequence-specificmanner, and the degradation product is separated andidentified by mass spectrometry. This will result in reliablesequence determination. Illustration kindly provided byC. Honisch (MassCLEAVE; Sequenom, San Diego, USA).
15
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

decade. An interesting innovative example isprovided by optical mapping of DNA molecules.This method enables one to really visualise DNAfragments of large size and it can already be usedfor bacterial comparison by looking at singlegenomic DNA molecules [153]. Another interest-ing MS approach is the identification of genotypesof bacteria in complex mixtures of clinical sam-ples using MS and base composition [154,155]. Itis anticipated that the combination of two-dimen-sional protein (or DNA) separation techniques, incombination with spectrometric technologies, willopen possibilities of new generations of typingsystems.
Different ‘‘-omics’’ approaches complete themodern phenotyping spectrum. Proteomics col-lectively describes the methods used for deci-phering the protein content of a bacterial cell.These range from ‘intelligent’ electrophoresistechnologies to high-throughput, automated,MS-based protein sequencing facilities.
Glycomics analyses the synthesis, precisemolecular features and diversity of polysaccha-rides, glycans, lipopolysaccharides and otherglyco- and lipid complexes, while metabolomicsencompasses the diverse metabolic activity ofcells. Phenotyping is thus resurfacing with theadvent of systems biology approaches [156].
Genotypic typing methods
Genotypic typing methods assess variation in thegenomes of bacterial isolates with respect tocomposition (e.g., presence or absence of plas-mids), overall structure (e.g., restriction endonu-clease profiles, number and positions ofrepetitive elements), or precise nucleotide se-quence (of one or more genes or intergenicregions). Basic genetic analysis of the molecularevent(s) (acquisition, multiplication, mutation,deletion, insertion) associated with pattern var-iation is the preferred approach to measuringinter-strain relatedness, but is neither alwaysrequired nor generally feasible [13,157]. A widevariety of genotypic methods has been pre-sented, of which the most widely used will bediscussed below in a ‘rational-historical’ order.The increasing availability of bacterial genomesequences has had, and is still exerting, a greatimpact on the evolution of these methods,by facilitating the choice of successful typingtargets.
Hybridisation-mediated methods.Direct (and reverse) hybridisation: Direct hy-bridisation testing of bacterial genomic DNA(without restriction enzyme treatment) is feasible.In all methods, the immobilised DNA to beinvestigated is probed with DNA molecules thatare selective; some templates are recognised, andothers are not. The technologies employed varywidely, but the core technology was developed bySouthern and colleagues [158] (hence ‘Southernhybridisation’). As a recent example, ‘binary’typing has been developed for S. aureus throughthe isolation of DNA probes that are specific forsome S. aureus strains [159,160]. The methodproved to be reproducible and easy to perform[161,162]. Similar systems have been developedfor other bacterial species [163,164]. Direct hy-bridisation tests can also be used to define thenature of mobile elements involved in methicillinresistance or to identify determinants of glyco-peptide resistance in S. aureus [165–167]. Thesame methodology can be used for typing ofDNA amplified by PCR [168] For instance,Mycobacterium tuberculosis ‘spoligotyping’ in-cludes amplification of a locus harbouring tan-dem repeats with some internal sequencevariation. These variants are then identified byhybridisation using repeat-specific DNA probes[169,170].
Ribotyping is a classic variant of a Southernhybridisation-mediated assay [171] that estimatesthe number of ribosomal gene loci and theirposition in the chromosome. It is reproducibleand applicable to (fast-growing) bacteria, but hasa discriminatory power that is usually lowerthan that of, for example, PFGE [12,26,172]. Fullyautomated robots for ribotyping have beenmade available, reducing hands-on time, albeitat a significant price [173,174]. The automatedmethod has been compared with a variety ofother genotyping methods [175–182] and,although it was demonstrated to be useful forvarious bacterial species, it did not always standout as a superior method [183] since its discrim-inatory power is relatively limited. Nevertheless,it is robust, and profiles can be compared amonglaboratories and be used for the generation ofdatabases; hence, it was adopted for somepathogens important in food microbiology[174]. Reproducibility has been documentedexperimentally during clinical microbiologicalusage [181,184].
16
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Genome analysis by array hybridisationArray systems currently represent state-of-the-arthybridisation-mediated testing. This method ca-pitalises on the technological possibility of immo-bilising up to several hundred thousands of DNAprobes per square centimetre of a solid matrix.For most of the clinically relevant microorgan-isms, whole genome arrays have been developed,based on the available whole genome sequences,and covering all of the genes identified. Probesmay be PCR products of defined length, butsynthetic oligonucleotides are more frequentlyused. These platforms facilitate bacterial typing inunprecedented detail. As the method is not yetsuited for day-to-day clinical application, carefulconsideration of target genes is necessary in orderto achieve optimal epidemiological concordance.Currently, costs and accessibility also remainproblematic. A recent comparison of multiplegenomes of strains of the same species has shownthat considerable gene variation exists within aspecies, and the term ‘pan genome’ was coined todenote the cumulative genome deduced from theindividual genome sequences [185]. Hence, it isemphasised that analyses based on single-straingenomes of a given species are not likely to besufficient to make generalisations about the spe-cies as a whole.
Fragment-based methodsPlasmid typing assesses the number size and/orrestriction endonuclease digestion profiles, afteragarose gel electrophoresis, of these bacterialextrachromosomal genetic elements. It has beenused for typing of many bacterial species [9].Typeability and discrimination are variable,depending on the bacterial species [9]. However,the lack of stability of plasmid content rendered itunsuitable for use as a reliable clonal marker insome studies [186]. It is best combined with othergenomic typing methods, to distinguish, forexample, between spread of a resistant cloneand that of a resistance plasmid [23]. Plasmidtyping is still used frequently in combination withtesting of antimicrobial susceptibility in modernclinical microbiology laboratories [187,188] toassess whether an antibiotic resistance gene isplasmid-borne and can be transferred.
Among restriction fragment length polymor-phism (RFLP) methods, restriction endonucleaseanalysis (REA) was the first to be widely used.The chromosome is digested by frequently cutting
restriction enzymes into several hundreds ofsmall fragments, which are separated by horizon-tal gel electrophoresis into complex patterns [10].It is rapid and, under standardised conditions,very reproducible and discriminatory. However,the complex patterns produced complicate inter-pretation and hinder data exchange among labo-ratories. In order to simplify the interpretation ofREA results, Southern blot and hybridisationsteps were added. A variant, which is veryimportant historically, is ribotyping (mentionedabove), a method that couples genome digestionby a ‘frequent-cutting’ restriction endonucleasewith a 4-bp recognition sequence, and hybridisa-tion with a probe complementary to rDNA. Someof the hybridisation probes used are restricted to asingle species; the most illustrious and popularexample is IS6110typing of M. tuberculosis [25].This method has been the agreed standard amongtuberculosis reference laboratories worldwideover the past 15 years. It has been applied duringhundreds of studies, and its output has beenshown to be communicable among institutionsand over the years, as thousands of profilesgenerated in different laboratories have beenintegrated in a central database [79,189,190].
A new electrophoresis technique, PFGE, madeit possible to separate large DNA fragments inagarose gels by periodic alternation of the angle ofthe electric field’s direction. These DNA ‘mac-rorestriction’ fragments are generated withrestriction endonucleases with six or more basepair recognition sites (‘rare cutters’), usuallyyielding fewer than 30 large fragments, normallyranging in size between 20 and 600 kbp. PFGEwas originally used for electrophoretic separationof the chromosomes of lower eukaryotes [191],and has enabled epidemiological studies of yeastsand fungi [192,193]. Only in the case of excessiveendogenous endonuclease or DNA methylationactivities has PFGE been problematic [194]. How-ever, even these technical problems can be over-come by the use of chemical endonucleaseinhibitors and alternative restriction endonucleas-es. PFGE has remarkable discriminatory powerand reproducibility, and has therefore become awidely applicable method for comparative typingof almost all bacterial species [8,11,195,196]. Withcareful standardisation, acceptable levels of inter-laboratory reproducibility can be achieved, whichhave allowed the creation and maintenance ofinternational databases, with the PulseNet effort
17
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

representing an important achievement (see alsolater sections) [28,197–204]. However, 2–4 daysare required to obtain results and relativelyexpensive PFGE equipment is required. Gels needto be analysed closely and carefully, even afterdigitalisation and computerised processing [205].To confirm the outcome of the mathematicalanalysis and to verify, establish or refute finerdiscrimination, quality control is essential (seealso the later section on PFGE data interpretation).
PCR fingerprinting relies on the amplificationof genomic fragments flanked by one or twooligonucleotide sequences used as primers.These primers should preferably be cognate tothe species being typed (e.g., BOX for S. pneu-moniae [206] or IS256 for S. aureus [207]). Cognateprimers allow for relatively high annealing tem-peratures, thus contributing to high reproduc-ibility, in contrast to non-cognate primers suchas the very widely used ‘arbitrary’, random-sequence primers that range between six and tennucleotides in length. Primer pairs are oftendesigned to be directed outwards from repetitiveelements, to amplify short spacer sequenceslying between these elements. It is a quasi-universal typing method, exhibiting an easilyadjustable level of discrimination [13,208]. Itsmajor advantages include flexibility, technicalsimplicity, wide availability of equipment andreagents, and rapid, same-day turnover. How-ever, interpretation of band differences, of neces-sity, remains biologically unfounded (it cannever be known, for example, if other unob-served ‘spacer sequences’ existed that werelonger than what could be amplified by theDNA polymerase used) and, as suggested, thismethod can rarely be considered a ‘library’method. PCR ‘fingerprinting’ data, in general,are considered to be non-exchangeable amonglaboratories [27,195,209,210], although commer-cial tests claim the contrary [211]. In order toincrease the resolution of PCR fingerprinting, anRFLP step is sometimes added. One example ofa PCR-RFLP method is amplified ribosomalDNA restriction analysis (ARDRA), which hasbeen used successfully for species identificationof various organisms, including acinetobacters[43,212], while there are numerous examples ofthis methodology for typing of other bacterialspecies. Essentially, PCR-RFLP monitors for avariety of mutations that can occur in restrictionsites and, as such, is a variant method for
detection of single nucleotide polymorphisms(SNPs) (see later in ‘Sequence-based methods’).Yet another PCR-based typing method is ampli-fied fragment length polymorphism (AFLP)analysis. AFLPTM is the patented name of amethod designed to selectively amplify subsetsof genomic fragments generated with one or tworestriction enzymes, usually a ‘rare’ and a ‘fre-quent cutter’ [213,214]. After ligation of adaptersto the restriction fragments, selective amplifica-tion is achieved by the use of primers thatconsist of the adapter-derived core sequence,including the 3¢-part of the restriction half-site,and an extension of one or more selective bases.Elongation will only take place if a nucleotidecomplementary to the selective base in theprimer sequence is present in the fragment.Products can be separated in agarose gels [215–217], but usually one primer is labelled andfragment separation is obtained using an auto-matic DNA sequencing instrument with auto-mated data capture. The digitised and complexDNA fingerprints are generally highly reproduc-ible and have been used very successfully for thehigh-throughput molecular typing of large num-bers of bacterial isolates [218,219] (Fig. 3). Essen-tially, nearly whole genome coverage can beattained.
For some bacterial species, databases havebeen developed and inter-centre reproducibilityassessed [220–222,224]. Recent and, as yet,unpublished studies have revealed that AFLPmay also suffer from the absence of inter-centrereproducibility, especially when different elec-trophoresis platforms are being employed[220,221,223]. For Acinetobacter spp., as illustratedin Fig. 4, the method is useful to identify species[217], clones within Acinetobacter baumannii[69,118] and epidemic strains [218]. The profilesgenerated with labelled primers and automatedsequencing equipment are highly complex, anddedicated software for cluster analysis is there-fore mandatory (Fig. 4).
Multilocus variable number tandem repeat(VNTR) analysis (MLVA) is also a PCR-basedtyping method that capitalises on the inherentvariability encountered in many regions of repet-itive DNA. Repetitive DNA is often incorrectlycopied in bacterial species, through slippedstrand mispairing (SSM) [225–227], thus resultingin shortening or lengthening of the repeat regiondue to deletion or insertion of repeat units,
18
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

respectively [226]. This type of DNA variation canbe simply assessed by performing repeat-span-ning PCRs and determining the length of the PCRproduct. In the case of large repeat units, theanalysis system can be simple (e.g., agarose gelelectrophoresis). However, for shorter repeats,more complex electrophoresis or MS methods arerequired. For each repeat locus, a digit can beassigned, representing the number of repeatsimplied (by electrophoresis) or demonstrated (bysequencing). When assessing the length of theproduct, normalisation of the migration distancesis required to guarantee accurate length measure-ment (Fig. 5). When several repeat loci are anal-ysed per isolate, several such digits are obtained,resulting in a multi-digit, specific strain code[226]. Dedicated MLVA systems have beendeveloped for a variety of species [227–236].When compared with other genotyping meth-ods, MLVA has, in general, performed well[229,237,238]. However, few multicentre studieshave been undertaken. Given its techical simplic-ity, MLVA may have a successful future. The
major drawback is that the evolution of repetitiveDNA may be too rapid, compromising epidemi-ological concordance. When the mutation fre-quency in a locus is known and the frequency ofcertain alleles in a population is documented, it ispossible to calculate whether two isolates areidentical on the basis of chance. This is notfeasible with other fragment-based methods.Also, as for all of the methods that rely on theestimation of molecular size based on standardcurves, the accurate sizing of fragments, evenusing fluorescent detection systems, is not asimple task, as it is mobility dependent onsequence composition as well as length.
Sequence-based methodsSingle-locus sequence typing (SLST) is anumbrella term for a variety of methods, in whichsequencing of a single genetic locus has beenshown to provide valuable typing results. Anal-ysing a single locus means that the amount ofDNA to be sequenced is limited, but it is imper-ative to select gene sequences that are (highly)variable. The best example of an established,epidemiologically significant SLST scheme is thatof emm typing for S. pyogenes, which is the‘genotypic descendant’ of M-serotyping, andrelies on DNA sequencing of only 150 nucleo-tides, coding for the N-terminal end of an isolate’sM protein [239]. An international database, incor-porating a query module, ensures the continuingenrichment of the type repertoire, and alreadyincludes over twice as many types as thoseacquired through the previous use of anti-sera (http://www.cdc.gov/ncidod/biotech/strep/strepindex.htm). Another more recent example isthat of the S. aureus protein A gene, spa, whoserepeats are variable in number and individualsequence [239]. This feature formed the basis forthe currently used sequencing system, which hasbeen further elaborated upon and validated[240,241] (Fig. 6). The development of dedicatedsoftware and the possibility of determiningsequences rapidly have now led to an automatedsystem, which is 100% reproducible among dif-ferent centres [242–244]. In the case of typingstudies performed on the basis of DNA sequencesin hypermutable regions, it should be noted thatgeneration of variation may exceed the speed ofspread; mutants may arise during an outbreakand thus falsely suggest that the outbreak hasmultiple sources rather than one.
MboI/Csp6I AFLP for Staphylococcus aureus
Figure 3. Example of high-throughput amplified fragmentlength polymorphism analysis for 12 strains of Staphylo-coccus aureus. For the same set of strains, various selectiveprimer pairs were employed, the terminal, selectivesequences of which are identified on top of the lanes.Courtesy of G. Simons (Pathofinder, Maastricht, TheNetherlands) and H. Witsenboer (Keygene, Wageningen,The Netherlands).
19
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46
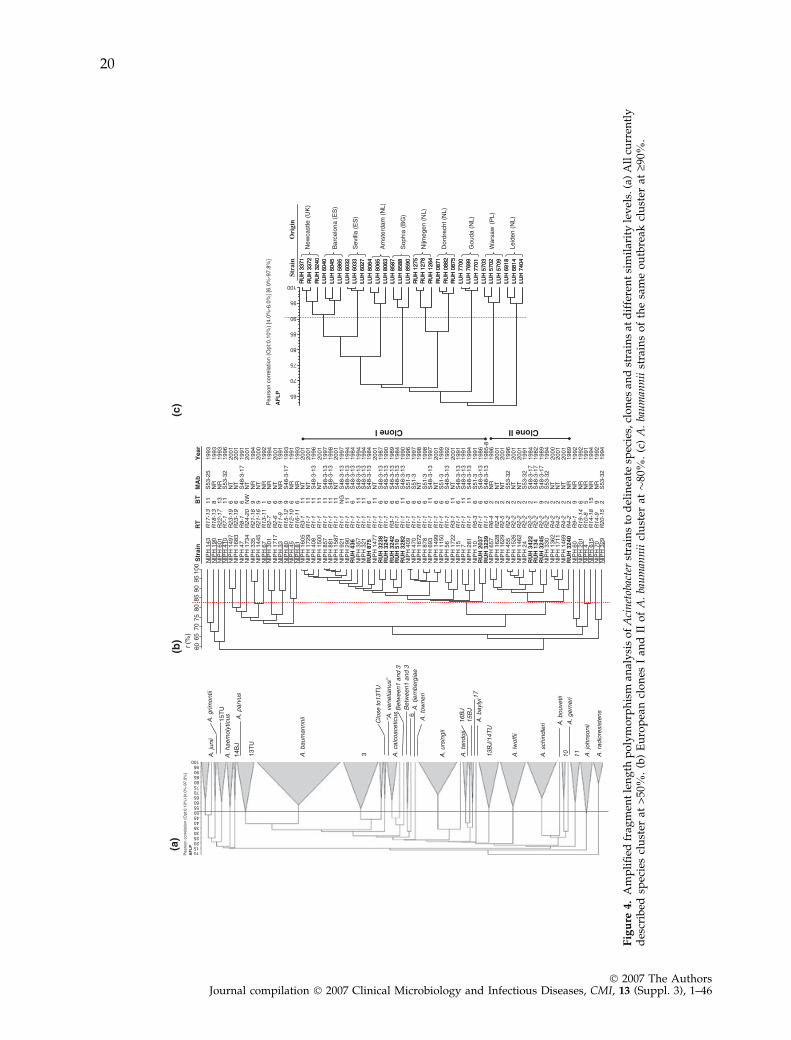
Fig
ure
4.
Am
pli
fied
frag
men
tle
ng
thp
oly
mo
rph
ism
anal
ysi
so
fA
cin
etob
acte
rst
rain
sto
del
inea
tesp
ecie
s,cl
on
esan
dst
rain
sat
dif
fere
nt
sim
ilar
ity
lev
els.
(a)
All
curr
entl
yd
escr
ibed
spec
ies
clu
ster
at>
50%
.(b
)E
uro
pea
ncl
on
esI
and
IIo
fA
.ba
um
ann
iicl
ust
erat�
80%
.(c
)A
.ba
um
ann
iist
rain
so
fth
esa
me
ou
tbre
akcl
ust
erat
‡90%
.
20
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46
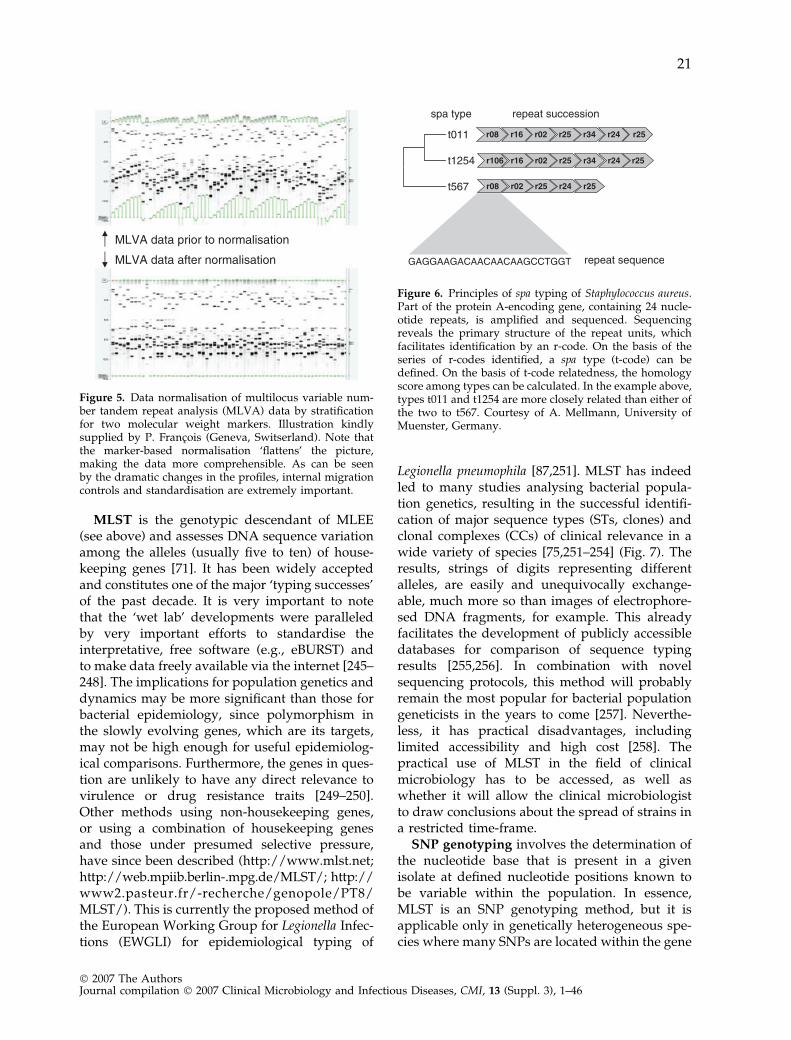
MLST is the genotypic descendant of MLEE(see above) and assesses DNA sequence variationamong the alleles (usually five to ten) of house-keeping genes [71]. It has been widely acceptedand constitutes one of the major ‘typing successes’of the past decade. It is very important to notethat the ‘wet lab’ developments were paralleledby very important efforts to standardise theinterpretative, free software (e.g., eBURST) andto make data freely available via the internet [245–248]. The implications for population genetics anddynamics may be more significant than those forbacterial epidemiology, since polymorphism inthe slowly evolving genes, which are its targets,may not be high enough for useful epidemiolog-ical comparisons. Furthermore, the genes in ques-tion are unlikely to have any direct relevance tovirulence or drug resistance traits [249–250].Other methods using non-housekeeping genes,or using a combination of housekeeping genesand those under presumed selective pressure,have since been described (http://www.mlst.net;http://web.mpiib.berlin-.mpg.de/MLST/; http://www2.pasteur.fr/-recherche/genopole/PT8/MLST/). This is currently the proposed method ofthe European Working Group for Legionella Infec-tions (EWGLI) for epidemiological typing of
Legionella pneumophila [87,251]. MLST has indeedled to many studies analysing bacterial popula-tion genetics, resulting in the successful identifi-cation of major sequence types (STs, clones) andclonal complexes (CCs) of clinical relevance in awide variety of species [75,251–254] (Fig. 7). Theresults, strings of digits representing differentalleles, are easily and unequivocally exchange-able, much more so than images of electrophore-sed DNA fragments, for example. This alreadyfacilitates the development of publicly accessibledatabases for comparison of sequence typingresults [255,256]. In combination with novelsequencing protocols, this method will probablyremain the most popular for bacterial populationgeneticists in the years to come [257]. Neverthe-less, it has practical disadvantages, includinglimited accessibility and high cost [258]. Thepractical use of MLST in the field of clinicalmicrobiology has to be accessed, as well aswhether it will allow the clinical microbiologistto draw conclusions about the spread of strains ina restricted time-frame.
SNP genotyping involves the determination ofthe nucleotide base that is present in a givenisolate at defined nucleotide positions known tobe variable within the population. In essence,MLST is an SNP genotyping method, but it isapplicable only in genetically heterogeneous spe-cies where many SNPs are located within the gene
Figure 5. Data normalisation of multilocus variable num-ber tandem repeat analysis (MLVA) data by stratificationfor two molecular weight markers. Illustration kindlysupplied by P. Francois (Geneva, Switserland). Note thatthe marker-based normalisation ‘flattens’ the picture,making the data more comprehensible. As can be seenby the dramatic changes in the profiles, internal migrationcontrols and standardisation are extremely important.
Figure 6. Principles of spa typing of Staphylococcus aureus.Part of the protein A-encoding gene, containing 24 nucle-otide repeats, is amplified and sequenced. Sequencingreveals the primary structure of the repeat units, whichfacilitates identification by an r-code. On the basis of theseries of r-codes identified, a spa type (t-code) can bedefined. On the basis of t-code relatedness, the homologyscore among types can be calculated. In the example above,types t011 and t1254 are more closely related than either ofthe two to t567. Courtesy of A. Mellmann, University ofMuenster, Germany.
21
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

portions considered. Strictly speaking, SNP geno-typing refers to the analysis of nucleotide poly-morphisms that are rare (e.g., less than one in 300bases) along the bacterial chromosome, renderingthe direct assessment of the base identity at thevariable position much more efficient than directsequencing of the surrounding region. Hence,SNP genotyping methods are primarily applied todefine the relationships among isolates of homo-geneous pathogens such as M. tuberculosis[259,260], Bacillus anthracis [261], E. coli O157:H7[262] or Salmonella enterica serotype Typhi [263].
Variable positions that are useful for typing orphylogenetic analysis must have been discoveredprior to the application of an SNP genotypingmethod. Mutation discovery can be achieved bygenome-wide approaches such as shotgunsequencing of several strains [264] or microarrayhybridisation-based comparative genomesequencing [262,265]. Polymorphisms can alsobe revealed by screening a number of definedtarget genes via sequencing, as in MLST-like
approaches or faster approaches such as denatur-ing High Performance Liquid Chromatography(dHPLC) [263]. In order to obtain a set of SNPsthat would represent the diversity of a species inan unbiased manner, it is crucial that mutationdiscovery be performed on a set of strains that arerepresentative of the breadth of diversity andphylogenetic lineages of that species. Indeed, setsof SNPs derived from comparison of strains thatare representative of a limited number of lineageswill mostly contain those SNPs that accumulatedduring the evolution of these lineages (but willignore SNPs that appeared in other lineages), aphenomenon called ‘discovery bias’ [259,266].
Once a set of SNPs has been selected, a givenbacterial sample can be screened by a variety ofSNP genotyping methods. Direct sequencing ofregions encompassing the SNPs either by Sangersequencing or by pyrosequencing is easy toimplement and reliable for determining the basepresent at the targeted SNPs [267]. However, asstated above, this approach is not efficient if theentire sequenced region contains only one orseveral SNPs. SNP genotyping methods, designedto be applicable to a high number of SNPs andsamples, are currently being developed at a fastpace, essentially because of the need for high-throughput SNP genotyping in human diversitystudies and pharmacogenetics. For example, themethod known as ‘mini-sequencing’ involves theuse of a mixture of all four dideoxynucleotides(without deoxynucleotides) to extend a primer bya single base. The identity of an SNP can bedetermined by using a primer ending just onebase upstream of the SNP. The incorporated basecan be determined by fluorescence after capillaryelectrophoresis [252] or by direct measurement ofthe mass of the resulting product by MS [268].Several reviews describe promising SNP geno-typing approaches [269–273].
Similar to the fact that using the same targetgenes in MLST studies allows international stan-dardisation and comparison of genotyping datafrom different users, the use of standard sets ofSNPs for given bacterial species or groups shouldfacilitate future collaboration.
I N T E R P R E T A T I O N O F T Y P I N GR E S U L T S
Theoretically, the ideal method with which todefine the genetic relatedness of bacterial isolates
Figure 7. The major clonal complexes of Staphylococcusaureus as defined by multilocus sequence typing (MLST)/eBURST. The figure was generated using eBURST on thewhole S. aureus MLST dataset, consisting of 1688 isolates(832 sequence types (STs)) as of October 2006 (http://saureus.mlst.net/eburst/). Singleton isolates and minorclusters were removed, and the remaining clonal com-plexes (CCs) arranged for clarity. Each circle representsone ST. The diameter of the circle reflects the frequency ofthat ST (i.e., the number of isolates). Linked STs differ atone locus out of the seven (single-locus variants (SLVs)).For each complex, a ‘founder’ ST is assigned, which is themost parsimoniously ‘central’ ST (shown in blue). ‘Sub-group founders’, which are STs from which at least twoSLVs have descended, are shown in yellow. Six major CCsare named (CC30, CC5, CC8, CC45, CC1, CC15)—in eachcase, these names refer to the ST of the founder (e.g., theblue founder of CC30 is ST30). Other common STs ofclinical relevance are indicated by the red arrows (e.g.,ST36 consists of EMRSA-16 strains). The arrangement ofthe CCs does not reflect the relatedness among them.
22
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

at the subspecies level would be completegenome sequencing [8]. Nevertheless, even whenit originated, the thresholds of epidemiologicallyuseful discrimination would have to be debatedaccording to, not only the species, but also therequirements of each epidemiological study set-ting. Therefore, several less comprehensive, butmore practical, methods are now used to assesspolymorphism in bacterial genomes, as outlinedabove. The data thus generated raise questionsabout interpretation, which is often complex.There are several general issues, however, thatare related to the quantitative analysis of bacterialgenomic polymorphisms obtained for typing. Anumber of ways of assigning types, building onpreviously published suggestions and the accu-mulated experience of numerous ‘typists’, areproposed below.
Interpreting DNA fragment patterns
Isolate relatedness is frequently inferred fromgenomic typing methods on the basis of DNAfragment size after separation by electrophoreticmethods, in terms of either absolute number ofband differences or percentage similarity ofbanding patterns. Percentage similarity scoresare generally preferred because they are indepen-dent of fingerprint complexity, require simplemathematics and can be generated by dedicatedsoftware programs. In addition, on the basis ofpercentage differences, categories of strainrelatedness can be defined in a concise manner.However, percentage similarity will be influencedby the level of tolerance in the differences inband position chosen for each analysis.
The absolute number of band differences is ameasure that needs to be interpreted with caution;its weight will be related to the denominator,which is the number of resolved DNA fragments.This number is related to inherent intra-speciesgenome variation rates, the choice and number ofgenomic sites probed (itself depending on thenumber and nature of restriction enzymes and/orprimers or probes used) and on the amplificationand/or separation conditions. Thus, genomicpattern similarity values must be based on asufficiently large number of genomic sites/bandsfor each isolate. If low-copy-number and variable-copy-number RFLP probes (e.g., IS sequences) areused, a composite similarity coefficient must beconstructed by adding the data obtained using
multiple probes. In addition, any inferred mea-sure of inter-strain relatedness is relative only tothe overall relatedness in that particular sample ofisolates. At any rate, genomic pattern similaritycan in no way be considered as a measure ofgenetic distance, because band positions are notindependent, and nor are they evolutionary units.Nevertheless, in practice, a concise set of simplerules for interpretation is obviously useful.
Assigning types by interpretingPFGE-generated patterns
When compared by PFGE, two isolates differingby one mutational event (from a single nucleotidesubstitution to insertions or deletions of longerDNA sequences) may differ by zero (when themutation alters neither a restriction endonucleasesite, nor the size of the resulting fragments—as asingle nucleotide mutation outside the restrictionendonuclease recognition site may do, for exam-ple) and up to four DNA fragments, or ‘bands’[274]. When there are no observed band differ-ences, the isolates should be termed ‘indistin-guishable’, rather than ‘identical’, and assigned tothe same type (e.g., A) and subtype (e.g., A1) ifother subtypes exist. Such subtypes (e.g., A1, A2,A3) will be assigned to isolates that differ by oneto four bands (Fig. 8). According to a similarcalculation, five to eight band differences could beattributable to at least two mutational events. Ithas been proposed that isolates putatively distantby two mutational events should also be assignedto subtypes, and that only isolates distant by threeor more mutations (therefore differing by at leastnine bands) should be assigned to distinct types(e.g., A, B, C) [90,273]. Tenover et al. [90] further-more suggested that, in the context of habitualhealthcare-associated outbreaks of limited dura-tion (usually restricted to episodes of half a yearor less), isolates differing by one to four bands beconsidered as ‘closely related’ and therefore‘probably part of the (same) outbreak’. In short-term outbreak typing, single band differencesshould be deemed important. Isolates differing byfive to eight bands would then be ‘possiblyrelated’ isolates and therefore ‘possibly part ofthe outbreak’. Unfortunately, these suggestionsare often misinterpreted as hard and fast ‘guide-lines’ and are applied outside the context that theauthors took great pains to delineate. We there-fore wish to emphasise that epidemiological
23
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

interpretation of differences in PFGE patternscannot blindly follow these suggestions.Examples of possible exceptions and points toconsider follow.
The relative validity of this simple ‘biologicalrule’ becomes immediately apparent if one con-siders that, according to the above, four banddifferences may arise from four independentmutational events, each giving rise to only asingle band difference, while five band differ-ences may arise from only two mutational events.In this case, two isolates with the former relation-ship would be less ‘related’ than two with thelatter. Once again, only the consideration ofepidemiological data would help to clarify theissue. On the other hand, even strains differing bya single band difference may have distinct bio-logical and epidemiological characteristics. Forexample, during an outbreak of S. enterica subsp.enterica serotype Blockley that lasted for severalmonths, two PFGE subtypes, named A2 and A4,differed not only in their resistance to nalidixicacid, but also in their temporal and geographicaldistribution [274]. In this case, the outbreak wasextended, and more band differences may havebeen expected. However, it needs to be borne inmind that the isolates belonged to the sameserotype of the same subspecies of the samespecies. Therefore, the inherent diversity of thestudy sample was already limited, which makes it
clear that it is, indeed, important to take theevolutionary mutation rate of a species (if known)into account.
The following recommendations follow fromthe above discussion:1. Isolates with patterns differing by one to four
bands should be assigned to subtypes of thesame type.
2. Isolates with patterns differing by five or morebands should be assigned to distinct types.
3. Inferring the epidemiological relationship oftwo or more isolates, according to PFGE typesor subtypes, requires careful thought in everycase, and consideration of the contribution ofother information (clinical, epidemiologicaland biological characteristics of the outbreakand the possibity of invader isolates beingintroduced during the outbreak).The foregoing applies to visual analysis of a
usually limited number of profiles. Computer-assisted cluster analysis based on the similarity ofprofiles requires the prior decision of a cut-offsimilarity level. The similarity of PFGE profilesrequires that only positional correspondence istaken into account; the Dice coefficient is the mostwidely used for this purpose. Computer-assistedcluster analysis is inevitable for the comparison oflarge numbers of profiles generated at differentmoments and—in the case of inter-laboratorynetworks—at different locations. A certain ‘simi-larity threshold’ has to be chosen to define types inthis situation [67]. An inter-laboratory study with arigorously standardised protocol investigatingoutbreak and non-outbreak strains of A. baumanniishowed that strains regarded as being the sametype clustered at 95–100% if processed in the samelaboratory. Central analysis of the data of the sameset of strains generated by the three participatinglaboratories showed that strains of the same typeclustered at 87% [28]. Identification of similar oridentical strains (types) by band-based patternanalysis at a similarity level of 80% was alsoinstrumental in delineating a clone of multidrug-resistant A. baumannii in Southeast England [276].Since percentages are usually calculated usingsoftware programs in which parameters (such asthe tolerance of band differences) can be set by theuser, the same patterns may yield different quan-titative relationships. Furthermore, the user mustalways check the software assignations, since, forexample, gel imperfections may be interpreted asbands by the program. Finally, when two or more
Figure 8. Example of a pulsed-field gel electrophoresis(PFGE) analysis. Lanes marked M display molecularweight markers, the sizes of which are indicated on theleft. Note that for normalisation purposes, markers areused every sixth lane. That such precautions are requiredis made obvious by the electrophoretic anomalies that canbe observed in lanes 1–3, where the fragments are notexactly vertically aligned. Red boxes identify indistin-guishable patterns, whereas the green box identifies a pairof related patterns differing by two bands, thereby tracingsubtypes. This suggests an insertion deletion event in onefragment or the presence of extrachromosomal elementsthat differ in molecular size. Gel picture provided by D.Horst-Kreft (Erasmus MC, Rotterdam, The Netherlands).
24
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

gels are compared, imperfect reproducibility ofelectrophoretic conditions may lead to, for exam-ple, systematic band shifts, which again the soft-ware program might interpret as differences,while the careful user can see them for what theyare. Confirmation of software groupings accordingto the user’s critical eye, and judgement based onadditional information, are therefore essential atall times. In conclusion, the following recommen-dations apply to assignment of types using algo-rithm-generated percentage differences (valid forall of the ‘band-based’ methods): (i) in the eventthat the ‘biological rule’ described above fits thealgorithm-generated grouping, the aforemen-tioned recommendations apply; (ii) in the contrarycase, i.e., when isolates differing by five or morebands cluster together in epidemiologically plau-sible groups, it is advisable to assign ‘clusters ofsimilarity’, rather than ‘types’.
As a final comment regarding interpretation ofPFGE-generated patterns, it is emphasised thatlarge studies assessing inherent intra-speciesvariability would have allowed a more rationaldesign of rules for the assignment of types.Unfortunately, such studies have not been under-taken in large numbers. However, most of theEuropean national health centres have developedvarious typing databases containing hundreds, ifnot thousands, of molecular fingerprints. Forexample, databases comprising over 1200 differ-ent fingerprints have been developed for the agentof whooping cough, Bordetella pertussis [277,278].
Assigning types by interpreting PCR-generatedpatterns
Type assignation to PCR-generated band patternsby visual analysis is even more problematic thanit is with PFGE-generated patterns, since thebiological explanation of band differences isusually unclear, and at any rate complex. Inaddition, it may be tempting to take band inten-sity into account. Thus, recommendations in thiscase would have to be limited to the following:1. Isolates differing by one or more bands should
be assigned to distinct types.2. Band intensity should only be taken into
account once it has been demonstratedunequivocally, by appropriate replicate exper-iments, that it is reproducible.However, essentially the same requirements
as those for PFGE apply to the inference of
strain relationships from PCR-generated typesthat are based on the comparison of bandingpatterns (e.g., AFLP). As discussed, whenassessing MLVA by gel electrophoresis, someinformation (e.g., point mutations) may be lost,in contrast to analysis by DNA sequencing.MLVA-generated banding patterns shouldtherefore be interpreted as though they werePCR-generated.
For computer-assisted pattern analysis of PCR-and AFLP-generated fingerprints, the Pearsonproduct moment correlation coefficient is themost objective and reliable similarity measure. It(i) is independent of relative intensities of patt-terns; (ii) is largely insensitive to differences inbackground; and (iii) does not suffer fromsubjective band detection and band-matchingcriteria, since it compares the entire profile ratherthan specific band characteristics and relativeband intensities. However, for simple PCR-RFLP-generated profiles, a band-based coefficient suchas Dice is therefore recommended.
Generally, if groups are robust, different sim-ilarity measures and clustering algorithms maylargely reveal the same grouping patterns.
Analysis of MLVA profiles
Analysis of MLVA profiles in potential outbreaksituations, as with other methods, is bestinformed by detailed population studies thathave been performed previously. These willindicate the likelihood of a change in repeatnumber at a particular locus during the time-frame of an outbreak. In most situations, isolateswithin an outbreak will (or should) have anidentical MLVA profile. However, whether thisprofile is common in isolates unrelated to thisoutbreak, i.e., the background distribution of theMLVA profile in the bacterial population as awhole, is critical information. Also essential,particularly with respect to microsatellites, is thefact that alterations in repeat numbers occur sorapidly that the profile could change during thecourse of an outbreak. Although MLVA schemeshave been proposed for many bacterial patho-gens, the prerequisites listed above have seldombeen met [278–280].
MLVA can be used for population studies ofmicroorganisms. An approach that is frequentlyapplied to MLVA profiles is an implementation ofthe minimum spanning tree, based on the same
25
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

principles as the eBURST algorithm. These ap-proaches yield maps of predicted relationshipsamong strains on the basis of single-locus (wherethe profile varies at one locus) and dual-locus(where the profile varies at two loci) variants. If itcan be assumed that variation in repeat number ata particular repeat locus is stepwise, i.e., an isolatewith six copies of a repeat at a given locus is moreclosely related to an isolate comprising ‘fiverepeats’ than one comprising ‘four repeats’, dis-tance methods that take the repeat number intoaccount can be applied. However, if this assump-tion cannot be made, categorical approaches thatconsider all allelic numbers as equally distant areappropriate.
Interpreting differences among DNA sequences
As mentioned previously, MLVA and sequence-based methods are likely to replace band-basedmethods, mainly because of the difficulties incomparing banding pattern profiles among lab-oratories, despite use of common protocols.Sequence-based methods are certainly moreportable, as the data are comparable regardlessof the platform used to generate them. The onlyprerequisite is that the data are of adequateaccuracy. The value of any sequence database isdetermined by the quality of the data within it,and the role of the database curator is thereforevery important. Most major sequence databases,e.g., MLST.net, require submission of the rawsequence trace files from the laboratory when anew allele type is proposed [281]. A personalcheck of the data by the curator, before accept-ing the submission, will ensure that the appar-ent new allele type is not due to an error in theDNA sequence. Although software (e.g., Phred/Phrap) can assess DNA sequence quality ofindividual traces or of contigs [282], mostcurators believe that manual curation remainsthe reference standard (PulseNet and SalmGeneare the best known representatives of thiscategory). The L. pneumophila SBT database[87], accessible via the EWGLI website (http://www.ewgli.org/), uses a combination of auto-mated sequence quality checks and manualcuration. If DNA sequences are submitted to adatabase in text format, no guarantee of thequality of the data is given, beyond checking forambiguous bases (the presence of non-A, G, T,C, such as N, R, W, Y), which may indicate that
the original sequence was not optimal. If thetarget used for typing is a coding sequence, itcan be confirmed that the open reading frameinvolved is not abrogated. As DNA sequencingis increasingly used in clinical applications,rigorous checking of the quality control pro-cesses that curators of such databases adopt isrecommended.
International efforts in standardisation of typenomenclature and typing protocols
International travel, migration and food com-merce are the main factors that have contributedto the worldwide spread of bacterial clones.Therefore, the need for international databases,including those with typing information concern-ing epidemiologically relevant strains, is strong.Building such databases, in turn, relies uponstandardisation of typing methods, and on regu-lar quality assessment ring trials for all partici-pating laboratories, to guarantee consistentlycomparable data. Currently, two types of suchdatabases have been developed. First, there areinternational catalogues of prototype strains, e.g.,MLST.net and the SeqNet.org spa sequence repos-itory (spaserver.ridom.de). Second, there are themolecular epidemiology databases. These includetyping data and information concerning the clin-ical and/or epidemiological features associatedwith the isolates analysed (e.g., PulseNet andSalmGene).
Inter-laboratory ‘ring trials’ are a relativelyrecent development, spurred on by the need forreliable data to be used in international surveil-lance. Unwillingness on the part of laboratories toabandon methods that have taken time and effortto develop and that produce good results hindersthe standardisation of methods. A way forward isto first seek harmonisation rather than standardi-sation, changing only those aspects of a protocolthat are shown to be critical to intra- or inter-laboratory reproducibility. An example of thiswas the HARMONY project [203], where PFGEprotocols were examined in several laboratories.DNA preparation methods, for example, were notfound to be an important factor, provided that allmethods produced good-quality DNA. Manyother aspects required direct standardisation,however. Pulsing conditions were particularlycritical and, by comparing gel results amonglaboratories, new electrophoresis conditions that
26
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

produced optimal separation within an accept-able run time with good inter-laboratory repro-ducibility were agreed. Several laboratories havealso found the protocol to be ideal for analysingcoagulase-negative staphylococci, thus reducingthe number of mandatory laboratory standardoperating procedures (SOPs).
Several validated databases exist, making itpossible to compare isolates and discover whethera given profile has been seen before, and in whatcontext. For MLST, an excellent website exists(http://pubmlst.org/) with updated databases ona range of microorganisms. This exemplifies thesuccessful use of a library typing method, wherethe sole sequence can be compared to well-known, validated MLST sequences and a typecan be assigned. The GENE network concentratedon exploring the use of the RiboPrinter technol-ogy (http://www.ewi.med.uu.nl/gene/), as usedfor database building, as this equipment is highlystandardised in itself. As already mentioned, thePulseNet USA database was the first databasebased on PFGE profiles of different foodbornepathogens (http://www.cdc.gov/pulsenet/), andwas followed by similar networks in Canada(http://www.cdc.gov/pulsenet/participants_pages/pulsenet_canada.htm), Latin America(http://www.panalimentos.org/pulsenet/), andEurope (http://www.pulsenet-europe.org/)[283]. Recently, a similar initiative was developedin Japan [284]. Other PFGE networks have beendeveloped, e.g., SalmGene, encompassing PFGEprofiles of Salmonella species (http://www.hpa-bioinformatics.org.uk/bionumerics/salm_gene/),HARMONY (http://www.harmony-microbe.net/index.htm), including a range of typing methodsfor S. aureus, with special attention given tomethicillin-resistant S. aureus (MRSA) isolates,and Listernet, including both PFGE and antibio-gram profiles of L. monocytogenes (http://www.eurosurveillance.org/em/v10n10/1010-225.asp).Such databases require an extremely high level ofstandardisation, simple protocols, educated pers-onnel, and continued quality control, to ensurethat the data can be trusted. These are a fewexamples of international cooperation in devel-oping databases for isolate comparison, as otherdatabases are being developed.
The use of typing has been extremely valuablein tracing foodborne outbreaks and pointing outreservoirs; some of the above databases areexcellent examples of this.
T R A N S L A T I N G T Y P I N G R E S U L T SI N T O C L I N I C A L L Y U S E F U LI N F O R M A T I O N A N D A P P L I C A T I O NF O R I N F E C T I O N C O N T R O L
Translating typing results into clinical practice isone of the most important endpoints of a typingexercise. Performing ‘real-time’ typing may now befeasible in the microbiology laboratory, but onceindistinguishable isolates are identified, appropri-ate clinical action must be taken. Prevention ofinfection should be the main goal, although inseveral settings, even the prevention of colonisa-tion (and its spread) is important. In the case of anti-MRSA policies in the northern European countrieswith low incidence rates, adequate typing plays animportant role. The results of typing isolates of(unexpected) MRSA strains should guide the clin-ical response; for example, in the case of twogenotypically indiscriminate isolates of MRSAoriginating from a single ward or department inDutch hospitals, the ward or department will beclosed. This implies that affected patients will becohorted, all exposed patients and personnel willbe screened for MRSA carriage (and treated whenfound to be positive), operations will be resched-uled or postponed, and a broad variety of hygienemeasures will be implemented. It is obvious thatthis strategy of ‘search and destroy ’ is costly, andtyping data need to be timely and accurate. In thecase of closure of intensive care units, false typingresults also have very expensive consequences.Ongoing quality assessment of method perfor-mance will ensure that results remain reliable.Standardised protocols, training of personnel anddetailed inventories of reagents are all absoluteprerequisites.
Reports of typing results represent an importantdiagnosticanddidactic tool for clinicians, includinginfection control staff and those involved in directpatient care. They should be written in an immedi-ately understandable format that both results from,and fosters, further interdisciplinary collaboration.These reports have to include typing and otherdata concerning isolates, enabling interpretationof results in the light of epidemiology (Fig. 9).
Typing should serve to identify clusters ofinfection in real time. The first indication ofidentical types should elicit alarm and lead toclinical action. However, in order to get the best outof typing, some prerequisites must be met evenbefore typing is undertaken. A clear working
27
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

hypothesis, or model, must have been formulated,on the basis of available clinical and epidemiolog-ical data (e.g., for an outbreak, putative transmis-sion routes and/or source(s). The hypothesis willthen be tested by typing; it will also guide thechoice of typing method, since different questionsmay require answers with different levels of
reproducibility and/or discrimination. For typingto contribute to infection control, all partiesinvolved (e.g., clinical and laboratory doctors andnurses) must be informed of what will be requiredof them (from sampling to performing the actualtyping), and what consequences the results willhave for their practice (e.g., in case one or more
Figure 9. An example of a result report. The report starts with a summary of logistic information, and then the hardlaboratory data are shown, usually as gel pictures or plain DNA sequences, followed by a sample identification and someform of data interpretation. This section may be complemented by a tree visualising the interrelatedness among the strainsisolated. Finally, concluding remarks are given and the form is authorised by the laboratory head, either a medical or amolecular microbiologist.
28
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

personnel are colonised with the outbreak strain).Similarly, feedback after typing is essential andmust include all those involved, not only to trans-late the results into practice, but also to guaranteecontinued motivation (Fig. 10). One of the mostimpressive published descriptions of the benefits,including financial, of molecular typing for infec-tion control outlined a scheme that relied on twosimple actions [285]: first, the introduction of REAas a typing method in the clinical microbiologylaboratory of a university hospital; and, second,weekly, 45-min meetings of everybody involved ininfection control within this hospital. While themethod may not have been everyone’s immediatefirst choice, the continuing feedback and the clear-cut aims guaranteed by the weekly meetings madethis scheme a success, leading to a 23% reduction ininfection rate, and consequent annual savings ofapproximately $2 000 000. Whether this conse-quence was fully dependent on the typing itself, orwhether increased awareness due to frequentdiscussions of infection control also played adecisive role, is not really important; it is the neteffect that remains important.
To summarise, collaboration at all stages of atyping exercise, clear aims and working hypoth-eses before typing is begun, reliable quality-
controlled data and adequate reporting and feed-back all contribute to the total value of typing inclinical practice.
T Y P I N G N E T W O R K S A N D Q U A L I T YC O N T R O L
A variety of scientific initiatives have led to theestablishment of typing networks, some of whichhave already been mentioned. Several Europeanscientists have made efforts towards standardisa-tion of typing technologies through the ESGEMnetwork (http://www.escmid.org). PFGE hasformed the basis for the development of severalof the international typing networks including theHARMONY/MRSA effort [203], and, of course,the PulseNet network, initiated in 1996 [286].Initially started in the USA, and now includingPulseNet Europe and other international off-shoots, it developed into one of the major typingnetworks in operation worldwide to date. Theinitial aim of PulseNet was to develop subtypingsynchronisation for food-related pathogens. Itstarted off with E. coli O157:H7, several non-typhoidal Salmonella serotypes, L. monocytogenesand Shigella [286]. Currently, Campylobacter hasbeen added to the list, which will expand further
Figure 10. Source tracking for Legionella pneumophila according to EWGLI. In cases where patients are identified fromremote regions and environmental investigations have been concluded, species will be typed using serogroup-specificantibodies. In cases of identical serogroup and monoclonal subgroup, multilocus sequence typing will be used to furthersubtype the strains, after which potential epidemiological associations can be derived.
29
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

in the foreseeable future [287]. For most of thespecies currently studied, several thousands ofPFGE fingerprints are stored and regularly anal-ysed in a cumulative fashion. It goes withoutsaying that PulseNet relies on extensive standardi-sation in order to enable fingerprint exchangeamong centres and centralised computerised anal-ysis. Standardisation involved the development ofa universal PFGE fragment size marker standard[288], refinement of the interpretation guidelines[289] and, of course, the development of robustexperimental protocols [290]. These efforts are alsocontrolled through annual accreditation of spe-cific, named laboratory personnel, rather thanentire laboratories, which has contributed hugelyto the current success of PulseNet.
However, there is currently no single specia-lised institution that focuses on the developmentof typing standards, both for protocols and fordata interpretation. In molecular diagnostics, suchinitiatives are far more advanced, and moleculartypists should profit from the experience gainedin this sector. Novel initiatives such as theexternal quality control assessment scheme devel-oped in Belgium [101] are urgently required.
M O L E C U L A R T Y P I N G S T R A T E G I E SI N A N U T S H E L L
Typing can be useful at different levels: (i)locally, at hospitals or other health institutions;(ii) regionally and nationally, in reference lab-oratories and research centres; and (iii) globally,through dedicated networks. The choice ofmethods and the concurrent quality assessmentdepend on the level at which typing is done.
Local typing
Local typing in clinical microbiology laborato-ries is undertaken mainly to assess whether anincrease in occurrence of particular organismsis due to the spread of a single strain. Cur-rently, the most obvious methods for localtyping are PCR fingerprinting and PFGE, andto a lesser extent AFLP, but it is likely thatsequence-based methods will soon be morewidely applicable at this level too. The choiceof typing method will be guided by conve-nience criteria and will depend on the mostcommon healthcare-associated pathogens(‘alert organisms’) to be studied.
For these species, collections of unrelatedcontrol strains and sets of isolates assumed tobe epidemiologically related, together withadditional data such as antibiogram and bio-type, should be set up. They will then be usedto assess precise test conditions for eachspecies to be typed. It is therefore advisablethat standardised protocols and qualifiedadvice from specialists in the field are soughtat the start. For each typing exercise it is usefulto include at least three to five unrelatedstrains of the same species to confirm discrim-inatory capacity, as well as a set of relatedisolates from a previous confirmed outbreak,to assess epidemiological concordance.
PCR fingerprinting
The most simple and rapid genotypic methodfor local application is PCR fingerprinting.PCR amplification and separation of frag-ments can be done in 1 or 2 days. For mostorganisms, crude DNA can be obtained bysimply boiling a colony in lysis solution.Every fifth or sixth lane should include areference sample for normalisation. This sam-ple must be carefully selected and shouldcontain fragments covering the size range ofthe fragments in the samples. Profiles should,preferably, be judged visually. Computer-assisted analysis of PCR profiles can also bedone, and for this purpose the Pearson prod-uct moment correlation coefficient (whichtakes into account band intensity) should beused, and clustering by the unweighted pair-group method using arithmetic averages (UP-GMA) is the recommended distance measure.Alternatively, Ward’s clustering algorithmcould be used. In the case of small numbersof samples on one gel, visual analysis ispreferable. In case of doubt about inter-isolaterelatedness, highly similar samples should bere-run in adjacent lanes to assess whetherthey are indistinguishable. Choice of primers,PCR amplification and electrophoresis condi-tions are important and depend on the micro-organism under investigation. Widely usedprimers, such as REP or ERIC, are of entero-bacterial origin and may not be truly uni-versal. They are, therefore, not ideally suitedto all species.
30
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

PCR typing is notorious for its susceptibilityto minor variations in experimental conditionsand reagents, and results may differ amongruns, even in one laboratory. Therefore, themethod is only suited for comparison of smallnumbers of samples processed simultaneouslyand run on one gel. For longitudinal compari-son, where large numbers of samples have to becompared over time, this method is not suitable.
PFGE
Today, for most organisms, protocols exist thatprovide results in 2–3 days. As with PCRfingerprinting, the protocols, although gener-ally similar, are organism-dependent. In elec-trophoresis, the use of reference samples isessential, as it is for PCR (see above). Thebuffers and reagents for lysis of cells in theagarose blocks, the enzymes used for DNAdigestion, and the electrophoresis conditionsare all important. For several alert organisms,well-established protocols are available. If alaboratory is confronted with another organ-ism for which, so far, no PFGE analysis hasbeen done, apart from seeking a protocol for a(closely) related species in the literature,advice can be sought from specialists in thefield. For comparison of a few isolates, visualanalysis of one gel is easy. ‘Fingerprints’ canalso be analysed by computer-assisted clusteranalysis, usually with the band-based Dicecoefficient. For local surveillance, it is feasibleand worthwhile to set up a database offingerprints for alert organisms. Every fifth orsixth lane should include a reference samplefor normalisation. This sample must be care-fully selected and should contain fragmentscovering the size range of the fragmentsincluded in the analysis.
Sequence-based methods
When the appropriate target sequences havebeen selected, sequence methods, whetherthey target single or multiple loci (SLST andMLST), are technically simple. After a selectiveamplification of (part of) the target, the ampli-fied product is sequenced using commerciallyavailable technology.
Sequenced mixtures are then read usingtools available in the laboratory, which mayvary from ‘old-fashioned’ radioactive slab gelsto high-throughput 96-capillary automatedsequencers. When sequences have been read,comparative assessment can be undertaken,again using a variety of software tools.Sequence-based methods do not need refer-ence samples, but they do need strict qualitycontrol in the sense that the sequence outputmust be compared with the experimental datafor correctness.
Other methods
Implementation and management criteria forthe other methods listed in previous sections ofthis article strongly overlap with those listedabove.
T H E I N T E R F A C E O F T Y P I N G A N DB A S I C S C I E N C E
Although the interrelatedness between basicmicrobiological science and bacterial typing is notthe main topic of this publication, the associationbetween the two is too important to be completelyignored. The study of bacterial pathogenicity andecology will continue to profit extensively fromstrain comparisons at the phenotypic or genotypiclevel. This has already resulted in a wealth ofinformation on specific virulence genes, theirvariability and epidemiology, and their involve-ment in the infectious disease process. A number offundamental science disciplines such as popula-tion biology interact with typing extensively.Among others, studies of the spread and diversityof types and genes in the population, and theselective pressures exerted on that diversity, havecontributed to the understanding that clonal andpanmictic species exist and thus enriched ourunderstanding of infectious disease epidemiology.An important recent development is the adoptionof a systems approach to biological studies(Fig. 11). The aim of this approach is to generate acomprehensive picture of not just the activity of asingle gene under a fixed condition, but of complexinteractions among multiple genes in an organism,and within a specific environment. Finally, onthe technical side, many novel miniature and
31
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

high-throughput technologies have been devel-oped recently, and these will certainly contribute totyping in the not too distant future [291].
A N E X A M P L E O F T H EI N T E R R E L A T E D N E S S O FM O L E C U L A R T Y P I N G( C a m p y l o b a c t e r j e j u n ia n d i t s i n f e c t i o u s p a t h o l o g y )
The Guillain–Barre Syndrome (GBS) is a post-infectious neuropathy, with the majority ofcases resulting from molecular mimicry be-tween human gangliosides and C. jejuni lipool-igosaccharides (LOS). This was initially shownto be correlated with O-serotypes of C. jejuni[292,293]. At a later stage, the molecular mim-icry hypothesis was refined on the basis ofbiophysical and serological studies of the cam-pylobacter LOS (see Koga et al. [294] and Moranet al. [295] for a review). However, recentepidemiological studies on the LOS composi-tion of larger numbers of GBS-associated strainsof C. jejuni definitely linked specific LOS genesto this mimicry phenomenon [296,297]. They
revealed that certain classes of LOS-encodinggene complexes were clearly associated withGBS-disease-invoking potential. Bacterial typ-ing has thus been instrumental in elucidatingthe pathogenic process in GBS patients.
A variety of genome sequences has beendetermined within the genus Campylobacter,including two for the species C. jejuni [298]. Onthe basis of these genome sequences, an entiregenome array has been developed by differentgroups (e.g., [299]). An array based on theC. jejuni 11168 genome sequence was used toconfirm that the overall genome plasticityamong C. jejuni strains was relatively low[300]. This approach resulted in the identifica-tion of several specific loci in the C. jejunigenome that showed enhanced evolutionarymutation rates, rendering them suitable forepidemiological studies.
Experimental validation of an extendedarray (including the RM1221 genome sequencedata) confirmed the results of the previousstudy and helped to define the levels ofreproducibility of this method of comparativegenomics [301]. A US group also developed
Figure 11. A general scheme showing the position of molecular typing technology in today’s microbiology laboratory andthe systems biology laboratory of the future. Pale blue: the classic microbiological core technology. Blue: the recentpossibilities facilitated by the introduction of molecular technology. Yellow: the integrated systems biology approach. Darkblue: the place of future personalised medical practice. Host-response based diagnosis is indicated where appropriate.
32
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

an array system that was primarily used fordefining transcription profiles [302,303].However, this array could also be used toidentify genes unique to certain C. jejuniisolates [304]. This establishes further levelsof Campylobacter diversity, and differentiallyoccurring genes or gene segments can be usedto develop binary typing approaches. A thirdarray was used to perform extensive phylog-enomics and to try and associate type withsource of infection [305]. It was demonstratedthat new reservoirs for C. jejuni can still beidentified and that comparative phylogeneo-mics is one of the methods of choice whentrying to define precise population structures.It has to be emphasised that the array technol-ogy at the full genome level is not yet suitedfor day-to-day clinical application. It is, how-ever, obvious that the output of array exper-iments is highly information-dense and can beused to define inter-isolate identity at thehighest possible level and that experimentaldata will identify novel targets for thedevelopment of more dedicated typing sys-tems.
C O N C L U D I N G R E M A R K S A N DP E R S P E C T I V E S
Although typing protocols and networks havecome a long way and are increasingly provingtheir value in the context of infection control andinternational infectious disease surveillance,there are several areas where future work wouldbe beneficial. Analysis of the molecular eventsleading to genomic polymorphism in naturaland experimental conditions should be under-taken to increase the understanding of theevolutionary mechanisms of bacterial clones asthey spread in human populations [26,157].Collections of extensively typed bacterial patho-gens should be assembled and made availablevia public culture collections [20,21,306]. Dedi-cated working groups should cooperate in opti-mising inter-laboratory standardisation andongoing and independent quality control ofgenomic typing methods for specific pathogens[25]. Nomenclature used within the variousdisciplines employing typing technologies
should be standardised as much as possible,and should be extended into the field of viraltyping [307,308], fungal typing [309], typing ofparasites [310], and perhaps even human geno-typing. Typing technologies should be madeavailable to those working in areas whereeconomic constraints currently prevent adequateimplementation. Appropriate training facilitiesshould be provided where needed and certifica-tion could be emphasised. In the end, theseinitiatives will lead to the establishment ofreference standard protocols amenable to multi-centre application.
For several organism–method combinations,this stage has already been reached and severalsuch combinations are listed in Table 1.However, there are still many challenges thatlie ahead. Significant funding and continuingsupport are required to sustain existing librariesand develop new methods with the objectiveof superior and/or more cost-effectiveapproaches.
We have not discussed the medico-legal impli-cations of some typing efforts, and nor have wediscussed applications to disease or colonisationsusceptibility of the human host. The linkage oftyping with disease manifestation has beentouched upon only superficially, but surelydeserves our undivided future attention. It iscurrently clear that banding pattern-based meth-ods are in decline and that more transportable,objective and technically simple sequence-basedtyping systems will be employed in the futureand may constitute a new reference standard.Although valuable information may be lost bychoosing pure sequence-based approaches,enlarging the number of sequencing targets perstrain will, in the end, generate sufficientamounts of data to allow confident deductionson inter-strain relatedness to be made. Reportsdescribing the assessment of hundreds ofsequences per organism have been used togenerate a phylogenetic tree [311]. Such reportsshow that, on the basis of pure sequence data,both taxonomically and epidemiologically signif-icant nucleotide variation can be monitored.Typing should be staged carefully and, prior tousing typing in the control of infectious disease,dissemination of a set of practical guidelinesshould be considered and put into practice (seebelow).
33
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Ta
ble
1.
Fre
qu
entl
yu
sed
bac
teri
alg
eno
typ
ing
met
ho
ds:
ali
tera
ture
-bas
edsu
rvey
of
mu
lti-
cen
tre
val
idat
edm
ole
cula
rm
eth
od
san
dp
roto
cols
(pu
bm
edsc
reen
Mar
ch20
07)
Path
og
en
s
Hy
bri
dis
ati
on
med
iate
d
typ
ing
tech
no
log
yP
FG
EM
LV
AS
ing
lelo
cus
seq
uen
cin
gM
ult
ilo
cus
seq
uen
cin
g
Hu
man
Bact
eri
al
Path
og
en
s
Aci
net
obac
ter
bau
man
nii
Bri
sse
S,
Mil
ato
vic
D,
Flu
itA
C,
etal
.M
ole
cula
rsu
rvei
llan
ceo
fE
uro
pea
nq
uin
olo
ne-
resi
stan
tcl
inic
alis
ola
tes
of
Pse
ud
om
on
asae
rug
ino
saan
dA
cin
eto
bac
ter
spp
.u
sin
gau
tom
ated
rib
oty
pin
g.
JC
lin
Mic
rob
iol.
2000
;38:
3636
-45.
Sei
fert
H,
Do
lzan
iL
,B
ress
anR
,et
al.
Sta
nd
ard
izat
ion
and
inte
rlab
ora
tory
rep
rod
uci
bil
ity
asse
ssm
ent
of
pu
lsed
-fiel
dg
elel
ectr
op
ho
resi
s-g
ener
ated
fin
ger
pri
nts
of
Aci
net
ob
acte
rb
aum
ann
ii.
JC
lin
Mic
rob
iol.
2005
;43:
4328
-35.
Yam
amo
toS
,B
ou
vet
PJ,
Har
ayam
aS
.P
hy
log
enet
icst
ruct
ure
so
fth
eg
enu
sA
cin
eto
bac
ter
bas
edo
ng
yrB
seq
uen
ces:
com
par
iso
nw
ith
the
gro
up
ing
by
DN
A-D
NA
hy
bri
diz
atio
n.
Int
JS
yst
Bac
teri
ol.
1999
;49:
87-9
5.
Bar
tual
SG
,S
eife
rtH
,H
ipp
ler
C,
etal
.D
evel
op
men
to
fa
mu
ltil
ocu
sse
qu
ence
typ
ing
sch
eme
for
char
acte
riza
tio
no
fcl
inic
alis
ola
tes
of
Aci
net
ob
acte
rb
aum
ann
ii.
JC
lin
Mic
rob
iol.
2005
;43:
4382
-90.
Clo
stri
diu
mdi
ffici
lev
and
enB
erg
RJ,
Sch
aap
I,T
emp
leto
nK
E,
etal
.T
yp
ing
and
Su
bty
pin
go
fC
lost
rid
ium
dif
fici
leIs
ola
tes
by
Usi
ng
Mu
ltip
le-L
ocu
sV
aria
ble
-Nu
mb
erT
and
em-R
epea
tA
nal
ysi
s.J
Cli
nM
icro
bio
l.20
07;4
5:10
24-8
.
Lem
eeL
,D
hal
luin
A,
Pes
tel-
Car
on
M,
etal
.M
ult
ilo
cus
seq
uen
cety
pin
gan
aly
sis
of
hu
man
and
anim
alC
lost
rid
ium
dif
fici
leis
ola
tes
of
var
iou
sto
xig
enic
typ
es.
JC
lin
Mic
rob
iol.
2004
;42:
2609
-17.
Co
agu
lase
neg
ativ
est
aph
ylo
cocc
i
Wan
gX
M,
No
ble
L,
Kre
isw
irth
BN
,et
al.
Ev
alu
atio
no
fa
mu
ltil
ocu
sse
qu
ence
typ
ing
syst
emfo
rS
tap
hy
loco
ccu
sep
ider
mid
is.
JM
edM
icro
bio
l.20
03;5
2:98
9-98
.E
nte
roco
cci
Bri
sse
S,
Fu
ssin
gV
,R
idw
anB
,et
al.
Au
tom
ated
rib
oty
pin
go
fv
anco
my
cin
-res
ista
nt
En
tero
cocc
us
faec
ium
iso
late
s.J
Cli
nM
icro
bio
l.20
02;4
0:19
77-8
4.
Tit
ze-d
e-A
lmei
da
R,
Wil
lem
sR
J,T
op
J,et
al.
Mu
ltil
ocu
sv
aria
ble
-nu
mb
erta
nd
em-r
epea
tp
oly
mo
rph
ism
amo
ng
Bra
zili
anE
nte
roco
ccu
sfa
ecal
isst
rain
s.J
Cli
nM
icro
bio
l.20
04;4
2(10
):48
79-8
1.
Ru
iz-G
arb
ajo
saP
,B
on
ten
MJ,
Ro
bin
son
DA
,et
al.
Mu
ltil
ocu
sse
qu
ence
typ
ing
sch
eme
for
En
tero
cocc
us
faec
alis
rev
eals
ho
spit
al-a
dap
ted
gen
etic
com
ple
xes
ina
bac
kg
rou
nd
of
hig
hra
tes
of
reco
mb
inat
ion
.J
Cli
nM
icro
bio
l.20
06;4
4:22
20-8
.H
aem
ophi
lus
infl
uen
zae
Sch
ou
lsL
M,
van
der
En
de
A,
van
de
Po
lI,
etal
.In
crea
sein
gen
etic
div
ersi
tyo
fH
aem
op
hil
us
infl
uen
zae
sero
typ
eb
(Hib
)st
rain
saf
ter
intr
od
uct
ion
of
Hib
vac
cin
atio
nin
Th
eN
eth
erla
nd
s.J
Cli
nM
icro
bio
l.20
05;4
3:27
41-9
.
Mea
tsE
,F
eil
EJ,
Str
ing
erS
,et
al.
Ch
arac
teri
zati
on
of
enca
psu
late
dan
dn
on
-ca
psu
late
dH
aem
op
hil
us
infl
uen
zae
and
det
erm
inat
ion
of
ph
ylo
gen
etic
rela
tio
nsh
ips
by
mu
ltil
ocu
sse
qu
ence
typ
ing
.J
Cli
nM
icro
bio
l.20
03;4
1:16
23-3
6.K
lebs
iell
asp
pB
riss
eS
,Is
sen
hu
th-J
ean
jean
S,
Gri
mo
nt
PA
.Mo
lecu
lar
sero
typ
ing
of
Kle
bsi
ella
spec
ies
iso
late
sb
yre
stri
ctio
no
fth
eam
pli
fied
cap
sula
ran
tig
eng
ene
clu
ster
.J
Cli
nM
icro
bio
l.20
04;4
2:33
88-9
8
Dra
no
urt
L,
Pas
set
V,
Ver
ho
efJ,
etal
.M
ult
ilo
cus
seq
uen
cety
pin
go
fK
leb
siel
lap
neu
mo
nia
en
oso
com
ial
iso
late
s.J
Cli
nM
icro
bio
l.20
05;4
3:41
78-8
2.
Myc
obac
teri
um
tube
rcu
losi
sv
anE
mb
den
JD,
Cav
eM
D,
Cra
wfo
rdJT
,D
ale
JW,
Eis
enac
hK
D,
Gic
qu
elB
,H
erm
ans
P,
Mar
tin
C,
McA
dam
R,
Sh
inn
ick
TM
,et
al.
Rel
ated
Art
icle
s,L
ink
sS
trai
nid
enti
fica
tio
no
fM
yco
-b
acte
riu
mtu
ber
culo
sis
by
DN
Afi
n-
ger
pri
nti
ng
:re
com
men
dat
ion
sfo
ra
stan
dar
diz
edm
eth
od
olo
gy
.J
Cli
nM
icro
bio
l.19
93F
eb;3
1(2)
:406
-9.
Oel
eman
nM
C,
Die
lR
,V
atin
V,
etal
.A
sses
smen
to
fan
op
tim
ized
my
cob
acte
rial
inte
rsp
erse
dre
pet
itiv
e-u
nit
-var
iab
le-n
um
ber
tan
dem
-rep
eat
typ
ing
syst
emco
m-
bin
edw
ith
spo
lig
oty
pin
gfo
rp
op
ula
tio
n-b
ased
mo
lecu
lar
epid
emio
log
yst
ud
ies
of
tub
ercu
losi
s.J
Cli
nM
icro
bio
l.20
07;4
5:69
1-7.
Pse
udo
mon
asae
rugi
nos
aS
arw
ari
A,
Has
anR
,L
imC
B,
etal
.P
CR
iden
tifi
cati
on
and
auto
mat
edri
bo
typ
ing
of
Pse
ud
om
on
asae
rug
ino
sacl
inic
alis
ola
tes
fro
min
ten
siv
eca
rep
atie
nts
.S
can
dJ
Infe
ctD
is.
2004
;36:
342-
9.
Cu
rran
B,
Jon
asD
,G
run
dm
ann
H,
etal
.D
evel
op
men
to
fa
mu
ltil
ocu
sse
qu
ence
typ
ing
sch
eme
for
the
op
po
rtu
nis
tic
pat
ho
gen
Pse
ud
om
on
asae
rug
ino
sa.
JC
lin
Mic
rob
iol.
2004
;42:
5644
-9.
34
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Ta
ble
1.
Co
nti
nu
ed
Path
og
en
s
Hy
bri
dis
ati
on
med
iate
d
typ
ing
tech
no
log
yP
FG
EM
LV
AS
ing
lelo
cus
seq
uen
cin
gM
ult
ilo
cus
seq
uen
cin
g
Sta
phyl
ococ
cus
aure
us
Lin
dsa
yJA
,M
oo
reC
E,
Day
NP
ET
AL
.M
icro
arra
ys
rev
eal
that
each
of
the
ten
do
min
ant
lin
eag
eso
fS
tap
hy
loco
ccu
sau
reu
sh
asa
un
iqu
eco
mb
inat
ion
of
surf
ace-
asso
ciat
edan
dre
gu
lato
ryg
enes
.J
Bac
teri
ol.
2006
;188
:669
-76.
Mu
rch
anS
,K
aufm
ann
ME
,D
epla
no
A,
etal
.H
arm
on
izat
ion
of
pu
lsed
-fiel
dg
elel
ectr
op
ho
resi
sp
roto
cols
for
epid
emio
log
ical
typ
ing
of
stra
ins
of
met
hic
illi
n-r
esis
tan
tS
tap
hy
loco
ccu
sau
reu
s:a
sin
gle
app
roac
hd
evel
op
edb
yco
nse
nsu
sin
10E
uro
pea
nla
bo
rato
ries
and
its
app
lica
tio
nfo
rtr
acin
gth
esp
read
of
rela
ted
stra
ins.
JC
lin
Mic
rob
iol.
2003
;41(
4):1
574-
85.
Mal
ach
ow
aN
,Sab
atA
,Gn
iad
ko
wsk
iM
,et
alC
om
par
iso
no
fm
ult
iple
-lo
cus
var
iab
le-n
um
ber
tan
dem
-rep
eat
anal
ysi
sw
ith
pu
lsed
-fiel
dg
elel
ectr
op
ho
resi
s,sp
aty
pin
g,
and
mu
ltil
ocu
sse
qu
ence
typ
ing
for
clo
nal
char
acte
riza
tio
no
fS
tap
hy
loco
ccu
sau
reu
sis
ola
tes.
JC
lin
Mic
rob
iol.
2005
;43:
3095
-100
.
Air
es-d
e-S
ou
saM
,B
oy
eK
,d
eL
enca
stre
H,
Dep
lan
oA
,E
nri
gh
tM
C,
Eti
enn
eJ,
Fri
edri
chA
,H
arm
sen
D,
Ho
lmes
A,
Hu
ijsd
ens
XW
,K
earn
sA
M,
Mel
lman
nA
,M
eug
nie
rH
,R
ash
eed
JK,
Sp
alb
urg
E,
Str
om
m-
eng
erB
,S
tru
elen
sM
J,T
eno
ver
FC
,T
ho
mas
J,V
og
elU
,W
esth
H,
Xu
J,W
itte
W.
Rel
ated
Art
icle
s,L
ink
sH
igh
inte
rlab
ora
tory
rep
rod
uci
bil
ity
of
DN
Ase
qu
ence
-bas
edty
pin
go
fb
acte
ria
ina
mu
ltic
ente
rst
ud
y.
JC
lin
Mic
rob
iol.
2006
Feb
;44(
2):6
19-2
1.G
En
rig
ht
MC
,D
ayN
P,
Dav
ies
CE
,et
al.
Mu
ltil
ocu
sse
qu
ence
typ
ing
for
char
acte
riza
tio
no
fm
eth
icil
lin
-re
sist
ant
and
met
hic
illi
n-s
usc
epti
ble
clo
nes
of
Sta
ph
ylo
cocc
us
aure
us.
JC
lin
Mic
rob
iol.
2000
;38:
1008
-15.
Str
epto
cocc
us
pneu
mon
iae
Han
age
WP
,K
aija
lain
enT
,H
erv
aE
,et
al.
Usi
ng
mu
ltil
ocu
sse
qu
ence
dat
ato
defi
ne
the
pn
eum
oco
ccu
s.J
Bac
teri
ol.
2005
;187
:622
3-30
.S
trep
toco
ccu
spy
ogen
esN
eal
S,
Bea
llB
,E
kel
un
dK
,H
enri
qu
es-N
orm
ark
B,
Jasi
rA
,Jo
hn
son
D,
Kap
lan
E,
Lo
vg
ren
M,
Rei
ner
tR
R,
Efs
trat
iou
A.
Rel
ated
Art
icle
s,L
ink
sIn
tern
atio
nal
Qu
alit
yA
ssu
ran
ceS
tud
yfo
rC
har
acte
riza
tio
no
fS
trep
toco
ccu
sp
yo
gen
es.
JC
lin
Mic
rob
iol.
2007
Ap
r;45
(4):
1175
-9.
En
rig
ht
MC
,S
pra
ttB
G,
Kal
iaA
,et
al.
Mu
ltil
ocu
sse
qu
ence
typ
ing
of
Str
epto
cocc
us
py
og
enes
and
the
rela
tio
nsh
ips
bet
wee
nem
mty
pe
and
clo
ne.
Infe
ctIm
mu
n.
2001
;69:
2416
-27.
Vib
rio
chol
erae
Ad
erK
L,
Lu
eyC
K,
Bir
dM
,T
eraj
ima
J,N
air
GB
,K
amK
M,
Ara
kaw
aE
,S
afa
A,
Ch
eun
gD
T,
Law
CP
,W
atan
abe
H,
Ku
bo
taK
,S
wam
inat
han
B,
Rib
ot
EM
.R
elat
edA
rtic
les,
Lin
ks
Dev
elo
pm
ent
and
val
idat
ion
of
aP
uls
eNet
stan
dar
diz
edp
uls
ed-fi
eld
gel
elec
tro
ph
ore
sis
pro
toco
lfo
rsu
bty
pin
go
fV
ibri
och
ole
rae.
Fo
od
bo
rne
Pat
ho
gD
is.
2006
Sp
rin
g;3
(1):
51-8
.F
oo
dA
sso
ciate
dP
ath
og
en
s
Cam
pylo
bact
erje
jun
iR
ibo
tE
M,
Fit
zger
ald
C,
Ku
bo
taK
,S
wam
inat
han
B,
Bar
rett
TJ.
Rel
ated
Art
icle
s,L
ink
sR
apid
pu
lsed
-fiel
dg
elel
ectr
op
ho
resi
sp
roto
col
for
sub
typ
ing
of
Cam
py
lob
acte
rje
jun
i.J
Cli
nM
icro
bio
l.20
01M
ay;3
9(5)
:188
9-94
.
Djo
rdje
vic
SP
,U
nic
om
bL
E,
Ad
amso
nP
J,M
ick
anL
,R
ios
R;
Au
stra
lian
Cam
py
lob
acte
rS
ub
typ
-in
gS
tud
yG
rou
p.
Rel
ated
Art
icle
s,L
ink
sC
lon
alco
mp
lex
eso
fC
amp
ylo
bac
ter
jeju
ni
iden
tifi
edb
ym
ult
ilo
cus
seq
uen
cety
pin
gar
ere
liab
lyp
red
icte
db
yre
stri
ctio
nfr
agm
ent
len
gth
po
lym
orp
his
man
aly
ses
of
the
flaA
gen
e.J
Cli
nM
icro
bio
l.20
07Ja
n;4
5(1)
:102
-8.
Din
gle
KE
,C
oll
esF
M,
War
ein
gD
R,
Ure
R,
Fo
xA
J,B
olt
on
FE
,B
oo
tsm
aH
J,W
ille
ms
RJ,
Urw
inR
,M
aid
enM
C.
Mu
ltil
ocu
sse
qu
ence
typ
ing
syst
emfo
rC
amp
ylo
bac
ter
jeju
ni.
JC
lin
Mic
rob
iol.
2001
;39:
14-2
3.
Esc
heri
chia
coli
Ger
ner
-Sm
idt
P,
Sch
eutz
F.
Sta
nd
ard
ized
pu
lsed
-fiel
dg
elel
ectr
op
ho
resi
so
fS
hig
ato
xin
-pro
du
cin
gE
sch
eric
hia
coli
:th
eP
uls
eNet
Eu
rop
eF
easi
bil
ity
Stu
dy
.F
oo
db
orn
eP
ath
og
Dis
.20
06;3
:74-
80.
Hy
yti
a-T
rees
E,
Sm
ole
SC
,F
ield
sP
A,
Sw
amin
ath
anB
,R
ibo
tE
M.
Sec
on
dg
ener
atio
nsu
bty
pin
g:
ap
rop
ose
dP
uls
eNet
pro
toco
lfo
rm
ult
iple
-lo
cus
var
iab
le-n
um
ber
tan
dem
rep
eat
anal
ysi
so
fS
hig
ato
xin
-pro
du
cin
gE
sch
eric
hia
coli
O15
7(S
TE
CO
157)
.F
oo
db
orn
eP
ath
og
Dis
.20
06;3
:118
-31.
No
ller
AC
,McE
llis
trem
MC
,Sti
ne
OC
,M
orr
isJG
Jr,
Bo
xru
dD
J,D
ixo
nB
,H
arri
son
LH
.M
ult
ilo
cus
seq
uen
cety
pin
gre
vea
lsa
lack
of
div
ersi
tyam
on
gE
sch
eric
hia
coli
O15
7:H
7is
ola
tes
that
are
dis
tin
ctb
yp
uls
ed-fi
eld
gel
elec
tro
ph
ore
sis.
JC
lin
Mic
rob
iol.
2003
;41:
675-
9.
35
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Ta
ble
1.
Co
nti
nu
ed
Path
og
en
s
Hy
bri
dis
ati
on
med
iate
d
typ
ing
tech
no
log
yP
FG
EM
LV
AS
ing
lelo
cus
seq
uen
cin
gM
ult
ilo
cus
seq
uen
cin
g
Lis
teri
am
onoc
ytog
enes
Lu
kin
maa
S,
Aar
nis
alo
K,
Su
ihk
oM
L,
Sii
ton
enA
.D
iver
sity
of
Lis
teri
am
on
ocy
tog
enes
iso
late
so
fh
um
anan
dfo
od
ori
gin
stu
die
db
yse
roty
pin
g,
auto
mat
edri
bo
typ
ing
and
pu
lsed
-fiel
dg
elel
ectr
op
ho
resi
s.C
lin
Mic
rob
iol
Infe
ct.
2004
;10:
562-
8.
Mar
tin
P,
Jacq
uet
C,
Go
ule
tV
,V
aill
ant
V,
De
Val
kH
;Par
tici
pan
tsin
the
Pu
lseN
etE
uro
pe
Fea
sib
ilit
yS
tud
y.
Rel
ated
Art
icle
s,L
ink
sP
uls
ed-fi
eld
gel
elec
tro
ph
ore
sis
of
Lis
teri
am
on
ocy
tog
enes
stra
ins:
the
Pu
lseN
etE
uro
pe
Fea
sib
ilit
yS
tud
y.
Fo
od
bo
rne
Pat
ho
gD
is.
2006
Fal
l;3(
3):3
03-8
.
Sal
ced
oC
,A
rrea
zaL
,A
lcal
aB
,d
ela
Fu
ente
L,
Vaz
qu
ezJA
.D
evel
op
men
to
fa
mu
ltil
ocu
sse
-q
uen
cety
pin
gm
eth
od
for
anal
ysi
so
fL
iste
ria
mo
no
cyto
gen
escl
on
es.
JC
lin
Mic
rob
iol.
2003
;41:
757-
62.
Sal
mon
ella
spp
Cla
rkC
G,
Kru
kT
M,
Bry
den
L,
Hir
vi
Y,
Ah
med
R,
Ro
dg
ers
FG
.S
ub
typ
ing
of
Sal
mo
nel
laen
teri
case
roty
pe
ente
riti
dis
stra
ins
by
man
ual
and
auto
mat
edP
stI-
Sp
hI
rib
oty
pin
g.
JC
lin
Mic
rob
iol.
2003
;41:
27-3
3.
pta
A,
Fo
nta
na
J,C
row
eC
,B
ols
torf
fB
,S
tou
tA
,V
anD
uy
ne
S,
Ho
ekst
raM
P,
Wh
ich
ard
JM,
Bar
rett
TJ,
An
gu
loF
J;T
he
Nat
ion
alA
nti
mic
rob
ial
Res
is-
tan
ceM
on
ito
rin
gS
yst
emP
uls
eNet
Wo
rkin
gG
rou
p.
Rel
ated
Art
icle
s,L
ink
sE
mer
gen
ceo
fm
ult
idru
g-
resi
stan
tS
alm
on
ella
ente
rica
sero
typ
eN
ewp
ort
infe
ctio
ns
resi
stan
tto
exp
and
ed-s
pec
tru
mce
ph
alo
spo
rin
sin
the
Un
ited
Sta
tes.
JIn
fect
Dis
.20
03D
ec1;
188(
11):
1707
-16.
Lin
dst
edt
BA
,T
orp
dah
lM
,N
iels
enE
M,
Var
du
nd
T,
Aas
L,
Kap
per
ud
G.H
arm
on
izat
ion
of
the
mu
ltip
le-l
ocu
sv
aria
ble
-nu
mb
erta
nd
emre
pea
tan
aly
sis
met
ho
db
etw
een
Den
mar
kan
dN
orw
ayfo
rty
pin
gS
alm
on
ella
Ty
ph
imu
riu
mis
ola
tes
and
clo
ser
exam
inat
ion
of
the
VN
TR
loci
.J
Ap
pl
Mic
rob
iol.
2007
;102
(3):
728-
35.
Ko
teti
shv
ili
M,
Sti
ne
OC
,K
reg
erA
,M
orr
isJG
Jr,
Su
lak
vel
idze
A.
Mu
ltil
ocu
sse
qu
ence
typ
ing
for
char
acte
riza
tio
no
fcl
inic
alan
den
vir
on
men
tal
salm
on
ella
stra
ins.
JC
lin
Mic
rob
iol.
2002
;40:
1626
-35.
Org
an
ism
sw
ith
bio
terr
ori
smp
ote
nti
al
Bac
illu
san
thra
cis
Van
Ert
MN
,E
aste
rday
WR
,S
imo
nso
nT
S,
U’R
enJM
,P
ears
on
T,K
enefi
cL
J,B
usc
hJD
,Hu
yn
hL
Y,
Du
ker
ich
M,
Tri
mC
B,
Bea
ud
ryJ,
Wel
ty-B
ern
ard
A,
Rea
dT
,F
rase
rC
M,
Rav
elJ,
Kei
mP
.S
trai
n-
spec
ific
sin
gle
-nu
cleo
tid
ep
oly
mo
rph
ism
assa
ys
for
the
Bac
illu
san
thra
cis
Am
esst
rain
.J
Cli
nM
icro
bio
l.20
07;4
5:47
-53.
Kim
K,C
heo
nE
,W
hee
ler
KE
,Yo
un
Y,
Lei
gh
ton
TJ,
Par
kC
,K
imW
,C
hu
ng
SI.
Det
erm
inat
ion
of
the
mo
stcl
ose
lyre
late
db
acil
lus
iso
late
sto
Bac
illu
san
thra
cis
by
mu
ltil
ocu
sse
qu
ence
typ
ing
.Y
ale
JB
iol
Med
.20
05;7
8:1-
14.
Bru
cell
am
elit
ensi
sG
arci
a-Y
old
iD
,L
eF
lech
eP
,M
arin
CM
,D
eM
igu
elM
J,M
un
oz
PM
,V
erg
nau
dG
,L
op
ez-G
on
iI.
Ass
essm
ent
of
gen
etic
stab
ilit
yo
fB
ruce
lla
mel
iten
sis
Rev
1v
acci
ne
stra
inb
ym
ult
iple
-lo
cus
var
iab
le-n
um
ber
tan
dem
rep
eat
anal
ysi
s.V
acci
ne.
2006
Oct
5;F
ran
sice
lla
tula
ren
sis
Gri
fK
,D
ieri
chM
P,
Mu
chP
,H
ofe
rE
,A
ller
ber
ger
F.
Iden
tify
ing
and
sub
typ
ing
spec
ies
of
dan
ger
ou
sp
ath
og
ens
by
auto
mat
edri
bo
typ
ing
.D
iag
nM
icro
bio
lIn
fect
Dis
.20
03;4
7:31
3-20
.
Far
low
J,W
agn
erD
M,
Du
ker
ich
M,
Sta
nle
yM
,C
hu
M,
Ku
bo
taK
,P
eter
sen
J,K
eim
P.
Fra
nci
sell
atu
lare
nsi
sin
the
Un
ited
Sta
tes.
Em
erg
Infe
ctD
is.
2005
;11:
1835
-41.
No
teto
the
tab
le:
Th
eli
stw
asd
evel
op
edb
ased
on
Pu
bM
edse
arch
es(M
arch
2007
)in
com
bin
atio
nw
ith
per
son
alex
per
ien
ceo
fth
eau
tho
rs.
Wh
ile
no
tal
lre
fere
nce
sh
ave
nec
essa
rily
add
ress
edal
lcr
iter
iad
etai
led
inth
eg
uid
lin
es,
they
hav
ed
on
eso
toa
gre
ater
deg
ree
than
pu
bli
cati
on
sn
ot
incl
ud
ed.
We
sele
cted
pap
ers
wit
ha
pre
fere
nce
for
mo
rere
cen
tp
ub
lica
tio
ns.
Inad
dit
ion
,o
rgan
ism
sfo
rw
hic
hn
ost
and
ard
ised
pro
toco
lsw
ere
avai
lab
lew
ere
excl
ud
edfr
om
the
tab
le.
Ob
vio
usl
y,
sim
ilar
list
sca
nb
em
ade
for
pla
nt
pat
ho
gen
s,v
eter
inar
yp
ath
og
ens
etc.
We
apo
log
ise
for
om
issi
on
san
dw
ron
gch
oic
es:
som
ete
chn
olo
gie
sh
ave
bee
nd
evel
op
edto
exce
llen
ceb
yv
ario
us
inv
esti
gat
ors
and
that
isw
her
ew
eh
adto
mak
ed
iffi
cult
cho
ices
.R
efer
ence
sli
sted
inth
ista
ble
are
no
tsy
stem
atic
ally
incl
ud
edin
the
refe
ren
celi
st.
36
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

T H E S E V E N P I L L A R SO F W I S E T Y P I N G
Formulation of test hypothesesInformed choice of method & control strainsUse of standardised protocolsCareful interpretation of resultsDatabase maintenanceFeedback to all involvedContinuous training and quality assessment
In conclusion, this position paper has endeav-oured to sketch the current state of affairs in thefield of molecular typing of bacteria. In theprocess, we have had to make some more orless bold choices and, although we hope thatthese guidelines will contribute to a fruitfuldiscussion and a rapprochement of all involvedin this thriving field, we would like to end bystating that we should be ready and willing toface many more challenges in the near future. Inaddition, it would be much appreciated iffunding agencies would remain open to supportof the field.
A C K N O W L E D G E M E N T S
We gratefully acknowledge A. Mellmann (University ofMuenster, Muenster, Germany), H. Witsenboer (Keygene,Wageningen, The Netherlands), C. Honisch (Sequenom, SanDiego, USA), G. Simons (Pathofinder, Maastricht, TheNetherlands), D. Horst-Kreft (Erasmus MC, Rotterdam, TheNetherlands) and P. Francois (CHUGE, Geneva, Switzerland)for making available several of the illustrations for thisarticle. The European Society of Clinical Microbiology andInfectious Diseases is thanked for making these guidelinesavailable as a supplement to Clinical Microbiology andInfection.
R E F E R E N C E S
1. Wentworth BB. Bacteriophage typing of the staphylo-cocci. Bacteriol Rev 1963; 27: 253–272.
2. Audurier A, Martin C. Phage typing of Listeria mono-cytogenes. Int J Food Microbiol 1989; 8: 251–257.
3. Uzzau S, Brown DJ, Wallis T et al. Host adapted serotypesof Salmonella enterica. Epidemiol Infect 2000; 125: 229–255.
4. Wolf MK. Occurrence, distribution and associations ofO and H serogroups, colonization factor antigens andtoxins of enterotoxigenic Escherichia coli. Clin MicrobiolRev 1997; 10: 569–584.
5. O’Hara CM, Brenner FW, Miller JM. Classification,identification and clinical significance of Proteus, Provi-dencia and Morganella. Clin Microbiol Rev 2000; 13: 534–546.
6. Burger R, Willensdorfer M, Nowak MA. Why are phe-notypic mutation rates much higher than genotypicmutation rates? Genetics 2006; 172: 197–206.
7. Graser Y, Volovsek M, Arrington J et al. Molecularmarkers reveal that population structure of the humanpathogen Candida albicans exhibits both clonality andrecombination. Proc Natl Acad Sci USA 1996; 93: 12473–12477.
8. Maslow JN, Ellis J, Mulligan M, Arbeit R. Molecularepidemiology: application of contemporary techniques tothe typing of microorganisms. Clin Infect Dis 1993; 17:153–164.
9. Mayer LW. Use of plasmid profiles in epidemiologicsurveillance of disease outbreaks and in tracing thetransmission of antibiotic resistance. Clin Microbiol Rev1988; 1: 228–243.
10. Owen RJ. Chromosomal DNA fingerprinting: a newmethod of species and strain identification applicable tomicrobial pathogens. J Med Microbiol 1989; 30: 89–99.
11. Goering RV. Molecular epidemiology of nosocomialinfection: analysis of chromosomal restriction fragmentpatterns by pulsed-field gel electrophoresis. Infect ControlHosp Epidemiol 1993; 14: 595–600.
12. Bingen E, Denamur E, Elion J. Use of ribotyping in epi-demiological surveillance of nosocomial outbreaks. ClinMicrobiol Rev 1994; 7: 311–327.
13. Van Belkum A. DNA fingerprinting of medically impor-tant microorganisms by use of PCR. Clin Microbiol Rev1994; 7: 174–184.
14. Goodfellow M, O’Donnell AG. Glossary of taxonomicterms. In: Goodfellow M, O’Donnell AG, (eds), Handbookof new bacterial systematics. London: Academic Press, 1993;29–40.
15. Colwell RR. Polyphasic taxonomy of bacteria. In: IizukaH, Hasegawa Y, (eds), Culture collections of microorganisms.Baltimore: University Press, 1968; 142–436.
16. Vandamme P, Pot B, Gillis M, De Vos P, Kersters K,Swings J. Polyphasic taxonomy, a consensus approach tobacterial classification. Microbiol Rev 1996; 60: 407–438.
17. Rossello-Mora R, Amann R. The species concept forprokaryotes. FEMS Microbiol Rev 2001; 25: 39–67.
18. Gevers D, Cohan FM, Lawrence JG et al. Opinion:re-evaluating prokaryotic species. Nat Rev Microbiol 2005;3: 733–739.
19. Vaneechoutte M, Van Eldere J. The possibilities and limi-tations of nucleic acid amplification technology in diag-nostic microbiology. J Med Microbiol 1997; 46: 188–194.
20. Pitt TL. Bacterial typing systems: the way ahead. J MedMicrobiol 1994; 40: 1–2.
21. The International P. aeruginosa Typing Study Group. Amulticenter comparison of methods for typing strains ofP. aeruginosa predominantly from patients with cysticfibrosis. J Infect Dis 1994; 169: 134–142.
22. Rabkin C, Jarvis W, Anderson R. P. cepacia typing sys-tems: collaborative study to assess their potential in epi-demiologic investigations. Rev Infect Dis 1989; 11: 600–607.
23. Tenover F, Arbeit RD, Archer G et al. Comparison oftraditional and molecular methods of typing isolates ofS. aureus. J Clin Microbiol 1994; 32: 407–415.
24. Dijkshoorn L, Aucken HM, Gerner-Schmidt P, KaufmanME, Ursing J, Pitt TL. Correlation of typing methods forAcinetobacter isolates from hospital outbreaks. J ClinMicrobiol 1993; 31: 702–705.
37
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

25. Van Embden JDA, Cave MD, Crawford JT et al. Strainidentification of M. tuberculosis by DNA fingerprinting:recommendations for a standardized methodology. J ClinMicrobiol 1993; 31: 406–409.
26. Monzo-Moreno C, Aubert S, Morvan A, El Solh N.Usefulness of three probes in typing isolates ofmethicillin-resistant S. aureus (MRSA). J Med Microbiol1991; 35: 80–88.
27. Grundmann HJ, Towner KJ, Dijkshoorn L et al. Multi-center study using standardised protocols and reagentsfor evaluation of reproducibility of PCr based finger-printing of Acinetobacter spp. J Clin Microbiol 1997; 35:3071–3077.
28. Seifert H, Dolzani L, Bressan R et al. Standardisation andinter-laboratory reproducibility assessment of pulsed-field generated fingerprints of Acinetobacter baumannii.J Clin Microbiol 2005; 43: 4328–4335.
29. Struelens MJ, Members of the European Study Group onEpidemiological Markers (ESGEM) of the EuropeanSociety for Clinical Microbiology and Infectious Diseases.Consensus guidelines for appropriate use and evaluationof microbial epidemiologic typing systems. Clin MicrobiolInfect 1996; 2: 2–11.
30. Webber HJ. New horticultural and agricultural terms.Science 1903; 18: 501–503.
31. Orskov F, Orskov O. Summary of a workshop on theclone concept in the epidemiology, taxonomy, and evo-lution of the Enterobacteriaceae. J Infect Dis 1983; 148:346–357.
32. Lan R, Reeves PR. When does a clone deserve a name? Aperspective on bacterial species based on populationgenetics. Trends Microbiol 2001; 9: 419–424.
33. Selander RK, Caugant DA, Whittam TS. Genetic struc-ture and variation in natural populations of E. coli. In:Neihardt FC, ed., E. coli and S. typhimurium. Cellularand molecular biology. Washington: ASM, 1987; 1625–1648.
34. Achtman M, Pluschke G. Clonal analysis of descent andvirulence among selected E. coli. Annu Rev Microbiol 1986;40: 185–210.
35. Maynard Smith J, Smith NH, O’Rourke M, Spratt BG.How clonal are bacteria? Proc Natl Acad Sci USA 1993; 90:4384–4388.
36. Van Dessel H, Kamp-Hopmans TE, Fluit AC et al.Outbreak of a susceptible strain of Acinetobacter species 13(sensu Tjernberg and Ursing) in an adult neurosurgicalintensive care unit. J Hosp Infect 2002; 51: 89–95.
37. Van Dessel H, Dijkshoorn L, Van der Reijden T et al.Identification of a new geographically widespread mul-tiresistant Acinetobacter baumannii clone from Europeanhospitals. Res Microbiol 2004; 155: 105–112.
38. Pitt TL, Livermore DM, Pitcher D, Vatopoulos AC, Leg-akis NJ. Multiresistant serotype O 12 P. aeruginosa; evi-dence for a common strain in Europe. Epidemiol Infect1989; 103: 565–567.
39. Dykhuizen DE, Green L. Recombination in Escherichia coliand the definition of biological species. J Bacteriol 1991;173: 7257–7268.
40. Tibayrenc M. Towards a unified evolutionary genetics ofmicroorganisms. Annu Rev Microbiol 1996; 50: 401–429.
41. Suerbaum S, Smith JM, Bapumia K et al. Free recombi-nation within Helicobacter pylori. Proc Natl Acad Sci USA1998; 95: 12619–12624.
42. Hamilton HL, Dillard JP. Natural transformation of Nei-sseria gonorrhoeae: from DNA donation to homologousrecombination. Mol Microbiol 2006; 59: 376–385.
43. Dijkshoorn L, Ursing BM, Ursing JB. Strain, clone andspecies: comments on three basic concepts of bacteriol-ogy. J Med Microbiol 2000; 49: 397–401.
44. Van Belkum A, Struelens MJ, De Visser A, Verbrugh HA,Tibayrenc M. Role of genomic typing in taxonomy, evo-lutionary genetics and microbial epidemiology. ClinMicrobiol Rev 2001; 14: 547–560.
45. Stackebrandt E, Frederiksen W, Garrity GM et al. Reportof the ad hoc committee for the re-evaluation of thespecies definition in bacteriology. Int J Syst Evol Bacteriol2002; 52: 1043–1047.
46. Miller JM. Molecular technology for hospital epidemiol-ogy. Diagn Microbiol Infect Dis 1993; 16: 153–157.
47. Jang TN, Fung CP, Yang TL, Shen SH, Huang CS, Lee SH.Use of pulsed field gel electrophoresis to investigate anoutbreak of Serratia marcescens infection in a neonatalintensive care unit. J Hosp Infect 2001; 48: 13–19.
48. Van Belkum A. High-throughput epidemiological typingin clinical microbiology. Clin Microbiol Infect 2003; 9: 86–100.
49. Struelens MJ, De Gheldre Y, Deplano A. Comparativeand library epidemiological typing systems: outbreakinvestigations versus surveillance systems. Infect ControlHosp Epidemiol 1998; 19: 565–569.
50. Singh A, Goering RV, Simjee S, Foley SL, Zervos MJ.Application of molecular techniques to the study of hos-pital infection. Clin Microbiol Rev 2006; 19: 512–530.
51. Garaizar J, Lopez Molina N, Laconcha I et al. Suitability ofPCR fingerprinting, infrequent restriction site PCR, andpulsed field gel electrophoresis combined with comput-erized gel analysis in library typing of Salmonelle entericaserovar enteritidis. Appl Environ Microbiol 2000; 66: 5273–5281.
52. De Gruijter JM, Gasser RB, Polderman AM, Asigri V,Dijkshoorn L. High resolution DNA fingerprinting byAFLP to study the genetic variation among Oesophagos-tomum bifurcum (Nematoda) from human and non-human primates from Ghana. Parasitology 2005; 130:229–237.
53. De Gruijter JM, Polderman AM, Dijkshoorn L et al. AFLPfingerprinting for the analysis of genetic diversity withinNecator americanus. Mol Cell Probes 2006; 20: 317–321.
54. Lessig R, Zoledziewska M, Fahr K et al. Y-SNP-genotyp-ing: a new approach in forensic analysis. Forensic Sci Int2005; 154: 128–136.
55. Calacal GC, Delfin FC, Tan MM et al. Identification ofexhumed remains of fire trgedy victims using conven-tional methods and autosomal ? Y chromosomal shorttandem repeat DNA profiling. Am J Forensic Med Pathol2005; 26: 285–291.
56. Budowle B, Johnson MD, Fraser CM, Leighton TJ, MurchRS, Chakraborty R. Genetic analysis and attribution ofmicrobial forensics evidence. Crit Rev Microbiol 2005; 31:233–254.
57. Nair GB, Shimada T, Kurazono H. Characterization ofphenotypic, serological and toxigenic traits of V. choleraeO139 Bengal. J Clin Microbiol 1994; 32: 2775–2779.
58. Urwin R, Maiden MC. Multi locus sequence typing: a toolfor global epidemiology. Trends Microbiol 2003; 11: 479–487.
38
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

59. Foxman B, Riley L. Molecular epidemiology: focus oninfection. Am J Epidemiol 2001; 153: 1135–1141.
60. Bernards AT, Harinck HI, Dijkshoorn L, Van der ReijdenT, Van den Broek PJ. Persistent Acinetobacter baumannii?Look inside your medical equipment. Infect Control HospEpidemiol 2004; 25: 1002–1004.
61. Weernink A, Severin WP, Tjernberg I, Dijkshoorn L. Pil-lows, an unexpected source of Acinetobacters. J HospInfect 1995; 43: 189–199.
62. Harbarth S, Albrich W, Goldmann DA, Huebner J. Con-trol of multiply resistant cocci: do international compar-isons help? Lancet Infect Dis 2001; 1: 251–261.
63. Zack JE, Garrison T, Trovillion E et al. Effect of an edu-cation program aimed at reducing the occurrence ofventilator-associated pneumonia. Crit Care Med 2002; 30:2593–2594.
64. McGuckin M, Porten LL. Handwashing education prac-tices: a descriptive survey. Clin Perform Qual Health Care1999; 7: 94–96.
65. Mermel LA, McCormick RD, Springman SR, Maki DG.The pathogenesis and epidemiology of catheter-relatedinfection with pulmonary artery Swan-Ganz catheters: aprospective study utilizing molecular subtyping. Am JMed 1991; 91: 197S–205S.
66. Grothues D, Tummler B. New approaches in genomeanalysis by pulsed-field gel electrophoresis: applicationto the analysis of P. aeruginosa. Mol Microbiol 1991; 5:2763–2776.
67. Struelens MJ, Schwam V, Deplano A, Baran D. Genomemacrorestriction analysis of diversity and variability ofP. aeruginosa infecting cystic fibrosis patients. J ClinMicrobiol 1993; 31: 2320–2326.
68. Janssen P, Maquelin K, Coopman R et al. Discriminationof Acinetobacter genomic species by AFLP fingerprinting.Int J Syst Bacteriol 1997; 47: 1179–1187.
69. Nemec A, Dijkshoorn L, van der Reijden TJ. Long-termpredominance of two pan-European clones among multi-resistant Acinetobacter baumannii strains in the CzechRepublic. J Med Microbiol 2004; 53: 147–153.
70. Dobrewski R, Savov E, Bernards AT et al. Genotypicdiversity and antibiotic susceptibility of Acinetobacterbaumannii isolates in a Bulgarian hospital. Clin MicrobiolInfect 2006; 12: 1135–1137.
71. Maiden MCJ, Bygraves JA, Feil E et al. Multilocussequence typing: a portable approach to the identificationof clones within populations of pathogenic microorgan-isms. Proc Natl Acad Sci USA 1998; 95: 3140–3145.
72. Leavis HL, Bonten MJ, Willems RJ. Identification ofhigh-risk enterococcal clonal complexes: global disper-sion and antibiotic resistance. Curr Opin Microbiol 2006;9: 454–460.
73. Hwang MS, Morgan RL, Sarkar SF, Wang PW, GuttmanDS. Phylogenetic characterization of virulence and resis-tance phenotypes of Pseudomonas syringae. Appl EnvironMicrobiol 2005; 71: 5182–5191.
74. Gomes AR, Vinga S, Zavolan M, De Lencastre H. Anal-ysis of the genetic variability of virulence-related loci inepidemic clones of methicillin-resistant Staphylococcusaureus. Antimicrob Agents Chemother 2005; 49: 366–379.
75. Feil EJ, Enright MC. Analysis of clonality and the evolu-tion of bacterial pathogens. Curr Opin Microbiol 2004; 7:308–313.
76. Marples RR, Rosdahl VT. International quality control ofphage typing of Staphylococcus aureus. International Unionof Microbial Societies Subcommittee. J Med Microbiol 1997;46: 511–516.
77. Laber TL, Iverson JT, Liberty JA, Giese SA. The evalua-tion and implementation of match criteria for forensicanalysis of DNA. J Forensic Sci 1995; 40: 1058–1064.
78. Olive DM, Bean P. Principles and applications of methodsfor DNA based-typing of microbial organisms. J ClinMicrobiol 1999; 37: 1661–1669.
79. Kanduma E, McHugh TD, Gillespie SH. Molecularmethods for Mycobacterium tuberculosis strain typing: auser’s guide. J Appl Microbiol 2003; 94: 781–791.
80. Hunter PR. A critical review of typing methods for Can-dida albicans and their applications. Crit Rev Microbiol1991; 17: 417–434.
81. Hunter PR. Reproducibility and indices of discriminatorypower of microbial typing methods. J Clin Microbiol 1990;28: 1903–1905.
82. Maslow JN, Mulligan ME. Epidemiologic typing systems.Infect Control Hosp Epidemiol 1996; 17: 595–604.
83. Mulligan ME, Arbeit RD. Epidemiologic and clinicalutility of typing systems for differentiating among strainsof methicillin-resistant Staphylococcus aureus. Infect ControlHosp Epidemiol 1991; 12: 20–28.
84. Mahony DE, Clow J, Atkinson L, Vakharia N, SchlechWF. Development and application of a multiple typingsystem for Clostridium difficile. Appl Environ Microbiol1991; 57: 1873–1879.
85. Gaston MA, Hunter PR. Efficient selection of tests forbacteriological typing schemes. J Clin Pathol 1989; 42: 763–766.
86. Hunter PR, Gaston MA. Numerical index of the dis-criminatory ability of typing systems: an application ofSimpson’s index of diversity. J Clin Microbiol 1988; 26:2465–2466.
87. Gaia V, Fry NK, Afshar B et al. Consensus sequence-based scheme for epidemiological typing of clinical andenvironmental isolates of Legionella pneumophila. J ClinMicrobiol 2005; 43: 2047–2052.
88. Tyler KD, Wang G, Tyler SD, Johnson WM. Factorsaffecting the reliability and reproducibility of amplifica-tion based DNA fingerprinting of representative bacterialpathogens. J Clin Microbiol 1997; 35: 339–346.
89. Tenover FC, Arbeit RD, Goering RV Molecular Typ-ing Workgroup of the Society of Healthcare Epidemi-ology of America, How to select and interpretmolecular strain typing methods for epidemiologicalstudies of bacterial infections: a review for healthcareepidemiologists. Infect Control Hosp Epidemiol 1997; 18:426–439.
90. Tenover FC, Arbeit RD, Goering RV et al. Interpretingchromosomal restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typ-ing. J Clin Microbiol 1995; 33: 2233–2239.
91. Konstantinidis KT, Tiedje JM. Towards a genome-basedtaxonomy for prokaryotes. J Bacteriol 2005; 187: 6255–6257.
92. Blanc DS, Struelens MJ, Deplano A et al. Epidemiologicalvalidation of pulsed-field gel electrophoresis patterns formethicillin resistant Staphylococcus aureus. J Clin Microbiol2001; 39: 3442–3445.
39
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

93. Kuhn G, Francioli P, Blanc DS. Evidence for clonal evo-lution among highly polymorphic genes in methicillin-resistant Staphylococcus aureus. J Bacteriol 2006; 188:169–178.
94. Tibayrenc M. Beyond strain typing and molecular epi-demiology: integrated genetic epidemiology of infectiousdiseases. Parasitol Today 1998; 14: 323–329.
95. Vogel L, Jones G, Triep S, Koek A, Dijkshoorn L. RAPDtyping of Klebsiella pneumoniae, Klebsiella oxytoca, Serratiamarcescens and Pseudomonas aeruginosa isolates using stan-dardized reagents. Clin Microbiol Infect 1999; 5: 270–276.
96. Carrico JA, Silva-Costa C, Melo-Cristino J et al. Illustra-tion of a common framework for relating multiple typingmethods by application to macrolide-resistant Streptococ-cus pyogenes. J Clin Microbiol 2006; 44: 2524–2532.
97. Vivanco AB, Alvarez J, Laconcha I, Lopez-Molina N,Rementeria A, Garaizar J. Molecular genotyping methodsand computerised analysis for the study of Salmonellaenterica. Methods Mol Biol 2004; 268: 49–58.
98. Ronaghi M, Elahi E. Pyrosequencing for bacterial typing.J Chromatogr B Analyt Technol Biomed Life Sci 2002; 25: 67–72.
99. Hinrikson HP, Hurst SF, De Aguirre L, Morison CJ.Molecular methods for the identification of Aspergillusspecies. Med Mycol 2005; 43: 129–137.
100. Garcia L, Kindt A, Bermudez H et al. Culture indepen-dent species typing of neotropical Leishmania for clinicalvalidation of a PCR based assay targeting heat shockprotein 70 genes. J Clin Microbiol 2004; 42: 2294–2297.
101. Deplano A, Mendonca R, De Ryck R, Struelens MJ.External quality assessment of molecular typing ofStaphylococcus aureus by a network of laboratories. J ClinMicrobiol 2006; 44: 3236–3244.
102. Achtman M. A surfeit of YATMs? J Clin Microbiol 1996;34: 1870.
103. Hallin M, Deplano A, Denis R, De Mendonca R, De RyckR, Struelens MJ. Validation of pulsed field gel electro-phoresis and spa typing for long-term national epidemi-ological surveillance studies of Staphylococcus aureusinfections. J Clin Microbiol 2007; 45: 127–133.
104. Yoshiyama K, Higuchi K, Matsumura H, Maki H.Directionality of DNA replication fork movementstrongly affects the generation of spontaneous mutationsin Escherichia coli. J Mol Biol 2001; 307: 1195–1206.
105. Kok J, Buist G, Zomer AL, Van Hijum SA, Kuipers OP.Comparative and functional genomics of lactococci.FEMS Microbiol Rev 2005; 29: 411–433.
106. Scarselli M, Giuliani MM, Adu-Bobie J, Pizza M, PappuoliR. The impact of genomics on vaccine design. TrendsBiotechnol 2005; 23: 84–91.
107. Kuroda M, Hiramatsu K. Genome sequencing andannotation: an overview. Methods Mol Biol 2004; 266:29–45.
108. Norris JR. Introduction. In: Goodfellow M, Board RG,(eds) Microbial classification and identification. London:Academic Press, 1980; 1–10.
109. Bouvet PJ, Grimont PA. Identification and biotyping ofclinical isolates of Acinetobacter. Ann Inst Pasteur Micro-biol 1987; 138: 569–578.
110. Kuhn I, Tullus K, Burman LG. The use of PhP-KE bio-chemical fingerprinting system in epidemiological stud-ies of faecal E. cloacae strains from infants in Swedishneonatal wards. Epidemiol Infect 1991; 107: 311–319.
111. Bochner BR. Sleuthing out bacterial identities. Nature1989; 339: 157–158.
112. Bochner BR. Advances in the identification of bacteriaand yeast. Am Clin Lab 1993; 12: 6.
113. Pitulle C, Citron DM, Bochner B, Barbers R, Appleman MD.Novel bacterium isolated from a lung transplant patientwith cystic fibrosis. J Clin Microbiol 1999; 37: 3851–3855.
114. Carnahan AM, Joseph SW, Janda JM. Species identifica-tion of Aeromonas strains based on carbon substrate oxi-dation profiles. J Clin Microbiol 1989; 27: 2128–2129.
115. Di Giovanni GD, Watrud LS, Seidler RJ, Widmer F. Fin-gerprinting of mixed bacterial strains and BIOLOG gram-negative (GN) substrate communities by enterobacterialrepetitive intergenic consensus sequence PCR (ERIC-PCR). Curr Microbiol 1999; 38: 217–223.
116. Bochner BR, Gadzinski P, Panomitros E. Phenotypemicroarrays for high throughput phenotypic testing andassay of gene function. Genome Res 2001; 11: 1246–1255.
117. Sloos JH, Horrevorts AM, Van Boven CP, Dijkshoorn L.Identification of multiresistant Staphylococcus epidermidisin neonates of a secondary care hospital using pulsedfield gel electrophoresis and quantitative antibiogramtyping. J Clin Pathol 1998; 51: 62–67.
118. Dijkshoorn L, Aucken H, Gerner-Smidt P et al. Compar-ison of outbreak and nonoutbreak Acinetobacter baumanniistrains by genotypic and phenotypic methods. J ClinMicrobiol 1996; 34: 1519–1525.
119. Fluit AC, Van der Bruggen JT, Aarestrup FM, Verhoef J,Jansen WT. Priorities for antibiotic resistance surveillancein Europe. Clin Microbiol Infect 2006; 12: 410–417.
120. Bronzwaer SL, Cars O, Buchholz U et al. A Europeanstudy on the relationship between antimicrobial use andantimicrobial resistance. Emerg Infect Dis 2002; 8: 278–282.
121. Frasch CE. Serogroup and serotype classification of bac-terial pathogens. Methods Enzymol 1994; 235: 159–174.
122. Vauterin L, Swings J, Kersters K. Protein electrophoresisand classification. In: Goodfellow M, O’Donnell AG,(eds), Handbook of bacterial systematics. London: AcademicPress, 1993; 251–280.
123. Stanley J, Linton D, Desai M, Efstratiou A, George R.Molecular subtyping of prevalent M serotypes of Strep-tococcus pyogenes causing invasive disease. J Clin Microbiol1995; 33: 2850–2855.
124. Nowakowska D, Colon I, Remington JS et al. Genotypingof Toxoplasm gondii by multiplex PCR and peptide basedserological testing of samples from infants in Polanddiagnosed with congenital toxoplasmosis. J Clin Microbiol2006; 44: 1382–1389.
125. Gimenez DF. Serological classification and typing ofClostridium botulinum. Dev Biol Stand 1976; 32: 175–183.
126. Coimbra RS, Grimont F, Grimont PA. Identification ofShigella serotypes by restriction of amplified O-antigengene cluster. Res Microbiol 1999; 50: 543–553.
127. Coimbra RS, Grimont F, Lenormand P, Burguiere P,Beutin L, Grimont PAD. Identification of Escherichia coliO-serogroups by restriction of the amplified O-antigengene cluster (rfb-RFLP). Res Microbiol 2000; 151: 639–654.
128. Weller TM. Methicillin resistant Staphylococcus aureustyping methods: which should be the international stan-dard? J Hosp Infect 2000; 44: 160–172.
129. Godovannyi BA, Litinskii IuI, Gerok GI. The role of col-icins in infectious pathology. Zh Mikrobiol Epidemiol Im-munobiol 1974; 1: 76–80.
40
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

130. Goerke C, Matias y Papenberg S, Dasbach S et al.Increased frequency of genomic alterations in Staphylo-coccus aureus during chronic infection is in part due tophage mobilisation. J Infect Dis 2004; 189: 724–734.
131. Dijkshoorn L, Van Vianen W, Degener JE, Michel MF.Typing of Acinetobacter calcoaceticus strains isolated fromhospital patients by cell envelope protein profiles. Epi-demiol Infect 1987; 99: 659–667.
132. Holmes B, Costas M, Wood AC. Typing of Proteus mira-bilis from clinical sources by computerized analysis ofelectrophoretic protein patterns. J Appl Bacteriol 1991; 71:467–476.
133. Van Alphen L, Bol P, Arends A, Riemens T, Geelen L.Comparison of antibiotic resistant and sensitive strains ofHaemophilus influenzae type b in The Netherlands byouter-membrane protein subtyping. Eur J Clin MicrobiolInfect Dis 1988; 7: 309–311.
134. Pot B, Hertel C, Ludwig W, Descheemaeker P, Kersters K,Schleifer KH. Identification and classification of Lacto-bacillus acidophilus, L. gasseri and L. johnsonii strains bySDS-PAGE and rRNA-targeted oligonucleotide probehybridization. J Gen Microbiol 1993; 139: 513–517.
135. Aucken HM, Pitt TL. Lipopolysaccharide profile typingas a technique for comparative typing of gram-negativebacteria. J Clin Microbiol 1993; 31: 1286–1289.
136. Pantophlet R, Brade L, Dijkshoorn L, Brade H. Specificityof rabbit antisera against lipopolysaccharide of Acineto-bacter. J Clin Microbiol 1998; 36: 1245–1250.
137. Garin Bastonje B, Bowden RA, Dubray G, Limet JN.Sodium dodecyl sulphate polyacrylamide gel electro-phoresis and immunoblotting analysis of smooth lipo-polysaccharide heterogeneity among Brucella biovarsrekated to A and M specificities. J Clin Microbiol 1990; 28:2169–2174.
138. Selander RK, Caugant DA, Ochman H, Musser J, GilmourMN, Whittam TS. Methods of multilocus enzyme elec-trophoresis for bacterial population genetics and sys-tematics. Appl Environ Microbiol 1986; 51: 873–884.
139. Duffield AM. The use of mass spectrometry for theidentification of metabolites of phenothiazines. Adv Bio-chem Psychopharmacol 1974; 9: 111–117.
140. Dworzanski JP, Snyder AP. Classification and identifica-tion of bacteria using mass spectrometry-based proteo-mics. Expert Rev Proteomics 2005; 2: 863–878.
141. Jackson KA, Edwards-Jones V, Sutton CW, Fox AJ.Optimisation of intact cell MALDI method for finger-printing of methicillin-resistant Staphylococcus aureus.J Microbiol Methods 2005; 62: 273–284.
142. Lechner M, Fille M, Hausdorfer J, Dierich MP, Rieder J.Diagnosis of bacteria by mass spectrometric fingerprint-ing: a pilot study. Curr Microbiol 2005; 51: 267–269.
143. Dieckmann R, Graeber I, Kaesler I, Szewzyk U, VonDohren H. Rapid screening and dereplication of bacterialisolates from marine sponges of the sula ridge by intactcell MALDI-TOF mass spectrometry (ICM-MS). ApplMicrobiol Biotechnol 2005; 67: 539–548.
144. Du Z, Yang R, Guo Z, Song Y, Wang J. Identification ofStaphylococcus aureus and determination of its methicillinresistance by matrix assisted laser desorption/ionizationtime-of-flight mass spectrometry. Anal Chem 2002; 74:5487–5491.
145. Kirschner C, Maquelin K, Pina P et al. Classification andidentification of enterococci: a comparative phenotypic,
genotypic and vibrational spectroscopic study. J ClinMicrobiol 2001; 39: 1763–1770.
146. Lefmann M, Honisch C, Bocker S et al. Novel massspectrometry-based tool for genotypic identification ofmycobacteria. J Clin Microbiol 2004; 42: 339–346.
147. Stanssens P, Zabeau M, Meersseman G et al. High-throughput MALDI-TOF discovery of genomic sequencepolymorphisms. Genome Res 2004; 14: 126–133.
148. Honisch C, Raghunathan A, Cantor CR, Palsson BO, Vanden Boom D. High throughput mutation detectionunderlying adaptive evolution of Escherichia coli K12.Genome Res 2004; 14: 2495–2502.
149. Goodacre R, Timmins EM, Burton R et al. Rapid identi-fication of urinary tract infection bacteria using hyper-spectral whole-organism fingerprinting and artificialnetworks. Microbiology 1998; 144: 1157–1170.
150. Maquelin K, Dijkshoorn L, Van der Reijden T, PuppelsGJ. Rapid epidemiological analysis of Acinetobacter strainsby Raman spectroscopy. J Microbiol Methods 2006; 64: 126–131.
151. Seltmann G, Voigt W, Beer W. Application of physico-chemical typing methods for the epidemiological analysisof Salmonella enteritidis strains of phage type 25/17. Epi-demiol Infect 1994; 113: 411–424.
152. Naumann D, Helm D, Labischinski H. Microbiologicalcharacterization by FT-IR spectroscopy. Nature 1991; 351:81–82.
153. Zhou S, Kile A, Bechner M et al. Single moleculeapproach to bacterial genomic comparisons via opticalmapping. J Bacteriol 2004; 186: 7773–7782.
154. Ecker DJ, Sampath R, Blyn LB et al. Rapid identificationand strain typing of respiratory pathogens for epidemicsurveillance. Proc Natl Acad Sci USA 2005; 102: 8012–8017.
155. Ecker JA, Massire C, Hall TA et al. Identification of Aci-netobacter species and genotyping Acinetabacter bauman-nii by multilocus PCR and mass spectrometry. J ClinMicrobiol 2006; 44: 2921–2932.
156. Bandow JE, Brotz H, Leichert LI, Labischinski H, HeckerM. Proteomic approach to understanding antibioticaction. Antimicrob Agents Chemother 2003; 47: 948–955.
157. Hall L. Are point mutations or DNA rearrangementsresponsible for the restriction fragment length polymor-phisms that are used to type bacteria? Microbiology 1994;140: 197–204.
158. Southern EM. Detection of specific sequences amongDNA fragments separated by gel electrophoresis. J MolBiol 1975; 98: 503–517.
159. Van Leeuwen WB, Verbrugh HA, Van der Velden J, VanLeeuwen N, Heck M, Van Belkum A. Validation of binarytyping for Staphylococcus aureus strains. J Clin Microbiol1999; 37: 664–674.
160. Van Leeuwen WB, Libregts C, Schalk M, Veuskens J,Verbrugh HA, Van Belkum A. Binary typing of Staphy-lococcus aureus strains through reversed hybridisationusing digoxigenin-universal linkage system-labeledbacterial genomic DNA. J Clin Microbiol 2001; 39: 328–331.
161. Van Leeuwen WB, Van Belkum A, Kreiswirth BN,Verbrugh HA. Genetic diversification of methicillinresistant Staphylococcus aureus as a function of prolongedgeographical dissemination and as measured by binarytyping and other genotyping methods. Res Microbiol 1998;149: 497–507.
41
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

162. Van Leeuwen WB, Snoeijers S, Van der Werken LibregtsC et al. Intercenter reproducibility of binary typing ofStaphylococcus aureus. J Microbiol Methods 2002; 51: 19–28.
163. Lemee L, Bourgeois I, Ruffin E, Collignon A, LemelandJF, Pons JL. Multilocus sequence analysis and compara-tive evolution of virulence associated genes and house-keeping genes of Clostridium difficile. Microbiology 2005;151: 3171–3180.
164. Shepard JR, Danin-Poleg Y, Kashi Y, Walt DR. Arraybased binary analysis for bacterial typing. Anal Chem2005; 77: 319–326.
165. Jansen WTM, Beitsma MM, Koeman CJ, Van Wamel WJB,Verhoef J, Fluit AC. Novel mobile variants of staphylo-coccal cassette chromosome mec in Staphylococcus aureus.Antimicrob Agents Chemother 2006; 50: 2072–2078.
166. Cui L, Lian J, Neoh H, Reyes E, Hiramatsu K. DNAmicro-array based identification of genes associated withglycopeptide resistance in Staphylococcus aureus. Antimic-rob Agents Chemother 2005; 49: 3404–3413.
167. Lindsay JA, Moore CE, Day NP et al. Microarrays revealthat each of ten dominant lineages of Staphylococcus aur-eus has a unique combination of surface associated andregulatory genes. J Bacteriol 2006; 188: 669–676.
168. Saunders NA, Underwood A, Kearns AM, Hallas G. Avirulence associated gene micro-array: a tool for investi-gating the evolution and pathogenic potential of Staphy-lococcus aureus. Microbiology 2004; 150: 3763–3771.
169. Glynn JR, Whiteley J, Bifani PJ, Kremer K, Van SoolingenD. Worldwide occurrence of Beijing/W strains of Myco-bacterium tuberculosis: a systematic review. Emerg InfectDis 2002; 8: 843–849.
170. Wootton SH, Gonzalez BE, Pawlak R et al. Epidemiologyor pediatric tuberculosis using traditional and moleculartechniques; Houston, Texas. Pediatrics 2005; 116: 1141–1147.
171. Grimont F, Grimont PAD. Ribosomal nucleic acid generestriction pattern as potential taxonomic tools. Ann InstPasteur Microbiol 1986; 137B: 165–175.
172. Blanc DS, Lugeon C, Wenger A, Siegrist HH, Francioli P.Quantitative antibiogram typing using inhibition zonediameters compared with ribotyping for epidemiologicaltyping of methicillin-resistant S. aureus. J Clin Microbiol1994; 32: 2505–2509.
173. Bruce JL. Automated system rapidly identifies and cha-racterises microorganisms in food. Food Technol 1996; 50:77–81.
174. Arvik T, Henick T, Gafner J. Automated genotyping ofSaccharomyces cerevisiae using the riboprinter. Int J FoodMicrobiol 2005; 104: 35–41.
175. Ito Y, Iinuma Y, Baba H et al. Evaluation of automatedribotyping system for characterisation and identificationof verocytotoxin-producing Escherichia coli isolated inJapan. Jpn J Infect Dis 2003; 56: 200–204.
176. Botes J, Williamson G, Sinickas V, Gurtler V. Genomictyping of Pseudomonas aerigunosa isolates by comparisonof riboprinting and PFGE: correlation of experimentalresults with those predicted from the complete genomesequence of isolate PAO1. J Microbiol Methods 2003; 55:231–240.
177. Grif K, Dierich MP, Much P, Hofer E, Allerberger F.Identifying and subtyping species of dangerous patho-gens by automated ribotyping. Diagn Microbiol Infect Dis2003; 47: 313–320.
178. Brisse S, Milatovic D, Fluit AC et al. Molecular surveil-lance of European quinolone resistant clinical isolates ofPseudomonas aeruginosa and Acinetobacter spp using auto-mated ribotyping. J Clin Microbiol 2000; 38: 3636–3645.
179. Brisse S, Verduin CM, Milatovic D et al. Distinguishingspecies of the Burkholderia cepacia complex and Burk-holderia gladioli by automated ribotyping. J Clin Microbiol2000; 38: 1876–1884.
180. Brisse S, Fussing V, Ridwan B, Verhoef J, Willems RJ.Automated ribotyping of vancomycin-resistant Entero-coccus faecium isolates. J Clin Microbiol 2002; 40: 1977–1984.
181. Pfaller MA, Wendt C, Hollis RJ et al. Comparative eval-uation of an automated ribotyping system versus pulsedfield gel electrophoresis for epidemiological typing ofclinical isolates of Escherichia coli and Pseudomonas aeru-ginosa from patients with recurrent Gram negative bac-teremia. Diagn Microbiol Infect Dis 2005; 25: 77–84.
182. Dalsgaard A, Forslund A, Fussing V. Traditional ribo-typing shows a higher discrimination than the automatedRiboPrinter system in typing Vibrio cholerae O1. Lett ApplMicrobiol 1999; 28: 327–333.
183. Skinner GE, Gendel SM, Fingerhut GA, Solomon HA,Ulaszek J. Differentiation between types and strains ofClostridium boltulinum by riboprinting. J Food Protect 2000;63: 1347–1352.
184. Tettelin H, Massignani V, Cieslewicz MJ et al. Genomeanalysis of multiple pathogenic isolates of Streptococcusagalactiae: implications for the microbial pan genome. ProcNatl Acad Sci USA 2005; 102: 13950–13955.
185. Eisgruber H, Wiedmann M, Stolle A. Use of plasmidprofiling as a typing method for epidemiologically relatedClostridium perfringens isolates from food poisoning casesand outbreaks. Lett Appl Microbiol 1995; 20: 290–294.
186. Pang JC, Chiu TH, Chiou CS et al. Pulsed field gel elec-trophoresis, plasmid profiles and phage types for thehuman isolates of Salmonella enterica serovar enteritidisobtained over 13 years in Taiwan. J Appl Microbiol 2005;99: 1472–1483.
187. Wei ZQ, Chen YG, Yu YS, Lu WX, Li LJ. Nosocomialspread of multi-resistant Klebsiella pneumoniae containinga plasmid encoding multiple beta-lactamases. J MedMicrobiol 2005; 54: 885–888.
188. Crawford JT. Genotyping in contact investigations: aCDC perspective. Int J Tuberc Lung Dis 2003; 7: S453–S457.
189. Dale JW. Mobile genetic elements in mycobacteria. EurRespir J Suppl 1995; 20: 633s–648s.
190. Wagner L, Lai E. Separation of large DNA molecules withhigh voltage pulsed field gel electrophoresis. Electropho-resis 1994; 15: 1078–1083.
191. Loidl J, Nairz K. Karyotype variability in yeast caused bynonallelic recombination in haploid meiosis. Genetics1997; 146: 79–88.
192. Beadle J, Wright M, NcNeely L, Bennett JW. Electropho-retic karyotype analysis in fungi. Adv Appl Microbiol 2003;53: 243–270.
193. Barney M, Volgyi A, Navarro A, Ryder D. Riboprintingand 16SrRNA gene sequencing for identification ofbrewery Pediococcus isolates. Appl Environ Microbiol 2001;67: 553–560.
194. Bens CCPM, Voss A, Klaassen CHW. Presence of anovel DNA methylation enzyme in methicillin resistantStaphylococcus aureus isolates associated with pig farm-ing leads to uninterpretable results in standard pulsed-
42
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

field gel electrophoresis. J Clin Microbiol 2006; 44: 1875–1876.
195. Coenye T, Spilker T, Martin A, LiPuma JJ. Comparativeassessment of genotyping methods for epidemiologicstudy of Burkholderia cepacia genomovar III. J Clin Micro-biol 2002; 40: 3300–3307.
196. De Zoysa AS, Harrison TG. Molecular typing of Legionellapneumophila serogroup 1 by pulsed field gel electropho-resis with SfiI and comparison of this method withrestriction fragment length polymorphism analysis. J MedMicrobiol 1999; 48: 269–278.
197. Tenover FC, McDougal LK, Goering RV et al. Charac-terisation of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in theUnited States. J Clin Microbiol 2006; 44: 108–118.
198. Cardinali G, Martini A, Preziosi R, Bistoni F, Baldelli F.Multicenter comparison of three different analytical sys-tems for evaluation of DNA banding patterns fromCryptococcus neoformans. J Clin Microbiol 2002; 40: 2095–2100.
199. Van Belkum A, Van Leeuwen WB, Kaufmann ME et al.Assessment of resolution and intercenter reproducibilityof results of genotyping Staphylococcus aureus by pulsedfield gel electrophoresis of SmaI macrorestriction frag-ments: a multicenter study. J Clin Microbiol 1998; 36: 1653–1659.
200. Samore M, Killgore G, Johnson S et al. Multicenter typingcomparison of sporadic and outbreak Clostridium difficileisolates from geographically diverse hospitals. J Infect Dis1997; 176: 1233–1238.
201. Foissaud V, Puyhardy JM, Chapalain JC et al. Inter-labo-ratory reproducibility of pulsed field gel electrophoresisfor the study of 12 types of Pseudomonas aeruginosa. PatholBiol 1999; 47: 1053–1059.
202. Walker J, Borrow R, Goering R, Egerton S, Fox AJ,Oppenheim BA. Subtyping of methicillin-resistantStaphylococcus aureus isolates from the North-West ofEngland: a comparison of standardised pulsed field gelelectrophoresis with bacteriophage typing including aninter laboratory reproducibility study. J Med Microbiol1999; 48: 297–301.
203. Zinn CS, Westh H, Rosdahl VT, Sarisa Study Group. Aninternational multicenter study of antimicrobial resis-tance and typing of hospital Staphylococcus aureus isolatesfrom 21 laboratories in 19 countries or states. Microb DrugResist 2004; 10: 160–168.
204. Murchan S, Kaufmann ME, Deplano A et al. Harmoni-sation of pulsed field gel electrophoresis protocols forepidemiological typing of strains of methicillin-resistantStaphylococcus aureus: a single approach developed byconsensus in 10 European laboratories and its applicationfor tracing the spread of related strains. J Clin Microbiol2003; 41: 1574–1585.
205. Chung M, de Lencastre H, Matthews P et al. Moleculartyping of methicillin-resistant Staphylococcus aureus bypulsed-field gel electrophoresis: comparison of resultsobtained in a multilaboratory effort using identical pro-tocols and MRSA strains. Microb Drug Res 2000; 6: 189–198.
206. Seward RJ, Ehrenstein B, Grundmann H, Towner KJ.Direct comparison of two commercially available com-puter programs for analysing DNA fingerprinting gels.J Med Microbiol 1997; 46: 314–320.
207. Van Belkum A, Sluijter M, De Groot R, Verbrugh HA,Hermans PW. Novel BOX repeat PCR assay for high-resolution typing of Streptococcus pneumoniae strains.J Clin Microbiol 1996; 34: 1176–1179.
208. Deplano A, Schuermans A, Van Eldere J et al. Multicenterevaluation of epidemiological typing of methicillin-resistant Staphylococcus aureus by repetitive-element PCRanalysis. J Clin Microbiol 2000; 38: 3527–3533.
209. Caetano-Anolles G. Scanning of nucleic acids by in vitroamplification: new developments and applications. NatBiotechnol 1996; 14: 1668–1674.
210. Al-Thawadi SI, Kessie G, Dela Cruz D, Al Ahdal MN. Acomparative study on the application of 3 molecularmethods in the epidemiological typing of bacterial iso-lates using MRSA as a prototype. Saudi Med J 2003; 24:1317–1324.
211. Krawczyk B, Naumiuk L, Lewandowski K et al. Evalua-tion and comparison of random amplification of poly-morphic DNA, pulsed field gel electrophoresis andADSRRS-fingerprinting for typing Serratia marcescensoutbreaks. . FEMS Immunol Med Microbiol 2003; 38: 241–248.
212. Healy M, Huong J, Bittner T et al. Microbial DNA typingby automated repetitive-sequence-based PCR. J ClinMicrobiol 2005; 43: 199–207.
213. Vaneechoutte M, Boerlin P, Tichy HV, Bannerman E,Jager B, Bille J. Comparison of PCR based DNA finger-printing techniques for the identification of Listeria spe-cies and their use for atypical Listeria isolates. Int J SystBacteriol 1998; 48: 127–139.
214. Vos P, Hogers R, Bleeker M et al. AFLP: a new techniquefor DNA fingerprinting. Nucleic Acids Res 1995; 23: 4407–4414.
215. Janssen P, Coopman R, Huys G et al. Evaluation of theDNA fingerprinting method AFLP as a new tool in bac-terial taxonomy. Microbiology 1996; 142: 1881–1893.
216. Valsangiacomo C, Baggi F, Gaia V, Balmelli T, Peduzzi R,Piffaretti JC. Use of AFLP in typing of Legionella pneu-mophila and application tp epidemiological studies.J Clin microbiol 1995; 33: 1716–1719.
217. Nemec A, De Baere T, Tjernberg I, Vaneechoutte M, vander Reijden TJ, Dijkshoorn L. Acinetobacter ursingii sp.nov. and Acinetobacter schindleri sp. nov., isolated fromhuman clinical specimens. Int J Syst Evol Microbiol 2001;51: 1891–1899.
218. Wroblewska MM, Dijkshoorn L, Marchel H et al. Out-break of nosocomial meningitis caused by Acinetobacterbaumannii in neurosurgical patients. J Hosp Infect 2004; 57:300–307.
219. Melles DC, Gorkink RFJ, Boelens HAM et al. Naturalpopulation dynamics and expansion of pathogenic clonesof Staphylococcus aureus. J Clin Invest 2004; 114: 1732–1740.
220. Van den Braak N, Simons G, Gorkink R et al. A new high-throughput AFLP approach for identification of newgenetic polymorphism in the genome of the clonalmicroorganism Mycobacterium tuberculosis. J MicrobiolMethods 2004; 56: 49–62.
221. Fry NK, Afshar B, Visca P et al. Assessment of fluorescentamplified fragment length polymorphism analysis forepidemiological genotyping of Legionella pneumophilaserogroup 1. Clin Microbiol Infect 2005; 11: 704–712.
222. Horvath R, Dendis M, Schlegelova J, Ruzicka F, Benedik J.A combined AFLP-multiplex PCR for molecular typing of
43
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

Escherichia coli strains using variable bacterial inter-spersed mosaic elements. Epidemiol Infect 2004; 132: 61–65.
223. Hong Y, Garcia M, Levinson S et al. Evaluation ofamplified fragment length polymorphism for differenti-ation of avian mycoplasma species. J Clin Microbiol 2005;43: 909–912.
224. Fry NK, Bangsborg JM, Bergmans A et al. Designation ofEuropean Working Group on Legionella infection(EWGLI) amplified fragment length polymorphism typesof Legionella pneumophila serogroup 1 and results ofintercentre proficiency testing using a standard protocol.Eur J Clin Microbiol Infect Dis 2002; 21: 722–728.
225. Van Belkum A, Scherer S, Van Alphen L, Verbrugh HA.Short sequence DNA repeats in prokaryotic genomes.Microbiol Mol Biol Rev 1998; 62: 275–293.
226. Hammerschmidt S, Muller A, Sillmann H et al. Capsulephase variation in Neisseria meningitidis serogroup B byslipped strand mispairing in the polysialyltransferasegene (siaD): correlation with bacterial invasion and theoutbreak of meningococcal disease. Mol Microbiol 1996;20: 1211–1220.
227. Lindstedt BA. Multiple locus variable number tandemrepeats analysis for genetic fingerprinting of pathogenicbacteria. Electrophoresis 2005; 26: 2567–2582.
228. Onteniente L, Brisse S, Tassios PT, Vergnaud G. Evalua-tion of the polymorphisms associated with tandemrepeats for Pseudomonas aeruginosa strain typing. J ClinMicrobiol 2003; 41: 4991–4997.
229. Francois P, Huyghe A, Charbonnier Y et al. Use of auto-mated multiple-locus variable number of tandem repeat-based method for rapid and high-throughput genotypingof Staphylococcus aureus isolates. J Clin Microbiol 2005; 43:3346–3355.
230. Malachowa N, Sabat A, Gniadkowski M et al. Compari-son of multiple locus variable number of tandem repeatanalysis with pulsed field gel electrophoresis, spa typingand multiple locus sequence typing for clonal charac-terisation of Staphylococcus aureus isolates. J Clin Microbiol2005; 43: 3095–3100.
231. Titze Almeida de R, Willems RJ, Top J et al. Multilocusvariable number of tandem repeat polymorphism amongBrazilian Enterococcus faecalis strains. J Clin Microbiol 2004;42: 4879–4881.
232. Johansson A, Koskiniemi S, Gottfridsson P, Wistrom J,Monsen T. Multiple locus variable number of tandemrepeat analysis for typing of Staphylococcus epidermidis.J Clin Microbiol 2006; 44: 260–265.
233. Noller AC, McEllistrem MC, Shutt KA, Harrison LH.Locus-specific mutational events in a multi-locus variablenumber of tandem repeat analysis of Escherichia coliO157:H7. J Clin Microbiol 2006; 44: 374–377.
234. Arricou-Bouvery N, Hauck Y, Bejaoui A et al. Molecularcharacterisation of Coxiella burnetii by infrequent restric-tion site PCR and MLVA typing. BMC Microbiol 2006; 26:38.
235. Sawires YS, Songer JG. Multiple-locus variable number oftandem repeat analysis for strain typing of Clostridiumperfringens. Anaerobe 2005; 11: 262–272.
236. Stratilo CW, Lewis CT, Bryden L, Mulvey MR, Bader D.Single nucleotide repeat analysis for subtyping of Bacillusanthracis. J Clin Microbiol 2006; 44: 777–782.
237. Kremer K, Arnold C, Cataldi A et al. Discriminatorypower and reproducibility of novel DNA typing methods
for Mycobacterium tuberculosis complex strains. J ClinMicrobiol 2005; 43: 5628–5638.
238. Schouls LM, Van der Ende A, Damen M, Van de Pol I.Multiple locus variable number of tandem repeat analysisof Neisseria meningitidis yields groupings similar to thoseobtained by multilocus sequence typing. J Clin Microbiol2006; 44: 1509–1518.
239. Facklam R, Beall B, Efstratiou A et al. emm Typing andvalidation of provisional M types for group A Strepto-cocci. Emerg Infect Dis 1999; 5: 247–253.
240. Frenay HM, Bunschoten AE, Schouls LM et al. Moleculartyping of methicillin resistant Staphylococcus aureus on thebasis of protein A gene polymorphisms. Eur J ClinMicrobiol Infect Dis 1996; 15: 768–770.
241. Shopsin B, Gomez M, Montgomery SO et al. Evaluation ofprotein A gene polymorphic region DNA sequencing fortyping of Staphylococcus aureus strains. J Clin Microbiol1999; 37: 3556–3563.
242. Koreen L, Ramaswamy SV, Graviss EA, Naidich S,Musser JM, Kreiswirth BN. Spa typing method fordiscriminating among Staphylococcus aureus isolates:implications for use of a single marker to detect geneticmicro- and macrovariation. J Clin Microbiol 2004; 42: 792–799.
243. Harmsen D, Claus H, Witte W et al. Typing of methicillinresistant Staphylococcus aureus in a university hospitalsetting by using novel software for spa repeat determi-nation and database management. J Clin Microbiol 2003;41: 5442–5448.
244. Mellmann A, Friedrich AW, Rosenkotter N et al. Auto-mated DNA sequence based early warning system for thedetection of methicillin-resistant Staphylococcus aureusoutbreaks. PLoS Med 2006; 3: e33.
245. Aires de Sousa M, Boye K, De Lencastre H et al. Highinterlaboratory reproducibility of DNA sequence basedtyping of bacteria in a multicenter study. J Clin Microbiol2006; 44: 619–621.
246. Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG.eBURST: inferring patterns of evolutionary descentamong clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 2004; 186: 1518–1530.
247. Enright MC, Robinson DA, Randle G, Feil EJ, Grund-mann H, Spratt BG. The evolutionary history of methi-cillin resistant Staphylococcus aureus (MRSA). Proc NatlAcad Sci USA 2002; 99: 7687–7692.
248. Serrano I, Melo-Cristino J, Carrico JA, Ramirez M. Char-acterisation of genetic lineages responsible for pneumo-coccal invasive disease in Portugal. J Clin Microbiol 2005;43: 1706–1715.
249. Brehony C, Jolley KA, Maiden MC. MLST for globalsurveillance of meningococcal disease. FEMS MicrobiolRev 2007; 31: 15–26.
250. Turner KME, Feil EJ. The secret life of the multi-locussequence type. Int J Antimicrob Agents 2007; 29: 129–135.
251. Jonas D, Meyer HG, Matthes P et al. Comparative eval-uation of three different genotyping methods for inves-tigation of nosocomial outbreaks of Legionnaires’ diseasein hospitals. J Clin Microbiol 2000; 38: 2284–2291.
252. Cooper JE, Feil EJ. Multi locus sequence typing: what isresolved? Trends Microbiol 2004; 12: 373–377.
253. Hommais F, Pereira S, Acquaviva C, Escobar Paramo P,Denamur E. Single nucleotide polymorphism phylotyping
44
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

of Escherichia coli. Appl Environ Microbiol 2005; 71: 4784–4792.
254. Kriz P, Kalmusova J, Felsberg J. Multilocus sequencetyping of Neisseria meningitidis directly from cerebrospi-nal fluid. Epidemiol Infect 2002; 128: 157–160.
255. Sa-Leao R, Tomasz A, De Lencastre H. Multilocussequence typing of Streptococcus pneumoniae clones withunusual drug resistance patterns: genetic backgroundsand relatedness to epidemic clones. J Infect Dis 2001; 184:1206–1210.
256. Aanensen DM, Spratt BG. The multi locus sequencingnetwork: mlst.net. Nucleic Acids Res 2005; 33: W728–W733.
257. Chan MS, Maiden MC, Spratt BG. Database driven multilocus sequence typing (MLST) of bacterial pathogens.Bioinformatics 2001; 17: 1077–1083.
258. Sullivan CB, Jefferies JM, Diggle MA, Clarke SC. Auto-mation of MLST using third generation liquid handlingtechnology. Mol Biotechnol 2006; 32: 219–226.
259. Clarke SC. Nucleotide sequence based typing of bacte-ria and the impact of automation. Bioessays 2002; 24:858–862.
260. Gutacker MM, Smoot JC, Migliaccio CA et al. Genome-wide analysis of synonymous single nucleotide poly-morphisms in Mycobacterium tuberculosis complexorganisms: resolution of genetic relationships amongclosely related microbial strains. Genetics 2002; 162: 1533–1543.
261. Baker L, Brown T, Maiden MC, Drobniewski F. Silentnucleotide polymorphisms, a phylogeny for Mycobacte-rium tuberculosis. Emerg Infect Dis 2004; 10: 1568–1577.
262. Van Ert MN, Easterday WR, Simonson TS et al. Strain-specific single-nucleotide polymorphism assays for theBacillus anthracis Ames strain. J Clin Microbiol 2007; 45:47–53.
263. Zhang W, Qi W, Albert TJ et al. Probing genomic diver-sity, evolution of Escherichia coli O157 by single nucleotidepolymorphisms. Genome Res 2006; 16: 757–767.
264. Roumagnac P, Weill FX, Dolecek C et al. Evolutionaryhistory of Salmonella typhi. Science 2006; 314: 1301–1304.
265. Read TD, Salzberg SL, Pop M et al. Comparative genomesequencing for discovery of novel polymorphisms inBacillus anthracis. Science 2002; 296: 2028–2033.
266. Albert TJ, Dailidiene D, Dailide G et al. Mutation dis-covery in bacterial genomes: metronidazole resistance inHelicobacter pylori. Nat Methods 2005; 2: 951–953.
267. Pearson T, Busch JD, Ravel J et al. Phylogenetic discoverybias in Bacillus anthracis using single-nucleotide poly-morphisms from whole-genome sequencing. Proc NatlAcad Sci USA 2004; 101: 13536–13541.
268. Sinclair A, Arnold C, Woodford N. Rapid detection,estimation by pyrosequencing of 23S rRNA genes with asingle nucleotide polymorphism conferring linezolidresistance in Enterococci. Antimicrob Agents Chemother2003; 47: 3620–3622.
269. Tost J, Gut IG. Genotyping single nucleotide polymor-phisms by mass spectrometry. Mass Spectrom Rev 2002;21: 388–418.
270. Syvanen AC. Accessing genetic variation: genotypingsingle nucleotide polymorphisms. Nat Rev Genet 2001; 2:930–942.
271. Kwok PY, Chen X. Detection of single nucleotide poly-morphisms. Curr Issues Mol Biol 2003; 5: 43–60.
272. Dunbar SA. Applications of Luminex xMAP technologyfor rapid, high-throughput multiplexed nucleic aciddetection. Clin Chim Acta 2006; 363: 71–82.
273. Sobrino B, Brion M, Carracedo A. SNPs in forensicgenetics: a review on SNP typing methodologies. ForensicSci Int 2005; 154: 181–194.
274. Goering RV. The molecular epidemiology of nosocomialinfection—an overview of principles, application, andinterpretation. In: Specter S, Bendinelli M, Friedman H.(eds), Rapid detection of infectious agents. New York: Ple-num Press, 1998; 131–157.
275. Tassios PT, Chadjichristodoulou C, Lambiri M et al.Molecular typing of multi-drug resistant Salmonellablockley outbreak isolates from Greece. Emerg Infect Dis2000; 6: 60–64.
276. Turton JF, Kaufmann ME, Warner M et al. A prevalentmultiresistant clone of Acinetobacter baumanii in SouthEast England. J Hosp Infect 2004; 58: 170–179.
277. Hallander HO, Advani A, Donnelly D, Gustafsson L,Carlsonn RM. Shifts of Bordetella pertussis variants inSweden from 1970 to 2003, during three periods markedby different vaccination programs. J Clin Microbiol 2005;43: 2856–2865.
278. Advani A, Donnely D, Hallander H. Reference system forcharacterization of Bordetella pertussis pulsed-field gelelectrophoresis profiles. J Clin Microbiol 2004; 4: 2890–2897.
279. Koeck JL, Njanpop-Lafourcade BM, Cade S et al. Evalu-ation and selection of tandem repeat loci for Strepotococ-cus pneumoniae MLVA strain typing. BMC Microbiol 2005;5: 66.
280. Schouls LM, Van der ende A, Van de Pol I et al. Increasein genetic diversity of Haemophilus influenzae serotype Bstrains after the introduction of the Hib vaccine in TheNetherlands. J Clin Microbiol 2005; 43: 2741–2749.
281. Johansson A, Farlow J, Larsson P et al. Worldwide geneticrelationships among Francisella tularensis isolates deter-mined by multiple locus variable number tandem repeatanalysis. J Bacteriol 2004; 186: 5808–5818.
282. Jolley KA, Chan MS, Maiden MC. mlstbNet—distributedmulti locus sequence typing (MLST) databases. BMCBioinformatics 2004; 5: 86.
283. Ricketts KD, McNaught B, Joseph CA. Travel associatedLegionnaires’ disease in Europe: 2004. Eurosurveillance2006; 11: 107–110.
284. Gerner Smidt P, Scheutz F. Standardised pulsed field gelelectrophoresis of Shiga toxin-producing Escherichia coli:the PulseNet Europe feasibility study. Foodborne PathogDis 2006; 3: 74–80.
285. Matsumoto M, Suzuki Y, Nagano H et al. Evaluation ofpulsed field gel electrophoresis analysis performed atselected prefectural institutes of public health for use inPulseNet Japan. Jpn J Infect Dis 2005; 58: 180–183.
286. Peterson LR, Noskin GA. New technology for detectingmultidrug-resistant pathogens in the clinical microbiol-ogy laboratory. Emerg Infect Dis 2001; 7: 306–311.
287. Swaminathan B, Barrett TJ, Hunter SB, Tauxe RV., CDCPulseNet Task Force PulseNet: the molecular subtypingnetwork foor foodborne bacterial disease surveillance,United States. Emerg Infect Dis 2001; 7: 382–389.
288. Gerner Smidt P, Hise K, Kincaid J et al. PulseNet USA: afive year update. Foodborne Pathog Dis 2006; 3: 9–19.
45
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46

289. Hunter SB, Vauterin P, Lambert Fair MA et al. Estab-lishment of a universal size standard for use with thePulseNet standardised pulsed field gel electrophoresisprotocols: converting the national databases to the newsize standard. J Clin Microbiol 2005; 43: 1045–1050.
290. Barrett TJ, Gerner Smidt P, Swaminathan B. Interpreta-tion of pulsed field gel electrophoresis patterns in food-borne disease investigations and surveillance. FoodbornePathog Dis 2006; 3: 20–31.
291. Cooper KL, Luey CK, Bird M et al. Development andvalidation of a PulseNet standardised pulsed field gelelectrophoresis protocol for subtyping of Vibrio cholerae.Foodborne Pathog Dis 2006; 3: 51–58.
292. Gemmell CG. Where is typing going? J Hosp Infect 1999;43: 89–92.
293. Prendergast MM, Lastovica AJ, Moran AP. Lipopolysac-charides from Campylobacter jejuni O:41 strains associatedwith Guillain–Barre syndrome exhibit molecular mimicryof GM1 ganglioside. Infect Immun 1998; 66: 3649–3655.
294. Tsang RS, Frosk P, Johnson WM. Heat labile serotyping oftwo Campylobacter jejuni strains isolated from patientswith Guillain Barre syndrome and belonging to serotypeO19 (Penner). J Clin Microbiol 2000; 38: 2021–2022.
295. Koga M, Gilbert M, Takahashi M et al. Comprehensiveanalysis of bacterial risk factors of Guillain Barre syn-drome after Campylobacter jejuni enteritis. J Infect Dis 2006;193: 547–555.
296. Moran AP, Prendergast MM, Appelmelk BJ. Molecularmimicry of host structures by bacterial lipopolysaccha-rides and its contribution to disease. FEMS Immunol MedMicrobiol 1996; 16: 105–115.
297. Van Belkum A, Van der Braak N, Godschalk P et al. ACampylobacter jejuni gene associated with immune-medi-ated neuropathy. Nature Med 2001; 7: 752–753.
298. Godschalk PC, Heikema AP, Gilbert M et al. The crucialrole of Campylobacter jejuni genes in antigangliosideantibody induction in Guillain Barre syndrome. J ClinInvest 2004; 114: 1659–1665.
299. Fouts DE, Mongodin EF, Mandrell RE et al. Major struc-tural differences and novel potential virulence mecha-nisms from the genomes of multiple Campylobacterspecies. PLoS Biol 2005; 3: e15.
300. Poly F, Threadgill D, Stinzi A. Genomic diversity inCampylobacter jejuni: identification of C. jejuni 81–176-specific genes. J Clin Microbiol 2005; 43: 2330–2338.
301. Taboada E, Acedillo RR, Carrillo CD et al. Large scalecomparative genomics meta-analysis of Campylobacterjejuni isolates reveals low level of genome plasticity. J ClinMicrobiol 2004; 42: 4566–4576.
302. Taboada E, Acedillo RR, Luebbert CC, Findlay WA, NashJHE. A new approach for the analysis of bacterial micro-array-based comparative genomic hybridisation: insightsfrom an empirical study. BMC Genomics 2005; 6: 78.
303. Stinzi A. Gene expression profile of Campylobacter jejuni inresponse to growth temperature variation. J Bacteriol 2003;185: 2009–2016.
304. Stinzi A, Whitworth L. Investigation of the Campylobacterjejuni coldshock response by global transcript profiling.Genome Lett 2003; 2: 18–27.
305. Poly F, Threadgill D, Stinzi A. Identification of Cam-pylobacter jejuni ATCC 43431-specific genes by wholemicrobial genome comparisons. J Bacteriol 2004; 186:4781–4795.
306. Champion OL, Gaunt MW, Gundogdu O et al. Compar-ative phylogenomics of the food borne pathogen Cam-pylobacter jejuni reveals genetic markers predicitive ofinfection source. Proc Natl Acad Sci USA 2005; 102: 16043–16048.
307. Van Belkum A. Molecular diagnostics in medical micro-biology: yesterday, today and tomorrow. Curr OpinPharmacol 2003; 3: 497–501.
308. Palacios G, Oberste MS. Enteroviruses as agents ofemerging infectious diseases. J Neurovirol 2005; 11: 424–433.
309. Thomson MM, Najera R. Molecular epidemiology of HIV-1 variants in the global AIDS pandemic: an update. AIDSRev 2005; 7: 210–224.
310. Vanittanakom N, Cooper CR Jr, Fisher MC, SirisanthanaT. Penicillium marneffi infection and recent advances in theepidemiology and molecular biology aspects. ClinMicrobiol Rev 2006; 19: 95–110.
311. Traub RJ, Monis PT, Robertson ID. Molecular epidemi-ology: a multi-disciplinary approach to understandingparasitic zoonoses. Int J Parasitol 2005; 35: 1295–1307.
312. Driskell AC, Ane C, Burleigh JG, McMahon MM,O’Meara BC, Sanderson MJ. Prospects for building thetree of life from large sequence databases. Science 2004;306: 1172–1174.
313. Cohan FM. Bacterial species and speciation. Syst Biol2001; 50: 513–524.
314. Christensen H, Bisgaard M, Frederiksen W, Mutters R,Kuhnert P, Olsen JE. Is characterisation of a single isolatesufficient for valid publication of a new genus or species?Proposal to modify recommendation 30b of the Bacteri-ological Code (1990 Revision). Int J Syst Evol Microbiol2001; 51: 2221–2225.
315. Dijkshoorn L, Towner KJ, Struelens M. New approaches forthe generation and analysis of microbial typing data.Amsterdam: Elsevier, 2001.
316. Gaia V, Fry NK, Harrison TG, Peduzzi R. Sequence-basedtyping of Legionella pneumophila serogroup 1 offers thepotential for true portability in legionellosis outbreakinvestigation. J Clin Microbiol 2003; 41: 1491–1502.
317. Neralla S, Glassroth J. Mycobacterium tuberculosis: thetreatment of active disease. Semin Respir Infect 2003; 18:292–306.
318. Van Belkum A, Kluytmans J, van Leeuwen W et al.Multicenter evaluation of arbitrarily primed PCR fortyping of S. aureus strains. J Clin Microbiol 1995; 33: 1537–1547.
319. Vaneechoutte M. DNA fingerprinting techniques formicroorganisms. A proposal for classification andnomenclature. Mol Biotechnol 1996; 6: 115–142.
320. Van Leeuwen JB, Jay C, Snijders S et al. Multilocussequence typing of Staphylococcus aureus with DNA arraytechnology. J Clin Microbiol 2003; 41: 3323–3326.
321. Van Ooyen A. Competition in the development of nerveconnections: a review of models. Network 2001; 12: 1–47.
46
� 2007 The AuthorsJournal compilation � 2007 Clinical Microbiology and Infectious Diseases, CMI, 13 (Suppl. 3), 1–46
Related Documents











