GSK3β inhibition promotes melanogenesis in mouse B16 melanoma cells and normal human melanocytes Barbara Bellei , Enrica Flori, Enzo Izzo, Vittoria Maresca, Mauro Picardo Laboratory of Cutaneous Physiopatology, San Gallicano Dermatological Institute, Rome, Italy ABSTRACT ARTICLE INFO Article history: Received 8 April 2008 Received in revised form 27 May 2008 Accepted 2 June 2008 Available online 12 June 2008 Keywords: β-catenin Melanogenesis Glycogen synthase kinase 3β Tyrosinase Microphthalmia-associated transcription factor Melanocytes Glycogen synthase kinase 3β (GSK3β) is implicated in many biological events, including embryonic development, cell differentiation, apoptosis, and the insulin response. GSK3β also plays a key role in the Wnt/ β-catenin pathway. The master regulator of the pigmentation microphthalmia-associated transcription factor (MITF) is a target for the Wnt pathway, however, to date, the regulatory role of GSK3β in the control of melanogenesis has not been elucidated. In this study, we evaluated the effect of inhibiting GSK3β activity on the regulation of melanocyte differentiation. Exposure of the murine melanoma cell line B16 and normal human melanocytes to GSK3β specic inhibitors (SB216763, SB415286, BIO, and LiCl) resulted in a dose- dependent accumulation of β-catenin. This is associated with the induction of melanocyte differentiation- associated markers such as melanin synthesis, tyrosinase activity, and expression of tyrosinase and the microphthalmia-associated transcription factor. Attenuation of GSK3β activity has an inhibitory effect on cell growth, and this was accompanied by morphological changes. Moreover, treatment of B16 cells with a siRNA targeted against β-catenin completely abolished the promelanogenic effect of GSK3β inhibition, however, the overexpression of a constitutively active mutant form of β-catenin (pCS2β-cat-mut) only slightly increased the degree of pigmentation. These results demonstrated that GSK3β is implicated in the regulation of melanogenesis and that pharmacological inhibition of its activity could increase melanin synthesis through mechanisms probably not restricted to Wnt/β-catenin pathway activation. © 2008 Elsevier Inc. All rights reserved. 1. Introduction Skin pigmentation serves a number of valuable functions, perhaps the most important being photoprotection of the underlying tissue from ultraviolet (UV) radiation. Therefore, numerous efforts have been devoted to understanding the molecular mechanisms that govern pigment production and their transfer to surrounding keratinocytes. Melanogenesis is subject to complex regulatory control by a large variety of intrinsic and extrinsic factors that may be produced by the environment, or by neighbouring cells in the skin. These factors include UV, melanocyte stimulating hormone (MSH), agouti signal protein (ASP), endothelin 1 (ET1), and a wide variety of growth factors and cytokines [1]. Melanin is synthesized via an enzymatic cascade controlled by tyrosinase, tyrosinase-related protein 1 (TRP1), and dopachrome tautomerase (DCT), leading to the conversion of tyrosine to melanin pigments. The most important transcription factor in the regulation of tyrosinase [2,3] and tyrosinase-related protein (TYRP) [4,5] gene expression is the microphtalmia-associated transcription factor (MITF). MITF expression is enhanced by activation of the melanocyte differentiation programme. Therefore, this cascade is consistent with the idea that MITF is a master regulator of pigmentation. In addition, MITF is a nuclear mediator of Wnt signals during melanocyte differentiation. MITF is a target for Wnt pathway [6,7] through activation of Frizzled receptor, inhibition of Glycogen synthase kinase 3β (GSK3β), and stimulation of β-catenin accumula- tion. In addition to MITF, the Wnt proteins play multiple roles in the process of neural crest formation, from induction to migration, proliferation and differentiation [8]. Mice decient in Wnt1 and Wnt3 lack pigment cells, and this phenotype is probably due to the failure of early expansion of neural crest cells [9]. Another hint at the importance of the link between Wnt signalling and MITF in melanocyte development is provided by the evidence that β-catenin is not only involved in Lef1-dependent control of Mitf gene transcription, but also functionally interacts with the MITF protein [10]. However, GSK3β, that is a negative regulator of Wnt signalling, could activate the function of MITF through phosphorylation at Ser298 [11]. Moreover, substitution of the MITF Ser298 has been associated with Waardenburg syndrome type 2 (WS2), a condition characterized by pigmentary disorders [12,13]. It is therefore conceivable that under certain conditions, GSK3β may contribute to maintaining the levels of functional MITF in melanocytes. For example, Khaled and co-workers [14] demonstrated that cAMP elevation leads to GSK3β activation and to an increased ability of MITF to bind to its target sequences. A variety Cellular Signalling 20 (2008) 1750–1761 Corresponding author. Istituto Dermatologico San Gallicano, Laboratory of Cutaneous Physiopathology, Via Elio Chianesi 53, 00144 Rome, Italy. Tel.: +39 0652666246; fax: +39 0652666247. E-mail address: [email protected] (B. Bellei). 0898-6568/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2008.06.001 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

GSK3! inhibition promotes melanogenesis in mouse B16 melanoma cells and normalhuman melanocytes
Barbara Bellei !, Enrica Flori, Enzo Izzo, Vittoria Maresca, Mauro PicardoLaboratory of Cutaneous Physiopatology, San Gallicano Dermatological Institute, Rome, Italy
A B S T R A C TA R T I C L E I N F O
Article history:Received 8 April 2008Received in revised form 27 May 2008Accepted 2 June 2008Available online 12 June 2008
Keywords:!-cateninMelanogenesisGlycogen synthase kinase 3!TyrosinaseMicrophthalmia-associatedtranscription factorMelanocytes
Glycogen synthase kinase 3! (GSK3!) is implicated in many biological events, including embryonicdevelopment, cell differentiation, apoptosis, and the insulin response. GSK3! also plays a key role in the Wnt/!-catenin pathway. The master regulator of the pigmentation microphthalmia-associated transcription factor(MITF) is a target for the Wnt pathway, however, to date, the regulatory role of GSK3! in the control ofmelanogenesis has not been elucidated. In this study, we evaluated the effect of inhibiting GSK3! activity onthe regulation of melanocyte differentiation. Exposure of the murine melanoma cell line B16 and normalhuman melanocytes to GSK3! speci!c inhibitors (SB216763, SB415286, BIO, and LiCl) resulted in a dose-dependent accumulation of !-catenin. This is associated with the induction of melanocyte differentiation-associated markers such as melanin synthesis, tyrosinase activity, and expression of tyrosinase and themicrophthalmia-associated transcription factor. Attenuation of GSK3! activity has an inhibitory effect on cellgrowth, and this was accompanied by morphological changes. Moreover, treatment of B16 cells with a siRNAtargeted against !-catenin completely abolished the promelanogenic effect of GSK3! inhibition, however, theoverexpression of a constitutively active mutant form of !-catenin (pCS2!-cat-mut) only slightly increasedthe degree of pigmentation. These results demonstrated that GSK3! is implicated in the regulation ofmelanogenesis and that pharmacological inhibition of its activity could increase melanin synthesis throughmechanisms probably not restricted to Wnt/!-catenin pathway activation.
© 2008 Elsevier Inc. All rights reserved.
1. Introduction
Skin pigmentation serves a number of valuable functions, perhapsthe most important being photoprotection of the underlying tissuefrom ultraviolet (UV) radiation. Therefore, numerous efforts have beendevoted to understanding the molecular mechanisms that governpigment production and their transfer to surrounding keratinocytes.Melanogenesis is subject to complex regulatory control by a largevariety of intrinsic and extrinsic factors that may be produced by theenvironment, or by neighbouring cells in the skin. These factorsinclude UV, melanocyte stimulating hormone (MSH), agouti signalprotein (ASP), endothelin 1 (ET1), and a wide variety of growth factorsand cytokines [1]. Melanin is synthesized via an enzymatic cascadecontrolled by tyrosinase, tyrosinase-related protein 1 (TRP1), anddopachrome tautomerase (DCT), leading to the conversion of tyrosineto melanin pigments. The most important transcription factor in theregulation of tyrosinase [2,3] and tyrosinase-related protein (TYRP)[4,5] gene expression is the microphtalmia-associated transcriptionfactor (MITF). MITF expression is enhanced by activation of the
melanocyte differentiation programme. Therefore, this cascade isconsistent with the idea that MITF is a master regulator ofpigmentation. In addition, MITF is a nuclear mediator of Wnt signalsduring melanocyte differentiation. MITF is a target for Wnt pathway[6,7] through activation of Frizzled receptor, inhibition of Glycogensynthase kinase 3! (GSK3!), and stimulation of !-catenin accumula-tion. In addition to MITF, the Wnt proteins play multiple roles in theprocess of neural crest formation, from induction to migration,proliferation and differentiation [8]. Mice de!cient in Wnt1 andWnt3 lack pigment cells, and this phenotype is probably due to thefailure of early expansion of neural crest cells [9]. Another hint at theimportance of the link between Wnt signalling and MITF inmelanocyte development is provided by the evidence that !-cateninis not only involved in Lef1-dependent control of Mitf genetranscription, but also functionally interacts with the MITF protein[10]. However, GSK3!, that is a negative regulator of Wnt signalling,could activate the function ofMITF through phosphorylation at Ser298[11]. Moreover, substitution of the MITF Ser298 has been associatedwith Waardenburg syndrome type 2 (WS2), a condition characterizedby pigmentary disorders [12,13]. It is therefore conceivable that undercertain conditions, GSK3!may contribute to maintaining the levels offunctional MITF in melanocytes. For example, Khaled and co-workers[14] demonstrated that cAMP elevation leads to GSK3! activation andto an increased ability of MITF to bind to its target sequences. A variety
Cellular Signalling 20 (2008) 1750–1761
! Corresponding author. Istituto Dermatologico San Gallicano, Laboratory of CutaneousPhysiopathology, Via Elio Chianesi 53, 00144 Rome, Italy. Tel.: +39 0652666246; fax: +390652666247.
E-mail address: [email protected] (B. Bellei).
0898-6568/$ – see front matter © 2008 Elsevier Inc. All rights reserved.doi:10.1016/j.cellsig.2008.06.001
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig

of extracellular stimuli inhibit cellular GSK3! activity, includinginsulin [15], growth factors [16,17], Wnt cell speci!cation proteins[18], and cell adhesion [19]. GSK3! phosphorylates and modulates theactivity of several key regulatory proteins involved in cell prolifera-tion, microtubule dynamics, cell motility, gene transcription, proteintranslation and glycogen metabolism [20]. However, there are noconclusive data that provide insights into the role of GSK3! inmelanocyte differentiation.
To provide further insights into the regulatory role of GSK3! in thecontrol of melanogenesis, we evaluated the effect of inhibiting GSK3!activity in the regulation of melanocyte differentiation. In this report,we clearly demonstrate that in B16 melanoma cells and in normalhuman melanocytes (NHM), activation of the Wnt pathway by GSK3!downregulation leads to an increase in the levels of differentiation-associated markers, such as melanin synthesis, tyrosinase activity, andprotein expression. These changes were also accompanied bymorphological changes in the cells.
2. Experimental procedures
2.1. Cell culture and reagents
The B16F0 murine melanoma cells were maintained in Dulbecco'smodi!ed Eagle's medium (DMEM) +7% heat-inactivated fetal bovineserum (FBS) and antibiotics (Gibco, Life Technologies Italia, Milan,Italy). For the induction studies, cells were plated, and 24 h later, themediumwas removed and the cells were then cultured in DMEMwith2% heat-inactivated FBS and antibiotics, with or without pharmaco-logical treatment. Normal humanmelanocytes were maintained inM-154 medium with Human Melanocyte Growth Supplements (HMGS)(Cascade Biologics, Inc. Mans!eld United Kingdom). SB216763,SB415286, BIO (2'Z,3'E)-6-bromoindirubin-3'-oxime), and LiCl werepurchased from Sigma (Sigma-Aldrich, Milan, Italy). The concentra-tions of the competitive speci!c inhibitors were selected according totheir IC50 values. For the less selective non-competitive inhibitor LiCl,we used doses according to previous studies that investigated thefunctional role of GSK3.
2.2. Cell proliferation analysis
Cells were plated in a 24-well plate at a density of 2!104 cells/cm2
and left to grow overnight. Cells were treated with increasingconcentrations of GSK3!' inhibitors in quadruplicate, and were leftto grow for 72 h before being incubated with 3-(4,5 dimethylthiazol)-2,5-diphenyl tetrazolium bromide (MTT) for 60 min. After this time,the medium was removed and the resulting crystals were solubilizedin DMSO. The absorbance was measured at 570 nm with a referencewavelength of 650 nm. Absorbance readings were subtracted from thevalue of blankwells, and the reduction in cell growthwas calculated asa percentage of control absorbance in the absence of any drug. Thenumber of viable cells exposed to LiCl, SB216367, SB415286, and BIOwas evaluated using a Trypan blue exclusion assay (!nal concentration0.1% w/v).
2.3. Melanin content determination
Extracellular melanin release was measured as previouslydescribed [21]. Brie"y, 2!105 B16 cells were seeded in 60 mm platesand incubated overnight, prior to GSK3 inhibitor administration. Theplates were then incubated for 72 h, after which 200 µl of the mediawas removed and the absorbance was measured spectrophotome-trically at 405 nm using a plate reader. The cells were then washedtwice with ice-cold PBS, lysed with RIPA buffer and centrifuged at10,000 !g for 10 min. Supernatants were analyzed for proteinconcentration, and pellets were solubilized in 200 µl of 1 M NaOH.Following an incubation period of 2 h at 60 °C, the absorbance was
measured spectrophotometrically at 405 nm using a plate reader.Standard curves using synthetic melanin (0-250 µg/ml) were preparedin duplicate for each experiment. Melanin production was calculatedby normalizing the total melanin values with protein content (µgmelanin/mg protein).
2.4. Tyrosinase assay
Tyrosinase enzyme activity was estimated bymeasuring the rate ofL-DOPA (3,4-dihydroxyphenylalanine) oxidation, as previouslydescribed [22] with slight modi!cations. Brie"y, cells were treatedwith different concentrations of GSK3' inhibitors for 72 h in DMEMcontaining 2% (v/v) FBS. At the end-point, the cells were solubilizedwith phosphate buffer (pH 6.8) containing 1% Triton X-100. The cellswere then disrupted by freezing and thawing, and the lysates wereclari!ed by centrifugation at 10,000 !g for 10 min. After proteinquanti!cation and adjusting protein concentrations with lysis buffer,90 µl of each lysate (each containing the same amount of protein) wasaliquoted into each well of a 96-well plate, and 10 µl of 10 mM L-DOPAwas then added to each well. Following a 90-min incubation at 37 °C,the end-point absorbance was measured spectrophotometrically at475 nm. In order to assess direct effects on tyrosinase activity,compounds were added to cell lysates at the highest concentrationused for treating whole-cell cultures and incubated for 5 min at roomtemperature. The cell lysates were then mixed with L-DOPA solutionand incubated for 2 h at 37 °C, as described above. In situ L-DOPAreactivity of B16 cells was assessed using cultures !xed in 4%paraformaldehyde in PBS for 30 min at room temperature. Afterpermeabilization in 0.1% Triton X-100 in PBS for 2 min, cells werewashed with PBS and incubated in 0.1% L-DOPA for 4 h at 37 °C. Cellswere then rinsed and images captured using an Axiovert 40C (Zeiss)inverted microscope equipped with a digital video camera system(PowerShot Digital Camera G5, Canon). In addition, absorbance wasmeasured spectrophotometrically at 475 nm.
2.5. Western Blot analysis
Whole cell extracts were prepared with RIPA buffer (Tris-bufferedsaline, 0.5% deoxycholate, 0.1% SDS, 1% Triton X-100) containingComplete Mini protease inhibitor cocktail. Cytoplasmic, nuclear,membrane and cytoskeleton proteins were sequentially isolatedusing a Compartmental protein extraction kit (Chemicon, Temecula,CA, USA). Aliquots of cell lysates were separated by electrophoresis onSDS-polyacrylamide gels, transferred to nitrocellulose membranesand then treated with the appropriate antibodies. Anti-!-catenin(Zymed, Laboratories Inc.) and anti-MITF (Exalpha Biologicals, Water-town,MA, USA) antibodies were used at 1:3000, anti-Tyrosinase (anti-TRP1 and anti-TRP2) antibodies were used at 1:2000 (Santa CruzBiotechnology Inc., Santa Cruz, CA, USA), and anti-Tubulin (Sigma-Aldrich, Milan, Italy) was used at 1:10,000. Horseradish peroxidase-conjugated goat anti-mouse and bovine anti-goat immunoglobulin(Santa Cruz Biotechnology) were used at 1:5000 and 1:3000,respectively. Antibody complexes were detected by chemilumines-cence (ECL; Amersham Life Science, Arlington Heights, IL, USA).Western blot assays were representative of at least three experiments.Densitometric analysis was performed using a GS-800 CalibratedImage Densitometer (BioRad).
2.6. siRNAs and transfection
The SignalSilenced™ GSK3"/! kit designed by Cell SignalingTechnology was used to speci!cally inhibit GSK3"/! expression. Anon-speci!c siRNA was used as a negative control. The siRNAtransfection protocol suggested by the manufacturer was optimizedas follows: 2!105 cells were plated on 60 mm plates and left to growovernight. The following day, cells were transfected with 100 pmol of
1751B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761

siRNA dimers (!nal concentration 50 nM) and 6 µl of TransfectionReagent. SiGENOME SMARTpool reagents against mouse MITF,designed by Dharmacon, Inc., and a !-catenin siRNA(m) duplex
(Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) were used tointerfere with MITF and !-catenin expression respectively. In bothcases, 100 pmol of the siRNA duplex (!nal concentration 50 nM) was
Fig. 1. GSK3! inhibition results in inhibition of cell growth in B16 cells. The effect of increasing concentrations of LiCl, (2'Z,3'E)-6-bromoindirubin-3-oxime (BIO), SB216367 andSB415286 on B16 melanoma cell viability (A and B) and on cell proliferation (C and D) was assessed in DMEM containing a high concentration of serum (7%) (A and C) and low serumconcentrations (2%) (B and D). AnMTT-based assaywas used to assess cytotoxic andmetabolic effects of GSK3! inhibition after 72 hours of treatment. Cell proliferationwas evaluatedby counting cells. Cell numbers were expressed as a percentage of the control cells maintained in medium for a comparable period of time. The addition of trypan blue 5 min beforecell counting indicated that there was a small (!5%) number of dead (stained) cells. The data show the mean±SD of three experiments performed in quadruplicate. !Pb0.05 and!!Pb0.01 versus ctrl. Changes in dendritic morphology following treatment with 20 mM LiCl, 2.5 µM BIO, 20 µM SB216367 and 50 µM SB415286 were assessed by phase-contrastmicroscopy. Original magni!cation 20! (C).
1752 B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761
mixed with 6 #l of Lipofectamine 2000 in Opti-MEM (Gibco, LifeTechnologies Italia, Milan, Italy), according to the manufacturer'sinstructions and then added to the cells. In all cases, 72 h aftertransfection, the cells were analyzed for melanin content, tyrosinaseactivity and protein expression by immunoblotting, as describedabove. In order to examine transactivation by the !-catenin/Lef/Tcfcomplex, the cells were transiently transfected with 1 µg pTopFlashluciferase reporter plasmid and 2 µg pCS2-!-cat-mut construct, or2 µg of empty vector using the Lipofectamine transfection reagent,according to the manufacturer's recommendations (Life Technolo-gies). The pFopFlash plasmid, which contains three copies of a mutantTcf/Lef consensus sequence, was used as a negative control. A pTK-Renilla-expressing vector was included as an internal control tonormalize the data. After 24 h, the cells were solubilized andresuspended in reporter lysis buffer. Fire"y and Renilla luciferaseactivity were determined by a Dual-Luciferase reporter assay(Promega, Milan, Italy) from duplicate plates. For melanogenesisstimulation experiments, the variability of the ef!ciency of transienttransfection of pCS2-!-cat-mut was evaluated by including the pCMV-!-galactosidase vector in the experiments.
2.7. Immuno!uorescence
For indirect immuno"uorescence experiments, cells were grownon coverslips that had previously been coated with 2% gelatine andplaced in 24-well plates. After treatment with GSK3! inhibitors, thecells were !xed in 4% paraformaldehyde in PBS for 30 min at 25 °C andpermeabilised with 0.05% Triton X-100 in PBS for 2min. The cells wererinsed in PBS, blocked for 30 min with PBS supplemented with 3%normal goat serum, and incubated for 2 h at room temperature with amouse monoclonal anti-!-catenin primary antibody (1:1000 in PBS)(Zymed, Laboratories Inc.) or an anti-MITF primary antibody (ExalfaBiologicals, Watertown, MA, USA). Cells were rinsed three times withPBS and incubated for 60 min at room temperature with an Alexa-Fluor-488-conjugated goat anti-mouse IgG (1:800 in PBS) (MolecularProbe, Leiden, Netherlands). After further rinsing, the nuclei werestained with DAPI (1:10,000 in PBS). Images were captured using aCCD camera (Zeiss, Oberkochen, Germany).
2.8. Reverse Transcription PCR
Semi-quantitative reverse transcription (RT-PCR) was performed inorder to characterize the expression level of tyrosinase mRNA. Afterincubation of the cells with the drugs, RNA was extracted using anRNeasymini kit (Qiagen, Inc., Valencia, CA, USA). cDNAwas synthesizedfrom 1 µg of total RNA in the presence of 0.5 µg/reaction of Oligo(dT)15primer using the Improm II™ Reverse Transcription System (Promega).First-strand cDNA was mixed with 25 pmol of forward and reverseprimers and 2.5 units of AmpliGold Taq polymerase (Perkin-Elmer,Wellesley, MA, USA). The PCR conditions were: 5 min of initialdenaturation at 94 °C and 25 cycles, consisting of 30 s for denaturationat 94 °C, 30 s at 56 °C for annealing and 1min at 72 °C for the elongationstep. Primer sequences and expected product sizes for tyrosinase andactin were: forward, 5'-GGCCAGCTTTCAGGCAGAGGT-3'; reverse 5'-TGGTGCTTCATGGGCAAAATC-3', 476 bp and forward, 5'-GACAGGATG-
CAGAAGGAGATTACT-3'; reverse, 5'- TGATCCACATCTGCTGGAAGGT-3',160 bp. The reactions were run in the exponential phase. The resultingPCR products were visualized by electrophoretic separation on 1.5%agarosegels. The gelswere stainedwith a0.5 µg/ml ethidiumbromide in1x TBE buffer. Densitometric analysis was performed using the Image Jsoftware.
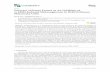
Fig. 2. Effects of GSK3! inhibitors on tyrosinase activity and on pigmentation in B16melanoma cells. Tyrosinase activity measured by L-DOPA oxidation using lysatesobtained from B16 cells treated for 72 h with GSK3! inhibitors (LiCl, BIO, SB216367 andSB415286) (A). The direct effect of GSK3! inhibitors on tyrosinase activity wasdetermined by adding 20mM LiCl, 2.5 µM BIO, 20 µM SB216367 and 50 µM SB415286 toB16 control cells lysates (B). Following incubation with increasing concentrations ofGSK3! inhibitors for 72 h, the extracellular and intracellular levels of melanin weredetermined separately by measuring the absorbance at 405 nm. Standard curves ofsynthetic melaninwere used to extrapolate the absolute values of melanin content. Thetotal amount of melanin was calculated for each experimental point by adding theextracellular and intracellular melanin values after normalization for protein content(C). The data show the mean±SD of three experiments performed in duplicate.
1753B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761
2.9. Statistical analysis
Statistical signi!cance was determined by Student's t-tests. Theminimal level of signi!cance was P=0.05
3. Results
3.1. GSK3 inhibition reduces growth of B16 melanoma cells in a dosedependent manner
The effects of GSK3 inhibition upon the proliferation and viability ofmouse melanoma B16 cells were examined in standard growth medium(7% FBS) and in low serum-medium (2%FBS). Treatment of cellswith LiCl,SB216367, SB415286 and BIO for 72 h led to a dose-dependent reductionin cell growth relative to control values (Fig. 1A and B). However,microscopic observation revealed that at the doses tested, the inhibitorsdid not compromise cell survival. Cells treated with 20 mM NaCl orvehicle alone (DMSO0.05%)were used as a control for lithiumsalt and fortreatment with speci!c inhibitors, respectively. As we never observedmodi!ed cell growth or other parameters, we used a unique generalcontrol in all of the !gures. Since the MTT assay cannot distinguishbetween reduction in cell growthandchanges in cellmetabolism,wealsoevaluated cell proliferation and membrane integrity with a trypan bluedye exclusion test and counted the cells after 72 h treatment (Fig. 1C andD). Both assays clearly demonstrated that GSK3 inhibition signi!cantlysuppressed growth (Pb0.05), and there was a slight effect upon cellviability (less than 5% trypan blue positive cells at all experimental
points). Comparison of the results obtained with theMTTassay andwithtrypan blue staining suggested that both a reduction in cell growth rateand a decrease in cell metabolism had occurred as a result of treatmentwith GSK3 inhibitors. As cell density is an important parameter that cancontribute to the activation of the differentiation programme, weperformed all of the following experiments in low serum-containingmedium to reduce cell proliferation, and to avoid cells reaching completecon"uence during the time frame of the experiments.
Melanocyte differentiation is normally coupled with an increase inmelanocyte dendrite production [23]. B16F0 cells have short dendriticprocesses under normal culture conditions (Fig. 1E). Inhibition ofGSK3 kinase activity resulted in a highly dendritic phenotype invirtually all of the cells within 24 h. Cells maintained their dose-dependent elongated cell processes during the entire duration of theexperiment (data not shown). While no differences in the timerequired to elicit morphological modi!cations were observedbetween the four inhibitors tested, an increase in the number ofthin and bipolar cells was observed with BIO treatment.
3.2. Effect of GSK3! inhibition on tyrosinase activity and melaninsynthesis
B16 cells were cultured for 3 days with DMEM containing 2% FBSand increasing doses of inhibitors. In order to evaluate the ability ofGSK3 inhibitors to stimulate melanogenesis, we assayed tyrosinaseactivity by quantifying 3,4-dihydroxyphenylalanine (DOPA) oxidation,since tyrosinase is known to be a rate-limiting enzyme in melanin
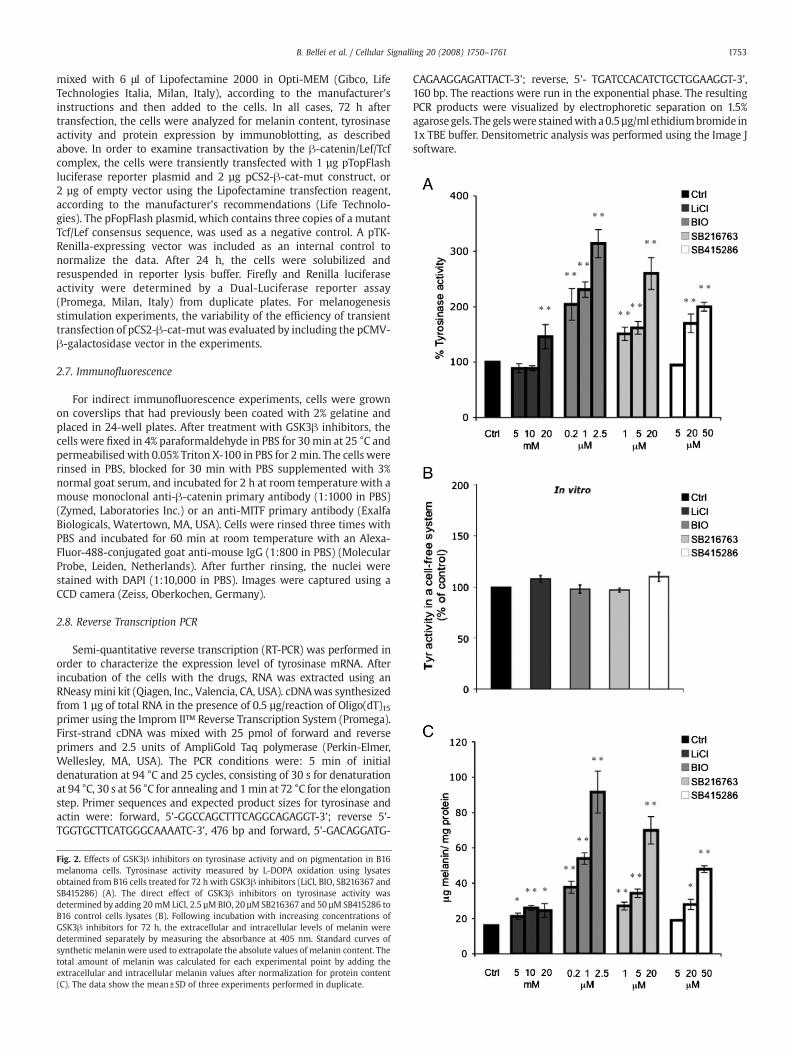
Fig. 3. GSK3! inhibition increases the expression of melanocyte differentiation markers. Expression of !-catenin, MITF, Tyrosinase, TRP1 and TRP2 proteins in B16 cells after 24 h oftreatment with increasing doses of BIO (0.25, 1 and 2.5 µM). Total cellular proteins (20 µg/lane) were subjected to 10% SDS-PAGE. Variation in protein loading was determined byblotting with an anti-!-tubulin antibody. Western blot assays are representative of at least three experiments. Densitometric scanning of band intensities obtained from threeseparate experiments were used to quantify change of proteins expression (control value taken as one-fold in each case). !Pb0.05, !!Pb0.01 (A). Semi-quantitative RT-PCR wasperformed to measure the expression of tyrosinase mRNA at various time points of GSK3 inhibition. The values shown represent the mean±SD of three independent experiments,quanti!ed by densitometry and normalized to actin mRNA levels (B).
1754 B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761
synthesis. The results obtained showed that in B16 cells, tyrosinaseactivity was signi!cantly upregulated (Pb0.05) by GSK3 inhibition in adose-dependent manner (Fig. 2A). To exclude a direct in"uence of GSK3inhibitors on tyrosinase activity, we carried out an in vitro study bydirectly adding compounds to untreated cell lysates and thenmeasuringtyrosinase activity (Fig. 2B). As shown in Fig. 2C, the melanin contentsmeasured in treated and control cells was also signi!cantly (Pb0.05)increased byGSK3 inhibition. Since compounds produced similar effectsand stimulated melanogenesis with comparable potency (with the
exception of the less speci!c inhibitor LiCl), in the second part of thisreport, we only present data obtained with BIO. Since the GSK3inhibitors did not directly regulate tyrosinase activity in a cell-freesystem, but stimulated tyrosinase activity in B16 cells, we hypothesizedthat increased melanogenic protein expression may be responsible foraugmentedmelaninproduction. Therefore,we examined theexpression
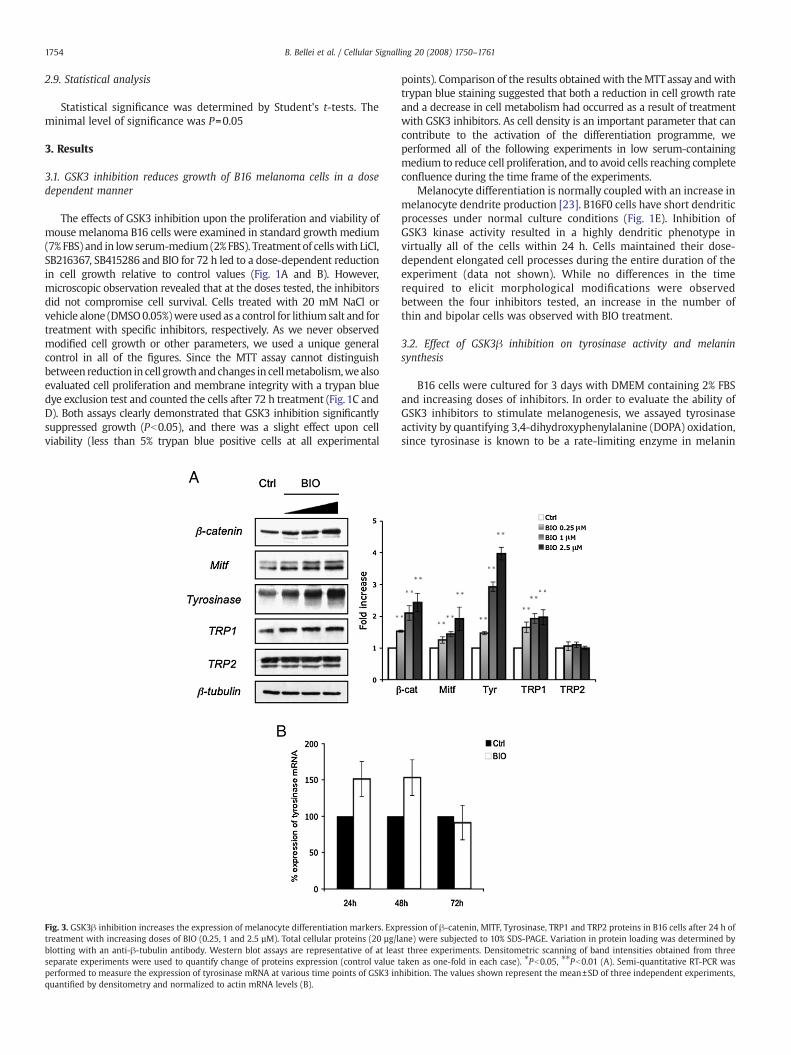
Fig. 5. The effect of siRNA on GSK3 expression and melanogenesis. B16 cells were seededinto 60 mm plates at a density of 2!105 per well, and the next day cells were transfectedwith 100 pmol of siRNA-GSK dimers and transfection reagent. Control cells were leftuntreated (transfection reagent alone), or treated by adding 1 µMBIO (transfection reagentplus BIO) 6 h after transfection. siRNA-GSK3 or a non-speci!c control siRNA (treated anduntreated) were transfected into the cells, and the cells were lysed 72 h after transfection.Cellular lysateswere analyzed by imunoblottingwith an anti-GSK3! antibodyand an anti-!-catenin antibody (A). Tyrosinase activity by L-DOPA oxidation (B) and melaninproduction (C) were measured using cell lysates obtained from cells treated as describedin A. Data show the mean±SD of two experiments performed in duplicate.
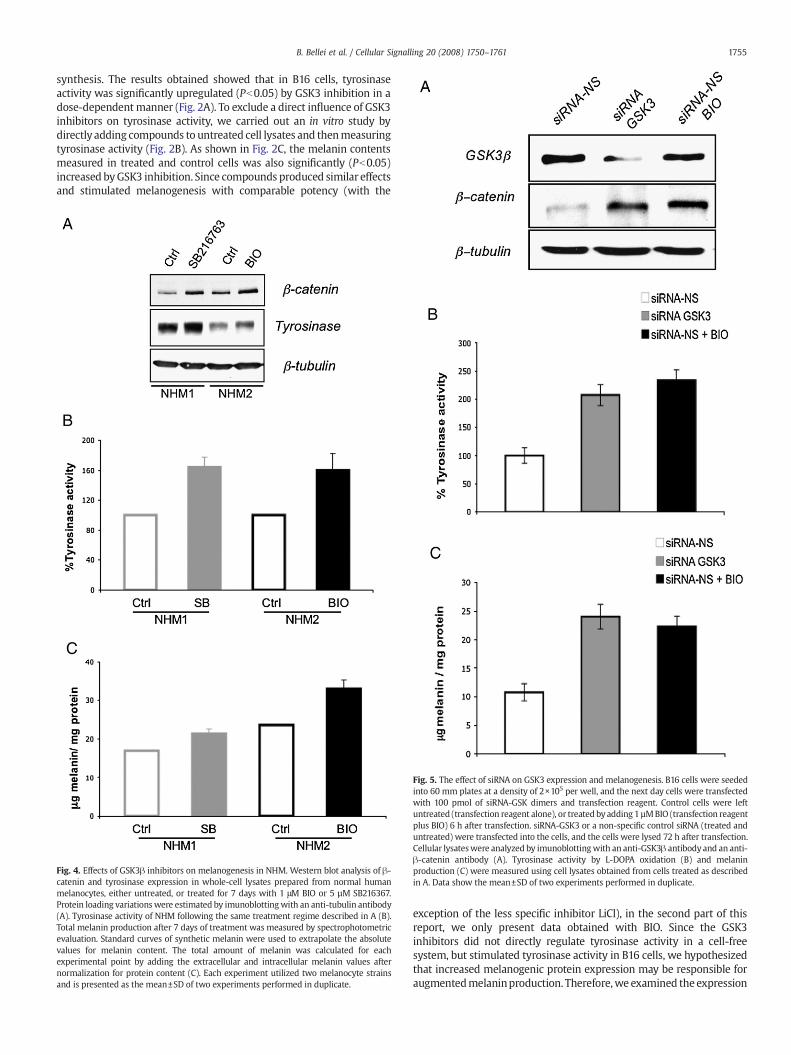
Fig. 4. Effects of GSK3! inhibitors on melanogenesis in NHM. Western blot analysis of !-catenin and tyrosinase expression in whole-cell lysates prepared from normal humanmelanocytes, either untreated, or treated for 7 days with 1 #M BIO or 5 µM SB216367.Protein loading variations were estimated by imunoblottingwith an anti-tubulin antibody(A). Tyrosinase activity of NHM following the same treatment regime described in A (B).Total melanin production after 7 days of treatment was measured by spectrophotometricevaluation. Standard curves of synthetic melanin were used to extrapolate the absolutevalues for melanin content. The total amount of melanin was calculated for eachexperimental point by adding the extracellular and intracellular melanin values afternormalization for protein content (C). Each experiment utilized two melanocyte strainsand is presented as the mean±SD of two experiments performed in duplicate.
1755B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761
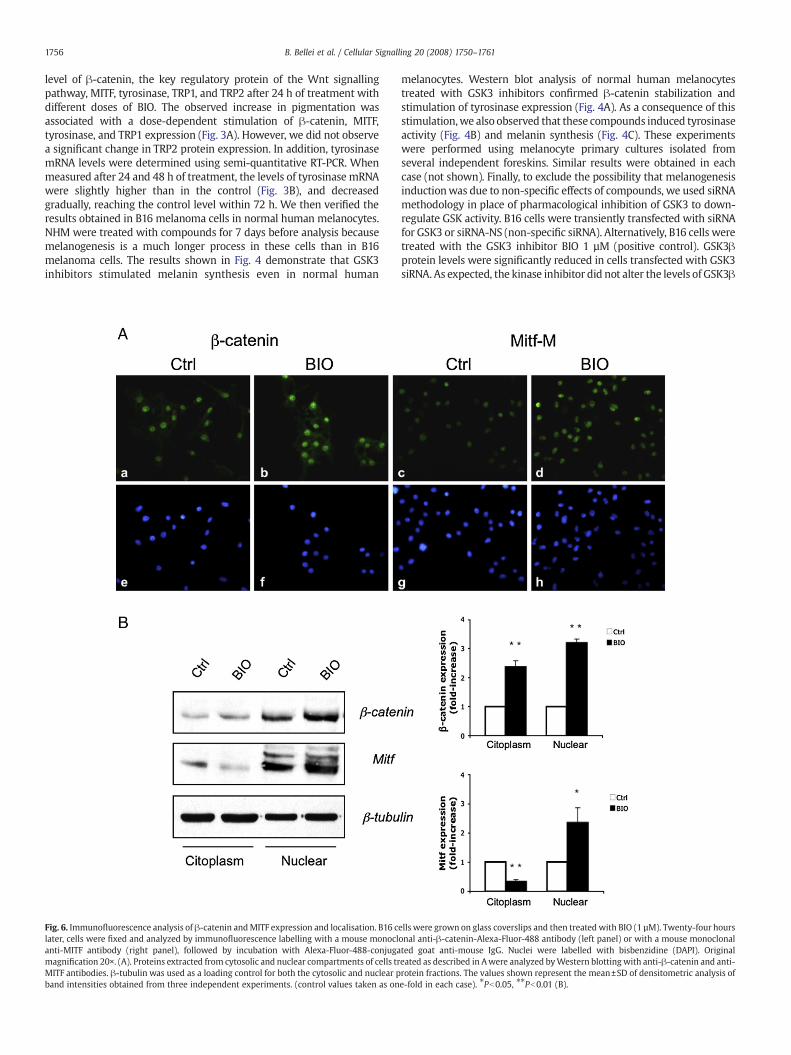
level of !-catenin, the key regulatory protein of the Wnt signallingpathway, MITF, tyrosinase, TRP1, and TRP2 after 24 h of treatment withdifferent doses of BIO. The observed increase in pigmentation wasassociated with a dose-dependent stimulation of !-catenin, MITF,tyrosinase, and TRP1 expression (Fig. 3A). However, we did not observea signi!cant change in TRP2 protein expression. In addition, tyrosinasemRNA levels were determined using semi-quantitative RT-PCR. Whenmeasured after 24 and 48 h of treatment, the levels of tyrosinase mRNAwere slightly higher than in the control (Fig. 3B), and decreasedgradually, reaching the control level within 72 h. We then veri!ed theresults obtained in B16 melanoma cells in normal human melanocytes.NHM were treated with compounds for 7 days before analysis becausemelanogenesis is a much longer process in these cells than in B16melanoma cells. The results shown in Fig. 4 demonstrate that GSK3inhibitors stimulated melanin synthesis even in normal human
melanocytes. Western blot analysis of normal human melanocytestreated with GSK3 inhibitors con!rmed !-catenin stabilization andstimulation of tyrosinase expression (Fig. 4A). As a consequence of thisstimulation,we also observed that these compounds induced tyrosinaseactivity (Fig. 4B) and melanin synthesis (Fig. 4C). These experimentswere performed using melanocyte primary cultures isolated fromseveral independent foreskins. Similar results were obtained in eachcase (not shown). Finally, to exclude the possibility that melanogenesisinductionwas due to non-speci!c effects of compounds, we used siRNAmethodology in place of pharmacological inhibition of GSK3 to down-regulate GSK activity. B16 cells were transiently transfected with siRNAfor GSK3 or siRNA-NS (non-speci!c siRNA). Alternatively, B16 cells weretreated with the GSK3 inhibitor BIO 1 µM (positive control). GSK3!protein levels were signi!cantly reduced in cells transfected with GSK3siRNA. As expected, the kinase inhibitor did not alter the levels of GSK3!
Fig. 6. Immuno"uorescence analysis of !-catenin andMITF expression and localisation. B16 cells were grown on glass coverslips and then treated with BIO (1 µM). Twenty-four hourslater, cells were !xed and analyzed by immuno"uorescence labelling with a mouse monoclonal anti-!-catenin-Alexa-Fluor-488 antibody (left panel) or with a mouse monoclonalanti-MITF antibody (right panel), followed by incubation with Alexa-Fluor-488-conjugated goat anti-mouse IgG. Nuclei were labelled with bisbenzidine (DAPI). Originalmagni!cation 20!. (A). Proteins extracted from cytosolic and nuclear compartments of cells treated as described in Awere analyzed byWestern blotting with anti-!-catenin and anti-MITF antibodies. !-tubulin was used as a loading control for both the cytosolic and nuclear protein fractions. The values shown represent the mean±SD of densitometric analysis ofband intensities obtained from three independent experiments. (control values taken as one-fold in each case). !Pb0.05, !!Pb0.01 (B).
1756 B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761

expression (Fig. 5A) As a consequence of reduced activity or expressionof GSK3!, we also observed !-catenin accumulation and stimulation ofmelanogenesis (Fig. 5B and C).
3.3. GSK3! inhibition results in increased expression and nuclearaccumulation of !-catenin and MITF
As the stimulation of the Wnt signal transduction pathway,including transcriptional activation of target genes, depends onelevation of !-catenin levels, and also on its nuclear accumulation,!-catenin expression and its cellular distributionwere investigated byimmunostaining after 24 h of treatment. In B16 melanoma cells, !-catenin proteins were localized in all cellular compartments, however,the strongest signals were observed in nuclei. As shown in Fig. 6A, BIOtreatment was associated with a marked increase in the amount of !-catenin protein in the nuclear compartment, con!rming that proteinstabilization was associated with nuclear translocation. Activation ofWnt signalling leads to an increase in MITF expression, since this is atarget gene of this pathway. In addition, the expression anddistribution of !-catenin and MITF proteins were quanti!ed usingsubcellular fractionation. Cytoplasmic, nuclear, membrane, andcytoskeleton protein fractions were prepared and investigatedseparately by western blot and densitometric analysis. As shown inFig. 6B, GSK3 inhibition was associated with a marked increase (3.2-fold increase) in the levels of !-catenin protein in the nuclearcompartment, con!rming that the Wnt pathway was activated.Consistent with GSK3! inhibition, a similar increase (2.4-foldincrease) in the free cytoplasmic pool of !-catenin protein wasobserved. In contrast, alteration in !-catenin levels in the membrane
and cytoskeleton compartments were less evident (data not shown).Interestingly, the immunoblots also showed that the increase in MITFin the nuclear compartment (2.4-fold increase) was associated with areduction in MITF in the cytoplasm (0.3-fold decrease), suggestingthat the activation of the melanogenic programme stimulates MITFexpression and also promotes its translocation from the cytoplasm tothe nuclear compartment.
3.4. Upregulation of!-catenin andMITF are required for themelanogenesisinduced by GSK3! inhibition
We then examined the effect of !-catenin depletion on melano-genesis in GSK3! inhibitor-treated cells. Lipid-mediated transfectionof a siRNA duplex corresponding to the !-catenin sequence (siRNA-!-catenin) was used to silence !-catenin expression. Western blotanalysis was performed to detect the !-catenin protein. As shown inFig. 7A, the intensity of the !-catenin signal was remarkably reducedafter treatment with siRNA-!-catenin, strongly suggesting that thesiRNA treatment successfully suppressed !-catenin expression. Thereduction of !-catenin protein in the immunoblots from cells thatwere simultaneously treated with siRNA-!-catenin and BIO wassmaller, probably because the silencing effect was balanced by proteinstabilization. Importantly, treatment of cells with siRNA-!-catenin,but not non-speci!c siRNA (siRNA-NS), signi!cantly reduced the pro-melanogenic effect of GSK3 inhibition as measured by a tyrosinaseactivity assay and measurement of total melanin content (Pb0.05)(Fig. 7B and C). At the same time, downregulation of MITF by a speci!csiRNA resulted in a dramatic block in stimulation of melanogenesis(Fig. 7C, D, and E). Taken together, these results suggested that
Fig. 7. The effect of !-catenin andMITF siRNA onmelanogenesis. B16 cells were seeded into 60mm plates at a density of 2!105 per well, and the next day, cells were transfected with100 pmol of siRNA-!-catenin dimers, siRNA-MITF plus Lipofectamine 2000 or Lipofectamine 2000 alone. Control cells were left untreated (Lipofectammine 2000 alone), or treated byadding 1 µM BIO 6 h after transfection. siRNAs or non-speci!c control siRNA (treated and untreated) were transfected into the cells, and the cells were lysed 72 h after transfection.Cellular lysates were analyzed by imunoblotting with anti-!-catenin (A) and anti-MITF antibodies (D). Tyrosinase activity by L-DOPA oxidation (B and E) and melanin production(C and F) measured in cell lysates obtained from cells treated as described above. The data show the mean±SD of two experiments performed in duplicate.
1757B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761

activated !-catenin was the mediator responsible for upregulation ofMITF expression, leading to induction of melanogenesis by GSK!inhibitors.
In order to determine whether the forced expression of !-cateninreproduced the induction of melanogenesis observed after molecularor pharmacological inhibition of GSK3!, we transiently transfectedB16 melanoma cells with a plasmid encoding a constitutively activemutant form of !-catenin (pCS2!-cat-mut) that cannot be phos-phorylated by GSK3, which is required for !-catenin accumulation.First, to compare the functionality of the mutated form of !-cateninprotein expressed by transient transfection with the overexpressedendogenous protein, we performed a gene reporter assay using a !-catenin-Lef/Tcf luciferase reporter plasmid (pTopFlash). For thisexperiment, 1!105 cells were co-transfected with 1 µg of the reporterplasmid and 2 µg pCS2-!-cat-mut construct, or empty vector (pCS2)and assayed after 24 h. Cells transfected with the reporter plasmidplus the pCS2 empty vector were also exposed to BIO for 24 h. Analysisof !-catenin-mediated transcriptional activity showed that bothtreatments produced a moderate increase in reporter gene transcrip-tion (Fig. 8A). As expected, transfection with a negative controlplasmid (pFopFlash) did not modulate luciferase activity. Since in thecurrent model of the Wnt signalling pathway, Tcf/Lef-dependenttranscription is largely dependent on the accumulation of !-catenin inthe nucleus [24], and given that we observed a moderate ("2.9-foldincrease) activation of luciferase transcription even in the presence ofa large amount of nuclear !-catenin (Fig. 8B), we postulated that inB16 cells, there might be some other limiting factors involved in theregulation of Wnt target genes. !-catenin expressed in cells
transfected with pCS2-!-cat-mut appeared as two bands at 92 and110 kDa, corresponding to the endogenous wild-type protein and tothe myc-tagged full-length mutant !-catenin, respectively. Animmunoblot with an anti-Tyr antibody and the results of assays ofmelanin production at 72 h, indicated that melanogenesis was slightlyinduced by !-catenin overexpression (Fig. 9A and B). Surprisingly, theincrease in tyrosinase protein expression did not result in adiscernable effect on protein activity as measured spectrophotome-trically by a L-DOPA oxidation assay (Fig. 9C). Finally, we evaluated thepossibility that the relatively mild increase in melanogenesis obtainedwith !-catenin transfection when compared to GSK3 inhibition wasdue to the limited transfection ef!ciency (50-60% !-galactosidase-positive cells, while BIO treatment affected thewhole cell population).To exclude the possibility that the observed upregulation ofmelanogenic enzyme expression and activity strongly occurred inonly a few cells, thus limiting melanin production, we performed an insitu tyrosinase activity assay. Measurement of L-DOPA oxidation after72 h transfection indicated that there was a modest increase intyrosinase activity in a large number of the transfected cells, and thissuggests that the observed differences in the induction of melanogen-esis between the BIO treatment and exogenous !-catenin expressionwere not related to the number of cells activated by the twotreatments (Fig. 9D). In addition, the products of the reaction werequanti!ed measuring absorbance at 475 nm (Fig. 9E). The apparentdiscordance between the two tyrosinase assays may be explained bythe higher concentration of melanin reaction products in the DOPAhistochemistry experiments due to the interference of melaninproduced during the entire duration of the experiment (72 hours).By contrast, measuring L-dopachrome formation at 475 nm in thecrude proteins sample the interference of endogenous melanin isavoided.
3.5. GSK3 inhibition promotes nuclear translocation of MITF
As the activation of the melanogenic programme by GSK3inhibition does not only stimulate MITF expression, but also promotesMITF translocation from the cytoplasm to the nuclear compartment,we explored the possibility that GSK3! controls MITF activity byregulating its cellular localization, in addition to activation of the Wntpathway. B16 cells were transfectedwith a pCS2-!-cat-mut plasmid ortreated with BIO, and protein fractions were extracted at 6 and24 hours. Immunoblots showed that !-catenin overexpression bytransient transfection increased MITF protein levels in the cytoplasm,but there was a limited change in MITF expression in the nuclearcompartment. In contrast, inhibition of GSK3! kinase activity not onlyupregulated MITF expression, but also rapidly led to translocation ofthis transcription factor to the nucleus (Fig. 10). This observation couldexplain the difference in the magnitude of stimulation of melanogen-esis observed with !-catenin overexpression and suppression of GSK3activity (Fig. 9B).
4. Discussion
The Wnt/!-catenin-signalling pathway is essential for neural crestinduction and melanocyte development. Mice lacking wild-typeWnt-1 and Wnt-3a have pigmentation defects [25]. WNT1 and WNT3Atrigger a canonical pathway resulting in !-catenin-induced transcrip-tion at Tcf/Lef (T-cell factor/lymphoid enhancer factor) promoter/enhancer elements. Numerous targets of Tcf/Lef have been identi!ed,including ubiquitous genes such as cyclin D1 [26] or c-myc [27] andmelanocyte-speci!c genes such as MITF-M [28] and Brn-2 [29].However, the importance of the Wnt pathway in the regulation ofpigmentation in normal humanmelanocytes, as well as in pigmentarydisorders remains to be determined. To provide further insight intothe regulatory role of GSK3! in intracellular signalling cascades, weinvestigated the effect of this enzyme on melanogenesis.
Fig. 8. GSK3 inhibition and!-cateninoverexpressionactivated theWnt signallingpathwayinB16 cells. B16 cellswere transfectedwith 1µgof theTopFlash luciferase reporterplasmidplus 2 µg of pCS2-!-cat-mut. The FopFlash insensitive reporter plasmid was used as anegative control. The total DNA was maintained at a constant of 3 µg per condition byaddition of empty pCS2 vector. After 6 h, the cells that were transfected with only thereporter plasmid were left untreated (control cells), or were exposed to 1 µM BIO for 24 h.Fire"y luciferase activity, normalized to the corresponding Renilla luciferase activity, isexpressed as -fold increase compared with the control cells (A). The values represent themeans±SD of two representative experiments performed in duplicate. Nuclear accumula-tion of !-catenin at the same time point was detected by Western blot analysis (B).
1758 B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761
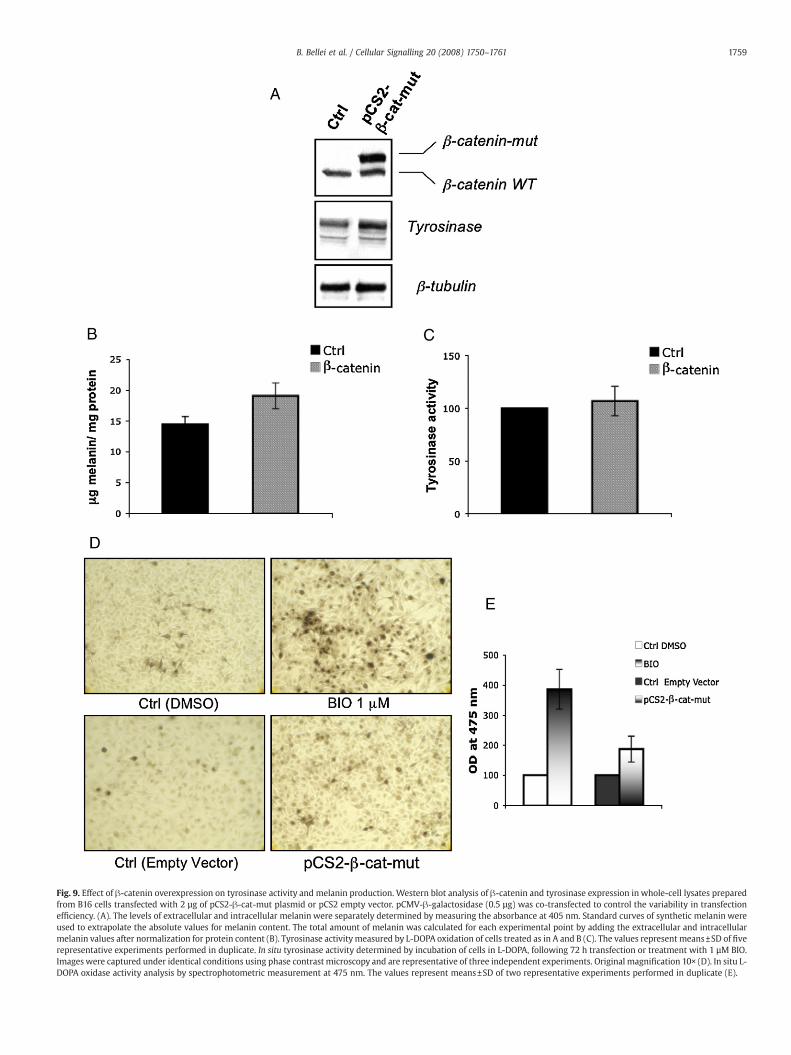
Fig. 9. Effect of !-catenin overexpression on tyrosinase activity and melanin production. Western blot analysis of !-catenin and tyrosinase expression in whole-cell lysates preparedfrom B16 cells transfected with 2 µg of pCS2-!-cat-mut plasmid or pCS2 empty vector. pCMV-!-galactosidase (0.5 µg) was co-transfected to control the variability in transfectionef!ciency. (A). The levels of extracellular and intracellular melanin were separately determined by measuring the absorbance at 405 nm. Standard curves of synthetic melanin wereused to extrapolate the absolute values for melanin content. The total amount of melanin was calculated for each experimental point by adding the extracellular and intracellularmelanin values after normalization for protein content (B). Tyrosinase activitymeasured by L-DOPA oxidation of cells treated as in A and B (C). The values represent means±SD of !verepresentative experiments performed in duplicate. In situ tyrosinase activity determined by incubation of cells in L-DOPA, following 72 h transfection or treatment with 1 µM BIO.Images were captured under identical conditions using phase contrast microscopy and are representative of three independent experiments. Original magni!cation 10! (D). In situ L-DOPA oxidase activity analysis by spectrophotometric measurement at 475 nm. The values represent means±SD of two representative experiments performed in duplicate (E).
1759B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761

We used an in vitro model of melanoma cells and primary humanmelanocyte differentiation to demonstrate that GSK3 plays a crucialrole in melanocyte differentiation. The !rst observable parameter ofmelanoma cell differentiation is morphological change [30,31]. Cellstreated with GSK3 inhibitors displayed a dendritic phenotype andrapidly acquired an elongated morphology. It is generally acceptedthat growth and differentiation are antinomic effects, meaning cellscannot proliferate and differentiate at the same time. This hypothesisis supported by the inverse correlation between cell growth andmelanogenesis observed in this study and in numerous other reports.Inhibition of GSK activity with four different inhibitors, as well asinterference with the GSK3 gene by siRNA, led to a dose-dependentreduction in cell growth and an increase in melanin synthesis andrelease. All of the compounds tested elicited cellular responsesassociated with activation of the Wnt pathway, and in addition,SB216763, SB415286, and BIO potently stimulated melanogenesis. Incontrast, LiCl slightly upregulated tyrosinase activity at all of the dosestested and signi!cantly increased melanin production only at higherdose (20 mM) only. However, LiCl is not a selective inhibitor of GSK3and has a number of additional activities that complicate interpreta-tion of the experimental data. Our experiments showed that althoughthe overexpression of the Wnt signalling pathway effector, !-catenin,is required for melanogenesis stimulation by GSK3! inhibition, it doesnot seem to satisfactorily explain the strong increase inmelanin that isobserved. In fact, a simple direct transcriptional control mechanism ofMITF gene expression due to !-catenin-mediated transcriptionalactivation is able to slightly stimulate melanin production, suggestingthat abrogation of GSK3! activity has other more direct effects on themelanogenic program. In agreement with our data, Gaggioli et al. [32]have reported that MITF is required, but is not suf!cient to induce theexpression of melanogenic genes. In fact, the forced expression ofMITF in B16 mouse melanoma cells or human melanocytes does notincrease the expression of endogenous melanogenic enzymes. Alongthe same lines, Vachtenheim et al. [33] have reported that theexpression of MITF in a set of melanoma cell lines that had lost MITF
endogenous expression did not induce tyrosinase or Tyrp1 expression.It is interesting that GSK3 inhibition promoted rapid nucleartranslocation of MITF. The predominant cytosolic distribution ofMITF in B16 cells transiently transfected with the stable form of !-catenin suggested that in addition to protein expression, other GSK3-dependent mechanisms must contribute to regulating the nuclearlocalization of MITF. As western blot analysis showed that the slowermigrating band of MITF protein was mainly localized in the nuclearextract, we cannot exclude the possibility that inhibition of GSK3 alsodirectly or indirectly causes post-transcriptional modi!cation of MITF.
GSK3 is a constitutively active kinase and phosphorylation at Ser9 inGSK3! and Ser21 in GSK3" inhibits the activity of these kinases.Phosphorylation of these serines is catalyzed by several kinases,including Akt, protein kinase A (PKA), and protein kinase C (PKC)amongothers [34]. GSK3 can also be phosphorylated at Ser9/Ser21 by themost downstream kinase of the classical mitogen-activated proteinkinase (MAPK) cascade, called MAPK-activated protein kinase-1(MAPKAP-K1, also called RSK) [35]. Growth factors [36] and tumour-promoting phorbol esters [37] inhibit GSK3 via the classical MAPKcascade. Since other agents that induce growth arrest and melanocytedifferentiation, such as UV light [38], "-MSH [39] stimulation, andhuman placental lipid [40] have also been shown to induce p38 MAPKactivation, it is possible that downregulation of GSK3 activity isphysiologically associated with melanogenesis. In line with thishypothesis, it has been shown that inhibitory phosphorylation ofGSK3 can also be induced by incubating cells with cAMP-elevatingagents or cell-permeant cAMP analogues [41,42]. In this respect, it isinteresting to note thatwe observed a UVA-induced transitory decreaseof GSK3! activity in B16 and NHM cells (unpublished data). Theapparent discrepancy of our results with previous reports demonstrat-ing that when melanogenesis stimulation involves inhibition of thephosphatidylinositol-3-kinase (PI3K) pathway, GSK3! is activated[14,43], reinforces the concept that regulation of melanin synthesisresults from cross-talk between several different signalling pathways.The data gathered in this study indicate that activation of Wnt
Fig. 10. GSK3 inhibition promotes MITF nuclear translocation. Subcellular localization of MITF was detected by Western blotting analysis of nuclear and cytoplasmic extracts after 6and 24 h of transfection with pCS2-!-cat-mut or treatment with BIO 1 µM. !-tubulin was used as a loading control. Data from densitometric scanning of band intensities obtainedfrom three separate experiments are presented as means±SD (control value taken as one-fold in each case). !Pb0.05, !!Pb0.01.
1760 B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761

signalling, through inhibition of GSK3!, may regulate pigmentation inmelanocyte and melanoma cells.
In conclusion, our results clearly demonstrated that GSK3!participates in the control of melanocyte differentiation, ensuringef!cient expression of MITF, and also regulating MITF function. Wepropose that an as yet unknown regulatory mechanism dependent onGSK3! inactivation exists, which synergizes with the Wnt canonicalpathway to control MITF function. The identi!cation of suchmechanisms will greatly improve our understanding of the processof melanocyte differentiation.
Acknowledgment
We are grateful for the gift of pCS2-!-cat-mut fromDr. Lionel Larue(Developmental Genetics of Melanocytes, Institut Curie, Cedex,France).
References
[1] J.J. Nordlund, R.E. Boissy, V.J. Hearing, B.A. King, J.P. Ortonne, The pigmentarysystem: physiology and pathophysiology, Oxford University Press, New York, 1998.
[2] K. Yasumoto, K. Yokoyama, K. Takahashi, Y. Tomita, S. Shibahara, J. Biol. Chem. 272(1997) 503.
[3] C. Bertolotto, P. Abbe, K. Bille, E. Aberdam, J.P. Ortonne, R. Ballotti, J. Cell Biol. 142(1998) 827.
[4] I. Aksan, C.R. Goding, Mol. Cell. Biol. 18 (1998) 6930.[5] C. Bertolotto, R. Busca, P. Abbe, K. Bille, E. Aberdam, J.P. Ortonne, R. Ballotti, Mol.
Cell Biol. 18 (1998) 694.[6] K. Takeda, K. Yasumoto, R. Takada, S. Takada, K. Watanabe, T. Udono, H. Saito, K.
Takahashi, S. Shibahara, J. Biol. Chem. 275 (2000) 14013.[7] H.R. Widlund, M.A. Horstmann, E.R. Price, J. Cui, S.L. Lessnick, M. Wu, X. He, D.E.
Fisher, Oncog. 22 (2002) 3035.[8] J. Wu, J.P. Saint-Jeannet, P.S. Klein, Trends Neurosci. 26 (2003) 40.[9] R.I. Dorsky, R.T. Moon, D.W. Raible, Nat. 396 (1998) 370.[10] A. Schepsky, K. Bruser, G.J. Gunnarsson, J. Goodall, J.H. Hallsson, C.R. Godin, E.
Steingrimsson, A. Hecht, Mol. Cell Biol. 26 (2006) 8914.[11] K. Takeda, C. Takemoto, I. Kobayashi, A. Watanabe, Y. Nobukuni, D.E. Fisher, M.
Tachibana, Hum. Mol. Genet. 9 (2000) 125.[12] A.E. Hughes, V.E. Newton, X.Z. Liu, P.A. Read, Nat. Genet. 7 (1994) 509.[13] M. Tassabehji, V.E. Newton, A.P. Read, Nat. genet. 8 (1994) 251.
[14] M. Khaled, L. Larribere, K. Bille, E. Aberdam, J.P. Ortonne, R. Ballotti, C. Bertolotto,J. Biol. Chem. 277 (2002) 33690.
[15] J. Ramon, J.R. Munoz-Montano, F.J. Moreno, J. Avila, J. Diaz-Nido, FEBS Lett. 411(1997) 183.
[16] H. Eldar-Finkelman, R. Seger, J.R. Vandenheede, E.G. Krebs, J. Biol. Chem. 270 (1995)987.
[17] D.A. Croos, D.R. Alessi, J.R. Vandenheede, H.E. McDowell, H.S. Hundal, P. Cohen,Biochem. J. 303 (1994) 21.
[18] J. Papkoff, M. Aikawa, Biochem. Biophys. Res. Commun. 247 (1998) 851.[19] A.A. Troussard, C. Tan, T.N. Yoganathan, S. Dedhar, Mol. Cell Biol. 19 (1999) 7420.[20] M.P. Coghlan, A.A. Culbert, D.A. Cross, S.L. Corcoran, J.W. Yates, N.J. Pearce, O.L.
Rausch, G.J. Murphy, P.S. Carter, L. Roxbee, Cox, D. Mills, M.J. Brown, D. Haigh, R.W.Ward, D.G. Smith, K.J. Murray, A.D. Reith, J.C. Holder, Chem. Biol. 7 (2000) 793.
[21] W. Siegrist, A.N. Eberle, Anal. Biochem. 159 (1986) 191.[22] H. Takahashi, P.G. Parsons, J. Invest. Dermatol. 98 (1992) 481.[23] R. Buscà, R. Ballotti, Pigment Cell Res. 13 (2000) 60.[24] A. Novak, S. Dedhar, Cell Mol. Life Sci. 56 (1999) 523.[25] M. Ikeya, S.M.K. Lee, J.E. Johnson, A.P. McMahon, S. Takada, Nat. 389 (1997) 966.[26] M. Shtutman, J. Zhurinsky, I. Simcha, C. Albanese, M. D'Amico, R. Pestell, A. Ben-
Ze'ev, Proc. Natl. Acad. Sci. U.S.A. 96 (1999) 5522.[27] T.C. He, A.B. Sparcks, C. Rago, H. Hermeking, L. Zawel, L.T. da Costa, P.J. Morin, B.
Vogelstein, K.W. Kinzler, Sci. 281 (1998) 1509.[28] R.I. Dorsky, D.W. Raible, R.T. Moon, Genes Dev. 14 (2000) 158.[29] J. Godall, S. Martinozzi, T.J. Dexter, D. Champeval, S. Carreira, L. Laure, C.R. Goding,
Mol. Cell Biol. 24 (2004) 2915.[30] J. Pawelek, M. Sansone, N. Koch, G. Christie, R. Halaban, J. Hendee, A.B. Lerner, J.M.
Varga, Proc Natl. Acad. Sci. U.S.A. 72 (1975) 951.[31] A. Busca, C. Bertolotto, J.P. Ortonne, R. Ballotti, J. Biol. Chem. 271 (1996) 31824.[32] C. Gaggioli, R. Buscà, P. Abbe, J.P. Ortonne, R. Ballotti, Pigment Cell Res. 16 (2003) 374.[33] J. Vachtenheim, H. Novotna, G. Ghanem, J. Invest. Dermatol. 117 (2001) 1505.[34] R.S. Jope, G.V. Johnson, Trends Biochem. Sci. 29 (2004) 95.[35] C. Sutherland, I.A. Leighton, P. Cohen, Biochem. J. 296 (1993) 15.[36] Y. Saito, J.R. Vanderheede, P. Cohen, Biochem. J. 303 (1994) 27.[37] V. Stambolic, J.R. Woodgett, Biochem. J. 303 (1994) 701.[38] H. Yanase, H. Ando, M. Horikawa, M. Watanabe, T. Mori, N. Matsuda, Pigment Cell
Res. 14 (2001) 103.[39] K. Smalley, T. Eisen, FEBS Lett. 476 (2000) 198.[40] S.K. Singh, C. Sarkar, S. Mallick, B. Saha, R. Bera, R. Bhadra, Pigment Cell Res. 18
(2005) 113.[41] X. Fang, S.X. Yu, R.C. Bast, J.R. Woodgett, G.B. Mills, Proc. Natl. Acad. Sci. U.S.A. 97
(2000) 11960.[42] P.C. Chin, N. Majdzadeh, S.R. D'Mello, Brain Res. Mol. Brain Res. 137 (2005) 193.[43] J. Lee, E. Jung, S. Hug, C.G. Hyun, Y.S. Kim, D. Park, Br. J. Dermatol. 157 (2007) 242.
1761B. Bellei et al. / Cellular Signalling 20 (2008) 1750–1761
Related Documents