GONADOBLASTOMA IN A PATIENT WITH 46XY GONADAL DYSGENESIS M. Coculescu *1 , N. Poiana 2 , Corina Rãducanu-Lichiardopol 3 , Mioara Ionescu 4 1 Department of Endocrinology, and 2 Department of Obstetrics and Gynecology, University of Medicine and Pharmacy “Carol Davila” Bucharest 3 Department of Endocrinology, University of Medicine and Pharmacy, Craiova, 4 Department of Pathology“Gh. Polizu” University Hospital, Bucharest, Romania We present a 18 year old phenotypic female patient who presented for primary amenorrhea. Pelvic ultrasound revealed a hypoplastic uterus and CT scan showed a hypoplastic right gonad and a left gonadal tumor with extrapelvic location. Karyotype was 46XY. Hormonal assessment indicated hypergonadotropic hypogonadism: FSH was 39.69 mUI/ml, estradiol was 28.07 pg/ml, testosterone was 0.17 ng/ml. DHEA level was high – 21 ng/ml. Gonadectomy was performed at 15 years and histologic examination diagnosed left gonadoblastoma and right teratoma in a dysgenetic gonad. The patient had a good postoperatory evolution. Menses were induced with estrogenic and then estroprogestogenic treatment. Plastic breast surgery was performed at 18 years. Establishing the genotypic sex in patients with primary amenorrhea represents a crucial step knowing that intersex disorders bearing Y chromosomal material have a high risk for gonadoblastoma and germ cell tumors. Key words: gonadoblastoma, gonadal dysgenesis. INTRODUCTION Gonadoblastoma is a rare tumor that occurs almost exclusively in patients with an underlying gonadal dissorder. It accounts for two thirds of gonadal tumors in women with abnormal gonadal development (1). Gonadoblastoma is a benign tumor occuring in intraabdominally located gonads with pure or mixed gonadal dysgenesys; A Y chromosome is detected in over 90% of cases. A differential diagnosis of patients with intersex disorders bearing Y chromosome material is necessary for exclusion of: male pseudohermaphroditism, complete androgen insensitivity, mixed gonadal dysgenesis 45XO/46XY and some patients with 227 Case Report *Correspondence to: Mihai Coculescu, Endocrinology Department, “Carol Davila” University of Medicine and Pharmacy, 34-36 Bd. Aviatorilor, 011863, Bucharest, Romania, Phone/Fax: + 4021 3198718, e-mail: [email protected] Acta Endocrinologica (Buc), vol. II, no. 2, 227-238, 2006 Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

GONADOBLASTOMA IN A PATIENT WITH 46XY GONADAL DYSGENESIS
M. Coculescu*1, N. Poiana2, Corina Rãducanu-Lichiardopol3, Mioara Ionescu4
1Department of Endocrinology, and 2Department of Obstetrics and Gynecology,University of Medicine and Pharmacy “Carol Davila” Bucharest
3Department of Endocrinology, University of Medicine and Pharmacy, Craiova, 4Department of Pathology“Gh. Polizu” University Hospital, Bucharest, Romania
We present a 18 year old phenotypic female patient who presented for primaryamenorrhea. Pelvic ultrasound revealed a hypoplastic uterus and CT scan showed a hypoplasticright gonad and a left gonadal tumor with extrapelvic location. Karyotype was 46XY.Hormonal assessment indicated hypergonadotropic hypogonadism: FSH was 39.69 mUI/ml,estradiol was 28.07 pg/ml, testosterone was 0.17 ng/ml. DHEA level was high – 21 ng/ml.Gonadectomy was performed at 15 years and histologic examination diagnosed leftgonadoblastoma and right teratoma in a dysgenetic gonad.
The patient had a good postoperatory evolution. Menses were induced with estrogenicand then estroprogestogenic treatment. Plastic breast surgery was performed at 18 years.
Establishing the genotypic sex in patients with primary amenorrhea represents acrucial step knowing that intersex disorders bearing Y chromosomal material have a highrisk for gonadoblastoma and germ cell tumors.
Key words: gonadoblastoma, gonadal dysgenesis.
INTRODUCTION
Gonadoblastoma is a rare tumor that occurs almost exclusively in patientswith an underlying gonadal dissorder. It accounts for two thirds of gonadal tumorsin women with abnormal gonadal development (1). Gonadoblastoma is a benigntumor occuring in intraabdominally located gonads with pure or mixed gonadaldysgenesys; A Y chromosome is detected in over 90% of cases. A differentialdiagnosis of patients with intersex disorders bearing Y chromosome material isnecessary for exclusion of: male pseudohermaphroditism, complete androgeninsensitivity, mixed gonadal dysgenesis 45XO/46XY and some patients with
227
Case Report
*Correspondence to: Mihai Coculescu, Endocrinology Department, “Carol Davila” University of
Medicine and Pharmacy, 34-36 Bd. Aviatorilor, 011863, Bucharest, Romania, Phone/Fax: + 4021
3198718, e-mail: [email protected]
Acta Endocrinologica (Buc), vol. II, no. 2, 227-238, 2006
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 1

Turner syndrome and molecular evidence of an Y chromosome. This tumor consistsof three types of cells: large germ cells (similar to dysgerminoma and seminoma),small cells (resembling immature Sertoli or granulosa cells) and in two thirds ofcases Leydig type cells without Reinke crystals and often contains calcifications (2).
Gonadoblastoma have potential risk of malignant transformation as germinalcomponent overgrows the stroma, 30% of them develop into dysgerminoma /seminoma, 10% give rise to other malignant germ cell neoplasms and 10% ofgerminomas / seminomas have demonstrated metastases at the time the diagnostic isestablished (3).
Most of gonadoblastoma arise in phenotypically females (80%), in the firsttwo decades of life and are billateral in 38,6% of cases (4).
The incidence of gonadoblastoma in patients with mixed gonadal dysgenesis45,XO/46XY, androgen insensitivity and male pseudohermaphroditism is 30-66%and 7-10% in Turner patients with evidence of Y chromosome material (5) or evenhigher – 33% as reported by others (6).
CASE REPORT
Patient DC, aged 15 years presented for primary amenorrhea. Phenotype wasfemale, sex identity – female, height 181 cm, clinical examination and usuallaboratory parameters were normal, excluding associated malformations or morbidity.
External genitalia had a normal female appearance. Transvaginal ultrasoundrevealed a small uterus (4 cm long) without endometrial echoes and could notidentify the gonads. Vaginoscopy showed a normal 80/23 mm vagina. CT scan wasable to identify the right gonad of 18/10 mm in the proximity of iliac bifurcation andthe left gonad enlarged, with extrapelvic location. Karyotype and G banding ofblood lymphocytes showed a normal 46XY chromosomial constitution. Hormonalassessment established the diagnostic of hypergonadotropic hypogonadism byincreased FSH (39.69 mUI/ml) and low testosterone (0.17ng/ml) level. Estradiollevel was in the normal male range – 28.07pg/ml and DHEA was increased(21ng/ml).
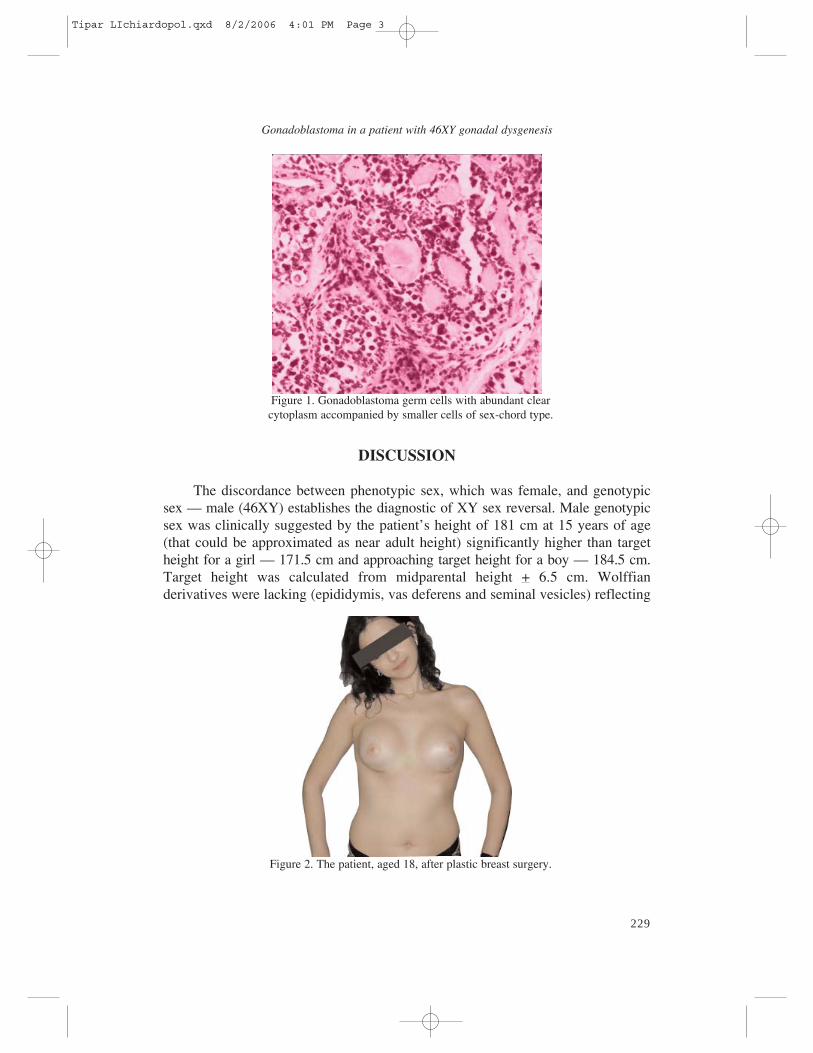
The patient underwent billateral gonadectomy and histologic examination ofthe gonads established the diagnostic of left gonadoblastoma (Fig. 1) and rightteratoma in a dysgenetic gonad.
Postoperatory evolution was uneventful. After transdermal estrogens a 3 daysspotting ensued. Unopposed estrogen therapy was given for three months and thenthe patient received transdermal combined therapy and experienced cyclic vaginalbleedings. Secondary female characteristics maintained. At 16 years, radiologicexamination revealed still active growth plates. At 18 years, breast plastic surgerywas performed (Fig. 2).
M. Coculescu et al
228
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 2
DISCUSSION
The discordance between phenotypic sex, which was female, and genotypicsex – male (46XY) establishes the diagnostic of XY sex reversal. Male genotypicsex was clinically suggested by the patient’s height of 181 cm at 15 years of age(that could be approximated as near adult height) significantly higher than targetheight for a girl – 171.5 cm and approaching target height for a boy – 184.5 cm.Target height was calculated from midparental height + 6.5 cm. Wolffianderivatives were lacking (epididymis, vas deferens and seminal vesicles) reflecting
Gonadoblastoma in a patient with 46XY gonadal dysgenesis
229
Figure 1. Gonadoblastoma germ cells with abundant clearcytoplasm accompanied by smaller cells of sex-chord type.
Figure 2. The patient, aged 18, after plastic breast surgery.
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 3

the absence of testosterone action during organogenesis of the male genital tract andthe presence of female external genitalia proved the lack of dihydrotestosteroneaction on the urogenital sinus. Mullerian derivatives (uterus, vagina, fallopiantubes) were present, though hypoplastic, which means the antimullerian hormone(AMH) secreted by Sertoli cells was deficient or AMH receptivity was altered andallowed exclusion of defects in testosterone biosynthesis and action causing XY sex
M. Coculescu et al
230
Figure 3. Genetic control of testis development.
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 4

reversal, disorders in which mullerian derivatives are absent (LH/hCG resistance,StAR deficiency, CYP17 deficiency, Smith Lemli Opitz syndrome, 17βhydroxysteroiddehydrogenase type 3 deficiency, complete androgen resistance) andATRX mutations.
Testicular dysgenesis cumulates both deficiencies – AMH and testosterone –and could be suggested by increased FSH, low testosterone, but was not confirmedhistologically in the right gonad. Often, because part of gonad is replaced by tumor,a diagnosis of the underlined gonadal abnormality may be impossible (1). In ourcase, the gonad in which the gonadoblastoma was found is of unknown nature. Theright gonad with teratoma seems to be a dysgenetic gonad. It was reported that insome extremely rare cases, an apparently normal ovary was found in patients withgonadoblastoma.
Whether endocrine disrupters can generate the gonadal dysgenesis syndromeis still a matter of debate (7); moreover, it would be possible that factors encodedby genes of certain Y chromosome haplogroups may be particularly susceptible toenvironmental influences that cause testicular dysgenesis syndrome (8).
In cases of XX or XY gonadal dysgenesis of undetermined origin gonadal sexchromosomal mosaicism may be the cause (9).
It is hypothesized also that numerical and structural aberrations of sexchromosomes are not a prerequisite for the appearance of testicular dysgenesiswhich is more frequently associated with the 46XY karyotype. The incidence ofneoplastic lesions is related more to the severity of testicular organogenesis
231
Gonadoblastoma in a patient with 46XY gonadal dysgenesis
Gene Locus Phenotype
SRY Yp11 XY sex reversal
WT1 11p13 – deletions – WAGR syndrome– Denys Drash syndrome– Frasier syndrome
SF1 9q33 – XY sex reversal associated or not with adrenal insufficiencySOX9 17q24 – campomelic dysplasia
DAX1 Xp21.3 Duplications: XY sex reversal (mutations cause adrenalhypoplasia congenita and hypogonadotropic hypogonadism)
DHH 12q12 – XY sex reversal– Partial testicular dysgenesis and minifascicular neuropathy
DMRT1 9p21-24 XY sex reversal, mental retardation craniofacial abnormalities
10q- 10q25-qter Urogenital anomalies
ATRX Xq13.3 Thalassemia, mental retardation, genital anomalies (absentmullerian derivatives; in all the other mutations are present)
TSPYL 6q22 Testicular dysgenesis and sudden infant death
Table 1. Mutations in genes involved in testicular development causing XY sex reversal in humans
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 5

disturbances than it is to aberrations in sex chromosomes, a less disturbed testicularstructuralization predisposing to germ cells neoplasms (10).
However, testicular dysgenesis is certainly the result of a perturbed testisdevelopment which occurs during a relatively narrow time window and it isgoverned by many genes and transcriptional regulators that act in a spatiotemporalcoordinated pattern. The developmental factors known to date may affect DNAbending, modulate chromatin remodelling, form complexes that activatetranscription or repress it, specify progenitor cell types and dictate cell fate.Crosstalk among intracellular signalling pathways mediate transcriptionalresponses (11). Reverse genetic approaches on human sex reversal syndromes andmouse gene knockout studies identified genes encoding transcription factors (SRY,SOX9, DMRT1, GATA4, WT1, LHX9), orphan nuclear receptors (SF1 and DAX1)and cell signalling molecules (AMH, WNT4, FGF9, DHH) that intervene in the sexdetermination cascade (Fig. 3).
Sex determination can be defined as the proliferation, migration anddifferentiation of supporting cells to become Sertoli cells, committing the fate of thebipotential gonad to the testis pathway (12). The Sertoli cells control differentiationof surrounding cells into Leydig or myoid cells, promote vasculogenesis and mitoticarrest in germ cells. This switch is accomplished by SRY – a key gene located onthe short arm of the Y chromosome which activates testis forming genes orrepresses ovary forming genes, or both.
SRY point mutations or deletions are found in only about 15% of XY sexreversed patients; most mutations are “de novo”, but there are cases in which thefertile father of an XY sex reversed individual carries, intriguingly, the samemutation that is probably compensated by other genes and does not occur (12). Intable 1 are shown the genes responsible for 46XY sex reversal in humans.
SRY expression occurs only in the Sertoli cell lineage and is increased in miceby GATA4 and Fog2 (friend of GATA), Ir (insulin receptor), Irr (insulin relatedreceptor), Igf1r (insulin growth factor 1 receptor) (13). Fgf9 (fibroblast growthfactor 9) acting on FgfR2 also stimulates proliferation of preSertoli cells, plays therole of a male specific chemoattractant for immigrant mesonephric cells that willcontribute to the structuralization of testes cords as endothelial and peritubularmyoid cells and, like SRY, induces SOX9 expression. These genes, when mutated,cause XY sex reversal in mice but mutations were not described in humans (11).SRY transcription is regulated by SF1, WT1 and an ubiquitous transcription factor,Sp1, which cooperates with SF1 (12).
WT1 mutations cause Denys Drash syndrome characterised by Wilms tumor,severe renal disease (mesangial sclerosis) and dysgenetic gonads leading toambigous genitalia in males and Frasier syndrome characterized by lack of +KTSisoforms generating XY sex reversal (complete testicular dysgenesis),predisposition to the development of a gonadoblastoma, late onset glomerulopathy(focal glomerular sclerosis) (14, 15). WT1 mutations are found in patients withisolated genital anomalies only in rare cases and recently a WT1 mutation (P181S)
M. Coculescu et al
232
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 6

was described in an XY patient with micropenis, severe hypospadias,cryptorchidism, normal androgen production, androgen resistance, absence of renaldisease and coarctation of the aorta (16).
WT1 controles kidney and gonad development by specifying coelomicepithelial cells and ensuring their survival. WT1 acts upstream two orphan nuclearreceptors – SF1 and DAX1; WT1 and SF1 synergize to enhance transcription ofAMH, this interaction being repressed by DAX1 (11).
WT1 encodes a transcription factor with four zinc finger motifs as aDNA/RNA binding or protein – protein interaction domain. WT1 binds and actssynergistically with SRY to activate transcription from a promoter containing a SRYbinding site. Mutant WT1 is not recruited efficiently to SRY binding sites andresults in an impaired testes development (17).
WT1 gene can give rise to 24 different isoforms by alternative splicing,alternative translational start sites and RNA editing. There are +KTS and –KTSisoformes distinguished by the presence or absence of three aminoacids – Lys, Thr,Ser (KTS) – between the third and the fourth zinc fingers. The +KTS isoforms actas a transcription factor and the –KTS isoforms are involved in mRNA productionand processing and have independent roles in sex determination, WT1 (-KTS)isoforms contributing to the full SRY expression and WT1 (+KTS) being imppliedin SRY processing and stability (12).
SF1 mutations are rare. There are two known cases of XY sex reversal andadrenal insufficiency (18) and recently two cases of testicular dysgenesis withoutadrenal insufficiency were described (19, 20) suggesting that the human testis is moresensitive to loss of SF1 function than the adrenal, or the adrenal may have a greatercapacity to undergo compensatory growth and function. SF1 gene dosage is of majorimportance, generating different phenotypes. SF1 codes for an orphan nuclearreceptor which acts as a key regulator of gonadal axis and adrenal steroidogenesis; inSertoli cells regulates AMH and in Leydig cells regulates the expression ofsteroidogenic enzymes; it is also expressed in the ventromedial hypothalamus and inpituitary gonadotropes. SF1 is necessary for survival of early progenitors of theadrenal and gonad by stimulating cell proliferation and its full expression isconditioned by Lhx9, m33 and Pod1 in mice (11). WT1 and SF1 synergize to increasethe expression of genes driven by SF1 and the interaction of SOX9 and SF1 appearsto have a role in the greater expression of SF1 in the fetal testis (14).
SOX9 mutations in humans cause campomelic dysplasia – a dominant lethaldisorder characterized by bowing of the long bones, narrow ilia, cleft palate,absence of olfactory bulbs and tracts, heart and renal malformations, hypoplasticlungs, narrow thorax, delayed bone age and in 75% of 46XY affected individuals –testicular dysgenesis and sex reversal (12).
Duplication of SOX9 was described in a SRY negative female to male sexreversed patient suggesting that overexpression of SOX9 can compensate for thelack of SRY (21). SOX9 upregulates expression of AMH by cooperative interactionwith SF1. Similarly, WT1 and GATA4 interact with SF1 to upregulate AMH. The
233
Gonadoblastoma in a patient with 46XY gonadal dysgenesis
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 7

ability of the SOX9 HMG box to bend DNA may bring SF1 and GATA4 in closerproximity to each other and along with WT1 and HSP70 form a tightly associatedprotein complex that activates AMH transcription (12) SOX9 binds as a dimer ingenes involved in chondrocyte differentiation (COL11A2, COL9A2) but binds as amonomer to the regulatory region of SF1; mutations that disrupt dimerization affectchondrogenesis but not sex determination explaining why campomelic dysplasianeeds not to be associated to XY sex reversal (22).
DAX1 duplications cause testicular dysgenesis with 46XY sex reversal. DAX1can antagonize the transcriptional activation function of SF1 by recruiting thenuclear corepressor, thus inhibiting SF1 mediated expression of SRY and/or by adirect competition for sites involved in male sex determination (12). SF1 and DAX1function as transcriptional antagonists for many target genes in vitro but they actindependently or cooperatively in vivo in male gonadal development.
Though it was thought that mutations of DAX1 permit a normal testicularorganogenesis and are characterized only by congenital adrenal hypoplasia andhypogonadotropic hypogonadism, some affected men showed unresponsiveness togonadotropin treatment (14). In Sf1 deficient mice Cyp17 and Cyp11a1 are reducedin a dose dependent manner but in Sf1/Dax1 double mutants are reduced furtherindicating that loss of Dax1 does not compensate for reduced Sf1 activity. MoreoverDhh and Amh expression was reduced transiently in single and double mutants butSOX9 was expressed suggesting that various Sertoli cell genes are differentiallysensitive to Sf1 and Dax1 (11) Dax1 null mice have gonadal defects in testis cordmorphogenesis and peritubular myoid cell proliferation.
DHH mutations were reported in three cases with complete testiculardysgenesis (23) and one patient with partial testicular dysgenesis andpolyneuropathy (24) proving that localization of mutations influences phenotypicexpression. DHH is secreted by Sertoli cells and induces Leydig cellsdifferentiation in a paracrine manner, acting on the receptor Ptc1 (patched 1). Pdgfrα (platelet derived growth factor receptor α) is believed to act upstream of Dhh andit is involved also in mesonephric cells migration and full Cyp11a1 expression. TheX linked gene Arx (aristaless) also influences Leydig cell development based onexpression of 3βHSD (11).
Haploinsufficiency or deletions of DMRT1 result in sex reversal. This gene hasa male specific expression in the early stages of gonadal differentiation (genitalridge, developing Sertoli cells). Several human DMRT genes exist and map to threewell defined regions on chromosome 1, 9 and 19, one gene on chromosome 19having an additional homologue on chromosome X. These regions harbor multiplesyntenic genes sharing highly specific paralogy relations. The 9p21-24.3 bandsrepresents the ancestral copy (paralogs are genes present in a single genome as aresult of gene duplication ) (25). In mice Dmrt1 is expressed in spermatogoniarelated to a putative role in mitotic or meiotic cell cycle; mutations cause abnormalSertoli cell morphology, overproliferation and subsequent apoptosis.
M. Coculescu et al
234
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 8

10q terminal deletions are associated with urogenital anomalies (renalhypoplasia, cryptorchidism, micropenis, hypospadias) and, rarely, XY sex reversalbut a candidate gene was not yet identified (14).
TSPYL mutations (testis specific Y like gene) cause an autosomal recessivesyndrome described in Amish population that associates testicular dysgenesis andsudden infant death (26).
The insulin signaling pathway is implied in male differentiation and triplemutants mice for Ir, Irr, Igf1r (insulin receptor, insulin related receptor, insulingrowth factor 1 receptor) exhibit male to female sex reversal (13). In humans INSL3(insulin like factor 3) and its receptor LGR8 (leucine rich repeat containing Gprotein coupled receptor 8) have a role in testicular descent and germ cell function.A pituitary – testicular axis involving LH and INSL3 was postulated (27) and it wasfound that LGR8 is expressed in male germ cells and binding of INSL3 preventsapoptosis which suggested a paracrine role in germ cell survival (28) but, till now,mutations of INSL3 and LGR8 have been described only in a minority of cases withcryptorchidism.
GATA transcription factors are involved in sex determination anddifferentiation; GATA4 plays an important role in testis development by regulatinggenes that mediate Mullerian duct regression and the onset of testosteroneproduction; GATA1 is expressed only in Sertoli cells at specific seminiferous tubulestages; GATA1 and GATA4 stimulate inhibin gene promoter constructs; GATA4 andGATA6 activate genes mediating gonadal steroidogenesis (29). Mutations of GATAgenes were not yet described in humans with altered testicular organogenesis.
Despite the fact that recent research revealed many aspects of testiculardifferentiation in most cases with 46XY sex reversal etiology remains unknown.Epigenetic factors (DNA methylation), protein misfolding and misrouting may alsobe involved.
Little is also known about the propensity of dysgenetic testes to developneoplastic lesions. Germ cells in gonadal dysgenesis exhibit a developmental delay andare prone to malignant transformation if they are able to survive in their inappropiateenvironment (30). In young subjects the number of germ cells is high, but withadvancing age decreases progressively. This loss is patchy – some gonadal areasmaintain an adequate number of germ cells while in others all cells disappear (31).
TSPY is a testis – specific multicopy gene family located in the GBY(gonadoblastoma on Y) critical region of the Y chromosome that is thought topromote gonadoblastoma formation; its X chromosome homologue (TSPX) is widelyexpressed, subject to X inactivation and is involved in cell cycle regulation (32).TSPY expression is upregulated in germ cells residing in an unfavorableenvironment (altered Sertoli cell function as a result of testicular dysgenesisgenerates such an environment) in an attempt to survive and proliferate. Thecombination of maturation delay, prolonged expression of OCT 3/4 and abundantTSPY expression can provide the surviving germ cell with an important proliferativeadvantage leading to clonal expansion (31). OCT 3/4 (POU5F1) is a transcription
Gonadoblastoma in a patient with 46XY gonadal dysgenesis
235
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 9

factor essential for maintainance of totipotency in embryonic stem cells andapoptosis prevention in migratory cells (33). In the testis its expression graduallydecreases until 20 weeks of gestation and thereafter persists in a few cells until 3-9 months postpartum when gonocytes finally differentiate into spermatogonia.OCT 3/4 positive cells are located almost exclussively in the central area of theseminiferous tubule. It is suggested that by reaching the basal lamina, early germcells lose their pluripotency and start to differentiate; if differentiation does notoccur and the cell is not removed by an apoptotic or other mechanism, clonalexpansion may lead to carcinoma in situ (31). In dysgenetic testes OCT 3/4 isexpressed until 14 months of age, thus increasing the risk for malignanttransformation.
Gonadoblastoma and carcinoma in situ (CIS) express abundantly OCT 3/4regardless of age (34). By modulating the level of OCT 3/4 expression in vitro inmice embryonic stem cell-derived tumors, the malignant phenotype of the cells waschanged, suggesting that OCT 3/4 is of pathogenetic relevance in the developmentof these tumors (35).
Considering these data, it is advisable to perform gonadectomy in XY sexreversed patients in order to prevent neoplastic transformation. In our casegonadoblastoma was already developped at presentation and made the diagnosticmore difficult because tumoral hormonal secretion changed both the clinical picture(breast development) and hormonal assessment. Suprinsingly, the estrogen levelwas in the normal male range but a weak androgen – DHEA was increased (whichwould suggest a 3βHSD deficiency if the mullerian derivatives were absent).
Establishing the etiologic diagnostic would be helpful in order to detect aputative associated morbidity suggested by a certain mutation (for example if a WT1mutation would be the cause, the patient should be monitored for a late onsetglomerulopathy) but is not essential for the patient’s management.
References
1.Sternberg SS (Ed), Diagnostic surgical pathology, Raven Press, New York, Vol 2, 1989
2.Rutgers JL, Scully RE. Pathology of the testis in intersex syndromes. Semin Diagn Pathol 1987;
4(4): 275-91.
3. Pena-Alonso R, Nieto K, Alvarez R, Palma J, Najera N, Erana L, Dorantes LM, Kofman-Alfaro S,
Queipo G. Distribution of Y chromosome – bearing cells in dysgenetic testis in 45,X/46XY infants.
Mod Pathol 2005; 18(3): 439-45.
4. Troche V, Hernandez E. Neoplasia arising in dysgenetic gonads. Obstet Gynecol Surv 1986; 41(2): 74-9.
5. Gravholt CH, Fedder J, Naeraa RW, Muller J. Occurence of gonadoblastoma in females with Turner
syndrome and Y chromosome material: a population study. J Clin Endocrinol Metab 2000; 85(9):
3199-202.
M. Coculescu et al
236
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 10

6. Canto P, Kofman Alfaro S, Jimenez AL, Soderlund D, Barron C, Reyes E, Mendez JP, Zenteno JC.
Gonadoblastoma in Turner syndrome patients with nonmosaic 45,X karyotype and Y chromosome
sequence. Cancer Genet Cytogenet 2004; 150(1): 70-72.
7. Fisher JS. Enviromental antiandrogens and male reproductive health: focus on phtalates and
testicular dysgenesis syndrome. Reproduction 2004; 127(3): 305-15
8. Mc Elreavy K, Quintana-Murci L. Y chromosome haplogroups: a correlation with testicular
dysgenesis syndrome. APMIS 2003; 111(1): 106-13.
9. Ropke A, Pelz AF, Volleth M, Schlosser HW, Morlot S, Wieacker PF. Sex chromosomal mosaicism
in the gonads of patients with gonadal dysgenesis, but normal female or male karyotype in
lymphocytes. Am J Obstet Gynecol 2004; 190(4): 1059-62.
10. Slowikowska-Hilczer J, Romer TE, Kula K. Neoplastic potential of germ cells in relation to
disturbances of gonadal organogenesis and changes in karyotype. J Androl 2003; 24(2): 270-8.
11. Park S, Jameson L. Transcriptional regulation of gonadal development and differentiation. J Clin
Endocrinol Metab 2005; 146(3): 1035-42.
12. Harley V, Clarkson M, Argentaro A. The molecular action and regulation of the testis determining
factors SRY and SOX9. Endocr Rev 2003; 24(4): 466-87.
13. Nef S, Verma-Kurvari S, Merenmies J. Testis determination requires insulin receptor family
function in mice. Nature 2003; 426: 291-5.
14. Grumbach M, Hughes I, Conte F. Disorders of sex differentiation. In Larsen R, Kronenberg H,
Melmed S, Polonski K (eds). Williams Textbook of Endocrinology tenth edition, WB Saunders,
Philadelphia, 2003: 842-1002.
15. Ruf RG, Schultheiss M, Lichtenberger A, Karle SM, Zalewski I, Mucha B. Prevalence of WT1
mutations in a large cohort of patients with steroid resistant and steroid sensitive nephrotic syndrome.
Kidney Int 2004; 66(2): 564-70.
16. Kohler B, Pienkowski C, Audran F, Delsol M, Tauber M, Paris F, Sultan C, Lumbroso S. An N-
terminal WT1 mutation (P1815) in an XY patient with ambigous genitalia, normal testosterone
production, absence of kidney disease and associated heart defect: enlarging the phenotypic spectrum
of WT1 defects. Eur J Endocrinol 2004; 150(6): 825-30.
17. Matsuqawa – Watanabe Y, Inoue J, Semba K. Transcriptional activity of SRY is modulated by
WT1. Oncogene 2003; 22(39): 5956-60.
18. Achermann J, Ozisik G, Ito M, Orun U, Harmanci K, Gurakan B, Jameson L. Gonadal
determination and adrenal development are regulated by the orphan nuclear receptor SF1 in a dose
dependent manner. J Clin Endocrinol Metab 2002; 87: 1829-33.
19. Hasegawa TR, Fukami M, Sato N, Katsumata N, Sasaki G, Fukutani K, Morohashi K, Ogata T.
Testicular dysgenesis without adrenal insufficiency in a 46XY patient with heterozygous inactive
mutation of SF1. J Clin Endocrinol Metab 2004; 89(12): 5930-5.
20. Mallet D, Bretones P, Michael-Calemard L, Dijoud F, David M, Morel Y. Gonadal dysgenesis
without adrenal insufficiency in a 46XY patient heterozygous for the nonsense C16X mutation: a case
of SF1 haploinsufficiency. J Clin Endocrinol Metab 2004; 89(10): 4829-32.
21. Huang B, Wang S, Ning Y, Lamb AN, Bartley J. Autosomal XX sex reversal caused by
duplication of SOX 9. Am J Med Genet 1999; 87: 349-353.
22. Bernard P, Tang P, Lin S, Dewing P, Harley VR, Vilain E. Dimerization of SOX9 is required for
chondrogenesis, but not for sex determination. Hum Mol Genet 2003; 12(14): 1755-65.
Gonadoblastoma in a patient with 46XY gonadal dysgenesis
237
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 11

23. Canto P, Soderlund D, Reyes E, Mendez JP. Mutations in the desert hedgehog (DHH) gene in
patients with 46XY complete pure gonadal dysgenesis. J Clin Endocrinol Metab 2004; 89(9): 4480-3.
24. Umehara F, Tate G, Itih K, Yamaguchi N, Douchi T, Mituya T, Osame M. A novel mutation of
DHH in a patient with 46XY partial gonadal dysgenesis accompanied by minifascicular neuropathy.
Am J Hum Genet 2000; 67: 1302-5.
25. Ottolenghi C, Fellous M, Barbieri M, Mc Elreavy K. Novel paralogy relations among human
chromosomes support a link between the phylogeny of doublesex – related genes and the evolution of
sex determination. Genomics 2002;79(3): 333-43.
26. Puffenberger EG, Hu-Lince D, Parod JM, Craig DW, Dobrin SE, Conway AR. Mapping of sudden
infant death with dysgenesis of the testes syndrome (SIDDT) by a SNP genome scan and identification
of TSPYL loss of function. Proc Natl Acad Sci USA 2004; 101(32): 11689-94.
27. Foresta C, Bettella A, Vinanzi C, Dabrilli P, Meriggiola MC, Garolla A, Ferlin A. Insulin – like
factor 3: a novel circulating hormone of testis origin in humans. J Clin Endocrinol Metab 2004; 89:
5952-8.
28. Kawamura K, Kumagai J, Sudo S, Chun SY, Pisarska M, Morita H, Toppari J, Fu P, Wade JD,
Bathgate RA, Hsueh AJ. Paracrine regulation of mammalian oocyte maturation and male germ cell
survival. Proc Natl Acad Sci USA 2004; 101: 7323-8.
29. LaVoie H. The role of GATA in mammalian reproduction. Exp Biol Med 2003; 228: 1282-90.
30. Rajpert-De Meyts E, Jorgensen N, Brondum-Nielsen K, Muller J, Skakkebaek NE. Developmental
arrest of germ cells in the pathogenesis of germ cell neoplasia. APMIS 1998; 106: 198-206
31. Cools M, van Aerde K, Kersemaekers AM, Boter M, Drop S, Wolffenbuttel K, Steyerberg E,
Oosterhuis W, Looijienga L. Morphological and immunohistochemical differences between gonadal
maturation delay and early germ cell neoplasia in patients with undervirilisation syndromes. J Clin
Endocrinol Metab 2005; 90: 5295-5303.
32. Delbridge ML, Longepied G, Depetris D, Mattei MG, Disteche CM, Marshall Graves JA, Mitchell
MJ. TSPY, the candidate gonadoblastoma gene on the human Y chromosome has a widely expressed
homologue on the X- implications for Y chromosome evolution. Chromosome Res 2004; 12(4): 345-56.
33. Kehler J, Tolkunova E, Koschorz B. OCT 3/4 is required for primordial germ cell survival EMBO
Rep 2004; 5: 1078-83.
34. Rajpert-DeMeyts E, Hanstein R, Jorgensen N, Graem N, Vogt PH, Skakkebaek NE.
Developmental expression of POU5F1 (OCT-3/4) in normal and dysgenetic human gonads. Hum
Reprod 2004; 19(6): 1338-44.
35. Gidekel S, Pizov G, Bergman Y, Pikarsky E. OCT 3/4 is a dose dependent oncogenic fate
determinant. Cancer Cell 2003; 4: 361-70.
M. Coculescu et al
238
Tipar LIchiardopol.qxd 8/2/2006 4:01 PM Page 12
Related Documents











