Gliomas in Neurofibromatosis Type 1: A Clinicopathologic Study of 100 Patients Fausto J. Rodriguez, MD, Arie Perry, MD, David H. Gutmann, MD, PhD, Brian Patrick O'Neill, MD, Jeffrey Leonard, MD, Sandra Bryant, MS, and Caterina Giannini, MD, PhD Departments of Laboratory Medicine and Pathology (FJR, CG), Neurology (BPON), and Biostatistics (SB), Mayo Clinic College of Medicine, Rochester, Minnesota; and Division of Neuropathology (AP), Departments of Neurology (DHG) and Neurosurgery (JL), Washington University School of Medicine, St. Louis, Missouri Abstract There are few pathologic studies of gliomas in patients with neurofibromatosis type 1. We analyzed clinical and pathologic features of gliomas from 100 neurofibromatosis type 1 patients (57 men; 43 women). The median age at tumor diagnosis was 13 years (range, 4 months to 68 years). Most tumors were typical pilocytic astrocytoma (PA) (49%) or diffusely infiltrating astrocytoma (DA) (27%) that included World Health Organization Grades II (5%), III (15%), and IV (7%); others were designated as low-grade astrocytoma, subtype indeterminate (LGSI; 17%). Two pilomyxoid astrocytomas, 1 desmoplastic infantile ganglioglioma and 1 conventional ganglioglioma, were also identified. The tumors in 24 cases arose in the optic pathways and included PA (n = 14), LGSI (n = 4), DA (n = 4), pilomyxoid astrocytoma (n = 1), and ganglioglioma (n = 1). The prognoses of the PA and LGSI gliomas overall were generally favorable; there were no survival differences between PA and LGSI groups based on site, tumor size, mitotic activity, or MIB-1 labeling index. In the combined PA and LGSI group, age younger than 10 years and gross total resection were associated with an increased overall survival rate (p = 0.047 and 0.002, respectively). Compared with the combined group (PA + LGSI), patients with DA at all sites had decreased overall and recurrence-free survival times (p < 0.001 and p = 0.003, respectively). This study emphasizes the wide histologic spectrum of gliomas that occur in patients with neurofibromatosis type 1. Classic PA and LGSI are the most common, and most have favorable prognoses. By contrast, DAs are more aggressive, similar to those that arise sporadically. Keywords Astrocytoma; Brain tumor; Central nervous system; Glioma; Neurofibromatosis; Pilocytic astrocytoma INTRODUCTION Neurofibromatosis type 1 (NF1) is an inherited tumor predisposition syndrome, and those affected with NF1 are prone to the development of both peripheral and central nervous system (CNS) tumors. The most common primary CNS tumors are gliomas that usually involve the optic pathways especially in children. In this regard, bilateral optic gliomas are Copyright © 2008 by the American Association of Neuropathologists, Inc. Send correspondence and reprint requests to: Fausto J. Rodriguez, MD, Mayo Clinic College of Medicine, 200 First Street SW, Mayo Clinic, Rochester, MN 55905; [email protected]. NIH Public Access Author Manuscript J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11. Published in final edited form as: J Neuropathol Exp Neurol. 2008 March ; 67(3): 240–249. doi:10.1097/NEN.0b013e318165eb75. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Gliomas in Neurofibromatosis Type 1: A Clinicopathologic Studyof 100 Patients
Fausto J. Rodriguez, MD, Arie Perry, MD, David H. Gutmann, MD, PhD, Brian PatrickO'Neill, MD, Jeffrey Leonard, MD, Sandra Bryant, MS, and Caterina Giannini, MD, PhDDepartments of Laboratory Medicine and Pathology (FJR, CG), Neurology (BPON), andBiostatistics (SB), Mayo Clinic College of Medicine, Rochester, Minnesota; and Division ofNeuropathology (AP), Departments of Neurology (DHG) and Neurosurgery (JL), WashingtonUniversity School of Medicine, St. Louis, Missouri
AbstractThere are few pathologic studies of gliomas in patients with neurofibromatosis type 1. Weanalyzed clinical and pathologic features of gliomas from 100 neurofibromatosis type 1 patients(57 men; 43 women). The median age at tumor diagnosis was 13 years (range, 4 months to 68years). Most tumors were typical pilocytic astrocytoma (PA) (49%) or diffusely infiltratingastrocytoma (DA) (27%) that included World Health Organization Grades II (5%), III (15%), andIV (7%); others were designated as low-grade astrocytoma, subtype indeterminate (LGSI; 17%).Two pilomyxoid astrocytomas, 1 desmoplastic infantile ganglioglioma and 1 conventionalganglioglioma, were also identified. The tumors in 24 cases arose in the optic pathways andincluded PA (n = 14), LGSI (n = 4), DA (n = 4), pilomyxoid astrocytoma (n = 1), andganglioglioma (n = 1). The prognoses of the PA and LGSI gliomas overall were generallyfavorable; there were no survival differences between PA and LGSI groups based on site, tumorsize, mitotic activity, or MIB-1 labeling index. In the combined PA and LGSI group, age youngerthan 10 years and gross total resection were associated with an increased overall survival rate (p =0.047 and 0.002, respectively). Compared with the combined group (PA + LGSI), patients withDA at all sites had decreased overall and recurrence-free survival times (p < 0.001 and p = 0.003,respectively). This study emphasizes the wide histologic spectrum of gliomas that occur inpatients with neurofibromatosis type 1. Classic PA and LGSI are the most common, and mosthave favorable prognoses. By contrast, DAs are more aggressive, similar to those that arisesporadically.
KeywordsAstrocytoma; Brain tumor; Central nervous system; Glioma; Neurofibromatosis; Pilocyticastrocytoma
INTRODUCTIONNeurofibromatosis type 1 (NF1) is an inherited tumor predisposition syndrome, and thoseaffected with NF1 are prone to the development of both peripheral and central nervoussystem (CNS) tumors. The most common primary CNS tumors are gliomas that usuallyinvolve the optic pathways especially in children. In this regard, bilateral optic gliomas are
Copyright © 2008 by the American Association of Neuropathologists, Inc.
Send correspondence and reprint requests to: Fausto J. Rodriguez, MD, Mayo Clinic College of Medicine, 200 First Street SW, MayoClinic, Rochester, MN 55905; [email protected].
NIH Public AccessAuthor ManuscriptJ Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
Published in final edited form as:J Neuropathol Exp Neurol. 2008 March ; 67(3): 240–249. doi:10.1097/NEN.0b013e318165eb75.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

nearly pathognomonic for NF1 (1). On pathologic examination, most are pilocyticastrocytomas (PAs), World Health Organization (WHO) Grade I (2, 3). However, patientswith NF1 may also develop diffusely infiltrating astrocytomas (DAs) (WHO grades II–IV),particularly with an onset later in life (4). In some instances, NF1-associated astrocytomasshow indistinct or overlapping features that prevent a precise histopathologic classification(5), thus precluding an accurate prediction of their biologic behavior.
Tumors involving the optic pathways in patients with NF1 have characteristic features onneuroimaging, exhibit largely indolent behavior, and may even regress without treatment(6). Therefore, pathologic specimens are generally not available for study. Even clinicallyprogressive optic gliomas are often treated without a tissue diagnosis. Consequently, mostmodern reports of gliomas in patients with NF1 either do not focus on their histopathologicfeatures (6–10) or are limited to small subsets and/or series (11, 12). Conversely, older seriesand reviews (13, 14) were written before the most recent WHO grading and classificationschemes and predate the formulation of diagnostic criteria for NF1.
The purpose of this study was to provide a detailed clinicopathologic description of thespectrum of gliomas in patients with NF1 using pathologic material available from 2 largereferral centers with dedicated NF1 clinics and to determine whether specific pathologic orclinical features predict clinical behavior.
MATERIALS AND METHODSThis study was approved by the institutional review boards at both the Mayo Clinic and atthe Washington University School of Medicine. A search was performed of the clinical andpathologic records of both medical centers for patients with a clinical diagnosis of NF1 anda glioma between 1950 and 2006. The patients for whom there was pathologic materialavailable for review and who satisfied established current clinical criteria for NF1 (1) wereincluded in this study. Clinical data were abstracted from retrospective chart review. Theextent of surgery was determined predominantly from operative reports and postoperativeimaging reports. All available hematoxylin and eosin–stained slides were reviewed by atleast 2 neuropathologists (C.G., A.P., F.J.R.), and the tumors were classified and gradedbased on the current WHO classification (2). Nuclear atypia was graded in a 3-tiered scale(mild, moderate, and severe) based on chromatin and nuclear irregularities independent ofdegenerative changes. Cellularity was rated as low, moderate, or high. Cellularity was highwhen there was frequent nuclear crowding (nuclei touching each other), moderate whenintercellular spaces equivalent to 1 to 2 nuclear diameters were present in most fields, andlow when there was significant acellular stroma/processes in between cells. Degree ofinvasiveness was classified as absent, partial, or diffuse based on neurofilament proteinimmunostains demonstrating underlying axons, when available, in combination withhematoxylin and eosin identification of entrapped neurons and/or axons when visible. Amitotic count was performed in 10 consecutive high-power fields (HPF) in the areas ofmaximum proliferation. The following histologic features were evaluated as being present orabsent: biphasic pattern, multinucleated cells, oligodendroglial-like cells, Rosenthal fibers,eosinophilic granular bodies, microcysts, chronic inflammation, hemosiderin-ladenmacrophages, calcification, glomeruloid vessels, vascular hyalinization, necrosis,perivascular pseudorosettes, fascicular pattern, and leptomeningeal extension.
Immunohistochemical stains were performed using antibodies directed against S100 protein(Dako, Carpinteria, CA; polyclonal; dilution, 1:1600), glial fibrillary acidic protein (Dako;polyclonal; 1:4000), p53 protein (Dako; clone DO7; 1:2000), neurofilament protein (Dako;clone 2F11; 1:800), synaptophysin (ICN, Costa Mesa, CA; clone SY38; 1:40),chromogranin (Chemicon, Temecula, CA; clone LK2H10; 1:500), Neu-N (Chemicon;
Rodriguez et al. Page 2
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

1:10000), and Ki-67 (MIB-1; Dako; 1:300) in selected cases. MIB-1 labeling indices wereevaluated in 51 cases at first resection by examining 20 consecutive tumor fields using theCAS200 computer imaging system (CAS 200; Bacus Laboratories, Lombard, IL). Nuclearp53 immunostaining was scored on a 4-tiered scale as follows: no staining (0); focal to lessthan 10% of cells (1+); 10% to 50% of cells or weak staining greater than 50% of cells (2+);and strong staining of greater than 50% of cells (3+).
Statistical MethodsMedians, interquartile ranges (IQRs), ranges, and frequencies were used to describe patientand tumor characteristics as appropriate. Only the first primary tumor for each patient withsurgically obtained material that was classified as PA WHO Grade I, low-gradeastrocytoma, subtype indeterminate (LGSI), or DA WHO Grades II to IV was included forsurvival analyses (n = 91). Overall and disease-specific survivals, as well as recurrence,were evaluated using the Kaplan-Meier method. The Fisher exact test was used to comparecategorical variables. In all analyses, a p value less than 0.05 was considered statisticallysignificant. All statistical analyses were performed using SAS software (SAS Institute Inc.,Cary, NC).
RESULTSGeneral
The initial search revealed approximately 300 patients. From these, 100 NF1 patients wereidentified. From these patients, there were 121 pathologic specimens available for reviewthat included biopsies, resections, and autopsies. There were 57 male and 43 female patientswith a median age at tumor diagnosis of 13 years (range, 4 months to 68 years). A familyhistory of NF1 was documented in 37 patients. Clinical features of NF1 included café au laitspots (97%), freckling (56%), neurofibromas (45%), Lisch nodules (35%), skeletalabnormalities (29%), intellectual impairment (25%), plexiform neurofibroma (23%), anddysmorphic features (16%). A total of 98 pathologic specimens of primary tumors wereidentified. Three patients had second primary tumors, 1 of which may have been induced byradiation (see succeeding sentences); 14 patients had recurrent tumors with availablepathologic material. Surgical procedures included biopsy (36%), subtotal resection (35%),gross total resection (24%), or they were not specified (9%). Autopsies were performed in 4cases, 2 of which did not have prior diagnostic surgical procedures.
Pathologic review of primary tumors revealed that WHO Grade I PAs were the predominanthistologic tumor type (49%), followed by infiltrating astrocytomas (27%) and indeterminateastrocytomas (19%). Although the latter were easily identifiable as having an astrocyticphenotype, they did not have specific features that permitted a definitive classification as PAor DA. Most (17/20) were low-grade gliomas based on low mitotic and MIB-1 labelingindices and a predominant solid architecture and were classified as LGSI. Two cases werehigh-grade, and an additional spinal cord tumor was also classified as an indeterminateastrocytoma because of limited biopsy material. The remaining diagnoses were desmoplasticganglioglioma (n = 1), ganglioglioma (n = 1), pilomyxoid astrocytoma (n = 1), and glialatypia suspicious but not diagnostic for glioma (n = 2).
Pathologic Features by GroupPA and Variants—The histologic features are summarized in Table 1 and illustrated inFigure 1. When the tumors involved the optic pathways, they tended to spread to theadjacent leptomeninges and surround the optic nerve in a circumferential manner. Sometumors exhibited prominent infiltration of optic nerve fascicles (Fig. 1A). Proliferative
Rodriguez et al. Page 3
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

activity (median [IQR] was low as assessed by mitotic count [0 {0–1.0}] or MIB-1 labelingindex [1.76 {0.4–5.3}]). The median (IQR) p53 immunopositivity score was 1 (0–2).
Perivascular pseudorosettes were identified in 3 of these tumors. However, only 1 case hadall the classic features of pilomyxoid astrocytoma at initial biopsy. A second case showedfeatures of a LGSI at initial spinal cord biopsy (Fig. 2A; see succeeding sentences), likelybecause of the very small size of the biopsy. The tumor, however, had diagnostic features ofpilomyxoid astrocytoma at autopsy with spread to supratentorial leptomeninges and caudaequina (Fig. 2B, C).
Low-Grade Astrocytoma, Subtype Indeterminate—Histologic features aresummarized in Table 1, and representative features are illustrated in Figure 3. Although thetumors were easily recognized as astrocytic, classic features of PA, including biphasicgrowth pattern, Rosenthal fibers, eosinophilic granular bodies, and glomeruloid vessels,were either absent or present in isolation, not allowing a confident diagnosis of PA. Mosttumors had less specific features, including refractile conspicuous cell processes, low tomoderate cellularity, and mild nuclear atypia (Fig. 3). Median (IQR) mitotic activity andMIB-1 labeling indices were 0 (0–1) and 3.7 (1.7–6.7). The median (IQR) p53immunopositivity score was 1 (0.5–1.5).
Pathologic material was available from a second time point in 4 cases, including thepilomyxoid astrocytoma mentioned previously. One of the tumors showed definite featuresof PA at resection for recurrence. In the other 2 cases, the histologic findings revealed LGSI.
Astrocytomas With Broad Cytoplasmic Processes and Prominent Nucleoli—Six tumors that were classified either as PA (n = 4) or LGSI (n = 2) showed distinctivemorphology (Fig. 4). They were composed of medium-sized cells with plump eosinophilic,nonglassy cytoplasm, broad processes, and prominent nucleoli (Fig. 4B, C) that oftenstained cherry-red. Rosenthal fibers were absent. Eosinophilic granular bodies were presentin 2 cases, although they were not conspicuous. The mitotic index in these tumors rangedfrom none to 4 per 10 HPF (median 1).
Diffusely Infiltrating Astrocytoma—The pathologic features of these tumors aresummarized in Table 2, and representative features are shown in Figure 5. Most werefibrillary astrocytomas (96%). Two tumors had a gemistocytic component, and 1 had smallcell undifferentiated features. One Grade III gemistocytic astrocytoma developed a smallcell undifferentiated component and progressed to glioblastoma. Four cases involved theoptic pathways (see succeeding sentences) and demonstrated nuclear atypia and conspicuousmitotic activity together with a diffusely infiltrating growth, rather than expanding opticnerve septae and leptomeninges (Fig. 6). A Grade IV fibrillary astrocytoma in a 21-year-oldfemale patient with a history of colon cancer with microsatellite instability demonstratedpreserved immunoexpression of the mismatch repair enzymes MLH1, MSH2, MSH6, andPMS2 (data not shown). The median (IQR) mitotic activity, MIB-1 labeling indices, andp53-immunopositive scores were 2 (1–5), 4.5 (2.8–12.9), and 2 (1–3), respectively.
Glioneuronal Tumors—One ganglioglioma (WHO Grade I) and a desmoplastic infantileganglioglioma (Fig. 7A) each were identified. The ganglioglioma was composed of clustersof dysmorphic neurons, some with binucleation. The desmoplastic infantile gangliogliomahad prominent desmoplasia (Fig. 7B) labeled strongly with antibodies directed against glialfibrillary acidic protein (Fig. 7C) and S100 in the predominant astrocytic component.Scattered small neurons showing immunoreactivity for synaptophysin (Fig. 7D) andchromogranin were observed. A biopsy of an enlarging cyst at the operative site 2 years laterrevealed gliotic cortex and microdysgenesis without evidence of recurrent tumor.
Rodriguez et al. Page 4
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Other Astrocytomas—One of the 2 tumors classified as high-grade astrocytoma, subtypeindeterminate, developed 4 years after radiation therapy for a right thalamic PA. It wascharacterized by a relatively solid architecture and the presence of granular bodies but, inaddition, had extensive necrosis, 6 mitoses per 10 HPF, and an MIB-1 labeling index of11.5%. The second tumor was composed of plump cells with a partial fasciculararrangement, but lacking obvious permeation. Increased mitotic activity (7/10 HPF) andpseudopalisading necrosis were present.
Another astrocytoma with a diffusely infiltrating pattern and strong and widespread p53immunostaining had a high mitotic index (13/10 HPF) despite the presence of palisadingaround hyalinized vessels and a loose stroma reminiscent of pilomyxoid astrocytoma (Fig.5C).
Clinical FeaturesThe clinical features of the different astrocytoma subtypes are summarized in Table 3.
Pilocytic Astrocytoma—The median age at pathologic diagnosis was 13 years (IQR, 9–21), with 31% younger than 10 years, 43% between 10 and 20 years, 10% between 20 and30 years, 10% between 30 and 40 years, and 4% between 40 and 50 years. A single patientwas older than 60 years at the time of diagnosis. Radiologic studies included magneticresonance imaging (27/50), computed tomographic scan (12/50), plain x-ray with or withoutoptic canal views (10/50), ventriculography/pneumoencephalography (5/50), andmyelogram (2/50). When visualized by computed tomographic or magnetic resonanceimaging after contrast, the tumors were usually well circumscribed, enhancing, andexhibited both cystic and solid components. Radiographic progression was noted in 15tumors after surgery. Postoperative treatment modalities included observation (36%),radiation (28%), chemotherapy (12%), both radiation and chemotherapy (5%), or unknown(19%). Multifocality was noted on imaging in 16 patients. Of the 2 pilomyxoid astrocytomasidentified, 1 was located in the hypothalamus of a 5-year-old boy (see succeedingsentences), and the second was located in the spinal cord of a 61-year-old man originallydiagnosed as LGSI. The latter demonstrated extensive leptomeningeal spread at autopsy, thepatient having died soon after surgery for a pheochromocytoma. No other casesdemonstrated clinical or pathologic evidence of leptomeningeal spread.
Low-Grade Astrocytoma, Subtype Indeterminate—Presenting symptoms andradiographic findings were also comparable to those of the PA group and are summarized inTable 3. Progression was noted on follow-up imaging in 6 of 17 patients.
Astrocytomas With Broad Cytoplasmic Processes and Prominent Nucleoli—These tumors were all in hemispheric, extraventricular locations; 4 of 6 occurred in thetemporal lobe.
Diffusely Infiltrating Astrocytoma—This group encompassed approximately a third ofthe cases (27%), with a median age of 28 (IQR, 9–38) and the following age distributions:28% between 0 and 9 years, 12% between 10 and 19 years, 12% between 20 and 29 years,24% between 30 and 39 years, 8% between 40 and 49 years, 4% between 50 and 59 years,and 12% older than 60 years. On neuroimaging studies, contrast enhancement was seen inmore than half of the tumors (16/28), and an infiltrative growth pattern was found in onefourth (7/28). Radiographic progression was documented in 14 of 28 tumors, 1 of which wasconsistent with subependymal spread.
Rodriguez et al. Page 5
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Glioneuronal Tumors—The patient with a desmoplastic infantile ganglioglioma was 4months old at the time of diagnosis. The tumor arose in the left temporal lobe, and no furthertreatment was given. Cystic enlargement at the operative site was noted 2 years after surgeryon magnetic resonance imaging, but a biopsy was negative. The tumor had not recurred 18years after the first surgery, although the patient underwent treatment (i.e. radiation andchemotherapy) for a rhabdomyosarcoma of the paranasal sinuses at 18 years of age. Thepatient is alive without evidence of disease 1 year later.
The 1 ganglioglioma developed in the hypothalamic region of a 34-year-old male patient.Subsequent growth required γ-knife treatment 13 months after resection. The patientdeveloped progressive neurologic deterioration and died 3.5 years after surgery.
Other CNS Tumors—One high-grade astrocytoma, subtype indeterminate, arose in a 50-year-old man 4 years after radiation of a recurrent thalamic PA (pathology discussed inprevious sentences). Magnetic resonance imaging demonstrated ring enhancement andmarked edema. Progression was noted, and the patient died 10 months after surgery. Thesecond such tumor arose de novo in the right frontal lobe of a 49-year-old woman who wastreated with radiation therapy and developed progressive neurologic deterioration. She died15 years after diagnosis, most likely secondary to sequelae of radiation.
One right frontal anaplastic astrocytoma with perivascular palisades developed in a 10-year-old boy. This patient received postoperative radiation and temozolomide and is stable withno evidence of disease 20 months after surgery.
One female patient who had undergone resection of a cerebellar PA included in this studydeveloped a pleomorphic spindle cell sarcoma after 12 years, at age 17. The tumor wassuperficially located in the left frontal lobe but recurred 1 year later and led to her death.
Optic Pathway/Hypothalamic Gliomas—Twenty-four cases involved the opticpathway/hypothalamic region; of these, 14 were classified histologically as PA. Of theremainder, 4 were classified as LGSI, 4 as anaplastic astrocytomas (WHO Grade III), 1 aspilomyxoid astrocytoma (WHO Grade II), and 1 as ganglioglioma. Among the anaplasticastrocytomas, 3 are alive without disease 8 to 16 years after surgery, whereas a fourthpatient died of unknown causes 9 years after. Conversely, the patient with the singlehypothalamic pilomyxoid astrocytoma underwent a recurrence 1.5 years after surgery butremains stable 15 years after the second surgery.
CNS Tumors Arising After Radiation Treatment and Additional Non-CNSTumors—Eight patients who had received irradiation, 7 for optic pathway gliomas and 1for a thalamic PA (age range, 2–31 years; mean, 13 years) developed a second tumor. Thesubsequent tumors included 4 PAs involving the lateral ventricles (n = 2), midbrain (n = 1),and temporoparietal lobe (n = 1), as well as fibrillary astrocytomas WHO Grades II and IIIin the brainstem (1 each), an LGSI also arising in the brainstem, and a large high-gradeastrocytoma, subtype indeterminate, of the right frontal lobe. It is uncertain whether all thesetumors arose in the prior radiation fields; therefore, it is unclear if some could bepostirradiation gliomas or, rather, second tumors secondary to the NF1 status. An additionalpatient developed an aggressive enhancing brainstem mass 5 years after irradiation treatmentfor a recurrent hypothalamic PA that was not biopsied.
Excluding neurofibromas, 13 patients developed 1 or more additional tumors outside of theCNS, including pheochromocytoma (3), malignant peripheral nerve sheath tumors (3),breast carcinoma (2), rhabdomyosarcoma (1), juvenile myelomonocytic leukemia (1),
Rodriguez et al. Page 6
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Burkitt lymphoma (1), colonic adenocarcinoma (1), pleomorphic spindle cell sarcoma (1),gastrointestinal stromal tumor (1), and prostate adenocarcinoma (1).
Statistical AnalysisOverall survival times at 5 and 10 years were 85% and 68% in the PA group, 74% for bothin the LGSI cases, and 39% and 24% in DA, respectively. Recurrence-free rates at 5 and 10years were 84% and 72% in the PA group, 64% for both in the LGSI cases, and 40% and32% in the DA, respectively. The DAs had a significantly decreased overall survival rateand an increased risk of recurrence when compared with the other groups (p < 0.001 and p =0.003, respectively) (Fig. 8).
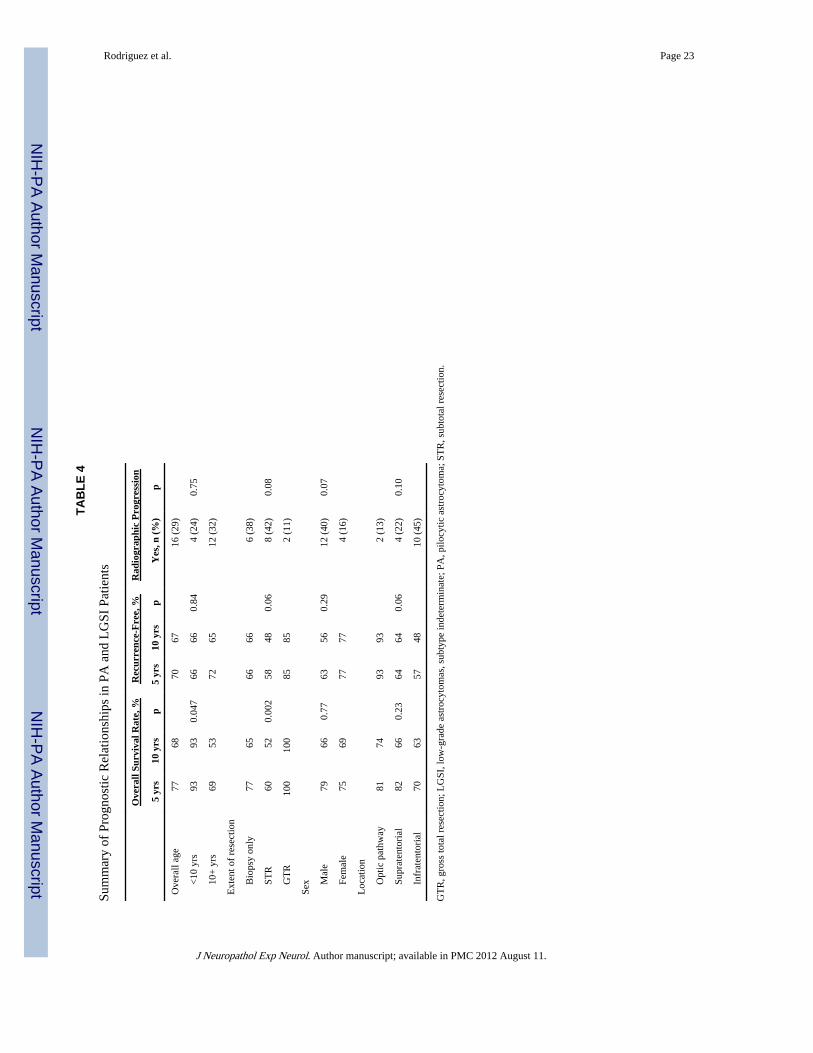
Pilocytic astrocytoma versus LGSI proliferative indices (mitoses/10 HPF; MIB-1), p53labeling, and degree of microscopic infiltration were not significantly different between thePA and LGSI cases. Moreover, there were no statistical significant differences betweenthese 2 groups for radiographic progression, time to recurrence, or overall and disease-specific survival times (p > 0.18). Prognostic variables in the combined PA/LGSI groups aresummarized in Table 4.
Age—Overall survival rate at 5 and 10 years was significantly better in patients youngerthan 10 years at diagnosis in the combined PA and LGSI groups (93% vs 69% and 93% vs53%, respectively) (p = 0.047) (Fig. 8A). Similar, although not significant, results were seenwith disease-specific survival (p = 0.07), with 5- and 10-year disease-specific survival ratesin patients younger than 10 years of 93% and 5- and 10-year disease-specific survival ratesin older patients of 84% and 72%, respectively. Increasing age was associated withradiographic progression in the PA-only group (p = 0.014) and marginally in the combinedPA and LGSI groups (p = 0.058). Patients with optic pathway tumors were significantlymore likely to be younger than 10 years of age (66%) than those with either infratentorial(32%) or extra-optic supratentorial tumors (10%) (p = 0.001).
Gross Total Resection—After subtotal resection or biopsy in the PA/LGSI combinedgroup, the patients had a significantly worse overall survival rate than those who hadundergone gross total resection (Table 4). The extent of resection was not, however,significantly associated with either recurrence or radiographic progression. The extent ofsurgery was not significantly related to tumor location (p = 0.09).
Postoperative radiation was associated with significantly decreased overall survival time inthe PA and LGSI combined groups (p = 0.009). No obvious prognostic differences werenoted among PA alone or combined PA and LGSI groups according to tumor size, mitoticactivity, cellularity, nuclear atypia, biphasic pattern, necrosis, or MIB-1 labeling index (p >0.07).
DISCUSSIONAlthough the predisposition for CNS tumor development in patients with NF1 is wellrecognized, and there are numerous studies focusing on the clinical features (4, 6–10),detailed pathologic studies on the histopathology of these brain tumors are lacking in theliterature. In 1985, Ilgren et al (14) reported a series of 87 gliomas in neurofibromatosispatients, with a comprehensive review of the literature at that time. However, this seriesincluded patients that almost certainly would today be classified as havingneurofibromatosis type 2, rather than type 1; histologic examination was not performed inall cases. Furthermore, the histologic studies were performed before the development ofcurrent histologic classification and grading schemes. Similar to the study of Ilgren et al(14), we found that gliomas in NF1 can arise in any location along the neuraxis, including
Rodriguez et al. Page 7
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

within the spinal cord. One of their main findings was that cerebellar tumors had a worseprognosis. However, their cases included glioblastomas and ependymomas. Although therewere only 5 cerebellar PAs in our study, they did not seem to behave more aggressively thanthose located in other brain regions.
Most of the tumors that arise in the setting of NF1 are PAs and, unlike their sporadiccounterparts, have a distinctive predilection to involve the optic nerve, chiasm, andhypothalamus. These optic gliomas are typically found in young children, afflicting 15% to20% of patients with NF1 (15). Although these tumors seem to have a more indolentbehavior than their sporadic counterparts and may even regress without treatment (6), theycan cause significant morbidity and progress in some patients (7).
There is considerable uncertainty in predicting which optic pathway tumors will behavemore aggressively. Cummings et al (16), in a study of 22 gliomas involving the optic nerve(19 PAs; 3 DAs), identified p53 and MIB-1 labeling index as potentially useful foridentifying tumors with either a worse behavior or more infiltrative histology. Most of theircases were, however, sporadic, rather than associated with NF1. In the present study, MIB-1was not a useful prognostic indicator in low-grade astrocytomas. Although strong diffusep53 staining (3+) was only encountered in DAs, the numbers were too small to draw anyfirm conclusion. Twenty-four cases in the current series involved the optic pathways/hypothalamic region, although the histology was more variable than in the study ofCummings (16), including 14 WHO Grade I PAs, 4 LGSI, 4 anaplastic astrocytomas, and 1example each of pilomyxoid astrocytoma and ganglioglioma. Pilocytic astrocytomas withmoderate to high mitotic activity (4–5/10 HPF) did not behave more aggressively than thosewith low proliferative rates. Furthermore, even the tumors that were classified as anaplasticastrocytomas of the optic pathways behaved in a clinically less aggressive fashion. Thesefindings support the observations from previous studies that suggest that the optic pathwayrepresents a permissive location for tumor growth in children with NF1. Recent reportsusing Nf1 genetically engineered mice suggest that specific growth cues from the tumormicroenvironment may uniquely promote the growth of Nf1-deficient glial cells to facilitateand limit glioma proliferation within the optic pathway (17, 18).
In addition to the astrocytomas that fall neatly into either PA or DA categories, some NF1-associated astrocytomas display indeterminate or overlapping features that precludedefinitive classification as one or the other histologic subtype. These gliomas are usuallylow grade (5), often with piloid cytologic features, but they lack the typical biphasicarchitecture, Rosenthal fibers, and eosinophilic granular bodies of classic PAs. Additionally,there is often an infiltrative growth pattern and/or increased proliferative activity providingadditional sources of concern. In this study, we referred to such cases as LGSI. Our datasuggest that there are no significant clinicopathologic or prognostic differences between thishistologic subtype and classic PA when they arise in patients with NF1. In small biopsies,however, the LGSI diagnosis may simply reflect sampling error. For example, 1 of 4 LGSIcases with multiple pathologic specimens showed morphologic features of pilomyxoidastrocytoma at autopsy. A second case showed classic PA after a second resection.Pilomyxoid astrocytoma has been regarded as a morphologic subtype of PA with apredilection for the hypothalamic area and worse clinical behavior (e.g. leptomeningealseeding) (19) and is therefore classified as a Grade II neoplasm in the most recent WHOclassification (2). Two of our cases can be placed in this category, and at least 1 isolatedcase of pilomyxoid astrocytoma in NF1 has been previously reported (20).
Glioneuronal tumors may also arise in NF1 (11, 21, 22), and we found 1 conventionalganglioglioma in the current study. Desmoplastic infantile ganglioglioma represents adistinct clinicopathologic subtype initially identified in young children and characterized by
Rodriguez et al. Page 8
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

large size, exuberant collagen deposition, and excellent overall prognosis (23). To ourknowledge, this unusual subtype has not been previously described in a patient with NF1.
Although most unusual tumors in our series were low-grade gliomas, caution is warrantedbecause tumors with "indeterminate" or "unusual" histologic diagnoses in NF1 can also behigh-grade tumors, as demonstrated by 2 of our cases. One was an anaplastic infiltrativeastrocytoma with a peculiar perivascular arrangement reminiscent of pilomyxoidastrocytoma, and the second was an unusual tumor arising after radiation for a conventionalPA. The latter tumor showed a solid architecture and eosinophilic granular bodies but hadremarkable proliferative activity and extensive necrosis.
The main prognostic indicator in our current series was histologic subtype. Patients with DAhad a reduced overall and recurrence free survival. Although these results may be expectedfrom previous data based on sporadic gliomas, it is important to highlight the importance ofaccurate histologic classification, when possible, for patients with NF1. In this regard,earlier reports have focused on tumor location and concluded that patients with nonopticpathway gliomas have a worse outcome (10). In the current study in which all tumors wereclassified histologically, we did not find significant prognostic differences when PAs or PAsand LGSIs were analyzed by tumor location. However, there was a trend for increasedrecurrence-free survival in optic pathway tumors. After adjusting for histology, patientsolder than 10 years had a reduced survival rate compared with younger patients.
Diffusely infiltrating astrocytoma as a group encompassed greater than one fourth of thepatients, a significant proportion of the tumors in this series. This is in contrast to previousclinical and epidemiologic studies that report or estimate a lower frequency (4). Theseresults might be related to our current series being comprised mostly of surgically obtainedspecimens at 2 large tertiary care centers. Diffusely infiltrating astrocytomas are more likelyto be progressive and demonstrate alarming radiographic features and therefore promptsurgery. Furthermore, PA of the optic pathways, the predominant clinicopathologic type, isnot usually biopsied or resected at the present time. Rather, it can be followed withoutintervention in most instances (6). Most DAs in our series seem to have arisen without ahistory of prior irradiation. This is consistent with prior publications (4).
In summary, we report the findings of a study with detailed histopathologic examination in alarge cohort of NF1 patients. The spectrum of morphologic subtypes is wide in thispopulation. A significant subset display unusual histologic features, but most tumors in thisgroup are low grade and behave similarly to PA. The diagnosis of a DA subtype is thestrongest prognostic indicator especially when arising outside of the optic pathways. For thisreason, careful histologic classification and grading should be attempted whenever possibleto provide an accurate diagnosis, prognostic information, and to guide appropriate therapyfor individuals with NF1-associated gliomas.
AcknowledgmentsSupported in part by training Grant No. T32 NS07494-04 from the National Institutes of Health (to F.J.R.).
REFERENCES1. Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary
management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997; 278:51–57. [PubMed:9207339]
2. Louis, D.; Ohgaki, H.; Wiestler, O.; Cavenee, W. WHO Classification of Tumours of the CentralNervous System. IARC Press; Lyon, France: 2007.
Rodriguez et al. Page 9
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

3. Stern J, DiGiacinto GV, Housepian EM. Neurofibromatosis and optic glioma: Clinical andmorphological correlations. Neurosurgery. 1979; 4:524–28. [PubMed: 113690]
4. Gutmann DH, Rasmussen SA, Wolkenstein P, et al. Gliomas presenting after age 10 in individualswith neurofibromatosis type 1 (NF1). Neurology. 2002; 59:759–61. [PubMed: 12221173]
5. Leonard JR, Perry A, Rubin JB, et al. The role of surgical biopsy in the diagnosis of glioma inindividuals with neurofibromatosis-1. Neurology. 2006; 67:1509–12. [PubMed: 17060590]
6. Parsa CF, Hoyt CS, Lesser RL, et al. Spontaneous regression of optic gliomas: Thirteen casesdocumented by serial neuroimaging. Arch Ophthalmol. 2001; 119:516–29. [PubMed: 11296017]
7. Thiagalingam S, Flaherty M, Billson F, et al. Neurofibromatosis type 1 and optic pathway gliomas:Follow-up of 54 patients. Ophthalmology. 2004; 111:568–77. [PubMed: 15019338]
8. Zeid JL, Charrow J, Sandu M, et al. Orbital optic nerve gliomas in children with neurofibromatosistype 1. J AAPOS. 2006; 10:534–39. [PubMed: 17189147]
9. King A, Listernick R, Charrow J, et al. Optic pathway gliomas in neurofibromatosis type 1: Theeffect of presenting symptoms on outcome. Am J Med Genet A. 2003; 122:95–99. [PubMed:12955759]
10. Guillamo JS, Creange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis1 (NF1): A retrospective study of 104 patients. Brain. 2003; 126:152–60. [PubMed: 12477702]
11. Vinchon M, Soto-Ares G, Ruchoux MM, et al. Cerebellar gliomas in children with NF1: Pathologyand surgery. Childs Nerv Syst. 2000; 16:417–20. [PubMed: 10958550]
12. Riffaud L, Vinchon M, Ragragui O, et al. Hemispheric cerebral gliomas in children with NF1:Arguments for a long-term follow-up. Childs Nerv Syst. 2002; 18:43–47. [PubMed: 11935243]
13. Rodriguez HA, Berthrong M. Multiple primary intracranial tumors in von Recklinghausen_sneurofibromatosis. Arch Neurol. 1966; 14:467–75. [PubMed: 4957904]
14. Ilgren E, Kinnier-Wilson L, Stiller C. Gliomas in neurofibromatosis: A series of 89 cases withevidence for enhanced malignancy in associated cerebellar astrocytomas. Pathol Annu. 1985;20:331–58. [PubMed: 3921930]
15. Listernick R, Ferner RE, Liu GT, et al. Optic pathway gliomas in neurofibromatosis-1:Controversies and recommendations. Ann Neurol. 2007; 61:189–98. [PubMed: 17387725]
16. Cummings TJ, Provenzale JM, Hunter SB, et al. Gliomas of the optic nerve: Histological,immunohistochemical (MIB-1 and p53), and MRI analysis. Acta Neuropathol (Berl). 2000;99:563–70. [PubMed: 10805102]
17. Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborateparacrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–1112. [PubMed: 17400655]
18. Warrington NM, Woerner BM, Daginakatte GC, et al. Spatiotemporal differences in CXCL12expression and cyclic AMP underlie the unique pattern of optic glioma growth inneurofibromatosis type 1. Cancer Res. 2007; 67:8588–95. [PubMed: 17875698]
19. Tihan T, Fisher PG, Kepner JL, et al. Pediatric astrocytomas with monomorphous pilomyxoidfeatures and a less favorable outcome. J Neuropathol Exp Neurol. 1999; 58:1061–68. [PubMed:10515229]
20. Khanani MF, Hawkins C, Shroff M, et al. Pilomyxoid astrocytoma in a patient withneurofibromatosis. Pediatr Blood Cancer. 2006; 46:377–80. [PubMed: 15800886]
21. Parizel PM, Martin JJ, Van Vyve M, et al. Cerebral ganglioglioma and neurofibromatosis type I.Case report and review of the literature. Neuroradiology. 1991; 33:357–59. [PubMed: 1922756]
22. Fedi M, Anne Mitchell L, Kalnins RM, et al. Glioneuronal tumours in neurofibromatosis type 1:MRI-pathological study. J Clin Neurosci. 2004; 11:745–47. [PubMed: 15337138]
23. VandenBerg SR, May EE, Rubinstein LJ, et al. Desmoplastic supratentorial neuroepithelial tumorsof infancy with divergent differentiation potential (“desmoplastic infantile gangliogliomas”).Report on 11 cases of a distinctive embryonal tumor with favorable prognosis. J Neurosurg. 1987;66:58–71. [PubMed: 3097276]
Rodriguez et al. Page 10
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
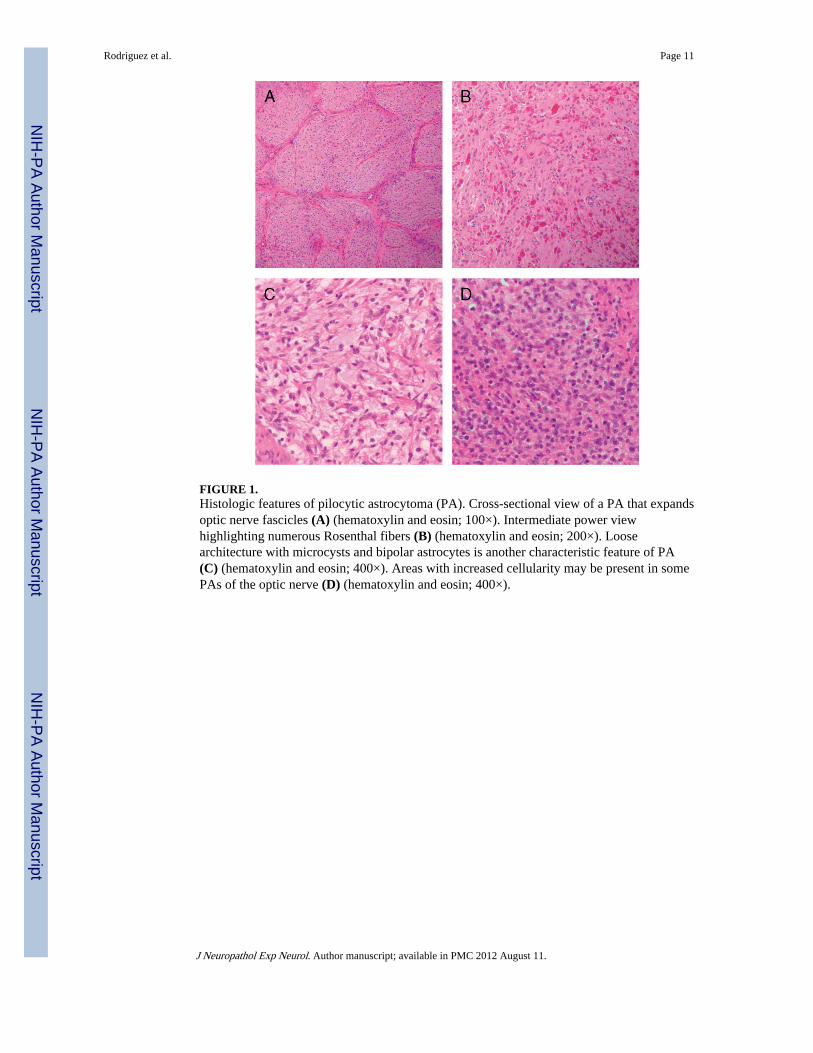
FIGURE 1.Histologic features of pilocytic astrocytoma (PA). Cross-sectional view of a PA that expandsoptic nerve fascicles (A) (hematoxylin and eosin; 100×). Intermediate power viewhighlighting numerous Rosenthal fibers (B) (hematoxylin and eosin; 200×). Loosearchitecture with microcysts and bipolar astrocytes is another characteristic feature of PA(C) (hematoxylin and eosin; 400×). Areas with increased cellularity may be present in somePAs of the optic nerve (D) (hematoxylin and eosin; 400×).
Rodriguez et al. Page 11
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
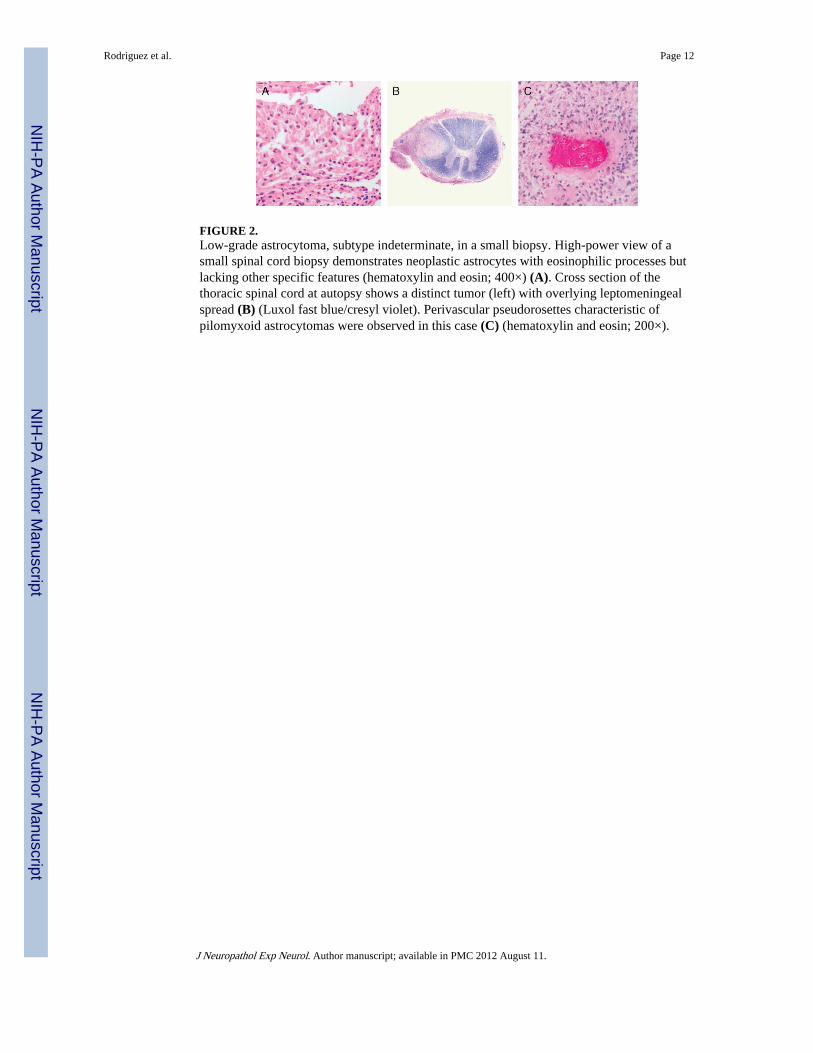
FIGURE 2.Low-grade astrocytoma, subtype indeterminate, in a small biopsy. High-power view of asmall spinal cord biopsy demonstrates neoplastic astrocytes with eosinophilic processes butlacking other specific features (hematoxylin and eosin; 400×) (A). Cross section of thethoracic spinal cord at autopsy shows a distinct tumor (left) with overlying leptomeningealspread (B) (Luxol fast blue/cresyl violet). Perivascular pseudorosettes characteristic ofpilomyxoid astrocytomas were observed in this case (C) (hematoxylin and eosin; 200×).
Rodriguez et al. Page 12
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
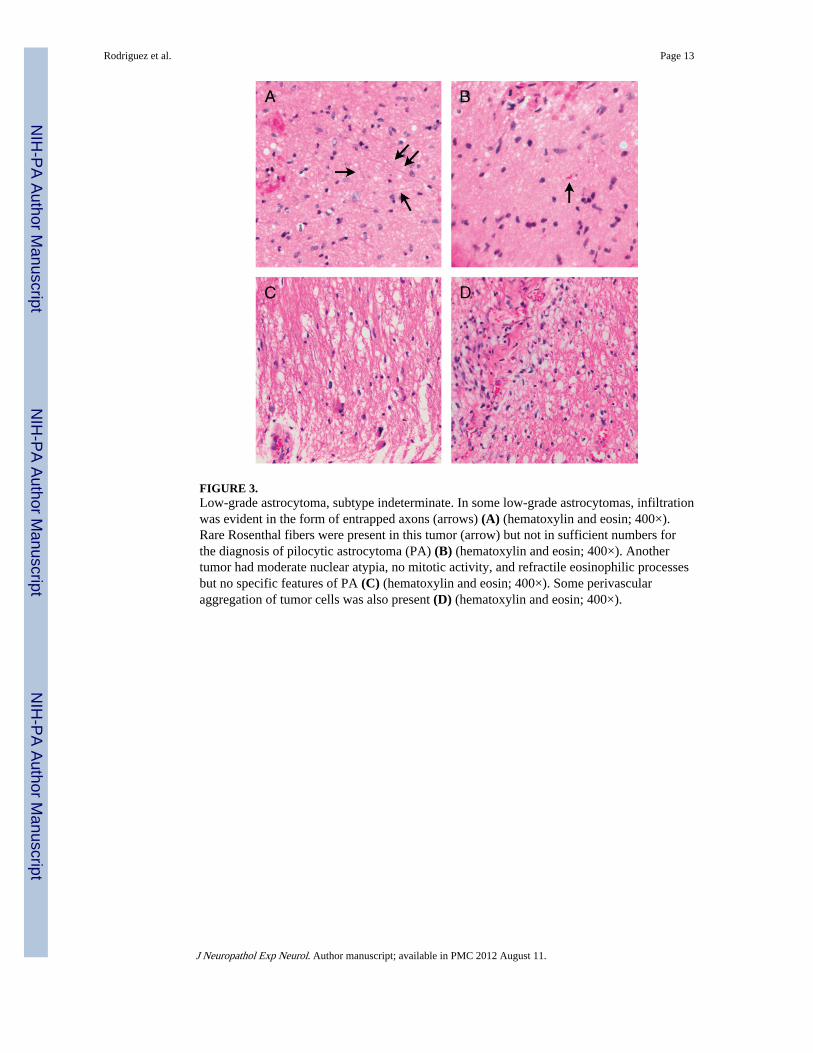
FIGURE 3.Low-grade astrocytoma, subtype indeterminate. In some low-grade astrocytomas, infiltrationwas evident in the form of entrapped axons (arrows) (A) (hematoxylin and eosin; 400×).Rare Rosenthal fibers were present in this tumor (arrow) but not in sufficient numbers forthe diagnosis of pilocytic astrocytoma (PA) (B) (hematoxylin and eosin; 400×). Anothertumor had moderate nuclear atypia, no mitotic activity, and refractile eosinophilic processesbut no specific features of PA (C) (hematoxylin and eosin; 400×). Some perivascularaggregation of tumor cells was also present (D) (hematoxylin and eosin; 400×).
Rodriguez et al. Page 13
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
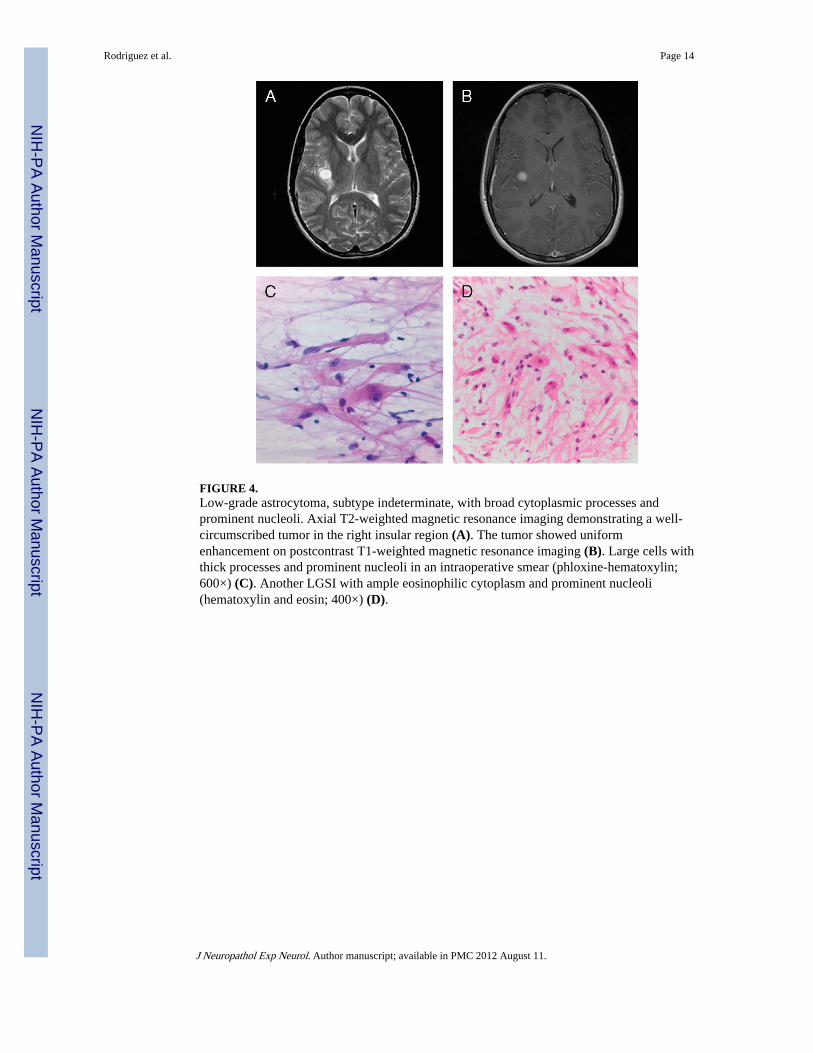
FIGURE 4.Low-grade astrocytoma, subtype indeterminate, with broad cytoplasmic processes andprominent nucleoli. Axial T2-weighted magnetic resonance imaging demonstrating a well-circumscribed tumor in the right insular region (A). The tumor showed uniformenhancement on postcontrast T1-weighted magnetic resonance imaging (B). Large cells withthick processes and prominent nucleoli in an intraoperative smear (phloxine-hematoxylin;600×) (C). Another LGSI with ample eosinophilic cytoplasm and prominent nucleoli(hematoxylin and eosin; 400×) (D).
Rodriguez et al. Page 14
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
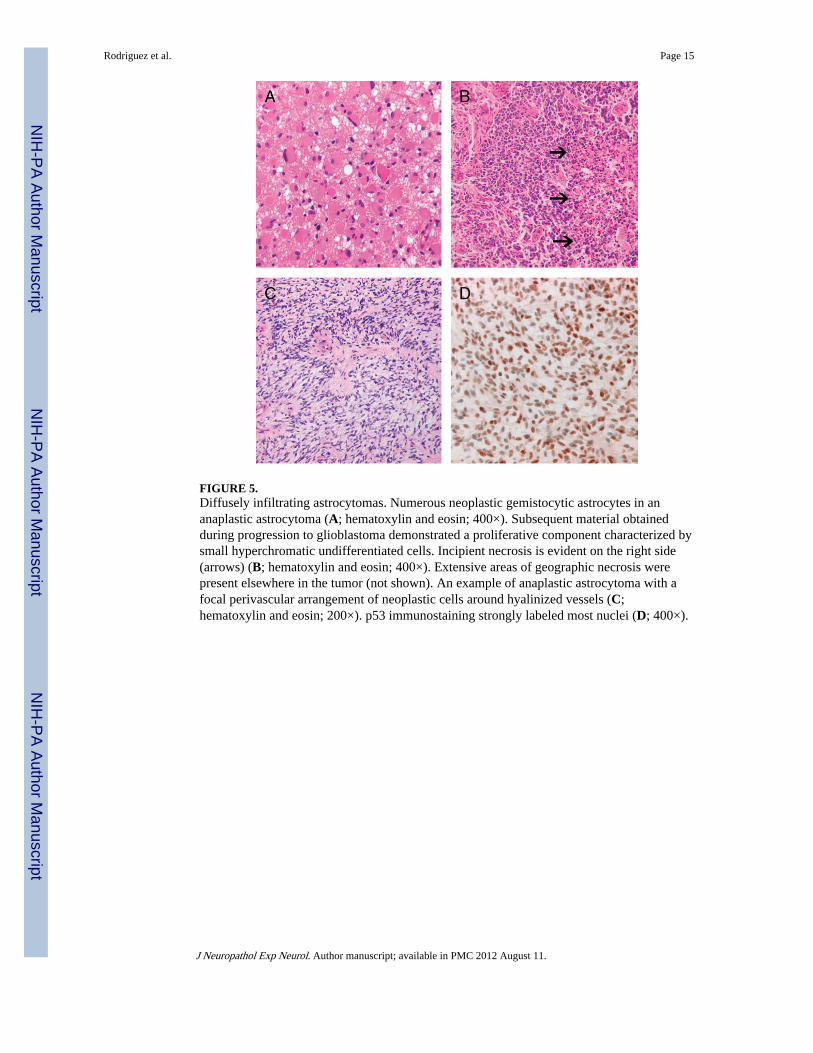
FIGURE 5.Diffusely infiltrating astrocytomas. Numerous neoplastic gemistocytic astrocytes in ananaplastic astrocytoma (A; hematoxylin and eosin; 400×). Subsequent material obtainedduring progression to glioblastoma demonstrated a proliferative component characterized bysmall hyperchromatic undifferentiated cells. Incipient necrosis is evident on the right side(arrows) (B; hematoxylin and eosin; 400×). Extensive areas of geographic necrosis werepresent elsewhere in the tumor (not shown). An example of anaplastic astrocytoma with afocal perivascular arrangement of neoplastic cells around hyalinized vessels (C;hematoxylin and eosin; 200×). p53 immunostaining strongly labeled most nuclei (D; 400×).
Rodriguez et al. Page 15
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
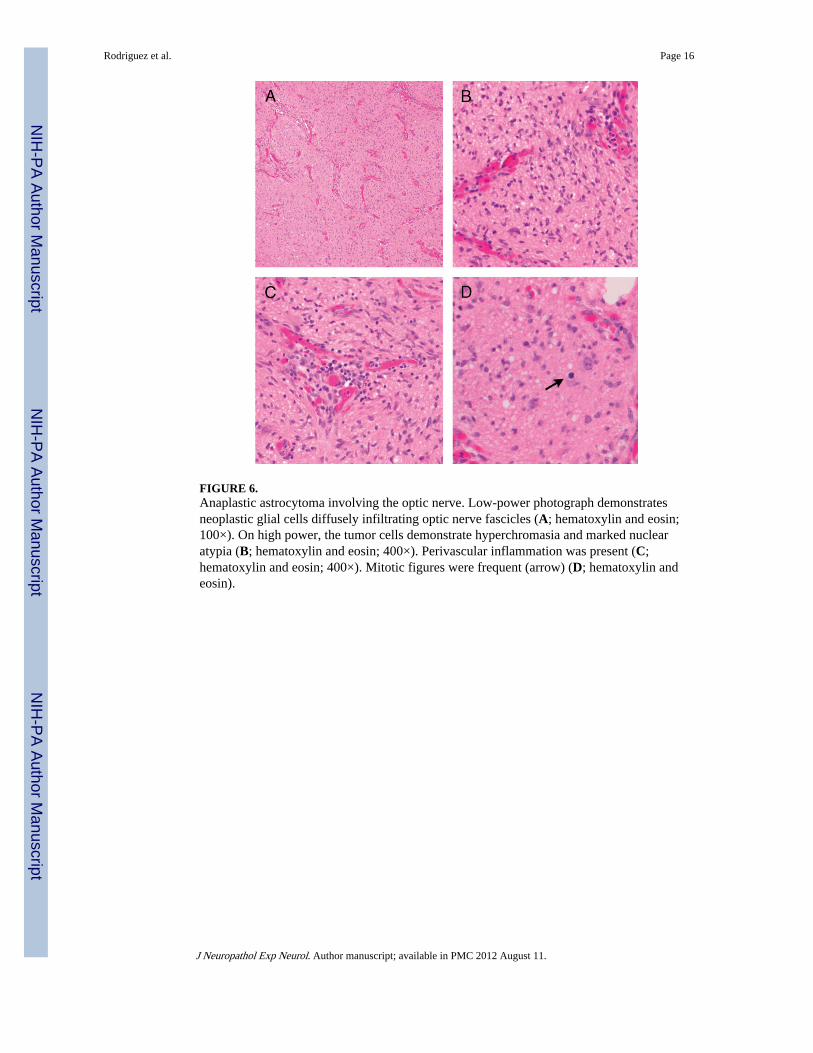
FIGURE 6.Anaplastic astrocytoma involving the optic nerve. Low-power photograph demonstratesneoplastic glial cells diffusely infiltrating optic nerve fascicles (A; hematoxylin and eosin;100×). On high power, the tumor cells demonstrate hyperchromasia and marked nuclearatypia (B; hematoxylin and eosin; 400×). Perivascular inflammation was present (C;hematoxylin and eosin; 400×). Mitotic figures were frequent (arrow) (D; hematoxylin andeosin).
Rodriguez et al. Page 16
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
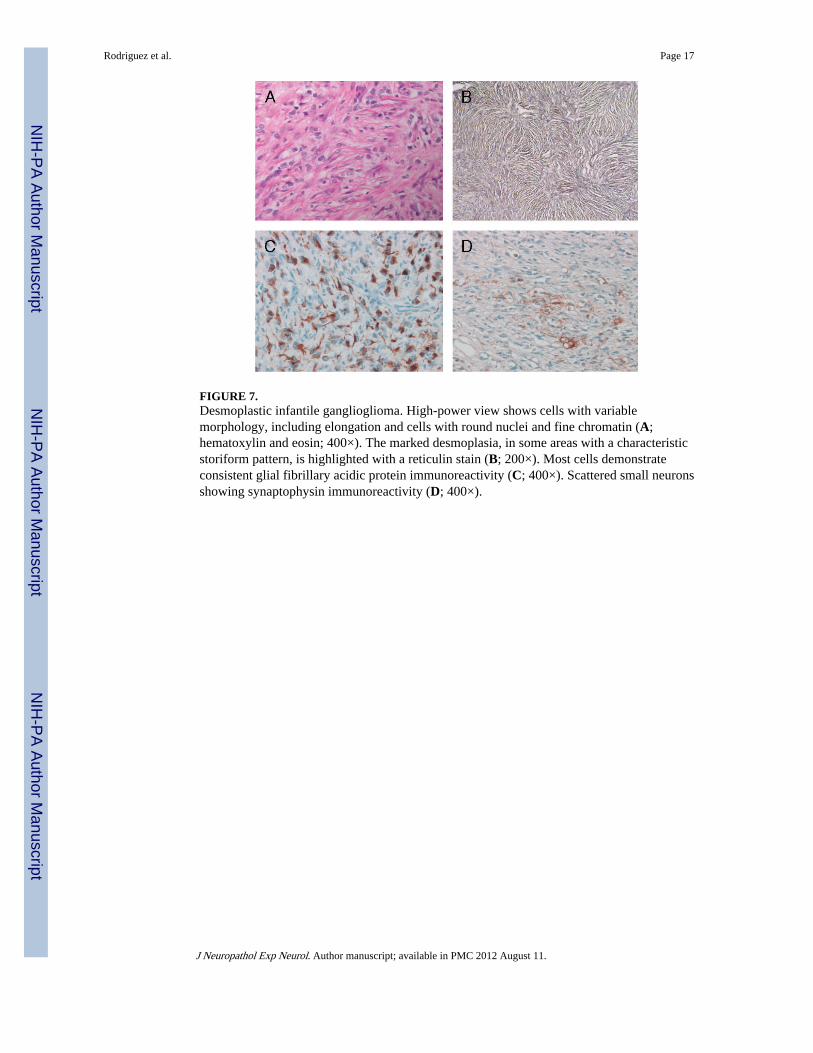
FIGURE 7.Desmoplastic infantile ganglioglioma. High-power view shows cells with variablemorphology, including elongation and cells with round nuclei and fine chromatin (A;hematoxylin and eosin; 400×). The marked desmoplasia, in some areas with a characteristicstoriform pattern, is highlighted with a reticulin stain (B; 200×). Most cells demonstrateconsistent glial fibrillary acidic protein immunoreactivity (C; 400×). Scattered small neuronsshowing synaptophysin immunoreactivity (D; 400×).
Rodriguez et al. Page 17
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
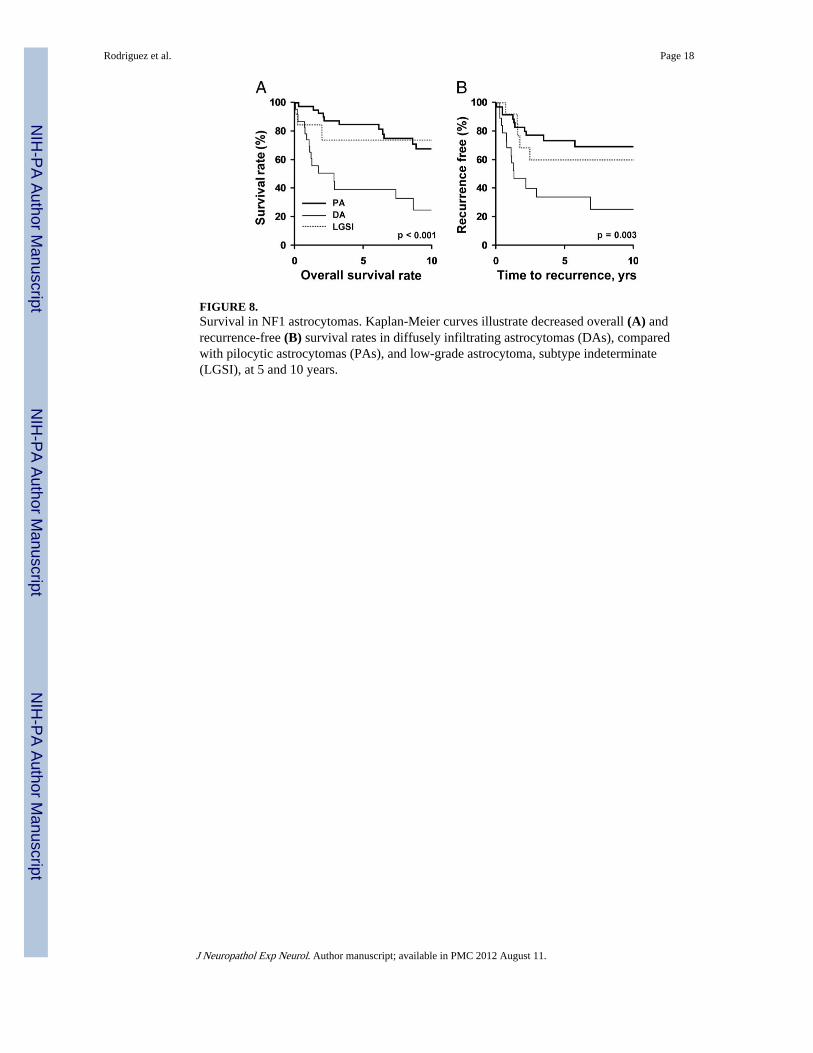
FIGURE 8.Survival in NF1 astrocytomas. Kaplan-Meier curves illustrate decreased overall (A) andrecurrence-free (B) survival rates in diffusely infiltrating astrocytomas (DAs), comparedwith pilocytic astrocytomas (PAs), and low-grade astrocytoma, subtype indeterminate(LGSI), at 5 and 10 years.
Rodriguez et al. Page 18
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Rodriguez et al. Page 19
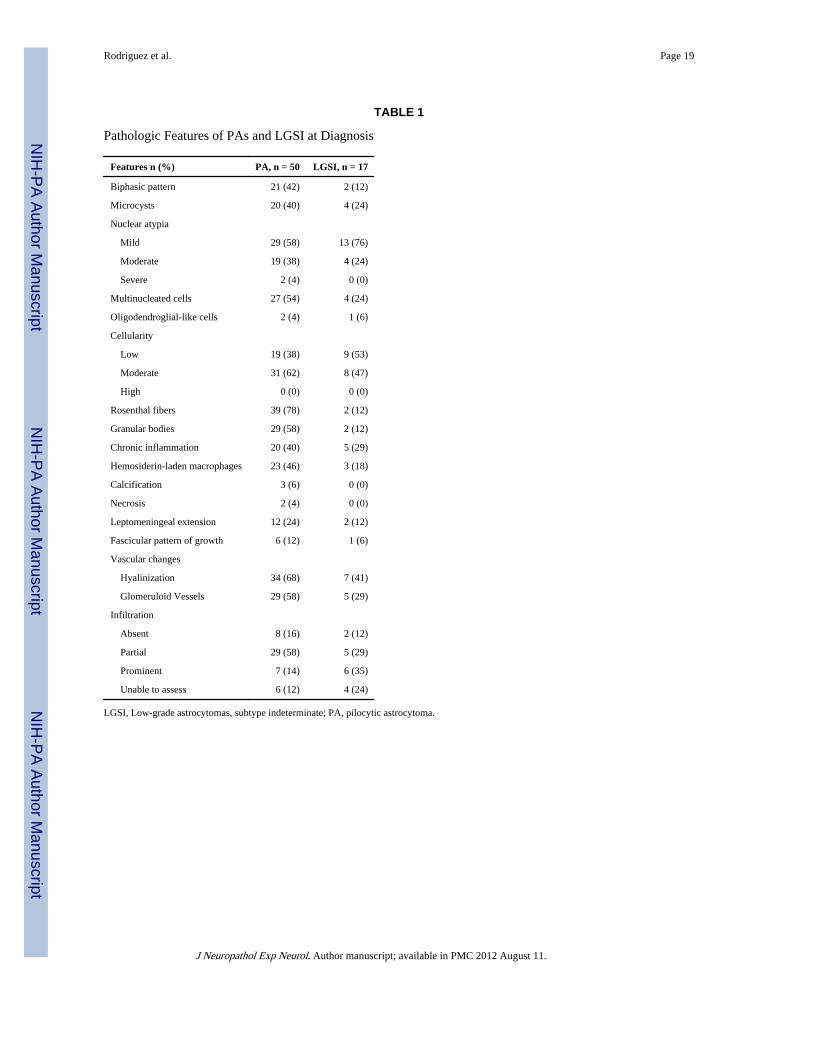
TABLE 1
Pathologic Features of PAs and LGSI at Diagnosis
Features n (%) PA, n = 50 LGSI, n = 17
Biphasic pattern 21 (42) 2 (12)
Microcysts 20 (40) 4 (24)
Nuclear atypia
Mild 29 (58) 13 (76)
Moderate 19 (38) 4 (24)
Severe 2 (4) 0 (0)
Multinucleated cells 27 (54) 4 (24)
Oligodendroglial-like cells 2 (4) 1 (6)
Cellularity
Low 19 (38) 9 (53)
Moderate 31 (62) 8 (47)
High 0 (0) 0 (0)
Rosenthal fibers 39 (78) 2 (12)
Granular bodies 29 (58) 2 (12)
Chronic inflammation 20 (40) 5 (29)
Hemosiderin-laden macrophages 23 (46) 3 (18)
Calcification 3 (6) 0 (0)
Necrosis 2 (4) 0 (0)
Leptomeningeal extension 12 (24) 2 (12)
Fascicular pattern of growth 6 (12) 1 (6)
Vascular changes
Hyalinization 34 (68) 7 (41)
Glomeruloid Vessels 29 (58) 5 (29)
Infiltration
Absent 8 (16) 2 (12)
Partial 29 (58) 5 (29)
Prominent 7 (14) 6 (35)
Unable to assess 6 (12) 4 (24)
LGSI, Low-grade astrocytomas, subtype indeterminate; PA, pilocytic astrocytoma.
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Rodriguez et al. Page 20
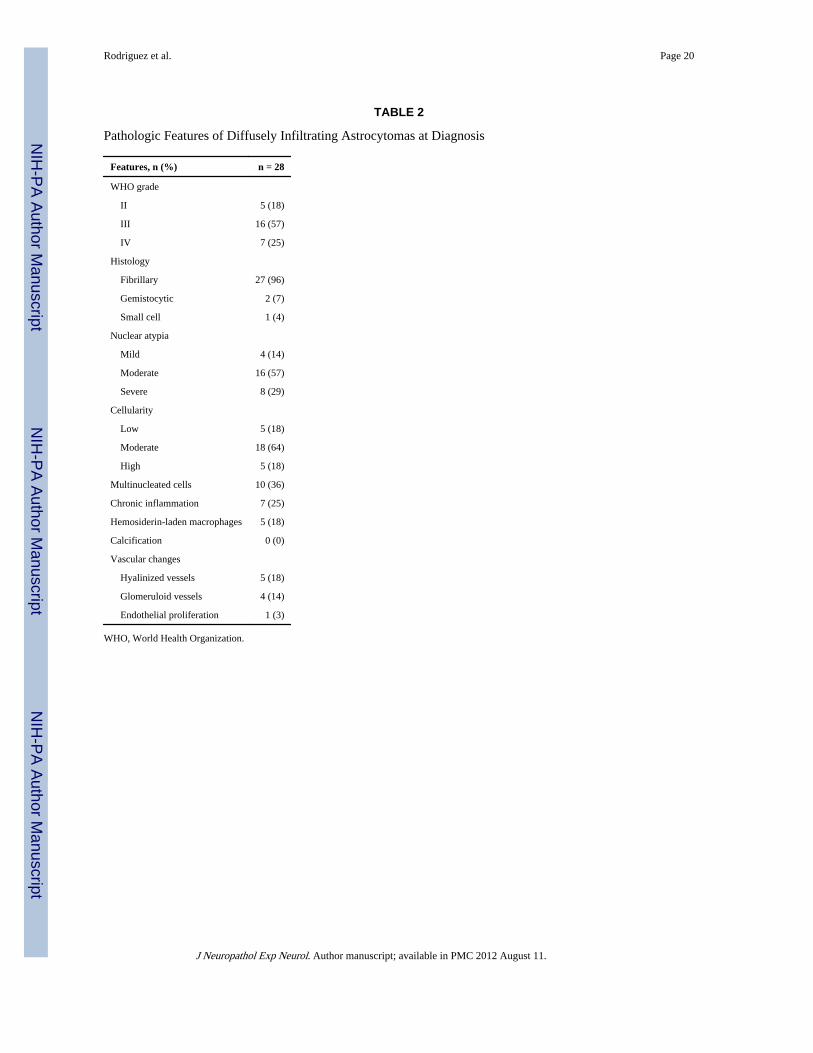
TABLE 2
Pathologic Features of Diffusely Infiltrating Astrocytomas at Diagnosis
Features, n (%) n = 28
WHO grade
II 5 (18)
III 16 (57)
IV 7 (25)
Histology
Fibrillary 27 (96)
Gemistocytic 2 (7)
Small cell 1 (4)
Nuclear atypia
Mild 4 (14)
Moderate 16 (57)
Severe 8 (29)
Cellularity
Low 5 (18)
Moderate 18 (64)
High 5 (18)
Multinucleated cells 10 (36)
Chronic inflammation 7 (25)
Hemosiderin-laden macrophages 5 (18)
Calcification 0 (0)
Vascular changes
Hyalinized vessels 5 (18)
Glomeruloid vessels 4 (14)
Endothelial proliferation 1 (3)
WHO, World Health Organization.
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Rodriguez et al. Page 21
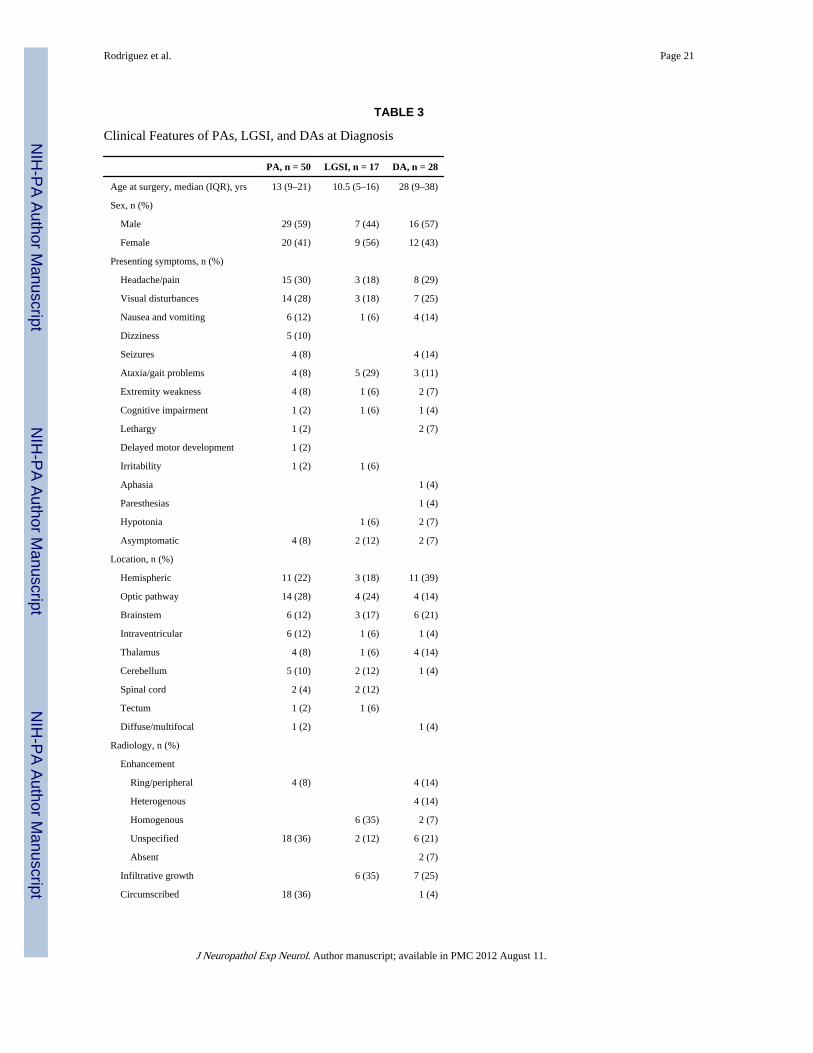
TABLE 3
Clinical Features of PAs, LGSI, and DAs at Diagnosis
PA, n = 50 LGSI, n = 17 DA, n = 28
Age at surgery, median (IQR), yrs 13 (9–21) 10.5 (5–16) 28 (9–38)
Sex, n (%)
Male 29 (59) 7 (44) 16 (57)
Female 20 (41) 9 (56) 12 (43)
Presenting symptoms, n (%)
Headache/pain 15 (30) 3 (18) 8 (29)
Visual disturbances 14 (28) 3 (18) 7 (25)
Nausea and vomiting 6 (12) 1 (6) 4 (14)
Dizziness 5 (10)
Seizures 4 (8) 4 (14)
Ataxia/gait problems 4 (8) 5 (29) 3 (11)
Extremity weakness 4 (8) 1 (6) 2 (7)
Cognitive impairment 1 (2) 1 (6) 1 (4)
Lethargy 1 (2) 2 (7)
Delayed motor development 1 (2)
Irritability 1 (2) 1 (6)
Aphasia 1 (4)
Paresthesias 1 (4)
Hypotonia 1 (6) 2 (7)
Asymptomatic 4 (8) 2 (12) 2 (7)
Location, n (%)
Hemispheric 11 (22) 3 (18) 11 (39)
Optic pathway 14 (28) 4 (24) 4 (14)
Brainstem 6 (12) 3 (17) 6 (21)
Intraventricular 6 (12) 1 (6) 1 (4)
Thalamus 4 (8) 1 (6) 4 (14)
Cerebellum 5 (10) 2 (12) 1 (4)
Spinal cord 2 (4) 2 (12)
Tectum 1 (2) 1 (6)
Diffuse/multifocal 1 (2) 1 (4)
Radiology, n (%)
Enhancement
Ring/peripheral 4 (8) 4 (14)
Heterogenous 4 (14)
Homogenous 6 (35) 2 (7)
Unspecified 18 (36) 2 (12) 6 (21)
Absent 2 (7)
Infiltrative growth 6 (35) 7 (25)
Circumscribed 18 (36) 1 (4)
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
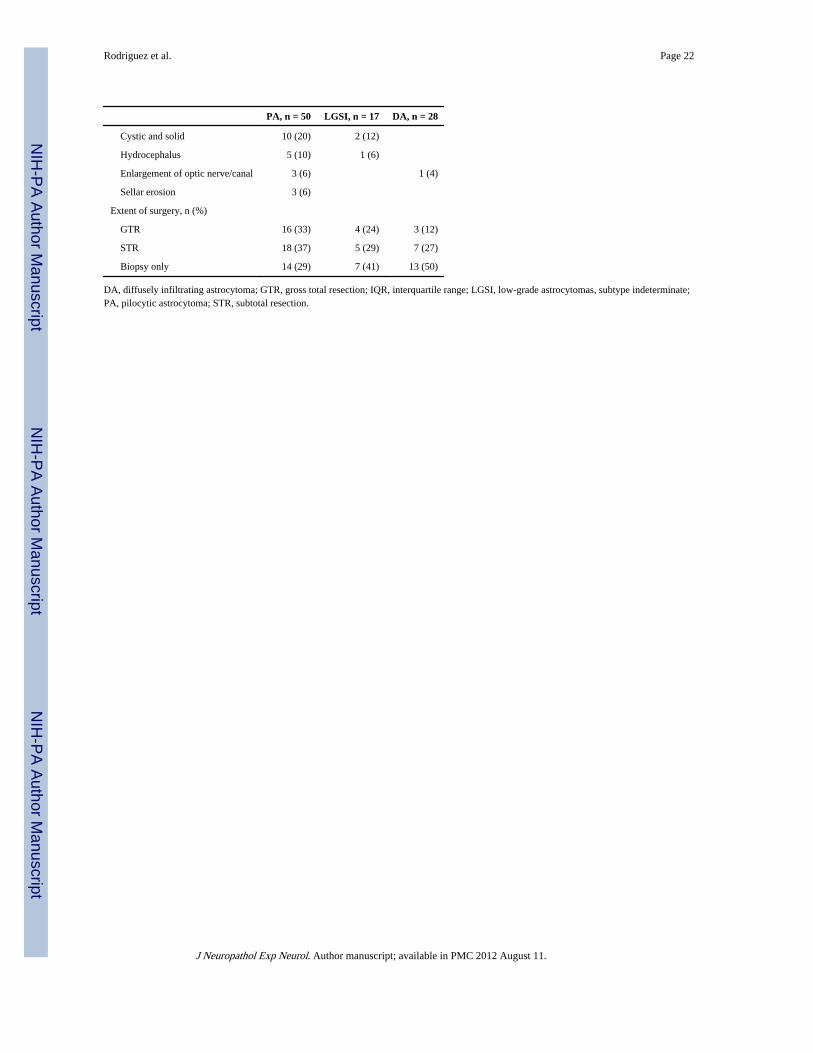
Rodriguez et al. Page 22
PA, n = 50 LGSI, n = 17 DA, n = 28
Cystic and solid 10 (20) 2 (12)
Hydrocephalus 5 (10) 1 (6)
Enlargement of optic nerve/canal 3 (6) 1 (4)
Sellar erosion 3 (6)
Extent of surgery, n (%)
GTR 16 (33) 4 (24) 3 (12)
STR 18 (37) 5 (29) 7 (27)
Biopsy only 14 (29) 7 (41) 13 (50)
DA, diffusely infiltrating astrocytoma; GTR, gross total resection; IQR, interquartile range; LGSI, low-grade astrocytomas, subtype indeterminate;PA, pilocytic astrocytoma; STR, subtotal resection.
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Rodriguez et al. Page 23
TAB
LE 4
Sum
mar
y of
Pro
gnos
tic R
elat
ions
hips
in P
A a
nd L
GSI
Pat
ient
s
Ove
rall
Surv
ival
Rat
e, %
Rec
urre
nce-
Fre
e, %
Rad
iogr
aphi
c P
rogr
essi
on
5 yr
s10
yrs
p5
yrs
10 y
rsp
Yes
, n (
%)
p
Ove
rall
age
7768
7067
16 (
29)
<
10 y
rs93
930.
047
6666
0.84
4 (2
4)0.
75
10
+ y
rs69
5372
6512
(32
)
Ext
ent o
f re
sect
ion
B
iops
y on
ly77
6566
666
(38)
ST
R60
520.
002
5848
0.06
8 (4
2)0.
08
G
TR
100
100
8585
2 (1
1)
Sex
M
ale
7966
0.77
6356
0.29
12 (
40)
0.07
Fe
mal
e75
6977
774
(16)
Loc
atio
n
O
ptic
pat
hway
8174
9393
2 (1
3)
Su
prat
ento
rial
8266
0.23
6464
0.06
4 (2
2)0.
10
In
frat
ento
rial
7063
5748
10 (
45)
GT
R, g
ross
tota
l res
ectio
n; L
GSI
, low
-gra
de a
stro
cyto
mas
, sub
type
inde
term
inat
e; P
A, p
ilocy
tic a
stro
cyto
ma;
ST
R, s
ubto
tal r
esec
tion.
J Neuropathol Exp Neurol. Author manuscript; available in PMC 2012 August 11.
Related Documents











