Genomic assessment of the evolution of the prion protein gene family in vertebrates Paul M. Harrison ⁎, Amit Khachane, Manish Kumar Dept. of Biology, McGill University, Stewart Biology Building, 1205 Dr. Penfield Ave., Montreal, QC, Canada H3A 1B1 abstract article info Article history: Received 22 June 2009 Accepted 24 February 2010 Available online 3 March 2010 Keywords: Prion PrP Doppel Shadoo Evolution Annotation Prion diseases are devastating neurological disorders caused by the propagation of particles containing an alternative β−sheet-rich form of the prion protein (PrP). Genes paralogous to PrP, called Doppel and Shadoo, have been identified, that also have neuropathological relevance. To aid in the further functional characterization of PrP and its relatives, we annotated completely the PrP gene family (PrP-GF), in the genomes of 42 vertebrates, through combined strategic application of gene prediction programs and advanced remote homology detection techniques (such as HMMs, PSI-TBLASTN and pGenThreader). We have uncovered several previously undescribed paralogous genes and pseudogenes. We find that current high-quality genomic evidence indicates that the PrP relative Doppel, was likely present in the last common ancestor of present-day Tetrapoda, but was lost in the bird lineage, since its divergence from reptiles. Using the new gene annotations, we have defined the consensus of structural features that are characteristic of the PrP and Doppel structures, across diverse Tetrapoda clades. Furthermore, we describe in detail a transcribed pseudogene derived from Shadoo that is conserved across primates, and that overlaps the meiosis gene, SYCE1, thus possibly regulating its expression. In addition, we analysed the locus of PRNP/PRND for significant conservation across the genomic DNA of eleven mammals, and determined the phylogenetic penetration of non-coding exons. The genomic evidence indicates that the second PRNP non-coding exon found in even-toed ungulates and rodents, is conserved in all high-coverage genome assemblies of primates (human, chimp, orang utan and macaque), and is, at least, likely to have fallen out of use during primate speciation. Furthermore, we have demonstrated that the PRNT gene (at the PRNP human locus) is conserved across at least sixteen mammals, and evolves like a long non- coding RNA, fashioned from fragments of ancient, long, interspersed elements. These annotations and evolutionary analyses will be of further use for functional characterisation of the PrP- GF, and will be updatable in a semi-automated fashion as more genomes accumulate. © 2010 Elsevier Inc. All rights reserved. Introduction Prions are the prime causative agent of transmissible spongiform encephalopathies (TSEs) in mammals [1]. The most notable of these are Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cows, and scrapie in sheep and goats. CJD is characterized by progressive dementia, and death within a year of diagnosis. TSEs can arise in inherited, sporadic or infectious forms. Infectious prions lack nucleic acids [1], and rely on the presence of a host prion-protein gene for propagation [2]. Whereas the normal cellular form of the prion protein (PrP-C) is mostly α−helical [3,4], the infectious form of the prion protein (PrP-Sc), is mostly β−sheet [5], indicating a profound conformational change coupled to disease progression and propagation. Different ‘strains’ of prion disease that cause different sets of symptoms and deposition patterns of PrP-Sc in the brain, appear to be linked to variant conformations/configurations of PrP-Sc [6]. Double knockouts of the PrP gene appear to have no effect on the viability of laboratory mice [2], and the function of PrP remains unclear, although this is likely linked to its ability to bind ions of copper and of other metals [7]. PrP-C appears to be involved in development of the central nervous system (CNS) [8], and has been shown to bind neural cell adhesion molecules [9]. While we now have extensive data on the structure of PrP-C, the structure of PrP-Sc remains more elusive. Several structures for monomeric PrP-C have been resolved for a variety of species, using 2D NMR spectroscopy [3,4]. The PrP-C structure comprises three α−helices and a small two-strand antiparallel β−sheet [3,4]. The N-terminal repetitive region is unstructured in the native state, if not bound to metal ions [3]. PrP 27–30, the proteinase K –resistant core of PrP-Sc, has an N-terminus at approximately residue 90 in the PrP amino-acid sequence. Evidence from a variety of antibodies that bind Genomics 95 (2010) 268–277 Abbreviations: CJD, Creutzfedlt-Jakob disease; BSE, bovine spongiform encepahalo- pathy; TSE, transmissible spongiform encephalopathy; PrP, prion protein; NMR, nuclear magnetic resonance; CNS, central nervous system; TM, transmembrane; PrP-GF, PrP gene family; GPI, glycosyl phosphatidyl inositol; GD, gene desert; NCE 1 , non-coding exon 1; NCE alt_1 , alternative first non-coding exon in humans; NCE 2 , non-coding exon 2; nt, nucleotides; MSA, multiple sequence alignment; mlncRNA, mRNA-like long non- coding RNA. ⁎ Corresponding author. Fax: +1 514 398 5069. E-mail address: [email protected] (P.M. Harrison). 0888-7543/$ – see front matter © 2010 Elsevier Inc. All rights reserved. doi:10.1016/j.ygeno.2010.02.008 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Genomics 95 (2010) 268–277
Contents lists available at ScienceDirect
Genomics
j ourna l homepage: www.e lsev ie r.com/ locate /ygeno
Genomic assessment of the evolution of the prion protein gene family in vertebrates
Paul M. Harrison ⁎, Amit Khachane, Manish KumarDept. of Biology, McGill University, Stewart Biology Building, 1205 Dr. Penfield Ave., Montreal, QC, Canada H3A 1B1
Abbreviations: CJD, Creutzfedlt-Jakob disease; BSE, bpathy; TSE, transmissible spongiform encephalopathy; Pmagnetic resonance; CNS, central nervous system; TMgene family; GPI, glycosyl phosphatidyl inositol; GD, gexon 1; NCEalt_1, alternative first non-coding exon in humnt, nucleotides; MSA, multiple sequence alignment; mcoding RNA.⁎ Corresponding author. Fax: +1 514 398 5069.
E-mail address: [email protected] (P.M. Harris
0888-7543/$ – see front matter © 2010 Elsevier Inc. Adoi:10.1016/j.ygeno.2010.02.008
a b s t r a c t
a r t i c l e i n f oArticle history:Received 22 June 2009Accepted 24 February 2010Available online 3 March 2010
Keywords:PrionPrPDoppelShadooEvolutionAnnotation
Prion diseases are devastating neurological disorders caused by the propagation of particles containing analternative β−sheet-rich form of the prion protein (PrP). Genes paralogous to PrP, called Doppel and Shadoo,have been identified, that also have neuropathological relevance. To aid in the further functionalcharacterization of PrP and its relatives, we annotated completely the PrP gene family (PrP-GF), in thegenomes of 42 vertebrates, through combined strategic application of gene prediction programs andadvanced remote homology detection techniques (such as HMMs, PSI-TBLASTN and pGenThreader). Wehave uncovered several previously undescribed paralogous genes and pseudogenes. We find that currenthigh-quality genomic evidence indicates that the PrP relative Doppel, was likely present in the last commonancestor of present-day Tetrapoda, but was lost in the bird lineage, since its divergence from reptiles. Usingthe new gene annotations, we have defined the consensus of structural features that are characteristic of thePrP and Doppel structures, across diverse Tetrapoda clades. Furthermore, we describe in detail a transcribedpseudogene derived from Shadoo that is conserved across primates, and that overlaps the meiosis gene,SYCE1, thus possibly regulating its expression.In addition, we analysed the locus of PRNP/PRND for significant conservation across the genomic DNA ofeleven mammals, and determined the phylogenetic penetration of non-coding exons. The genomic evidenceindicates that the second PRNP non-coding exon found in even-toed ungulates and rodents, is conserved inall high-coverage genome assemblies of primates (human, chimp, orang utan and macaque), and is, at least,likely to have fallen out of use during primate speciation. Furthermore, we have demonstrated that the PRNTgene (at the PRNP human locus) is conserved across at least sixteen mammals, and evolves like a long non-coding RNA, fashioned from fragments of ancient, long, interspersed elements.These annotations and evolutionary analyses will be of further use for functional characterisation of the PrP-GF, and will be updatable in a semi-automated fashion as more genomes accumulate.
ovine spongiform encepahalo-rP, prion protein; NMR, nuclear, transmembrane; PrP-GF, PrPene desert; NCE1, non-codingans; NCE2, non-coding exon 2;lncRNA, mRNA-like long non-
on).
ll rights reserved.
© 2010 Elsevier Inc. All rights reserved.
Introduction
Prions are the prime causative agent of transmissible spongiformencephalopathies (TSEs) in mammals [1]. The most notable of theseare Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiformencephalopathy (BSE) in cows, and scrapie in sheep and goats. CJD ischaracterized by progressive dementia, and death within a year ofdiagnosis. TSEs can arise in inherited, sporadic or infectious forms.Infectious prions lack nucleic acids [1], and rely on the presence of ahost prion-protein gene for propagation [2]. Whereas the normalcellular form of the prion protein (PrP-C) is mostly α−helical [3,4],
the infectious form of the prion protein (PrP-Sc), is mostly β−sheet[5], indicating a profound conformational change coupled to diseaseprogression and propagation. Different ‘strains’ of prion disease thatcause different sets of symptoms and deposition patterns of PrP-Sc inthe brain, appear to be linked to variant conformations/configurationsof PrP-Sc [6]. Double knockouts of the PrP gene appear to have noeffect on the viability of laboratory mice [2], and the function of PrPremains unclear, although this is likely linked to its ability to bind ionsof copper and of other metals [7]. PrP-C appears to be involved indevelopment of the central nervous system (CNS) [8], and has beenshown to bind neural cell adhesion molecules [9].
While we now have extensive data on the structure of PrP-C, thestructure of PrP-Sc remains more elusive. Several structures formonomeric PrP-C have been resolved for a variety of species, using 2DNMR spectroscopy [3,4]. The PrP-C structure comprises threeα−helices and a small two-strand antiparallel β−sheet [3,4]. TheN-terminal repetitive region is unstructured in the native state, if notbound to metal ions [3]. PrP 27–30, the proteinase K –resistant core ofPrP-Sc, has an N-terminus at approximately residue 90 in the PrPamino-acid sequence. Evidence from a variety of antibodies that bind

269P.M. Harrison et al. / Genomics 95 (2010) 268–277
to both PrP-C and PrP-Sc indicates that α−helices 2 and 3 have notconverted in the PrP-Sc structure [10]. Recent spectroscopic data alsosuggest that α−helix 1 remains unchanged in PrP-Sc [11]. Thus,conformational conversion to β−sheet is most likely to occurbetween PrP residues 90 and 145. Electron crystallographic analysishas also indicated constraints on the possible configuration of PrP-Scmonomers within multimeric assemblies [12]. In contradiction to theantibody binding information (summarized in ref. [10]), hydrogen/deuterium exchange data indicates that helix 2 and part of helix 3maybe converted to β−sheet in the PrP-Sc form [13].
PrP is highly conserved across mammals, typically with N50%sequence identity relative to human [14,15], and maintains the metalion–binding octarepeat region (whose copy number is implicated insome human prion diseases [16]), and very high sequence conserva-tion (N95%) in other regions associated with disease [17]. PrP is lessconserved in birds relative to human (down to 25% sequenceidentity), with hexarepeats replacing the mammalian octarepeats[15].
Mammalian paralogs of PrP have continued to be discovered andhave been demonstrated to be of neurological relevance andfunctionally linked to PrP [18]. Doppel is a divergent homolog of PrP(∼25% sequence identity), that is mainly expressed in the testis,and can cause neurodegeneration in the CNS [18]. It conserves the C-terminal globular domain, but does not contain a repetititive metalion –binding region, or the intervening alanine-rich stretch. A secondparalog of PrP, dubbed Shadoo, only reported four years ago, is morehighly conserved across vertebrates, than either Doppel or PrP; itintriguingly shares a high degree of homology with PrP in the shortalanine-rich stretch that forms a transmembrane (TM) α−helix insome disease-associated PrP products [19]. Shadoo is expressed in theCNS, and both Shadoo and PrP-C can counteract Doppel neurotoxicityin a similar ways [20]. Further PrP homologs have also been found infish [21].
To aid in the further functional characterization of PrP and itsknown relatives, we filtered comprehensively in 42 vertebrategenome assemblies for other unknown paralogs and orthologs withinthe PrP gene family (PrP-GF), and have uncovered several undescribedgenes and pseudogenes. This current accumulated genomic evidenceindicates that Doppel was likely present in the last common ancestorof Tetrapoda, but was lost in the bird lineage since divergence fromreptiles. Our annotation procedures are semi-automated and will beapplied regularly for updates on the expanding database of vertebrategenomes.
Results & discussion
Terminology
PRNP, PRND and SPRN are the names of the genes that encode theproteins Prion Protein (PrP), Doppel, and Shadoo respectively. The PrPgene family (PrP-GF) is the set of all orthologous and paralogous genesof PrP.
Determining the extent of the PrP-GF in vertebrate genomes
We annotated undiscoveredmembers of the PrP gene family (PrP-GF) in forty-two vertebrates (representing the taxa Mammalia, Birds,Reptiles, Amphibia and Fish) using the pipeline described in Methods.Our findings are summarized in Table 1, and graphically in Fig. 1. Wedetected the three core members of the PrP-GF (PrP, Doppel andShadoo) in all of the high-coverage mammalian genome assemblies,with one exception: Shadoo was not detected in the horse (Equuscaballus). Various gene annotations for PrP, Doppel and Shadoo aremissing in several low-coverage assemblies (Table 1). We havediscovered twenty-seven new vertebrate PrP-GF members in thepresent analysis, and have updated the annotations for nine others
(Table 1). Multiple sequence alignments and genomic coordinates forthe complete PrP-GF including the novel/adjusted annotations arelisted in Suppl. Fig. 1 and Suppl. Tabs. 1-2, with further observations/comments listed in Table 1.
The current genomic evidence indicates that Doppel was lost in an earlyancestor of birds
Firstly, we investigated a key question for the PrP-GF, theevolutionary history of Doppel. We did not find any Doppel duplicatesin either of the high-coverage (≥6X) bird genome assemblies, chickenand zebra finch. This is despite using a range of parameters for psi-tblastn [22], and also using psi-blast with translated genomic DNA as aprotein database (see Methods). In addition, for further sensitivity, weemployed other remote homolog detection techniques (HiddenMarkovModels and pGenThreader [63]) in combination, but found no furtherhomologs (see Methods). The best protein match detected in eithergenome is a non-significant homology (psi-tblastn e-value=0.01) onchicken chromosome 25, that cannot be formed into a predicted gene(using FgenesH [23]). Also, throughpsi-tblastn searches against genomicDNA, using as queries the genes flankingmammalian PRNPs and PRNDs,we could not detect any segmental duplications in either bird, thatmight harbour the missing Doppel.
In addition,wefind that there is a likely ortholog of Doppel situatednear PRNP in the X. tropicalis genome (we term this Frog Doppel). FrogDoppel is located syntenically between the RASSF2 and PRNP genes onscaffold#143 of theX. tropicalis genome, and has a single-exon coding-sequence. (It is at present un-annotated in the X. tropicalis genome.)Out of all known PrP-GF homologymatches to this sequence, themosthighly similar are mammalian Doppels (matches with marginalsignificance [e-value=1×10−4]) to Doppels from cow, sheep andother even-toed ungulates). With psi-tblastn, this e-value decreases to2×10−7 for alignment to bovineDoppel [identity 27%over 152 amino-acid residues]). Interestingly, FrogDoppel alignsmost significantly [psi-tblastn e-value=2×10−9; 25% over 162 residues] to the Doppelortholog in Anolis caroliensis (see below). Frog Doppel has no seconddisulphide bridge in its structure (as in other Doppels). Weconstructed a comparative model of Frog Doppel using the programMODELLER [24], based on a pairwise alignment to human Doppelextracted from a Doppel multiple sequence alignment. We note thatthere is a single cysteine residue that may form an inter-moleculardisulphide. There is one conserved N-glycosylation site and one in adistinct solvent-exposed position. In addition, Frog Doppel has a likelysignal peptide and N-terminal disordered region, as in other knownDoppels (Suppl. Figs. 2-3). (The programs used for such protein-levelannotations are listed in the Methods section.)
The putative Doppel in the lizard Anolis caroliensis is also placedsyntenically, directly beside the putative PRNP ortholog in this species,anddisplays a similar profile of species homologies (i.e., it ismost similarto Doppels from even-toed ungulates [e.g., cow Doppel, e-value=1×10−14]). It maintains the universally conserved N-glycosylationsite of the PrP-GF on helix 2, and has two other distinct solvent-exposed N-glycosylation sites. Also, it is strongly predicted to have asignal peptide and the characteristic disordered region of Doppel(Suppl. Fig. 3). It has two more cysteines, that are placed in its com-parative protein model structure (made using MODELLER [24],based on human Doppel, as above) in such a way that they couldnot bond to each other mutually. They are thus likely intermoleculardisulphides.
In summary, current genomic evidence indicates, in aggregate,that Doppel was present in the last common ancestor of tetrapods, butwas lost in birds since the divergence of this lineage from reptiles.
Further gene and pseudogene paralogs of PrPThe PrP ortholog in A. caroliensiswas designated as that most closely
matching to known PrPs (specifically, these PrPs are from aardvark,elephant and opossum, using psi-tblastn). We named it PrP-1
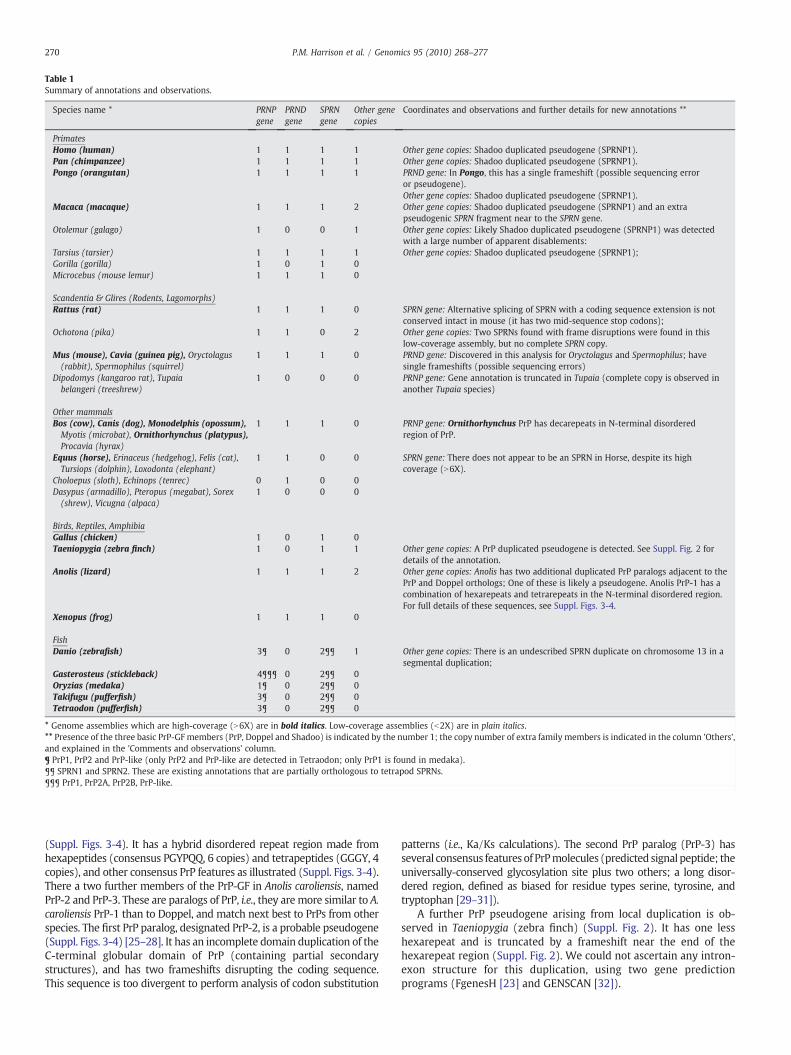
Table 1Summary of annotations and observations.
Species name * PRNPgene
PRNDgene
SPRNgene
Other genecopies
Coordinates and observations and further details for new annotations **
PrimatesHomo (human) 1 1 1 1 Other gene copies: Shadoo duplicated pseudogene (SPRNP1).Pan (chimpanzee) 1 1 1 1 Other gene copies: Shadoo duplicated pseudogene (SPRNP1).Pongo (orangutan) 1 1 1 1 PRND gene: In Pongo, this has a single frameshift (possible sequencing error
or pseudogene).Other gene copies: Shadoo duplicated pseudogene (SPRNP1).
Macaca (macaque) 1 1 1 2 Other gene copies: Shadoo duplicated pseudogene (SPRNP1) and an extrapseudogenic SPRN fragment near to the SPRN gene.
Otolemur (galago) 1 0 0 1 Other gene copies: Likely Shadoo duplicated pseudogene (SPRNP1) was detectedwith a large number of apparent disablements:
Tarsius (tarsier) 1 1 1 1 Other gene copies: Shadoo duplicated pseudogene (SPRNP1);Gorilla (gorilla) 1 0 1 0Microcebus (mouse lemur) 1 1 1 0
Scandentia & Glires (Rodents, Lagomorphs)Rattus (rat) 1 1 1 0 SPRN gene: Alternative splicing of SPRN with a coding sequence extension is not
conserved intact in mouse (it has two mid-sequence stop codons);Ochotona (pika) 1 1 0 2 Other gene copies: Two SPRNs found with frame disruptions were found in this
low-coverage assembly, but no complete SPRN copy.Mus (mouse), Cavia (guinea pig), Oryctolagus(rabbit), Spermophilus (squirrel)
1 1 1 0 PRND gene: Discovered in this analysis for Oryctolagus and Spermophilus; havesingle frameshifts (possible sequencing errors)
Dipodomys (kangaroo rat), Tupaiabelangeri (treeshrew)
1 0 0 0 PRNP gene: Gene annotation is truncated in Tupaia (complete copy is observed inanother Tupaia species)
Other mammalsBos (cow), Canis (dog), Monodelphis (opossum),Myotis (microbat), Ornithorhynchus (platypus),Procavia (hyrax)
1 1 1 0 PRNP gene: Ornithorhynchus PrP has decarepeats in N-terminal disorderedregion of PrP.
Equus (horse), Erinaceus (hedgehog), Felis (cat),Tursiops (dolphin), Loxodonta (elephant)
1 1 0 0 SPRN gene: There does not appear to be an SPRN in Horse, despite its highcoverage (N6X).
Choloepus (sloth), Echinops (tenrec) 0 1 0 0Dasypus (armadillo), Pteropus (megabat), Sorex(shrew), Vicugna (alpaca)
1 0 0 0
Birds, Reptiles, AmphibiaGallus (chicken) 1 0 1 0Taeniopygia (zebra finch) 1 0 1 1 Other gene copies: A PrP duplicated pseudogene is detected. See Suppl. Fig. 2 for
details of the annotation.Anolis (lizard) 1 1 1 2 Other gene copies: Anolis has two additional duplicated PrP paralogs adjacent to the
PrP and Doppel orthologs; One of these is likely a pseudogene. Anolis PrP-1 has acombination of hexarepeats and tetrarepeats in the N-terminal disordered region.For full details of these sequences, see Suppl. Figs. 3-4.
Xenopus (frog) 1 1 1 0
FishDanio (zebrafish) 3¶ 0 2¶¶ 1 Other gene copies: There is an undescribed SPRN duplicate on chromosome 13 in a
segmental duplication;Gasterosteus (stickleback) 4¶¶¶ 0 2¶¶ 0Oryzias (medaka) 1¶ 0 2¶¶ 0Takifugu (pufferfish) 3¶ 0 2¶¶ 0Tetraodon (pufferfish) 3¶ 0 2¶¶ 0
* Genome assemblies which are high-coverage (N6X) are in bold italics. Low-coverage assemblies (b2X) are in plain italics.** Presence of the three basic PrP-GF members (PrP, Doppel and Shadoo) is indicated by the number 1; the copy number of extra family members is indicated in the column 'Others',and explained in the 'Comments and observations' column.¶ PrP1, PrP2 and PrP-like (only PrP2 and PrP-like are detected in Tetraodon; only PrP1 is found in medaka).¶¶ SPRN1 and SPRN2. These are existing annotations that are partially orthologous to tetrapod SPRNs.¶¶¶ PrP1, PrP2A, PrP2B, PrP-like.
270 P.M. Harrison et al. / Genomics 95 (2010) 268–277
(Suppl. Figs. 3-4). It has a hybrid disordered repeat region made fromhexapeptides (consensus PGYPQQ, 6 copies) and tetrapeptides (GGGY, 4copies), and other consensus PrP features as illustrated (Suppl. Figs. 3-4).There a two further members of the PrP-GF in Anolis caroliensis, namedPrP-2 and PrP-3. These are paralogs of PrP, i.e., they are more similar to A.caroliensis PrP-1 than to Doppel, and match next best to PrPs from otherspecies. The first PrP paralog, designated PrP-2, is a probable pseudogene(Suppl. Figs. 3-4) [25–28]. It has an incomplete domain duplication of theC-terminal globular domain of PrP (containing partial secondarystructures), and has two frameshifts disrupting the coding sequence.This sequence is too divergent to perform analysis of codon substitution
patterns (i.e., Ka/Ks calculations). The second PrP paralog (PrP-3) hasseveral consensus features of PrPmolecules (predicted signal peptide; theuniversally-conserved glycosylation site plus two others; a long disor-dered region, defined as biased for residue types serine, tyrosine, andtryptophan [29–31]).
A further PrP pseudogene arising from local duplication is ob-served in Taeniopygia (zebra finch) (Suppl. Fig. 2). It has one lesshexarepeat and is truncated by a frameshift near the end of thehexarepeat region (Suppl. Fig. 2). We could not ascertain any intron-exon structure for this duplication, using two gene predictionprograms (FgenesH [23] and GENSCAN [32]).

Fig. 1. New structural consensuses for PrP and Doppel.
(A) PrP: Consensuses are mapped onto the human PrP sequence, with the following colour coding:
all tetrapods = blue_double_underline;mammals+birds+reptiles = blue_underline;mammals+birds+anolis_lizard = red_double_underline;mammals+birds = red_single_underline;mammals = red (placental)+black bold (egg-laying).
(B) Doppel: Consensuses are mapped onto the human Doppel sequence, with the following colour coding:Mammals+reptiles+amphibia = green_underline;Mammals+reptiles = green;Mammals = red (placental)+egg-laying (black bold).
(C) Absolutely conserved residues in PrP: hydrophobic residues in green; disulphide (cystine) in yellow; aspartate in red. These are labeled on the Human PrP structure (PDB code1qm0). All those that could be mapped to non-disordered parts of the structure (omitting the N and T from the glycosylation sequon) are indicated. The same colour coding isindicated on part (A).(D) Absolutely conserved residues in Doppel: hydrophobic residues in green; disulphide (cystine) in yellow; aspartate in red; proline in grey; polar residues in purple. These arelabeled on the Human PrP structure (PDB code 1qm0). All those that could be mapped to non-disordered parts of the structure (omitting the N and T from the glycosylationsequon) are indicated. The same colour coding is indicated on part (B).
271P.M. Harrison et al. / Genomics 95 (2010) 268–277
Definition of tetrapod consensus features for PrP and DoppelThe PrP-GF annotations across new tetrapod clades (egg-laying
mammals, reptiles and amphibia) enable us to derive a structuralconsensus of PrP and Doppel features, at different maximumevolutionary distances from human (Fig. 1). Over all tetrapod groupsexamined, the only absolutely-conserved features in both PrP andDoppel combined are: (i) the disulphide that bridges helices 2 and 3;(ii) the adjacent N-glycosylation site in helix 2, (iii) an aromaticresidue (either Y of F) packing against helix 1 (this arises at differentsequential positions in PrP and Doppel). Also deeply conserved in PrPacross all Tetrapoda are two valines that pack tightly on either side ofthe disulphide bridge connecting helices 2 and 3, and the aspartateadjacent to this disulphide (Figs. 1A and C). Valines have high βpropensity, and the deep conservation of the valine in helix 2 for thislocal packing role is one of the contributing factors to the secondary-structure 'discordance' of helix 2 [33] ('Discordance' occurs whensecondary-structure prediction algorithms consistently predict thewrong regular secondary structure [33]). The only distinct covariationof two amino-acid residue sites, arising from the new sequence data,is for two salt-bridge residues (inter-changing between K and E) inhelix 3 of platypus PrP. In Doppel, the first β-strand is the most highlyconserved part of the structure (Figs. 1B and D), with a universally
conserved proline-aspartate unit maintained as the core of a likelyfunctional site.
Alternative splicingsWedetected no novel alternative splicings of PrP-GFmembers. We
observe that a known alternative splicing of Shadoo in rat (thatextends the coding sequence of the rat Shadoo sequence, so thatthere is no GPI-anchor signal sequence) is not conserved as a coding-sequence extension in mouse (Table 1), and acquires several stopcodon mutations.
Genomic evolutionary history of ShadooWe have observed the following in our PrP-GF annotation, in
relation to Shadoo:
(i) Platypus Shadoo: This previously undiscovered gene likely hastwo exons (Suppl. Fig. 2A). It maintains the N-terminal {ARGX}nrepeat region, but no central {AG}-rich region could be detectedin this contig of the high-coverage (N6X) Platypus genome.
(ii) Shadoo duplications in placental mammals: A SPRN pseudogeneresulting from a segmental duplication is found in multipleprimates (Fig. 2A). This pseudogene yields a full-length cDNA

272 P.M. Harrison et al. / Genomics 95 (2010) 268–277
in human (accession # FLJ44653; Fig. 2B), and is conserved infive other primates (chimpanzee [Pan troglodytes], orang utan[Pongo pygmaeus], macaque [Macaca mulatta], Otolemur gar-nettii and Tarsius syrichta) (Fig. 2C). In humans, this transcribedpseudogene overlaps a 5' non-coding exon of the SYCE1 gene,which is essential for meiosis in mammals [34]. This SPRNP1sequence thus may be a source of transcriptional interferencefor the SYCE1 gene. By using the intermediate sequence align-ment procedure [35], with the complete SPRNP1 sequences inconceptual translation, we determined that this SYCE1-overlapregion is conserved in chimp, macaque, and Tarsius (Fig. 2C).However, there is no transcriptional information for this locusin the other primates. Calculations of Ka/Ks (the relative rate ofcodon substitution at (non-)synonymous sites) cannot sensiblybe performed for these sequences since the two longest ORFsoverlap by b40 codons, and are formed via multiple distinctdisablements. Also, the sequence fragment found in orang utanand chimpanzee is truncated and only comprises the part of thegene that overlaps a SYCE1 exon in humans. This sequence isthus clearly a human transcribed pseudogene that mayfunction in meiosis control; the region of this transcript thatoverlaps SYCE1 is conserved in multiple other primates, andmay have a conserved regulatory function (Fig. 2C).
In addition, in macaque there is a small pseudogenic fragment ofShadoo next to the Shadoo gene, that presumably resulted frompartial local duplication (Table 1).
Fig. 2. SPRN duplicated transcribed pseudogene (SPRNP1). (A) A schematic picture of the lo(B) Alignment of human SPRNP1 with human SPRN, (FASTA E()–VALUE=6.2×105 68% idenbases inserted/deleted. (C) SPRNP1multiple sequence alignment. A composite alignmentmautan),Macaca (macaque), Tarsius and Otolemur. Frameshifts and stop codons are labelled as iafter an initial alignment leaving it out of the calculation (to show the degree of conservationexon is in black bold.
The PRNP/PRND locus
We performed genomic conservation analysis of the PRNP/PRNDlocus in eleven species (human, chimpanzee, orang utan, macaque,mouse, rat, cow, sheep, dog, false killer whale and horse), using anovel pipeline (described in Methods). We identified areas underrelative purifying selection that may have a regulatory function orcomprise additional non-coding exons.
Conservation across human, chimpanzee, macaque, cow, dog and mouseFirstly, we performed an analysis of the conservation of the PRNP-
PRND locus using the windowing procedure outlined in Methods.There are numerous small conservation islands that are as well-conserved as the exons of PRNP and PRND, that may be regulatoryregions for these genes (Suppl. Fig. 5). These conservation islands alsoarise in the small gene desert (GD) that is 5' to the PRNP-PRND locus[from ∼4200000➔∼4600000 on chromosome 20]. There are no full-length human cDNA mappings in this GD, that are not transposon- orpseudogene-derived. Also, however, we found no areas in thegenomic region between genes ADRA1D and RASSF2 (the PRNP-PRND-PRNT gene locus's immediate neighbours) outside of the PRNPand PRND genes themselves, that are conserved amongst all of themammals that are known to be susceptible to prion diseases (human,chimp [36], macaque [37], mouse, rat [38], cow, sheep).
For detailed further analysis, we defined a suitable gene neighbour-hood of the PRNP and PRND genes, as indicated in Fig. 3A. To define
cus with gene as arrows, and the position on the chromosome labelled in megabases.tity in 151 residue alignment). Frameshifts are labelled in red text with the number ofdewith human SPRN and the SPRNP1 sequences from human, Pan (chimp), Pongo (orangn previous figures. The Otolemur alignment was added from anMSA of all of the species,with(out) Otolemur). The part of each SPRNP1 that is homologous to the human SYCE1
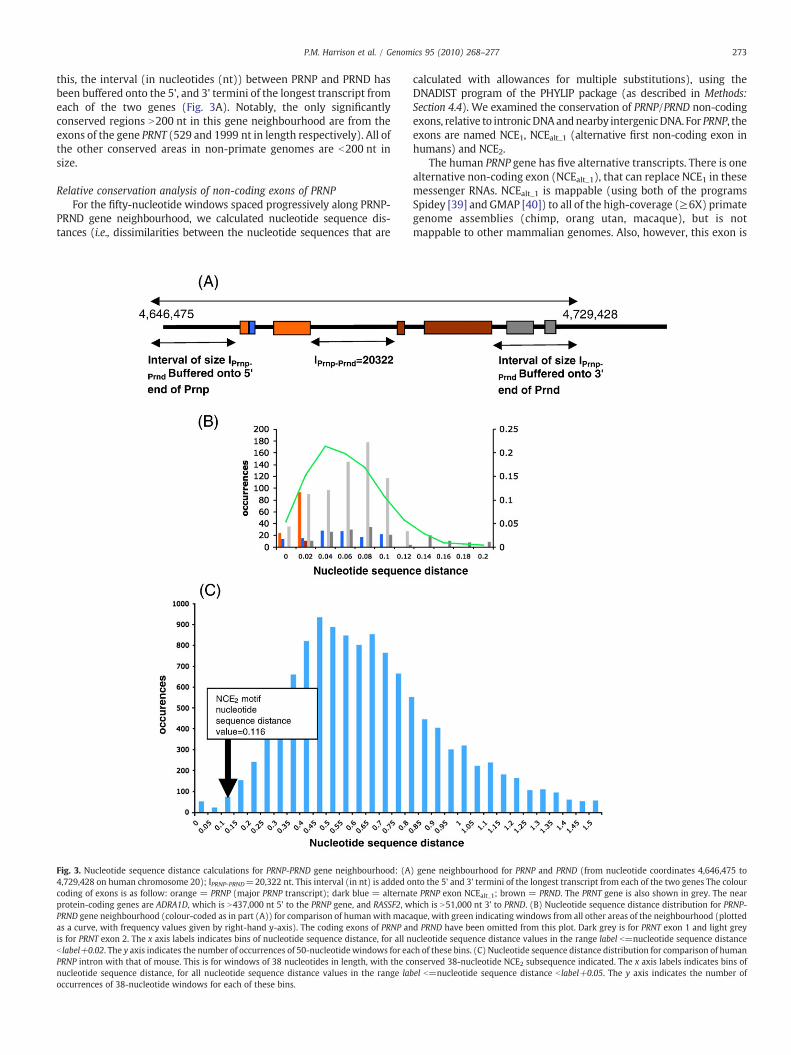
273P.M. Harrison et al. / Genomics 95 (2010) 268–277
this, the interval (in nucleotides (nt)) between PRNP and PRND hasbeen buffered onto the 5', and 3' termini of the longest transcript fromeach of the two genes (Fig. 3A). Notably, the only significantlyconserved regions N200 nt in this gene neighbourhood are from theexons of the gene PRNT (529 and 1999 nt in length respectively). All ofthe other conserved areas in non-primate genomes are b200 nt insize.
Relative conservation analysis of non-coding exons of PRNPFor the fifty-nucleotide windows spaced progressively along PRNP-
PRND gene neighbourhood, we calculated nucleotide sequence dis-tances (i.e., dissimilarities between the nucleotide sequences that are
Fig. 3. Nucleotide sequence distance calculations for PRNP-PRND gene neighbourhood: (A4,729,428 on human chromosome 20); IPRNP-PRND=20,322 nt. This interval (in nt) is added ocoding of exons is as follow: orange = PRNP (major PRNP transcript); dark blue = alternaprotein-coding genes are ADRA1D, which is N437,000 nt 5' to the PRNP gene, and RASSF2, wPRND gene neighbourhood (colour-coded as in part (A)) for comparison of human with macaas a curve, with frequency values given by right-hand y-axis). The coding exons of PRNP anis for PRNT exon 2. The x axis labels indicates bins of nucleotide sequence distance, for all nb label+0.02. The y axis indicates the number of occurrences of 50-nucleotide windows for eaPRNP intron with that of mouse. This is for windows of 38 nucleotides in length, with the conucleotide sequence distance, for all nucleotide sequence distance values in the range laboccurrences of 38-nucleotide windows for each of these bins.
calculated with allowances for multiple substitutions), using theDNADIST program of the PHYLIP package (as described in Methods:Section 4.4). We examined the conservation of PRNP/PRND non-codingexons, relative to intronicDNAandnearby intergenicDNA. For PRNP, theexons are named NCE1, NCEalt_1 (alternative first non-coding exon inhumans) and NCE2.
The human PRNP gene has five alternative transcripts. There is onealternative non-coding exon (NCEalt_1), that can replace NCE1 in thesemessenger RNAs. NCEalt_1 is mappable (using both of the programsSpidey [39] and GMAP [40]) to all of the high-coverage (≥6X) primategenome assemblies (chimp, orang utan, macaque), but is notmappable to other mammalian genomes. Also, however, this exon is
) gene neighbourhood for PRNP and PRND (from nucleotide coordinates 4,646,475 tonto the 5' and 3' termini of the longest transcript from each of the two genes The colourte PRNP exon NCEalt_1; brown = PRND. The PRNT gene is also shown in grey. The nearhich is N51,000 nt 3' to PRND. (B) Nucleotide sequence distance distribution for PRNP-que, with green indicating windows from all other areas of the neighbourhood (plottedd PRND have been omitted from this plot. Dark grey is for PRNT exon 1 and light greyucleotide sequence distance values in the range label b=nucleotide sequence distancech of these bins. (C) Nucleotide sequence distance distribution for comparison of humannserved 38-nucleotide NCE2 subsequence indicated. The x axis labels indicates bins ofel b=nucleotide sequence distance b label+0.05. The y axis indicates the number of

Table 2PRNT RNA analysis.
Longest ORFs †
PRNT RNA (gi | 210032049 | ref | NR_024267.1)
ORF name Number of disablements†† Ka/Ks
ORF_939_1223(95 codons)
Chimp: 1#; Macaque: 0; Cow:2#; Horse: 2#; Dog: 2#, 2*;Mouse: 1#
human-dog: 0.71human-horse: 0.04human-cow: 0.56
ORF_790_972(61 codons)
Chimp: 0; Macaque: 1*; Cow:1#; Horse: 1*
Not calculable
ORF_2142_2288(49 codons)
Chimp: 0; Macaque:0; Horse:0;Dog: 1#; Mouse: 1#
Not calculable
ORF_1949_2068(40 codons)
Chimp: 0; Macaque: 1*; Cow:1#; Horse: 1*
human-dog: 1.25human-horse: 0.37human-cow: 0.01
† The ORF names are in the format ORF_x_y, where x is the start coordinate and y is theend coordinate. Previously we have shown that, in analyses of genome-scalepopulations of ribosomal processed pseudogenes, in comparison to known validatedprotein-coding genes, that 95% of the sequences with Ka/Ks ≥0.5 are pseudogenes arenot genes (Harrison, et al. (2005) Nucleic Acids. Res.). Each of the four ORF here forwhich Ka/Ks is calculable, demonstrates Ka/Ks values ≥0.5; all twelve ORFs harbour atleast one disablement in multiple mammal species.†† Number of disablements in ORF in other species (if the ORF is not detectable, thespecies is not listed). Aligned with TFASTX (e-value≤0.02) (Pearson, et al., (2000),Methods Mol. Biol.).
274 P.M. Harrison et al. / Genomics 95 (2010) 268–277
not especially conserved, relative to other non-coding regions of thePRNP-PRND gene neighbourhood (Fig. 3B). In comparisons withmacaque, there are N20 other tracts that are at least as long, thatare conserved to the same extent (i.e., same mean nucleotide se-quence distance value or better).
In stark contrast, NCE1 is highly conserved relative to other non-coding sequences. It arises in the top 3% of tracts in the nucleotidesequence distance distribution for the PRNP–PRND gene neighbour-hood, for comparison of human and macaque (Fig. 3B). In addition, asmall piece of PRNP NCE1 (19 nt long) from each of the four high-coverage primates is detected as 100% completely conserved in thegenomic DNA of these primates, and also in sheep and cow (but notrat and mouse) (Suppl. Fig. 6).
The known rodent (mouse, rat) and even-toed ungulate (cow,sheep) PRNP mRNAs have a three-exon structure, with a shorter NCE1and an extra NCE2.Was NCE2 found in the common ancestor of rodents,even-toed ungulates and primates? We set about to answer thisquestion definitively. Firstly, we found weak blastn homology to thisexon (in a 38-nucleotide stretch) in all of the high-coverage primates ina precisely syntenic position for alignments of the PRNP intronic DNAto all of the known three-exon PRNPmessenger RNAs, with the higheste-value for rat [e()=5×10−7] (Suppl. Fig. 6).Noothermatches to othercomplete eukaryotic genomes (from any source) can be detected tosuch e-values. These suggestive homology hits prompted furtherinvestigation. So, to tackle the question of the evolution of NCE2definitively, we derived multiple sequence alignments (with theprogram MUSCLE [41]) of the introns of primate PRNPs, and theequivalent regions of other high-coverage mammal assemblies, includ-ing those that are known to make three-exon messenger RNAs. Weperformed a nucleotide sequence distance windowing analysis withDNADIST (see Methods for details) on the MSAs, to find out how wellconserved this region is relative to the rest of the primate PRNP intron.We observed that the 38-nucleotide NCE2 conserved element isamongst the 0.8-1.5% most conserved regions in primate intronicDNA, for comparisons of the intronic DNAs to the equivalent regions ofeach of the species with three-exon PRNP mRNAs. All of the windowswith smaller nucleotide sequence distance are b100 nt from the exonsplice sites. An example is shown for the human-mouse comparison inFig. 3C. We could not discern any small RNA motifs or RNA-secondarystructure for these conserved NCE1 and NCE2 elements, using the RNAzserver [http://rna.tbi.univie.ac.at/cgi-bin/RNAz.cgi]. Chimeric primatePRNP mRNAs containing inserted NCE2s from the three-exon speciescould not be mapped successfully (using either Spidey or GMAP[39,40]). Also, using GENSCAN [32], we could not find any strong splicesites around this homology in the primate introns.
In summary, these findings suggest that NCE2 was possibly at leaststill a component of PRNP mRNAs earlier in primate evolution, andmay have fallen out of use during primate speciation.
PRNTThe PRNT gene is the nearest gene to PRNP and PRND in humans,
and like PRNP and PRND, it has a two-exon structure [42], but has nodetectable sequence homology to them. It is 48% comprised oftransposon homologies (LINE-2 and MER elements) [42,43]. Inhuman, there are three alternately-spliced transcripts [42]; we tookthe longest transcript for further analysis. In contrast to previousreports [43–45], we find that the PRNT gene is widely conserved inmammals, including a partial unique homology in mouse in a regionderived from an ancient LINE-2 (Suppl. Fig. 7). Complete two-exonRNAs can bemapped in the high-coverage primate assemblies (chimp,orang utan, macaque). Likely two-exon RNA structures that align toN90% of human PRNT can also be predicted in cow, sheep, horse, dog(Suppl. Fig. 7). PRNT is also detectable in twelve low-coverage (b6X)mammal assemblies (i.e., in bushbaby, tarsier, cat, dolphin, elephant,hyrax, megabat, microbat, rabbit, squirrel, tenrec and treeshrew, withblastn e-value ≤1×10−10, with appropriate repeat-masking [46]),
and the PrP locus from the killer whale Pseudorca [44]. AnMSA of PRNTputative two-exon RNAs is illustrated (Suppl. Fig. 7). For this align-ment, we constructed RNA for primates frommapping PRNT onto theirgenomic sequences; for other species, the best exons were estimatedfrom blastn alignments and GENSCAN output.
We examinedwhether PRNT is a protein-coding gene or anmRNA-like long non-coding RNA (mlncRNA). There are hundreds ofmlncRNAs in mammals that have been shown to be conserved ingenomic DNA and have specific patterns of tissue expression [47].Well-documented cases includeH19, which regulates expression of itsneighbouring gene IGF2 during embryogenesis and can act as a tumoursuppressor [48], and XIST, which functions in X-chromosome silencingas part of heterogametic dosage compensation during development[49]. To assess PRNT for protein-coding ability, we analysed the fourORFs that are ≥40 codons, since, where there are multiple largetransposon insertions, the longest ORFsmay overlap a transposon, andmaynot be the true cellular coding sequences. (All of theseORFs donotstraddle the exon splice area.) In each case, we find that the ORFs areeither mutated away beyond detectability, or they have disablements(frameshifts and stop codons) in more than one mammal species(Table 2). Also, the patterns of Ka/Ks values [50], where calculable,indicate a general lack of purifying selection (Table 2). Finally, in thePRNT gene neighbourhood, the only homology to a knownprotein thatis not assignable to a transposon is a sequence in its intron. Thissequence is a likely processed pseudogene of isopentenyl-diphosphateisomerase, that has multiple coding-sequence disablements, and Ka/Ks (=1.3 for human-macaque comparison) indicating lack of protein-coding ability (Suppl. Fig. 8). This sequence is only found in primates.Thus, in summary, PRNT is unlikely to be a protein-coding gene, and ismore likely to be an mlncRNA.
Conclusions
As genome sequencing efforts expand, there is a pressing need forannotation efforts to keep track of biological sequence families ofwide research interest, such as the PrP gene family (PrP-GF). To thisend, we have annotated the PrP-GF in 42 complete eukaryotic genomeassemblies, uncovering new genes and pseudogenes that give usfurther insight into the evolutionary history of the PrP-GF. Thiscurrent genomic evidence indicates that the Doppel gene was likelypresent in the last common ancestor of present-day Tetrapoda, butwas lost in the bird lineage, since its divergence from reptiles.

275P.M. Harrison et al. / Genomics 95 (2010) 268–277
Furthermore, we describe in detail a transcribed pseudogene derivedfrom Shadoo that may interfere with transcription of SYCE1, amammalianmeiosis gene, since its overlapwith this gene is conservedacross multiple primates.
In addition, we analysed the locus of PRNP/PRND for significantconservation across the genomic DNA of eleven mammals, anddetermined the phylogenetic penetration of their non-coding exons.The genomic evidence indicates that the second PRNP non-codingexon found in even-toed ungulates and rodents, is conserved in allhigh-coverage genome assemblies of primates (human, chimp, orangutan and macaque), and thus, at least, may have fallen out of useduring primate speciation. Furthermore, we have demonstrated thatthe PRNT gene (proximal to PRNP/PRND) is conserved across at leastsixteen mammals, and is likely a mRNA-like long non-coding RNA.
Methods
Nucleotide sequence data
We obtained the following genome assembly versions in January2009, from the Ensembl website (ftp.ensembl.org): Anolis_carolinensis(AnoCar1.0.53); Bos_Taurus (Btau_4.0.53); Canis_familiaris(BROADD2.53); Cavia_porcellus (GUINEAPIG.53); Choloepus_hoffmanni(choHof1.53); Danio_rerio (ZFISH7.49); Dasypus_novemcinctus (dasNov2.53); Dipodomys_ordii (dipOrd1.53); Echinops_telfairi (TENREC.53); Equus_caballus (EquCab2.53); Erinaceus_europaeus (HEDGEHOG.53); Felis_catus (CAT.53); Gallus_gallus (WASHUC2.49); Gasterosteus_aculeatus (BROADS1.53); Gorilla_gorilla (gorGor1.53); Homo_sapiens (NCBI36.53); Loxodonta_Africana (loxAfr2.53); Macaca_mulatta(MMUL_1.53); Microcebus_murinus (micMur1.53); Monodelphis_domestica (BROADO5.53); Mus_musculus (NCBIM37.53); Myotis_ luci-fugus (MICROBAT1.53); Ochotona_princeps (pika.53); Ornithorhynchu-s_anatinus (OANA5.53); Oryctolagus_cuniculus (RABBIT.53);Oryzias_latipes (MEDAKA1.53); Otolemur_garnettii (BUSHBABY1.53);Pan_troglodytes (CHIMP2.1.53); Pongo_pygmaeus (PPYG2.53); Procavia_capensis (proCap1.53); Pteropus_vampyrus (pteVam1.53); Rattus_norvegicus (RGSC3.4.53); Sorex_araneus (COMMON_SHREW1.53);Spermophilus_tridecemlineatus (SQUIRREL.52); Taeniopygia_guttata(taeGut3.2.4.53); Takifugu_rubripes (FUGU4.53); Tarsius_syrichta (tarSyr1.53); Tetraodon_nigroviridis (TETRAODON8.53); Tupaia_belangeri(TREESHREW.53); Tursiops_truncates (turTru1.53); Vicugna_pacos (vicPac1.53); Xenopus_tropicalis (JGI4.1.53).
Additional PrP locus genomic sequence for Pseudorca false killerwhale [44] and for sheep [51], and all mRNA sequences examined inthis paper, were taken from the NCBI website.
Examining genomic DNA for undiscovered PrP gene family members
All known PrP, Doppel and Shadoo protein sequences were col-lated from the Uniprot and NCBI websites. These were then embeddedin the Swissprot protein database [52] (to make sure that there arenonemissing from Swissprot). Tomake a comprehensive set of profilematrices, each PrP, Doppel and Shadoo protein sequence was thensearched against this augmented Swissprot database using psiblast[22], for a range of values for j, the maximum number of iterations(j=3 up to j=11), with other parameters set to default. Then, thesematrices were used to run psi-tblastn, using the same protein querysequences [22], and e-value threshold=1×10−4. Psi-tblastn is a ver-sion of tblastn that uses a psi-blast-derived position-specific scoringmatrix to search in nucleotide sequences for protein homology [22]. Asmall number of proteins produced large numbers of false-positivematches to repetitive genomic DNA, with a non-phylogenetic patternof distribution; these were readily discarded through several roundsof visual curation.
For further sensitivity, we performed the following analysis on thehuman, chicken and zebra finch (Taeniopygia guttata) genomes.
Hidden Markov Models (HMMs) for the globular domains of PrP andDoppel were constructed using HMMER (http://hmmer.janelia.org/),and these HMMs were searched against six-frame translations of thethree genomes, using the default expectation value threshold of 10.0.All of these hits were then assessed for compatibility to the PrP andDoppel protein structures using pGenThreader [63] with defaultparameters, and curated visually for conserved disulphides bridges.However, we found no further PrP-GF homologs, using this furtherthorough technique.
For all significant novel protein homologies, gene structures werepredicted using FGenesH [23] and GeneWise [64]. Where appropriate,the program GENSCAN [32] was also used for ab initio inference ofexons and splice signals. Pseudogenes were annotated using previ-ously developed procedures, wherein initial BLAST hits in genomicDNA are grouped and extended to longer alignments using TFASTX/Y[25–28].
Protein-level analysis
Protein MSAs were made using CLUSTALW [53]. Signal peptides,transmembrane helices and glycosylations sites were annotated usingthe collated machine-learning programs on the CBS Prediction Serverwebsite [54]. For glycosylation sites, wewere stringent in that we onlylabeled N-glycosylation sequons that receive a neural-network juryvote of 9/9 and have a potential score ≥0.7 [55]. GPI anchors wereannotated using the big-PI server [56]. Comparative models of pro-teins were constructed using the most similar protein structure andthe programMODELLER, using default parameters [24]. Protein struc-tures were visualized using the SwissPDB-Viewer [57]. Disorderedregions were predicted using the consensus of the two methodsRONN [58] and PrDOS [59]. Compositionally-biased regions weredelimited using a published algorithm developed in our laboratory[29–31].
Genomic conservation analysis & nucleotide sequence distancecalculations
We performed a genomic conservation analysis centred on thehuman genome, as follows. Nucleotide sequence MSAs were con-structed using MUSCLE [41] (parameters -maxiters=1, -diags), afterinitial blastn pairwise alignments (with e-value threshold e=1×10−10
and default parameters, or the alternative parameter set, '-e 1×10−10
-r 1 -q -1 -E 1 -G 2'). MSAs of introns were made by aligning the intronsalong with the two exons from either end of the intron, and thendiscarding the parts of the alignments for the exons. These sequenceswere thendivided intowindows starting at eachbase position (windowlength=50ntwas found to be suitable). Nucleotide sequence distancesare dissimilarities between the nucleotide sequences (i.e., thenumber ofsubstitutions per site) that are calculated to allow for multiplesubstitutions at the same site in a sequence. The distances werecalculated for each window subalignment using DNADIST from thepackage PHYLIP, with default parameters [60], and distributions of thedistances were studied. This analysis was performed, where appropri-ate, for comparisons of human genomic DNA to genomic DNA of theeleven mammals listed above.
Codon substitution rates
The relative rates of substitution at non-synonymous and syn-onymous codon sites were calculated by using PAL2NAL [61] togenerate pairwise alignments, which were then used as input for thePAML package for pairwise Ka/Ks analysis, using default parameters[50].

276 P.M. Harrison et al. / Genomics 95 (2010) 268–277
Acknowledgments
This work was support by the PrioNet Canada Network of Centresof Excellence, and by les Fonds Quebecois de la Recherche sur la Natureet les Technologies.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.ygeno.2010.02.008.
References
[1] S.B. Prusiner, Prions, Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 13363–13383.[2] S.B. Prusiner, D. Groth, A. Serban, R. Koehler, D. Foster, M. Torchia, D. Burton, S.L.
Yang, S.J. DeArmond, Ablation of the prion protein (PrP) gene in mice preventsscrapie and facilitates production of anti-PrP antibodies, Proc. Natl. Acad. Sci. U. S. A.90 (1993) 10608–10612.
[3] D.G. Donne, J.H. Viles, D. Groth, I.Mehlhorn, T.L. James, F.E. Cohen, S.B. Prusiner, P.E.Wright, H.J. Dyson, Structure of the recombinant full-length hamster prion proteinPrP(29-231): the N terminus is highly flexible, Proc. Natl. Acad. Sci. U. S. A. 94(1997) 13452–13457.
[4] R. Riek, S. Hornemann, G. Wider, M. Billeter, R. Glockshuber, K. Wuthrich, NMRstructure of the mouse prion protein domain PrP(121-321), Nature 382 (1996)180–182.
[5] K. Pan, M. Baldwin, J. Nguyen, M. Gasset, A. Serban, D. Groth, I. Mehlhorn, Z.Huang, R.J. Fletterick, F.E. Cohen, S.B. Prusiner, Conversion of alpha-helices intobeta-sheets features in the formation of the scrapie prion proteins, Proc. Natl.Acad. Sci. U. S. A. 90 (1993) 10962–10966.
[6] G.C. Telling, P. Parchi, S.J. DeArmond, P. Cortelli, P. Montagna, R. Gabizon, J.Mastrianni, E. Lugaresi, P. Gambetti, S.B. Prusiner, Evidence for the conformationof the pathologic isoform of the prion protein enciphering and propagating priondiversity, Science 274 (1996) 2079–2082.
[7] J. Stockel, J. Safar, A.C. Wallace, F.E. Cohen, S.B. Prusiner, Prion protein selectivelybinds copper(II) ions, Biochemistry 37 (1998) 7185–7193.
[8] W. Hu, B. Kieseier, E. Frohman, T.N. Eagar, R.N. Rosenberg, H.P. Hartung, O. Stuve,Prion proteins: Physiological functions and role in neurological disorders, J.Neurol. Sci. 264 (2008) 1–8.
[9] A. Santuccione, V. Sytnyk, I. Leshchyns'ka, M. Schachner, Prion protein recruits itsneuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neuriteoutgrowth, J. Cell Biol. 169 (2005) 341–354.
[10] M. DeMarco, J. Silveira, B. Caughey, V. Daggett, Structural properties of prionprotein protofibrils and fibrils, Biochemistry 45 (2006) 15573–15582.
[11] J. Watzlawik, L. Skora, D. Frense, C. Griesinger, M. Zweckstetter, W. Schulz-Schaeffer, M. Kramer, Prion protein helix1 promotes aggregation but is notconverted into -sheet, J. Biol. Chem. 281 (2006) 30242–30250.
[12] H. Wille, M.D. Michelitsch, V. Guenebaut, S. Supattapone, A. Serban, F.E. Cohen, D.A.Agard, S.B. Prusiner, Structural studies of the scrapie prion protein by electroncrystallography, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 3563–3568.
[13] X. Lu, P. Wintrode, W. Surewicz, Beta-sheet core of human prion protein amyloidfibrils as determined by hydrogen/deuterium exchange, PNAS 104 (2007)1510–1515.
[14] H.M. Schatzl, M. Da Costa, L. Taylor, F.E. Cohen, S.B. Prusiner, Prion protein genevariation among primates, J. Mol. Biol. 245 (1995) 362–374.
[15] F. Wopfner, G. Weidenhofer, R. Schneider, A. von Brunn, S. Gilch, T.F. Schwarz, T.Werner, H.M. Schatzl, Analysis of 27 mammalian and 9 avian PrPs reveals highconservation of flexible regions of the prion protein, J. Mol. Biol. 289 (1999)1163–1178.
[16] L.G. Goldfarb, P. Brown,W.R. McCombie, D. Goldgaber, G.D. Swergold, P.R. Wills, L.Cervenakova, H. Baron, C.J. Gibbs Jr., D.C. Gajdusek, Transmissible familialCreutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptidecoding repeats in the PRNP gene, Proc. Natl. Acad. Sci. U. S. A. 88 (1991)10926–10930.
[17] http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=176640.[18] R. Moore, I. Lee, G. Silverman, P. Harrison, R. Strome, C. Heinrich, A. Karunaratne, S.
Pasternak, M. Chishti, Y. Liang, P. Mastrangelo, K. Wang, A. Smit, S. Katamine, G.Carlson, F. Cohen, S. Prusiner, D. Melton, P. Tremblay, L. Hood, D. Westaway,Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of thenovel PrP-like protein doppel, J. Mol. Biol. 292 (1997) 797–817.
[19] R.S. Hegde, J.A. Mastrianni, M.R. Scott, K.A. DeFea, P. Tremblay, M. Torchia, S.J.DeArmond, S.B. Prusiner, V.R. Lingappa, A transmembrane form of the prionprotein in neurodegenerative disease, Science 279 (1998) 827–834.
[20] J. Watts, B. Drisaldi, V. Ng, J. Yang, B. Strome, P. Horne, M. Sy, L. Yoong, R. Young, P.Mastrangelo, C. Bergeron, P. Fraser, G. Carlson, H. Mount, G. Schmitt-Ulms,W.D., TheCNS glycoprotein Shadoo has PrP(C)-like protective properties and displays reducedlevels in prion infections, EMBO J. 26 (2007) 4038–4050.
[21] E. Rivera-Milla, C. Stuermer, E. Malaga-Trillo, An evolutionary basis for scrapiedisease: identification of a fish prion mRNA, Trends Genet. 19 (2003) 72–75.
[22] S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller, D.J. Lipman,Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms, Nucleic Acids Res. 25 (1997) 3389–3402.
[23] http://linux1.softberry.com/berry.phtml. [ http://linux1.softberry.com/berry.phtml ].
[24] A. Sali, T.L. Blundell, Comparative protein modelling by satisfaction of spatialrestraints, J. Mol. Biol. 234 (1993) 779–815.
[25] P. Harrison, M. Gerstein, Studying genomes through the aeons: protein families,pseudogenes and proteome evolution, J. Mol. Biol. 318 (2002) 1155–1174.
[26] P.M. Harrison, H. Hegyi, S. Balasubramanian, N.M. Luscombe, P. Bertone, N. Echols,T. Johnson, M. Gerstein, Molecular fossils in the human genome: identificationand analysis of the pseudogenes in chromosomes 21 and 22, Genome Res. 12(2002) 272–280.
[27] Z. Zhang, P. Harrison, Y. Liu, M. Gerstein, Millions of years of evolution preserved:a comprehensive catalog of the processed pseudogenes in the human genome,Genome Res. 13 (2003) 2541–2558.
[28] Z.L. Zhang, P.M. Harrison, M. Gerstein, Digging deep for ancient relics: a survey ofprotein motifs in the intergenic sequences of four eukaryotic genomes, J. Mol. Biol.323 (2002) 811–822.
[29] P.M. Harrison, Exhaustive assignment of compositional bias reveals universallyprevalent biased regions: analysis of functional associations in human andDrosophila, BMC Bioinformatics 7 (2006) 441.
[30] P. Harrison, M. Gerstein, A method to assess compositional bias in biologicalsequences and its application to prion-like glutamine/asparagine -rich domains ineukaryotic proteomes, Genome Biol. 4 (2003) R40.
[31] L. Harrison, Z. Yu, J. Stajich, F. Dietrich, P. Harrison, Evolution of budding yeast prion-determinant sequences across diverse fungi, J. Mol. Biol. 368 (2007) 273–282.
[32] C. Burge, S. Karlin, Prediction of complete gene structures in human genomic DNA,J. Mol. Biol. 268 (1997) 78–94.
[33] R.I. Dima, D. Thirumalai, Exploring the propensities of helices in PrP(C) to formbeta sheet using NMR structures and sequence alignments, Biophys. J. 83 (2002)1268–1280.
[34] Y. Costa, R. Speed, R. Ollinger, M. Alsheimer, C.A. Semple, P. Gautier, K. Maratou, I.Novak, C. Hoog, R. Benavente, H.J. Cooke, Two novel proteins recruited bysynaptonemal complex protein 1 (SYCP1) are at the centre of meiosis, J. Cell Sci.118 (2005) 2755–2762.
[35] J. Park, S.A. Teichmann, T. Hubbard, C. Chothia, Intermediate sequences increasethe detection of homology between sequences, J. Mol. Biol. 273 (1997) 349–354.
[36] C.J. Gibbs Jr., D.M. Asher, A. Kobrine, H.L. Amyx, M.P. Sulima, D.C. Gajdusek,Transmission of Creutzfeldt-Jakob disease to a chimpanzee by electrodescontaminated during neurosurgery, J. Neurol. Neurosurg. Psychiatry 57 (1994)757–758.
[37] M.B. Gardner, P.A. Luciw, Macaque models of human infectious disease, Ilar J. 49(2008) 220–255.
[38] N. Jorn, D. Hansen, N.J. Hansen, Human spongiform encephalopathies. Diseasescaused by prions, Ugeskr Laeger 158 (1996) 4066–4072.
[39] S.J. Wheelan, D.M. Church, J.M. Ostell, Spidey: a tool for mRNA-to-genomicalignments, Genome Res. 11 (2001) 1952–1957.
[40] T.D. Wu, C.K. Watanabe, GMAP: a genomic mapping and alignment program formRNA and EST sequences, Bioinformatics 21 (2005) 1859–1875.
[41] R.C. Edgar, MUSCLE: a multiple sequence alignment method with reduced timeand space complexity, BMC Bioinformatics 5 (2004) 113.
[42] E. Makrinou, J. Collinge, M. Antoniou, Genomic characterization of the humanprion protein (PrP) gene locus, Mamm. Genome 13 (2002) 696–703.
[43] M. Premzl, V. Gamulin, Comparative genomic analysis of prion genes, BMCGenomics 8 (2007) 1.
[44] D.W. Kim, S.H. Chae, B.R. Kang, S.H. Choi, A. Kim, S. Woo, H.S. Park, Comparativegenomic analysis of the whale (Pseudorca crassidens) PRNP locus, Genome 51(2008) 452–464.
[45] S.H. Choi, I.C. Kim, D.S. Kim, D.W. Kim, S.H. Chae, H.H. Choi, I. Choi, J.S. Yeo, M.N.Song, H.S. Park, Comparative genomic organization of the human and bovinePRNP locus, Genomics 87 (2006) 598–607.
[46] Repeatmasker. [ http://www.repeatmasker.org ].[47] T.R. Mercer, M.E. Dinger, S.M. Sunkin, M.F. Mehler, J.S. Mattick, Specific expression
of long noncoding RNAs in the mouse brain, Proc. Natl. Acad. Sci. U. S. A. 105(2008) 716–721.
[48] A. Gabory, M.A. Ripoche, T. Yoshimizu, L. Dandolo, The H19 gene: regulation andfunction of a non-coding RNA, Cytogenet. Genome Res. 113 (2006) 188–193.
[49] G. Kay, S. Barton, M. Surani, S. Rastan, Imprinting and X chromosome countingmechanisms determine Xist expression in early mouse development, Cell 77 (1994)639–650.
[50] Z. Yang, PAML: a program package for phylogenetic analysis by maximumlikelihood, Comput. Appl. Biosci. 13 (1997) 555–556.
[51] I.Y. Lee, D. Westaway, A.F. Smit, K. Wang, J. Seto, L. Chen, C. Acharya, M. Ankener,D. Baskin, C. Cooper, H. Yao, S.B. Prusiner, L.E. Hood, Complete genomic sequenceand analysis of the prion protein gene region from three mammalian species,Genome Res. 8 (1998) 1022–1037.
[52] R. Apweiler, A. Bairoch, C. Wu, W. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H.Huang, R. Lopez, M. Magrane, C. O'Donovan, N. Redaschi, L. Yeh, UniProt: theUniversal Protein knowledgebase, Nucleic Acids Res. 32 (2004) D115–D119.
[53] J. Thompson, D. Higgins, T. Gibson, CLUSTAL W: improving the sensitivity ofprogressive multiple sequence alignment through sequence weighting, positionspecific gap penalties and weight matrix choice, Nucleic Acids Res. 22 (1994)4673–4680.
[54] http://www.cbs.dtu.dk/services/. [http://www.cbs.dtu.dk/services/].[55] J.D. Bendtsen, H. Nielsen, G. von Heijne, S. Brunak, Improved prediction of signal
peptides: SignalP 3.0, J. Mol. Biol. 340 (2004) 783–795.[56] F. Eisenhaber, B. Eisenhaber, W. Kubina, S. Maurer-Stroh, G. Neuberger, G.
Schneider, M. Wildpaner, Prediction of lipid posttranslational modifications and

277P.M. Harrison et al. / Genomics 95 (2010) 268–277
localization signals from protein sequences: big-Pi, NMT and PTS1, Nucleic AcidsRes. 31 (2003) 3631–3634.
[57] http://spdbv.vital-it.ch/. [http://spdbv.vital-it.ch/].[58] Z.R. Yang, R. Thomson, P. McNeil, R.M. Esnouf, RONN: the bio-basis function neural
network technique applied to the detection of natively disordered regions inproteins, Bioinformatics 21 (2005) 3369–3376.
[59] T. Ishida, K. Kinoshita, PrDOS: prediction of disordered protein regions fromamino acid sequence, Nucleic Acids Res. 35 (2007) W460–W464.
[60] J. Felsenstein, PHYLIP – Phylogeny Inference Package (Version 3.2), Cladistics 5(1989) 164–166.
[61] M. Suyama, D. Torrents, P. Bork, PAL2NAL: robust conversion of protein sequencealignments into the corresponding codon alignments, Nucleic Acids Res. 34(2006) W609–W612.
[62] W.R. Pearson, Flexible sequence similarity searching with the FASTA3 programpackage, Methods Mol. Biol. 132 (2000) 185–219.
[63] A. Lobley, M.I. Sadowski, D.T. Jones, pGenTHREADER and pDomTHREADER: newmethods for improved protein fold recognition and superfamily discrimination,Bioinformatics 25 (14) (2009) 1761–1767.
[64] E. Birney, M. Clamp, R. Durbin, GeneWise and Genomewise, Genome Res. 14 (5)(2004) 988–995.
Related Documents











