1 Ioana Borze Department of Pathology Haartman Institute and HUSLAB University of Helsinki and Helsinki University Central Hospital Academic Dissertation To be publicly presented, with the permission of the Faculty of Medicine, University of Helsinki, for public discussion in the large lecture hall of the Haartman Institute on October 7 th , at 12:00 noon. Helsinki 2011

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
Ioana Borze
Department of Pathology
Haartman Institute and HUSLAB
University of Helsinki and Helsinki University Central Hospital
Academic Dissertation
To be publicly presented, with the permission of the Faculty of Medicine, University of
Helsinki, for public discussion in the large lecture hall of the Haartman Institute on
October 7th, at 12:00 noon.
Helsinki 2011

2
Supervised by
Professor Sakari Knuutila, PhD
Department of Pathology
Haartman Institute and HUSLAB
University of Helsinki and
Helsinki University Central Hospital
Helsinki, Finland
Reviewed by:
Docent Freja Ebeling, MD, PhD
Department of Medicine, Division of Haematology
Helsinki University Central Hospital
Helsinki, Finland
Docent Heli Nevanlinna, PhD
Department of Obstetrics and Gynecology
Helsinki University Central Hospital
Helsinki, Finland
Official Opponent:
Docent Maija Itälä-Remes, MD, PhD
Department of Medicine, Division of Hematological Diseases
Turku University Central Hospital
Turku, Finland
ISBN 978-952-10-7182-9 (paperback)
ISBN 978-952-10-7183-6 (PDF)
http://ethesis.helsinki.fi/
Helsinki 2011
Yliopistopaino

3
CONTENTS
LIST OF ORIGINAL PUBLICATIONS ............................................................................. 5
ABBREVIATIONS ................................................................................................................ 6
INTRODUCTION................................................................................................................ 10
REVIEW OF THE LITERATURE ................................................................................... 11
Normal hematopoiesis ...................................................................................................................... 11
Myeloid neoplasms .......................................................................................................................... 13
Myeloproliferative neoplasms .......................................................................................................... 15
Myelodysplastic syndromes ............................................................................................................. 22
Methods for studying genetic alteration in hematological malignancies ......................................... 26
MicroRNA expression profiling ....................................................................................................... 29
AIMS OF THE STUDY....................................................................................................... 31
MATERIALS ....................................................................................................................... 32
Patients ............................................................................................................................................. 32
Sample preparation ........................................................................................................................... 32
Myeloproliferative neoplasms (I-II) ................................................................................................. 33
Leukemia samples (III) .................................................................................................................... 34
Myelodysplastic syndromes (IV, V) ................................................................................................ 34
Ethical Permission ............................................................................................................................ 34
METHODS ........................................................................................................................... 35
Nucleic acid extraction ..................................................................................................................... 35
Mutation analysis (I, II) .................................................................................................................... 35
Methylation-specific PCR (IV) ........................................................................................................ 36
In vitro cultures of hematopoietic progenitors (I, II) ........................................................................ 36
Oligonucleotide-based array comparative genomic hybridization (II, IV) ...................................... 36
MicroRNA array (III, V) .................................................................................................................. 37
RESULTS AND DISCUSSION .......................................................................................... 39

4
Association of the JAK2V617F mutation status and the in vitro growth of hematopoietic
progenitors (I) ................................................................................................................................... 39
Copy number alterations in ET and PV (II) ..................................................................................... 40
Core biopsy paraffin-embedded core bone marrow biopsy samples as a reliable source of miRNA
(III) ................................................................................................................................................... 42
Copy number alterations in MDS (IV) ............................................................................................. 44
MicroRNA expression profiling in MDS (V) .................................................................................. 48
CONCLUSIONS .................................................................................................................. 52
ACKNOWLEDGEMENTS ................................................................................................ 55
REFERENCES ..................................................................................................................... 56

5
LIST OF ORIGINAL PUBLICATIONS This thesis is based on the following original publications, which are referred to in the text by their Roman numerals.
I Mustjoki S, Borze I, Lasho TL, Alitalo R, Pardanani A, Knuutila S, Juvonen E. JAK2V617F mutation and spontaneous megakaryocytic or erythroid colony formation in patients with essential thrombocythaemia (ET) or polycythaemia vera (PV). Leuk Res 2009,33:54-59.
II Borze I, Mustjoki S, Juvonen E, Knuutila S. Oligoarray comparative genomic
hybridization in polycythemia vera and essential thrombocythemia. Haematologica 2008,93:1098-1100 (Letter).
III Borze I*, Guled M*, Musse S, Raunio A, Elonen E, Saarinen-Pihkala U,
Karjalainen-Lindsberg ML, Lahti L, Knuutila S: MicroRNA microarrays on archive bone marrow core biopsies of leukemias – method validation. Leuk Res 2011;35:188-195.
IV Borze I, Juvonen E, Ninomiya S, Jee KJ, Elonen E, Knuutila S. High-resolution
oligonucleotide array comparative genomic hybridization study and methylation status of the RPS14 gene in de novo myelodysplastic syndromes. Cancer Genet Cytogenet 2010,197:166-173.
V Borze I, Scheinin I, Siitonen S, Elonen E, Juvonen E, Knuutila S. miRNA
expression profiles in myelodysplastic syndromes reveal Epstein-Barr virus miR-BART13 dysregulation. Leuk Lymphoma 2011,52:1567-1573.
*These authors contributed equally to the study. These original publications are reprinted with the permission of their copyright holders.

6
ABBREVIATIONS ALL acute lymphoblastic leukemia
AML acute myeloid leukemia
ASXL1 additional sex combs like 1
array CGH array comparative genomic hybridization
B blood
BA bone marrow aspirate
BAC bacterial artificial chromosome
BCR breakpoint cluster region gene
BFU-E erythroid burst-forming unit
BM bone marrow
bp base pair
CB core biopsy
CBL Casitas B-lineage Lymphoma
CML chronic myelogenous (or myeloid) leukemia
CNA copy number alterations
CSF colony-stimulating factor
CFU-Meg megakaryocyte colony-forming unit
CGH comparative genomic hybridization
CTNNA1 catenin (cadherin-associated protein), alpha 1
DNA deoxyribonucleic acid
EGR1 early growth response 1
ET essential thrombocythemia
EZH2 enhancer of zeste homolog 2
FGFR1 fibroblast growth factor receptor 1
FF fresh frozen (tissue)
FFPE formalin-fixed, paraffin-embedded (tissue)
FISH fluorescent in situ hybridization
HSPA9 heat shock 70kda protein 9 (mortalin)
i isochromosome
IL interleukin
IDH1 and IDH2 isocitrate dehydrogenase 1 and 2

7
IKZF1 IKAROS family zinc finger 1
Mb megabase (pair)
MDS myelodysplastic syndromes
miRNA micro-ribonucleic acid
MPD myeloproliferative diseases
MPL myeloproliferative leukemia virus oncogene
MPN myeloproliferative neoplasm
mRNA messenger RNA
MSPCR methylation-specific polymerase chain reaction
nt nucleotide
p short arm of the chromosome
PCR polymerase chain reaction
PDGFRA, PDGFRB platelet-derived growth factor receptors alpha and beta
PMF primary myelofibrosis
PV polycythemia vera
q long arm of the chromosome
RISC RNA-induced silencing complex
RNA ribonucleic acid
RMA robust multichip average
RUNX1 runt-related transcription factor 1
STAT signal transducer and activator of transcription
TET2 ten-eleven translocation 2 gene
TP53 tumor protein 53
TPO thrombopoietin
WHO World Health Organization
Any gene symbols not listed here can be found at hhttp://www.ncbi.nml.nih.gov/

8
ABSTRACT
Myeloproliferative neoplasms (MPN) and myelodysplastic syndromes (MDS) are a
heterogeneous group of clonal hematopoietic disorders whose etiology and molecular
pathogenesis are poorly understood. During the past decade, enormous developments in
microarray technology and bioinformatics methods have made it possible to mine novel
molecular alterations in a large number of malignancies, including MPN and MDS,
which has facilitated the detection of new prognostic, predictive and therapeutic
biomarkers for disease stratification.
By applying novel microarray techniques, we profiled copy number
alterations and microRNA (miRNA) expression changes in bone marrow aspirate and
blood samples. In addition, we set up and validated an miRNA expression test for bone
marrow core biopsies in order to utilize the large archive material available in many
laboratories. We also tested JAK2 mutation status and compare it with the in vitro growth
pattern of hematologic progenitors cells.
In the study focusing on 100 MPN cases, we detected a Janus kinase 2 (JAK2)
mutation in 71 cases. We observed spontaneous erythroid colony growth in all mutation-
positive cases in addition to nine mutation negative cases. Interestingly, seven
JAK2V167F negative ET cases showed spontaneous megakaryocyte colony formation,
one case of which also harbored a myeloproliferative leukemia virus oncogene (MPL)
mutation.
We studied copy number alterations in 35 MPN and 37 MDS cases by using
oligonucleotide-based array comparative hybridization (array CGH). Only one essential
thrombocythemia (ET) case presented copy number alterations in chromosomes 1q and

9
13q. In contrast, MDS cases were characterized by numerous novel cryptic chromosomal
aberrations with the most common copy number losses at 5q21.3q33.1 and 7q22.1q33,
while the most common copy number gain was trisomy 8.
As for the study of the bone marrow core biopsy samples, we showed that
even though these samples were embedded in paraffin and underwent decalcification,
they were reliable sources of miRNA and suitable for array expression analysis.
Further, when studying the miRNA expression profiles of the 19 MDS cases,
we found that, compared to controls, two miRNAs (one human Epstein-Barr virus (miR-
BART13) miRNA and one human (has-miR-671-5p) miRNA) were downregulated,
whereas two other miRNAs (hsa-miR-720 and hsa-miR-21) were upregulated. However,
we could find no correlation between copy number alterations and microRNA expression
when integrating these two data.
This thesis brings to light new information about genomic changes implicated
in the development of MPN and MDS, and also underlines the power of applying
genome-wide array screening techniques in neoplasias. Rapid advances in molecular
techniques and the integration of different genomic data will enable the discovery of the
biological contexts of many complex disorders, including myeloid neoplasias.

10
INTRODUCTION
Myeloid neoplasias are defined as clonal hematological diseases with abnormal maturation
and proliferation of myeloid cell lineages in bone marrow. Genomic aberrations are
considered to be present in all myeloid neoplasms, and we now know that genetic changes
are involved in leukemogenesis as well as in neoplastic progression. As for the diagnosis
of hematological neoplasias, molecular genetics techniques are now routinely used in
laboratories to reveal genetic alterations that have proved to be good biomarkers (Cotta &
Tubbs 2008).
Neoplasia is a complex phenomenon involving interaction between different
genes and environmental factors. Although cytogenetic and molecular genetics techniques
have revealed a considerable number of chromosomal and gene defects, the target genes
for many malignancies, including many of the myeloid neoplasm, remain unknown.
Microarray technology has recently yielded a detailed analysis of the entire
genome in an experiment that sought copy number alterations (array comparative genomic
hybridization) or expression profiling (gene or microRNA expression profiling). Collecting
and combining data from cytogenetic and genome-wide molecular analyses provides a
more detailed understanding of the pathogenesis and progression of a particular
malignancy/disease.
This thesis focused on the genetic analysis of two groups of myeloid neoplasms
- myeloproliferative neoplasms (MPN) and myelodysplastic syndromes (MDS) – as well as
on microRNA profiling (in MDS) by applying microarray techniques. The purpose of these
techniques was not only to detect copy number alterations and miRNA expression profiling,
but also to report the feasibility of the archival paraffin-embedded core biopsy samples for
microRNA profiling.

11
REVIEW OF THE LITERATURE
Normal hematopoiesis
Hematopoiesis is the formation of blood cells where the bone marrow is the main organ
(Hoffbrand et al. 2006). All blood cells derive from pluripotent hematopoietic stem cells
that have the capacity to self-renew and also give rise to myeloid and lymphoid cell
lineages (Figure 1).
The proliferation, differentiation and maturation of hematopoietic progenitor
cells are regulated by various hematopoietic growth factors, in addition to
microenvironment and the interactions between the cells. Growth factors influence
transcription factors and implicit associated genes that determine the differentiation and
function of mature blood cells.
The hematopoietic growth factors include a group of cytokines, which
comprise colony stimulating factors (CSFs), hormones, interleukins (IL) and glycoproteins
(Wadhwa & Thorpe 2008). Hematopoietic growth factors have been the subject of
extensive study in the past two decades and the biological role of most of them has been
elucidated. Some act on early progenitor cells and lead to multilineage differentiation,
whereas others act on late differentiated cells; in addition, some factors are lineage specific,
whereas others have a broad lineage action.
Erythropoietin (EPO) is a hormone-like glycoprotein with the most lineage-
specific activity that promotes the maturation and proliferation of erythroid cell lineages
(by interacting with the receptors on erythroid burst-forming units and erythroid colony-
forming units) (Bociek & Armitage 1996).

12
Thrombopoietin (TPO), known as megakaryocyte growth and development
factor, specifically regulates the production and differentiation of megakaryocytes, which
also produce platelets. Beside the primary action in megakaryocytopoiesis, TPO also plays
an important role in the growth and survival of hematopoietic stem cells (Bociek &
Armitage 1996).
The CSFs include granulocyte-macrophage colony-stimulating factor (GM-
CSF), granulocyte colony-stimulating factor (G-CSF) and macrophage colony-
stimulating factor (M-CSF). GM-CSF stimulates the production of neutrophils,
eosinophils, basophils, monocytes and their progenitor cells. G-CSF stimulates the
production of granulocytes, and M-CSF stimulates the production of macrophages
(Wadhwa & Thorpe 2008).
Interleukins (IL) are secreted by different types of body cells, regulate the
immune system and participate in the development and differentiation of hematopoietic
cells.

13
Figure 1. Hematopoiesis (Figure 1 has been modified from Wadhwa & Thorpe 2008).
Myeloid neoplasms
Hematological malignancies stem from disordered maturation and often uncontrolled
proliferation of hematopoietic cells due to dysregulation in their proliferation and life span.
According to cell lineage, hematologic malignancies can be classified into two major
groups: myeloid and lymphoid malignancies. Hematological malignancies represent
approximately 9% of all new cancer diagnoses.
In compliance with the 2008 World Health Organization (WHO) classification,
myeloid neoplasms include: myeloproliferative neoplasms (MPN) (previously known as

14
myeloproliferative diseases, MPDs), myeloid and lymphoid neoplasms associated with
eosinophilia and abnormalities of platelet-derived growth factor receptors alpha and beta
(PDGFRA, PDGFRB) or fibroblast growth factor receptor (FGFR1),
myelodysplastic/myeloproliferative neoplasms (MDS/MPN), myelodysplastic syndromes
(MDS) and acute myeloid leukemia (AML), and related precursor neoplasms (Table 1).
Table 1. The 2008 World Health Organization classification for myeloid neoplasms. Data modified from Tefferi & Vardiman (2008).
1. Myeloproliferative neoplasms (MPN) 1.1 Chronic myelogenous leukemia 1.2 Polycythemia vera 1.3 Essential thrombocythemia 1.4 Primary myelofibrosis 1.5 Chronic neutrophilic leukemia 1.6 Chronic eosinophilic leukemia, not otherwise categorized 1.7 Hypereosinophilic syndrome 1.8 Mast cell disease 1.9 MPN, unclassifiable
2. Myeloid neoplasms associated with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1
2.1 Myeloid neoplasms associated with PDGFRA rearrangement 2.2 Myeloid neoplasms associated with PDGFRB rearrangement 2.3 Myeloid neoplasms associated with FGFR1 rearrangement
3. Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) 3.1 Chronic myelomonocytic leukemia 3.2 Juvenile myelomonocytic leukemia 3.3 Atypical chronic myeloid leukemia 3.4 MDS/MPN, unclassifiable
4. Myelodysplastic syndromes (MDS) 5. Acute myeloid leukemia (AML)

15
Myeloproliferative neoplasms
Myeloproliferative neoplasms are characterized by the clonal expansion of one or more
myeloid cell lineages (erythroid, granulocytic, megakaryocytic, monocyte/macrophage, or
mast cell) (Verstovsek & Tefferi 2011). Polycythemia vera (PV), essential
thrombocythemia (ET) and primary myelofibrosis (PMF), along with chronic myeloid
leukemia (CML), belong to classic myeloproliferative neoplasms (MPN) (Table 2).
Table 2. Classification of the classic MPN.
1. BCR-ABL-positive
2. BCR-ABL-negative Chronic myeloid leukemia (CML)
a. Polycythemia vera (~90-95% JAK2V617F+) b. Essential thrombocythemia (~50% JAK2V617F+) c. Myelofibrosis (~50% JAK2V617F+)
Data based on Tefferi (2006) and Klco et al. (2010).
MPN affect adults in their fifth to seventh decade of life. The etiology of these
diseases is incompletely known, and their diagnosis can be challenging and sometimes
based on exclusion criteria. However, correct diagnosis and appropriate treatment are
crucial for the clinical course of these diseases since, even if their onset is insidious, any
MPN entity can progress to bone marrow failure and/or evolve into AML.
A hallmark of the classic BCR-ABL-negative MPN is the in vitro formation of
an endogenous colonies in the absence of exogenous hematopoietic growth factors (Prchal
& Axelrad 1974). MPN are frequently characterized by dysregulated tyrosine kinases,
within the BCR-ABL-negative group in particular the dysregulated Janus kinase 2 (JAK2)
gene, which seems to indicate that kinase activation is involved in the etiology of these
diseases. A typical example of this dysregulation is CML, where a unique chromosomal
rearrangement (the Philadelphia chromosome resulting from a reciprocal translocation
between chromosomes 9 and 22) leads to constitutive activation of BCR-ABL tyrosine
kinase.

16
The key feature of PV is persistent erythrocytosis (Hoffbrand et al. 2006), often
accompanied by the increased production of granulocytes and megakaryocytes. According
to the 2008 WHO classification, the diagnosis of PV requires either two major criteria and
one minor criterion or the first major criterion and 2 minor criteria (Table 3) (Thiele et al.
2008a). The clinical picture of PV is a direct consequence of elevated number of red cell
mass and is mostly characterized by hyperviscosity of the blood, thus predisposing patients
to venous and arterial thrombosis. Patients may present with neurological symptoms such
as headache, visual disturbances, paresthesias as well as pruritus after bathing. Physical
examination reveals that as many as 70% of patients have an enlarged spleen, while 40%
of patients present with hepatomegaly (Spivak 2002; McMullin et al. 2005). Sometimes,
the disease is found after routine laboratory blood tests with an elevated hemoglobin and/or
hematocrit levels.
ET is characterized by a persistently elevated number of platelets. Most ET
patients are asymptomatic, but a few clinically present with thrombosis or hemorrhage
often with neurological symptoms. The diagnosis of ET requires meeting of all four criteria
(Table 3) (Thiele et al. 2008b).
As for PMF, abnormalities in blood cell production due to bone marrow
fibrosis are typical. At diagnosis, most PMF patients are asymptomatic, but physical
examination can reveal an enlarged spleen in 80% of patients. In addition, patients may
present with some non-specific symptoms such as fatigue, weight loss and night sweats
(Ansell 2008). PMF diagnosis requires meeting all three major and two minor criteria
(Table 3) (Thiele et al. 2008c).

17
Table 3. WHO 2008 diagnosis criteria of PV, ET and PMF.
PV ET PMF Major criteria
1. Hemoglobin values should be >18.5g/dL (men) and >16.5g/dL (women) or elevated red cell mass >25% above normal value.
2. Presence of JAK2V617F mutation or other similar mutation.
1. Platelet count ≥450x109/L.
2. Bone marrow biopsy showing megakaryocyte proliferation with large and mature morphology.
3. Not meeting WHO diagnostic criteria for PV, PMF, CML, MDS or other MPN.
4. Presents of JAK2V617F mutation or other clonal marker, or no evidence of reactive thrombocytosis in the absence of JAK2V167F.
1. Megakaryocyte proliferation and atypia associated either with reticulin and/or collagen fibrosis, or in the absence of reticulin fibrosis, the megakaryocyte changes must be associated with increased bone marrow cellularity, granulocytic proliferation and often decreased erythropoiesis (i.e., prefibrotic disease).
2. Not meeting WHO diagnostic criteria for PV, CML, MDS or other MPN.
3. Presents of JAK2V617For other clonal markers or in the absents of these no evidence of reactive bone marrow fibrosis.
Minor criteria
1. Hypercellular bone marrow with prominent erythroid, granulocytic and megakaryocytic proliferation.
2. Serum erythropoietin below normal levels.
3. In vitro erythroid colony formation.
1. Leukoerythroblastosis.
2. Increased serum lactate dehydrogenase level.
3. Anemia.
4. Splenomegaly.
Data has been modified from Tefferi et al. (2009c).

18
Spontaneous colony formation (in the absence of added cytokines/growth
factors) of hematopoietic progenitor cell cultures in vitro is a key feature of MPN. Almost
three decades ago erythroid progenitor cells from PV patients, lacking erythropoietin in
vitro, were observed to form endogenous erythroid colonies (EECs), unlike progenitor cells
from secondary erythrocytosis patients or normal healthy controls (Zanjani et al. 1977).
Erythroid and megakaryocyte colony formations have proven to be quite useful in
diagnostics (Juvonen et al. 1993; Kralovics et al. 2005). Endogenous erythroid colony
formation is currently a minor diagnostic criterion of PV according to the WHO 2008
classification. Initially, EEC formation was considered an effective and specific diagnostic
tool mainly for PV (Mossuz & Groupe d'Etudes Multicentriques des Syndrome
MyeloProliferatifs 2006), but spontaneous EEC formation was later also observed in ET and
PMF. Similarly, reports indicated that some PV patients also presented with endogenous
megakaryocyte colony (EMC) formation in the absence of exogenous growth factors, even
if this feature was thought to be specific to ET (Westwood & Pearson 1996). Hibbin et al.
(1984) has found increased number of circulating BFU-E, granulocyte-macrophage
progenitor (CFU-GM) and megakaryocyte progenitors cells (CFU-Meg) in PMF patients
compared with controls. In the absence of specific diagnostic markers for MPN, the culture
assay, although it is laborious and not widely accessible, has served for several decades, but
has nowadays been largely replaced by the Janus kinase 2 (JAK2) mutation assay.
The description of the JAK2 and myeloproliferative leukemia virus oncogene
(MPL) mutations in these disorders opened a new chapter in our understanding of the
pathogenesis and our search for diagnostic and prognostic markers of these disorders. Both
JAK2 and MPL mutations confer hypersensitivity to cytokines and the independent in vitro
colony formation of hematopoietic cells in cases of MPN (Bruchova et al. 2008; Passamonti
et al. 2010; Rumi et al. 2010).

19
PV usually develops slowly; if left untreated, average survival is only 1.5 years,
but current therapy strategies have extended it to over 10 years. However, it can progress to
secondary myelofibrosis, or the abnormal clone may begin to grow uncontrollably, thus
leading to AML. On the other hand, ET is more indolent, and the course of the disease is
often more benign, but associated sometimes with thrombotic or hemorrhagic complications.
Leukemic transformation occurs in less than 5% of cases and usually associated with
previous cytotoxic treatment (Passamonti et al. 2008). PMF, however, has the worst
prognosis among MPN (Mesa et al. 2006). The median survival time is approximately 3 to 7
years in patients diagnosed in the fibrotic stage.
In a minor fraction of MPN cases, cytogenetic abnormalities are found at
diagnosis. With disease progression, an abnormal karyotype is more frequently detected
(Heim & Mitelman 2010), even in the absence of disease-specific genetic defects. The most
common chromosomal abnormalities detected in PV with conventional cytogenetic
techniques are deletions located in the long arm of chromosomes 20 and 13, the duplication
of 1q, and trisomies of chromosomes 8 and 9 (Bench & Pahl 2005). Thanks to high
throughput and array technologies, researchers have identified new regions involved in
disease progression and new candidate genes (Stegelmann et al. 2010; Thoennissen et al.
2010). Chromosome aberrations are unusual findings in ET, but are still observed in 5-10%
of these patients (Kralovics & Skoda 2005), while in PMF patients, deletions of 13q and 20q,
+8 and chromosomal abnormalities of 1, 7 and 9 are present in 40-50% of cases (Reilly
2002).
JAK2 gene, which plays a major role in cytokine signal transduction and carries
an activating point mutation (JAK2V617F), strongly correlates with MPN (Baxter et al.
2005), although it can occur in other myeloid neoplasms as well (Webersinke & Rumpold
2009). Actually, the JAK2V617F mutation is present in approximately 95% of PV patients

20
and half of ET and PMF cases (Bruchova et al. 2008; Passamonti et al. 2010; Rumi et al.
2010). On the contrary, JAK2 exon 12 mutations, which seem to be specific to the
JAK2V617F mutation-negative PV, are absent from other cases of MPN or from healthy
individuals (Scott et al. 2007). The JAK2V617F allele burden (the ratio of JAK2V617F
mutant to non-mutant alleles) influences disease progression and correlates with the clinical
phenotype. Actually, the PV cases with high allele burden levels present more severe
disease. By contrast, the allele burden role in ET is unknown, but the JAK2V617F mutation
in a homozygous state is associated with a high risk for thrombosis in ET patients (Lussana
et al. 2009).
Additionally, MPLW515L/K mutations were reported in JAK2 wild-type ET
cases (Pardanani et al. 2006) and in 5% of patients with PMF (Mesa et al. 2006). Several
other mutations (in TET2, ASXL1, IDH, CBL, EZH2, NF1 and IKZF1 genes) have been
described in MPN, but their importance in the pathogenesis of MPN remains unclear
because they are present in other myeloid neoplasms as well (Tefferi 2010). Using X-
chromosome inactivation clonality analysis, Levine et al. (2006) showed, that development
of a clonal MPN may precede acquisition of JAK2V617F and that there exist a subset of ET
and PMF patients with JAK2V617F-negative clonal disease. All aforementioned mutations
affect the hematopoietic stem cell and result in cytokine-independent activation of the JAK-
STAT signal transduction pathway. Interestingly, this pathway also appears to be
hyperactive in patients with none of these mutations, which indicates that these mutation-
free cases may have hitherto unidentified mutation(s) functionally similar to a JAK2
mutation.
Similarly, chronic myelogenous leukemia (CML) is an MPN, characterized by
the increased and uncontrolled growth of myeloid cells, mainly of the mature granulocytes
(neutrophils, eosinophils, and basophils) and of their precursors in bone marrow and the

21
accumulation of these cells in the blood. The hallmark of CML is the Philadelphia
chromosome. The disease affects the middle aged and elderly people, and is rare in children
(Millot et al. 2005). Although the etiology of CML remains unknown, some studies have
implicated radiation exposure (Vardiman et al. 2008).
CML is usually detected when an elevated white blood cell count is observed
during a routine laboratory examination. An enlarged spleen, weight loss and fatigue may
also be present. The natural course of the untreated disease includes three phases: an
indolent phase followed by an accelerated phase and a blast phase. At diagnosis,
approximately 90-95% of CML cases have a standard Philadelphia translocation
(9;22)(q34;q11.2) (Vardiman et al. 2008), while 5-10% of CML cases have some variant
translocation and a small number of patients have a cryptic translocation (Huret 1990).
However the Philadelphia chromosome does not occur exclusively in CML; it has also been
observed in B-cell acute lymphoblastic leukemia (Gleissner et al. 2002).
The BCR-ABL1 fusion gene, which arises from the Philadelphia translocation,
encodes an aberrant tyrosine kinase which is constitutively activated and favors cell
proliferation. This can, however, be inhibited by imatinib (a selective inhibitor of ABL1
kinase) (Capdeville et al. 2002; De Keersmaecker et al. 2005). Actually, 70-90% of patients
treated with imatinib achieve a complete cytogenetic response (Vardiman et al. 2008).
Lately, the second generation of BCR-ABL kinase inhibitors have been introduced being
more potent and providing a complete response even more rapidly than does imatinib
(Kantarjian et al. 2010; Saglio et al. 2010).

22
Myelodysplastic syndromes
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal stem cell diseases
characterized by peripheral blood cytopenias, bone marrow dysplasia and by a strong
predisposition to become acute myeloid leukemia (AML); for this reason, MDS were
previously considered to be preleukemias (Silverman 2003). The bone marrow is usually
normo- or hypercellular but sometimes hypocellular. Peripheral blood cytopenias appear as
a results of ineffective hematopoiesis, different degrees of maturation disturbances and
apoptosis.
MDS affect mainly the elderly, the median age at diagnosis being 70 years
(Jadersten & Hellstrom-Lindberg 2010). The clinical features of MDS are non-specific;
rather, patients present general symptoms stemming from their cytopenias, such as pallor
and weakness, infections, bruises, bleeding and petechiae. Many patients are asymptomatic,
and routine blood counts identify anemia, neutropenia and/or thrombocytopenia.
The etiology of MDS is poorly understood, but has in some primary, de novo
MDS cases been associated with exposure to certain chemicals (benzene, pesticides,
solvents, etc), infections, and a family history of primary disease. Similarly, the etiology can
be associated with previous chemo/radiotherapy in cases of secondary (therapy-related)
MDS (Brunning et al. 2008).
Several classifications exist for MDS. In 1982, a French-American-British (FAB)
consensus classified MDS into five entities based on bone marrow morphology: refractory
anemia (RA), refractory anemia with ring sideroblasts (RARS), refractory anemia with
excess blasts (RAEB), refractory anemia with excess blasts in transformation (RAEB-T),
and chronic myelomonocytic leukemia (CMML) (Bennett et al. 1982). The World Health
Organization, in turn, modified and complemented the FAB classification in 2001 and

23
updated this classification in 2008 (Brunning et al. 2008) (see Table 4). The WHO 2008
classification, which is, currently, the most widely used classification of MDS, considers the
blast levels of 20% to be the line separating myelodysplasia from AML.
Table 4. The 2008 WHO classification of MDS.
Refractory cytopenia with unilineage dysplasia (RCUD) (Refractory anemia, Refractory neutropenia, and Refractory thrombocytopenia)
Refractory anemia with ring sideroblasts (RARS)
Refractory cytopenia with multilineage dysplasia (RCMD), which includes the subset Refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS). RCMD includes patients with pathological changes not limited to red cells (i.e., prominent white cell precursor and platelet precursor (megakaryocyte) dysplasia.
Refractory anemia with excess blasts 1 and 2. RAEB was divided into RAEB-1 (5-9% blasts) and RAEB-2 (10-19% blasts), which has a poorer prognosis than RAEB-1. Auer rods may appear in RAEB-2, which may be difficult to distinguish from acute myeloid leukemia.
Myelodysplastic syndrome – unclassifiable (MDS-U) (observed in cases of megakaryocyte dysplasia with fibrosis and others)
Myelodysplastic syndrome with isolated del(5q)
Childhood myelodysplastic syndrome(dysplasia in childhood) Refractory anemia with ring sideroblasts - thrombocytosis (RARS-T), which is in essence a MDS/MPN disorder and usually has a JAK2 mutation. CMML was removed from the MDS and placed in a separate category of MDS/MPN.
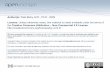
In MDS, prognosis varies considerably; the propensity for leukemic
transformation is almost absent in some categories and very high in others. The International
Prognostic Scoring System (Greenberg et al. 1997) evaluates survival and risk of
progression to leukemia based on the percentage of blasts in the marrow, cytogenetic
aberrations, and the number of cytopenias (Table 5). Based on these four risk groups are
recognized: low, 0; intermediate-1 (INT-1), 0.5-1.0; intermediate-2 (INT-2), 1.5-2; and high
≥2.5. The median survival in time and the evolution to AML of MDS patients is shown in
Table 6 (Greenberg et al. 1997).
24
Table 5. The International Prognostic Scoring System in MDS.
Score 0 0,5 1 1,5 2 Prognostic variables Bone marrow blasts (%) < 5% 5-10% 11-19% 20-30% Karyotype a Good Intermediate Poor Cytopenias b 0-1 2-3
aGood: normal,-Y, deletion of chromosomes 5q and 20q; Poor: complex karyotype (≥3 chromosomal aberrations) or chromosome 7 abnormalities; Intermediate: other abnormalities. bHemoglobin <10g/dl; Neutrophils <1.8x109/L, Platelets <100x109/L. Data retrieved from Brunning et al. (2008).
Table 6. Evolution to AML and median survival in time of MDS patients in each IPSS group.
Age ≤ 60 > 60 > 70 Median survival (years) low INT-1 INT-2 high
11.8 5.2 1.8 0.3
4.8 2.7 1.1 0.5
3.9 2.4 1.2 0.4
25% AML evolution (years) low INT-1 INT-2 high
> 9.4 6.9 0.7 0.2
> 9.4 2.7 1.3 0.2
> 5.8 2.2 1.4 0.4
Data obtained from Greenberg et al. (1997).
Cyto- and molecular genetics greatly impact the pathogenesis, diagnosis and
evaluation of the prognosis as well as the treatment of MDS patients. A wide range of
cytogenetic aberrations occur in 20-70% of patients, varying according to MDS type
(Tefferi & Vardiman 2009). The loss of chromosomal material is the most common
cytogenetic alteration found in these disorders, especially the loss of the whole/long arm of
chromosomes 5 or 7, or partial deletions of the long arm of chromosome 20; in contrast,
gained material frequently manifests as trisomy 8. Similarly, aberrations of the short arm of
chromosomes 17 and 12, as well deletion in 11q23 and the loss of chromosome Y also occur
(Nimer 2008; Bejar & Ebert 2010). As for prognosis, cases with more than three

25
chromosomal aberrations often have a poor prognosis, whereas a normal karyotype is
associated with a more favorable prognosis, similar to that of 5q- syndrome, the loss of 20q
or Y. Translocations are rare and non-specific to MDS.
Recently, several mutations have been detected in MDS, among them a point
mutation of the TET2 (Ten-Eleven Translocation 2) gene; but it is not limited only to MDS,
however, and also occur in other myeloid malignances (Tefferi et al. 2009a). Similarly, the
activating JAK2V617F mutation can occur in 5% of MDS, even if it is characteristic of
MPN. Other mutated genes which have also been involved in MDS and other myeloid
malignances include: ASXL1, CBL, RAS family of genes, TP53, RUNX1, and IDH
(Heinrichs et al. 2009; Jadersten & Hellstrom-Lindberg 2010).
Epigenetic events greatly impact the pathogenesis and treatment of MDS. CpG
island aberrant methylation is the most common, and was observed to be a progressive
process in MDS (Jiang et al. 2009). All in all, the hypermethylation of genes involved in
apoptosis, cell cycle regulation and histone deacethylation is frequently observed in these
disorders. Nowadays, both hypermethylation and deacethylation are targets for MDS
treatment. The first agent in MDS to prolong survival is the hypomethylating pyrimidine
analogue azacitidine (Fenaux et al. 2009).
The application of genome-wide array technologies has described various
cryptic genomic changes (Gondek et al. 2008; Starczynowski et al. 2008; Heinrichs et al.
2009). In recent years, however, research has added new insight into the pathogenesis and
progression of MDS by finding new mutations (Tefferi et al. 2009a; Tefferi 2010),
deregulated miRNAs, however, the primary genetic events involved in MDS are still
unidentified. The lack of biological and clinical markers as well as of a specific targeted
therapy makes MDS a target for intense study.

26
Methods for studying genetic alteration in hematological malignancies
Many efforts sought to identify the molecular basis and the importance of chromosomal
imbalances in the pathogenesis of various malignancies (Mitelman 2000). Cytogenetic and
molecular genetics analyses have become an important tool in the diagnosis, prognosis
evaluation, and monitoring of the residual disease in hematologic neoplasms, and are
essential component of the latest WHO classification of hematologic malignancies
(Swerdlow et al. 2008).
The great diversity of myeloid malignancies implies the use of various
techniques (Table 7). The most frequently used cytogenetic technique in routine clinical
practice is standard karyotype analysis (“G banding”) (Speicher & Carter 2005), which
easily detects gross chromosomal deletions, inversions, insertions, translocations and other
complex chromosomal rearrangements. However, karyotype analyses require actively
dividing cells and have relatively low resolution. These limitations were overcome in part
by the development of fluorescent in situ hybridization (FISH) (van Prooijen-Knegt et al.
1982), polymerase chain reaction (PCR) (Bartlett & Stirling 2003) and comparative
genomic hybridization (CGH) (Kallioniemi et al. 1992), thereby revealing many previously
unknown chromosomal rearrangements.
Table 7. Comparison of various cytogenetic and molecular techniques comparison.
Method Resolution Tissue Karyotype 5-10 Mb at 550-850 band bone marrow, blood
FISH 5Mb–10 Kb bone marrow, blood, FFPE PCR several Kb bone marrow, blood, FFPE
conventional CGH 3-5 Mb bone marrow, blood, FFPE array CGH > 1Mb–a few Kb bone marrow, blood, FFPE
The FISH technique is rapid, provides targeted analysis, and is suitable for non-
dividing cells also. This technique can reveal chromosomal aberrations undetectable by

27
conventional cytogenetic methods and is especially appropriate for diverse sample types and
for the detection of smaller clones, evaluation of for instance prognostically significant
known aberrations and monitoring of treatment response. The chromosomal regions
evaluated by FISH are usually larger than those studied with PCR, which, in turn, is used in
diagnostics and monitoring of minimal residual disease. The PCR method may be used to
detect mutations as well as very small malignant clones in the patient sample, permits
quantification of the transcript, and assesses disease status. Over time, different variations of
the basic PCR method have been developed and have become the method of choice for gene
copy number quantification, gene expression analysis, and the detection of single nucleotide
polymorphisms (SNPs) and mutations in the test samples.
CGH detects copy number changes (gains/losses) in the test DNA (often in a
tumor test sample), but the balanced structural changes (balanced translocation, inversions)
cannot be detected. In short, the DNA from a test sample and from a control (normal tissue)
is labeled with different fluorescent dyes. The equal amounts of labeled test (red) and
reference sample (green) DNA are combined and hybridized to normal metaphase
chromosomes (Figure 2). CGH radically changed the cytogenetic field, even though the
resolution was still too low and the normal cell contamination of the sample influenced the
results. To overcome these limitations, metaphase preparates, such as the hybridization
targets, have been replaced with genomic DNA clones spotted onto microscope slides
(microarray) (Solinas-Toldo et al. 1997; Pinkel et al. 1998). For this fluorescent labeled test
and reference DNA samples are hybridized against the array slide, which contains DNA
oligonucleotide probes (Figure 2). During hybridization, the samples compete for
complementary regions on the array slide, and the binding ratio of the probes will depend on
the genetic material in the test sample compared with the control sample. After
hybridization, the array slides are washed to remove any unbound DNA, and then scanned.

28
The image obtained is further processed and the ratios of both dyes are calculated for each
probe to determine the chromosomal aberrations in the test sample. A gained or amplified
chromosomal region in the test sample will have an increased fluorescence ratio (appearing
in red), whereas a loss/deleted region has a reduced ratio (appearing in green). The array
CGH can be applied in cases where the amount of DNA is very small, (e.g. analysis of a
single cell) (Gangnus et al. 2004).
Figure 2. Schematic overview of the metaphase CGH and array CGH.

29
Knowledge of the human genome sequence (Lander et al. 2001) has enhanced
the development of different high throughput techniques to map defective genes and to
identify single nucleotide alterations, thereby increasing the resolution from megabase to
kilobase levels (Vissers et al. 2005).
MicroRNA expression profiling
In the past few years, even smaller genomic units, such as microRNAs (miRNA), have been
extensively studied in human cancers. MiRNAs are small non-coding, phylogenetically well
conserved RNA molecules (20-25 nt in length) that regulate gene expression by degrading
or repressing target messenger RNA (mRNA) (Calin & Croce 2006). Some miRNA genes
are located in exons and others in non-coding regions of the genome (Negrini et al. 2009).
Thus, over 700 miRNAs have been identified in the human genome (Zhang et al. 2011). The
abundance of studies has pointed out that miRNAs greatly impact not only normal cell and
tissue development, but different pathological states as well (e.g. cancers, diabetes, heart
disease) (Zhang 2009; Santarpia et al. 2010).
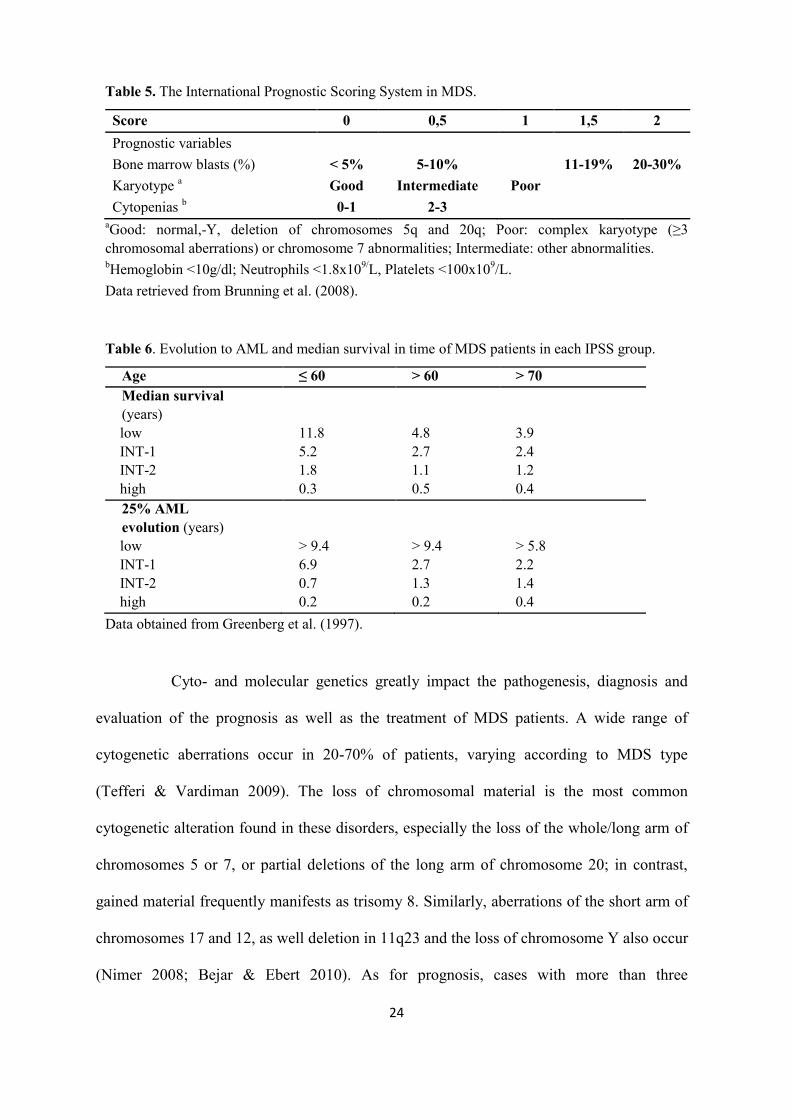
The biogenesis of miRNA originates in the nucleus by the transcription of DNA
to long transcripts known as primary miRNA (can be several thousand nucleotides long),
which is cleaved by Drosha endonuclease into pre-miRNA (double stranded RNA of about
70 nt in length). The pre-miRNA is transferred to the cytoplasm, where it is cleaved by
Dicer ribonuclease into 20 to 25 nucleotide long, double-stranded RNA, and is then
separated into one mature strand (miRNA) and one passenger strand (miRNA*). The mature
strand is incorporated into the RNA-induced silencing complex (RISC), and the passenger
strand is preferentially destroyed. Because miRNAs require no perfect complementarity for
their target mRNA, one miRNA can target multiple genes and, many miRNA can regulate
one gene.
30
Figure 3. MicroRNA biosynthesis (Figure 3 has been modified from Santarpia et al. 2010).
Recent studies have revealed that the aberrant expression of miRNAs is
involved in many diseases, including malignancies. Also, some studies reported that
miRNAs can function as an oncogene or as a tumor suppressor gene (Chen 2005; Hammond
2007; Wang et al. 2010), and their expression profiling can serve to classify human cancers
more accurately than gene expression does (Lu et al. 2005). In cancer, the dysregulated
miRNAs are evidently located in or near altered chromosomal regions breaking points
(gains, deletions or translocations). Promisingly, altered miRNA expression (disease-,
tissue- and cell-specific expression) can serve in the future in diagnostics and as potential
therapeutic targets.

31
AIMS OF THE STUDY The aim of the study was to identify genomic imbalances in MPN and MDS using the latest
microarray techniques (array CGH and miRNA) in order to identify the clinically relevant
genes involved in these disorders.
The specific aims were:
● To screen the JAK2 gene point mutation status of the clinical samples and to
compare it to the in vitro growth pattern of hematologic progenitor cells in MPN.
● To screen for novel/cryptic copy number alterations and to identify clinically
relevant genes in MPN and MDS.
● To show the feasibility of miRNA array profiling of formalin-fixed, paraffin-
embedded (FFPE) and decalcified bone marrow core biopsy specimens.
● To integrate array CGH and miRNA expression data and to identify molecular
markers to aid in the diagnosis and prognosis of MDS.
32
MATERIALS
Patients For the present study, we selected 155 bone marrow and blood samples from MPN, MDS
and leukemia patients. The patients were diagnosed and treated at the Helsinki University
Central Hospital between 1998 and 2007. Information about the sample and methods used
for this study appear summarized in Table 8.
Table 8. Overview of the sample material and methods.
Study Disease Number of patients
Focus of genetic changes
Method Sample material
I II
ET/ PV ET/PV
100 (58/42) 35 (14/21)
Mutation CNA
PCR array CGH
FF BM, B FF BM, B, C
III ALL/CML 18 (9/9) Gene Expression miRNA array FFPE IV MDS 37 CNA array CGH FF BM, B V MDS 19 Gene Expression miRNA array BM smears Abbreviations: CNA, copy number alterations; FF, fresh frozen; BM, bone marrow; B, blood; C, colonies of hematopoietic progenitors; FFPE, formalin-fixed paraffin embedded.
Sample preparation
Bone marrow aspirates and peripheral blood samples Bone marrow aspirates were taken at the time of diagnosis at the Helsinki University
Central Hospital. About two to three millilitres of bone marrow aspirate or five milliliters of
peripheral blood were collected in heparin tubes and stored at -70°C.

33
Bone marrow smear samples
According to standard procedures, two representative smears from aspirated bone marrow
were stained with May-Grünwald-Giemsa staining and air dried, and cover slides with
Permount® mounting medium (Fisher Scientific, Fair Lawn, NJ, USA) were then applied.
Core biopsy preparations
Bone marrow core biopsies were fixed in formalin at the Central Laboratory of Pathology,
Helsinki University Central Hospital for at least 24 hours before decalcification in a neutral
solution. Following standard protocols, the sample was also embedded in paraffin.
In vitro cultures of hematopoietic progenitors
As a part of routine diagnostic work, erythroid (BFU-E) and megakaryocytic (CFU-Meg)
progenitors from BM aspirates were cultured as previously described (Juvonen et al. 1993).
The cultures of BFU-E and CFU-Meg were counted on the 14th day of culture.
Myeloproliferative neoplasms (I-II)
Bone marrow, blood, BM mononucleated cells and hematopoietic progenitor colony cells
from ET/PV patient were collected during diagnosis in the Helsinki University Central
Hospital and stored at the Pathology Department, Helsinki University Central Hospital. The
samples were mainly fresh and fresh frozen, but 9 were collected and kept in stabilization
liquid.

34
Leukemia samples (III)
ALL and CML formalin-fixed paraffin-embedded (FFPE) bone marrow core biopsies and
frozen bone marrow aspirates were retrieved from the archives of the Pathology Department,
Helsinki University Central Hospital. Karyotype analyses were available for all leukemia
samples, as were array CGH data for ALL cases.
Myelodysplastic syndromes (IV, V)
Frozen bone marrow aspirates samples from MDS patients (Study IV) were retrieved from
the archives of the Pathology Department, Helsinki University Central Hospital. Bone
marrow smears from diagnosis were obtained from the Medicine Department, Helsinki
University Central Hospital (Study V).
Ethical Permission
The patients provided their informed consent before collection of the samples. The Ethical
Review Board of the Helsinki University Central Hospital (§303/2006) had approved the
study protocols.

35
METHODS
Nucleic acid extraction
Studies I, II, and IV used standard methods for DNA extraction (Lahiri & Nurnberger 1991)
from fresh frozen BM and blood, whereas a Puregene DNA purification kit was used for
mononucleated cells and for the hematopoietic progenitor colony cells (Studies I and II). As
for the samples in stabilization liquid, a Boehringer Mannheim DNA extraction kit
(Promega, Madison, USA) was applied (Study I). DNA from pooled healthy male and
female peripheral blood samples served as references in the array CGH experiments. Total
RNA was extracted from the bone marrow samples in Studies III and V using the Qiagen
miRNeasy FFPE Kit and miRNeasy Mini Kit (Qiagen, CA, USA) protocols in accordance
with the sample types. RNAs from eight healthy donors were used as control samples in the
miRNA expression experiments.
Mutation analysis (I, II)
For the ET and PV samples, we determined JAK2 (Studies I and II) and MPLW515L
(Study I only) point mutations status. For the JAK2V617F was used an allele-specific PCR
method as described Baxter et al. 2005. Exon 12 mutations of the Janus kinase 2 gene were
screened with allele-specific PCR using forward primer 5´CTC CTC TTT GGA GCA ATT
CA3´ and reverse primer 5´GAG AAC TTG GGA GTT ATA3´. Genotyping of the
MPLW515L allele was performed on a LighCycler (Roche Applied Biosience, Indianapolis,
IN, USA) and verified using direct DNA sequencing.

36
Methylation-specific PCR (IV)
We used three different primer sets to check the methylation status of the RPS14 gene
(Tasheva & Roufa 1995). The DNA was treated with sodium bisulfide using an Imprint
DNA Modification Kit (Sigma-Aldrich, MO, USA) prior to amplification. Positive control
DNA was selected from the CpGenome universal methylated DNA (Milipore, MA, USA),
and negative control DNA from the pooled peripheral blood of healthy individuals.
In vitro cultures of hematopoietic progenitors (I, II)
The hematopoietic progenitor cells were cultured as a part of the routine diagnostic work
previously described (Juvonen et al. 1993). Briefly, for the cultures of megakaryocytic
(CFU-Meg) and erythroid (BFU-E) progenitors, mononuclear cells from bone marrow or
blood were separated using Ficoll gradient centrifugation and cultured in a methyl
cellulose-containing medium. Growth of CFU-Meg was considered spontaneous when 10
colonies/1 x 105 mononuclear cells were present. BFU-E progenitors were cultured with the
addition of erythropoietin (EPO 2 U/ml, Janssen-Cilag AG, Schaffhausen, Switzerland) or
without EPO (spontaneous growth). The cultures were scored on day 14, and BFU-E, CFU-
Meg and granulocyte-macrophage colonies were then collected from selected cases (2
patients).
Oligonucleotide-based array comparative genomic hybridization (II, IV)
Chromosomal copy number alterations were studied in ET, PV (Study II) and MDS (Study
IV) using 44K (containing 44 000 probes) and 244K arrays (with 236 000 probes) from
Agilent (Agilent Technologies, Santa Clara, CA, USA). As a reference, pooled DNA from

37
healthy donors was sex matched with the samples. Test and reference samples were digested,
then labeled and hybridized in conformance with Agilent`s protocol (v2.0 for Study II and
v4.0 for Study IV). After 40 hours, the arrays were washed and scanned with an Agilent
microarray scanner (G265BA in Study II and G2505B in Study IV). Output images were
processed with Feature extraction (v8.1 and v9.5) and analyzed with Agilent CGH analytics
software (v3.2 and v3.5) (Agilent Technologies). The chromosomal aberrations were scored
using the Z-score (Study II) and the ADM-1 (Study IV) algorithms (Feature extraction
reference guide) with threshold set at 2.5 and 6.0, respectively; in addition, an aberration
filter was set to minimize small aberrations/variations and to show only regions with a
minimum of five aberrant probes. Polymorphic variations, according to the Database of
Genomic Variants (Iafrate et al. 2004), were excluded from the analysis.
MicroRNA array (III, V)
MicroRNA expression profiling was studied using an Human miRNA Microarray Kit V2
(representing 723 human and 76 human viral miRNAs from Sanger miRBase release 10.1)
in ALL and CML (Study III), and an Human miRNA Microarray Kit V3 (representing 886
human and 89 human viral miRNAs from Sanger miRBase release 12.0) in MDS (Study V)
(Agilent Technologies). The RNA was dephosphorylated, labeled and hybridized according
to the miRNA complete labeling and hyb kit protocol v2.1. The arrays were washed after 20
hours and scanned with a high resolution Agilent scanner (G2505B, Agilent Technologies);
the images were then processed with Feature extraction software (v9.5, Agilent
Technologies). Data and statistical analyses were carried out with Chipster software
(http://chipster.csc.fi/) (Studies III and V) using default settings for the Agilent miRNA
array and with SPSS v16.0 (Study III).

38
The poor-quality arrays were excluded from further analysis. The log2
transformed data were normalized using the default setting for the Agilent miRNA array
platform V2 (Study III) and quantile normalized with no background subtraction (Study V)
using Chipster software. We summarized the multiple probes of miRNA into a single value
with a robust multichip average (RMA) algorithm. To identify significantly differentially
expressed miRNA, we used an empirical Bayes procedure and the Benjamini-Hochberg
method (Benjamini & Hochberg 1995) as multiple corrected testings with a p-value under
0.05 and a more than two-fold change. For Study V, we used Chipster annotation tools to
identify the target genes of the altered miRNA and to further pathway analysis using a gene
ontology enrichment statistical test.

39
RESULTS AND DISCUSSION
Association of the JAK2V617F mutation status and the in vitro growth of hematopoietic progenitors (I)
To find a possible correlation between the spontaneous megakaryocytic and erythroid
colony formation of hematopoietic progenitor cells and the JAK2V617F mutation status, we
analyzed samples from 100 patients: 42 PV and 58 ET (Study I). The JAK2V617F mutation
was detected in 38 (91%) of the PV and 33 (57%) of the ET patients. Spontaneous colony
growth was observed in 85% of all the MPN patient samples. All JAK2V617F mutation-
positive patients had spontaneous erythroid growth, with one exception: an ET patient
whose apparent diagnosis was RARS-T. The 2008 WHO classification proposed RARS-T
(previously described as ET with ring sideroblasts) as unclassified MDS/MPN, which may
explain the different behaviours regarding the in vitro culture of the hematopoietic
progenitors. We detected spontaneous BFU-E growth as well in nine patients (4 PV, 5 ET)
with JAK2V617F wild-type. Six patients (2 PV, 4 ET) from these cases were tested for the
JAK2 exon 12 mutation; none of them tested positive for this mutation. Overall, we can
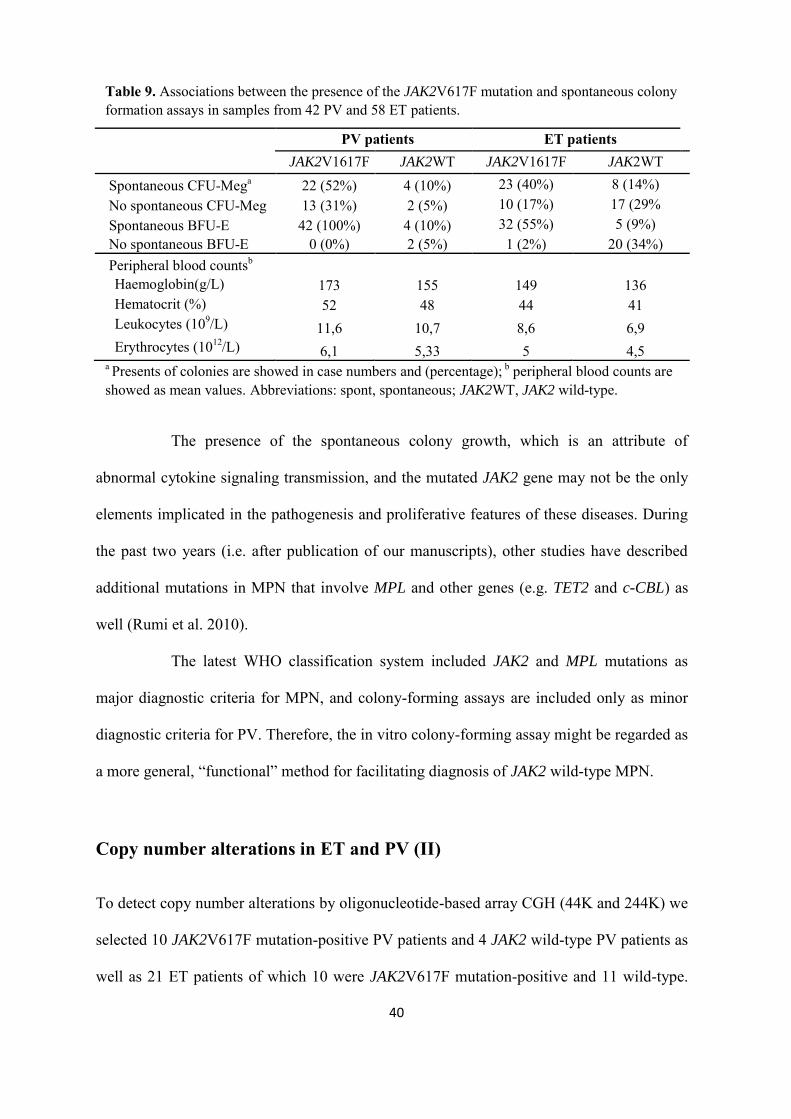
conclude that the presence of a JAK2 mutation was associated with spontaneous erythroid
colony formation in both PV and ET patients, but in ET patients was associated with
spontaneous CFU-Meg also (Table 9).
Similarly, Boissinot et al. (2006) found that the JAK2V617F mutation was
present in megakaryocytes as well. In our study, all patients with spontaneous CFU-Meg
growth, but without spontaneous BFU-E growth, were diagnosed as ET with wild-type
JAK2. Therefore, we looked for a MPLW515L mutation in seven of these cases, but only
one case was found to have this mutation. These results suggest that other pathogenetic
events may be implicated in the spontaneous growth of hematopoietic progenitors in MPN.
40
Table 9. Associations between the presence of the JAK2V617F mutation and spontaneous colony formation assays in samples from 42 PV and 58 ET patients.
PV patients ET patients JAK2V1617F JAK2WT JAK2V1617F JAK2WT Spontaneous CFU-Mega 22 (52%) 4 (10%) 23 (40%) 8 (14%) No spontaneous CFU-Meg 13 (31%) 2 (5%) 10 (17%) 17 (29% Spontaneous BFU-E 42 (100%) 4 (10%) 32 (55%) 5 (9%) No spontaneous BFU-E 0 (0%) 2 (5%) 1 (2%) 20 (34%) Peripheral blood countsb Haemoglobin(g/L) 173 155 149 136 Hematocrit (%) 52 48 44 41 Leukocytes (109/L) 11,6 10,7 8,6 6,9 Erythrocytes (1012/L) 6,1 5,33 5 4,5
a Presents of colonies are showed in case numbers and (percentage); b peripheral blood counts are showed as mean values. Abbreviations: spont, spontaneous; JAK2WT, JAK2 wild-type.
The presence of the spontaneous colony growth, which is an attribute of
abnormal cytokine signaling transmission, and the mutated JAK2 gene may not be the only
elements implicated in the pathogenesis and proliferative features of these diseases. During
the past two years (i.e. after publication of our manuscripts), other studies have described
additional mutations in MPN that involve MPL and other genes (e.g. TET2 and c-CBL) as
well (Rumi et al. 2010).
The latest WHO classification system included JAK2 and MPL mutations as
major diagnostic criteria for MPN, and colony-forming assays are included only as minor
diagnostic criteria for PV. Therefore, the in vitro colony-forming assay might be regarded as
a more general, “functional” method for facilitating diagnosis of JAK2 wild-type MPN.
Copy number alterations in ET and PV (II)
To detect copy number alterations by oligonucleotide-based array CGH (44K and 244K) we
selected 10 JAK2V617F mutation-positive PV patients and 4 JAK2 wild-type PV patients as
well as 21 ET patients of which 10 were JAK2V617F mutation-positive and 11 wild-type.

41
The analysis revealed copy number alterations in chromosomes 1q (1q21.1q32.1 duplication)
and 13q (13q12.3q21.32 deletion) in one patient (diagnosed as ET with JAK2V617F wild-
type) (Figure 4). Both the 1q partial duplication and the 13q deletion (Pastore et al. 1995;
Kawamata et al. 2008) have been described in MPN, but no specific cytogenetic
abnormalities have been reported to date. Although the deletion of 13q can occur in ET, it is
more frequent in PV (Panani 2007). We found no deletion leading to a loss of genetic
material at the 9p loss of heterozygosity (LOH) locus/JAK2 locus. The 9pLOH has been
found to be present in 30% of the PV patients (Kralovics & Skoda 2005).
Figure 4. Aberration summary of chromosomes 1 and 13 in the ET case (Duplication is indicated in red and deletion in green).
Due to limited quantity of DNA we performed high resolution (244K) array for
five samples that included two hematopoietic progenitor cell colony samples: one BFU-E
and one CFU-Meg sample. Overall, 244K array CGH served to verify the results obtained
with 44K array CGH. The normal results from progenitor cells were in agreement with the
results obtained from whole bone marrow cells from the same patients.
No previous reports were available on array CGH using high-density
oligonucleotide-based microarrays, but previous results with array CGH on BAC (bacterial
artificial chromosome) platforms had indicated no chromosomal imbalances in MPN
(Espinet et al. 2006). After our study, one report showed that array CGH revealed copy

42
number alterations in 35% of PV and 15% of ET cases in their study (Tefferi et al. 2009b).
About half of the patients in this study had undergone myelosuppressive treatment, and the
sample material included purified granulocytes, which could account for the discrepancies
between their and our study (Study II).
Our results suggest that copy number changes are rare in PV or ET; even though
we used sensitive high resolution microarray analysis to unearth submicroscopic genomic
imbalances. The frequency of CNA in MPN will probably increase with the development of
more sensitive and higher density arrays.
Core biopsy paraffin-embedded core bone marrow biopsy samples as a reliable source of miRNA (III)
Formalin-fixed and paraffin-embedded (FFPE) is the standard method for
preserving and storing biopsies and surgical specimens; it is the most abundant archival
material in the world and is available in many pathology laboratories. Although FFPE
specimens are widely available, their use in molecular profiling experiments is significantly
limited due to formalin fixation, which causes crosslinkage between proteins and nucleic
acids, and partial degradation of RNA and DNA (Srinivasan et al. 2002). Consequently,
microarray expression analysis using these specimens is challenging (Farragher et al. 2008).
MiRNAs are, however, more stable in the samples due to their integration into the RNA-
induced silencing complex (in which miRNA are incorporated after synthesis).
In our study (Study III), besides the formalin fixation, bone marrow core
biopsies had been exposed to decalcification, which also affects the integrity of the nucleic
acids, and especially that of RNA (Liu et al. 2002). In Study III, 18 matched (9 ALL, 9
CML) core biopsy (CB) and bone marrow aspirate (BA) samples were investigated using
miRNA array V2 (Agilent Technologies).

43
RNA quantification permitted us to continue with the hybridizations. First, we
performed technical replicates (duplicates) of three samples to evaluate their reproducibility.
The average Pearson correlation between replicates was higher (coefficient above 0.94 in all
cases) than between unrelated samples, and the number of probes detected (present
according to the Feature extraction analysis algorithm) in the CB samples was comparable
to that in the matched BAs, but on average fewer in CB. Technical replicates generally
grouped together when unsupervised hierarchical clustering was used (Figure 5). All these
facts suggest that core biopsies are suitable for the microarray profiling of miRNA. Even
though the number of expressed miRNAs between the CB and BA samples differed slightly,
this result can be attributed to the sample contents and their cellularity as well as additional
bone and connective tissues.
Figure 5. Hierarchical clustering of the technical replicates. Abbreviations: Dup, duplication; CB, core biopsy; BA, bone marrow aspirate; 2, 4,9,10 represent the patient number.
We separately profiled the two leukemia types against the controls. We
detected differentially expressed miRNAs, common between CBs and BAs, for each
leukemia type: 43 in CML and 21 in ALL. Among these, miR-142-3p and miR-15b were
present in both leukemias; the only difference was that miR-142-3p was upregulated in ALL
and downregulated in CML, but miR-15b was downregulated in both leukemia types. In

44
agreement with other ALL reports (Zanette et al. 2007; Fulci et al. 2009; Ju et al. 2009;
Schotte et al. 2009), miR-222 and miR-181b were also upregulated in our study. After
combining the all BAs and CBs for each leukemia type using an unsupervised clustering
method, we obtained two separate/different groups of miRNAs, which actually
corresponded to the two leukemia types.
The inconsistency, in CML, of the miRNA profiles of miR-15, miR-16, miR-
221, miR-155, miR-10a, miR-150 and miR-96 (reported previously) (Ramkissoon et al.
2006; Agirre et al. 2008) as well as those obtained by our studies was probably due to the
types of sample used (we used FFPE samples and in the other studies used hematopoietic
cell lines, mononuclear and CD34+ cells).
Our results, like those of others, indicated that miRNAs are stable in FFPE (Li
et al. 2007; Doleshal et al. 2008) samples, even if the samples used in our study (Study III)
were additionally subjected to decalcification. In conclusion, bone marrow core biopsy is a
reliable starting material for miRNA expression profiling.
Copy number alterations in MDS (IV)
In our aCGH analysis numerous submicroscopic copy number alterations were observed in
the bone marrow samples taken at diagnosis from 37 de novo MDS patients, but none
correlated with the MDS entities or stages (the cohort comprised of 2 RAEB-1, 7 RAEB-2,
17 RCMD, 2 CMML, 5 RARS and 4 cases of 5q– syndrome). Karyotype analysis revealed
that 18 of the 37 samples had chromosomal aberrations, while array CGH analysis refined
the breakpoint of known aberrations and revealed new ones. Overall, these sizes ranged
from a few kb (> 65 kb) to the loss/gain of a whole chromosome (Table 10).

45
Table 10. Summary of array CGH results on 37 MDS cases. Number of patients/
Diagnosis Array CGH results (number of cases/ %)
a1 b2 c3 17 RCMD 5 RARS 2 RAEB 1 7 RAEB 2 2 CMML* 4 5q– syndrome
6 (35%) 4 (80%) 1 (50%) 4 (57%) 1 (50%) –
9 (53%) – 1 (50%) 1 (14%) 1 (50%) 4 (100%)
2 (12%) 1 (20%) – 2 (29%) – –
1No copy number changes; 2 with one or two copy number changes, and 3 with complex copy number alterations. *The 2008 WHO classification place CMML as a mixed myelodysplastic/myeloproliferative neoplasms.
Due to the genetic heterogeneity of MDS, we detected small aberrations in
nearly all chromosomes; deletions were observed more frequently than gains. The most
frequently observed aberrations were deletions at 5q and 7q with a common region at
5q21.3q32 and 7q22.1q33, respectively, as well as trisomy 8. In addition, losses commonly
recurred at 11q, 12p, 17p, 18q and 20q, as did gains at 8p; we detected no high-level
amplifications (Figure 6).

46
Figure 6. The most frequent copy number alteration in 37 de novo MDS cases. The image, generated with DNA analytics software (Agilent Technologies), shows along the X axis the percentage of each chromosome aberration across all samples (green indicates losses, and red indicates gains).

47
The commonly deleted region in MDS, 5q, contains about 40 genes, but neither
tumor suppressor genes nor mutations were reported in this region until Ebert et al. (2008)
showed the haploinsufficiency of the RPS14 gene. They proposed RPS14 as a candidate
gene for MDS 5q– syndrome. However, this gene has been shown not to be responsible for
all clinical characteristics in MDS 5q– syndrome, which indicates that one or more genes
are implicated in the pathogenesis and progression of MDS to leukemia (Ebert 2009). To
determine whether epigenetic mechanisms inactivate, in addition to deletion, the RPS14
gene, we tested its methylation status in 24 MDS patient samples, including four patients
with 5q– syndrome. Our results provided no evidence of hypermethylation of this gene,
which is in concordance with results of a previous report (Valencia et al. 2008).
Alongside RSP14 gene in the 5q deleted region, SPARC tumor suppressor gene
was reported to show haploinsufficiency also and may be implicated in pathogenesis of
MDS (Boultwood et al. 2007). SPARC gene was deleted in all our cases showing 5q
deletion. In addition, at chromosome 5q31.2 region, haploinsufficiency of CTNNA1, HSPA9,
CTNNA1 and EGR1 genes, have been previously described and together, greatly impact the
pathogenesis of the disease, but individually, these genes cannot account for the clinical
features of MDS (Graubert et al. 2009).
Most of the previous studies have shown that in MDS, the 7q deletion has
several breakpoints. Although these studies have postulated that the breakpoints may
implicate some tumor suppressor genes, they reported no evident target genes (Wong et al.
2008).
Some of the drawbacks of array CGH include its inability to detect balanced
translocations, inversions or mosaicism, and besides, to be detectable, a malignant clone
should be present as an aberration in at least 35% of the cells (Evers et al. 2007). In our
study (IV) however, one patient, with mosaic monosomy 7 detected with array CGH

48
analysis, was not observed with karyotype analysis. After re-examination of the metaphases,
we noticed that the patient actually had one subline with der(1;7)(q10;p10) and another with
monosomy 7. These may stem from the fact that the in vitro proliferation of the der(1;7)
clone was superior to the monosomic clone (Knuutila et al. 1981).
Also, possibly due to the high sample heterogeneity and lesser content in
malignant cells in the samples from four patients, the array CGH was unable to detect the
copy number alteration already observed with karyotype analysis. Two of these samples
contained many clones with complex karyotype changes, and for the other two, the
proportion of cells containing aberrations was probably below the detection limits of array
CGH (~35%).
Previous studies report inconsistent results for copy number alterations with
microarray CGH due to the great heterogeneity of MDS (between subtypes, group risk or
categories) as well as to the array technologies applied (array CGH or SNP). In Study IV,
57% of cases showed CNA, which is in agreement with the 41% reported by Heinrichs et al.
(2009), although other report indicated CNA up to 80% (Slovak et al. 2010).
The losses at 12p (TEL discussed target gene), 17p (TP53 discussed target gene)
and 20q (no target gene known) that we detected with our sensitive array CGH may be more
frequent in MDS than previously thought; and identifying the target genes will require
further study.
MicroRNA expression profiling in MDS (V)
To assess whether miRNAs are involved in the pathogenesis of MDS, we first compared
miRNA expression profiles from both MDS and healthy bone marrow samples and,
secondly, we integrated the array CGH with miRNA expression profiling data.
49
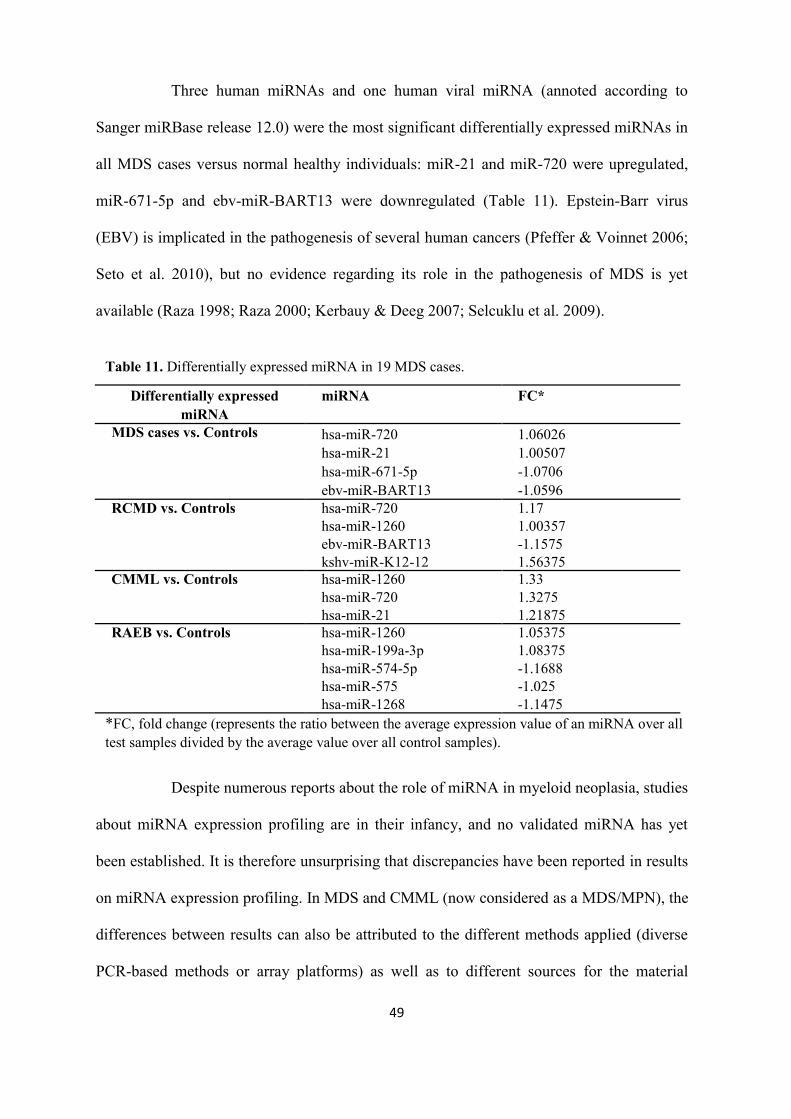
Three human miRNAs and one human viral miRNA (annoted according to
Sanger miRBase release 12.0) were the most significant differentially expressed miRNAs in
all MDS cases versus normal healthy individuals: miR-21 and miR-720 were upregulated,
miR-671-5p and ebv-miR-BART13 were downregulated (Table 11). Epstein-Barr virus
(EBV) is implicated in the pathogenesis of several human cancers (Pfeffer & Voinnet 2006;
Seto et al. 2010), but no evidence regarding its role in the pathogenesis of MDS is yet
available (Raza 1998; Raza 2000; Kerbauy & Deeg 2007; Selcuklu et al. 2009).
Table 11. Differentially expressed miRNA in 19 MDS cases.
Differentially expressed miRNA
miRNA FC*
MDS cases vs. Controls hsa-miR-720 1.06026 hsa-miR-21 1.00507 hsa-miR-671-5p -1.0706 ebv-miR-BART13 -1.0596
RCMD vs. Controls hsa-miR-720 1.17 hsa-miR-1260 1.00357 ebv-miR-BART13 -1.1575 kshv-miR-K12-12 1.56375
CMML vs. Controls hsa-miR-1260 1.33 hsa-miR-720 1.3275 hsa-miR-21 1.21875
RAEB vs. Controls hsa-miR-1260 1.05375 hsa-miR-199a-3p 1.08375 hsa-miR-574-5p -1.1688 hsa-miR-575 -1.025 hsa-miR-1268 -1.1475
*FC, fold change (represents the ratio between the average expression value of an miRNA over all test samples divided by the average value over all control samples).
Despite numerous reports about the role of miRNA in myeloid neoplasia, studies
about miRNA expression profiling are in their infancy, and no validated miRNA has yet
been established. It is therefore unsurprising that discrepancies have been reported in results
on miRNA expression profiling. In MDS and CMML (now considered as a MDS/MPN), the
differences between results can also be attributed to the different methods applied (diverse
PCR-based methods or array platforms) as well as to different sources for the material

50
(CD34+ cells, bone marrow or blood mononuclear cells, bone marrow smears, FFPE
trephine biopsies); the type of controls used also varies (CD34+ cells, bone marrow or bone
marrow smears from healthy individuals, peripheral blood total nucleated cells) (Bousquet
et al. 2008; Gaken et al. 2008; Kumar et al. 2009; Pons et al. 2009; Dickstein et al. 2010;
Dostalova Merkerova et al. 2010; Hussein et al. 2010a; Hussein et al. 2010b; Nassiri et al.
2010; Starczynowski et al. 2010; Hussein et al. 2011).
Has been shown that miR-145 and miR-146 are correlate with 5q- syndrome
(Starczynowski et al. 2010). However Hussein et al. (2010a; 2010b) could not see the
deletion of these two miRNAs. Dostalova Merkerova et al. (2010) reported normal
expression levels of miR-145 and miR-146 in 5q- cases. In our study (Study V) we could
not see any of these miRNAs to have significantly differentiated expression in MDS
samples comparing with controls.
Apoptosis is predominant in the early stages of MDS, and proliferation in the
advanced stages, when leukemic transformation occurs. MiR-34b*, one of the upregulated
(but not significantly) miRNAs in our MDS 5q- cases, is one of the regulators of apoptosis;
it may therefore play a crucial role in preventing the evolution of the disease and leukemic
transformation. In contrast, miR-21, known to be implicated in cell proliferation, was
upregulated in RAEB, which has a strong tendency to develop AML. Both miR-21 and
miR-34 are known to have altered expression in many cancers (Kerbauy & Deeg 2007;
Cazzola 2008; Hermeking 2010).
The predicted target genes for the four most differentially expressed miRNAs
were fetched using two databases (in order to minimize the false positive results) (Krek et al.
2005; Lewis et al. 2005), which enumerated 86 common genes. Pathway analysis was
applied to see the biological and functional networks for these genes. The results showed
that miR-21 is involved in the processes which regulate myeloid and lymphoid cell

51
differentiation into particular kinds of cells, maturation and specific cell fate. Is well
established that miR-21 modulates cell proliferation and is high expressed in different
cancers. In fact in MDS the balance between apoptosis and cell proliferation have a great
impact during leukemic transformation and disease evolution; therefore miR-21 may have a
central role in MDS. Even though miR-21 is one of the most studied miRNA its functions
are still not well understood.
Combining the array CGH and miRNA expression data, we were unable to
detect whether copy number alterations influenced miRNA expression in our MDS cases.
Various reports demonstrate that copy number alterations do in fact correlate with miRNA
gene expression (Bray et al. 2009), but on the other hand, other reports could not
demonstrate this (Lamy et al. 2006). The discrepancy may stem from the differences
between the sample types used in the two studies, or from the obscure correlation between
copy numbers and miRNA, unlike copy numbers and mRNA expression. The miRNAs
regulate more than one mRNA target (Pillai 2005), which can be found in several different
genomic regions and can be associated with several copy number alterations.
Although our study contributes new information about miRNA expression in
MDS. We can conclude that in the future, the analysis of different genome-wide data sets
and different sample types should be integrated in order to obtain reliable results and to
provide a deeper understanding of the molecular defects and pathogenesis not only of MDS,
but also of other myeloid neoplasms.

52
CONCLUSIONS The genome-wide screening of copy number alterations using microarray-based
technologies has proved to be an efficient tool for detecting de novo cryptic chromosome
imbalances in many myeloid malignancies. Thus far, the majority of these aberrations
appear as single submicroscopic microdeletions or duplications. In this thesis, we applied a
microarray technique in a subset of patients diagnosed with PV, ET and MDS, and also
studied the JAK2V617F mutation as well as the in vitro colony formation of hematopoietic
progenitors and their importance in the diagnosis of PV and ET.
The discovery of the involvement of JAK2 mutation in MPN has revolutionized
our understanding of these diseases. We studied 100 patients (58 ET/42 PV) to determine
whether any correlation exists between JAK2 gene mutation status and the in vitro growth
pattern of hematologic progenitor cells. Of all patients, 71% had the JAK2V617F mutation,
while 56% showed spontaneous megakaryocyte colony growth (CFU-Meg) and 79%,
spontaneous erythroid colony growth (BFU-E). All patients with the JAK2V617F mutation
exhibited BFU-E spontaneous growth, but nine patients with JAK2 wild-type also showed
BFU-E spontaneous growth. Eight patients (6 ET and 2 PV) with JAK2 wild-type presented
only CFU-Meg spontaneous growth. Therefore in vitro culture assay showed to be a good
and useful diagnostic tool, in particularly for the JAK2 wild-type MPN cases showing only
spontaneous CFU-Meg growth.
In addition, we analyzed DNA copy number alterations in 14 ET and 21 PV
cases, but only one ET case had a partial deletion of the long arm of chromosome 13 and a
partial duplication of 1q. The importance of these array-based technologies are currently
being recognized in the detection and characterization of altered gene expression profiles

53
and copy number alterations with an essential role in the pathogenesis of MPN alongside
searching for new gene mutation analysis.
In MDS, 57% (21/37) of the cases showed chromosome alterations. The most
frequent copy number alterations were deletions at 5q21.3q32 and 7q22.1q33, as well as
trisomy 8. Overall, copy number deletions were more common than copy number gains. We
detected some recurrent deletions at 11q, 12p, 17p, 18q and 20q, and gains at 8p. Besides
these copy number alteration findings, we aimed to identify MDS subtype-specific
differentially expressed miRNAs (Study V) by profiling 19 MDS cases using microarray
technology, which resulted in the detection of four differentially expressed miRNAs (ebv-
miR-BART13, hsa-miR-720, hsa-miR-21, hsa-miR-671-5p), as compared to normal
individuals. By integrating the array CGH and miRNA expression data, we could not
observe the influence of copy number alterations on the miRNA expression pattern.
Nevertheless, we contributed new information about the involvement of miRNAs in MDS,
especially that of human Epstein-Barr virus miRNA (ebv-miR-BART13). To understand the
biological processes behind these alterations, we carried out pathway analysis; some of
these pathways were related to apoptosis, cell fate and myeloid cell differentiation.
Formalin-fixed paraffin-embedded tissue (FFPE) samples are widely available,
however, performing gene expression and even array CGH from FFPE samples is
challenging due to nucleic acid fragmentation/degradation. In Study III, we performed
microarray-based profiles for 19 FFPE leukemia samples and compared them to fresh frozen
bone marrow aspirate samples, which revealed that FFPE bone marrow biopsies (formalin-
fixed paraffin-embedded and decalcified specimens) are reliable sources of miRNA, and
that these samples are suitable for miRNA array profiling.
In the future, using the latest chip techniques and integrating various kinds of
genomic profiling data will confidently reveal more detailed information about the

54
pathogenesis of MPN and MDS. Observed genetic and epigenetic changes or an miRNA can
serve as biomarkers or targets for new drugs. It is important to identify clinically relevant
genes and, thus, more accurate diagnostic subgroups to be able to treat diseases more
effectively and to develop targeted therapies. In this way, the effectiveness of the treatments
can be improved considerably, and drug-related side effects as well as unnecessary medical
costs can be avoided.

55
ACKNOWLEDGEMENTS
This thesis work was carried out at the Department of Pathology, Haartman Institute,
University of Helsinki, during 2006-2011 and was supported financially by the Helsinki
University Central Hospital research founds and Finnish Association of Haematology. I
would like to address my sincere gratitude to everyone who helped me and made this work
possible, especially to:
Professor Sakari Knuutila, my supervisor, for giving me the opportunity to carry
on this works and for introducing me to the field of molecular genetics. I am grateful for his
guidance and for giving me the opportunity to learn so many things, and showed me the ins
and outs of the scientific research.
Docent Eeva Juvonen for providing the research material and her valuable
contribution and knowledge during this project. Satu Mustjoki, MD for her significant
contribution to this work and I am grateful for having the opportunity to work together.
The official reviewers, Docent Freja Ebeling and Docent Heli Nevanlinna, for
their suggestions and critical remarks were invaluable to me.
Pirjo Pennanen and Tarja Nieminen for their support in language revision and
help with many practical issues. Stephen Stalter for critical language revision of this book.
To all my co-authors for fruitful collaboration during these years. To my
friends outside the laboratory, especially Dalia, and colleagues of the CMG group: Neda,
Pamela, Anne, Ilari, Katja, Kowan, Linda, Leo, Penny, Shinsuke, Tarja, Virinder and
many others for creating such a nice working atmosphere and helping in solving any
problem, not just scientific, for many good advice and tips during these years. Tiina
Wirtanen is especially thanked for introducing and guiding me in the laboratory work at
the beginning of my PhD study.
I express my gratitude to my family for their support. Especially to my parents,
my sister Loredana, and my mother-in-law. My deepest gratitude goes to my husband
Marius for his love and support during these years and believing that I could finish this
project. To our lovely son Paul, to whom I dedicate this work, for his patience and
understanding. I am so lucky to have you in my life.
Helsinki, September 2011

56
REFERENCES Agirre, X., Jimenez-Velasco, A., San Jose-Eneriz, E., Garate, L., Bandres, E., Cordeu, L., Aparicio, O., Saez, B., Navarro, G., Vilas-Zornoza, A., Perez-Roger, I., Garcia-Foncillas, J., Torres, A., Heiniger, A., Calasanz, M.J., Fortes, P., Roman-Gomez, J. & Prosper, F. 2008. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Molecular cancer research : MCR 6: 1830-1840.
Ansell, S.M. 2008. Rare Hematological Malignancies. Vol. 142: 433.
Bartlett, J.M. & Stirling, D. 2003. A short history of the polymerase chain reaction. Methods in molecular biology (Clifton, N.J.) 226: 3-6.
Baxter, E.J., Scott, L.M., Campbell, P.J., East, C., Fourouclas, N., Swanton, S., Vassiliou, G.S., Bench, A.J., Boyd, E.M., Curtin, N., Scott, M.A., Erber, W.N., Green, A.R. & Cancer Genome Project. 2005. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365: 1054-1061.
Bejar, R. & Ebert, B.L. 2010. The genetic basis of myelodysplastic syndromes. Hematology/oncology clinics of North America 24: 295-315.
Bench, A.J. & Pahl, H.L. 2005. Chromosomal abnormalities and molecular markers in myeloproliferative disorders. Seminars in hematology 42: 196-205.
Benjamini, Y. & Hochberg, Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. 57: 289-300.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A., Gralnick, H.R. & Sultan, C. 1982. Proposals for the classification of the myelodysplastic syndromes. British journal of haematology 51: 189-199.
Bociek, R.G. & Armitage, J.O. 1996. Hematopoietic growth factors. CA: a cancer journal for clinicians 46: 165-184.
Boissinot, M., Lippert, E., Girodon, F., Dobo, I., Fouassier, M., Masliah, C., Praloran, V. & Hermouet, S. 2006. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood 108: 3223-3224.
Boultwood, J., Pellagatti, A., Cattan, H., Lawrie, C.H., Giagounidis, A., Malcovati, L., Della Porta, M.G., Jadersten, M., Killick, S., Fidler, C., Cazzola, M., Hellstrom-Lindberg, E. & Wainscoat, J.S. 2007. Gene expression profiling of CD34+ cells in patients with the 5q- syndrome. British journal of haematology 139: 578-589.
Bousquet, M., Quelen, C., Rosati, R., Mansat-De Mas, V., La Starza, R., Bastard, C., Lippert, E., Talmant, P., Lafage-Pochitaloff, M., Leroux, D., Gervais, C., Viguie, F., Lai, J.L., Terre, C., Beverlo, B., Sambani, C., Hagemeijer, A., Marynen, P., Delsol, G., Dastugue, N., Mecucci, C. & Brousset, P. 2008. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. The Journal of experimental medicine 205: 2499-2506.
Bray, I., Bryan, K., Prenter, S., Buckley, P.G., Foley, N.H., Murphy, D.M., Alcock, L., Mestdagh, P., Vandesompele, J., Speleman, F., London, W.B., McGrady, P.W., Higgins, D.G., O'Meara, A., O'Sullivan, M. & Stallings, R.L. 2009. Widespread dysregulation of MiRNAs by MYCN amplification and chromosomal imbalances in neuroblastoma: association of miRNA expression with survival. PloS one 4: e7850.
Bruchova, H., Merkerova, M. & Prchal, J.T. 2008. Aberrant expression of microRNA in polycythemia vera. Haematologica 93: 1009-1016.

57
Brunning, R.D., Orazi, A., Germing, U. & Le Beau, M.M. 2008. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. & Vardiman, J.W. (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. IARC.
Calin, G.A. & Croce, C.M. 2006. MicroRNA signatures in human cancers. Nature reviews.Cancer 6: 857-866.
Capdeville, R., Buchdunger, E., Zimmermann, J. & Matter, A. 2002. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nature reviews.Drug discovery 1: 493-502.
Cazzola, M. 2008. Myelodysplastic syndrome with isolated 5q deletion (5q- syndrome). A clonal stem cell disorder characterized by defective ribosome biogenesis. Haematologica 93: 967-972.
Chen, C.Z. 2005. MicroRNAs as oncogenes and tumor suppressors. The New England journal of medicine 353: 1768-1771.
Cotta, C.V. & Tubbs, R.R. 2008. Mutations in myeloid neoplasms. Diagnostic molecular pathology : the American journal of surgical pathology, part B 17: 191-199.
De Keersmaecker, K., Graux, C., Odero, M.D., Mentens, N., Somers, R., Maertens, J., Wlodarska, I., Vandenberghe, P., Hagemeijer, A., Marynen, P. & Cools, J. 2005. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32). Blood 105: 4849-4852.
Dickstein, J., Senyuk, V., Premanand, K., Laricchia-Robbio, L., Xu, P., Cattaneo, F., Fazzina, R. & Nucifora, G. 2010. Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proceedings of the National Academy of Sciences of the United States of America 107: 9783-9788.
Doleshal, M., Magotra, A.A., Choudhury, B., Cannon, B.D., Labourier, E. & Szafranska, A.E. 2008. Evaluation and validation of total RNA extraction methods for microRNA expression analyses in formalin-fixed, paraffin-embedded tissues. The Journal of molecular diagnostics : JMD 10: 203-211.
Dostalova Merkerova, M., Krejcik, Z., Votavova, H., Belickova, M., Vasikova, A. & Cermak, J. 2010. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. European journal of human genetics : EJHG.
Ebert, B.L. 2009. Deletion 5q in myelodysplastic syndrome: a paradigm for the study of hemizygous deletions in cancer. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 23: 1252-1256.
Ebert, B.L., Pretz, J., Bosco, J., Chang, C.Y., Tamayo, P., Galili, N., Raza, A., Root, D.E., Attar, E., Ellis, S.R. & Golub, T.R. 2008. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451: 335-339.
Espinet, B., Puigdecanet, E., Florensa, L., González, L., Salido, M. & Bellosillo, B. 2006. Array comparative genomic hybridization reveals an absence of recurrent genomic copy number changes in essential thrombocythemia. EHA Annual Meeting Abstract: 37.
Evers, C., Beier, M., Poelitz, A., Hildebrandt, B., Servan, K., Drechsler, M., Germing, U., Royer, H.D. & Royer-Pokora, B. 2007. Molecular definition of chromosome arm 5q deletion end points and detection of hidden aberrations in patients with myelodysplastic syndromes and isolated del(5q) using oligonucleotide array CGH. Genes, chromosomes & cancer 46: 1119-1128.
Farragher, S.M., Tanney, A., Kennedy, R.D. & Paul Harkin, D. 2008. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochemistry and cell biology 130: 435-445.
Fenaux, P., Mufti, G.J., Hellstrom-Lindberg, E., Santini, V., Finelli, C., Giagounidis, A., Schoch, R., Gattermann, N., Sanz, G., List, A., Gore, S.D., Seymour, J.F., Bennett, J.M., Byrd, J., Backstrom, J., Zimmerman, L., McKenzie, D., Beach, C., Silverman, L.R. & International Vidaza High-Risk MDS Survival Study Group. 2009. Efficacy of azacitidine compared with that of conventional care

58
regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. The lancet oncology 10: 223-232.
Fulci, V., Colombo, T., Chiaretti, S., Messina, M., Citarella, F., Tavolaro, S., Guarini, A., Foa, R. & Macino, G. 2009. Characterization of B- and T-lineage acute lymphoblastic leukemia by integrated analysis of MicroRNA and mRNA expression profiles. Genes, chromosomes & cancer 48: 1069-1082.
Gaken, J., Mohamedali, A.Z., Twine, N. & et al. 2008. MicroRNA Expression Profiling of High and Low Risk MDS. 112: 3645.
Gangnus, R., Langer, S., Breit, E., Pantel, K. & Speicher, M.R. 2004. Genomic profiling of viable and proliferative micrometastatic cells from early-stage breast cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research 10: 3457-3464.
Gleissner, B., Gokbuget, N., Bartram, C.R., Janssen, B., Rieder, H., Janssen, J.W., Fonatsch, C., Heyll, A., Voliotis, D., Beck, J., Lipp, T., Munzert, G., Maurer, J., Hoelzer, D., Thiel, E. & German Multicenter Trials of Adult Acute Lymphoblastic Leukemia Study Group. 2002. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood 99: 1536-1543.
Gondek, L.P., Tiu, R., O'Keefe, C.L., Sekeres, M.A., Theil, K.S. & Maciejewski, J.P. 2008. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood 111: 1534-1542.
Graubert, T.A., Payton, M.A., Shao, J., Walgren, R.A., Monahan, R.S., Frater, J.L., Walshauser, M.A., Martin, M.G., Kasai, Y. & Walter, M.J. 2009. Integrated genomic analysis implicates haploinsufficiency of multiple chromosome 5q31.2 genes in de novo myelodysplastic syndromes pathogenesis. PloS one 4: e4583.
Greenberg, P., Cox, C., LeBeau, M.M., Fenaux, P., Morel, P., Sanz, G., Sanz, M., Vallespi, T., Hamblin, T., Oscier, D., Ohyashiki, K., Toyama, K., Aul, C., Mufti, G. & Bennett, J. 1997. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89: 2079-2088.
Hammond, S.M. 2007. MicroRNAs as tumor suppressors. Nature genetics 39: 582-583.
Heim, S. & Mitelman, F. 2010. Cytogenetic Nomenclature, in Cancer Cytogenetics.
Heinrichs, S., Kulkarni, R.V., Bueso-Ramos, C.E., Levine, R.L., Loh, M.L., Li, C., Neuberg, D., Kornblau, S.M., Issa, J.P., Gilliland, D.G., Garcia-Manero, G., Kantarjian, H.M., Estey, E.H. & Look, A.T. 2009. Accurate detection of uniparental disomy and microdeletions by SNP array analysis in myelodysplastic syndromes with normal cytogenetics. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 23: 1605-1613.
Hermeking, H. 2010. The miR-34 family in cancer and apoptosis. Cell death and differentiation 17: 193-199.
Hibbin, J.A., Njoku, O.S., Matutes, E., Lewis, S.M. & Goldman, J.M. 1984. Myeloid progenitor cells in the circulation of patients with myelofibrosis and other myeloproliferative disorders. British journal of haematology 57: 495-503.
Hoffbrand, A.V., Moss, P.A.H. & Pettit, J.E. 2006. Essential haematology.
Huret, J.L. 1990. Complex translocations, simple variant translocations and Ph-negative cases in chronic myelogenous leukaemia. Human genetics 85: 565-568.
Hussein, K., Busche, G., Muth, M., Gohring, G., Kreipe, H. & Bock, O. 2011. Expression of myelopoiesis-associated microRNA in bone marrow cells of atypical chronic myeloid leukaemia and chronic myelomonocytic leukaemia. Annals of Hematology 90: 307-313.

59
Hussein, K., Theophile, K., Busche, G., Schlegelberger, B., Gohring, G., Kreipe, H. & Bock, O. 2010a. Aberrant microRNA expression pattern in myelodysplastic bone marrow cells. Leukemia research 34: 1169-1174.
Hussein, K., Theophile, K., Busche, G., Schlegelberger, B., Gohring, G., Kreipe, H. & Bock, O. 2010b. Significant inverse correlation of microRNA-150/MYB and microRNA-222/p27 in myelodysplastic syndrome. Leukemia research 34: 328-334.
Iafrate, A.J., Feuk, L., Rivera, M.N., Listewnik, M.L., Donahoe, P.K., Qi, Y., Scherer, S.W. & Lee, C. 2004. Detection of large-scale variation in the human genome. Nature genetics 36: 949-951.
Jadersten, M. & Hellstrom-Lindberg, E. 2010. New clues to the molecular pathogenesis of myelodysplastic syndromes. Experimental cell research 316: 1390-1396.
Jiang, Y., Dunbar, A., Gondek, L.P., Mohan, S., Rataul, M., O'Keefe, C., Sekeres, M., Saunthararajah, Y. & Maciejewski, J.P. 2009. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 113: 1315-1325.
Ju, X., Li, D., Shi, Q., Hou, H., Sun, N. & Shen, B. 2009. Differential microRNA expression in childhood B-cell precursor acute lymphoblastic leukemia. Pediatric hematology and oncology 26: 1-10.
Juvonen, E., Ikkala, E., Oksanen, K. & Ruutu, T. 1993. Megakaryocyte and erythroid colony formation in essential thrombocythaemia and reactive thrombocytosis: diagnostic value and correlation to complications. British journal of haematology 83: 192-197.
Kallioniemi, A., Kallioniemi, O.P., Sudar, D., Rutovitz, D., Gray, J.W., Waldman, F. & Pinkel, D. 1992. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science (New York, N.Y.) 258: 818-821.
Kantarjian, H., Shah, N.P., Hochhaus, A., Cortes, J., Shah, S., Ayala, M., Moiraghi, B., Shen, Z., Mayer, J., Pasquini, R., Nakamae, H., Huguet, F., Boque, C., Chuah, C., Bleickardt, E., Bradley-Garelik, M.B., Zhu, C., Szatrowski, T., Shapiro, D. & Baccarani, M. 2010. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. The New England journal of medicine 362: 2260-2270.
Kawamata, N., Ogawa, S., Yamamoto, G., Lehmann, S., Levine, R.L., Pikman, Y., Nannya, Y., Sanada, M., Miller, C.W., Gilliland, D.G. & Koeffler, H.P. 2008. Genetic profiling of myeloproliferative disorders by single-nucleotide polymorphism oligonucleotide microarray. Experimental hematology 36: 1471-1479.
Kerbauy, D.B. & Deeg, H.J. 2007. Apoptosis and antiapoptotic mechanisms in the progression of myelodysplastic syndrome. Experimental hematology 35: 1739-1746.
Klco, J.M., Vij, R., Kreisel, F.H., Hassan, A. & Frater, J.L. 2010. Molecular pathology of myeloproliferative neoplasms. American Journal of Clinical Pathology 133: 602-615.
Knuutila, S., Vuopio, P., Elonen, E., Siimes, M., Kovanen, R., Borgstrom, G.H. & de la Chapelle, A. 1981. Culture of bone marrow reveals more cells with chromosomal abnormalities than the direct method in patients with hematologic disorders. Blood 58: 369-375.
Kralovics, R., Passamonti, F., Buser, A.S., Teo, S.S., Tiedt, R., Passweg, J.R., Tichelli, A., Cazzola, M. & Skoda, R.C. 2005. A gain-of-function mutation of JAK2 in myeloproliferative disorders. The New England journal of medicine 352: 1779-1790.
Kralovics, R. & Skoda, R.C. 2005. Molecular pathogenesis of Philadelphia chromosome negative myeloproliferative disorders. Blood reviews 19: 1-13.
Krek, A., Grun, D., Poy, M.N., Wolf, R., Rosenberg, L., Epstein, E.J., MacMenamin, P., da Piedade, I., Gunsalus, K.C., Stoffel, M. & Rajewsky, N. 2005. Combinatorial microRNA target predictions. Nature genetics 37: 495-500.

60
Kumar, M., Narla, A., Nonami, A. & et al. 2009. Coordinate Loss of a MicroRNA Mir 145 and a Protein-Coding Gene RPS14 Cooperate in the Pathogenesis of 5q- Syndrome. 114: Abstract 947.
Lahiri, D.K. & Nurnberger, J.I.,Jr. 1991. A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic acids research 19: 5444.
Lamy, P., Andersen, C.L., Dyrskjot, L., Torring, N., Orntoft, T. & Wiuf, C. 2006. Are microRNAs located in genomic regions associated with cancer? British journal of cancer 95: 1415-1418.
Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., McKernan, K., Meldrim, J., Mesirov, J.P., Miranda, C., Morris, W., Naylor, J., Raymond, C., Rosetti, M., Santos, R., Sheridan, A., Sougnez, C., Stange-Thomann, N., Stojanovic, N., Subramanian, A., Wyman, D., Rogers, J., Sulston, J., Ainscough, R., Beck, S., Bentley, D., Burton, J., Clee, C., Carter, N., Coulson, A., Deadman, R., Deloukas, P., Dunham, A., Dunham, I., Durbin, R., French, L., Grafham, D., Gregory, S., Hubbard, T., Humphray, S., Hunt, A., Jones, M., Lloyd, C., McMurray, A., Matthews, L., Mercer, S., Milne, S., Mullikin, J.C., Mungall, A., Plumb, R., Ross, M., Shownkeen, R., Sims, S., Waterston, R.H., Wilson, R.K., Hillier, L.W., McPherson, J.D., Marra, M.A., Mardis, E.R., Fulton, L.A., Chinwalla, A.T., Pepin, K.H., Gish, W.R., Chissoe, S.L., Wendl, M.C., Delehaunty, K.D., Miner, T.L., Delehaunty, A., Kramer, J.B., Cook, L.L., Fulton, R.S., Johnson, D.L., Minx, P.J., Clifton, S.W., Hawkins, T., Branscomb, E., Predki, P., Richardson, P., Wenning, S., Slezak, T., Doggett, N., Cheng, J.F., Olsen, A., Lucas, S., Elkin, C., Uberbacher, E., Frazier, M., Gibbs, R.A., Muzny, D.M., Scherer, S.E., Bouck, J.B., Sodergren, E.J., Worley, K.C., Rives, C.M., Gorrell, J.H., Metzker, M.L., Naylor, S.L., Kucherlapati, R.S., Nelson, D.L., Weinstock, G.M., Sakaki, Y., Fujiyama, A., Hattori, M., Yada, T., Toyoda, A., Itoh, T., Kawagoe, C., Watanabe, H., Totoki, Y., Taylor, T., Weissenbach, J., Heilig, R., Saurin, W., Artiguenave, F., Brottier, P., Bruls, T., Pelletier, E., Robert, C., Wincker, P., Smith, D.R., Doucette-Stamm, L., Rubenfield, M., Weinstock, K., Lee, H.M., Dubois, J., Rosenthal, A., Platzer, M., Nyakatura, G., Taudien, S., Rump, A., Yang, H., Yu, J., Wang, J., Huang, G., Gu, J., Hood, L., Rowen, L., Madan, A., Qin, S., Davis, R.W., Federspiel, N.A., Abola, A.P., Proctor, M.J., Myers, R.M., Schmutz, J., Dickson, M., Grimwood, J., Cox, D.R., Olson, M.V., Kaul, R., Raymond, C., Shimizu, N., Kawasaki, K., Minoshima, S., Evans, G.A., Athanasiou, M., Schultz, R., Roe, B.A., Chen, F., Pan, H., Ramser, J., Lehrach, H., Reinhardt, R., McCombie, W.R., de la Bastide, M., Dedhia, N., Blocker, H., Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J.A., Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D.G., Burge, C.B., Cerutti, L., Chen, H.C., Church, D., Clamp, M., Copley, R.R., Doerks, T., Eddy, S.R., Eichler, E.E., Furey, T.S., Galagan, J., Gilbert, J.G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L.S., Jones, T.A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W.J., Kitts, P., Koonin, E.V., Korf, I., Kulp, D., Lancet, D., Lowe, T.M., McLysaght, A., Mikkelsen, T., Moran, J.V., Mulder, N., Pollara, V.J., Ponting, C.P., Schuler, G., Schultz, J., Slater, G., Smit, A.F., Stupka, E., Szustakowski, J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf, Y.I., Wolfe, K.H., Yang, S.P., Yeh, R.F., Collins, F., Guyer, M.S., Peterson, J., Felsenfeld, A., Wetterstrand, K.A., Patrinos, A., Morgan, M.J., de Jong, P., Catanese, J.J., Osoegawa, K., Shizuya, H., Choi, S., Chen, Y.J. & International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921.
Levine, R.L., Belisle, C., Wadleigh, M., Zahrieh, D., Lee, S., Chagnon, P., Gilliland, D.G. & Busque, L. 2006. X-inactivation-based clonality analysis and quantitative JAK2V617F assessment reveal a strong association between clonality and JAK2V617F in PV but not ET/MMM, and identifies a subset of JAK2V617F-negative ET and MMM patients with clonal hematopoiesis. Blood 107: 4139-4141.
Lewis, B.P., Burge, C.B. & Bartel, D.P. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15-20.
Li, J., Smyth, P., Flavin, R., Cahill, S., Denning, K., Aherne, S., Guenther, S.M., O'Leary, J.J. & Sheils, O. 2007. Comparison of miRNA expression patterns using total RNA extracted from

61
matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC biotechnology 7: 36.
Liu, H., Huang, X., Zhang, Y., Ye, H., El Hamidi, A., Kocjan, G., Dogan, A., Isaacson, P.G. & Du, M.Q. 2002. Archival fixed histologic and cytologic specimens including stained and unstained materials are amenable to RT-PCR. Diagnostic molecular pathology : the American journal of surgical pathology, part B 11: 222-227.
Lu, J., Getz, G., Miska, E.A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B.L., Mak, R.H., Ferrando, A.A., Downing, J.R., Jacks, T., Horvitz, H.R. & Golub, T.R. 2005. MicroRNA expression profiles classify human cancers. Nature 435: 834-838.
Lussana, F., Caberlon, S., Pagani, C., Kamphuisen, P.W., Buller, H.R. & Cattaneo, M. 2009. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thrombosis research 124: 409-417.
McMullin, M.F., Bareford, D., Campbell, P., Green, A.R., Harrison, C., Hunt, B., Oscier, D., Polkey, M.I., Reilly, J.T., Rosenthal, E., Ryan, K., Pearson, T.C., Wilkins, B. & General Haematology Task Force of the British Committee for Standards in Haematology. 2005. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. British journal of haematology 130: 174-195.
Mesa, R.A., Powell, H., Lasho, T., Dewald, G., McClure, R. & Tefferi, A. 2006. JAK2(V617F) and leukemic transformation in myelofibrosis with myeloid metaplasia. Leukemia research 30: 1457-1460.
Millot, F., Traore, P., Guilhot, J., Nelken, B., Leblanc, T., Leverger, G., Plantaz, D., Bertrand, Y., Bordigoni, P. & Guilhot, F. 2005. Clinical and biological features at diagnosis in 40 children with chronic myeloid leukemia. Pediatrics 116: 140-143.
Mitelman, F. 2000. Recurrent chromosome aberrations in cancer. Mutation research 462: 247-253.
Mossuz, P. & Groupe d'Etudes Multicentriques des Syndrome MyeloProliferatifs (GEMSMP). 2006. Influence of the assays of endogenous colony formation and serum erythropoietin on the diagnosis of polycythemia vera and essential thrombocythemia. Seminars in thrombosis and hemostasis 32: 246-250.
Nassiri, M., Olczyk, J., knapp, S., Vance, G., Tewari, A., Kemp, S. & Czader, M. 2010. MicroRNA Expression Profiles in Chronic Myelomonocytic Leukemia. 50th ASH annual meeting.
Negrini, M., Nicoloso, M.S. & Calin, G.A. 2009. MicroRNAs and cancer--new paradigms in molecular oncology. Current opinion in cell biology 21: 470-479.
Nimer, S.D. 2008. Myelodysplastic syndromes. Blood 111: 4841-4851.
Panani, A.D. 2007. Cytogenetic and molecular aspects of Philadelphia negative chronic myeloproliferative disorders: clinical implications. Cancer letters 255: 12-25.
Pardanani, A.D., Levine, R.L., Lasho, T., Pikman, Y., Mesa, R.A., Wadleigh, M., Steensma, D.P., Elliott, M.A., Wolanskyj, A.P., Hogan, W.J., McClure, R.F., Litzow, M.R., Gilliland, D.G. & Tefferi, A. 2006. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108: 3472-3476.
Passamonti, F., Rumi, E., Arcaini, L., Boveri, E., Elena, C., Pietra, D., Boggi, S., Astori, C., Bernasconi, P., Varettoni, M., Brusamolino, E., Pascutto, C. & Lazzarino, M. 2008. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica 93: 1645-1651.
Passamonti, F., Rumi, E., Pietra, D., Elena, C., Boveri, E., Arcaini, L., Roncoroni, E., Astori, C., Merli, M., Boggi, S., Pascutto, C., Lazzarino, M. & Cazzola, M. 2010. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on

62
fibrotic or leukemic disease transformation and vascular complications. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 24: 1574-1579.
Pastore, C., Nomdedeu, J., Volpe, G., Guerrasio, A., Cambrin, G.R., Parvis, G., Pautasso, M., Daglio, C., Mazza, U. & Saglio, G. 1995. Genetic analysis of chromosome 13 deletions in BCR/ABL negative chronic myeloproliferative disorders. Genes, chromosomes & cancer 14: 106-111.
Pfeffer, S. & Voinnet, O. 2006. Viruses, microRNAs and cancer. Oncogene 25: 6211-6219.
Pillai, R.S. 2005. MicroRNA function: multiple mechanisms for a tiny RNA? RNA (New York, N.Y.) 11: 1753-1761.
Pinkel, D., Segraves, R., Sudar, D., Clark, S., Poole, I., Kowbel, D., Collins, C., Kuo, W.L., Chen, C., Zhai, Y., Dairkee, S.H., Ljung, B.M., Gray, J.W. & Albertson, D.G. 1998. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature genetics 20: 207-211.
Pons, A., Nomdedeu, B., Navarro, A., Gaya, A., Gel, B., Diaz, T., Valera, S., Rozman, M., Belkaid, M., Montserrat, E. & Monzo, M. 2009. Hematopoiesis-related microRNA expression in myelodysplastic syndromes. Leukemia & lymphoma 50: 1854-1859.
Prchal, J.F. & Axelrad, A.A. 1974. Letter: Bone-marrow responses in polycythemia vera. The New England journal of medicine 290: 1382.
Ramkissoon, S.H., Mainwaring, L.A., Ogasawara, Y., Keyvanfar, K., McCoy, J.P.,Jr, Sloand, E.M., Kajigaya, S. & Young, N.S. 2006. Hematopoietic-specific microRNA expression in human cells. Leukemia research 30: 643-647.
Raza, A. 1998. Hypothesis: myelodysplastic syndromes may have a viral etiology. International journal of hematology 68: 245-256.
Raza, A. 2000. Myelodysplastic syndromes may have an infectious etiology. Journal of toxicology and environmental health.Part A 61: 387-390.
Reilly, J.T. 2002. Cytogenetic and molecular genetic aspects of idiopathic myelofibrosis. Acta Haematologica 108: 113-119.
Rumi, E., Elena, C. & Passamonti, F. 2010. Mutational status of myeloproliferative neoplasms. Critical reviews in eukaryotic gene expression 20: 61-76.
Saglio, G., Kim, D.W., Issaragrisil, S., le Coutre, P., Etienne, G., Lobo, C., Pasquini, R., Clark, R.E., Hochhaus, A., Hughes, T.P., Gallagher, N., Hoenekopp, A., Dong, M., Haque, A., Larson, R.A., Kantarjian, H.M. & ENESTnd Investigators. 2010. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. The New England journal of medicine 362: 2251-2259.
Santarpia, L., Nicoloso, M. & Calin, G.A. 2010. MicroRNAs: a complex regulatory network drives the acquisition of malignant cell phenotype. Endocrine-related cancer 17: F51-75.
Schotte, D., Chau, J.C., Sylvester, G., Liu, G., Chen, C., van der Velden, V.H., Broekhuis, M.J., Peters, T.C., Pieters, R. & den Boer, M.L. 2009. Identification of new microRNA genes and aberrant microRNA profiles in childhood acute lymphoblastic leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 23: 313-322.
Scott, L.M., Tong, W., Levine, R.L., Scott, M.A., Beer, P.A., Stratton, M.R., Futreal, P.A., Erber, W.N., McMullin, M.F., Harrison, C.N., Warren, A.J., Gilliland, D.G., Lodish, H.F. & Green, A.R. 2007. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. The New England journal of medicine 356: 459-468.
Selcuklu, S.D., Donoghue, M.T. & Spillane, C. 2009. miR-21 as a key regulator of oncogenic processes. Biochemical Society transactions 37: 918-925.

63
Seto, E., Moosmann, A., Gromminger, S., Walz, N., Grundhoff, A. & Hammerschmidt, W. 2010. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS pathogens 6: e1001063.
Silverman, L.R. 2003. Myelodysplastic Syndrome. In: Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F. & Frei, E. (eds.) Cancer Medicine, 6th edition. ed. BC Decker, Hamilton (ON).
Slovak, M.L., Smith, D.D., Bedell, V., Hsu, Y.H., O'Donnell, M., Forman, S.J., Gaal, K., McDaniel, L., Schultz, R., Ballif, B.C. & Shaffer, L.G. 2010. Assessing karyotype precision by microarray-based comparative genomic hybridization in the myelodysplastic/myeloproliferative syndromes. Molecular cytogenetics 3: 23.
Solinas-Toldo, S., Lampel, S., Stilgenbauer, S., Nickolenko, J., Benner, A., Dohner, H., Cremer, T. & Lichter, P. 1997. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes, chromosomes & cancer 20: 399-407.
Speicher, M.R. & Carter, N.P. 2005. The new cytogenetics: blurring the boundaries with molecular biology. Nature reviews.Genetics 6: 782-792.
Spivak, J.L. 2002. Polycythemia vera: myths, mechanisms, and management. Blood 100: 4272-4290.
Srinivasan, M., Sedmak, D. & Jewell, S. 2002. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. The American journal of pathology 161: 1961-1971.
Starczynowski, D.T., Kuchenbauer, F., Argiropoulos, B., Sung, S., Morin, R., Muranyi, A., Hirst, M., Hogge, D., Marra, M., Wells, R.A., Buckstein, R., Lam, W., Humphries, R.K. & Karsan, A. 2010. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nature medicine 16: 49-58.
Starczynowski, D.T., Vercauteren, S., Telenius, A., Sung, S., Tohyama, K., Brooks-Wilson, A., Spinelli, J.J., Eaves, C.J., Eaves, A.C., Horsman, D.E., Lam, W.L. & Karsan, A. 2008. High-resolution whole genome tiling path array CGH analysis of CD34+ cells from patients with low-risk myelodysplastic syndromes reveals cryptic copy number alterations and predicts overall and leukemia-free survival. Blood 112: 3412-3424.
Stegelmann, F., Bullinger, L., Griesshammer, M., Holzmann, K., Habdank, M., Kuhn, S., Maile, C., Schauer, S., Dohner, H. & Dohner, K. 2010. High-resolution single-nucleotide polymorphism array-profiling in myeloproliferative neoplasms identifies novel genomic aberrations. Haematologica 95: 666-669.
Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. & Vardiman, J.W. 2008. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.
Tasheva, E.S. & Roufa, D.J. 1995. A densely methylated DNA island is associated with a chromosomal replication origin in the human RPS14 locus. Somatic cell and molecular genetics 21: 369-383.
Tefferi, A. 2006. Classification, diagnosis and management of myeloproliferative disorders in the JAK2V617F era. Hematology / the Education Program of the American Society of Hematology.American Society of Hematology.Education Program: 240-245.
Tefferi, A. 2010. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 24: 1128-1138.
Tefferi, A., Lim, K.H., Abdel-Wahab, O., Lasho, T.L., Patel, J., Patnaik, M.M., Hanson, C.A., Pardanani, A., Gilliland, D.G. & Levine, R.L. 2009a. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 23: 1343-1345.

64
Tefferi, A., Sirhan, S., Sun, Y., Lasho, T., Finke, C.M., Weisberger, J., Bale, S., Compton, J., LeDuc, C.A., Pardanani, A., Thorland, E.C., Shevchenko, Y., Grodman, M. & Chung, W.K. 2009b. Oligonucleotide array CGH studies in myeloproliferative neoplasms: comparison with JAK2V617F mutational status and conventional chromosome analysis. Leukemia research 33: 662-664.
Tefferi, A., Thiele, J. & Vardiman, J.W. 2009c. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer 115: 3842-3847.
Tefferi, A. & Vardiman, J.W. 2008. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K 22: 14-22.
Tefferi, A. & Vardiman, J.W. 2009. Myelodysplastic syndromes. The New England journal of medicine 361: 1872-1885.
Thiele, J., Kvasnicka, H.M., Orazi, A., Tefferi, A. & Birgegard, G. 2008a. Polycythemia vera. In: Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. & Vardiman, J.W. (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. International Agency for Research on Cancer (IARC), Lion, France.
Thiele, J., Kvasnicka, H.M., Orazi, A., Tefferi, A. & Glisslinger, H. 2008b. Essential Thrombocythemia. In: Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. & Vardiman, J.W. (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. International Agency for Research on Cancer (IARC), Lyon, France.
Thiele, J., Kvasnicka, H.M., Tefferi, A., Barosi, G., Orazi, A. & Vardiman, J.W. 2008c. Primary myelofibrosis . In: Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. & Vardiman, J.W. (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. International Agency for Research on Cancer (IARC), Lion, France.
Thoennissen, N.H., Krug, U.O., Lee, D.H., Kawamata, N., Iwanski, G.B., Lasho, T., Weiss, T., Nowak, D., Koren-Michowitz, M., Kato, M., Sanada, M., Shih, L.Y., Nagler, A., Raynaud, S.D., Muller-Tidow, C., Mesa, R., Haferlach, T., Gilliland, D.G., Tefferi, A., Ogawa, S. & Koeffler, H.P. 2010. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood 115: 2882-2890.
Valencia, A., Cervera, J., Such, E., Sanz, M.A. & Sanz, G.F. 2008. Lack of RPS14 promoter aberrant methylation supports the haploinsufficiency model for the 5q- syndrome. Blood 112: 918.
van Prooijen-Knegt, A.C., Raap, A.K., van der Burg, M.J., Vrolijk, J. & van der Ploeg, M. 1982. Spreading and staining of human metaphase chromosomes on aminoalkylsilane-treated glass slides. The Histochemical journal 14: 333-344.
Vardiman, J.W., Melo, J.V., Baccarani, M. & Thiele, J. 2008. Chronic myelogenous leukaemia, BCR-ABL1 positive. In: Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J. & Vardiman, J.W. (eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. IARC.
Verstovsek, S. & Tefferi, A. 2011. Myeloproliferative neoplasms Biology and therapy.: 227.
Vissers, L.E., Veltman, J.A., van Kessel, A.G. & Brunner, H.G. 2005. Identification of disease genes by whole genome CGH arrays. Human molecular genetics 14 Spec No. 2: R215-23.
Wadhwa, M. & Thorpe, R. 2008. Haematopoietic growth factors and their therapeutic use. Thrombosis and haemostasis 99: 863-873.
Wang, D., Qiu, C., Zhang, H., Wang, J., Cui, Q. & Yin, Y. 2010. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: from functions to targets. PloS one 5: e13067.

65
Webersinke, G. & Rumpold, H. 2009. Pathogenetic and clinical impact of JAK2 mutations in chronic myeloproliferative diseases. 2: 89-93.
Westwood, N.B. & Pearson, T.C. 1996. Diagnostic applications of haemopoietic progenitor culture techniques in polycythaemias and thrombocythaemias. Leukemia & lymphoma 22 Suppl 1: 95-103.
Wong, J.C., Le Beau, M.M. & Shannon, K. 2008. Tumor suppressor gene inactivation in myeloid malignancies. Best practice & research.Clinical haematology 21: 601-614.
Zanette, D.L., Rivadavia, F., Molfetta, G.A., Barbuzano, F.G., Proto-Siqueira, R., Silva-Jr, W.A., Falcao, R.P. & Zago, M.A. 2007. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Brazilian journal of medical and biological research 40: 1435-1440.
Zanjani, E.D., Lutton, J.D., Hoffman, R. & Wasserman, L.R. 1977. Erythroid colony formation by polycythemia vera bone marrow in vitro. Dependence on erythropoietin. The Journal of clinical investigation 59: 841-848.
Zhang, C. 2009. Novel functions for small RNA molecules. Current opinion in molecular therapeutics 11: 641-651.
Zhang, L., Jamaluddin, M.S., Weakley, S.M., Yao, Q. & Chen, C. 2011. Roles and Mechanisms of MicroRNAs in Pancreatic Cancer. World journal of surgery.
Related Documents