770–784 Nucleic Acids Research, 2008, Vol. 36, No. 3 Published online 15 December 2007 doi:10.1093/nar/gkm1105 Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells Jairo Rodriguez 1 , Laura Vives 2,3 , Mireia Jorda ` 1 , Cristina Morales 1 , Mar Mun ˜ oz 2,3 , Elisenda Vendrell 1 and Miguel A. Peinado 1,2, * 1 Institut d’Investigacio ´ Biome ` dica de Bellvitge (IDIBELL), L’Hospitalet, 2 Institut de Medicina Predictiva i Personalitzada del Ca ` ncer (IMPPC), Badalona and 3 Institut Catala ` d’Oncologia (ICO), L’Hospitalet, Catalonia, Spain Received September 19, 2007; Revised October 19, 2007; Accepted November 27, 2007 ABSTRACT Methylation of the cytosine is the most frequent epigenetic modification of DNA in mammalian cells. In humans, most of the methylated cytosines are found in CpG-rich sequences within tandem and interspersed repeats that make up to 45% of the human genome, being Alu repeats the most common family. Demethylation of Alu elements occurs in aging and cancer processes and has been asso- ciated with gene reactivation and genomic instabil- ity. By targeting the unmethylated SmaI site within the Alu sequence as a surrogate marker, we have quantified and identified unmethylated Alu elements on the genomic scale. Normal colon epithelial cells contain in average 25 486 10 157 unmethylated Alu’s per haploid genome, while in tumor cells this figure is 41 995 17 187 (P = 0.004). There is an inverse relationship in Alu families with respect to their age and methylation status: the youngest elements exhibit the highest prevalence of the SmaI site (AluY: 42%; AluS: 18%, AluJ: 5%) but the lower rates of unmethylation (AluY: 1.65%; AluS: 3.1%, AluJ: 12%). Data are consistent with a stronger silencing pressure on the youngest repetitive ele- ments, which are closer to genes. Further insights into the functional implications of atypical unmethy- lation states in Alu elements will surely contribute to decipher genomic organization and gene regula- tion in complex organisms. INTRODUCTION Progress in large-scale sequencing projects is critical to identify and decipher gene organization and regulation in many species including human. Nevertheless, cumulated evidences indicate that the complexity of living organisms is not just a direct outcome of the number of coding sequences and that the presence of multiple regulatory mechanisms accounts for a significant part of biological complexity (1,2). Among these mechanisms, repetitive elements may play a key role in gene regulation and geno- mic structure. Active transposable elements are involved in genome rearrangement and illegitimate recombination and can also influence gene expression by altering splicing or by acting as enhancers or promoters (3–7). Advances in the understanding of epigenetic mechanisms that regulate these repetitive elements may contribute to elucidate their specific participation in biological processes (8). Silenced regions in mammals and other vertebrates are differentiated, although not exclusively, by the presence of DNA methylation (9). Methylation of the cytosine is an epigenetic modification of DNA that plays an impor- tant role in the control of gene expression and chromo- some structure in mammalian cells (10–13). Most of the 5-methylcytosines are found in CpG-rich sequences within tandem and interspersed repeats (9,12) of which the previous estimates indicate that constitute up to 45% of the human genome (14). Among these repeats, Alu’s, with more than one million copies per haploid genome, are considered the most successful family (15). Interestingly, Alu’s are not randomly distributed within the human genome, as they tend to accumulate in gene-rich regions (14,16,17). Previous works have estimated that Alu ele- ments harbor up to 33% of the total number of CpG sites in the genome (18) and have been reported to be highly methylated in most somatic tissues (18–20). Methylation represents the primary mechanism of transposon suppres- sion and active transposons are demethylated in mamma- lian genomes (12). It has been proposed that regions of the genome containing repetitive elements might be masked by compartmentalization of the chromatin, resulting in a reduction of the effective size of the genome (21). Noteworthy, even though a vast number of CpG dinucle- otides are provided by the collection of repetitive sequences in the human genome, this dinucleotide is greatly under- represented throughout the genome, but it can be found *To whom correspondence should be addressed. Tel: +34 934978693; Fax: +34 934978697; Email: [email protected] ß 2007 The Author(s) This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/ by-nc/2.0/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. by guest on November 8, 2015 http://nar.oxfordjournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript
770ndash784 Nucleic Acids Research 2008 Vol 36 No 3 Published online 15 December 2007doi101093nargkm1105
Genome-wide tracking of unmethylated DNAAlu repeats in normal and cancer cellsJairo Rodriguez1 Laura Vives23 Mireia Jorda1 Cristina Morales1 Mar Munoz23
Elisenda Vendrell1 and Miguel A Peinado12
1Institut drsquoInvestigacio Biomedica de Bellvitge (IDIBELL) LrsquoHospitalet 2Institut de Medicina Predictiva iPersonalitzada del Cancer (IMPPC) Badalona and 3Institut Catala drsquoOncologia (ICO) LrsquoHospitaletCatalonia Spain
Received September 19 2007 Revised October 19 2007 Accepted November 27 2007
ABSTRACT
Methylation of the cytosine is the most frequentepigenetic modification of DNA in mammalian cellsIn humans most of the methylated cytosinesare found in CpG-rich sequences within tandemand interspersed repeats that make up to 45 of thehuman genome being Alu repeats themost commonfamily Demethylation of Alu elements occurs inaging and cancer processes and has been asso-ciated with gene reactivation and genomic instabil-ity By targeting the unmethylated SmaI site withinthe Alu sequence as a surrogate marker we havequantified and identified unmethylated Alu elementson the genomic scale Normal colon epithelial cellscontain in average 25 486 10157 unmethylatedAlursquos per haploid genome while in tumor cells thisfigure is 41 995 17187 (P=0004) There is aninverse relationship in Alu families with respect totheir age and methylation status the youngestelements exhibit the highest prevalence of the SmaIsite (AluY 42 AluS 18 AluJ 5) but the lowerrates of unmethylation (AluY 165 AluS 31AluJ 12) Data are consistent with a strongersilencing pressure on the youngest repetitive ele-ments which are closer to genes Further insightsinto the functional implications of atypical unmethy-lation states in Alu elements will surely contributeto decipher genomic organization and gene regula-tion in complex organisms
INTRODUCTION
Progress in large-scale sequencing projects is critical toidentify and decipher gene organization and regulation inmany species including human Nevertheless cumulatedevidences indicate that the complexity of living organisms
is not just a direct outcome of the number of codingsequences and that the presence of multiple regulatorymechanisms accounts for a significant part of biologicalcomplexity (12) Among these mechanisms repetitiveelements may play a key role in gene regulation and geno-mic structure Active transposable elements are involvedin genome rearrangement and illegitimate recombinationand can also influence gene expression by altering splicingor by acting as enhancers or promoters (3ndash7) Advances inthe understanding of epigenetic mechanisms that regulatethese repetitive elements may contribute to elucidate theirspecific participation in biological processes (8)
Silenced regions in mammals and other vertebrates aredifferentiated although not exclusively by the presenceof DNA methylation (9) Methylation of the cytosine isan epigenetic modification of DNA that plays an impor-tant role in the control of gene expression and chromo-some structure in mammalian cells (10ndash13) Most of the5-methylcytosines are found in CpG-rich sequences withintandem and interspersed repeats (912) of which theprevious estimates indicate that constitute up to 45 ofthe human genome (14) Among these repeats Alursquos withmore than one million copies per haploid genome areconsidered the most successful family (15) InterestinglyAlursquos are not randomly distributed within the humangenome as they tend to accumulate in gene-rich regions(141617) Previous works have estimated that Alu ele-ments harbor up to 33 of the total number of CpG sitesin the genome (18) and have been reported to be highlymethylated in most somatic tissues (18ndash20) Methylationrepresents the primary mechanism of transposon suppres-sion and active transposons are demethylated in mamma-lian genomes (12) It has been proposed that regions of thegenome containing repetitive elements might be maskedby compartmentalization of the chromatin resulting ina reduction of the effective size of the genome (21)
Noteworthy even though a vast number of CpGdinucle-otides are provided by the collection of repetitive sequencesin the human genome this dinucleotide is greatly under-represented throughout the genome but it can be found
To whom correspondence should be addressed Tel +34 934978693 Fax +34 934978697 Email mapimppcorg
2007 The Author(s)
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (httpcreativecommonsorglicenses
by-nc20uk) which permits unrestricted non-commercial use distribution and reproduction in any medium provided the original work is properly cited
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at close to its expected frequency in small genomic regions(200 bp to a few kb) known as CpG islands (22) Theseareas are lsquoprotectedrsquo from methylation and are locatedin the proximal promoter regions of 75 of human genes(121322) Methylated CpG islands are strongly andhereditably repressed (12) Hence DNA methylation isusually considered as a sign of long-term inactivation(91012)
Cancer cells are characterized by the accumulation ofboth genetic and epigenetic changes Widespread genomichypomethylation is an early alteration in carcinogenesisand has been associated with genomic disruption andgenetic instability (23ndash27) Repeats unmasked by demeth-ylation are likely to facilitate rearrangements due to mito-tic recombination and unwanted transcription (28ndash30)Alternatively aberrant de novo methylation of CpGislands is a hallmark of human cancers and is associatedwith epigenetic silencing of multiple tumor suppressorgenes (31ndash37) Therefore the screening for differentiallymethylated sequences in tumors appears as a key toolto further understand the molecular mechanisms under-lying malignant transformation of cells Although therepertoire of methylation screening methodologies hasexpanded widely (37ndash39) and different approaches havebeen used to make bulk estimates of methylation inrepetitive elements (4041) there is still a lack of screeningstrategies that specifically allow a feasible identification ofDNA methylation alterations in repetitive elements (21)
Here we report two variants of a novel methodologyto quantify and identify unmethylated Alu sequencesThe CpG site within the consensus Alu sequence AACCCGGG is used as a surrogate reporter of methylationUnmethylated sites are cut with the methylation-sensitiverestriction endonuclease SmaI (CCCGGG) and an adap-tor is ligated to the DNA ends Quantification ofUnMethylated Alus (QUMA) is performed by real-timeamplification of the digested and adaptor-ligated DNAusing an Alu consensus primer that anneals upstreamof the SmaI site and an adaptor primer extended with theTT dinucleotide in its 30 end (Figure 1A) The productgenerated by this approach is completely inside theAlu element and hence it is not possible to make aunique identification As an alternative approach we havealso performed restrained amplification of digested andadaptor-ligated DNA fragments that are flanked by twoclose SmaI sites In this case the same primer homologousto the adaptor with the additional TT nucleotides at the30 end to enrich for Alu sequences is used in absence of theAlu consensus primer (Figure 1B) This second approachis named Amplification of UnMethylated Alursquos (AUMA)and results in a complex representation of unique DNAsequences flanked by two unmethylated SmaI sites Whenresolved by high-resolution electrophoresis the AUMAgenerated sequences appear as a fingerprint characteristicof each sample (Figure 2) and individual scoring andidentification of each band can be performed BecauseAUMArsquos stringency is based on a short sequence(AACCCGGG) that is found preferentially but not exclu-sively in Alu elements other unmethylated sequences arealso present in AUMA fingerprints
Application of QUMA and AUMA to a series of colo-rectal carcinomas and their paired normal mucosa hasoffered global estimates of unmethylation of Alu elementsin normal and cancer cells and has revealed a large collec-tion of unique sequences that undergo highly recurrenthypomethylation and hypermethylation in colorectaltumors
MATERIALS AND METHODS
Tissues and cell lines
Fifty colorectal carcinomas and their paired non-adjacent areas of normal colonic mucosa were included
Figure 1 Schematic diagram of the QUMA and AUMA methods DNAis depicted by a solid line Alu elements are represented by dashed boxesThe QUMA and AUMA recognition sites (AACCCGGG) are repre-sented by dashedgray boxes CpGs at SmaI sites are shown as full circleswhen methylated and as open circles when unmethylated The methyla-tion-sensitive restriction endonuclease SmaI can only digest unmethylatedtargets leaving blunt ends to which adaptors can be ligated (A) QUMA isperformed by real-time PCR of an inner Alu fragment using a primercomplementary to the Alu consensus sequence upstream of the SmaI siteand the primer complementary to the adaptor to which two Aluhomologous nucleotides (TT) have been added (B) In AUMA sequencesflanked by two ligated adaptors are amplified by PCR using a singleprimer the same adaptor primer plus the TT nucleotidesWhen only a fewnucleotides are added to the primer ie TT as illustrated here other non-Alu sequences may be amplified This allows the amplification of a largenumber of sequences that typically range from 100 to 2000 bp
Nucleic Acids Research 2008 Vol 36 No 3 771
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
in this analysis Samples were collected simultaneously asfresh specimens and snap-frozen within 2 h of removal andthen stored at 808C All samples were obtained from theCiutat Sanitaria i Universitaria de Bellvitge (BarcelonaSpain) The study protocol was approved by the EthicsCommittee Human colon cancer cell lines (HT29 SW480HCT116 LoVo DLD-1 CaCo-2 and LS174T) wereobtained from the American Type Culture Collection(ATCCManassas VA) KM12C and KM12SM cells weregenerously provided by A Fabra DNA from tumorndashnormal pairs was obtained by conventional organic extrac-tion and ethanol precipitation DNA purity and qualitywas checked in a 08 agarose gel electrophoresis RNAfrom cell lines was obtained by phenolndashchloroformextraction and ethanol precipitation following standardprocedures
Bioinformatic analysis
The distribution of SmaI sites putative amplification hitsPCR homologies CpG islands and repetitive elementswas assessed using the human genome assembly 361 fromNCBI Data were obtained from the Repbase (httpwwwgirinstorgrepbaseindexhtml) and the GenomeBrowser Databases (httphgdownloadcseucscedugoldenPathhg18database) Only assembled chromo-some fragments were considered A Perl routine wasused to score all positions containing the target sequencesin all chromosomes (available from the authors uponrequest) Data were analyzed using Excel spreadsheetsTo calculate the proportion of unmethylated Alu
elements at the genomic level the number of AUMAhits identified in bioinformatic analysis were correctedaccording to the distribution of experimentally generated
AUMA products performing Monte Carlo simulationsOne thousand Monte Carlo simulations were performedusing an Excel Add-in (available at wwwwabashedueconometrics) In Monte Carlo simulations it wasassumed that 80ndash100 of SmaI sites at CpG islands areunmethylated and that 50ndash100 of SmaI sites in othergenomic regions different from Alursquos and CpG islandsare unmethylated
Quantification of QUMA
One microgram of DNA was digested with 20U ofthe methylation sensitive restriction endonucleaseSmaI (Roche Diagnostics GmbH Mannheim Germany)for 16 h at 308C leaving cleaved fragments with bluntends (CCCGGG) Adaptors were prepared incubatingthe oligonucleotides Blue (CCGAATTCGCAAAGCTCTGA) and the 50 phosphorylated MCF oligonucleotide(TCAGAGCTTTGCGAAT) at 658C for 2min and thencooling to room temperature for 30ndash60min One micro-gram of the digested DNA was ligated to 2 nmol ofadaptor using T4 DNA ligase (New England BiolabsBeverly MA USA) Subsequent digestion of the ligatedproducts with the methylation insensitive restrictionendonuclease XmaI (New England Biolabs) was per-formed to avoid amplifications from non-digested methy-lated Alursquos The products were purified using the GFXKit (Amersham Biosciences Buckinghamshire UK) andeluted in 250 ml of sterile water
Quantitative real-time PCR was performed using 1 ng(the equivalent of 333 genomes) of DNA in a LightCycler480 real-time PCR system with Fast Start Master SYBRGreen I kit (Roche) Mastermix was prepared to a finalconcentration of 35mM MgCl2 and 1 mM of each primerThe downstream BAu-TT primer (constituted by the 30
end of Blue primer and the GGGTT sequence includingthe GGG 30 side of the cut SmaI site and the Aluhomologous TT dinucleotide ATTCGCAAAGCTCTGAGGGTT) and the upstream primer was an Aluconsensus sequence (CCGTCTCTACTAAAAATACA)(see Supplementary Data) Magnitudes were expressedas number of unmethylated Alus per haploid genomeafter DNA input normalization The number of haploidgenomes present in the test tube was determined in thesame multiwell plate by quantification of Alu sequencesirrespectively of the methylation state A real-time PCRusing Alu consensus primers upstream of the CCCGGGsite was performed (see Supplementary Data) and thenumber of genomes was calculated against a standardcurve constructed with a reference genomic DNA mea-sured by UV spectrophotometry
To determine the efficiency of the assay and to performabsolute quantification an external Alu product generatedby PCR from a DNA fragment containing an AluSxelement was used as standard (Supplementary Methods)The number of copies of the external control were spectro-photometrically quantified and dilution curves were gene-rated and treated as samples Comparison of dilutioncurves before and after sample processing indicated thatthe mean recovery was 73 DNA samples overdigested
Figure 2 AUMA of normal (N)ndashtumor (T) pairs of two differentpatients performed using primer BAu-TT A highly reproducible bandpatterning is observed among the four replicates Representative bandsshowing gains (hypomethylations) and losses (hypermethylations) aremarked with up and down arrowheads respectively
772 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
with the methylation insensitive XmaI endonuclease werespiked with different amounts of the external standard andprocessed The sensitivity of the QUMA detection was 100unmethylated Alursquos per haploid genome (SupplementaryFigure 1) using 1 ng of genomic DNA per PCR A linearresponse was observed between 1000 and 100 000 unmethy-lated Alursquos per haploid genome (Supplementary Data)
Amplification of AUMA
DNAdigestion with SmaI enzyme and ligation to the linkerwas performed as described above for QUMA except forthe XmaI digestion that was skipped The product waspurified using the GFX Kit (Amersham Biosciences) andeluted in 250 ml of sterile water Six different chimericprimers constituted by the 30 end of the Blue primersequence (ATTCGCAAAGCTCTGA) the cut SmaI site(GGG) and two four or seven additional nucleotideshomologous to the Alu consensus sequence were used toenrich for Alu sequences (see Supplementary Methods)Three primers were designed to amplify lsquoupstreamrsquo of theSmaI site (towards ALUpromoter) BAu-TT BAu-TTCABAu-TTCAAGC Three other primers were designed toamplify lsquodownstreamrsquo (towards ALU poly-A) BAd-AGBAd-AGGC BAd-AGGCGGA Letters after the dashcorrespond to the 30 sequence of the primer (see Supple-mentary Data) Data reported here were obtained by usingthe BAu-TT primer
In each PCR reaction only one primer was used ata time Products were resolved on denaturing sequencinggels Although bands can be visualized by silver stainingof the gels radioactive AUMArsquos were performed fornormalndashtumor comparisons A more detailed descriptionof the PCR and the visualization of the bands are givenas Supplementary Data
Only sharp bands that were reproducible and clearlydistinguishable from the background were tagged andincluded in the analysis Faint bands with inconsistentdisplay due to small variations in gel electrophoresisresolution were not considered Band reproducibility wasassessed with the analysis of PCR duplicates of threeindependent sample digests from two different samplesand PCR replicates from the same digest from four pairedtumorndashnormal samples AUMA fingerprints were visuallychecked for methylation differences between bands in thetumor with regard to its paired normal mucosa Underthese premises a given band was scored according to threepossible behaviors hypomethylation (increased intensityin the tumor) hypermethylation (decreased intensity inthe tumor) and no change (no substantial difference inintensity between normal and tumor samples) (Figure 2)Only those bands showing clear changes in their intensitiesin the fingerprint were considered to represent methylationchanges This is consistent with previous studies doneusing a related technique (4243)
Competitive hybridization of AUMA products to metaphasechromosomes and BAC arrays
The origin and chromosomal distribution of sequencesgenerated by AUMA was analyzed using procedures
analogous to CGH Briefly an AUMA product obtainedfrom a normal tissue DNA was purified using Jet quickPCR product purification kit (Genomed LohneGermany) and labeled with SpectrumRed dUTP (VysisDowners Grove IL USA) using a Nick Translation kit(Vysis) Similarly genomic DNA of the same normalsample was labeled with SpectrumGreen dUTP (Vysis)and both probes were cohybridized to metaphase chromo-somes Procedures and image analysis were performed asdescribed (44)Differential normalndashtumor representation of AUMA
at the genomic scale was performed by competitivehybridization of AUMA products to BAC arraysAUMA products from two normalndashtumor pairs werepurified using Jet quick PCR product purification kit(Genomed Lohne Germany) and 1 mg was labeled withdCTP-Cy3 or dCTP-Cy5 (Amersham Biosciences UK) byuse of the Bioprime DNA Labeling System (InvitrogenCarlsbad CA USA) Probes were hybridized to Spectral-Chip 2600 BAC arrays (Spectral Genomics Houston TXUSA) following the manufacturerrsquos instructions Arrayswere scanned with a ScanArray 4000 (GSI LumonicsWatertown MA USA) and processed with GenePixsoftware (Axon Instruments Union City CA USA) Theresulting data were processed to filter out low-qualityspots based on spot area and similarity of readingsbetween the two replicates of each BAC Data manipula-tion was performed using Excel spreadsheets BecauseAUMA products are not evenly distributed along chro-mosomes only BACs with intensities above the 10 ofmaximum intensity in at least one of the two channelswere considered for ratio calculations The pattern ofchromosomal alterations in these two tumors was deter-mined by conventional CGH as described (44)
Isolation and cloning of AUMA tagged bands
DNA excised from gels was directly amplified with thesame primer used in AUMA (BAu-TT) (SupplementaryFigure 2) The amplified product was cloned into plasmidvectors using the pGEM-T easy vector System I cloningkit (Promega Madison WI USA) Automated sequenc-ing of multiple colonies was performed using the Big DyeTerminator v31 Cycle Sequencing kit (Applied Biosys-tems Foster City CA USA) to ascertain the uniqueidentity of the isolated band Sequence homologies weresearched for using the Blat engine (httpgenomeucscedu) Selected clones corresponding to AUMA isolatedbands were radioactively labeled and used as a probe toconfirm the identity of the excised band by hybridizationto AUMA fingerprints as previously described (45)
Bisulfite genomic sequencing
Differential methylation observed in some AUMAtagged bands was confirmed by direct sequencing ofbisulfite treated normal and tumor DNA as previouslydescribed (46) Prior to sequencing DNA was amplifiedusing a nested or semi-nested PCR approach as appro-priate Three independent PCRs were done and productswere pooled to ensure a representative sequencing
Nucleic Acids Research 2008 Vol 36 No 3 773
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
The sequence of PCR primers is described inSupplementary Data
Histone modification analysis by chromatinimmunoprecipitation (ChIP)
Briefly 6 106 cells were washed twice with PBS and cross-linked on the culture plate for 15min at room temperaturein the presence of 05 formaldehyde Cross-linkingreaction was stopped by adding 0125M glycine All subse-quent steps were carried out at 48C All buffers were pre-chilled and contained protease inhibitors (Complete MiniRoche) Cells were washed twice with PBS and thenscraped Collected pellets were dissolved in 1ml lysis buffer(1 SDS 5mM EDTA 50mM Tris pH 8) and weresonicated in a cold ethanol bath for 10 cycles at 100amplitude using a UP50H sonicator (Hielscher TeltowGermany) Chromatin fragmentation was visualized in 1agarose gel Obtained fragments were in the 200ndash500 pbrange Soluble chromatin was obtained by centrifuging thesonicated samples at 14 000g for 10min at 48C The solublefraction was diluted 110 in dilution buffer (1 TritonX-100 2mM EDTA 20mM Tris pH 8 150mM NaCl)then aliquoted and stored at 808C until useImmunoprecipitation was carried out at 48C by adding
5ndash10mg of the desired antibody to 1ml of chromatinChromatinndashantibody complexes were immunoprecipitatedwith specific antibodies using a protein AG 50 slurry(Upstate Millipore Billerica MA USA) and subse-quently washed and eluted according to the manufac-turerrsquos instructions Antibodies against acetylated H3K9K14 (Upstate) dimethylated H3 K79 and trimethyl-ated H3 K9 (Abcam Cambridge UK) were used Enrich-ment for a given chromatin modification was quantified asa fold enrichment over the input using quantitative real-time PCR (Roche) For every PCR a standard curve wasobtained to assess amplification efficiency All quantifica-tions were performed in duplicate
RESULTS
Genomic estimation of the targets and evaluation of theadequacy of the approach by computational analysis
The availability of the human genome map has allowedus to make a detailed estimation of the frequency anddistribution of the sites targeted by our approaches onthe genomic scale A Perl routine was used to score allpositions containing the target sequences in all chromo-somes and was also applied to perform a virtual AUMA(see Material and Methods section) Some of the mostimportant data derived from the bioinformatic analysisare shown in Table 1 and Figure 3
Because of the C to T mutational bias at CpG sites (47)any amplification method relying on the consensussequence (see Supplementary Data) will only cover afraction of all the Alursquos Therefore it is important toestimate the degree of representativity of the methodsused here if genome-wide estimations are to be made Alurepeats constitute 74 of the human genome but accu-mulate 407 of all SmaI sites (Table 1) Nearly 200 000Alursquos (18 of all Alursquos) contain a SmaI site and 155 000retain the AACCCGGG consensus sequence (Table 1 andFigure 3) and are therefore potential targets of QUMAand AUMA While 380 of the youngest AluY elementscontain this sequence the proportions drop to 148 and04 in AluS and AluJ families respectively (Table 1 andFigure 3) These frequencies are consistent with a higher Cto T transition trend at CpG sites in older Alursquos (47)
The representativity of AUMA was analyzed by avirtual bioinformatic assay of the human genomesequence A total of 168 309 AACCCGGG (or CCCGGGTT) hits were identified throughout the genome with929 of all hits within Alu elements (Table 1 andFigure 3) This implies that 142 of all Alu elementscontained the AACCCGGG sequence Another 10 ofthe hits were in CpG islands and 68 in the rest of thegenome (including unique sequences and other repeats)(Table 1 and Figure 3) As expected Alu elements con-taining the target sequence mostly belonged to the AluS
Table 1 Content and distribution of QUMA and AUMA hits in the human genome
Sequence Mba Number ofelementsb
SmaI sites(CCCGGG)
AACCCGGGhitsc
VirtualAUMA hitsd
AUMAhitse
Unmethylatedhitsf
Unmethylatedhits ()g
Total 30804 1 118 195 486 835 168 309 5498 201 14332 2418 852 14Alu (S+J+Y) 2273 1 091 110 198 201 155 226 5109 59 (293) 4104 688 264 044AluS 1412 660 415 122 459 97 951 3382 45 (224) 3028 510 309 051AluJ 540 283 104 14 017 1235 38 2 (10) 151 25 1225 197AluY 321 147 591 61 725 56 040 1689 12 (60) 925 156 165 027CpG islands 162 27 085 49 430 1673 63 55 (274) 1501 97 905 579Rest 28369 ndash 239 204 11 410 326 87 (433) 8530 1650 759 1463
aGenome Mb represented by each type of element Total number corresponds to the number of megabases analyzed for the presence of hitsOnly assembled chromosome fragments were consideredbElements considered in the analysis as obtained from the Repbase and the Genome Browser Databases (see Material and Methods section)cNumber of occurrences of the sequence AACCCGGG (or CCCGGGTT) within each type of elementdNumber of AUMA hits present in virtual PCR products of up to 1000 bpeHits of actual AUMA products Only bands appearing in normal tissue were considered Eighty-seven bands contributed two hits each (174 hits)and 27 bands contributed only one due to poor sequence or incomplete homology with the NCBI Build 361 of the human genome (hg18 assemblyMarch 2006) Twenty-three additional bands were detected mainly in tumor tissue and were not considered to perform calculationsfEstimated number of unmethylated sites using Monte Carlo simulations (Material and Methods section)gIn respect to the total number of AACCCGGG (or CCCGGGTT) hits
774 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
(23) and AluY families (13) with a minimal representa-tion of the older family AluJ (lt1) Virtual AUMAdetermined the presence of 5498 putative products oflt1Kb (the sequence AACCCGGG in the up strand andthe sequence CCCGGGTT in the down strand at adistance of lt1 kb) Although actual AUMA PCRproducts may reach 2 kb length (see below) we used the1 kb limit to compare with AUMA-isolated bands whichwere shorter than 1 kb Most virtual AUMA productscontained an Alu element at one of the ends at least(93)
Quantification of unmethylated Alu in normaland tumor tissues
The QUMA approach was applied to quantify unmethy-lated Alursquos in a series of 18 colorectal carcinomas and theirpaired normal colonic mucosa An external DNA frag-ment containing an AluSx element was used as a standard(see Materials and Methods section and SupplementaryData) in order to make an absolute quantification of thenumber of unmethylated Alursquos Replicates and dilutioncurves of the samples and standard were performedto assess reproducibility sensitivity and accuracy(Supplementary Figure 1) Results were normalized byassessment of the number of haploid genomes per test tube(see Material and Methods section) The average numberof unmethylated Alursquos per haploid genome was25 486 10 157 in normal mucosa and 41 995 17 187in tumor samples (P=0004 paired t-test) (Figure 4)We computationally identified a total of 168 309 Aluelements containing the AACCCGGG sequence (potentialtargets of QUMA) (Table 1) Therefore we estimatethat 151 of Alu repeats with a AACCCGGG site areunmethylated in the average normal colonic mucosa cell
while this figure is 249 in the cancer cell Consideringthat the human genome contains 11 million Alu ele-ments these estimates indicate that unmethylated Alursquosconstitute the 23 and 38 of all Alursquos in the normal andtumor tissues respectively
Set-up and optimization of AUMA fingerprinting
Because QUMA products are fully contained within theAlu sequence it is not possible to identify and positionin the genome the unmethylated Alu elements To achievethis it is necessary to amplify the targeted unmethylatedAlu element together with an adjacent unique sequenceThis was attained through the use of the second methodthe Amplification of UnMethylated Alus (AUMA)AUMA also targets the unmethylated AACCCGGGsequence as in QUMA but in this case a single primeris used in the PCR (BAu-TT) (see Figure 1 and Supple-mentary Data) Moreover the product is resolved ona high-resolution gel electrophoresis resulting in a band-rich fingerprint AUMA bands correspond to sequencesflanked by two unmethylated target sequences in oppositestrands and sufficiently close to allow PCR amplification(Figure 1 and Supplementary Data) Since 92 of theAACCCGGG occurrences in the human genome are inAlursquos (Table 1) the approach is largely biased towardsthe amplification of unmethylated Alursquos The presenceof non-Alu sequences at one of the ends or between tworepetitive elements allows the positioning within thegenome map of all productsAUMA products generated using the BAu-TT primer
produced highly reproducible fingerprints consisting ofbands ranging from 100 to 2000 bp when resolved inhigh-resolution sequencing gels (Figure 2 and Supplemen-tary Figure 2) Some well-identifiable bands (up to 5 perexperiment) showed random display in both intra-assayand inter-assay replicates (Supplementary Figure 2) andwere not considered for analysis A subset of 110 bandswith consistent display among all the experiments weretagged and selected for comparative analysis betweensamples (see Materials and Methods section)It should be noted that different fingerprints containing
alternative representations may be obtained by AUMA
Figure 3 Relative distribution the Alu elements and sequence targetsconsidered in bioinformatic and experimental QUMA and AUMAMb number of megabases occupied by each type of element elementsnumber of elements considered (lsquoRestrsquo has been set arbitrarily to 50)SmaI site CCCGGG sequence vQUMA hits AACCCGGG (or GGGCCCTT) sites in Alu elements vAUMA hits AACCCGGG (or GGGCCCTT) sites vAUMA ends vAUMA hits considering only putativeAUMA products of lt1 kb (see Material and Methods section)AUMA elements at each one of the two ends of actual AUMAproducts
Figure 4 Quantitation of unmethylated Alursquos in 17 paired normalmucosa and colorectal carcinoma by QUMA The values represent theestimated number of unmethylated Alursquos per haploid genome Mosttumors exhibited a higher level of hypomethylation when comparedwith the respective normal
Nucleic Acids Research 2008 Vol 36 No 3 775
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

at close to its expected frequency in small genomic regions(200 bp to a few kb) known as CpG islands (22) Theseareas are lsquoprotectedrsquo from methylation and are locatedin the proximal promoter regions of 75 of human genes(121322) Methylated CpG islands are strongly andhereditably repressed (12) Hence DNA methylation isusually considered as a sign of long-term inactivation(91012)
Cancer cells are characterized by the accumulation ofboth genetic and epigenetic changes Widespread genomichypomethylation is an early alteration in carcinogenesisand has been associated with genomic disruption andgenetic instability (23ndash27) Repeats unmasked by demeth-ylation are likely to facilitate rearrangements due to mito-tic recombination and unwanted transcription (28ndash30)Alternatively aberrant de novo methylation of CpGislands is a hallmark of human cancers and is associatedwith epigenetic silencing of multiple tumor suppressorgenes (31ndash37) Therefore the screening for differentiallymethylated sequences in tumors appears as a key toolto further understand the molecular mechanisms under-lying malignant transformation of cells Although therepertoire of methylation screening methodologies hasexpanded widely (37ndash39) and different approaches havebeen used to make bulk estimates of methylation inrepetitive elements (4041) there is still a lack of screeningstrategies that specifically allow a feasible identification ofDNA methylation alterations in repetitive elements (21)
Here we report two variants of a novel methodologyto quantify and identify unmethylated Alu sequencesThe CpG site within the consensus Alu sequence AACCCGGG is used as a surrogate reporter of methylationUnmethylated sites are cut with the methylation-sensitiverestriction endonuclease SmaI (CCCGGG) and an adap-tor is ligated to the DNA ends Quantification ofUnMethylated Alus (QUMA) is performed by real-timeamplification of the digested and adaptor-ligated DNAusing an Alu consensus primer that anneals upstreamof the SmaI site and an adaptor primer extended with theTT dinucleotide in its 30 end (Figure 1A) The productgenerated by this approach is completely inside theAlu element and hence it is not possible to make aunique identification As an alternative approach we havealso performed restrained amplification of digested andadaptor-ligated DNA fragments that are flanked by twoclose SmaI sites In this case the same primer homologousto the adaptor with the additional TT nucleotides at the30 end to enrich for Alu sequences is used in absence of theAlu consensus primer (Figure 1B) This second approachis named Amplification of UnMethylated Alursquos (AUMA)and results in a complex representation of unique DNAsequences flanked by two unmethylated SmaI sites Whenresolved by high-resolution electrophoresis the AUMAgenerated sequences appear as a fingerprint characteristicof each sample (Figure 2) and individual scoring andidentification of each band can be performed BecauseAUMArsquos stringency is based on a short sequence(AACCCGGG) that is found preferentially but not exclu-sively in Alu elements other unmethylated sequences arealso present in AUMA fingerprints
Application of QUMA and AUMA to a series of colo-rectal carcinomas and their paired normal mucosa hasoffered global estimates of unmethylation of Alu elementsin normal and cancer cells and has revealed a large collec-tion of unique sequences that undergo highly recurrenthypomethylation and hypermethylation in colorectaltumors
MATERIALS AND METHODS
Tissues and cell lines
Fifty colorectal carcinomas and their paired non-adjacent areas of normal colonic mucosa were included
Figure 1 Schematic diagram of the QUMA and AUMA methods DNAis depicted by a solid line Alu elements are represented by dashed boxesThe QUMA and AUMA recognition sites (AACCCGGG) are repre-sented by dashedgray boxes CpGs at SmaI sites are shown as full circleswhen methylated and as open circles when unmethylated The methyla-tion-sensitive restriction endonuclease SmaI can only digest unmethylatedtargets leaving blunt ends to which adaptors can be ligated (A) QUMA isperformed by real-time PCR of an inner Alu fragment using a primercomplementary to the Alu consensus sequence upstream of the SmaI siteand the primer complementary to the adaptor to which two Aluhomologous nucleotides (TT) have been added (B) In AUMA sequencesflanked by two ligated adaptors are amplified by PCR using a singleprimer the same adaptor primer plus the TT nucleotidesWhen only a fewnucleotides are added to the primer ie TT as illustrated here other non-Alu sequences may be amplified This allows the amplification of a largenumber of sequences that typically range from 100 to 2000 bp
Nucleic Acids Research 2008 Vol 36 No 3 771
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
in this analysis Samples were collected simultaneously asfresh specimens and snap-frozen within 2 h of removal andthen stored at 808C All samples were obtained from theCiutat Sanitaria i Universitaria de Bellvitge (BarcelonaSpain) The study protocol was approved by the EthicsCommittee Human colon cancer cell lines (HT29 SW480HCT116 LoVo DLD-1 CaCo-2 and LS174T) wereobtained from the American Type Culture Collection(ATCCManassas VA) KM12C and KM12SM cells weregenerously provided by A Fabra DNA from tumorndashnormal pairs was obtained by conventional organic extrac-tion and ethanol precipitation DNA purity and qualitywas checked in a 08 agarose gel electrophoresis RNAfrom cell lines was obtained by phenolndashchloroformextraction and ethanol precipitation following standardprocedures
Bioinformatic analysis
The distribution of SmaI sites putative amplification hitsPCR homologies CpG islands and repetitive elementswas assessed using the human genome assembly 361 fromNCBI Data were obtained from the Repbase (httpwwwgirinstorgrepbaseindexhtml) and the GenomeBrowser Databases (httphgdownloadcseucscedugoldenPathhg18database) Only assembled chromo-some fragments were considered A Perl routine wasused to score all positions containing the target sequencesin all chromosomes (available from the authors uponrequest) Data were analyzed using Excel spreadsheetsTo calculate the proportion of unmethylated Alu
elements at the genomic level the number of AUMAhits identified in bioinformatic analysis were correctedaccording to the distribution of experimentally generated
AUMA products performing Monte Carlo simulationsOne thousand Monte Carlo simulations were performedusing an Excel Add-in (available at wwwwabashedueconometrics) In Monte Carlo simulations it wasassumed that 80ndash100 of SmaI sites at CpG islands areunmethylated and that 50ndash100 of SmaI sites in othergenomic regions different from Alursquos and CpG islandsare unmethylated
Quantification of QUMA
One microgram of DNA was digested with 20U ofthe methylation sensitive restriction endonucleaseSmaI (Roche Diagnostics GmbH Mannheim Germany)for 16 h at 308C leaving cleaved fragments with bluntends (CCCGGG) Adaptors were prepared incubatingthe oligonucleotides Blue (CCGAATTCGCAAAGCTCTGA) and the 50 phosphorylated MCF oligonucleotide(TCAGAGCTTTGCGAAT) at 658C for 2min and thencooling to room temperature for 30ndash60min One micro-gram of the digested DNA was ligated to 2 nmol ofadaptor using T4 DNA ligase (New England BiolabsBeverly MA USA) Subsequent digestion of the ligatedproducts with the methylation insensitive restrictionendonuclease XmaI (New England Biolabs) was per-formed to avoid amplifications from non-digested methy-lated Alursquos The products were purified using the GFXKit (Amersham Biosciences Buckinghamshire UK) andeluted in 250 ml of sterile water
Quantitative real-time PCR was performed using 1 ng(the equivalent of 333 genomes) of DNA in a LightCycler480 real-time PCR system with Fast Start Master SYBRGreen I kit (Roche) Mastermix was prepared to a finalconcentration of 35mM MgCl2 and 1 mM of each primerThe downstream BAu-TT primer (constituted by the 30
end of Blue primer and the GGGTT sequence includingthe GGG 30 side of the cut SmaI site and the Aluhomologous TT dinucleotide ATTCGCAAAGCTCTGAGGGTT) and the upstream primer was an Aluconsensus sequence (CCGTCTCTACTAAAAATACA)(see Supplementary Data) Magnitudes were expressedas number of unmethylated Alus per haploid genomeafter DNA input normalization The number of haploidgenomes present in the test tube was determined in thesame multiwell plate by quantification of Alu sequencesirrespectively of the methylation state A real-time PCRusing Alu consensus primers upstream of the CCCGGGsite was performed (see Supplementary Data) and thenumber of genomes was calculated against a standardcurve constructed with a reference genomic DNA mea-sured by UV spectrophotometry
To determine the efficiency of the assay and to performabsolute quantification an external Alu product generatedby PCR from a DNA fragment containing an AluSxelement was used as standard (Supplementary Methods)The number of copies of the external control were spectro-photometrically quantified and dilution curves were gene-rated and treated as samples Comparison of dilutioncurves before and after sample processing indicated thatthe mean recovery was 73 DNA samples overdigested
Figure 2 AUMA of normal (N)ndashtumor (T) pairs of two differentpatients performed using primer BAu-TT A highly reproducible bandpatterning is observed among the four replicates Representative bandsshowing gains (hypomethylations) and losses (hypermethylations) aremarked with up and down arrowheads respectively
772 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
with the methylation insensitive XmaI endonuclease werespiked with different amounts of the external standard andprocessed The sensitivity of the QUMA detection was 100unmethylated Alursquos per haploid genome (SupplementaryFigure 1) using 1 ng of genomic DNA per PCR A linearresponse was observed between 1000 and 100 000 unmethy-lated Alursquos per haploid genome (Supplementary Data)
Amplification of AUMA
DNAdigestion with SmaI enzyme and ligation to the linkerwas performed as described above for QUMA except forthe XmaI digestion that was skipped The product waspurified using the GFX Kit (Amersham Biosciences) andeluted in 250 ml of sterile water Six different chimericprimers constituted by the 30 end of the Blue primersequence (ATTCGCAAAGCTCTGA) the cut SmaI site(GGG) and two four or seven additional nucleotideshomologous to the Alu consensus sequence were used toenrich for Alu sequences (see Supplementary Methods)Three primers were designed to amplify lsquoupstreamrsquo of theSmaI site (towards ALUpromoter) BAu-TT BAu-TTCABAu-TTCAAGC Three other primers were designed toamplify lsquodownstreamrsquo (towards ALU poly-A) BAd-AGBAd-AGGC BAd-AGGCGGA Letters after the dashcorrespond to the 30 sequence of the primer (see Supple-mentary Data) Data reported here were obtained by usingthe BAu-TT primer
In each PCR reaction only one primer was used ata time Products were resolved on denaturing sequencinggels Although bands can be visualized by silver stainingof the gels radioactive AUMArsquos were performed fornormalndashtumor comparisons A more detailed descriptionof the PCR and the visualization of the bands are givenas Supplementary Data
Only sharp bands that were reproducible and clearlydistinguishable from the background were tagged andincluded in the analysis Faint bands with inconsistentdisplay due to small variations in gel electrophoresisresolution were not considered Band reproducibility wasassessed with the analysis of PCR duplicates of threeindependent sample digests from two different samplesand PCR replicates from the same digest from four pairedtumorndashnormal samples AUMA fingerprints were visuallychecked for methylation differences between bands in thetumor with regard to its paired normal mucosa Underthese premises a given band was scored according to threepossible behaviors hypomethylation (increased intensityin the tumor) hypermethylation (decreased intensity inthe tumor) and no change (no substantial difference inintensity between normal and tumor samples) (Figure 2)Only those bands showing clear changes in their intensitiesin the fingerprint were considered to represent methylationchanges This is consistent with previous studies doneusing a related technique (4243)
Competitive hybridization of AUMA products to metaphasechromosomes and BAC arrays
The origin and chromosomal distribution of sequencesgenerated by AUMA was analyzed using procedures
analogous to CGH Briefly an AUMA product obtainedfrom a normal tissue DNA was purified using Jet quickPCR product purification kit (Genomed LohneGermany) and labeled with SpectrumRed dUTP (VysisDowners Grove IL USA) using a Nick Translation kit(Vysis) Similarly genomic DNA of the same normalsample was labeled with SpectrumGreen dUTP (Vysis)and both probes were cohybridized to metaphase chromo-somes Procedures and image analysis were performed asdescribed (44)Differential normalndashtumor representation of AUMA
at the genomic scale was performed by competitivehybridization of AUMA products to BAC arraysAUMA products from two normalndashtumor pairs werepurified using Jet quick PCR product purification kit(Genomed Lohne Germany) and 1 mg was labeled withdCTP-Cy3 or dCTP-Cy5 (Amersham Biosciences UK) byuse of the Bioprime DNA Labeling System (InvitrogenCarlsbad CA USA) Probes were hybridized to Spectral-Chip 2600 BAC arrays (Spectral Genomics Houston TXUSA) following the manufacturerrsquos instructions Arrayswere scanned with a ScanArray 4000 (GSI LumonicsWatertown MA USA) and processed with GenePixsoftware (Axon Instruments Union City CA USA) Theresulting data were processed to filter out low-qualityspots based on spot area and similarity of readingsbetween the two replicates of each BAC Data manipula-tion was performed using Excel spreadsheets BecauseAUMA products are not evenly distributed along chro-mosomes only BACs with intensities above the 10 ofmaximum intensity in at least one of the two channelswere considered for ratio calculations The pattern ofchromosomal alterations in these two tumors was deter-mined by conventional CGH as described (44)
Isolation and cloning of AUMA tagged bands
DNA excised from gels was directly amplified with thesame primer used in AUMA (BAu-TT) (SupplementaryFigure 2) The amplified product was cloned into plasmidvectors using the pGEM-T easy vector System I cloningkit (Promega Madison WI USA) Automated sequenc-ing of multiple colonies was performed using the Big DyeTerminator v31 Cycle Sequencing kit (Applied Biosys-tems Foster City CA USA) to ascertain the uniqueidentity of the isolated band Sequence homologies weresearched for using the Blat engine (httpgenomeucscedu) Selected clones corresponding to AUMA isolatedbands were radioactively labeled and used as a probe toconfirm the identity of the excised band by hybridizationto AUMA fingerprints as previously described (45)
Bisulfite genomic sequencing
Differential methylation observed in some AUMAtagged bands was confirmed by direct sequencing ofbisulfite treated normal and tumor DNA as previouslydescribed (46) Prior to sequencing DNA was amplifiedusing a nested or semi-nested PCR approach as appro-priate Three independent PCRs were done and productswere pooled to ensure a representative sequencing
Nucleic Acids Research 2008 Vol 36 No 3 773
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
The sequence of PCR primers is described inSupplementary Data
Histone modification analysis by chromatinimmunoprecipitation (ChIP)
Briefly 6 106 cells were washed twice with PBS and cross-linked on the culture plate for 15min at room temperaturein the presence of 05 formaldehyde Cross-linkingreaction was stopped by adding 0125M glycine All subse-quent steps were carried out at 48C All buffers were pre-chilled and contained protease inhibitors (Complete MiniRoche) Cells were washed twice with PBS and thenscraped Collected pellets were dissolved in 1ml lysis buffer(1 SDS 5mM EDTA 50mM Tris pH 8) and weresonicated in a cold ethanol bath for 10 cycles at 100amplitude using a UP50H sonicator (Hielscher TeltowGermany) Chromatin fragmentation was visualized in 1agarose gel Obtained fragments were in the 200ndash500 pbrange Soluble chromatin was obtained by centrifuging thesonicated samples at 14 000g for 10min at 48C The solublefraction was diluted 110 in dilution buffer (1 TritonX-100 2mM EDTA 20mM Tris pH 8 150mM NaCl)then aliquoted and stored at 808C until useImmunoprecipitation was carried out at 48C by adding
5ndash10mg of the desired antibody to 1ml of chromatinChromatinndashantibody complexes were immunoprecipitatedwith specific antibodies using a protein AG 50 slurry(Upstate Millipore Billerica MA USA) and subse-quently washed and eluted according to the manufac-turerrsquos instructions Antibodies against acetylated H3K9K14 (Upstate) dimethylated H3 K79 and trimethyl-ated H3 K9 (Abcam Cambridge UK) were used Enrich-ment for a given chromatin modification was quantified asa fold enrichment over the input using quantitative real-time PCR (Roche) For every PCR a standard curve wasobtained to assess amplification efficiency All quantifica-tions were performed in duplicate
RESULTS
Genomic estimation of the targets and evaluation of theadequacy of the approach by computational analysis
The availability of the human genome map has allowedus to make a detailed estimation of the frequency anddistribution of the sites targeted by our approaches onthe genomic scale A Perl routine was used to score allpositions containing the target sequences in all chromo-somes and was also applied to perform a virtual AUMA(see Material and Methods section) Some of the mostimportant data derived from the bioinformatic analysisare shown in Table 1 and Figure 3
Because of the C to T mutational bias at CpG sites (47)any amplification method relying on the consensussequence (see Supplementary Data) will only cover afraction of all the Alursquos Therefore it is important toestimate the degree of representativity of the methodsused here if genome-wide estimations are to be made Alurepeats constitute 74 of the human genome but accu-mulate 407 of all SmaI sites (Table 1) Nearly 200 000Alursquos (18 of all Alursquos) contain a SmaI site and 155 000retain the AACCCGGG consensus sequence (Table 1 andFigure 3) and are therefore potential targets of QUMAand AUMA While 380 of the youngest AluY elementscontain this sequence the proportions drop to 148 and04 in AluS and AluJ families respectively (Table 1 andFigure 3) These frequencies are consistent with a higher Cto T transition trend at CpG sites in older Alursquos (47)
The representativity of AUMA was analyzed by avirtual bioinformatic assay of the human genomesequence A total of 168 309 AACCCGGG (or CCCGGGTT) hits were identified throughout the genome with929 of all hits within Alu elements (Table 1 andFigure 3) This implies that 142 of all Alu elementscontained the AACCCGGG sequence Another 10 ofthe hits were in CpG islands and 68 in the rest of thegenome (including unique sequences and other repeats)(Table 1 and Figure 3) As expected Alu elements con-taining the target sequence mostly belonged to the AluS
Table 1 Content and distribution of QUMA and AUMA hits in the human genome
Sequence Mba Number ofelementsb
SmaI sites(CCCGGG)
AACCCGGGhitsc
VirtualAUMA hitsd
AUMAhitse
Unmethylatedhitsf
Unmethylatedhits ()g
Total 30804 1 118 195 486 835 168 309 5498 201 14332 2418 852 14Alu (S+J+Y) 2273 1 091 110 198 201 155 226 5109 59 (293) 4104 688 264 044AluS 1412 660 415 122 459 97 951 3382 45 (224) 3028 510 309 051AluJ 540 283 104 14 017 1235 38 2 (10) 151 25 1225 197AluY 321 147 591 61 725 56 040 1689 12 (60) 925 156 165 027CpG islands 162 27 085 49 430 1673 63 55 (274) 1501 97 905 579Rest 28369 ndash 239 204 11 410 326 87 (433) 8530 1650 759 1463
aGenome Mb represented by each type of element Total number corresponds to the number of megabases analyzed for the presence of hitsOnly assembled chromosome fragments were consideredbElements considered in the analysis as obtained from the Repbase and the Genome Browser Databases (see Material and Methods section)cNumber of occurrences of the sequence AACCCGGG (or CCCGGGTT) within each type of elementdNumber of AUMA hits present in virtual PCR products of up to 1000 bpeHits of actual AUMA products Only bands appearing in normal tissue were considered Eighty-seven bands contributed two hits each (174 hits)and 27 bands contributed only one due to poor sequence or incomplete homology with the NCBI Build 361 of the human genome (hg18 assemblyMarch 2006) Twenty-three additional bands were detected mainly in tumor tissue and were not considered to perform calculationsfEstimated number of unmethylated sites using Monte Carlo simulations (Material and Methods section)gIn respect to the total number of AACCCGGG (or CCCGGGTT) hits
774 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
(23) and AluY families (13) with a minimal representa-tion of the older family AluJ (lt1) Virtual AUMAdetermined the presence of 5498 putative products oflt1Kb (the sequence AACCCGGG in the up strand andthe sequence CCCGGGTT in the down strand at adistance of lt1 kb) Although actual AUMA PCRproducts may reach 2 kb length (see below) we used the1 kb limit to compare with AUMA-isolated bands whichwere shorter than 1 kb Most virtual AUMA productscontained an Alu element at one of the ends at least(93)
Quantification of unmethylated Alu in normaland tumor tissues
The QUMA approach was applied to quantify unmethy-lated Alursquos in a series of 18 colorectal carcinomas and theirpaired normal colonic mucosa An external DNA frag-ment containing an AluSx element was used as a standard(see Materials and Methods section and SupplementaryData) in order to make an absolute quantification of thenumber of unmethylated Alursquos Replicates and dilutioncurves of the samples and standard were performedto assess reproducibility sensitivity and accuracy(Supplementary Figure 1) Results were normalized byassessment of the number of haploid genomes per test tube(see Material and Methods section) The average numberof unmethylated Alursquos per haploid genome was25 486 10 157 in normal mucosa and 41 995 17 187in tumor samples (P=0004 paired t-test) (Figure 4)We computationally identified a total of 168 309 Aluelements containing the AACCCGGG sequence (potentialtargets of QUMA) (Table 1) Therefore we estimatethat 151 of Alu repeats with a AACCCGGG site areunmethylated in the average normal colonic mucosa cell
while this figure is 249 in the cancer cell Consideringthat the human genome contains 11 million Alu ele-ments these estimates indicate that unmethylated Alursquosconstitute the 23 and 38 of all Alursquos in the normal andtumor tissues respectively
Set-up and optimization of AUMA fingerprinting
Because QUMA products are fully contained within theAlu sequence it is not possible to identify and positionin the genome the unmethylated Alu elements To achievethis it is necessary to amplify the targeted unmethylatedAlu element together with an adjacent unique sequenceThis was attained through the use of the second methodthe Amplification of UnMethylated Alus (AUMA)AUMA also targets the unmethylated AACCCGGGsequence as in QUMA but in this case a single primeris used in the PCR (BAu-TT) (see Figure 1 and Supple-mentary Data) Moreover the product is resolved ona high-resolution gel electrophoresis resulting in a band-rich fingerprint AUMA bands correspond to sequencesflanked by two unmethylated target sequences in oppositestrands and sufficiently close to allow PCR amplification(Figure 1 and Supplementary Data) Since 92 of theAACCCGGG occurrences in the human genome are inAlursquos (Table 1) the approach is largely biased towardsthe amplification of unmethylated Alursquos The presenceof non-Alu sequences at one of the ends or between tworepetitive elements allows the positioning within thegenome map of all productsAUMA products generated using the BAu-TT primer
produced highly reproducible fingerprints consisting ofbands ranging from 100 to 2000 bp when resolved inhigh-resolution sequencing gels (Figure 2 and Supplemen-tary Figure 2) Some well-identifiable bands (up to 5 perexperiment) showed random display in both intra-assayand inter-assay replicates (Supplementary Figure 2) andwere not considered for analysis A subset of 110 bandswith consistent display among all the experiments weretagged and selected for comparative analysis betweensamples (see Materials and Methods section)It should be noted that different fingerprints containing
alternative representations may be obtained by AUMA
Figure 3 Relative distribution the Alu elements and sequence targetsconsidered in bioinformatic and experimental QUMA and AUMAMb number of megabases occupied by each type of element elementsnumber of elements considered (lsquoRestrsquo has been set arbitrarily to 50)SmaI site CCCGGG sequence vQUMA hits AACCCGGG (or GGGCCCTT) sites in Alu elements vAUMA hits AACCCGGG (or GGGCCCTT) sites vAUMA ends vAUMA hits considering only putativeAUMA products of lt1 kb (see Material and Methods section)AUMA elements at each one of the two ends of actual AUMAproducts
Figure 4 Quantitation of unmethylated Alursquos in 17 paired normalmucosa and colorectal carcinoma by QUMA The values represent theestimated number of unmethylated Alursquos per haploid genome Mosttumors exhibited a higher level of hypomethylation when comparedwith the respective normal
Nucleic Acids Research 2008 Vol 36 No 3 775
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

in this analysis Samples were collected simultaneously asfresh specimens and snap-frozen within 2 h of removal andthen stored at 808C All samples were obtained from theCiutat Sanitaria i Universitaria de Bellvitge (BarcelonaSpain) The study protocol was approved by the EthicsCommittee Human colon cancer cell lines (HT29 SW480HCT116 LoVo DLD-1 CaCo-2 and LS174T) wereobtained from the American Type Culture Collection(ATCCManassas VA) KM12C and KM12SM cells weregenerously provided by A Fabra DNA from tumorndashnormal pairs was obtained by conventional organic extrac-tion and ethanol precipitation DNA purity and qualitywas checked in a 08 agarose gel electrophoresis RNAfrom cell lines was obtained by phenolndashchloroformextraction and ethanol precipitation following standardprocedures
Bioinformatic analysis
The distribution of SmaI sites putative amplification hitsPCR homologies CpG islands and repetitive elementswas assessed using the human genome assembly 361 fromNCBI Data were obtained from the Repbase (httpwwwgirinstorgrepbaseindexhtml) and the GenomeBrowser Databases (httphgdownloadcseucscedugoldenPathhg18database) Only assembled chromo-some fragments were considered A Perl routine wasused to score all positions containing the target sequencesin all chromosomes (available from the authors uponrequest) Data were analyzed using Excel spreadsheetsTo calculate the proportion of unmethylated Alu
elements at the genomic level the number of AUMAhits identified in bioinformatic analysis were correctedaccording to the distribution of experimentally generated
AUMA products performing Monte Carlo simulationsOne thousand Monte Carlo simulations were performedusing an Excel Add-in (available at wwwwabashedueconometrics) In Monte Carlo simulations it wasassumed that 80ndash100 of SmaI sites at CpG islands areunmethylated and that 50ndash100 of SmaI sites in othergenomic regions different from Alursquos and CpG islandsare unmethylated
Quantification of QUMA
One microgram of DNA was digested with 20U ofthe methylation sensitive restriction endonucleaseSmaI (Roche Diagnostics GmbH Mannheim Germany)for 16 h at 308C leaving cleaved fragments with bluntends (CCCGGG) Adaptors were prepared incubatingthe oligonucleotides Blue (CCGAATTCGCAAAGCTCTGA) and the 50 phosphorylated MCF oligonucleotide(TCAGAGCTTTGCGAAT) at 658C for 2min and thencooling to room temperature for 30ndash60min One micro-gram of the digested DNA was ligated to 2 nmol ofadaptor using T4 DNA ligase (New England BiolabsBeverly MA USA) Subsequent digestion of the ligatedproducts with the methylation insensitive restrictionendonuclease XmaI (New England Biolabs) was per-formed to avoid amplifications from non-digested methy-lated Alursquos The products were purified using the GFXKit (Amersham Biosciences Buckinghamshire UK) andeluted in 250 ml of sterile water
Quantitative real-time PCR was performed using 1 ng(the equivalent of 333 genomes) of DNA in a LightCycler480 real-time PCR system with Fast Start Master SYBRGreen I kit (Roche) Mastermix was prepared to a finalconcentration of 35mM MgCl2 and 1 mM of each primerThe downstream BAu-TT primer (constituted by the 30
end of Blue primer and the GGGTT sequence includingthe GGG 30 side of the cut SmaI site and the Aluhomologous TT dinucleotide ATTCGCAAAGCTCTGAGGGTT) and the upstream primer was an Aluconsensus sequence (CCGTCTCTACTAAAAATACA)(see Supplementary Data) Magnitudes were expressedas number of unmethylated Alus per haploid genomeafter DNA input normalization The number of haploidgenomes present in the test tube was determined in thesame multiwell plate by quantification of Alu sequencesirrespectively of the methylation state A real-time PCRusing Alu consensus primers upstream of the CCCGGGsite was performed (see Supplementary Data) and thenumber of genomes was calculated against a standardcurve constructed with a reference genomic DNA mea-sured by UV spectrophotometry
To determine the efficiency of the assay and to performabsolute quantification an external Alu product generatedby PCR from a DNA fragment containing an AluSxelement was used as standard (Supplementary Methods)The number of copies of the external control were spectro-photometrically quantified and dilution curves were gene-rated and treated as samples Comparison of dilutioncurves before and after sample processing indicated thatthe mean recovery was 73 DNA samples overdigested
Figure 2 AUMA of normal (N)ndashtumor (T) pairs of two differentpatients performed using primer BAu-TT A highly reproducible bandpatterning is observed among the four replicates Representative bandsshowing gains (hypomethylations) and losses (hypermethylations) aremarked with up and down arrowheads respectively
772 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
with the methylation insensitive XmaI endonuclease werespiked with different amounts of the external standard andprocessed The sensitivity of the QUMA detection was 100unmethylated Alursquos per haploid genome (SupplementaryFigure 1) using 1 ng of genomic DNA per PCR A linearresponse was observed between 1000 and 100 000 unmethy-lated Alursquos per haploid genome (Supplementary Data)
Amplification of AUMA
DNAdigestion with SmaI enzyme and ligation to the linkerwas performed as described above for QUMA except forthe XmaI digestion that was skipped The product waspurified using the GFX Kit (Amersham Biosciences) andeluted in 250 ml of sterile water Six different chimericprimers constituted by the 30 end of the Blue primersequence (ATTCGCAAAGCTCTGA) the cut SmaI site(GGG) and two four or seven additional nucleotideshomologous to the Alu consensus sequence were used toenrich for Alu sequences (see Supplementary Methods)Three primers were designed to amplify lsquoupstreamrsquo of theSmaI site (towards ALUpromoter) BAu-TT BAu-TTCABAu-TTCAAGC Three other primers were designed toamplify lsquodownstreamrsquo (towards ALU poly-A) BAd-AGBAd-AGGC BAd-AGGCGGA Letters after the dashcorrespond to the 30 sequence of the primer (see Supple-mentary Data) Data reported here were obtained by usingthe BAu-TT primer
In each PCR reaction only one primer was used ata time Products were resolved on denaturing sequencinggels Although bands can be visualized by silver stainingof the gels radioactive AUMArsquos were performed fornormalndashtumor comparisons A more detailed descriptionof the PCR and the visualization of the bands are givenas Supplementary Data
Only sharp bands that were reproducible and clearlydistinguishable from the background were tagged andincluded in the analysis Faint bands with inconsistentdisplay due to small variations in gel electrophoresisresolution were not considered Band reproducibility wasassessed with the analysis of PCR duplicates of threeindependent sample digests from two different samplesand PCR replicates from the same digest from four pairedtumorndashnormal samples AUMA fingerprints were visuallychecked for methylation differences between bands in thetumor with regard to its paired normal mucosa Underthese premises a given band was scored according to threepossible behaviors hypomethylation (increased intensityin the tumor) hypermethylation (decreased intensity inthe tumor) and no change (no substantial difference inintensity between normal and tumor samples) (Figure 2)Only those bands showing clear changes in their intensitiesin the fingerprint were considered to represent methylationchanges This is consistent with previous studies doneusing a related technique (4243)
Competitive hybridization of AUMA products to metaphasechromosomes and BAC arrays
The origin and chromosomal distribution of sequencesgenerated by AUMA was analyzed using procedures
analogous to CGH Briefly an AUMA product obtainedfrom a normal tissue DNA was purified using Jet quickPCR product purification kit (Genomed LohneGermany) and labeled with SpectrumRed dUTP (VysisDowners Grove IL USA) using a Nick Translation kit(Vysis) Similarly genomic DNA of the same normalsample was labeled with SpectrumGreen dUTP (Vysis)and both probes were cohybridized to metaphase chromo-somes Procedures and image analysis were performed asdescribed (44)Differential normalndashtumor representation of AUMA
at the genomic scale was performed by competitivehybridization of AUMA products to BAC arraysAUMA products from two normalndashtumor pairs werepurified using Jet quick PCR product purification kit(Genomed Lohne Germany) and 1 mg was labeled withdCTP-Cy3 or dCTP-Cy5 (Amersham Biosciences UK) byuse of the Bioprime DNA Labeling System (InvitrogenCarlsbad CA USA) Probes were hybridized to Spectral-Chip 2600 BAC arrays (Spectral Genomics Houston TXUSA) following the manufacturerrsquos instructions Arrayswere scanned with a ScanArray 4000 (GSI LumonicsWatertown MA USA) and processed with GenePixsoftware (Axon Instruments Union City CA USA) Theresulting data were processed to filter out low-qualityspots based on spot area and similarity of readingsbetween the two replicates of each BAC Data manipula-tion was performed using Excel spreadsheets BecauseAUMA products are not evenly distributed along chro-mosomes only BACs with intensities above the 10 ofmaximum intensity in at least one of the two channelswere considered for ratio calculations The pattern ofchromosomal alterations in these two tumors was deter-mined by conventional CGH as described (44)
Isolation and cloning of AUMA tagged bands
DNA excised from gels was directly amplified with thesame primer used in AUMA (BAu-TT) (SupplementaryFigure 2) The amplified product was cloned into plasmidvectors using the pGEM-T easy vector System I cloningkit (Promega Madison WI USA) Automated sequenc-ing of multiple colonies was performed using the Big DyeTerminator v31 Cycle Sequencing kit (Applied Biosys-tems Foster City CA USA) to ascertain the uniqueidentity of the isolated band Sequence homologies weresearched for using the Blat engine (httpgenomeucscedu) Selected clones corresponding to AUMA isolatedbands were radioactively labeled and used as a probe toconfirm the identity of the excised band by hybridizationto AUMA fingerprints as previously described (45)
Bisulfite genomic sequencing
Differential methylation observed in some AUMAtagged bands was confirmed by direct sequencing ofbisulfite treated normal and tumor DNA as previouslydescribed (46) Prior to sequencing DNA was amplifiedusing a nested or semi-nested PCR approach as appro-priate Three independent PCRs were done and productswere pooled to ensure a representative sequencing
Nucleic Acids Research 2008 Vol 36 No 3 773
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
The sequence of PCR primers is described inSupplementary Data
Histone modification analysis by chromatinimmunoprecipitation (ChIP)
Briefly 6 106 cells were washed twice with PBS and cross-linked on the culture plate for 15min at room temperaturein the presence of 05 formaldehyde Cross-linkingreaction was stopped by adding 0125M glycine All subse-quent steps were carried out at 48C All buffers were pre-chilled and contained protease inhibitors (Complete MiniRoche) Cells were washed twice with PBS and thenscraped Collected pellets were dissolved in 1ml lysis buffer(1 SDS 5mM EDTA 50mM Tris pH 8) and weresonicated in a cold ethanol bath for 10 cycles at 100amplitude using a UP50H sonicator (Hielscher TeltowGermany) Chromatin fragmentation was visualized in 1agarose gel Obtained fragments were in the 200ndash500 pbrange Soluble chromatin was obtained by centrifuging thesonicated samples at 14 000g for 10min at 48C The solublefraction was diluted 110 in dilution buffer (1 TritonX-100 2mM EDTA 20mM Tris pH 8 150mM NaCl)then aliquoted and stored at 808C until useImmunoprecipitation was carried out at 48C by adding
5ndash10mg of the desired antibody to 1ml of chromatinChromatinndashantibody complexes were immunoprecipitatedwith specific antibodies using a protein AG 50 slurry(Upstate Millipore Billerica MA USA) and subse-quently washed and eluted according to the manufac-turerrsquos instructions Antibodies against acetylated H3K9K14 (Upstate) dimethylated H3 K79 and trimethyl-ated H3 K9 (Abcam Cambridge UK) were used Enrich-ment for a given chromatin modification was quantified asa fold enrichment over the input using quantitative real-time PCR (Roche) For every PCR a standard curve wasobtained to assess amplification efficiency All quantifica-tions were performed in duplicate
RESULTS
Genomic estimation of the targets and evaluation of theadequacy of the approach by computational analysis
The availability of the human genome map has allowedus to make a detailed estimation of the frequency anddistribution of the sites targeted by our approaches onthe genomic scale A Perl routine was used to score allpositions containing the target sequences in all chromo-somes and was also applied to perform a virtual AUMA(see Material and Methods section) Some of the mostimportant data derived from the bioinformatic analysisare shown in Table 1 and Figure 3
Because of the C to T mutational bias at CpG sites (47)any amplification method relying on the consensussequence (see Supplementary Data) will only cover afraction of all the Alursquos Therefore it is important toestimate the degree of representativity of the methodsused here if genome-wide estimations are to be made Alurepeats constitute 74 of the human genome but accu-mulate 407 of all SmaI sites (Table 1) Nearly 200 000Alursquos (18 of all Alursquos) contain a SmaI site and 155 000retain the AACCCGGG consensus sequence (Table 1 andFigure 3) and are therefore potential targets of QUMAand AUMA While 380 of the youngest AluY elementscontain this sequence the proportions drop to 148 and04 in AluS and AluJ families respectively (Table 1 andFigure 3) These frequencies are consistent with a higher Cto T transition trend at CpG sites in older Alursquos (47)
The representativity of AUMA was analyzed by avirtual bioinformatic assay of the human genomesequence A total of 168 309 AACCCGGG (or CCCGGGTT) hits were identified throughout the genome with929 of all hits within Alu elements (Table 1 andFigure 3) This implies that 142 of all Alu elementscontained the AACCCGGG sequence Another 10 ofthe hits were in CpG islands and 68 in the rest of thegenome (including unique sequences and other repeats)(Table 1 and Figure 3) As expected Alu elements con-taining the target sequence mostly belonged to the AluS
Table 1 Content and distribution of QUMA and AUMA hits in the human genome
Sequence Mba Number ofelementsb
SmaI sites(CCCGGG)
AACCCGGGhitsc
VirtualAUMA hitsd
AUMAhitse
Unmethylatedhitsf
Unmethylatedhits ()g
Total 30804 1 118 195 486 835 168 309 5498 201 14332 2418 852 14Alu (S+J+Y) 2273 1 091 110 198 201 155 226 5109 59 (293) 4104 688 264 044AluS 1412 660 415 122 459 97 951 3382 45 (224) 3028 510 309 051AluJ 540 283 104 14 017 1235 38 2 (10) 151 25 1225 197AluY 321 147 591 61 725 56 040 1689 12 (60) 925 156 165 027CpG islands 162 27 085 49 430 1673 63 55 (274) 1501 97 905 579Rest 28369 ndash 239 204 11 410 326 87 (433) 8530 1650 759 1463
aGenome Mb represented by each type of element Total number corresponds to the number of megabases analyzed for the presence of hitsOnly assembled chromosome fragments were consideredbElements considered in the analysis as obtained from the Repbase and the Genome Browser Databases (see Material and Methods section)cNumber of occurrences of the sequence AACCCGGG (or CCCGGGTT) within each type of elementdNumber of AUMA hits present in virtual PCR products of up to 1000 bpeHits of actual AUMA products Only bands appearing in normal tissue were considered Eighty-seven bands contributed two hits each (174 hits)and 27 bands contributed only one due to poor sequence or incomplete homology with the NCBI Build 361 of the human genome (hg18 assemblyMarch 2006) Twenty-three additional bands were detected mainly in tumor tissue and were not considered to perform calculationsfEstimated number of unmethylated sites using Monte Carlo simulations (Material and Methods section)gIn respect to the total number of AACCCGGG (or CCCGGGTT) hits
774 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
(23) and AluY families (13) with a minimal representa-tion of the older family AluJ (lt1) Virtual AUMAdetermined the presence of 5498 putative products oflt1Kb (the sequence AACCCGGG in the up strand andthe sequence CCCGGGTT in the down strand at adistance of lt1 kb) Although actual AUMA PCRproducts may reach 2 kb length (see below) we used the1 kb limit to compare with AUMA-isolated bands whichwere shorter than 1 kb Most virtual AUMA productscontained an Alu element at one of the ends at least(93)
Quantification of unmethylated Alu in normaland tumor tissues
The QUMA approach was applied to quantify unmethy-lated Alursquos in a series of 18 colorectal carcinomas and theirpaired normal colonic mucosa An external DNA frag-ment containing an AluSx element was used as a standard(see Materials and Methods section and SupplementaryData) in order to make an absolute quantification of thenumber of unmethylated Alursquos Replicates and dilutioncurves of the samples and standard were performedto assess reproducibility sensitivity and accuracy(Supplementary Figure 1) Results were normalized byassessment of the number of haploid genomes per test tube(see Material and Methods section) The average numberof unmethylated Alursquos per haploid genome was25 486 10 157 in normal mucosa and 41 995 17 187in tumor samples (P=0004 paired t-test) (Figure 4)We computationally identified a total of 168 309 Aluelements containing the AACCCGGG sequence (potentialtargets of QUMA) (Table 1) Therefore we estimatethat 151 of Alu repeats with a AACCCGGG site areunmethylated in the average normal colonic mucosa cell
while this figure is 249 in the cancer cell Consideringthat the human genome contains 11 million Alu ele-ments these estimates indicate that unmethylated Alursquosconstitute the 23 and 38 of all Alursquos in the normal andtumor tissues respectively
Set-up and optimization of AUMA fingerprinting
Because QUMA products are fully contained within theAlu sequence it is not possible to identify and positionin the genome the unmethylated Alu elements To achievethis it is necessary to amplify the targeted unmethylatedAlu element together with an adjacent unique sequenceThis was attained through the use of the second methodthe Amplification of UnMethylated Alus (AUMA)AUMA also targets the unmethylated AACCCGGGsequence as in QUMA but in this case a single primeris used in the PCR (BAu-TT) (see Figure 1 and Supple-mentary Data) Moreover the product is resolved ona high-resolution gel electrophoresis resulting in a band-rich fingerprint AUMA bands correspond to sequencesflanked by two unmethylated target sequences in oppositestrands and sufficiently close to allow PCR amplification(Figure 1 and Supplementary Data) Since 92 of theAACCCGGG occurrences in the human genome are inAlursquos (Table 1) the approach is largely biased towardsthe amplification of unmethylated Alursquos The presenceof non-Alu sequences at one of the ends or between tworepetitive elements allows the positioning within thegenome map of all productsAUMA products generated using the BAu-TT primer
produced highly reproducible fingerprints consisting ofbands ranging from 100 to 2000 bp when resolved inhigh-resolution sequencing gels (Figure 2 and Supplemen-tary Figure 2) Some well-identifiable bands (up to 5 perexperiment) showed random display in both intra-assayand inter-assay replicates (Supplementary Figure 2) andwere not considered for analysis A subset of 110 bandswith consistent display among all the experiments weretagged and selected for comparative analysis betweensamples (see Materials and Methods section)It should be noted that different fingerprints containing
alternative representations may be obtained by AUMA
Figure 3 Relative distribution the Alu elements and sequence targetsconsidered in bioinformatic and experimental QUMA and AUMAMb number of megabases occupied by each type of element elementsnumber of elements considered (lsquoRestrsquo has been set arbitrarily to 50)SmaI site CCCGGG sequence vQUMA hits AACCCGGG (or GGGCCCTT) sites in Alu elements vAUMA hits AACCCGGG (or GGGCCCTT) sites vAUMA ends vAUMA hits considering only putativeAUMA products of lt1 kb (see Material and Methods section)AUMA elements at each one of the two ends of actual AUMAproducts
Figure 4 Quantitation of unmethylated Alursquos in 17 paired normalmucosa and colorectal carcinoma by QUMA The values represent theestimated number of unmethylated Alursquos per haploid genome Mosttumors exhibited a higher level of hypomethylation when comparedwith the respective normal
Nucleic Acids Research 2008 Vol 36 No 3 775
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

with the methylation insensitive XmaI endonuclease werespiked with different amounts of the external standard andprocessed The sensitivity of the QUMA detection was 100unmethylated Alursquos per haploid genome (SupplementaryFigure 1) using 1 ng of genomic DNA per PCR A linearresponse was observed between 1000 and 100 000 unmethy-lated Alursquos per haploid genome (Supplementary Data)
Amplification of AUMA
DNAdigestion with SmaI enzyme and ligation to the linkerwas performed as described above for QUMA except forthe XmaI digestion that was skipped The product waspurified using the GFX Kit (Amersham Biosciences) andeluted in 250 ml of sterile water Six different chimericprimers constituted by the 30 end of the Blue primersequence (ATTCGCAAAGCTCTGA) the cut SmaI site(GGG) and two four or seven additional nucleotideshomologous to the Alu consensus sequence were used toenrich for Alu sequences (see Supplementary Methods)Three primers were designed to amplify lsquoupstreamrsquo of theSmaI site (towards ALUpromoter) BAu-TT BAu-TTCABAu-TTCAAGC Three other primers were designed toamplify lsquodownstreamrsquo (towards ALU poly-A) BAd-AGBAd-AGGC BAd-AGGCGGA Letters after the dashcorrespond to the 30 sequence of the primer (see Supple-mentary Data) Data reported here were obtained by usingthe BAu-TT primer
In each PCR reaction only one primer was used ata time Products were resolved on denaturing sequencinggels Although bands can be visualized by silver stainingof the gels radioactive AUMArsquos were performed fornormalndashtumor comparisons A more detailed descriptionof the PCR and the visualization of the bands are givenas Supplementary Data
Only sharp bands that were reproducible and clearlydistinguishable from the background were tagged andincluded in the analysis Faint bands with inconsistentdisplay due to small variations in gel electrophoresisresolution were not considered Band reproducibility wasassessed with the analysis of PCR duplicates of threeindependent sample digests from two different samplesand PCR replicates from the same digest from four pairedtumorndashnormal samples AUMA fingerprints were visuallychecked for methylation differences between bands in thetumor with regard to its paired normal mucosa Underthese premises a given band was scored according to threepossible behaviors hypomethylation (increased intensityin the tumor) hypermethylation (decreased intensity inthe tumor) and no change (no substantial difference inintensity between normal and tumor samples) (Figure 2)Only those bands showing clear changes in their intensitiesin the fingerprint were considered to represent methylationchanges This is consistent with previous studies doneusing a related technique (4243)
Competitive hybridization of AUMA products to metaphasechromosomes and BAC arrays
The origin and chromosomal distribution of sequencesgenerated by AUMA was analyzed using procedures
analogous to CGH Briefly an AUMA product obtainedfrom a normal tissue DNA was purified using Jet quickPCR product purification kit (Genomed LohneGermany) and labeled with SpectrumRed dUTP (VysisDowners Grove IL USA) using a Nick Translation kit(Vysis) Similarly genomic DNA of the same normalsample was labeled with SpectrumGreen dUTP (Vysis)and both probes were cohybridized to metaphase chromo-somes Procedures and image analysis were performed asdescribed (44)Differential normalndashtumor representation of AUMA
at the genomic scale was performed by competitivehybridization of AUMA products to BAC arraysAUMA products from two normalndashtumor pairs werepurified using Jet quick PCR product purification kit(Genomed Lohne Germany) and 1 mg was labeled withdCTP-Cy3 or dCTP-Cy5 (Amersham Biosciences UK) byuse of the Bioprime DNA Labeling System (InvitrogenCarlsbad CA USA) Probes were hybridized to Spectral-Chip 2600 BAC arrays (Spectral Genomics Houston TXUSA) following the manufacturerrsquos instructions Arrayswere scanned with a ScanArray 4000 (GSI LumonicsWatertown MA USA) and processed with GenePixsoftware (Axon Instruments Union City CA USA) Theresulting data were processed to filter out low-qualityspots based on spot area and similarity of readingsbetween the two replicates of each BAC Data manipula-tion was performed using Excel spreadsheets BecauseAUMA products are not evenly distributed along chro-mosomes only BACs with intensities above the 10 ofmaximum intensity in at least one of the two channelswere considered for ratio calculations The pattern ofchromosomal alterations in these two tumors was deter-mined by conventional CGH as described (44)
Isolation and cloning of AUMA tagged bands
DNA excised from gels was directly amplified with thesame primer used in AUMA (BAu-TT) (SupplementaryFigure 2) The amplified product was cloned into plasmidvectors using the pGEM-T easy vector System I cloningkit (Promega Madison WI USA) Automated sequenc-ing of multiple colonies was performed using the Big DyeTerminator v31 Cycle Sequencing kit (Applied Biosys-tems Foster City CA USA) to ascertain the uniqueidentity of the isolated band Sequence homologies weresearched for using the Blat engine (httpgenomeucscedu) Selected clones corresponding to AUMA isolatedbands were radioactively labeled and used as a probe toconfirm the identity of the excised band by hybridizationto AUMA fingerprints as previously described (45)
Bisulfite genomic sequencing
Differential methylation observed in some AUMAtagged bands was confirmed by direct sequencing ofbisulfite treated normal and tumor DNA as previouslydescribed (46) Prior to sequencing DNA was amplifiedusing a nested or semi-nested PCR approach as appro-priate Three independent PCRs were done and productswere pooled to ensure a representative sequencing
Nucleic Acids Research 2008 Vol 36 No 3 773
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
The sequence of PCR primers is described inSupplementary Data
Histone modification analysis by chromatinimmunoprecipitation (ChIP)
Briefly 6 106 cells were washed twice with PBS and cross-linked on the culture plate for 15min at room temperaturein the presence of 05 formaldehyde Cross-linkingreaction was stopped by adding 0125M glycine All subse-quent steps were carried out at 48C All buffers were pre-chilled and contained protease inhibitors (Complete MiniRoche) Cells were washed twice with PBS and thenscraped Collected pellets were dissolved in 1ml lysis buffer(1 SDS 5mM EDTA 50mM Tris pH 8) and weresonicated in a cold ethanol bath for 10 cycles at 100amplitude using a UP50H sonicator (Hielscher TeltowGermany) Chromatin fragmentation was visualized in 1agarose gel Obtained fragments were in the 200ndash500 pbrange Soluble chromatin was obtained by centrifuging thesonicated samples at 14 000g for 10min at 48C The solublefraction was diluted 110 in dilution buffer (1 TritonX-100 2mM EDTA 20mM Tris pH 8 150mM NaCl)then aliquoted and stored at 808C until useImmunoprecipitation was carried out at 48C by adding
5ndash10mg of the desired antibody to 1ml of chromatinChromatinndashantibody complexes were immunoprecipitatedwith specific antibodies using a protein AG 50 slurry(Upstate Millipore Billerica MA USA) and subse-quently washed and eluted according to the manufac-turerrsquos instructions Antibodies against acetylated H3K9K14 (Upstate) dimethylated H3 K79 and trimethyl-ated H3 K9 (Abcam Cambridge UK) were used Enrich-ment for a given chromatin modification was quantified asa fold enrichment over the input using quantitative real-time PCR (Roche) For every PCR a standard curve wasobtained to assess amplification efficiency All quantifica-tions were performed in duplicate
RESULTS
Genomic estimation of the targets and evaluation of theadequacy of the approach by computational analysis
The availability of the human genome map has allowedus to make a detailed estimation of the frequency anddistribution of the sites targeted by our approaches onthe genomic scale A Perl routine was used to score allpositions containing the target sequences in all chromo-somes and was also applied to perform a virtual AUMA(see Material and Methods section) Some of the mostimportant data derived from the bioinformatic analysisare shown in Table 1 and Figure 3
Because of the C to T mutational bias at CpG sites (47)any amplification method relying on the consensussequence (see Supplementary Data) will only cover afraction of all the Alursquos Therefore it is important toestimate the degree of representativity of the methodsused here if genome-wide estimations are to be made Alurepeats constitute 74 of the human genome but accu-mulate 407 of all SmaI sites (Table 1) Nearly 200 000Alursquos (18 of all Alursquos) contain a SmaI site and 155 000retain the AACCCGGG consensus sequence (Table 1 andFigure 3) and are therefore potential targets of QUMAand AUMA While 380 of the youngest AluY elementscontain this sequence the proportions drop to 148 and04 in AluS and AluJ families respectively (Table 1 andFigure 3) These frequencies are consistent with a higher Cto T transition trend at CpG sites in older Alursquos (47)
The representativity of AUMA was analyzed by avirtual bioinformatic assay of the human genomesequence A total of 168 309 AACCCGGG (or CCCGGGTT) hits were identified throughout the genome with929 of all hits within Alu elements (Table 1 andFigure 3) This implies that 142 of all Alu elementscontained the AACCCGGG sequence Another 10 ofthe hits were in CpG islands and 68 in the rest of thegenome (including unique sequences and other repeats)(Table 1 and Figure 3) As expected Alu elements con-taining the target sequence mostly belonged to the AluS
Table 1 Content and distribution of QUMA and AUMA hits in the human genome
Sequence Mba Number ofelementsb
SmaI sites(CCCGGG)
AACCCGGGhitsc
VirtualAUMA hitsd
AUMAhitse
Unmethylatedhitsf
Unmethylatedhits ()g
Total 30804 1 118 195 486 835 168 309 5498 201 14332 2418 852 14Alu (S+J+Y) 2273 1 091 110 198 201 155 226 5109 59 (293) 4104 688 264 044AluS 1412 660 415 122 459 97 951 3382 45 (224) 3028 510 309 051AluJ 540 283 104 14 017 1235 38 2 (10) 151 25 1225 197AluY 321 147 591 61 725 56 040 1689 12 (60) 925 156 165 027CpG islands 162 27 085 49 430 1673 63 55 (274) 1501 97 905 579Rest 28369 ndash 239 204 11 410 326 87 (433) 8530 1650 759 1463
aGenome Mb represented by each type of element Total number corresponds to the number of megabases analyzed for the presence of hitsOnly assembled chromosome fragments were consideredbElements considered in the analysis as obtained from the Repbase and the Genome Browser Databases (see Material and Methods section)cNumber of occurrences of the sequence AACCCGGG (or CCCGGGTT) within each type of elementdNumber of AUMA hits present in virtual PCR products of up to 1000 bpeHits of actual AUMA products Only bands appearing in normal tissue were considered Eighty-seven bands contributed two hits each (174 hits)and 27 bands contributed only one due to poor sequence or incomplete homology with the NCBI Build 361 of the human genome (hg18 assemblyMarch 2006) Twenty-three additional bands were detected mainly in tumor tissue and were not considered to perform calculationsfEstimated number of unmethylated sites using Monte Carlo simulations (Material and Methods section)gIn respect to the total number of AACCCGGG (or CCCGGGTT) hits
774 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
(23) and AluY families (13) with a minimal representa-tion of the older family AluJ (lt1) Virtual AUMAdetermined the presence of 5498 putative products oflt1Kb (the sequence AACCCGGG in the up strand andthe sequence CCCGGGTT in the down strand at adistance of lt1 kb) Although actual AUMA PCRproducts may reach 2 kb length (see below) we used the1 kb limit to compare with AUMA-isolated bands whichwere shorter than 1 kb Most virtual AUMA productscontained an Alu element at one of the ends at least(93)
Quantification of unmethylated Alu in normaland tumor tissues
The QUMA approach was applied to quantify unmethy-lated Alursquos in a series of 18 colorectal carcinomas and theirpaired normal colonic mucosa An external DNA frag-ment containing an AluSx element was used as a standard(see Materials and Methods section and SupplementaryData) in order to make an absolute quantification of thenumber of unmethylated Alursquos Replicates and dilutioncurves of the samples and standard were performedto assess reproducibility sensitivity and accuracy(Supplementary Figure 1) Results were normalized byassessment of the number of haploid genomes per test tube(see Material and Methods section) The average numberof unmethylated Alursquos per haploid genome was25 486 10 157 in normal mucosa and 41 995 17 187in tumor samples (P=0004 paired t-test) (Figure 4)We computationally identified a total of 168 309 Aluelements containing the AACCCGGG sequence (potentialtargets of QUMA) (Table 1) Therefore we estimatethat 151 of Alu repeats with a AACCCGGG site areunmethylated in the average normal colonic mucosa cell
while this figure is 249 in the cancer cell Consideringthat the human genome contains 11 million Alu ele-ments these estimates indicate that unmethylated Alursquosconstitute the 23 and 38 of all Alursquos in the normal andtumor tissues respectively
Set-up and optimization of AUMA fingerprinting
Because QUMA products are fully contained within theAlu sequence it is not possible to identify and positionin the genome the unmethylated Alu elements To achievethis it is necessary to amplify the targeted unmethylatedAlu element together with an adjacent unique sequenceThis was attained through the use of the second methodthe Amplification of UnMethylated Alus (AUMA)AUMA also targets the unmethylated AACCCGGGsequence as in QUMA but in this case a single primeris used in the PCR (BAu-TT) (see Figure 1 and Supple-mentary Data) Moreover the product is resolved ona high-resolution gel electrophoresis resulting in a band-rich fingerprint AUMA bands correspond to sequencesflanked by two unmethylated target sequences in oppositestrands and sufficiently close to allow PCR amplification(Figure 1 and Supplementary Data) Since 92 of theAACCCGGG occurrences in the human genome are inAlursquos (Table 1) the approach is largely biased towardsthe amplification of unmethylated Alursquos The presenceof non-Alu sequences at one of the ends or between tworepetitive elements allows the positioning within thegenome map of all productsAUMA products generated using the BAu-TT primer
produced highly reproducible fingerprints consisting ofbands ranging from 100 to 2000 bp when resolved inhigh-resolution sequencing gels (Figure 2 and Supplemen-tary Figure 2) Some well-identifiable bands (up to 5 perexperiment) showed random display in both intra-assayand inter-assay replicates (Supplementary Figure 2) andwere not considered for analysis A subset of 110 bandswith consistent display among all the experiments weretagged and selected for comparative analysis betweensamples (see Materials and Methods section)It should be noted that different fingerprints containing
alternative representations may be obtained by AUMA
Figure 3 Relative distribution the Alu elements and sequence targetsconsidered in bioinformatic and experimental QUMA and AUMAMb number of megabases occupied by each type of element elementsnumber of elements considered (lsquoRestrsquo has been set arbitrarily to 50)SmaI site CCCGGG sequence vQUMA hits AACCCGGG (or GGGCCCTT) sites in Alu elements vAUMA hits AACCCGGG (or GGGCCCTT) sites vAUMA ends vAUMA hits considering only putativeAUMA products of lt1 kb (see Material and Methods section)AUMA elements at each one of the two ends of actual AUMAproducts
Figure 4 Quantitation of unmethylated Alursquos in 17 paired normalmucosa and colorectal carcinoma by QUMA The values represent theestimated number of unmethylated Alursquos per haploid genome Mosttumors exhibited a higher level of hypomethylation when comparedwith the respective normal
Nucleic Acids Research 2008 Vol 36 No 3 775
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
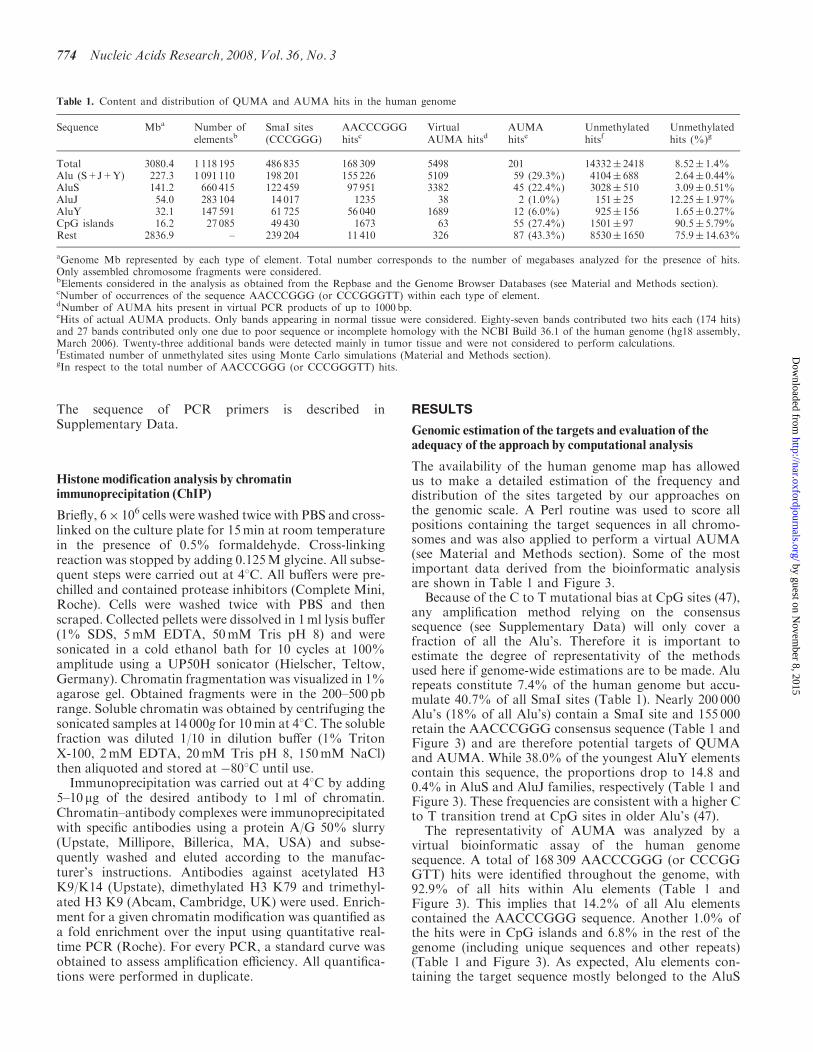
The sequence of PCR primers is described inSupplementary Data
Histone modification analysis by chromatinimmunoprecipitation (ChIP)
Briefly 6 106 cells were washed twice with PBS and cross-linked on the culture plate for 15min at room temperaturein the presence of 05 formaldehyde Cross-linkingreaction was stopped by adding 0125M glycine All subse-quent steps were carried out at 48C All buffers were pre-chilled and contained protease inhibitors (Complete MiniRoche) Cells were washed twice with PBS and thenscraped Collected pellets were dissolved in 1ml lysis buffer(1 SDS 5mM EDTA 50mM Tris pH 8) and weresonicated in a cold ethanol bath for 10 cycles at 100amplitude using a UP50H sonicator (Hielscher TeltowGermany) Chromatin fragmentation was visualized in 1agarose gel Obtained fragments were in the 200ndash500 pbrange Soluble chromatin was obtained by centrifuging thesonicated samples at 14 000g for 10min at 48C The solublefraction was diluted 110 in dilution buffer (1 TritonX-100 2mM EDTA 20mM Tris pH 8 150mM NaCl)then aliquoted and stored at 808C until useImmunoprecipitation was carried out at 48C by adding
5ndash10mg of the desired antibody to 1ml of chromatinChromatinndashantibody complexes were immunoprecipitatedwith specific antibodies using a protein AG 50 slurry(Upstate Millipore Billerica MA USA) and subse-quently washed and eluted according to the manufac-turerrsquos instructions Antibodies against acetylated H3K9K14 (Upstate) dimethylated H3 K79 and trimethyl-ated H3 K9 (Abcam Cambridge UK) were used Enrich-ment for a given chromatin modification was quantified asa fold enrichment over the input using quantitative real-time PCR (Roche) For every PCR a standard curve wasobtained to assess amplification efficiency All quantifica-tions were performed in duplicate
RESULTS
Genomic estimation of the targets and evaluation of theadequacy of the approach by computational analysis
The availability of the human genome map has allowedus to make a detailed estimation of the frequency anddistribution of the sites targeted by our approaches onthe genomic scale A Perl routine was used to score allpositions containing the target sequences in all chromo-somes and was also applied to perform a virtual AUMA(see Material and Methods section) Some of the mostimportant data derived from the bioinformatic analysisare shown in Table 1 and Figure 3
Because of the C to T mutational bias at CpG sites (47)any amplification method relying on the consensussequence (see Supplementary Data) will only cover afraction of all the Alursquos Therefore it is important toestimate the degree of representativity of the methodsused here if genome-wide estimations are to be made Alurepeats constitute 74 of the human genome but accu-mulate 407 of all SmaI sites (Table 1) Nearly 200 000Alursquos (18 of all Alursquos) contain a SmaI site and 155 000retain the AACCCGGG consensus sequence (Table 1 andFigure 3) and are therefore potential targets of QUMAand AUMA While 380 of the youngest AluY elementscontain this sequence the proportions drop to 148 and04 in AluS and AluJ families respectively (Table 1 andFigure 3) These frequencies are consistent with a higher Cto T transition trend at CpG sites in older Alursquos (47)
The representativity of AUMA was analyzed by avirtual bioinformatic assay of the human genomesequence A total of 168 309 AACCCGGG (or CCCGGGTT) hits were identified throughout the genome with929 of all hits within Alu elements (Table 1 andFigure 3) This implies that 142 of all Alu elementscontained the AACCCGGG sequence Another 10 ofthe hits were in CpG islands and 68 in the rest of thegenome (including unique sequences and other repeats)(Table 1 and Figure 3) As expected Alu elements con-taining the target sequence mostly belonged to the AluS
Table 1 Content and distribution of QUMA and AUMA hits in the human genome
Sequence Mba Number ofelementsb
SmaI sites(CCCGGG)
AACCCGGGhitsc
VirtualAUMA hitsd
AUMAhitse
Unmethylatedhitsf
Unmethylatedhits ()g
Total 30804 1 118 195 486 835 168 309 5498 201 14332 2418 852 14Alu (S+J+Y) 2273 1 091 110 198 201 155 226 5109 59 (293) 4104 688 264 044AluS 1412 660 415 122 459 97 951 3382 45 (224) 3028 510 309 051AluJ 540 283 104 14 017 1235 38 2 (10) 151 25 1225 197AluY 321 147 591 61 725 56 040 1689 12 (60) 925 156 165 027CpG islands 162 27 085 49 430 1673 63 55 (274) 1501 97 905 579Rest 28369 ndash 239 204 11 410 326 87 (433) 8530 1650 759 1463
aGenome Mb represented by each type of element Total number corresponds to the number of megabases analyzed for the presence of hitsOnly assembled chromosome fragments were consideredbElements considered in the analysis as obtained from the Repbase and the Genome Browser Databases (see Material and Methods section)cNumber of occurrences of the sequence AACCCGGG (or CCCGGGTT) within each type of elementdNumber of AUMA hits present in virtual PCR products of up to 1000 bpeHits of actual AUMA products Only bands appearing in normal tissue were considered Eighty-seven bands contributed two hits each (174 hits)and 27 bands contributed only one due to poor sequence or incomplete homology with the NCBI Build 361 of the human genome (hg18 assemblyMarch 2006) Twenty-three additional bands were detected mainly in tumor tissue and were not considered to perform calculationsfEstimated number of unmethylated sites using Monte Carlo simulations (Material and Methods section)gIn respect to the total number of AACCCGGG (or CCCGGGTT) hits
774 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
(23) and AluY families (13) with a minimal representa-tion of the older family AluJ (lt1) Virtual AUMAdetermined the presence of 5498 putative products oflt1Kb (the sequence AACCCGGG in the up strand andthe sequence CCCGGGTT in the down strand at adistance of lt1 kb) Although actual AUMA PCRproducts may reach 2 kb length (see below) we used the1 kb limit to compare with AUMA-isolated bands whichwere shorter than 1 kb Most virtual AUMA productscontained an Alu element at one of the ends at least(93)
Quantification of unmethylated Alu in normaland tumor tissues
The QUMA approach was applied to quantify unmethy-lated Alursquos in a series of 18 colorectal carcinomas and theirpaired normal colonic mucosa An external DNA frag-ment containing an AluSx element was used as a standard(see Materials and Methods section and SupplementaryData) in order to make an absolute quantification of thenumber of unmethylated Alursquos Replicates and dilutioncurves of the samples and standard were performedto assess reproducibility sensitivity and accuracy(Supplementary Figure 1) Results were normalized byassessment of the number of haploid genomes per test tube(see Material and Methods section) The average numberof unmethylated Alursquos per haploid genome was25 486 10 157 in normal mucosa and 41 995 17 187in tumor samples (P=0004 paired t-test) (Figure 4)We computationally identified a total of 168 309 Aluelements containing the AACCCGGG sequence (potentialtargets of QUMA) (Table 1) Therefore we estimatethat 151 of Alu repeats with a AACCCGGG site areunmethylated in the average normal colonic mucosa cell
while this figure is 249 in the cancer cell Consideringthat the human genome contains 11 million Alu ele-ments these estimates indicate that unmethylated Alursquosconstitute the 23 and 38 of all Alursquos in the normal andtumor tissues respectively
Set-up and optimization of AUMA fingerprinting
Because QUMA products are fully contained within theAlu sequence it is not possible to identify and positionin the genome the unmethylated Alu elements To achievethis it is necessary to amplify the targeted unmethylatedAlu element together with an adjacent unique sequenceThis was attained through the use of the second methodthe Amplification of UnMethylated Alus (AUMA)AUMA also targets the unmethylated AACCCGGGsequence as in QUMA but in this case a single primeris used in the PCR (BAu-TT) (see Figure 1 and Supple-mentary Data) Moreover the product is resolved ona high-resolution gel electrophoresis resulting in a band-rich fingerprint AUMA bands correspond to sequencesflanked by two unmethylated target sequences in oppositestrands and sufficiently close to allow PCR amplification(Figure 1 and Supplementary Data) Since 92 of theAACCCGGG occurrences in the human genome are inAlursquos (Table 1) the approach is largely biased towardsthe amplification of unmethylated Alursquos The presenceof non-Alu sequences at one of the ends or between tworepetitive elements allows the positioning within thegenome map of all productsAUMA products generated using the BAu-TT primer
produced highly reproducible fingerprints consisting ofbands ranging from 100 to 2000 bp when resolved inhigh-resolution sequencing gels (Figure 2 and Supplemen-tary Figure 2) Some well-identifiable bands (up to 5 perexperiment) showed random display in both intra-assayand inter-assay replicates (Supplementary Figure 2) andwere not considered for analysis A subset of 110 bandswith consistent display among all the experiments weretagged and selected for comparative analysis betweensamples (see Materials and Methods section)It should be noted that different fingerprints containing
alternative representations may be obtained by AUMA
Figure 3 Relative distribution the Alu elements and sequence targetsconsidered in bioinformatic and experimental QUMA and AUMAMb number of megabases occupied by each type of element elementsnumber of elements considered (lsquoRestrsquo has been set arbitrarily to 50)SmaI site CCCGGG sequence vQUMA hits AACCCGGG (or GGGCCCTT) sites in Alu elements vAUMA hits AACCCGGG (or GGGCCCTT) sites vAUMA ends vAUMA hits considering only putativeAUMA products of lt1 kb (see Material and Methods section)AUMA elements at each one of the two ends of actual AUMAproducts
Figure 4 Quantitation of unmethylated Alursquos in 17 paired normalmucosa and colorectal carcinoma by QUMA The values represent theestimated number of unmethylated Alursquos per haploid genome Mosttumors exhibited a higher level of hypomethylation when comparedwith the respective normal
Nucleic Acids Research 2008 Vol 36 No 3 775
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
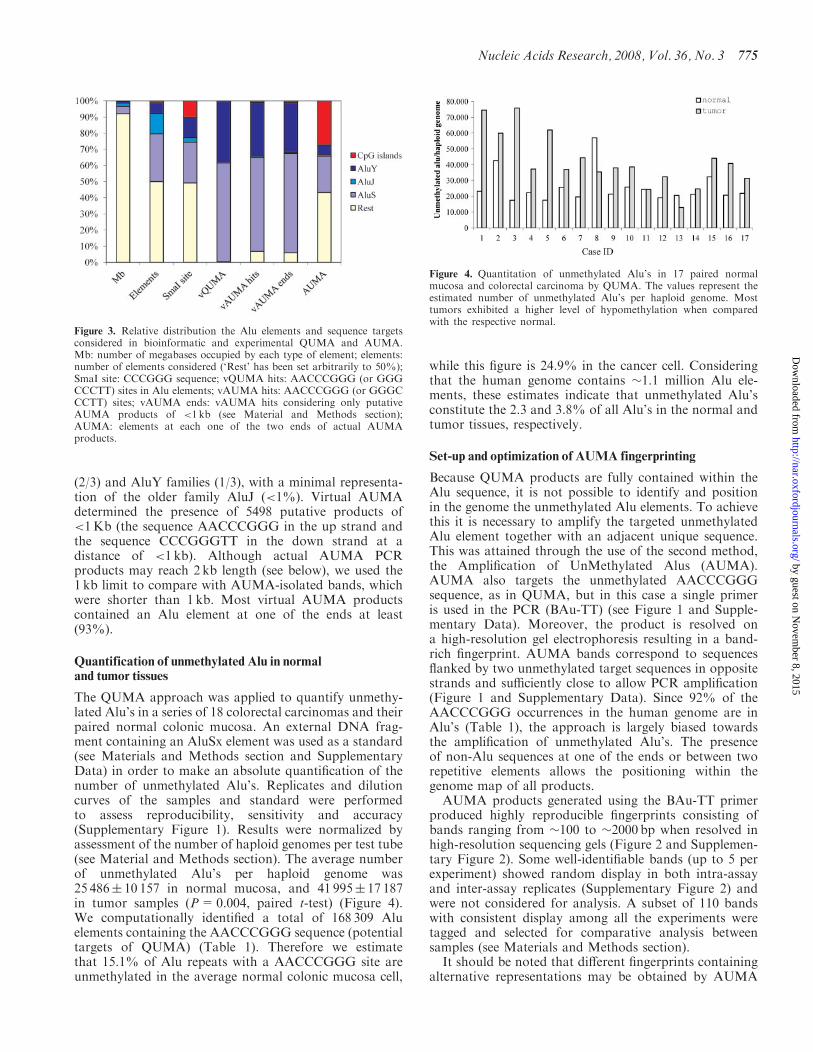
(23) and AluY families (13) with a minimal representa-tion of the older family AluJ (lt1) Virtual AUMAdetermined the presence of 5498 putative products oflt1Kb (the sequence AACCCGGG in the up strand andthe sequence CCCGGGTT in the down strand at adistance of lt1 kb) Although actual AUMA PCRproducts may reach 2 kb length (see below) we used the1 kb limit to compare with AUMA-isolated bands whichwere shorter than 1 kb Most virtual AUMA productscontained an Alu element at one of the ends at least(93)
Quantification of unmethylated Alu in normaland tumor tissues
The QUMA approach was applied to quantify unmethy-lated Alursquos in a series of 18 colorectal carcinomas and theirpaired normal colonic mucosa An external DNA frag-ment containing an AluSx element was used as a standard(see Materials and Methods section and SupplementaryData) in order to make an absolute quantification of thenumber of unmethylated Alursquos Replicates and dilutioncurves of the samples and standard were performedto assess reproducibility sensitivity and accuracy(Supplementary Figure 1) Results were normalized byassessment of the number of haploid genomes per test tube(see Material and Methods section) The average numberof unmethylated Alursquos per haploid genome was25 486 10 157 in normal mucosa and 41 995 17 187in tumor samples (P=0004 paired t-test) (Figure 4)We computationally identified a total of 168 309 Aluelements containing the AACCCGGG sequence (potentialtargets of QUMA) (Table 1) Therefore we estimatethat 151 of Alu repeats with a AACCCGGG site areunmethylated in the average normal colonic mucosa cell
while this figure is 249 in the cancer cell Consideringthat the human genome contains 11 million Alu ele-ments these estimates indicate that unmethylated Alursquosconstitute the 23 and 38 of all Alursquos in the normal andtumor tissues respectively
Set-up and optimization of AUMA fingerprinting
Because QUMA products are fully contained within theAlu sequence it is not possible to identify and positionin the genome the unmethylated Alu elements To achievethis it is necessary to amplify the targeted unmethylatedAlu element together with an adjacent unique sequenceThis was attained through the use of the second methodthe Amplification of UnMethylated Alus (AUMA)AUMA also targets the unmethylated AACCCGGGsequence as in QUMA but in this case a single primeris used in the PCR (BAu-TT) (see Figure 1 and Supple-mentary Data) Moreover the product is resolved ona high-resolution gel electrophoresis resulting in a band-rich fingerprint AUMA bands correspond to sequencesflanked by two unmethylated target sequences in oppositestrands and sufficiently close to allow PCR amplification(Figure 1 and Supplementary Data) Since 92 of theAACCCGGG occurrences in the human genome are inAlursquos (Table 1) the approach is largely biased towardsthe amplification of unmethylated Alursquos The presenceof non-Alu sequences at one of the ends or between tworepetitive elements allows the positioning within thegenome map of all productsAUMA products generated using the BAu-TT primer
produced highly reproducible fingerprints consisting ofbands ranging from 100 to 2000 bp when resolved inhigh-resolution sequencing gels (Figure 2 and Supplemen-tary Figure 2) Some well-identifiable bands (up to 5 perexperiment) showed random display in both intra-assayand inter-assay replicates (Supplementary Figure 2) andwere not considered for analysis A subset of 110 bandswith consistent display among all the experiments weretagged and selected for comparative analysis betweensamples (see Materials and Methods section)It should be noted that different fingerprints containing
alternative representations may be obtained by AUMA
Figure 3 Relative distribution the Alu elements and sequence targetsconsidered in bioinformatic and experimental QUMA and AUMAMb number of megabases occupied by each type of element elementsnumber of elements considered (lsquoRestrsquo has been set arbitrarily to 50)SmaI site CCCGGG sequence vQUMA hits AACCCGGG (or GGGCCCTT) sites in Alu elements vAUMA hits AACCCGGG (or GGGCCCTT) sites vAUMA ends vAUMA hits considering only putativeAUMA products of lt1 kb (see Material and Methods section)AUMA elements at each one of the two ends of actual AUMAproducts
Figure 4 Quantitation of unmethylated Alursquos in 17 paired normalmucosa and colorectal carcinoma by QUMA The values represent theestimated number of unmethylated Alursquos per haploid genome Mosttumors exhibited a higher level of hypomethylation when comparedwith the respective normal
Nucleic Acids Research 2008 Vol 36 No 3 775
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

just by using primers that either amplify from the SmaIsite towards the Alu promoter (upstream Alu amplifica-tion) or towards the Alu poly-A tail (downstream Aluamplification) Also the stringency of the Alu selectionmay be increased by using longer primers containingadditional nucleotides corresponding to the Alu consensussequence (see Material and Methods section) An illus-trative example of AUMA fingerprints generated withdifferent Alu-upstream and Alu-downstream primers isshown in Supplementary Figure 2 All the data reportedin this article regarding AUMA were obtained using theBAu-TT primer
Chromosomal origin of AUMA products
Competitive hybridization between AUMA productsand genomic DNA on metaphase chromosomes yieldeda characteristic hybridization pattern demonstratingthe unequal distribution of AUMA products along thehuman genome (Figure 5A) Competitive hybridization ofAUMA products to BAC arrays showed profiles consis-tent with those obtained on metaphase chromosomes(Figure 5B) The highest AUMA signal was detectedin whole chromosomes 16 17 and 19 in contrast withchromosomes 2 13 18 and X which were mainly labeledby genomic DNA Other chromosomes showed a discretepattern of AUMA product hybridization in which telo-meric bands in chromosomes 1 4 5 9 12 and X andinterstitial bands in chromosomes 1 3 7 11 and 12 are themost prominent examples
Identification of AUMA amplified DNA products
To determine the identity of bands displayed by AUMA38 tagged bands were isolated and cloned Multiple clonesfrom each band were sequenced resulting in a total of49 different sequences due to the coincidence of morethan one sequence in some bands Characterized bandsincluded bands displaying no changes in the normalndashtumor comparisons and bands recurrently altered in thetumor Table 2 summarizes the main features of a subsetof the bands showing recurrent alterations A list of all thesequences isolated from AUMA fingerprints is providedas Supplementary Table 1 All sequenced bands containeda region of non-repetitive sequence and matched with theBLAST reference sequence allowing the assignment ofa unique chromosomal localization The BLAST referencesequence corresponding to the 49 sequences isolated fromthe AUMA fingerprint presented the target sequenceCCCGGGTT including the SmaI at both ends Southernblot analysis of selected cloned sequences showing coinci-dental size was performed to confirm its correspondencewith the band displayed in AUMA fingerprints (Supple-mentary Figure 4)To obtain a more representative collection of AUMA
bands 200 clones obtained from normal tissue AUMAproducts were sequenced The analysis revealed 88 addi-tional sequences This resulted in a total of 137 differentloci represented in AUMA (Supplementary Table 1)Most sequences obtained by random cloning were alsoflanked by two AACCCGGG sequences in opposite DNA
strands Nevertheless in 27 sequences the AACCCGGGsite was only present at one of the ends with the otherend showing high homology with the primer althoughit was not a perfect match The presence of thesesequences suggests that in some instances a single cutin the sequence may be enough to produce an amplifiablefragment This is not considered an artifact since thesebands still represent an unmethylated AACCCGGG site
Genome-wide estimations of unmethylation in Alursquosand distribution by subfamily
Of the 137 identified loci represented in AUMA 114 wereisolated from normal tissue DNA and 23 from tumorDNA Half of the sequences contained an Alu sequence atone of the ends and two were flanked by two invertedAlursquos AUMA sequences isolated from tumor tissue andnot present in normal tissue (this corresponds to a tumor-specific hypomethylation) showed a higher proportionof Alu elements (16 out of 23 70) and included onesequence flanked by two inverted Alursquos Globally 78unmethylated Alu elements were identified and positionedin the human genome map
To study the genomic distribution of unmethylatedsequences in normal colon mucosa we only considered the114 sequences obtained from normal tissue This resultedin a total of 201 unmethylated hits characterized through-out the genome The nature of the sequences representedin actual AUMA showed striking differences with thedistribution expected from the virtual AUMA analysisThe methylation status of the sequence is likely to be themain (if not the only) source of these differences becausethe virtual AUMA did not consider this state Thereforewe can use these differences to estimate the degree ofunmethylation of the Alu repeats Only 294 of theAUMA ends consisted of Alursquos as compared with theexpected 929 resulting from the bioinformatic analysisThe highest downrepresentation corresponded to theyoungest AluY family which was present in 60 of theAUMA ends while it was expected to add up to 331 invirtual AUMA AluS representation in actual and virtualAUMA was 223 and 60 respectively InterestinglyAluJ representations in both actual and virtual AUMAwere closer (10 and 07 respectively) (Figure 3 andTable 1) These results suggest that there is a strongerpressure to methylate younger Alus Alternatively the hitscorresponding to CpG islands were overrepresented inactual versus virtual AUMA by a factor of nearly 25-fold(274 versus 11) consistently with the unmethylatedstatus of most CpG islands (Figure 3 and Table 1) Therest of the hits were located in different types of repetitiveelements (MIR MER LTR LINE etc) and uniquesequences (Supplementary Table 1) The miscellaneouscollection of sequences (lsquoRestrsquo) was over-represented byabout 7-fold (observed hits 433 expected hits 59Table 1) The 46 AUMA hits represented by the 23 bandsspecific of tumor tissue showed a higher proportion ofAlursquos compared with those obtained from normal tissue(41 versus 29 respectively) but similar distribution
776 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
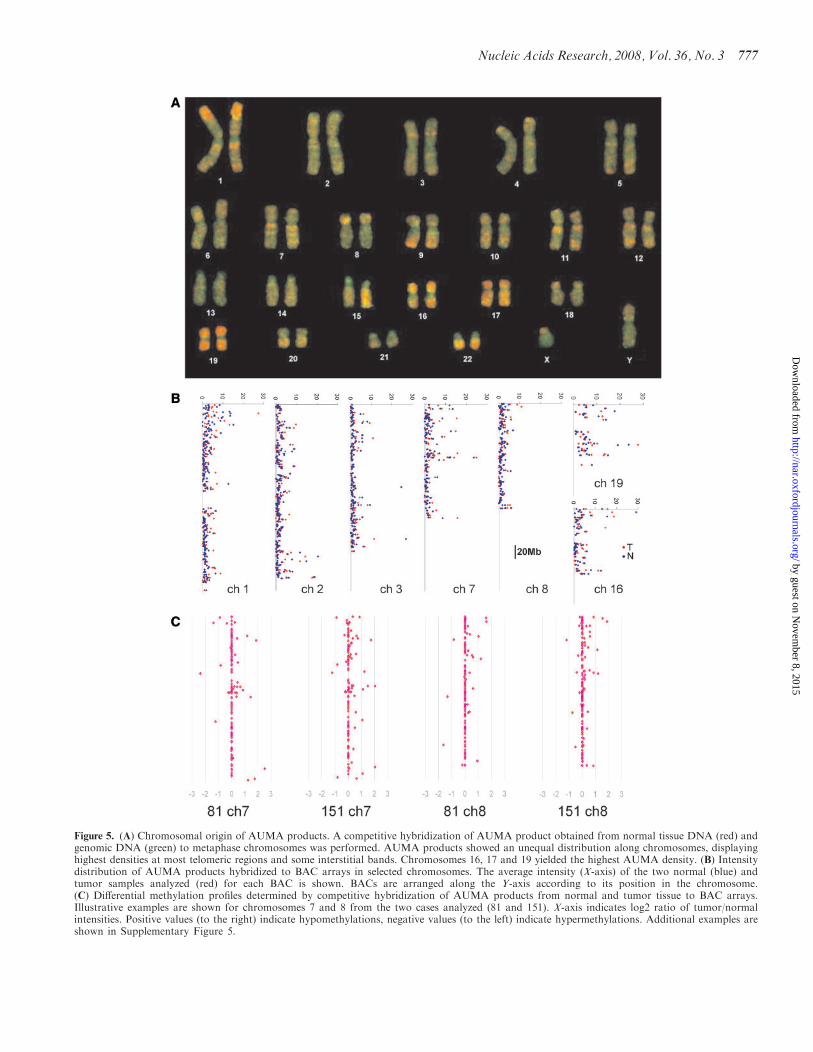
Figure 5 (A) Chromosomal origin of AUMA products A competitive hybridization of AUMA product obtained from normal tissue DNA (red) andgenomic DNA (green) to metaphase chromosomes was performed AUMA products showed an unequal distribution along chromosomes displayinghighest densities at most telomeric regions and some interstitial bands Chromosomes 16 17 and 19 yielded the highest AUMA density (B) Intensitydistribution of AUMA products hybridized to BAC arrays in selected chromosomes The average intensity (X-axis) of the two normal (blue) andtumor samples analyzed (red) for each BAC is shown BACs are arranged along the Y-axis according to its position in the chromosome(C) Differential methylation profiles determined by competitive hybridization of AUMA products from normal and tumor tissue to BAC arraysIllustrative examples are shown for chromosomes 7 and 8 from the two cases analyzed (81 and 151) X-axis indicates log2 ratio of tumornormalintensities Positive values (to the right) indicate hypomethylations negative values (to the left) indicate hypermethylations Additional examples areshown in Supplementary Figure 5
Nucleic Acids Research 2008 Vol 36 No 3 777
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

by Alu family (10 AluS 5 AluY 1 AluJ 4 in CpG islandsand 26 in other sequences)To calculate the proportion and distribution of
unmethylated Alu elements on a genomic scale weperformed Monte Carlo simulations taking into accountthe observed and expected distribution of hits in eachAlu family and CpG islands and the rest of sequences(see Material and Methods section) We estimate thatat least 4104 Alu elements are unmethylated or partiallyunmethylated in normal colonic mucosa This correspondsto 264 of all Alu elements containing the targetsequence AACCCGGG (Table 1) Although AluS andAluY represent the majority of these sequences it shouldbe noted that the methylation pressure is inverse to theconservation of the SmaI site That is the most conservedand younger AluY family shows the lowest relative rateof unmethylation and the older and more degeneratedAluJ family exhibits the highest unmethylation
Application of AUMA to detect differential DNAmethylation in colorectal carcinomas
In order to test the usefulness of the method for thedetection of new altered methylation targets we appliedAUMA to a series of 50 colorectal carcinomas and theirpaired normal mucosa Two cases were excluded fromthe analysis due to recurring experimental failure of thenormal or tumor tissue DNA For the rest of 48 normalndashtumor pairs consistent and fully readable fingerprintswere generated and evaluated for normalndashtumor differ-ential representation A given case presented on average107 29 informative bands (range 98ndash110) The variationwas due to polymorphic display or variable resolutionpower of gel electrophoresisIn this study only those bands showing clear intensity
differences between normal and tumor tissue fingerprints(Figure 2) have been scored as methylation changes sincethey are more likely to reflect tumor-wide alterationsBecause the fingerprints represent sequences flanked bytwo unmethylated sites a decreased intensity in a givenband in the tumor in regard to the paired normal tissueis indicative of hypermethylation while an increasedintensity corresponds to hypomethylation (Figure 2)All tumors displayed changes in regard to the paired
normal tissue The average tumor showed 19 7 (range6ndash37) hypomethylations and 22 10 (range 1ndash39) hyper-methylations It is of note that hypomethylations couldeither be seen as an increase in the intensity of a pre-existingband in the normal tissue or as the appearance of an non-existent band in the normal tissue This contrasts withhypermethylation events which rarely showed the com-plete loss of a band in the tumor sample most likely due tothe unavoidable contamination of normal tissue
Virtually all tagged bands (109 out of 110) were foundto be altered in at least one tumor when compared to itsnormal paired mucosa AUMA tagged bands presented awide distribution in the hypomethylationhypermethyla-tion rates (proportion of tumors showing differentialdisplay compared to the paired normal tissue) (Figure 6)Hypomethylation and hypermethylation showed a strongnegative correlation (r=055 and Plt 00001) indicat-ing that most bands tended to be either hypomethylated
Table 2 A selection of characterized AUMA bands
Band ID Size(bp)
GC Chromosome map(Locationa)
Gene CpG islandb Repetitiveelements inband ends (5030)
Methylation statusin tumorc
Ai1 c3 509 55 17p112 (18206453ndash18206961) SHMT1 Yes Alu SxMIR HypermethylatedAj2 c1 458 49 1q322 (206389082ndash206389539) MGC29875 Yes Alu SqNone HypomethylatedAo1 c4 365 50 19q1332 (53550179ndash53550543) AK001784 No Alu SxMIRb HypomethylatedAp1 c6 358 56 5q352 (175157321ndash175157674) CPLX2 Yes NoneMIR HypermethylatedAq3 c6 339 58 8p233 (2007343ndash2007682) MYOM2 No LTRAlu Y HypomethylatedAr3 c3 329 57 2q143 (127875178ndash127875506) AF370412 Yes NoneMIRb HypomethylatedAs3 c6 306 63 16p133 (3160477ndash3160782) None Yes NoneNone HypermethylatedAu4 c1 268 57 16p133 (3162099ndash3162366) None No tRNANone Hypermethylated
aNucleotide position within the contig (strand +) NCBI Build 361 of the human genomebThe whole sequence or a fragment of the sequence lays not further than 200 bp of a predicted CpG islandcAs compared to the paired normal tissue
Figure 6 Distribution of hypermethylation and hypomethylation ratesin the 110 AUMA tagged bands Rates were obtained by comparisonof the AUMA fingerprints obtained in 50 colorectal tumors ascompared to their respective matched normal tissue
778 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

or hypermethylated A large proportion of tagged bands(78 bands) were recurrently altered in over 25 of thecases included in this series
In order to determine whether normalndashtumor differenceswere limited to isolated independent loci or changes thatmight affect larger chromosomal regions we compared thedistribution of AUMAproducts generated from two pairednormal and tumor tissues and hybridized to BAC arraysDifferential hybridization was observed in many BACssuggesting that relatively large regions encompassing fromseveral hundredKbs to a fewMbsmay undergo concurrenthypomethylation or hypermethylation Telomeric regionsof many chromosomes contained most of the differentialdisplay (Figure 5C) The differential methylation profileswere unaffected by chromosomal dosage as demonstratedby its independence of chromosomal losses and gains (asdetected by Comparative Genomic Hybridization (CGH)(Supplementary Figure 5)
Validation of methylation changes detected by AUMA
To confirm that the changes observed in AUMA finger-prints corresponded to actual changes in the methylationstatus of the sequence eight different sequences obtainedfrom AUMA fingerprints were analyzed in normal andtumor tissues by direct sequencing of sodium bisulfite-treated DNAs (Table 2) Moreover it was demonstratedthat methylation changes affected not only the CpG in atleast one of the two flanking SmaI sites (whose methyla-tion prevents AUMA representation) but also neighboringCpGs (Supplementary Figure 6) In two samples hyper-methylationshypomethylations detected by AUMA couldnot be confirmed by bisulfite sequencing suggesting thatthe change could affect only a small fraction of tumor cellsand that both methods may exhibit different sensitivitiesThe presence of minor subpopulations can be detectedusing more sensitive techniques ie the MethylationSpecific PCR or by sequencing of multiples clones
Functional implications of changes detected by AUMA
Next we wondered if DNA methylation changes detectedby AUMA may have any functional consequences Wechose one of the most recurrent hypomethylated AUMAsequences (Aq3) and performed an insightful epigeneticcharacterization of the region in a series of normalndashtumorpairs and in colon cancer cell lines
Aq3 band is recurrently hypomethylated in tumorsaccording to AUMA fingerprints (Figure 7A) It repre-sents a sequence situated in the eighth intron of theMYOM2 gene (Table 2) and does not fall inside or close toany CpG island The SmaI sites are located in a MLT1Arepeat and an AluYd3 element The methylation status thetwo flanking regions of the AUMA band (465 bp and213 bp long spanning 20 and 11 CpGs respectively) wasanalyzed by bisulfite direct sequencing (Figure 7B)Confirmation of AUMA data was performed in threenormalndashtumor pairs exhibiting differential display of theAq3 band in AUMA fingerprints (cases 17 63 and 74)and two cases lacking this band in both normal and tumorpair (cases 53 and 99) (Figure 7A) as well as five cell lines(HCT116 DLD-1 LoVo HT29 and CaCo2) All normal
tissues as well as tumors 53 and 99 showed heavy methy-lation of this region (Figure 7C) In contrast to this andin agreement with AUMA results tumors 17 63 and74 exhibited hypomethylation at most CpGs Cell linesshowed variable profiles of DNA methylation withCaCo2 exhibiting unmethylation of the MLT1A elementbut heavy methylation of the AluYd3 element whichwas also heavily methylated in HCT116 cells but not in therest of the cell lines tested MYOM2 expression levelsanalyzed by real-time RT-PCR were not affected by themethylation status of this sequence (data not shown)Further 45 normalndashtumor pairs were analyzed for methy-lation of the AluYd3 element by real-time dissociationanalysis (Supplementary Figure 7) and it was foundhypomethylated in 26 tumors (58)Next we wondered whether the DNA methylation
status of the AluYd3 element was associated with alter-native chromatin states We performed ChromatinImmunoPrecipitation (ChIP) analysis of histone 3 (H3)modifications indicative of active chromatin acetylationof lysines 9 and 14 (AcH3K9K14) and dimethylationof lysine 79 (2mH3K79) and silent chromatin trimethy-lation of lysine 9 (3mH3K9) These histone marks werecompared between cell lines HCT116 and LoVo (with100 and 30 methylation of the AluY element respec-tively) The silencing mark 3mH3K9 was 35-fold higher inHCT116 cells compared to the LoVo cell line (Figure 7D)No differences in active marks were observed and thesewere significantly lower than the silencing mark 3mH3K9When HCT116 cells were treated with the demethy-lating agent 5-aza-20-deoxycytidine (5AzaC) and theinhibitor of histone deacetylase trichostatin A (TSA)a moderate decrease in the amount of the 3mH3K9 markwas observed (Figure 7E) As a whole these data suggestthat DNA methylation changes in this AluYd3 elementare accompanied by alternated chromatin states Themolecular consequences of such epigenetic changes remainto be identified
DISCUSSION
Epigenetic states of Alu elements
Full genome sequencing has provided precise mapsof repetitive elements and several studies have investigatedtheir distribution and relationship with genome structure(48ndash51) More recently a few studies have exploredsequence-dependent associations between repetitive ele-ments and the epigenetic landscape There is a character-istic distribution of interspersed elements along methylatedand unmethylated domains with most elements in themethylated compartment of the genome (21) NeverthelessSINEs which include Alu elements are the repetitivesequences most commonly found in unmethylated domains(21) and some Alu elements may contain discriminatorymotifs associated with methylation-resistant CpG islands(52) Somatic cells show unstable epigenetic profiles inrepetitive elements as demonstrated by global measure-ments of either DNA methylation (18204041) or histonemodifications (5354) Recent studies have revealedinterindividual variability in DNA methylation profiles
Nucleic Acids Research 2008 Vol 36 No 3 779
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
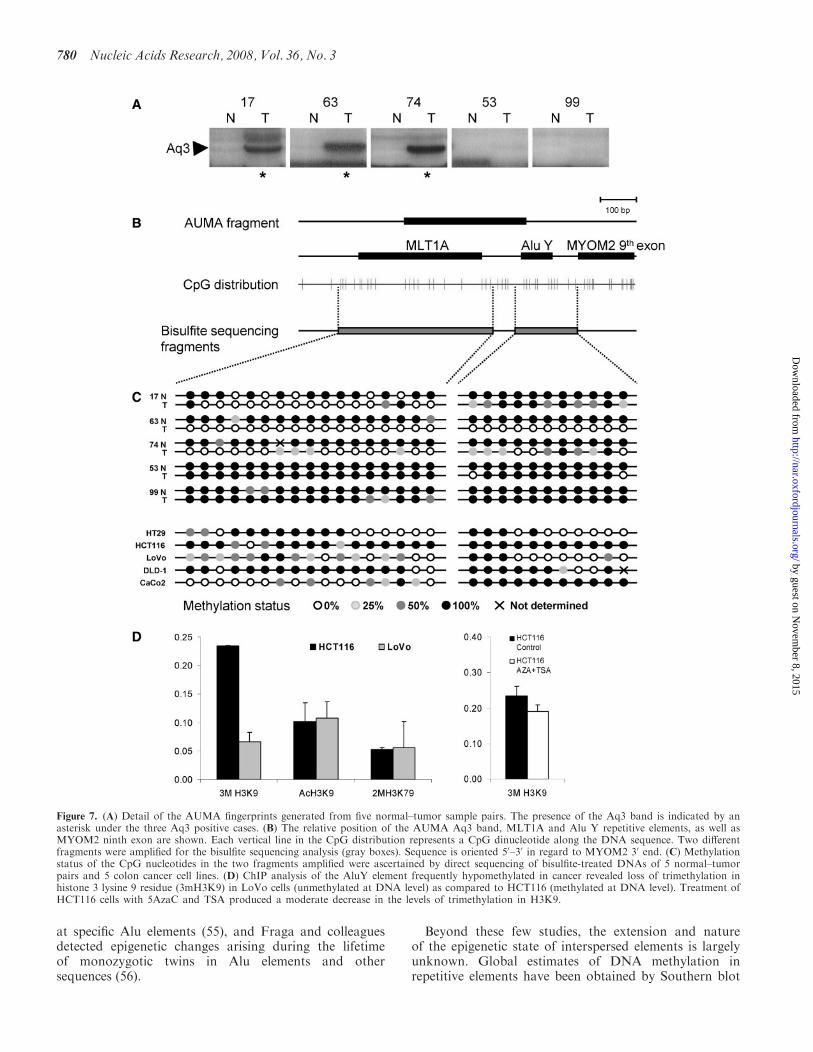
at specific Alu elements (55) and Fraga and colleaguesdetected epigenetic changes arising during the lifetimeof monozygotic twins in Alu elements and othersequences (56)
Beyond these few studies the extension and natureof the epigenetic state of interspersed elements is largelyunknown Global estimates of DNA methylation inrepetitive elements have been obtained by Southern blot
Figure 7 (A) Detail of the AUMA fingerprints generated from five normalndashtumor sample pairs The presence of the Aq3 band is indicated by anasterisk under the three Aq3 positive cases (B) The relative position of the AUMA Aq3 band MLT1A and Alu Y repetitive elements as well asMYOM2 ninth exon are shown Each vertical line in the CpG distribution represents a CpG dinucleotide along the DNA sequence Two differentfragments were amplified for the bisulfite sequencing analysis (gray boxes) Sequence is oriented 50ndash30 in regard to MYOM2 30 end (C) Methylationstatus of the CpG nucleotides in the two fragments amplified were ascertained by direct sequencing of bisulfite-treated DNAs of 5 normalndashtumorpairs and 5 colon cancer cell lines (D) ChIP analysis of the AluY element frequently hypomethylated in cancer revealed loss of trimethylation inhistone 3 lysine 9 residue (3mH3K9) in LoVo cells (unmethylated at DNA level) as compared to HCT116 (methylated at DNA level) Treatment ofHCT116 cells with 5AzaC and TSA produced a moderate decrease in the levels of trimethylation in H3K9
780 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

analyses (30) and more recently by using approachesbased on bisulfite conversion of the unmethylated cytosine(404157) These studies have confirmed the globalhypomethylation of most tumors but they do not providedetailed information on the nature and localization of theunmethylated elements In silico analysis has revealed thata number of Alu elements close to CpG islands retain ahigh proportion of CpG sites and this is presumed to bea sign of unmethylation (58) but no experimental proofhas been provided In our point of view the lack ofsimple specific and sensitive methodologies to screen forepigenetic changes in repetitive elements on the genomicscale has precluded a clearer understanding of the natureand implications of these sequences in cell biology
Properties of QUMA and AUMA
Here we report a systematic screening of unmethylatedAlus as a tool to determine the extent of DNA hypo-methylation to identify specifically unmethylated ele-ments and to detect epigenetic alterations in cancer cellsQUMA is a very simple and specific method and providesaccurate relative estimates of the number of unmethylatedelements QUMA is specially appropriate for compara-tive studies but also provides a raw quantitation of thenumber of unmethylated elements per haploid genomeoutlining the extent of hypomethylated Alursquos in normaland pathologic cells QUMA analysis indicates that about1 out of 6 Alu elements containing the AACCCGGGsite are unmethylated while in tumors this figure nearlydoubles in agreement with previous studies (23) Althoughthese analyses are likely to generate good estimates at thecomparative level (between samples) absolute valuesshould be treated with caution because the determinationrefers to a single CpG site within the Alu element
To date there is still a lack of proper methodologiesallowing genome-wide screenings for recurrent hypo-methylated regions that may have some impact ontumor biology Even though QUMA and other method-ologies (4159) allow quantitation of unmethylatedrepeats they do not provide a straightforward approachto identify and map the amplified targets At this pointAUMA takes us a step further allowing the undoubtfulidentification of hypomethylated sequences in additionto hypermethylated targets Although AUMA is speciallysuited to determine the nature of the unmethylatedelements it also allows the calculation of global unmethy-lation in Alu elements Nevertheless it should be takeninto account that this is an indirect measure because itrelies in the extent of methylation in CpG islands andother sequences Moreover unmethylation of a secondSmaI site near the Alu is also required to generate theAUMA band and hence to be detected While AUMAshares many technical steps with other techniques namelyMCA (60) and AIMS (43) its design is conceptuallyunique since AUMA scans for the atypically unmethy-lated Alu sequences unlike the other approaches that areenriched for typical methylated sequences
Due to sequence degeneration both QUMA andAUMA are more effective in screening for unmethylationin younger elements This trend is more clearly seen
in AUMA with only 9 of the Alu elements of the old Jsubfamily containing the SmaI site retain the AA dinu-cleotide needed for their amplification while this figure is91 and 80 in the younger AluY and AluS subfamiliesrespectively (Table 1) making clear that younger Aluelements tend to retain the SmaI site nearly as much asthey retain the AA dinucleotide required for their ampli-fication This bias is not a handicap since unmethylatedAlu sequences revealed by AUMA are likely to representthe most relevant events of this kind because spuriousunmethylation of old Alu elements retaining a single or afew CpG sites is expected to have less biological signif-icance than unmethylation of younger Alu elements thatare usually closer to active chromatin regions (21) andretain more CpGs The stronger methylation pressureobserved in the AluY class is consistent with thispostulateAUMA was designed to amplify DNA fragments
containing the target sequence (AACCCGGG) which ispresent in Alu and other repetitive elements Because asingle primer was used for PCR amplification the targetsequence must appear in both strands of the DNA atrelatively nearby positions As expected Alu elementswith more than one million copies per human genome(15) were the most frequent repeat in AUMA bands(50 in sequences isolated from non-tumor tissue) butonly two sequenced bands contained two inverted Alurepeats (Supplementary Table 1) This observation isin concordance with previous works reporting on theinstability of this inverted repeats which might havecaused their exclusion from the human genome (6162)More restrictive conditions to select for Alu or any otherrepeat of interest may be achieved by extending the 30 endof the primer specific sequence (see Supplementary Data)however the number of sequences we obtained was consi-dered appropriate to accomplish the original aim of thestudy which is to screen for differentially methylatedrepetitive elements in colorectal cancerIt is worth noting that AUMA patterns are highly
reproducible not only in replicates but also among differ-ent samples which indicates that the unmethylated statusof these repeats is tightly controlled probably by theepigenetic status of nearby regions This is strengthened bythe confirmation that unmethylation extends many CpGsites beyond the SmaI cut site Moreover about 50 ofthe bands tagged in AUMA fingerprints exhibited variabledisplay among normal tissues (data not shown) suggest-ing the usefulness of this technique to investigate epigene-tic polymorphismsAlursquos and other repetitive elements tend to be highly
methylated in most somatic tissues (894063) Here wehave identified 78 lsquoatypicalrsquo Alu elements exhibitingfull or partial unmethylation in normal colonic mucosacells Different evidences underscore the adequacy of thisapproach to track changes with possible functional impli-cations (i) a significant portion of the characterized bandsare located inside or nearby CpG islands and genes(ii) AUMA products show a characteristic distributionin R bands coincidentally with the distribution of Alusequences (15) indicating a bias toward the gene-richestportion of the genome known as the H3 isochore (64)
Nucleic Acids Research 2008 Vol 36 No 3 781
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

DNAmethylation along Alu families
Alu families showed striking differences in their methyla-tion level Most of the Alu elements characterized here arefrom the younger families AluS and AluY (74 and 22respectively) Nevertheless this observation is mainly dueto the depletion of CpG sites in older Alu elementsHence only 1 out 230 AluJ elements maintains theAUMA target site (AACCCGGG) while younger ele-ments show higher rates of maintenance in accordancewith their age (AluS 1 out of 7 AluY 2 out of 5)(Table 1) Interestingly the rate of unmethylation is higherin older elements (AluJ 122 AluS 31 AluY 16)(Table 1) As noted by Rollins et al (21) the boundariesof unmethylated domains tend to be occupied by methy-lated Alu transposons of the younger AluS and AluYfamilies Other studies have noted that CpG island-associated Alursquos retain a higher proportion of CpG sitessuggesting that these elements are unmethylated in thegerm line (58) These unmethylated elements that can beeasily revealed by AUMA are likely to play a regulatoryrole in a significant number of genes (65) AUMA oftumors was enriched in Alu sequences as compared withnormal tissue (41 versus 29 respectively) suggestinga favored hypomethylation of Alu elements within theglobal genomic hypomethylation associated with tumor-igenesis (23)
Application of AUMA to detect epigenetic changesin cancer cells
Cancer-related hypomethylation is well documented (23)and different studies have demonstrated the demethylationof Alursquos and other repetitive elements in different typesof neoplasias (66) Our data are consistent with previousestimates and go one step forward in the characterizationof unmethylated repeats The AUMA approach wasconceived as a straightforward DNA methylation screen-ing strategy targeting specific interspersed repeats andsuitable to be applied to large series of samples orexperimental conditions as reported hereThe application of AUMA to a series of colorectal
carcinomas and their paired matched normal tissue hasrevealed a high rate of alterations This indicates theplasticity of epigenetic control of the elements screenedby AUMA in colorectal carcinogenesis Although somebands show bidirectional changes (hypomethylations andhypermethylations) which have been also reported inother sequences (4267) most bands display an alternativetrend either toward hypermethylations or hypomethyla-tions Some of these changes are highly recurrent (in morethan 50 of tumors) suggesting that they may representrelevant alterations related to mechanisms frequentlydisturbed in colon cancer Because the default status ofrepetitive elements is methylation hypomethylationsare readily detected as the emergence of a new band inthe tumor AUMA fingerprint Since all amplified bandsinclude a unique sequence it has been possible to identifyall of the isolated bands and pinpoint them in thegenomic mapAs an example we have investigated the Aq3 sequence
one of the most recurrent hypomethylations in this study
Aq3 band is flanked by two repeats a LTR and anAluY which map within an intron between exons 8 and9 of the MYOM2 gene at 8p233 Both elements areheavily methylated in normal tissue and partially to fullyunmethylated in tumor tissue Interestingly we havefound moderately high levels of the heterochromatinassociated mark 3mH3K9 in the fully methylated AluYd3element in the HCT116 cell line while the levels weresignificantly lower (35-fold) in the partially demethylatedLoVo cell line Furthermore none of the classical activemarks AcH3K9K14 and 2mH3K79 have been foundenriched in the LoVo cell line These data are in concor-dance with preliminary data showing that the hypomethy-lation does not affect the expression of MYOM2 (data notshown) but could rather affect chromatin structure in theregion In agreement with these observations this genomicregion undergoes frequent losses (68ndash70) and is rearrangedin many different types of tumors (7172) which hints ata role for DNA hypomethylation in genomic instability(24ndash2773) The specific functional consequences of thishypomethylation deserve further investigations
Another application of AUMA is the detection ofgenomic regions that have been silenced in cancer Inter-spersed elements are concentrated in gene-rich regions anddue to the intended selection of unmethylated repetitiveelements in AUMA it appears reasonable to postulatethat normally unmethylated sequences are likely to pin-point active genomic regions In this context AUMAprovides a large collection of genomic regions undergoinghypermethylation which are readily seen as bands recur-rently loss in the fingerprints DNA methylation asso-ciated epigenetic silencing is probably one of the mostprevalent mechanisms of tumor suppression inactivationin cancer (333774) Therefore AUMA can also be usedto screen for differential methylation not only in Aluelements but also in unique sequences and repetitiveelements other than Alu
In summary QUMA and AUMA methodologies area simple and novel approach to explore and gain insightsinto the functional significance of interspersed genomicelements and neighboring sequences Due to its distinctivefeatures (bias for unmethylated elements in gene-richregions and detection of both hypomethylation and hyper-methylation) we think that these techniques constitutea new and unique tool that should complement globaldeterminations and high-resolution genome-wide scanningstrategies Beyond unmethylated repetitive elementsAUMA can be also used to detect recurrent epigeneticchanges associated with tumorigenesis including geneepigenetic inactivation
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online
ACKNOWLEDGEMENTS
We thank Gemma Aiza for technical support and JessicaHalow for critical review of the manuscript JR wasa fellow of the Generalitat de Catalunya EV was a fellow
782 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

of the Fondo de Investigacion Sanitaria (FIS) This workwas supported by a grant from the Ministry of Educationand Science (SAF2006351) and the Consolider-Ingenio2010 Program (CSD2006-49) Funding to pay the OpenAccess publication charges for this article was providedby the Institute of Predictive and Personalized Medicine ofCancer (IMPPC)
Conflict of interest statement None declared
REFERENCES
1 MattickJS (2003) Challenging the dogma the hidden layer ofnon-protein-coding RNAs in complex organisms Bioessays 25930ndash939
2 ZuckerkandlE (2002) Why so many noncoding nucleotides Theeukaryote genome as an epigenetic machine Genetica 115 105ndash129
3 KreahlingJ and GraveleyBR (2004) The origins and implicationsof Aluternative splicing Trends Genet 20 1ndash4
4 JasinskaA and KrzyzosiakWJ (2004) Repetitive sequences thatshape the human transcriptome FEBS Lett 567 136ndash141
5 JordanIK RogozinIB GlazkoGV and KooninEV (2003)Origin of a substantial fraction of human regulatory sequences fromtransposable elements Trends Genet 19 68ndash72
6 HaslerJ and StrubK (2006) Alu elements as regulators of geneexpression Nucleic Acids Res 34 5491ndash5497
7 SlotkinRK and MartienssenR (2007) Transposable elements andthe epigenetic regulation of the genome Nat Rev Genet 8272ndash285
8 SchmidCW (1998) Does SINE evolution preclude Alu functionNucleic Acids Res 26 4541ndash4550
9 YoderJA WalshCP and BestorTH (1997) Cytosine methyla-tion and the ecology of intragenomic parasites Trends Genet 13335ndash340
10 JaenischR and BirdA (2003) Epigenetic regulation of geneexpression how the genome integrates intrinsic and environmentalsignals Nat Genet 33(Suppl) 245ndash254
11 WeissmannF and LykoF (2003) Cooperative interactions betweenepigenetic modifications and their function in the regulation ofchromosome architecture Bioessays 25 792ndash797
12 GollMG and BestorTH (2005) Eukaryotic cytosine methyl-transferases Annu Rev Biochem 74 481ndash514
13 FazzariMJ and GreallyJM (2004) Epigenomics beyond CpGislands Nat Rev Genet 5 446ndash455
14 LanderES LintonLM BirrenB NusbaumC ZodyMCBaldwinJ DevonK DewarK DoyleM et al (2001) Initialsequencing and analysis of the human genome Nature 409860ndash921
15 BatzerMA and DeiningerPL (2002) Alu repeats and humangenomic diversity Nat Rev Genet 3 370ndash379
16 KorenbergJR and RykowskiMC (1988) Human genomeorganization Alu lines and the molecular structure of metaphasechromosome bands Cell 53 391ndash400
17 ChenC GentlesAJ JurkaJ and KarlinS (2002) Genespseudogenes and Alu sequence organization acrosshuman chromosomes 21 and 22 Proc Natl Acad Sci USA 992930ndash2935
18 SchmidCW (1991) Human Alu subfamilies and theirmethylation revealed by blot hybridization Nucleic Acids Res 195613ndash5617
19 Gama-SosaMA WangRY KuoKC GehrkeCW andEhrlichM (1983) The 5-methylcytosine content of highlyrepeated sequences in human DNA Nucleic Acids Res 113087ndash3095
20 KochanekS RenzD and DoerflerW (1993) DNA methylation inthe Alu sequences of diploid and haploid primary human cellsEMBO J 12 1141ndash1151
21 RollinsRA HaghighiF EdwardsJR DasR ZhangMQJuJ and BestorTH (2006) Large-scale structure of genomicmethylation patterns Genome Res 16 157ndash163
22 BirdA (2002) DNA methylation patterns and epigenetic memoryGenes Dev 16 6ndash21
23 EhrlichM (2002) DNA methylation in cancer too much but alsotoo little Oncogene 21 5400ndash5413
24 EdenA GaudetF WaghmareA and JaenischR (2003)Chromosomal instability and tumors promoted by DNA hypo-methylation Science 300 455
25 ChenRZ PetterssonU BeardC Jackson-GrusbyL andJaenischR (1998) DNA hypomethylation leads to elevatedmutation rates Nature 395 89ndash93
26 SuzukiK SuzukiI LeodolterA AlonsoS HoriuchiSYamashitaK and PeruchoM (2006) Global DNA demethylationin gastrointestinal cancer is age dependent and precedes genomicdamage Cancer Cell 9 199ndash207
27 RodriguezJ FrigolaJ VendrellE RisquesRA FragaMFMoralesC MorenoV EstellerM CapellaG et al (2006)Chromosomal instability correlates with genome-wide DNAdemethylation in human primary colorectal cancers Cancer Res66 8462ndash9468
28 LiuWM MaraiaRJ RubinCM and SchmidCW (1994)Alu transcripts cytoplasmic localisation and regulation by DNAmethylation Nucleic Acids Res 22 1087ndash1095
29 Roman-GomezJ Jimenez-VelascoA AgirreX CervantesFSanchezJ GarateL BarriosM CastillejoJA NavarroG et al(2005) Promoter hypomethylation of the LINE-1 retrotransposableelements activates senseantisense transcription and marks theprogression of chronic myeloid leukemia Oncogene 24 7213ndash7223
30 EhrlichM (2006) Cancer-linked DNA hypomethylation and itsrelationship to hypermethylation Curr Top Microbiol Immunol310 251ndash274
31 PlassC (2002) Cancer epigenomics Hum Mol Genet 112479ndash2488
32 JonesPA and BaylinSB (2002) The fundamental role ofepigenetic events in cancer Nat Rev Genet 3 415ndash428
33 HermanJG and BaylinSB (2003) Gene silencing in cancer inassociation with promoter hypermethylation N Engl J Med 3492042ndash2054
34 LundAH and van LohuizenM (2004) Epigenetics and cancerGenes Dev 18 2315ndash2335
35 FeinbergAP and TyckoB (2004) The history of cancer epige-netics Nat Rev Cancer 4 143ndash153
36 LairdPW (2005) Cancer epigenetics Hum Mol Genet 14R65ndashR76
37 EstellerM (2007) Cancer epigenomics DNA methylomes andhistone-modification maps Nat Rev Genet 6 6
38 LairdPW (2003) The power and the promise of DNA methylationmarkers Nat Rev Cancer 3 253ndash266
39 CottrellSE (2004) Molecular diagnostic applications of DNAmethylation technology Clin Biochem 37 595ndash604
40 YangAS EstecioMR DoshiK KondoY TajaraEH andIssaJP (2004) A simple method for estimating global DNAmethylation using bisulfite PCR of repetitive DNA elementsNucleic Acids Res 32 e38
41 WeisenbergerDJ CampanM LongTI KimM WoodsCFialaE EhrlichM and LairdPW (2005) Analysis of repetitiveelement DNA methylation by MethyLight Nucleic Acids Res 336823ndash6836
42 FrigolaJ SoleX PazMF MorenoV EstellerM CapellaGand PeinadoMA (2005) Differential DNA hypermethylation andhypomethylation signatures in colorectal cancer Hum Mol Genet14 319ndash326
43 FrigolaJ RibasM RisquesRA and PeinadoMA (2002)Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS) Nucleic Acids Res 30 e28
44 VendrellE RibasM VallsJ SoleX GrauM MorenoVCapellaG and PeinadoMA (2007) Genomic and transcriptomicprognostic factors in R0 Dukes B and C colorectal cancer patientsInt J Oncol 30 1099ndash1107
45 PeruchoM WelshJ PeinadoMA IonovY and McClellandM(1995) Fingerprinting of DNA and RNA by arbitrarily primedpolymerase chain reaction applications in cancer research MethodsEnzymol 254 275ndash290
46 StirzakerC SongJZ DavidsonB and ClarkSJ (2004)Transcriptional gene silencing promotes DNA hypermethylationthrough a sequential change in chromatin modifications in cancercells Cancer Res 64 3871ndash3877
Nucleic Acids Research 2008 Vol 36 No 3 783
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from

47 XingJ HedgesDJ HanK WangH CordauxR andBatzerMA (2004) Alu element mutation spectra Molecular clocksand the effect of DNA methylation J Mol Biol 344 675ndash682
48 MedstrandP van de LagemaatLN and MagerDL (2002)Retroelement distributions in the human genome variationsassociated with age and proximity to genes Genome Res 121483ndash1495
49 GroverD MajumderPP RaoCB BrahmachariSK andMukerjiM (2003) Nonrandom distribution of alu elements in genesof various functional categories insight from analysis of humanchromosomes 21 and 22 Mol Biol Evol 20 1420ndash1424
50 GroverD MukerjiM BhatnagarP KannanK andBrahmachariSK (2004) Alu repeat analysis in the complete humangenome trends and variations with respect to genomic compositionBioinformatics 20 813ndash817
51 PriceAL EskinE and PevznerPA (2004) Whole-genomeanalysis of Alu repeat elements reveals complex evolutionaryhistory Genome Res 14 2245ndash2252
52 FeltusFA LeeEK CostelloJF PlassC and VertinoPM(2006) DNA motifs associated with aberrant CpG island methyla-tion Genomics 15 15
53 FragaMF BallestarE Villar-GareaA Boix-ChornetMEspadaJ SchottaG BonaldiT HaydonC RoperoS et al(2005) Loss of acetylation at Lys16 and trimethylation at Lys20 ofhistone H4 is a common hallmark of human cancer Nat Genet 1313
54 KondoY and IssaJP (2003) Enrichment for histone H3 lysine 9methylation at Alu repeats in human cells J Biol Chem 27827658ndash27662
55 SandoviciI Kassovska-BratinovaS Loredo-OstiJC LeppertMSuarezA StewartR BautistaFD SchiraldiM and SapienzaC(2005) Interindividual variability and parent of origin DNAmethylation differences at specific human Alu elements Hum MolGenet 14 2135ndash2143
56 FragaMF BallestarE PazMF RoperoS SetienFBallestarML Heine-SunerD CigudosaJC UriosteM et al(2005) Epigenetic differences arise during the lifetime of mono-zygotic twins Proc Natl Acad Sci USA 102 10604ndash10609
57 XiongZ and LairdPW (1997) COBRA a sensitive and quanti-tative DNA methylation assay Nucleic Acids Res 25 2532ndash2534
58 BrohedeJ and RandKN (2006) Evolutionary evidence suggeststhat CpG island-associated Alus are frequently unmethylated inhuman germline Hum Genet 119 457ndash458
59 ChoiIS EstecioMR NaganoY Kim doH WhiteJAYaoJC IssaJP and RashidA (2007) Hypomethylation ofLINE-1 and Alu in well-differentiated neuroendocrine tumors(pancreatic endocrine tumors and carcinoid tumors) Mod Pathol20 802ndash810
60 ToyotaM HoC AhujaN JairKW LiQ Ohe-ToyotaMBaylinSB and IssaJP (1999) Identification of differentiallymethylated sequences in colorectal cancer by methylated CpGisland amplification Cancer Res 59 2307ndash2312
61 LobachevKS StengerJE KozyrevaOG JurkaJGordeninDA and ResnickMA (2000) Inverted Alu repeatsunstable in yeast are excluded from the human genome EMBO J19 3822ndash3830
62 StengerJE LobachevKS GordeninD DardenTA JurkaJand ResnickMA (2001) Biased distribution of inverted and directAlus in the human genome implications for insertion exclusionand genome stability Genome Res 11 12ndash27
63 Hellmann-BlumbergU HintzMF GatewoodJM andSchmidCW (1993) Developmental differences in methylation ofhuman Alu repeats Mol Cell Biol 13 4523ndash4530
64 SacconeS CaccioS KusudaJ AndreozziL and BernardiG(1996) Identification of the gene-richest bands in human chromo-somes Gene 174 85ndash94
65 OeiSL BabichVS KazakovVI UsmanovaNMKropotovAV and TomilinNV (2004) Clusters of regulatorysignals for RNA polymerase II transcription associated with Alufamily repeats and CpG islands in human promoters Genomics 83873ndash882
66 WilsonAS PowerBE and MolloyPL (2007) DNA hypo-methylation and human diseases Biochim Biophys Acta 1775138ndash162
67 NishiyamaR QiL TsumagariK WeissbeckerK DubeauLChampagneM SikkaS NagaiH and EhrlichM (2005) A DNARepeat NBL2 Is Hypermethylated in Some Cancers butHypomethylated in Others Cancer Biol Ther 4 4
68 EmiM FujiwaraY NakajimaT TsuchiyaE TsudaHHirohashiS MaedaY TsurutaK MiyakiM et al (1992)Frequent loss of heterozygosity for loci on chromosome 8p inhepatocellular carcinoma colorectal cancer and lung cancerCancer Res 52 5368ndash5372
69 SunwooJB SunPC GuptaVK SchmidtAP El-MoftyS andScholnickSB (1999) Localization of a putative tumor suppressorgene in the sub-telomeric region of chromosome 8p Oncogene 182651ndash2655
70 MuscheckM SukosdF PestiT and KovacsG (2000) Highdensity deletion mapping of bladder cancer localizes the putativetumor suppressor gene between loci D8S504 and D8S264 atchromosome 8p233 Lab Invest 80 1089ndash1093
71 WongN WongKF ChanJK and JohnsonPJ (2000)Chromosomal translocations are common in natural killer-celllymphomaleukemia as shown by spectral karyotyping HumPathol 31 771ndash774
72 JinY JinC WennerbergJ HoglundM and MertensF (2001)Cytogenetic and fluorescence in situ hybridization characterizationof chromosome 8 rearrangements in head and neck squamous cellcarcinomas Cancer Genet Cytogenet 130 111ndash117
73 KarpfAR and MatsuiS (2005) Genetic disruption of cytosineDNA methyltransferase enzymes induces chromosomal instability inhuman cancer cells Cancer Res 65 8635ndash8639
74 EggerG LiangG AparicioA and JonesPA (2004) Epigeneticsin human disease and prospects for epigenetic therapy Nature 429457ndash463
784 Nucleic Acids Research 2008 Vol 36 No 3
by guest on Novem
ber 8 2015httpnaroxfordjournalsorg
Dow
nloaded from
Related Documents











