This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Virus Research 151 (2010) 118–130
Contents lists available at ScienceDirect
Virus Research
journa l homepage: www.e lsev ier .com/ locate /v i rusres
Genome analysis of an orange stem pitting citrus tristeza virus isolate reveals anovel recombinant genotype
Avijit Roy, R.H. Brlansky ∗
University of Florida, IFAS, Plant Pathology Department, Citrus Research and Education Center, 700 Experiment Station Road, Lake Alfred, FL 33850, United States
a r t i c l e i n f o
Article history:Received 14 January 2010Received in revised form 26 March 2010Accepted 30 March 2010Available online 8 April 2010
Keywords:ClosterovirusGenotypePhylogenetic analysisRecombination
a b s t r a c t
An orange stem pitting citrus tristeza virus (CTV) isolate CTV-B165 was found to be symptomatically simi-lar to other known CTV-VT isolates however molecular methods failed to classify it as an identifiable CTVgenotype. The sequence variation of the Indian CTV-B165 isolate was compared to the three well knownCTV genotypes, T36, T30, and VT. The genome of the predominant component of CTV-B165 was 19,247 ntin length with 12 open reading frames (ORFs) and was structurally identical to the other CTV isolates. Allthe completely sequenced CTV isolates except the VT isolate were 2–55 nt longer than the CTV-B165. Incomparison to the other fully sequenced T36, T30 and VT genotypic isolates, CTV-B165 had nucleotideidentity of 72–86% in ORF1 and 92–99% in ORFs 2–11. Sequence data of independent overlapping clonesfrom the CTV-B165 genome showed highly divergent sequences of the overlapping region of 5′-UTR andORF1a, the inter-domain region of ORF1a and the partial regions of ORF2. Phylogenetic analysis of fivedomains of ORF1a, ORF1b, and ORF2 revealed that CTV-B165 isolate distinctly segregates from the existingthree genotypes in the dendrograms and was supported by high bootstrap values and robust tree topol-ogy. The PHYLPRO graphical analysis showed multiple recombination signals with significant correlationvalues. The precise detection of recombination sites for different genomic regions in CTV sequences wassupported by several recombination-detecting methods. Collectively, the phylogenetic and recombina-tion analyses suggest that the observed CTV-B165 genotype variation is an outcome of inter-genotyperecombination. To determine the presence of the CTV-B165 genotype a pair of genome specific primerswas designed and standardized for reliable detection of the novel CTV genotype by reverse-transcriptionpolymerase chain reaction.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Citrus tristeza virus (CTV), a member of the genus Closteroviruswithin the family Closteroviridae, is one of the more complex singlestranded RNA plant viruses. CTV is semi-persistently transmitted byseveral aphid species, graft transmitted, and is limited to phloem ofinfected plants species of the Rutaceae (Bar-Joseph and Lee, 1989).Isolates of CTV noticeably differ in their pathogenicity on varioushost species and cultivars (Moreno and Guerri, 1997). CTV usuallyoccurs as a complex mixture of distinct viral genotypes, subge-nomic RNAs (sgRNAs), and defective RNAs (dRNAs) with one ofthe genotypes often dominating the population (Ayllón et al., 2001,1999; Roy and Brlansky, 2009; Roy et al., 2005b). The filamentousflexuous virions are 2000 × 10–12 nm in size (Bar-Joseph and Lee,1989) and the viral genome is encapsidated by two coat proteinsof 25 and 27 kDa (Febres et al., 1996). The single stranded positivesense genomic RNA (gRNA) of CTV varies from 19,226 nucleotides
∗ Corresponding author. Tel.: +1 863 956 1151; fax: +1 863 956 4631.E-mail address: [email protected] (R.H. Brlansky).
(nt) (GenBank accession U16304) to 19,302 (GenBank accessionAB046398). The virus genome is organized into 12 open readingframes (ORFs) and encodes at least 19 protein products with twountranslated regions (UTRs) of about 107 and 273 nt at the 5′ and 3′
termini, respectively (Karasev, 2000; Mawassi et al., 1996; Pappuet al., 1994). The CTV genome exhibits over 30% diversity in the 5′
proximal half and less than 10% in the 3′ proximal half of the dif-ferent known genotypes (Mawassi et al., 1996; Roy et al., 2005b).Computer assisted sequence analysis showed that CTV as well asother closterovirus genomes have two conserved groups or blocksof genes. The first group (ORFs 1a and 1b) includes replication asso-ciated genes encoding RNA depending RNA polymerase (RdRp),helicase (HEL), methyltransferase (MET), and two papain like pro-teases (LProI and II) processing enzymes (Dolja et al., 1994). Thereplication associated genes of all closteroviruses are conservedwhich also is found throughout the entire super group of Sindbislike viruses (Dolja et al., 1994; Koonin and Dolja, 1993). The secondgroup of five gene products (ORFs 3–7), the hall-mark proteins ofClosteroviridae are present in all reported members of the family.
Hilf et al. (1999) developed multiple molecular markers(MMMs) primers to four CTV genotypes which have been used in
0168-1702/$ – see front matter © 2010 Elsevier B.V. All rights reserved.doi:10.1016/j.virusres.2010.03.017

Author's personal copy
A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130 119
classifying the genetic mixture of CTV in various isolates (Hilf etal., 1999; Roy and Brlansky, 2004). MMM analysis of a worldwidecollection of CTV isolates found that 55 isolates were unassigned(Hilf et al., 2005) and includes 2 isolates from India. In a previousstudy (Roy and Brlansky, 2004) determining the presence of mixedinfections of 21 Indian CTV isolates, we found that even though CTV-B165 was amplified with VT genotype specific MMMs the sequenceof the overlapping region of ORF1a and ORF1b was not related tothe CTV-VT. However in further studies involving CTV-B165, a par-tial region of the ORF1a was found to be totally distinct from otherCTV genotypes (Roy et al., 2005b). Because of this, we began stud-ies to determine the complete sequence of CTV-B165, to compareit with other known 10 complete CTV sequences (Albiach-Martíet al., 2000; Harper et al., 2009; Karasev et al., 1995; Mawassi etal., 1996; Pappu et al., 1994; Ruiz-Ruiz et al., 2006; Suastika et al.,2001; Vives et al., 1999; Yang et al., 1999) and to determine if anyrecombination has occurred.
Recombination is one of the major causes of virus evolution (Lai,1992; Posada et al., 2002; Roossinck, 1997; Simon and Bujarski,1994) but it requires two parental sequences in the same cell.Recombination occurs in mixed genotype infections and after aphidtransmission from mixed infections (Roy and Brlansky, 2009).Long-term persistent infections and multiple aphid transmissionsof CTV from one tree to another tree may increase the accumu-lation of multiple CTV genotypes in a host that then generatesabundant viral genetic variants (Weng et al., 2007). Multiple CTVgenotypic variants are evolved either by subsequent homologous(Vives et al., 1999) or nonhomologous recombination (Mawassi etal., 1995b). The most direct evidence for recombination in CTV isthe presence of dRNAs (Ayllón et al., 2001; Mawassi et al., 1995a,b)or exchange of viral gene with sgRNA in the process of replica-tion (Yang et al., 1997). In an earlier study, multiple recombinationevents were observed within CTV isolate SY568 and multiple vari-ants with divergent sequence identities were described (Vives etal., 1999, 2005).
We report here the complete genomic sequence of a severeseedling yellows (SY) and orange stem pitting (SP) isolate, CTV-B165, from southern India. This is the first complete genomesequence of a CTV isolate from India. The genome sequencevariability was used to determine the phylogenetic evolutionaryrelatedness between CTV-B165 and 10 other completely sequencedgeographical isolates. To better understand the impact of recom-bination on genomic evolution, bioinformatics and statisticalapproaches were utilized to identify the new recombinant geno-type. The possibility that the CTV-B165 isolate may have a commonorigin with T30, T36 and VT genotypes is discussed. A CTV-B165genotype specific primer pair was developed and standardized toidentify the novel genotype.
2. Materials and methods
2.1. Virus isolates
CTV-B165 was obtained from the Exotic Pathogens of Citrus Col-lection (EPCC) at the USDA, ARS, MPPL, Beltsville, MD, USA butit was originally collected from Gonikopal, Bangalore in southernIndia and sent to the EPCC by K.L. Manjunath in 1988. The originalsource of this isolate was from an infected Ellendale Mandarin (Cit-rus reticulata, Blanco) which tested serologically positive for CTV.Biologically, CTV-B165 induced a severe vein clearing leaf reactionin Mexican lime (C. aurantifolia (Christm.) Swing.), and Coorg man-darin (C. reticulata, Blanco), strong foliar SY symptoms in both sourorange (C. aurantium L.) and Duncan grapefruit (C. paradisi Macf.)seedlings as well as some bark thickening with wood bristles inthe grapefruit. The symptom of severe SP was produced in sweet
orange (C. sinensis (L.) Osb.) plants however no SP symptoms werefound in Duncan grapefruit. Reduced growth of sweet orange wasseen when grafted on sour orange rootstock.
CTV isolates T30, T36, and VT were used as positive controls inall the reverse-transcription polymerase chain reaction (RT-PCR)tests in order to standardize the detection method of CTV-B165genotype. Healthy Mexican lime and sweet orange (cultivar Madamvinous) tissues were used as negative controls.
2.2. Nucleic acid extractions, primer designing, and RT-PCRamplification
The RNeasy Plant Mini Kit (QIAGEN, Valencia, CA) was used toextract the total RNA from CTV-B165 infected and healthy plantsfollowing the manufacturer’s protocol. The 5′ rapid amplificationof cDNA ends (RACE) (Roche, Nutley, NJ) was used to allow theamplification of unknown sequences at the 5′ end of the mRNA.This was done using the 5′ CTV gene specific primer, transcriptorreverse transcriptase, and the deoxyribonucleotide mixture follow-ing the manufacturer’s protocol. The 3′ RACE (Roche) also was usedto initiate cDNA synthesis using the PCR anchor primer and 3′ CTV-B165 gene specific primer. Multiple sets of degenerate and specificoverlapping primers were designed for RT-PCR amplification of12 ORFs of CTV-B165. Nucleotide sequences of all the GenBankisolates were aligned using the program Clustal X (Thompson etal., 1997) followed by GeneDoc (Nicholas and Nicholas, 1997) andwere examined for the conserved regions of the different ORFs ofCTV. Five domains of ORF1a and ORF1b to ORF11 were separatelyamplified using RT-PCR. Further, specific nucleotide regions alsowere selected to design the 27 pairs of overlapping PCR primersfor completion and reconfirmation of the full genome sequence ofCTV-B165. To facilitate RT-PCR, both the primers were designedbased on specificity, stability, and compatibility utilizing the pro-gram Oligo Analyzer 3.1 of Integrated DNA Technology. RT-PCRprimers for CTV-B165 ORF1a, ORF1b, and ORF2 are listed in Table 1.The overlapping RT-PCR amplicons of CTV-B165 were amplified byspecific and degenerate primers following a previously describedprotocol (Roy et al., 2003, 2005b). The production of approximately1.5–2.0 kb amplicons, with an overlap of 200–300 bp between eachfragment eliminated the possibility of the assembly of mosaicsequences. The LProII domain of ORF1a was selected for detection ofthe CTV-B165 genotype. The specific forward and reverse primers(1885B165-F: GTC AGG ATT TTG ATG ATT TGT GCC ACT C1912 andB165-R: 2633AAA ATG CAC TGT AAC AAG ACC CGA CTC2607) for CTV-B165 isolate were designed and utilized for differentiation of theCTV-B165 genotype from existing T30, T36 and VT genotypes byconventional RT-PCR. The generic p18 gene (ORF7) primer pair wasused that detects all CTV isolates unequivocally (Roy et al., 2005a).
2.3. Cloning, single strand conformation polymorphism andsequencing
A QIAGEN gel purification kit (QIAGEN, Valencia, CA) was usedto purify the amplified RT-PCR products from the agarose gels andwere directly ligated into a pGEM-T Easy Vector (Promega, Madi-son, WI) according to the manufacturer’s protocol and followed bytransformation into Escherichia coli JM109 cells. The PCR amplifiedproducts of 25 clones of each cDNA segment were analyzed by sin-gle strand conformation polymorphism (SSCP) using a previouslyestablished protocol (Roy and Brlansky, 2009). The sequence ofclones with differing SSCP patterns was determined in an ABI Prism3100 automatic DNA Sequencer at the University of Florida (DNASequencing Core Laboratory, Gainesville). In all cases, at least two tothree clones were sequenced for each amplified overlapping regionto analyze the sequence variation within the isolate. The sequenceswere analyzed using the computer program Clustal X (Thompson
Author's personal copy
120 A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130
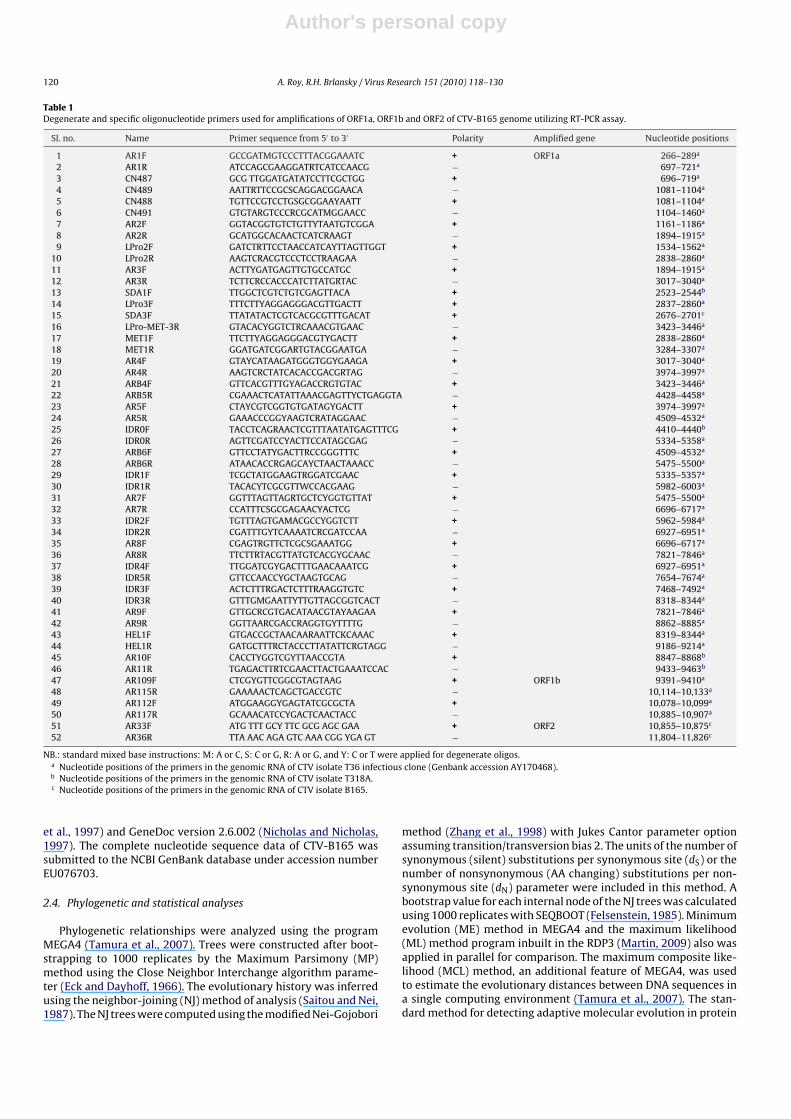
Table 1Degenerate and specific oligonucleotide primers used for amplifications of ORF1a, ORF1b and ORF2 of CTV-B165 genome utilizing RT-PCR assay.
Sl. no. Name Primer sequence from 5′ to 3′ Polarity Amplified gene Nucleotide positions
1 AR1F GCCGATMGTCCCTTTACGGAAATC + ORF1a 266–289a
2 AR1R ATCCAGCGAAGGATRTCATCCAACG − 697–721a
3 CN487 GCG TTGGATGATATCCTTCGCTGG + 696–719a
4 CN489 AATTRTTCCGCSCAGGACGGAACA − 1081–1104a
5 CN488 TGTTCCGTCCTGSGCGGAAYAATT + 1081–1104a
6 CN491 GTGTARGTCCCRCGCATMGGAACC − 1104–1460a
7 AR2F GGTACGGTGTCTGTTYTAATGTCGGA + 1161–1186a
8 AR2R GCATGGCACAACTCATCRAAGT − 1894–1915a
9 LPro2F GATCTRTTCCTAACCATCAYTTAGTTGGT + 1534–1562a
10 LPro2R AAGTCRACGTCCCTCCTRAAGAA − 2838–2860a
11 AR3F ACTTYGATGAGTTGTGCCATGC + 1894–1915a
12 AR3R TCTTCRCCACCCATCTTATGRTAC − 3017–3040a
13 SDA1F TTGGCTCGTCTGTCGAGTTACA + 2523–2544b
14 LPro3F TTTCTTYAGGAGGGACGTTGACTT + 2837–2860a
15 SDA3F TTATATACTCGTCACGCGTTTGACAT + 2676–2701c
16 LPro-MET-3R GTACACYGGTCTRCAAACGTGAAC − 3423–3446a
17 MET1F TTCTTYAGGAGGGACGTYGACTT + 2838–2860a
18 MET1R GGATGATCGGARTGTACGGAATGA − 3284–3307a
19 AR4F GTAYCATAAGATGGGTGGYGAAGA + 3017–3040a
20 AR4R AAGTCRCTATCACACCGACGRTAG − 3974–3997a
21 ARB4F GTTCACGTTTGYAGACCRGTGTAC + 3423–3446a
22 ARB5R CGAAACTCATATTAAACGAGTTYCTGAGGTA − 4428–4458a
23 AR5F CTAYCGTCGGTGTGATAGYGACTT + 3974–3997a
24 AR5R GAAACCCGGYAAGTCRATAGGAAC − 4509–4532a
25 IDR0F TACCTCAGRAACTCGTTTAATATGAGTTTCG + 4410–4440b
26 IDR0R AGTTCGATCCYACTTCCATAGCGAG − 5334–5358a
27 ARB6F GTTCCTATYGACTTRCCGGGTTTC + 4509–4532a
28 ARB6R ATAACACCRGAGCAYCTAACTAAACC − 5475–5500a
29 IDR1F TCGCTATGGAAGTRGGATCGAAC + 5335–5357a
30 IDR1R TACACYTCGCGTTWCCACGAAG − 5982–6003a
31 AR7F GGTTTAGTTAGRTGCTCYGGTGTTAT + 5475–5500a
32 AR7R CCATTTCSGCGAGAACYACTCG − 6696–6717a
33 IDR2F TGTTTAGTGAMACGCCYGGTCTT + 5962–5984a
34 IDR2R CGATTTGYTCAAAATCRCGATCCAA − 6927–6951a
35 AR8F CGAGTRGTTCTCGCSGAAATGG + 6696–6717a
36 AR8R TTCTTRTACGTTATGTCACGYGCAAC − 7821–7846a
37 IDR4F TTGGATCGYGACTTTGAACAAATCG + 6927–6951a
38 IDR5R GTTCCAACCYGCTAAGTGCAG − 7654–7674a
39 IDR3F ACTCTTTRGACTCTTTRAAGGTGTC + 7468–7492a
40 IDR3R GTTTGMGAATTYTTGTTAGCGGTCACT − 8318–8344a
41 AR9F GTTGCRCGTGACATAACGTAYAAGAA + 7821–7846a
42 AR9R GGTTAARCGACCRAGGTGYTTTTG − 8862–8885a
43 HEL1F GTGACCGCTAACAARAATTCKCAAAC + 8319–8344a
44 HEL1R GATGCTTTRCTACCCTTATATTCRGTAGG − 9186–9214a
45 AR10F CACCTYGGTCGYTTAACCGTA + 8847–8868b
46 AR11R TGAGACTTRTCGAACTTACTGAAATCCAC − 9433–9463b
47 AR109F CTCGYGTTCGGCGTAGTAAG + ORF1b 9391–9410a
48 AR115R GAAAAACTCAGCTGACCGTC − 10,114–10,133a
49 AR112F ATGGAAGGYGAGTATCGCGCTA + 10,078–10,099a
50 AR117R GCAAACATCCYGACTCAACTACC − 10,885–10,907a
51 AR33F ATG TTT GCY TTC GCG AGC GAA + ORF2 10,855–10,875c
52 AR36R TTA AAC AGA GTC AAA CGG YGA GT − 11,804–11,826c
NB.: standard mixed base instructions: M: A or C, S: C or G, R: A or G, and Y: C or T were applied for degenerate oligos.a Nucleotide positions of the primers in the genomic RNA of CTV isolate T36 infectious clone (Genbank accession AY170468).b Nucleotide positions of the primers in the genomic RNA of CTV isolate T318A.c Nucleotide positions of the primers in the genomic RNA of CTV isolate B165.
et al., 1997) and GeneDoc version 2.6.002 (Nicholas and Nicholas,1997). The complete nucleotide sequence data of CTV-B165 wassubmitted to the NCBI GenBank database under accession numberEU076703.
2.4. Phylogenetic and statistical analyses
Phylogenetic relationships were analyzed using the programMEGA4 (Tamura et al., 2007). Trees were constructed after boot-strapping to 1000 replicates by the Maximum Parsimony (MP)method using the Close Neighbor Interchange algorithm parame-ter (Eck and Dayhoff, 1966). The evolutionary history was inferredusing the neighbor-joining (NJ) method of analysis (Saitou and Nei,1987). The NJ trees were computed using the modified Nei-Gojobori
method (Zhang et al., 1998) with Jukes Cantor parameter optionassuming transition/transversion bias 2. The units of the number ofsynonymous (silent) substitutions per synonymous site (dS) or thenumber of nonsynonymous (AA changing) substitutions per non-synonymous site (dN) parameter were included in this method. Abootstrap value for each internal node of the NJ trees was calculatedusing 1000 replicates with SEQBOOT (Felsenstein, 1985). Minimumevolution (ME) method in MEGA4 and the maximum likelihood(ML) method program inbuilt in the RDP3 (Martin, 2009) also wasapplied in parallel for comparison. The maximum composite like-lihood (MCL) method, an additional feature of MEGA4, was usedto estimate the evolutionary distances between DNA sequences ina single computing environment (Tamura et al., 2007). The stan-dard method for detecting adaptive molecular evolution in protein
Author's personal copy
A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130 121
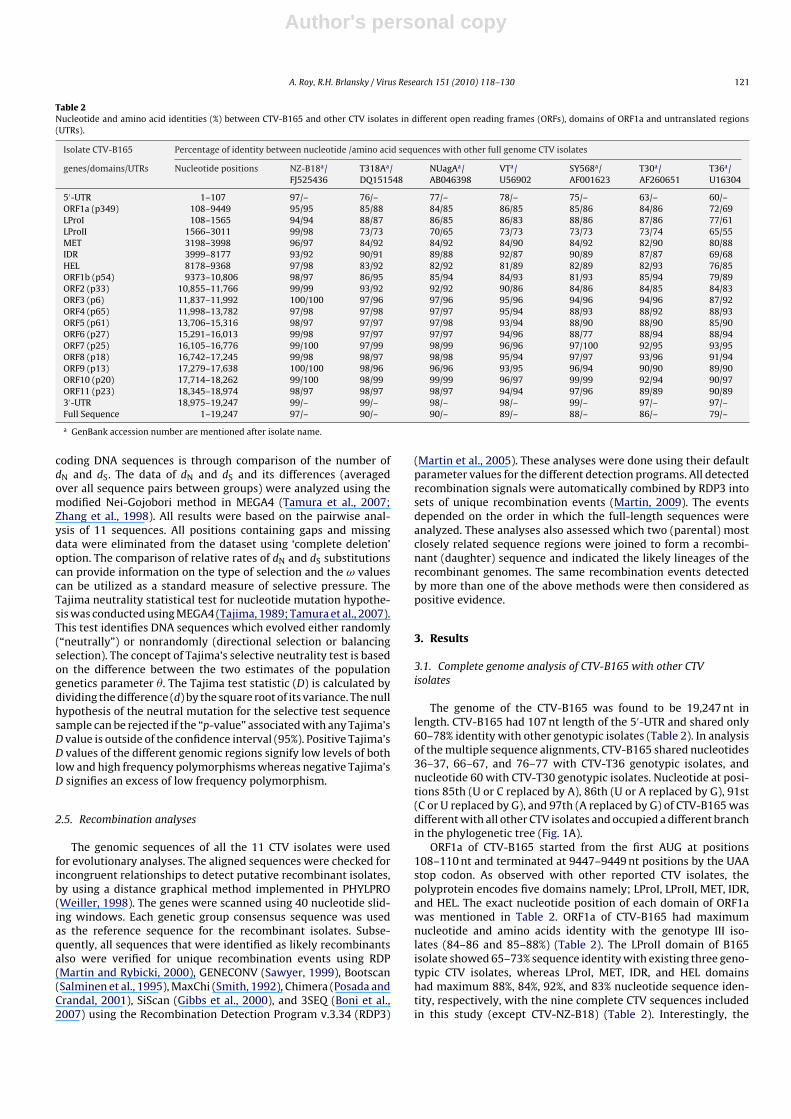
Table 2Nucleotide and amino acid identities (%) between CTV-B165 and other CTV isolates in different open reading frames (ORFs), domains of ORF1a and untranslated regions(UTRs).
Isolate CTV-B165 Percentage of identity between nucleotide /amino acid sequences with other full genome CTV isolates
genes/domains/UTRs Nucleotide positions NZ-B18a/FJ525436
T318Aa/DQ151548
NUagAa/AB046398
VTa/U56902
SY568a/AF001623
T30a/AF260651
T36a/U16304
5′-UTR 1–107 97/– 76/– 77/– 78/– 75/– 63/– 60/–ORF1a (p349) 108–9449 95/95 85/88 84/85 86/85 85/86 84/86 72/69LProI 108–1565 94/94 88/87 86/85 86/83 88/86 87/86 77/61LProII 1566–3011 99/98 73/73 70/65 73/73 73/73 73/74 65/55MET 3198–3998 96/97 84/92 84/92 84/90 84/92 82/90 80/88IDR 3999–8177 93/92 90/91 89/88 92/87 90/89 87/87 69/68HEL 8178–9368 97/98 83/92 82/92 81/89 82/89 82/93 76/85ORF1b (p54) 9373–10,806 98/97 86/95 85/94 84/93 81/93 85/94 79/89ORF2 (p33) 10,855–11,766 99/99 93/92 92/92 90/86 84/86 84/85 84/83ORF3 (p6) 11,837–11,992 100/100 97/96 97/96 95/96 94/96 94/96 87/92ORF4 (p65) 11,998–13,782 97/98 97/98 97/97 95/94 88/93 88/92 88/93ORF5 (p61) 13,706–15,316 98/97 97/97 97/98 93/94 88/90 88/90 85/90ORF6 (p27) 15,291–16,013 99/98 97/97 97/97 94/96 88/77 88/94 88/94ORF7 (p25) 16,105–16,776 99/100 97/99 98/99 96/96 97/100 92/95 93/95ORF8 (p18) 16,742–17,245 99/98 98/97 98/98 95/94 97/97 93/96 91/94ORF9 (p13) 17,279–17,638 100/100 98/96 96/96 93/95 96/94 90/90 89/90ORF10 (p20) 17,714–18,262 99/100 98/99 99/99 96/97 99/99 92/94 90/97ORF11 (p23) 18,345–18,974 98/97 98/97 98/97 94/94 97/96 89/89 90/893′-UTR 18,975–19,247 99/– 99/– 98/– 98/– 99/– 97/– 97/–Full Sequence 1–19,247 97/– 90/– 90/– 89/– 88/– 86/– 79/–
a GenBank accession number are mentioned after isolate name.
coding DNA sequences is through comparison of the number ofdN and dS. The data of dN and dS and its differences (averagedover all sequence pairs between groups) were analyzed using themodified Nei-Gojobori method in MEGA4 (Tamura et al., 2007;Zhang et al., 1998). All results were based on the pairwise anal-ysis of 11 sequences. All positions containing gaps and missingdata were eliminated from the dataset using ‘complete deletion’option. The comparison of relative rates of dN and dS substitutionscan provide information on the type of selection and the ω valuescan be utilized as a standard measure of selective pressure. TheTajima neutrality statistical test for nucleotide mutation hypothe-sis was conducted using MEGA4 (Tajima, 1989; Tamura et al., 2007).This test identifies DNA sequences which evolved either randomly(“neutrally”) or nonrandomly (directional selection or balancingselection). The concept of Tajima’s selective neutrality test is basedon the difference between the two estimates of the populationgenetics parameter �. The Tajima test statistic (D) is calculated bydividing the difference (d) by the square root of its variance. The nullhypothesis of the neutral mutation for the selective test sequencesample can be rejected if the “p-value” associated with any Tajima’sD value is outside of the confidence interval (95%). Positive Tajima’sD values of the different genomic regions signify low levels of bothlow and high frequency polymorphisms whereas negative Tajima’sD signifies an excess of low frequency polymorphism.
2.5. Recombination analyses
The genomic sequences of all the 11 CTV isolates were usedfor evolutionary analyses. The aligned sequences were checked forincongruent relationships to detect putative recombinant isolates,by using a distance graphical method implemented in PHYLPRO(Weiller, 1998). The genes were scanned using 40 nucleotide slid-ing windows. Each genetic group consensus sequence was usedas the reference sequence for the recombinant isolates. Subse-quently, all sequences that were identified as likely recombinantsalso were verified for unique recombination events using RDP(Martin and Rybicki, 2000), GENECONV (Sawyer, 1999), Bootscan(Salminen et al., 1995), MaxChi (Smith, 1992), Chimera (Posada andCrandal, 2001), SiScan (Gibbs et al., 2000), and 3SEQ (Boni et al.,2007) using the Recombination Detection Program v.3.34 (RDP3)
(Martin et al., 2005). These analyses were done using their defaultparameter values for the different detection programs. All detectedrecombination signals were automatically combined by RDP3 intosets of unique recombination events (Martin, 2009). The eventsdepended on the order in which the full-length sequences wereanalyzed. These analyses also assessed which two (parental) mostclosely related sequence regions were joined to form a recombi-nant (daughter) sequence and indicated the likely lineages of therecombinant genomes. The same recombination events detectedby more than one of the above methods were then considered aspositive evidence.
3. Results
3.1. Complete genome analysis of CTV-B165 with other CTVisolates
The genome of the CTV-B165 was found to be 19,247 nt inlength. CTV-B165 had 107 nt length of the 5′-UTR and shared only60–78% identity with other genotypic isolates (Table 2). In analysisof the multiple sequence alignments, CTV-B165 shared nucleotides36–37, 66–67, and 76–77 with CTV-T36 genotypic isolates, andnucleotide 60 with CTV-T30 genotypic isolates. Nucleotide at posi-tions 85th (U or C replaced by A), 86th (U or A replaced by G), 91st(C or U replaced by G), and 97th (A replaced by G) of CTV-B165 wasdifferent with all other CTV isolates and occupied a different branchin the phylogenetic tree (Fig. 1A).
ORF1a of CTV-B165 started from the first AUG at positions108–110 nt and terminated at 9447–9449 nt positions by the UAAstop codon. As observed with other reported CTV isolates, thepolyprotein encodes five domains namely; LProI, LProII, MET, IDR,and HEL. The exact nucleotide position of each domain of ORF1awas mentioned in Table 2. ORF1a of CTV-B165 had maximumnucleotide and amino acids identity with the genotype III iso-lates (84–86 and 85–88%) (Table 2). The LProII domain of B165isolate showed 65–73% sequence identity with existing three geno-typic CTV isolates, whereas LProI, MET, IDR, and HEL domainshad maximum 88%, 84%, 92%, and 83% nucleotide sequence iden-tity, respectively, with the nine complete CTV sequences includedin this study (except CTV-NZ-B18) (Table 2). Interestingly, the

Author's personal copy
122 A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130
Fig. 1. Evolutionary relationship of CTV-B165 and other CTV isolates were inferred using the unrooted Maximum Parsimony phylogeny method based on Close NeighborInterchange algorithm utilizing (A) the 5′-UTR, (B) the ORF1a and (B1–B5) its five domains, (C) the ORF1b, (D) the ORF2, and (E) the complete genome sequences. The treewas constructed using the MEGA4 program. A 1000 replicates of bootstrap phylogeny test support the particular phylogenetic group. Branch lengths are proportional to thegenetic distances. Isolates within the same genotype group are circled.
265 nt amplicon within this domain (nt 2133–2397) had only a42–56% sequence identity with the other three genogroups (a groupof CTV isolates belonging to a specific genotype). In the BLAST(http://blast.ncbi.nlm.nih.gov) analysis these 265 nt did not alignwith any of the genotypic CTV isolates except the New ZealandCTV isolate NZ-B18 which is a similar genotype.
Six phylogenetic trees were constructed using the nucleotidesequences of the total ORF1a and its five different domains (LProI,LProII, MET, IDR, and HEL) from all CTV sequences available inthe database using Clustal X followed by MEGA4 and RDP3 pro-grams. The phylogenetic analysis of the five domains of CTV-B165suggested that the studied isolate could be clustered in all of thedendrograms into a distinct genotype branch (Fig. 1B and B1–B5).As previously shown (Moreno et al., 2008), genotype I includessevere decline isolates from Florida (T36), Egypt (Qaha), and Mex-ico. Genotype II includes mild isolate T30 from Florida and T385from Spain, whereas the SY and SP isolates (VT, T318A, SY568, andNUagA) clustered into the genotype III. The new cluster, genotype IVgroup, is represented by CTV-B165 and -NZ-B18. Sequence analysisof the individual domains of LProI, MET, IDR and HEL of CTV-B165ORF1a showed the nearest phylogenetic relationship with geno-type III, whereas the LProII was most closely related with genotypeII isolates (Table 2).
ORF1b encodes the RdRp-like domain. The downstream ORF1bstarted at nucleotide 9373 and ended at nucleotide10,806 andoverlapped ORF1a by 77 nt. ORF1b of CTV-B165 had 79–86%nucleotide sequence identity and 89–95% amino acid sequenceidentity with the corresponding sequence of other CTV geno-typic isolates (Table 2). A phylogenetic analysis using ORF1bsequences of all the CTV isolates displayed a distinct branchfor CTV-B165 in the dendrogram (Fig. 1C). This evidence fur-
ther supports the establishment of a new genotype for the CTVcomplex.
ORFs 2–11 are expressed via 3′ co-terminal sgRNAs (16). Thesedownstream ORFs encoding 10 protein products ranging from 6 to65 kDa were described in detail previously (Karasev et al., 1995;Pappu et al., 1994). The total length of ORF 2–11 is 8120 nt as com-pared to 8119–8159 nt for the other 10 CTV sequences compared inthis study. ORF2 encodes for the p33 and showed a 93% nucleotideand 92% amino acid sequence identity with Spanish T318A iso-late followed by a 92% sequence identity with the NUagA Japaneseisolate (Table 2). A phylogenetic tree was generated using ORF2nucleotide sequences from the CTV-B165 and the other CTV isolatesand displayed four main clusters (Fig. 1D). Isolate B165 was wellseparated from all the other characterized CTV genotypic isolatesand occupied a new branch in the dendrogram as a type memberof the novel genotype IV (Fig. 1D).
ORF3–ORF11 had a maximum 97–99% nucleotide and aminoacid sequence identity with other CTV sequences (Table 2). Ninephylogenetic trees were constructed using the nucleotide andamino acid sequences of ORF3–ORF11 (p6, p65, p61, p27, p25,p18, p13, p20, and p23) from the CTV-B165 and the other stud-ied CTV isolates. The phylogenetic analyses suggested that all theisolates could be clustered into three distinct genotypes of CTV andwas displayed in all of the dendrograms generated based on ninedifferent ORF sequences (data not shown). These results indicatethat CTV-B165 is more closely related to SY and SP isolates andbelongs to genotype III. The length of the 3′-UTRs of the CTV-B165 is273 nucleotides and revealed ∼97–99% identity among the isolates(Table 2). Overall, the phylogenetic analysis revealed four distinctbranches utilizing the total nucleotide sequences of CTV-B165 andthe other 10 completely sequenced CTV isolates (Fig. 1E).
Author's personal copy
A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130 123
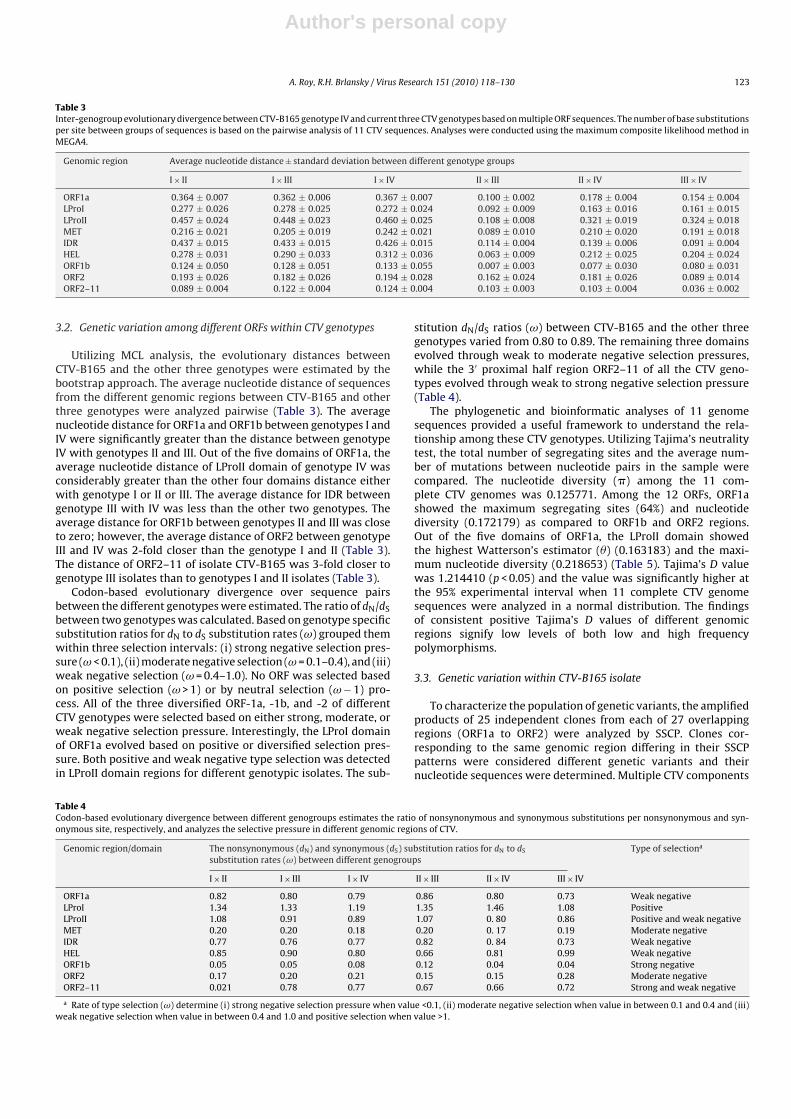
Table 3Inter-genogroup evolutionary divergence between CTV-B165 genotype IV and current three CTV genotypes based on multiple ORF sequences. The number of base substitutionsper site between groups of sequences is based on the pairwise analysis of 11 CTV sequences. Analyses were conducted using the maximum composite likelihood method inMEGA4.
Genomic region Average nucleotide distance ± standard deviation between different genotype groups
I × II I × III I × IV II × III II × IV III × IV
ORF1a 0.364 ± 0.007 0.362 ± 0.006 0.367 ± 0.007 0.100 ± 0.002 0.178 ± 0.004 0.154 ± 0.004LProI 0.277 ± 0.026 0.278 ± 0.025 0.272 ± 0.024 0.092 ± 0.009 0.163 ± 0.016 0.161 ± 0.015LProII 0.457 ± 0.024 0.448 ± 0.023 0.460 ± 0.025 0.108 ± 0.008 0.321 ± 0.019 0.324 ± 0.018MET 0.216 ± 0.021 0.205 ± 0.019 0.242 ± 0.021 0.089 ± 0.010 0.210 ± 0.020 0.191 ± 0.018IDR 0.437 ± 0.015 0.433 ± 0.015 0.426 ± 0.015 0.114 ± 0.004 0.139 ± 0.006 0.091 ± 0.004HEL 0.278 ± 0.031 0.290 ± 0.033 0.312 ± 0.036 0.063 ± 0.009 0.212 ± 0.025 0.204 ± 0.024ORF1b 0.124 ± 0.050 0.128 ± 0.051 0.133 ± 0.055 0.007 ± 0.003 0.077 ± 0.030 0.080 ± 0.031ORF2 0.193 ± 0.026 0.182 ± 0.026 0.194 ± 0.028 0.162 ± 0.024 0.181 ± 0.026 0.089 ± 0.014ORF2–11 0.089 ± 0.004 0.122 ± 0.004 0.124 ± 0.004 0.103 ± 0.003 0.103 ± 0.004 0.036 ± 0.002
3.2. Genetic variation among different ORFs within CTV genotypes
Utilizing MCL analysis, the evolutionary distances betweenCTV-B165 and the other three genotypes were estimated by thebootstrap approach. The average nucleotide distance of sequencesfrom the different genomic regions between CTV-B165 and otherthree genotypes were analyzed pairwise (Table 3). The averagenucleotide distance for ORF1a and ORF1b between genotypes I andIV were significantly greater than the distance between genotypeIV with genotypes II and III. Out of the five domains of ORF1a, theaverage nucleotide distance of LProII domain of genotype IV wasconsiderably greater than the other four domains distance eitherwith genotype I or II or III. The average distance for IDR betweengenotype III with IV was less than the other two genotypes. Theaverage distance for ORF1b between genotypes II and III was closeto zero; however, the average distance of ORF2 between genotypeIII and IV was 2-fold closer than the genotype I and II (Table 3).The distance of ORF2–11 of isolate CTV-B165 was 3-fold closer togenotype III isolates than to genotypes I and II isolates (Table 3).
Codon-based evolutionary divergence over sequence pairsbetween the different genotypes were estimated. The ratio of dN/dSbetween two genotypes was calculated. Based on genotype specificsubstitution ratios for dN to dS substitution rates (ω) grouped themwithin three selection intervals: (i) strong negative selection pres-sure (ω < 0.1), (ii) moderate negative selection (ω = 0.1–0.4), and (iii)weak negative selection (ω = 0.4–1.0). No ORF was selected basedon positive selection (ω > 1) or by neutral selection (ω − 1) pro-cess. All of the three diversified ORF-1a, -1b, and -2 of differentCTV genotypes were selected based on either strong, moderate, orweak negative selection pressure. Interestingly, the LProI domainof ORF1a evolved based on positive or diversified selection pres-sure. Both positive and weak negative type selection was detectedin LProII domain regions for different genotypic isolates. The sub-
stitution dN/dS ratios (ω) between CTV-B165 and the other threegenotypes varied from 0.80 to 0.89. The remaining three domainsevolved through weak to moderate negative selection pressures,while the 3′ proximal half region ORF2–11 of all the CTV geno-types evolved through weak to strong negative selection pressure(Table 4).
The phylogenetic and bioinformatic analyses of 11 genomesequences provided a useful framework to understand the rela-tionship among these CTV genotypes. Utilizing Tajima’s neutralitytest, the total number of segregating sites and the average num-ber of mutations between nucleotide pairs in the sample werecompared. The nucleotide diversity (�) among the 11 com-plete CTV genomes was 0.125771. Among the 12 ORFs, ORF1ashowed the maximum segregating sites (64%) and nucleotidediversity (0.172179) as compared to ORF1b and ORF2 regions.Out of the five domains of ORF1a, the LProII domain showedthe highest Watterson’s estimator (�) (0.163183) and the maxi-mum nucleotide diversity (0.218653) (Table 5). Tajima’s D valuewas 1.214410 (p < 0.05) and the value was significantly higher atthe 95% experimental interval when 11 complete CTV genomesequences were analyzed in a normal distribution. The findingsof consistent positive Tajima’s D values of different genomicregions signify low levels of both low and high frequencypolymorphisms.
3.3. Genetic variation within CTV-B165 isolate
To characterize the population of genetic variants, the amplifiedproducts of 25 independent clones from each of 27 overlappingregions (ORF1a to ORF2) were analyzed by SSCP. Clones cor-responding to the same genomic region differing in their SSCPpatterns were considered different genetic variants and theirnucleotide sequences were determined. Multiple CTV components
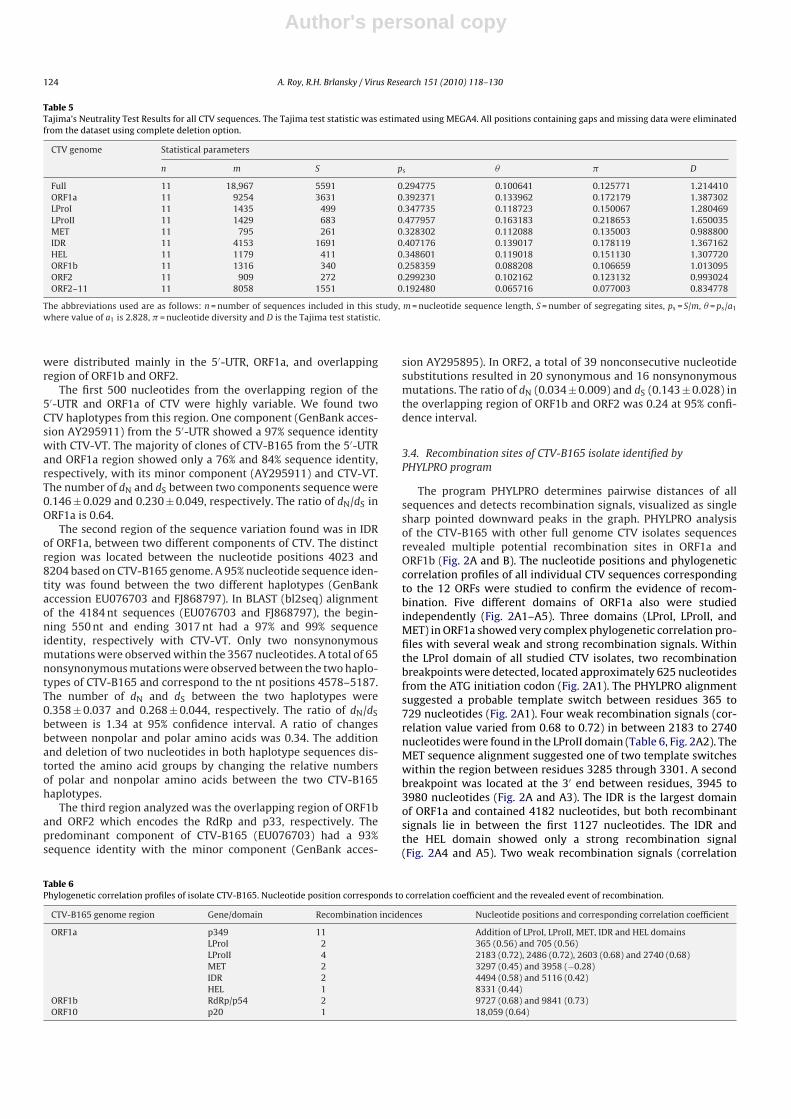
Table 4Codon-based evolutionary divergence between different genogroups estimates the ratio of nonsynonymous and synonymous substitutions per nonsynonymous and syn-onymous site, respectively, and analyzes the selective pressure in different genomic regions of CTV.
Genomic region/domain The nonsynonymous (dN) and synonymous (dS) substitution ratios for dN to dS
substitution rates (ω) between different genogroupsType of selectiona
I × II I × III I × IV II × III II × IV III × IV
ORF1a 0.82 0.80 0.79 0.86 0.80 0.73 Weak negativeLProI 1.34 1.33 1.19 1.35 1.46 1.08 PositiveLProII 1.08 0.91 0.89 1.07 0. 80 0.86 Positive and weak negativeMET 0.20 0.20 0.18 0.20 0. 17 0.19 Moderate negativeIDR 0.77 0.76 0.77 0.82 0. 84 0.73 Weak negativeHEL 0.85 0.90 0.80 0.66 0.81 0.99 Weak negativeORF1b 0.05 0.05 0.08 0.12 0.04 0.04 Strong negativeORF2 0.17 0.20 0.21 0.15 0.15 0.28 Moderate negativeORF2–11 0.021 0.78 0.77 0.67 0.66 0.72 Strong and weak negative
a Rate of type selection (ω) determine (i) strong negative selection pressure when value <0.1, (ii) moderate negative selection when value in between 0.1 and 0.4 and (iii)weak negative selection when value in between 0.4 and 1.0 and positive selection when value >1.
Author's personal copy
124 A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130
Table 5Tajima’s Neutrality Test Results for all CTV sequences. The Tajima test statistic was estimated using MEGA4. All positions containing gaps and missing data were eliminatedfrom the dataset using complete deletion option.
CTV genome Statistical parameters
n m S ps � � D
Full 11 18,967 5591 0.294775 0.100641 0.125771 1.214410ORF1a 11 9254 3631 0.392371 0.133962 0.172179 1.387302LProI 11 1435 499 0.347735 0.118723 0.150067 1.280469LProII 11 1429 683 0.477957 0.163183 0.218653 1.650035MET 11 795 261 0.328302 0.112088 0.135003 0.988800IDR 11 4153 1691 0.407176 0.139017 0.178119 1.367162HEL 11 1179 411 0.348601 0.119018 0.151130 1.307720ORF1b 11 1316 340 0.258359 0.088208 0.106659 1.013095ORF2 11 909 272 0.299230 0.102162 0.123132 0.993024ORF2–11 11 8058 1551 0.192480 0.065716 0.077003 0.834778
The abbreviations used are as follows: n = number of sequences included in this study, m = nucleotide sequence length, S = number of segregating sites, ps = S/m, � = ps/a1
where value of a1 is 2.828, � = nucleotide diversity and D is the Tajima test statistic.
were distributed mainly in the 5′-UTR, ORF1a, and overlappingregion of ORF1b and ORF2.
The first 500 nucleotides from the overlapping region of the5′-UTR and ORF1a of CTV were highly variable. We found twoCTV haplotypes from this region. One component (GenBank acces-sion AY295911) from the 5′-UTR showed a 97% sequence identitywith CTV-VT. The majority of clones of CTV-B165 from the 5′-UTRand ORF1a region showed only a 76% and 84% sequence identity,respectively, with its minor component (AY295911) and CTV-VT.The number of dN and dS between two components sequence were0.146 ± 0.029 and 0.230 ± 0.049, respectively. The ratio of dN/dS inORF1a is 0.64.
The second region of the sequence variation found was in IDRof ORF1a, between two different components of CTV. The distinctregion was located between the nucleotide positions 4023 and8204 based on CTV-B165 genome. A 95% nucleotide sequence iden-tity was found between the two different haplotypes (GenBankaccession EU076703 and FJ868797). In BLAST (bl2seq) alignmentof the 4184 nt sequences (EU076703 and FJ868797), the begin-ning 550 nt and ending 3017 nt had a 97% and 99% sequenceidentity, respectively with CTV-VT. Only two nonsynonymousmutations were observed within the 3567 nucleotides. A total of 65nonsynonymous mutations were observed between the two haplo-types of CTV-B165 and correspond to the nt positions 4578–5187.The number of dN and dS between the two haplotypes were0.358 ± 0.037 and 0.268 ± 0.044, respectively. The ratio of dN/dSbetween is 1.34 at 95% confidence interval. A ratio of changesbetween nonpolar and polar amino acids was 0.34. The additionand deletion of two nucleotides in both haplotype sequences dis-torted the amino acid groups by changing the relative numbersof polar and nonpolar amino acids between the two CTV-B165haplotypes.
The third region analyzed was the overlapping region of ORF1band ORF2 which encodes the RdRp and p33, respectively. Thepredominant component of CTV-B165 (EU076703) had a 93%sequence identity with the minor component (GenBank acces-
sion AY295895). In ORF2, a total of 39 nonconsecutive nucleotidesubstitutions resulted in 20 synonymous and 16 nonsynonymousmutations. The ratio of dN (0.034 ± 0.009) and dS (0.143 ± 0.028) inthe overlapping region of ORF1b and ORF2 was 0.24 at 95% confi-dence interval.
3.4. Recombination sites of CTV-B165 isolate identified byPHYLPRO program
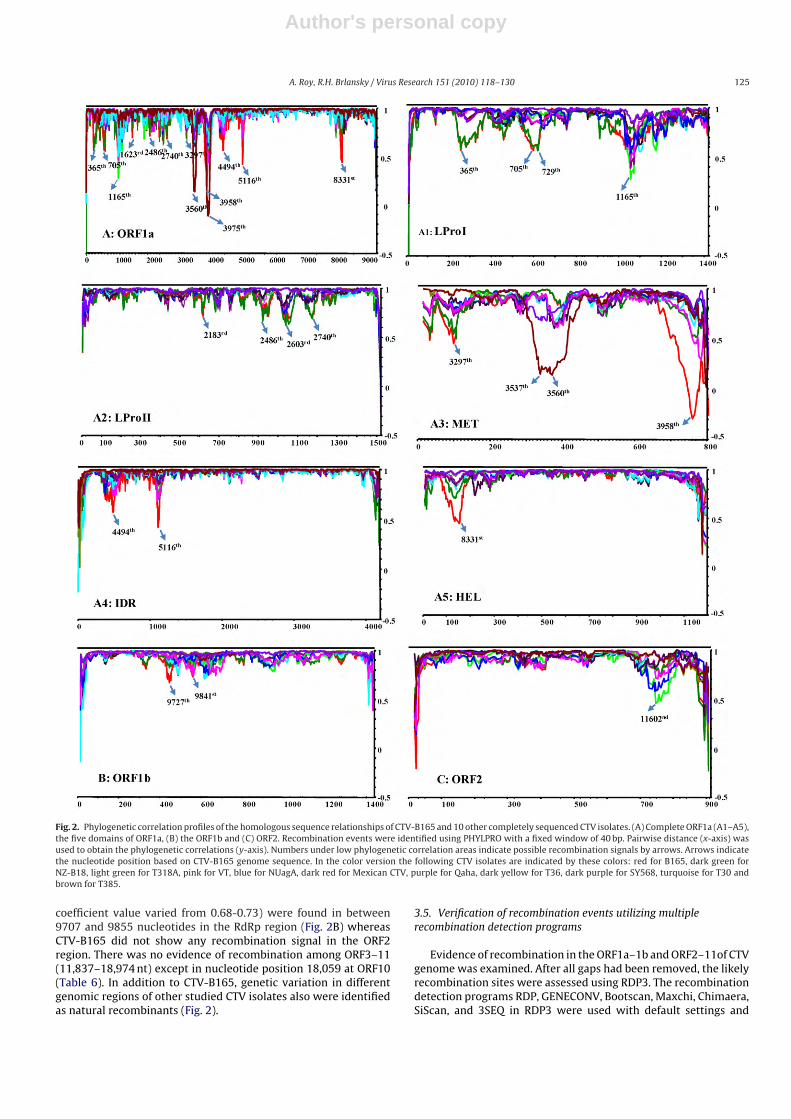
The program PHYLPRO determines pairwise distances of allsequences and detects recombination signals, visualized as singlesharp pointed downward peaks in the graph. PHYLPRO analysisof the CTV-B165 with other full genome CTV isolates sequencesrevealed multiple potential recombination sites in ORF1a andORF1b (Fig. 2A and B). The nucleotide positions and phylogeneticcorrelation profiles of all individual CTV sequences correspondingto the 12 ORFs were studied to confirm the evidence of recom-bination. Five different domains of ORF1a also were studiedindependently (Fig. 2A1–A5). Three domains (LProI, LProII, andMET) in ORF1a showed very complex phylogenetic correlation pro-files with several weak and strong recombination signals. Withinthe LProI domain of all studied CTV isolates, two recombinationbreakpoints were detected, located approximately 625 nucleotidesfrom the ATG initiation codon (Fig. 2A1). The PHYLPRO alignmentsuggested a probable template switch between residues 365 to729 nucleotides (Fig. 2A1). Four weak recombination signals (cor-relation value varied from 0.68 to 0.72) in between 2183 to 2740nucleotides were found in the LProII domain (Table 6, Fig. 2A2). TheMET sequence alignment suggested one of two template switcheswithin the region between residues 3285 through 3301. A secondbreakpoint was located at the 3′ end between residues, 3945 to3980 nucleotides (Fig. 2A and A3). The IDR is the largest domainof ORF1a and contained 4182 nucleotides, but both recombinantsignals lie in between the first 1127 nucleotides. The IDR andthe HEL domain showed only a strong recombination signal(Fig. 2A4 and A5). Two weak recombination signals (correlation
Table 6Phylogenetic correlation profiles of isolate CTV-B165. Nucleotide position corresponds to correlation coefficient and the revealed event of recombination.
CTV-B165 genome region Gene/domain Recombination incidences Nucleotide positions and corresponding correlation coefficient
ORF1a p349 11 Addition of LProI, LProII, MET, IDR and HEL domainsLProI 2 365 (0.56) and 705 (0.56)LProII 4 2183 (0.72), 2486 (0.72), 2603 (0.68) and 2740 (0.68)MET 2 3297 (0.45) and 3958 (−0.28)IDR 2 4494 (0.58) and 5116 (0.42)HEL 1 8331 (0.44)
ORF1b RdRp/p54 2 9727 (0.68) and 9841 (0.73)ORF10 p20 1 18,059 (0.64)
Author's personal copy
A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130 125
Fig. 2. Phylogenetic correlation profiles of the homologous sequence relationships of CTV-B165 and 10 other completely sequenced CTV isolates. (A) Complete ORF1a (A1–A5),the five domains of ORF1a, (B) the ORF1b and (C) ORF2. Recombination events were identified using PHYLPRO with a fixed window of 40 bp. Pairwise distance (x-axis) wasused to obtain the phylogenetic correlations (y-axis). Numbers under low phylogenetic correlation areas indicate possible recombination signals by arrows. Arrows indicatethe nucleotide position based on CTV-B165 genome sequence. In the color version the following CTV isolates are indicated by these colors: red for B165, dark green forNZ-B18, light green for T318A, pink for VT, blue for NUagA, dark red for Mexican CTV, purple for Qaha, dark yellow for T36, dark purple for SY568, turquoise for T30 andbrown for T385.
coefficient value varied from 0.68-0.73) were found in between9707 and 9855 nucleotides in the RdRp region (Fig. 2B) whereasCTV-B165 did not show any recombination signal in the ORF2region. There was no evidence of recombination among ORF3–11(11,837–18,974 nt) except in nucleotide position 18,059 at ORF10(Table 6). In addition to CTV-B165, genetic variation in differentgenomic regions of other studied CTV isolates also were identifiedas natural recombinants (Fig. 2).
3.5. Verification of recombination events utilizing multiplerecombination detection programs
Evidence of recombination in the ORF1a–1b and ORF2–11of CTVgenome was examined. After all gaps had been removed, the likelyrecombination sites were assessed using RDP3. The recombinationdetection programs RDP, GENECONV, Bootscan, Maxchi, Chimaera,SiScan, and 3SEQ in RDP3 were used with default settings and

Author's personal copy
126 A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130
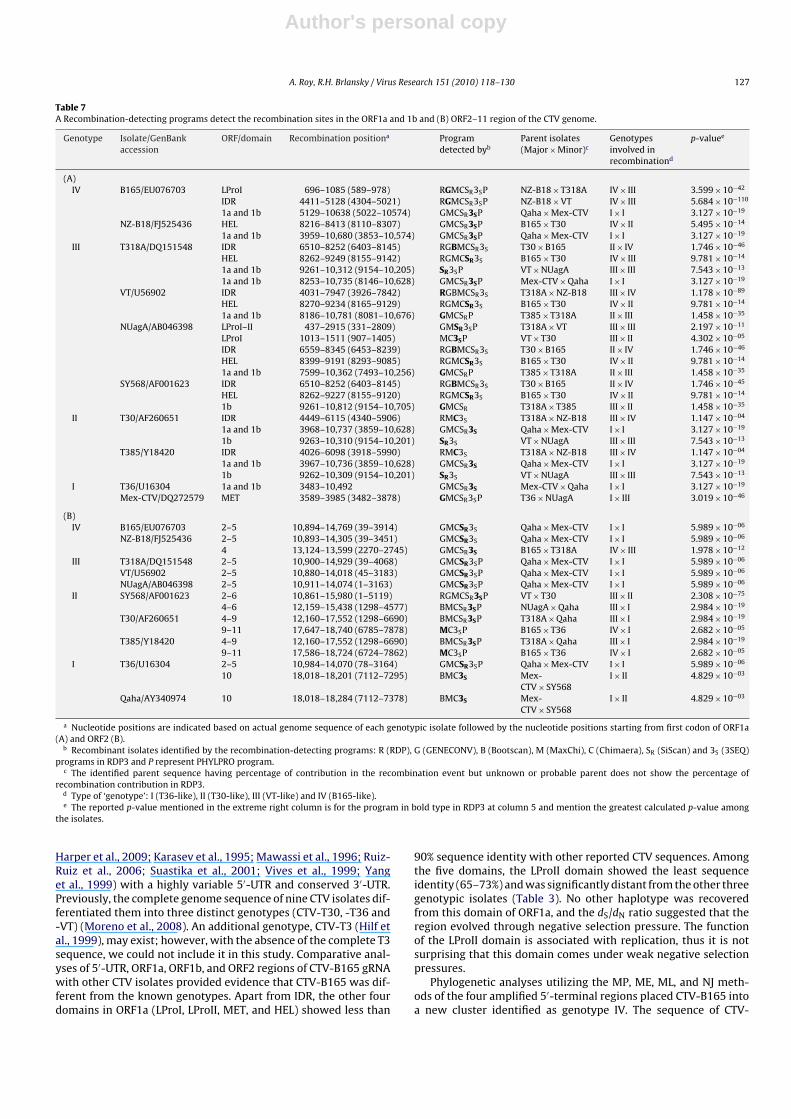
Fig. 3. Recombination analysis by RDP3 identified positions of unique recombination events in relation to the 5′ and 3′ proximal half of 11 CTV sequences alignment. Eachhalf of the full-length CTV genome is represented by a long green bar. The corresponding isolate and the minor parents name are given to the top and side of the each colorbox, respectively. (A and B) shows a total of 24 and 7 unique recombination events, respectively, that are demarcated by the boxes below the genome of each CTV isolate.
a Bonferroni corrected p-value cutoff of 0.05 or 0.01 for the 11aligned CTV sequences. CTV-B165 individual genes were ranked asORF1a (LProII > LProI = IDR = MET > HEL) > ORF1b > ORF2 and ORF5based on the number of potential recombination sites identified.This rank also is not true for the other genotypic CTV isolates. A sig-nificant p-value was detected by each program was in the order of3SEQ > SiScan > GENCONV > Bootscan > Chimaera > Maxchi > RDP.The multi recombination detection approach was applied to findthe actual recombination sites and also to avoid the evidenceof false recombination sites. Two sequences from the major andminor parents are joined to form a recombinant daughter sequenceand the recombination signal(s) was detected in all descendantsof the parental and daughter isolates as well as other related CTVisolates (Fig. 3A and B). Phylogenetic relationship between all the11 CTV isolates sequence utilizing ML method confirmed the majorand minor parent and daughter sequences (data not shown).
Twenty-four recombination sites were found at the 5′ prox-imal half of the CTV genome sequences (Table 7A). Out of the24, fifteen recombination sites were found in multiple domainsof ORF1a, and only one in the ORF1b region. Eight recombina-tion sites were found in the overlapping regions of ORF1a andORF1b (Fig. 3A). All recombination sites were identified by morethan one method (Table 7A). At the 5′ proximal half no recom-bination event was found in the CTV-Qaha isolate. Combined MLphylogeny and recombinant detection analysis of ORF1a and ORF1bsupported Qaha as a parent isolate and that the T36 and Mex-ican CTV isolates of genogroup I originated through intra- andinter-genotype recombination, respectively. In the evolution ofgenotype IV, isolates CTV-B165 and -NZ-B18 are both assignedas major parents (Table 7A). Isolate CTV-B165 (genotype IV) con-tributed as a probable major and minor parent for genotype IIIand isolate NZ-B18 (genotype IV) contributes as a minor parentof genotype II (Table 7A). CTV isolates from genogroup I played arole of major parent in the event of recombination at the overlap-ping region of ORF1a and ORF1b, in the development of genotypesII and IV.
Seven recombination sites were found in between ORF2–11 atthe 3′ proximal half (Fig. 3B). All recombination sites were identifiedby more than three methods. No recombination event was foundin Mexican CTV isolate and the isolate acts as one of the parentalsources for evolution of genotypes III and IV. The ML phylogeneticanalysis of ORF2–11 CTV genome sequences also reveals that theisolates of genogroup I are the major parents of genotype III and IVisolates and the minor parents of genotype II, III and IV isolates. TheCTV isolates of genogroup III and IV played a role of major parentfor the evolution of the genogroup II isolates (Table 7B).
3.6. Standardization and detection of CTV-B165 genotypic isolate
Previously the overlapping regions of the 5′-UTR and ORF1a, par-tial IDR of ORF1a and the overlapping regions of ORF1b and ORF2were used for CTV genotyping (Hilf et al., 1999; Roy and Brlansky,2004; Roy et al., 2005b). Since these regions were unsuitable toidentify CTV-B165, we investigated the complete CTV genome todetermine alternative regions that could be used to distinguishbetween CTV-B165 and the existing three CTV genotypes. TheLProII domain of ORF1a was selected. The primer pair amplified a749 nt amplicon from ORF1a of CTV-B165 whereas the type mem-bers of the other three genotypes (CTV-T30, -T36 and -VT) failedto amplify (Fig. 4). The presence of the new genotype was con-firmed by sequencing analysis (data not shown). The LProII regionof each genotype includes multiple nucleotide polymorphisms thatare specifically associated with that genotype.
4. Discussion
In this study, the complete nucleotide sequence of CTV-B165was determined, making it the first fully sequenced Indian CTVisolate, and phylogenic analysis proved that the isolate formed anew CTV genotype. Comparative analysis of the complete CTV-B165sequence indicated that its genome organization is structurally thesame as other tristeza virus isolates (Albiach-Martí et al., 2000;
Author's personal copy
A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130 127
Table 7A Recombination-detecting programs detect the recombination sites in the ORF1a and 1b and (B) ORF2–11 region of the CTV genome.
Genotype Isolate/GenBankaccession
ORF/domain Recombination positiona Programdetected byb
Parent isolates(Major × Minor)c
Genotypesinvolved inrecombinationd
p-valuee
(A)IV B165/EU076703 LProI 696–1085 (589–978) RGMCSR3SP NZ-B18 × T318A IV × III 3.599 × 10−42
IDR 4411–5128 (4304–5021) RGMCSR3SP NZ-B18 × VT IV × III 5.684 × 10−110
1a and 1b 5129–10638 (5022–10574) GMCSR3SP Qaha × Mex-CTV I × I 3.127 × 10−19
NZ-B18/FJ525436 HEL 8216–8413 (8110–8307) GMCSR3SP B165 × T30 IV × II 5.495 × 10−14
1a and 1b 3959–10,680 (3853–10,574) GMCSR3SP Qaha × Mex-CTV I × I 3.127 × 10−19
III T318A/DQ151548 IDR 6510–8252 (6403–8145) RGBMCSR3S T30 × B165 II × IV 1.746 × 10−46
HEL 8262–9249 (8155–9142) RGMCSR3S B165 × T30 IV × III 9.781 × 10−14
1a and 1b 9261–10,312 (9154–10,205) SR3SP VT × NUagA III × III 7.543 × 10−13
1a and 1b 8253–10,735 (8146–10,628) GMCSR3SP Mex-CTV × Qaha I × I 3.127 × 10−19
VT/U56902 IDR 4031–7947 (3926–7842) RGBMCSR3S T318A × NZ-B18 III × IV 1.178 × 10−89
HEL 8270–9234 (8165–9129) RGMCSR3S B165 × T30 IV × II 9.781 × 10−14
1a and 1b 8186–10,781 (8081–10,676) GMCSRP T385 × T318A II × III 1.458 × 10−35
NUagA/AB046398 LProI–II 437–2915 (331–2809) GMSR3SP T318A × VT III × III 2.197 × 10−11
LProI 1013–1511 (907–1405) MC3SP VT × T30 III × II 4.302 × 10−05
IDR 6559–8345 (6453–8239) RGBMCSR3S T30 × B165 II × IV 1.746 × 10−46
HEL 8399–9191 (8293–9085) RGMCSR3S B165 × T30 IV × II 9.781 × 10−14
1a and 1b 7599–10,362 (7493–10,256) GMCSRP T385 × T318A II × III 1.458 × 10−35
SY568/AF001623 IDR 6510–8252 (6403–8145) RGBMCSR3S T30 × B165 II × IV 1.746 × 10−45
HEL 8262–9227 (8155–9120) RGMCSR3S B165 × T30 IV × II 9.781 × 10−14
1b 9261–10,812 (9154–10,705) GMCSR T318A × T385 III × II 1.458 × 10−35
II T30/AF260651 IDR 4449–6115 (4340–5906) RMC3S T318A × NZ-B18 III × IV 1.147 × 10−04
1a and 1b 3968–10,737 (3859–10,628) GMCSR3S Qaha × Mex-CTV I × I 3.127 × 10−19
1b 9263–10,310 (9154–10,201) SR3S VT × NUagA III × III 7.543 × 10−13
T385/Y18420 IDR 4026–6098 (3918–5990) RMC3S T318A × NZ-B18 III × IV 1.147 × 10−04
1a and 1b 3967–10,736 (3859–10,628) GMCSR3S Qaha × Mex-CTV I × I 3.127 × 10−19
1b 9262–10,309 (9154–10,201) SR3S VT × NUagA III × III 7.543 × 10−13
I T36/U16304 1a and 1b 3483–10,492 GMCSR3S Mex-CTV × Qaha I × I 3.127 × 10−19
Mex-CTV/DQ272579 MET 3589–3985 (3482–3878) GMCSR3SP T36 × NUagA I × III 3.019 × 10−46
(B)IV B165/EU076703 2–5 10,894–14,769 (39–3914) GMCSR3S Qaha × Mex-CTV I × I 5.989 × 10−06
NZ-B18/FJ525436 2–5 10,893–14,305 (39–3451) GMCSR3S Qaha × Mex-CTV I × I 5.989 × 10−06
4 13,124–13,599 (2270–2745) GMCSR3S B165 × T318A IV × III 1.978 × 10−12
III T318A/DQ151548 2–5 10,900–14,929 (39–4068) GMCSR3SP Qaha × Mex-CTV I × I 5.989 × 10−06
VT/U56902 2–5 10,880–14,018 (45–3183) GMCSR3SP Qaha × Mex-CTV I × I 5.989 × 10−06
NUagA/AB046398 2–5 10,911–14,074 (1–3163) GMCSR3SP Qaha × Mex-CTV I × I 5.989 × 10−06
II SY568/AF001623 2–6 10,861–15,980 (1–5119) RGMCSR3SP VT × T30 III × II 2.308 × 10−75
4–6 12,159–15,438 (1298–4577) BMCSR3SP NUagA × Qaha III × I 2.984 × 10−19
T30/AF260651 4–9 12,160–17,552 (1298–6690) BMCSR3SP T318A × Qaha III × I 2.984 × 10−19
9–11 17,647–18,740 (6785–7878) MC3SP B165 × T36 IV × I 2.682 × 10−05
T385/Y18420 4–9 12,160–17,552 (1298–6690) BMCSR3SP T318A × Qaha III × I 2.984 × 10−19
9–11 17,586–18,724 (6724–7862) MC3SP B165 × T36 IV × I 2.682 × 10−05
I T36/U16304 2–5 10,984–14,070 (78–3164) GMCSR3SP Qaha × Mex-CTV I × I 5.989 × 10−06
10 18,018–18,201 (7112–7295) BMC3S Mex-CTV × SY568
I × II 4.829 × 10−03
Qaha/AY340974 10 18,018–18,284 (7112–7378) BMC3S Mex-CTV × SY568
I × II 4.829 × 10−03
a Nucleotide positions are indicated based on actual genome sequence of each genotypic isolate followed by the nucleotide positions starting from first codon of ORF1a(A) and ORF2 (B).
b Recombinant isolates identified by the recombination-detecting programs: R (RDP), G (GENECONV), B (Bootscan), M (MaxChi), C (Chimaera), SR (SiScan) and 3S (3SEQ)programs in RDP3 and P represent PHYLPRO program.
c The identified parent sequence having percentage of contribution in the recombination event but unknown or probable parent does not show the percentage ofrecombination contribution in RDP3.
d Type of ‘genotype’: I (T36-like), II (T30-like), III (VT-like) and IV (B165-like).e The reported p-value mentioned in the extreme right column is for the program in bold type in RDP3 at column 5 and mention the greatest calculated p-value among
the isolates.
Harper et al., 2009; Karasev et al., 1995; Mawassi et al., 1996; Ruiz-Ruiz et al., 2006; Suastika et al., 2001; Vives et al., 1999; Yanget al., 1999) with a highly variable 5′-UTR and conserved 3′-UTR.Previously, the complete genome sequence of nine CTV isolates dif-ferentiated them into three distinct genotypes (CTV-T30, -T36 and-VT) (Moreno et al., 2008). An additional genotype, CTV-T3 (Hilf etal., 1999), may exist; however, with the absence of the complete T3sequence, we could not include it in this study. Comparative anal-yses of 5′-UTR, ORF1a, ORF1b, and ORF2 regions of CTV-B165 gRNAwith other CTV isolates provided evidence that CTV-B165 was dif-ferent from the known genotypes. Apart from IDR, the other fourdomains in ORF1a (LProI, LProII, MET, and HEL) showed less than
90% sequence identity with other reported CTV sequences. Amongthe five domains, the LProII domain showed the least sequenceidentity (65–73%) and was significantly distant from the other threegenotypic isolates (Table 3). No other haplotype was recoveredfrom this domain of ORF1a, and the dS/dN ratio suggested that theregion evolved through negative selection pressure. The functionof the LProII domain is associated with replication, thus it is notsurprising that this domain comes under weak negative selectionpressures.
Phylogenetic analyses utilizing the MP, ME, ML, and NJ meth-ods of the four amplified 5′-terminal regions placed CTV-B165 intoa new cluster identified as genotype IV. The sequence of CTV-

Author's personal copy
128 A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130
Fig. 4. Detection of CTV-B165 genotype by reverse-transcription polymerase chainreaction utilizing genotype specific primer pair. Lanes 1 and 11: 100 bp molecu-lar marker (Promega, USA). Lane 2: healthy citrus phloem tissue used as negativecontrol. Lanes 3, 5, 7 and 9: CTV-T36, -T30, -VT and -B165 isolates, respectively,amplified by generic CTV-p18 primer pair. Lanes 4, 6 and 8: no amplification by CTV-B165 genotype specific primer pair for CTV-T36, -T30, and -VT isolates, respectively.Lane 10: genotype specific amplification using CTV-B165 specific primer pair.
B165 ORF1a and its domains were distinctive and showed morerelatedness towards SY and SP (genotype III) isolates than decline(genotype I) and mild (genotype II) symptomatic isolates. Phy-logenies generated from the translated nucleotide sequences ofthe LProI, LProII, MET, HEL, RdRp, and p33 of other CTVs showedcomparable branching and suggested that the genotype IV possi-bly evolved as a single unit from long-term persistent infectionsof existing CTV genotypic isolates. Similar kinds of evolutionaryevents have been recorded for the other members of Closteroviri-dae (Karasev, 2000). Utilizing the distance analyses, interpretationof the phylogenetic trees based on conserved proteins of ORF3 toORF11 also suggested that there was a stronger relation betweengenotype III with CTV-B165 than genotype II isolates. Analysis ofinter-genogroup evolutionary divergence by the MCL method con-firmed that genotype I isolates were probably evolved from a verydistantly related ancestor, whereas genotypes II, III, and IV mayhave a common ancestral origin.
Alternatively, the positive D value (1.214410) in the Tajima neu-trality selection test indicated that CTV-B165 and the other threedescribed CTV genotypes might have naturally originated by bal-ancing selection with low or high frequency polymorphisms withina population. Increased levels of genetic variation that are com-mon among CTV haplotypes might be the result of genetic drift inthe 5′ proximal region, whereas the conservation of the consen-sus sequence in the 3′ proximal region (ORF2–11) might be theresult of negative selection. In this type of selection, observablephenotypic differences will not always be comparable to the majorCTV component genotype presence in the population. The positiveD value from all the ORFs also indicated that the rate of geneticdrift is directly proportional to the population size and that all thegenotypes follow similar type of replications.
Higher levels of genetic variation of CTV-B165 were foundwithin the 5′-UTR, ORF1a, and ORF2 regions. The nucleotidesequence of the minor components of CTV-B165 was closely relatedto the CTV-VT (genotype III). Divergent sequence variants and mul-tiple recombinants have been reported from ORF1a, ORF1b andORF2 region in CTV-SY568 from California, and CTV isolates fromIndia and Spain (Roy and Brlansky, 2004; Roy et al., 2005b; Rubioet al., 2001; Vives et al., 2005; Yang et al., 1999). The nucleotidedifferences in the above mentioned three regions changes multipleamino acids in CTV-B165. In the study, a low dN/dS ratio of 0.64for LProI domain of ORF1a and 0.24 for ORF2 suggested stabilizingor negative selection, whereas a high ratio of 1.34 for partial IDR ofORF1a suggested that the gene sequence of the two CTV-B165 com-ponents were evolved through positive selection for diversification.The changes in the relative numbers of polar and nonpolar amino
acids were 34–40% in ORF1a and 54% in ORF2 regions between thetwo components of CTV-B165. The mutational exchange of a non-polar group for a polar one and vice-versa has been reported tobe more critical than the transition from one nonpolar group toanother or from a polar to another polar one (Volkenstein, 1965).
In the field, citrus trees often are co-infected with nonrecombi-nant and recombinant CTV of different genotypes. Recombinationhas played a significant role in the evolution of all the known CTVgenotypes (Rubio et al., 2001; Susana et al., 2009; Weng et al.,2007). A very recent study on the genetic variation and phyloge-netic analysis of the seven genomic regions from 3′ proximal halfof 18 CTV isolates showed diverse patterns of molecular evolutioncaused by variable selection pressure and frequent recombinationevents (Susana et al., 2009). In our study, multiple numbers ofunique recombination events were consistently detected by pro-gram RDP3. This program provided a more complete evolutionaryhistory since full-length sequences were analyzed rather than par-tial sequences. Analysis of CTV-B165 and the other 10 CTV isolatesby the RDP3 program suggested the possibility of frequent inter-and intra-recombination events. Four major putative breakpointswere estimated for CTV-B165 and three were located in the 5′ prox-imal half sequence. The detection of highly variable nucleotides inLProII domain (2133–2397 nt) implies intertype genetic exchangevia RNA recombination (nt 2183) in the natural CTV populations(Fig. 2A2). Further analysis of the ORF1b (9280–10,806 nt) alsoshowed two moderate recombination signals and suggests that therecombination event at the RdRP region might be involved in theevolution of CTV-B165.
Genogroups II and IV CTV isolates were identified as inter-recombinants. Isolates from genogroup I and III were identified asinter- and intra-recombinants. Recombination involving genotypeIII isolates was more frequent than genotype I and II CTV isolates.Twenty-four recombination events were found in multiple CTV iso-lates at the 5′ proximal half, whereas only seven recombinationsignals were found at the 3′ proximal half. This is the first time thatall eleven full sequenced CTV isolates were analyzed for recom-bination events. Genomic regions ORF1a showed several strongand weak recombination signals for most of the isolates, suggest-ing strong recombination events have accumulated, while weakrecombination events might be rendered indistinct by high selec-tion pressure and/or high mutation frequencies over time.
We further analyzed the complete genome of the previouslyidentified CTV genotypic isolates to identify sequence variationsfor use in the detection of the CTV-B165 genotype. On the basis ofthese analyses, we suggest the partial region of the LProII domainof ORF1a as an alternative region for CTV genotyping. This regionis highly variable among the genotypes and includes several poly-morphic sites that can be used to distinguish between CTV-B165(genotype IV) and the other genotypes. We also found severalshorter subregions (<700 bp) that belong to ORF1a to ORF2, and aresuitable for single RT-PCR genotyping (A. Roy and R.H. Brlansky,Unpublished). Even though these four genotypes are the only onesdescribed, we cannot exclude that other genotypes will be revealedas more isolates are sequenced. The availability of the completesequences of CTV-B165 and the other CTV genotypes should facili-tate future comprehensive analyses of CTV.
A total of 11 different CTV biological symptom patterns havebeen described (Garnsey et al., 2005). CTV-B165 belongs to pat-tern 6 along with 41 other CTV isolates from the EPCC (Garnsey etal., 2005). CTV-B165 produced severe symptoms in Mexican lime,SY in sour orange and Duncan grapefruit and severe SP in sweetorange but did not show any SP in grapefruit. Recently a CTV-B165genotypic isolate, NZ-B18 from New Zealand was reported (Harperet al., 2009). It also produced severe SP in sweet orange and SY insour orange but was asymptomatic in Mexican lime. Even though ahigh level of variability in symptom expression patterns was found

Author's personal copy
A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130 129
among CTV-T36, -T30 and -VT genotypes, the CTV-B165 genotypewas symptomatically close to other Indian CTV-VT genotypic iso-lates (Roy and Brlansky, 2004).
Comparative analysis of CTV-B165 was determined, but nocorrelation was observed between its geographic origin and thenucleotide distance through sequence and phylogenetic analysisof overlapping cDNA clones corresponding to different genomicregions. Genetically, CTV-B165 should be considered as a novelrecombinant CTV genotype that evolved through low to high selec-tion pressure of negative selection. We further suggest, based onthe symptoms, phylogenies, recombinant analyses, and statisticalanalyses that CTV-B165 sequence variants originated by multiplerecombination events. The striking observation from this work isthe sheer number of events and the apparent participation of mul-tiple genotypes in the recombination history of this virus. ClearlyCTV populations are not just isolated mixed infections, where onlya single genotype occupies any given cell. Rather, the multiple com-ponents so regularly found in CTV mixed infections must interactregularly during the course of their life cycle, particularly duringreplication. The opportunities for modular evolution in such a set-ting abound. The recombinant CTV-B165 isolate established itselfin the CTV complex through inter-genotypic exchange betweendiverged sequence variants from genotype I, II and III. Future stud-ies on CTV may further demonstrate the role of recombination inthe evolution of CTV genotypes. Information is important on thepresence of new CTV genotypes that may cause problems to cit-rus industries impacting existing cultivars and the development ofnew ones.
Acknowledgements
Thanks to Dr. S. Gowda, University of Florida/IFAS, CREC, LakeAlfred, Florida; Dr. Satyanarayana Tatineni, Research Plant Pathol-ogist, USDA-ARS, Lincoln, Nebraska; and Dr. William Schneider,USDA, ARS, Fort Detrick, Maryland (MD) for useful discussion andcritical comments on the manuscript. The authors are grateful toDr. John Hartung USDA-ARS, Molecular Plant Pathology Laboratory,Beltsville, MD for providing the CTV-B165 extracts from the ExoticCitrus Disease Collection, Beltsville, MD. The authors acknowledgethe financial assistance from the USDA, CREES, Special TristezaGrant Project 62506.
References
Albiach-Martí, M.R., Mawassi, M., Gowda, S., Satyanarayana, T., Hilf, M.E., Shanker,S., Almira, E.C., Vives, M.C., López, C., Guerri, J., Flores, R., Moreno, P., Garnsey,S.M., Dawson, W.O., 2000. Sequences of citrus tristeza virus separated in timeand space are essentially identical. J. Virol. 74, 6856–6865.
Ayllón, M.A., López, C., Navas-Castillo, J., Garnsey, S.M., Guerri, J., Flores, R., Moreno,P., 2001. Polymorphism of the 5′ terminal region of citrus tristeza virus (CTV) RNA:incidence of three sequence types in isolates of different origin and pathogenic-ity. Arch. Virol. 146, 27–40.
Ayllón, M.A., López, C., Navas-Castillo, J., Mawassi, M., Dawson, W.O., Guerri, J., Flores,R., Moreno, P., 1999. New defective RNAs from citrus tristeza virus: evidence fora replicase-driven template switching mechanism in their generation. J. Gen.Virol. 80, 817–821.
Bar-Joseph, M., Lee, R.F., 1989. Citrus tristeza virus. AAB Descriptions of Plant VirusesNo. 353, 7 pp.
Boni, M.F., Posada, D., Feldman, M.W., 2007. An exact nonparametric method forinferring mosaic structure in sequence triplets. Genetics 176, 1035–1047.
Dolja, V.V., Karasev, A.V., Koonin, E.V., 1994. Molecular biology and evolution ofclosteroviruses: sophisticated build-up of large RNA genomes. Annu. Rev. Phy-topathol. 32, 261–285.
Eck, R.V., Dayhoff, M.O., 1966. Atlas of Protein Sequence and Structure. NationalBiomedical Research Foundation, Silver Springs, MD.
Febres, V.J., Ashoulin, L., Mawasi, M., Frank, A., Bar-Joseph, M., Manjunath, K.L., Lee,R.F., Niblett, C.L., 1996. The p27 protein is present at one end of citrus tristezaparticles. Phytopathology 86, 1331–1335.
Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the boot-strap. Evolution 39, 783–791.
Garnsey, S.M., Civerolo, E., Gumpf, D.J., Paul, C., Hilf, M.E., Lee, R.F., Brlansky, R.H.,Yokomi, R.K., Hartung, J.S., 2005. Biological characterization of an international
collection of citrus tristeza virus (CTV) isolates. In: Hilf, M.E., Duran-Vila, N.,Rocha-Pena, M.A. (Eds.), Proc. 16th Conf. Intl. Org. Citrus Virologists. IOCV, River-side, CA, pp. 75–93.
Gibbs, M.J., Armstrong, J.S., Gibbs, A.J., 2000. Sister-scanning: a Monte Carlo pro-cedure for assessing signals in recombinant sequences. Bioinformatics 16,573–582.
Harper, S.J., Dawson, T.E., Pearson, M.N., 2009. Complete genome sequences of twodistinct and diverse Citrus tristeza virus isolates from New Zealand. Archives ofVirology 154, 1505–1510.
Hilf, M.E., Karasev, A.V., Albiach-Marti, M.R., Dawson, W.O., Garnsey, S.M., 1999. Twopaths of sequence divergence in the citrus tristeza virus complex. Phytopathology89, 336–342.
Hilf, M.E., Mavrodieva, V.A., Garnsey, S.M., 2005. Genetic marker analysis of a globalcollection of isolates of Citrus tristeza virus: characterization and distribution ofCTV genotypes and association with symptoms. Phytopathology 95, 909–917.
Karasev, A.V., Boyko, V.P., Gowda, S., Nikolaeva, O.V., Hilf, M.E., Koonin, E.V., Nibblet,C.L., Cline, K., Gumpf, D.J., Lee, R.F., Garnsey, S.M., Dawson, W.O., 1995. Completesequence of the citrus tristeza virus RNA genome. Virology 208, 511–520.
Karasev, A.V., 2000. Genetic diversity and evolution of closteroviruses. Annu. Rev.Phytopathol. 38, 293–324.
Koonin, E.V., Dolja, V.V., 1993. Evolution and taxonomy of positive-strand RNAviruses: implications of comparative analysis of amino acid sequences. Crit. Rev.Biochem. Mol. Biol. 28, 375–430.
Lai, M.M.C., 1992. RNA recombination in animal and plant viruses. Microbiol. Rev.56, 61–79.
Martin, D.P., 2009. Recombination detection and analysis using RDP3. Meth. Mol.Biol. 537, 185–205.
Martin, D.P., Rybicki, E., 2000. RDP: detection of recombination amongst alignedsequences. Bioinformatics 16, 562–563.
Martin, D.P., Williamson, C., Posada, D., 2005. RDP2: recombination detection andanalysis from sequence alignments. Bioinformatics 21, 260–262.
Mawassi, M., Karasev, A.V., Mietkiewska, E., Gafny, R., Lee, R.F., Dawson, W.O., Bar-Joseph, M., 1995a. Defective RNA molecules associated with citrus tristeza virus.Virology 208, 383–387.
Mawassi, M., Mietkiewska, E., Gofman, R., Yang, G., Bar-Joseph, M., 1996. Unusualsequence relationship between two isolates of citrus tristeza virus. J. Gen. Virol.77, 2359–2364.
Mawassi, M., Mitkiewska, E., Hilf, M.E., Ashoulin, L., Karasev, A.V., Gafny, R., Lee, R.F.,Garnsey, S.M., Dawson, W.O., Bar-Joseph, M., 1995b. Multiple species of defectiveRNAs in plants infected with citrus tristeza virus. Virology 214, 264–268.
Moreno, P., Guerri, J., 1997. Variability of citrus tristeza closterovirus (CTV): methodsto differentiate isolates. In: Monette, P. (Ed.), Filamentous Viruses of WoodyPlants. Research Signpost, Trivandrum, India, pp. 97–107.
Moreno, P., Ambrós, S., Albiach-Martí, M.R., Guerri, J., Pená, L., 2008. Citrus tristezavirus: a pathogen that changed the course of the citrus industry. Mol. PlantPathol. 9, 251–268.
Nicholas, K.B., Nicholas, H.B., Jr., 1997. GeneDoc: a tool for editing and annotatingmultiple sequence alignments. www.psc.edu/biomed/genedoc.
Pappu, H.R., Karasev, A.V., Anderson, E.J., Pappu, S.S., Hilf, M.E., Febres, V.J., Eckloff,R.M.G., McCaffery, M., Boyko, V., Gowda, S., Dolja, V.V., Koonin, E.V., Gumpf, D.J.,Cline, K.C., Garnsey, S.M., Dawson, W.O., Lee, R.F., Niblett, C.L., 1994. Nucleotidesequence and organization of eight open reading frames of the citrus tristezaclosterovirus genome. Virology 199, 35–46.
Posada, D., Crandal, K.A., 2001. Evaluation of methods for detecting recombina-tion from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. U.S.A.98, 13757–13762.
Posada, D., Crandal, K.A., Holmes, E.C., 2002. Recombination in evolutionarygenomics. Annu. Rev. Genet. 36, 75–97.
Roossinck, M.J., 1997. Mechanisms of plant virus evolution. Annu. Rev. Phytopathol.35, 191–209.
Roy, A., Brlansky, R.H., 2004. Genotype classification and molecular evidence for thepresence of mixed infections in Indian citrus tristeza virus isolates. Arch. Virol.149, 1911–1929.
Roy, A., Brlansky, R.H., 2009. Population dynamics of a Florida Citrus tristeza virus iso-late and aphid transmitted subisolates: identification of three genotypic groupsand recombinants after aphid transmission. Phytopathology 99, 1297–1306.
Roy, A., Fayad, A., Barthe, G., Brlansky, R.H., 2005a. A multiplex polymerase chainreaction method for reliable, sensitive and simultaneous detection of multipleviruses in citrus trees. J. Virol. Methods 129, 47–55.
Roy, A., Manjunath, K.L., Brlansky, R.H., 2005b. Assessment of sequence diversityin the 5′ terminal region of Indian citrus tristeza virus isolates. Virus Res. 113,132–142.
Roy, A., Ramachandran, P., Brlansky, R.H., 2003. Grouping and comparison of Indiancitrus tristeza virus isolates based on coat protein gene sequences and restrictionanalysis patterns. Arch. Virol. 148, 707–722.
Rubio, L., Ayllon, M.A., Kong, P., Fernandez, A., Polek, M.L., Guerri, J., Moreno, P., Falk,B.W., 2001. Genetic variation of citrus tristeza virus isolates from California andSpain: evidence for mixed infections and recombination. J. Virol. 75, 8054–8062.
Ruiz-Ruiz, S., Moreno, P., Guerri, J., Ambrós, S., 2006. The complete nucleotidesequence of a severe stem pitting isolate of citrus tristeza virus from Spain:comparison with isolates from different origins. Arch. Virol. 151, 387–398.
Saitou, N., Nei, M., 1987. The neighbor-joining method: a new method for recon-structing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Salminen, M.O., Carr, J.K., Burke, D.S., McCutchan, F.E., 1995. Identification of break-points in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res.Hum. Retroviruses 11, 1423–1425.

Author's personal copy
130 A. Roy, R.H. Brlansky / Virus Research 151 (2010) 118–130
Sawyer, S.A., 1999. GENECONV: A computer package for the statistical detectionof gene conversion. Distributed by the author. Department of Mathemat-ics, Washington University in St. Louis, USA. Available at: http://www.math.wustl.edu/∼sawyer.
Simon, A.E., Bujarski, J.J., 1994. RNA–RNA recombination and evolution in virusinfected plants. Annu. Rev. Phytopathol. 32, 337–362.
Smith, J.M., 1992. Analyzing the mosaic structure of genes. J. Mol. Evol. 34, 126–129.Suastika, G., Natsuaki, T., Terui, H., Kano, T., Ieki, H., Okuda, S., 2001. Nucleotide
sequence of citrus tristeza virus seedling yellows isolate. J. Gen. Plant Pathol. 67,73–77.
Susana, M., Sambade, A., Rubio, L., Vives, M.C., Moya, P., Guerri, J., Elena, S.F., Moreno,P., 2009. Contribution of recombination and selection to molecular evolution ofCitrus tristeza virus. J. Gen. Virol. 90, 1527–1538.
Tajima, F., 1989. Statistical methods to test for nucleotide mutation hypothesis byDNA polymorphism. Genetics 123, 585–595.
Tamura, K., Dudley, J., Nei, M., Kumar, S., 2007. MEGA4: Molecular EvolutionaryGenetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599.
Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G., 1997. TheClustal X windows interface: flexible strategies for multiple sequence alignmentaided by quality analysis tools. Nucleic Acids Res. 24, 4876–4882.
Vives, M.C., Rubio, L., Sambade, A., Mirkov, T.E., Moreno, P., Guerri, J., 2005. Evidenceof multiple recombination events between two RNA sequence variants within acitrus tristeza virus isolate. Virology 331, 232–237.
Vives, M.C., Rubio, L., Lopez, C., Navas-Castillo, J., Albiach-Martí, M.R., Dawson, W.O.,Guerri, J., Flores, R., Moreno, P., 1999. The complete genome sequence of themajor component of a mild citrus tristeza isolate. J. Gen. Virol. 80, 811–816.
Volkenstein, M.V., 1965. Coding of polar and nonpolar amino acids. Nature 207,294–295.
Weiller, G.F., 1998. Phylogenetic profiles: a graphical method for detecting geneticrecombinations in homologous sequences. Mol. Biol. Evol. 15, 326–335.
Weng, Z., Barthelson, R., Gowda, S., Hilf, M.E., Dawson, W.O., Galbraith, D.W., Xiong,Z., 2007. Persistent infection and promiscuous recombination of multiple geno-types of an RNA virus within a single host generate extensive diversity. PLoSOne 2 (9), e917.
Yang, G., Mawassi, M., Gofman, R., Gafny, R., Bar-Joseph, M., 1997. Involvement of asubgenomic mRNA in the generation of a variable population of defective citrustristeza virus molecules. J. Virol. 71, 9800–9802.
Yang, Z.N., Mathews, D.H., Dodds, J.A., Mirkov, T.E., 1999. Molecular characterizationof an isolate of citrus tristeza virus that causes severe symptoms in sweet orange.Virus Genes 19, 131–142.
Zhang, J., Rosenberg, H.F., Nei, M., 1998. Positive Darwinian selection after geneduplication in primate ribonuclease genes. Proc. Natl. Acad. Sci. U.S.A. 95,3708–3713.
Related Documents











