
Genetics Spring 2015 Molecular Genetics of the Cell Cycle and Cancer.
Dec 19, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript


Genetics Spring 2015
Molecular Genetics of the Cell Cycle and Cancer
Outline
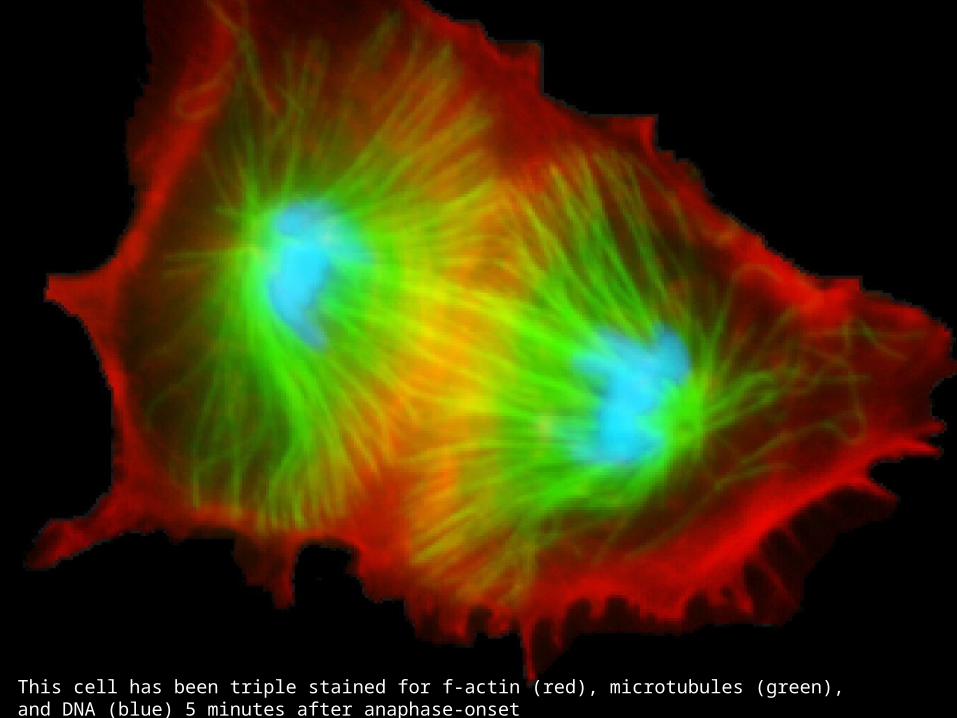
This cell has been triple stained for f-actin (red), microtubules (green), and DNA (blue) 5 minutes after anaphase-onset
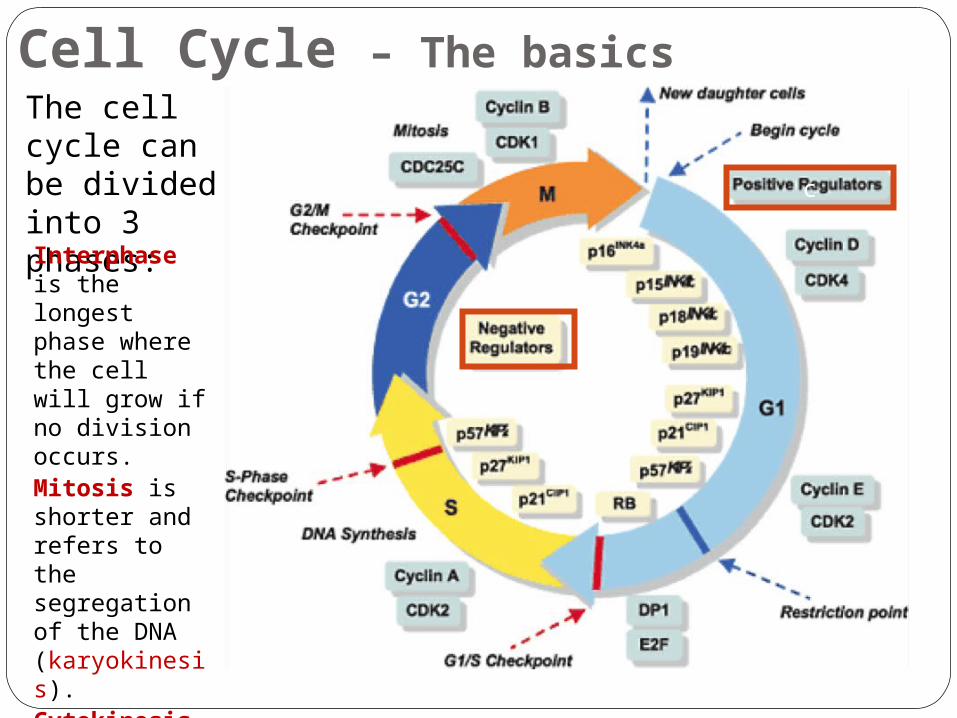
Cell Cycle – The basicsThe cell cycle can be divided into 3 phases:Interphase is the longest phase where the cell will grow if no division occurs.Mitosis is shorter and refers to the segregation of the DNA (karyokinesis).Cytokinesis is the shortest phase and refers to the division of the cytoplasm.
c
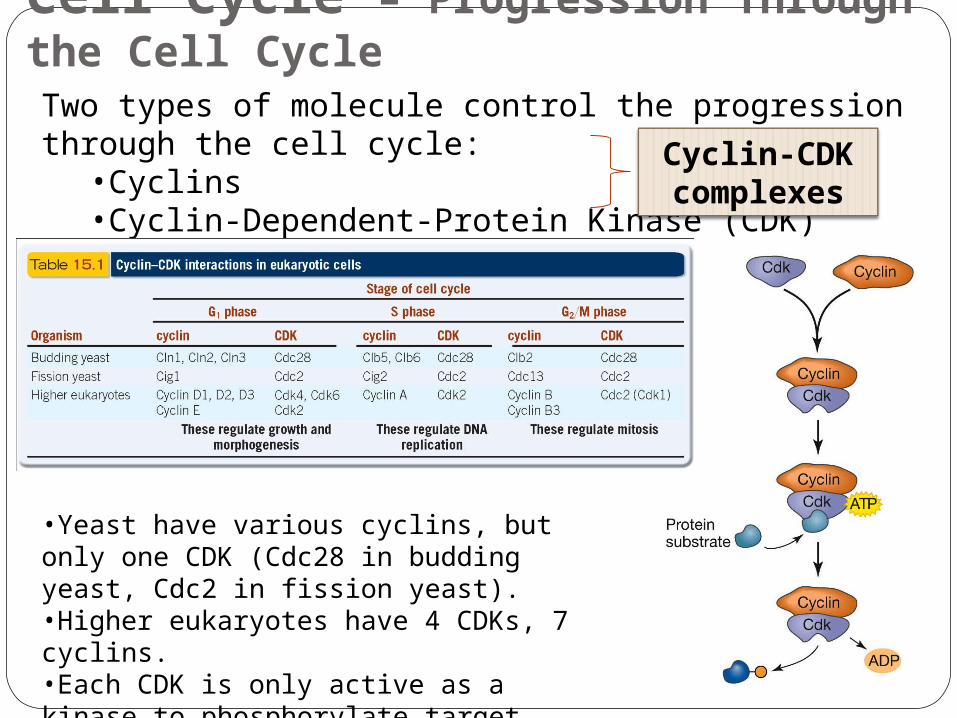
Cell Cycle – Progression Through the Cell CycleTwo types of molecule control the progression through the cell cycle:
•Cyclins•Cyclin-Dependent-Protein Kinase (CDK)
Cyclin-CDK complexes
•Yeast have various cyclins, but only one CDK (Cdc28 in budding yeast, Cdc2 in fission yeast).•Higher eukaryotes have 4 CDKs, 7 cyclins.•Each CDK is only active as a kinase to phosphorylate target proteins when it is bound to specific cyclin(s).
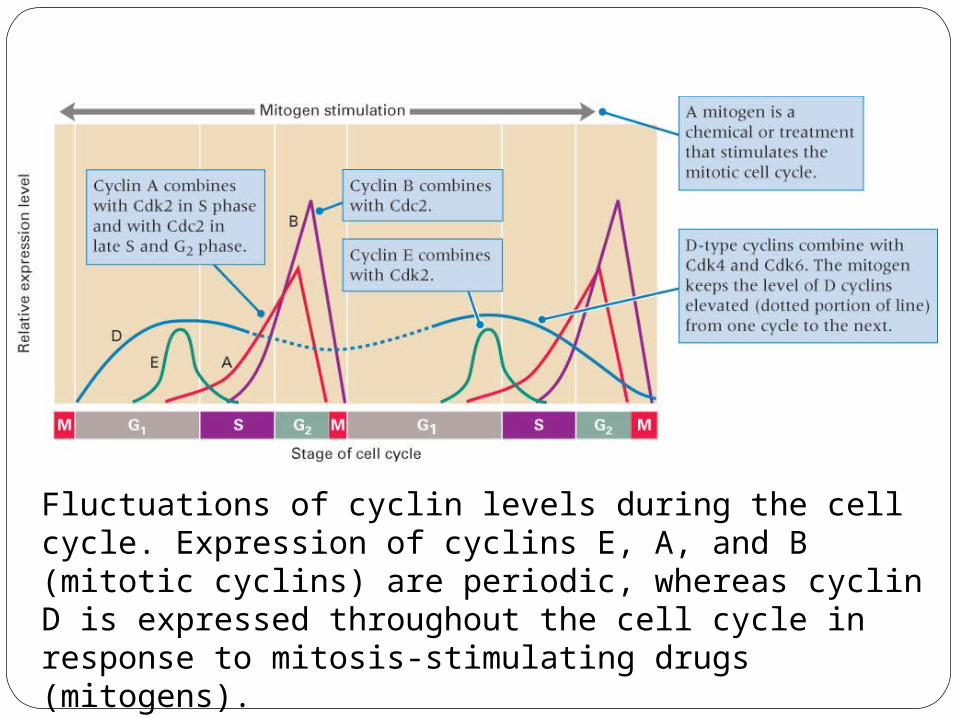
Fluctuations of cyclin levels during the cell cycle. Expression of cyclins E, A, and B (mitotic cyclins) are periodic, whereas cyclin D is expressed throughout the cell cycle in response to mitosis-stimulating drugs (mitogens).
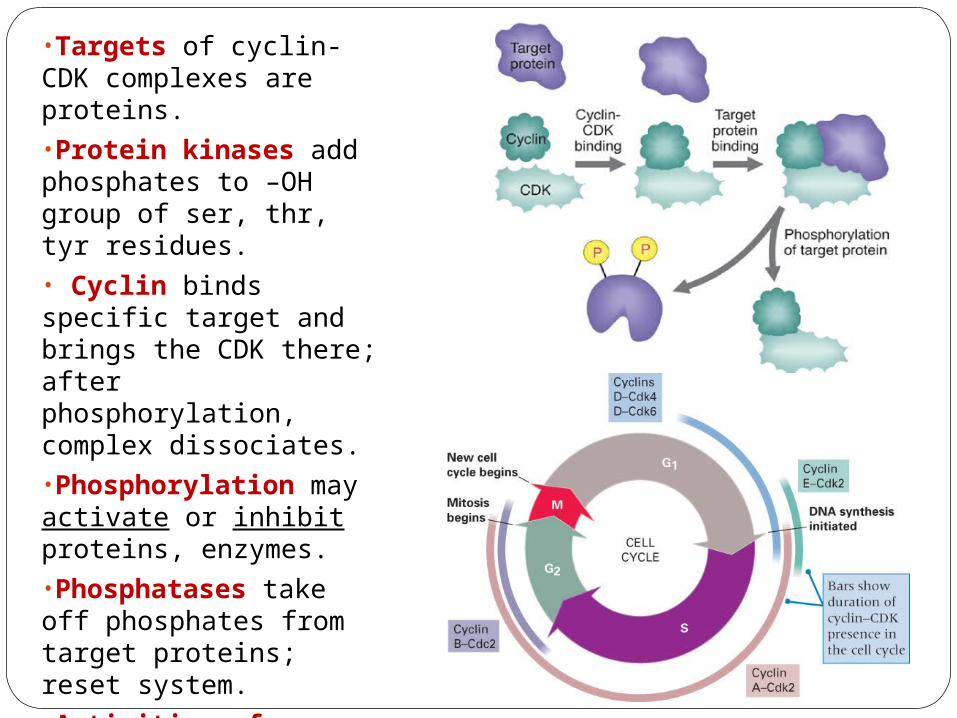
•Targets of cyclin-CDK complexes are proteins.•Protein kinases add phosphates to –OH group of ser, thr, tyr residues.• Cyclin binds specific target and brings the CDK there; after phosphorylation, complex dissociates.•Phosphorylation may activate or inhibit proteins, enzymes.•Phosphatases take off phosphates from target proteins; reset system.•Activities of cyclin-CDK are controlled by phosphorylation: Cyclin D-CDK complexes are controlled by inhibitor p16 protein and by dephosphorylation.
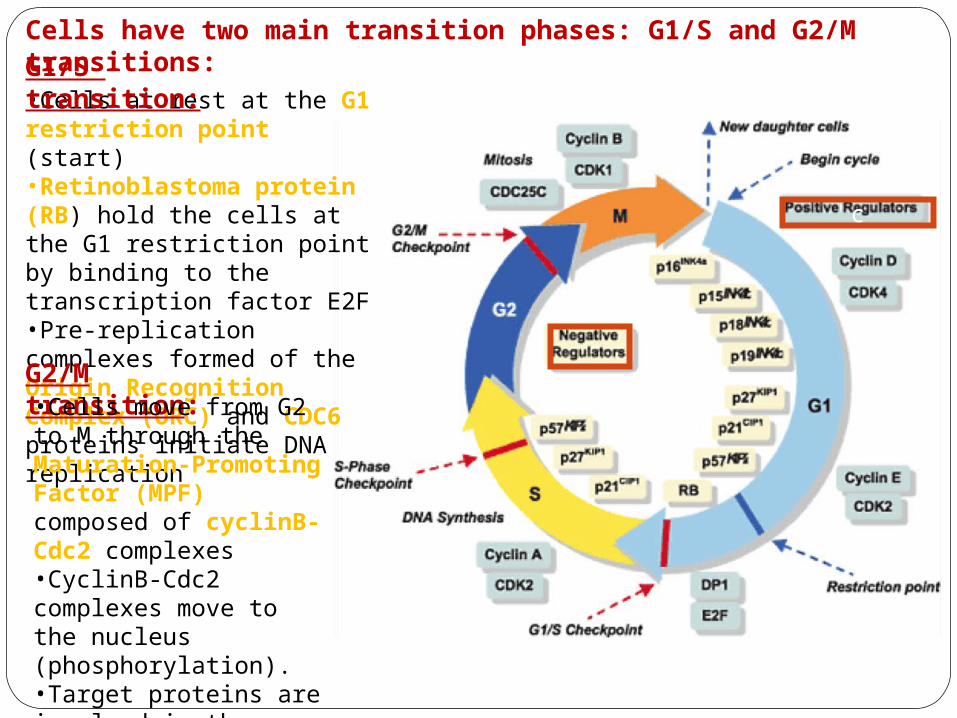
•Cells at rest at the G1 restriction point (start)•Retinoblastoma protein (RB) hold the cells at the G1 restriction point by binding to the transcription factor E2F•Pre-replication complexes formed of the Origin Recognition Complex (ORC) and CDC6 proteins initiate DNA replication
Cells have two main transition phases: G1/S and G2/M transitions:G1/S transition:
G2/M transition:•Cells move from G2 to M through the Maturation-Promoting Factor (MPF) composed of cyclinB-Cdc2 complexes•CyclinB-Cdc2 complexes move to the nucleus (phosphorylation).•Target proteins are involved in the duplication of the spindle and breakdown of nuclear envelope
c
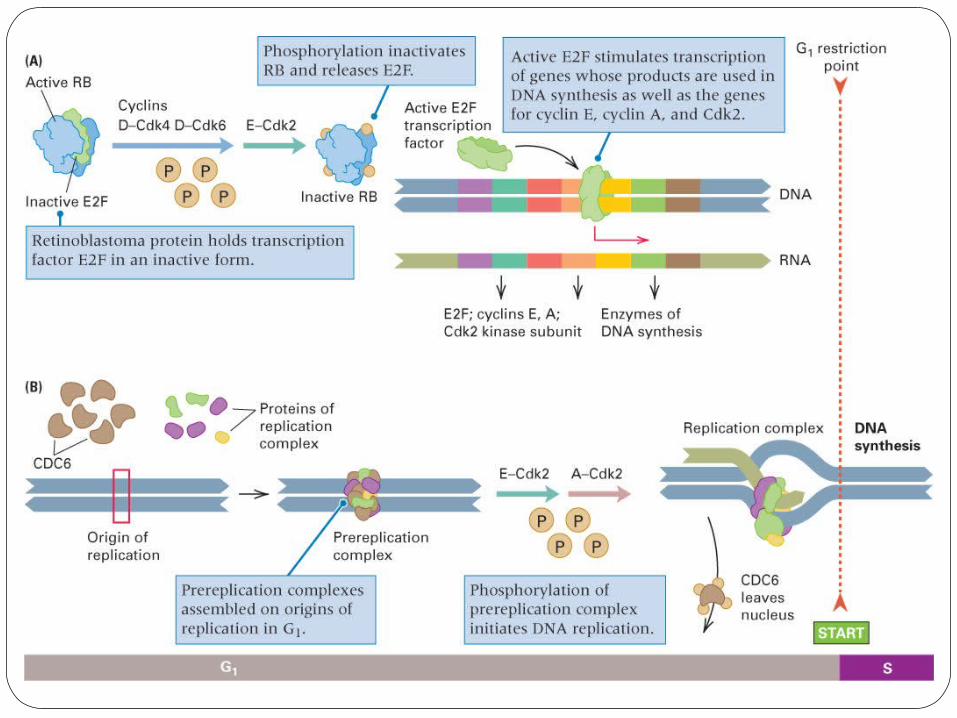
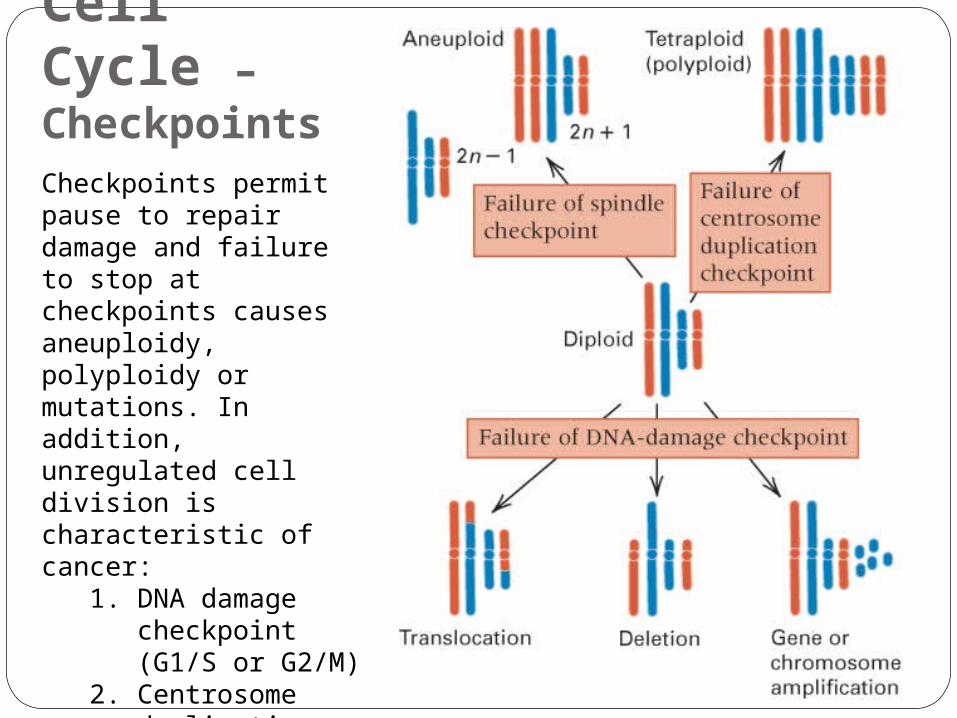
Cell Cycle – CheckpointsCheckpoints permit pause to repair damage and failure to stop at checkpoints causes aneuploidy, polyploidy or mutations. In addition, unregulated cell division is characteristic of cancer:
1. DNA damage checkpoint (G1/S or G2/M)
2. Centrosome duplication checkpoint (G2/M)
3. Spindle checkpoint (metaphase/anaphase)
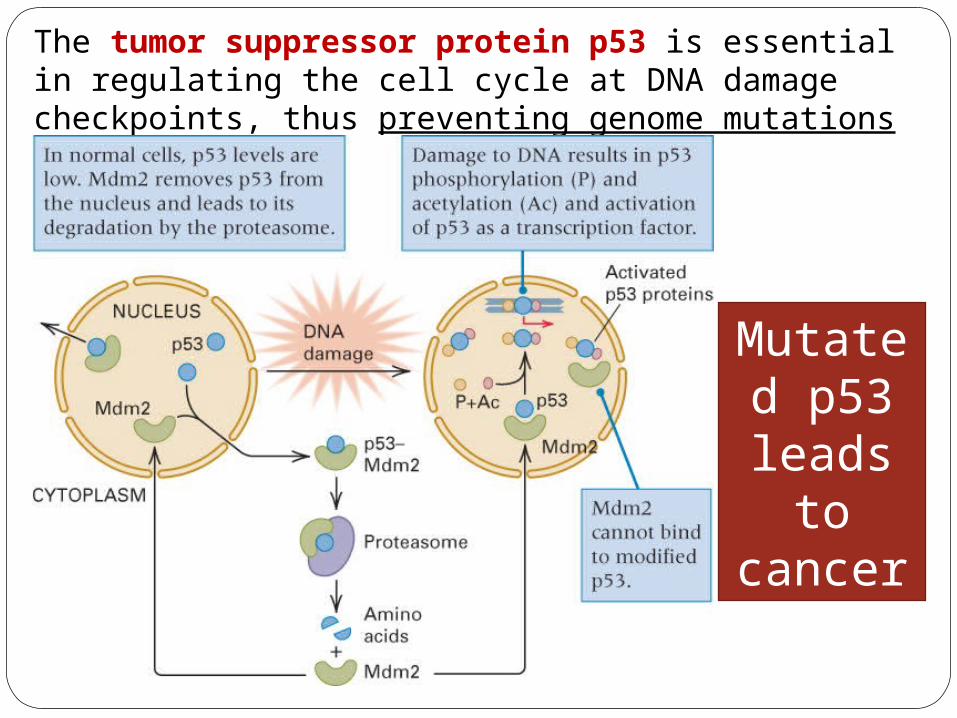
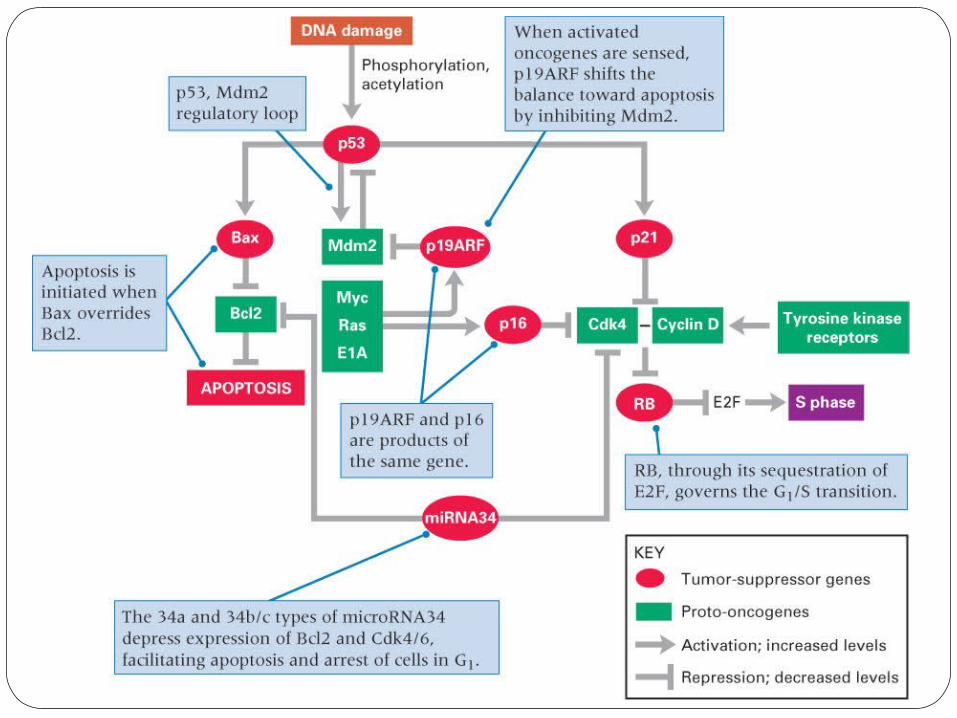
The tumor suppressor protein p53 is essential in regulating the cell cycle at DNA damage checkpoints, thus preventing genome mutations that can lead to cancer.
Mutated p53 leads
to cancer
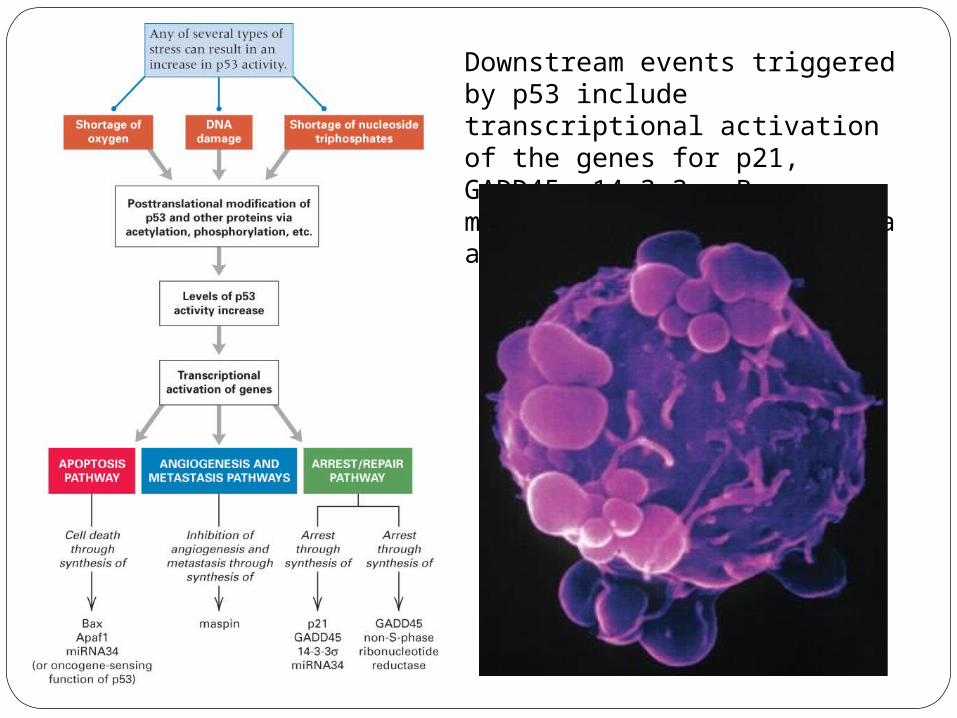
Downstream events triggered by p53 include transcriptional activation of the genes for p21, GADD45, 14-3-3σ, Bax, maspin, Apaf1, and miRNA34a and b/c.
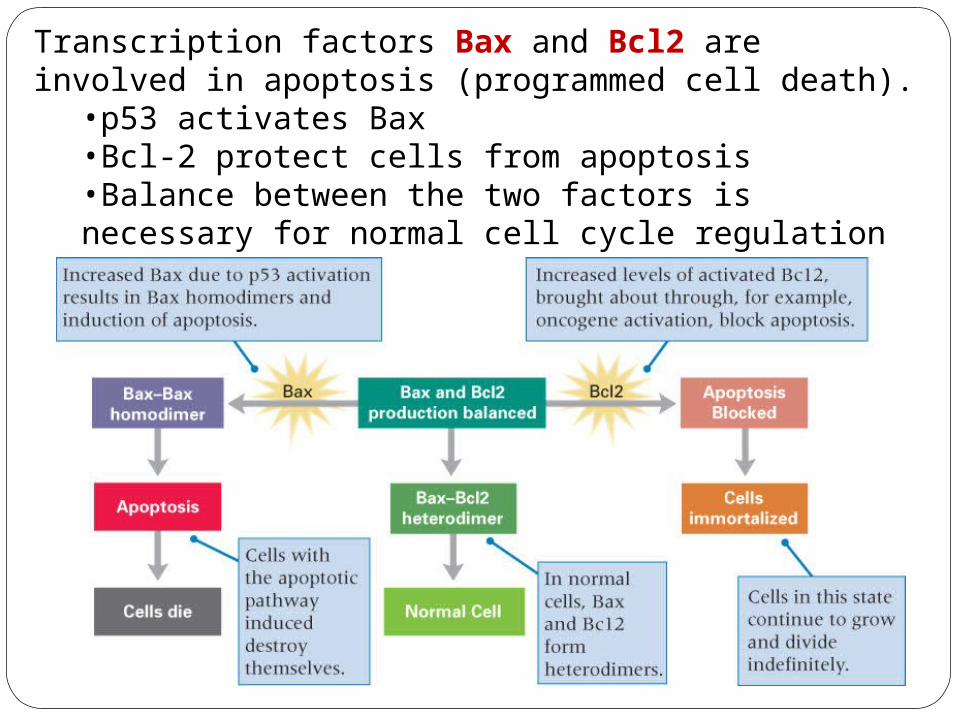
Transcription factors Bax and Bcl2 are involved in apoptosis (programmed cell death).
•p53 activates Bax•Bcl-2 protect cells from apoptosis•Balance between the two factors is necessary for normal cell cycle regulation
Cancer Cells
Familial breast cancer, BRCA1 was positionally cloned in 1994 (21 years ago)
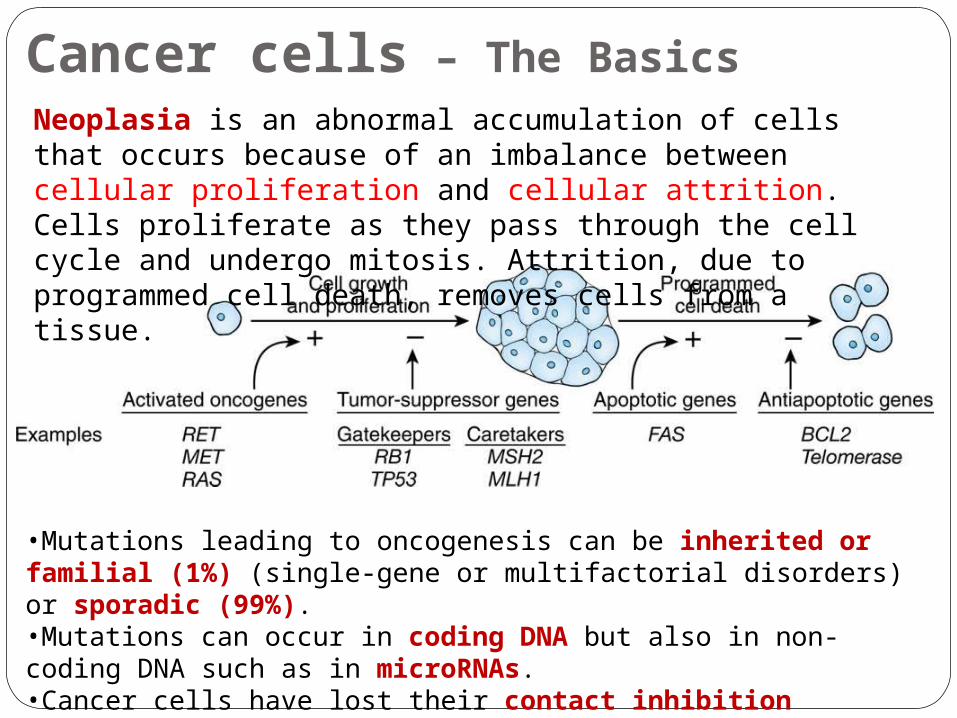
Cancer cells – The Basics Neoplasia is an abnormal accumulation of cells that occurs because of an imbalance between cellular proliferation and cellular attrition. Cells proliferate as they pass through the cell cycle and undergo mitosis. Attrition, due to programmed cell death, removes cells from a tissue.
•Mutations leading to oncogenesis can be inherited or familial (1%) (single-gene or multifactorial disorders) or sporadic (99%).•Mutations can occur in coding DNA but also in non-coding DNA such as in microRNAs. •Cancer cells have lost their contact inhibition properties.
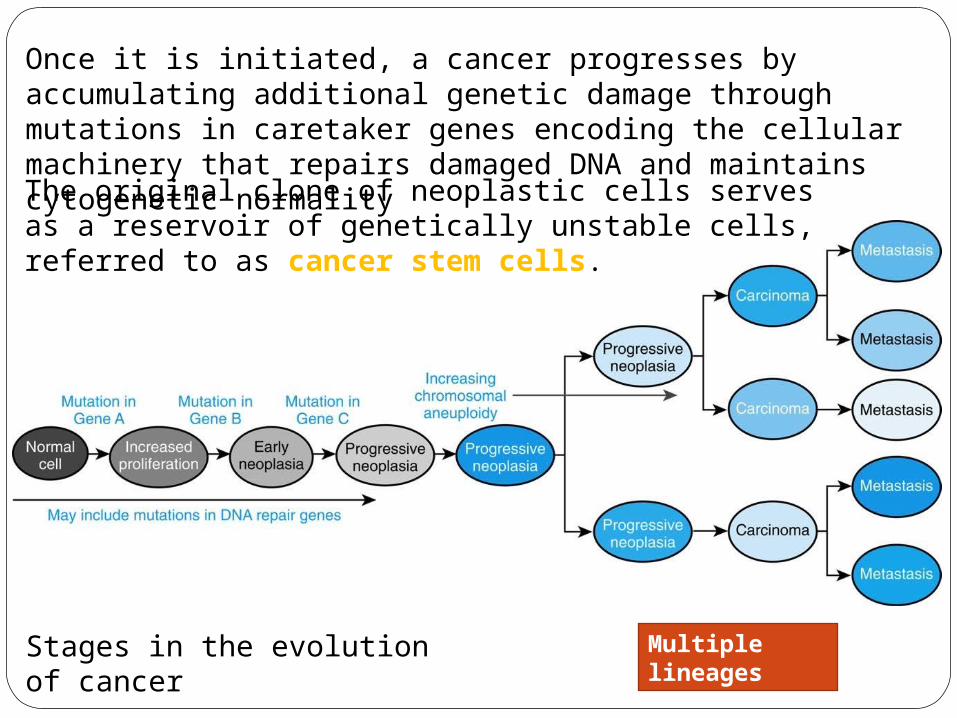
Once it is initiated, a cancer progresses by accumulating additional genetic damage through mutations in caretaker genes encoding the cellular machinery that repairs damaged DNA and maintains cytogenetic normality The original clone of neoplastic cells serves as a reservoir of genetically unstable cells, referred to as cancer stem cells.
Stages in the evolution of cancer
Multiple lineages
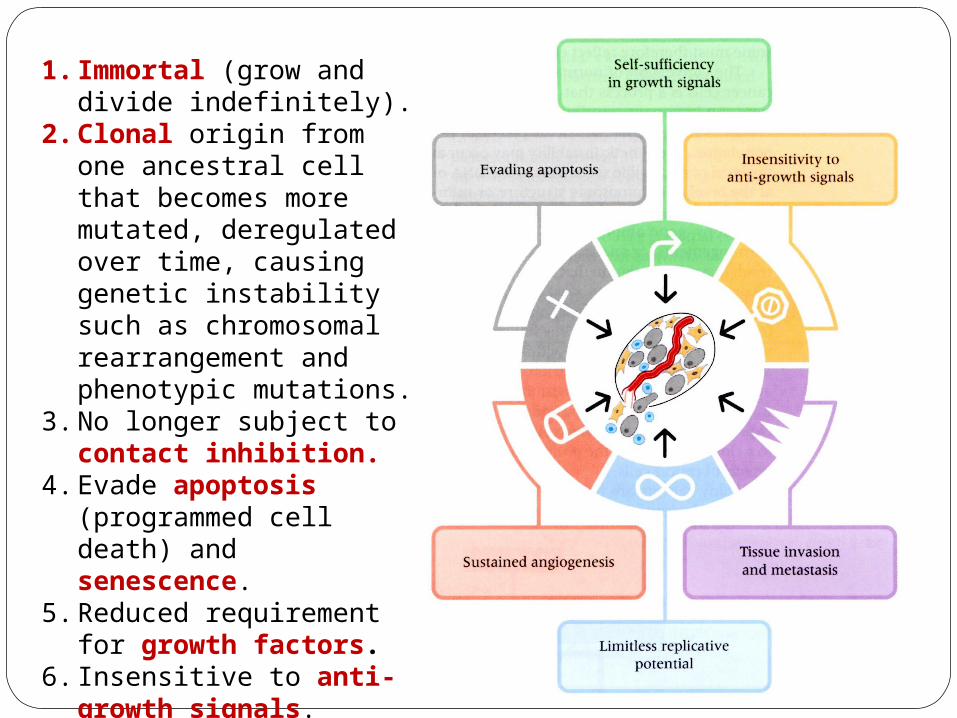
1. Immortal (grow and divide indefinitely).
2. Clonal origin from one ancestral cell that becomes more mutated, deregulated over time, causing genetic instability such as chromosomal rearrangement and phenotypic mutations.
3. No longer subject to contact inhibition.
4. Evade apoptosis (programmed cell death) and senescence.
5. Reduced requirement for growth factors.
6. Insensitive to anti-growth signals.
7. Ability to metastasize (invade other tissues).
8. Sustained angiogenesis (formation blood vessels).

Cancer Cells -Types of Genes
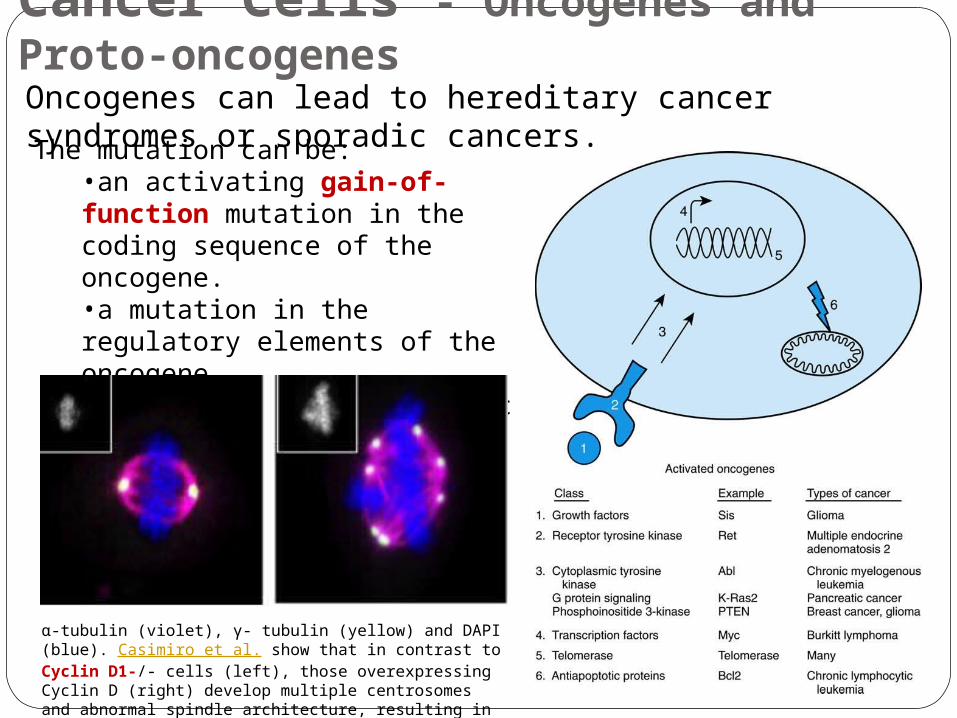
The genes implicated in cancer development are classified into three types: •Oncogenes stimulate cell division excessively. They are mutated from normal genes called proto-oncogenes that encode components of the cell's normal growth, such as growth factors. •Tumor Suppressor Genes inhibit cell division and/or cause apoptosis. Mutations in tumor suppressor genes would promote cell division or allow genetically damaged cell to grow out of control. •DNA Repair Genes correct mutation of a gene.
Cancer Cells - Oncogenes and Proto-oncogenes
The mutation can be:•an activating gain-of-function mutation in the coding sequence of the oncogene.•a mutation in the regulatory elements of the oncogene.•an increase in the genomic copy number of the oncogene.
Oncogenes can lead to hereditary cancer syndromes or sporadic cancers.
-α tubulin (violet), - γ tubulin (yellow) and DAPI (blue). Casimiro et al. show that in contrast to Cyclin D1-/- cells (left), those overexpressing Cyclin D (right) develop multiple centrosomes and abnormal spindle architecture, resulting in chromosomal instability.
Cancer Cells – Tumor-suppressor Genes
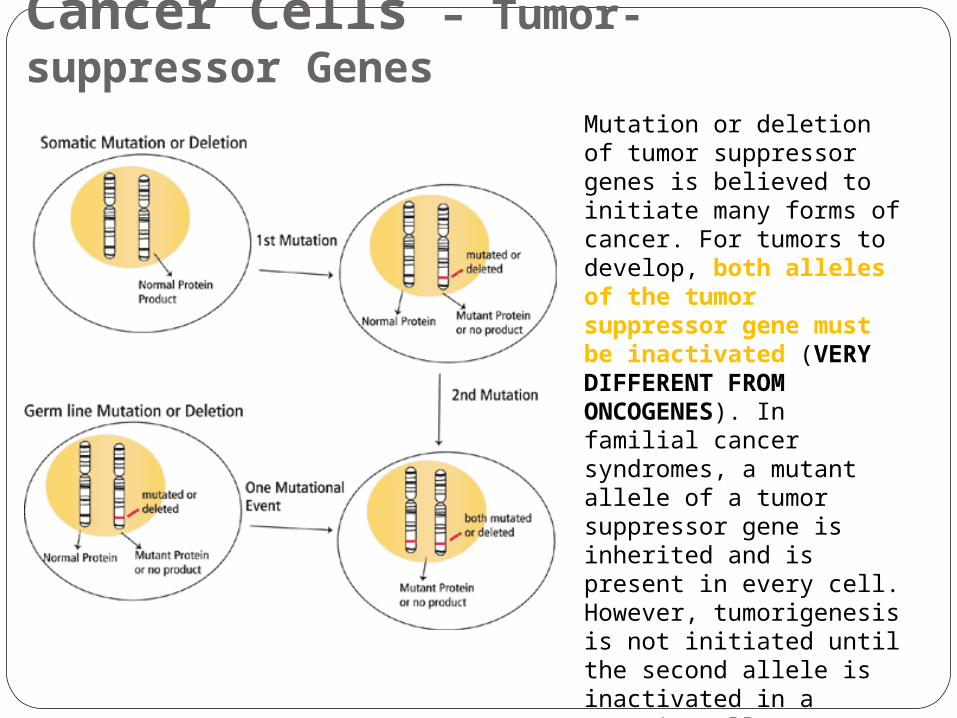
Mutation or deletion of tumor suppressor genes is believed to initiate many forms of cancer. For tumors to develop, both alleles of the tumor suppressor gene must be inactivated (VERY DIFFERENT FROM ONCOGENES). In familial cancer syndromes, a mutant allele of a tumor suppressor gene is inherited and is present in every cell. However, tumorigenesis is not initiated until the second allele is inactivated in a somatic cell. In non-familial cases, inactivation of both alleles occurs via somatic mutation or deletion. The end result is the same in both cases, the lack of a functional tumor suppressor gene leads to tumor development.
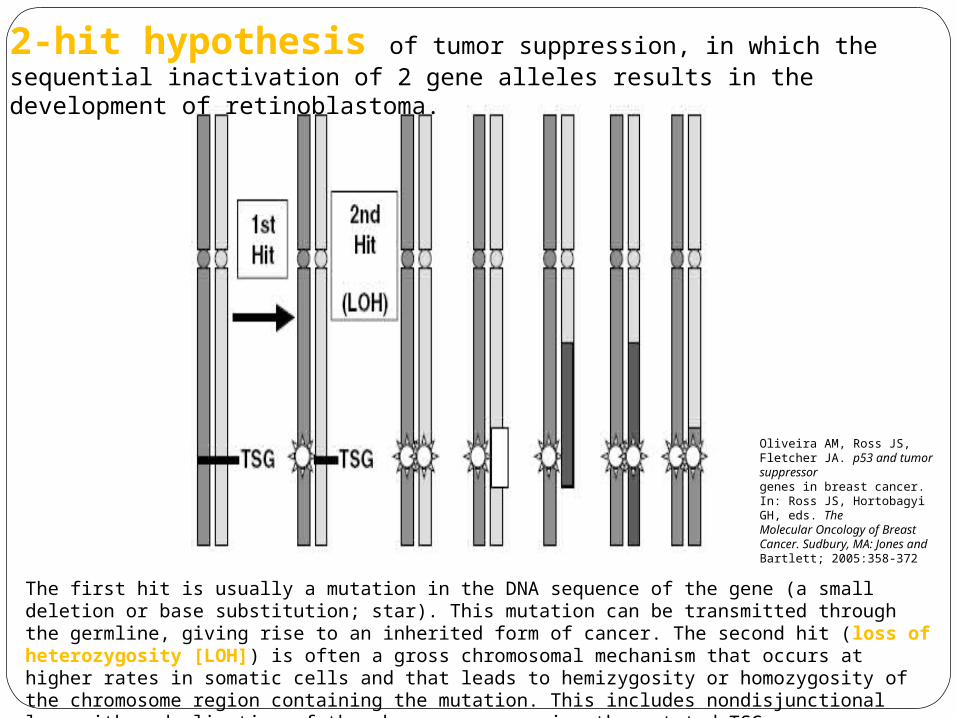
2-hit hypothesis of tumor suppression, in which the sequential inactivation of 2 gene alleles results in the development of retinoblastoma.
The first hit is usually a mutation in the DNA sequence of the gene (a small deletion or base substitution; star). This mutation can be transmitted through the germline, giving rise to an inherited form of cancer. The second hit (loss of heterozygosity [LOH]) is often a gross chromosomal mechanism that occurs at higher rates in somatic cells and that leads to hemizygosity or homozygosity of the chromosome region containing the mutation. This includes nondisjunctional loss with reduplication of the chromosome carrying the mutated TSG, subchromosomal deletion, unbalanced translocation, and mitotic recombination.
Oliveira AM, Ross JS, Fletcher JA. p53 and tumor suppressorgenes in breast cancer. In: Ross JS, Hortobagyi GH, eds. TheMolecular Oncology of Breast Cancer. Sudbury, MA: Jones andBartlett; 2005:358-372
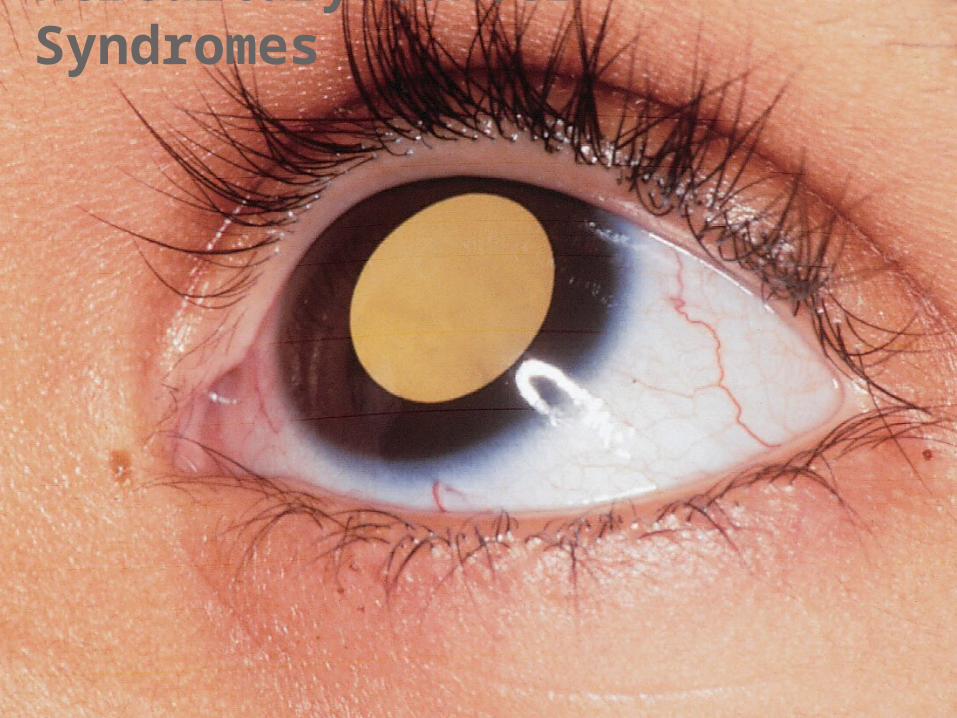
Hereditary Cancer Syndromes
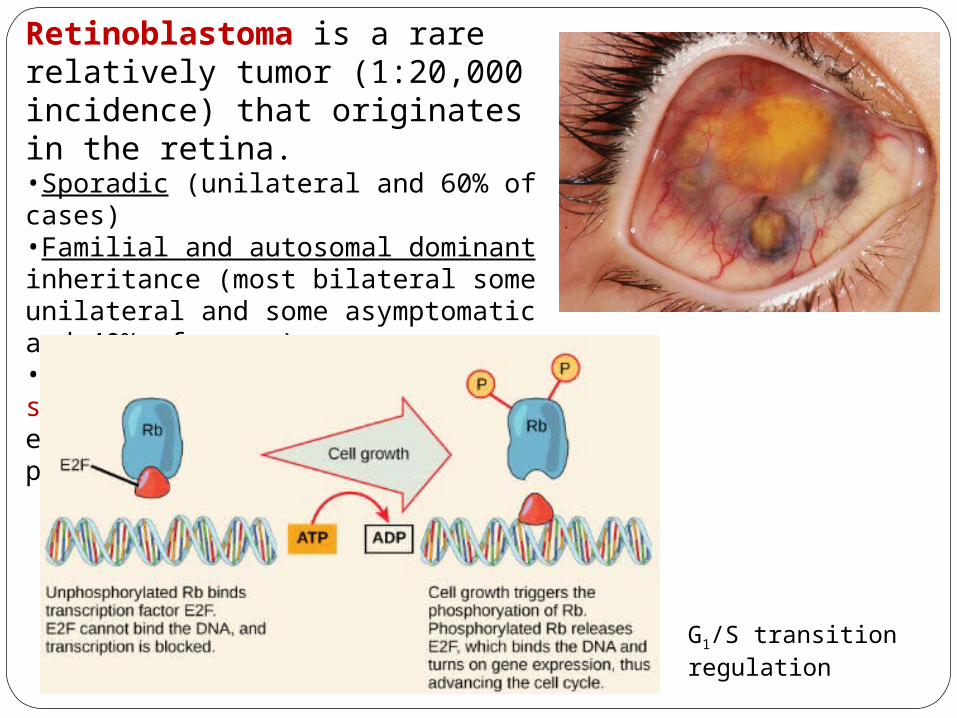
Retinoblastoma is a rare relatively tumor (1:20,000 incidence) that originates in the retina.•Sporadic (unilateral and 60% of cases)•Familial and autosomal dominant inheritance (most bilateral some unilateral and some asymptomatic and 40% of cases)•Induced by mutation in the tumor suppressor gene Rb (ch 13) encoding the Retinoblastoma protein (RB).
G1/S transition regulation
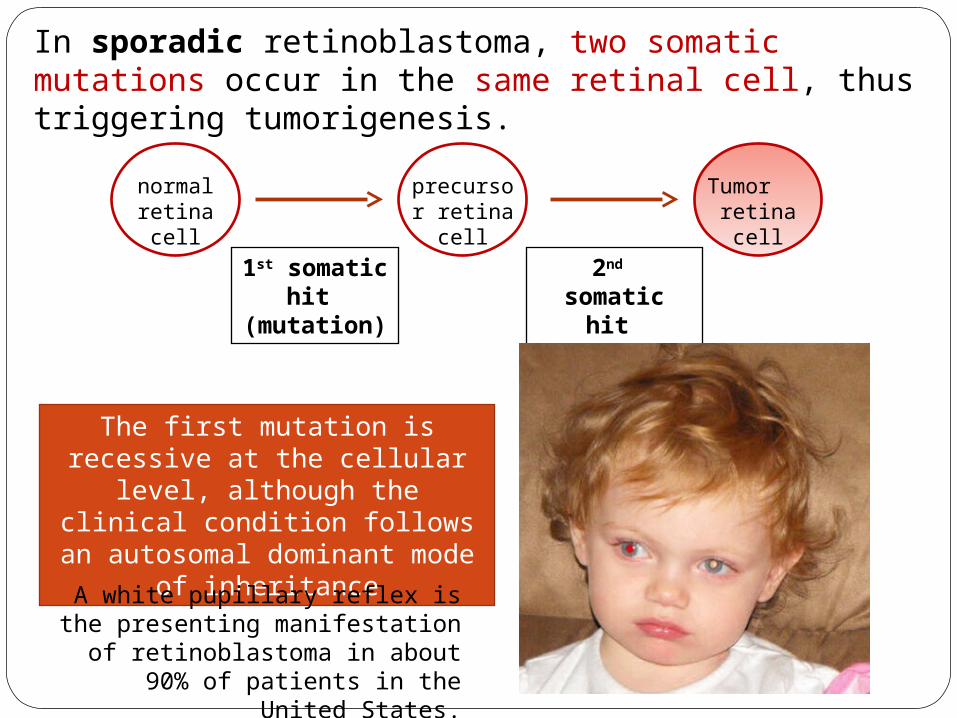
In sporadic retinoblastoma, two somatic mutations occur in the same retinal cell, thus triggering tumorigenesis.
normal retina cell
precursor retina cell
Tumor retina cell
1st somatic hit
(mutation)
2nd somatic hit (mutation)
The first mutation is recessive at the cellular level, although
the clinical condition follows an autosomal dominant mode of
inheritance
A white pupillary reflex is the presenting manifestation of
retinoblastoma in about 90% of patients in the United States.
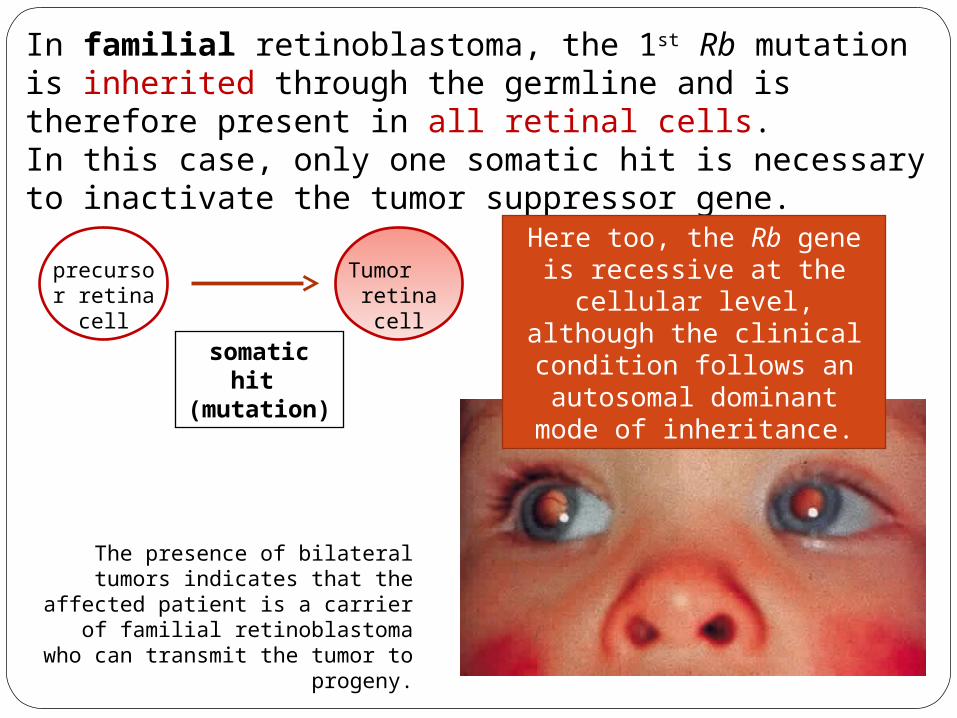
The presence of bilateral tumors indicates that the affected patient
is a carrier of familial retinoblastoma who can transmit
the tumor to progeny.
precursor retina cell
Tumor retina cell
somatic hit
(mutation)
In familial retinoblastoma, the 1st Rb mutation is inherited through the germline and is therefore present in all retinal cells.In this case, only one somatic hit is necessary to inactivate the tumor suppressor gene.
Here too, the Rb gene is recessive at the cellular
level, although the clinical condition follows an
autosomal dominant mode of inheritance.
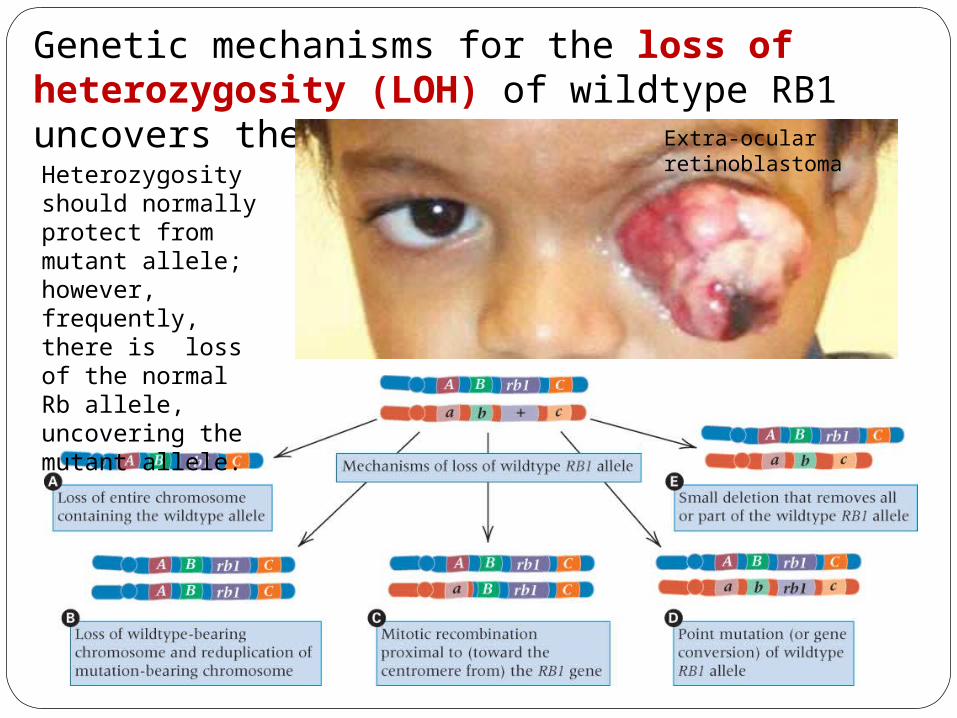
Heterozygosity should normally protect from mutant allele; however, frequently, there is loss of the normal Rb allele, uncovering the mutant allele.
Genetic mechanisms for the loss of heterozygosity (LOH) of wildtype RB1 uncovers the recessive allele. Extra-ocular
retinoblastoma
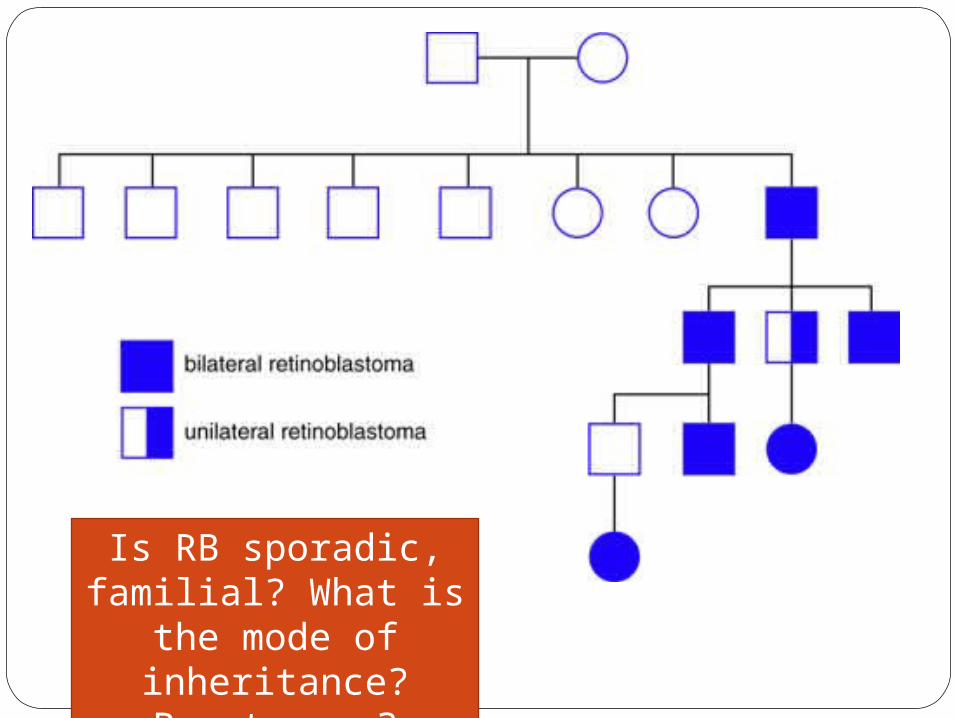
Is RB sporadic, familial? What is the
mode of inheritance? Penetrance?
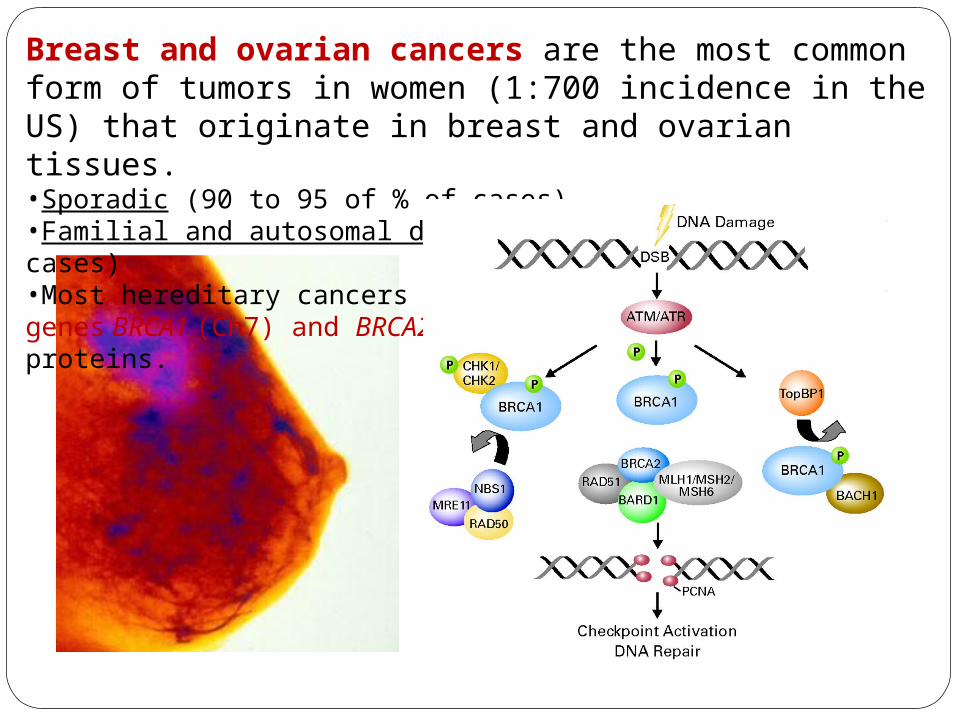
Breast and ovarian cancers are the most common form of tumors in women (1:700 incidence in the US) that originate in breast and ovarian tissues.•Sporadic (90 to 95 of % of cases)•Familial and autosomal dominant inheritance (5-10% of cases)•Most hereditary cancers involves the tumor suppressor genes BRCA1 (Ch7) and BRCA2 (Ch13) encoding DNA repair proteins.
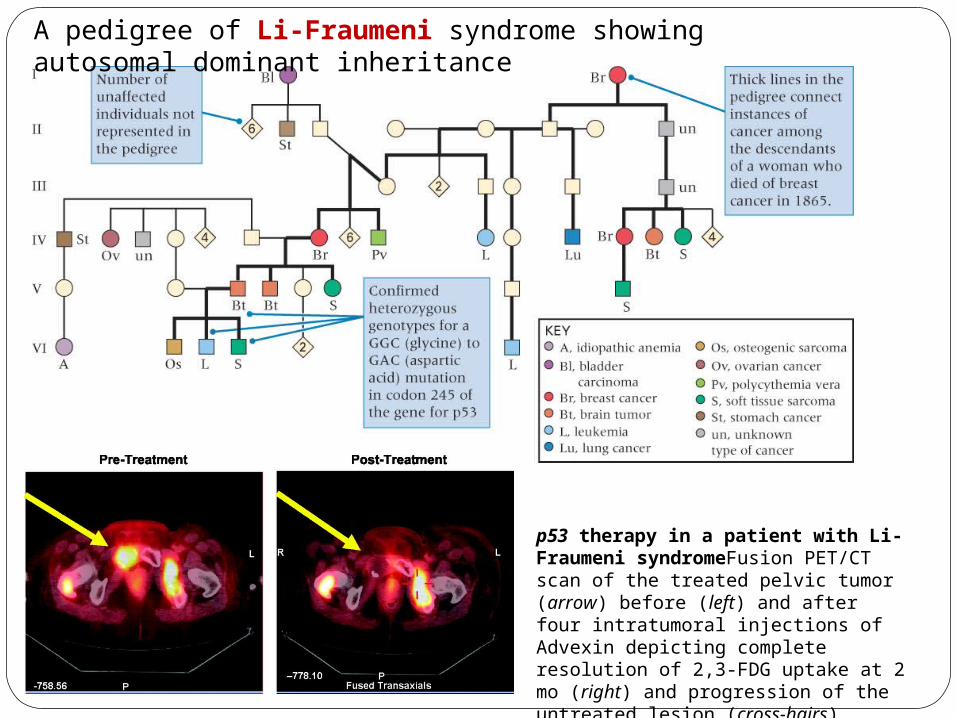
A pedigree of Li-Fraumeni syndrome showing autosomal dominant inheritance
p53 therapy in a patient with Li-Fraumeni syndromeFusion PET/CT scan of the treated pelvic tumor (arrow) before (left) and after four intratumoral injections of Advexin depicting complete resolution of 2,3-FDG uptake at 2 mo (right) and progression of the untreated lesion (cross-hairs)
Genetic of the Acute LeukemiasAcute leukemias often involve translocations:
Cells of hematopoietic system rapidly divide: bone marrow stem cells leading to red blood cells and white blood cells.
Ex. promoter fusions of an Immunoglobulin promoter to a proto-oncogene will overexpress the normal protein in B lymphocytes - promote excessive cell division.
Ex. gene fusions: CML = chronic myeloid leukemia: fusion of Bcr and abl genes by chromosome 9-22 translocation overactive tyrosine kinase.
Ex. PMLRAR = acute promyelocytic leukemia = fusion of PML and RAR (retinoic acid receptor); fusion protein defective.
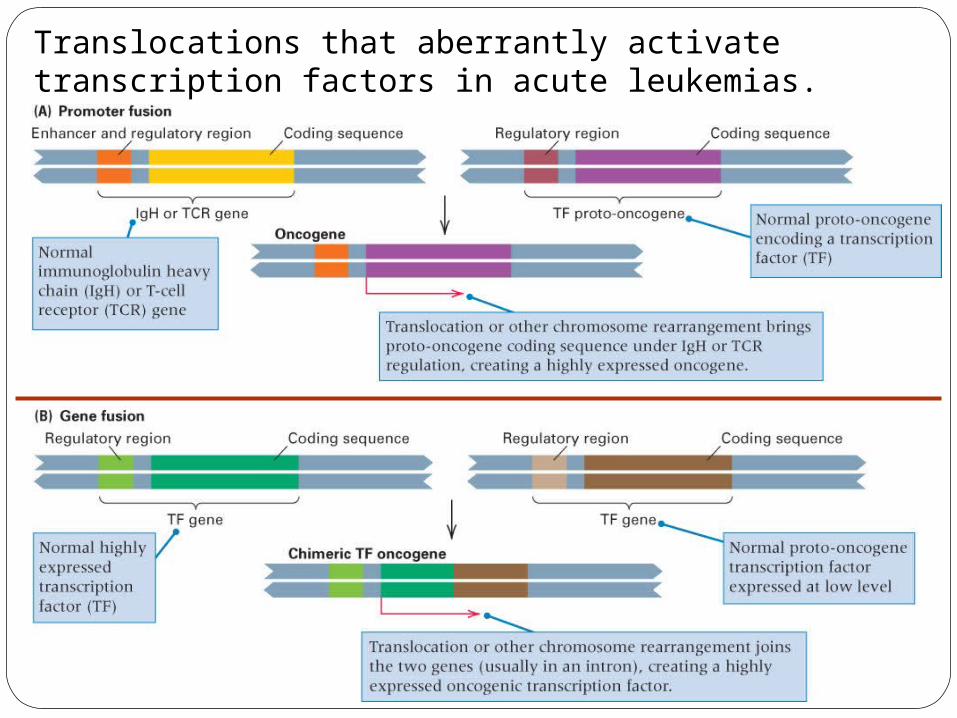
Translocations that aberrantly activate transcription factors in acute leukemias.
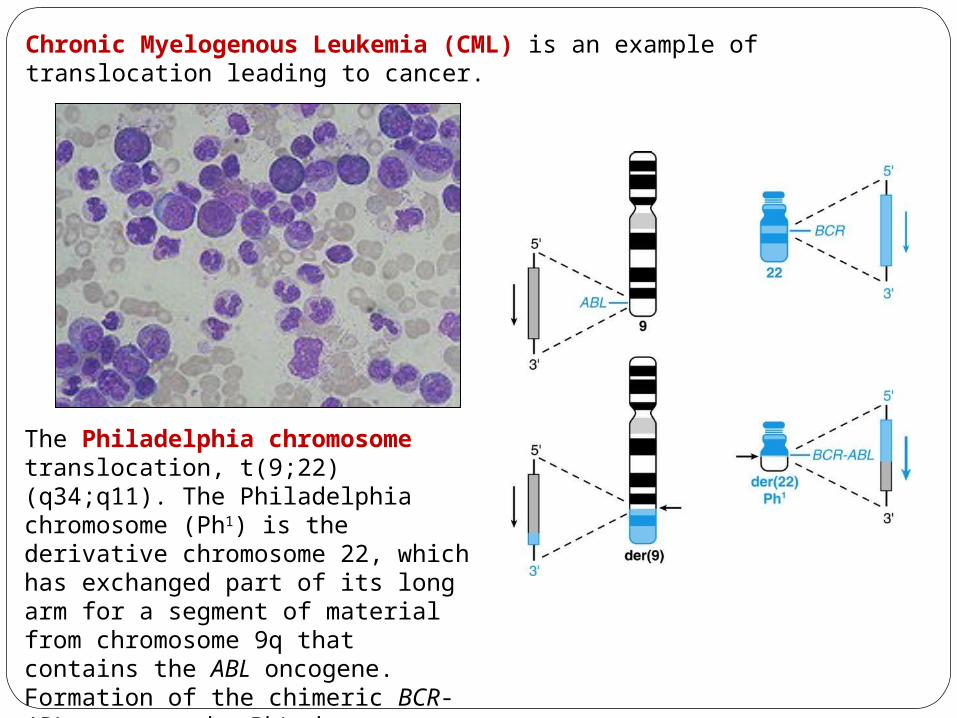
Chronic Myelogenous Leukemia (CML) is an example of translocation leading to cancer.
The Philadelphia chromosome translocation, t(9;22)(q34;q11). The Philadelphia chromosome (Ph1) is the derivative chromosome 22, which has exchanged part of its long arm for a segment of material from chromosome 9q that contains the ABL oncogene. Formation of the chimeric BCR-ABL gene on the Ph1 chromosome is the critical genetic event in the development of chronic myelogenous leukemia.

The Cancer Genome Atlas (TCGA) began as a 3-year pilot in 2006 with an investment of $50 million each from the National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI).
Creation of an atlas of changes for specific cancer typesCharacteriz-ation of 33 tumor typesExome and whole genome sequencing Free access to all

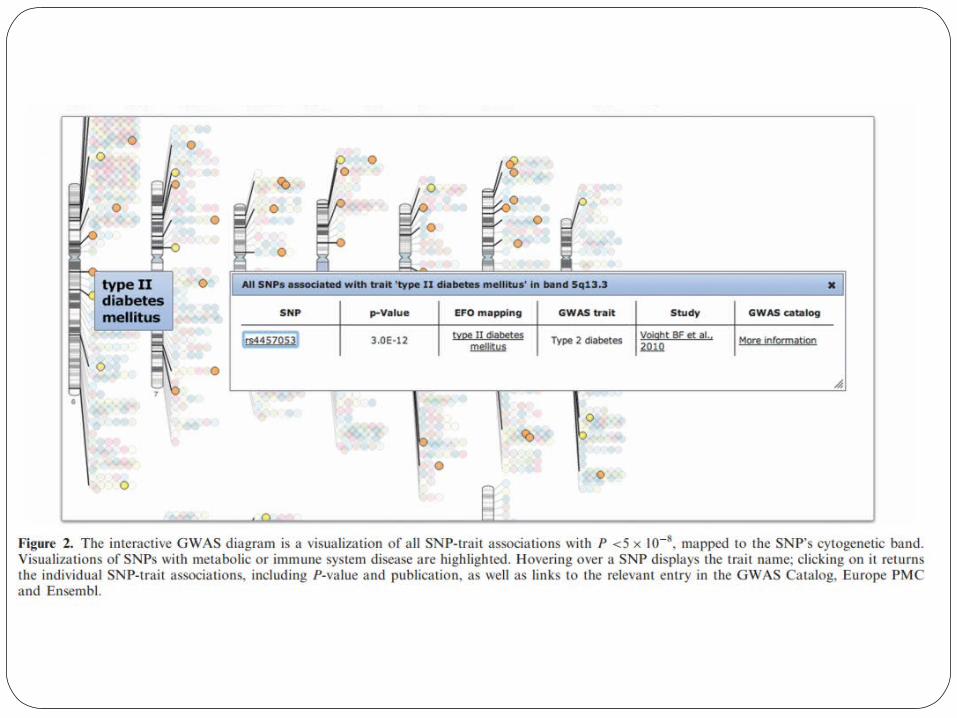
Published Genome-Wide Associations through 12/2013 Published GWA at p≤5X10-8 for 17 trait categories
NHGRI GWA Catalog Genome-wide association studies (GWAS) assay at minimum hundreds of thousands of single-nucleotide polymorphisms (SNPs) to identify associations with complex clinical conditions and phenotypic traits
Related Documents











