Genetically Altered Mutant Mouse Models of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Exhibit the Cardiac Expression of Proinflammatory Mediators in a Gene-Dose-Dependent Manner Elangovan Vellaichamy, Subhankar Das, Umadevi Subramanian, Nobuyo Maeda, and Kailash N. Pandey Department of Physiology Health Sciences Center (E.V., S.D., U.S., K.N.P.), Tulane University School of Medicine, New Orleans, Louisiana 70112; and Department of Pathology and Laboratory of Medicine (N.M.), University of North Carolina, Chapel Hill, North Carolina 27516 The objective of this study was to examine whether genetically determined differences in the guanylyl cyclase/natriuretic peptide receptor-A gene (Npr1) affect cardiac expression of proin- flammatory cytokines, hypertrophic markers, nuclear factor-B (NF-B), and activating protein-1 (AP-1) in am Npr1 gene-dose– dependent manner. In the present studies, adult male Npr1 gene- disrupted (Npr1 / ), wild-type (Npr1 / ), and gene-duplicated (Npr1 / ) mice were used. The Npr1 / mice showed 41 mm Hg higher systolic blood pressure and 60% greater heart weight to body weight (HW/BW) ratio; however, Npr1 / mice exhibited 15 mm Hg lower systolic blood pressure and 12% reduced HW/BW ratio compared with Npr1 / mice. Significant upregulation of gene expression of proinflammatory cytokines and hypertrophic markers along with enhanced NF-B/AP-1 binding activities were observed in the Npr1 / mouse hearts. Conversely, hypertrophic markers and proinflammatory cytokines gene expression as well as NF-B/AP-1 binding activities were markedly decreased in Npr1 / mouse hearts compared with wild-type mice. The ventric- ular guanylyl cyclase activity and cGMP levels were reduced by 96% and 87%, respectively, in Npr1 / mice; however, these parameters were amplified by 2.8-fold and 3.8-fold, respectively, in Npr1 / mice. Echocardiographic analysis revealed significantly increased fractional shortening in Npr1 / mice (P .05) but greatly decreased in Npr1 / mice (P .01) hearts compared with Npr1 / mice. The present findings suggest that Npr1 represses the expression of cardiac proin- flammatory mediators, hypertrophic markers, and NF-B/AP-1–mediated mechanisms, which seem to be associated in an Npr1 gene-dose– dependent manner. (Endocrinology 155: 1045–1056, 2014) A trial natriuretic peptide (ANP) elicits natriuretic, di- uretic, vasorelaxant, and antiproliferative responses and thereby lowers blood pressure (BP) and blood volume (1–5). ANP binds to guanylyl cyclase-A/natriuretic pep- tide receptor-A (GC-A/NPRA), which is considered the principal natriuretic peptide hormone receptor that syn- thesizes the intracellular second-messenger cGMP (6 – 8). Recent investigations have suggested that GC-A/NPRA signaling is involved not only in maintaining BP homeo- stasis but also in antagonizing cardiac growth responses to hypertrophic stimuli (9 –12). Mice carrying targeted dis- ruption of the Npr1 gene (coding for GC-A/NPRA) de- velop hypertension and congestive heart failure (13, 14). On the other hand, targeted gene duplication of Npr1 reduces BP, attenuates hypertrophic agonist-induced myocyte growth, and lowers cardiac angiotensin II (Ang II) and aldosterone levels (15–17). ANP gene delivery at- tenuates cardiac hypertrophy in spontaneously hyperten- ISSN Print 0013-7227 ISSN Online 1945-7170 Printed in U.S.A. Copyright © 2014 by the Endocrine Society Received May 6, 2013. Accepted December 20, 2013. First Published Online January 3, 2014 Abbreviations: Ang II, angiotensin II; ANP, trial natriuretic peptide; AP-1, activating pro- tein-1; BP, blood pressure; C T , threshold cycle; DTT, dithiothreitol; GC-A/NPRA, guanylyl cyclase-A/natriuretic peptide receptor-A; HW/BW, heart weight to body weight; LT-, lymphotoxin-; LV, left ventricular; -MHC, -myosin heavy chain; MIF, macrophage in- hibitory factor; NF, nuclear factor; PMSF, phenylmethylsulfonyl fluoride; SBP, systolic BP; SERCA-2a, sarcolemal endoplasmic reticulum Ca 2 ATPase-2. RENAL-CARDIAC-VASCULAR doi: 10.1210/en.2013-1416 Endocrinology, March 2014, 155(3):1045–1056 endo.endojournals.org 1045 The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Genetically Altered Mutant Mouse Models ofGuanylyl Cyclase/Natriuretic Peptide Receptor-AExhibit the Cardiac Expression of ProinflammatoryMediators in a Gene-Dose-Dependent Manner
Elangovan Vellaichamy, Subhankar Das, Umadevi Subramanian, Nobuyo Maeda,and Kailash N. Pandey
Department of Physiology Health Sciences Center (E.V., S.D., U.S., K.N.P.), Tulane University School ofMedicine, New Orleans, Louisiana 70112; and Department of Pathology and Laboratory of Medicine(N.M.), University of North Carolina, Chapel Hill, North Carolina 27516
The objective of this study was to examine whether genetically determined differences in theguanylyl cyclase/natriuretic peptide receptor-A gene (Npr1) affect cardiac expression of proin-flammatory cytokines, hypertrophic markers, nuclear factor-�B (NF-�B), and activating protein-1(AP-1) in am Npr1 gene-dose–dependent manner. In the present studies, adult male Npr1 gene-disrupted (Npr1�/�), wild-type (Npr1�/�), and gene-duplicated (Npr1��/��) mice were used. TheNpr1�/� mice showed 41 mm Hg higher systolic blood pressure and 60% greater heart weight tobody weight (HW/BW) ratio; however, Npr1��/�� mice exhibited 15 mm Hg lower systolic bloodpressure and 12% reduced HW/BW ratio compared with Npr1�/� mice. Significant upregulation ofgene expression of proinflammatory cytokines and hypertrophic markers along with enhancedNF-�B/AP-1 binding activities were observed in the Npr1�/� mouse hearts. Conversely, hypertrophicmarkers and proinflammatory cytokines gene expression as well as NF-�B/AP-1 binding activitieswere markedly decreased in Npr1��/�� mouse hearts compared with wild-type mice. The ventric-ular guanylyl cyclase activity and cGMP levels were reduced by 96% and 87%, respectively, inNpr1�/� mice; however, these parameters were amplified by 2.8-fold and 3.8-fold, respectively, inNpr1��/�� mice. Echocardiographic analysis revealed significantly increased fractional shorteningin Npr1��/�� mice (P � .05) but greatly decreased in Npr1�/� mice (P � .01) hearts compared withNpr1�/� mice. The present findings suggest that Npr1 represses the expression of cardiac proin-flammatory mediators, hypertrophic markers, and NF-�B/AP-1–mediated mechanisms, which seemto be associated in an Npr1 gene-dose–dependent manner. (Endocrinology 155: 1045–1056, 2014)
Atrial natriuretic peptide (ANP) elicits natriuretic, di-uretic, vasorelaxant, and antiproliferative responses
and thereby lowers blood pressure (BP) and blood volume(1–5). ANP binds to guanylyl cyclase-A/natriuretic pep-tide receptor-A (GC-A/NPRA), which is considered theprincipal natriuretic peptide hormone receptor that syn-thesizes the intracellular second-messenger cGMP (6–8).Recent investigations have suggested that GC-A/NPRAsignaling is involved not only in maintaining BP homeo-
stasis but also in antagonizing cardiac growth responses tohypertrophic stimuli (9–12). Mice carrying targeted dis-ruption of the Npr1 gene (coding for GC-A/NPRA) de-velop hypertension and congestive heart failure (13, 14).On the other hand, targeted gene duplication of Npr1reduces BP, attenuates hypertrophic agonist-inducedmyocyte growth, and lowers cardiac angiotensin II (AngII) and aldosterone levels (15–17). ANP gene delivery at-tenuates cardiac hypertrophy in spontaneously hyperten-
ISSN Print 0013-7227 ISSN Online 1945-7170Printed in U.S.A.Copyright © 2014 by the Endocrine SocietyReceived May 6, 2013. Accepted December 20, 2013.First Published Online January 3, 2014
Abbreviations: Ang II, angiotensin II; ANP, trial natriuretic peptide; AP-1, activating pro-tein-1; BP, blood pressure; CT, threshold cycle; DTT, dithiothreitol; GC-A/NPRA, guanylylcyclase-A/natriuretic peptide receptor-A; HW/BW, heart weight to body weight; LT-�,lymphotoxin-�; LV, left ventricular; �-MHC, �-myosin heavy chain; MIF, macrophage in-hibitory factor; NF, nuclear factor; PMSF, phenylmethylsulfonyl fluoride; SBP, systolic BP;SERCA-2a, sarcolemal endoplasmic reticulum Ca2� ATPase-2.
R E N A L - C A R D I A C - V A S C U L A R
doi: 10.1210/en.2013-1416 Endocrinology, March 2014, 155(3):1045–1056 endo.endojournals.org 1045
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

sive rats (18, 19). Furthermore, a significant positive as-sociation between the NPRA gene variants and both leftventricular (LV) mass index and LV septal wall thicknesswas found in patients with essential hypertension, sug-gesting that the ANP/NPRA system significantly contrib-utes to ventricular remodeling in human essential hyper-tension (20–24). Nevertheless, the cellular mechanisms bywhich the GC-A/NPRA system blocks hypertrophicgrowth and prevents cardiac events are not wellunderstood.
Several lines of evidence suggest that proinflammatorycytokines have a central pathophysiological function inthe development of endothelial dysfunction, cardiac hy-pertrophy, and heart failure in experimental animal mod-els and humans (4, 25–28). Elevated circulating and myo-cardial levels of inflammatory cytokines, includingTNF-�, IL-1�, and IL-6, have been reported in cardio-myopathic patients, with plasma levels correlating withdisease severity (27, 28). Evidence indicates that myocar-dial cells are capable of producing substantial amounts ofinflammatory cytokines in response to ischemia and ex-perimental load-induced stress (29, 30). The aforemen-tioned cytokines are reported to play a dual role, activat-ing apoptosis in myocytes while also functioningcytoprotectively (31). In addition, several agonists thatprovoke hypertrophic changes, including Ang II and nor-epinephrine, induce TNF-� and TGF-�1 expression inboth myocytes and non-muscle cells (32, 33). Further-more, inappropriate activation of TNF-�, IL-1�, and IL-6has been shown to induce myocardial effects similar to thephenotypic changes in heart failure, including myocytehypertrophy and induction of a fetal gene program, myo-cardial apoptosis, extracellular matrix alterations, andcontractile depression (34, 35).
Previous studies have suggested that the ANP/NPRAsystem acts as a negative regulator of inflammation andhypertrophic growth (11, 36, 37). ANP has been shown toinhibit TNF-� production in interferon (IFN)-�–activatedmacrophages and to suppress the TNF-�–induced adhe-sion molecule expression in endothelial cells. However, invivo studies on the function of NPRA signaling in regu-lating the expression of proinflammatory cytokines andstructural changes in heart function in an Npr1 gene-dose–dependent manner have been limited. In the present study,we have used Npr1 gene-disrupted (Npr1�/�), wild-type(Npr1�/�), and gene-duplicated (Npr1��/��) mice to ex-amine whether genetically determined differences in Npr1gene copy numbers affect the expression of cardiac pro-inflammatory cytokines, hypertrophic markers, activat-ing protein-1 (AP-1), and nuclear factor (NF)-�B alongwith the functional parameters in a gene-dose–dependentmanner.
Materials and Methods
MaterialsThe RNeasy mini-kit for total RNA isolation, RT2 First
Strand cDNA kit, and RT2 SYBR Green/ROX Master Mix wasobtained from QIAGEN. Sequence-specific oligonucleotideswere purchased from Eurofins MWG Operon. NF-�B/AP-1 oli-gonucleotides and antibodies of �-tubulin (sc-8035), c-fos (sc-52), gp130 (sc-656), histone HI (sc-8030), IL-6 (sc-1266), p-NF-�B/p65 (sc-101749), TGF-�1 (sc-146), TGF-�R1 (sc-398),TNF-� (sc-8301), and TNF-R1 (sc-1069) were obtained fromSanta Cruz Biotechnology. T4 polynucleotide kinase, proteinA-agarose, NAP-5 columns, and [�-32P]ATP (3000 Ci/mmol)were purchased from Amersham Biosciences. All other reagentsused were analytical grade.
Generation of Npr1 gene-targeted miceNpr1 gene-targeted mice were generated by homologous re-
combination in embryonic stem cells as previously described (13,15). Animals were bred and maintained at the Tulane UniversityHealth Sciences Center animal facility and handled according toprotocols approved by the Institutional Animal Care and UseCommittee. The mice were housed at 25°C in a 12-hour light,12-hour dark cycle and fed regular chow (Purina Laboratory)and tap water ad libitum. Npr1 mouse genotypes were littermateprogeny of C57BL6 genetic background; they have been desig-nated as follows: Npr1 gene-disrupted null mutant allele(Npr1�/�; 0-copy), wild-type allele (Npr1�/�; 2-copy), andgene-duplicated allele (Npr1��/��; 4-copy). In this study, weused adult (24–26 weeks) male 0-copy, 2-copy, and 4-copy mice.The breeding of 1-copy (�/�) heterozygous mice produced prog-eny consisting of 0-, 1-, and 2-copy mice. On the other hand, thebreeding of 3-copy heterozygous (��/�) mice generated 2-, 3-,and 4-copy animals. The mice were genotyped by PCR analysesof DNA isolated from tail biopsies as previously reported (14,38).
Analyses of BP, cardiac hypertrophy, and fibrosisSystolic BP (SBP) was measured by a noninvasive computer-
ized tail-cuff method as described previously (39). BPs were cal-culated as the average of 6 to 7 sessions per day for 6 consecutivedays. Animals were killed by cervical dislocation, and the heartswere removed and weighed. The heart weight to body weight(HW/BW) ratio was calculated as an index of cardiac hypertro-phy. Heart tissue sections were stained with Masson’s trichromefor the presence of perivascular collagen fiber accumulation as amarker of cardiac fibrosis. The ratio of perivascular fibrosis tothe total LV area was calculated from 20 randomly selected fieldsin 5 individual sections per heart, using image analysis software(Image-Pro Plus; Media Cybernetics).
Assessment of cardiac functionCardiac functions of Npr1�/�, Npr1�/�, and Npr1��/��
mice were analyzed using 2-dimensional echocardiography. An-imals were lightly sedated using 0.2 mL of Avertin (Aldrich) andwere evaluated using M-mode transthoracic views to measurethe LV dimensions, interventricular septal wall thickness, LVposterior wall thickness, and fractional shortening. Digitized M-mode images were obtained using an ultrasound system (ToshibaPower Vision) with a 7-mHz transducer at a sweep speed of 100
1046 Vellaichamy et al Functional Analyses of Npr1 Gene Copies Endocrinology, March 2014, 155(3):1045–1056
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

mm/s. For each measurement, 4 consecutive cardiac cycles weretraced and averaged.
Real-time RT-PCR analysesTotal RNA was extracted using the RNeasy plus mini-kit.
Heart tissues (30 mg) were homogenized, and the RNA wasextracted according to the manufacturer’s instructions. First-strand cDNA was synthesized from 1 �g of total RNA in a finalvolume of 20 �L using RT2 First Strand kit. Real-time RT-PCRwas performed using the Mx3000P real-time PCR system, anddata were analyzed with MxPro software (Stratagene) as previ-ously described (40, 41) The following primers (forward andreverse, respectively) primers were used: ANP, 5�-gcaggggccg-cacttagctc-3� and 5�-tcgtaggctccgagggccag-3�; c-fos, 5�-ccac-cgacctgcctgcaaga-3� and 5�-tggcttgggctcagggtcgt-3�; c-myc, 5�-tctggatcacctt ctgctgg-3� and 5�-cctcttgacatttctcctcgg-3�;�-myosin heavy chain (�-MHC), 5�-tccgcaaggtgcagcacgag-3�and 5�-cacggg cacccttggagctg-3�; sarcolemal endoplasmic retic-ulum Ca2� ATPase-2 (SERCA-2a), 5�-tcacaccgctgaatctgacc-3�and 5�-tgctaacaacgcacatgcac-3�; lymphotoxin-� (LT-�), 5�-ggtttgccaaagagaccttgc-3� and 5�-attacagtgccctctcccca-3�; TNF-�,5�-caacgccctcctggccaacg-3� and 5�-tcggggcagccttgtccctt-3�;IL-6, 5�-cacggccttccctacttcac-3� and 5�-tgcaagtgcatcatcgttgt-3�;TGF-�1, 5�-tacagggctttcgattcagc-3� and 5�-gtga gctgtgcaggtgct-3�; IFN-�, 5�-agacaatca ggccatcagca-3� and 5�-tggacctgtgggttgt-tgac-3�; and macrophage inhibitory factor (MIF), 5�-ttccac-cttcgcttgagtcc-3� and 5�-atttct cccggctggaaggt-3�; IL-10, 5�-aggcgctgtcatcgatttct-3� and 5�-atggccttgtagacaccttgg-3�; andglyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5�-tc-cctcaagattgtcagcaa-3� and 5�-agatccacaaacggatacatt-3�. PCRamplifications (in triplicates) were carried out in a 20-�L reac-tion volume using RT2 real-time SYBR Green/ROX PCR MasterMix. The reaction conditions were 95°C for 10 minutes followedby 45 cycles at 95°C for 15 seconds and 60°C for 1 minute andfinally 1 cycle at 95°C for 1 minute, 55°C for 30 seconds, and95°C for 30 seconds for the dissociation curve. The reactionmixture without template cDNA was used as negative control.Threshold cycle (CT) numbers were determined with MxProQPCR Software and transformed using the �CT comparativemethod. The mRNA expression was normalized to expressionvalues of GAPDH endogenous control within each sample andrelative to positive and negative controls. The level of gene ex-pression was determined by the comparative CT method (��CT).
Ventricular GC activity and cGMP assayVentricular plasma membranes were prepared as previously
described (42). GC activity was assayed according to Leitman etal (43) with some modification (38, 42). An aliquot of plasmamembrane (25 �g) was added to a 100-�L reaction mixturecontaining 50mM Tris-HCl buffer (pH 7.6), 4mM MnCl2,0.2mM GTP, 1 mg/mL BSA, 7.5mM creatine phosphate, 2mM3-isobutyl-1-methylxanthine, and 3 U creatine phosphokinase.The reaction mixture was incubated at 37°C, and the reactionwas stopped by the addition of 900 �L sodium acetate buffer(50mM, pH 6.2) and boiling the samples for 3 minutes. Theamount of cGMP generated was determined with a direct cGMPenzyme immunoassay kit (Assay Designs). To determine the in-tracellular cGMP content, frozen ventricular tissue samples werehomogenized in 10 vol 0.1M HCl containing 1% Triton X-100.Homogenate was heated at 95°C for 5 minutes and centrifuged
at 600g for 20 minutes at 22°C, after which the supernatantswere collected and cGMP was assayed using a direct cGMP im-munoassay kit.
Western blot analysis of cytokines and receptorsLV tissue homogenate (20 �g proteins) was mixed with sam-
ple loading buffer and separated under reducing conditions using10% SDS-PAGE. The separated proteins were electrotransferredonto a polyvinylidene difluoride membrane. The membrane wasblocked with 1� Tris-buffered saline-Tween 20 (25mM Tris,500mM NaCl, and 0.05% Tween 20 [pH 7.5]) containing 5%fat-free milk and then incubated overnight in Tris-buffered sa-line-Tween 20 containing 3% fat-free milk at 4°C with specificprimary antibodies to TNF-�, IL-6, TGF-�1, and �-tubulin. An-tibodies for IL-6 and TNF-� were used at a dilution of 1:1000;those for TGF-� were used at a dilution of 1:2000. Antibodies forcytokine receptor glycoprotein 130 (gp130), TNF-�1 receptor,and TGF-�1 receptor were used at a dilution of 1:1000. Anti-bodies for p-65 (sc-114), c-fos (sc-52), and histone (sc-1086)were used at a dilution of 1:1000. The membrane was thentreated with corresponding secondary antirabbit or antimousehorseradish peroxidase-conjugated antibodies (1:20 000 dilu-tion). Protein bands were visualized using the ChemGlow West-ern blot detection reagent kit (Alpha Innotech).
Quantification of plasma and tissue levels ofcytokines
The concentration of proinflammatory cytokines, includingTNF-�, IL-6, IL-1�, and profibrotic cytokine TGF-�1, were de-termined in plasma and heart tissues by a multiplex bead arrayformat (Millipore), using a Bio-Plex instrument (Bio-Rad) ac-cording to the manufacturer’s guidelines as previously described(40). Spectrally addressed polystyrene beads coated with cyto-kine-specific monoclonal antibodies were used to capture thecytokine of interest. The instrument sorted out and measured thefluorescent signal from each bead by dual excitation.
Histopathology and immunohistochemistryHeart tissues were fixed in 4% paraformaldehyde solution,
after which 5-�m-thick paraffin-embedded tissue sections werecut and stained with hematoxylin and eosin. Sections were ex-amined with Image-Pro Plus software (Media Cybernetics, Inc)to determine the myocyte cross-sectional area and vascular wallthickening. For immunohistochemistry, heart sections were per-fused without fixation. The tissues were immediately embeddedin tissue-freezing medium (O.C.T. compound; Marivac), frozen,and cut into 5-�m-thick slices. The sections were preincubatedwith blocking rabbit serum for 20 minutes and then treated for90 minutes with specific primary NF-�B subunit-p65-rabbitpolyclonal antibody diluted in PBS containing 1% BSA. Thesections were washed and incubated with secondary biotin-con-jugated rabbit antimouse IgG for 30 minutes, after which per-oxidase activity was visualized using the ABC Vectastain kit(Vector Laboratories). The slides were then counterstained withhematoxylin and mounted. To determine the infiltration of in-flammatory cells, sections were stained with toluidine blue toidentify the monocytes or mast cells. Toluidine blue is a basicthiazine metachromatic dye that which has high affinity for mastcell granular glycosaminoglycans and displays a purple metach-romatic color. The NF-�B–positive cells and monocyte-positive
doi: 10.1210/en.2013-1416 endo.endojournals.org 1047
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

cells were calculated from 20 randomly selected microscopicfields of 5 individual sections from heart using Image Pro plusimage analysis software.
Preparation of cytosolic and nuclear extractsNuclear and cytosolic proteins were extracted from the LV
tissues of Npr1�/�, Npr1�/�, and Npr1��/�� mice as previ-ously described (44). Tissues were homogenized in an ice-cold10mM Tris-HCl buffer (pH 8.0) containing 0.32M sucrose,3mM CaCl2, 2mM magnesium acetate, 0.1mM EDTA, 0.5%Nonidet P-40, 1mM dithiothreitol (DTT), 0.5mM phenylmeth-ylsulfonyl fluoride (PMSF), and 4.0 �g/mL each of leupeptin,aprotinin, and pepstatin. The homogenate was centrifuged at800g, and the supernatant was separated and saved as a cytosolicfraction. The nuclear fraction was resuspended in a low-salt buf-fer containing 20mM HEPES (pH 7.9), 1.5mM MgCl2, 20mMKCl, 0.2mM EDTA, 25% glycerol, 0.5mM DTT, and 0.5mMPMSF; incubated on ice for 5 minutes; and mixed with an equalvolume of high-salt buffer containing 20mM HEPES, 1.5mMMgCl2, 800mM KCl, 0.2mM EDTA, 25% glycerol, 1% NonidetP-40, 0.5mM DTT, 0.5mM PMSF, and 4.0 �g/mL each of leu-peptin, aprotinin, and pepstatin. The mixture was incubated onice for 30 minutes and centrifuged at 14 000g for 15 minutes.The supernatant was separated and stored at �80°C until used.
Electrophoretic mobility shift assayEMSA was performed as described previously (14). Double-
stranded oligonucleotides containing the consensus binding sitefor NF-�B were end-labeled using [�-32P]ATP and T4 polynu-cleotide kinase. A binding reaction was initiated by incubating 5�g of nuclear proteins in 5 �L of binding buffer containing50mM Tris-HCl (pH 8.0), 750mM KCl, 2.5mM EDTA, 0.5%Triton X-100, 62.5% glycerol, and 1mM DTT containing 2 �gof poly deoxyinosinic-deoxycytidylic acid (dI-dC) and radiola-beled oligonucleotide (50 000 cpm) at 22°C for 20 minutes. Coldcompetitor assays were performed by adding 100-fold excessmolar concentrations of unlabeled NF-�B. The DNA-proteincomplex was resolved using 4% native polyacrylamide gel elec-trophoresis and autoradiography.
Statistical analysisThe statistical significance was evaluated by two-way
ANOVA, followed by Bonferroni post hoc multiple-comparisontests using GraphPad Prism version 5 software. The results arepresented as mean � SEM. A P value � .05 was consideredsignificant.
Results
The results demonstrate that the LV chamber volume wasgreatly reduced in the Npr1�/� mouse hearts as comparedwith age-matched Npr1�/� and Npr1 ��/�� mouse hearts(Figure 1 A). The individual myocyte cross-sectional areawas significantly increased in null mutant mice (Npr1�/�,785 � 62) and reduced in gene-duplicated mice; (Npr1��/
��, 250 � 15) compared with wild-type mice (Npr1�/�,402 � 23) (Figure 1, B and C). The gene-disrupted
Npr1�/� mice displayed significantly increased vascularwall remodeling, including increased vascular wall thick-ening and perivascular fibrosis, as compared with theirwild-type and gene-duplicated counterparts (Figure 2 A,panels a–d). Masson’s trichrome staining showed in-creased perivascular fibrosis in the vascular media ofNpr1-null mutant mouse hearts as compared with wild-type and gene-duplicated mouse hearts (Figure 2A, panelb). The immunoreactivity of NF-�B (p65) was greatly in-creased in the vascular media of the Npr1�/� mousehearts, whereas it was markedly decreased in the vascularmedia of Npr1�/� and Npr1��/�� mouse hearts (Figure2A, panel c). Toluidine blue staining of infiltrating mastand monocyte cells showed that these cells were increasedin the arteries of Npr1 gene-disrupted mice; however, bothwild-type and gene-duplicated mice exhibited signifi-cantly reduced levels of these cells (Figure 2A, panel d).The percentages of perivascular fibrosis, the wall-to-lu-
A
Npr1-/- Npr1+/+ Npr1++/++B
C
-/- +/+ ++/++
Npr1-/- Npr1+/+ Npr1++/++
Myo
cyte
Cro
ss
Sect
iona
l Are
a (μ
m2 )
Adult Npr1
Figure 1. Comparative analysis of LV chamber volume (hypertrophicgrowth) and myocyte cross-sectional area in adult Npr1�/�, Npr1�/�,and Npr1��/�� mouse hearts. A, Representative heart sections ofadult mice. LV chamber volume was reduced in the Npr1-null mutantmouse hearts as compared with age-matched wild-type mouse hearts.B, Representative heart sections of Npr1 mice showing the myocytecross-sectional area of individual myocytes. C, Myocyte cross-sectionalarea analysis of individual myocytes in Npr1 mouse hearts. Values areexpressed as means � SEM; n 8 mice per group. *, P � .05;***, P � .001, Npr1�/� vs Npr1�/� and Npr1�/� vs Npr1��/��.
1048 Vellaichamy et al Functional Analyses of Npr1 Gene Copies Endocrinology, March 2014, 155(3):1045–1056
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

men ratios, the densitometric analyses of immunoreactiv-ity of NF-�B, and the average mast- and monocyte-posi-tive cell infiltration per section are presented in Figure 2 B(panels a–d). The Npr1�/� mice displayed an increasedwall-to-lumen ratio and NF-�B protein immunoreactivityin the vascular walls with less infiltration of mast or mono-cyte cells as compared with the hearts of wild-type mice.
In contrast, significant reductionswere observed in myocyte cross-sec-tional area, wall to lumen ratio, andNF-�B protein immunoreactivity ingene-duplicated mice as comparedwith wild-type mice.
The data presented in Table 1show that Npr1�/� mice exhibited41 mm Hg higher SBP, whereas gene-duplicated Npr1��/�� mice showed15 mm Hg lower SBP than age-matched Npr1�/� mice. TheHW/BW ratio (indicative of hyper-trophic growth) was increased by60% (decompensated hypertrophy)in adult Npr1�/� mice and reducedby 12% in Npr1��/�� mice com-pared with age-matched wild-typemice (6.79 � 0.63 vs 4.25 � 0.23,P � .01; 3.82 � 0.12 vs 4.25 � 0.23,P � .05). M-mode echocardio-graphic analysis revealed that adultNpr1�/� mice showed increased sep-tal wall thickness (P � .01) and pos-terior ventricular wall thickness (P �.001) and significantly elevated LVend diastolic and systolic dimensions(P � .01) as compared with Npr1�/�
mice. Functional parameters such asfractional shortening (P � .05) wassignificantly reduced in adultNpr1�/� mice; however, the gene-duplicated Npr1��/�� mice dis-played higher (P � .05) fractionalshortening (Table 1). The LV GC ac-tivity and the intracellular cGMPconcentrations were drastically re-duced, by almost 96% and 87%, re-spectively, in Npr1�/� mice as com-pared with Npr1�/� wild-type mice.On the other hand, Npr1��/�� miceexhibited increased ventricular GCactivity and cGMP levels by 2.8-foldand 3.8-fold, respectively, as com-pared with wild-type mice (Table 1).
The expression of various hypertrophic marker genes inthe heart tissues of gene-disrupted, wild-type, and gene-duplicated mice is presented in Figure 3 A. The mRNAexpression of hypertrophic markers such as ANP (5.4-fold), c-fos (3.7-fold), c-myc (3-fold), and �-MHC (4-fold)were markedly upregulated (P � .001) in Npr1�/� mutantmouse hearts compared with wild-type Npr1�/� mice.
A a b c d
Npr1-/-
Npr1+/+
Npr1++/++
B
Wal
l to
Lum
en r
atio
Npr1-/- Npr1+/+ Npr1++/++
Npr1-/- Npr1+/+ Npr1++/++ Npr1-/- Npr1+/+ Npr1++/++
Npr1-/- Npr1+/+ Npr1++/++
% N
F-κB
prot
ein
ratio
% P
eriv
ascu
lar
fibro
sis
% M
onoc
yte
posi
tive c
ells
/sec
tion
a) b)
d)c)
Figure 2. Comparative analyses of vascular remodeling, perivascular fibrosis,immunohistochemical analysis of NF-�B (p65 subunits) protein expression, and mast cell stainingin adult Npr1�/�, Npr1�/�, and Npr1��/�� mouse hearts. A, Representative heart sections ofNpr1�/�, Npr1�/�, and Npr1��/�� mice show coronary vessel wall thickening (panel a),perivascular fibrosis in the coronary vessels (panel b), NF-�B protein expression in the vessel walls(panel c), and the number of inflammatory mast and monocyte cells infiltrating coronary vessels(panel d). B, Analysis of wall-to-lumen ratio (panel a), percentage of fibrosis (panel b),quantitative analysis of NF-�B protein expression (panel c), and the average of the mast- andmonocyte-positive cell infiltration per section (panel d) in coronary vessels of mouse hearts.Values are expressed as means � SEM; n 8 mice per group. *, P � .05; **, P � .01; Npr1�/�
vs Npr1�/� and Npr1�/� vs Npr1��/��.
doi: 10.1210/en.2013-1416 endo.endojournals.org 1049
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

However, the expression of hypertrophic mRNA levelswas significantly suppressed (P � .01) in gene-duplicatedNpr1��/�� mouse hearts. In contrast, a decreased level ofSERCA-2a expression was observed in Npr1�/� mice,which was upregulated (3-fold; P � .01) in the heart tis-sues of 4-copy mice. The expression levels of proinflam-matory cytokines, including LT-� (3.5-fold), TNF-� (4.0-fold), IL-6 (5.3-fold), TGF-�1 (4-fold), IFN-� (2.7-fold),and MIF (2-fold) were markedly increased in Npr1�/�
mutant mouse hearts and significantly suppressed (P �.05) in Npr1��/�� gene-duplicated mouse hearts com-pared with age-matched wild-type littermates. ThemRNA expression levels of inhibitory cytokine IL-10 werereduced by 4-fold in 0-copy mice but increased by almost2.0-fold (P � .05) in 4-copy mice compared with 2-copywild-type mouse hearts (Figure 3B).
We determined both plasma and cardiac proinflamma-tory cytokines, including TNF-�, IL-6, IL-1�, and profi-brotic cytokine TGF-�1 in Npr1 gene-disrupted, wild-type, and gene-duplicated mice. As shown in Figure 4A,plasma TNF-� levels were increased significantly inNpr1�/� (0-copy) mice by 3.4-fold (28.45 � 2.81 pg/mL;P � .001) and decreased by 67% (2.79 � 0.87 pg/mL; P �.05) when compared with 2-copy (8.42 � 1.41 pg/mL)mice. Plasma IL-6 was also increased in 0-copy mice com-pared with 2-copy mice (2.9-fold; 63.13 � 6.15 vs 21.74 �2.77 pg/mL; P � .001). On the other hand, Npr1��/��
(4-copy) mice showed a reduced level of plasma IL-6(40%; 13.02 � 1.47 pg/mL). Similarly, plasma IL-1� andTGF-�1 levels were increased in 0-copy mice by 2.4-fold(30.21 � 2.70 pg/mL; P � .001) and 3.4-fold (52.66 �3.92 pg/mL; P � .001), respectively, compared with2-copy mice. However, 4-copy mice exhibited a markedlyreduced levels of plasma IL-1� (6.17 � 1.14; P � .05) andTGF-�1 (4.39 � 1.10; P � .05) than 2-copy mice. Asshown in Figure 4 B, cardiac TNF-�, IL-6, IL-1�, andTGF-�1 levels were greatly (P � .001) elevated in 0-copymice (4.4-fold, 60.34 � 4.11 pg/mg protein; 2.8-fold,80.91 � 5.50 pg/mg protein; 3.7-fold, 73.86 � 9.17 pg/mgprotein; and 3.4-fold, 152.80 � 19.86 pg/mg protein)compared with 2-copy mice. In contrast, 4-copy miceshowed a significantly reduced levels of cardiac TNF-�(80.2%, 2.72 � 0.80 pg/mg protein), IL-6 (70.6%, 8.35 �2.27 pg/mg protein), IL-1� (68.2%, 6.29 � 1.33 pg/mgprotein), and TGF-�1 (75%, 11.19 � 2.03 pg/mg protein)
ANPc-f
osc-m
yc-M
HC
β SERCA20
2
4
6 Npr1-/-Npr1+/+Npr1++/++
***
***
***
***
**** ** *** **
***
Fold
Incr
ease
A
B
βLT- α
TNF- IL-6 1β
TGF-γ
IFN-
MIF
IL-10
0
2
4
6Npr1-/-Npr1+/+Npr1++/++
*****
***
***
**
*
*** ** ** ** ** *
**Fo
ld In
crea
se
Figure 3. Gene expression profiles of hypertrophy markers andproinflammatory cytokines in adult Npr1�/�, Npr1�/�, and Npr1��/��
mouse hearts. A, Analysis of hypertrophic marker genes by real-timequantitative RT-PCR in the heart tissue of gene-disrupted (Npr1�/�),wild-type (Npr1�/�), and gene-duplicated (Npr1��/��) mice. B, Realtime quantitative RT-PCR analysis of proinflammatory andnoninflammatory cytokine gene expression in the heart tissues ofgene-disrupted (Npr1�/�), wild-type (Npr1�/�), and gene-duplicated(Npr1��/��) mice. Bars represent the means � SEM; n 8 mice pergroup. *, P � .05; ***, P � .001, Npr1�/� vs Npr1�/� and Npr1�/� vsNpr1��/��.
Table 1. Comparisons of SBP, Cardiac FunctionalAnalyses, and GC Activity in Adult Npr1�/�, Npr1�/�,and Npr1��/�� Mouse Heartsa
Parameters Npr1�/� Npr1�/� Npr1��/��
SBP, mm Hg 136 � 4b 95 � 5 80 � 3d
HW/BW ratio 6.79 � 0.63b 4.25 � 0.23 3. 82 � 0.12d
LVEDS, mm 2.84 � 0.05b 2.22 � 0.04 2.18 � 0.03LVEDD, mm 4.42 � 0.07c 3.95 � 0.05 3.63 � 0.42IVSTD, mm 0.97 � 0.07b 0.66 � 0.02 0.62 � 0.02PWT, mm 1.32 � 0.05c 0.95 � 0.04 0.85 � 0.03FS, % 34 � 0.07b 48 � 2.5 56 � 2.2d
cGMP, pmol/mgprotein
5 � 1.2c 36 � 5.1 140 � 12.9e
GC activity,pmol/mgprotein/min
4 � 2.2c 93 � 5.2 262 � 16e
Abbreviations: FS, fractional shortening; IVSTD, interventricular septalwall thickness (diastolic); LVEDD, LV end diastolic dimension; LVEDS,LV end systolic dimension; PWT, posterior ventricular wall thickness(systolic).a Systolic tail-cuff blood pressure, cardiac structure and functionanalysis, cGMP concentration, and GC activity assays were performedas described in Materials and Methods. Digitized M-mode images wereobtained using an ultrasound system (Toshiba Power Vision) with a7-mHz transducer at a sweep speed of 100 mm/s. Values areexpressed as means � SEM (n 8 animals per group).b,c Npr1�/� vs Npr1�/�: b P � 0.01; c P � 0.001.d,e Npr1�/� vs Npr1��/��: d P � 0.05; e P � 0.001.
1050 Vellaichamy et al Functional Analyses of Npr1 Gene Copies Endocrinology, March 2014, 155(3):1045–1056
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.
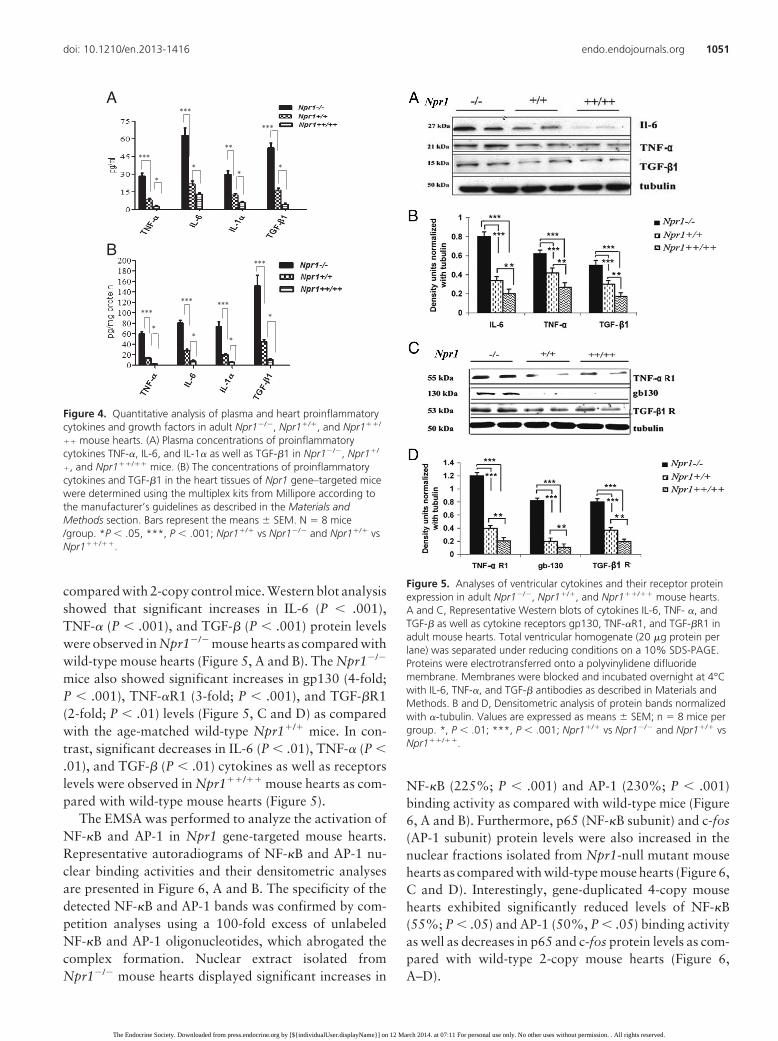
compared with 2-copy control mice. Western blot analysisshowed that significant increases in IL-6 (P � .001),TNF-� (P � .001), and TGF-� (P � .001) protein levelswere observed in Npr1�/� mouse hearts as compared withwild-type mouse hearts (Figure 5, A and B). The Npr1�/�
mice also showed significant increases in gp130 (4-fold;P � .001), TNF-�R1 (3-fold; P � .001), and TGF-�R1(2-fold; P � .01) levels (Figure 5, C and D) as comparedwith the age-matched wild-type Npr1�/� mice. In con-trast, significant decreases in IL-6 (P � .01), TNF-� (P �.01), and TGF-� (P � .01) cytokines as well as receptorslevels were observed in Npr1��/�� mouse hearts as com-pared with wild-type mouse hearts (Figure 5).
The EMSA was performed to analyze the activation ofNF-�B and AP-1 in Npr1 gene-targeted mouse hearts.Representative autoradiograms of NF-�B and AP-1 nu-clear binding activities and their densitometric analysesare presented in Figure 6, A and B. The specificity of thedetected NF-�B and AP-1 bands was confirmed by com-petition analyses using a 100-fold excess of unlabeledNF-�B and AP-1 oligonucleotides, which abrogated thecomplex formation. Nuclear extract isolated fromNpr1�/� mouse hearts displayed significant increases in
NF-�B (225%; P � .001) and AP-1 (230%; P � .001)binding activity as compared with wild-type mice (Figure6, A and B). Furthermore, p65 (NF-�B subunit) and c-fos(AP-1 subunit) protein levels were also increased in thenuclear fractions isolated from Npr1-null mutant mousehearts as compared with wild-type mouse hearts (Figure 6,C and D). Interestingly, gene-duplicated 4-copy mousehearts exhibited significantly reduced levels of NF-�B(55%; P � .05) and AP-1 (50%, P � .05) binding activityas well as decreases in p65 and c-fos protein levels as com-pared with wild-type 2-copy mouse hearts (Figure 6,A–D).
B
***
*
***
**
*
***
**
***
*
***
*
***
*
***
*
A
Figure 4. Quantitative analysis of plasma and heart proinflammatorycytokines and growth factors in adult Npr1�/�, Npr1�/�, and Npr1��/
�� mouse hearts. (A) Plasma concentrations of proinflammatorycytokines TNF-�, IL-6, and IL-1� as well as TGF-�1 in Npr1�/�, Npr1�/
�, and Npr1��/�� mice. (B) The concentrations of proinflammatorycytokines and TGF-�1 in the heart tissues of Npr1 gene–targeted micewere determined using the multiplex kits from Millipore according tothe manufacturer’s guidelines as described in the Materials andMethods section. Bars represent the means � SEM. N 8 mice/group. *P � .05, ***, P � .001; Npr1�/� vs Npr1�/� and Npr1�/� vsNpr1��/��.
Figure 5. Analyses of ventricular cytokines and their receptor proteinexpression in adult Npr1�/�, Npr1�/�, and Npr1��/�� mouse hearts.A and C, Representative Western blots of cytokines IL-6, TNF- �, andTGF-� as well as cytokine receptors gp130, TNF-�R1, and TGF-�R1 inadult mouse hearts. Total ventricular homogenate (20 �g protein perlane) was separated under reducing conditions on a 10% SDS-PAGE.Proteins were electrotransferred onto a polyvinylidene difluoridemembrane. Membranes were blocked and incubated overnight at 4°Cwith IL-6, TNF-�, and TGF-� antibodies as described in Materials andMethods. B and D, Densitometric analysis of protein bands normalizedwith �-tubulin. Values are expressed as means � SEM; n 8 mice pergroup. *, P � .01; ***, P � .001; Npr1�/� vs Npr1�/� and Npr1�/� vsNpr1��/��.
doi: 10.1210/en.2013-1416 endo.endojournals.org 1051
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

Discussion
The results of the present study demonstrate that SBP andcardiac functional efficacy and structural parameters are
regulated in an Npr1 gene-dose–dependent manner. InNpr1 gene-disrupted mutant mice, the ventricular dilata-tion and functional shortening were greatly reduced andinflammatory cytokines along with NF-�B and AP-1 bind-ing activities were enhanced; however, in gene-duplicatedmouse hearts, the inflammatory pathways were signifi-cantly reduced and cardiac functional parameters weregreatly improved compared with wild-type mice. Evidenceindicates that inflammation is a key component in thepathogenesis of hypertension and cardiac diseases (26–30, 34, 45, 46). Our data demonstrate that ventricularmRNA expression of proinflammatory cytokines, includ-ing LT-�, IL-6, TNF-�, IFN-�, and MIF were significantlyupregulated in Npr1�/� mutant mice as compared withage-matched Npr1�/� wild-type mice. On the other hand,Npr1��/�� gene-duplicated mice exhibited significantlydecreased expression levels of these cytokines as comparedwith wild-type mice, suggesting that the ANP/NPRA sys-tem seems to play a critical role in regulating the expres-sion of proinflammatory cytokines in the heart in an Npr1gene-dose–dependent manner. Similarly, the expressionof profibrotic cytokines such as TGF-�1 was markedlyincreased in Npr1�/� mice but greatly reduced inNpr1��/�� mice compared with Npr1�/� mouse hearts.Previous reports have also suggested that sustained acti-vation of proinflammatory cytokines contributes to thepathological form of ventricular remodeling and heartfailure in experimental animals (17, 25, 26, 29, 30). Ourfindings indicate that an enhanced ANP/NPRA system canprotect the heart by inhibiting the ventricular expressionof proinflammatory cytokines in relation to increasingNpr1 gene copy numbers. The present results advance thenotion that theNPRA/cGMP signaling pathway can aug-ment the cardiac function leading to the mechanisms thatprevent extracellular remodeling, pathological hypertro-phy, and heart failure. In support of this hypothesis, recentevidence suggests that the ANP/NPRA signaling systemexerts anti-inflammatory activity and inhibits the bacte-rial toxins lipopolysaccharide- and IFN-�–induced proin-flammatory cytokines gene expression in macrophages bysuppressing the signal transduction pathway leading toNF-�B activation (4, 36).
The present findings indicate that the expression ofboth proinflammatory and profibrotic cytokines aregreatly induced in null mutant Npr1�/� mouse hearts butmarkedly reduced in gene-duplicated Npr1��/�� micecompared with wild-type Npr1�/� mouse hearts. Expres-sion of myocardial cytokine genes such as TNF-�, IL-1�,and IL-6 has been found to be elevated in patients withcompensated heart failure conditions. Moreover, it hasbeen suggested that increased cytokine gene expressionhas an adaptive role in the LV remodeling process (27, 47).
Figure 6. NF-�B and AP-1 binding activity in adult Npr1�/�, Npr1�/�,and Npr1��/�� mouse hearts. A, Representative autoradiograph ofNF-�B and AP-1 binding activity in the nuclear extract isolated fromadult mouse hearts. Double-stranded oligonucleotides containing theconsensus binding site for NF-�B and AP-1 were end-labeled with [�-32P]ATP using T4 polynucleotide kinase. Cold competitor assays weredone by adding 100-fold molar excess of unlabeled NF-�B and AP-1oligonucleotides. The DNA-protein complex was resolved from the freelabeled DNA by electrophoresis in 4% (wt/vol) native polyacrylamidegel and autoradiographed. B, Densitometric analysis of NF-�B and AP-1 binding activity. C, Western blots of p65 and c-fos protein levels inthe nuclear extract isolated from adult mouse heart. D, Densitometricanalysis of p65 and c-fos protein bands from C. Values are expressedas means � SEM; n 8 mice per group. *, P � .05; ***, P � .001,Npr1�/� vs Npr1�/� and Npr1�/� vs Npr1��/��.
1052 Vellaichamy et al Functional Analyses of Npr1 Gene Copies Endocrinology, March 2014, 155(3):1045–1056
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

Experimental induction of hemody-namic overload in the adult mamma-lian hearts has been shown to pro-voke a transient increase inproinflammatory cytokines (29, 30).Cardiac-specific overexpression ofIL-6 and TNF-� in mouse heart hasbeen shown to develop cardiac hy-pertrophy and ventricular dysfunc-tion similar to that seen in humans,suggesting that proinflammatory cy-tokines are critically involved in thecardiac remodeling (25, 31, 48). Thespecificity of the cytokines, includingIL-6, has already been demonstratedusing specific antibodies availablecommercially (49–52). Further-more, blocking the action of theaforementioned cytokines has beenshown to prevent hypertension, car-diac hypertrophy, and diastolic dys-function in experimental animalmodels (31, 53–56). In the presentstudies, both plasma and cardiac tis-sue concentrations of proinflamma-tory and profibrotic cytokines werequantified in Npr1 mouse geno-types, which strongly complementedthe analysis of cytokines at the genelevels. Our data indicate that thegene regulation of cytokines has in-deed affected the cytokine release aswell as their protein levels in the cir-culation in an Npr1 gene-dose–de-pendent manner.
Activation of proto-oncogenesand re-expression of fetal genes suchas c-fos, �-MHC, and skeletal actinin adult ventricular tissues is consid-ered to be a sensitive indicator of thepathological form of cardiac hyper-trophy (57, 58). In the present study,Npr1�/� mutant mice displayed sig-nificantly increased levels of c-fos, c-myc, and �-MHC mRNA expres-sion, whereas decreased level ofSERCA-2a mRNA expression wasobserved. On the contrary,Npr1��/�� gene-duplicated micedisplayed significantly suppressedlevels of c-fos, c-myc, and �-MHCmRNAs as compared with wild-type
GTP
cGMP PKG
Growth factorsTGF-β, TNF-α
IKK
p65655 p50
Transcription
AP-1p65 p5065 p5
Inflammatory cytokines
Hypertension/Ventricular remodeling/Cardiac Hypertrophy
Cardiac Myocyte
c-fos, c-jun
P-1
ff
A
GTP
cGMP PKG
Growth factorsTGF-β, TNF-α
IKK
p656555 p50
Transcription
AP-1p65 p50p65 p
Inflammatory cytokines
Hypertension/Ventricular remodeling/Cardiac Hypertrophy
Cardiac Myocyte
c-fos, c-jun
P 1
f
B
Figure 7. Schematic representation of cardiac remodeling in Npr1 gene-disrupted and gene-duplicated mice. A, Disruption of Npr1 leads to an unbalanced activation of NF-�B and AP-1 (c-fos and c-jun) signaling cascades that triggers the expression of proinflammatory cytokines andhypertrophic marker genes and thereby promotes specific molecular and structural changes inNpr1�/� mouse heart. As a result, medial thickening and perivascular fibrosis occurs, which inturn promotes and leads to cardiac remodeling, hypertrophy, and heart failure. B, Overexpressionof Npr1 leads to an increased synthesis of cGMP and subsequent activation of protein kinase G(PKG) in cardiomyocytes, in turn, inhibits NF-�B and AP-1 signaling cascades that suppresses theexpression of inflammatory cytokines and hypertrophic marker genes, thereby attenuatesabnormal vascular and cardiac remodeling and hypertrophic growth in Npr1��/�� mouse heart.IKK, inhibitory kappa B kinase.
doi: 10.1210/en.2013-1416 endo.endojournals.org 1053
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

mice, suggesting that the ANP/NPRA signaling greatly re-presses the hypertrophic genes, in parallel with the sup-pression of proinflammatory cytokines. Previous studieshave reported that cardiac myocytes become significantlylarger in Npr1�/� mutant mice at birth than at later ages,suggesting that hypertrophic genes are activated at anearly age, before the onset of high BP (14, 59). It is con-sidered that a decreased level of SERCA-2a is associatedwith impaired uptake of Ca2� into sarcoplasmic reticu-lum, which contributes to slowing of relaxation and isassociated with contractile dysfunction. Furthermore, ithas been shown that normalization of systolic BP did nothave any salutary effect in reversing hypertrophy and fi-brosis in Npr1�/� mice, suggesting that NPRA/cGMP sig-naling has a direct regulatory function in the expression ofearly genes involved in cardiac hypertrophic growth andfibrosis (37, 60). The present study provides evidence thatat least in part, parallel activation of both proinflamma-tory cytokines and hypertrophic genes is a leading cause ofhigh BP in progressive pathological hypertrophy and heartfailure in Npr1-null mutant mice; however, the expressionof these genes are greatly suppressed in Npr1 gene-dupli-cated mice. The GC activity and intracellular cGMP levelswere decreased by 96% and 87%, respectively in Npr1�/�
mice but increased by 2.8-fold and 3.8-fold, respectively,in gene-duplicated mice compared with wild-type mice.Recent studies have demonstrated that augmentation ofsecond messenger cGMP via chronic inhibition of cGMP-specific phosphodiesterase 5A attenuates load-inducedhypertrophy, fibrosis, and myocardial dysfunction inde-pendent of changes in load, suggesting that local NPRA/cGMP signaling antagonizes cardiac hypertrophic medi-ators (61, 62). Furthermore, ANP/NPRA/cGMP signalinghas been shown to oppose the deleterious cardiovasculareffects mediated by hypertrophic agonists such as Ang II,endothelin-1, and vascular endothelial growth factor (10,63, 64).
Significant increases occurred in cardiac NF-�B andAP-1 binding activities in null mutant mice, whereas thehearts of Npr1 gene-duplicated mice exhibited a signifi-cantly suppressed level of NF-�B and AP-1 binding activ-ity as compared with wild-type mice. It is implicated thatboth NF-�B and AP-1 binding activities were positivelycorrelated with the observed changes in expression of cy-tokine genes in Npr1�/� mice but negatively correlated inNpr1��/�� mouse hearts. Both NF-�B and AP-1 havebeen shown to regulate the expression of many genes thatare involved in inflammation, the immune response, viralinfection, and cell survival (65). Previous studies have alsoindicated that NF-�B and AP-1 mediate expression of var-ious cytokine genes as well as the development of cardiachypertrophy in vivo and in vitro (66, 67). It has been sug-
gested that ANP pretreatment improves the survival oflipopolysaccharide-treated mice by inhibiting TNF-�–de-pendent NF-�B activation (68). Interestingly, ANP andcGMP analogs have been reported to suppress NF-�B ac-tivation by inhibiting the phosphorylation of inhibitorykappa B � (I�B�) and its subsequent degradation (40, 69).ANP has also been reported to inhibit the agonist -inducedactivation of AP-1 (c-fos and c-jun) signaling pathways,implicating the inhibitory role of ANP/NPRA signaling inAP-1 and NF-�B transactivation (40, 64).
The comparative schematic representations of the ab-sence or enhanced activation of NPRA/cGMP signaling ingene-disrupted and gene-duplicated mouse hearts areshown in Figure 7, A and B, respectively. The disruptionof the NPRA/cGMP signaling pathway abolishes the localgrowth-modulating effects, leading to unbalanced en-hanced activation of NF-�B– and AP-1–mediated signal-ing cascades and thereby increases the activation of pro-inflammatory mediators. It is implicated that in theabsence of NPRA/cGMP signaling, the transcriptional ac-tivity of AP-1 (c-fos and c-jun) and NF-�B is increased,which leads to the expression of proinflammatory cyto-kine and hypertrophic marker gene expression, therebypromoting medial thickening and perivascular fibrosis,which in turn leads to cardiac remodeling, hypertrophy,and heart failure in Npr1-null mutant mice (Figure 7A).Conversely, overexpression of NPRA leads to increasedsynthesis of cGMP and subsequent activation of proteinkinase G-mediated downstream signaling, resulting in areduced activation and translocation of NF-�B and AP-1and then decreasing the expression levels of proinflam-matory cytokine and hypertrophic marker genes. It isimplicated that the decreased NF-�B and AP-1 signalingpathways attenuate the abnormal vascular and cardiacremodeling and hypertrophic growth in Npr1 gene-du-plicated mouse hearts (Figure 7B). Together, the presentresults demonstrate that an absence of NPRA/cGMPsignaling leads to the activation of proinflammatorycytokine gene expression and decreased cardiac func-tion, whereas an enhanced activation of ANP/NPRAsystem contributes toward the control of cardiac hy-pertrophy and remodeling in a Npr1 gene-dose– depen-dent manner.
Acknowledgments
We thank Ms Gevoni Bolden for excellent technical assistanceand Mrs Kamala Pandey for assistance during the preparation ofthis manuscript. We are indebted to Dr Oliver Smithies for pro-viding initial breeding pairs of Npr1 gene-targeted mice colonies.
1054 Vellaichamy et al Functional Analyses of Npr1 Gene Copies Endocrinology, March 2014, 155(3):1045–1056
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

Address all correspondence and requests for reprints to: DrKailash N. Pandey, Department of Physiology, SL-39, TulaneUniversity School of Medicine, 1430 Tulane Avenue, New Or-leans, LA 70112. E-mail: [email protected].
This work was supported by National Institutes of HealthGrant HL62147 and IDeA Program.
Disclosure Summary: The authors have nothing to disclose.
References
1. de Bold AJ. Atrial natriuretic factor: a hormone produced by theheart. Science. 1985;230(4727):767–770.
2. Garbers DL. Guanylyl cyclase receptors and their endocrine, para-crine, and autocrine ligands. Cell. 1992;71(1):1–4.
3. Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N EnglJ Med. 1998;339(5):321–328.
4. Pandey KN. The functional genomics of guanylyl cyclase/natriureticpeptide receptor-A: perspectives and paradigms. FEBS J. 2011;278(11):1792–1807.
5. Pandey KN. Biology of natriuretic peptides and their receptors. Pep-tides. 2005;26(6):901–932.
6. Drewett JG, Garbers DL. The family of guanylyl cyclase receptorsand their ligands. Endocr Rev. 1994;15(2):135–162.
7. Pandey KN, Singh S. Molecular cloning and expression of murineguanylate cyclase/atrial natriuretic factor receptor cDNA. J BiolChem. 1990;265(21):12342–12348.
8. Garg R, Oliver PM, Maeda N, Pandey KN. Genomic structure, or-ganization, and promoter region analysis of murine guanylyl cy-clase/atrial natriuretic peptide receptor-A gene. Gene. 2002;291(1–2):123–133.
9. Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS.Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit thegrowth-promoting effects of norepinephrine in cardiac myocytesand fibroblasts. J Clin Invest. 1998;101(4):812–818.
10. Pandey KN, Nguyen HT, Li M, Boyle JW. Natriuretic peptide re-ceptor-A negatively regulates mitogen-activated protein kinase andproliferation of mesangial cells: role of cGMP-dependent proteinkinase. Biochem Biophys Res Commun. 2000;271(2):374–379.
11. Ellmers LJ, Scott NJ, Piuhola J, et al. Npr1-regulated gene pathwayscontributing to cardiac hypertrophy and fibrosis. J Mol Endocrinol.2007;38(1–2):245–257.
12. Li P, Wang D, Lucas J, et al. Atrial natriuretic peptide inhibits trans-forming growth factor �-induced Smad signaling and myofibroblasttransformation in mouse cardiac fibroblasts. Circ Res. 2008;102(2):185–192.
13. Oliver PM, Fox JE, Kim R, et al. Hypertension, cardiac hypertrophy,and sudden death in mice lacking natriuretic peptide receptor A.Proc Natl Acad Sci U S A. 1997;94(26):14730–14735.
14. Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of theNF-�B/matrix metalloproteinase pathway in cardiac fibrosis of micelacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem.2005;280(19):19230–19242.
15. Oliver PM, John SW, Purdy KE, et al. Natriuretic peptide receptor1 expression influences blood pressures of mice in a dose-dependentmanner. Proc Natl Acad Sci U S A. 1998;95(5):2547–2551.
16. Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF.Expression of constitutively active guanylate cyclase in cardiomy-ocytes inhibits the hypertrophic effects of isoproterenol and aorticconstriction on mouse hearts. J Biol Chem. 2003;278(48):47694–47699.
17. Zhao D, Das S, Pandey KN. Interactive roles of NPR1 gene-dosageand salt diets on cardiac angiotensin II, aldosterone and pro-inflam-matory cytokines levels in mutant mice. J Hypertens. 2013;31(1):134–144.
18. He JG, Chen YL, Chen BL, et al. B-type natriuretic peptide atten-uates cardiac hypertrophy via the transforming growth factor-�1/smad7 pathway in vivo and in vitro. Clin Exp Pharmacol Physiol.2010;37(3):283–289.
19. Lin KF, Chao J, Chao L. Atrial natriuretic peptide gene deliveryattenuates hypertension, cardiac hypertrophy, and renal injury insalt-sensitive rats. Hum Gene Ther. 1998;9(10):1429–1438.
20. Knowles JW, Erickson LM, Guy VK, Sigel CS, Wilder JC, Maeda N.Common variations in noncoding regions of the human natriureticpeptide receptor A gene have quantitative effects. Hum Genet. 2003;112(1):62–70.
21. Rubattu S, Bigatti G, Evangelista A, et al. Association of atrial na-triuretic peptide and type a natriuretic peptide receptor gene poly-morphisms with left ventricular mass in human essential hyperten-sion. J Am Coll Cardiol. 2006;48(3):499–505.
22. Karayannis G, Tsezou A, Giannatou E, Papanikolaou V, GiamouzisG, Triposkiadis F. Polymorphisms of renin-angiotensin system andnatriuretic peptide receptor A genes in patients of Greek origin witha history of myocardial infarction. Angiology. 2010;61(8):737–743.
23. Newton-Cheh C, Larson MG, Vasan RS, et al. Association of com-mon variants in NPPA and NPPB with circulating natriuretic pep-tides and blood pressure. Nat Genet. 2009;41(3):348–353.
24. Del Greco MF, Pattaro C, Luchner A, et al. Genome-wide associa-tion analysis and fine mapping of NT-proBNP level provide novelinsight into the role of the MTHFR-CLCN6-NPPA-NPPB gene clus-ter. Hum Mol Genet. 2011;20(8):1660–1671.
25. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activationof gp130, a signal-transducing receptor component for interleukin6-related cytokines, causes myocardial hypertrophy in mice. ProcNatl Acad Sci U S A. 1995;92(11):4862–4866.
26. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated cir-culating levels of tumor necrosis factor in severe chronic heart fail-ure. N Engl J Med. 1990;323(4):236–241.
27. Testa M, Yeh M, Lee P, et al. Circulating levels of cytokines and theirendogenous modulators in patients with mild to severe congestiveheart failure due to coronary artery disease or hypertension. J AmColl Cardiol. 1996;28(4):964–971.
28. Vanderheyden M, Paulus WJ, Voss M, et al. Myocardial cytokinegene expression is higher in aortic stenosis than in idiopathic dilatedcardiomyopathy. Heart. 2005;91(7):926–931.
29. Baumgarten G, Knuefermann P, Kalra D, et al. Load-dependent and-independent regulation of proinflammatory cytokine and cytokinereceptor gene expression in the adult mammalian heart. Circulation.2002;105(18):2192–2197.
30. Palmieri EA, Benincasa G, Di Rella F, et al. Differential expressionof TNF-�, IL-6, and IGF-1 by graded mechanical stress in normal ratmyocardium. Am J Physiol Heart Circ Physiol. 2002;282(3):H926–H934.
31. Krown KA, Page MT, Nguyen C, et al. Tumor necrosis factor �-in-duced apoptosis in cardiac myocytes. Involvement of the sphingo-lipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98(12):2854–2865.
32. Sano M, Fukuda K, Kodama H, et al. Interleukin-6 family of cyto-kines mediate angiotensin II-induced cardiac hypertrophy in rodentcardiomyocytes. J Biol Chem. 2000;275(38):29717–29723.
33. Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. AngiotensinII stimulates cardiac myocyte hypertrophy via paracrine release ofTGF-� 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40(2):352–363.
34. Sekiguchi K, Li X, Coker M, et al. Cross-regulation between therenin-angiotensin system and inflammatory mediators in cardiachypertrophy and failure. Cardiovasc Res. 2004;63(3):433–442.
35. Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1�
modulates the growth and phenotype of neonatal rat cardiac myo-cytes. J Clin Invest. 1995;96(2):1093–1099.
36. Tsukagoshi H, Shimizu Y, Kawata T, et al. Atrial natriuretic peptide
doi: 10.1210/en.2013-1416 endo.endojournals.org 1055
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.

inhibits tumor necrosis factor-� production by interferon-�-acti-vated macrophages via suppression of p38 mitogen-activated pro-tein kinase and nuclear factor-�B activation. Regul Pept. 2001;99(1):21–29.
37. Vellaichamy E, Zhao D, Somanna N, Pandey KN. Genetic disrup-tion of guanylyl cyclase/natriuretic peptide receptor-A upregulatesACE and AT1 receptor gene expression and signaling: role in cardiachypertrophy. Physiol Genomics. 2007;31(2):193–202.
38. Pandey KN, Oliver PM, Maeda N, Smithies O. Hypertension asso-ciated with decreased testosterone levels in natriuretic peptide re-ceptor-A gene-knockout and gene-duplicated mutant mouse mod-els. Endocrinology. 1999;140(11):5112–5119.
39. Shi SJ, Vellaichamy E, Chin SY, Smithies O, Navar LG, Pandey KN.Natriuretic peptide receptor A mediates renal sodium excretory re-sponses to blood volume expansion. Am J Physiol Renal Physiol.2003;285(4):F694–F702.
40. Das S, Periyasamy R, Pandey KN. Activation of IKK/NF-�B pro-vokes renal inflammatory responses in guanylyl cyclase/natriureticpeptide receptor-A gene-knockout mice. Physiol Genomics. 2012;44(7):430–442.
41. Kumar P, Garg R, Bolden G, Pandey KN. Interactive roles of Ets-1,Sp1, and acetylated histones in the retinoic acid-dependent activa-tion of guanylyl cyclase/atrial natriuretic peptide receptor-A genetranscription. J Biol Chem. 2010;285(48):37521–37530.
42. Khurana ML, Pandey KN. Catalytic activation of guanylate cyclase/atrial natriuretic factor receptor by combined effects of ANF andGTP�S in plasma membranes of Leydig tumor cells: involvement ofG-proteins. Arch Biochem Biophys. 1995;316(1):392–398.
43. Leitman DC, Agnost VL, Tuan JJ, Andresen JW, Murad F. Atrialnatriuretic factor and sodium nitroprusside increase cyclic GMP incultured rat lung fibroblasts by activating different forms of guan-ylate cyclase. Biochem J. 1987;244(1):69–74.
44. Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription ini-tiation by RNA polymerase II in a soluble extract from isolatedmammalian nuclei. Nucleic acids research. 1983;11(5):1475–1489.
45. Prabhu SD. Cytokine-induced modulation of cardiac function. CircRes. 2004;95(12):1140–1153.
46. Das S, Au E, Krazit ST, Pandey KN. Targeted disruption of guanylylcyclase-A/natriuretic peptide receptor-A gene provokes renal fibro-sis and remodeling in null mutant mice: role of proinflammatorycytokines. Endocrinology. 2010;151(12):5841–5850.
47. Kong X, Wang X, Xu W, et al. Natriuretic peptide receptor a as anovel anticancer target. Cancer Res. 2008;68(1):249–256.
48. Oral H, Sivasubramanian N, Dyke DB, et al. Myocardial proin-flammatory cytokine expression and left ventricular remodeling inpatients with chronic mitral regurgitation. Circulation. 2003;107(6):831–837.
49. Chandrasekar B, Mitchell DH, Colston JT, Freeman GL. Regula-tion of CCAAT/enhancer binding protein, interleukin-6, interleu-kin-6 receptor, and gp130 expression during myocardial ischemia/reperfusion. Circulation. 1999;99(3):427–433.
50. Ghorbel MT, Sharman G, Leroux M, et al. Microarray analysisreveals interleukin-6 as a novel secretory product of the hypo-thalamo-neurohypophyseal system. J Biol Chem. 2003;278(21):19280–19285.
51. Lemke R, Härtig W, Rossner S, Bigl V, Schliebs R. Interleukin-6 isnot expressed in activated microglia and in reactive astrocytes inresponse to lesion of rat basal forebrain cholinergic system as dem-onstrated by combined in situ hybridization and immunocytochem-istry. J Neurosci Res. 1998;51(2):223–236.
52. Ma F, Li Y, Jia L, et al. Macrophage-stimulated cardiac fibroblastproduction of IL-6 is essential for TGF �/Smad activation and car-diac fibrosis induced by angiotensin II. PloS One. 2012;7(5):e35144.
53. Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokinegene expression after myocardial infarction in rat hearts: possibleimplication in left ventricular remodeling. Circulation. 1998;98(2):149–156.
54. Kurrelmeyer KM, Michael LH, Baumgarten G, et al. Endogenoustumor necrosis factor protects the adult cardiac myocyte againstischemic-induced apoptosis in a murine model of acute myocardialinfarction. Proc Natl Acad Sci U S A. 2000;97(10):5456–5461.
55. Li YY, Feng YQ, Kadokami T, et al. Myocardial extracellular ma-trix remodeling in transgenic mice overexpressing tumor necrosisfactor � can be modulated by anti-tumor necrosis factor � therapy.Proc Natl Acad Sci U S A. 2000;97(23):12746–12751.
56. Villarreal FJ, Dillmann WH. Cardiac hypertrophy-induced changesin mRNA levels for TGF-�1, fibronectin, and collagen. Am JPhysiol. 1992;262(6 Pt 2):H1861–H1866.
57. Morgan HE, Baker KM. Cardiac hypertrophy. Mechanical, neural,and endocrine dependence. Circulation. 1991;83(1):13–25.
58. Sadoshima J, Izumo S. The cellular and molecular response of car-diac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571.
59. Ellmers LJ, Knowles JW, Kim HS, Smithies O, Maeda N, CameronVA. Ventricular expression of natriuretic peptides in Npr1�/� micewith cardiac hypertrophy and fibrosis. Am J Physiol Heart CircPhysiol. 2002;283(2):H707–H714.
60. Knowles JW, Esposito G, Mao L, et al. Pressure-independent en-hancement of cardiac hypertrophy in natriuretic peptide receptorA-deficient mice. J Clin Invest. 2001;107(8):975–984.
61. Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclicGMP phosphodiesterase 5A prevents and reverses cardiac hyper-trophy. Nat Med. 2005;11(2):214–222.
62. Lee DI, Kass DA. Phosphodiesterases and cyclic GMP regulation inheart muscle. Physiology (Bethesda). 2012;27(4):248–258.
63. Kumar R, Cartledge WA, Lincoln TM, Pandey KN. Expression ofguanylyl cyclase-A/atrial natriuretic peptide receptor blocks the ac-tivation of protein kinase C in vascular smooth muscle cells. Role ofcGMP and cGMP-dependent protein kinase. Hypertension. 1997;29(1 Pt 2):414–421.
64. Tripathi S, Pandey KN. Guanylyl cyclase/natriuretic peptide recep-tor-A signaling antagonizes the vascular endothelial growth factor-stimulated MAPKs and downstream effectors AP-1 and CREB inmouse mesangial cells. Mol Cell Biochem. 2012;368(1–2):47–59.
65. Purcell NH, Molkentin JD. Is nuclear factor �B an attractive ther-apeutic target for treating cardiac hypertrophy? Circulation. 2003;108(6):638–640.
66. Frantz S, Fraccarollo D, Wagner H, et al. Sustained activation ofnuclear factor �B and activator protein 1 in chronic heart failure.Cardiovasc Res. 2003;57(3):749–756.
67. Li Y, Ha T, Gao X, et al. NF-�B activation is required for the de-velopment of cardiac hypertrophy in vivo. Am J Physiol Heart CircPhysiol. 2004;287(4):H1712–H1720.
68. Ladetzki-Baehs K, Keller M, Kiemer AK, et al. Atrial natriureticpeptide, a regulator of nuclear factor-�B activation in vivo. Endo-crinology. 2007;148(1):332–336.
69. Kiemer AK, Weber NC, Vollmar AM. Induction of I�B: atrial na-triuretic peptide as a regulator of the NF-�B pathway. BiochemBiophys Res Commun. 2002;295(5):1068–1076.
1056 Vellaichamy et al Functional Analyses of Npr1 Gene Copies Endocrinology, March 2014, 155(3):1045–1056
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 12 March 2014. at 07:11 For personal use only. No other uses without permission. . All rights reserved.
Related Documents











