Genetic inhibition of protein kinase Cε attenuates necrosis in experimental pancreatitis Yannan Liu, 1,2 Jingzhen Yuan, 1 Tanya Tan, 1,3 Wenzhuo Jia, 1,2 Aurelia Lugea, 1,4 Olga Mareninova, 1 Richard T. Waldron, 1,4 and Stephen J. Pandol 1,4 1 Veterans Affairs Greater Los Angeles Healthcare System, University of California at Los Angeles, and South California Research Center for Alcoholic Liver and Pancreatic Diseases, California; 2 Beijing Hospital, Beijing, China, 3 St. George’s University School of Medicine, St. George’s, Grenada; and 4 Cedars-Sinai Medical Center, Los Angeles, California Submitted 13 December 2013; accepted in final form 21 May 2014 Liu Y, Yuan J, Tan T, Jia W, Lugea A, Mareninova O, Waldron RT, Pandol SJ. Genetic inhibition of protein kinase Cε attenuates necrosis in experimental pancreatitis. Am J Physiol Gas- trointest Liver Physiol 307: G550 –G563, 2014. First published July 17, 2014; doi:10.1152/ajpgi.00432.2013.—Understanding the regula- tion of death pathways, necrosis and apoptosis, in pancreatitis is important for developing therapies directed to the molecular patho- genesis of the disease. Protein kinase Cε (PKCε) has been previously shown to regulate inflammatory responses and zymogen activation in pancreatitis. Furthermore, we demonstrated that ethanol specifically activated PKCε in pancreatic acinar cells and that PKCε mediated the sensitizing effects of ethanol on inflammatory response in pancreatitis. Here we investigated the role of PKCε in the regulation of death pathways in pancreatitis. We found that genetic deletion of PKCε resulted in decreased necrosis and severity in the in vivo cerulein- induced pancreatitis and that inhibition of PKCε protected the acinar cells from CCK-8 hyperstimulation-induced necrosis and ATP reduc- tion. These findings were associated with upregulation of mitochon- drial Bak and Bcl-2/Bcl-xL, proapoptotic and prosurvival members in the Bcl-2 family, respectively, as well as increased mitochondrial cytochrome c release, caspase activation, and apoptosis in pancreatitis in PKCε knockout mice. We further confirmed that cerulein pancre- atitis induced a dramatic mitochondrial translocation of PKCε, sug- gesting that PKCε regulated necrosis in pancreatitis via mechanisms involving mitochondria. Finally, we showed that PKCε deletion downregulated inhibitors of apoptosis proteins, c-IAP2, survivin, and c-FLIPs while promoting cleavage/inactivation of receptor-interacting protein kinase (RIP). Taken together, our findings provide evidence that PKCε activation during pancreatitis promotes necrosis through mechanisms involving mitochondrial proapoptotic and prosurvival Bcl-2 family proteins and upregulation of nonmitochondrial pathways that inhibit caspase activation and RIP cleavage/inactivation. Thus PKCε is a potential target for prevention and/or treatment of acute pancreatitis. apoptosis; necrosis; inhibitors of apoptotic proteins (IAPs), Bcl-2 family proteins; receptor-interacting protein kinase (RIP) PANCREATIC ACINAR CELL NECROSIS is a major pathological re- sponse of acute pancreatitis associated with significant mor- bidity and mortality from the disease (16, 39). Currently, there are no therapies directed to the molecular pathogenesis of necrosis in pancreatitis (39). Thus elucidating the molecular signals that mediate acinar cell necrosis is important for de- veloping new therapeutic strategies. Apoptosis and necrosis are two major forms of acinar cell death in acute pancreatitis and associated with specific mor- phological and biochemical features (16, 32). Apoptosis is a tightly regulated process involving the caspase family of cys- teine proteases and manifests as chromatin condensation, nu- clear shrinkage, and reduction in cell volume but with intact plasma membranes; thus there is no leakage of cellular con- tents to the extracellular environment. Conversely, necrosis leads to early plasma membrane permeabilization and rupture with release of intracellular constituents to the extracellular space and no sign of nuclear shrinkage (16, 39). Necrosis has been considered a passive, unregulated form of cell death; however, recent studies indicate that necrosis can occur in a tightly regulated fashion, called programmed necrosis (13, 20, 25, 32, 33, 42). Although the consequences of apoptosis and necrosis are distinct in pancreatitis, the mechanisms underlying these two types of cell death are interrelated and they both involve mitochondria (6, 7, 16, 21, 39, 48). Apoptosis is mediated by the release of mitochondrial cytochrome c into the cytosol through outer membrane permeabilization (OMP) followed by caspase activation, whereas necrosis is associ- ated with injury of inner membrane or opening of the mitochondrial permeability transition pore (mPTP) resulting in mitochondrial depolarization and subsequent ATP deple- tion (26, 48, 37, 56, 1, 34). Bcl-2 proteins are known important regulators of mitochon- drial permeabilization (1, 26). Proapoptotic Bax and Bak form channels in the outer membrane through which mitochondrial cytochrome c is released into the cytosol (1, 26); BH3-only proteins, such as Bim and Puma, activate Bax/Bak channels. In contrast, prosurvival Bcl-2 proteins such as Bcl-2 and Bcl-xL inhibit apoptosis by sequestering BH3-only proteins and Bax/ Bak (1, 26). Notably, the prosurvival Bcl-2 proteins can also stabilize inner membrane and block mPTP opening, thus main- taining energy production and preventing necrosis (52, 53). Our recent studies demonstrated that the predominant effect of Bcl-2/Bcl-xL proteins is to stabilize mitochondrial inner mem- brane integrity rather than to prevent OMP opening-caused cytochrome c release in pancreatitis (48). Thus the prosurvival Bcl-2 proteins are now recognized to play an important role in protection of acinar cells from necrosis by stabilizing mito- chondria against death signals. Inhibitors of apoptosis proteins (IAPs) are an important family of endogenous proteins that inhibit the caspase sys- tem, the essential mediators of apoptotic death pathways (11, 12). The importance of IAPs in regulating the type of death in pancreatitis has been reported by our group (32, 39, Address for reprint requests and other correspondence: J. Yuan, UCLA/VA Greater Los Angeles Healthcare System, West Los Angeles VA Healthcare Center, South California Research Center for Alcoholic Liver and Pancreatic Diseases, 11301 Wilshire Blvd., Bldg. 258, Rm. 340, Los Angeles, CA 90073 (e-mail: [email protected]). Am J Physiol Gastrointest Liver Physiol 307: G550–G563, 2014. First published July 17, 2014; doi:10.1152/ajpgi.00432.2013. 0193-1857/14 Copyright © 2014 the American Physiological Society http://www.ajpgi.org G550

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Genetic inhibition of protein kinase Cε attenuates necrosis in experimentalpancreatitis
Yannan Liu,1,2 Jingzhen Yuan,1 Tanya Tan,1,3 Wenzhuo Jia,1,2 Aurelia Lugea,1,4 Olga Mareninova,1
Richard T. Waldron,1,4 and Stephen J. Pandol1,4
1Veterans Affairs Greater Los Angeles Healthcare System, University of California at Los Angeles, and South CaliforniaResearch Center for Alcoholic Liver and Pancreatic Diseases, California; 2Beijing Hospital, Beijing, China, 3St. George’sUniversity School of Medicine, St. George’s, Grenada; and 4Cedars-Sinai Medical Center, Los Angeles, California
Submitted 13 December 2013; accepted in final form 21 May 2014
Liu Y, Yuan J, Tan T, Jia W, Lugea A, Mareninova O,Waldron RT, Pandol SJ. Genetic inhibition of protein kinase Cεattenuates necrosis in experimental pancreatitis. Am J Physiol Gas-trointest Liver Physiol 307: G550–G563, 2014. First published July17, 2014; doi:10.1152/ajpgi.00432.2013.—Understanding the regula-tion of death pathways, necrosis and apoptosis, in pancreatitis isimportant for developing therapies directed to the molecular patho-genesis of the disease. Protein kinase Cε (PKCε) has been previouslyshown to regulate inflammatory responses and zymogen activation inpancreatitis. Furthermore, we demonstrated that ethanol specificallyactivated PKCε in pancreatic acinar cells and that PKCε mediated thesensitizing effects of ethanol on inflammatory response in pancreatitis.Here we investigated the role of PKCε in the regulation of deathpathways in pancreatitis. We found that genetic deletion of PKCεresulted in decreased necrosis and severity in the in vivo cerulein-induced pancreatitis and that inhibition of PKCε protected the acinarcells from CCK-8 hyperstimulation-induced necrosis and ATP reduc-tion. These findings were associated with upregulation of mitochon-drial Bak and Bcl-2/Bcl-xL, proapoptotic and prosurvival members inthe Bcl-2 family, respectively, as well as increased mitochondrialcytochrome c release, caspase activation, and apoptosis in pancreatitisin PKCε knockout mice. We further confirmed that cerulein pancre-atitis induced a dramatic mitochondrial translocation of PKCε, sug-gesting that PKCε regulated necrosis in pancreatitis via mechanismsinvolving mitochondria. Finally, we showed that PKCε deletiondownregulated inhibitors of apoptosis proteins, c-IAP2, survivin, andc-FLIPs while promoting cleavage/inactivation of receptor-interactingprotein kinase (RIP). Taken together, our findings provide evidencethat PKCε activation during pancreatitis promotes necrosis throughmechanisms involving mitochondrial proapoptotic and prosurvivalBcl-2 family proteins and upregulation of nonmitochondrial pathwaysthat inhibit caspase activation and RIP cleavage/inactivation. ThusPKCε is a potential target for prevention and/or treatment of acutepancreatitis.
apoptosis; necrosis; inhibitors of apoptotic proteins (IAPs), Bcl-2family proteins; receptor-interacting protein kinase (RIP)
PANCREATIC ACINAR CELL NECROSIS is a major pathological re-sponse of acute pancreatitis associated with significant mor-bidity and mortality from the disease (16, 39). Currently, thereare no therapies directed to the molecular pathogenesis ofnecrosis in pancreatitis (39). Thus elucidating the molecularsignals that mediate acinar cell necrosis is important for de-veloping new therapeutic strategies.
Apoptosis and necrosis are two major forms of acinar celldeath in acute pancreatitis and associated with specific mor-phological and biochemical features (16, 32). Apoptosis is atightly regulated process involving the caspase family of cys-teine proteases and manifests as chromatin condensation, nu-clear shrinkage, and reduction in cell volume but with intactplasma membranes; thus there is no leakage of cellular con-tents to the extracellular environment. Conversely, necrosisleads to early plasma membrane permeabilization and rupturewith release of intracellular constituents to the extracellularspace and no sign of nuclear shrinkage (16, 39). Necrosis hasbeen considered a passive, unregulated form of cell death;however, recent studies indicate that necrosis can occur in atightly regulated fashion, called programmed necrosis (13, 20,25, 32, 33, 42).
Although the consequences of apoptosis and necrosis aredistinct in pancreatitis, the mechanisms underlying thesetwo types of cell death are interrelated and they both involvemitochondria (6, 7, 16, 21, 39, 48). Apoptosis is mediatedby the release of mitochondrial cytochrome c into thecytosol through outer membrane permeabilization (OMP)followed by caspase activation, whereas necrosis is associ-ated with injury of inner membrane or opening of themitochondrial permeability transition pore (mPTP) resultingin mitochondrial depolarization and subsequent ATP deple-tion (26, 48, 37, 56, 1, 34).
Bcl-2 proteins are known important regulators of mitochon-drial permeabilization (1, 26). Proapoptotic Bax and Bak formchannels in the outer membrane through which mitochondrialcytochrome c is released into the cytosol (1, 26); BH3-onlyproteins, such as Bim and Puma, activate Bax/Bak channels. Incontrast, prosurvival Bcl-2 proteins such as Bcl-2 and Bcl-xLinhibit apoptosis by sequestering BH3-only proteins and Bax/Bak (1, 26). Notably, the prosurvival Bcl-2 proteins can alsostabilize inner membrane and block mPTP opening, thus main-taining energy production and preventing necrosis (52, 53).Our recent studies demonstrated that the predominant effect ofBcl-2/Bcl-xL proteins is to stabilize mitochondrial inner mem-brane integrity rather than to prevent OMP opening-causedcytochrome c release in pancreatitis (48). Thus the prosurvivalBcl-2 proteins are now recognized to play an important role inprotection of acinar cells from necrosis by stabilizing mito-chondria against death signals.
Inhibitors of apoptosis proteins (IAPs) are an importantfamily of endogenous proteins that inhibit the caspase sys-tem, the essential mediators of apoptotic death pathways(11, 12). The importance of IAPs in regulating the type ofdeath in pancreatitis has been reported by our group (32, 39,
Address for reprint requests and other correspondence: J. Yuan, UCLA/VAGreater Los Angeles Healthcare System, West Los Angeles VA HealthcareCenter, South California Research Center for Alcoholic Liver and PancreaticDiseases, 11301 Wilshire Blvd., Bldg. 258, Rm. 340, Los Angeles, CA 90073(e-mail: [email protected]).
Am J Physiol Gastrointest Liver Physiol 307: G550–G563, 2014.First published July 17, 2014; doi:10.1152/ajpgi.00432.2013.
0193-1857/14 Copyright © 2014 the American Physiological Society http://www.ajpgi.orgG550

54). NF-�B activation is a key early event in acute pancre-atitis (39, 55). A wealth of evidence indicates that NF-�Bactivation plays an important role in regulation of IAPs suchas c-IAP1, c-IAP2, survivin, XIAP, as well as antiapoptoticprotein FLICE-inhibitory protein (c-FLIP) (12, 16, 22, 23,39, 47, 57).
A number of reports indicate that the programmed necrosisrequires the receptor-interacting protein kinase (RIP or RIP1)(10, 14, 20, 28, 33, 49). RIP forms a death signaling complexwith the Fas-associated death domain and caspases in responseto death domain receptor stimulation (10, 28, 49). Duringapoptosis, RIP is cleaved and inactivated by caspase-3 and -8(10, 28, 33). The regulation of RIP by caspases has beensuggested to be one of mechanisms underlying the protectiverole of caspases from necrosis in cerulein-induced pancreatitis(20, 32, 54).
Protein kinase Cs (PKCs) are a family of serine/threoninekinases comprising 10 isoforms, namely conventional PKCisoforms (�, �I, �II, and �), novel PKC isoforms (�, ε, �, and�), and atypical PKC isoforms ( and /�) (35). Each PKCisoform can be activated independently by specific stimuli andmediates distinct biological functions (36, 4, 19, 8). In pancre-atic acinar cell, four PKC isoforms, �, �, ε, and , have beendetected (3), which have been increasingly implicated in theregulation of pathological responses of pancreatitis in pancre-atic acinar cells (15). PKCε and � have been shown to play arole in zymogen activation in pancreatitis (50). Our groupdemonstrated that PKCε and � are key regulators of proinflam-matory transcription factor NF-�B activation induced byCCK-8 in pancreatitis (43). Furthermore, we demonstrated thatethanol causes specific activation of PKCε in acinar cellsand that PKCε mediated the sensitizing effects of ethanol oninflammatory response in alcoholic pancreatitis, suggestingthat PKCε activation plays a key role in alcoholic pancre-atitis (44).
To better understand the role PKCε in pancreatitis involv-ing necrosis, we designed the present study to explore themolecular mechanisms through which PKCε regulates celldeath responses in in vivo and in vitro experimental modelsof acute pancreatitis. Our findings here show that geneticdeletion of PKCε decreases inflammation, necrosis, and theseverity of acute pancreatitis. PKCε mediates necrosis inpancreatitis through mechanisms involving mitochondrialproapoptotic and prosurvival Bcl-2 family proteins andupregulation of nonmitochondrial pathways inhibitingcaspase activation and RIP cleavage. Thus we suggest thatPKCε is a potential target for prevention and/or treatment ofnecrotizing pancreatitis.
MATERIALS AND METHODS
Reagents. CCK-8 (CCK) was from American Peptide (Sunnyvale,CA); cerulein was from Peninsula Laboratories (Belmont, CA). Me-dium 199 was from GIBCO (Grand Island, NY). �-32P ATP was fromPerkin Elmer (Torrance, CA). Nitrocellulose membranes were fromSchleicher and Schuell BioScience. Caspase-3 fluorogenic substratesAc-DEVD-AMC were from Biomol (Plymouth Meeting, PA). Anti-bodies against PKCε, c-IAP1, c-IAP2, c-FLIP, survivin, or Bcl-2 werefrom Santa Cruz Biotechnology (Santa Cruz, CA). Cytochrome c andRIP antibodies were from BD Science (San Diego, CA). COX IVantibody was from Molecular Probes (Eugene, OR); antibodiesagainst Bax, Bak, XIAP, Bcl-xL, GAPDH, or ERK1/2 were from Cell
Signaling (Beverly, MA). VDAC1 antibody was from Abcam(Cambridge, MA). The specific PKCε translocation inhibitor pep-tide was synthesized as described previously (43, 44, 55). ProteinA-agarose was from Roche Applied Science (Mannheim, Ger-many) and the specific PKCε peptide substrate was from Calbio-chem (La Jolla, CA). Other items were from standard suppliers oras indicated in text.
Experimental pancreatitis. PKCε-deficient (PKCε�/�) and wild-type (WT, PKCε / ) littermate mice were both C57BL/6J back-ground and obtained by breeding PKCε�/ mice generously pro-vided by Dr. Robert O. Messing (University of California, SanFrancisco, CA). The phenotype of these mice was described (24).PKCε�/� mice were viable, normal in size/body weight, and withoutdisplay of any gross physical or behavioral abnormalities. There wereno gross differences in pancreatic fat. A problem observed by theprovider (24) and us was that homozygous females rarely producedlitters (0–2 pups). Thus we obtained PKCε�/� by breedingPKCε�/ mice. Breeding of the PKCε-deficient mice and handlingof the animals were approved by the Animal Research Committee ofthe VA Greater Los Angeles Healthcare System, in accordance withthe National Institutes of Health guidelines.
Pancreatitis was induced in WT and PKCε�/� mice (25–30 g) byup to 7 hourly intraperitoneal (IP) injections of 50 �g/kg cerulein, aCCK analog used for experimental pancreatitis models. Control ani-mals received similar injections of physiological saline. The animalswere euthanized by CO2-induced asphyxiation at 30 min, 4 h, and 7h after the first injection, and the blood and pancreas were harvestedfor measurements. Tissues were immediately removed, frozen inliquid nitrogen, and stored at �80°C.
Preparation of tissue lysates, Western blot analysis, PKC� immu-noprecipitation, and in vitro kinase assay. Pancreatic tissue lysateswere prepared and Western blot analyses were performed asdescribed previously (43, 54). PKCε in pancreatic tissue lysateswas immunoprecipitated at 4°C overnight with the PKCε antibody(1:100) and protein A-agarose. In vitro kinase assay was performedas described (43).
Preparation and treatments of dispersed pancreatic acini. Pancre-atic acini were isolated from WT and PKCε�/� mice and cultured asdescribed (54, 55). For experimental purposes, the acini were prein-cubated in medium 199 with or without inhibitors and then treatedfurther with CCK, as described (43, 54, 55).
Enzymatic assays. Animal serum amylase and lipase activities weredetermined by Antech Diagnostics (Irvine, CA) Custom Service.Active trypsin in pancreatic tissue or acini homogenates was mea-sured by using Boc-Gln-Ala-Arg-AMC as a substrate by a fluoroenicassay as described previously (29). Caspase-3 activities in pancreatictissue homogenates were measured by using a fluorogenic assay withsubstrates specific for caspase-3 (Ac-DEVDAMC) as we previouslydescribed (32, 48). ATP level in pancreatic acini samples was mea-sured as described (48) with a luciferin/luciferase-based ATP deter-mination kit (Molecular Probes).
Quantification of necrosis. Necrosis in pancreatic acini was deter-mined by the release of lactate dehydrogenase (LDH) into the incu-bation medium, as described previously (16, 17, 48, 54). LDH activitywas measured by use of a cytotoxicity detection kit (Roche Diagnos-tics, Indianapolis, IN).
Quantification of necrosis in in vivo pancreatitis was performed onpancreatic tissue (collected after 7 hourly cerulein injections) sectionsstained with hematoxylin and eosin (H&E). Cells with swollen cyto-plasm, loss of plasma membrane integrity, and leakage of organellesinto interstitium were considered necrotic. A total of at least 2,000acinar cells were counted on tissue sections from each animal andthree to five animals per condition were counted.
Quantification of apoptosis. Apoptosis was quantified in pancreatictissue sections (7 hourly cerulein injections) stained with TdT-medi-ated dUTP nick end-labeling (TUNEL) to measure DNA breaks, as
G551PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
we described previously (16, 32) by use of the DeadEnd FluorometricTUNEL System kit (Promega, Madison, WI). For these and otherquantifications of histological measurements, a total of at least 3,000acinar cells were counted on tissue sections from each animal andthree to five animals per condition were counted.
Histological analysis for pancreas inflammatory cell infiltrationand vacuolization and measurement of edema. Quantification ofinflammatory cell infiltration and vacuolization was performed onH&E-stained pancreatic tissue (7 hourly cerulein injections) sectionsfrom at least four mice per group and expressed as the number ofinflammatory cells or vacuoles per 100 acinar cells. Neutrophil infil-tration in pancreas was further quantified by measuring myeloperox-idase (MPO) activity by using a MPO Activity Assay kit (Sigma, no.MAK068) according to the manufacturer’s instructions. Pancreaticedema was evaluated by measuring the wet-to-dry weight ratio asdescribed (18). The results were calculated and expressed as a waterindex (wet wt/dry wt).
Tissue and cell fractioning and measurement of cytochrome crelease. Cytosolic and mitochondria-enriched membrane fractionsfrom pancreatic tissue or isolated pancreatic acini were prepared asdescribed in detail (32, 48) and used as samples for Western blotanalysis.
Preparation of nuclear extracts and NF-�B DNA binding activitymeasurement. Pancreatic tissue nuclear protein extracts were preparedby using ActiveMotif nuclear extract kit (Carlsbad, CA) following themanufacturer’s instructions. NF-�B DNA binding activities in pan-creas tissue collected 30 min after 1 injection of cerulein weremeasured with EMSA as described previously (43, 44, 55).
Immunofluorescence. For immunolabeling, pancreas was dissectedand fixed for 6 h in formalin. Sections were stained with primaryantibodies against PKCε and mitochondrial marker protein voltage-dependent anion channel (VDAC1), followed by incubation withsecondary antibodies conjugated with Alexa 488 or 594 (Life Tech-nologies, Grand Island, NY). Nuclei were counterstained with 4=,6-diamidino-2-phenylindole (DAPI). Images were acquired with a ZeissLSM 710 confocal microscope using a �63 objective.
Measurements of mRNA expression by RT-PCR. RNA isolation andconventional quantitative RT-PCR using gene specific, intron-span-ning primers (Table 1) were performed as we described previously(48). Mouse 18S ribosomal RNA (m18S) was used as a reference(housekeeping) control. The m18S and our target sequences wereamplified at the annealing temperature 57°C during 29 or 32 cycles,respectively, to yield visible products within linear amplificationrange. PCR products were visualized and quantified with a Fluo-rChem-HD2 imager (Alpha Innotech).
Statistical analysis of data. Results are means � SE and representdata from at least three independent experiments. The experimentaldata were evaluated by the analysis of variance (ANOVA) followedby Bonferroni multiple comparison post hoc tests with the GraphPadPrism software (GraphPad Software, La Jolla, CA), and t-tests wereused to analyze differences between two groups. P � 0.05 wasconsidered statistically significant.
RESULTS
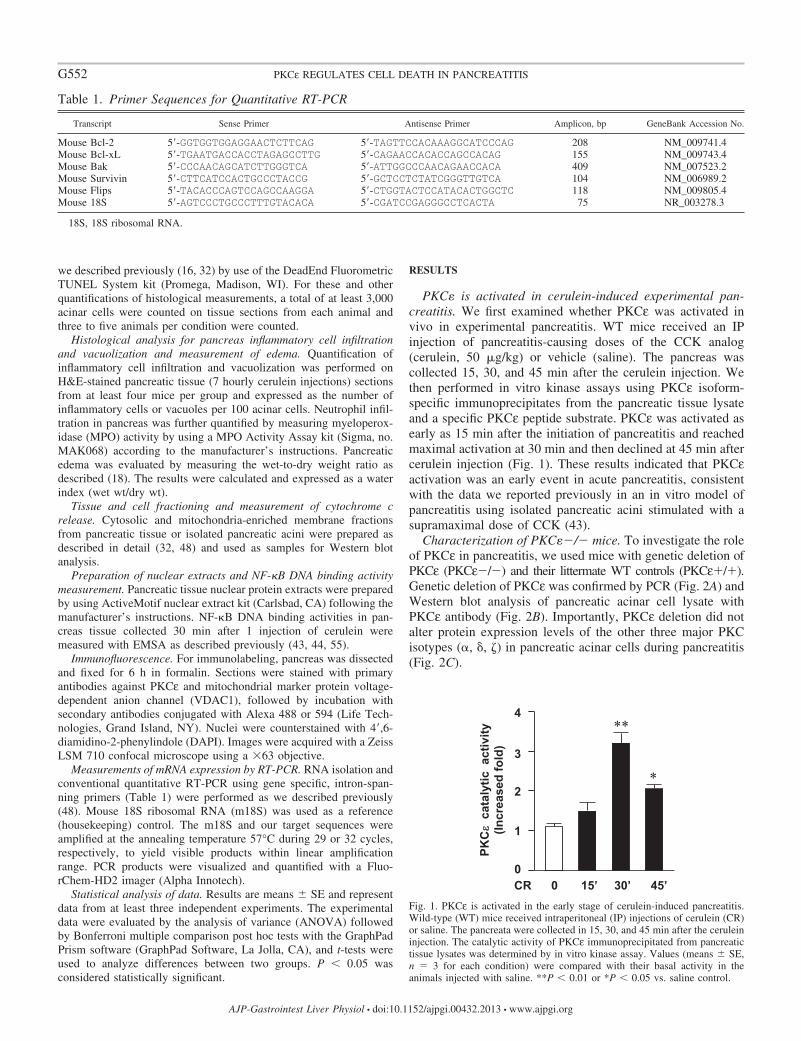
PKC� is activated in cerulein-induced experimental pan-creatitis. We first examined whether PKCε was activated invivo in experimental pancreatitis. WT mice received an IPinjection of pancreatitis-causing doses of the CCK analog(cerulein, 50 �g/kg) or vehicle (saline). The pancreas wascollected 15, 30, and 45 min after the cerulein injection. Wethen performed in vitro kinase assays using PKCε isoform-specific immunoprecipitates from the pancreatic tissue lysateand a specific PKCε peptide substrate. PKCε was activated asearly as 15 min after the initiation of pancreatitis and reachedmaximal activation at 30 min and then declined at 45 min aftercerulein injection (Fig. 1). These results indicated that PKCεactivation was an early event in acute pancreatitis, consistentwith the data we reported previously in an in vitro model ofpancreatitis using isolated pancreatic acini stimulated with asupramaximal dose of CCK (43).
Characterization of PKC��/� mice. To investigate the roleof PKCε in pancreatitis, we used mice with genetic deletion ofPKCε (PKCε�/�) and their littermate WT controls (PKCε / ).Genetic deletion of PKCε was confirmed by PCR (Fig. 2A) andWestern blot analysis of pancreatic acinar cell lysate withPKCε antibody (Fig. 2B). Importantly, PKCε deletion did notalter protein expression levels of the other three major PKCisotypes (�, �, ) in pancreatic acinar cells during pancreatitis(Fig. 2C).
Table 1. Primer Sequences for Quantitative RT-PCR
Transcript Sense Primer Antisense Primer Amplicon, bp GeneBank Accession No.
Mouse Bcl-2 5=-GGTGGTGGAGGAACTCTTCAG 5=-TAGTTCCACAAAGGCATCCCAG 208 NM_009741.4Mouse Bcl-xL 5=-TGAATGACCACCTAGAGCCTTG 5=-CAGAACCACACCAGCCACAG 155 NM_009743.4Mouse Bak 5=-CCCAACAGCATCTTGGGTCA 5=-ATTGGCCCAACAGAACCACA 409 NM_007523.2Mouse Survivin 5=-CTTCATCCACTGCCCTACCG 5=-GCTCCTCTATCGGGTTGTCA 104 NM_006989.2Mouse Flips 5=-TACACCCAGTCCAGCCAAGGA 5=-CTGGTACTCCATACACTGGCTC 118 NM_009805.4Mouse 18S 5=-AGTCCCTGCCCTTTGTACACA 5=-CGATCCGAGGGCCTCACTA 75 NR_003278.3
18S, 18S ribosomal RNA.
0
1
2
3
4
PKCεε
cata
lytic
act
ivity
(In
crea
sed
fold
)
CR 0 15’ 30’ 45’
**
*
Fig. 1. PKCε is activated in the early stage of cerulein-induced pancreatitis.Wild-type (WT) mice received intraperitoneal (IP) injections of cerulein (CR)or saline. The pancreata were collected in 15, 30, and 45 min after the ceruleininjection. The catalytic activity of PKCε immunoprecipitated from pancreatictissue lysates was determined by in vitro kinase assay. Values (means � SE,n � 3 for each condition) were compared with their basal activity in theanimals injected with saline. **P � 0.01 or *P � 0.05 vs. saline control.
G552 PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org

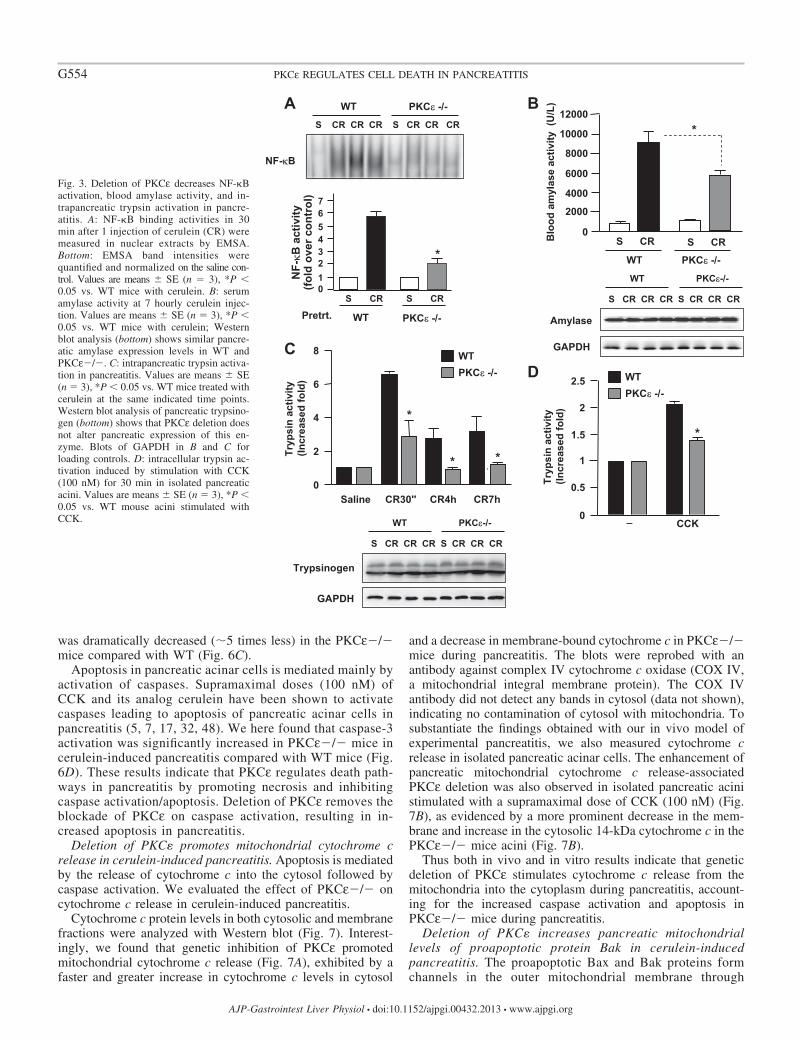
Deletion of PKC� decreases NF-�B activation in cerulein-induced pancreatitis. Our previous studies using specific PKCεpharmacological inhibitor in an in vitro pancreatitis model inisolated pancreatic acini demonstrated that PKCε was requiredfor CCK-induced NF-�B activation (43, 44). Here, using the invivo pancreatitis model, we showed that genetic deletion ofPKCε largely prevented NF-�B activation in cerulein-inducedpancreatitis (Fig. 3A), supporting the critical role of PKCε inNF-�B activation in acute pancreatitis.
Deletion of PKC� ameliorates necrosis and other pancre-atitis parameters in cerulein-induced pancreatitis. Next, weevaluated the effect of PKCε�/� on the parameters of ceru-lein-induced pancreatitis. Cerulein caused a marked increase inblood amylase activity after 7 hourly IP injections of ceru-lein in WT mice (Fig. 3B). This increase was significantlyattenuated in PKCε�/� mice. Cerulein-induced intracellu-lar trypsinogen conversion to trypsin (as measured by tryp-sin activity) was markedly reduced in PKCε�/� mice (Fig.3C). Reduction of trypsin conversion in PKCε�/� micewas also observed in an in vitro experimental pancreatitismodel (Fig. 3D) using isolated pancreatic acini stimulatedwith a supramaximal dose (100 nM) of CCK, which was
known to cause cell pathologies of pancreatitis in vitro (17,43, 48). Western blot analyses of pancreatic tissue lysatesshowed similar protein expression levels of amylase andtrypsinogen between PKCε�/� mice and WT mice (Fig. 3Band 3C, bottom), which excluded the possibility that thedecreased activities of blood amylase and intracellular tryp-sin were caused by the alteration of their expression levelsin the pancreas of PKCε�/� mice.
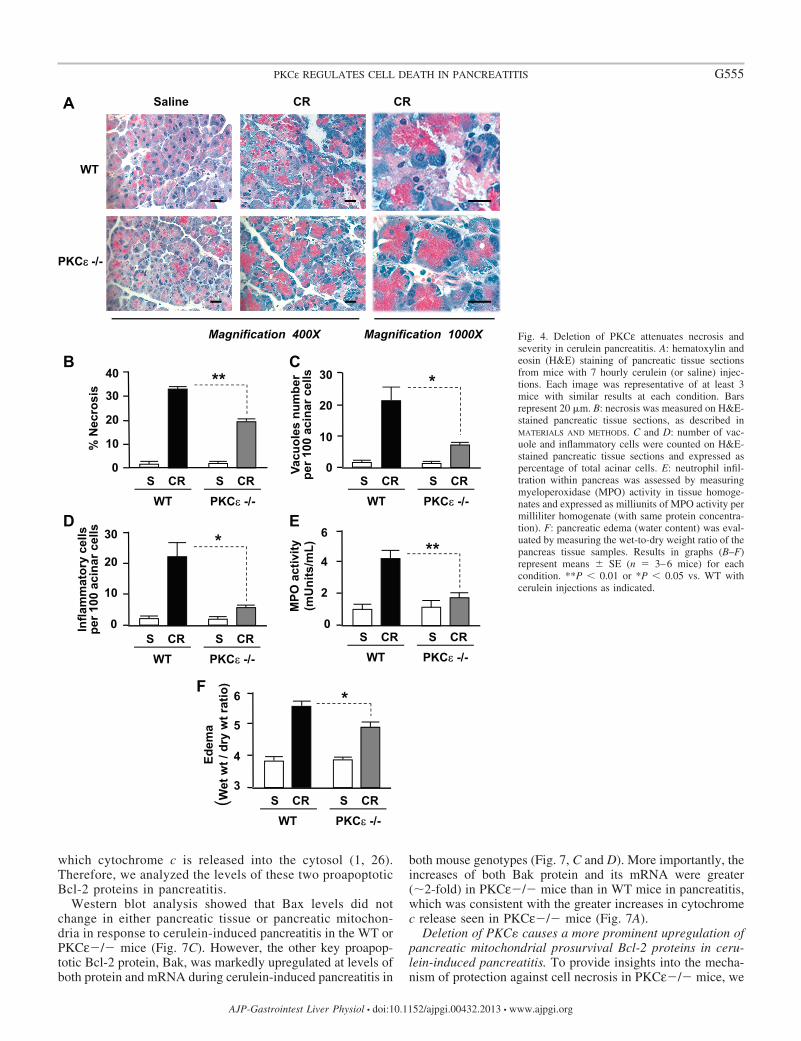
In addition to the above findings, PKCε deletion dramati-cally ameliorated the histological damage in cerulein-inducedpancreatitis (Fig. 4A). The first salient feature of the pancreatichistological changes in PKCε�/� mice compared with WTmice was that cerulein-induced acinar cell necrosis was signif-icantly attenuated (Fig. 4B). Other histopathological features ofpancreatitis including accumulation of cytoplasmic vacuolesand inflammatory cell infiltration were also greatly attenuatedin PKCε�/� mice (Fig. 4, C and D). Measurements of MPOactivities further confirmed the attenuated inflammatory cellinfiltration in pancreatitis in PKCε�/� mice (Fig. 4E). Mea-surements of the wet-to-dry weight ratio of pancreatic tissue(Fig. 4E) indicated that the edema was significantly reduced inpancreatitis in PKCε�/� mice. All these results demonstratethat PKCε deletion ameliorates necrosis, inflammation, and theseverity of acute pancreatitis.
Inhibition of PKC� attenuates acinar cell necrosis and ATPreduction in the in vitro model of pancreatitis. To corroboratethe findings that PKCε promoted necrosis in the in vivo modelof pancreatitis, we performed experiments on isolated pancre-atic acinar cells. The acinar cells were stimulated with supra-maximal (100 nM) CCK, which was known to induce pancre-atitis responses in acinar cells, such as activation of trypsino-gen and NF-�B, necrosis, and apoptosis (17, 43, 48, 54, 55).Therefore, this system was considered in vitro model of acutepancreatitis.
We applied two approaches to inactivate PKCε in acinarcells in this study: using the pancreatic acini isolated fromPKCε�/� mice and using a pharmacological isoform-spe-cific peptide PKCε inhibitor to treat WT acinar cells. WTpancreatic acini were preincubated with a cell-permeablespecific PKCε translocation inhibitor peptide or a scrambledpeptide (10 �M, 1 h). Then PKCε�/� acini or the pre-treated WT acini were hyperstimulated with CCK (100 nM).Changes in cellular ATP levels and LDH release into theextracellular medium were measured after 3-h incubationwith CCK. We observed that CCK hyperstimulation causedincreased LDH release, an indicator of cell necrosis (Fig.5A), which was associated with a dramatic decrease incellular ATP (Fig. 5B), but either genetic or pharmacolog-ical inhibition of PKCε protected the acinar cells fromCCK-induced necrosis and ATP reduction.
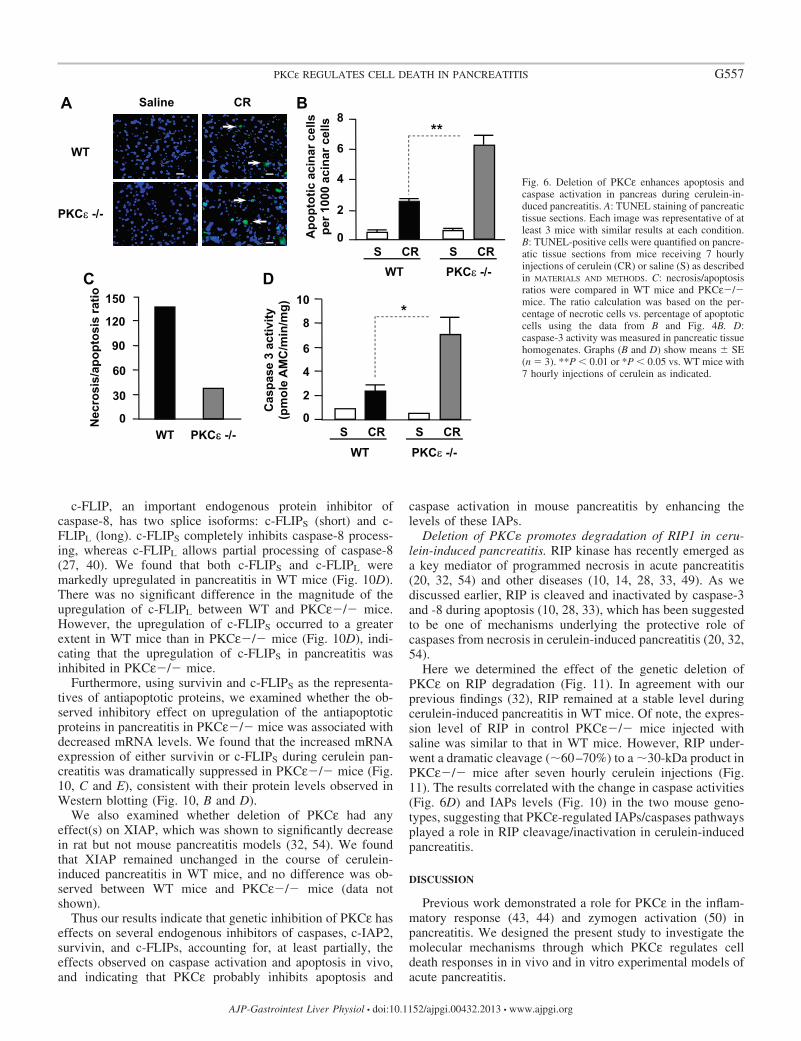
Deletion of PKC� promotes apoptosis and caspase activa-tion in cerulein-induced pancreatitis. We investigated the ef-fects of PKCε deletion on apoptosis in the tissue slides with insitu TUNEL assays. Our results showed that, in contrast to theinhibitory effect of PKCε deletion on necrosis in pancreatitis,cerulein-induced apoptosis increased twofold in PKCε�/�mice compared with WT controls (Fig. 6, A and B). Theseresults demonstrate the antiapoptotic effect of PKCε on acinarcell death in pancreatitis. Analysis of the effect of PKCε on thetwo death pathways (necrosis vs. apoptosis) showed that theratio of necrosis to apoptosis in cerulein-induced pancreatitis
A WT PKCε+/-
PKCε-/-
PCR
C PKCε -/-WT
S CR CR CR S CR CR CR
PKCε
W. Blot
GAPDH
PKCε
PKCδ
PKCε -/-WTB W. Blot
PKCα
PKCδ
PKCζ
Fig. 2. Genetic deletion of PKCε does not affect expression of the other 3 PKCisoforms presenting in pancreatic acinar cells. A: genotyping of PKC� mousecolonies with PCR. B: Western blot (W. Blot) analysis of PKCε and PKC� inlysates of pancreatic acini isolated from WT mice and PKCε�/� mice.Samples were run in a single gel but were not continuous, as indicated by a linebetween lanes. C: Western blot analysis of PKC isoforms, ε, �, �, and in thelysates of pancreatic tissue collected from WT mice and PKCε�/� mice IPinjected with saline (S) or cerulein (CR). GAPDH: for loading controls.
G553PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
was dramatically decreased (�5 times less) in the PKCε�/�mice compared with WT (Fig. 6C).
Apoptosis in pancreatic acinar cells is mediated mainly byactivation of caspases. Supramaximal doses (100 nM) ofCCK and its analog cerulein have been shown to activatecaspases leading to apoptosis of pancreatic acinar cells inpancreatitis (5, 7, 17, 32, 48). We here found that caspase-3activation was significantly increased in PKCε�/� mice incerulein-induced pancreatitis compared with WT mice (Fig.6D). These results indicate that PKCε regulates death path-ways in pancreatitis by promoting necrosis and inhibitingcaspase activation/apoptosis. Deletion of PKCε removes theblockade of PKCε on caspase activation, resulting in in-creased apoptosis in pancreatitis.
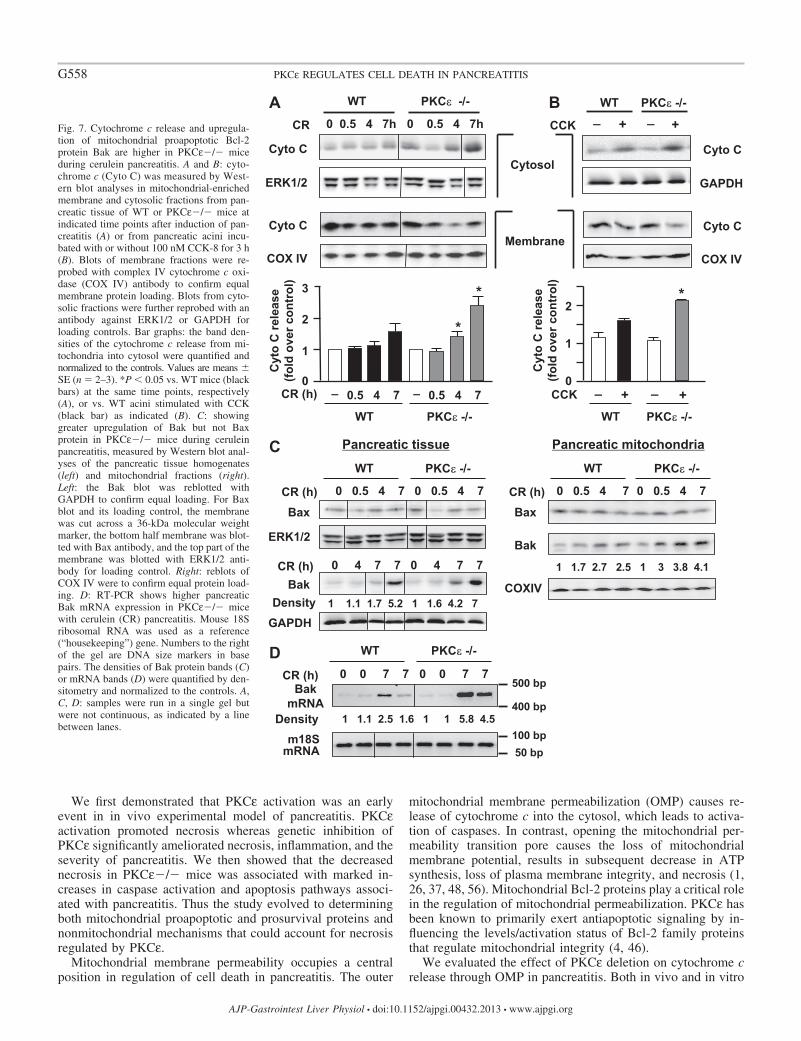
Deletion of PKC� promotes mitochondrial cytochrome crelease in cerulein-induced pancreatitis. Apoptosis is mediatedby the release of cytochrome c into the cytosol followed bycaspase activation. We evaluated the effect of PKCε�/� oncytochrome c release in cerulein-induced pancreatitis.
Cytochrome c protein levels in both cytosolic and membranefractions were analyzed with Western blot (Fig. 7). Interest-ingly, we found that genetic inhibition of PKCε promotedmitochondrial cytochrome c release (Fig. 7A), exhibited by afaster and greater increase in cytochrome c levels in cytosol
and a decrease in membrane-bound cytochrome c in PKCε�/�mice during pancreatitis. The blots were reprobed with anantibody against complex IV cytochrome c oxidase (COX IV,a mitochondrial integral membrane protein). The COX IVantibody did not detect any bands in cytosol (data not shown),indicating no contamination of cytosol with mitochondria. Tosubstantiate the findings obtained with our in vivo model ofexperimental pancreatitis, we also measured cytochrome crelease in isolated pancreatic acinar cells. The enhancement ofpancreatic mitochondrial cytochrome c release-associatedPKCε deletion was also observed in isolated pancreatic acinistimulated with a supramaximal dose of CCK (100 nM) (Fig.7B), as evidenced by a more prominent decrease in the mem-brane and increase in the cytosolic 14-kDa cytochrome c in thePKCε�/� mice acini (Fig. 7B).
Thus both in vivo and in vitro results indicate that geneticdeletion of PKCε stimulates cytochrome c release from themitochondria into the cytoplasm during pancreatitis, account-ing for the increased caspase activation and apoptosis inPKCε�/� mice during pancreatitis.
Deletion of PKC� increases pancreatic mitochondriallevels of proapoptotic protein Bak in cerulein-inducedpancreatitis. The proapoptotic Bax and Bak proteins formchannels in the outer mitochondrial membrane through
CD
Saline CR30'' CR4h CR7h0
2
4
6
8 WTPKCε -/-
Tryp
sin
activ
ity(In
crea
sed
fold
)
*
* *
_ CCK0
0.5
1.5
2
2.5
Tryp
sin
activ
ity(In
crea
sed
fold
)
*
1
B
Blo
od a
myl
ase
activ
ity (
U/L
)
0
4000
10000
2000
8000
12000*
S CR
WT PKCε -/-
S CR
6000
PKCε-/-WT
S CR CR CR S CR CR CR
Amylase
GAPDH
WTPKCε -/-
PKCε -/-WTAS CR CR CR S CR CR CR
NF-κ B
activ
ity(fo
ld o
ver c
ontr
ol)
0
23
5
WT
S CR S CR
*1
4
76
Pretrt. PKCε -/-
NF-κB
PKCε-/-WT
S CR CR CR S CR CR CR
GAPDH
Trypsinogen
Fig. 3. Deletion of PKCε decreases NF-�Bactivation, blood amylase activity, and in-trapancreatic trypsin activation in pancre-atitis. A: NF-�B binding activities in 30min after 1 injection of cerulein (CR) weremeasured in nuclear extracts by EMSA.Bottom: EMSA band intensities werequantified and normalized on the saline con-trol. Values are means � SE (n � 3), *P �0.05 vs. WT mice with cerulein. B: serumamylase activity at 7 hourly cerulein injec-tion. Values are means � SE (n � 3), *P �0.05 vs. WT mice with cerulein; Westernblot analysis (bottom) shows similar pancre-atic amylase expression levels in WT andPKCε�/�. C: intrapancreatic trypsin activa-tion in pancreatitis. Values are means � SE(n � 3), *P � 0.05 vs. WT mice treated withcerulein at the same indicated time points.Western blot analysis of pancreatic trypsino-gen (bottom) shows that PKCε deletion doesnot alter pancreatic expression of this en-zyme. Blots of GAPDH in B and C forloading controls. D: intracellular trypsin ac-tivation induced by stimulation with CCK(100 nM) for 30 min in isolated pancreaticacini. Values are means � SE (n � 3), *P �0.05 vs. WT mouse acini stimulated withCCK.
G554 PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
which cytochrome c is released into the cytosol (1, 26).Therefore, we analyzed the levels of these two proapoptoticBcl-2 proteins in pancreatitis.
Western blot analysis showed that Bax levels did notchange in either pancreatic tissue or pancreatic mitochon-dria in response to cerulein-induced pancreatitis in the WT orPKCε�/� mice (Fig. 7C). However, the other key proapop-totic Bcl-2 protein, Bak, was markedly upregulated at levels ofboth protein and mRNA during cerulein-induced pancreatitis in
both mouse genotypes (Fig. 7, C and D). More importantly, theincreases of both Bak protein and its mRNA were greater(�2-fold) in PKCε�/� mice than in WT mice in pancreatitis,which was consistent with the greater increases in cytochromec release seen in PKCε�/� mice (Fig. 7A).
Deletion of PKC� causes a more prominent upregulation ofpancreatic mitochondrial prosurvival Bcl-2 proteins in ceru-lein-induced pancreatitis. To provide insights into the mecha-nism of protection against cell necrosis in PKCε�/� mice, we
WT
PKCεε -/-
A CR CRSaline
Magnification 400X Magnification 1000X
D
0
20
30 *
S CR S CRWT PKCε -/-
Vacu
oles
num
ber
per 1
00 a
cina
r cel
ls
10
0
10
20
30 *
S CR S CRWT PKCε -/-
Infla
mm
ator
y ce
lls
per 1
00 a
cina
r cel
ls
C
F
Edem
a(W
et w
t / d
ry w
t rat
io)
S CR S CRWT PKCε -/-
3
4
5
6 *
B
0
10
20
30
40
% N
ecro
sis **
S CR S CRWT PKCε -/-
E
S CR S CRWT PKCε -/-
MPO
act
ivity
(m
Uni
ts/m
L)
0
2
4
6**
Fig. 4. Deletion of PKCε attenuates necrosis andseverity in cerulein pancreatitis. A: hematoxylin andeosin (H&E) staining of pancreatic tissue sectionsfrom mice with 7 hourly cerulein (or saline) injec-tions. Each image was representative of at least 3mice with similar results at each condition. Barsrepresent 20 �m. B: necrosis was measured on H&E-stained pancreatic tissue sections, as described inMATERIALS AND METHODS. C and D: number of vac-uole and inflammatory cells were counted on H&E-stained pancreatic tissue sections and expressed aspercentage of total acinar cells. E: neutrophil infil-tration within pancreas was assessed by measuringmyeloperoxidase (MPO) activity in tissue homoge-nates and expressed as milliunits of MPO activity permilliliter homogenate (with same protein concentra-tion). F: pancreatic edema (water content) was eval-uated by measuring the wet-to-dry weight ratio of thepancreas tissue samples. Results in graphs (B–F)represent means � SE (n � 3–6 mice) for eachcondition. **P � 0.01 or *P � 0.05 vs. WT withcerulein injections as indicated.
G555PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org

next analyzed Bcl-2 and Bcl-xL, two mitochondrial prosur-vival Bcl-2 proteins. Recent studies from our group indicatedthat these two Bcl-2 proteins stabilized pancreatic mitochon-dria and protected against necrosis in pancreatitis and thatupregulation of Bcl-2 and Bcl-xL was a key protective mech-anism against necrosis in pancreatitis (38, 48).
We found that Bcl-2 and Bcl-xL protein levels increasedboth in the whole pancreatic homogenates (Fig. 8A, left) andpancreatic mitochondria in cerulein-induced pancreatitis inWT mice (Fig. 8B), in accord with previous results from ourgroup (48). More importantly, the upregulation of Bcl-2 andBcl-xL proteins in pancreatitis was more pronounced inPKCε�/� mice than in WT mice (Fig. 8). Consistent withtheir greater protein levels, the mRNA expression of Bcl-2and Bcl-xL was also more increased in PKCε�/� micecompared with WT mice (Fig. 8A, right). The greater extentof upregulation of the prosurvival Bcl-2 and Bcl-xL proteinsin PKCε�/� mice may account, in part, for the attenuationof necrosis in pancreatitis in PKCε�/� mice and suggeststhat the negative regulation of Bcl-2 and Bcl-xL proteins byPKCε might be a mechanism whereby PKCε promotesnecrosis in pancreatitis.
PKC� translocates to mitochondria in cerulein-inducedpancreatitis. Translocation from cytosol to membranes is acharacteristic of activated PKCs. Activated PKCε has beenreported to translocate to intracellular membranes in previousstudies (44, 50). The effect of PKCε on mitochondrial proteinsprompted us to address the possibility of mitochondrial trans-location of PKCε in cerulein pancreatitis. Again we used twoapproaches: subcellular fractionation, and immunostaining ofthe pancreatic tissue collected in cerulein pancreatitis (60 minafter one IP injection) in WT mice. Comparing PKCε level incytosolic fractions and mitochondria-enriched membrane frac-tions (Fig. 9A), we found that PKCε localized predominantly inthe cytosolic fraction of pancreas from control mice injectedwith saline and that cerulein treatment dramatically decreasedthe presence of PKCε in the cytosolic fraction and increasedPKCε in the membrane fraction, indicating mitochondrialmembrane translocation of this kinase (Fig. 9A). The sameblots were also probed with antibody against COX IV or LDHfor loading controls and to assess the quality of our separationof membrane from cytosolic fractions.
The mitochondrial translocation of PKCε was further con-firmed by immunostaining of the pancreatic tissue sections.Confocal immunofluorescence (Fig. 9B) showed that in salinecontrol mice, the staining of PKCε (shown in green) distributedthroughout the cytosol. Mitochondrial marker (VDAC1) wasseen as sharp punctate structures (shown in red) and distributedaround the nuclei, granules, and cell periphery [see DIC (dif-ferential interference contrast) image in Fig. 9B], as describedbefore (41). Little colocalization (shown in yellow) of PKCεand VDAC1 was observed in the pancreatic tissue sectionsfrom saline control mice. Upon cerulein administration, weobserved a condensed, ringlike pattern of staining for bothPKCε and VDAC1, indicating the redistribution of PKCε andVDAC1-positive compartments. More importantly, we ob-served a dramatically increased colocalization of PKCε andVDAC1-positive compartments (around granules, Fig. 9B),supporting that PKCε might translocate to mitochondria toeffect death regulating proteins directly during pancreatitis.Similar results were also obtained when we used anothermitochondrial marker protein, Tom20, instead of VDAC1 inimmunostaining (data not shown).
Deletion of PKC� decreases pancreatic levels of antiapop-totic proteins c-IAP2, survivin, and c-FLIPs in cerulein-in-duced pancreatitis. Next, we determined whether PKCε regu-lated cell death also through nonmitochondrial pathways. IAPsare potent endogenous suppressors of caspases and play apivotal role in regulation of cell death (11, 12, 16, 22, 23, 32,39, 47, 55).
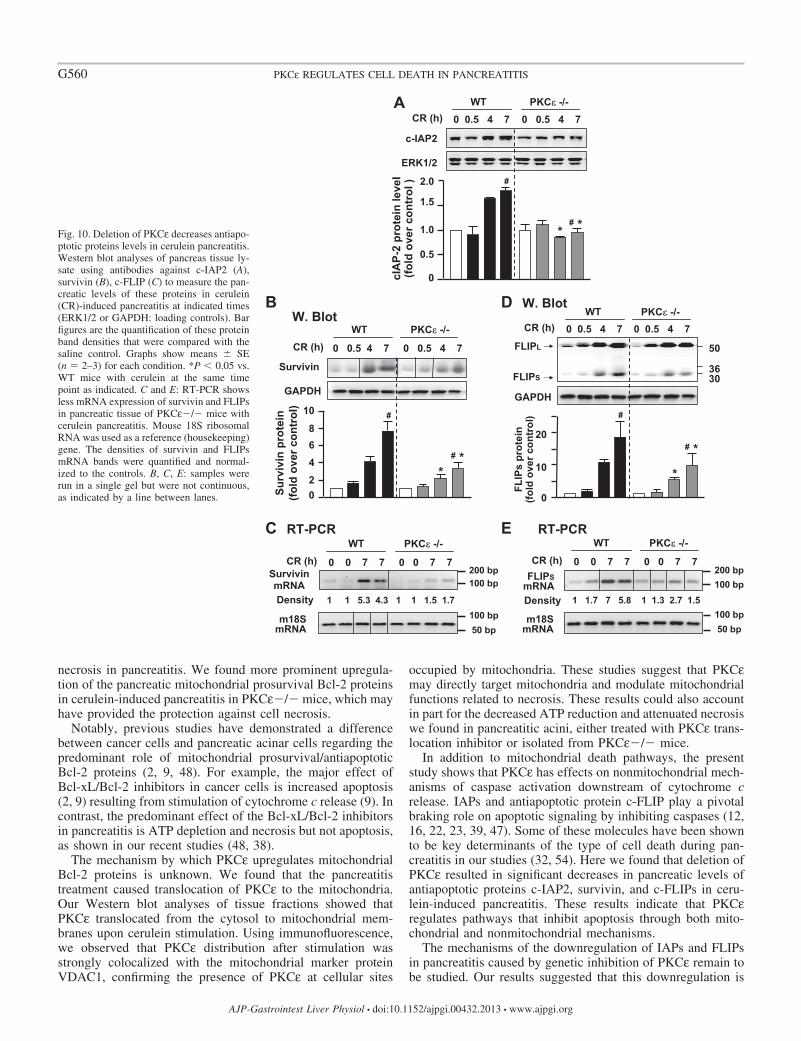
We first determined whether PKCε regulates c-IAP1 andc-IAP2, two key members of the eight mammalian IAPs andendogenous inhibitors of caspase-3, 7, and 9 (12, 57), inpancreatitis. We did not detect any alteration of c-IAP1 inpancreatitis in WT and PKCε�/� mice (data not shown).However, we found that c-IAP2 was upregulated markedly andtime dependently in pancreatitis in WT mice (Fig. 10A), andimportantly, this upregulation of c-IAP2 was significantlysuppressed in PKCε�/� mice. We next examined the level ofsurvivin, an endogenous inhibitor of caspase-3 (22). Similar toc-IAP2, the time-dependent upregulation of survivin duringcerulein-induced pancreatitis was also dramatically inhibited inPKCε�/� mice (Fig. 10B).
% o
f LD
H r
elea
seA
0
10
25
20
5
15
_ CCKPKCε -/-
*
_ CCKWT +PKCεi
WT
_ CCKWT+SP
_ CCK
#
# *
_ CCKPKCε -/-
**
_ CCKWT +PKCεi
ATP
(rel
ativ
e to
con
trol
w
ithou
t CC
K )
0
0.4
1.0
0.6
B
0.2
0.8
WT
_ CCKWT+SP
_ CCK
# **#
Fig. 5. Inhibition of PKCε prevents ATP depletion and necrosis in CCK-hyperstimulated pancreatic acinar cells. Pancreatic acini isolated fromPKCε�/� mice or WT mice were incubated for 3 h without or with 10 �MPKCε translocation inhibitor (PKCεi) or a scrambled peptide (SP) followed bystimulation with 100 nM CCK-8. A: percentage of lactate dehydrogenase(LDH) released into the extracellular medium. B: cellular ATP was determinedvia ATP determination kit. ATP values in PKCε�/� or WT acinar cells werecompared with their own control cells incubated without CCK. Values aremeans � SE (n � 3). **P � 0.01 and *P � 0.05 vs. WT acinar cells pretreatedwithout or with a scrambled peptide (WT SP, #) and then incubated withCCK (black bar) as indicated.
G556 PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
c-FLIP, an important endogenous protein inhibitor ofcaspase-8, has two splice isoforms: c-FLIPS (short) and c-FLIPL (long). c-FLIPS completely inhibits caspase-8 process-ing, whereas c-FLIPL allows partial processing of caspase-8(27, 40). We found that both c-FLIPS and c-FLIPL weremarkedly upregulated in pancreatitis in WT mice (Fig. 10D).There was no significant difference in the magnitude of theupregulation of c-FLIPL between WT and PKCε�/� mice.However, the upregulation of c-FLIPS occurred to a greaterextent in WT mice than in PKCε�/� mice (Fig. 10D), indi-cating that the upregulation of c-FLIPS in pancreatitis wasinhibited in PKCε�/� mice.
Furthermore, using survivin and c-FLIPS as the representa-tives of antiapoptotic proteins, we examined whether the ob-served inhibitory effect on upregulation of the antiapoptoticproteins in pancreatitis in PKCε�/� mice was associated withdecreased mRNA levels. We found that the increased mRNAexpression of either survivin or c-FLIPS during cerulein pan-creatitis was dramatically suppressed in PKCε�/� mice (Fig.10, C and E), consistent with their protein levels observed inWestern blotting (Fig. 10, B and D).
We also examined whether deletion of PKCε had anyeffect(s) on XIAP, which was shown to significantly decreasein rat but not mouse pancreatitis models (32, 54). We foundthat XIAP remained unchanged in the course of cerulein-induced pancreatitis in WT mice, and no difference was ob-served between WT mice and PKCε�/� mice (data notshown).
Thus our results indicate that genetic inhibition of PKCε haseffects on several endogenous inhibitors of caspases, c-IAP2,survivin, and c-FLIPs, accounting for, at least partially, theeffects observed on caspase activation and apoptosis in vivo,and indicating that PKCε probably inhibits apoptosis and
caspase activation in mouse pancreatitis by enhancing thelevels of these IAPs.
Deletion of PKC� promotes degradation of RIP1 in ceru-lein-induced pancreatitis. RIP kinase has recently emerged asa key mediator of programmed necrosis in acute pancreatitis(20, 32, 54) and other diseases (10, 14, 28, 33, 49). As wediscussed earlier, RIP is cleaved and inactivated by caspase-3and -8 during apoptosis (10, 28, 33), which has been suggestedto be one of mechanisms underlying the protective role ofcaspases from necrosis in cerulein-induced pancreatitis (20, 32,54).
Here we determined the effect of the genetic deletion ofPKCε on RIP degradation (Fig. 11). In agreement with ourprevious findings (32), RIP remained at a stable level duringcerulein-induced pancreatitis in WT mice. Of note, the expres-sion level of RIP in control PKCε�/� mice injected withsaline was similar to that in WT mice. However, RIP under-went a dramatic cleavage (�60–70%) to a �30-kDa product inPKCε�/� mice after seven hourly cerulein injections (Fig.11). The results correlated with the change in caspase activities(Fig. 6D) and IAPs levels (Fig. 10) in the two mouse geno-types, suggesting that PKCε-regulated IAPs/caspases pathwaysplayed a role in RIP cleavage/inactivation in cerulein-inducedpancreatitis.
DISCUSSION
Previous work demonstrated a role for PKCε in the inflam-matory response (43, 44) and zymogen activation (50) inpancreatitis. We designed the present study to investigate themolecular mechanisms through which PKCε regulates celldeath responses in in vivo and in vitro experimental models ofacute pancreatitis.
C
0
30
60
Nec
rosi
s/ap
opto
sis
ratio
120
150
WT PKCεε -/-
90
WT PKCε -/-
A
Apo
ptot
ic a
cina
r cel
lspe
r 100
0 ac
inar
cel
ls
0
2
6
8
S CR S CR
**
4
D
0
2
6
8
S CR S CRWT PKCε -/-
*
4
10
Cas
pase
3 a
ctiv
ity
(pm
ole
AM
C/m
in/m
g)
BCR
PKCε -/-
WT
Saline
Fig. 6. Deletion of PKCε enhances apoptosis andcaspase activation in pancreas during cerulein-in-duced pancreatitis. A: TUNEL staining of pancreatictissue sections. Each image was representative of atleast 3 mice with similar results at each condition.B: TUNEL-positive cells were quantified on pancre-atic tissue sections from mice receiving 7 hourlyinjections of cerulein (CR) or saline (S) as describedin MATERIALS AND METHODS. C: necrosis/apoptosisratios were compared in WT mice and PKCε�/�mice. The ratio calculation was based on the per-centage of necrotic cells vs. percentage of apoptoticcells using the data from B and Fig. 4B. D:caspase-3 activity was measured in pancreatic tissuehomogenates. Graphs (B and D) show means � SE(n � 3). **P � 0.01 or *P � 0.05 vs. WT mice with7 hourly injections of cerulein as indicated.
G557PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
We first demonstrated that PKCε activation was an earlyevent in in vivo experimental model of pancreatitis. PKCεactivation promoted necrosis whereas genetic inhibition ofPKCε significantly ameliorated necrosis, inflammation, and theseverity of pancreatitis. We then showed that the decreasednecrosis in PKCε�/� mice was associated with marked in-creases in caspase activation and apoptosis pathways associ-ated with pancreatitis. Thus the study evolved to determiningboth mitochondrial proapoptotic and prosurvival proteins andnonmitochondrial mechanisms that could account for necrosisregulated by PKCε.
Mitochondrial membrane permeability occupies a centralposition in regulation of cell death in pancreatitis. The outer
mitochondrial membrane permeabilization (OMP) causes re-lease of cytochrome c into the cytosol, which leads to activa-tion of caspases. In contrast, opening the mitochondrial per-meability transition pore causes the loss of mitochondrialmembrane potential, results in subsequent decrease in ATPsynthesis, loss of plasma membrane integrity, and necrosis (1,26, 37, 48, 56). Mitochondrial Bcl-2 proteins play a critical rolein the regulation of mitochondrial permeabilization. PKCε hasbeen known to primarily exert antiapoptotic signaling by in-fluencing the levels/activation status of Bcl-2 family proteinsthat regulate mitochondrial integrity (4, 46).
We evaluated the effect of PKCε deletion on cytochrome crelease through OMP in pancreatitis. Both in vivo and in vitro
Cyto C
COX IV
Cyto C
ERK1/2
0 0.5 4 7h 0 0.5 4 7hCR
PKCε -/-WTA
Cytosol
Membrane
PKCε -/-WT
CCK _ _ ++
Cyto C
COX IV
Cyto C
GAPDH
B
Cyt
o C
rele
ase
(fold
ove
r con
trol
)
Cyt
o C
rele
ase
(fold
ove
r con
trol
)
0
1
2
3 *
*
0
1
2*
_ _0.5 4 7 CR (h) 0.5 4 7
WT PKCε -/- WT PKCε -/-
CCK _ _ ++
0 0.5 4 7 0 0.5 4 7
WT PKCε -/-
Bak
Pancreatic mitochondria
1 1.7 2.7 2.5 1 3 3.8 4.1
C
CR (h)
WT PKCε -/-
Bak1 1.1 1.7 5.2 1 1.6 4.2 7
0 0.5 4 7 0 0.5 4 7
Pancreatic tissue
Density
BaxBax
COXIV
GAPDH
CR (h)
CR (h) 0 4 7 7 0 4 7 7
m18SmRNA
Bak mRNA
1 1.1 2.5 1.6 1 1 5.8 4.5
CR (h)
WT PKCε -/-
0 0 7 7 0 0 7 7D
500 bp
400 bpDensity
100 bp50 bp
ERK1/2
Fig. 7. Cytochrome c release and upregula-tion of mitochondrial proapoptotic Bcl-2protein Bak are higher in PKCε�/� miceduring cerulein pancreatitis. A and B: cyto-chrome c (Cyto C) was measured by West-ern blot analyses in mitochondrial-enrichedmembrane and cytosolic fractions from pan-creatic tissue of WT or PKCε�/� mice atindicated time points after induction of pan-creatitis (A) or from pancreatic acini incu-bated with or without 100 nM CCK-8 for 3 h(B). Blots of membrane fractions were re-probed with complex IV cytochrome c oxi-dase (COX IV) antibody to confirm equalmembrane protein loading. Blots from cyto-solic fractions were further reprobed with anantibody against ERK1/2 or GAPDH forloading controls. Bar graphs: the band den-sities of the cytochrome c release from mi-tochondria into cytosol were quantified andnormalized to the controls. Values are means �SE (n � 2–3). *P � 0.05 vs. WT mice (blackbars) at the same time points, respectively(A), or vs. WT acini stimulated with CCK(black bar) as indicated (B). C: showinggreater upregulation of Bak but not Baxprotein in PKCε�/� mice during ceruleinpancreatitis, measured by Western blot anal-yses of the pancreatic tissue homogenates(left) and mitochondrial fractions (right).Left: the Bak blot was reblotted withGAPDH to confirm equal loading. For Baxblot and its loading control, the membranewas cut across a 36-kDa molecular weightmarker, the bottom half membrane was blot-ted with Bax antibody, and the top part of themembrane was blotted with ERK1/2 anti-body for loading control. Right: reblots ofCOX IV were to confirm equal protein load-ing. D: RT-PCR shows higher pancreaticBak mRNA expression in PKCε�/� micewith cerulein (CR) pancreatitis. Mouse 18Sribosomal RNA was used as a reference(“housekeeping”) gene. Numbers to the rightof the gel are DNA size markers in basepairs. The densities of Bak protein bands (C)or mRNA bands (D) were quantified by den-sitometry and normalized to the controls. A,C, D: samples were run in a single gel butwere not continuous, as indicated by a linebetween lanes.
G558 PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org

results suggested that genetic inhibition of PKCε stimulatedcytochrome c release from the mitochondria to the cytoplasmduring pancreatitis. More interestingly, the increased cyto-chrome c release in PKCε�/� mice was closely associatedwith marked increases in Bak levels in both whole pancreatictissue homogenate and mitochondria in PKCε�/� mice. Theseresults suggest that inhibition of PKCε promotes OMP open-ing, leading to cytochrome c release and caspase activation, atleast partially via upregulation of mitochondrial Bak.
Next, we assessed whether PKCε inhibition had an effect oncellular ATP and cell necrosis induced by supramaximal CCKstimulation in an in vitro pancreatitis. Our results showed thatinhibition of PKCε with either genetic deletion or PKCεtranslocation inhibitor prevented supramaximal CCK-inducedATP decrease and cell necrosis in pancreatitis.
Of note, it has been controversial whether ATP depletionplays a causal or primary role in the pathophysiology of acutepancreatitis in earlier studies (30, 31). For example, an equiv-
alent ATP decrease was observed in response to physiologicaldoses of cerulein (30) in mice and in rats with supraphysiologi-cal cerulein (31), which did not develop necrosis. Recentevidence (1, 26, 48, 56) indicated, however, that necrosis inpancreatitis was associated with ATP depletion. The presentstudy showing that inhibition of PKCε prevented ATP reduc-tion and necrosis suggested a key role for PKCε in regulatingATP production and necrosis. The decrease in ATP itself isimportant but is not the only effect mediating the actions ofPKCε in pancreatitis as shown in this report.
We further investigated other factors mediating the effect ofPKCε on necrosis.
We analyzed expression of Bcl-2 and Bcl-xL, which weremitochondrial prosurvival Bcl-2 proteins in pancreatitis. Therecent studies in our group (38, 48) indicated that these twoBcl-2 proteins could stabilize pancreatic mitochondria andprotect against necrosis in pancreatitis and that upregulation ofBcl-2 and Bcl-xL was a key protective mechanism against
Bcl-2
1 1 1.9 2.1 0.8 1.7 2.8 3.6
Bcl-XL
ERK1/2
1 2.2 2.4 2.8 1.5 2 3.2 3.8
A
CR (h)
WT PKCε -/-
0 0.5 4 7 0 0.5 4 7 0 0.5 4 7 0 0.5 4 7
WT PKCε -/-
COX IV
B
Bcl-2
Bcl-XL
Pancreatic tissue Pancreatic mitochondria
Density 1 1 2.6 2.2 1.1 1.8 2.2 3.7
1 0.8 2.2 4.8 1.2 1.8 1.8 5.8Density
Density
Density
Bcl-2mRNA
CR (h)
WT PKCε -/-
0 0 7 7 0 0 7 7
m18SmRNA
Bcl-XLmRNA
1 1 2.4 2.6 0.8 1.2 3.8 3.4
0.8 1 3.2 2.4 0.8 1.4 4.4 3.8
200 bp
100 bp
300 bp200 bp
100 bp50 bp
CR (h)
Fig. 8. Deletion of PKCε leads to a higher upregulation of pancreatic prosurvival Bcl-2 proteins in cerulein pancreatitis. Protein levels of Bcl-2 and Bcl-xL weremeasured by Western blot analysis for pancreatic tissue homogenates (A, left) and mitochondrial membrane fractions (B) at indicated times after the inductionof pancreatitis. The densities of Bcl-2 and Bcl-xL bands were quantified and normalized to the control. Blots of ERK1/2 or COX IV were for loading controls.RT-PCR (A, right) shows higher Bcl-2 and Bcl-xL mRNA expression in pancreatic tissue of PKCε�/� mice with cerulein (CR) pancreatitis. Mouse 18Sribosomal RNA was used as a reference (housekeeping) gene. For either Bcl-xL or m18S PCR, samples were run in a single gel but were not continuous, asindicated by a line between lanes. The densities of Bcl-2 and Bcl-xL mRNA bands were quantified and normalized to the controls. Numbers to the right of thegel are DNA size markers in base pairs.
Fig. 9. PKCε translocates to mitochondria in cerulein-induced pancreatitis. A: subcellular distribution of PKCε in early stage (30 min) of pancreatitis wasdetermined in cytosolic and mitochondrial-enriched membrane fractions by Western blot analysis. The densities of PKCε bands were quantified and normalizedto the saline controls. The blots were reprobed with an antibody against COX IV or LDH for loading controls and to assess the quality of separating membranefractions from cytosolic fractions. Shown are representative Western blots from 3 independent experiments. B: colocalization of PKCε with VDAC1, amitochondria marker in CR pancreatitis. Pancreatic tissue sections were double immunostained for PKCε (green) and VDAC1 (red), and secondary antibodieswere conjugated with Alexa 488 or 594. Nuclei were counterstained with DAPI. Images were visualized under confocal microscope. The original magnificationis �63. Bar represents 10 �m. DIC, differential interference contrast.
G559PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
necrosis in pancreatitis. We found more prominent upregula-tion of the pancreatic mitochondrial prosurvival Bcl-2 proteinsin cerulein-induced pancreatitis in PKCε�/� mice, which mayhave provided the protection against cell necrosis.
Notably, previous studies have demonstrated a differencebetween cancer cells and pancreatic acinar cells regarding thepredominant role of mitochondrial prosurvival/antiapoptoticBcl-2 proteins (2, 9, 48). For example, the major effect ofBcl-xL/Bcl-2 inhibitors in cancer cells is increased apoptosis(2, 9) resulting from stimulation of cytochrome c release (9). Incontrast, the predominant effect of the Bcl-xL/Bcl-2 inhibitorsin pancreatitis is ATP depletion and necrosis but not apoptosis,as shown in our recent studies (48, 38).
The mechanism by which PKCε upregulates mitochondrialBcl-2 proteins is unknown. We found that the pancreatitistreatment caused translocation of PKCε to the mitochondria.Our Western blot analyses of tissue fractions showed thatPKCε translocated from the cytosol to mitochondrial mem-branes upon cerulein stimulation. Using immunofluorescence,we observed that PKCε distribution after stimulation wasstrongly colocalized with the mitochondrial marker proteinVDAC1, confirming the presence of PKCε at cellular sites
occupied by mitochondria. These studies suggest that PKCεmay directly target mitochondria and modulate mitochondrialfunctions related to necrosis. These results could also accountin part for the decreased ATP reduction and attenuated necrosiswe found in pancreatitic acini, either treated with PKCε trans-location inhibitor or isolated from PKCε�/� mice.
In addition to mitochondrial death pathways, the presentstudy shows that PKCε has effects on nonmitochondrial mech-anisms of caspase activation downstream of cytochrome crelease. IAPs and antiapoptotic protein c-FLIP play a pivotalbraking role on apoptotic signaling by inhibiting caspases (12,16, 22, 23, 39, 47). Some of these molecules have been shownto be key determinants of the type of cell death during pan-creatitis in our studies (32, 54). Here we found that deletion ofPKCε resulted in significant decreases in pancreatic levels ofantiapoptotic proteins c-IAP2, survivin, and c-FLIPs in ceru-lein-induced pancreatitis. These results indicate that PKCεregulates pathways that inhibit apoptosis through both mito-chondrial and nonmitochondrial mechanisms.
The mechanisms of the downregulation of IAPs and FLIPsin pancreatitis caused by genetic inhibition of PKCε remain tobe studied. Our results suggested that this downregulation is
*
# *
*# *
C RT-PCR
FLIPL
GAPDH
FLIPS
50
3630
WT PKCε -/-0 0.5 4 7CR (h) 0 0.5 4 7
D W. Blot
Survivin
GAPDH
BW. Blot
WT PKCε -/-
Surv
ivin
pro
tein
(fo
ld o
verc
ontr
ol)
0 0.5 4 7 CR (h) 0 0.5 4 7
0
4
8
2
6
10
*
#
# *
FLIPSmRNA
m18SmRNA
WT PKCε -/-
0 0 7 7CR (h) 0 0 7 7
E RT-PCR
1 1.7 7 5.8 1 1.3 2.7 1.5
0
10
20
#
FLIP
s pr
otei
n (fo
ld o
ver c
ontr
ol)
c-IAP2
ERK1/2
A WT PKCε -/-
cIA
P-2
prot
ein
leve
l (fo
ld o
verc
ontr
ol )
0
0.5
1.5
1.0
2.0 #
0 0.5 4 7CR (h) 0 0.5 4 7
Density
200 bp100 bp
100 bp50 bp
SurvivinmRNA
WT PKCε -/-
0 0 7 7CR (h) 0 0 7 7
m18SmRNA
1 1 5.3 4.3 1 1 1.5 1.7 Density
200 bp100 bp
100 bp50 bp
Fig. 10. Deletion of PKCε decreases antiapo-ptotic proteins levels in cerulein pancreatitis.Western blot analyses of pancreas tissue ly-sate using antibodies against c-IAP2 (A),survivin (B), c-FLIP (C) to measure the pan-creatic levels of these proteins in cerulein(CR)-induced pancreatitis at indicated times(ERK1/2 or GAPDH: loading controls). Barfigures are the quantification of these proteinband densities that were compared with thesaline control. Graphs show means � SE(n � 2–3) for each condition. *P � 0.05 vs.WT mice with cerulein at the same timepoint as indicated. C and E: RT-PCR showsless mRNA expression of survivin and FLIPsin pancreatic tissue of PKCε�/� mice withcerulein pancreatitis. Mouse 18S ribosomalRNA was used as a reference (housekeeping)gene. The densities of survivin and FLIPsmRNA bands were quantified and normal-ized to the controls. B, C, E: samples wererun in a single gel but were not continuous,as indicated by a line between lanes.
G560 PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org

mediated at least in part through transcriptional mechanism,possibly through NF-�B inhibition. A wealth of evidenceindicates that transcriptional activation of survivin and c-FLIPthrough NF-�B pathways is crucial in regulating cell death (12,22, 23, 39, 47, 57). PKCε was shown to be required for NF-�Bactivation in isolated pancreatic acini (43, 44). Here we dem-onstrated that NF-�B activation in cerulein-induced pancreati-tis model was potently inhibited in PKCε�/� mice (Fig. 3A).These findings support the potential role of PKCε activation inregulation of IAPs through NF-�B activation. Nevertheless, thecomplete mechanism of reduction in IAPs with PKCε inhibi-tion needs further study.
Of additional importance, our study showed that deletion ofPKCε promoted degradation/inactivation of RIP in cerulein-induced pancreatitis. RIP, a key mediator of programmednecrosis in many diseases including acute pancreatitis (10, 14,20, 28, 33, 49), was previously found to be cleaved/inactivatedby caspase-3 and -8. Our findings suggest that increased RIPcleavage/inactivation may be a mechanism through whichdeletion of PKCε ameliorates necrosis in cerulein-inducedpancreatitis.
In summary, our studies present the first in vivo evidence thatPKCε is a critical mediator of necrosis in experimental pancreati-tis through its effects on mitochondrial and nonmitochondrialdeath pathways. Genetic inhibition of PKCε decreases necrosisand the severity of pancreatitis. As we summarized in Fig. 12,activation of PKCε in pancreatitis promotes necrosis throughregulating mitochondrial death pathways involving Bcl-2 familyproteins, OMP and cytochrome c release, and ATP depletion; andnonmitochondrial pathways involving IAPs, c-FLIPs, caspaseactivation/apoptosis, and RIP.
Although our findings on the role of PKCε in pancreatitiswere based on cerulein pancreatitis models, evidence sup-ports a role for PKCε in other models of experimentalpancreatitis and human pancreatitis. PKCε activation medi-ates key pathobiological processes in alcohol-induced pan-creatitis (44). Furthermore, the cholinergic system is knownto mediate exocrine pancreas functions and pancreatitisresponses in humans, which we have recapitulated in amodel of alcoholic pancreatitis (29). Hyperstimulation withcholinergic agonist carbachol also induces pancreatitis re-sponses such as NF-�B activation and zymogen activationin rat pancreatic acinar cells (51, 55), and carbachol-inducedstimulation of PKC� and PKCε activities are required forthese pathological responses (51, 55). In addition, PKCεactivation is observed in an early stage of arginine model ofpancreatitis (our unpublished data). PKC inhibitors signifi-cantly reduce biliary acute pancreatitis-induced tissue injury(45). These findings indicate a potential role for PKCεacross animal models and human pancreatitis. Thus wesuggest that targeting PKCε may be a novel approach forprevention or treatment of necrosis in acute pancreatitis.
ACKNOWLEDGMENTS
We thank Dr. Robert O. Messing (University of California, San Francisco)for providing the initial breeding pairs of PKCε deficiency mice. We also thankDr. Anna Gukovskaya for discussion in this study.
GRANTS
This study was supported by the Southern California Research Center forAlcoholic Liver and Pancreatic Diseases (NIH Grant P50-A11999); UCLACenter of Excellence in Pancreatic Diseases (NIH Grant 1P01AT0003960-01);the Department of Veterans Affairs Merits Grant; and Lee Summer StudentResearch Award from Southern California Research Center for Liver andPancreatic Diseases (to T. Tan).
RIP
ERK1/2
0 4 4 7 7CR (h)
WT PKCε -/-0 4 4 7 7
RIP
pro
tein
leve
l ( %
of c
ontr
ol )
0
40
80100
60
20
# *
#
*
0 4 7CR (h) 0 4 7
WT PKCε -/-
CleavedRIP
6436
22
98
Fig. 11. Deletion of PKCε promotes receptor-interacting protein kinase (RIP)degradation/inactivation in cerulein-induced pancreatitis. Western blot analysis ofpancreas tissue lysate using antibodies against RIP to measure the protein levels ofthe protein in cerulein (CR) pancreatitis at indicated times. The same samples wereused to rerun gel and blotted with ERK1/2 antibody to confirm equal proteinloading. Samples were run in a single gel but were not continuous, as indicated bya line between lanes. Bar figures are the quantification of these protein banddensities that were compared with saline (S) control. Values are means � SE (n �2–3) for each condition. *P � 0.05 vs. WT mice with CR at the same time pointas indicated; #, the 2 bars are compared statistically. Numbers to the right ofthe gels are protein size (kDa).
CCK/CR
PKCε
Mitochondrial Non-mitochondrial • Bak upregulation
• OMP opening
• Cytochrome crelease
• Bcl-2/Bcl-xLupregulation
• ATP depletion
• IAPs/FLIPs
• Caspaseactivation
• Apoptosis
• RIP cleavage
NecrosisNecrosisFig. 12. Scheme illustrating the regulation of necrosis in pancreatitis by PKCεthrough mitochondrial and nonmitochondrial pathways. OMP, outer mem-brane permeabilization.
G561PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org

DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.L., J.Y., T.T., W.J., O.A.M., and R.T.W. performed experiments; Y.L.,J.Y., T.T., W.J., A.L., O.A.M., and S.J.P. analyzed data; Y.L., J.Y., T.T., A.L.,and S.J.P. interpreted results of experiments; Y.L., J.Y., T.T., W.J., A.L.,O.A.M., R.T.W., and S.J.P. approved final version of manuscript; J.Y. andS.J.P. conception and design of research; J.Y. and O.A.M. prepared figures;J.Y. drafted manuscript; J.Y., R.T.W., and S.J.P. edited and revised manu-script.
REFERENCES
1. Armstrong JS. Mitochondrial membrane permeabilization: the sine quanon for cell death. BioEssays 28: 253–260, 2006.
2. Azmi AS, Mohammad RM. Non-peptidic small molecule inhibitorsagainst Bcl-2 for cancer therapy. J Cell Physiol 218: 13–21, 2009.
3. Bastani B, Yang L, Baldassare JJ, Pollo DA, Gardner JD. Cellulardistribution of isoforms of protein kinase C (PKC) in pancreatic acini.Biochim Biophys Acta 1269: 307–315, 1995.
4. Basu A, Sivaprasad U. Protein kinase Cepsilon makes the life and deathdecision. Cell Signal 19: 1633–1642, 2007.
5. Beil M, Leser J, Lutz MP, Gukovskaya A, Seufferlein T, Lynch G,Pandol SJ, Adler G. Caspase 8-mediated cleavage of plectin precedesF-actin breakdown in acinar cells during pancreatitis. Am J PhysiolGastrointest Liver Physiol 282: G450–G460, 2002.
6. Bhatia M, Wallig MA, Hofbauer B, Lee HS, Frossard JL, Steer ML,Saluja AK. Induction of apoptosis in pancreatic acinar cells reduces theseverity of acute pancreatitis. Biochem Biophys Res Commun 246: 476–483, 1998.
7. Bhatia M. Apoptosis vs. necrosis in acute pancreatitis. Am J PhysiolGastrointest Liver Physiol 286: G189–G196, 2004.
8. Cacace AM, Guadagno SN, Krauss RS, Fabbro D, Weinstein IB. Theepsilon isoform of protein kinase C is an oncogene when overexpressed inrat fibroblasts. Oncogene 8: 2095–2104, 1993.
9. Campas C, Cosialls AM, Barragán M, Iglesias-Serret D, SantidriánAF, Coll-Mulet L, de Frias M, Domingo A, Pons G, Gil J. Bcl-2inhibitors induce apoptosis in chronic lymphocytic leukemia cells. ExpHematol 34: 1663–1669, 2006.
10. Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M,Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factorreceptor-2 and receptor-interacting protein in programmed necrosis andantiviral responses. J Biol Chem 278: 51613–51621, 2003.
11. Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW.Suppression of tumor necrosis factor-induced cell death by inhibitor ofapoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci USA94: 10057–10062, 1997.
12. Deveraux QL, Reed JC. IAP family proteins-suppressors of apoptosis.Genes Dev 13: 239–252, 1999.
13. Edinger AL, Thompson CB. Death by design: apoptosis, necrosis andautophagy. Curr Opin Cell Biol 16: 663–669, 2004.
14. Galluzzi L, Kepp O, Kroemer GI. RIP kinases initiate programmednecrosis. J Mol Cell Biol 1: 8–10, 2009.
15. Gorelick F, Pandol S, Thrower E. Protein kinase C in the pancreaticacinar cell. J Gastroenterol Hepatol Suppl 1: S37–S41, 2008.
16. Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis andpancreatic cancer. Pancreatology 4: 567–586, 2004.
17. Gukovskaya AS, Gukovsky I, Jung Y, Mouria M, Pandol SJ. Chole-cystokinin induces caspase activation and mitochondrial dysfunction inpancreatic acinar cells. Roles in cell injury processes of pancreatitis. J BiolChem 277: 22595–22604, 2002.
18. Hackert T, Sperber R, Hartwig W, Fritz S, Schneider L, Gebhard M,Werner J. P-selectin inhibition reduces severity of acute experimentalpancreatitis. Pancreatology 9: 369–374, 2009.
19. Hafeez BB, Zhong W, Weichert J, Dreckschmidt NE, Jamal MS,Verma AK. Genetic ablation of PKC epsilon inhibits prostate cancerdevelopment and metastasis in transgenic mouse model of prostate ade-nocarcinoma. Cancer Res 71: 2318–2327, 2011.
20. He S, Wang L, Miao L, Wang T, Du F, Wang X. Receptor interactingprotein kinase-3 determines cellular necrotic response to TNF-alpha. Cell137: 1100–1111, 2009.
21. Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationshipbetween severity, necrosis, and apoptosis in five models of experimentalacute pancreatitis. Am J Physiol Cell Physiol 269: C1295–C1304, 1995.
22. Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M,Hatano M, Tokuhisa T, Mori N. Transcriptional activation of survivinthrough the NF-kappaB pathway by human T-cell leukemia virus type Itax. Int J Cancer 115: 967–974, 2005.
23. Kerbauy DM, Lesnikov V, Abbasi N, Seal S, Scott B, Deeg HJ.NF-kappaB and FLIP in arsenic trioxide (ATO)-induced apoptosis inmyelodysplastic syndromes (MDSs). Blood 106: 3917–3925, 2005.
24. Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D,Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW,Levine JD, Messing RO. A novel nociceptor signaling pathway revealedin protein kinase C epsilon mutant mice. Neuron 24: 253–260, 1999.
25. Krantic S, Mechawar N, Reix S, Quirion R. Apoptosis-inducing factor:a matter of neuron life and death. Prog Neurobiol 81: 179–196, 2007.
26. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permea-bilization in cell death. Physiol Rev 87: 99–163, 2007.
27. Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S.Cellular FLICE-inhibitory protein splice variants inhibit different steps ofcaspase-8 activation at the CD95 death-inducing signaling complex. J CellBiochem 276: 20633–20640, 2001.
28. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domainkinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13:2514–2526, 1999.
29. Lugea A, Gong J, Nguyen J, Nieto J, French SW, Pandol SJ. Cholin-ergic mediation of alcohol-induced experimental pancreatitis. Alcohol ClinExp Res 34: 1768–1781, 2010.
30. Lüthen RE, Niederau C, Ferrell LD, Grendell JH. Energy metabolismin mouse pancreas in response to different dosages of a CCK analogue.Pancreas 11: 141–146, 1995.
31. Lüthen RE, Niederau C, Grendell JH. Intrapancreatic zymogen activa-tion and levels of ATP and glutathione during caerulein pancreatitis inrats. Am J Physiol Gastrointest Liver Physiol 268: G592–G604, 1995.
32. Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I,Gukovskaya AS. Cell death in pancreatitis: caspases protect from necro-tizing pancreatitis. J Biol Chem 281: 3370–3381, 2006.
33. Moquin D, Chan FK. The molecular regulation of programmed necroticcell injury. Trends Biochem Sci 35: 434–441, 2010.
34. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K,Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D depen-dent mitochondrial permeability transition regulates some necrotic but notapoptotic cell death. Nature 434: 652–658, 2005.
35. Newton AC. Protein kinase C: structure, function, and regulation. J BiolChem 270: 28495–28498, 1995.
36. Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids andactivation of protein kinase C. Science 258: 607–614, 1992.
37. Odinokova IV, Sung KF, Mareninova OA, Hermann K, EvtodienkoY,Andreyev A, Gukovsky I, Gukovskaya AS. Mechanisms regulatingcytochrome c release pancreatic mitochondria. Gut 58: 431–442, 2009.
38. Odinokova IV, Sung KF, Mareninova OA, Hermann K, EvtodienkoY,Andreyev A, Gukovsky I, Gukovskaya AS. Bcl-xL and Bcl-2 prosur-vival proteins protect against necrosis in pancreatitis. Pancreas 35: 420,2007.
39. Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: benchto the bedside. Gastroenterology 132: 1127–11251, 2007.
40. Peter ME. The flip side of FLIP. Biochem J 382: e1–e3, 2004.41. Petersen OH. Specific mitochondrial functions in separate sub-cellular
domains of pancreatic acinar cells. Pflügers Arch 464: 77–87, 2012.42. Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: a specific
form of programmed cell death? Exp Cell Res 283: 1–16, 2003.43. Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR
Jr, Shimosegawa T, Pandol SJ. PKC-� and -ε regulate NF-�B activationinduced by cholecystokinin and TNF-� in pancreatic acinar cells. Am JPhysiol Gastrointest Liver Physiol 287: G582–G591, 2004.
44. Satoh A, Gukovskaya AS, Reeve JR Jr, Shimosegawa T, Pandol SJ.Ethanol sensitizes NF-�B activation in pancreatic acinar cells througheffects on protein kinase C-ε. Am J Physiol Gastrointest Liver Physiol291: G432–G438, 2006.
45. Shi C, Zhao X, Wang X, Zhao L, Andersson R. Potential effects of PKCor protease inhibitors on acute pancreatitis-induced tissue injury in rats.Vascul Pharmacol 46: 406–411, 2007.
G562 PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org

46. Soh JW, Lee YS, Weinstein IB. Effects of regulatory domains of specificisoforms of protein kinase C on growth control and apoptosis in MCF-7breast cancer cells. J Exp Ther Oncol 3: 115–126, 2003.
47. Stehlik C, Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J.NF-�B regulated X-chromosome-linked IAP gene expression protectsendothelial cells from TNF-� induced apoptosis. J Exp Med 188: 211–216,1998.
48. Sung KF, Odinokova IV, Mareninova OA, Rakonczay Z Jr, Hegyie P,Pandol SJ, Gukovsky I, Gukovskaya AS. Prosurvival Bcl-2 proteinsstabilize pancreatic mitochondria and protect against necrosis in experi-mental pancreatitis. Exp Cell Res 315: 1975–1989, 2009.
49. Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayamaa M,Debouckc CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interact-ing protein kinases mediate retinal detachment-induced photoreceptornecrosis and compensate for inhibition of apoptosis. Proc Natl Acad SciUSA 107: 21695–21700, 2010.
50. Thrower EC, Osgood S, Shugrue CA, Kolodecik TR, Chaudhuri AM,Reeve JR Jr, Pandol SJ, Gorelick FS. The novel protein kinase Cisoforms -� and -ε modulate caerulein-induced zymogen activation inpancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol 294:G1344–G1353, 2008.
51. Thrower EC, Yuan J, Usmani A, Liu Y, Jones C, Minervini SN,Alexandre M, Pandol SJ, Guha S. A novel protein kinase D inhibitorattenuates early events of experimental pancreatitis in isolated rat acini.Am J Physiol Gastrointest Liver Physiol 300: G120–G129, 2011.
52. Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane per-meability transition and cell death. Biochim Biophys Acta 1757: 1297–1300, 2006.
53. Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permea-bility transition in cell death. Apoptosis 12: 835–840, 2007.
54. Yuan J, Liu Y, Tan T, Guha S, Gukovsky I, Gukovskaya A, PandolSJ. Protein kinase D regulates cell death pathways in experimentalpancreatitis. Front Physiol 3: 60, 2012.
55. Yuan J, Lugea A, Zheng L, Gukovsky I, Edderkaoui M, Rozengurt E,Pandol SJ. Protein kinase D1 mediates NF-�B activation induced bycholecystokinin and cholinergic signaling in pancreatic acinar cells. Am JPhysiol Gastrointest Liver Physiol 295: G1190–G1201, 2008.
56. Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev 20:1–15, 2006.
57. Zou T, Rao JN, Guo X, Liu L, Zhang HM, Strauch ED, Bass BL,Wang JY. NF-�B-mediated IAP expression induces resistance of intesti-nal epithelial cells to apoptosis after polyamine depletion. Am J PhysiolCell Physiol 286: C1009–C1018, 2004.
G563PKCε REGULATES CELL DEATH IN PANCREATITIS
AJP-Gastrointest Liver Physiol • doi:10.1152/ajpgi.00432.2013 • www.ajpgi.org
Related Documents











