Genetic and Molecular Basis of Cardiac Arrhythmias: Impact on Clinical Management Parts I and II* Silvia G. Priori, MD, PhD; Jacques Barhanin, MD, PhD; Richard N.W. Hauer, MD; Wilhelm Haverkamp, MD; Habo J. Jongsma, MD, PhD; Andre ´ G. Kleber, MD, PhD; William J. McKenna, MD; Dan M. Roden, MD, PhD; Yoram Rudy, PhD; Ketty Schwartz, PhD; Peter J. Schwartz, MD; Jeffrey A. Towbin, MD; Arthur M. Wilde, MD, PhD Abstract—Genetic approaches have succeeded in defining the molecular basis of an increasing array of heart diseases, such as hypertrophic cardiomyopathy and the long-QT syndromes, associated with serious arrhythmias. Importantly, the way in which this new knowledge can be applied to managing patients and to the development of syndrome-specific antiarrhythmic strategies is evolving rapidly because of these recent advances. In addition, the extent to which new knowledge represents a purely research tool versus the extent to which it can be applied clinically is also evolving. The present article represents a consensus report of a meeting of the European Working Group on Arrhythmias. The current state of the art of the molecular and genetic basis of inherited arrhythmias is first reviewed, followed by practical advice on the role of genetic testing in these and other syndromes and the way in which new findings have influenced current understanding of the molecular and biophysical basis of arrhythmogenesis. (Circulation. 1999;99:518-528.) Key Words: death, sudden n genetics n arrhythmia n molecular biology n electrophysiology C linical cardiologists who manage arrhythmias are in- creasingly faced with new complexities in management decisions. The once obscure science and jargon of medical genetics is assuming a much more prominent position in the mainstream medical literature, with almost weekly reports of new mutations to explain what once seemed to be very obscure diseases. This rapidly expanding knowledge base places the clinician (who usually trained when the concepts were not a major component of the medical school or fellowship training curricula) at a disadvantage in making day-to-day decisions with respect to managing common symptoms, such as unexplained syncope or heart failure. Even entertaining a diagnosis such as the congenital long-QT syndrome (LQTS) or hypertrophic cardiomyopathy used to be a medical curiosity. Now, with increased public and physician awareness of these and even more esoteric condi- tions, the questions in patient management have become more common and more complex. They include not only broad questions such as “How can I establish (or better yet, rule out) a diagnosis?” but also more specific issues such as “Should this patient undergo genetic testing? Where? How? And how can I interpret the results?” The first and second parts of this article attempt to answer these questions. They neither teach molecular genetics nor provide an exhaustive review of the current state of the art of molecular and genetic cardiology relevant to arrhythmias. Rather, they try to put into a very practical perspective the ways in which ongoing progress in genetics may affect day-to-day clinical management. The recognition that diversity in cardiac electrophysiology, and indeed in many aspects of cardiac function, can be attributed to variable expression of specific genes or variabil- ity in the function of their protein products has the potential to alter the way in which we think about normal and abnormal electrical heart function. The third part of the article reviews the potential for a genetic approach to understanding diversity Received July 15, 1998; revision received October 28, 1998; accepted November 18, 1998. *Part III of this article will be published in Circulation. 1999;99(5):February 9 issue. The article is also published in Eur Heart J. 1999;20:179 –195. From the Molecular Cardiology and Electrophysiology Laboratory (S.G.P.), Fondazione S. Maugeri, IRCCS, Pavia, Italy; Institut de Pharmacologie Moleculaire et Cellulaire (J.B.), Laboratoire de Genetique de la Neurotrasmission, CNRS, Valbonne, France; University Hospital of Utrecht, Heart Lung Institute (R.N.W.H.), Netherlands; Medizinische Klinik und Poliklinik (W.H.), Innere Medizin C-Universitat Munster, Germany; Physiologic Laboratory (H.J.J.), University of Utrecht, Netherlands; Department of Physiology (A.G.K.), University of Bern, Switzerland; Department of Cardiological Sciences (W.J.M.), St. George’s Hospital Medical School, London, UK; Division of Medicine and Pharmacology (D.M.R.), Vanderbilt University Medical Center, Nashville, Tenn; Department of Biomedical Engineering (Y.R.), Case Western Reserve University, Cleveland, Ohio; UR 153 INSERM (K.S.), Pavillon Rambuteau, Groupe Hopitalier Pitie-Salpetriere, Paris, France; Dipartimento di Cardiologia (P.J.S.), Policlinico S. Matteo, IRCCS, Pavia, Italy; Ped Molecular Cardiology (J.A.T.), Baylor College of Medicine, Texas Children’s Hospital, Houston, Tex; and Department of Clinical and Experimental Cardiology (A.M.W.), Academic Medical Centre, Amsterdam, Netherlands. Correspondence to Silvia G. Priori, MD, PhD, Molecular Cardiology and Electrophysiology Laboratory, Fondazione “S. Maugeri” IRCCS, Via Ferrata, 8, 27100 Pavia, Italy. E-mail [email protected] © 1999 American Heart Association, Inc. Circulation is available at http://www.circulationaha.org 518 by guest on August 15, 2015 http://circ.ahajournals.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Genetic and Molecular Basis of Cardiac Arrhythmias:Impact on Clinical Management
Parts I and II*Silvia G. Priori, MD, PhD; Jacques Barhanin, MD, PhD; Richard N.W. Hauer, MD;Wilhelm Haverkamp, MD; Habo J. Jongsma, MD, PhD; Andre G. Kleber, MD, PhD;
William J. McKenna, MD; Dan M. Roden, MD, PhD; Yoram Rudy, PhD; Ketty Schwartz, PhD;Peter J. Schwartz, MD; Jeffrey A. Towbin, MD; Arthur M. Wilde, MD, PhD
Abstract—Genetic approaches have succeeded in defining the molecular basis of an increasing array of heart diseases, suchas hypertrophic cardiomyopathy and the long-QT syndromes, associated with serious arrhythmias. Importantly, the wayin which this new knowledge can be applied to managing patients and to the development of syndrome-specificantiarrhythmic strategies is evolving rapidly because of these recent advances. In addition, the extent to which newknowledge represents a purely research tool versus the extent to which it can be applied clinically is also evolving. Thepresent article represents a consensus report of a meeting of the European Working Group on Arrhythmias. The currentstate of the art of the molecular and genetic basis of inherited arrhythmias is first reviewed, followed by practical adviceon the role of genetic testing in these and other syndromes and the way in which new findings have influenced currentunderstanding of the molecular and biophysical basis of arrhythmogenesis.(Circulation. 1999;99:518-528.)
Key Words: death, suddenn geneticsn arrhythmian molecular biologyn electrophysiology
Clinical cardiologists who manage arrhythmias are in-creasingly faced with new complexities in management
decisions. The once obscure science and jargon of medicalgenetics is assuming a much more prominent position in themainstream medical literature, with almost weekly reports ofnew mutations to explain what once seemed to be veryobscure diseases. This rapidly expanding knowledge baseplaces the clinician (who usually trained when the conceptswere not a major component of the medical school orfellowship training curricula) at a disadvantage in makingday-to-day decisions with respect to managing commonsymptoms, such as unexplained syncope or heart failure.Even entertaining a diagnosis such as the congenital long-QTsyndrome (LQTS) or hypertrophic cardiomyopathy used tobe a medical curiosity. Now, with increased public andphysician awareness of these and even more esoteric condi-tions, the questions in patient management have become morecommon and more complex. They include not only broad
questions such as “How can I establish (or better yet, rule out)a diagnosis?” but also more specific issues such as “Shouldthis patient undergo genetic testing? Where? How? And howcan I interpret the results?”
The first and second parts of this article attempt to answerthese questions. They neither teach molecular genetics norprovide an exhaustive review of the current state of the art ofmolecular and genetic cardiology relevant to arrhythmias.Rather, they try to put into a very practical perspective theways in which ongoing progress in genetics may affectday-to-day clinical management.
The recognition that diversity in cardiac electrophysiology,and indeed in many aspects of cardiac function, can beattributed to variable expression of specific genes or variabil-ity in the function of their protein products has the potentialto alter the way in which we think about normal and abnormalelectrical heart function. The third part of the article reviewsthe potential for a genetic approach to understanding diversity
Received July 15, 1998; revision received October 28, 1998; accepted November 18, 1998.*Part III of this article will be published inCirculation. 1999;99(5):February 9 issue. The article is also published inEur Heart J.1999;20:179–195.From the Molecular Cardiology and Electrophysiology Laboratory (S.G.P.), Fondazione S. Maugeri, IRCCS, Pavia, Italy; Institut de Pharmacologie
Moleculaire et Cellulaire (J.B.), Laboratoire de Genetique de la Neurotrasmission, CNRS, Valbonne, France; University Hospital of Utrecht, Heart LungInstitute (R.N.W.H.), Netherlands; Medizinische Klinik und Poliklinik (W.H.), Innere Medizin C-Universitat Munster, Germany; Physiologic Laboratory(H.J.J.), University of Utrecht, Netherlands; Department of Physiology (A.G.K.), University of Bern, Switzerland; Department of Cardiological Sciences(W.J.M.), St. George’s Hospital Medical School, London, UK; Division of Medicine and Pharmacology (D.M.R.), Vanderbilt University Medical Center,Nashville, Tenn; Department of Biomedical Engineering (Y.R.), Case Western Reserve University, Cleveland, Ohio; UR 153 INSERM (K.S.), PavillonRambuteau, Groupe Hopitalier Pitie-Salpetriere, Paris, France; Dipartimento di Cardiologia (P.J.S.), Policlinico S. Matteo, IRCCS, Pavia, Italy; PedMolecular Cardiology (J.A.T.), Baylor College of Medicine, Texas Children’s Hospital, Houston, Tex; and Department of Clinical and ExperimentalCardiology (A.M.W.), Academic Medical Centre, Amsterdam, Netherlands.
Correspondence to Silvia G. Priori, MD, PhD, Molecular Cardiology and Electrophysiology Laboratory, Fondazione “S. Maugeri” IRCCS, Via Ferrata,8, 27100 Pavia, Italy. E-mail [email protected]
© 1999 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org
518 by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

in cardiac function, focusing in particular on ion channels andgap junction proteins as the central players in normal andabnormal electrophysiology. Moreover, integration of molec-ular function into a single cell and of single cells into cellularnetworks reveals a multitude of interactions that eventuallydetermine the generation and conduction of the cardiac actionpotential and therefore arrhythmogenesis.
This text is the outcome of a workshop convened by theStudy Group on Molecular Basis of Arrhythmias of theWorking Group on Arrhythmias of the European Society ofCardiology.
Part I: Inherited Arrhythmogenic DisordersLong-QT SyndromeLQTS is a familial disease1,2 characterized by abnormallyprolonged ventricular repolarization and high risk of malig-nant ventricular tachyarrhythmias, often but not always oc-curring in the setting of high adrenergic activity, ie, physicalor emotional stress. Two major clinical syndromes have beencharacterized on the basis of the pattern of transmission of thedisease: a more common autosomal dominant form with apure cardiac phenotype (Romano-Ward3) and a rarer autoso-mal recessive form characterized by the coexistence ofcardiac abnormalities and congenital deafness (Jervell andLange-Nielsen4).
LQTS GenesFive loci5–8 have been associated with the Romano-WardLQTS, and they are located on chromosomes 3, 4, 7, 11, and21 (Table). As illustrated in Figure 1, 4 LQTS disease genes,each encoding an ion channel protein, have been identified:SCN5A, encoding the cardiac sodium channel (chromosome3)6,9–11; HERG, encoding theIKr potassium channel protein(chromosome 7)6,12; KvLQT1, encoding thea-subunit of theIKs potassium channel protein (chromosome 11)5,13,14; andKCNE1, encodingminK, an ancillary subunit for theIKs
channel complex (chromosome 21).15,16 The gene at thechromosome 4 locus (LQT4) has not been identified. Fami-lies linked to none of these 5 loci have been described, sothere are other disease genes. The recognition that LQTS isactually a group of ion channel diseases with a similarphenotype has led to the new terminology for mutations:(1) LQT1 on KvLQT1, (2) LQT2 onHERG, (3) LQT3 onSCN5A, and (4) LQT5 onminK. Although the prevalence ofeach variant of LQTS has not been precisely defined, LQT1is the most frequently encountered form, whereas LQT3 andLQT5 are rare.
Mutations in LQTS GenesMost of the mutations identified to date in LQTS genes aremissense mutations. These mutations are not confined to asingle location but rather are located at various positionswithin each gene in different families. Thus, in most affectedfamilies, LQTS is due to a distinctive, or “private,” mutation.This remarkable genetic heterogeneity probably contributesto variability in the clinical presentation.
A few mutational “hot spots” (ie, specific positions withina gene mutated in multiple families) have been identified inKvLQT117 andHERG.18 Unrelated kindreds worldwide with
Genetics of Arrhythmogenic Disorders
Disease Locus Gene Reference
X-linked DCM Xp.21.2 Dystropin Muntoni et al70
Muntoni et al71
Milasin et al72
Ortiz-Lopez et al68
Barth syndrome Xq28 G.4.5 Bione et al69
ADDCM 1q32 ? Durand et al73
2p31 ? Siu et al74
9q13-q21 ? Krajinovic et al75
10q21-q23 ? Bowles et al76
3p22-p25 ? Olson et al78
15q14 Actin Olson et al79
CDDC 1p1-1q1 ? Kass et al77
FHCM 1q3 cTnT Thierfelder et al39
Watkins et al54
3p MELC Poetter et al38
7q3 ? MacRae et al43
11p11.2 MyPBC Bonne et al40
Watkins et al41
12q23-q24.3 MRLC Poetter et al38
14q11-q12 BetaMHC Geisterfer et al37
15q2 AlfaTM Thierfelder et al39
Watkins et al54
19p13.2-q13.2 cTnI Kimura et al42
FAfib 10q22-q24 ? Brugada et al89
PFHB-I 19q13.2-q13.3 ? Brink et al90
De Meeus et al92
LQTS (R-W) 3p21-p23 SCN5A Wang et al9
4q25-q27 ? Schott et al8
7q35-q36 HERG
Curran et al12
11p15.5 KvLQT1 Wang et al5
21q22.1-p22 MinK Splawski et al15
LQTS (JLN) 11p15.5 KvLQT1 Neyroud et al19
Splawski et al20
21q22.1-q22 MinK Schultze-Bahr et al16
ARVD* 1q42-q43 ? Rampazzo et al61
14q12-q22 ? Severini et al60
14q23-q24 ? Rampazzo et al59
2q32.1-q32.2 ? Rampazzo et al62
Naxos disease 17q21 ? Coonar et al58
F-IVF 3p21-p23 SCN5A Chen et al82
CDDC, indicates conduction defect and dilated cardiomyopathy; Fafib,familial Afib; LQTS (R-W), LQTS, Romano-Ward type; LQTS (JLN), LQTS, Jervelland Lange-Nielsen type; ARVD, arrhythmogenic right ventricular cardiomyop-athy; and F-IVF, familial idiopathic ventricular fibrillation.
*Note added in proof: A new locus for ARVD has been identified onchromosome 3p23 (Ahmad F, Li D, Karibe A, Gonzalez O, Topscott T, Hill R,Weilbaecher D, Blackie P, Furey M, Gardner M, Bachinski LL, Roberts R.Localization of a gene responsible for arrhythmogenic right ventricular dyspla-sia to chromosome 3p23. Circulation. 1998;98:2791–2795).
Priori et al February 2, 1999 519
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

the same mutation can therefore be studied to test the logicalhypothesis that they share common clinical or epidemiolog-ical features. Contrary to expectations, initial studies indicatethat substantial phenotypic heterogeneity remains even withan identical LQTS gene abnormality. This, in turn, suggeststhat variable expression of as-yet-unidentified “modifiergenes” contributes to the clinical manifestations of thedisease.
The Jervell and Lange-Nielsen (autosomal recessive)variant of LQTS (in which affected subjects have espe-cially long QT intervals) arises in individuals who inheritabnormalKvLQT1or minK alleles from both parents. Theabnormal allele can be the same (usually in consanguine-ous families)19,20 or different (compound heterozygosi-ty).16 Thus, parents of subjects with Jervell and Lange-Nielsen carry LQTS mutations, although most (but not all)are asymptomatic. Recently, a family with apparentlyautosomal recessive LQTS without deafness has also beenidentified.21 These findings all suggest an effect of genedosage to determine phenotype (2 abnormal alleles appearto be worse than 1) and also highlight the extraordinaryvariability in LQTS phenotype.22–25 The location of themutations within the gene (eg, close to the regions encod-ing specific structures such as the pore, the voltage sensor,the S1-S6 region, or the N- or C-terminal portions) or thetype of mutation (the nature of the amino acid substitution,missense mutation versus deletions or insertions) may alsoplay a role.
Functional Consequences of MutationsThe channels carryingIKr and IKs are multimeric; that is,alleles from both parents are thought to contribute to thechannel complexes. When mutations inKvLQT1,KCNE1, orHERG are expressed alone or with wild-type alleles inoocytes or in other cell lines, they exhibit “loss of function,”ie, the total current carried by the defective channel com-plexes is reduced. Some of the mutations not only reducecurrent but also modify channel kinetics. ManyHERG andKvLQT1mutations have been defined as “dominant negative”because when the mutant protein is coexpressed with the
native protein,13,14,26,27the resulting defect in current exceeds50%. One explanation for this phenomenon is that incorpo-ration of a single abnormal protein subunit into the tetramericchannel structure is sufficient to alter the overall behavior ofthe current.
By contrast, mutations in theSCN5Achannels cause a“gain of function.”10,11 These mutations produce a persistentlate INa that is not present physiologically and that is due todefective inactivation. In most described mutations, theINa isincreased because of late reopenings of the channels, whereasin the 3-amino-acid deletion (DKPQ), long-lasting bursts ofchannel activity are also present. These mutations also differin severity, with theDKPQ deletion being associated with aquantitatively larger increase in late sodium inward current.11
It is generally difficult to develop specific therapies for loss offunction (eg, the K1 channel defects described above). Bycontrast, the gain of abnormal function exhibited by mutantSCN5Agene products raises the possibility that a cure couldbe accomplished by pharmacological agents that inhibit thegained function, ie, block the lateINa. Indeed, some datasuggest that these currents are especially sensitive to block bymexiletine or lidocaine.10,11
Genotype-Phenotype CorrelationsThe different time- and voltage-dependence of the ioniccurrents involved in LQTS may help explain some aspects ofthe variable phenotype and raise the possibility of gene-specific treatment. Indeed, available data on several hundredgenotyped patients indicate the existence of gene-specificdifferences in the triggers for cardiac events.28 Exercise-related events dominate the clinical picture inIKs-relatedLQTS (LQT1).28 IKs is the predominant K1 current in condi-tions of high sympathetic activity, particularly at shorter cyclelengths. Thus, reducedIKs will be predicted to lead toinadequate action potential shortening with adrenergic stressand thereby account for the high prevalence of arrhythmicevents in these patients during exercise. By contrast, mostLQT3 patients experience events during sleep or at rest; theyare also able to markedly shorten their QT interval duringexercise.29 In this case, it seems likely that the presence of
Figure 1. LQTS genes. Chromosomal loca-tions of genes and predicted topology of ionchannel proteins associated with geneticvariants of LQTS.
520 Molecular Biology and Arrhythmias
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from
normal K1 currents produces normal action potential short-ening during exercise; however, at rest, defective inactivationof INa will result in an increase in the plateau inward Na1
current. This apparently nice distinction between LQT1 andLQT3, however, is complicated by the reality that LQT2patients also tend to display events both at rest and duringexercise, thus pointing to the persistent limitations in currentunderstanding.
There is an emerging sense that gene-specific therapy maybe feasible for some forms of LQTS. This relates both topharmacological therapy and to advice regarding lifestyle. Adisorder based on disturbed inactivation kinetics of thesodium channel (LQT3) seems likely to respond to a sodiumchannel blocker. Indeed, in LQT3 patients, the QT intervalseems to shorten more than in LQT1 and LQT2 patients inresponse to mexiletine, but individual exceptions do exist,29
and significant shortening of QT intervals by sodium channelblockers has been reported in some LQT2 patients.30 It is alsopossible that although mexiletine or similar drugs shorten QTin LQT3, b-blockade might still be necessary to suppressarrhythmias. Because the amplitude ofIKr increases whenextracellular potassium concentration is increased, attemptshave been undertaken to increase K1 levels in LQTS patients.To date, QT interval has been shown to shorten significantlyin LQT2 patients,31 but neither LQT1 nor LQT3 patients haveyet been tested with this approach. BecauseIKr function isnormal in the latter subjects, elevating potassium to increaseIKr should shorten QT in them as well. The putative role ofIKs
in cardiac physiology suggests an especially favorable effectof b-blockade and the avoidance of vigorous increase in heartrate (ie, competitive sports) in LQT1 and LQT5. Theseexamples demonstrate that gene-specific therapy may befeasible in LQTS. However, it should be emphasized thatlong-term trials are not yet available and that at present,b-blockers remain the first-choice therapy.
Drug-Induced LQTSIt has long been postulated that drug-induced LQTS mightrepresent a genetically mediated “forme fruste” of LQTS.32
Recent studies have identified relatively large numbers ofindividuals who carry “silent” mutations on LQTS genes.22–25
Thus, these persons, whose LQTS mutations by themselvesproduce an alteration in repolarizing currents that is insuffi-cient to prolong the QT interval at rest, may be especiallysensitive to any drug that affects K1 currents. The combina-tion of even a modest degree ofIKr blockade, induced by avariety of drugs used for multiple purposes,33 and such silentmutations could thereby produce the major prolongation inaction potential that triggers the onset of torsades de pointes.Indeed, occasional patients with typical drug-induced LQTSand underlying mutations on LQTS genes have now beenidentified. However, this phenomenon is sufficiently rare thatgenetic testing in patients with drug-induced LQTS is not yetwarranted in the absence of other indications (eg, familyhistory, long baseline QT).34,35
Familial Hypertrophic CardiomyopathyHypertrophic cardiomyopathy36 is transmitted as an autoso-mal dominant disease. Its clinical phenotype is characterizedby unexplained and inappropriate clinical left and/or rightventricular hypertrophy, which may be severe (4 to 5 cm),mild, or even absent. Characterization of the distribution ofleft ventricular hypertrophy is arbitrary, but by convention,hypertrophy is considered to be either asymmetrical septal,concentric, or predominantly distal ventricular hypertrophy.Any pattern of hypertrophy may be seen, however, includinghypertrophy confined to the posterior or free wall. Charac-teristic histological features include myocyte disarray sur-rounding areas of increased loose connective tissue.
Clinically, there is marked hemodynamic heterogeneityamong patients with familial hypertrophic cardiomyopathy(FHCM). Systolic function may be hyperdynamic (with or
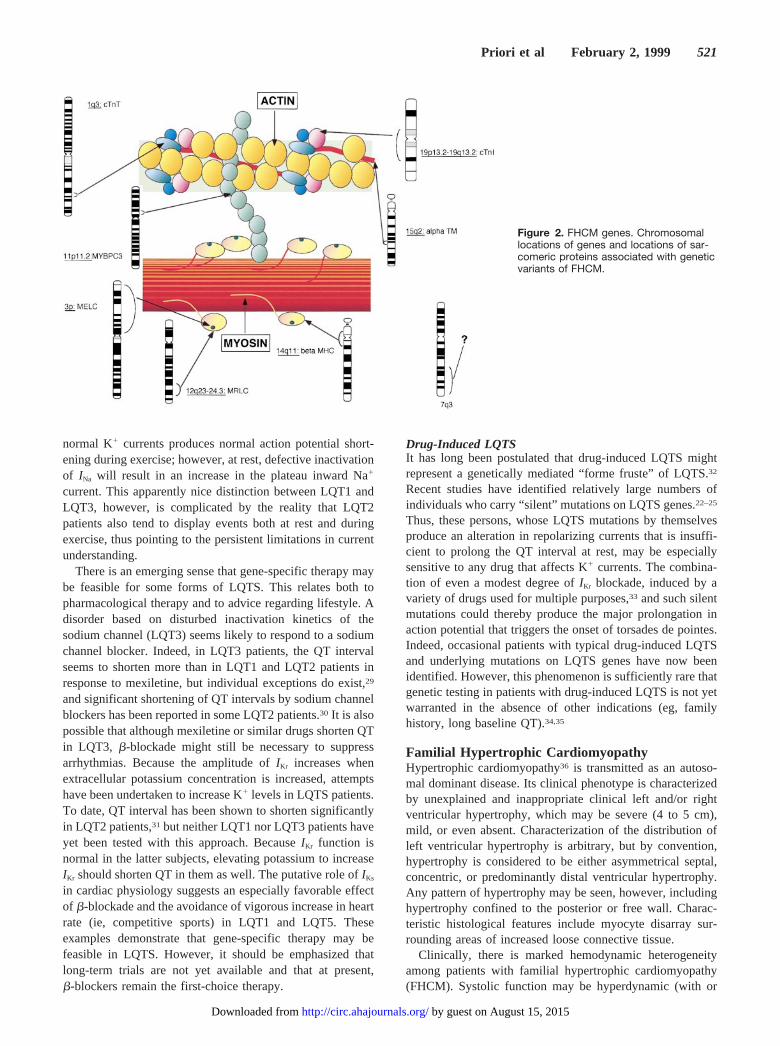
Figure 2. FHCM genes. Chromosomallocations of genes and locations of sar-comeric proteins associated with geneticvariants of FHCM.
Priori et al February 2, 1999 521
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

without obstruction), “normal,” or impaired (10% to 15%).Diastolic dysfunction is the usual physiological abnormality,although the precise abnormality of ventricular filling andcompliance is extremely variable.
FHCM-related arrhythmias occur both at the ventricularand at the atrial levels. Importantly, sudden cardiac death inFHCM is not necessarily caused by ventricular arrhythmias.Atrial fibrillation (Afib) in the presence of an accessorypathway, bradyarrhythmias, and ischemia all may lead tosudden death. In patients withb-myosin heavy chain–relatedFHCM (likely the majority), hypertrophy itself does not seemto be the main determinant of malignant ventricular arrhyth-mia. One caveat in interpreting electrophysiological changesin these settings is that a common secondary response toinjury (such as pressure overload or coronary occlusion) iscardiac hypertrophy, which in diseased hearts then producesfurther functional changes, notably in calcium handling.Thus, the extent to which any of the observed electrophysi-ological alterations are primary or secondary to the responseto the disease process requires further study.
FHCM GenesAs illustrated in Figure 2, there is considerable geneticheterogeneity in FHCM. Mutations in 7 sarcomeric proteingenes have been identified in families with FHCM (Table).These are (1)b-myosin heavy chain on chromosome 14,37
(2) cardiac essential myosin light chain on chromosome 3,38
(3) cardiac regulatory myosin light chain on chromosome12,38 (4) cardiac troponin T on chromosome 1,39 (5)a-tropomyosin on chromosome 15,39 (6) cardiac myosin-binding protein C on chromosome 11,40,41 and (7) cardiactroponin I on chromosome 19.42 An additional locus has beenidentified on chromosome 7 in a large family with bothFHCM and cardiac preexcitation (Wolff-Parkinson-Whitesyndrome [WPW]).43
The prevalence of the different gene abnormalities inFHCM is being delineated. To date, information in,100genotyped families suggests that mutations inb-myosinheavy chains and myosin-binding protein C are more com-mon than the others. In addition to this locus heterogeneity,there is, as in LQTS, marked allelic heterogeneity for all therecognized disease genes, and to date,.85 different muta-tions have been reported (for reviews see References 34, 44,and 45). The majority of mutations are missense mutations,although for the cardiac myosin-binding protein C gene, mostof the mutations lead to an early stop codon resulting intruncated mutant proteins.46
Functional studies of mutant myosin indicate that sarco-meric contractile performance is depressed.47–49This, in turn,suggests that myocyte hypertrophy characteristic of FHCMreflects a compensatory response. The molecular (or other)determinants of myocyte disarray and myocardial fibrosis(interstitial and replacement) remain unclear. It may well bethat these latter responses relate to the type of mutation (eg,greater with troponin-related disease) and that sudden deathand clinical arrhythmia are the clinical consequences ofextensive disarray and fibrosis.
Genotype-Phenotype CorrelationsInformation on the genotype-phenotype relation in FHCM isstill preliminary, because the published data on genotyped
patients relate to only a few hundred individuals from centersthat may reflect different referral biases. It is neverthelessclear that the phenotype varies not only with the type ofmutation but also within individuals bearing the same muta-tion. The 403 codon inb-myosin is a hot spot for mutations;the arginine-to-glutamine mutation is associated with a poorprognosis, whereas the arginine-to-tryptophan mutation ap-pears to be more benign.50–52Current practice suggests that ifECG and 2-dimensional echocardiography are normal by age25 years, then the patient can be safely reassured that he orshe will not develop clinical FHCM. However, myosin-binding protein C mutations appear to be associated withage-related penetrance during adult life.40,41,53Further infor-mation confirming the impression that adult onset of diseaseis an important feature seen with myosin-binding protein Cmutations would thus have a significant impact on manage-ment and counseling. The disease caused by troponin Tmutations appears to be associated with mild or absenthypertrophy, a 20% to 25% incidence of nonpenetrance, anda high incidence of premature sudden death (possibly greaterin young men, although the numbers are small), which canoccur even in the absence of significant clinical left ventric-ular hypertrophy.54–56
Arrhythmogenic Right Ventricular DysplasiaArrhythmogenic right ventricular dysplasia (ARVD) is arecently recognized familial cardiomyopathy.57 The disease ischaracterized by fibrofatty replacement of the right ventric-ular myocardium and life-threatening ventriculartachyarrhythmias originating from the right ventricle. Occa-sionally, the left ventricular myocardium is involved as well.Disease progression is associated with left ventricular in-volvement (50%), atrial dilatation, and arrhythmias withembolic risk. Malignant ventricular arrhythmias are a com-mon manifestation of the disease. Inducibility and reproduc-ibility in the clinical electrophysiological laboratory is high,suggesting that reentrant mechanisms related to the distinc-tive structural changes are likely. The disease appears to beespecially common in Northeastern Italy (prevalence,1:1000), with an autosomal dominant inheritance (30%). Anautosomal recessive variant of ARVD that is associated witha distinctive extracardiac phenotype (woolly hair and palmo-plantar keratoderma) has been reported from the island ofNaxos in Greece.58
Molecular Basis of ARVDTo date, 4 loci for autosomal dominant ARVD have beenidentified, 2 of which are in close proximity on chromosome14 (14q23-q24 and 14q12-q22).59,60A third locus was locatedon chromosome 1 (1q42-q43),61 and the fourth on chromo-some 2 (2q32.1-q32.2)62 (Table). The autosomal recessivesyndromic variant of ARVD has been linked to a locus onchromosome 17 (17q21), within the gene encoding a keratin,a reasonable candidate for the entity.58 Further advances willfacilitate recognition of the nonarrhythmic clinical presenta-tions and the broader phenotype of ARVD/Naxos disease(Table).
Dilated CardiomyopathyDilated cardiomyopathy (DCM) is a genetically and clinicallyheterogeneous disease63 that can affect newborns, children,
522 Molecular Biology and Arrhythmias
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

adolescents, adults, and the elderly. The disease may beassociated with other organ or muscle abnormalities orpresent as a pure disorder. Malignant life-threatening ventric-ular arrhythmia and atrial arrhythmia with serious impact oncardiac function are frequently associated with the disorder.As in FHCM, sudden death in DCM may also be caused notonly by ventricular arrhythmias but also by bradyarrhythmias.Whenever spontaneous ventricular arrhythmias have beenclinically documented, the inducibility and reproducibility ofthe arrhythmia in electrophysiological studies is usually low,favoring the possibility of a predominant role for nonreen-trant mechanisms.64,65 At least 30% of cases of DCM areinherited (ie, familial DCM), with a significant percentage ofthe remaining cases being acquired (ie, myocarditis, ischemicheart disease, etc). Inherited DCM may have autosomaldominant, autosomal recessive, X-linked, or mitochondrialtransmission (Table).
Molecular Basis of DCMTo date, genes for X-linked DCM and autosomal dominantDCM (ADDCM) have been mapped, demonstrating geneticheterogeneity.66 The genes for 2 X-linked cardiomyopathieshave been identified: the dystrophin gene, which is alsoresponsible for Duchenne and Becker muscular dystro-phy,67,68and G4.5 in Barth syndrome (X-linked cardioskeletalmyopathy with neutropenia, abnormal mitochondria, and3-methylglutaconic aciduria).69 Multiple mutations in bothgenes have been reported as well.68–72
Dystrophin is a large cytoskeletal protein that is found onthe inner face of the sarcolemma and attaches at itsN-terminal domain to F-actin in the matrix and to thedystrophin-associated glycoprotein (DAG) complex (an oli-gomeric transmembrane protein) at its C-terminal domain.The protein encoded by the G4.5 gene is called “tafazzin,”but its function is still unknown.
Genes for autosomal dominant DCM have been mapped to6 different loci thus far. “Pure” DCM has been localized to1q32, 2p31, 9q13, and 10q21-q23,73–76 whereas DCM withconduction defects has been mapped to 1p1-1q177 and 3p22-3p25.78 Recently, mutations in cardiac actin79 located onchromosome 15q14 have been identified; therefore, actin isthus far the only known gene for ADDCM. On the basis ofthis finding, Olson et al79 have now proposed that DCM is aconsequence of defective transmission of force in cardiacmyocytes leading to heart failure.
Idiopathic Ventricular Fibrillation and theBrugada SyndromeAnother interesting group of patients that has become a targetfor genetic studies is represented by those individuals withso-called idiopathic ventricular fibrillation (ie, patients with anormal heart who experience cardiac arrest with documentedVF).80 A subgroup of these patients experience sudden death(which may occur in families), apparently have no structuralheart disease, and have right precordial ST-segment eleva-tion, sometimes with right bundle-branch block (RBBB;Brugada syndrome).81 These ECG characteristics may de-pend on exaggerated transmural differences in action poten-tial configuration, especially in the right ventricular outflow
tract. This could arise from dysfunction of a number of ioncurrents, such asI to, L-type Ca21 current [ICa(L)], and INa.
At least 1 variant of the Brugada syndrome is caused bydefects in the sodium channel gene (SCN5A), ie, the samegene implicated in LQT3.82 In the Brugada syndrome, themutations identified apparently lead to a loss of function,whereas in LQT3, most cause a gain of function. Thus, LQTSand the Brugada syndromes appear to be separate allelicdisorders.
The evidence that not all patients with Brugada syndromehave defects on the cardiac sodium channel (S.G. Priori et al,1998, unpublished observations) suggest that, in analogy withthe other inherited cardiac diseases, genetic heterogeneity isalso present in Brugada syndrome.
Atrial FibrillationPerhaps the most common arrhythmia requiring interventionis Afib. Data are now emerging from a number of laboratorieson the potential molecular basis of electrophysiologicalchanges observed in atria that have been fibrillating for hoursto days and those that have been fibrillating for weeks tomonths.83–87 They all share a marked shortening of refracto-riness, most likely reflecting decreased action potential dura-tion early during Afib. Available data suggest that a majormechanism is decreased inward current through L-type cal-cium channels and possibly sodium channels.88 Later duringthe “remodeling” that appears to accompany chronic Afib,changes in expression and/or distribution of connexin pro-teins and/or other ion channel proteins, as well as changes incellular ultrastructure, may play a role.
Inherited Afib is considered uncommon and has beenreported with autosomal dominant transmission. Recently,familial Afib has been mapped to 10q22-q24 (a region of'11 cM) in 3 families.89 Expansion of the previouslyidentified kindreds has allowed further refining of the mapposition and limitation of the gene critical region.
A fascinating issue concerning Afib is its association withother disorders, such as DCM, FHCM, and LQT4 and thepossibility that a mutation in a gene responsible for 1 of theseassociated disorders could cause familial Afib. For instance,is it simply circumstantial that a familial DCM locus76 and themapped Afib locus are within the same relatively smallregion of 10q21-q24? Is there something different about theclinical course, and thus the causative gene responsible forLQT4,8 in which prolonged QTc appears to be associatedwith a high incidence of Afib and slower heart rates thantypically seen in LQTS? Could this be a different type of gene(ie, not an ion channel) or a new channel disorder?
Progressive Familial Heart BlockTwo forms of progressive familial heart block (PFHB),90
which differ in their ECG characteristics, have been reported.The first, PFHB-I, is defined on ECG by evidence ofbundle-branch disease such as RBBB, left anterior hemi-block, left posterior hemiblock, or complete heart block withbroad QRS complexes. Progression of disease occurs withchanges in the ECG going from a normal ECG to RBBB tocomplete heart block. Typical manifestations of the diseaseare syncope, sudden death, and Stokes-Adams attacks. The
Priori et al February 2, 1999 523
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

second form of PFHB, known as PFHB-II, presents withcomplete heart block and narrow QRS complexes and isbelieved to occur as a result of AV nodal disease with AVblock and an idionodal escape rhythm. Typically, thesepatients present with sinus bradycardia and left posteriorhemiblock and develop syncope and Stokes-Adams attacks.
Genetically, PFHB-I is better studied than PFHB-II andappears to be inherited in autosomal dominant fashion. Brinkand Torrington91 studied 3 South African families withPFHB-I, including one 9-generation kindred, for linkageanalysis. In 86 family members (39 affected), linkage wasidentified on chromosome 19 at 19q13.2-q13.3, and the genewas localized to within 10 cM of the kallikrein locus.Confirmation of this localization was subsequently reportedby De Meeus et al92 in a large Lebanese family. Othercandidate genes within the mapped region include apoli-poprotein C2, creatine kinase–MM, myotonic dystrophy,troponin T, and the histidine-rich Ca21-binding protein (aluminal sarcoplasmic reticulum protein). Myotonic dystro-phy, creatine kinase–MM, and apolipoprotein C2 have beenexcluded as the causative genes.
Familial WPW SyndromeFamilial WPW has rarely been reported, but an inherited formof WPW associated with FHCM has been described and itslocus mapped to chromosome 7q3.43 It is unknown whether asingle defect is responsible for both aspects of the syndromeor whether 2 genes are located in close proximity (ie,contiguous gene syndrome) and thus frequently cosegregate.In the latter case, familial WPW could be caused by a singlegene defect on chromosome 7. However, other associationsof FHCM and WPW have also been identified. For example,Kimura et al42 found mutations in the cardiac troponin I gene(on chromosome 19) in patients with FHCM and WPW.Furthermore, some children with mitochondrial abnormalitiesand metabolic disease (Pompe disease) associated withFHCM also have been noted to have WPW. Therefore, itcurrently appears that WPW may have multiple differentgenetic pathogeneses.
Part II: Molecular Diagnosis of InheritedArrhythmogenic Disorders
Role of DNA Screening in Diagnosis of InheritedArrhythmogenic DiseasesThe potential of making a genetic diagnosis makes it possibleto consider the role of genetic testing in routine clinicalpractice. Applications could include preclinical diagnosis andidentification of patients who might benefit from prophylactictreatment for sudden death. For this approach to become areality, however, several conditions must be met: the devel-opment of routine clinical DNA diagnostic testing facilities; asufficiently large database to determine risk in relation togenotype, as well as the recognized heterogeneity in pheno-type; an estimate of the efficacy of available treatments; anda consideration of the cost implications.
Molecular diagnosis has the potential to define with 100%sensitivity and 100% specificity the genetic status of anymember of an affected family. However, for this potential to
become fully expressed, it is necessary that all the genes andall the mutations within these genes causing a given diseasebe identified. This is not yet even close to reality for any ofthe inherited arrhythmogenic diseases discussed here. As aconsequence, physicians still generally have to rely onclinical criteria to establish these diagnoses.
For some diseases, not even the specific affected gene(s)are known. In these cases (eg, familial Afib, progressivefamilial AV block), the available genetic information isderived from linkage studies and provides only data as towhich chromosomal region the disease gene is located on. Ifthis region is large, it may take years before the generesponsible for the disease is located. Thus, at this stage ofknowledge, molecular screening for these entities is limited toresearch activities; it is not possible to consider genotype-phenotype correlations, and most importantly, the nature ofthe defect underlying the disease remains undefined. Linkagestudies can nonetheless provide the important information onwhether only 1 gene is associated with the disease or whethergenetic heterogeneity exists (ie, several genes accounting fora disease).
When a gene responsible for a disease is identified, it thenbecomes possible to search for specific mutations. Theorganization and the sequence of the disease genes are oftennot entirely known, and thus, mutations are usually searchedfor (at least initially) only in portions of the gene. As aconsequence, a positive finding (ie, the identification of amutation) is diagnostic, whereas a negative finding in a linkedgene suggests that mutations may be present in an unexploredregion of the gene (or that the linkage is incorrect). Thediagnostic power of molecular screening is further limited(for all arrhythmogenic disorders discussed here) by thepresence of genetic heterogeneity and the lack of identifica-tion of all of the genes responsible for the disease.
Implications of Molecular Diagnosis onPatient ManagementWhen the genetic bases of FHCM and LQTS were elucidated,one hope of molecular biologists and clinicians alike was thatit would have become possible to reach, in a relatively shorttime, some important goals in the establishment of genotype/phenotype correlations. In this respect, valuable informationwould be the ability to categorize mutations as mild versussevere to guide the therapeutic approach on the basis of thepredicted risk. For the time being, this goal has not beenachieved, and we are still far from being able to predictadverse or favorable prognosis on the basis of the geneticdefect.
A major goal in LQTS and FHCM is to have sufficientgenotyped patients to understand the diagnostic, functional,and prognostic implications of the different mutations. Aproblem in genetic testing in LQTS and FHCM is that thedisease-associated gene and specific mutations are still beingidentified. This research information is not yet widely imple-mented in commercial laboratories, and the resource demandsfor such an effort on a routine (or “service”) basis aregenerally beyond those available to the research laboratoriesengaged in the problem.
524 Molecular Biology and Arrhythmias
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

Genetic Testing for LQTSWhen should genetic testing be considered in dealing withLQTS patients93? The cardiologist will confront 3 clinicalscenarios.
The first situation is the patient who has a definitediagnosis based on established clinical diagnostic criteria.Here, genetic testing is not absolutely necessary, because thecardiologist has most of the elements necessary to decideabout initiation of therapy. However, genetic testing could beuseful, because, depending on the gene (and ultimately eventhe specific mutation) identified as responsible for the dis-ease, modifications in management29,94 may be suggested.Examples discussed above include the addition of mexiletinein LQT3 or lifestyle modifications such as limitation ofstrenuous or competitive exercise in LQT1. It should bepointed out, however, that in symptomatic patients with anestablished diagnosis of LQTS, implementation of therapywith b-blockers should not be delayed while waiting forresults of genetic screening.
A second scenario occurs when the diagnosis of LQTS isonly suspected or the patient has a borderline diagnosis basedon clinical criteria. Under these circumstances, genetic testingcould be very useful in establishing the diagnosis, becauseidentification of a mutated LQTS gene would convert asuspected diagnosis into a certain one and would remove thecardiologist’s hesitation in making therapeutic choices. How-ever, the failure to identify a mutation does not rule out thediagnosis (because only a minority of mutations have beenidentified to date). Although genetic testing in this situation isnot yet widely available, techniques to automate screening forthe hundreds of possible known mutations are now beingdeveloped and will probably be available in the next 5 to 10years.
A third scenario is an apparently asymptomatic relative ofa patient with LQTS. Here, genetic testing can be especiallyuseful if the disease-causing mutation has previously beenidentified in the proband. Otherwise, the same issues arise asthose in evaluating the borderline LQTS diagnosis.
Genetic Testing for Hypertrophic CardiomyopathySimilar considerations apply in FHCM. From the clinicalperspective, comprehensive screening of the disease-causinggenes would be both inappropriate and impractical at thistime. Specific clinical situations exist in which DNA diagno-sis is likely to have an important impact on management. Forexample, sudden death/resuscitated VF in association withnormal or near-normal heart weight and/or mild morpholog-ical features in the young should lead to testing for mutationsin the cardiac troponin T gene. Premature sudden death inassociation with obvious morphological features in the younghas been associated with the Arg403Glu and Arg453Cysmutation in the b-myosin heavy chain gene, and thesemutations could be tested in this clinical context. Identifica-tion in the proband of troponin or myosin heavy chainmutations that are associated with poor prognosis wouldpermit an early or even a preclinical diagnosis in familymembers with the potential for lifestyle modifications (avoid-ance of competitive exercise) and prophylactic treatment
(amiodarone or implantable cardioverter-defibrillator) to pre-vent sudden death.
Genetic Testing for Autosomal Dominant DCM,Arrhythmogenic Right Ventricular Dysplasia, FamilialAfib, and Progressive Familial AV BlockUntil specific genes are discovered and characterized, molec-ular diagnosis should be considered a research tool only inlarge families in which linkage analysis may be performed.
Genetic Testing for DCMIn both X-linked DCM and Barth syndrome, definite diagno-sis at the molecular level may be useful clinically, becauseboth are rapidly progressive and severe disorders. In the caseof X-linked DCM, in which anticongestive and antiarrhyth-mic management initially and cardiac transplantation shortlythereafter are lifesaving, determination of a mutation couldhelp diagnose presymptomatic male gene carriers. In Barthsyndrome, therapeutic options are less clear-cut, but a defin-itive diagnosis in family members and potentially in fetusescould be similarly useful.
The recent identification of mutations in the actin geneopens the opportunity to perform family screening for muta-tions; however, until the prevalence of actin-related ADDCMis defined, the cost/benefit ratio of actin gene screeningcannot be defined.
Genetic Testing for Brugada SyndromeThe identification of mutations in the cardiac sodium channelin families with Brugada syndrome opens the possibility ofscreening patients with the disease. The importance of theidentification of the defect obviously consists of the ability toidentify the carriers before they become symptomatic. This isparticularly important for a disease in which the first mani-festation is often cardiac arrest. However, because not allpatients with Brugada syndrome have mutations in thesodium channel (S.G. Priori et al, 1998, unpublished obser-vations), the cost/benefit ratio of mutation screening in thesodium channel gene cannot be defined until the prevalenceof the genetic variant of the form associated with sodiumchannel defects is defined.
Ethical Aspects of Molecular ScreeningImportant ethical aspects are involved when DNA screeningis considered in families affected by a congenital disease.Discussion of the information that could be provided bygenetic testing with families is a most important first step.The experience of the team, which should include an appro-priately trained genetic counselor, in caring for patients withsimilar disorders is an important component for patientacceptance. One specific objective of counseling in arrhyth-mogenic disorders is to help the patient decide whether he orshe should undergo genetic screening at all. The consider-ations involved when an individual is deciding whether or notbe tested are as follows.
The patient should be given information on (1) the samplerequired, (2) use of the sample, (3) results of the testperformed, (4) implications of these results for managementof the patient and family, and (5) who will have access to theresults.
Priori et al February 2, 1999 525
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

The patient should use this information to decide whetherto give or withhold consent.
There should be no coercion by anyone (healthcare team,family members, insurance companies).
The consent form that patients sign should include state-ments that (1) all blood samples are coded to preventidentification and (2) the results of screening will be com-municated only to the patient and that no disclosure will bemade to third parties (not even family members) without thepatient’s written consent. Asymptomatic patients should havethe option of providing samples (eg, for family study) but notbeing informed of the results.
Reimbursement and Cost IssuesAt the present time, DNA screening for arrhythmogenicdisorders is not considered a routine test, and therefore, costsare not usually covered by insurance. Linkage to isolate thedisease gene can be performed in large families. When smallpedigrees or single patients in whom linkage cannot beapplied are studied, the only approach for DNA screening isthe systematic search for known mutations in any disease-linked gene. As discussed above, clinical evaluation may helpin selecting gene(s) to be screened first. Depending on thesize of the gene and on the number of genes to be screened,costs may be substantial (.$1000 US per gene screened ineach family). Currently, costs are covered almost exclusivelyby research funding of the laboratories involved in the field.The development of automated screening and identificationof more mutations may change this in the near (?) future.
AppendixThis article summarizes the outcome of a workshop held at theEuropean Heart House C, Sophia Antipolis, France, October 2–5,1997. The need for the workshop was proposed by Silvia G. Priori.It was organized by the Study Group on Molecular Basis ofArrhythmias of the Working Group on Arrhythmias of the EuropeanSociety of Cardiology, and its funding was administered by theWorking Group itself. The workshop was cochaired by Silvia G.Priori and Andre G. Kleber. The final preparation and organizationof the manuscript were the responsibility of Andre G. Kleber, SilviaG. Priori, Dan M. Roden, and Peter J. Schwartz.
References1. Schwartz PJ. Idiopathic long QT syndrome: progress and questions.Am
Heart J. 1985;109:399–411.2. Schwartz PJ, Locati EH, Napolitano C, Priori SG. The long QT
syndrome. In: Zipes DP, Jalife J, eds.Cardiac Electrophysiology: FromCell to Bedside. Philadelphia, Pa: WB Saunders Co; 1995:788–811.
3. Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell’etapediatrica.Clin Pediatr. 1963;45:658–683.
4. Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heartdisease with prolongation of the Q-T interval and sudden death.AmHeart J. 1957;54:59–68.
5. Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, Van RaayTJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, TowbinJA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT.Positional cloning of a novel potassium channel gene: KVLQT1mutations cause cardiac arrhythmias.Nat Genet. 1996;12:17–23.
6. Jiang C, Atkinson D, Towbin JA, Splawski I, Lehmann MH, Li H,Timothy K, Taggart RT, Schwartz PJ, Vincent GM, Moss AJ, KeatingMT. Two long QT syndrome loci map to chromosomes 3 and 7 withevidence for further heterogeneity.Nat Genet. 1994;8:141–147.
7. Towbin JA, Li H, Taggart RT, Lehmann MH, Schwartz PJ, Satler CA,Ayyagari R, Robinson JL, Moss A, Hejtmancik JF. Evidence of geneticheterogeneity in Romano-Ward long QT syndrome: analysis of 23families.Circulation. 1994;90:2635–2644.
8. Schott JJ, Peltier S, Foley P, Drouin E, Bouhour JB, Donnely P, VergnaudG, Moisan JP, Le Marec H, Pascal O. Mapping of a new gene for theLQTS syndrome.Am J Hum Genet. 1995;57:1114–1122.
9. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ,Towbin JA, Keating MT. SCN5A mutations associated with an inheritedcardiac arrhythmia, long QT syndrome.Cell. 1995;80:805–811.
10. Bennett PB, Yazawa K, Makita N, George AJ. Molecular mechanism foran inherited cardiac arrhythmia.Nature. 1995;376:683–685.
11. Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, BrownAM, Kirsch GE. Multiple mechanisms of Na1 channel–linked long-QTsyndrome.Circ Res. 1996;78:916–924.
12. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, KeatingMT. A molecular basis for cardiac arrhythmia: HERG mutations causelong QT syndrome.Cell. 1995;80:795–803.
13. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL,Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to formcardiac I(Ks) potassium channel.Nature. 1996;384:80–83.
14. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G.K(V)LQT1 and IsK (minK) proteins associate to form the I(Ks) cardiacpotassium current.Nature. 1996;384:78–80.
15. Splawski I, Tristani-Firouzi, M, Lehmannn MH, Sanguinetti MC, KeatingMT. Mutations in the hminK gene cause long-QT syndrome and suppressIKs function.Nat Genet. 1997;17:338–340.
16. Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y.KCNE1 mutations cause Jervell and Lange-Nielsen syndrome.Nat Genet.1997;17:267–268.
17. De Jager T, Corbett CH, Badenhorst JC, Brink PA, Corfield VA.Evidence of a long QT founder gene with varying phenotypic expressionin South African families.J Med Genet. 1996;33:567–573.
18. Napolitano C, Priori SG, Schwartz PJ, Timothy K, Paganini V, Cantu` F,Bloise R, De Fusco M, Casari G. Identification of a mutational hot spotin HERG-related long QT-syndrome (LQT2): phenotypic implications.Circulation. 1997;96(suppl I):I-212. Abstract.
19. Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J,Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novelmutation in the potassium channel gene KVLQT1 causes the Jervell andLange-Nielsen cardioauditory syndrome.Nat Genet. 1997;15:186–189.
20. Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT.Molecular basis of the long-QT syndrome associated with deafness.N Engl J Med. 1997;336:1562–1567.
21. Priori SG, Schwartz PJ, Napolitano C, Bianchi L, Dennis A, De Fusco M,Brown AM, Casari G. A recessive variant of the Romano-Ward long QTsyndrome?Circulation. 1998;97:2420–2425.
22. Vincent GM, Timothy KM, Leppert M, Keating M. The spectrum ofsymptoms and QT intervals in carriers of the gene for long QT syndrome.N Engl J Med. 1992;327:846–852.
23. Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M,Chivoret G, Schwartz K, Coumel P, Guicheney P. KVLQT1 C-terminalmissense mutation causes a forme fruste long-QT syndrome.Circulation.1997;96:2778–2781.
24. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QTsyndrome: clinical impact.Circulation. 1999;99:529–533.
25. Neyroud N, Denjoy I, Donger C, Gary F, Villain E, Leenhardt A, BenaliK, Schwartz K, Coumel P, Guicheney P. Heterozygous mutation in thepore of potassium channel geneKvLQT1 causes an apparent normalphenotype in long QT syndrome.Eur J Hum Genet. 1998;6:129–133.
26. Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum ofHERG K1-channel dysfunction in an inherited cardiac arrhythmia.ProcNatl Acad Sci U S A. 1996;93:2208–2212.
27. Chouabe C, Neyroud N, Guiceney P, Lazdunski M, Romey G, BarhaninJ. Properties of KvLQT1 K channel mutations in Romano-Ward andJervell and Lange-Nielsen inherited cardiac arrhythmias.EMBO J. 1997;17:5472–5479.
28. Schwartz PJ, Moss AL, Priori SG, Wang Q, Lehmann MH, Timothy K,Denjoy IF, Haverkamp W, Guicheney P, Paganini V, Scheinman MM,Karnes PS. Gene-specific influence on the triggers for cardiac arrest in thelong QT syndrome.Circulation. 1997;96(suppl I):I-212. Abstract.
29. Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA,Keating MT, Hammoude H, Brown AM, Chen LS, Colatsky TJ. Long QTsyndrome patients with mutations of the SCN5A and HERG genes havedifferential responses to Na1 channel blockade and to increases in heartrate: implications for gene-specific therapy.Circulation. 1995;92:3381–3386.
526 Molecular Biology and Arrhythmias
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

30. Compton SJ. QT interval shortening by lidocaine in chromosome 7-linkedlong QT syndrome (LQTS).Pacing Clin Electrophysiol. 1997;20:1060.Abstract.
31. Compton SJ, Lux RL, Ramsey MR, Strelich KR, Sanguinetti MC, GreenLS, Keating MT, Mason JW. Genetically defined therapy of inheritedlong-QT syndrome: correction of abnormal repolarization by potassium.Circulation. 1996;94:1018–1022.
32. Schwartz PJ, Moss AJ. QT interval prolongation: what does it mean?J Cardiovasc Med. 1982;7:1317–1330.
33. Priori SG, Diehl L, Schwartz PJ. Torsade de pointes. In: Podrid PJ,Kowey PR, eds.Cardiac Arrhythmia, Mechanisms, Diagnosis and Man-agement. Baltimore, Md: Williams & Wilkins; 1995:951–963.
34. Schulze-Bahr E, Haverkamp W, Hordt M, Borggrefe M, Wedekind H,Assmann G, Breithardt G, Funke H. A genetic basis for quinidine-induced(acquired) long QT syndrome.Eur Heart J. 1997;18:29. Abstract.
35. Napolitano C, Priori SG, Schwartz PJ, Cantu F, Paganini V, De Fusco M,Pinnavaia A, Aquaro G, Casari G. Identification of a long QT syndromemolecular defect in drug-induced torsade de pointes.Circulation. 1997;96(suppl I):I-211. Abstract.
36. Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management ofhypertrophic cardiomyopathy.N Engl J Med. 1997;336:775–785.
37. Geisterfer LA, Kass S, Tanigawa G, Vosberg HP, McKenna W, SeidmanCE, Seidman JG. A molecular basis for familial hypertrophic cardiomy-opathy: a beta cardiac myosin heavy chain gene missense mutation.Cell.1990;62:999–1006.
38. Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC,Rayment I, Sellers JP, Fananapazir L, Epstein ND. Mutation in either theessential or regulatory light chains of myosin are associated with a raremyopathy in human heart and skeletal muscle.Nat Genet. 1996;13:63–69.
39. Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, VosbergHP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponinT mutations cause familial hypertrophic cardiomyopathy: a disease of thesarcomere.Cell. 1994;77:701–712.
40. Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, GautelM, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP,Fiszman M, Komajda M, Schwartz K. Cardiac myosin binding protein-Cgene splice acceptor site mutation is associated with familial hypertrophiccardiomyopathy.Nat Genet. 1995;11:438–440.
41. Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKennaWJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiacmyosin binding protein-C gene on chromosome 11 cause familial hyper-trophic cardiomyopathy.Nat Genet. 1995;11:434–437.
42. Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S,Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA,Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, MatsuzakiM, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T. Mutationsin the cardiac troponin I gene associated with hypertrophic cardiomyop-athy.Nat Genet. 1997;16:379–382.
43. MacRae CA, Ghaisas N, Kass S, Donnelly S, Basson CT, Watkins HC,Anan R, Thierfelder LH, McGarry K, Rowland E, McKenna WJ,Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy withWolff-Parkinson-White syndrome maps to a locus on chromosome 7p3.J Clin Invest. 1995;96:1216–1220.
44. Schwartz K, Carrier L, Guicheney P, Komajda M. Molecular basis offamilial cardiomyopathies.Circulation. 1995;91:532–540.
45. Vikstrom KL, Leinwand LA. Contractile protein mutations and heartdisease.Curr Opin Cell Biol. 1996;8:97–105.
46. Carrier L, Bonne G, Bahrend E, Yu B, Richard P, Niel F, Hainque B,Cruaud C, Gary F, Labeit S, Bouhour JB, Dubourg O, Desnos M, HagegeAA, Trent RJ, Komajda M, Fiszman M, Schwartz K. Organization andsequence of human cardiac myosin binding protein C gene (MYBPC3)and identification of mutations predicted to produce truncated proteins infamilial hypertrophic cardiomyopathy.Circ Res. 1997;80:427–434.
47. Cuda G, Fananapazir L, Zhu WS, Sellers JR, Epstein ND. Skeletal muscleexpression and abnormal function ofb-myosin in hypertrophic cardio-myopathy.J Clin Invest. 1993;91:2861–2865.
48. Lankford EB, Epstein ND, Fananapazir L, Sweeney HL. Abnormal con-tractile properties of muscle fibers expressingb-myosin heavy chain genemutations in patients with hypertrophic cardiomyopathy.J Clin Invest.1995;95:1409–1414.
49. Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heter-ologous expression of a cardiomyopathic myosin that is defective in itsactin interaction.J Biol Chem. 1994;269:1603–1605.
50. Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, SeidmanCE, Seidman JG. Characteristics and prognostic implications of myosinmissense mutations in familial hypertrophic cardiomyopathy.N EnglJ Med. 1992;326:1108–1114.
51. Dausse E, Komajda M, Fetler L, Dubourg O, Dufour C, Carrier L,Wisnewsky C, Bercovici J, Hengstenberg C, al-Mahdawi S, Isnard R,Hagege A, Bouhour J-B, Desnos M, Beckmann J, Weissenbach J,Schwartz K,Guicheney P, et al. Familial hypertrophic cardiomyopathy:microsatellite haplotyping and identification of a hot spot for mutations inthe b-myosin heavy chain gene.J Clin Invest. 1993;92:2807–2813.
52. Epstein ND, Cohn GM, Cyran F, Fananapazir L. Differences in clinicalexpression of hypertrophic cardiomyopathy associated with two distinctmutations in theb-myosin heavy chain gene: a 908Leu-Val mutation anda 403Arg-Gln mutation.Circulation. 1992;86:345–352.
53. Charron P, Dubourg O, Desnos M, Isnard R, Hagege A, Bonne G, CarrierL, Tesson F, Bouhour JB, Buzzi JC, Feingold J, Schwartz K, Komajda M.Genotype-phenotype correlations in familial hypertrophic cardiomyopa-thy: a comparison between mutations in the cardiac protein-C and theb-myosin heavy chain genes.Eur Heart J. 1998;19:1377–1382.
54. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’DonoghueA, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE.Mutations in the genes for cardiac troponin T and tropomyosin in hyper-trophic cardiomyopathy.N Engl J Med. 1995;332:1058–1064.
55. Forissier JF, Carrier L, Farza H, Bonne G, Bercovici J, Richard P,Hainque B, Townsend PJ, Yacoub MH, Faure S, Dubourg O, Millaire A,Hagege AA, Desnos M, Komajda M, Schwartz K. Codon 102 of thecardiac troponin T gene is a putative hot spot for mutations in familialhypertrophic cardiomyopathy.Circulation. 1996;94:3069–3073.
56. Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA,Watkins H. Sudden death due to troponin T mutations.J Am Coll Cardiol.1997;29:549–555.
57. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhyth-mogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myo-carditis?Circulation. 1996;94:983–991.
58. Coonar AS, Protonotarios N, Tsatsopoulou A, Needham EWA, HoulstonRS, Cliff S, Otter MI, Murday VA, Mattu RK, McKenna WJ. Gene forarrhythmogenic right ventricular cardiomyopathy with diffuse nonepider-molytic palmoplantar keratoderma and woolly hair (Naxos disease) mapsto chromosome 17q21.Circulation. 1998;97:2049–2058.
59. Rampazzo A, Nava A, Danieli GA, Buja G, Daliento L, Fasoli G,Scognamiglio R, Corrado D, Thiene G. The gene for arrhythmogenicright ventricular cardiomyopathy maps to chromosome 14q23–q24.HumMol Genet. 1994;3:959–962.
60. Severini GM, Krajinovic M, Pinamonti B, Sinagra G, Fioretti P, BrunazziMC, Falaschi A, Camerini F, Giacca M, Mestroni L. A new locus forarrhythmogenic right ventricular dysplasia on the long arm of chro-mosome 14.Genomics. 1996;31:193–200.
61. Rampazzo A, Nava A, Erne P, Eberhard M, Vian E, Slomp P, Tiso N,Thiene G, Danieli GA. A new locus for arrhythmogenic right ventricularcardiomyopathy (ARVD2) maps to chromosome 1q42–q43.Hum MolGenet. 1995;4:2151–2154.
62. Rampazzo A, Nava A, Miorin M, Fonderigo P, Pope B, Tiso N, LivolsiB, Zimbello R, Thiene G, Danieli GA. ARVD4, a new locus for arrhyth-mogenic right ventricular cardiomyopathy maps to chromosome 2 longarm.Genomics. 1997;45:259–263.
63. Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, BurnettJC, Rodeheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familialdilated cardiomyopathy in a series of patients with idiopathic dilatedcardiomyopathy.N Engl J Med. 1992;326:77–82.
64. Stamato NJ, O’Connell JB, Murdock DK, Moran JF, Loeb HS, ScanlonPJ. The response of patients with complex ventricular arrhythmias sec-ondary to dilated cardiomyopathy to programmed electrical stimulation.Am Heart J. 1986;112:505–508.
65. Stevenson WG, Stevenson LW, Weiss J, Tillisch JH. Inducible ventric-ular arrhythmias and sudden death during vasodilator therapy of severeheart failure.Am Heart J. 1988;116:1447–1454.
66. Schultz KR, Gajarski RJ, Pignatelli R, Goytia V, Roberts R, Bachinski L,Towbin JA. Genetic heterogeneity in familial dilated cardiomyopathy.Biochem Mol Med. 1995;56:87–93.
67. Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS,McCabe ER, Swift M. X-linked dilated cardiomyopathy: moleculargenetic evidence of linkage to the Duchenne muscular dystrophy (dys-trophin) gene at the Xp21 locus.Circulation. 1993;87:1854–1865.
Priori et al February 2, 1999 527
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

68. Ortiz-Lopez R, Li H, Su J, Goytia V, Towbin JA. Evidence for adystrophin missense mutation as a cause of X-linked dilated cardiomy-opathy.Circulation. 1997;95:2434–2440.
69. Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D.A novel X-linked gene, G4.5, is responsible for Barth syndrome.NatGenet. 1996;12:385–389.
70. Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, Mateddu A, MarrosuMG, Cianchetti C, Realdi G, Cao A, Melis MA. Deletion of the dys-trophin muscle-promoter region associated with X-linked dilated cardio-myopathy.N Engl J Med. 1993;329:921–925.
71. Muntoni F, Wilson L, Marrosu G, Marrosu MG, Cianchetti C, MestroniL, Ganau A, Dubowitz V, Sewry C. A mutation in the dystrophin geneselectively affecting dystrophin expression in the heart.J Clin Invest.1995;96:693–699.
72. Milasin J, Muntoni F, Severini GM, Bartoloni L, Vatta M, Krajinovic M,Mateddu A, Angelini C, Camerini F, Falaschi A, Mestroni L, Giacca M.A point mutation in the 59 splice site of the dystrophin gene first intronresponsible for X-linked dilated cardiomyopathy.Hum Mol Genet. 1996;5:73–79.
73. Durand JB, Bachinski LL, Bieling LC, Czernuszewicz GZ, Abchee AB,Yu QT, Tapscott T, Hill R, Ifegwu J, Marian AJ, Brugada R, Daiger S,Gregoritch JM, Anderson JL, Quinones M, Towbin JA, Roberts R. Local-ization of a gene responsible for familial dilated cardiomyopathy tochromosome 1q32.Circulation. 1995;92:3387–3389.
74. Siu BL. A novel locus for familial dilated cardiomyopathy on chro-mosome 2p31. Proc Second World Congress of Pediatric Cardiology andCardiac Surgery. 1997.
75. Krajinovic M, Pinamonti B, Sinagra G, Vatta M, Severini GM, Milasin J,Falaschi A, Camerini F, Giacca M, Mestroni L, Heart Muscle DiseaseStudy Group. Linkage of familial dilated cardiomyopathy to chromosome9. Am J Hum Genet. 1995;57:846–852.
76. Bowles KR, Gajarski R, Porter P, Goytia V, Bachinski L, Roberts R,Pignatelli R, Towbin JA. Gene mapping of familial autosomal dominantdilated cardiomyopathy to chromosome 10q21–23.J Clin Invest. 1996;98:1355–1360.
77. Kass S, MacRae C, Graber HL, Sparks EA, McNamara D, Boudoulas H,Basson CT, Baker PR, Cody RJ, Fishman MC, Cox N, Kong A, WooleyCF, Seidman JG, Seidman CE. A gene defect that causes conductionsystem disease and dilated cardiomyopathy maps to chromosome1p1–1q1.Nat Genet. 1994;7:546–551.
78. Olson TM, Keating MT. Mapping a cardiomyopathy locus to chro-mosome 3p22–p25.J Clin Invest. 1996;97:528–532.
79. Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT. Actinmutations in dilated cardiomyopathy: a heritable form of heart failure.Science. 1998;280:750–753.
80. Consensus statement of the Joint Steering Committees of UCARE and ofIVF-US (Bardy GH, Bigger JT Jr, Borggrefe M, Camm AJ, Cobb LA,Ewy GA, Hauer RNW, Kuck KH, Lane RD, Lazzara R, Marcus FI,Muller JE, Myerburg RJ, Priori SG, Schwartz PJ, Touboul P, Verrier RL,
Wellens HJJ, Zipes DP). Survivors of out-of-hospital cardiac arrest withapparently normal heart: need for definition and standardized clinicalevaluation.Circulation. 1997;95:265–272.
81. Brugada P, Brugada J. Right bundle branch block, persistent ST segmentelevation and sudden cardiac death: a distinct clinical and electrocardio-graphic syndrome: a multicenter report.J Am Coll Cardiol. 1992;20:1391–1396.
82. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, PotenzaD, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z,Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA,Wang Q. Genetic basis and molecular mechanism for idiopathic ventric-ular fibrillation. Nature. 1998;392:293–294.
83. Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillationbegets atrial fibrillation: a study in awake chronically instrumented goats.Circulation. 1995;92:1954–1968.
84. van der Velden HMW, van Zijverden M, van Kempen MJA, WijffelsMCEF, Groenewegen WA, Allessie MA, Jongsma HJ. Abnormalexpression of the gap junction protein connexin 40 during chronic atrialfibrillation in the goat.Circulation. 1996;94(suppl I):I-593. Abstract.
85. Yue L, Feng J, Gaspo R, Li GR, Nattel S. Ionic remodeling underlyingtachycardia-induced atrial fibrillation in dogs.Circulation. 1996;94(supplI):I-592. Abstract.
86. Tieleman RG, De LC, Van GI, de KP, Grandjean J, Bel KJ, Wijffels MC,Allessie MA, Crijns HJ. Verapamil reduces tachycardia-induced elec-trical remodeling of the atria.Circulation. 1997;95:1945–1953.
87. Satoh T, Zipes DP. Unequal atrial stretch in dogs increases dispersion ofrefractoriness conducive to developing atrial fibrillation.J CardiovascElectrophysiol. 1996;7:833–842.
88. Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodelingunderlying action potential changes in a canine model of atrial fibrillation.Circ Res. 1997;81:512–525.
89. Brugada R, Tapscott T, Czernuszewicz GZ, Marian AJ, Iglesias A, MontL, Brugada J, Girona J, Domingo A, Bachinski LL, Roberts R. Identifi-cation of a genetic locus for familial atrial fibrillation.N Engl J Med.1997;336:905–911.
90. Brink PA, Ferreira A, Moolman JC, Weymar HW, van der Merwe PL,Corfield VA. Gene for progressive familial heart block type I maps tochromosome 19q13.Circulation. 1995;91:1633–1640.
91. Brink AJ, Torrington M. Progressive familial heart block: two types.S AfrMed J. 1977;52:53–59.
92. De Meeus A, Stephan E, Debrus S, Jean MK, Loiselet J, Weissenbach J,Demaille J, Bouvagnet P. An isolated cardiac conduction disease maps tochromosome 19q.Circ Res. 1995;77:735–740.
93. Priori SG. Is long QT syndrome entering the era of molecular diagnosis?Heart. 1997;77:5–6.
94. Priori SG, Napolitano C, Cantu F, Brown AM, Schwartz PJ. Differentialresponse to Na1 channel blockade,b-adrenergic stimulation, and rapidpacing in a cellular model mimicking the SCN5A and HERG defectspresent in the long-QT syndrome.Circ Res. 1996;78:1009–1015.
528 Molecular Biology and Arrhythmias
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from

Peter J. Schwartz, Jeffrey A. Towbin and Arthur M. WildeJongsma, André G. Kleber, William J. McKenna, Dan M. Roden, Yoram Rudy, Ketty Schwartz,
Silvia G. Priori, Jacques Barhanin, Richard N. W. Hauer, Wilhelm Haverkamp, Habo J.Parts I and II
Genetic and Molecular Basis of Cardiac Arrhythmias: Impact on Clinical Management
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 1999 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.99.4.518
1999;99:518-528Circulation.
http://circ.ahajournals.org/content/99/4/518World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on August 15, 2015http://circ.ahajournals.org/Downloaded from
Related Documents











