Generation of induced progenitor-like (iPL) cells using interrupted reprogramming A novel strategy to produce highly specified functional therapeutic cell populations for lung regeneration By Li Guo A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by Li Guo 2018

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Generation of induced progenitor-like (iPL) cells using
interrupted reprogramming
A novel strategy to produce highly specified functional therapeutic
cell populations for lung regeneration
By
Li Guo
A thesis submitted in conformity with the requirements
for the degree of Doctor of Philosophy
Institute of Medical Science
University of Toronto
© Copyright by Li Guo 2018

ii
Generation of induced progenitor-like (iPL) cells using
interrupted reprogramming
A novel strategy to produce highly specified functional therapeutic
cell populations for lung regeneration
Li Guo
Doctor of Philosophy
Institute of Medical Science
University of Toronto
2018
Abstract
Regenerative medicine is constrained by suboptimal cell sources with either limited
or uncontrolled proliferation and/or incompletely restricted differentiation. We developed a
novel "interrupted reprogramming" strategy without traversing the pluripotent state, to
generate "induced Progenitor-Like (iPL) cells" using carefully timed transient expression of
induced Pluripotent Stem (iPS) cell reprogramming factors (Oct4, Sox2, Klf4 and c-Myc;
OSKM). Interrupted reprogramming is not only able to achieve controlled expansion of the
selected cell type, but results in the "de-differentiation" of the cells to a progenitor-like state
while preserving the parental lineage commitment to generate a limited range of functional
progeny. Lineage-specific iPL cells can be derived from distinct airway epithelial
populations and function as region-specific progenitor cells. For example, bronchiolar
progenitor-like iPL cells can be derived from mature Club cells (Club-iPL cells) and give
rise to Club cells, goblet cells and functional CFTR-expressing ciliated epithelium.
Embryonic bipotent progenitor-like iPL cells can be derived from adult alveolar type II cells

iii
(AEC-II-iPL cells) and the iPL process results in the rescue of the in vitro limited
clonogenic capacity of AEC-II cells.
These highly specified iPL populations are functional and therapeutic that are
capable of directly contributing to lung regeneration through engraftment and differentiation.
In vivo, Club-iPL cells were able to repopulate CFTR-deficient bronchiolar epithelium and
AEC-II-iPL cells showed utility in ameliorating bleomycin-induced pulmonary fibrosis. This
interrupted reprogramming process could be metronomically applied both in vitro and in
vivo, to achieve controlled progenitor-like proliferation.
Furthermore, harnessing the residual epigenetic “memory” and the plasticity existing
in the early stage of the reprogramming process, interrupted reprogramming is able to
rejuvenate aged endogenous progenitor AEC-II cells to a youthful state by ameliorating
multiple hallmarks of aging, including impaired self-renewal, declined telomerase activity,
mitochondrial DNA damages and the epigenome histone alteration. Importantly, cellular
identity and function of parental AEC-II cells were maintained, thereby generating large
numbers of younger AEC-II cells.
In summary, this novel interrupted reprogramming strategy will allow the production
of highly specified functional therapeutic populations which can potentially have significant
implications for regenerative medicine, specifically for, cell replacement therapy, biohybrid
devices, disease modelling, and drug screening for human diseases.

iv
To
Mom and Dad

v
Acknowledgments
I am most grateful to my supervisor Dr. Thomas Waddell, for his kindness, continuous
guidance, constructive criticism, inspiration, encouragement, and patience over the course of
my PhD program. Thank you for teaching me how to become a qualified scientist. I am very
thankful for having the opportunity to learn from you, such a great intelligent knowledgeable
mentor. Without you, I would not be able to have these achievements.
I am grateful to my committee members Dr. Andras Nagy, Dr. Ian Rogers and Dr. Martin
Post for acting as secondary supervisor. Their continuous guidance, advice, constant
encouragement and consideration have played an integral part in my training. Thank you for
your support throughout this long process.
I am also grateful to Dr. Mingyao Liu and Dr. Shaf Keshavjee for providng invaluable
insights into this project and guidance throughout my PhD program.
Many thanks to Dr. Golnaz Karoubi, my dear friend, for the great guidance during my
writing process and the precise mental support.
To Pascal Duchesneau, who contributes to this project tremendously. I appreciate your great
help with all the animal studies. To John Soleas and Dr. Siba Haykal, who were big supports
while we were working at late night and weekends. To Dr. Sherry Zhao and Dr. Xu Feng,
who have been providing me great technical support for years.

vi
A huge thank you to the Institute of Medical Science, for providing me this great
opportunity to pursue my PhD degree.
I am forever grateful to my family (especially mom and dad) and my best friend-Susannah
Moore, who have been giving me unconditional love and support.

vii
List of contributors
Li Guo is the main contributor on this thesis and was involved in project design, performing
all experiments, analyzing the results and writing the papers.
Dr. Thomas K Waddell is the supervisor and contributed to project design, provided
facilities and resources and input in paper writing.
Dr. Andras Nagy and Dr. Ian Rogers provided animal resources, contributed to project
design and input in paper editing.
Dr. Golnaz Karoubi contributed to project design and input in paper editing.
Pascal Duchesneau-performed in vivo animal injury models and cell delivery for Chapter 3
and 4, and Flexivent lung functional measurement for Chapter 4.
Dr. Hoon-Ki Sung-performed teratoma assay for Chapter 3.
Dr. Maria Shutova and Dr. Peter Tonge-analysed the microarray data for Chapter 3.
Dr. Fabio Gava Aoki-performed Flexivent lung functional measurement and analysed
pressure-volume (PV) curves for Chapter 4.
Dr. Christine Bear- provided instrument and reagents for iodide efflux assay for Chapter 3

viii
Table of Contents
ACKNOWLEDGMENTS ................................................................................................................................. V
LIST OF CONTRIBUTORS .......................................................................................................................... VII
TABLE OF CONTENTS .............................................................................................................................. VIII
LIST OF TABLES ........................................................................................................................................ XIV
LIST OF FIGURES ........................................................................................................................................ XV
LIST OF APPENDICES ............................................................................................................................... XIX
LIST OF ABBREVIATIONS ......................................................................................................................... XX
CHAPTER 1 INTRODUCTION AND LITERATURE REVIEW ................................................................. 1
1.1 RESPIRATORY EPITHELIUM ...................................................................................................................... 1
1.1.1 The physiological function of respiratory epithelium .................................................................... 1
1.1.2 Epithelial organization of adult mouse and human lungs ............................................................. 1
1.1.2.1 The size and structure ......................................................................................................................... 1
1.1.2.2 Cellular composition ........................................................................................................................... 2
1.1.3 Epithelial lineages in adult lungs .................................................................................................. 2
1.1.3.1 Conducting airways ............................................................................................................................ 2
1.1.3.1.1 Basal cells ...................................................................................................................................... 2
1.1.3.1.2 Pulmonary neuroendocrine cells.................................................................................................... 3
1.1.3.1.3 Club cells ....................................................................................................................................... 4
1.1.3.1.4 Goblet cells .................................................................................................................................... 4
1.1.3.1.5 Ciliated cells .................................................................................................................................. 5
1.1.3.2 Distal airways ..................................................................................................................................... 5
1.1.3.2.1 Alveolar type I (AEC-I) cells ........................................................................................................ 5
1.1.3.2.2 Alveolar type II (AEC-II) cells ...................................................................................................... 6
1.2 EPITHELIAL BIOLOGY IN THE PATHOGENESIS OF PULMONARY DISEASE ................................................... 6
1.2.1 Obstructive lung diseases .............................................................................................................. 6
1.2.1.1 Chronic obstructive pulmonary diseases ............................................................................................. 6
1.2.1.1.1 Airway epithelium remodeling ...................................................................................................... 7
1.2.1.1.2 Epithelial dysfunction .................................................................................................................... 8
1.2.1.2 Asthma .............................................................................................................................................. 10
1.2.1.2.1 Epithelium alteration and dysfunction ......................................................................................... 11
1.2.1.2.2 Dysregulation of epithelium-mesenchymal network ................................................................... 11
1.2.1.2.3 Current treatments of asthmatic epithelium ................................................................................. 12
1.2.1.3 Cystic fibrosis ................................................................................................................................... 13
1.2.1.3.1 Defects of CFTR gene and protein .............................................................................................. 13

ix
1.2.1.3.2 Epithelium dysfunction attributes to the adaptive and innate immune defect .............................. 14
1.2.1.3.3 Cystic fibrosis disease models ..................................................................................................... 15
1.2.2 Restrictive lung diseases ............................................................................................................. 16
1.2.2.1 Interstitial lung disease-idiopathic pulmonary fibrosis ..................................................................... 16
1.2.2.1.1 Profoundly altered differentiation of alveolar epithelial cells in IPF ........................................... 17
1.2.2.1.2 Epithelial cells contribute to the profibrotic microenvironment .................................................. 17
1.2.2.1.3 Mechanisms of epithelium damage ............................................................................................. 18
1.2.2.1.4 Cell-based therapeutic approaches for treating IPF ..................................................................... 20
1.2.3 Acute respiratory distress syndrome (ARDS) .............................................................................. 21
1.2.3.1 Damage in epithelial tight junctions ................................................................................................. 22
1.2.3.2 Apoptosis of alveolar epithelial cells ................................................................................................ 22
1.2.3.3 Direct injury to alveolar epithelium .................................................................................................. 23
1.2.3.4 Inflammatory-induced epithelial injury ............................................................................................ 24
1.2.3.5 Epithelium repair in ARDS ............................................................................................................... 24
1.3 STEM CELLS IN LUNG EPITHELIAL REGENERATION ................................................................................. 26
1.3.1 Stem cells category ...................................................................................................................... 26
1.3.2 Adult stem cell hierarchy ............................................................................................................. 26
1.3.3 Endogenous lung stem cells......................................................................................................... 27
1.3.3.1 Basal stem cells ................................................................................................................................ 27
1.3.3.2 Trp63+/Krt5+ basal-like stem cells .................................................................................................. 28
1.3.3.3 Variant Club cells ............................................................................................................................. 29
1.3.3.4 Bronchioalveolar stem cells (BACs) ................................................................................................. 30
1.3.3.5 Neuroendocrine cells ........................................................................................................................ 30
1.3.3.6 Alveolar type II (AEC-II) cells ......................................................................................................... 31
1.3.3.7 Alveolar type I (AEC-I) cells ............................................................................................................ 32
1.3.4 Derivation of pulmonary epithelium from pluripotency stem cells ............................................. 32
1.3.4.1 ESCs and iPS cells ............................................................................................................................ 33
1.3.4.2 Differentiation approaches ................................................................................................................ 33
1.4 INDUCED PLURIPOTENT STEM (IPS) CELL REPROGRAMMING .................................................................. 36
1.4.1 Reprogramming methods ............................................................................................................ 36
1.4.1.1 Reprogramming factors .................................................................................................................... 37
1.4.1.2 Delivery systems ............................................................................................................................... 38
1.4.1.3 Target cell type ................................................................................................................................. 39
1.4.1.4 Derivation conditions ........................................................................................................................ 40
1.4.1.5 Identification of pluripotency ........................................................................................................... 41
1.4.2 Roadmap of faithful reprogramming ........................................................................................... 42
1.4.2.1 The early phase ................................................................................................................................. 43
1.4.2.2 The intermediate phase ..................................................................................................................... 44
1.4.2.3 The stabilization phase...................................................................................................................... 45
1.4.3 Epigenetic alteration during reprogramming ............................................................................. 46

x
1.4.4 Cooperation of reprogramming transcription factors ................................................................. 47
1.4.5 Molecular and functional similarities and differences between ESCs and iPS cells ................... 49
1.5 AGING AND IPS REPROGRAMMING......................................................................................................... 51
1.5.1 Molecular and cellular hallmarks of aging ................................................................................. 51
1.5.1.1 Stem cell exhaustion ......................................................................................................................... 51
1.5.1.2 Telomere attrition/dysfunction.......................................................................................................... 51
1.5.1.3 Mitochondrial dysfunction ................................................................................................................ 52
1.5.1.4 Epigenetic alterations ........................................................................................................................ 53
1.5.2 Aging in the lung ......................................................................................................................... 55
1.5.3 Rejuvenation using iPS reprogramming strategy ........................................................................ 56
CHAPTER 2 RATIONALE, HYPOTHESES AND OBJECTIVES............................................................. 58
CHAPTER 3 GENERATION OF INDUCED PROGENITOR-LIKE (IPL) CELLS FROM MATURE
EPITHELIAL CELLS USING INTERRUPTED REPROGRAMMING .................................................... 61
3.1 RATIONALE, OBJECTIVES, HYPOTHESES AND SPECIFIC AIMS ................................................................ 61
3.2 ABSTRACT ............................................................................................................................................. 62
3.3 INTRODUCTION ...................................................................................................................................... 63
3.4 MATERIAL AND METHODS ..................................................................................................................... 65
3.4.1 Animal Husbandry ....................................................................................................................... 65
3.4.2 Naphthalene Administration and Cell Delivery .......................................................................... 65
3.4.3 Teratoma Assay ........................................................................................................................... 65
3.4.4 Cell Isolation and Culture ........................................................................................................... 66
3.4.4.1 Isolation of Club Cells from Mouse Lung ........................................................................................ 66
3.4.4.2 Cell Culture ...................................................................................................................................... 66
3.4.4.3 Matrigel-based iPL Induction ........................................................................................................... 67
3.4.4.4 In Vitro Differentiation Assays ......................................................................................................... 67
3.4.4.5 Air-liquid Interface (ALI) Differentiation Assay .............................................................................. 67
3.4.4.6 In Vitro Pluripotency Assay .............................................................................................................. 67
3.4.4.7 Neuron Differentiation Assay ........................................................................................................... 68
3.4.5 Fluorescence Activated Cell Sorting and Analysis ...................................................................... 68
3.4.6 Bottom-feeder conditioned CFSE assay ...................................................................................... 68
3.4.7 Immunofluorescence .................................................................................................................... 69
3.4.8 Iodide Efflux Assay ...................................................................................................................... 71
3.4.9 Western Blot ................................................................................................................................ 71
3.4.10 Real-time PCR Analysis .......................................................................................................... 71
3.4.11 Microarray and Data Analysis ............................................................................................... 73
3.4.12 Statistics .................................................................................................................................. 73
3.5 RESULTS ................................................................................................................................................ 74
3.5.1 Isolation and Identification of Terminally Differentiated Club Cells .......................................... 74

xi
3.5.2 Interrupted Reprogramming Allows OSKM-dependent Proliferation of EpCAMhigh
-Club Cells 74
3.5.3 Interrupted Reprogramming Results in Clonal Expansion of Quiescent Mature Club Cells
without Traversing the Pluripotent State ................................................................................................... 79
3.5.4 Interrupted Reprogramming Allows Club-iPL Cells to Return to Their Original Epithelial
Phenotype upon Withdrawal of Factors .................................................................................................... 84
3.5.5 Interrupted Reprogramming Allows Preservation of Lineage Commitment ............................... 88
3.5.6 Club-iPL Cells Function as Multipotent Bronchiolar Progenitor-Like Cells ............................. 92
3.5.7 Club-iPL Cells are Able to Generate Functional CFTR-Expressing Ciliated Epithelium .......... 94
3.5.8 Club-iPL cells Are Useful in Cell Replacement Therapy for Cystic Fibrosis in Vivo ................. 94
3.5.9 Cyclical Interrupted Reprogramming Enables Greater Expansion of Club-iPL Cells ............... 99
3.6 DISCUSSION ......................................................................................................................................... 103
CHAPTER 4 INTERRUPTED REPROGRAMMING OF ALVEOLAR TYPE II CELLS INDUCES
PROGENITOR-LIKE CELLS THAT AMELIORATE PULMONARY FIBROSIS ............................... 108
4.1 RATIONALE, OBJECTIVES, HYPOTHESES AND SPECIFIC AIMS .............................................................. 108
4.2 ABSTRACT ........................................................................................................................................... 109
4.3 INTRODUCTION .................................................................................................................................... 109
4.4 MATERIALS AND METHODS ................................................................................................................. 111
4.4.1 Animal husbandry...................................................................................................................... 111
4.4.2 Bleomycin administration and cell delivery .............................................................................. 112
4.4.3 Measurement of respiratory mechanics .................................................................................... 112
4.4.4 Isolation of AEC-II cell from mouse lung .................................................................................. 112
4.4.5 Cell culture ................................................................................................................................ 113
4.4.6 Fluorescence activated cell sorting and analysis ...................................................................... 113
4.4.7 Matrigel-based iPL cell induction ............................................................................................. 113
4.4.8 Immunofluorescence .................................................................................................................. 114
4.4.9 Real-time PCR analysis ............................................................................................................. 115
4.4.10 Statistics ................................................................................................................................ 116
4.5 RESULTS .............................................................................................................................................. 117
4.5.1 Interrupted reprogramming rescues the limited clonogenic capacity of AEC-II cells while
achieving expansion ................................................................................................................................ 117
4.5.2 Interrupted reprogramming allows preservation of AEC-II lineage commitment without
traversing the pluripotent state ................................................................................................................ 119
4.5.3 Interrupted reprogramming induces expression of alveolar progenitor markers Hopx and α6β4
121
4.5.4 AEC-II-iPL cells ameliorate bleomycin-mediated pulmonary fibrosis ..................................... 123
4.5.5 AEC-II-iPL cells are able to engraft and differentiate to mature AEC-II and AEC-I cells in
severely injured alveolar epithelium of male mice .................................................................................. 127

xii
4.6 DISCUSSION ......................................................................................................................................... 133
CHAPTER 5 REJUVENATION OF AGED AEC-II CELLS TO A YOUTHFUL STATE USING
INTERRUPTED REPROGRAMMING ....................................................................................................... 136
5.1 RATIONALE, OBJECTIVES, HYPOTHESES AND SPECIFIC AIMS .............................................................. 136
5.2 ABSTRACT ........................................................................................................................................... 137
5.3 INTRODUCTION .................................................................................................................................... 138
5.4 MATERIALS AND METHODS ................................................................................................................. 140
5.4.1 Animal Husbandry ..................................................................................................................... 140
5.4.2 Cell Isolation and Culture ......................................................................................................... 140
5.4.2.1 Isolation of AEC-II Cells from Mouse Lung .................................................................................. 140
5.4.2.2 Matrigel-based 3D condition .......................................................................................................... 140
5.4.2.3 Mix-culture of young and aged AEC-II cells .................................................................................. 141
5.4.3 Fluorescence activated cell sorting and analysis ...................................................................... 141
5.4.4 Immunofluorescence .................................................................................................................. 141
5.4.5 Real-time PCR analysis ............................................................................................................. 142
5.4.6 Statistics .................................................................................................................................... 142
5.5 RESULTS .............................................................................................................................................. 142
5.5.1 The clonogenic capacity of AEC-II cells declines with age ....................................................... 142
5.5.2 Interrupted reprogramming is able to “rejuvenate” aged AEC-II cells to a younger state to form
large numbers of alveolar-like colonies .................................................................................................. 144
5.5.3 ROCK inhibitor failed to rescue the aged-related decline of clonogenic capacity in AEC-II cells
146
5.5.4 Interrupted reprogramming of aged AEC-II cells results in controlled expansion of alveolar-like
colonies 148
5.5.5 Interrupted reprogramming allows controlled restoration of telomerase gene expression ...... 149
5.5.6 Interrupted reprogramming ameliorates mitochondrial DNA damage ..................................... 150
5.5.7 Interrupted reprogramming allows restoration of age-related expression of H3K4me3,
H3K9me3 and H3K27me3 ....................................................................................................................... 152
5.6 DISCUSSION ......................................................................................................................................... 154
CHAPTER 6 SUMMARY AND DISCUSSION ........................................................................................... 157
6.1 SUMMARY OF KEY FINDINGS ................................................................................................................ 157
6.2 DISCUSSION ......................................................................................................................................... 158
6.2.1 What are iPL cells? ................................................................................................................... 158
6.2.2 The regenerative capacity of iPL cells ...................................................................................... 159
6.2.3 The mechanisms underlying the iPL phenomenon .................................................................... 160
6.2.4 Target cell type in iPL induction ............................................................................................... 162

xiii
6.2.5 Derivation condition for iPL cells ............................................................................................. 164
6.2.6 The heterogeneity observed in iPL induction ............................................................................ 166
6.2.7 Highlights of interrupted reprogramming-compared to other approaches ............................... 166
CHAPTER 7 FUTURE DIRECTIONS AND CONCLUSION ................................................................... 169
7.1 CHARACTERIZATION OF THE PROGENITOR CELL MARKERS EXPRESSED BY IPL CELLS ......................... 169
7.2 EPIGENETIC CHARACTERIZATION OF IPL INDUCTION ........................................................................... 170
7.3 IDENTIFICATION OF EPIGENETIC TARGETS RESULTING REJUVENATION VIA INTERRUPTED
REPROGRAMMING.......................................................................................................................................... 170
7.4 EVALUATION OF THE REPARATIVE CAPACITY OF AGED AEC-II-IPL CELLS IN REVERSAL OF PULMONARY
FIBROSIS INDUCED IN AGED MICE .................................................................................................................. 171
7.5 GENERATION OF HUMAN AEC-II-IPL CELLS AND AMELIORATION OF AGED-RELATED DETERIORATION
172
7.6 CONCLUSION ....................................................................................................................................... 173
CHAPTER 8 APPENDIX ............................................................................................................................... 174
REFERENCES ................................................................................................................................................ 182

xiv
List of Tables
Chapter 3:
Table 1. Tabulated list of all antibodies used in the studies.............................................69
Table 2. Tabulated list of all qPCR primers used in the studies......................................72
Chapter 4:
Table 1. Tabulated list of all antibodies used in the studies...........................................114
Table 2. Tabulated list of all qPCR primers used in the studies....................................116

xv
List of Figures
Chapter 1:
Figure 1.
Schematic graph depicting the generation of lung epithelial cells in development................34
Chapter 3:
Figure 1.
CD31-CD45
-EpCAM
high epithelial cells are a highly purified naphthalene-sensitive Club cell
population in which regulation of inductive factors results in controlled proliferation.........76
Figure 2.
Carefully defined length of interrupted reprogramming results in efficient clonal expansion
of quiescent mature Club cells without traversing the pluripotent state in vitro....................81
Figure 3.
Interrupted reprogramming allows EpCAMhigh
-Club cell derived iPL colonies to return to
their original epithelial phenotype upon factor-withdrawal...................................................85
Figure 4.
Interrupted reprogramming allows preservation of lineage preference and commitment......89
Figure 5.
Club-iPL cells function as multipotent bronchiolar progenitor-like cells..............................93
Figure 6.
Club-iPL cells are able to generate functional CFTR-expressing ciliated epithelium in vitro
which are useful as a component of cell replacement therapy for cystic fibrosis in vivo.......96
Figure 7.
Cyclical interrupted reprogramming enables further expansion of Club-iPL cells..............102

xvi
Supplementary Figures
Figure S1.
Characterization of freshly isolated EpCAMhigh
population; 2D bottom-feeder culture
condition.................................................................................................................................78
Figure S2.
Hollow-luminal colony formation during iPL induction; H&E staining of teratomas
developed in NOD/SCID mice 3 weeks post R1-ESC and 5w+Dox
cells transplant...............83
Figure S3.
Relative gene expression of Oct4, Sox2, klf4, c-Myc, EpCAM and E-Cadherin; hierarchical
clustering analysis of Club cell and pluripotency-related gene expression in different groups
of cells....................................................................................................................................87
Figure S4.
Lineage commitment of Club-iPL cells at the single cell level..............................................91
Figure S5.
Club-iPL cells are able to engraft and differentiate to mature Club cells and Ciliated cells in
CFTR-deficient injured airway; Club-iPL cell lines lack of tumorigenicity..........................98
Figure S6.
Preliminary study of interrupted reprogramming on human epithelial cells........................107
Chapter 4:
Figure 1.
Interrupted reprogramming rescues the in vitro limited clonogenic capacity of AEC-II cells
while achieving expansion in cell numbers..........................................................................118

xvii
Figure 2.
A defined period of interrupted reprogramming allows preservation of AEC-II phenotype
without traversing pluripotency............................................................................................120
Figure 3.
The expression of alveolar progenitor markers in AEC-II-iPL cells....................................122
Figure 4.
AEC-II-iPL cells are able to engraft and contribute to alveolar epithelial lineage in vivo...125
Figure 5.
AEC-II-iPL cells are able to engraft and differentiate to mature AEC-II and AEC-I cells in
severely fibrotic lungs...........................................................................................................129
Figure 6.
Treatment with AEC-II-iPL cells ameliorates severe pulmonary fibrosis...........................132
Supplementary Figures
Figure S1.
Time course of expression of Hopx and SPC in embryonic lungs.......................................123
Figure S2.
Transplant of AEC-II-iPL cells ameliorates bleomycin-mediated pulmonary fibrosis........126
Chapter 5:
Figure 1.
The clonogenic capacity of AEC-II cells declines with age.................................................143
Figure 2.

xviii
Aged AEC-II cells exhibit a limited clonal proliferation of epithelial colony forming
units.......................................................................................................................................144
Figure 3.
Interrupted reprogramming ameliorates the aged-related decline of clonogenic capacity in
aged AEC-II cells..................................................................................................................146
Figure 4.
Comparison of clonal proliferation of young and aged AEC-II cells under OSKM iPL
induction and ROCK inhibitor treatment..............................................................................147
Figure 5.
Interrupted reprogramming allows controlled expansion of rejuvenated AEC-II cells........148
Figure 6.
Interrupted reprogramming allows controlled restoration of telomerase gene expression...150
Figure 7.
Mitochondrial DNA damage presents in cultured AEC-II cells that accumulate with the
aging process.........................................................................................................................151
Figure 8.
Interrupted reprogramming ameliorates the aged-related mitochondrial DNA damage......152
Figure 9.
Interrupted reprogramming restores the age-associated expression of H3K4me3 and
H3K9me3 in AEC-IIs...........................................................................................................153
Figure 10.
Interrupted reprogramming restores the age-associated expression of H3K27me3 in AEC-IIs
..............................................................................................................................................154

xix
List of Appendices
Appendix 1
Comparison of OSKM iPL induction and N-myc induction alone in mature Club cells
Appendix 2
The proliferative response of EpCAMhigh
and EpCAMlow
cells to the inductive factors
Appendix 3
Characterization of induced colonies derived from EpCAMhigh
and EpCAMlow
populations in
2D conditions
Appendix 4
Early induction failed to efficiently expand AEC-IIs and preserve AEC-II phenotype
Appendix 5
Transient activation of inductive factors results an enhanced clonal proliferation of
EpCAMlow
population.
Appendix 6
Comparison of the proliferative response of EpCAMhigh
-Club cells to the OSKM inductive
factors and ROCK inhibitor treatment.

xx
List of Abbreviations
8-OHdG 8-hydroxy-guanosine
ARDS Acute respiratory distress syndrome
ATP adenosine triphosphatase
ARGLS age-associated gland-like structures
ALI air-liquid Interface
AEC alveolar epithelial cell
AEC-I alveolar epithelial type I
AEC-II alveolar epithelial type II
BCs Basal cells
BLPCs basal luminal precursor cells
BSCs basal stem cells
BMC bone marrow cell
BMP bone morphogenetic protein
BADJ bronchioalveolar duct junctions
BAL bronchioalveolar lavage
BASCs bronchioalveolar stem cells
CGRP calcitonin gene-related peptide
CFSE carboxyfluorescein diacetate, succinimidyl ester
COPD chronic obstructive pulmonary disease
Cldn10 Claudin10
CCSP Club cell secretory protein
CFU% colony forming-efficiency
CHARM comprehensive high-throughput array-based relative methylation
COX cyclooxygenase
CF cystic fibrosis
CFTR cystic fibrosis transmembrane conductance regulator
Krt14 cytokeratins 14
Krt5 cytokeratins 5
Dppa3 developmental pluripotency associated 3
Dppa5 developmental pluripotency associated 5
DAPI diamidino-2-phenylindole
DASCs distal alveolar stem cells
DNMT1 DNA methyltransferase
ESC embryonic stem cell
ER endoplasmic reticulum
EGF epidermal growth factor
EGFR epidermal growth factor receptor

xxi
EMT epithelial-mesenchymal transition
EpiS epithelial-specific
ECM extracellular-matrix
FGF-10 fibroblast growth factor-10
FoxJ1 forkhead transcription factor
H3K27me3 H3K27 trimethylation
H3K4me3 H3K4 trimethylation
H3K9me3 H3K9 methylation
H4K20me3 H4K20 trimethylation
HA hemagglutinin
HGF hepatocyte growth factor
HSVtk herpes simplex virus thymidine kinase
H4K16Ac histone H4K16 acetylation
Hopx homeodomain only protein x
HHV human herpesvirus
HGPS Hutchinson–Gilford progeria syndrome
IPF Idiopathic pulmonary fibrosis
iPS induced pluripotent stem cells
iPL induced Progenitor-Like
IMM inner mitochondrial membrane
IGF insulin growth factor
KGF keratinocyte growth factor
LTA4 leukotriene A4
LNEP lineage negative epithelial precursor
MET mesenchymal epithelial transition
MSC mesenchymal stromal cell
mtDNA mitochondrial DNA
MEFs mouse embryonic fibroblasts
Muc mucin
PNECs neuroendocrine cells
NEBs neuroepithelial bodies
ncRNAs noncoding RNAs
OSKM Oct4, Sox2, Klf4 and c-Myc
PFA paraformaldehyde
PBS phosphate buffered saline
PEG poly ethyleneglycol
PcG polycomb group protein
ROS reactive oxygen species
RA retinoic acid
ROCK Rho-associated kinase
Pol II RNA polymerase II

xxii
Scgb1a1 secretoglobin family 1a member 1
SCLC small cell lung carcinoma
SSEA1 stage-specific embryonic antigen 1
SPC/Sftpc surfactant protein C
Trf2 telomeric repeat-binding factor 2
TGF transforming growth factor
P63 Trp-63
Utf1 undifferentiated embryonic cell transcription factor 1
Zfp42 zinc finger protein 42
ZO zona occludens

1
Chapter 1
Introduction and Literature Review
1.1 Respiratory epithelium
1.1.1 The physiological function of respiratory epithelium
The respiratory system, develop from the primary bud stage, is composed of tree-like
branched tubes where gases exchange takes place. The maintenance of this function relies on
the proper organization and specification of airway epithelium and the surrounding capillary
network lining the entire airspace (Knight and Holgate, 2003). The gas exchange is carried
out and maximized by hundreds of millions of sacs known as alveoli and the airway
epithelium acts as the conduit for air to and from the alveoli. The airway epithelium not only
serves as a physical barrier, it is also composed of various types of epithelial cells that play a
critical role in maintaining normal airway function from the trachea to the alveoli. It
regulates fluid balance, modulates metabolism and provides host defense against
environmental insults, such as pathogens, inhaled noxious gases and particulates, and
through the production of biologically active factors and cellular self-renewal. The airway
epithelium functions interdependently with other cellular components in the lung, including
mesenchymal cells, endothelial cells, and the extracellular matrix (Crystal et al., 2008).
Dysregulation of airway epithelium function contribute to the pathogenesis of major
respiratory disorders, such as chronic obstructive pulmonary disease (COPD), asthma, cystic
fibrosis, IPF and cancer (Thompson et al., 1995; Puchelle et al., 2006).
1.1.2 Epithelial organization of adult mouse and human lungs
1.1.2.1 The size and structure
Interspecies differences are found between rodents and human respiratory systems.
The average internal diameter of human trachea is ~12 mm.The mouse trachea has an
internal diameter of ~1.5 mm that is equivalent to the size of small peripheral airways in the

2
human lung. Human lungs have more generations of intralobar branches (6-8 branches) than
the mouse lungs (Metzger et al., 2008).
In mice, the trachea, bronchi and most proximal intralobar airways are lined with a
pseudostratified columnar epithelium, while the remaining airways are lined by a simple
cuboidal or columnar epithelium. In humans, the pseudostratified epithelium lines to the
very distal airways (Rock et al., 2010).
1.1.2.2 Cellular composition
In both mouse and human lungs, the pseudostratified epithelium contains basal
(30%), ciliated (55% in mouse; 30% in human), secretory (goblet, serous and Club cells)
and neuroendocrine cells, and the alveolar epithelium consists of type I and type II cells.
However, differences are found in the cellular composition of the airways between mouse
and human along the proximal-distal axis. Human basal cells are contained in the
pseudostratified epithelium which extends to the terminal bronchioles, but not in part of the
bronchioles which are lined with a simple cuboidal epithelium. In contrast, mouse basal cells
only exist in the pseudostratified epithelium restricted to trachea, but not in the columnar
bronchi epithelium. Moreover, mucin-producing goblet cells are relatively abundant in
humans, whereas these cells are rare in mouse lungs (Mercer et al., 1994).
1.1.3 Epithelial lineages in adult lungs
1.1.3.1 Conducting airways
1.1.3.1.1 Basal cells
Basal cells (BCs) are present in the pseudostratified epithelium of mouse trachea and
human airways including the bronchioles (Boers et al., 1998; Evans et al., 2001). They
appear to be the only cells that have abundant hemidesmosomes (Evans et al., 2001) and are
attached to the basement membrane by integrin α64. The number of BCs attached to the
basement membrane is correlated with the thickness of the epithelium and progressively
decreases in smaller airways (Evans and Plopper, 1988; Knight and Holgate, 2003). In
human lungs, BCs form a monolayer in larger airways and are distributed in clusters or as
individual cells in the smaller bronchioles (Nakajima et al., 1998). Despite the interspecies

3
differences between rodents and human airways, BCs in both species are similar with
respect to histology and molecular function.
Basal cells play a critical structural and functional role in conducting airway
epithelium. They are relatively undifferentiated and characterized by high expression levels
of transcription factor Trp-63 (P63) and cytokeratins 5 and 14 (Krt5/14). In mouse trachea,
there is a differential expression of Krt5 and Krt14 within the basal population. In
homeostasis, Krt14 is co-expressed in a subset of Krt5+
BCs which is upregulated in
response to epithelial injury (e.g. naphthalene-induced injury) (Hong, 2003a; Hong et al.,
2004).
Studies have demonstrated that BCs possess stem cell properties that are capable of
self-renewal and repopulating secretory and ciliated lineages during homeostasis and repair
after injury (Hong et al., 2004; Rock et al., 2009a). In addition to their critical role in
maintaining epithelium structural and repopulating injured epithelium, BCs involve in host
defense by their production of a number of bioactive molecules, such as cytokines, neutral
endopeptidase and 15-lipoxygenase products (Knight and Holgate, 2003).
1.1.3.1.2 Pulmonary neuroendocrine cells
During lung development, neuroendocrine cells (PNECs) are the first cells to form
and differentiate within the respiratory epithelium (Ito et al., 2000; Shan et al., 2006). In
morphology, PNECs are tall and pyramidal in shape and have apical microvilli (Gustafsson
et al., 2008). They extend from the basal lamina of the airway epithelium and are located
within the trachea, bronchi, and bronchiole alveolar junctions (Lauweryns et al., 1985). The
number of PNECs increases after birth and reach the peak in neonatal stage (Gustafsson et
al., 2008). In adult lungs, PNEC distribute as individual cells in proximal airways or in
clusters, termed neuroepithelial bodies (NEBs), in intralobar airways representing the lung
stem cell niche (Cutz et al., 2007; Van Lommel, 2001). PNECs play a critical role in the
maintenance of immune function, flow of air and blood by secreting serotonin, calcitonin
(CGRP or Calca), and bombesin. In addition, PNECs can transmit stimuli to the central
nervous system upon the innervations by sensory nerve fibers (Van Lommel et al., 1998).
Importantly, the PNECs system contributes to airway epithelium regeneration and involes in
the pathogenesis of small cell lung cancer (Ito et al., 2003; Cutz et al., 2007).

4
1.1.3.1.3 Club cells
Mucociliary clearance, to clear inhaled microorganisms and particulates from the
lung, is primarily driven by Club and ciliated cells located within the columnar epithelium.
Club cells are secretory cells that are prominently found in the small bronchioles (Knight
and Holgate, 2003). These cells contain electron-dense granules and produce secretoglobin
Scgb1a1 and/or Scgb3a2 and Scgb3a1 (in proximal airways) (Reynolds et al., 2002). Club
cells regulate bronchiolar epithelial integrity and immunity by secretion of bronchiolar
surfactants and specific antiproteases, such as secretory leukocyte protease inhibitor. In
addition to their secretory role, Club cells shown to metabolize xenobiotic compounds such
as aromatic hydrocarbons by production of p450 mono-oxygenases (De Water et al., 1986).
The mature Club cells possess limited proliferative capacity. After injury, they are replaced
from a pool of “variant” Club cells, which are resistant to toxins such as naphthalene. This
population also then act as stem cells for ciliated and mucous cell populations (Rawlins et
al., 2009; Reynolds and Malkinson, 2010).
1.1.3.1.4 Goblet cells
Mucin-producing goblet cells are sparse in the mouse airways but are relatively
abundant in human airways. Goblet cells are characterized by electron-lucent acidic-mucin
granules that secrete mucous into the airway from the level of the trachea to the bronchioles
to trap pathogens and dust particles (Jeffery, 1983). The amount and viscoelasticity of
mucus are critical for maintaining efficient mucociliary clearance. In a healthy condition,
there is a fine equilibrium between mucous production and clearance (Evans and Koo,
2009). However, mucous cells become hyperplasic and metaplasic in chronic airway
inflammatory diseases, such as chronic bronchitis and asthma, which result in excessive
sputum production (Lumsden et al., 1984). Goblet cells can self-renew and
transdifferentiate into ciliated cells (Evans and Plopper, 1988). Evidence from pathological
remodeling in human airways and mouse models of lung disease showed the abundance of
goblet cells is regulated by the Notch pathway (Evans et al., 2004; Williams et al., 2006).
Activation of Notch in developing airways transgenically in vivo or by stimulated molecular
pathways in vitro results in expansion of goblet cell population at the expense of ciliated

5
cells. Conversely, inhibition of Notch signaling blocks the transdifferention of goblet cells
and results in an increased proportion of ciliated cell lineage (Guseh et al., 2009).
1.1.3.1.5 Ciliated cells
Ciliated cells are predominant cell type in the airways, which account for over 50%
of all epithelial cells (Spina, 1998). These cells typically contain 300 cilia per cell and
numerous mitochondria that provide energy to the cilia for ciliary beating and the transport
of mucus from the lung to the throat for mucociliary clearance. Ciliated cells are terminally
differentiated cells which arise from either basal or Club cells (Ayers and Jeffery, 1988).
The differentiation and maturation of ciliated cells are dependent on their expression of
forkhead transcription factor (FoxJ1) (Brody et al., 2000).
1.1.3.2 Distal airways
Gas exchange is carried out in the alveolus. Alveolar walls cover more than 99% of
the internal surface area of the lung (Dobbs and Johnson, 2007). The thin layer of liquid
lining the epithelial surface of alveolus contains surfactant phospholipids and an aqueous
subphase (Bastacky et al., 1995). In additions to regulating gas exchange within the
alveolus, surfactants are responsible for the maintenance of surface tension which is
essential for elastic recoil of the lung. The alveolar ion and liquid transport is tightly
regulated by local alveolar epithelial cells (Olivera et al., 1994). In normal adult lung, the
alveolar epithelium facing the air-filled compartment is composed of two major cell types,
the alveolar epithelial type I (AEC-I) and alveolar epithelial type II (AEC-II) cells.
1.1.3.2.1 Alveolar type I (AEC-I) cells
AEC-I cells are squamous in shape. They interface with pulmonary capillaries and
cover more than 90% of the alveolar surface providing a physical barrier between the air and
blood compartments (Whitsett et al., 2010). AEC-I cells are the primary sites of gas
exchange and regulate fluid homoeostasis through the expression of ion channels and pores,
including AQP5 and CFTR. The presence of of caveolae (Gumbleton, 2001) and small
intracellular vacuoles in AEC-I cells suggests they possess endocytic function and metabolic
activities. In addition, AEC-I cells have active biosynthetic functions in regards to their

6
cellular components-microvilli, abundant mitochondria, and both rough and smooth
endoplasmic reticulum (Dobbs and Johnson, 2007).
Although it is widely assumed that AEC-I cells are terminally differentiated cells
derived from AEC-II cells during postnatal growth, a recent lineage-tracing study showed
Hopx+ AEC-I cells are able to proliferate and give rise to AEC-II cells following partial
pneumonectomy in adult mouse lungs (Jain et al., 2015).
1.1.3.2.2 Alveolar type II (AEC-II) cells
Unlike AEC-I cells, AEC-II cells are small cuboidal cells located in the corners of
alveoli that cover only 5% of the alveolar surface. They are characterized by lamellar bodies
and intracellular storage organelles for pulmonary surfactants (Dobbs and Johnson, 2007b).
AEC-II cells are multifunctional cells that produce, secrete and recycle pulmonary
surfactants, regulate alveolar fluid balance, and synthesize and secrete a number of immune-
modulatory proteins involved in host defense (Fehrenbach, 2001). Importantly, a subset of
surfactant protein C-positive (SPC+) AEC-II cells act as regional stem cells in the alveoli
and differentiate into AEC-I cells, playing a crucial role in replenishing the alveolar
epithelial barrier during both homeostasis and repair after injury (Barkauskas et al., 2013a;
Rock and Hogan, 2011a; Whitsett and Alenghat, 2014).
1.2 Epithelial biology in the pathogenesis of pulmonary disease
The airway epithelium plays a crucial role in providing host defense against
environmental insults. Therefore, to better understand the development and progression of
pulmonary diseases, one must consider how epithelial dysfunction could lead to pathologic
outcomes.
1.2.1 Obstructive lung diseases
1.2.1.1 Chronic obstructive pulmonary diseases
Chronic obstructive pulmonary disease (COPD), a critical condition with high
morbidity and mortality, is characterized by non-reversible progressive airflow limitation.
Although cigarette smoke is the main cause, air pollution, biomass fuel, and occupational
exposures have also been linked to the development of COPD (Eisner et al., 2010).

7
Emphysema and chronic bronchitis are the two major pathologic subjects with COPD.
Emphysema involves the destruction of alveolar structure. Chronic bronchitis refers to
chronic inflammation and the resultant airway remodeling. To date, although numbers of
medications with bronchodilating and/or anti-inflammatory effects have been prescribed in
clinical practice, no definite treatments are able to successfully repair the severely injured
lungs of patients with COPD (Hochberg and Sidhaye, 2017).
The respiratory epithelium, as the first line of exposure to the noxious particles and
gases, provides a protective barrier of which functions include physical, chemical, and
immunological roles. It plays a central role in the pathophysiology of COPD that the
abnormal response of epithelial cells to these insults results in the remodeling of airway and
air spaces which are the major characteristic of COPD.
In COPD, the large airways and the alveoli present distinct pathological changes. In
the airways, the remodeling process results in an altered epithelial cell lining, fibrosis,
smooth muscle hypertrophy, and inflammatory infiltration (Hogg and Timens, 2009). In
alveoli, the remodeling is characterized as emphysema showing abnormal permanent
enlargement of air spaces and the destruction of epithelium walls without obvious fibrosis
(1985).
1.2.1.1.1 Airway epithelium remodeling
In normal lungs, the larger airways are pseudostratified mucociliated epithelium
comprising of ciliated cells in the majority and mucin-producing goblet cells and basal cells.
The number of both goblet and basal cells declines towards the distal branching and the
pseudostratified epithelium is gradually replaced with cubodial epithelium consisting of
serous secreting cells and Club cells.
In COPD, the morphology and function of airways epithelium are altered. Goblet cell
hyperplasia and metaplasia occur as a result of transdifferentation of other cell types into
goblet cells. In addition, metaplasia is seen in the transdifferentiation of epithelial cells into
mesenchymal and squamous cell-types (Gohy et al., 2016). These pathological changes lead
to ciliary dysfunction, fibrosis, and inflammatory cell infiltration seen in the airway wall and
the airway lumen (Hogg and Timens, 2009). In the chronic bronchitis which is a subject

8
with COPD, mucous cells become hyperplasic and hypersecretory which lead to chronic
cough and sputum production.
1.2.1.1.2 Epithelial dysfunction
A healthy airway epithelium serves as a physical barrier, as well as restores proper
barrier function responding to injury. The breakdown in these functions attributes to the
development of COPD.
1.2.1.1.2.1 Physical barrier dysfunction
Epithelial barrier dysfunction has been observed in airway epithelium injury by
cigarette smoke and in the disease state of COPD (Jones et al., 1980; Gangl et al., 2009;
Heijink et al., 2012).
The junctional proteins and ion channels are involved in the physical barrier function
of the airway epithelium by regulating epithelium permeability. The integrity of the airway
epithelium is maintained by tight junctions, adherent junctions, and desmosomes (Brune et
al., 2015). There are multiple tight junction proteins involved in regulating ion transport,
including claudins, occludins, junctional adhesion molecules, and the zona occludens (ZO).
The adherent junction composed of E-cadherin which regulates tight junction formation and
cell adhesion (Hartsock and Nelson, 2008a). E-cadherin interacts with α-catenin and β-
catenin to form a complex. This cadherin/catenin complex plays a critical role in cellular
signaling pathways to regulate cell proliferation, differentiation and the reparative response
of epithelium to injury (Ganesan et al., 2013; Hartsock and Nelson, 2008). Importantly, E-
cadherin also interacts with epidermal growth factor (EGF) receptor (EGFR) to maintain
normal polarity and function of epithelium. In COPD, the EGFR pathway is altered and the
increased EGFR activity contributes to pathologic consequences, including barrier
dysfunction, goblet cell hyperplasia/metaplasia, mucous hypersecretion, and epithelial-
mesenchymal transition (EMT) (Gohy et al., 2016b; Petecchia et al., 2009; Shaykhiev et al.,
2013). Furthermore, the oxidative stress from smoking and chronic injury result in protein
modification and disruption of epithelium tight junctions (Rao, 2008).

9
1.2.1.1.2.2 Mucociliary clearance defect
The efficient mucociliary clearance is regulated by mucin (Muc) production, and the
transmembrane ion channels such as the cystic fibrosis transmembrane conductance
regulator (CFTR), and the apical membrane Na+ channel (Boucher, 2003).
Abnormal mucociliary clearance occurs in COPD, causing increased mucous which
leads to mucous plugging and airflow obstruction. This mucociliary dysfunction is a
consequence of mucous hypersecretion from goblet cell hyperplasia, metaplasia and
subsequent stimulus for mucous release, altered types of mucin expressed, and the decrease
of ciliary function from the shortening and loss of cilia. The number of mucin-producing
cells is markedly increased in COPD airways as a result of goblet cell hyperplasia and
metaplasia, as well as the trans-differentiation of both ciliated and Club cells into the
secretory cell type (Rogers, 2005). In addition, an increased quantity and altered
composition of mucous are seen in COPD. Under the normal condition, Muc5AC and
Muc5B are the predominant mucins that are expressed in goblet cells and mucosal gland,
respectively. In COPD, the ratio of Muc5B/Muc5AC increases as Muc5B is expressed in
both goblet and glandular cells (Rose and Voynow, 2006). As seen in cystic fibrosis (CF),
there is an increase of viscosity of mucus found in COPD, which is partially due to CFTR
dysfunction (Cantin et al., 2006). Furthermore, the dysfunction of ciliated epithelium as a
result of the decreased number of ciliated cells and shortened cilia contributes to mucociliary
dysfunction in COPD (Lam et al., 2013).
1.2.1.1.2.3 Aberrant repair of airway epithelium
In response to injury, the airway epithelium initiates endogenous repair process to
replenish the pool of epithelial cells and restore lung function. EMT is seen in normal tissue
repair process, characterized by the loss of epithelial markers (e.g. E-cadherin) and gain of
mesenchymal markers including vimentin, alpha smooth muscle actin and fibroblastic-
specific protein 1/S100A4 (Kalluri and Weinberg, 2009). A proper EMT, which is mainly
driven by TGF-β, EGF, and fibroblast growth factors, allows cell migration and the secretion
of the extracellular-matrix (ECM) products to restore the damaged epithelial barrier.
However, EMT is also involved in the development of fibrosis due to the excessive
propagation of ECM materials produced in response to the constitutive inflammatory insults

10
(Kalluri and Weinberg, 2009). Studies showed EMT plays a crucial role in the pathogenesis
of COPD that persistent EMT presents in the large and small airways of COPD subjects
(Milara et al., 2013; Sohal et al., 2010) and contributes to the development of airway fibrosis
in the COPD subjects with severity of airway obstruction (Gohy et al., 2015). Furthermore,
squamous metaplasia is observed in the bronchial epithelium of COPD lungs. These
mataplasic squamous cells produce large amount of IL-1β which results in the EMT-
associated fibrosis and small airways remodeling via the TGF-β pathway (Araya et al.,
2007). Importantly, both squamous metaplasia and EMT alter cell polarity and adhesion,
causing fibroblast proliferation, secretory-cell hyperplasia, and smooth muscle hypertrophy.
Taken together, the airway epithelium plays a crucial role in the pathogenesis of COPD due
to the dysregulation of the epithelium barrier resulting in the failure to protect airways from
prolonged pathogen insults and tissue repair. Thus, a better understanding of the underlying
pathophysiology of COPD and the restoration of a functional epithelium in COPD will be
the targets in future studies.
1.2.1.2 Asthma
Asthma, a chronic lung disease, features respiratory distress with airway
inflammation and airflow obstruction. The increased incidence of asthma over the past
decades is correlated with the increased inhaled environmental stimuli. As the host defense
barrier, healthy airway epithelium effectively clears out the majority of inhaled substances.
Once this epithelium barrier is broken-down, the following inflammatory response is
initiated to protect the internal milieu of the lungs. In asthma, the structure and function of
the airway epithelium become abnormal. Similar to that of COPD, the disruption of
epithelium structure is found in asthma, accompanied with an increased number of mucin-
producing cells compared to normal airways. Functionally, the airway epithelium barrier in
asthma lungs is breached and become more permeable and susceptible to the oxidative
products, resulting a deficient immune response that fails to protect the lungs from virus
infection. Therefore, the disruption and dysfunction of airway epithelium play a crucial role
in the development and progression asthma.

11
1.2.1.2.1 Epithelium alteration and dysfunction
In asthmatic airways, the epithelium structure is altered with marked loss of
columnar epithelial cells and goblet cells hyperplasia and metaplasia (Ordoñez et al., 2001).
In addition, the barrier function of asthmatic epithelium changes so the epithelium becomes
more permeable and susceptible to the oxidative stress from inhaled stimuli, including
allergens, airborne particulates, infectious agents, and noxious gases. These barrier function
defects in asthma are caused by genetic or /and the alteration of junctional complex proteins.
Studies of epithelial junctional complexes in the biopsies from subjects with asthma showed
the remarkable reductions of α-catenin, β-catenin, E-cadherin, and ZO-1 compared with lung
tissues from non-asthmatic controls (Hackett et al., 2013). In addition, Claudin 18, one of
the key tight junction proteins, was also significantly down-regulated in epithelial cells
brushed from asthmatic airways (Sweerus et al., 2017). Moreover, studies have identified
PCHD1, IL-33, and ORMDL3, genes that are expressed in the airway epithelial cells, as
being closely associated with the increased susceptibility of asthmatic lungs to inhaled
substances.
The airway epithelium is also a key component of the immune system with the
ability of regulating the protective activities of inflammatory cells (Swindle et al., 2009).
Thus, the breakdown of the epithelium barrier is also attributed to the deficient innate
immune response to virus infection in asthma (Xiao et al., 2011). In addition, the
dysregulation of the stem cell population is observed in asthmatic epithelium reflecting an
abnormal repair response. Although stem cells increase up to 40 times in number in
asthmatic lungs compared to that in normal lungs, asthmatic lungs still fail to efficiently
restore the function of injured lungs (Hackett et al., 2008).
1.2.1.2.2 Dysregulation of epithelium-mesenchymal network
In the airways, epithelium and the underlying fibroblast sheath are functionally
accompanied to control the microenvironment during lung growth and development, in
response to injury and during tissue repair. During normal repair process, epithelial cell
located at the edges of the wound undergo EMT and these epithelial-derived mesenchymal-
like cells migrate into the matrix to temporally restore the barrier function. Meanwhile, the
underlying fibroblasts proliferate and differentiate to myofibroblasts to support the injured

12
epithelium surface. Following the efficient restoration of the epithelial barrier, the remaining
epithelial cells differentiate to mucin-producing cells and later to ciliated cells to restore the
epithelial surface and mucoclilary clearance. Thereafter, the myofibroblasts undergo
apoptosis and the extracellular matrix is remodeled to normal architecture (Crosby and
Waters, 2010).
In asthma, this epithelial-mesenchymal network is dysregulated and results in
aberrant repair and airway remodeling. As mentioned before, the activation of EGFR
regulates cell proliferation and migration which plays a critical role during normal tissue
repair (Xiao et al., 2012). However, in asthmatic lungs, there is a markedly increased
expression of EGFR which is responsible for the impeded reparative response (Fedorov et
al., 2005). Concurrently, the cell cycle inhibitor genes (e.g. P21 waf1) are upregulated in
asthmatic lungs (Puddicombe et al., 2003). These may directly contribute to
hyperprolifeation of the stem cell population seen in the asthmatic epithelium. Furthermore,
studies using bronchial biopsy specimens from children with asthma showed the correlation
of the overexpression of EGFR with the extensive collagen deposition indicating the
abnormal communication between asthmatic epithelium with the underlying fibroblasts
(Fedorov et al., 2005).
1.2.1.2.3 Current treatments of asthmatic epithelium
Epithelium dysfunction has been considered as a main driver in the initiation and
progression of asthma. Therefore, current approaches to treating asthma focus on the
restoration of the barrier function of the epithelium to inhaled insults. As asthmatic
epithelium suffers from cycled oxidative stress, tight control of pollution while exercising
and antioxidant therapy would be beneficial to protect the epithelial barrier. Studies of the
response of asthmatic lungs to viral infection suggest that IFN replacement might decrease
the severity of viral infection and regulate immune pathway (Kroegel et al., 2009). To
reestablish the damaged tight junction of injured epithelium, studies showed apical
application of supplemental epidermal growth factor (EGF) which can increase tight
junction formation of columnar epithelial cells and maintain epithelial integrity, maybe
beneficial. Although EGF stimulates cell proliferation, a tight control of the amount and

13
length of apical delivery of EGF could be ideal as the intact tight junctions could prevent
excessive EGF penetrating across the epithelium (Sinha et al., 2003).
In future studies, a better understanding of the underlying mechanisms of epithelial
barrier dysfunction in asthma will be an important advance and will help the development of
specific therapies to maintain and restore the protective function of asthmatic epithelium is
required.
1.2.1.3 Cystic fibrosis
Cystic fibrosis (CF) is a lethal genetic disease characterized by obstructive
pulmonary disorder and pancreatic insufficiency (Davis, 2006). The progressive obstructive
pulmonary disease, which is responsible for the high mortality in CF patients, features
airway obstruction, thick mucus production, extensive inflammation and defects in host
defense to pathogen (Elborn, 2016). CF is caused by the defect of the CF transmembrane
conductance regulator protein (CFTR). In the airways, defect of CFTR causes aberrant
chloride ion transport (Quinton, 1983; Welsh and Liedtke, 1986) and alterations in surface
fluid, mucus and host defenses resulting in chronic airway infection and occlusion. Thus,
CFTR has been a target gene for development of gene therapies treating CF, although no
successful approach has yet been proven. In addition to the CFTR gene itself, the epigenetic
and microbiological interactions are closely associated with CFTR defects and contribute to
the progression and this complex multi-systemic disorder (Elborn, 2016).
1.2.1.3.1 Defects of CFTR gene and protein
Cystic fibrosis transmembrane conductance regulator (CFTR), mainly expressed in
the apical surface of the ciliated epithelium in the lung, encodes a cAMP-regulated chloride
channel that plays a critical role in regulating chloride and water transport. There is also
evidence suggesting that CFTR may transport bicarbonate and glutathione (Schwiebert et
al., 1999) and interact with ENaC to maintain the airway surface liquid (ASL) (Knowles et
al., 1983).
To date, mutation of the CFTR gene has been extensively studied showing over 1800
mutations, though the effects on CFTR protein function with many of these mutations have
not yet been fully understood (Castellani et al., 2008). These effects of these mutations range

14
from a completely deficient CFTR protein to a functional CFTR protein with insufficient
quantities. It is known that the prevalence of mutation depends on the race and region. In
North America, the most common CFTR mutation is the loss of phenylalanine at codon 508-
ΔF508. Approximately, 90% of CF patients only have one copy of ΔF508 with 50% of them
homozygous for this mutation. The ΔF508 mutation results in misfolded CFTR protein that
fails to reach the apical surface to function normally (Rohlfs et al., 2011). Notably, there is
significant individual variability in lung functions of the patients homozygous for ΔF508.
Nevertheless, manipulation of CFTR modulators and application of supplemental functional
CFTR protein are the important aspect in the development of therapies treating CF.
1.2.1.3.2 Epithelium dysfunction attributes to the adaptive and
innate immune defect
In addition to causing a failure of the host defense, the dysregulation of airway
epithelium contributes to the innate immune dysfunction seen in CF.
The epithelium interacts with inhaled infection substances, modulates airway fluid
clearance and is able to resists infection and recruit immune cells to infected areas by the
secretion of anti-infective proteins, including defensins and anti-microbial enzymes and
chemotactic products (Hiemstra et al., 2015). The neutralization of infectious insults relies
on the neurophils migrating through the epithelium to the airway (Whitsett and Alenghat,
2015). Neutrophil is the main component of the adaptive immune system in the airways
which function to clear infective substances by producing reactive oxygen species, protease
and cytokines (Gifford and Chalmers, 2014; Twigg et al., 2015). To the airway epithelium,
elastase secreted by neurophils is a destructive protease which decreases ciliary beating
frequency, increases permeability of epithelial barrier (Peterson et al., 1995), and stimulates
epithelium to produce cytokines (Taggart et al., 2000). Therefore, the activity of neutrophils
could be a source of epithelial dysfunction in CF. For example, the pro-inflammatory
cytokines secreted by CF neutrophils, including IL-1, IL-6, IL-8, IL-17, CCL10, and
CXCL10, affect the ability of airway epithelial cells to recruit other types of immune cells
(Taylor et al., 2016). Importantly, studies showed many of these cytokines destroy epithelial
tight junctions and cause epithelial barrier dysregulation in ion and fluid balance, as well as
metabolites in response to infection in CF lungs (Molina et al., 2015).

15
1.2.1.3.3 Cystic fibrosis disease models
Disease models are critical in understanding the pathophysiological mechanisms
underlying the disease and to develop therapeutic strategies.
1.2.1.3.3.1 In vivo CF models
Mouse models are the most commonly used in in vivo CF studies. There are over 15
strains of mice have developed, including the simple deletion of the mouse CFTR and ENaC
genes (Fisher et al., 2011). Although mouse models could present many of the
manifestations of human CF, including epithelium dysfunction and exaggerated immune
response, they fail to faithfully replicate the human CF pathophysiology (Scholte et al.,
2004).
Thus, great efforts have been placed on creating new in vivo models in larger
animals, including rats, ferrets, and pigs (Rogers et al., 2008). Pigs are specifically important
for studying CF as pigs have a longer lifespan compared to other species; the size and
structure of pig lungs resemble that of human lungs; and CF pigs develop CF-related
pancreatic disease similar to that in human CF patients (Lavelle et al., 2016). One of the
major differences between mice and human airways is that mouse airways lack of
submucosal glands. CF pig studies are able to show submucosal glands contribute to CF
airway dysfunction by aberrant mucus production (Hoegger et al., 2014). In addition, the
recurrent and progression of infected bacteria colonization is one of the phenotypic
pathological changes in human CF that is absent in mouse models. The increased
colonization of bacterial infection is aided once the airway surface liquid PH is acidified.
Mouse models fail to replicate the acidification of human CF airways because of the lack of
the nongastric H+/K+ adenosine triphosphatase (ATP12A) protein, which is the key
hydrogen-potassium exchanger. In contrast, the CF pig model is able to replicate the airway
acidification in human CF and show CFTR deficiency contributes to the increased bacterial
colonization in the airways (Shah et al., 2016).
1.2.1.3.3.2 In vitro CF models
In vitro, models of epithelial cells and immune cells are important for understanding
the cell molecular mechanisms underlying CF and identification of potential therapeutic

16
targets. Primary epithelial cells isolated from human bronchial, tracheal and nasal epithelial
cells have been used in in vitro CF studies (Fulcher and Randell, 2013). Many of the initial
studies of CFTR used epithelial cell lines generated from cells isolated from donor lungs or
cancerous tissue upon viral transduction to overexpress mutant CFTR genes. However, the
cells of origin and their deviance from normal cell physiology limit their ability to faithfully
replicate the physiology/pathophysiology of epithelial cells in CF (Molenda et al., 2014).
Human epithelial cells are poorly grown under regular cell culture condition. To date, much
progress has been made in derivation of human epithelial cells by conditional
reprogramming (Reynolds et al., 2016) or multi-step differentiation of pluripotent cells
(Huang et al., 2013a; Zhou et al., 2014). However, in regards to the significant individual
variability in CF patients, identification of the effective drug combination for individual
patients relays on responses of their own cells to the drugs. Thus, the progress of
personalized medicines (individual patient-directed therapy) acquires new techniques that
allow larger scale expansion of epithelial cells while retaining the genotype of each patient.
1.2.2 Restrictive lung diseases
1.2.2.1 Interstitial lung disease-idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is an irreversible, fatal interstitial lung disease
characterized by progressive decline in lung function with death occurring in most patients
within 5 years of diagnosis. The precise molecular mechanisms underlying the progressive
fibrosis which leads to the relentless destruction of the lungs are still not completely
understood. However, increasing evidence suggests that the pathogenesis of IPF is driven by
alveolar epithelial cell dysfunction, followed by aberrant regeneration of epithelium,
persistent activation of fibroblasts and alterations in epithelial-mesenchymal communication
with the extracellular matrix (ECM), together resulting in the disruption of architecture and
progressive loss of lung function (King et al., 2011; Yanagi et al., 2015; Zoz et al., 2011a).
Currently, medical therapy for IPF is limited and lung transplantation is the only option for
patients with end-stage IPF (Akram et al., 2014; Lomas et al., 2012). Clinical studies of
Pirfenidone and Nintedanib, the two anti-fibrotic drug approved by FDA for IPF, showed no
evidence on arresting or reversing fibrosis (King et al., 2014; Richeldi et al., 2014).

17
1.2.2.1.1 Profoundly altered differentiation of alveolar epithelial
cells in IPF
In response to injury, AEC-II stem cells undergo self-renewal and differentiate to
AEC-I cells to replenish the damaged alveolar epithelial pool. However, differentiation of
AEC-II to AEC-I cells is profoundly altered in IPF airway, as a result of the vast
abnormalities in ECM, epithelial basement membrane damages and destruction of the
airway architecture. The ECM is not only a supportive structure but also functions to
regulate cell proliferation and differentiation. It has been shown that changes in ECM
component influence the morphology, the phenotype of alveolar epithelial cells and the
differentiation of AEC-II to AEC-I-like cells (Olsen et al., 2005). In addition, mechanical
changes have been shown to regulate AEC phenotype. For instance, mechanical distension
in favor of the AEC-I cell phenotype and inhibit the AEC-II phenotype, while contraction
promotes AEC-II phenotype (Dobbs and Gutierrez, 2001).
Furthermore, many growth factors and cytokines produced in response to injury can
influence AEC-II differentiation. For instance, keratinocyte growth factor (KGF) which is an
epithelial mitogen can induce apoptosis and differentiation of hyperplasic AEC-II cells to
restore the normal alveolar epithelium in vivo (Fehrenbach et al., 1999). In addition, insulin
growth factor (IGF) signaling has been found to regulate the differentiation of AEC-II cells
to AEC-I-like cells. The inhibition of IGF receptors in AEC-II cells derived from hyperoxia-
exposed lung is able to maintain the AEC-II phenotype without the transdifferentiation to
AEC-I-like cells (Narasaraju et al., 2006). The activity of leukotriene A4 (LTA4) hydrolase
has also been linked to AEC-II to AEC-I differentiation. In normal AEC-IIs and the ones
located in the fibrotic regions of IPF airways, LTA4 hydrolase is predominantly expressed in
the nucleus. While in AEC-I and AEC-II-derived AEC-I-like cells, LTA4 hydrolase
accumulates in the cytoplasm, suggesting nuclear export of LTA4 hydrolase influences
AEC-II differentiation (Brock et al., 2005).
1.2.2.1.2 Epithelial cells contribute to the profibrotic
microenvironment
Epithelial cells can induce fibroblast activation, proliferation, and migration as well
as ECM accumulation in IPF by secreting platelet-derived growth factor, TGF-, connective

18
tissue growth factor, tumor necrosis factor α, endothelin-1, and osteopontin (Pardo et al.,
2005). Polarized T cells regulate lung fibrosis by secreting cytokines, including interleukin 4
(IL-4) and IL-13, which induce fibroblast proliferation and accumulation of ECM. There is
evidence showing that AEC-II cells can also produce IL-4 in IPF potentially causing the
imbalance of profibrogenic cytokines. In addition to providing activation signals to
mesenchymal cells, injured epithelium fail to suppress fibroblasts due to the decreased
production of PGE2 and two cyclooxygenase (COX) enzymes, COX-1 and COX-2 (Lama et
al., 2002; Petkova et al., 2003).
1.2.2.1.3 Mechanisms of epithelium damage
In IPF, the combination of various types of injuries repeatedly acts on AEC cells and
results in epithelium dysfunction and aberrant repair.
1.2.2.1.3.1 Viral infection
Viral infection has been considered to be one of the major sources causing repetitive
injury to alveolar epithelium. While the involvement of some specific virus in IPF is not
universal, the etiologic significance needs to be further evaluated.
The Epstein-Barr virus (EBV) has been identified in the human IPF lung tissues that
mainly infects AECs and promotes the development of pulmonary fibrosis (Egan et al.,
1995; Lok et al., 2002). A study on the presence of eight herpesviruses in human IPF lung
tissues showed one or more of four herpesviruses, including cytomegalovirus, EBV, human
herpesvirus (HHV)-7 and HHV-8, were detected in all IPF lung tissues. Furthermore,
detection of viral antigens expression in lung sections showed EBV and HHV-8 infection
localized to AECs (Tang et al., 2003).
1.2.2.1.3.2 Induced apoptosis
Epithelial cell apoptosis is an essential feature of IPF. Studies showed that the up-
regulation of Fas signaling and the angiotensin peptides produced by fibroblasts induce
alveolar epithelial cell apoptosis (Selman and Pardo, 2003). Oxidative stress also induces
damages in epithelial cells. In response to injury, H2O2 produced by myofibroblasts acts as
an apoptosis signal on epithelial cells. Moreover, oxidative stress and reactive oxygen

19
species cause DNA modifications in AECs and result in cell damage and apoptosis
(Waghray et al., 2005).
1.2.2.1.3.3 Telomerase mutation and telomere dysfunction
To date, an increasing body of evidence has shown the intrinsic abnormalities in
alveolar epithelial cells contributing to the development and progression of IPF. Mutations
in telomerase have been identified to be the most common genetic risk factor for IPF.
Telomeres protect the chromosome ends for DNA repair and degradation activities
and telomerase is the enzyme responsible for maintaining telomere length and for cell self-
renewal (Blackburn et al., 2006). Telomerase has two major components: hTERT, the
telomerase reverse transcriptase, and hTR, a specialized RNA has a template for telomere
repeat addition. The genetic defect caused by mutations in hTERT and hTR accounts for 8-
15% of IPF familial cases (Armanios, 2012-37,38).
IPF is an age-related disease in that the majority of patients are age 60 or older
(Raghu et al., 2006). Telomeres and telomerase are susceptible to age-related deterioration.
Studies using genetically modified animal models have demonstrated the causal links of
telomere dysfunction, cellular senescence and aging. Although telomere attrition manifests
during normal physiological aging, pathological telomere dysfunction provokes aging
(Blackburn et al., 2006; Flores and Blasco, 2010) and plays a causal role in premature
development of various human diseases, including IPF (Tsakiri et al., 2007; Cronkhite et al.,
2008). The mechanisms of telomere defects provoking IPF are not fully understood, but
numbers of evidence has revealed the causal links of telomere defects with epithelial
dysfunction observed in IPF (Alder et al., 2008; Zoz et al., 2011b). Importantly, mutations in
telomerase and telomere genes cause abnormal telomere shortening. The shortening of
telomere length is a common feature of IPF observed in lymphocytes, granulocytes as well
as alveolar epithelial cells, even when no mutations in telomerase genes are detected (Alder
et al., 2008). AEC-II cells are the distal stem cells that response to injury and replenish the
alveolar epithelial pool. Recent studies using telomeric repeat-binding factor 2 (Trf2) mutant
mice model (de Lange, 2009) demonstrated that the telomere dysfunction restricted to AEC-
II cells caused their stem cell function failure by inducing senescence and contributed to the
pathogenesis of IPF (Alder et al., 2015; Chen et al., 2015).

20
1.2.2.1.4 Cell-based therapeutic approaches for treating IPF
To date, numerous putative distal stem cell populations have been described that are
able to self-renew and repair damaged epithelium in response to certain types of injury
(Barkauskas et al., 2013b; Chapman et al., 2011; Jain et al., 2015; Kim et al., 2005;
McQualter et al., 2010; Teisanu et al., 2011; Vaughan et al., 2014). However, these cells are
rare, which limits their expansion, and they usually change rapidly upon in vitro culture
(Bertoncello and McQualter, 2010; Kosmider et al., 2011; Liebler et al., 2016; Raiser and
Kim, 2009). Importantly, these endogenous stem cell populations are limited in number and
function in certain pathological conditions (Randell, 2006). Thus, recent focus has been
placed on using exogenous cell-based therapeutic approaches for ameliorating pulmonary
fibrosis. Tremendous efforts have been made in application of bone marrow (BMC) cells
(Ghadiri et al., 2016; Kotton et al., 2005; Weiss, 2014), mesenchymal stromal (MSC) cells
(Cargnoni et al., 2009; Ortiz et al., 2003; Tashiro et al., 2015; Toonkel et al., 2013) and
respiratory epithelial cells derived from pluripotent sources such as embryonic stem (ESC)
and induced pluripotent stem (iPS) cells (Ghaedi et al., 2013; Gotoh et al., 2014; Huang et
al., 2013a; Zhou et al., 2014).
Studies argued against the direct contribution of bone marrow derived circulating
cells to epithelial lineage. Kotton et. al. (Kotton et al., 2005) showed there was no detectable
reconstitution of injured epithelium by unfractionated bone marrow cells or purified
hematopoietic stem cells transplanted to the recipients subjected to bleomycin-induced lung
injury.
Amongst these cell sources, MSCs have advantages as a practical source for use in
cell-based therapies for lung disease. The vast majority of studies report some biological
effects after MSC delivery during the early inflammatory phase of bleomycin-induced
pulmonary fibrosis. However, low levels of cell engraftment or retention suggest a
paracrine-based mechanisms of action responsible for repair (Kumamoto et al., 2009; Lee et
al., 2012; Ortiz et al., 2003).
In contrary, freshly isolated AEC-II cells appear to be effective even after
administration in later stages of IPF where fibrosis is prevalent (Serrano-Mollar et al.,
2007),(Serrano-Mollar et al., 2016). Nevertheless, the practical usages of freshly isolated

21
AEC-IIs are limited by donor availability and maintenance in culture (Kosmider et al., 2011;
Liebler et al., 2016).
Numerous studies have shown the successful derivation of distal epithelial cells from
directed differentiation of ESC and iPS cells (Ghaedi et al., 2013; Gotoh et al., 2014; Huang
et al., 2013a; Zhou et al., 2014). However, current differentiation protocols remain limited
by the yield and purity of AEC-II cells and the potential risk of tumorigenicity of the
contaminated undifferentiated cells needs to be addressed for clinic-relevant applications
(Kimbrel and Lanza, 2015; Shi et al., 2016).
1.2.3 Acute respiratory distress syndrome (ARDS)
ARDS, firstly described in 1967 (Ashbaugh et al., 1967), is clinically characterized
by bilateral infiltrates and hypoxemia attributed to acute non-cardiogenic pulmonary edema
(Matthay et al., 2012; Spragg et al., 2010). ARDS, with a morality of 22%-40%, is a
heterogeneous syndrome as a result of diverse etiologies, including pneumonia, sepsis,
trauma, pancreatitis, aspiration, transfusion related acute lung injury, and ventilation induced
lung injury (Matthay et al., 2012).
One of the pathophysiologic hallmarks of ARDS is the alveolar epithelium injury.
The extent of epithelium injury correlates with severity of the disease (Nakamura et al.,
2011) and its repair is a predictive factor of clinical recovery (Ware and Matthay, 2001)
During ARDS, the injury to alveolar epithelium results in the impairment of barrier
function with an enhanced permeability that results in the airspaces being flooded with
protein fluid (Wiener-Kronish et al., 1991). In addition, other impairments of epithelial
functions are observed, including surfactant deficiency (Gregory et al., 1991; Ingenito et al.,
2001) causing alveolar atelectasis and impaired ion and fluid transport resulting in failure in
edema fluid clearance (Ware and Matthay, 2001). While the direct injury to the alveolar
epithelium occurs in some of the cases of ARDS by certain insults, the inflammatory
induced epithelium injury is universally implicated in ARDS. Although no specific
pharmacologic treatments to restore injured epithelium has yet been developed, the
supportive management therapy including low tidal volume ventilation and fluid restriction
has been reported to improve the survival of ARDS patients (National Heart, Lung, and

22
Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network et al.,
2006).
1.2.3.1 Damage in epithelial tight junctions
The expression of tight junction proteins in epithelium correlates with barrier
function and is critical to barrier integrity. Studies using animal models of ARDS showed
decreased expression levels of claudins and occludins in alveolar epithelial cells after injury
(Ohta et al., 2012). Specifically, the downregulation of claudin 4 and 5 results in an impaired
interaction with claudin 18 and ZO-1 that decreases epithelial barrier function and
contributes to the development of ARDS (Moss et al., 1996). In addition, the altered
expression of PTEN responding to severe injury has been proposed as another underlying
mechanism disassembly of epithelial tight junctions. Targeted deletion of PTEN in AEC-II
cells causes marked downregulation of claudin 4 and E-cadherin and results in aberrant
alveolar flooding (Miyoshi et al., 2013).
1.2.3.2 Apoptosis of alveolar epithelial cells
The loss of functional alveolar epithelial pool is another dominant feature of ARDS.
In addition to the alteration in epithelial tight junction protein expression, the injury-induced
alveolar epithelial cell death contributes to the barrier dysfunction. Studies of epithelial
injury during ARDS showed AEC-I cells are particularly susceptible to injury while AEC-
IIs damages are also observed (Bachofen and Weibel, 1982).
Apoptosis of alveolar epithelial cells has been observed in the lungs of ARDS
patients and animal models of ARDS induced by LPS, bacterial, virus and so on (Imai et al.,
2003). It's known that apoptosis occurs by the activation of extrinsic (e.g. death receptors) or
intrinsic (e.g. mitochondrial damage) pathways. Fas, one of the classic extrinsic death
receptors, binds cell surface by its ligand FasL to induce apoptosis. High levels of FasL have
been detected in the alveolar epithelial cells from ARDS patients and animal models of
ARDS. The beneficial effect of prevention of apoptosis has been demonstrated that
inhibition of apoptosis pathways attenuates lung injury in animal models of ARDS
(Budinger et al., 2011).

23
1.2.3.3 Direct injury to alveolar epithelium
Direct injury to alveolar epithelium is found in some causes of ARDS depending on
the types of the insults. Mechanical stress, such as mechanical ventilation, can cause direct
injury to epithelium. Mechanical stress induces necrosis of epithelial cells and activation of
some specific inflammatory signaling pathways in epithelial cells which in turn causes the
secondary inflammatory injury (Slutsky and Ranieri, 2013). Mechanical ventilation can
cause lung overdistention and disrupt epithelial tight junctions and adherent junctions (Wang
et al., 2011). The correlation of ventilation and induced epithelial injury has been examined
in animal models showing high tidal volume ventilation provokes epithelial injury while low
tidal volume ventilation is able to reduce the damage in epithelium (Slutsky and Ranieri,
2013). Likewise, decreased epithelium injury is observed in ARDS patients treated with low
tidal volume ventilation.
Viral infection can cause pneumonia which is the most common etiology of ARDS.
Particularly, influenza virus infection results in both direct and inflammatory injury to
epithelium. The type of epithelial cells which influenza viruses target is related to the
specificity of the hemagglutinin (HA) expressed on the surface of the virus and the specific
sialic acid residing on the epithelial cell surface (Herold et al., 2015). Avian influenza
viruses preferentially infect AEC-II cells and cause pneumonia. Once HA binds to sialic
acid, virus enter AEC-II cells by endocytosis, replicate, and release viral particles causing
AEC-II cell death (Hashimoto et al., 2007).
Bacterial infection can cause direct injury to epithelial cells by their exotoxins, in
addition to inducing inflammatory injury. For instance, pseudomonas produces exoenzymes
resulting in lysis of epithelial cells. These exoenzymes disrupt integrin survival signalling
and activate intrinsic apoptosis pathways (Wood et al., 2015) causing epithelial cell death.
Gram-positive infection also induces alveolar epithelial cells lysis by their toxins (e.g.
pneumolysin) forming transmembrane pores in host epithelial cells (Rubins and Janoff,
1998). Besides exoenzymes, hydrogen peroxide released from bacterial causes DNA damage
in alveolar epithelial cells and results in apoptosis (Rai et al., 2015).

24
1.2.3.4 Inflammatory-induced epithelial injury
The inflammatory-induced damage in epithelium is broadly implicated in the
pathogenesis of ARDS. Among all types of inflammatory cells, the most strongly implicated
in ARDS are neutrophils, of which the number correlates with the mortality of ARDS
patients. In response to injury, neutrophils are recruited to airway spaces and activated to
participate in host defense via phagocytosis and secretion of mediators killing pathogens
(Zemans et al., 2009). A growing body of evidence from animal studies showed that the
dysregulation of neutrophils, as the case seen in ARDS, results in injury to the alveolar
epithelium. Furthermore, in various animal models of ARDS (e.g. LPS, virus, bacteria,
surfactant depletion, VILI-induced ARDS), inhibition of neutrophil migration or depletion
of neutrophils can prevent the progression of epithelial injury showing a reduction of fluid
and protein content in the bronchioalveolar lavage (BAL) fluid (José et al., 2015; Koma et
al., 2014).
In addition to neutrophils, monocyte-derived cells can affect the injury to epithelium.
Studies showed a sub-population of monocyte-derived dendritic cells, which express antigen
to cytotoxic CD8+ T cells, contributes to epithelium injury during viral infection (Aldridge
et al., 2009). The effects of monocyte-derived macrophages on epithelium injury are
conflicting. In animal model of ARDS induced by LPS, recruited monocyte-derived
macrophages act as a protective role which can significantly reduce epithelial cell apoptosis
by producing IL-1 receptor antagonist production (Herold et al., 2011). However, monocyte-
derived macrophages can also induce apoptosis of epithelial cells by interacting with death
receptors on the surface of epithelial cells. During influenza infection, blockage of the
recruited macrophages protects epithelium against injury induced by virus (Lin et al., 2011).
Thus, future studies need to dissect the distinct subpopulations of macrophages based on
their response to specific injuries.
1.2.3.5 Epithelium repair in ARDS
In alveolar epithelium, AEC-I cells are more susceptible to injury while AEC-II cells
are also damaged in severe injury, particularly when pathogens preferentially infect AEC-II
cells (e.g. Avian influenza viruses). In mild lung injury, when AEC-IIs largely survive,
surviving AEC-II cells function as distal stem cells that are able to proliferate and

25
transdifferentiate to AEC-I cells to effectively replenish the alveolar epithelium (Barkauskas
et al., 2013).
The ability of AEC-IIs-derived AEC-I cells to replace lost AEC-I cells requires
sufficient cell spreading. Cell spreading is limited in certain circumstances (e.g. ventilation
induced acute lung injury) (Crosby et al., 2011) due to the altered expression of factors
regulating cell spreading, such as β-catenin (Zemans et al., 2013), KGF (Waters and Savla,
1999), and PTEN inhibition (Mihai et al., 2012).
In more severe cases of injury, with the loss of huge numbers of AEC-IIs as seen in
influenza infection (Zuo et al., 2014) or genetic disorder (Lawson et al., 2011), the
regeneration of injured alveolar epithelium relies on the mobilization of an alternative stem
cell population and their ability to differentiate to AEC-I and AEC-II cells (Barkauskas et
al., 2013). Particularly, recent animal studies of influenza virus infection specifically targets
AEC-II cells showed that a Krt5+ basal-like population contributes to the repair of injured
alveolar epithelium which may migrate from upper airways or derived from a rare lineage
negative epithelial stem/progenitor (LNEP) cells (Chapman et al., 2011; Vaughan et al.,
2014). However, the existence of these putative stem cells in human lungs and their
reparative capacity in human ARDS still remain to be determined.
A subset of ARDS patients with high mortality developed excessive fibrosis and
collagen deposition in their lung caused by the aberrant repair (Horowitz et al., 2006;
Marshall et al., 2000).
In summary, alveolar epithelial cells play a crucial role in the pathogenesis of ARDS.
During ARDS, the disruption of the epithelial barrier is attributed to damage to tight
junctions and the loss of functional epithelial cells. The direct and inflammatory injury to the
epithelium can result in impaired epithelium regeneration and patient mortality. Although
the supportive management therapy, including low tidal volume ventilation and fluid
restriction, has been reported to improve the outcome of ARDS patients, effective
therapeutic approaches to attenuate inflammatory injury and repair damaged alveolar
epithelium need to be the goal of future studies.

26
1.3 Stem cells in lung epithelial regeneration
Lung stem cells play a crucial role in maintaining lung homeostasis and regeneration.
The cellular turnover of the adult lung during homeostasis is remarkably slow. However,
facultative endogenous lung stem cells which are normally quiescent but become activated
in response to injury or insult, undergo self-renew and differentiate to desired progeny to
regenerate the damaged lungs (Beers and Morrisey, 2011). Disruption of this reparative
process leads to fibroproliferation, aberrant tissue remodeling and lung dysfunction
contributing to the pathogenesis of many pulmonary diseases (Beers and Morrisey, 2011;
Wansleeben et al., 2013a). Recent advances in the identification of specialised putative lung
stem cell populations by in vivo lineage tracing approach and development of organiod
cultures have increased our knowledge about the presence and regenerative potential of lung
stem cells.
1.3.1 Stem cells category
Stem cells are characterized by their ability to divide (self-renew) and differentiate
into progeny. All stem cells can be categorized into five groups according to their
differentiation potential: totipotent, pluripotenty, multipotent, oligopotent and unipotent.
Totipotent cells, like zygote and early embryonic cells, are able to give rise to any cell type
during development. Pluripotent cells have the ability to give rise to all of the cell types of
three germ layers (ectoderm, mesoderm and endoderm), such as embryonic stem cells
(ESCs) and induced pluripotent stem (iPS) cells. Multipotent cells, which have a more
limited differentiation potential, can development to more than one cell types. Oligopotent
cells, such as lineage stem cells, differentiate to two or more mature cell types. Unipotent
cells can only give rise to one cell type (Smith, 2006). Most of the defined endogenous lung
stem cells are multipotent or oligopotent cells, while AEC-II stem cells are unipotent that
only give rise to AEC-I cells (Barkauskas et al., 2013).
1.3.2 Adult stem cell hierarchy
In the classical stem cell hierarchy, the adult stem cells function to maintain tissue
homeostasis and can give rise to transit-amplifying cells which are relatively
undifferentiated and able to self-renew. In nonclassical stem cell hierarchy, the adult stem

27
cells only participate in tissue repair process in response to injury, but not in the
maintenance of tissue under normal conditions.
1.3.3 Endogenous lung stem cells
A number of studies in both mouse lineage tracing models and human lung tissues
describe multiple specialized stem cell populations in distinct anatomic locations of the adult
lung (Rock and Hogan, 2011; Wansleeben et al., 2013). These region-specific stem cell
populations reside in trachea, bronchi, bronchiolar, and alveoli to maintain lung homeostasis
or/ and participate in repair.
1.3.3.1 Basal stem cells
Basal cells, located in tracheobronchial region, are considered as one of the major
tissue-specific stem cells in the upper airways marked by the expression of transcription
factors Trp63, surface receptor NGFR, GSIβ4 lectin and cytokeratin5 (Krt5). They are able
to self-renew and give rise secretory and ciliated cells (Tata et al., 2013). In addition to
maintaining local epithelial homeostasis, basal stem cells can respond to sulfur dioxide
injury (Rock et al., 2009) or naphthalene injury (Hong, 2003), and migrate to repopulate
alveolar epithelium after H1N1 influenza infection (Kumar et al., 2011). The differentiation
of basal cells is regulated by Notch signaling which is altered in certain injury conditions.
Recent lineage tracing studies further dissect the distinct subpopulations of basal
cells and their functional differences in homeostatic and injury conditions. During
homeostasis, rare expression of Krt14 is found in either basal stem cells (BSCs) or basal
luminal precursor cells (BLPCs) both expressing Trp63 and Krt5. After injury, the
expression of Krt14 is remarkably up-regulated and the proportion of Krt5+Krt14+ cells
increases (Cole et al., 2010; Ghosh et al., 2011), suggesting Krt14 could serve as a maker for
activated basal stem cells. BLPCs are the progeny of BSCs with a lower rate of self-renewal
and differentiation, and can be distinguished from BSCs by its Krt8 expression (Watson et
al., 2015). A study using Notch transgeneic mice showed Notch3 signaling regulates the
expansion and differentiation of a basal population that the deletion of Notch3 results in an
increased number of both BSCs and Krt5+Krt8 basal cells, but not in other epithelial
lineages (Mori et al., 2015). Additionally, a new study showed Trp63+ basal cells specific

28
segregate into two distinct subpopulations-N2ICD+ (the active Notch2 intracellular domain)
and c-Myb+ cells after injury, but not in homeostatic conditions. These two basal
populations exhibit distinct differentiation potentials that N2ICD+ cells generate mature
secretory lineage cells whereas c-Myb+ cells only give rise to ciliated cells (Pardo-Saganta
et al., 2015).
1.3.3.2 Trp63+/Krt5+ basal-like stem cells
In addition to basal stem cells residing in the upper airways, other putative
Trp63+/Krt5+ basal-like stem cell populations have been found to present in the distal
airway under certain injury conditions.
Clusters of Trp63+/Krt5+ cells, termed distal alveolar stem cells (DASCs), present in
the distal airways after H1N1 influenza virus infection. In response to influenza-induced
lung injury, these DASCs are able to proliferate and differentiate to alveolar epithelial cells
to replace the damaged alveolar epithelium. Although these DASCs can express basal cell
marker, Trp63 and Krt5, they exhibit a different cell fate from the tracheal basal stem cells.
In both in vitro and in vivo, tracheal basal cells only commit to a proximal epithelial lineage
whereas the DASCs can generate alveolar epithelial cells and secretory cells (Zuo et al.,
2014). Furthermore, Krt5 lineage tracing studies demonstrate that these cells are not pre-
existing but only appear in response to injury. However, the reparative capacity of this stem
cell population for alveolar epithelium seems limited to the alveolar architecture of influenza
infected lungs, which it can only partially restore.
Another Trp63+/Krt5+ stem cell population, the lineage negative epithelial
stem/progenitor (LNEP) cells, have been proposed that are capable of regenerating
bleomycin-injured alveolar epithelium. The differentiation of this population is regulated by
Notch signaling. The activation of Notch signalling maintains their expression of Trp63 and
Krt5 and prevents the differentiation to AEC-II cells (Vaughan et al., 2014).
In conclusion, basal/basal-like cells function as tissue-specific stem cells that are able
to maintain homeostasis of the airway epithelium and/or response to injury. Future studies
need to further investigate the heterogeneity in the basal population, the underlying
molecular mechanisms and signaling pathways regulating the stem cell property of basal

29
cells, and to validate the existence and function of different subsets of basal cells in human
lungs.
1.3.3.3 Variant Club cells
After injury to Club cells in the trachea, basal cells serve as stem cells to repopulate
airways. However, if injury targets ciliated cells, Club cells become the stem cell population
responsible for repair (Hong et al., 2001).
Naphthalene is a Club cell-specific toxicant that can induce airway injury in mice.
The majority of Club cells are sensitive to naphthalene and ablated after naphthalene
exposure, while a subset of Club cells expressing secretoglobin family 1a member 1
(Scgb1a1) but not cytochrome p450, are resistant to naphthalene. Distinguished from mature
Club cells, these naphthalene-resistant Club cells are associated with neuroepithelial bodies
(NEBs) and within the bronchioalveolar duct junctions (BADJ), express lower levels of
CCSP protein, and are negative for the phase I xenobiotic enzyme Cyp2f2 (Rawlins et al.,
2009; Reynolds et al., 2000a, 2000b). These cells, termed as "variant club cells", are
designated as facultative stem cells of bronchiolar airways and are capable to self-renew to
repopulate the damaged airways by differentiating into Cyp2f2+ mature Club cells, goblet
cells and ciliated cells. The proliferation of these variant Club cells can be stimulated by
injury and is associated with NEB hyperplasia and hypertrophy (Reynolds et al., 2000b). In
addition to naphthalene models, transgenic (CCtk) mice expressing herpes simplex virus
thymidine kinase (HSVtk) under CCSP promoter have been used to control the depletion of
CCSP-expressing cells by gangcyclovir (GCV) administration. Upon GCV treatment, the
number of CCSP-expressing cells dramatically decreased whereas the number of variant
Club cell significantly increased, suggestive of a proliferative response to injury and during
repair (Hong et al., 2001; Reynolds et al., 2000a). Moreover, studies using various animal
injury models suggest that the extent and degree of injury influences the reparative response
of CCSP+ Club cells. Responding to mild injury, Club cells are activated and differentiate to
ciliated cells. In severe injury conditions, variant Club cells located around NEBs and within
the BADJ become activated to regenerate injured epithelium, which is regulated by Notch
and FGF-10 signaling (Giangreco et al., 2009).

30
1.3.3.4 Bronchioalveolar stem cells (BACs)
In the mouse lungs, another subset of Scgb1a1+ cells coexpressing the AEC-II cell
marker surfactant protein C (Sftpc) are found within the BADJ. These Scgb1a1+ / Sftpc +
cells are termed as bronchioalveolar stem cells (BASCs) which are able to expand in vivo
after bleomycin-induced lung injury. In vitro, these cells can be clonally expanded and
function as multipotent stem cells to differentiate into both bronchiolar and alveolar lineages
(Kim et al., 2005).
The differentiation potential of these cells has been extensively studied using various
lineage tracing approaches and injury models. In response to influenza and bleomycin-
induced injury, BACs are capable of differentiating AEC-II cells, although their expansion
and contribution to repair seem limited. In contrast, BACs don't contribute in alveolar
lineage reconstitution during development or hyperoxia-induced alveolar injury (Rawlins et
al., 2009b; Zheng et al., 2012). These conflicting results could attribute to different subsets
of Scgb1a1+ cells being labeled, or the different signaling pathways been activated under
certain injury conditions.
In future, advanced lineage tracing tools allowing cell-specific labelling are required
to clarify the correlation or distinction of BACs with variant club cells located within the
BADJ. Moreover, BACs are not the only cells co-expressing Scgb1a1and Sftpc. In the
alveolar regions, a subset of AEC-II, although it is rare, has been found co-expressing both
markers (Kotton and Morrisey, 2014). Thus, further evaluations are required to distinguish
BACs from those rare AEC-II cells.
1.3.3.5 Neuroendocrine cells
The airway epithelium of both human and mouse lungs contains pulmonary
neuroendocrine cells (PNEC), which are distributed as single cells and as clusters,
neuroepithelial bodies (NEB). These cells are marked by expression of calcitonin gene-
related peptide (CGRP). PENC/NEB can function as the pulmonary stem cell niche to
provide the microenvironment for stem cell populations to participate tissue repair.
Moreover, CGRP-expressing PNEC have proposed to be the stem cells in the lung. Lineage
tracing studies show these cells are able to proliferate in homeostasis and contribute to
airway epithelium regeneration by differentiating into Club and ciliated cells after

31
naphthalene injury (Song et al., 2012). However, the reparative capacity of PNEC seems
limited because they were not able to, by default, restore normal airway architecture after
depletion of Scgb1a1+ cells in the CCtk model (Hong et al., 2001).
Studies showed that PENC/NEB are invovled in the pathogenesis of small cell lung
carcinoma (SCLC) (Rawlins and Hogan, 2006) and pediatric lung diseases, such as
congenital lung disorders, bronchopulmonary dysplasia, cystic fibrosis, and pulmonary
hypertension (Cutz et al., 2007).
1.3.3.6 Alveolar type II (AEC-II) cells
Alveolar sacs where the gas exchange takes place are lined with elongated alveolar
type I (AEC-I) and cuboidal type II (AEC-II) cells. The stem cell role of AEC-II cells has
been known since the 1970s (Miller and Hook, 1990). Classically, a fraction of Sftpc-
expressing AEC-II cells is categorized as the progenitors in the distal airways. These AEC-II
cells are able to self-renew and generate AEC-I cells to replenish alveolar epithelium during
homeostasis and during repair after injury. A growing body of evidence from in vivo lineage
tracing and injury studies shows the reparative capacity of AEC-II cells in response to injury
are to proliferate and differentiate into AEC-I progeny. Even in some case of severe injury,
when large numbers of AEC-II cells are ablated, the surviving AEC-II cells can function as
stem cells to rapidly expand and spread to repair. In vitro, AEC-II cells show clonogenic
capacity to self-renew and give rise to alveolar-like spheres composed of cells expressing
AEC-I and AEC-II markers (Barkauskas et al., 2013). Recent studies using single-cell RNA
sequencing technique demonstrate that AEC-I and AEC-II cells arise from a bipotent
progenitor cell marked by the expression of homeodomain only protein x (Hopx) during
lung development; whereas, in postnatal stage, AEC-I cells are only derive from a subset of
mature AEC-II cells (Treutlein et al., 2014).
Numbers of growth factors and signaling pathways have been shown to regulate
AEC-II cell proliferation. For instance, TGF-, heparin-binding EGF, and NRG1, these
EGFR ligands can induce AEC-II cell proliferation in vitro, but do not affect the
differentiation of AEC-II to AEC-I cells. The activation of the kras signalling pathway
contributes to the proliferative response of AEC-II cells to injury in vivo (Desai et al.,
2014). Thus, the EGFR-kras pathway regulates AEC-II cell proliferation, but not the

32
differentiation. Although the molecular mechanisms underlying differentiation of AEC-II
cells into AEC-I cells are not completely understood, it has been suggested that β-catenin
signaling is involved by regulating cell spreading and flattening, which is a key component
of the regenerative capacity of AEC-II cells (Liu et al., 2015; Mutze et al., 2015; Rieger et
al., 2016).
Future comprehensive studies on different pathways and their alterations in response
to injury are required in order to better harness the therapeutic potential of AEC-IIs.
1.3.3.7 Alveolar type I (AEC-I) cells
Alveolar type I (AEC-I) cells are long assumed to be a terminally differentiated
epithelial cell type. A recent study using lineage tracing and injury model suggests that a
subset of Hopx-expressing AEC-I cells can function as alveolar stem cells to generate new
AEC-I and AEC-II cells after pneumonectomy (Jain et al., 2015). Similar to AEC-II stem
cells, isolated Hopx+ AEC-I cells are able to give rise to alveolar-like colonies comprising
of cells positive for makers of AEC-I and AEC-II cells in vitro. However, it is still unknown
whether these putative AEC-I stem-cell-like cells directly differentiate to AEC-II cells or
they firstly dedifferentiate to bipotent progenitor-like cells then give rise to new AEC-I and
AEC-II cells. Moreover, the stem cell property of these AEC-I progenitors needs to be
further evaluated in different disease model
In conclusion, numbers of epithelial populations have been designated as
endogenous lung stem cells. Specifically, their activation and regenerative capacity seem to
depend on the stem cell niches and the injury been induced. Whether these stem cell
populations are intrinsically different or arise from a common precursor but exhibit different
phenotypes due to the specific type of injury induced, needs to be further elucidated.
1.3.4 Derivation of pulmonary epithelium from pluripotency
stem cells
A major obstacle of lung regeneration is the lack of a suitable source of large
numbers of epithelial cells. Primary cultures of airway epithelium possess limited
proliferative capacity and gradually lose differentiation. Although numbers of putative
endogenous lung stem cell populations have been described, these cells are rare, which

33
limits their expansion in vitro (Bertoncello and McQualter, 2010; Kosmider et al., 2011;
Liebler et al., 2016; Raiser and Kim, 2009). Importantly, the number and function of
endogenous stem cells are limited in certain pathological conditions (Randell, 2006).
Pluripotent embryonic stem cells (ESCs) and induced pluripotent stem (iPS) cells, capable
of generating cells of all three embryonic germ layers, offer enormous potential for lung
regeneration. Thus, tremendous efforts have been placed on the derivation of pulmonary
epithelial cells by differentiation of pluripotent stem cells.
1.3.4.1 ESCs and iPS cells
ESCs are derived from the inner cell mass of preimplantation blastocysts of an
embryo. In vitro, their undifferentiated state can be maintained, with the ability to
differentiate into specialized cell types upon the application of specific signalling
transduction mimicking tissue development (Odorico et al., 2001). Classically, iPS cells are
generated from fibroblasts by induction of exogenous reprogramming factors Klf4, Sox2,
Oct4, and c-Myc. Unlike ESCs, iPS cells can be derived from most of somatic cell types and
all these iPS cell lines express pluripotency genes and are able to generate chimeric mice.
They have a great differentiation potential as ESCs to differentiation to desired cell types
(Takahashi and Yamanaka, 2006a; Takahashi et al., 2007a; Yu et al., 2007). In addition to
avoiding the ethical issue of destroying embryos to obtain ESCs, iPS cells can be derived
from individual patients, known as patient-specific iPS cells, to generate genetically
matched somatic cells. This phenomenal feature of iPS cells offers great hope for the
developing patient-specific cell therapy, drug screening, and in vitro human diseases models
(Leeman et al., 2014).
1.3.4.2 Differentiation approaches
During development, definitive endoderm cells give rise to tissue of thyroid, lung,
liver, and pancreas. Anterior foregut endoderm which derived from definitive endoderm
generates lung endoderm. The lung epithelium development then processes a primordial
progenitor stage at which Nkx2.1 expression initiates whereas Sox2 expression is
downregulated. The proximal airway (trachea, bronchus, and bronchioles) epithelial cells
arise from the Nkx2.1+ embryonic progenitors with the upregulated expression of Sox2

34
(Que et al., 2009). Distal airway epithelium is derived from Sox9+Id2+ multipotent
embryonic distal progenitors (Perl et al., 2005; Rawlins et al., 2009).
Figure 1. Schematic graph depicting the generation of lung epithelial cells in development
The initial studies showed successful generation of functional airway epithelium
from ESCs, but the contamination of undifferentiated cells in the ending products still
remain the risk of teratoma formation (Van Haute et al., 2009). How to obtain pure, fully
differentiated lung epithelial population from is one of the major obstacles in differentiation
of pluripotent stem cells. Therefore, large amount of efforts have been placed on how to
increase the efficiency at each steps in differentiation.
In most of the differentiation protocols, the initial specification of definitive
endoderm from ESCs is directed by activin treatment and subsequent dual inhibition of
TGF- and BMP signalling to enrich anterior foregut endoderm (Green et al., 2011).
Recently, significant progress has been made to generate different airway epithelial
populations by carefully timed signaling induction. Herein, we will review some of the
major findings. Mou et al. (Mou et al., 2012) described a step-wise differentiation protocol
that was able to generate multipotent embryonic lung progenitor and airway progenitors
from ESCs. They found that carefully-timed TGF-, BMP-4, FGF-2, and Wnt signalling
treatments after conversion of mouse ESCs to foregut endoderm, allowed the generation of
Nkx2.1-expressing lung endoderm. These mouse Nkx2.1+ lung endoderm were capable of

35
generating lung progenitors with the ability to form tracheospheres after transplantation to
NOD-SCID mice. They also demonstrate this mouse differentiation protocol can be applied
to human cystic fibrosis iPS cells to obtain disease-specific lung progenitor population. A
similar approach described by Longmire et al. (Longmire et al., 2012) showed that definitive
endodermal precursors can be generated via the inhibition of TGF- and BMP signalling
with subsequent stimulation of BMP-4 and FGF-2 signalling in ESC differentiation. These
precursors show their utility in the recellularization of a decellularized lung scaffold. Huang
and colleagues are able to improve the differentiation efficiency to obtain lung progenitor
cells up to 80% by the refinement of differentiation protocol. As they described, definitive
endoderm is derived from hESC and iPS cells by Activin/BMP-4/bFGF treatment;
subsequent derivation of anterior foregut endoderm was through the dual inhibition of TGF-
(SB431542) and BMP (dorsomorphin) signalling, activation of Wnt signalling
(CHIR99021), and sequential treatment of exogenous growth factors (FGF-10, KGF, BMP-4
and retinoic acid). The obtained lung progenitor population is capable of differentiating to
both airway epithelial lineage (basal, Club, goblet and ciliated cells) and alveolar epithelial
lineage (AEC-I and AEC-II cells) in vivo and in vitro (Huang et al., 2013).
Furthermore, the potential of developing iPS patient-specific epithelial cells from iPS
for drug screening, and in vitro human lung diseases models cells has been demonstrated.
Ciliated cells are the major epithelial cells expressing cystic fibrosis transmembrane
conductance regulator (CFTR) in the lung. Especially for CF, the generation of patient-
specific ciliated epithelium in vitro is extremely important for testing CF drugs and studying
the precise cellular molecular mechanism underlying the disease. Wong et al. described a
direct differentiation protocol for generating functional CFTR-expressing airway epithelium
from human ESCs and CF-derived iPS cells. To achieve this, they used precisely-timed
treatment of exogenous growth factors (FGF-7, FGF-10, FGF-18 and BMP-4) with
subsequent air-liquid interface culture which can induce the maturation of ciliated cells.
In summary, both ESCs and iPS cells with superior self-renewal and differentiation
capacity offer enormous potential for lung regeneration. However, some protocols remain
limited by low yield and purity of the desired mature cell types; heterogeneous final
products (Plath and Lowry, 2011; Schwartz et al., 2014) and contamination by potentially
tumorigenic undifferentiated cells (Ben-David and Benvenisty, 2011; Tapia and Schöler,

36
2016). A variety of approaches may overcome these problems but producing each unique
cell that may be desirable will require the development of hundreds of unique protocols.
Therefore, efforts need to be placed on the standardization of differentiation protocols in
future studies.
1.4 Induced pluripotent stem (iPS) cell reprogramming
In 2006, the group of Yamanaka introduced their milestone strategy to generate cells
with pluripotency properties from somatic cells, so called induced pluripotent stem (iPS)
cells. This is achieved by ectopic expression of four reprogramming factors: Oct4 (also
known as POU5F1), Sox2, KLF4, and c-Myc (Takahashi and Yamanaka, 2006a).
In the last decade, using novel experimental strategies, tremendous progress has been
made on understanding the mechanisms underlying somatic cell fate changes during the
multi-step reprogramming and the factor-induced ESC-like transcriptional network
established to confer pluripotency. Many studies have demonstrated the similarities of both
mouse and human iPS cells and their respective ESCs counterparts, including morphology,
self-renew capacity, differentiation property, and the molecular levels of transcription and
genome-wide chromatin modifications. Notably, differences are found between ESCs and
iPS cells, such as the epigenetic memory of the cell of origin presenting both in human and
mouse iPS cells which greatly influences the molecular epigenetic profile and functional
differentiation potential of the iPS cells (Kim et al., 2010; Polo et al., 2010; Shipony et al.,
2014). Therefore, the generation of iPS cells from various somatic cell types constuties a
route to derive patient-specific iPS cells and opens new paths to model diseases and develop
cell therapy for tissue regeneration.
1.4.1 Reprogramming methods
There are several variables need to be considered to obtain faithful reprogrammed
iPS cell lines, including the choice of reprogramming factors; the delivery methods; target
cell type; the induction conditions and identification of the pluripotency of generated iPS
cells.

37
1.4.1.1 Reprogramming factors
iPS reprogramming was initially performed in mouse fibroblasts through retroviral
transduction of four transcription factors: Oct4 (also known as POU5F1), Sox2, KLF4, and
c-Myc (Takahashi and Yamanaka, 2006a). This core set of factors has been shown to
successfully generate iPS cell lines across many types of both human and mouse somatic
cells (Aoi et al., 2008; Eminli et al., 2008; Hanna et al., 2008; Kim et al., 2008; Stadtfeld et
al., 2008a, 2008b).
Various combinations of transcription factor cocktails have been described that are
capable of successfully reprogramming somatic cells to iPS cells. In the reprogramming of
mouse fibroblasts, Sox2 can be replaced by Sox1 and Sox3, but results in a lower
reprogramming efficiency; Klf4 can be replaced by Klf2; and c-Myc can be replaced by L-
Myc and N-Myc (Blelloch et al., 2007; Nakagawa et al., 2008). In addition, a partially
different combination of factors, Oct4, Sox2, Nanog and Lin28, is able to reprogramming
human fibroblasts (Kastenberg and Odorico, 2008). It has also been shown that the certain
reprogramming factors can be excluded from the reprogramming cocktail when the target
cell type can express them endogenously, although with a lower reprogramming efficiency
compared to that with Yamanaka factors. For instance, neural precursors expressing
endogenous Sox2 and c-Myc can be reprogrammed using only Oct4/Klf4 or Oct/c-Myc.
Exogenous c-Myc can be omitted for the reprogramming of both mouse and human
fibroblasts expressing endogenous c-Myc (Nakagawa et al., 2008; Wernig et al., 2008).
Numbers of small molecules and soluble factors have been reported that are able to
replace the need of the transcription factors and/or enhance the reprogramming efficiency,
including Valproic Acid (Huangfu et al., 2008), 5-azacytidine/shRNA against Dnmt
(Mikkelsen et al., 2008), BayK8644 (Shi et al., 2008), BIX01294 (Silva et al., 2008), Wnt3a
(Marson et al., 2008), and siRNA against p53 / Utf1 cDNA (Zhao et al., 2008). These non-
integrative, non-viral methods of reprogramming avoid permanent geneome modification in
contrast to the use of retrovirus or lentivirus. However, it still remains to be determined if
these small molecules and soluble factors can recapitulate the gradient transcriptional and
epigenetic changes occurring during the reprogramming by the four transcription factors.

38
1.4.1.2 Delivery systems
Retroviral and lentiviral approaches were employed in the initial iPS reprogramming
efforts (Blelloch et al., 2007; Brambrink et al., 2008; Hockemeyer et al., 2008; Takahashi
and Yamanaka, 2006b; Takahashi et al., 2007b), which have been criticized for their risk of
causing insertional mutagenesis that potentially limits clinical application.
Other delivery systems have been developed to limit the genomic integration,
especially for the derivation of human iPS cells. Low-risk reprogramming using non-viral
methods, including adenovirus (Stadtfeld et al., 2008a), plasmids (Okita et al., 2008),
excision of reprogramming factors using Cre/LoxP 19252477(Soldner et al., 2009) and
piggyBAC transposition (Woltjen et al., 2009) have been reported, but suffers from relative
low reprogramming efficiencies and may require intense screening of excised lines due to
the left-behind residual vector sequences. The non-integrating DNA-based system including
adenoviral, episomal plasmids and minicircle vector has been used in reprogramming
various somatic cell types. Studies showed adenovirus carrying the reprogramming factors is
able to reprogram mouse fibroblast, hepatocytes and human fibroblasts without genomic
integration, but also suffer from low reprogramming efficiency (Stadtfeld et al., 2008a).
Episomal vectors, which are derived from the Epstein-Barr virus, have been reported in the
successful derivation of iPS cells from human neonatal foreskin fibroblasts with a low
genomic integration (Yu et al., 2009). However, this method is technically challenging in
that three individual plasmids are required to carrying a total of seven factors, including
oncogene SV40. Minicircle vector, a plasmid (P2PhiC31-LGNSO) contains a single cassette
of four reprogramming factors (Oct4, Sox2, Lin28, Nanog), has been used in reprogramming
of human adipose stem cells and fibroblasts showing no footprint (Jia et al., 2010). Although
minicircles benefits from a higher transfection efficiency and longer ectopic expression, the
reprogramming efficiency is still fairly low.
In addition, non-integrating DNA-free system, including sendai virus, protein,
modified mRNA, microRNA and small molecules, allows no genomic integration or
footprint. Sendai virus and modified mRNA reprogramming both have a good efficiency,
but the usage is limited by the high cost. Protein-based iPS cells have been successfully
generated from both mouse and human
fetal and neonatal cells, but require multiple
applications due to the short half-life of protein and high handling-skills (Kim et al., 2009;

39
Zhou et al., 2009). In addition, it has been shown that small molecules are able to reprogram
mouse embryonic fibroblast to iPS cells. Nevertheless, this approach needs to be validated in
human cells (Liao et al., 2011).
1.4.1.3 Target cell type
Since the successful generation of iPS cells from fibroblasts of both mouse and
human, many other somatic cells types have been reprogrammed, including stomach cells
(Aoi et al., 2008), liver cells (Aoi et al., 2008; Stadtfeld et al., 2008a), pancreatic ß cells
(Stadtfeld et al., 2008b), lymphocytes (Hanna et al., 2008), neural progenitor cells (Eminli et
al., 2008; Kim et al., 2011), keratinocytes (Aasen et al., 2008; Maherali et al., 2008), and
others. Genetic labeling methods have been employed in many of these studies to confirm
the cell of origin of derived-iPS cells, ruling out the possibility of iPS cells derive from the
residual fibroblasts contaminated in the starting population.
There is the notion that the cell type influences reprogrammability, including the
efficiency and kinetics of the reprogramming process, and which reprogramming factors can
be delivered. For instance, mouse stomach cells, hepatocytes (Aoi et al., 2008) and human
keratinocytes (Aasen et al., 2008; Maherali et al., 2008) were reprogrammed to iPS cells
much faster and more efficiently than fibroblasts. In addition, variation of the effective
delivery of reprogramming factors is found among different cell types. For example, 100- to
200-fold higher titer of adenoviral vectors is required in reprogramming of mouse fibroblasts
than that in liver cells (Stadtfeld et al., 2008a).
Although it is still debated whether the degree of differentiation of cells within a
lineage influences the efficiency and kinetics of the reprogramming process, the former
hypothesis that only adult stem cells are amenable for reprogramming has been discarded
since terminally differentiated cells, such as pancreatic islets are capable of generating iPS
cells (Stadtfeld et al., 2008b). In addition, given time, all cells in a pre-B-cell population
showed the ability to undergo reprogramming with more than 90% of the wells contained
cells expressing pluripotency markers (Hanna et al., 2009). Although it seems every cell is
reprogrammable to iPS cells, the correlation of the heterogeneous reprogrammability and the
degree of differentiation of cells within a lineage needs to be precisely studied.

40
1.4.1.4 Derivation conditions
Both mouse and human iPS cells are derived under the same culture conditions used
to support the growth and maintenance of ESCs (Lerou et al., 2008). While ESC conditions
are sufficient to maintain iPS cells derived from various somatic cell types, the conditions
that enhance the derivation of ESCs may also enhance the derivation of iPS cells. For
example, knockout serum replacement used to enhance the ESC derivation can improve the
reprogramming of mouse fibroblasts (Blelloch et al., 2007). Knockout serum replacement
could be used in the reprogramming of some somatic cell types for which the standard
serum (e.g. fetal bovine serum) is not suitable.
In addition, the feeder cells or fibroblast-derived factors have been used to support
the growth of ESCs. As mouse ESCs can be maintained on gelatin, a feeder-free condition,
mouse iPS cells been reported can be derived in the absence of feeder cells (Wernig et al.,
2008). To date, majority of the human iPS cells were derived under feeder conditions. Thus,
for clinical relevant applications, culture conditions for human iPS derivation remain to be
defined to avoid the use of animal products.
Furthermore, when iPS cells are derived from some non-fibroblast somatic cells
types, the culture conditions need to be optimized to satisfy both somatic cells and the
derived iPS cells. This has been reported in the reprogramming of lymphocytes that both B
lineage growth factors and LIF were used to support the growth of both lymphocytes and
iPS cells, respectively (Hanna et al., 2008).
To note, there are some unique cases with human iPS reprogramming. When using
inducible systems in human cell reprogramming, one needs to consider that human iPS cells
are more sensitive to doxycycline exposure compared to their mouse counterparts. In
addition, human iPS cells and ESCs poorly grow as single cells so the additional chemical
compounds are required to enhance single-cell survival, such as Rho-associated kinase
(ROCK) inhibitor (Watanabe et al., 2007).
While most of the reprogramming studies were preformed under two-dimensional
(2D) cell-culture system, a recent study demonstrated the successful derivation of both
mouse and human iPS cells using a chemical defined 3D ECMs (Caiazzo et al., 2016). As
described, fibroblasts were reprogrammed in enzymatically crosslinked poly
(ethyleneglycol) (PEG)-based hydrogels inserted with the fibronectin-derived adhesion

41
peptide RGDSP which mimic the biochemical features of native ECMs. They demonstrated
that this defined 3D condition can boost induction of pluripotency by accelerating the two
key events for the initiation of iPS programming, MET and chromatin remodelling. As
known, the stem cell niche plays a crucial role in manipulating stem cell fate, including self-
renewal, differentiation, as well as the response to injury. In this first proof-of-principle 3D
reprogramming study, they raised a novel "reprogramming niche" concept that
microenvironment can modulate reprogramming process by manipulating somatic cell fate.
1.4.1.5 Identification of pluripotency
Pluripotent stem cells are able to give rise to all cells of an organism. The definition
of pluripotency is predicated on functional assays of differentiation potential and diagnostic
transcriptional, epigenetic and metabolic signatures.
A set of core transcription factors have been selected as the molecular hallmarks of
pluripotency: Oct4, Sox2 and Nanog, which play essential roles in establishing pluripotency
network in both mice and humans (Masui et al., 2007; Takahashi et al., 2007b; Wang et al.,
2012).
A range of functional assays have been employed to examine the developmental
potential of iPS cells: in vitro differentiation assay; in vivo teratoma formation and
chimaeras formation.
In vitro analysis is the basic layer of pluripotency characterization, including colony
morphology, self-renewal /expansion capacity, and differentiation capacity of generating
cells of all three embryonic germ layers. In vitro differentiation assay is typically preceded
by replacing conditions for maintaining undifferentiated state with different cocktails
containing lineage- specific inducers, and characterization of the expression of specific
makers of target tissues.
In vivo assays measuring differentiation potential have been regarded as more robust
parameters of pluripotency. Teratoma formation assay measuring developmental potency at
a population-based level is the gold standard for define plutipotency in human iPS cells.
Cells are injected into immune-compromised NOD-SCID mice and the generation of
differentiated tissues of all the three germ layers is assessed by histology analysis of possible
cell types. To note, cells injected with matrices or scaffold could provoke inflammatory

42
response which can be mistaken as tissue differentiation. In this case, lineage-tracing should
be applied to distinguish injected cells from reactive recipient tissues. Blastocyst chimaera
formation is used to examine the ability of mouse pluripotency cells to re-enter development
upon introduction into host embryos (Nagy et al., 1990). Cells with good kinetic
development potency are able to generate high-grade chimaeras with colonization of all
embryonic tissues including the germ line. Chimaeras assay in humans is ethically
impermissible.
1.4.2 Roadmap of faithful reprogramming
Since the innovation of iPS technology a decade ago, enormous progress has been
made in understanding the molecular mechanisms underlying somatic cell reprogramming to
pluripotency.
Although iPS cells can be derived from many somatic cell types by various
combinations of transcription factors and small molecules, our knowledge of the
mechanisms underlying the steps leading to faithful reprogramming is mainly learned from
the studies performed with the original Yamanaka factors on mouse embryonic fibroblasts
(MEFs).
During iPS reprogramming, only a small subset of cells within the starting somatic
cell population are able to proceed to the next steps of reprogramming and eventually
commit to pluripotency within a certain timeframe. Evidences from the studies on the events
that occur between the initial expression of the reprogramming factors in somatic cells and
the establishment of faithful reprogramming suggest that successful reprogramming of
fibroblasts is a stepwise process, through key intermediate steps (Brambrink et al., 2008;
Mikkelsen et al., 2008; Smith et al., 2010; Sridharan et al., 2009).
Studies using gene expression profiling and RNAi screening revealed the three
reprogramming phases: initiation, maturation, and stabilization (Li et al., 2010; Samavarchi-
Tehrani et al., 2010). A rapid induced cell proliferation and suppression of somatic cell-
specific transcription occurs initially, followed by acquisition of epithelial characteristics
and activation of some markers of ESCs, including alkaline phosphatase and stage-specific
embryonic antigen 1 (SSEA1). The final step is marked by the activation of pluripotency
genes, such as Nanog (Mikkelsen et al., 2008; Wernig et al., 2007). The ectopic expression

43
of reprogramming factors is required until the iPS state is established. The maintenance of
iPS cells is factor-independent, representing a stable conversion of somatic cell fate
(Stadtfeld et al., 2008c; Brambrink et al., 2008).
1.4.2.1 The early phase
A high-resolution time-lapse imaging approach tracking the reprogramming process
demonstrated that the first noticeable changes are the rapid induction of cell proliferation
and a decrease in cell size immediately upon induction of the reprogramming factors (Smith
et al., 2010). Concomitantly, there are transcriptional changes including downregulation of
somatic genes which occurs in the majority of cells and upregulation of cell proliferation
genes (e.g. Ccdn1, Ccnd2) (Stadtfeld et al., 2008c).
Although the vast majority of cells are able to initiate reprogramming, only a small
subset of the cells are able to successfully induce the initial morphological change while the
remaining cells undergo apoptosis, senescence and cell-cycle arrest which are thought to be
the one of the roadblocks of reprogramming and associated with reprogramming efficiency.
Indeed, evidence showed silencing of genes regulating these responses (e.g. p53 and
CDKN2A) (Hanna et al., 2009; Hong et al., 2009; Utikal et al., 2009) or promotion of the
cell cycle allowing more cells to transit through S phase (Hanna et al., 2009; Ruiz et al.,
2011) could enhance the reprogramming efficiency.
Another proposed barrier to reprogramming is the extinction of the somatic program
that the efficient silencing of somatic fate is required to en route to the iPS state. Supportive
studies showed that the expression of lineage-specific genes blocks reprogramming
(Mikkelsen et al., 2008; Pereira et al., 2010).
Both ESCs and iPS cells have epithelial cell characteristics, with the expression of
the key epithelial markers, including E-cadherin and EpCAM. Therefore, the loss of somatic
mesenchymal signature (e.g. Snail1/2; Zeb1/2) and acquisition of epithelial characteristics
(e.g. E-cadherin, EpCAM) are required during reprogramming of fibroblasts to iPS cells,
and the signalling pathways regulating MET or EMT would affect the reprogramming
efficiency (Li et al., 2010). For example, Samavarchi-Tehrani et al. demonstrated that BMP
signaling drives the mesenchymal-to-epithelial (MET) in the initiation phase of
reprogramming of fibroblasts and the activation of bone morphogenetic protein (BMP)

44
signalling enhances reprogramming through the upregulation of pro-MET microRNAs
(Samavarchi-Tehrani et al., 2010). This may explain the higher reprogramming efficiency
observed in epithelial cells, such as keratinocytes and hepatocytes that their already existing
epithelial character allows them to avoid the MET transition.
1.4.2.2 The intermediate phase
After MET, a subset of E-cadherin+ cells proceed to the next reprogramming phase
that larger colonies are formed, many embryonic genes regulating housekeeping functions
are upregulated (Samavarchi-Tehrani et al., 2010; Sridharan et al., 2009), and ESC makers
(e.g. SSEA-1) are induced (Li et al., 2010; Stadtfeld et al., 2008b). Studies tracing the
SSEA-1-positive and SSEA-1-negative populations during reprogramming process
demonstrate that only SSEA-1-positive cells give rise to faithfully iPS cells and only a few
of them can make the final transition.
Lujan et al. (Lujan et al., 2015) preformed a high-dimensional mass cytometry to
identify the surface markers of cells at different phases of reprogramming and demonstrated
that cells reprogrammed to faithful iPS cells acquire surface marker expression in a stepwise
manner. In this study, they performed a systematic functional surface maker screening on the
cell lines representing the early, intermediate and late pluripotency stages and identified 21
candidate surface markers, of which the expression was then characterized in primary cells
by mass cytometry over the reprogramming process. Consistent with previous reports, they
also demonstrate that the transient, intermediate stage is a general property of iPS
reprogramming that presents across multiple reprogramming systems. They showed that the
intermediate cells can be identified by a unique set of surface markers, including CD73,
CD49d and CD200, which are absent in both parental fibroblasts and the generated iPS cells.
Partially reprogrammed (pre-iPS) cells, which have already efficiently silenced
somatic genes but have not induced the endogenous pluripotency program, arise from this
intermediate stage. These pre-iPS cells have ESC-like colonies morphology, but not yet
gained Nanog expression or induced pluripotency-related genes. However, pre-iPS cells are
capable of transition to iPS cells with additional treatments, such as inhibition of MAPK and
GSK3; TGF inhibition and Nanog overexpression (Silva et al., 2009; Theunissen et al.,
2011).

45
Studies mapping the binding sites of 4 reprogramming factors in pre-iPS cells
revealed that c-Myc is largely engaged at this intermediate phase and many of its target
genes have already been bound, while the pluripotency network hasn't yet been established
(Sridharan et al., 2009).
1.4.2.3 The stabilization phase
The stabilization phase is characterized by the activation of the core plutipotency
network which marks the acquisition of a stable pluripotent stage. At this final step of
reprogramming, transcriptional or developmental regulators which are highly expressed in
ESCs have been activated, including endogenous Oct4, Sox2 and Nanog, and many other
pluripotency-related genes (Sancho-Martinez and Izpisua Belmonte, 2013; Zhang et al.,
2012).
Aside from iPS cells, alternative pluripotent stem cells obtained from reprogramming
have been documented, specifically the F-class cells described by Tonge et al. (Tonge et al.,
2014), and Nanoglow
Sox2low
Lin28high
population identified by Zunder et al. (Zunder et al.,
2015).
The F-class (fuzzy colony forming) cells, which do not morphologically resemble
ESCs, are derived from the cells which skip the MET during the early reprogramming.
These Nanog-expressing F-class cells are pluripotent, can be stably maintained in cell
culture, but their existence is dependent on the high expression of reprogramming factors. In
addition, although both F-class cells and intermediate pre-iPS cells require the continuous
expression of reprogramming factors, F-class cells are transcriptionally and epigenetically
distinct from pre-iPS cells (Tonge et al., 2014).
To construct the molecular roadmap of iPS reprogramming, Zunder et al. (Zunder et
al., 2015) developed a novel combination of high-dimensional mass cytometry and time-
resolved progression analysis that allowed for the comprehensive measurement of the
protein expression at the single cell level. They analyzed three different MEF
reprogramming systems (Oct4-GFP primary MEFs, Nanog-GFP secondary MEFs, and
Nanog-Neo secondary MEFs) and identified a group of proteins of which the expression can
represent reprogramming landmarks. They found that cells express high levels of both Oct4
and Klf4 (Oct4high
Klf4high
cells) at the early phase of reprogramming of which a subset

46
transitioned to a CD73high
CD104high
CD54low
population at the intermediate phase. Further
analysis of Ki67 expression in this intermediate population revealed two distinct
subpopulations: the Ki67low
cells which revert to the parental MEF-like cells, and the
Ki67high
cells which undergo MET and then to pluripotency. During the late stage of
reprogramming, Ki67high
cells segregated into two subpopulations: an ESC-like
Nanoghigh
Sox2high
CD54high
population and a mesendoderm-like Nanoglow
Sox2low
Lin28high
CD24high
PDGFR-αhigh
population representative of an alternative stem cell state.
1.4.3 Epigenetic alteration during reprogramming
Epigenetic alteration is the major underlying mechanism driving somatic cells from
the differentiated to the pluripotent state during the reprogramming process. The epigenetic
feature of the starting somatic cells has been erased to some extent during the conversion to
adopt a pluripotent stem cell epigenome. The epigenetic alterations during reprogramming
have been extensively studied, including genome-wide resetting of histone posttranslational
modifications, DNA demethylation of promoter regions of pluripotency genes, chromatin
reorganization, and reactivation of the somatically silenced X chromosome (Fussner et al.,
2011; Takahashi et al., 2007b).
According to several independent reports describing the molecular mechanisms of
iPS reprogramming at bulk population or single-cell levels, epigenetic remodeling is
observed during the distinct transcriptional phases.
Changes in histone modifications occur immediately after induction of
reprogramming factors. Accumulation of H3K4me2 is observed at the promoters of many
pluripotency genes (e.g. Sall4 and Fgf4) and enhancer regions (Koche et al., 2011). Changes
in H3K4me2 occur in the majority of starting fibroblasts in response to the induction of
reprogramming factors, even before cell division is initiated (Koche et al., 2011). The loss of
H3K4me2 at the promoters of somatic cells is associated with the suppression of somatic
MEF markers (Thy1 and Postn) (Sridharan et al., 2009; Stadtfeld et al., 2008c). In parallel,
somatic gene enhancer also loses H3K4me2 which leads to hypermethylation and silencing
at the late phase of reprogramming. The gradual depletion of H3K27me3 and promoter
hypomethylation in the regions responsible for cell fate conversion also occurs during the
early phase of reprogramming. However, at this early phase, chromatin remodeling has not

47
yet completed, including the correlation of H3Kme2 with H3k26me3, occupancy of RNA
PolII, and transcriptional activity (Koche et al., 2011). More histone makers are robustly
modified in later stages, including activation of H3K4me3 and repression of H3K27me3.
DNA demethylations and X reactivation occur during late stages of reprogramming
process (Polo et al., 2012). The expression of microRNA, such as let-7, miR34c, miR-294
and miR-106a changes during the whole reprogramming process. DNA hypermethylation is
observed in the regions of many pluripotency-related genes, including Oct4, Nanog,
undifferentiated embryonic cell transcription factor 1 (Utf1), developmental pluripotency
associated 3 (Dppa3) and 5 (Dppa5), and zinc finger protein 42 (Zfp42) in both somatic cells
and pre-iPSCs, concomitant with the repression of H3K4me3 (Maherali et al., 2007). Loss
of repressive histone methylation markers, such as H3K27me3 and H3K9me3, at many
pluripotency-related genes and binding of the reprogramming factors Oct4, Sox2 and Klf4
occur at the end of reprogramming process (Mikkelsen et al., 2008).
1.4.4 Cooperation of reprogramming transcription factors
Understanding of the contribution of individual reprogramming factors to the
different phases of reprogramming is required to yield mechanistic insights into
reprogramming and the induced pluripotent state. A growing body of evidence reveals that
each individual reprogramming factor plays a distinct role during stepwise reprogramming
process (Li et al., 2010; Nakagawa et al., 2008; Sridharan et al., 2009). The initial
transcriptional wave is mainly mediated by c-Myc whereas the later wave is mediated by
Oct4 and Sox2 which facilitate the expression of pluriptency-related genes and establish
pluripotnecy network. Klf4 is involved in both stages including repression of somatic genes
at the early stage and activation of pluripotency genes at the second stage (Polo et al., 2012).
Studies have mapped the binding sites of 4 reprogramming factors during fibroblast
reprogramming and shown OSKM binds promoters of genes which are active or repressed
immediately after induction (Koche et al., 2011). During MET, that occurs in the initial
phase of reprogramming, both Oct4 and Sox2 suppress pro-mesenchymal genes (e.g. Snail);
Klf4 is involved in the activation of epithelial-related genes, including E-cadherin; c-Myc
repress the expression of Tgfb1 and Tgfbr1 to reduce TGFβ signalling (Li et al., 2010). This
collaboration of 4 reprogramming factors in suppression of pro-MET regulators and

48
promotion of pro-MET signals enable the MET process during fibroblast reprogramming.
During the later stage of reprogramming, c-Myc, unlike Oct4, Sox2 and Klf4, is not
involved in the upregulation of pluripotency-related genes (Sridharan et al., 2009). Oct4 and
Sox2 co-occupy promoters of pluripotency-related genes, and Klf4 shares nearly half of its
targets with Oct4 and Sox2, together to form the pluripotency network. By contrast, c-Myc
only targets the promoters involved in regulating cell proliferation, metabolism and
biosynthetic pathways. Recently, it has been demonstrated that c-Myc also acts as an
amplifier of gene expression. For instance, c-Myc binds to promoter regions associated with
H3K4me3 and H3K27ac and promotes the production of promoter-proximal pausing of
RNA polymerase II (Pol II) to enhance transcriptional elongation (Lin et al., 2012; Rahl et
al., 2010). Therefore, although c-Myc is not absolutely required for reprogramming, it is still
able to enhance transdifferentiation, the kinetics of reprogramming process, and build a
foundation for the other reprogramming factors to activate the pluripotency network
(Nakagawa et al., 2008; Rahl et al., 2010).
Moreover, studies of the binding site of reprogramming factors in pre-iPS cells
demonstrate c-Myc is largely engaged at the intermediate stage of reprogramming and that
many of the c-Myc target genes in iPS cells have already been bound in pre-iPS cells. In
contrast to that in iPS cells, many pluripotency-related genes targeted by Oct4, Sox2 and
Klf4 lack of binding and are not activated in pre-iPSCs (Buganim et al., 2012). This can
explain why additional transcription factors with the ability to cooperatively bind with Oct4,
Sox2 and Klf4 are required to enable the transition of pre-iPS cells to faithful pluripotenct
iPS cells. For example, Nanog can interact with Oct4 and Sox2 and other pluripotency genes
to promote their function and overexpression of Nanog in pre-iPS cells can enhance the
induction of pluripotency by lowering the intrinsic barriers (Hanna et al., 2009; Silva et al.,
2009; Theunissen et al., 2011).
Furthermore, taking the advantages of the collaboration of the 4 reprogramming
factors to open chromatin and induce cell plasticity during the early reprogramming process,
studies show that transient induction of these factors is sufficient to open the chromatin and
enable direct conversion of fibroblasts to other somatic cell types, such as neural progenitors
and cardiomyocytes (Efe et al., 2011; Kim et al., 2011).

49
1.4.5 Molecular and functional similarities and differences
between ESCs and iPS cells
ESCs and iPS cells share similarities in morphology, self-renew capacity,
differentiation potential, and age-affected cellular systems such as telomeres and
mitochondria. However, numbers studies comparing ESCs and iPS cells at the epigenetic,
transcriptional, proteomic and metabolic levels have demonstrated the molecular differences
between ESCs and iPS cells (Bock et al., 2011; Chin et al., 2009, 2010; Doi et al., 2009;
Ghosh et al., 2010; Kim et al., 2010b; Lister et al., 2011; Loewer et al., 2010; Marchetto et
al., 2009; Polo et al., 2010b).
At the transcriptional level, human iPS cells and ESCs can be distinguished by their
differential expression of protein-coding RNAs which mainly attributes to the residual
expression of somatic genes but dissipate upon extended passaging (Chin et al., 2009, 2010).
In addition, ten large intergenic non-coding RNAs (lincRNAs) are differentially expressed
between human iPS cells and ESCs. Some of these lincRNAs participate in the
reprogramming process that their overexpression enhances and the downregulation inhibits
reprogramming (Loewer et al., 2010).
These transcriptional differences can result in functional differences between iPS
cells and ESCs. For example, the repression of a small group of non-coding RNAs encoded
in the Dlk1–Dio3 gene cluster could affect the functionality of mouse iPS cells. Although
some iPS cells lacking of expression at this locus are capable of generating chimaeras, fail
the tetraploid complementation assay (the gold standard for examining mouse pluripotency)
to show animals are entirely derived from these cells (Stadtfeld et al., 2010).
The chromatin state of iPSCs and ESCs has been extensively examined to date and
consistent differences are observed. Among histone modifications, genome-wide studies
showed the expression patterns for H3K4me3 and H3K27me3 are indistinguishable between
both mouse and human iPS cells and their respective ESCs counterparts (Maherali et al.,
2007; Mikkelsen et al., 2008). However, the expression patterns of H3K9me3 within
promoter regions are different (Hawkins et al., 2010) and its overexpression was found
among the genes which are differentially expressed between human iPS cells and ESCs.
Notably, many of the transcriptional and chromatin differences described are
observed in the early-passage iPS cells and disappeared at a later passage, suggestive of a

50
residual ‘epigenetic memory’ persisting in the early-passage iPS cells, reflecting the cell of
origin (Chin et al., 2009; Ghosh et al., 2010; Marchetto et al., 2009).
Specifically, Kim et.al and Polo et al. provide functional evidence showing that an
epigenetic memory of cell of origin persist in mouse iPS cells which is linked to the residual
DNA methylation within lineage-specific genes. This persisting DNA methylation pattern
influences the functionality and differentiation potential of derived iPS cells (Kim et al.,
2010; Polo et al., 2010). For example, iPS cells derived from blood cells are more easily
differentiated to their original blood cell lineage than fibroblast-derived iPS. This may due to
the DNA hypermethylation of blood cell markers in fibroblast-derived iPS cells that
potentially prevent their upregulation under the induction of blood lineage differentiation.
Reversely, treatments of non-blood cell-derived iPS cells with DNA methylation inhibitors
enable a more efficient differentiation towards blood lineage. Notably, this residual
epigenetic memory in mouse iPS cells could be erased upon extended passaging.
Furthermore, this epigenetic memory has been uncovered in human iPS cells. Single-
cell based whole-genome DNA methylation mapping studies reveal that the somatic DNA
methylome was only partially erased during reprogramming and an epigenetic memory of
the somatic DNA methylation pattern persists in human iPS cells. In addition, some iPS cells
fail to establish ESC-like methylation pattern which is associated with transcriptional and
functional differences found between late-passage iPS cells and ESCs (Lister et al., 2011).
Nevertheless, genetic abnormalities have been seen in some iPS cells. One study
suggests that these abnormalities might due to the oncogenic stress induced by
reprogramming factors. They observed a higher level of phosphorylated histone H2AX, one
of the earliest indicators of DNA double-strand breaks, in the cells induced with OSKM or
OSK. They demonstrated that the homologous recombination pathway is essential for repair
DNA double-strand breaks to maintain genomic integrity during reprogramming process
(González et al., 2013). Nevertheless, more evidence are required to settle whether these
defects are as a result of reprogramming process or due to the genetic and epigenetic
differences existing within the individual parental fibroblasts (Abyzov et al., 2012; Cheng et
al., 2012).

51
1.5 Aging and iPS reprogramming
1.5.1 Molecular and cellular hallmarks of aging
Aging is generally characterized by a progressive loss of physiological fecundity,
increased impaired tissue regeneration and susceptibility to diseases (e.g., cancer, diabetes,
cardiovascular disorders and pulmonary diseases), leading to increased risk of mortality
(Hayflick, 2007; Kirkwood, 2005; Kirkwood and Shanley, 2010). Aging is caused by time-
dependent accumulation of cellular damages. A large body of evidence describes molecular
and cellular hallmarks of aging that mediate the aging process and phenotype. The nine
proposed pivotal hallmarks include stem cell exhaustion, telomere attrition, mitochondrial
dysfunction, epigenetic alterations, genomic instability, cellular senescence/apoptosis,
deficient proteostasis, dysregulated nutrient sensing and distorted intercellular
communication (Gems and Partridge, 2013; López-Otín et al., 2013; Vijg and Campisi,
2008).
1.5.1.1 Stem cell exhaustion
One of the most obvious characteristics of aging is tissue degeneration. Adult stem
cells, dividing throughout an organism’s lifespan for maintenance of the structure and
function of adult tissues, experience both chronological (e.g., neurons and myofibers) and
replicative exhaustion (e.g. the epithelia of the skin, gut and lung) during aging (Rando,
2006; Liu and Rando, 2011; Charville and Rando, 2011). The age-related progressive
decline in the number and cell-cycle activity of stem cells, leading to depletion of the
functional stem cell pool and tissue dysfunction, unfolds as a consequence of multiple types
of damages, such as telomere shortening, DNA mutations occurring during cell division, and
the imbalance of stem cell quiescence and proliferation (Flores, 2005; Rando, 2006;
Sharpless and DePinho, 2007; Liu and Rando, 2011).
1.5.1.2 Telomere attrition/dysfunction
Telomeres protect the chromosome ends for DNA repair and degradation activities
and telomerase is the enzyme responsible for maintaining telomere length and cell self-
renewal (Blackburn et al., 2006). Many types of in-vitro-cultured cells experience telomere

52
exhaustion and exhibit limited proliferative capacity, a process termed replicative
senescence (or Hayflick limit) (Hayflick and MOORHEAD, 1973).
Telomeres and telomerase are susceptible to age-related deterioration. Studies using
genetically modified animal models have demonstrated the causal links of telomere
dysfunction, cellular senescence and aging. Although telomere attrition manifests during
normal physiological aging, pathological telomere dysfunction provokes aging (Blackburn et
al., 2006; Flores and Blasco, 2010) and plays a causal role in premature development of
various human diseases, such as idiopathic pulmonary fibrosis (Tsakiri et al., 2007;
Cronkhite et al., 2008), aplastic anaemia (Yamaguchi et al., 2005; Du et al., 2008) and
dyskeratosis congenital (Armanios et al., 2005; Calado et al., 2009).
Importantly, telomere and telomerase are the main components of stem cell
"ignition" mechanism, referring to uncontrolled proliferation resulting in tumor formation
or, inversely, impaired self-renewal as happens in age-related tissue degeneration (Flores
and Blasco, 2010). Thus, tremendous efforts have been drawn on the manipulation of
telomere and telomerase to restrain cancer and delay aging. Among these, Armanios et al.,
and Toma´s-Loba et al., demonstrated that shortened telomere length caused tissue
degeneration whereas lengthened telomere increased lifespan in mice (Armanios et al., 2009;
Tomás-Loba et al., 2008). Moreover, normal physiological aging in mice can be delayed by
systemic viral transduction of telomerase (Bernardes de Jesus et al., 2012) and reactivation
of telomerase in aged telomerase-deficient mice showing reverted premature aging
phenotypes in testes, spleens, and intestines (Jaskelioff et al., 2011).
1.5.1.3 Mitochondrial dysfunction
As cellular power centers, the main function of mitochondria is to produce ATP via
the process of oxidative phosphorylation, which is carried out by the four respiratory chain
complexes and ATP synthase located in the inner mitochondrial membrane (IMM). Other
biochemical functions of mitochondria include the regulation of metabolic (both catabolic
and anabolic), signaling pathways and apoptosis (Nunnari and Suomalainen, 2012).
Mitochondria are unique cellular organelles that contain their own genetic information, the
mitochondrial DNA (mtDNA), a double-stranded and circular molecule of 16.5 kb in size
(Yue et al., 2015).

53
Emerging evidence has elucidated the precise relationship between mitochondrial
function, and signaling pathways regulating lifespan and aging. During aging, decline in the
number of mitochondrial, mtDNA copy number and protein levels of mitochondria were
observed both in human and in mice (Bratic and Larsson, 2013). Damage in mtDNA with
aging causes defects in the encoding proteins for the respiratory chain which in turn
amplifies ROS production and mitochondrial dysfunction (Van Houten et al., 2006).
Furthermore, this age-related progressive mitochondrial dysfunction results in further
increased levels of ROS production causing severe mitochondrial deterioration and
extensive cellular damage.
Accumulating evidence has shown mitochondrial dysfunction works in conjuction
with several other aging hallmarks (including loss of proteostasis, cellular senescence, and
stem cell exhaustion) to accelerate the aging process (Vermulst et al., 2008). Aggravated
mitochondrial dysfunction and DNA are associated with a variety age-related human
diseases, including cancer, diabetes and COPD (Ahmad et al., 2015; Boland et al., 2013;
Montgomery and Turner, 2014; Rera et al., 2012). Therefore, improvement of mitochondrial
function is a potential therapeutic strategy to delay the onset of age-related diseases.
1.5.1.4 Epigenetic alterations
All cells in a multicellular organism are genetically identical as they share the same
DNA sequence, but have variable gene expression patterns as the result of epigenetic
mechanisms. Epigenetic alterations affects all cells, both dividing and non-dividing cells,
and tissues throughout life (Talens et al., 2012). The essence of epigenetics is the
maintenance of not only the biological function and fate of all cells and tissues, but also their
response to environmental influences (Rando and Chang, 2012). Connecting the genotype
with the phenotype, epigenetics is a reversible heritable mechanism that changes either
spontaneously or driven by external or internal influences, but without altering the
underlying DNA sequence (O’Sullivan and Karlseder, 2012; Zane et al., 2014). Epigenome
composes multiple types of epigenetic information, including histones on DNA sequence,
DNA methylation, chromatin remodeling, variable structural and functional in histones,
posttranslational modifications of the histone proteins, and transcription of noncoding RNAs
(ncRNAs) (Brunet and Berger, 2014; Feser and Tyler, 2011).

54
Chromatin, which carries much of the epigenetic information, is the polymer of
nucleosomes that consists of 147 bp DNA wrapping around core histone proteins, H2A,
H2B, H3, and H4 (Luger et al., 1997). Epigenetic mechanisms are primarily regulated by a
series of enzymes that modify DNA directly or the core histones (methyltransferases,
demethylases, acetyltransferases, deacetylases) to regulate gene expression (Rando and
Chang, 2009). Epigenetic regulation proceeds by direct methylation and demethylation of
DNA bases referring to “Cis-epigenetics” (Bonasio et al., 2010). Histones can be altered by
modifications of methylation and acetylation which are closely associated with the
expression [e.g., histone 3 trimethylated at lysine 4 (H3K4me3)] and repression of genes
[e.g., histone 3 trimethylated at lysine 27 (H3K27me3)] (Wang et al., 2008). Particular
modifications on DNA and histones can result in alteration or ease of chromatin states.
Growing evidence from aging research show progressive loss in epigenome
configuration results in changes in the chromosomal architecture, genomic integrity, and
gene expression patterns during aging and in age-related disorders (O’Sullivan and
Karlseder, 2012; Brunet and Berger, 2014; Pal and Tyler, 2016).
DNA methylation plays a crucial role during development and functions to silence
genes no longer needed. Global alterations in DNA methylation patterns at specific sites
have been observed during aging. In young cells, the majority of CpGs are methylated
leading to transcriptional repression while the rest of CpGs referring to CpG islands are
demethylated resulting in upregulated gene expression (Jung and Pfeifer, 2015). During
aging, CpG hypomethylation occurs and increases the risk of genomic instability (Bormann
et al., 2016; Zampieri et al., 2015). In addition, the progressive decline in the levels of the
DNA methyltransferase (DNMT1) also contributes to the decreased DNA methylation
during aging (Jung and Pfeifer, 2015). A study using a DNMT1+/− mouse model showed
DNA methylation contributes to age-dependent impaired learning and memory function (Liu
et al., 2011b). The contribution of DNA methylation to aging is further demonstrated in a
recent study of aged pancreatic cells showing loss of DNA methylation and decreased
activity of DNMT1 resulting in aberrant gene expression (Avrahami et al., 2015). Among
the histone modifications known to associate with aging and affect longevity, the most
conspicuous ones are acetylation and methylation of lysine residues. Studies have
demonstrated the age-associated histone modifications including increased levels of histone

55
H4K16 acetylation (H4K16Ac), H4K20 trimethylation (H4K20me3), or H3K4
trimethylation (H3K4me3), and decreased levels of H3K9 methylation (H3K9me3) or
H3K27 trimethylation (H3K27me3) (Fraga and Esteller, 2007; Han and Brunet, 2012).
DNA methylation and histone modifications function interdependently and both
exert changes during aging. During development in both mice and human, the repression of
polycomb group protein (PcG)-mediated target genes in specific DNA hypermethylated loci
is regulated by H3K27me3 and reversed by H3K4me3. In contrast, during aging, DNA
hypermethylation is enriched at the regions carrying bivalent histone marks-both H3K4me3
and H3K27me3 (Weidner and Wagner, 2014; Weidner et al., 2014). Moreover, DNA
hypomethylation couples with the histone marks H3K9Ac, H3K27Ac, H3K4me1~3 found in
enhancer regions (Raddatz et al., 2013).
1.5.2 Aging in the lung
Like other organs, the lungs also age, manifesting degenerative changes during
aging. At the cellular level, age-related deterioration in the lungs includes the depletion of
stem cell pool, increased oxidative stress and accumulated DNA damages leading to
telomere shortening and mitochondrial dysfunction (Sahin et al., 2011). As a global
phenomenon in the lung, aging affects various cell types, including epithelial, endothelial,
mesenchymal and immune cells.
The lung harbors distinct stem cell populations which reside in the supporting niches
to maintain tissue homeostasis and respond to injury (Kim et al., 2005a; Rawlins et al.,
2009a; Rock et al., 2009a; Barkauskas et al., 2013a; Hogan et al., 2014; Vaughan et al.,
2014; Jain et al., 2015a). During aging, the number and function of lung stem cells decline
and gradually lose their regenerative capacity. Age-related replicative exhaustion, depletion
of stem cell pool, and alterations in stem cell niches result in lung dysfunction. Growing
evidence reveals that the dysfucntion of epithelial cell populations is a key component of the
aging process. For instance, a recent study showed age-associated changes in the cellular
composition, organization and local microenvironment of the mouse tracheal epithelium.
They found that the number and proportion of basal cells in the airways of aged mice
decreased and the appearance of age-associated gland-like structures (ARGLS) in the

56
submucosa may due to a response to chronic changes in the microenvironment (Wansleeben
et al., 2014).
Lung aging is closely associated with the development and pathogenesis of chronic
respiratory diseases, of which the prevalence and diagnosis increase with age. These include
chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF),
emphysema, submucosal gland hypertrophy and cancer (Chilosi et al., 2013; Faner et al.,
2012). Studies in tissues from emphysema patients showed that the depletion of stem cell
pool in aged lungs was due to the inability of turnover and increased cellular senescence,
resulting in the loss of normal alveolar structures and diminished tissue regeneration (Tsuji
et al., 2006; Yokohori et al., 2004). Telomere shortening, cellular senescence and stem cell
exhaustion, have been found to be closely associated with the development of IPF. For
instance, mutations in the enzyme telomerase and the consequential telomere shortening are
involved in the pathogenesis of both familial and sporadic IPF, which also link to the
development of COPD and emphysema (Alder et al., 2011; Armanios et al., 2007).
Furthermore, age also causes extracellular changes in the lung that influence the activation
of signaling pathways of proliferation, differentiation and senescence in the seeded cells
(Sokocevic et al., 2013). Overall, aging alters the lungs at both cellular and extracellular
levels and these age-associated changes cause tissue dysfunction and the development of
chronic pulmonary disorders.
1.5.3 Rejuvenation using iPS reprogramming strategy
Great efforts have been placed on rejuvenation of aged cells in regenerative medicine
research. Numerous studies demonstrate that the iPS reprogramming is not only able to alter
cell fate, from terminally differentiated to pluripotent state, but also enable aged cells to
adopt a more youthful state. The reprogramming process ameliorates many cellular
hallmarks of aging, such as mitochondrial dysfunction, telomere length shortening, stem cell
exhaustion, cellular senescence, and changes in histone marks.
During reprogramming of aged somatic cells, the expression of telomerase gene can
be reactivated that attributes to the rejuvenation consequence (Marión and Blasco, 2010). In
addition, the restoration of telomere length is observed during reprogramming aged
fibroblasts to iPS cells (Lapasset et al., 2011a; Marion et al., 2009). Notably, certain types of

57
genetic defects in telomerase components hindered the full restoration of telomerase activity
and its ability to lengthen telomeres during reprogramming (Batista et al., 2011).
Furthermore, studies in reprogramming of aged fibroblasts show that the aging
phenotypic mitochondrial dysfunction and the associated production of reactive oxygen
species (ROS) are restored to a rejuvenated state in iPS cells and in their derivatives at early
passages (Lapasset et al., 2011b; Prigione et al., 2011; Suhr et al., 2010)
Epigenetic dysregulation is a major driver of cellular damage observed during aging
and in age-related disorders (Benayoun et al., 2015; Pollina and Brunet, 2011). The
restoration of aging hallmarks to the youthful state during iPS reprogramming is mainly
driven by epigenetic remodeling. For instance, iPS reprogramming of fibroblasts from
Hutchinson–Gilford progeria syndrome (HGPS) patients results in restoration of age-
associated epigenetic marks to wild-type levels (e.g., H3K9me3) (Liu et al., 2011a).
Moreover, a recent study showed that iPS reprogramming erases age-associated epigenetic
memory in aged cells showing the efficient restoration of epigenetic markers (e.g., 5hmC,
H3K4me3, H3K9me3, and H3K27me3) and achieves enhanced rejuvenation in iPS-derived
neurons compared with the neurons derived via direct conversion process (Yang et al.,
2015).
Thus, based on the plastic and reversible nature of epigenetic mechanisms,
epigenetic reprogramming/remodeling which referrers to changes in the stable
transcriptional profile of a cell without alteration in DNA sequences could provide a
promising avenue for therapeutics against age-related decline and diseases.

58
Chapter 2
Rationale, Hypotheses and Objectives
Introduction
Regenerative medicine approaches for airway diseases such as cell replacement
therapy of lung allograft rejection and tissue engineering (such as the ultimate goal of
recellularizing a decellularized lung scaffold) all require a source of proliferative epithelial
cells with restricted differentiation. As a source for cell therapies, primary cultures of
airway epithelium possess limited proliferative capacity and gradual loss of differentiation is
seen, at least, in vitro. Endogenous progenitor cells are rare, which limits their expansion,
and their number and function decline in certain pathological conditions. Furthermore,
directed differentiation protocols of ES and iPS cells to generate large numbers of pure, fully
differentiated cells are still imperfect and derivation of each unique cell will require the
development of hundreds of unique protocols. Therefore, we aimed to develop an alternative
approach to obtain large number of progenitor-like cells which can theoretically be applied
to almost any cell type that can be isolated and purified.
This thesis describes the generation of induced Progenitor-Like (iPL) cells using a
novel interrupted reprogramming strategy. The approaches described in this thesis are 3-
fold: 1. Generation of bronchiolar progenitor-like iPL cells from quiescent mature Club cells
which are useful in cell replacement therapy for cystic fibrosis; 2. Generation of bipotent
progenitor-like iPL cells from adult AEC-IIs capable of ameliorating bleomycin-induced
pulmonary fibrosis; and 3. Rejuvenation of aged AEC-II cells to a youthful state using
interrupted reprogramming.
Rationale
The generation of iPS cells is a multistep process

59
Reprogramming is a multistep process comprising of initiation, maturation and
stabilization phases. One of the phenotypic changes in the early reprogramming process is
the rapid induction of proliferation with upregulatios of proliferation genes such as Ccnd1,
Ccnd2 and DNA replication genes. Moreover, ectopic expression of the four reprogramming
factors (Oct4, Sox2, Klf4 and c-Myc) is required until the iPS state is established, otherwise
cells return to a differentiated state.
The epigentic fingerprint of iPS cells
Induced pluripotent cells can be derived from not only fibroblasts, but also other
somatic cell types, such as blood, stomach and liver cells, keratinocytes, melanocytes,
pancreatic B cells and neural progenitors. All these iPS cell lines express pluripotency genes
and are able to generate chimeric mice. However, reprogrammed cells retain epigenetic
“memory” during the process for a significant number of doublings, with the cell of origin
influencing the molecular epigenetic profile and functional differentiation potential of the
iPS cells.
Herein, to create therapeutically valuable intermediate cellular products, we took
advantage of this early proliferation, and speculated that even greater residual epigenetic
“memory” exists early in the reprogramming process.
Hypothesis
Carefully timed transient expression of induced Pluripotent Stem (iPS) cell
reprogramming factors OSKM (termed "interrupted reprogramming") allows controlled
expansion yet preservation of lineage commitment.
Objectives
To isolate and purify distinct lung epithelial populations
To determine if define length of interrupted reprogramming leads to controlled
expansion of purified epithelial cells;

60
To determine if interrupted reprogramming allows preservation of lineage
commitment;
To characterize iPL cells derived from distinct epithelial populations;
To determine if iPL cells are able to engraft, survive and repopulate injured
epithelium in vivo.

61
Chapter 3
Generation of induced progenitor-like (iPL) cells from mature
epithelial cells using interrupted reprogramming
Contents of this chapter have been modified from Guo, L. et al. Generation of Induced
Progenitor-like Cells from Mature Epithelial Cells Using Interrupted Reprogramming. Stem
Cell Reports 9, 1780–1795 (2017).
The published paper can be found at the following link
http://www.cell.com/stem-cell-reports/fulltext/S2213-6711(17)30477-0
3.1 Rationale, Objectives, Hypotheses and Specific Aims
Rationale:
Induction of cells to iPS cells through delivery of 4 reprogramming transcription
factors (OSKM) activates proliferation far before full pluripotency is obtained. Residual
‘epigenetic memory’ persists in the early-passage iPS cells, reflecting the cell of origin, and
influences the molecular epigenetic profile and functional differentiation potential of the iPS
cells.
Objective:
To evaluate the potential of induced progenitor-like (iPL) cells to repopulate the
airway epithelium in lung injury models in mice.
Hypothesis:
Transient expression of iPS reprogramming transcription factors leads to expansion
of epithelial populations which return to their original phenotype after withdrawal.

62
Specific Aims:
To isolate and purify mature Club cells from mouse lung;
To determine if transient expression of iPS reprogramming factors leads to expansion
of purified mature Club cell population;
To determine if Club-iPL cells return to quiescent upon withdrawal of
reprogramming factors;
To determine if Club-iPL cells return to parental lineage epithelial cells upon factors-
withdrawal;
To characterize Club-iPL cells
To evaluate if Club-iPL cells are able to engraft, survive and repopulate injured
bronchiolar epithelium in vivo
3.2 Abstract
We present a novel "interrupted reprogramming" strategy to generate "induced
Progenitor-Like (iPL) cells" using carefully timed transient expression of induced
Pluripotent Stem (iPS) cell reprogramming factors (Oct4, Sox2, Klf4 and c-Myc; OSKM)
from highly purified mature Club cells. Interrupted reprogramming allowed controlled
expansion yet preservation of lineage commitment. Under clonogenic conditions, iPL cells
expanded and functioned as a bronchiolar progenitor-like population to generate mature
bronchiolar Club cells, mucin-producing goblet cells, and CFTR-expressing ciliated
epithelium in vitro. In vivo, iPL cells were able to repopulate CFTR-deficient epithelium.
This interrupted reprogramming process could be metronomically applied, to achieve
controlled progenitor-like proliferation. By carefully controlling the duration of transient
expression of OSKM, iPL cells do not become pluripotent and maintain their memory of
origin and retain their ability to efficiently return to their original phenotype. A generic
technique to produce highly specified populations may have significant implications for
regenerative medicine.

63
3.3 Introduction
A major block in the critical path of regenerative medicine is the lack of suitable cells
to restore function or repair damage. An ‘ideal’ cell for cell therapy will possess the
following attributes: (1) non-immunogenic, ideally patient-derived; (2) controllable
proliferation, allowing directed expansion; and (3) regulatable differentiation, with intrinsic
restriction to the appropriate lineage. Primary cultures of somatic cells are not ideal as they
possess limited proliferative capacity and gradually lose their differentiated properties.
Endogenous progenitor cells exist in various organs, including the lung (Kim et al., 2005;
Rawlins et al., 2009; Rock et al., 2009; Barkauskas et al., 2013; Vaughan et al., 2014; Jain et
al., 2015). However, these endogenous progenitor populations are often difficult to identify
and isolate, and usually rapidly change upon in vitro culture (Bertoncello and McQualter,
2010; Raiser and Kim, 2009). In several disease or injury states, they may be limited in
number and function (Rock, 2012; Randell, 2006). Therefore, great efforts have been placed
on exogenous cell sources. Despite significant progress to generate mature cell types using
embryonic stem cells (ESCs) and induced pluripotent stem (iPS) cells (Christodoulou et al.,
2011; Longmire et al., 2012; Mou et al., 2012; Wong et al., 2012; Ghaedi et al., 2013; Huang
et al., 2013; Firth et al., 2014), some protocols remain limited by low yield and purity of the
desired mature cell types. There is no standardized approach applicable to all cell types.
Development of personalized therapies based on autologous pluripotent cells remains very
expensive. Moreover, for many cell therapy applications, the cells will need externally
controllable proliferative capacity to maintain homeostasis or respond to injury.
The early steps of reprogramming have recently been the subject of intense
investigations (Clancy et al., 2014; Hussein et al., 2014; Lujan et al., 2015; Shakiba et al.,
2015). Two papers have documented additional alternative pluripotent stem cell states,
specifically the F-class cells described by Tonge et al. (Tonge et al., 2014), and separate
populations expressing identified as either Lin28high
or Nanoghigh
by Zunder et al. (Zunder et
al., 2015). In this work, we explore the earlier stages of iPS reprogramming for potentially
useful intermediate cell states. Reprogramming is a multi-step process comprising initiation,
maturation and stabilization phases (Samavarchi-Tehrani et al., 2010) during continuous
expression of exogenous reprogramming factors until the iPS cell state is established

64
(Sridharan et al., 2009). There is a rapid induction of cell proliferation during the early
phase of reprogramming (Woltjen et al., 2009). Reprogrammed cells retain epigenetic
“memory” in the process for significant number of doubling, with the cell of origin
influencing the molecular epigenetic profile and functional differentiation potential of the
iPS cells (Kim et al., 2010; Polo et al., 2010; Shipony et al., 2014). Herein, to create
therapeutically valuable intermediate cellular products, we took advantage of this early
proliferation, and speculated that even greater residual epigenetic “memory” exists early in
the reprogramming process.
For proof of principle, we chose the lung, in which there has been recent progress in
the identification of progenitor cells and their hierarchical relationships (Raiser and Kim,
2009; Rawlins et al., 2009; Rock et al., 2009; McQualter et al., 2010; Chapman et al., 2011;
Teisanu et al., 2011; Barkauskas et al., 2013; Lee et al., 2014; Treutlein et al., 2014;
Vaughan et al., 2014; Zuo et al., 2014; Jain et al., 2015) but there remains an unmet need to
produce highly purified epithelial populations. For specificity, we selected mature Club
cells, which possess limited proliferative capacity in vitro. Interrupted reprogramming
resulted in the generation of large numbers of epithelial progenitor-like cells that
demonstrated controlled proliferation and restricted differentiation. This was achieved by
optimized, controlled, transient induction of reprogramming factors, turning off their
expression prior to reaching independent pluripotency (Figure 1A). We term these cells
"induced Progenitor-Like (iPL)" cells.
Compared to the difficulty of developing directed differentiation protocols for each
cell type required, isolation of highly purified populations of adult cells from most organs is
already possible using flow cytometric sorting and cell culture techniques. These
populations, if bestowed controllable proliferative capacity and limited differentiation
potential, would be extremely useful in a variety of regenerative medicine practices,
including cell replacement therapy, biohybrid devices, disease modelling, and drug screening
for human diseases.

65
3.4 Material and Methods
3.4.1 Animal Husbandry
ROSA26-rtTA and Col1a1: tetO-4F2A mice (Jackson Labs, Cat#011004) were used
to generate inducible lung epithelial cells. For in vivo studies, ROSA26-rtTA/Col1a1:: tetO-
4F2A double transgenic mice were bred to actin GFP mice. There were no significant
differences between heterozygotes and homozygotes with respect to inductive factor gene
expression in dox-treated Club cells. Adult (6- to 8-week-old) female FABP/CFTR-
knockout mice (where the FABP [human fatty acid binding protein 1 liver] promoter drives
expression of hCFTR, to facilitate growth and fertility) (Jackson Labs, Cat#002364) were
used for naphthalene treatment studies. Animals were maintained as an in-house breeding
colony under specific pathogen–free conditions. All procedures involving animals were
approved by the Institutional Animal Care and Use Committee of the University Health
Network (Toronto, Ontario, Canada).
3.4.2 Naphthalene Administration and Cell Delivery
Naphthalene (>99% pure; Sigma-Aldrich, St Louis, MO) was dissolved in Mazola
corn oil and injected as described before (Stripp et al., 1995). Busulfan (Otsuka America
Pharmaceutical, Rockville, MD) was given by intra-peritoneal injection 1 day after
naphthalene treatment at a dose of 20–50mg/kg and donor cells were transplanted the
following day (106 cells in 50l PBS) transtracheally using sterile gel-loading tips. The
mice receiving donor cells were rotated to ensure equal dispersion of cell suspension to both
lungs.
3.4.3 Teratoma Assay
Cells were suspended in Matrigel (BD Bioscience) diluted 1:2 (vol/vol) with PBS.
Appropriate numbers of cells (0.25 ~ 2 × 106 cells in 100 μL) were injected subcutaneously
into both dorsal flanks of nude mice (CByJ.Cg-Foxn1nu/J) anaesthetized with isoflurane.
Four to five weeks after injection, mice were killed and teratoma tissues were harvested. For
long-term evaluation for tumorigenic potential of the cells, recipient mice were kept for 6
months. Teratoma tissues were fixed overnight in 10% buffered formalin phosphate, and

66
embedded in paraffin. Three-to-four-micrometre-thick sections were deparaffinized and
hydrated in distilled water. Sections were stained with haematoxylin and eosin for regular
histological examination.
3.4.4 Cell Isolation and Culture
3.4.4.1 Isolation of Club Cells from Mouse Lung
Mice were injected intra-peritoneally with heparin (250U/mouse) and by CO2
narcosis. Lungs were perfused through the right ventricle with cold phosphate buffered
saline (PBS) to remove blood by directing the catheter towards the main pulmonary artery.
Endo-bronchial lavage was then performed to remove alveolar leukocytes. Club cells were
isolated using a previously described protocol (Atkinson et al., 2008) with modifications.
Briefly, lungs were instilled with 0.5mL of 1% low melting temperature agarose in PBS
through the trachea then placed on ice for 2min. For lung digestion, 0.5~1mL of 0.25%
trypsin was instilled into the lung followed by ligation of the trachea with a suture. Lungs
were incubated for 10min at 37°C, then lung tissue was teased away from the large airways,
finely minced to 1mm2 pieces and placed in 250μg/mL of DNAse I in DMEM containing
antibiotic for 10 minutes. The suspension was transferred to a 50mL tube, and FBS was
added to 10% of final volume. The suspension was sieved through 100 and 40μm nylon
meshes and centrifuged at 200g for 10 minutes. The cell pellet was re-suspended in red
blood cell lyses buffer for 3min and the lysis was stopped by addition of an equal volume of
PBS. Cells were centrifuged at 40g for 6min then re-suspended in 10% FBS-DMEM and
centrifuged 2 more times at 40g for 6min. The final pellet was suspended in 0.5 % vol/vol
FBS-PBS for all subsequent procedures.
3.4.4.2 Cell Culture
Epithelial-specific medium comprised of DMEM/F12 (Invitrogen) supplemented
with 10% FBS, penicillin/streptomycin, 10mg/ml insulin, 5mg/ml transferrin-selenium
(Sigma), epidermal growth factor (EGF, 20ng/mL; Sigma), fibroblast growth factor-10
(FGF-10, 50ng/mL; R&D Systems) and hepatocyte growth factor (HGF, 30ng/mL; R&D
Systems).

67
3.4.4.3 Matrigel-based iPL Induction
Feeders (MEF) were seeded on 0.1% gelatin coated 24-well transwell filter inserts
(Corning) one day prior to the addition of epithelial cells. Sorted epithelial cells
resuspended in 100μL of Matrigel (BD Biosciences) prediluted 1:1 (vol/vol) with epithelial-
specific (EpiS) media were added to a MEF-coated 24-well transwell filter inserts in a 24-
well tissue culture plate containing 500μL of epithelial media for 3-5 days then replaced
with ES (embryonic stem cell) medium containing 1.5ug/mL doxycycline (Sigma). Media
was replenished three times per week. For bulk passaging, whole cultures were dissociated
in Collagenase (1mg/mL; Sigma) Dispase (3mg/mL; BD Biosciences) in PBS to generate a
single-cell suspension. For clonal passaging, single colonies were picked and dissociated.
3.4.4.4 In Vitro Differentiation Assays
To examine the in vitro potential of these cells to differentiate along certain lineages
a variety of differentiation assays were performed. iPL cells induced for 3 weeks were
compared to a positive control group consisting of cells exposed to reprogramming factors
for 8 weeks.
3.4.4.5 Air-liquid Interface (ALI) Differentiation Assay
Prior to the ALI assay, induced cells were cultured and recovered in ES medium for
2 weeks. For ALI culture, the ES medium from upper chamber was removed to expose cells
to the air while medium in lower chamber was replaced with ALI-specific medium (Lonza).
Media was replenished 2 times per week and cells were maintained under ALI conditions for
23 weeks.
3.4.4.6 In Vitro Pluripotency Assay
An in vitro pluripotency assay to assess the potential of these cells to develop into a
variety of cell lineages was performed (Levenberg et al., 2003). Briefly, induced cells were
dissociated digested to single cell suspension then re-suspended in 50% (Matrigel) and 20%
FBS containing medium supplemented with: activin-A (20ng/mL), transforming growth
factor (TGF)-1 (2ng/mL), 10g/mL insulin, 5g/mL transferrin and retinoic acid (RA)

68
(300ng/mL) for 2-3 weeks. Lineage differentiation was assessed by immunostaining of Pan-
Cytokeratin, -actinin and -tubulin III.
3.4.4.7 Neuron Differentiation Assay
To determine the lineage commitment of induced cells, we performed a defined
neuron differentiation assay (Millipore) with slight modifications. Briefly, 3-week and >8-
week induced cells from Matrigel cultures were digested to single cell suspension and
differentiated under neuron-specific conditions for 2-3 weeks following the manufacturer’s
protocol. Generation of neurons was accessed by immunostaining of -tubulin III.
3.4.5 Fluorescence Activated Cell Sorting and Analysis
For purification of epithelial cells, fresh isolated cells were suspended and incubated
in 0.5 % vol/vol FBS-PBS containing an optimally pre-titered mixture of antibodies [anti-
CD45, anti-CD31 (BD Biosciences), anti-EpCAM (Abcam) and relevant isotype controls]
for approximately 30min on ice. Labeled cells were washed and re-suspended at 3~5 × 106
cells/mL in 0.5 % vol/vol FBS-PBS. Cell viability was accessed by propidium iodide
(1μg/mL) staining. For intra-cellular antigen analysis, cells were fixed and stained using a
Fix and Perm kit (Invitrogen) as per manufacturer instructions. Sorting was performed using
a Moflo BRU cell sorter (Becton Dickinson), aquisition was performed using a BD LSRII
analyzer (Becton Dickinson) and data was analyzed using FlowJo software.
3.4.6 Bottom-feeder conditioned CFSE assay
CFSE (carboxyfluorescein diacetate, succinimidyl ester) cell proliferation assay was
used to evaluate proliferative capacity. Mitomycin treated in-activated mouse embryonic
fibroblast (MEF) feeders were seeded and allowed to attach to the bottom of the transwell
(Corning) membrane one day prior to addition of sorted cells on the top of the membrane.
CFSE working solution (10-15M106 cells; Invitrogen) was prepared and applied to cells
according to the manufacturer’s protocol. Cell were labelled with CFSE and analyzed using
flow cytometry.
69
3.4.7 Immunofluorescence
Samples were fixed with 4% paraformaldehyde (PFA) for 30min and blocked with
5% goat serum and 2% BSA in PBS containing 0.5% Triton X-100 for 1 hour. Primary
antibodies (Table 1) were diluted in BSA/PBS, applied to samples and incubated overnight
at 4°C. Secondary antibodies AlexaFluors 488, 532, 546, 633 or 647 (Invitrogen) were
applied according to the species in which the primary antibody was used for 2 hours at room
temperature. Nuclear staining was performed using 2mg/ml 4, 6, diamidino-2-phenylindole
(DAPI; Sigma). Stained samples were mounted with immunofluorescent mounting medium
(DAKO). Appropriate non-specific IgG isotypes were used as controls. Immunoreactivities
of antigens were visualized as single optical planes using an Olympus Fluoview confocal
microscope and analyzed using FV10-ASW 2.0 Viewer software.
Antibody name Company Cat. Num
CD45 BD phamingen 553081
CD31 BD phamingen 553373
EpCAM Abcam ab95641
CCSP Cedarlanes/Upstate 07-623
Santa cruz biotechnology sc-9772
Claudin10 Invitrogen 388400
pro-SPC Chemicon AB3786
T1a Santa Cruz sc-53533
-tubulin IV SIGMA-Aldrich T7941
pro-collagen Santa Cruz sc-166007
a-SMA SIGMA-Aldrich A2547
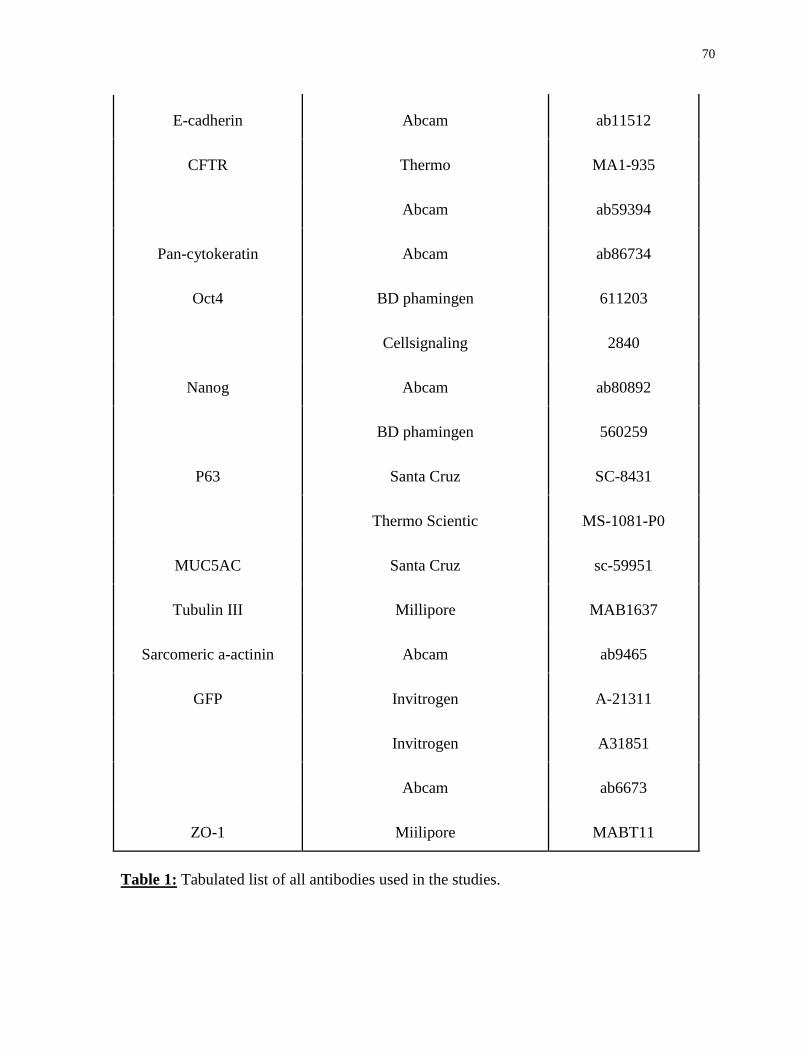
70
E-cadherin Abcam ab11512
CFTR Thermo MA1-935
Abcam ab59394
Pan-cytokeratin Abcam ab86734
Oct4 BD phamingen 611203
Cellsignaling 2840
Nanog Abcam ab80892
BD phamingen 560259
P63 Santa Cruz SC-8431
Thermo Scientic MS-1081-P0
MUC5AC Santa Cruz sc-59951
Tubulin III Millipore MAB1637
Sarcomeric a-actinin Abcam ab9465
GFP Invitrogen A-21311
Invitrogen A31851
Abcam ab6673
ZO-1 Miilipore MABT11
Table 1: Tabulated list of all antibodies used in the studies.

71
3.4.8 Iodide Efflux Assay
ALI-conditioned cells cultured on transwell membranes were loaded with 500 µl NaI
solution [3.0mM KNO3, 2.0mM Ca(NO3)2, 11mM glucose, 20mM HEPES, 136mM NaI]
from the bottom chamber and incubated at 37°C for 1h. To remove the redundant iodide,
cultures were washed out with 5 times ml of washing buffer comprised of nitrate (3.0mM
KNO3, 2.0mM Ca(NO3)2, 11mM glucose, 2 mM Hepes, 136mM NaNO3) and 100μM
amiloride. For time course measurement, 200µl of washing buffer was added to the top
chamber of transwells at one-minute intervals for 3 minutes followed by adding 200µl of
cAMP agonists which contain forskolin (10μM), 3-isobutyl-1-methylxanthine (100μM,
IBMX), and genistein (50μM) in washing buffer at one-minute intervals for 6 minutes.
Reacted solutions at each minute were transferred to a 96-well plate to measure the absolute
iodide electrode value (mV) using halide-selective microelectrode (Lazar Research
Laboratories, Los Angeles, CA). Measured mV values were converted to iodide
concentrations using a standard curve measuring the mV values of 1μM to 1mM iodide.
3.4.9 Western Blot
Lung tissues (~25mg) were lysed in RIPA buffer (Cell Signaling) with one Complete
Mini EDTA-free protease inhibitor tablet (Roche) for 30 minutes on ice then centrifuged at
10 000g and the supernatants were resolved by SDS-PAGE. For detection on PVDF
membrane (Roche), anti-CFTR (rabbit polyclonal, abcam # ab59394) and anti--actin (Cell
signaling #4970) antibodies were used.
3.4.10 Real-time PCR Analysis
Total RNA was prepared using the RNeasy Kit (Qiagen) as per manufacturer’s
instructions. cDNA was prepared and assayed using Superscript III (Sigma) according to
manufacturer’s protocol. Differential gene expression was determined using SYBR green
detection (Roche). All Real-time PCR reactions were done in triplicate for each sample.
GAPDH was used as a housekeeping gene to normalize gene expression levels using
LightCycler 480 software (Roche). Normalized mRNA levels were shown as relative to the
control samples (day 0 fresh isolated cells or adult lung) (Table 2).

72
qPCR primers
Gene name Primer sequence 5’-3’ length
4F2A Forward GGCTGGAGATGTTGAGAGCAA 21
Reverse AAAGGAAATCCAGTGGCGC 19
c-Myc Forward ACCACCAGCAGCGACTCTGA 20
Reverse TGCCTCTTCTCCACAGACACC 21
K1f4 Forward GCACACCTGCGAACTCACAC 20
Reverse CCGTCCCAGTCACAGTGGTAA 21
Oct4 Forward ACATCGCCAATCAGCTTGG 19
Reverse AGAACCATACTCGAACCACATCC 23
Sox2 Forward ACAGATGCAACCGATGCACC 20
Reverse TGGAGTTGTACTGCAGGGCG 20
CyclinD1 Forward CCTGACACCAATCTCCTCAACG 22
Reverse TCTTCGCACTTCTGCTCCTCAC 22
Cyp2f2 Forward GCATACCCCGTCTTTTTCAA 20
Reverse TCATCGTCATAGTCGAAGCG 20
Cld10 Forward CCAGGGTCTGTGGATGAACT 20
Reverse GCCAAGCAAGCAATTTTAGC 20
Aox3 Forward AGAGCCCTCCACAGAACAGA 20
Reverse TGTATTTTGGGACTCCTCGG 20
Pon1 Forward GAACAAGAAGGAGCCAGCAG 20
Reverse AACAACATTGGACCACGACA 20
EpCAM Forward GCTGGCAACAAGTTGCTCTCTGAA 24
Reverse CGTTGCACTGCTTGGCTTTGAAGA 24

73
E-Cadherin Forward AACAACTGCATGAAGGCGGGAATC 24
Reverse CCTGTGCAGCTGGCTCAAATCAAA 24
CCSP Forward ATCTGCCCAGGATTTCTTCA 20
Reverse TCTTGCTTACACAGAGGACTTGTT 24
Sftpc Forward GCAAAGAGGTCCTGATGGAG 20
Reverse GCAGTAGGTTCCTGGAGCTG 20
AQP5 Forward ATGAACCCAGCCCGATCTTT 20
Reverse ACGATCGGTCCTACCCAGAAG 21
CFTR Forward CTGGACCACACCAATTTTGAGG 22
Reverse GCGTGGATAAGCTGGGGAT 19
Foxj1 Forward GCCGGCAGTTCAACTGCCCT 20
Reverse CAGTCCTGCAGGTCAGCGGC 20
Table 2: Tabulated list of all qPCR primers used in the studies.
3.4.11 Microarray and Data Analysis
Total RNA was extracted using RNeasy kit (Qiagen, Canada). Equal amounts of
RNA from three separate samples in each group were used for microarray. Microarray
expression profiling using Mouse Gene 2.0 ST chips was performed by The Centre for
Applied Genomics, (The Hospital for Sick Children, Toronto, Canada). GO term analysis
was performed by the DAVID Bioinformatics tool. Self-organizing map analysis of gene
expression was performed with the use of the analysis tool - MultiExperiment Viewer.
3.4.12 Statistics
Statistical analysis was performed using GraphPad Prism 5.0 statistical software (San
Diego, CA, USA). The statistical significance of multiple groups was compared to each
other using Tukey’s multiple comparison test ANOVA. A p value of <0.05 was considered
significant.

74
3.5 Results
3.5.1 Isolation and Identification of Terminally Differentiated
Club Cells
Club cells are secretory cells prominently found in the small bronchioles,
identifiable by the presence of Club cell secretory protein (CCSP) (Hackett and Gitlin,
1992). The mature cells have very limited proliferative capacity. After injury, they are
replaced from a pool of “variant” Club cells, which are resistant to toxins such as
naphthalene. This population also then serves as progenitors for ciliated and mucous cell
populations (Rawlins et al., 2009; Reynolds and Malkinson, 2010). To demonstrate that the
value of the iPL cell approach, we chose to isolate this terminally differentiated population.
Following a modified Club cell isolation protocol (Atkinson et al., 2008), a
CD45neg
CD31neg
EpCAMpos
profile defined two distinct epithelial populations, namely
EpCAMhigh
and EpCAMlow
cells (Figure 1B). The EpCAMhigh
population was exclusively
Club cells expressing CCSP (Figure 1C) and Claudin10 (Zemke et al., 2008) (Cldn10)
(Figure S1A), whereas the EpCAMlow
population contained both Club cells and type II
alveolar epithelial (AEC-II) cells (Figure 1C, S1B). Club cell-related genes CCSP, Cyp2f2,
Cldn10, Aox3, and Pon1 were detected within both populations with higher levels in the
EpCAMhigh
population (Figure 1D, E). Naphthalene administration results in selective loss
of mature Club cells (Stripp et al., 1995). To evaluate whether EpCAMpos
populations
contained functionally different subtypes of Club cells, we compared EpCAM expression in
cells isolated from naphthalene-treated and non-treated mice. EpCAMhigh
cells were nearly
completely ablated with naphthalene treatment, confirming that they are naphthalene-
sensitive, mature Club cells (Figure 1F).
3.5.2 Interrupted Reprogramming Allows OSKM-dependent
Proliferation of EpCAMhigh
-Club Cells
EpCAMpos
cells were isolated from R26-rtTA/Col1a1::tetO-4F2A double transgenic
mice (Carey et al., 2009) enabling expression of Oct4, Sox2, Klf4 and c-Myc (OSKM)
following treatment with doxycycline (Dox). To measure the proliferative response of
EpCAMpos
cells to inductive factors, we used a specific 2D culture system allowing

75
separation of seeded cells from a feeder population (Kim et al., 2007) (Figure S1C). Control
non-treated and Dox-treated EpCAMhigh
and EpCAMlow
cells were labelled with
carboxyfluorescein diacetate, succinimidyl ester (CFSE) dye at day 7 and maintained in
culture for an additional week in the presence or absence of Dox. Non-treated EpCAMhigh
cells showed very limited proliferation consistent with lower expression of Cyclin D1 while
dox treatment resulted in significant proliferation of the majority of cells (Figure 1G, H).
After treatment with Dox, EpCAMhigh
cells showed significant upregulation of the 4F2A
construct transgene and Cyclin D1 (Figure 1J, K). Withdrawal of Dox stopped proliferation
in the EpCAMhigh
group, while EpCAMlow
cells continued to proliferate (Figure 1H, I). Note
the spontaneous loss of EpCAM in the EpCAMlow
population (Figure 1I). Importantly,
EpCAM and CCSP expression re-emerged following withdrawal of Dox (Figure 1L, M).
Thus, for these proof of principle studies, we selected the EpCAMhigh
population, where, as
non-proliferative mature Club cells, induced proliferation could be easily distinguished from
endogenous proliferation.
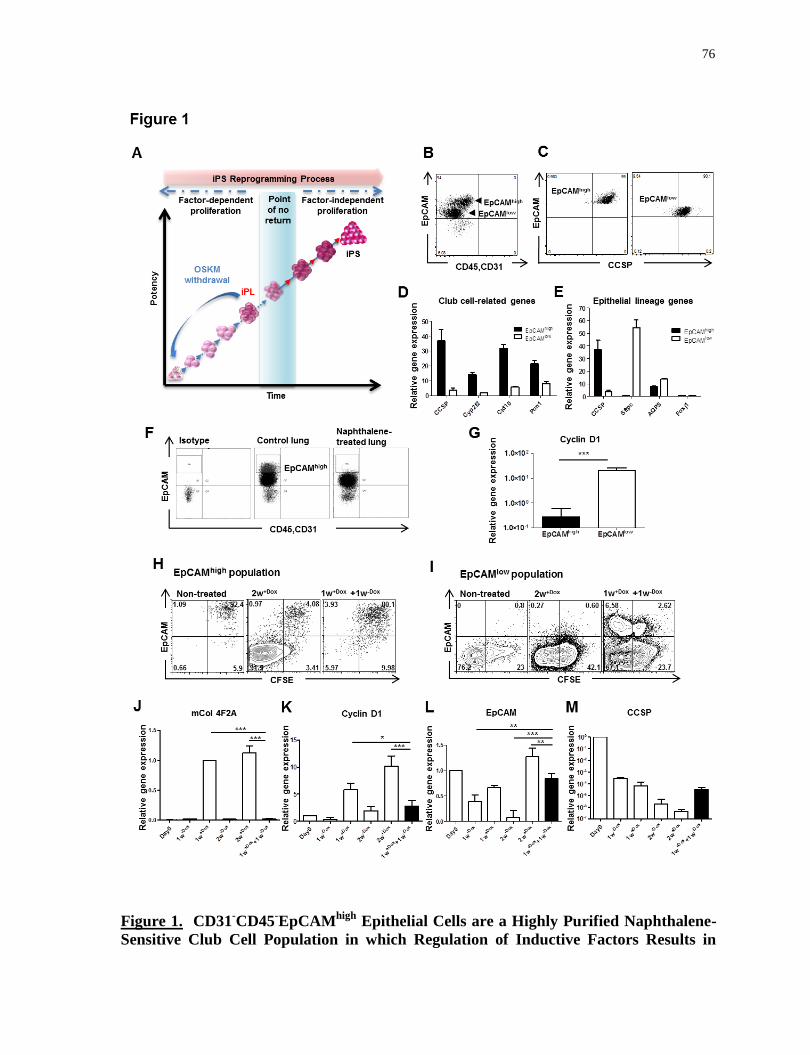
76
Figure 1. CD31-CD45
-EpCAM
high Epithelial Cells are a Highly Purified Naphthalene-
Sensitive Club Cell Population in which Regulation of Inductive Factors Results in

77
Controlled Proliferation. (A) Schematic graph depicting generation of iPL cells using
interrupted reprogramming. (B) Representative flow cytometry dot-plots showing
EpCAMhigh
and EpCAMlow
cells in a parental population of CD31-CD45
- fresh isolated lung
tissue digested cells. (C) Dot plots showing EpCAMhigh
cells are exclusive Club cells
showing immunoactivity of CCSP. Relative expression of (D) Club cell and (E) other
epithelial lineage-related genes to adult lung, comparing fold-differences in gene expression
in EpCAMhigh
(solid black bars) and EpCAMlow
(open bars) cells. (F) Representative dot
plots comparing EpCAM expression in CD45-CD31
- freshly isolated lung cells from non-
treated and naphthalene treated mice (n=3). (G) Comparison of Cyclin D1 expression in
EpCAMhigh
cells and EpCAMlow
cells. Representative flow cytometery dot plots showing
CFSE-labeled cells at day 7 (untreated and Dox-treated). (H) EpCAMhigh
cells and (I)
EpCAMlow
cells maintained in feeder-separated semi-supportive culture for an additional 7
days with and without Dox treatment. Control untreated cells were cultured without Dox for
the entire 2 weeks. Expression of (J) mCol4F2A, (K) CyclinD1, (L) EpCAM, and (M)
CCSP, comparing fold-differences in gene expression of EpCAMhigh
freshly isolated (Day0),
EpCAMhigh
one-week Dox-treated (1w+Dox
), induced cells cultured for an additional week in
the presence (2w+Dox
) and absence (1w+Dox
+1w-Dox
) of Dox. In B, C, F, H and I, data are
representative of a minimum of three biological replicates. For D, E, G and J-M, values are
mean S.D. of three independent biological replicates. *, p<0.05; **, p<0.001; ***,
p<0.0001.
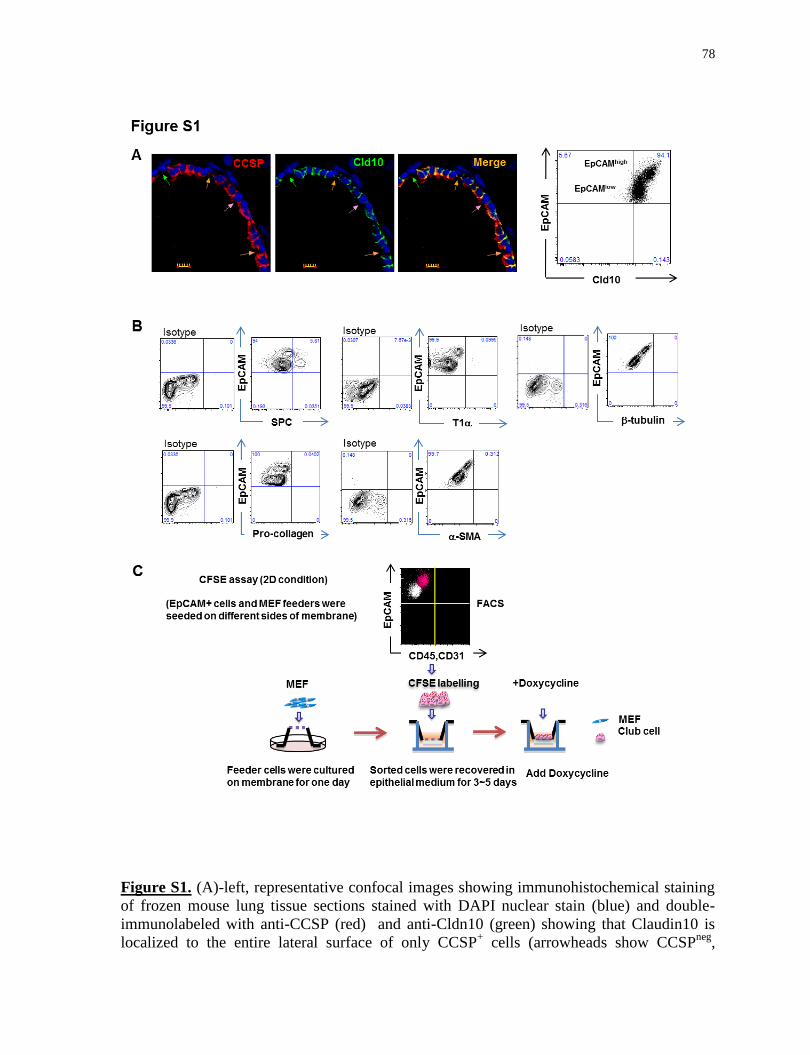
78
Figure S1. (A)-left, representative confocal images showing immunohistochemical staining
of frozen mouse lung tissue sections stained with DAPI nuclear stain (blue) and double-
immunolabeled with anti-CCSP (red) and anti-Cldn10 (green) showing that Claudin10 is
localized to the entire lateral surface of only CCSP+ cells (arrowheads show CCSP
neg,

79
Cldn10neg
cells). (A)-right, flow cytometry dot-plots showing freshly isolated lung cells,
stained with antibodies against Cldn10 and EpCAM. (B) Flow cytometry analysis of freshly
isolated lung cells, showing EpCAM-positive epithelial cells marked with antibodies
specific for SPC, T1, -tubulin, Pro-collagen, and -SMA. EpCAMhigh
cells are
exclusively Club cells whereas the EpCAMlow
population is composed largely of Club cells
(>90% CCSP+) with a small number of AEC-II cells (<10%), which are positive for SPC,
the classic marker for AEC-II cells. Both populations are negative for T1 (a marker for
AT-I cells) and -tubulin (a marker for ciliated cells). (C) Schematic graph of bottom-feeder
culture condition which enables the separation of seeded cells from supporting feeder cells.
In A-right and B, data are representative of a minimum of three independent biological
replicates. Scale bar, (A-left) 100 µm.
3.5.3 Interrupted Reprogramming Results in Clonal Expansion
of Quiescent Mature Club Cells without Traversing the
Pluripotent State
To better support epithelial cell growth, EpCAMhigh
Club cells were cultured in
Matrigel-based 3D conditions and seeded on inactivated mouse embryonic fibroblast (MEF)
feeder cells prior to Dox treatment. No colonies were formed in the absence of Dox,
confirming that native EpCAMhigh
cells lack clonogenic ability. Strikingly, Dox-treated
cells exhibited clonogenic growth (Figure 2A) showing epithelial colonies with hollow
lumens (Figure 2B, C). Time course analysis indicated that lumen formation occurred as
early as day4 (Figure S2A). This lumen formation occurs at least in part via apoptosis
(Grant et al., 2006), marked by caspase-3 expression (Figure S2B), and apical-basal
polarization (Martin-Belmonte et al., 2008), as shown by basolateral distribution of EpCAM
(Figure 2C) and apical expression of ZO-1 (Figure S2C). Dox induction for 3 weeks
(3w+Dox
) resulted in suppression of CCSP without activation of the pluripotency marker,
Nanog (Figure 2D, 3E). An additional 2 weeks of Dox treatment (5w+Dox
) resulted in
22.9±8.3% of colonies becoming Nanog-positive (Figure 2E). In vivo, 3w+Dox
cells injected
into NOD/SCID mice failed to form tumors or teratomas over a 6-month observation
period. In contrast, mice developed teratomas 2-3 weeks after injection with either R1-ES
cells (Nagy et al., 1993) or 5w+Dox
cells (Figure 2G, S2D). Thus, we selected a 3-week
induction period during which we achieved the greatest level of expansion without
activation of Nanog expression and hereafter term the 3w+Dox
cells Club-iPL cells. During

80
this 3-week period of expansion, cells were serially passaged in the presence of Dox,
retained colony-forming potential (Figure 2H, right Y axis), and showed an exponential
increase in total cell number (~ 30-fold expansion) (Figure 2H, left Y axis). Microarray
analysis of genome-wide transcriptional changes showed Club-iPL cells have minimal
expression of ES cell-related genes (e.g. Nanog, Utf1, Dapp4) and maintained significant
overlapping expression of Club cell-related genes (Figure 2I).
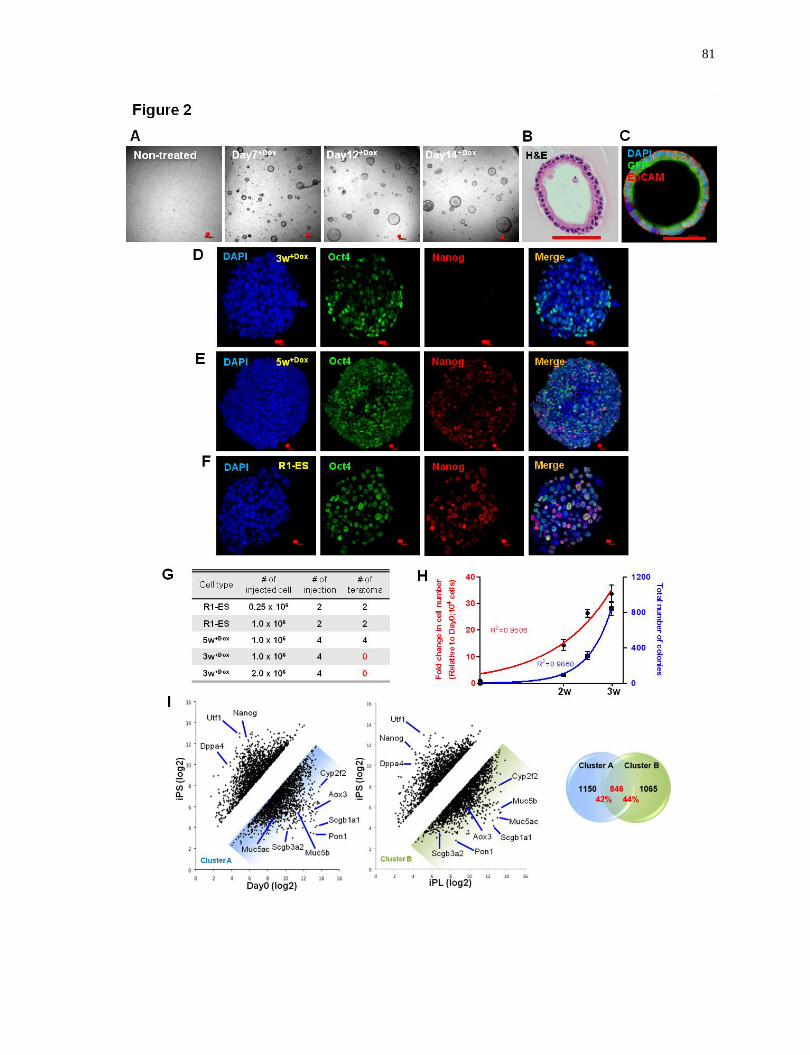
81

82
Figure 2. Carefully Defined Length of Interrupted Reprogramming Results in
Efficient Clonal Expansion of Quiescent Mature Club Cells without Traversing the
Pluripotent State In Vitro. (A) Light microscopy bright-field images showing the
generation of hollow-luminal colonies in the presence of doxycycline in Matrigel-based
clonogenic 3D conditions. (B) H&E staining of sectioned EpCAMhigh
cell-derived colonies
under Dox-treatment showing hollow-luminal morphology. (C) Confocal microscopy
images of induced colonies derived from GFP-Club cells stained with nuclear stain DAPI
(blue), GFP (green) and EpCAM (red). (D) Confocal microscopy images depicting 3-week
induced colonies (3w+Dox
) stained with nuclear stain DAPI (blue), Oct4 (green) and Nanog
(red). Immunostaining of 5 week-induced colonies (5w+Dox
) (E) showing Nanog
immunactivity as that seen in R-1 ESCs (F). In vivo teratoma assay (G), R1-ESCs (positive
control), 3w+Dox
Club-iPL cells and 5w+Dox
cells were transplanted into NOD/SCID mice.
(N=2 mice for each group). (H) Bulk serial passage of induced colonies during the 3 weeks
of induction. Left Y axis represents the folds change in total cell number relative to day0
seeded cells (10,000 cells/ well). Right Y axis represents the total number of colony forming
units (CFU) generated. (I) Microarray analysis scatter plot showing comparison of iPS
(generated from EpCAMhigh
-Club cells) to day0 and Club-iPL cells (left panel); pie charts
depicting number of genes overlap in cluster A and B (right panel). For H, values are mean
S.D. of three independent biological replicates. Scale bar, 100 µm (A-C), 10µm (D-F).
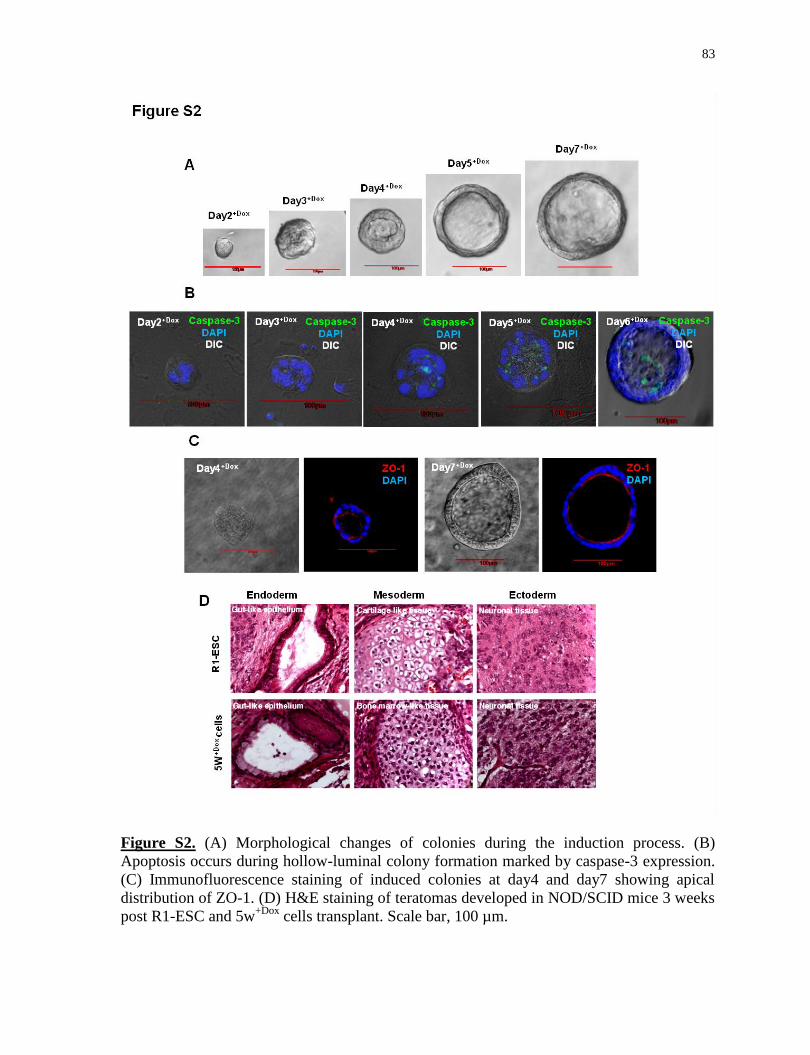
83
Figure S2. (A) Morphological changes of colonies during the induction process. (B)
Apoptosis occurs during hollow-luminal colony formation marked by caspase-3 expression.
(C) Immunofluorescence staining of induced colonies at day4 and day7 showing apical
distribution of ZO-1. (D) H&E staining of teratomas developed in NOD/SCID mice 3 weeks
post R1-ESC and 5w+Dox
cells transplant. Scale bar, 100 µm.

84
3.5.4 Interrupted Reprogramming Allows Club-iPL Cells to
Return to Their Original Epithelial Phenotype upon
Withdrawal of Factors
We assessed the ability of Club-iPL cells to return to their original epithelial
phenotype following withdrawal of Dox. Two weeks after withdrawal of Dox (3w+Dox
+2w-
Dox) cells turned off transgene 4F2A expression and down-regulated Cyclin D1 (Figure 3A,
B). Expression of the 4 individual factors was also significantly down-regulated (Figure
S3A). Some epithelial genes, such as EpCAM and E-cadherin, were well maintained both
with treatment and upon withdrawal of Dox (Figure S3B). CCSP, which was markedly
suppressed after 3 weeks of Dox, was expressed at robust levels upon withdrawal of Dox
(Figure 3C, E, F, I). Importantly, Nanog expression was not observed in either Club-iPL
cells or 3w+Dox
+2w-Dox
cells (Figure 3D, F) whereas in 4w+Dox
+1w-Dox
cells, a few Nanog+
colonies were observed (Figure 3G, H). Unbiased clustering analysis of the genes with a
change 3-fold (5966/24779 genes) demonstrated that the induced cell groups clustered
with the day0 group (Figure 3J). Further analysis showed that the expression of large
numbers of genes temporally changed during Dox-driven expansion of Club cells but
returned upon withdrawal of the factors. These gene clusters were enriched for epithelial
genes by Gene Ontology analysis (Figure 3K, top). Other genes demonstrating progressive
changes were enriched for metabolism and cell adhesion functions (Figure 3K, bottom),
suggestive merely of adaptation to the in vitro environment. Figure S3C shows a heat map of
the genes shown in Figures 2I across all 4 groups.
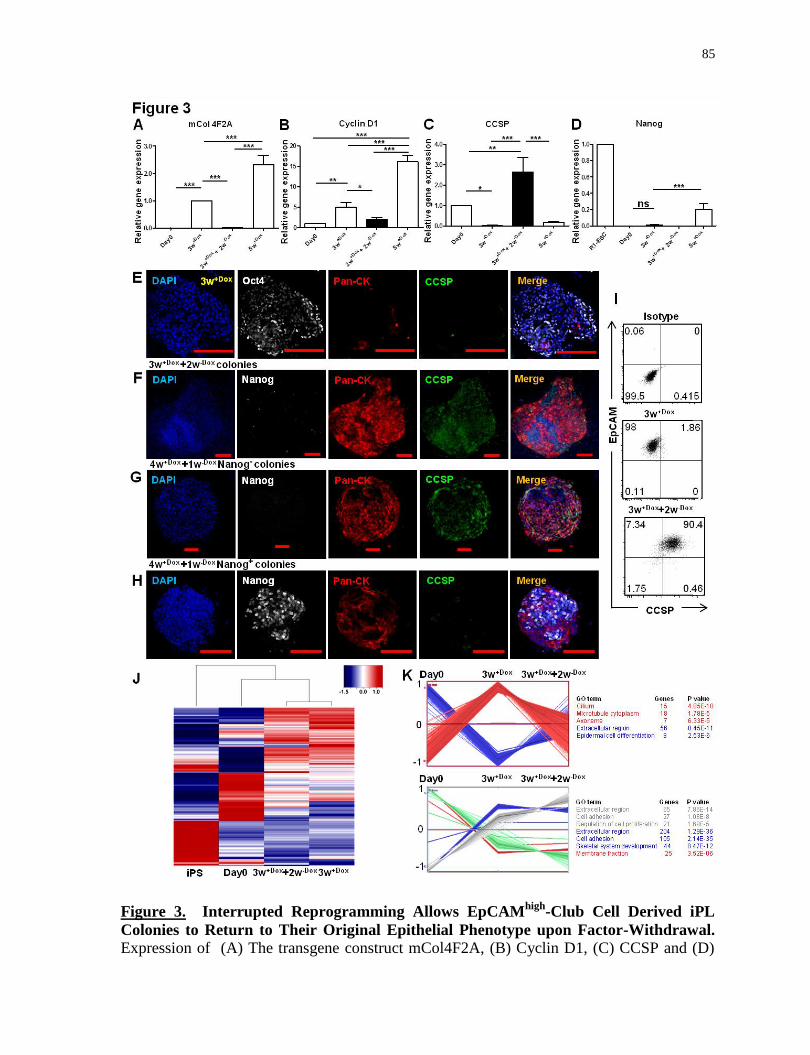
85
Figure 3. Interrupted Reprogramming Allows EpCAMhigh
-Club Cell Derived iPL
Colonies to Return to Their Original Epithelial Phenotype upon Factor-Withdrawal.
Expression of (A) The transgene construct mCol4F2A, (B) Cyclin D1, (C) CCSP and (D)

86
Nanog, as measured by qRT-PCR comparing fold-differences in gene expression in freshly
isolated cells (Day0), 3w+Dox
(Club-iPL cells), Club-iPL cells with subsequent 2-week
culture in Dox-free media (3w+Dox
+2w-Dox
), and 5-week induced cells (5w+Dox
). Confocal
microscopy images of (E) 3w+Dox
(Club-iPL cells) colonies, (F) Colonies obtained from
3w+Dox
(Club-iPL cells) maintained in culture for 2 weeks in the absence of Dox (3w+Dox
+
2w-Dox
), (G) Nanog-negative and (H) Nanog–positive colonies obtained from 4-week
induced cells (4w+Dox
) cultured without Dox for one subsequent week (4w+Dox
+1w-Dox
),
showing cells stained with nuclear stain DAPI (blue), Oct4Nanog (grey), Pan-CK (red) and
CCSP (green). (I) Representative flow cytometry dot-plots showing expression of EpCAM
and CCSP in 3w+Dox
Club-iPL cells, and iPL cells cultured without Dox for 2 subsequent
weeks (3w+Dox
+2w-Dox
). (J) Unbiased clustering analysis of all genes expressing 3-fold
change (5966/24779). (K) Self-organizing map analysis of genes that exhibited a >3 fold
gene expression difference between cell samples. For A-D, values are mean S.D. of three
independent biological replicates. For I, data are representative of a minimum of three
independent biological replicates. *, p<0.05; **, p<0.001; ***, p<0.0001. Scale bar, 100
µm (E-H).

87

88
Figure S3. Expression of (A) Oct4, Sox2, klf4, c-Myc, and (B) EpCAM and E-Cadherin, as
measured by qRT-PCR comparing fold-differences in gene expression in R1-ESC cells,
freshly isolated cells (day0), 3-week induced cells (3w+Dox
), 3-week induced cells with
subsequent 2-week culture in doxycycline-free media (3w+Dox
+ 2w-Dox
), and 5-week
induced cells (5w+Dox
). (C) Hierarchical clustering analysis of Club cell and pluripotency-
related gene expression of iPS (derived from Club cells), day0, 3w+Dox
and 3w+Dox
+ 2w-Dox
cells. For A and B, values are mean S.D. of three independent biological replicates.*,
p<0.05; **, p<0.001; ***, p<0.0001.
3.5.5 Interrupted Reprogramming Allows Preservation of
Lineage Commitment
To ensure that Club-iPL cells have restricted differentiation, lineage preference was
accessed by in vitro differentiation assays. Immunostaining of bulk colonies for Pan-CK
(endodermal epithelial cells), -actinin (mesodermal cardiomyocytes) and -tubulin III
(ectodermal neuron cells) demonstrated that Club-iPL cells only developed along an
epithelial lineage (Figure 4A). In contrast, cells treated with Dox for 8 weeks (8w+Dox
), in
addition to showing Pan-CK expression (Figure 4B), were able to generate -actinin+
(Figure 4C) and -tubulin III+ cells (Figure 4E). Nanog
+ undifferentiated cells were also
observed (Figure 4D). Moreover, Club-iPL cells exhibited a higher tendency for generation
of ciliated cells (-tubulin IV+: 66.7%7.6% cells) compared to 8w
+Dox cells (-tubulin IV
+:
25.2%5.1% cells) (Figure 4F). The existence of mixed populations of differentiated cells
and undifferentiated cells from the long-term induced cell group suggests that prolonged
induction results in greater divergence from the original lineage and enhanced pluripotency,
if not full creation of traditional iPS cells. Lineage commitment at the single cell level was
assessed by culturing independent colonies in 3 different media, reported to direct
differentiation to either neuronal, cardiomyocyte, or epithelial lineages (12
coloniescondition) (Figure S4A). Club-iPL colonies remain committed to an epithelial fate
generating Pan-CK+ cells but no cardiac troponin T (cTnT) or -tubulin III-expressing cells
(Figure 4G-I, S4B).
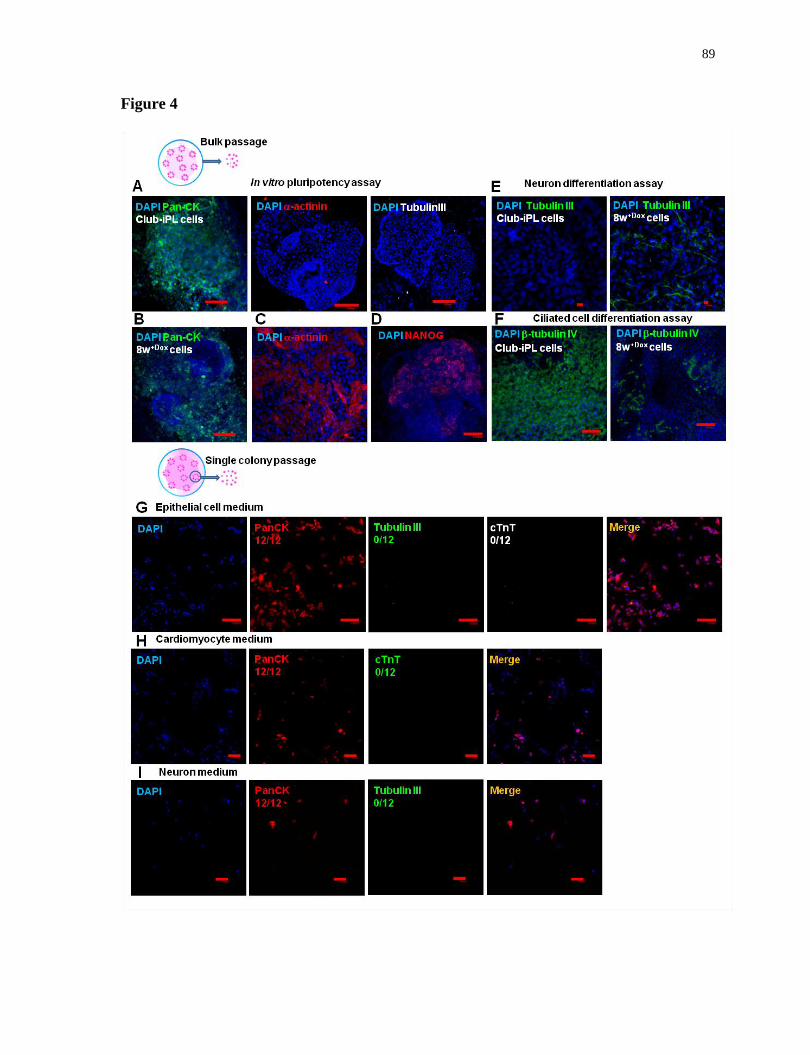
89
Figure 4

90
Figure 4. Interrupted Reprogramming Allows Preservation of Lineage Preference and
Commitment. Confocal microscopy images of bulk-passaged (A) Club-iPL (3w+Dox
) cells,
following in vitro culture under pluripotency assay conditions. Images show staining for
nuclear stain DAPI (blue), Pan-CK (green), -actinin (red), and tubulin III (grey). Cells
induced for 8 weeks (8w+Dox
), under in vitro pluripotency assay conditions with (B) Pan-CK
(green), (C) -actinin (red), and (D) Nanog (red) respectively. Confocal microscopy images
of Club-iPL (3w+Dox
) cells and 8 week induced (8w+Dox
) cells under (E) neuron cell
differentiation assay conditions showing tubulin III (green) staining and (F) under ALI
ciliated cell differentiation conditions showing -tubulin IV (green) staining. Confocal
microscopy images of cells dissociated from single Club-iPL colonies cultured on 10%
Matrigel-coated plate under conditions reported for (G) epithelial cells, (H) cardiomyocytes
and (I) neurons. Images showing staining for nuclear stain DAPI (blue), Pan-CK (red),
tubulin III (green) and cTnT (top panel-grey; middle and bottom panel-green ) (n=12
colonies/each condition). Scale bar, 10µm (C and E), 100 µm (A, B and D-I).

91
Figure S4. Schematic (A) showing different conditions used in single colony assay.
Confocal microscopy images of cells dissociated from single Club-iPL colonies cultured on
(B) gelatin-coated plate under conditions reported for epithelial, cardiomyocyte and neuronal
cells. Images showed staining for nuclear stain DAPI (blue), Pan-CK (red), tubulin III
(green) and cTnT (top panel-grey; middle and bottom panel-green ) (n=12 colonies/each
condition). Scale bar, 100 µm.

92
3.5.6 Club-iPL Cells Function as Multipotent Bronchiolar
Progenitor-Like Cells
We evaluated the differentiation potential of Club-iPL cells using a 2-step protocol
(Figure 5A) in which Club-iPL cells were cultured without Dox for 2 weeks (1st step)
followed by air-liquid interface (ALI) conditions for 2~3 weeks (2nd
step). The 1st step of
differentiation resulted in upregulation of CCSP (Figure 5B, D). Under 2nd
step ALI
conditions, iPL cells spontaneously differentiated to Muc5AC-expressing mucin-producing
goblet cells (Figure 5E) and β-tubulin IV-expressing ciliated cells (Figure 5F) coinciding
with downregulation of CCSP (Figure 5B) and upregulation of Foxj1 mRNA levels (Figure
5C). Thus, Club-iPL cells can function as multipotent bronchiolar progenitor-like cells,
giving rise to Club, goblet and ciliated cells (Figure 5G).

93
Figure 5. Club-iPL Cells Function as Multipotent Bronchiolar Progenitor-Like Cells.
(A) Schematic graph representing a 2-step differentiation protocol. Comparing fold-
differences in CCSP (B) and Foxj1(C) gene expression in adult lung, freshly isolated day0
cells, 3w+Dox
+2w-Dox
cells (pre-ALI), and cells subsequently cultured in ALI conditions for
2-3 weeks (ALI) by qRT-PCR. (D) Confocal microscopy images showing immunostaining

94
of iPL cells after the 1st
step in the differentiation protocol, with nuclear stain DAPI (blue)
Pan-CK (red) and CCSP (green). Immunostaining of cells after the 2nd
step of
differentiation protocol (E) with nuclear stain DAPI (blue), E-cadherin (red) and Muc5AC
(green); (F) with nuclear stain DAPI (blue) and -tubulin IV (green). (G) Schematic graph
depicting the differentiation capacity of 3w+Dox
(Club-iPL) cells. For B-C, values are mean
S.D. of three independent biological replicates. *, p<0.05; **, p<0.001; ***, p<0.0001.
Scale bar, 10 µm (D-F).
3.5.7 Club-iPL Cells are Able to Generate Functional CFTR-
Expressing Ciliated Epithelium
Cystic fibrosis transmembrane conductance regulator (CFTR), mainly expressed in
ciliated epithelium in the lung, encodes a cAMP-regulated chloride channel that plays a
critical role in regulating chloride and water transport. Ciliated epithelium derived from iPL
cells expressed functional CFTR protein. We observed that 61.9±6.1% of E-cadherin+ cells
expressed CFTR, consistent with the formation of functional ciliated epithelium with tight
junctions (Figure 6A). We confirmed the apical membrane localization of CFTR while E-
cadherin staining was visualized at the lateral membranes (Figure 6B). ALI-conditioning
resulted in CFTR-expressing cells (Figure 6C). Gene expression analysis of ALI-
conditioned cells compared to pre-ALI cells showed a reduction of CCSP expression
(Figure 5B) and marked up-regulation of CFTR (Figure 6D), suggesting the appropriate
differentiation of expanded Club cells to CFTR-expressing ciliated cells. An iodide efflux
assay measuring cAMP agonist stimulation of the CFTR channel suggests that CFTR seen
in Club-iPL-derived ciliated epithelium was functional and appropriately regulated (Figure
6E).
3.5.8 Club-iPL cells Are Useful in Cell Replacement Therapy for
Cystic Fibrosis in Vivo
The engraftment of Club-iPL cells into CFTR-deficient epithelium was assessed in
vivo. For tracking purposes, ROSA26-rtTA/Col1a1:: tetO-4F2A double transgenic mice
were bred to actin GFP mice and GFP+ Club-iPL cells were delivered to CFTR-knockout
mice transtracheally following our previously reported conditioning regimen (Duchesneau et
al., 2010, 2017) to augment retention of delivered cells. The engraftment and differentiation

95
capacity of Club-iPL cells in recipient lungs were assessed via staining for ZO-1 or E-
cadherin, anti-GFP and epithelial-type specific markers at 7 and 21 days after cell delivery.
CFTR expression in airways of wildtype mice was used as a positive control (Figure 6F)
while non-treated and naphthalene-treated CFTR-knockout mice served as negative controls
(Figure 6G-I). Injected GFP cells expressed ZO-1 and E-cadherin suggestive of their
incorporation in recipient epithelium. CFTR-expressing GFP cells can be found at day 7
and 21 (Figure 6J, K) after iPL treatment. Western blotting (Figure 6L) confirmed
expression of CFTR protein. A faint band of immature hypoglycosylated CFTR seen in the
untreated CFTR-knockout controls as expected (in this transgenic animal where the lung is
known to have low level expression of human CFTR) (Zhou et al., 1994). They also
increased expression of CCSP and partially restored expression of CFTR in CFTR-deficient
lungs (Figure 6M, N). Consistent with in vitro results, CCSP and α-tubulin staining
confirmed the differentiation of engrafted Club-iPL cells to Club cells and ciliated cells in
vivo (Figure S5A-E). No SPC+ or AQP5
+ GFP
+ cells were found, in vitro or in vivo (data
not shown), consistent with restricted commitment to the bronchiolar lineage. To quantify
retention, genomic DNA for GFP was measured using a standard curve. Donor iPL cells
were retained in recipient lungs (Figure S5F) with no significant difference between 7 days
(27.8±12.2% of the initial injected cell number) and 21 days (25.8±12.1%) after cell
delivery. We also examined the long-term tumorigenicity of iPL cell lines in vivo (n=3).
Whole body micro-CT scans taken at 9 months after iPL cell delivery showed no tumors
(Figure S5G).
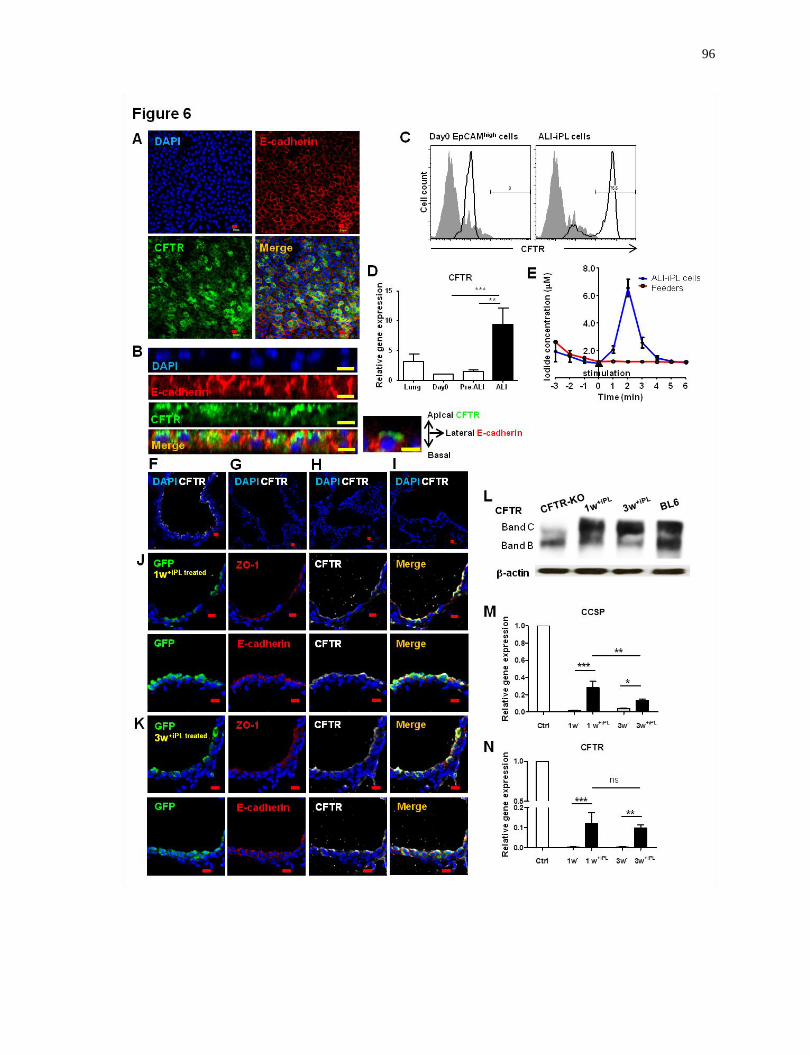
96

97
Figure 6. Club-iPL Cells are Able to Generate Functional CFTR-Expressing Ciliated
Epithelium in Vitro which are Useful as a Component of Cell Replacement Therapy for
Cystic Fibrosis in Vivo. (A) Confocal microscopy images showing immunostaining of
ALI-conditioned iPL cells, with nuclear stain DAPI (blue), E-cadherin (red) and CFTR
(green). (B) Reconstruction of X-Z projections of horizontal sections showing nuclear stain
DAPI (blue), the apical membrane staining of CFTR (green) and the lateral membrane
staining of E-cadherin (red). (C) Flow cytometry analysis of CFTR expression in freshly
isolated (day0) cells and ALI-conditioned cells. (D) Comparing fold-differences in CFTR
gene expression in adult lung, freshly isolated (day0) cells, pre-ALI, and ALI-conditioned
cells. (E) Iodide efflux assay showing CFTR activity in ALI-iPL cells (blue line) induced
by cyclic AMP agonist. Confocal microscopy images of (F) native B57/L6 airway control,
(G) native CFTR-KO airway epithelium, injured airway epithelium of CFTR-KO mice at
(H) 1 week and (I) 3 weeks post naphthalene treatment showing nuclear stain DAPI (blue)
and CFTR (grey). Confocal microscopy images of iPL cell-treated injured airway sections,
(J) 1 week and (K) 3 weeks post cell delivery, showing nuclear stain DAPI (blue). GFP
(green), ZO-1/E-cadherin (red) and CFTR (grey). (L) Western blot showing the presence of
CFTR protein band appearing at approximately 170 kDa representative of the complex
glycosylated functional form of CFTR in homogenized lung tissue from iPL cell-treated
CFTR-knockout injured mice. Expression levels of (M) CCSP and (N) CFTR in recipient
lungs, as measured by qRT-PCR comparing fold-differences in expression in wildtype
lungs. In C, data are representative of a minimum of three biological replicates. For D-E
and M-N, values are mean S.D. of three independent biological replicates. *, p<0.05; **,
p<0.001; ***, p<0.0001. Scale bar, 10µm (A, B and F-K).
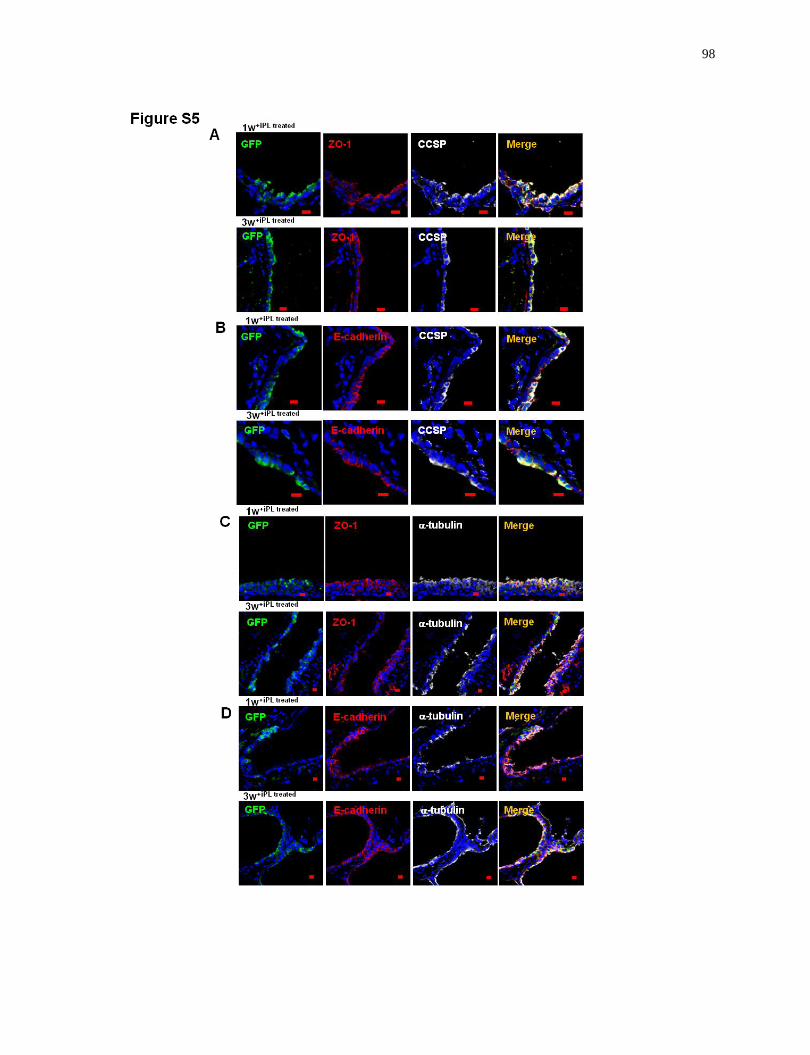
98

99
Figure S5. Confocal microscopy images of CFTR-deficient injured airway treated with
Club-iPL cells at 1 week (1w+iPL treated
, top panel) and 3 weeks (3w+iPL treated
, bottom panel).
(A, B) Immunohistochemistry showing nuclear stain DAPI (blue), GFP (green), ZO-1/E-
cadherin (red) and CCSP (grey). (C, D) Immunohistochemistry showing nuclear stain DAPI
(blue), GFP (green), ZO-1/E-cadherin (red) and α-tubulin (grey). (E) Expression levels of
Foxj1 in recipient lungs, as measured by qRT-PCR comparing fold-differences in expression
in wildtype lungs. (F) Cell retention rate of engrafted cells in recipient lungs (% of day0
injected cells), was calculated using genomic GFP expression at 1 week (1w+iPL treated
) and 3
weeks (3w+iPL treated
) (relative to β-actin GFP lungs) measured by qRT-PCR. (G) Whole body
micro-CT scans on mice injected with Club-iPL cells for 9 months showing no tumors. For
E and F, values are mean S.D. of three independent biological replicates *, p<0.05; **,
p<0.001; ***, p<0.0001. Scale bar, 10µm.
3.5.9 Cyclical Interrupted Reprogramming Enables Greater
Expansion of Club-iPL Cells
To evaluate the similarities of iPL cells to known endogenous progenitors, we
evaluated a number of putative lung progenitor cell markers. We found that P63, absent in

100
normal adult bronchiolar epithelium and in the EpCAMhigh
Club cell starting population
(Figure 7B), is highly expressed in iPL cells (Figure 7C, D). Unlike tracheal basal
progenitors (Tata et al., 2013) and H1N1 infection-induced progenitor pods (Kumar et al.,
2011), we found no expression of CK5 in iPL cells (Figure 7E). Nor was there any
expression of the alveolar progenitor marker-Sox9 (Figure 7F). After careful comparison
using available specific marker expression and microarray data for known lung progenitor
populations (Rock et al., 2009; Vaughan et al., 2014) (Figure 7G, H), Club-iPL cells do not
resemble any currently identified progenitors. However, the two fundamental properties of
endogenous progenitors, that is, proliferation and limited multipotency, could be
recapitulated. Moreover, the ability of progenitor cells to traverse to a transit amplifying
population can also be reproduced. In vitro, cyclical interrupted reprogramming using
intermittent Dox treatment in which iPL-derived Club cells (3w+Dox
+2w-Dox
) were re-induced
with Dox for 1 week (3w+Dox
+2w-Dox
+1w+Dox
; iPL2nd
) followed by an additional 2 week
culture in the absence of factors (3w+Dox
+2w-Dox
+1w+Dox
+2w-Dox
; iPL2nd
-Club cells), showed
that iPL-Club cells can respond to a 2nd
cycle (Figure 7I). Using this approach we were able
to achieve greater expansion in total cell (95±5 fold) and CFU number (Figure 7J). As with
the 1st generation iPL cells, 2
nd generation iPL cells expressed P63 and maintained their
ability to give rise to Club cells upon withdrawal of the inductive factors (Figure 7I, K).
Importantly, no Nanog expression was detected (data not shown).
We also evaluated the value of cyclical interrupted reprogramming in vivo. GFP+
iPL-derived Club cells (3w+Dox
+2w-Dox
) were delivered to naphthalene-busulfan (NAB)
conditioned BL6 mice transtracheally. Recipient mice were administrated with doxycycline
food (625µg/g) for 1 week (+cell 1w+Dox
) followed by an additional 1 week of food without
doxycycline (+cell 1w+Dox
+1w-Dox
) (Figure 7L). These iPL-derived Club cells (3w+Dox
+2w-
Dox), which are non-proliferative, do not engraft as well as iPL cells (3w
+Dox) themselves
(Figure 7M-1 week vs. Figure 5SF-1 week). More importantly, they are not retained (Figure
7M +cell 2w-Dox
). Consistent with in vitro results, iPL-Club cells are able to respond to re-
induction in that the number of retained cells significantly increased after in vivo Dox-
treatment (+cell 1w+Dox
). Subsequent withdrawal of Dox (+cell 1w+Dox
+1w-Dox
) resulted in a
decrease in the number of retained cells, suggesting factor-regulated controlled proliferation
(Figure 7M). The expression of transgene 4F2A was activated in vivo under Dox-treatment

101
and significantly down-regulated upon Dox-withdrawal (Figure 7N). The expression of
CCSP, markedly down-regulated as a result from the loss of mature Club cells in
naphthalene-busulfan-treated lungs, was only partially restored without in vivo induction of
iPL-derived Club cells (+cell 1w-Dox
) (Figure 7O), but, consistent with the increased cell
number (Figure 7M), Dox-treatment for 1 week increased CCSP expression. Strikingly,
CCSP expression was dramatically recovered in induced lungs upon the subsequent removal
of Dox (+cell 1w+Dox
+1w-Dox
). We speculated this is due to the re-differentiation of
expanded iPL cells back to CCSP-expressing Club cells (Figure 7O). Immnostaining result
confirmed the sufficient restoration of CCSP-expressing cells in recipient epithelium by iPL
cells after in vivo interrupted reprogramming (Figure 7P). Together, these results highlight
the potential of cyclical interrupted reprogramming for greater expansion of iPL cells and in
repopulating injured airway epithelium.
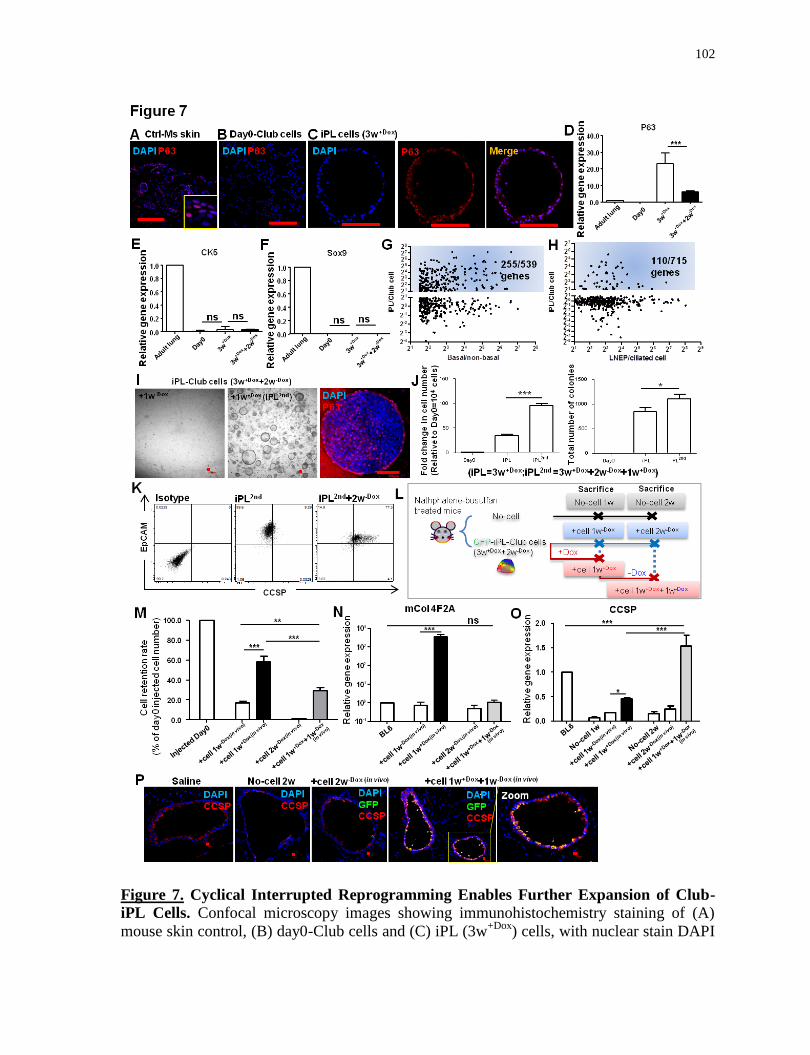
102
Figure 7. Cyclical Interrupted Reprogramming Enables Further Expansion of Club-
iPL Cells. Confocal microscopy images showing immunohistochemistry staining of (A)
mouse skin control, (B) day0-Club cells and (C) iPL (3w+Dox
) cells, with nuclear stain DAPI

103
(blue) and P63 (red). Fold-differences in P63 (D), CK5 (E) and Sox9 (F) gene expression in
adult lung, freshly isolated EpCAMhigh
cells (day0), iPL cells (3w+Dox
), and Club-iPL cells
cultured for 2 weeks in the absence of factors (3w+Dox
+2w-Dox
). (G) Comparison of Club-
iPL cells with basal cell progenitors. (H) Comparison of Club-iPL cells with lineage-
negative epithelial stem/progenitor (LNEP) cells. (I) Brightfield images showing the
generation of iPL2nd
(3w+Dox
+2w-Dox
+1w+Dox
) and stained with nuclear stain DAPI (blue),
P63 (red). (J) Fold changes in total cell number of iPL (3w+Dox
) and iPL2nd
(3w+Dox
+2w-
Dox+1w
+Dox) relative to day0 seeded cells (10,000 cells/well) and the total number of colony
forming units (CFU) generated. (K) Representative flow cytometry dot-plots showing
expression of EpCAM and CCSP in iPL2nd
cells and iPL2nd
cells cultured for 2 weeks in the
absence of factors (iPL2nd
+2w-Dox
). (L) Schematic depicting the methodology of in vivo
study. (M) Cell retention rate of engrafted cells in recipient lungs (% of day0 injected cells)
at 1 week and 2 weeks with or without in vivo induction measured by qRT-PCR. Expression
levels of 4F2A construct transgene (N) and CCSP (O) in recipient lungs, as measured by
qRT-PCR comparing fold-differences in expression in wildtype lungs. (P) Confocal
microscopy images of lung sections of the saline control, nathphalene-busulfan treated
nontransplanted (No-cell 2w) and transplanted (+cell 2w-Dox (in vivo)
, +cell 1w
+Dox+1w
-Dox (in
vivo)) showing nuclear stain DAPI (blue) and CCSP (red) and GFP (green). In D-F, J and M-
O, values are mean S.D. of three independent biological replicates. For K, data are
representative of a minimum of three biological replicates. *, p<0.05; **, p<0.001; ***,
p<0.0001. Scale bar, 100 µm (A-C and I), 10 µm (P).
3.6 Discussion
Carefully defined period of interrupted reprogramming enabled quiescent mature
Club cells to proliferate, start the reprogramming process but not pass the point of no-return
(Nagy and Nagy, 2010), thereby generating large numbers of iPL cells. Upon withdrawal of
the inductive factors, P63-expressing iPL cells give rise to CCSP+ Club cells, mucin
+ goblet
cells, and functional CFTR-expressing ciliated epithelium in vitro. They show in vivo utility
by repopulating CFTR-knockout epithelium after a recipient conditioning regimen. Our
results suggest that interrupted reprogramming is not only able to achieve controlled
expansion of the selected cell type, but results in the "de-differentiation" of the cells to a
progenitor-like state while preserving the differentiation potential of the parental population
to generate a limited range of functional progeny.
Recent advances in the understanding of the mechanisms involved in iPS cell
reprogramming have demonstrated that epigenetic memory” found both in human and
mouse iPS cells renders them permissive to preferential differentiation to the original lineage

104
(Ghosh et al., 2010; Kim et al., 2010; Bar-Nur et al., 2011; Shipony et al., 2014; De Los
Angeles et al., 2015). In our study, we harnessed this “residual epigenetic memory” of the
starting cells and generated large numbers of relatively pure progenitor-like populations with
intrinsic restriction to the appropriate lineage.
Despite recent progress in delineating a number of lung progenitor populations and
some of their associated markers (Chapman et al., 2011; Jain et al., 2015; Kim et al., 2005;
McQualter et al., 2010; Rawlins et al., 2009; Rock et al., 2009; Treutlein et al., 2014;
Vaughan et al., 2014), we are still limited in our grasp of the number and characteristics of
the lung stem cell hierarchy. After careful comparison using available specific marker
expression and microarray data for known lung progenitor populations (Rock et al., 2009;
Vaughan et al., 2014), Club-iPL cells do not resemble any currently identified progenitors.
It remains a possibility that they represent a yet unidentified progenitor cell population.
Alternatively, transient expression of OSKM in mature lung epithelial cells could result in
the generation of novel progenitor cells not existing in the developing or mature lung. That
is, bearing no clear relation to cells in the natural state but nonetheless capable of controlled
expansion and differentiation to a limited and appropriate range of progeny. Recent reports
highlight the existence of multiple unique cell states en route to pluripotency (Hussein et al.,
2014; Tonge et al., 2014; Zunder et al., 2015; Treutlein et al., 2016). Whether or not iPL
cells recapitulate a specific developmental stage, enhanced proliferation and restricted
differentiation makes them useful especially given this technology can theoretically be
applied to almost any cell type that can be isolated and purified.
Superior self-renewal and differentiation capacity of ES and iPS cells make both
very useful for regenerative medicine but protocols to generate large numbers of pure, fully
differentiated cells are still imperfect. Problems include low differentiation efficiency,
heterogeneous final products (Plath and Lowry, 2011; Schwartz et al., 2014) and
contamination by potentially tumorigenic undifferentiated cells (Ben-David and Benvenisty,
2011; Tapia and Schöler, 2016). A variety of approaches may overcome these problems but
producing each unique cell that may be desirable will require the development of hundreds
of unique protocols. Direct reprogramming through ectopic expression of combinations of
specific transcription factors (Caiazzo et al., 2011; Pfisterer et al., 2011; Sancho-Martinez et
al., 2012) and/or microRNA (Ambasudhan et al., 2011; Leonardo et al., 2012), resulting in

105
direct conversion from one cell type to another, has also been extensively studied. Direct
conversion does not require a pluripotent intermediate state and thus may raise fewer safety
concerns. Our novel interrupted reprogramming strategy similarly avoids pluripotency but
may be easier to apply to a broad range of cells types, only requiring that they can be
isolated in relative purity.
Recent studies showed in vitro differentiation of iPS and ES cells to lung epithelium,
but were not able to generate large numbers of either Club cells or ciliated cells. Importantly
none of these groups evaluated the in vivo contribution of resultant CFTR-expressing cells in
an animal model. On the contrary, our iPL cells hold great promise for treating respiratory
diseases. Our proof of concept in vivo studies showed successful retention and incorporation
in CFTR-deficient epithelium with injected iPL cells being able to give rise to both Club
cells and ciliated cells.
The novelty of this concept results in a number of key issues that need to be
addressed. Firstly, since iPL cells are never reprogrammed to pluripotency, they are likely
less tumorigenic. However, the sensitivity of our current methodology is not absolute.
Secondly, although we have selected a highly purified population of cells as the starting
material, interrupted reprogramming still produces clones that are variable in size suggesting
some degree of heterogeneity in the system. While proliferation during iPL induction seems
relatively uniform (Figure 1H, middle panel) and the observed return to phenotype upon dox
withdrawal (Figure 2I) is also homogeneous, potential pluripotency (as suggested by Nanog
expression) is more heterogeneous, varying between colonies and amongst cells within an
individual colony after a prolonged induction period (> 5 weeks). This will need further
exploration but is not surprising as evidence suggests higher levels of heterogeneity in the
early phases of iPS induction (Lujan et al., 2015; Treutlein et al., 2016; Zunder et al., 2015).
There is also some heterogeneity in the differentiation capacity of the Club cells after return
from the iPL state although this can be attributed to the normal biology of Club cells
(Rawlins et al., 2009; Reynolds et al., 2000). Finally, for therapeutic use, the current data
was obtained from Col1a1-4F2A mice and it remains to be shown that this technique can be
efficiently applied in human cells. In preliminary studies, we isolated normal human lung
epithelial cells containing Club cells from excess tissue remaining from lung transplant
donors. Cells were transfected with a polycistronic lentiviral vectors driving dox-regulatable

106
OSKM. Although the induction conditions require further optimization, dox-induction
resulted in increased cell numbers compared to that of non-treated native epithelial cells
(Figure S6A). Importantly, consistent with our observation in mouse system, dox-treated
cells lost their somatic CCSP gene expression, but regained it following dox-withdrawal
suggestive of ability to return to their original phenotype (Figure S6B). The iPL cell
induction process will need to be optimized to obtain maximum expansion and scale-up of
cells, but should theoretically benefit from the advances driving iPS cell research towards
non-integrative, non-viral methods of reprogramming. Our studies showed intermittent
Dox-treatment cycles resulted in second generation iPL cells. The number of generations
resulting in maximal expansion of cells without loss of iPL function remains to be
determined. Further refinement is needed to widen the relatively narrow time window
where iPL cell induction and expansion is maximized but before potential factor-
independent pluripotency is possible. This may require optimization of exogenous growth
factors and/or changes to culture conditions. In addition, the precise requirement for each
individual transcription factor remains unknown. Singovski and colleagues evaluated the
effect of transient activation of OSKM, OSK and M alone (Singovski et al., 2016). While c-
Myc was important for driving proliferation, only the full 4 factors resulted in acquisition of
a robust ‘cancer stem cell” phenotype, including increase in colony forming efficiency.
Whether this observation is analogous to our non-malignant “progenitor-like” population
remains to be studied. Theoretically, our iPL concept is broadly applicable and could be
extended to other somatic cell types giving rise to numerous progenitor cell populations.
Future investigations will be needed but these proof-of-concept in vivo studies showed iPL
cells hold great promise for treating respiratory diseases by true engraftment as “induced”
progenitors.
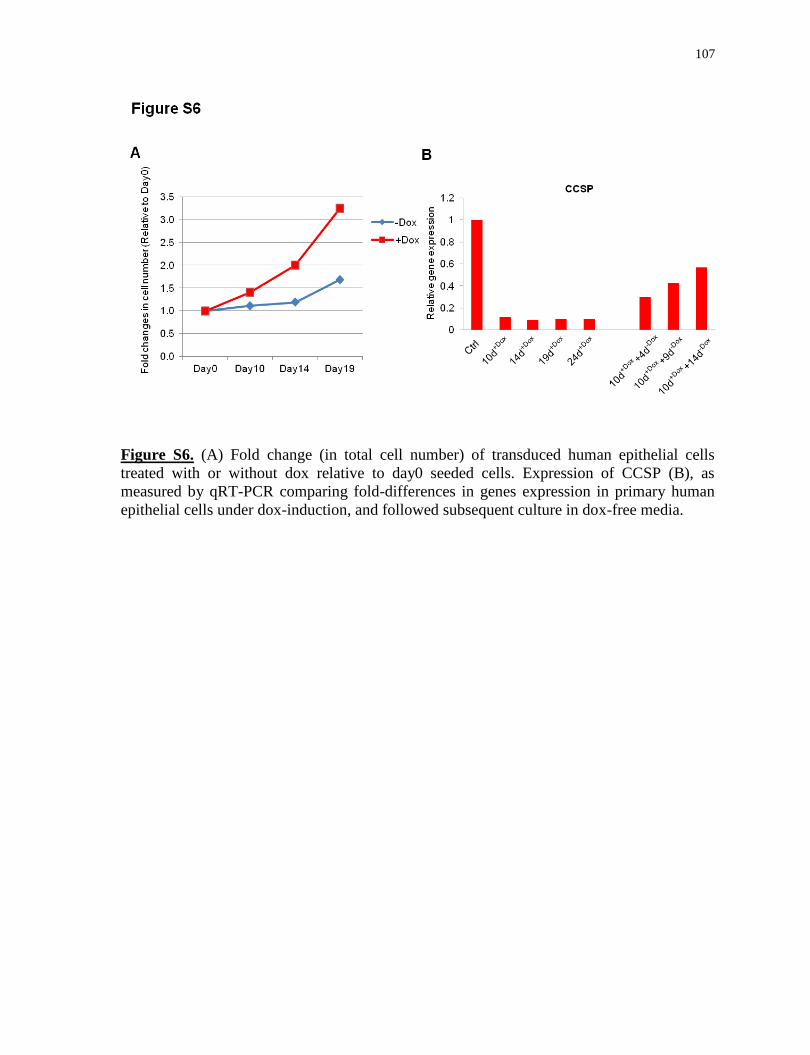
107
Figure S6. (A) Fold change (in total cell number) of transduced human epithelial cells
treated with or without dox relative to day0 seeded cells. Expression of CCSP (B), as
measured by qRT-PCR comparing fold-differences in genes expression in primary human
epithelial cells under dox-induction, and followed subsequent culture in dox-free media.

108
Chapter 4
Interrupted reprogramming of Alveolar Type II cells induces
progenitor-like cells that ameliorate pulmonary fibrosis
4.1 Rationale, Objectives, Hypotheses and Specific Aims
Rationale:
Carefully timed interrupted reprogramming allowed controlled expansion of mature
Club cells yet preservation of bronchiolar epithelial lineage commitment.
SftPC+ AEC-II cells serve as progenitor cells in the alveoli, but their number and
function decrease or deplete in certain pathological conditions and in age-related
decline;
Although AEC-II cells can give rise to alveolar-like colonies in vitro, they possess
limited passaging capacity
Objective:
To evaluate the potential of AEC-II derived iPL cells to ameliorate bleomycin-
induced pulmonary fibrosis
Hypothesis:
Interrupted reprogramming can rescue the in vitro limited clonogenic capacity of AEC-II
cells
Specific Aims:
To isolate and purify AEC-II cells from mouse lungs;
To optimize the iPL induction conditions of AEC-II cells;
To define the length of iPL induction of AEC-II cells which allows expansion yet
preservation of AEC-II-lineage commitment prior to reaching pluripotency;

109
To characterize AEC-II derived iPL cells;
To determine if AEC-II-iPL cells engraft, survive and repopulate bleomycin-induced
injured alveolar epithelium in vivo
4.2 Abstract
We describe here an interrupted reprogramming strategy to generate "induced
Progenitor-Like (iPL) cells" from Alveolar Epithelial Type II (AEC-II) cells. A carefully
defined period of transient expression of reprogramming factors (Oct4, Sox2, Klf4 and c-
Myc; OSKM) is able to rescue the limited in vitro clonogenic capacity of AEC-II cells,
potentially by activation of a bipotential progenitor-like state. Importantly, our results
demonstrate that interupted reprogramming results in controlled expansion of cell numbers
yet preservation of the differentiation pathway to the alveolar epithelial lineage. When
transplanted to injured lungs, AEC-II-iPL cells are retained in the lung and ameliorate
bleoomycin-induced pulmonary fibrosis. Interrupted reprogramming can be used as an
alternative approach to produce highly specified functional therapeutic cell populations and
may lead to significant advances in regenerative medicine.
4.3 Introduction
In normal adult lung, the alveolar epithelium is composed of two major cell types,
the alveolar epithelial type I (AEC-I) and alveolar epithelial type II (AEC-II) cells. It is
generally thought that the squamous AEC-I cells are terminally differentiated cells that
interface with pulmonary capillaries and cover more than 90% of the alveolar surface where
the exchange of CO2/O2 takes place(Whitsett et al., 2010). In contrast, AEC-II cells are
small cuboidal cells located in the corners of alveoli which cover only 5% of the alveolar
surface. They are multifunctional cells that produce, secrete and recycle pulmonary
surfactants, regulate alveolar fluid balance, and synthesize and secrete a number of immune-
modulatory proteins involved in host defense(Fehrenbach, 2001). Importantly, a subset of
surfactant protein C-positive (SPC+) AEC-II cells serve as regional progenitors in the alveoli
and differentiate into AEC-I cells, playing a crucial role in replenishing the alveolar
epithelial barrier during both homeostasis and repair after injury(Barkauskas et al., 2013a;
Rock and Hogan, 2011; Whitsett and Alenghat, 2014). Impaired regeneration of injured

110
alveolar epithelium has been observed in fibrotic interstitial lung diseases, including
idiopathic pulmonary fibrosis (IPF). IPF is an irreversible, fatal interstitial lung disease with
death occurring in most patients within 5 years of diagnosis. While not completely
understood, increasing evidence suggests that the pathogenesis of IPF may be driven by
alveolar epithelial cell dysfunction, followed by aberrant regeneration of epithelium,
persistent activation of fibroblasts and alterations in epithelial-mesenchymal communication
with the extracellular matrix (ECM), together resulting in disruption of architecture and
progressive loss of lung function(King et al., 2011; Yanagi et al., 2015; Zoz et al., 2011).
Currently, medical therapy for IPF is limited and lung transplantation is the only option for
patients with end-stage IPF(Akram et al., 2014; Lomas et al., 2012).
A growing body of evidence describes putative progenitor cell populations in the
distal lung that function to replenish or repair damaged epithelium(Barkauskas et al., 2013b;
Chapman et al., 2011; Jain et al., 2015; Kim et al., 2005; McQualter et al., 2010a; Teisanu et
al., 2011; Vaughan et al., 2014). However, these cells are rare, which limits their expansion,
and they usually change rapidly upon in vitro culture(Bertoncello and McQualter, 2010;
Kosmider et al., 2011; Liebler et al., 2016; Raiser and Kim, 2009). Importantly, in several
disease or injury states, endogenous progenitors are limited in number and function(Randell,
2006). Thus, recent focus has been placed on using cell-based therapeutic approaches for
ameliorating fibrosis via a cell replacement strategy. Tremendous efforts have been made in
application of bone marrow (BMC) cells(Ghadiri et al., 2016; Kotton et al., 2005; Weiss,
2014), mesenchymal stromal (MSC) cells(Cargnoni et al., 2009; Ortiz et al., 2003; Tashiro
et al., 2015; Toonkel et al., 2013) and respiratory epithelial cells differentiated from
pluripotent sources such as embryonic stem (ESC) and induced pluripotent stem (iPS)
cells(Ghaedi et al., 2013; Gotoh et al., 2014; Huang et al., 2013; Zhou et al., 2014).
Amongst these, MSCs have advantages as a practical source for use in cell-based therapies
for lung disease. The vast majority of studies report some biological effects after MSC
delivery during the early inflammatory phase of bleomycin-induced pulmonary fibrosis.
However, low levels of cell engraftment or retention suggest a paracrine-based mechanisms
of action responsible for repair(Kumamoto et al., 2009; Lee et al., 2012; Ortiz et al., 2003).
In contrast, freshly isolated AEC-II cells appear to be effective even after administration in
later stages of IPF where fibrosis is prevalent(Serrano-Mollar et al., 2007),(Serrano-Mollar

111
et al., 2016). However, the practical usages of freshly isolated AEC-IIs are limited by donor
availability and maintenance in culture(Kosmider et al., 2011; Liebler et al., 2016). Despite
recent progress in obtaining distal epithelial cells from directed differentiation of ESC and
iPS cells(Ghaedi et al., 2013; Gotoh et al., 2014; Huang et al., 2013; Zhou et al., 2014),
protocols remain limited by yield and purity of AEC-II cells. Furthermore, the pluripotent
nature of ESC and iPS cells, still present a potential risk of tumorigenicity which must be
addressed for clinical applicability(Kimbrel and Lanza, 2015; Shi et al., 2016). Regardless of
cell source, for most cell therapy applications, the cells will need externally controllable
proliferative capacity to maintain homeostasis or respond to injury.
We present a novel interrupted reprogramming strategy which provides an
alternative approach to generate a functional AEC-II population with high purity. We took
advantage of the rapid induction of cell proliferation and residual epigenetic “memory”
retained during the early phase of reprogramming(Clancy et al., 2014; Hussein et al., 2014;
Lujan et al., 2015; Samavarchi-Tehrani et al., 2010; Shipony et al., 2014; Tonge et al., 2014;
Woltjen et al., 2009; Zunder et al., 2015) to create cells we have termed "induced
Progenitor-Like (iPL) cells". We achieved this by optimizing and carefully controlling the
duration of transient expression of induced Pluripotent Stem (iPS) cell reprogramming
factors (Oct4, Sox2, Klf4 and c-Myc; OSKM), turning off their expression prior to reaching
independent pluripotency. Interrupted reprogramming allows controlled expansion yet
preservation of AEC-II lineage commitment and rescues the limited in vitro clonogenic
capacity of AEC-II cells. Importantly, iPL cells derived from AEC-II cells ameliorate
bleomycin-induced pulmonary fibrosis in vivo. The ability to produce highly specified
populations, retaining critical functions may have significant implications for cell-based
therapies.
4.4 Materials and Methods
4.4.1 Animal husbandry
ROSA26-rtTA and Col1a1: tetO-4F2A mice (Jackson Labs, Cat#011004) and R26-
rtTA/Col1a1::tetO-4F2A;Oct4-GFP mice ( A gift from Dr. Andras Nagy) were used to
generate inducible lung epithelial cells. For in vivo studies, ROSA26-rtTA/Col1a1:: tetO-
4F2A double transgenic mice were bred to actin GFP mice. There were no significant

112
differences between heterozygotes and homozygotes with respect to inductive factor gene
expression in Dox-treated AEC-II cells. Adult (6 to 8-week-old) female C57BL/6 mice were
used for AEC-II-iPL cell replacement therapy studies. Animals were maintained as an in-
house breeding colony under specific pathogen–free conditions. All procedures involving
animals were approved by the Institutional Animal Care and Use Committee of the
University Health Network (Toronto, Ontario, Canada).
4.4.2 Bleomycin administration and cell delivery
For BLM injury, BLM 2.5 U kg-1
body weight was administered intratracheally.
Donor AEC-II-iPL cells or MSCs (106 cells in 50l PBS) were delivered intratracheally 7
days or 14 days after injury. Control animals received the same volume of PBS without any
cells. The mice receiving cells were rotated to ensure equal dispersion of the cell suspension
to both lungs.
4.4.3 Measurement of respiratory mechanics
C57BL/6 male mice (4-5 month old) were anesthetized with ketamine (125 mg/kg,
ip) and xylazine (5 mg/kg, ip) and paralyzed with rocuronium (5 mg/kg, ip). Animals were
tracheostomized with a blunt 18G metal cannula, and supplementary anesthesia was
employed when necessary. Conventional mechanical ventilation was maintained with a
small animal ventilator (FlexiVent, SCIREQ Inc., Canada) using a tidal volume of 10
mL/kg, a frequency of 150 breaths per minute, and a positive end-expiratory pressure
(PEEP) set at 3 cmH2O. PV loops were obtained with a quasi-static pressure-controlled PV
ramp.
4.4.4 Isolation of AEC-II cell from mouse lung
AEC-II cells were isolated using a previously described protocol(Berndt-Weis et al.,
2009) with modifications. Before lung digestion, mice were injected intra-peritoneally with
heparin (250U/mouse) and euthanized by CO2 narcosis. Lungs were flushed through the
right ventricle with cold phosphate buffered saline (PBS). Endobronchial lavage was then
performed to remove alveolar leukocytes. Mouse lung was filled with porcine elastase 10 U

113
/mL and incubated for 20 min at 37C. Trachea and bronchi were cut away. The remaining
tissues were finely minced and incubated in 100μg/mL of DNAse I in DMEM containing
antibiotics for 10 minutes. The suspension was mixed with FBS and sieved through 100 µm,
40 µm and 20 µm nylon meshes. Cells were centrifuged at 32g for 12 min then re-suspended
in red blood cell lysis buffer for 3min and the lysis was stopped by addition of an equal
volume of PBS. Cells were centrifuged at 100g for 12 min then re-suspended in 0.5 %
vol/vol FBS-PBS for all subsequent procedures.
4.4.5 Cell culture
Epithelial-specific medium comprised of DMEM/F12 (Invitrogen) supplemented
with 10% FBS, penicillin/streptomycin, 10mg/mL insulin, 5mg/mL transferrin-selenium
(Sigma), epidermal growth factor (EGF, 20ng/mL; Sigma), fibroblast growth factor-10
(FGF-10, 50ng/mL; R&D Systems) and hepatocyte growth factor (HGF, 30ng/mL; R&D
Systems).
4.4.6 Fluorescence activated cell sorting and analysis
For purification of epithelial cells, fresh isolated cells were suspended and incubated
in 0.5 % vol/vol FBS-PBS containing an optimally pre-titered mixture of antibodies [anti-
CD45, anti-CD31 (BD Biosciences), anti-EpCAM (Abcam) and relevant isotype controls]
for approximately 30min on ice. Labeled cells were washed and re-suspended at 3~5 × 106
cells/mL in 0.5 % vol/vol FBS-PBS. Cell viability was accessed by propidium iodide
(1μg/mL) staining. For intra-cellular antigen analysis, cells were fixed and stained using a
Fix and Perm kit (Invitrogen) as per manufacturer instructions. Sorting was performed using
a MoFlo BRU cell sorter (Becton Dickinson), acquisition was performed using a BD LSRII
analyzer (Becton Dickinson) and data was analyzed using FlowJo software.
4.4.7 Matrigel-based iPL cell induction
Feeders (MEF) were seeded on 0.1% gelatin coated 24-well transwell filter inserts
(Corning) one day prior to the addition of epithelial cells. Sorted epithelial cells resuspended
in 100μL of Matrigel (BD Biosciences) prediluted 1:1 (vol/vol) with epithelial-specific

114
(EpiS) media were added to a MEF-coated 24-well transwell filter inserts in a 24-well tissue
culture plate containing 500μL of epithelial media. To study the clonogenic capacity of
passaged AEC-II cells, AEC-IIs were cultured in EpiS media for 2 weeks, passaged then
exposed to Dox. Media was replenished three times per week. For bulk passaging, whole
cultures were dissociated in Collagenase (1mg/mL; Sigma) Dispase (3mg/mL; BD
Biosciences) in PBS to generate a single-cell suspension. For clonal passaging, single
colonies were picked and dissociated.
4.4.8 Immunofluorescence
Samples were fixed with 4% paraformaldehyde (PFA) for 30min and blocked with
5% goat serum and 2% BSA in PBS containing 0.5% Triton X-100 for 1 hour. Primary
antibodies (Table 1) were diluted in BSA/PBS, applied to samples and incubated overnight
at 4°C. Secondary antibodies AlexaFluors 488, 532, 546, 633 or 647 (Invitrogen) were
applied according to the species in which the primary antibody was used for 2 hours at room
temperature. Nuclear staining was performed using 2mg/mL 4, 6, diamidino-2-phenylindole
(DAPI; Sigma). Stained samples were mounted with immunofluorescent mounting medium
(DAKO). Appropriate non-specific IgG isotypes were used as controls. Immunoreactivities
of antigens were visualized as single optical planes using an Olympus Fluoview confocal
microscope and analyzed using FV10-ASW 2.0 Viewer software.
Antibody name Company Cat. Num
CD45 BD phamingen 553081
CD31 BD phamingen 553373
EpCAM Abcam ab95641
SPC Chemicon AB3786
Santa Cruz sc-7706
Hopx Santa Cruz sc-398703

115
a-SMA SIGMA-Aldrich A2547
Integrin α6 R&D FAB13501G
Integrin 4 Abcam ab182120
Biolegend 123608
CD74 BD phamingen 561941
LAMP3 Novusbio DDX0192P-100
Nanog
Abcam ab80892
BD phamingen 560259
GFP Invitrogen A-21311
AQP5 Abcam ab104751
SSEA-1 Santa Cruz sc-21702
E-cadherin Abcam ab11512
Table 1: Tabulated list of all antibodies used in the studies
4.4.9 Real-time PCR analysis
Total RNA was prepared using the RNeasy Kit (Qiagen) as per manufacturer’s
instructions. cDNA was prepared and assayed using Superscript III (Sigma) according to
manufacturer’s protocol. Differential gene expression was determined using SYBR green
detection (Roche). Real-time PCR reactions were done in triplicate for each sample.
GAPDH was used as a housekeeping gene to normalize gene expression levels using
LightCycler 480 software (Roche). Normalized mRNA levels were shown as relative to the
control samples (day 0 fresh isolated cells or adult lung) (Table 2).
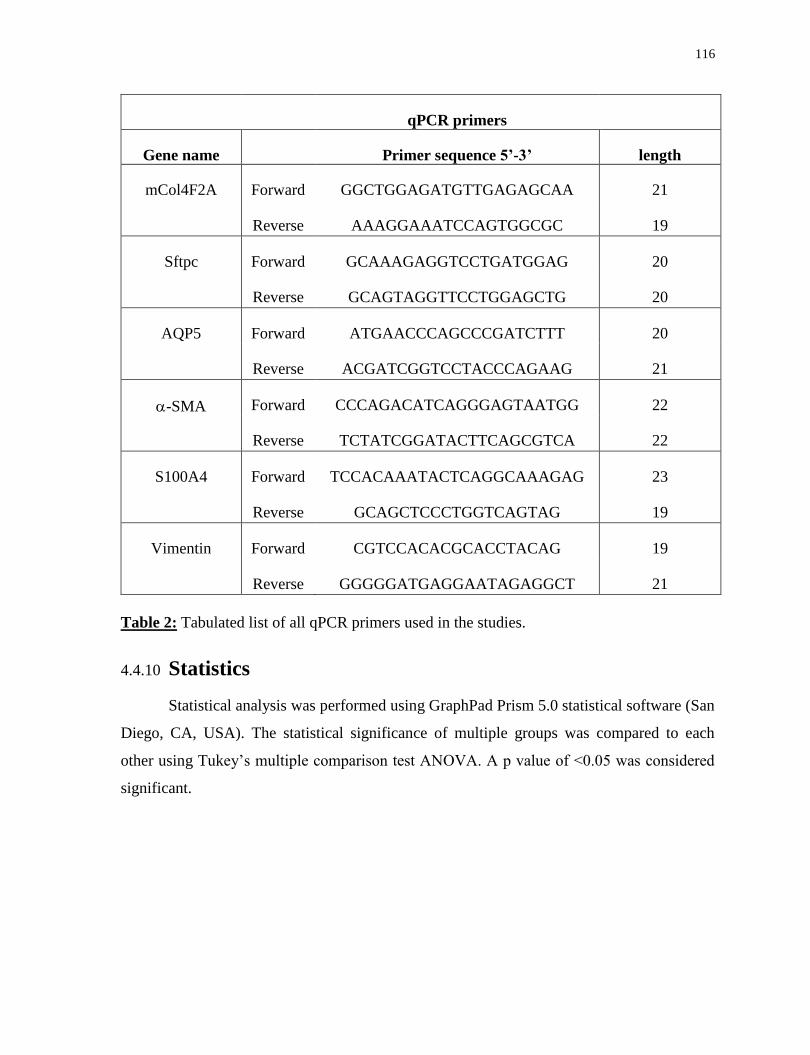
116
qPCR primers
Gene name Primer sequence 5’-3’ length
mCol4F2A Forward GGCTGGAGATGTTGAGAGCAA 21
Reverse AAAGGAAATCCAGTGGCGC 19
Sftpc Forward GCAAAGAGGTCCTGATGGAG 20
Reverse GCAGTAGGTTCCTGGAGCTG 20
AQP5 Forward ATGAACCCAGCCCGATCTTT 20
Reverse ACGATCGGTCCTACCCAGAAG 21
-SMA Forward CCCAGACATCAGGGAGTAATGG 22
Reverse TCTATCGGATACTTCAGCGTCA 22
S100A4 Forward TCCACAAATACTCAGGCAAAGAG 23
Reverse GCAGCTCCCTGGTCAGTAG 19
Vimentin Forward CGTCCACACGCACCTACAG 19
Reverse GGGGGATGAGGAATAGAGGCT 21
Table 2: Tabulated list of all qPCR primers used in the studies.
4.4.10 Statistics
Statistical analysis was performed using GraphPad Prism 5.0 statistical software (San
Diego, CA, USA). The statistical significance of multiple groups was compared to each
other using Tukey’s multiple comparison test ANOVA. A p value of <0.05 was considered
significant.

117
4.5 Results
4.5.1 Interrupted reprogramming rescues the limited clonogenic
capacity of AEC-II cells while achieving expansion
AEC-II cells play a central role in alveolar epithelial repair and regeneration. In vitro,
although AEC-IIs can give rise to alveolar-like colonies, they possess limited clonogenic
capacity which decreases with passaging(Lee et al., 2014a). Importantly, their number and
function decline with age and in certain pathological conditions(Chen et al., 2015; Navarro
and Driscoll, 2017), including IPF(Liang et al., 2016). To determine if interrupted
reprogramming is able to rescue the in vitro limited clonogenic capacity of AEC-II cells, we
isolated AEC-IIs from R26-rtTA/Col1a1::tetO-4F2A double transgenic mice(Carey et al.,
2010) enabling expression of Oct4, Sox2, Klf4 and c-Myc (OSKM) following treatment
with doxycycline (Dox). AEC-II cells were isolated using a modified elastase-based protocol
and characterized using anti-CD74(Marsh et al., 2009) in addition to the classical AEC-II
marker anti-SPC. Flow-cytometric analysis of freshly isolated cells showed that over 95% of
CD45neg
CD31neg
EpCAMpos
AEC-II cells expressed both markers (Figure 1a, b). Isolated
AEC-II cells gave rise to colonies when cultured in a Matrigel-based 3D culture system
(Figure 1c). Consistent with previous reports(Lee et al., 2014a), there was a gradual
reduction in colony forming-efficiency (CFU%) with passaging (Figure 1d-ND groups). We
optimized iPL induction conditions for AEC-II cells and found an initial two weeks of
culture without Dox (ND) prior to Dox-treatment (+D) (named "late induction") appeared
advantageous. Induction of AEC-II cells (2WND
+2W+D
) significantly increased the CFU%
and total number of cells (Figure 1d-f). Subsequent withdrawal of Dox for 2 weeks
(2WND
+2W+D
+2W-D
) resulted in a decrease in transgene 4F2A expression (Figure 1g), a
decrease in CFU%, while the colonies that did develop were larger and numerous adherent
epithelial-like cells were seen (Figure 1d, e). Overall, this resulted in a 100-fold cell
expansion (Figure 1f). Non-treated AEC-II cells gradually lost their phenotype with
passaging, possibly due to the differentiation to AEC-I-like cells. In contrast, the AEC-II
phenotype was well preserved in the 2WND
+2W+D
induced cells, even after withdrawal of
Dox for 2 weeks (2WND
+2W+D
+2W-D
) (Figure 1d, h). This data showed that interrupted

118
reprogramming of AEC-II is able to rescue the limited passaging capacity of AEC-II
colonies and efficiently expand cells while preserving AEC-II lineage commitment.

119
Figure 1. Interrupted reprogramming rescues the in vitro limited clonogenic capacity
of AEC-II cells while achieving expansion in cell numbers (a) Flow cytometry analysis of
freshly isolated lung cells using a modified elastase-based protocol, showing epithelial cells
marked with antibodies specific for SPC and CD74. Over 95% of EpCAM+, CD45
-, CD31
-
cells are AEC-II cells co-expressing SPC and CD74. (b) Freshly isolated AEC-II cells
express EpCAM (red), CD74 (green) and SPC (grey); nuclei are stained with DAPI (blue).
(c) Isolated AEC-II cells are able to give rise to colonies in a Matrigel-based 3D culture
system. Light microscopy bright-field image and H&E staining are shown. (d) The left panel
shows bright-field images depicting the generation of colonies in the absence (ND), presence
of Dox (+D), and upon subsequent Dox-withdrawal (-D). The right panel shows confocal
microscopy images of colonies obtained from each condition, showing nuclear stain DAPI
(blue), SPC (green) and EpCAM (red). The colony forming efficiency (e) and the fold
changes in total cell number (f) showed relative to day0 seeded cells (5,000 cells/ well).
Expression levels of (g) mCol4F2A and (h) SPC in cells obtained from each condition, as
measured by qRT-PCR comparing fold-differences in expression in day0 freshly isolated
AEC-II cells. In a, data are representative of a minimum of three biological replicates. For e-
h, values are mean S.D. of three independent biological replicates. *, p<0.05; **, p<0.001;
***, p<0.0001. Scale bar, 10 µm (b and d); 50 µm (c-H&E); 100 µm (c-bright field and d).
4.5.2 Interrupted reprogramming allows preservation of AEC-II
lineage commitment without traversing the pluripotent
state
To evaluate the extent of reprogramming of the cells, we cultured AEC-II cells
derived from R26-rtTA/Col1a1::tetO-4F2A;Oct4-GFP mice in which the activation of
endogenous Oct4 could be monitored by GFP expression. AEC-II cells treated with Dox for
3 weeks resulted in the presence of Dox-independent Oct4-GFP cells expressing the
pluripotency markers; Nanog and SSEA-1 (Figure 2b, c). In contrast, AEC-II cells cultured
in the presence of Dox for only 2 weeks (2WND
+2W+D
) cells did not contain any Oct4-GFP+
cells and showed no expression of other pluripotency markers (Figure 2a). We assessed the
ability of induced AEC-II cells to return to their original AEC-Il phenotype following
withdrawal of Dox. Withdrawal of Dox after a 2-week induction (2WND
+2W+D
+2W-D
)
resulted in Oct4-GFP-SPC
+ colonies showing the return to the AEC-II phenotype (Figure
2d). In contrast, colonies obtained upon Dox-withdrawal after 3 weeks induction
(2WND
+3W+D
+2W-D
, 2WND
+4W+D
+2W-D
) contained Oct4-GFP cells and fewer SPC+ cells
suggesting that subsets of 3 week induced cells had already passed the point-of-no return
(Figure 2e, f). Taken together, our results showed that, with our defined length of interrupted
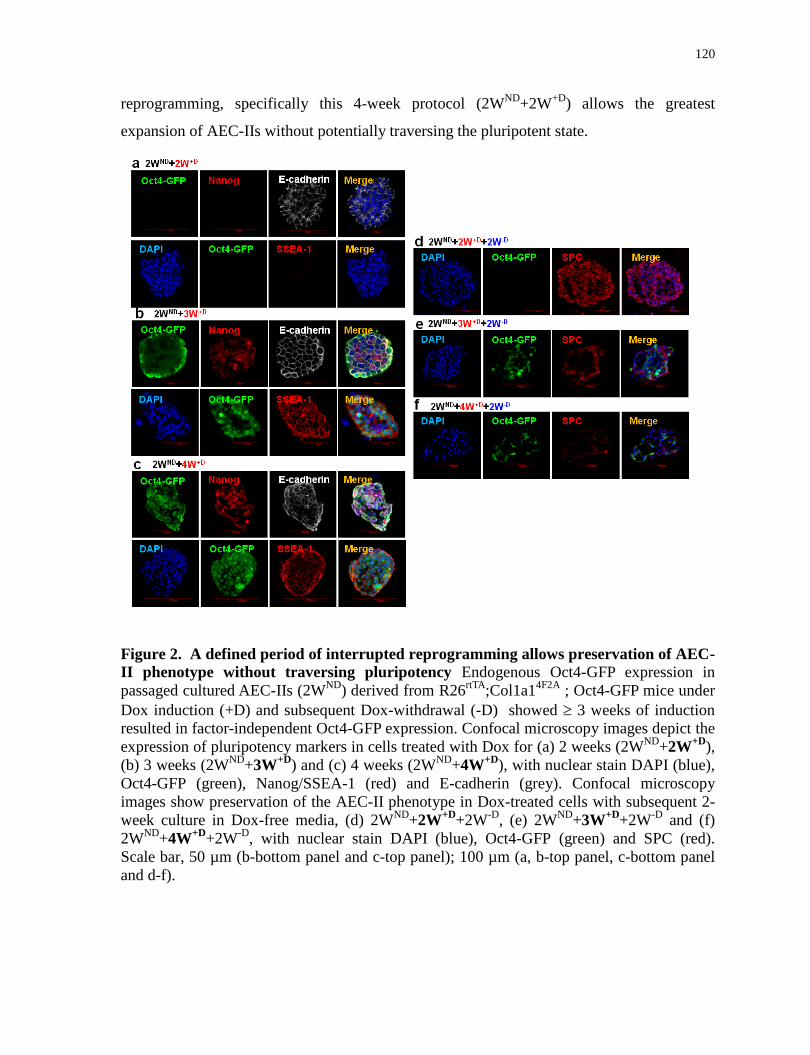
120
reprogramming, specifically this 4-week protocol (2WND
+2W+D
) allows the greatest
expansion of AEC-IIs without potentially traversing the pluripotent state.
Figure 2. A defined period of interrupted reprogramming allows preservation of AEC-
II phenotype without traversing pluripotency Endogenous Oct4-GFP expression in
passaged cultured AEC-IIs (2WND
) derived from R26rtTA
;Col1a14F2A
; Oct4-GFP mice under
Dox induction (+D) and subsequent Dox-withdrawal (-D) showed 3 weeks of induction
resulted in factor-independent Oct4-GFP expression. Confocal microscopy images depict the
expression of pluripotency markers in cells treated with Dox for (a) 2 weeks (2WND
+2W+D
),
(b) 3 weeks (2WND
+3W+D
) and (c) 4 weeks (2WND
+4W+D
), with nuclear stain DAPI (blue),
Oct4-GFP (green), Nanog/SSEA-1 (red) and E-cadherin (grey). Confocal microscopy
images show preservation of the AEC-II phenotype in Dox-treated cells with subsequent 2-
week culture in Dox-free media, (d) 2WND
+2W+D
+2W-D
, (e) 2WND
+3W+D
+2W-D
and (f)
2WND
+4W+D
+2W-D
, with nuclear stain DAPI (blue), Oct4-GFP (green) and SPC (red).
Scale bar, 50 µm (b-bottom panel and c-top panel); 100 µm (a, b-top panel, c-bottom panel
and d-f).

121
4.5.3 Interrupted reprogramming induces expression of alveolar
progenitor markers Hopx and α6β4
Lineage tracing studies have demonstrated that both AEC-I and AEC-II cells arise
from a bipotent progenitor cell during lung development; whereas AEC-I cells derive from
subsets of AEC-IIs after birth and in adult lung. Hopx (homeodomain only protein x), first
expressed in the embryonic lung at day 15.5 marks a bipotent alveolar progenitor in
developing lung (Treutlein et al., 2014). Hopx is turned off in maturing AEC-II cells, and
becomes restricted to AEC-I cells in the mature lung (Figure 3a). We observed that alveolar-
like colonies, generated by freshly isolated AEC-II cells, cultured in Matrigel conditions for
2 weeks without Dox (2WND
), contained SPC+ AEC-II cells as well as Hopx
+ AEC-I cells.
Notably, no co-expression of SPC and Hopx was observed (Figure 3b). In contrast, the
majority of AEC-II cells after interrupted reprogramming expressed SPC with a large
proportion (85.5±4.0% of SPC+ cells) co-expressing Hopx (Fig 3d, f) with a pattern similar
to that seen in E15.5 lungs (Figure 3c, S1). Withdrawal of Dox (2WND
+2W+D
+2W-D
)
resulted in a reduction in the number of SPC+/Hopx
+ cells (Figure 3e, f). While integrin α6 is
ubiquitously expressed in lung epithelium(Lee et al., 2014b), coexpression of integrin α6β4
marks a rare progenitor sub-population within normal distal lung involved in maintenance of
AEC-IIs during lung repair(Chapman et al., 2011; McQualter et al., 2010b). Flow cytometry
showed 4 to be absent in the native α6+ AEC-II starting population but expressed in over
60% of AEC-II cells after interrupted reprogramming (Figure 3g). Immunostaining
confirmed β4 expression (Figure 3h, i) and showed that β4 was co-expressed with Hopx in
these cells, similar to that seen in distal epithelium of E15.5 lungs (Figure 3j, k).
Collectively, our results suggest that interrupted reprogramming rescues the limited
clonogenic capacity of AEC-II cells in vitro as a result of activation of an alveolar progenitor
state. We have therefore termed these cells, induced Progenitor-Like cells (AEC-II-iPL
cells) (Figure 3l).

122
Figure 3. The expression of alveolar progenitor markers in AEC-II-iPL cells Confocal
microscopy images depict the expression of Hopx and SPC in: (a) adult lung; (b) colonies
derived from AEC-II cells after a 2-week culture period in the absence of Dox (2WND
); (c)
E15.5 lungs; (d) passaged 2WND
colonies induced with Dox for 2 weeks (2WND
+2W+D
); (e)
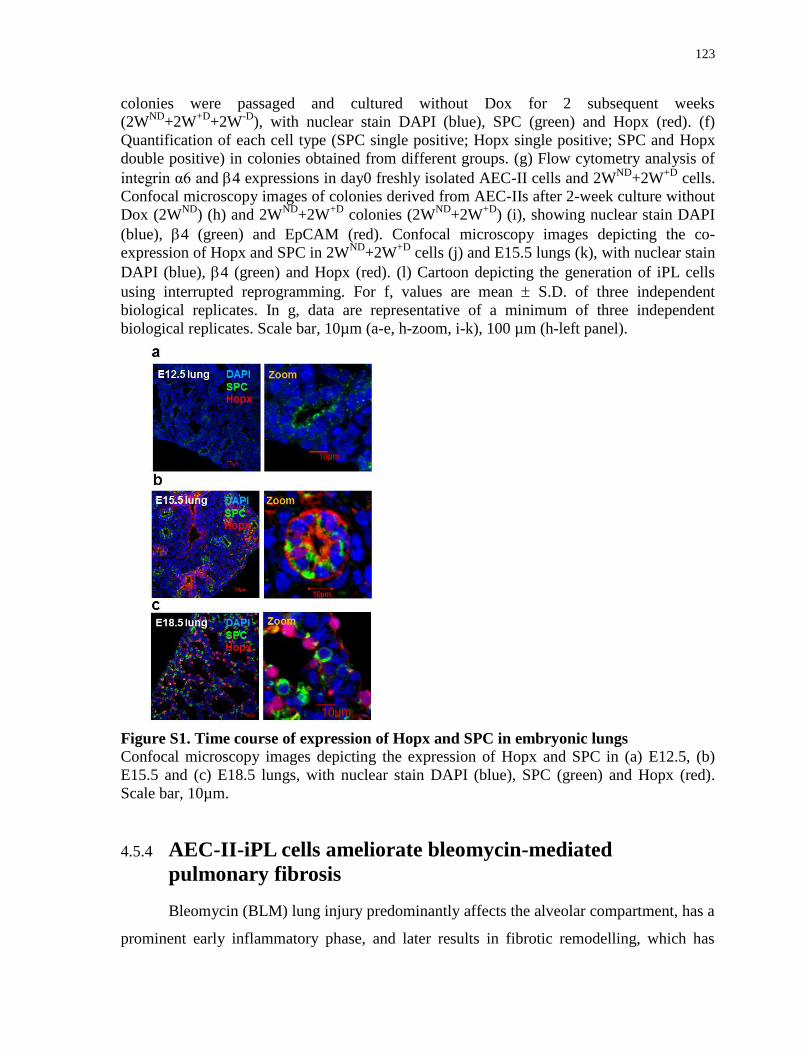
123
colonies were passaged and cultured without Dox for 2 subsequent weeks
(2WND
+2W+D
+2W-D
), with nuclear stain DAPI (blue), SPC (green) and Hopx (red). (f)
Quantification of each cell type (SPC single positive; Hopx single positive; SPC and Hopx
double positive) in colonies obtained from different groups. (g) Flow cytometry analysis of
integrin α6 and 4 expressions in day0 freshly isolated AEC-II cells and 2WND
+2W+D
cells.
Confocal microscopy images of colonies derived from AEC-IIs after 2-week culture without
Dox (2WND
) (h) and 2WND
+2W+D
colonies (2WND
+2W+D
) (i), showing nuclear stain DAPI
(blue), 4 (green) and EpCAM (red). Confocal microscopy images depicting the co-
expression of Hopx and SPC in 2WND
+2W+D
cells (j) and E15.5 lungs (k), with nuclear stain
DAPI (blue), 4 (green) and Hopx (red). (l) Cartoon depicting the generation of iPL cells
using interrupted reprogramming. For f, values are mean S.D. of three independent
biological replicates. In g, data are representative of a minimum of three independent
biological replicates. Scale bar, 10µm (a-e, h-zoom, i-k), 100 µm (h-left panel).
Figure S1. Time course of expression of Hopx and SPC in embryonic lungs
Confocal microscopy images depicting the expression of Hopx and SPC in (a) E12.5, (b)
E15.5 and (c) E18.5 lungs, with nuclear stain DAPI (blue), SPC (green) and Hopx (red).
Scale bar, 10µm.
4.5.4 AEC-II-iPL cells ameliorate bleomycin-mediated
pulmonary fibrosis
Bleomycin (BLM) lung injury predominantly affects the alveolar compartment, has a
prominent early inflammatory phase, and later results in fibrotic remodelling, which has

124
some similarities to idiopathic pulmonary fibrosis (IPF) in humans(Reinert et al., 2013).
Animal studies have demonstrated that MSCs reduce acute inflammation and protect against
subsequent development of fibrosis in BLM-induced injury, only when cells were
administered in the early inflammatory phase(Ortiz et al., 2003). Therefore, we examined
the engraftment and therapeutic benefit of AEC-II-iPL cell treatment in ameliorating
pulmonary fibrosis at both the inflammation and fibrotic phases of BLM-induced injury
(Figure 4a). Female recipients were sacrificed 2 weeks after cell delivery. Lungs of animals
treated with AEC-II-iPL cells (with a constitutive GFP marker) either during the
inflammatory (BLM day7
) or fibrotic phase (BLM day14
) showed a dramatically improved
external appearance (Figure 4b). Retention of AEC-II-iPL cells was greater when cells were
administered during the inflammatory phase (39.1±3.1% of the initial injected cell number)
compared to cells delivered during the fibrotic phase (27.3±4.4%) (Figure 4c). SPC mRNA
levels were also restored in AEC-II-iPL cell-treated lungs (Figure 4d). Bleomycin treatment
results in aberrant differentiation of AEC-II cells into AEC-I cells due to the ECM
abnormalities and interruption of the epithelial basement membrane, with abundant AQP5-
expressing cells and up-regulated expression of AQP5 mRNA. In AEC-II-iPL cell-treated
lungs, AQP5 mRNA was expressed at a normal level (Fig 4e). Compared to control lungs,
there were few AEC-II cells (as marked by LAMP3(Barkauskas et al., 2013) following BLM
treatment, whereas numerous LAMP3+ GFP
+ cells were seen in lungs treated with AEC-II-
iPL cells (Figure 4f). AQP5-expressing GFP cells were found, suggesting the delivered
AEC-II-iPL cells contributed directly to this lineage as well (Figure 4g).
There was evidence of inflammation and epithelial damage, with infiltration of
inflammatory cells in peribronchiolar and interstitial regions 7 days after BLM. Figure 4h
shows Masson’s trichrome stained lung sections. Hematoxylin–eosin staining is shown in
Figure S2a. Two weeks later, without cell therapy, lungs showed extensive fibrosis and
collagen deposition. In contrast, AEC-II-iPL cell–treated lungs showed fewer fibrotic lesions
and less collagen in the parenchyma with normal alveolar architecture, suggesting AEC-II-
iPL cells can prevent the progression to fibrosis. In lungs treated with AEC-II-iPL cells 14
days after BLM administration (fibrotic phase), there was still a marked reduction in the
number of -SMA+ cells in the interstitium (Figure S2b). In addition, we observed a
significant reduction in the expression of mesenchymal cell related genes in AEC-II-iPL

125
treated lungs (Figure S2c, d). Taken together, AEC-II-iPL cells can engraft as epithelial cells
and may be useful as a cell replacement therapy for ameliorating pulmonary fibrosis, even
when administrated at the established fibrotic phase.
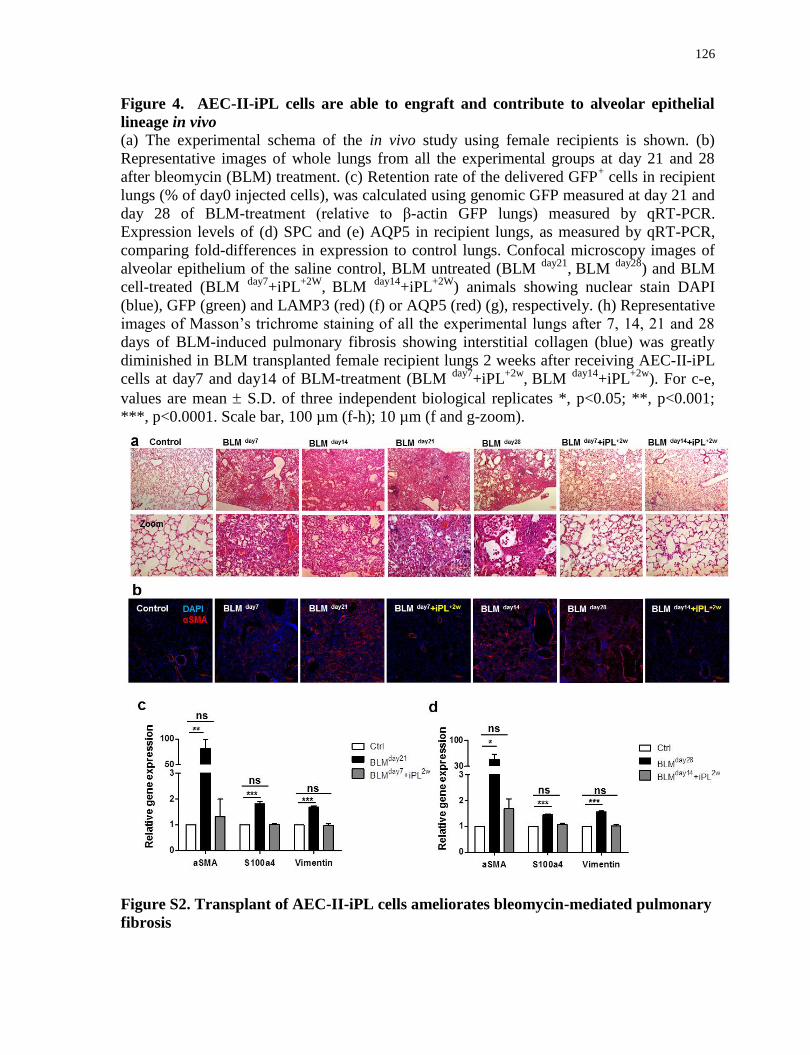
126
Figure 4. AEC-II-iPL cells are able to engraft and contribute to alveolar epithelial
lineage in vivo
(a) The experimental schema of the in vivo study using female recipients is shown. (b)
Representative images of whole lungs from all the experimental groups at day 21 and 28
after bleomycin (BLM) treatment. (c) Retention rate of the delivered GFP+ cells in recipient
lungs (% of day0 injected cells), was calculated using genomic GFP measured at day 21 and
day 28 of BLM-treatment (relative to β-actin GFP lungs) measured by qRT-PCR.
Expression levels of (d) SPC and (e) AQP5 in recipient lungs, as measured by qRT-PCR,
comparing fold-differences in expression to control lungs. Confocal microscopy images of
alveolar epithelium of the saline control, BLM untreated (BLM day21
, BLM
day28) and BLM
cell-treated (BLM day7
+iPL+2W
, BLM
day14+iPL
+2W) animals showing nuclear stain DAPI
(blue), GFP (green) and LAMP3 (red) (f) or AQP5 (red) (g), respectively. (h) Representative
images of Masson’s trichrome staining of all the experimental lungs after 7, 14, 21 and 28
days of BLM-induced pulmonary fibrosis showing interstitial collagen (blue) was greatly
diminished in BLM transplanted female recipient lungs 2 weeks after receiving AEC-II-iPL
cells at day7 and day14 of BLM-treatment (BLM day7
+iPL+2w
, BLM
day14+iPL
+2w). For c-e,
values are mean S.D. of three independent biological replicates *, p<0.05; **, p<0.001;
***, p<0.0001. Scale bar, 100 µm (f-h); 10 µm (f and g-zoom).
Figure S2. Transplant of AEC-II-iPL cells ameliorates bleomycin-mediated pulmonary
fibrosis

127
(a) Representative images of hematoxylin–eosin stained lung sections in all the experimental
groups after 7, 14, 21 and 28 days of BLM-induced pulmonary fibrosis. (b) Immunostaining
of lung sections in all experimental groups with nuclear stain DAPI (blue) and αSMA (red).
Expression of mesenchymal genes (αSMA, S100a4 and vimentin) in BLM non-treated (c,
BLMday21
)
(d, BLMday28
) and BLM-treated (c,
BLMday7
+iPL+2w
)
(d,
BLMday14
+iPL+2w
)
recipient lungs, as measured by qRT-PCR comparing fold-differences in expression to saline
treated control (Ctrl) lung. For c and d, values are mean S.D. of three independent
biological replicates. *, p<0.05; **, p<0.001; ***, p<0.0001.
4.5.5 AEC-II-iPL cells are able to engraft and differentiate to
mature AEC-II and AEC-I cells in severely injured alveolar
epithelium of male mice
Male sex is associated with a significant increase in the prevalence of fibrotic
interstitial lung diseases, including IPF(Gribbin et al., 2006; Olson et al., 2007). In BLM
model of pulmonary fibrosis, male mice develop more prominent fibrotic disease compared
with female mice, and the pulmonary functional alterations can be detected by FlexiVent
measurement(Voltz et al., 2008). We therefore selected a male mouse BLM model to do a
direct comparison of AEC-II-iPL cells with MSCs in improving lung function. As no
significant pulmonary dysfunction could be detected 28 days after BLM compared to saline
control (data not shown), we performed lung function analysis using a FlexiVent system 21
days after BLM. The engraftment and therapeutic benefit of AEC-II-iPL cells and MSCs
were examined following cell delivery at both the inflammatory and fibrotic phases of
BLM-induced injury. Animals were sacrificed at BLM day21
(Fig. 5a). Pressure-volume (PV)
loops were obtained using a quasi-static pressure-controlled PV ramp perturbation. Lungs
treated with both AEC-II-iPL and MSC cells during the inflammatory phase (BLM day7
)
showed improved lung function, closer to that of control lungs. (Fig. 5b). No significant
improvement of lung function was found in animals treated with either cell types during the
fibrotic phase (Fig. 5c). This may due to short in situ period of delivered cells (injected on
BLM day 14
and measurement performed on BLM day21
). Similarly, lungs of animals treated
with AEC-II-iPL cells during the inflammatory (BLM day7
) showed an improved external
appearance compared to other groups (Fig. 5d). Compared to the poor cell retention seen in
MSC-treated lungs, AEC-II-iPL cells were retained in higher numbers when delivered
during either the inflammatory or fibrotic phase (Fig. 5e). The contribution of engrafted

128
cells to the alveolar epithelial lineage in recipient lungs was assessed by immunostaining.
Compared to control lungs, there were few AEC-II cells (as marked by LAMP3) following
BLM treatment, whereas numerous LAMP3+ GFP
+ cells were seen in lungs treated with
AEC-II-iPL cells at either the inflammatory or fibrotic phases. The LAMP+ cells found in
MSC-treated lungs were not GFP+
suggesting that MSCs do not directly contribute to the
AEC-II lineage (Fig. 5f). SPC mRNA levels were also greatly restored in AEC-II-iPL cell-
treated lungs (Fig. 5g). AQP5-expressing GFP+ cells were found in AEC-II-iPL cell-treated
lungs, whereas AQP5+ cells found in MSC-treated lungs were not GFP
+ (Fig. 5h, i).
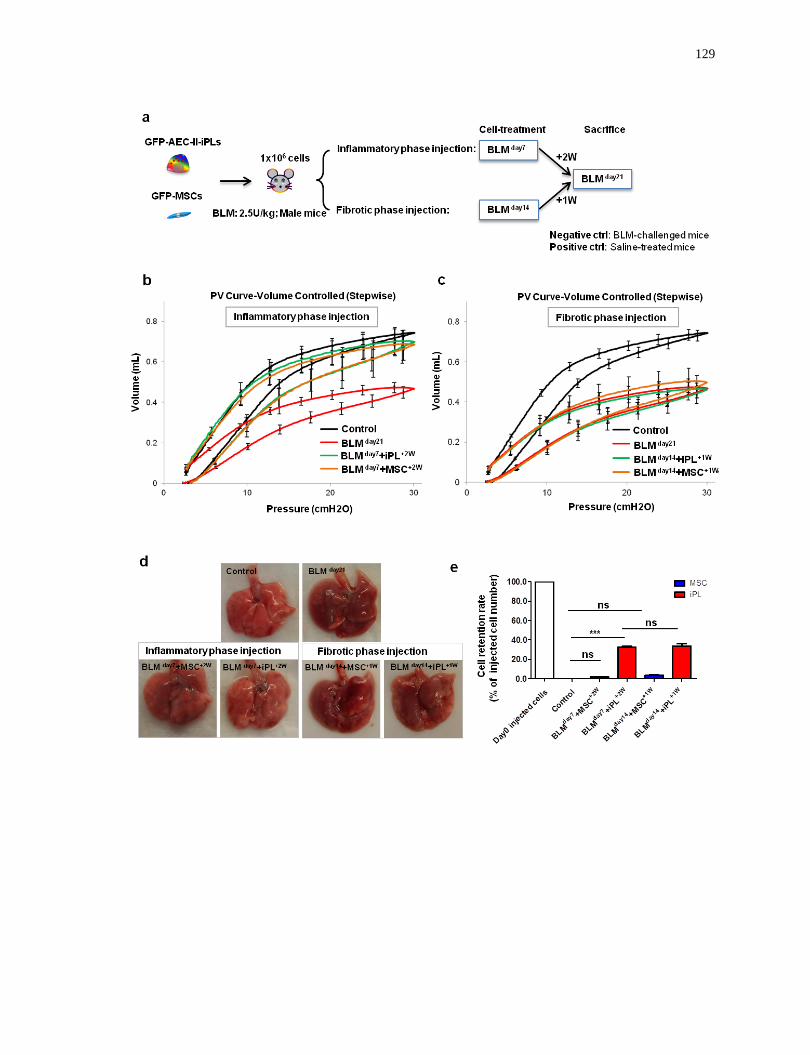
129
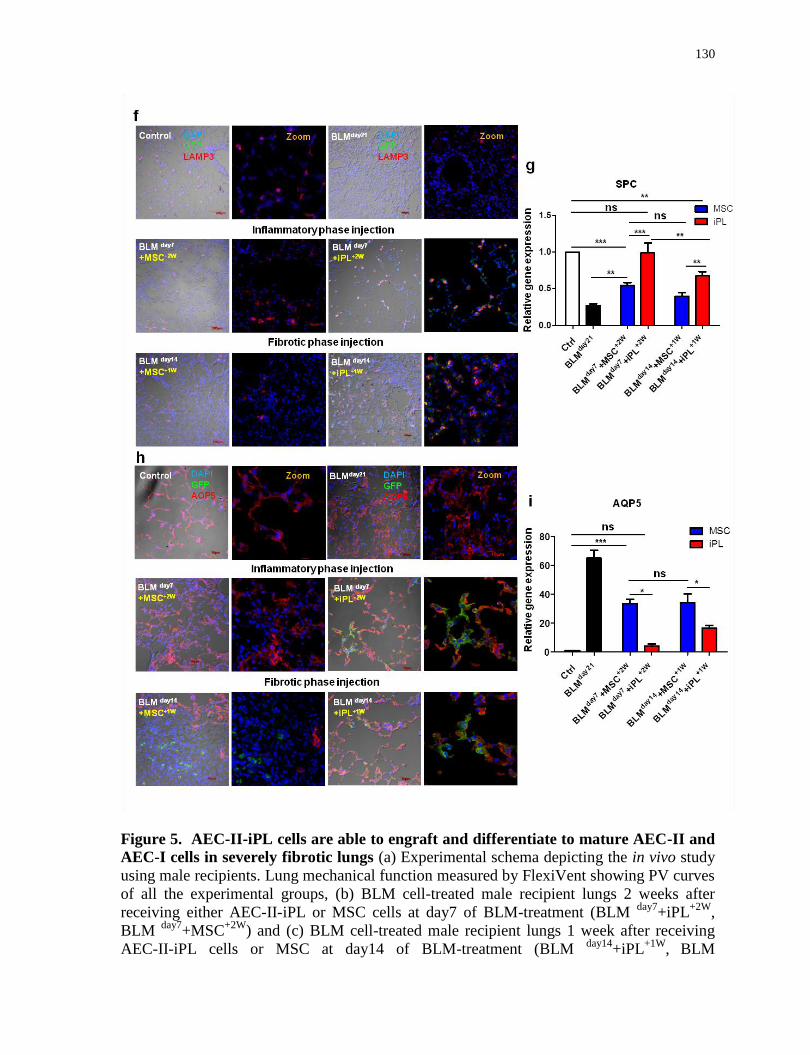
130
Figure 5. AEC-II-iPL cells are able to engraft and differentiate to mature AEC-II and
AEC-I cells in severely fibrotic lungs (a) Experimental schema depicting the in vivo study
using male recipients. Lung mechanical function measured by FlexiVent showing PV curves
of all the experimental groups, (b) BLM cell-treated male recipient lungs 2 weeks after
receiving either AEC-II-iPL or MSC cells at day7 of BLM-treatment (BLM day7
+iPL+2W
,
BLM day7
+MSC+2W
) and (c) BLM cell-treated male recipient lungs 1 week after receiving
AEC-II-iPL cells or MSC at day14 of BLM-treatment (BLM day14
+iPL+1W
,
BLM

131
day14+MSC
+1W). (d) Representative images of whole lungs from all the experimental groups
at day 21 of bleomycin (BLM) treatment. (e) The cell retention rate of engrafted GFP cells
in recipient lungs (% of day0 injected cells), was calculated using genomic GFP at day 21 of
BLM-treatment (relative to β-actin GFP lungs) measured by qRT-PCR. (f) Confocal
microscopy images of alveolar epithelium of the saline control, BLM untreated (BLM day21
),
BLM cell-treated at day 7 (BLM day7
+iPL+2W
, BLM
day7+MSC
+2W) or at day 14 (BLM
day14+iPL
+1W, BLM
day14+MSC
+1W) animals showing nuclear stain DAPI (blue) and LAMP3
(red) and GFP (green) expression. (g) Expression levels of SPC in recipient lungs, as
measured by qRT-PCR, comparing fold-differences in expression to control lungs. (h)
Confocal microscopy images of alveolar epithelium of the saline control, BLM untreated
(BLM day21
), BLM cell-treated at day 7 (BLM day7
+iPL+2W
, BLM
day7+MSC
+2W) and at day
14 (BLM day14
+iPL+1W
, BLM
day14+MSC
+1W) showing nuclear stain DAPI (blue) and AQP5
(red) and GFP (green). (i) Expression levels of AQP5 in recipient lungs, as measured by
qRT-PCR, comparing fold-differences in expression to control lungs. For b, c, e, g and i,
values are mean S.D. of three independent biological replicates *, p<0.05; **, p<0.001;
***, p<0.0001. Scale bar, 100 µm (f); 10 µm (f-zoom and h).
Hematoxylin–eosin (Figure 6a) and Masson’s trichrome (Figure 6b) staining showed
evidence of severe inflammation, extensive fibrosis and collagen deposition developed in
BLM-treated male lungs. In contrast, AEC-II-iPL cell–treated lungs showed fewer fibrotic
lesions and less collagen in the parenchyma compared to MSC-treated lungs, suggesting
AEC-II-iPL cells can reduce the progression to fibrosis. Compared to the lungs treated with
MSC, AEC-II-iPL cell-treated lungs, when cells were administered during the fibrotic phase,
showed a marked reduction in the number of -SMA+ cells in the interstitium (Figure 6c).

132
Figure 6. Treatment with AEC-II-iPL cells ameliorates severe pulmonary fibrosis.
Representative images of lung histopathology are shown for all the experimental groups
after 21 days of BLM-induced pulmonary fibrosis, the saline control, BLM untreated (BLM day21
), BLM cell-treated at day 7 (BLM day7
+iPL+2W
, BLM
day7+MSC
+2W) or day 14(BLM
day14+iPL
+1W, BLM
day14+MSC
+1W), (a) lung sections were stained with hematoxylin–eosin,

133
(b) Masson’s trichrome staining of all the experimental lungs showing a remarkable
decrease of interstitial collagen deposition (the blue stain) in BLM-injured lungs treated with
AEC-II-iPL cells compared with the ones treated with MSC. (c) Immunostaining of lung
sections of all experimental groups with nuclear stain DAPI (blue) and αSMA (red). Scale
bar, 4 mm (a and b-whole section tile scan); 600 µm (a and b-4x); 200 µm (a and b-20x) and
100 µm(c).
4.6 Discussion
We show here that a carefully defined period of interrupted reprogramming, where
the reprogramming process is initiated but not allowed to pass the point of no-return(Nagy
and Nagy, 2010), is able to rescue the limited in vitro clonogenic capacity of AEC-II cells.
Importantly, our results demonstrate that using interupted reprogramming we can achieve
controlled expansion of an otherwise difficult to maintain AEC-II phenotype in
vitro(Kosmider et al., 2011; Liebler et al., 2016; Wang et al., 2007). AEC-II-iPL cells, when
delivered to injured lungs, are able to engraft and contribute to both the AEC II and AEC-I
cell lineage. They ameliorated bleomycin-induced pulmonary fibrosis. Our result showed
that interrupted reprogramming is not only able to achieve controlled expansion of AEC-IIs,
but also may result in the “de-differentiation” of the cells to a bipotent progenitor-like state.
While it remains to be determined if AEC-iPL cells truly recapitulate a distinct lung
epithelial progenitor population, our data suggests at least a phenotypic likeness (SPC+
Hopx+ α6β4
+)
to an embryonic bipotent alveolar progenitor(Treutlein et al., 2014).
Interrupted reprogramming may be useful as an alternative approach to generate large
numbers of functional AEC-IIs with high purity.
There continues to be an interest in cell-based therapeutic approaches for lung diseases
including IPF(Ghadiri et al., 2016). A variety of cell types have been investigated with the
majority of studies focusing on mesenchymal stromal cells (MSC) isolated from bone
marrow(Ortiz et al., 2003; Toonkel et al., 2013), umbilical cord(Moodley et al., 2009),
placenta(Cargnoni et al., 2009) and adipose(Tashiro et al., 2015) tissues. A limitation to
using these cells types is they act mainly through paracrine signalling with little contribution
to lung regeneration through engraftment and/or differentiation. Numerous animal studies
have demonstrated that paracrine signalling by MSC reduces acute inflammation and protect

134
against subsequent development of fibrosis in BLM-induced injury(Ortiz et al., 2003; Rojas
et al., 2015; Weiss and Ortiz, 2013). However, these beneficial effects were only observed
when cells were administered in the early inflammatory phase of lung injury, but not in the
later fibrotic phase(Ortiz et al., 2003) where in fact there is even a possibility of stimulating
detrimental fibroblast proliferation and extracellular matrix deposition(Epperly et al., 2003).
Nevertheless, a number of clinical studies using MSC are currently underway for IPF
(NCT02013700, NCT02135380, NCT01919827, NCT01385644, NCT02277145)(Ghadiri et
al., 2016). Low MSC retention suggests that the therapeutic effects are likely via a paracrine
mechanism rather than engraftment. Unlike MSCs, we found that AEC-II derived iPL cells
are retained at a high rate, potentially even engraft, and appear to contribute directly to the
alveolar epithelial lineage. AEC-II-iPL cells, once engrafted as alveolar epithelial cells,
could limit the deleterious effects of BLM by restoring the pool of alveolar epithelial
progenitor cells for the resolution of disrupted alveolar surfaces. Delivery of AEC-II-iPL
cells during the established fibrotic phase, by analogy, when most patients are diagnosed
with IPF, also showed beneficial effects on fibrosis. Importantly, MSC transplanted during
the fibrotic phase were not able to restore the alveolar epithelium barrier or reduce fibrosis,
suggesting that cells capable of directly contributing to tissue mass, such as the AEC-II-iPL
cells are preferred over paracrine signalling of MSC. Our findings are in keeping with recent
studies showing the efficacy of freshly isolated AEC-II cells in ameliorating
fibrosis(Serrano-Mollar et al., 2007, 2016). Our results suggest that the observed therapeutic
effect is at least in part due to localization of AEC-II-iPL cells within the alveolar milieu and
due to their subsequent differentiation to restore damaged alveolar epithelium through the
dual mechanism of engraftment and likely through paracrine or autocrine effects. Future
studies, such as evaluation of cytokine antagonists which can disrupt fibrosis signaling
pathways at cell engraftment sites; and AEC-II-iPL cell secreted chemokine screens for
fibroblast proliferation inhibiting factors or factors promoting collagen degradation(Serrano-
Mollar et al., 2007); are needed to elucidate the underlying mechanisms whereby AEC-II-
iPL cells promote the recovery of injured alveolar epithelium.
Although the BLM model is not an optimal model for human IPF, it is very
commonly used and thus our results can be compared against a large body of experimental
data. One of the limitations of the BLM injury model is the spontaneous repair following

135
the initial development of fibrosis, in contrast to the progressive pathological remodeling
seen in human IPF. In our BLM-injury study in male mice, no significant changes of
pulmonary function were detected at day 28 compared to that of saline control. We
therefore assessed lung mechanics at day 21. This resulted in a shortened in situ period for
cells delivered to male recipients at the fibrotic phase (male recipients: BLMday14
+iPL cells,
sacrificed at day 21 vs. female recipients: BLMday14
+iPL cells, sacrificed at day 28). We
speculate that the reduced efficacy observed in male recipients may be due to both the
increased severity of fibrosis in male lungs as well as the shorter in situ period of the
delivered cells.
Preclinical studies by Serrano-Mollar et al.(Serrano-Mollar et al., 2016)
demonstrated that the therapeutic potential of transplantation of AEC-IIs in treating human
IPF. However, the usage of freshly isolated AEC-IIs is limited by donor lung availability.
Although our current data was generated from transgenic mice as a proof of principle, our
technique may provide an alternative source of AEC-IIs which could be applied in human
cells using non-integrative, non-viral methods of reprogramming(Hou et al., 2013; Zhou et
al., 2009). The iPL cell induction process can be optimized to obtain maximum expansion
and scale-up of any cell. This may require precise regulation of expression of individual
reprogramming factors to widen the time window of interrupted reprogramming, or
optimization of exogenous growth factors, and/or changes to culture conditions.
Combined with other rapidly developing areas of genome editing, addressing issues
such as safety of cell therapy and tolerance of allogeneic cells, the era of “designer cells” is
just beginning. Our method of creating progenitor-like cells for in vitro expansion, and their
ability to integrate and functionally contribute to the repair of the diseased lung could be a
significant component of an efficient cell-based therapy for this vital organ.

136
Chapter 5
Rejuvenation of aged AEC-II cells to a youthful state using
interrupted reprogramming
5.1 Rationale, Objectives, Hypotheses and Specific Aims
Rationale:
The lungs manifest degenerative changes during aging which is closely associated
with the development and pathogenesis of chronic respiratory diseases, including
idiopathic pulmonary fibrosis (IPF). In human IPF, many of the implicated age-related
deteriorations are mainly observed in endogenous progenitor AEC-II cells, including
epigenetic alteration, DNA damage, telomere dysfunction, mitochondria-mediated
(intrinsic) apoptosis and activated endoplasmic reticulum (ER) stress response in apoptotic
AECs. However, there is yet no direct evidence showing the impact of aging on the
regenerative capacity of AEC-II cells and rejuvenation of aged AEC-II cells in vitro.
Amelioration of age-related deterioration in aged cells is critical for reestablishment
of homeostasis and tissue regeneration. Reprogramming enables aged cells to adopt a
pluripotent state associated with a youthful state which can be maintained in iPS cells and
their derivatives at early passages. A recent report showed in vivo partial reprogramming
through transient cyclic expression of iPS reprogramming factors ameliorates hallmarks of
aging and extends the lifespan in premature aging mouse models, and improves recovery
from metabolic disease and muscle injury in older mice. In Chapter 3 and 4, we
demonstrate that interrupted reprogramming allows controlled expansion yet preservation
of lineage commitment without gaining tumorigenic pluripotency.
Objective:
To evaluate if age-related deterioration in aged AEC-II cells can be ameliorated via
in vitro interrupted reprogramming.

137
Hypothesis:
Interrupted reprogramming allows rejuvenation of aged AEC-II cells to a youthful
state while preserving AEC-II identity and function.
Specific Aims:
To examine the influence of aging on the clonogenic capacity of AEC-II cells;
To determine if interrupted reprogramming can rescue the age-related defective
clonogenic capacity of AEC-II cells while preserving cellular identity and function;
To evaluate if other age-related deteriorations in aged AEC-IIs, inlcuding declined
telomerase activity, mitochondrial DNA damge and epigenetic alteration, can be
ameliorated by interrupted reprogramming.
5.2 Abstract
We present a novel "interrupted reprogramming" strategy to achieve rejuvenation of
aged stem cells using carefully timed transient expression of induced Pluripotent Stem (iPS)
cell reprogramming factors (Oct4, Sox2, Klf4 and c-Myc; OSKM) without traversing the
pluripotent stage. Interrupted reprogramming is able to rejuvenate aged alveolar progenitor
AEC-II cells to a youthful state by ameliorating age-related deterioration in stem cell self-
renewal capacity, telomerase activity, mitochondrial DNA and the epigenetic alterations. By
carefully controlling the duration of transient expression of OSKM, interrupted
reprogramming resulted in controlled expansion of younger cells yet preservation of AEC-II
identity and function as that of young AEC-II cells.
Harnessing the residual epigenetic memory and age plasticity existing in the early
reprogramming process, interrupted reprogramming allows controlled expansion of
functional younger cells retaining lineage identity of cell of origin. This novel approach may
provide a promising avenue for therapeutics against age-related decline and diseases.

138
5.3 Introduction
Idiopathic pulmonary fibrosis (IPF) is an irreversible, fatal disease with mortality up
to 50% at 3-4 years (Selman and Pardo, 2014). Although the molecular and cellular
mechanisms underlying IPF are not fully understood, a growing body of evidence shows that
the extent of alveolar epithelial cell (AEC) injury, repair process, and aging are the key
determinants in the pathogenesis of IPF.
At cellular level, many of the proposed pivotal hallmarks of aging have been
implicated in humans with IPF, including epigenetic alterations, DNA damage, stem cell
exhaustion, telomere dysfunction (shortened telomeres), mtDNA damage-mediated alveolar
epithelial cell apoptosis, and activated endoplasmic reticulum (ER) stress response (Liu et
al., 2013; Mossman et al., 2011; Noble et al., 2012; Thannickal, 2013; Uhal and Nguyen,
2013; Weiss et al., 2010). Notably, these age-related deteriorations leading to impaired lung
regeneration and progressive loss of lung function are mainly observed in AEC-II cells,
suggesting a prominent role in triggering the pathogenesis of IPF.
Like other tissue-specific stem cells, AEC-IIs, the endogenous stem cells in alveoli,
may also suffer from a progressive loss in their regenerative capacity during aging.
Mutations in telomerase is the most common distinguishable risk factor for IPF causing
abnormal telomere shortening and dysfunction (Alder et al., 2008, 2011; Armanios, 2013).
Recent studies using genetic animal models demonstrated that telomere dysfunction is
restricted to AEC-IIs and causes stem cell failure/exhaustion as a result of induced cellular
senescence (Alder et al., 2015; Chen et al., 2015). While telomeres are susceptible to age-
related deterioration, the age-associated telomere dysfunction in AEC-IIs could result in
stem cell exhaustion leading to impaired tissue regeneration. Although there is no direct
evidence showing age-related functional decline in AEC-IIs in vitro, a dramatic reduction in
the number and clonogenic capacity has been illustrated in AEC-IIs isolated from human
lung tissue of IPF patients (Liang et al., 2016). Mitochondrial DNA (mtDNA) damage and
consequent dysfunction in AECs are also linked to the pathogenesis of IPF. Evidence
suggests that AEC mtDNA damage induces apoptosis and promotes the development of IPF
and other age-related lung diseases (e.g., lung cancer and COPD) (Schumacker et al., 2014;
Wallace, 2013). Further, the accumulation of mtDNA mutations arising from age-related

139
mtDNA damage is crucial in depleting the longevity of lung stem cells (Held and
Houtkooper, 2015; Schumacker et al., 2014; Wallace, 2013). Therefore, amelioration of age-
related deterioration in aged AEC-II cells is critical for reestablishment of homeostasis and
tissue regeneration in aged lungs.
Rejuvenation as a consequence of iPS reprogramming has recently been the subject
of intense investigation. Reprogramming enables aged cells to adopt a youthful state which
is maintained in iPS cells and their derivatives at early passages. Epigenetic remodeling
during the reprogramming process is the key driver of the restoration of aging hallmarks to a
more youthful state, including telomere shortening, stem cell exhaustion, mitochondrial
dysfunction and cellular senescence. Recent studies yield insight to molecular mechanisms
underlying iPS reprogramming and describe distinct transcriptional phases during this
progressive process (Buganim et al., 2012; Hansson et al., 2012; Polo et al., 2012a).
Notably, one of epigenomic dynamics during the early phase of reprogramming is the
activation and repression of age-associated histone marks, such as H3K4me3 and
H3K27me3 (Polo et al., 2012a). We speculated that rejuvenation of aged lung stem cells can
be achieved by carefully timed transient reprogramming (termed "interrupted
reprogramming”). Interrupted reprogramming is able to rejuvenate aged lung progenitor
AEC-II cells and age-related deterioration in their stem cell self-renewal capacity,
telomerase activity, mitochondrial DNA and the epigenome histone methylation marks
(‘epigenetic clock’) can be reset to a younger state. Furthermore, interrupted reprogramming
resulted in the controlled expansion of youthful induced Progenitor-Like (iPL) cells
retaining AEC-II identity and function. We achieved this by optimized, controlled, transient
induction of reprogramming factors, turning off their expression prior to reaching
independent pluripotency.
To the best of our knowledge, this is the first study demonstrating that the aged
phenotype of endogenous lung progenitor AEC-IIs can be reverted to a youthful state in
vitro. In contrast to the complexity of directed differentiation protocols for each cell type
required, interrupted reprogramming can theoretically be applied to rejuvenate almost any
type of aged cells that can be isolated and purified. Therefore, this novel interrupted
reprogramming approach which results in the generation of large numbers of younger cells

140
may provide a promising avenue for cell-based therapy approaches where age-related
disease is prevalent.
5.4 Materials and Methods
5.4.1 Animal Husbandry
Young (8- to 12-week-old) and old (12- to 16-month-old) ROSA26-rtTA and
Col1a1: tetO-4F2A mice (Jackson Labs, Cat#011004) were used to generate inducible lung
epithelial cells. Animals were maintained as an in-house breeding colony under specific
pathogen–free conditions. All procedures involving animals were approved by the
Institutional Animal Care and Use Committee of the University Health Network (Toronto,
Ontario, Canada).
5.4.2 Cell Isolation and Culture
5.4.2.1 Isolation of AEC-II Cells from Mouse Lung
AEC-II cells were isolated using a previously described protocol (Berndt-Weis et al.,
2009) with modifications. Before lung digestion, mice were injected intra-peritoneally with
heparin (250U/mouse) and euthanized by CO2 narcosis. Lungs were flushed through the
right ventricle with cold phosphate buffered saline (PBS). Endobronchial lavage was then
performed to remove alveolar leukocytes. Mouse lung was filled with porcine elastase 10 U
/mL and incubated for 20 min at 37C. Trachea and bronchi were cut away. The remaining
tissues were finely minced and incubated in 100μg/mL of DNAse I in DMEM containing
antibiotics for 10 minutes. The suspension was mixed with FBS and sieved through 100 µm,
40 µm and 20 µm nylon meshes. Cells were centrifuged at 32g for 12 min then re-suspended
in red blood cell lysis buffer for 3min and the lysis was stopped by addition of an equal
volume of PBS. Cells were centrifuged at 100g for 12 min then re-suspended in 0.5 %
vol/vol FBS-PBS for all subsequent procedures.
5.4.2.2 Matrigel-based 3D condition
Feeders (MEF) were seeded on 0.1% gelatin coated 24-well transwell filter inserts
(Corning) one day prior to the addition of epithelial cells. Sorted epithelial cells

141
resuspended in 100μL of Matrigel (BD Biosciences) prediluted 1:1 (vol/vol) with epithelial-
specific (EpiS) media were added to a MEF-coated 24-well transwell filter inserts in a 24-
well tissue culture plate containing 500μL of epithelial media for 2 weeks then replaced with
ES (embryonic stem cell) medium containing 1.5ug/mL doxycycline (Sigma). For ROCK
inhibitor treatment, 10 μM Y-27632 (Enzo Life Sciences) was added to medium. Media was
replenished three times per week. For bulk passaging, whole cultures were dissociated in
Collagenase (1mg/mL; Sigma) Dispase (3mg/mL; BD Biosciences) in PBS to generate a
single-cell suspension.
5.4.2.3 Mix-culture of young and aged AEC-II cells
Young AEC-IIs were isolated from GFP; ROSA26-rtTA/Col1a1:: tetO-4F2A
transgenic mice. Aged AEC-II cells were derived from non-GFP ROSA26-rtTA/Col1a1::
tetO-4F2A transgenic mice and labeled with CMTMR dye (Thermo Fisher, C2927). Equal
numbers of young and old AEC-II cells were mixed and cultured under Matrigel-based 3D
condition.
5.4.3 Fluorescence activated cell sorting and analysis
For purification of AEC-IIl cells, fresh isolated cells were suspended and incubated
in 0.5 % vol/vol FBS-PBS containing an optimally pre-titered mixture of antibodies [anti-
CD45, anti-CD31 (BD Biosciences), anti-EpCAM (Abcam) and relevant isotype controls]
for approximately 30min on ice. Labeled cells were washed and re-suspended at 3~5 × 106
cells/mL in 0.5 % vol/vol FBS-PBS. Cell viability was accessed by propidium iodide
(1μg/mL) staining. For intra-cellular antigen analysis, cells were fixed and stained using a
Fix and Perm kit (Invitrogen) as per manufacturer instructions. Sorting was performed using
a MoFlo BRU cell sorter (Becton Dickinson), acquisition was performed using a BD LSRII
analyzer (Becton Dickinson) and data was analyzed using FlowJo software.
5.4.4 Immunofluorescence
Samples were fixed with 4% paraformaldehyde (PFA) for 30min and blocked with
5% goat serum and 2% BSA in PBS containing 0.5% Triton X-100 for 1 hour. Primary

142
antibodies were diluted in BSA/PBS, applied to samples and incubated overnight at 4°C.
Secondary antibodies AlexaFluors 488, 532, 546, 633 or 647 (Invitrogen) were applied
according to the species in which the primary antibody was used for 2 hours at room
temperature. Nuclear staining was performed using 2mg/mL 4, 6, diamidino-2-phenylindole
(DAPI; Sigma). Stained samples were mounted with immunofluorescent mounting medium
(DAKO). Appropriate non-specific IgG isotypes were used as controls. Immunoreactivities
of antigens were visualized as single optical planes using an Olympus Fluoview confocal
microscope and analyzed using FV10-ASW 2.0 Viewer software.
5.4.5 Real-time PCR analysis
Total RNA was prepared using the RNeasy Kit (Qiagen) as per manufacturer’s
instructions. cDNA was prepared and assayed using Superscript III (Sigma) according to
manufacturer’s protocol. Differential gene expression was determined using SYBR green
detection (Roche). Real-time PCR reactions were done in triplicate for each sample.
GAPDH was used as a housekeeping gene to normalize gene expression levels using
LightCycler 480 software (Roche). Normalized mRNA levels were shown as relative to the
control samples (day 0 fresh isolated cells).
5.4.6 Statistics
Statistical analysis was performed using GraphPad Prism 5.0 statistical software (San
Diego, CA, USA). The statistical significance of multiple groups was compared to each
other using Tukey’s multiple comparison test ANOVA. A p value of <0.05 was considered
significant.
5.5 Results
5.5.1 The clonogenic capacity of AEC-II cells declines with age
Age-dependent progressive decline in stem cell number and functionality has been
reported in various tissues, such as skeletal muscle stem cells (Cerletti et al., 2012; Sousa-
Victor et al., 2014), neural stem cells (Molofsky et al., 2006) and some lung epithelial stem
cells (Navarro and Driscoll, 2017). The reduction of self-renewal activity in aged stem cells
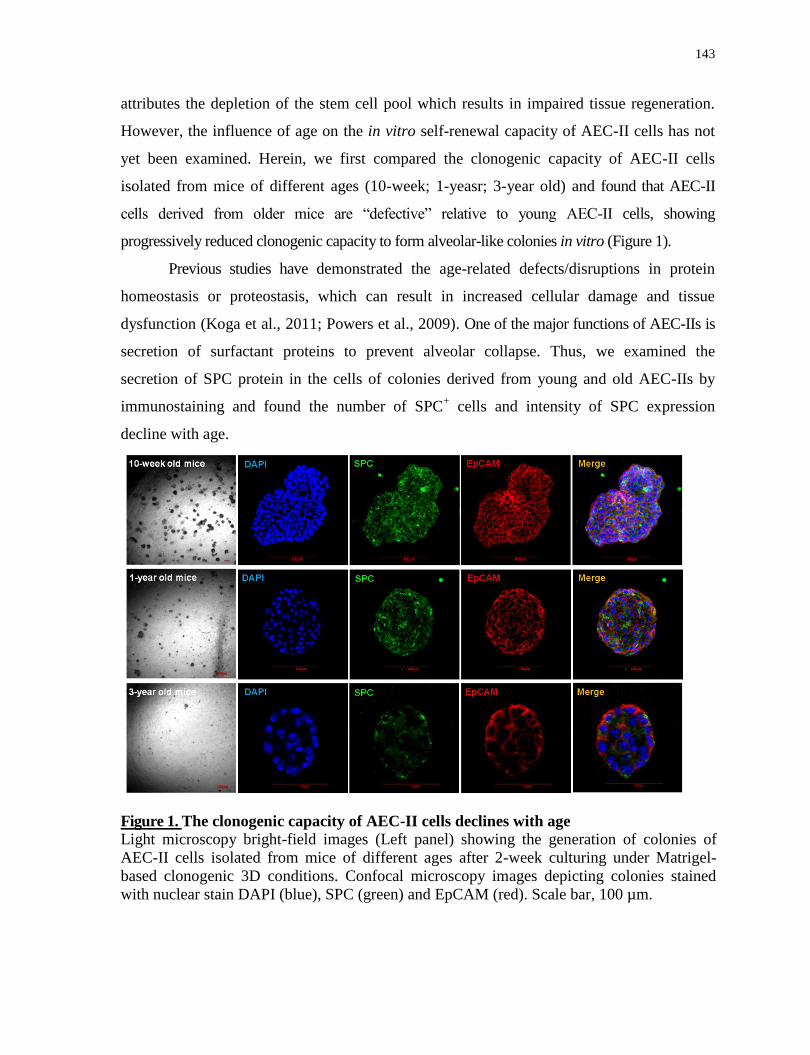
143
attributes the depletion of the stem cell pool which results in impaired tissue regeneration.
However, the influence of age on the in vitro self-renewal capacity of AEC-II cells has not
yet been examined. Herein, we first compared the clonogenic capacity of AEC-II cells
isolated from mice of different ages (10-week; 1-yeasr; 3-year old) and found that AEC-II
cells derived from older mice are “defective” relative to young AEC-II cells, showing
progressively reduced clonogenic capacity to form alveolar-like colonies in vitro (Figure 1).
Previous studies have demonstrated the age-related defects/disruptions in protein
homeostasis or proteostasis, which can result in increased cellular damage and tissue
dysfunction (Koga et al., 2011; Powers et al., 2009). One of the major functions of AEC-IIs is
secretion of surfactant proteins to prevent alveolar collapse. Thus, we examined the
secretion of SPC protein in the cells of colonies derived from young and old AEC-IIs by
immunostaining and found the number of SPC+ cells and intensity of SPC expression
decline with age.
Figure 1. The clonogenic capacity of AEC-II cells declines with age
Light microscopy bright-field images (Left panel) showing the generation of colonies of
AEC-II cells isolated from mice of different ages after 2-week culturing under Matrigel-
based clonogenic 3D conditions. Confocal microscopy images depicting colonies stained
with nuclear stain DAPI (blue), SPC (green) and EpCAM (red). Scale bar, 100 µm.

144
We also performed a mixed-culture experiment using equal numbers of CMTMR-
labeled aged AEC-IIs (1-year old) and GFP+ young AEC-IIs (8-12-week old) co-culturing
under Matrigel-based 3D condition (Figure 2a). All colonies were exclusively
monochromatic (CMTMR+
or GFP+) demonstrating that epithelial colonies are clonally
derived from the proliferation of single AEC-II progenitors rather than by cell aggregation,
which would generate colonies containing both CMTMR+
and GFP+
cells. Importantly, we
found fewer numbers of CMTMR+
colonies compared to that of GFP+ colonies, further
confirming the age-related decline of clonogenic capacity in aged AEC-IIs, which cannot
even be rescued by culturing in the microenvironment provided by young AEC-II cells
(Figure 2b).
a b
Figure 2. Aged AEC-II cells exhibit a limited clonal proliferation of epithelial colony
forming units (a) Schematic graph representing potential outcomes of mix-culture
experiment. (b) Bright field,green, red and overlay of fluorescence from coculture of GFPpos
young AEC-II cells and CMTMR-labeled aged AEC-II cells. Scale bar, 100 µm.
5.5.2 Interrupted reprogramming is able to “rejuvenate” aged
AEC-II cells to a younger state to form large numbers of
alveolar-like colonies
To study the effect of interrupted programming on the clonogenic capacity of AEC-II
cells, we isolated AEC-II cells from young (8-12-week old) and old (1-year old) R26-

145
rtTA/Col1a1::tetO-4F2A double transgenic mice (Carey et al., 2010) enabling expression of
Oct4, Sox2, Klf4 and c-Myc (OSKM) following treatment with doxycycline (dox, D).
Consistent with our previous findings, although both young and aged AEC-II cells gave rise
to alveolar-like colonies when cultured in Matrigel-based 3D culture system, native aged
AEC-II cells possess a greater limited colony forming ability, which further decreased over
passages.
Following our previously defined AEC-II-iPL induction protocol (2WND
+2W+D
),
which allows the greatest expansion without traversing the pluripotent state, both young and
aged AEC-II cells were cultured in Matrigel-based 3D conditions for 2 weeks prior to dox
treatment (ND). We found that iPL induction is able to "rejuvenate" aged AEC-II cells with an
enhanced self-renewal capacity to form larger numbers of alveolar-like colonies. For example, the
colony forming efficiency (CFU %) of aged AEC-II cells without dox-treatment (2WND
) was
1.2%±0.34% but increased up to 4.96%±0.8% after iPL induction (2WND
+2W+D
) which is similar
to that of non-treated young AEC-II cells (2WND
: 5.2%±0.8%). Importantly, the AEC-II
phenotypic expression of SPC was restored in the aged AEC-II-iPL cells (2WND
+2W+D
), as
seen in young AEC-II cells, even after withdrawal of dox for 2 weeks (2WND
+2W+D
+2W-D
)
(Figure 3). This date showed that interrupted reprogramming is able to restore youthfulness
to aged AEC-II cells, firstly characterized by enhanced capacity of self-renewal and
secretion of surfactant C.

146
Figure 3. Interrupted reprogramming ameliorates the aged-related decline of
clonogenic capacity in aged AEC-II cells Stereomicroscopy images (left panel) showing the
generation of colonies in the absence (ND), present of dox (+D) or dox-withdrawal (-D) in
Matrigel-based clonogenic 3D conditions (seeding density 5,000 cells/well, cells were
passaged every 2 weeks); confocal microscopy images (right panel) of colonies obtained
under different conditions from aged AEC-II cells, showing nuclear stain DAPI (blue), SPC
(green) and EpCAM (red). Scale bar, 100 µm.
5.5.3 ROCK inhibitor failed to rescue the aged-related decline of
clonogenic capacity in AEC-II cells
Recent published work has shown the ability of human keratinocytes (Chapman et
al., 2010) and epithelial cells (e.g. prostate, breast and lung) (Bove et al., 2014; Liu et al.,
2012; Suprynowicz et al., 2012) to proliferate in vitro when co-cultured with inactivated
fibroblast feeder cells in the presence of a Rho kinase (ROCK) inhibitor (Y-27632, “Y”).
Yet no studies have demonstrated the impact of age on the cell proliferation response to
ROCK inhibitor. Thus, we determined whether the age difference would have an impact on
the induced proliferation in AEC-IIs by ROCK inhibitor treatment and whether ROCK

147
inhibitor could rejuvenate the limited self-renewal capacity of aged AEC-IIs. Both young
and aged AEC-IIs were cultured in Matrigel-based 3D conditions for 2 weeks with or
without Rock-inhibitor (Figure 4). For iPL induction, cells were cultured without the
presence of Rock-inhibitor for 2 weeks prior to dox treatment. Induced proliferation was
observed in young AEC-IIs treated Rock-inhibitor showing slightly increased CFU%, but
not as robust as that seen with iPL induction. In contrast, no significant improvement of self-
renewal was observed in aged AEC-IIs treated with Rock-inhibitor, suggesting that the
aged-related endogenous proliferative capacity of the starting AEC-II population influences
their response to Rock-inhibitor treatment.
Figure 4. Comparison of clonal proliferation of young and aged AEC-II cells under
OSKM iPL induction and ROCK inhibitor treatment. The experimental schema of the
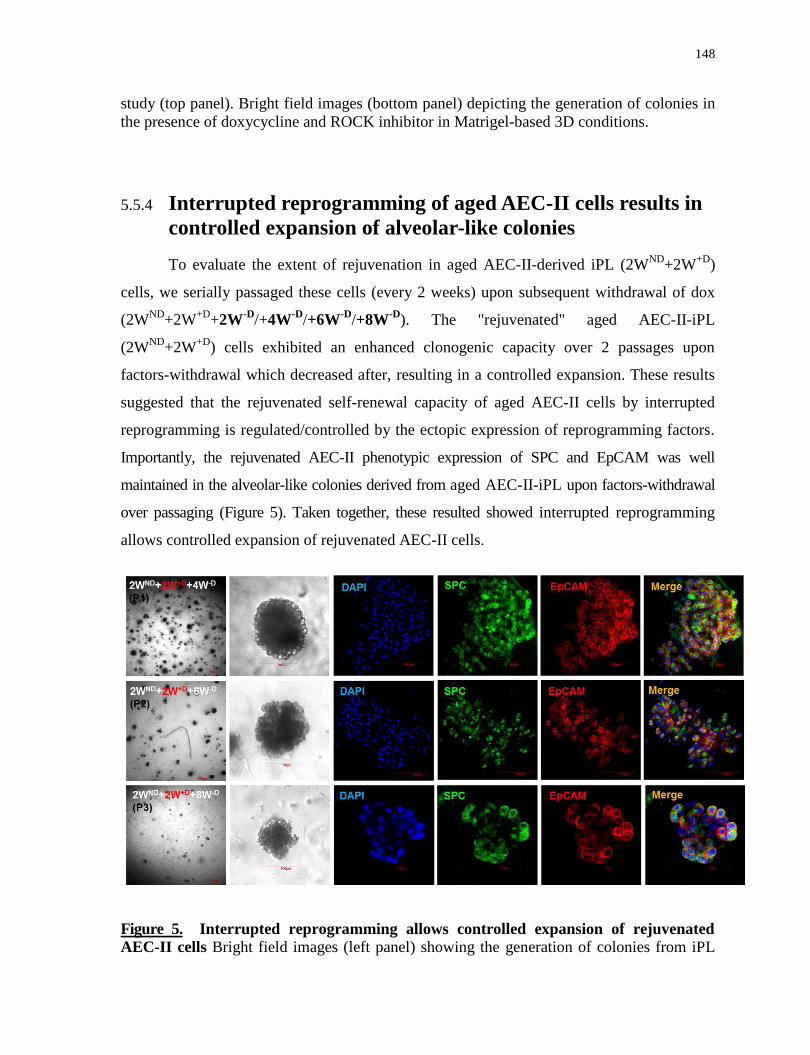
148
study (top panel). Bright field images (bottom panel) depicting the generation of colonies in
the presence of doxycycline and ROCK inhibitor in Matrigel-based 3D conditions.
5.5.4 Interrupted reprogramming of aged AEC-II cells results in
controlled expansion of alveolar-like colonies
To evaluate the extent of rejuvenation in aged AEC-II-derived iPL (2WND
+2W+D
)
cells, we serially passaged these cells (every 2 weeks) upon subsequent withdrawal of dox
(2WND
+2W+D
+2W-D
/+4W-D
/+6W-D
/+8W-D
). The "rejuvenated" aged AEC-II-iPL
(2WND
+2W+D
) cells exhibited an enhanced clonogenic capacity over 2 passages upon
factors-withdrawal which decreased after, resulting in a controlled expansion. These results
suggested that the rejuvenated self-renewal capacity of aged AEC-II cells by interrupted
reprogramming is regulated/controlled by the ectopic expression of reprogramming factors.
Importantly, the rejuvenated AEC-II phenotypic expression of SPC and EpCAM was well
maintained in the alveolar-like colonies derived from aged AEC-II-iPL upon factors-withdrawal
over passaging (Figure 5). Taken together, these resulted showed interrupted reprogramming
allows controlled expansion of rejuvenated AEC-II cells.
Figure 5. Interrupted reprogramming allows controlled expansion of rejuvenated
AEC-II cells Bright field images (left panel) showing the generation of colonies from iPL

149
cells upon dox-withdrawal over passaging (seeding density 5,000 cells/well, cells were
passaged every 2 weeks); confocal microscopy images (right panel) of colonies at different
passages, showing nuclear stain DAPI (blue), SPC (green) and EpCAM (red). Scale bar, 100
µm (P0-P2 and P3 bright field), 10 µm (P3 confocal images).
5.5.5 Interrupted reprogramming allows controlled restoration of
telomerase gene expression
Telomeres are particularly susceptible to age-related deterioration (Blackburn et al.,
2006). Telomere dysfunction is involved in the loss of the tissue regeneration and associated
with premature development diseases (Armanios and Blackburn, 2012), including idiopathic
pulmonary fibrosis (IPF) (Armanios, 2012), the most common manifestation of telomere-
medicated disease. Studies using aged telomerase-deficient mice showed the premature
aging phenotypes across multiple organs including testes, spleens, and intestines can be
reverted upon the reactivation of telomerase (Jaskelioff et al., 2011). Most recent studies
showed that induction of telomere dysfunction in AEC-II cells provoke impaired self-
renewal and cellular senescence resulting in lung function abnormalities and secondary
inflammation, as seen in telomere-mediated lung diseases (Alder et al., 2015b; Chen et al.,
2015b). Aging hallmarks can be erased during iPS reprogramming including restoration of
telomere length and telomerase activity to a youthful state (Rando and Chang, 2012;
Ocampo et al., 2016a). Thus, we wondered if interrupted programming is able to restore
telomerase activity in aged AEC-II cells. As a pilot study, we compared the expression level
of telomerase gene in non-treated aged AEC-II cells, aged AEC-II-iPL cells and cells
derived upon subsequent dox-withdrawal at different passages to that in freshly isolated
young AEC-II cells. Without induction, telomerase gene is remarkably down-regulated in
native aged AEC-II cells compared to that in young AEC-II cells, which further decreased
over passaging. Strikingly, significant up-regulation of telomerase gene was observed in
aged-AEC-II-iPL (2WND
+2W+D
) cells which progressively decreased over passaging upon
dox-withdrawal (P0: 2WND
+2W+D
+2W-D
; P1:+4W-D
; P2: +6W-D
) (Figure 6), suggesting the
restoration of telomerase gene expression is regulated the inductive factors. This date
suggest that the previous observed controlled expansion of rejuvenated AEC-II cells results
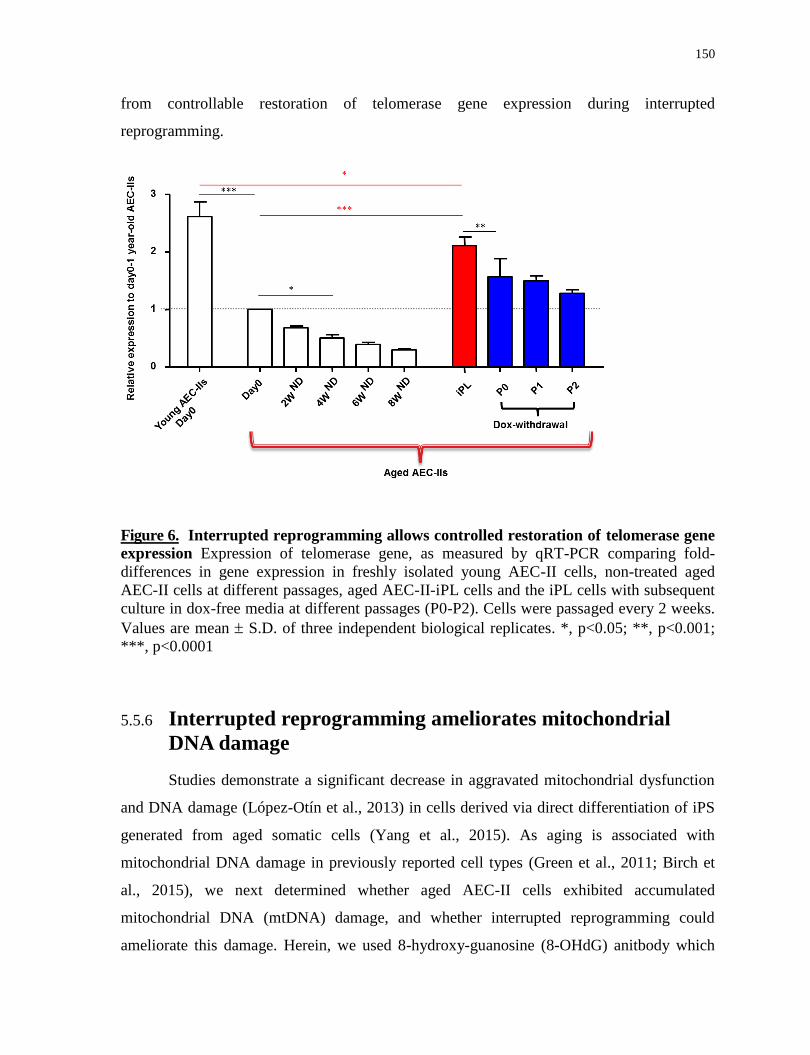
150
from controllable restoration of telomerase gene expression during interrupted
reprogramming.
Figure 6. Interrupted reprogramming allows controlled restoration of telomerase gene
expression Expression of telomerase gene, as measured by qRT-PCR comparing fold-
differences in gene expression in freshly isolated young AEC-II cells, non-treated aged
AEC-II cells at different passages, aged AEC-II-iPL cells and the iPL cells with subsequent
culture in dox-free media at different passages (P0-P2). Cells were passaged every 2 weeks.
Values are mean S.D. of three independent biological replicates. *, p<0.05; **, p<0.001;
***, p<0.0001
5.5.6 Interrupted reprogramming ameliorates mitochondrial
DNA damage
Studies demonstrate a significant decrease in aggravated mitochondrial dysfunction
and DNA damage (López-Otín et al., 2013) in cells derived via direct differentiation of iPS
generated from aged somatic cells (Yang et al., 2015). As aging is associated with
mitochondrial DNA damage in previously reported cell types (Green et al., 2011; Birch et
al., 2015), we next determined whether aged AEC-II cells exhibited accumulated
mitochondrial DNA (mtDNA) damage, and whether interrupted reprogramming could
ameliorate this damage. Herein, we used 8-hydroxy-guanosine (8-OHdG) anitbody which
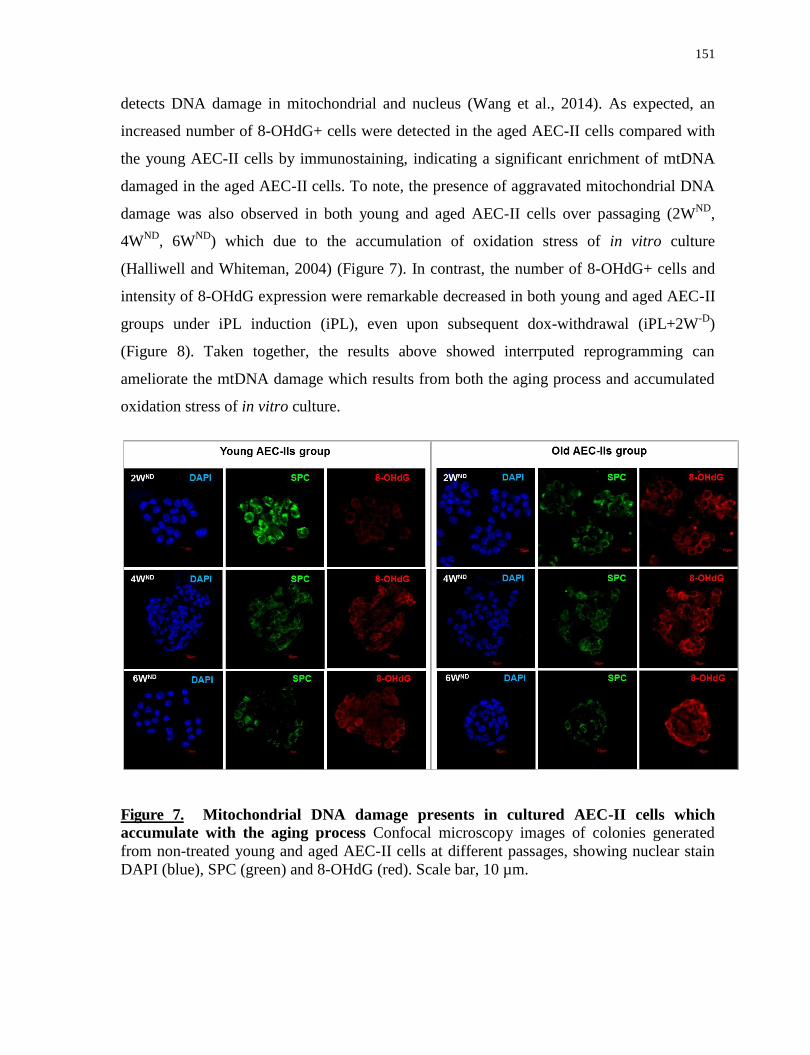
151
detects DNA damage in mitochondrial and nucleus (Wang et al., 2014). As expected, an
increased number of 8-OHdG+ cells were detected in the aged AEC-II cells compared with
the young AEC-II cells by immunostaining, indicating a significant enrichment of mtDNA
damaged in the aged AEC-II cells. To note, the presence of aggravated mitochondrial DNA
damage was also observed in both young and aged AEC-II cells over passaging (2WND
,
4WND
, 6WND
) which due to the accumulation of oxidation stress of in vitro culture
(Halliwell and Whiteman, 2004) (Figure 7). In contrast, the number of 8-OHdG+ cells and
intensity of 8-OHdG expression were remarkable decreased in both young and aged AEC-II
groups under iPL induction (iPL), even upon subsequent dox-withdrawal (iPL+2W-D
)
(Figure 8). Taken together, the results above showed interrputed reprogramming can
ameliorate the mtDNA damage which results from both the aging process and accumulated
oxidation stress of in vitro culture.
Figure 7. Mitochondrial DNA damage presents in cultured AEC-II cells which
accumulate with the aging process Confocal microscopy images of colonies generated
from non-treated young and aged AEC-II cells at different passages, showing nuclear stain
DAPI (blue), SPC (green) and 8-OHdG (red). Scale bar, 10 µm.

152
Figure 8. Interrupted reprogramming ameliorates the aged-related mitochondrial
DNA damage Confocal microscopy images of 2WND
+2W+D
(iPL) colonies and colonies
obtained from iPL cells maintained in culture for 2 weeks in the absence of Dox (iPL+2W-D
)
from young and aged AEC-II cells, showing nuclear stain DAPI (blue), SPC (green) and 8-
OHdG (red). Scale bar, 10 µm.
5.5.7 Interrupted reprogramming allows restoration of age-
related expression of H3K4me3, H3K9me3 and H3K27me3
Cellular damage resulting from aging (e.g., genome instability, telomere dysfunction
and mitochondrial dysfunction) are primarily regulated through epigenomic mechanisms
(Blasco, 2007; Issa, 2014). Meeting the criteria for a hallmark of aging, epigenetic alteration
in DNA methylation and histone methylation patterns has been described showing increases
in the levels of histone H4K16 acetylation (H4K16ac), H4K20 trimethylation (H4K20me3),
or H3K4 trimethylation (H3K4me3), together with decreases in the levels of H3K9
trimethylation (H3K9me3) or H3K27 trimethylation (H3K27me3) (Fraga and Esteller,
2007b; Han and Brunet, 2012b). During iPS reprogramming, aging hallmarks are restored to
a youthful state primarily driven by epigenetic remodeling (Ocampo et al., 2016a; Yang et
al., 2015). While changes in histone marks are mainly detected in the early phase of
reprogramming (Polo et al., 2012), we determined whether aging-associated epigenetic
alteration in histone modification could be restored by interrupted reprogramming. The
expression of histone markers, including H3K4me3, H3K9me3, and H3K27me3, in young
AEC-II cells, aged AEC-II cells and their counterpart iPL cells were examined by
immunostaining. Consistent with previously described age-associated histone modification
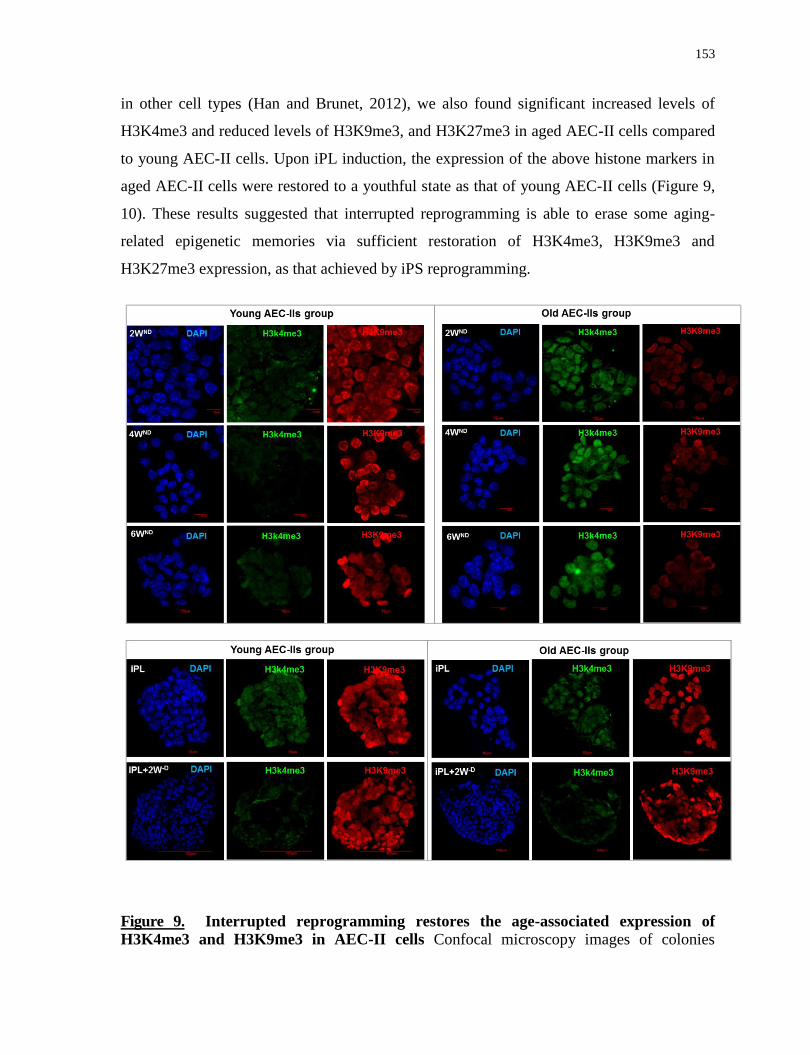
153
in other cell types (Han and Brunet, 2012), we also found significant increased levels of
H3K4me3 and reduced levels of H3K9me3, and H3K27me3 in aged AEC-II cells compared
to young AEC-II cells. Upon iPL induction, the expression of the above histone markers in
aged AEC-II cells were restored to a youthful state as that of young AEC-II cells (Figure 9,
10). These results suggested that interrupted reprogramming is able to erase some aging-
related epigenetic memories via sufficient restoration of H3K4me3, H3K9me3 and
H3K27me3 expression, as that achieved by iPS reprogramming.
Figure 9. Interrupted reprogramming restores the age-associated expression of
H3K4me3 and H3K9me3 in AEC-II cells Confocal microscopy images of colonies

154
obtained under different conditions from young and aged AEC-II cells, showing nuclear
stain DAPI (blue), H3K4me3 (green) and H3k9me3 (red). Scale bar, 10 µm (2WND
, 4WND
,
6WND
, iPL), 100 µm (iPL+2W-D
).
Figure 10. Interrupted reprogramming restores the age-associated expression of
H3K27me3 in AEC-II cells Confocal microscopy images of colonies obtained under
different conditions from young and aged AEC-II cells, showing nuclear stain DAPI (blue)
and H3K27me3 (green). Scale bar, 10 µm (2WND
, 4WND
, 6WND
, iPL), 100 µm (iPL+2W-D
).
5.6 Discussion
We demonstrated that carefully defined period of interrupted reprogramming allows
rejuvenation of aged lung progenitor AEC-II cells to a youthful state yet preservation of
AEC-II identity and function of cell of origin. The age-related deterioration observed in aged
AEC-II cells, including impaired self-renewal, declined telomerase activity, mitochondrial
DNA damages and the epigenome histone alteration, was reset to a younger state in aged-

155
AEC-II-iPL and their progeny. Meanwhile, controlled expansion of younger cells retaining
the parental AEC-II cellular identity and function was achieved.
A major goal in lung therapy research is to sufficiently maintain or restore lung
function in aged individuals. Lung stem cells play crucial roles in maintaining tissue
homeostasis and tissue regeneration. However, even without extrinsic insults, lung stem
cells experience replicative exhaustion and gradually loss their regenerative capacity during
aging, resulting in lung dysfunction and chronic lung diseases (Meiners et al., 2015). Despite
the recent progress in identifying a number of specialized stem cell populations within the
lung that function to replenish or repair damaged epithelium (Kim et al., 2005; McQualter et
al., 2010; Teisanu et al., 2011; Chapman et al., 2011; Barkauskas et al., 2013; Vaughan et
al., 2014; Jain et al., 2015), all of these studies were performed on young animals. No direct
evidence has been shown on the impact of aging on in vitro behavior and on regenerative
response to injury. Ours is the first in vitro study demonstrating the age-related dysfunction
of airway progenitors. Most recently and in support of our hypothesis, Ocampo et al
demonstrated that in vivo partial reprogramming ameliorates hallmarks of aging and extends
the lifespan in premature aging mouse models. They also show that partial reprogramming
improves recovery from metabolic disease and muscle injury in older mice (Ocampo et al.,
2016a, 2016b). However, the rejuvenation consequence of transient reprogramming on
tissue specific stem cells has not been evaluated in this study. In contrast, we are the first
demonstrated that aged tissue stem cells can be reversed to a youthful state by carefully
timed transient reprogramming in vitro .
It is clear that a number of key issues needed to be addressed in our future studies.
Firstly, although aged-AEC-II cells were rejuvenated to a younger state, whether these
generated iPL cells are consistent with bipotent alveolar progenitors expressing Hopx and
α64 requires further characterization. Secondly, interrupted reprogramming seems to able
to replicate the rejuvenation effects seen in iPS reprogramming, but how aging-related
epigenetic memory has been erased and how the epigenome is remodeled, needs further
exploration. In addition, the role of each individual reprogramming factor on the aging
process or reversal of needs to be determined.
Notch signaling pathway plays a crucial role in regulating the regenerative capacity
of numerous stem cell populations, including basal stem cells and lineage-negative epithelial

156
stem/progenitor population (LNEP) (Vaughan et al., 2014) that excessive Notch signaling
dysregulates their ability to repair. Altered levels of Notch signaling have been observed in
human IPF, suggesting the age-associated alteration in pathways could be one of the
underlying factors responsible for the failure of endogenous stem cells to repair injured
tissue or even accelerate the development of disease. Therefore, our future studies need to
yield insight into the pathways, including Notch signaling, to determine their roles in
regulating AEC-II aging and rejuvenation consequence induced by interrupted
reprogramming.
The limited regenerative capacity of aged stem cells has been demonstrated in in vivo
disease models of other tissues, but not yet in the lungs. Although the age-related impaired
reparative capacity has been observed in bleomycin-induced fibrosis in aged mice (Hecker et
al., 2014), the contribution of aged AEC-II stem cells to the impaired tissue regeneration
needs to be evaluated by transplantation into the microenvironment of young recipients.
Moreover, the rejuvenated regenerative capacity of aged-AEC-II-iPL cells and the derived
younger AEC-II-like cells to ameliorate age-related pulmonary fibrosis needs to be
examined in vivo by cell transplantation to belomycin-treated aged recipients.
Finally, for therapeutic use, the current data was obtained from transgenic mice and
it remains to be shown that this technique can be efficiently applied in human cells to rescue
the aging phenotype.
Theoretically, this technique is broadly applicable and could be extended to other
aged stem cell populations giving rise to large numbers of highly specified younger cell
populations, holding great promise for treating age-related disorders.

157
Chapter 6
Summary and Discussion
6.1 Summary of key findings
Regenerative medicine is constrained by suboptimal cell sources with either limited
or uncontrolled proliferation and/or incompletely restricted differentiation. Seeking an
alternative approach to produce highly specified functional cell sources for tissue
regeneration, we have developed a novel "interrupted reprogramming" strategy, harnessing
the residual epigenetic memory in combination with the rapid induction of cell proliferation
existing early in the reprogramming process.
We show that this strategy, using a carefully timed transient expression of iPS cell
reprogramming factors (OSKM), allows the generation of lineage-specific induced
progenitor-like (iPL) cells from both mature and progenitor epithelial populations without
gaining tumorigenic pluripotency. Both our in vitro and in vivo results support our
hypothesis that interrupted reprogramming, where the reprogramming process is initiated
but not passing the point of no-return (Nagy and Nagy, 2010), allows controlled expansion
and preservation of the cell’s memory of origin and thus their ability to efficiently return to
their original phenotype.
In chapter 3, we showed that iPL cells derived from bronchiolar mature Club cells
function as bronchiolar progenitor-like cells to generate mature bronchiolar Club cells,
goblet cells and functional CFTR-expressing ciliated epithelium and show in vivo utility in
repopulating CFTR-deficient epithelium. In chapter 4, we further explored the utility and
generalizability of interrupted reprogramming to a lung progenitor cell population and
showed that interrupted reprogramming is able to rescue the limited in vitro clonogenic
capacity of AEC-II cells and generate large numbers of AEC-II-iPL cells which
phenotypically and functionally resemble a bipotent distal epithelial progenitor seen in
embryonic lung. Importantly, these AEC-II-iPL cells can ameliorate bleomycin-induced
pulmonary fibrosis in vivo via cell engraftment and subsequent differentiation to the alveolar
epithelial lineage.

158
In chapter 4, we demonstrated that interrupted reprogramming is able to rejuvenate
aged AEC-II cells to a youthful state by ameliorating multiple aging hallmarks, including
impaired self-renewal, declined telomerase activity, mitochondrial DNA damages and the
epigenome histone alteration. These results are consistent with ‘age’-related plasticity in the
early stages of reprogramming. Concomitantly, the cellular identity of AEC-II is well
preserved thereby generating large number of younger AEC-II cells.
6.2 Discussion
We showed that interrupted reprogramming can be used as an alternative approach to
produce highly specified functional therapeutic cell populations that may lead to significant
advances in regenerative medicine. The novelty of this concept and the relevant issues will
be further discussed with perceived future directions. The key focus in this discussion
includes the progenitor features of iPL cells; variables need to be considered to obtain iPL
cells-the target cell types and induction conditions; the heterogeneity observed during iPL
induction and cellular mechanisms underlying iPL phenomenon.
6.2.1 What are iPL cells?
We designate these intermediate products "induced progenitor-like cells" which
takes into account their controlled proliferation and restricted differentiation to a limited
range of progeny, which resemble the fundamental functional characteristic of adult stem
cells. We think “progenitor-like” is a fair description and gets to the heart of our findings –
an approach to create cells which can function as progenitors from any mature, readily
available cell type. However, whether the reprogramming is a stepwise de-differentiation
process that multipotent iPL cells could de-differentiate to foregut endoderm, and then into
definitive endoderm upon continuous reprogramming is yet to be determined. To do this,
further refinement of precise regulation of expression of individual reprogramming factors
is needed to widen the relatively narrow time window between where factor-dependent iPL
cells are created and factor-independent pluripotency is established.

159
We selected two distinct populations from different regions in the lung: (1) mature
bronchiolar Club cells, and (2) AEC-II cells, which possess limited proliferative or
clonogenic capacity in vitro, respectively.
Club-iPL cells function as bronchiolar progenitor-like cells that are able to give rise
to Club, goblet and ciliated lineage in both in vitro and in vivo. This induced multipotency
resembles that of basal stem cells and variant Club cells. Scgb1a1+ variant Club cells can
respond to naphthalene or allergen challenge and give rise to ciliated and goblet cells
(Rawlins et al., 2009a; Pardo-Saganta et al., 2013); basal stem cells respond to sulfur
dioxide injury (Rock et al., 2009a) or naphthalene injury (Hong, 2003) and differentiate into
club and ciliated cells, but also able to migrate and repopulate alveolar epithelium after
H1N1 influenza infection (Kumar et al., 2011). At the molecular level, Club-iPL cells are
distinguished from variant Club cells and basal stem cells by their expression of P63,
lacking of CK5 activity, respectively. Furthermore, comparison of microarray data for
known facultative progenitor populations suggests that Club-iPL cells are not identical to
any currently identified progenitors. Nevertheless, it remains a possibility that Club-iPL
cells are similar to a yet unidentified naturally existing progenitor cell population.
Alternatively, Club-iPL cells function as a novel "artificial" progenitor cell population not
existing in the developing or mature lung. In contrast, we found that AEC-II-iPL cells may
recapitulate an identified distal lung epithelial progenitor population. Their phenotypic
expression of SPC, Hopx, and intergin α6β4
bares likeness to an embryonic bipotent
alveolar progenitor (Treutlein et al., 2014).
Although multiple unique cell states en route to pluripotency during iPS
reprogramming have been documented, whether iPL cells represent a unique intermediate
stage and/or recapitulate a specific developmental stage requires further characterization and
dissection of the early reprogramming process.
6.2.2 The regenerative capacity of iPL cells
As proof of concept, we evaluated the utility of iPL cells for lung cell-based
therapeutic applications by assessing the retention and differentiation of the cells in (1) a
CFTR-knockout mouse; and (2) a bleomycin-induced pulmonary fibrosis model. We found
that lineage-specific iPL cells can directly contribute to the repair of injured lungs by

160
engraftment and incorporation in the epithelium and differentiation into the desired
functional progeny.
Although the in vivo models we applied are very commonly used and have many of
the manifestations of human lung diseases, including epithelium dysfunction and
exaggerated immune response, they fail to faithfully replicate the pathophysiology in
humans (Scholte et al., 2004). Naphthalene treatment of recipient animals was used simply
as a conditioning regimen to augment retention of delivered iPL cells and is not a model or
airway lung injury. The dose of naphthalene is not lethal and animals fully recover after a
period of time. The BLM injury model is not an optimal model for human IPF in that the
spontaneous repair following the initial development of fibrosis is in contrast to the
progressive pathological remodeling seen in human IPF. Thus, in vivo models that better
recapitulating the pathophysiology in human pulmonary diseases should be employed in
future evaluation of the therapeutic potential of iPL cells. For instance, the use of Club-iPL
cells in reversing the susceptibility of CFTR-deficient animals to Pseudomonas infection; as
well as AEC-II-iPL for functional restoration of alveolar architecture in H1N1 influenza
virus infected lungs.
6.2.3 The mechanisms underlying the iPL phenomenon
Since the innovation of iPS technology a decade ago, tremendous progress has been
made on understanding the mechanisms underlying somatic cell fate changes during the
multi-step reprogramming and the factor-induced ESC-like transcriptional network
established to confer pluripotency. Whether the mechanisms underlying interrupted
reprogramming preformed on epithelial populations share similarity with or differ from what
we learn from the studies on fibroblasts reprogramming needs to be explored.
It has been demonstrated that certain reprogramming factors can be omitted from the
reprogramming cocktail when the target cell type can express them endogenously
(Nakagawa et al., 2008; Wernig et al., 2008). In our case, Club cells can express endogenous
Sox2, but at a much lower level compared to that in ESCs. It remains to be determined if
Sox2 can be excluded from the OSKM cocktail to generate Club-iPL cells.
Numerous studies of iPS reprogramming have demonstrates that each individual
reprogramming factor plays a distinct role during the stepwise reprogramming process (Li et

161
al., 2010; Nakagawa et al., 2008; Sridharan et al., 2009). Although c-Myc is the main factor
mediating the initial transcriptional wave, the successful MET occurring in the initial phase
of fibroblast reprogramming relies on the collaboration of all four factors. The second
transcriptional wave is mainly mediated by Oc4, Sox2 and Klf4, whereas c-Myc acts as an
amplifier of gene expression to build a foundation for the other reprogramming factors to
activate the pluripotency network (Nakagawa et al., 2008; Rahl et al., 2010). Notably, this
knowledge is mainly learned from the studies performed with the Yamanaka factors on
mouse embryonic fibroblasts (MEFs). Therefore, whether the role of each individual factor
in lung epithelial reprogramming process is similar to that seen in fibroblast reprogramming
remains to be determined.
Furthermore, the contribution of each of the individual reprogramming factors to the
iPL phenomenon during the interrupted programming process needs to be investigated in the
future. In our proof-of-principle study, all experiments were performed on cells carrying a
doxy-inducible polycistronic 4F (OSKM) 2A cassette. We suspect that the permutations of
the 4 factors and the relative expression levels of the individual factors play a crucial role in
iPL induction. However, we cannot assess all four single gene transgenic mice and siRNA
techniques may not fully knockdown genes. Similarly, if cells are virally transduced with
individual factors, the expression levels of factors will be different from that in 4F2A cells.
In all of these approaches, distinguishing between quantitative differences based on
expression levels of individual factors versus synergistic effects in any of the combinations
would be extremely difficult. To overcome these difficulties, other reprogramming systems
should be employed in future studies to examine the effects of different combinations of the
various 4 factors in iPL induction.
During iPS reprogramming of fibroblasts, c-Myc targets the promoters involved in
regulating cell proliferation, metabolism and biosynthetic pathways. In addition,
overexpression of oncogenic c-Myc has been shown to induce proliferation of mature cells
and lead to partial transformation. In contrast to c-Myc which is transcribed only in
mesenchyme, N-myc is specifically expressed in the epithelium and is essential for
maintaining undifferentiated and proliferating progenitor cells in the lung (Okubo, 2005).
Thus, we wondered if the generation of iPL from epithelial populations is distinct from
simple proliferation induced by over-expression of oncogenic N-myc. We compared four-

162
factor iPL induction to induction with N-myc alone using mature Club cells derived from
Rosa-rtTA-EGFP; teto-Nmyc transgenic mice. Although N-myc induction alone was able to
generate colonies, they were much smaller in number and showed morphological
heterogeneity (Appendix 1). In contrast, OSKM iPL induction resulted in a larger number of
hollow-luminal colonies with greater homogeneity. Transient induction of OSKM was able
to achieve a greater expansion of cells than N-myc induction alone. Our results with N-myc
transgenic mice are not surprising and show that colonies can be formed. However, these are
far less homogeneous and fewer in number in comparison to colonies obtained from OSKM
transgenic mice suggesting that the presence of all 4 factors is optimal for the iPL cell
generation process. It is important to stress that iPL cells have not committed to pluripotency
and long-term in vivo assays have shown no teratoma or tumor formation suggesting that it
is not partial transformation.
Meanwhile, a recent study evaluated the effect of transient activation of OSKM,
OSK and M alone in cancer cell system (Singovski et al., 2016). Although c-Myc was
important for driving proliferation of tested cancer cells, only the full four factors can result
in acquisition of a robust ‘cancer stem cell” phenotype, including increase in colony forming
efficiency. Whether this observation is analogous to our non-malignant iPL population
remains to be studied.
6.2.4 Target cell type in iPL induction
Numerous factors, including cell proliferation, target cell types and epigenetic
remodelling, have been identified that could influence iPS reprogramming and are
associated with reprogramming efficiency and therefore they may also be involved in the
generation of iPL cells.
Promotion of the cell cycle allowing more cells to transit through S phase (Hanna et
al., 2009; Ruiz et al., 2011) could enhance the iPS reprogramming efficiency. Although it is
still debating whether the degree of proliferation of starting population influences the effi-
ciency and kinetics of the iPS reprogramming process, we found that the proliferative
property of the starting epithelial population influences the induced proliferation at early
stage of reprogramming and the ability to obtain pluripotency at the later stage.

163
In our attempt to isolate and purify Club cells, we have defined two distinct epithelial
populations, EpCAMhigh
and EpCAMlow
cells. Compared to EpCAMhigh
mature Club cells,
non-treated EpCAMlow
cells exhibited a greater endogenous proliferative capacity showing
larger portion of CFSEnegative
proliferative cells. Although dox-induction results in more
CFSEnegative
proliferative cells within both groups, a higher induced proliferation was
observed in EpCAMlow
population, suggesting the proliferative status of starting epithelial
population influences their response to the inductive factors to proliferate (Appendix 2).
Regarding the induction of pluripotency, both EpCAMhigh
and EpCAMlow
cells were
seeded on transwell membrane pre-coated with MEF feeders (2D condition) and treated with
doxycycline. Activation of the inductive factors was monitored by immunostaining of Oct4
and pluripotency was accessed by expression of alkaline phosphatase, SSEA-1, and Nanog.
The induced colonies derived from EpCAMhigh
mature Club cell population failed to express
pluripotency markers and were unable to gain pluripotency up to 12~13 weeks under dox-
treatment. In contrast, dox-independent induced colonies expressing all the classical
pluripotency markers can be derived from EpCAMlow
population after 3-4 week of induction
(Appendix 3). These observations suggest that the proliferative property of the starting cells
could influence their ability to obtain pluripotency at the later stage.
Evidence from previous studies revealed that cell type could influence the
reprogrammability (Aoi et al., 2008; Aasen et al., 2008; Maherali et al., 2008). Although it
seems every cell is able to be reprogrammed to iPS cells (Hanna et al., 2009), whether the
degree of differentiation of starting cells within the same lineage influences the efficiency
and kinetics of the reprogramming process is still under debate.
In our study, both mature and progenitor epithelial cells were selected for iPL
induction. Progenitor AEC-II cells can give rise to Nanog-expressing Dox-independent
endogenous Oct4+ cells 1 week earlier than mature Club cells during reprogramming. In
addition, the expansion is much higher in iPL induction of AEC-II cells. These results
suggest that the proliferative capacity and the degree of differentiation of the starting cells
within the lung epithelial lineage could influence the reprogramming process and the
generation of iPL cells.
Epigenetic remodelling is the major underlying mechanism driving somatic cells
from the differentiated to the pluripotent state during the reprogramming process. The

164
epigenetic features of the starting somatic cells need to be erased to some extent to adopt a
pluripotent stem cell epigenome. Supportive studies showed that the expression of lineage-
specific genes blocks reprogramming (Mikkelsen et al., 2008; Pereira et al., 2010). Thus, we
speculate that the extent of epigenetic memory of the cell of origin linked to the residual
DNA methylation within lineage-specific genes persisting in mature and progenitor
epithelial cells is different, and may influence the extinction of the somatic program during
the early stage of reprogramming and attribute to the variation seen in their iPL induction.
Changes in histone modifications occur immediately after induction of
reprogramming factors. The loss of H3K4me2 at the promoters of somatic cells and the
gradual depletion of H3K27me3 and promoter hypomethylation in the regions responsible
for cell fate conversion all occurs during the early phase of reprogramming (Sridharan et al.,
2009; Stadtfeld et al., 2008). Whether these epigenetic alternations are responsible for the
variation seen in iPL induction of mature and progenitor lung epithelial cells remains to be
studied.
6.2.5 Derivation condition for iPL cells
One of the advantages of the iPL process is the resulting expansion of epithelial
cells. There are several factors that could influence the transgene activation and the induced
proliferative capacity. Thus, interrupted reprogramming culture conditions need to be
optimized to satisfy both starting epithelial populations and the derived iPL cells.
While most of the reprogramming studies were preformed under two-dimensional
(2D) cell-culture system, our initial experiments were carried out in 2D conditions. We
tested multiple factors and found that the cell seeding density, doxycycline concentration
and the differentiation status of starting cells could influence the transgene activation and the
induced proliferative capacity. Moreover, a greater induced proliferation was observed when
cells were induced under the conditions of transwells coated with supporting MEF feeders.
Nevertheless, optimized 2D conditions result in the loss of epithelial phenotype which can
only be partial restored upon factor-withdrawal.
It is well known that Matrigel can better support epithelial growth and maintain
epithelial phenotype in vitro. Furthermore, iPS cells can be maintained in Matrigel
conditions as embryonic bodies. Thus, we developed a Matrigel-based 3D culture system

165
and examined its ability to support iPL induction. Compared to the 2D reprogramming
condition, we found this Matrigel-based 3D condition can boost induction of iPL cells and
pluripotency. During interrupted reprogramming, large numbers of luminal iPL colonies can
be obtained from mature Club cells in 3D conditions. Importantly, induced colonies obtained
under 2D conditions were not able to gain pluripotency even after 12~13 weeks of dox-
treatment, whereas Nanog+ colonies can be obtained in 3D conditions after 4 weeks of
induction. Although the precise underlying mechanisms are unknown, we speculate that this
Matrigel-based 3D condition provides a more supportive "reprogramming niche" and
modulates the reprogramming process of epithelial cells. Meanwhile, a recent report
highlights the importance of mimicking the biochemical features of native
microenvironment during reprogramming and demonstrates that a chemical defined 3D
ECMs can accelerate the two key events for initiation of iPS programming, MET and
chromatin remodeling (Caiazzo et al., 2016).
Furthermore, the timing of induction may require some optimization to "satisfy"
target cell type to obtain maximum expansion of iPL cells. In our study of iPL induction of
AEC-II cells, we first adopted the induction conditions used in Club cells that AEC-II cells
were immediately induced after cell isolation (named "early induction") in Matrigel-based
3D conditions. We found that AEC-II cells treated with Dox for 4weeks (4W+D
, 6W+D
)
resulted in the generation of type A (epithelial-like) and type B (mesenchymal-like) colonies
with distinct morphologies. Importantly, there is no significant increase in colony forming
efficiency and the resultant cell expansion was not satisfactory (6W+D
results in 19.73± 2.4
fold expansion relative to day0 seeded cells). In terms of the ability of the induced cells to
return to their original phenotype, SPC, which was suppressed after 2 weeks of Dox, was
only expressed in type A colonies upon withdrawal of Dox but not in type B colonies
(Appendix 4). This result suggests some cells under 2-week early induction have already
passed the point-of-no return and fail to return to AEC-II phenotype. Therefore, we
developed a slightly different condition for AEC-II cells, which is an initial two weeks of
culture without Dox prior to Dox-treatment (named "late induction"), and found it appears
advantageous for greater expansion and preservation of lineage commitment.

166
6.2.6 The heterogeneity observed in iPL induction
The heart of interrupted reprogramming is to create controllable conditions that
maintain the adult stem cell-like state but when removed, allow the cells to then
spontaneously revert to the mature cell of origin. Interrupted reprogramming was performed
on highly purified starting populations and resulted in relatively homogeneous induced cell
proliferation and returning ability upon withdrawal of reprogramming factors. Similar to the
higher levels of heterogeneity observed in the early phases of iPS induction (Lujan et al.,
2015; Treutlein et al., 2016; Zunder et al., 2015), some degree of heterogeneity has also been
observed during our iPL induction. Although the vast majority of cells are able to initiate
reprogramming, individual cells within the starting population differ in their response to
inductive factors and generate iPL colonies which are variable in size, as well as their ability
to obtain pluripotency (marked by Nanog expression). Whether this heterogeneity is
attributed to the various levels of endogenous Sox2 expression representing the normal
biology of Club cells or the variance in the pre-existing epigenetic fingerprint regulating
individual cell fate within the starting Club cell population needs further exploration.
6.2.7 Highlights of interrupted reprogramming-compared to
other approaches
We exploit recent efforts to dial back reprogramming technologies for somatic cells
to a potentially useful intermediate factor-dependent state rather than factor-independent
state of partially reprogrammed iPS and iPS cells. Harnessing the “residual epigenetic
memory” of the starting cells, carefully timed interrupted reprogramming enables generation
of large numbers of lineage-specific progenitor-like cells with restricted differentiation to a
limited and appropriate range of functional progeny. Our proof of concept in vivo studies
showed lineage-specific iPL cells hold great promise for treating a variety of diseases by
true engraftment as “induced” progenitors. Furthermore, intermittent induction cycles
resulted in second generation iPL cells with a greater expansion without loss of lineage
committement. Further studies to maximize the expansion of cells require the precise

167
regulation of expression of individual reprogramming factors, potential use of growth
factors, and/or changes to culture conditions.
Endogenous progenitor cells exist in various organs, including the lung (Kim et al.,
2005; McQualter et al., 2010; Rawlins et al., 2009b; Rock et al., 2009b). However, these
cells are rare, which limits their expansion. In our initial work with Club cells, we found the
native EpCAMlow
population containing some endogenous progenitor cells which can give
rise to colonies in 3D condition. We have performed a co-culture experiment to examine the
effect of iPL induction on the EpCAMlow
population. In this study, EpCAMlow
cells were
isolated from both GFP-Col1a14F2 and RFP mice, which lack the inducible 4F2A construct.
Cells were mixed in a 1:1 ratio and treated with doxycycline for 2 weeks. Co-cultures
resulted in 100% monochromatic colonies supporting the idea colonies are clonally derived.
Importantly, although both colors of cells were able to generate colonies, higher CFU% was
found in the inducible GFP+
group (Appendix 5), consistent with our interpretation that
interrupted reprogramming has converted additional cells to iPL cells.
Recent published work has shown that conditional reprogramming using feeder cells
and ROCK inhibitor (Y-27632) is able to maintain upper airway P63+ stem cells in their
immature, proliferative forms (Bove et al., 2014; Liu et al., 2012). Despite fundamental
differences from our approach, we have compared iPL induction of EpCAMhigh
-mature Club
cells to conditional reprogramming. We found that cell proliferation induced by iPL
induction was 12.3±1.6% fold greater than using the ROCK inhibitor in 2D CFSE assay. In
a 3D assay, the morphology of the colonies formed using the ROCK inhibitor was
significantly different from those obtained after iPL induction. Unlike iPL colonies, ROCK-
inhibitor treatment alone doesn’t contribute to the generation of luminal colonies but
resulted in few small dense cell clusters/ colonies (Appendix 6). A comparison experiment
was also performed on AEC-II population. As shown in chapter 4, the expansion induced by
Rock-inhibitor relies on endogenous proliferative capacity of the starting populations and no
improvement in self-renewal was observed in aged AEC-II cells treated with Rock-inhibitor.
In contrast, a robust increase in CFU% was seen in both young and aged AEC-II cells under
iPL induction. Thus we feel our interrupted reprogramming approach is significantly
different and quantitatively much more powerful.

168
Great effort have been placed on overcoming the low differentiation efficiency,
heterogeneous final products (Plath and Lowry, 2011; Schwartz et al., 2014) and
contamination by potentially tumorigenic undifferentiated cells (Ben-David and Benvenisty,
2011; Tapia and Schöler, 2016) in differentiation of ES and iPS cells to desired cell types.
There is no standardized approach applicable to all cell types, thus derivation of each unique
cell type from ES and iPS cells may require the development of hundreds of unique
protocols. In contrast, interrupted reprogramming was relatively straightforward to extend to
2 distinct populations of lung cells. This strategy is theoretically broadly applicable and
could be extended to other somatic cell types giving rise to numerous progenitor cell
populations with enhanced proliferation and restricted differentiation. This technology only
requires the starting population to be isolated in relative purity and current isolation of very
specific populations of adult cells is already possible using advanced flow cytometric sorting
and cell culture techniques from most organs. Nevertheless, the induction conditions may
require some optimization to satisfy specific cell type to obtain maximum expansion of iPL
cells.

169
Chapter 7
Future directions and Conclusion
As discussed above, we recognize that there are many questions surrounding iPL
cells need to be addressed in future studies. Firstly, the progenitor markers expressed by iPL
cells and epigenetic remodeling during interrupted reprogramming require further
characterization. Secondly, regarding to the rejuvenation consequence of interrupted
reprogramming, the epigenetic targets need to be explored. Thirdly, the utility of rejuvenated
progenitor-like cells to regenerate injured aged lungs needs to be examined in vivo. Finally,
for therapeutic use, the efficient application of this technique in human cells requires
evaluation.
7.1 Characterization of the progenitor cell markers expressed
by iPL cells
Some of the markers of current defined stem cell populations, which are absent in the
starting cells, have been found to be activated in iPL cells. Club-iPL cells can express basal
stem cell marker-P63, while AEC-II-iPL cells express markers of bipotent progenitor,
including Hopx, SPC and α64. Are these markers paralogous with the ones expressed in
endogenous airway stem cells? Do those corresponding proteins display similar or distinct
roles in regulating cell self-renewal and differentiation compared to the endogenous ones?
Future studies to examine the gene sequence and the protein structure will provide us
information for better understanding the mechanisms underlying the progenitor-like
functionality of lineage-specific iPL cells.

170
7.2 Epigenetic characterization of iPL induction
Epigenetic mechanisms, DNA and histone methylation in particular, are responsible
for the maintenance of cellar memory and the regulation of cell-type-specific gene
expression.
Reprogramming through forced expression of OSKM transcription factors occurs through
major epigenetic remodeling driving somatic cells from the differentiated to the pluripotent
state. The epigenetic feature of the starting somatic cells has been erased to some extent
during the conversion to adopt a pluripotent stem cell epigenome.
Interrupted reprogramming, which occurs during the early stage of reprogramming,
allows induction of a progenitor-like state yet preservation of cellular identity to
spontaneously return to their differentiated state upon factors-withdrawal. Therefore,
understanding of the contribution of epigenetic regulation to the iPL process is required.
Which parts of the somatic epigenetic memory have been erased and which have been
maintained during interrupted reprogramming? How they specifically induce de-
differentiation state without affecting cellular identity? Experiments to examine the changes
of histone modification and DNA methylation during iPL induction will be performed.
7.3 Identification of epigenetic targets resulting rejuvenation
via interrupted reprogramming
Epigenetic dysregulation is a major driver of the aging process and is proposed as a
novel biomarker of biological aging.
Despite the fundamental differences, both interrupted reprogramming and direct
conversion/reprogramming employ a short period of induction of the 4 reprogramming
factors, avoiding pluripotency. Studies have demonstrated that transient expression of the 4
reprogramming factors to open chromatin and induce cell plasticity during the early
reprogramming process, fibroblasts can be converted to other somatic cell types, such as
neural progenitors and cardiomyocytes (Efe et al., 2011; Kim et al., 2011). This is done by
applying combinations of specific transcription factors (Caiazzo et al., 2011; Pfisterer et al.,
2011; Sancho-Martinez et al., 2012) and/or microRNA (Ambasudhan et al., 2011; Leonardo

171
et al., 2012) after a transient induction of these factors. However, rejuvenation cannot be
achieved by direct reprogramming and cells derived from aged fibroblast-iPS cells still
retain age-related epigenetic memory, including the age-specific transcriptional profiles.
In contrast, reprogramming of iPS cells can erase age-related epigenetic memory.
Epigenetic remodeling during reprogramming process is the key driver of the restoration of
aging hallmarks to the youthful state, including telomere shortening, stem cell exhaustion,
mitochondrial dysfunction and cellular senescence.
Interrupted reprogramming is not only able to recapitulate the rejuvenation
consequence of iPS reprogramming, but also allows the maintenance of cellular identity to
generate large numbers of younger cells. We will determine if cells undergo progressive and
continuous rejuvenation during the reprogramming process, how aging-related epigenetic
memory has been erased and epigenome is remodeled during interrupted reprogramming. To
do this, we will compare the genome-wide transcriptional profiles of young AEC-IIs, aged
AEC-IIs and their iPL counterparts, and analyze the expression of aged-related genes,
including the ones related to DNA damage, stress response, and apoptosis. In addition, we
will employ comprehensive high-throughput array-based relative methylation (CHARM)
microarray analysis to compare the patterns of genomic DNA methylation in these cells.
7.4 Evaluation of the reparative capacity of aged AEC-II-iPL
cells in reversal of pulmonary fibrosis induced in aged mice
Aging plays a prominent role in triggering the pathogenesis of IPF. Studies have
demonstrated the age-related impaired reparative capacity in bleomycin-induced fibrosis
model (Hecker et al., 2014). Two months after bleomycin injury, young mice were able to
recover from fibrosis, whereas aged mice failed to resolve fibrotic injury showing persistent
fibrosis characterized by extensive accumulation of senescent and apoptosis-resistant
myofibroblasts in the lungs. Importantly, many of the hallmarks of aging implicated in
humans IPF, including epigenetic alterations, DNA damage, stem cell exhaustion, telomere
dysfunction and(shortened telomeres), mtDNA damage-mediated AEC apoptosis, and
activated endoplasmic reticulum (ER) stress response (Liu et al., 2013; Mossman et al.,

172
2011; Noble et al., 2012; Thannickal, 2013; Uhal and Nguyen, 2013; Weiss et al., 2010) are
mainly observed in AEC-II cells.
Thus, we will evaluate the therapeutic benefit of aged-AEC-II-iPL cells in reversal of
fibrosis in aged recipients. Four groups of cells, including young AEC-IIs, aged AEC-IIs and
their iPL counterparts will be transtracheally delivered to aged recipients at both the
inflammatory and fibrotic phases of BLM-induced injury. Previous studies of BLM model
of pulmonary fibrosis showed that aged male mice develop more prominent fibrotic disease
compared with aged female mice, and the pulmonary functional alterations can be detected
by FlexiVent measurement. We will therefore select an aged-male mouse BLM model to do
a direct comparison of all four groups of cells in improving lung function. A pilot study will
be performed to determine the in situ period for cells where a significant pulmonary
dysfunction could be detected in BLM mice compared to saline control. The therapeutic
benefit of injected cells will be examined following cell delivery. In addition to mortality,
development and severity of fibrosis will be assessed by measurements of static compliance,
leukocyte infiltration, levels of collagen deposition, and stereological quantification of
fibrotic areas in histological sections. We will also quantify proinflammatory and profibrotic
chemokine and cytokine levels in the bronchoalveolar lavage fluid. The cell retention and
differentiation will be examined by PCR and immunostaining of relevant markers.
7.5 Generation of human AEC-II-iPL cells and amelioration of
aged-related deterioration
Finally, for therapeutic use, we will examine if this technique can be efficiently
applied in human cells. Normal human lung cells will be isolated from excess tissue
remaining from both young and aged lung transplant donors and further purified by flow
cytometry to sort CD45neg
CD31neg
EpCAMpos
T1αneg
population which is enrich for AEC-II
cells. To better understand the aged-related decline in self-renewal capacity in human cells,
both young and aged AEC-II cells will be cultured in Matrigel-3D condition to access their
clonogenic capacity over passaging. For iPL induction, cells will be transfected with a
polycistronic lentiviral vectors driving dox-regulatable OSKM, and induced under a
underdetermined optimized condition allowing greater expansion of iPL cells before

173
reaching pluripotency. We will evaluate if interrupted reprogramming allows the
rejuvenation of aged AEC-II cells to a more youthful state yet preservation of AEC-II
identity and function. Specifically, to determine if cells are reset to a younger state, we will
measure the age-related deterioration in stem cell self-renewal capacity, telomerase activity,
telomere length, mitochondrial DNA and the epigenome histone methylation marks
(‘epigenetic clock’). The maintenance of AEC-II cell identity in aged-AEC-II-iPL and their
derivatives upon factors-withdrawal will be evaluated by their ability to generate alveolar-
like colonies composed of both AEC-I and AEC-II cells, and synthesis and secretion of
surfactant.
7.6 Conclusion
We developed a novel "interrupted reprogramming" strategy, to generate "induced
Progenitor-Like (iPL) cells" using carefully timed transient expression of induced
Pluripotent Stem (iPS) cell reprogramming factors. Lineage-specific iPL cells can be derived
from distinct epithelial populations and function as region-specific progenitor cells to
repopulate injured epithelium. Furthermore, interrupted reprogramming has the capacity to
rejuvenate aged AEC-II cells to a youthful state by ameliorating multiple age-associated
hallmarks, including self-renew exhaustion of stem cells, telomere attrition, accumulation of
DNA damage and epigenetic dysregulation.
Collectively, distinct from other approaches, interrupted reprogramming enables the
production of progenitor-like cells with controlled proliferation and lineage-restricted
differentiation in high purity. This would be extremely useful in a variety of regenerative
medicine practices, including cell replacement therapy, biohybrid devices, disease
modelling, and drug screening for human diseases.
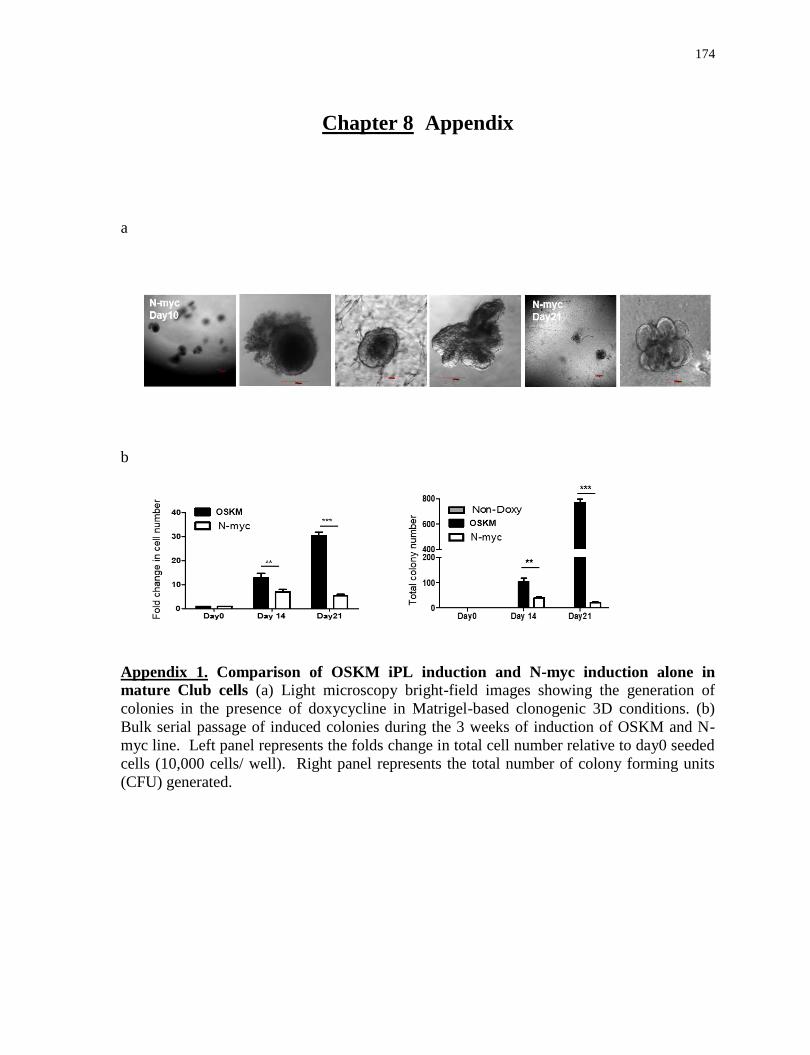
174
Chapter 8 Appendix
a
b
Appendix 1. Comparison of OSKM iPL induction and N-myc induction alone in
mature Club cells (a) Light microscopy bright-field images showing the generation of
colonies in the presence of doxycycline in Matrigel-based clonogenic 3D conditions. (b)
Bulk serial passage of induced colonies during the 3 weeks of induction of OSKM and N-
myc line. Left panel represents the folds change in total cell number relative to day0 seeded
cells (10,000 cells/ well). Right panel represents the total number of colony forming units
(CFU) generated.
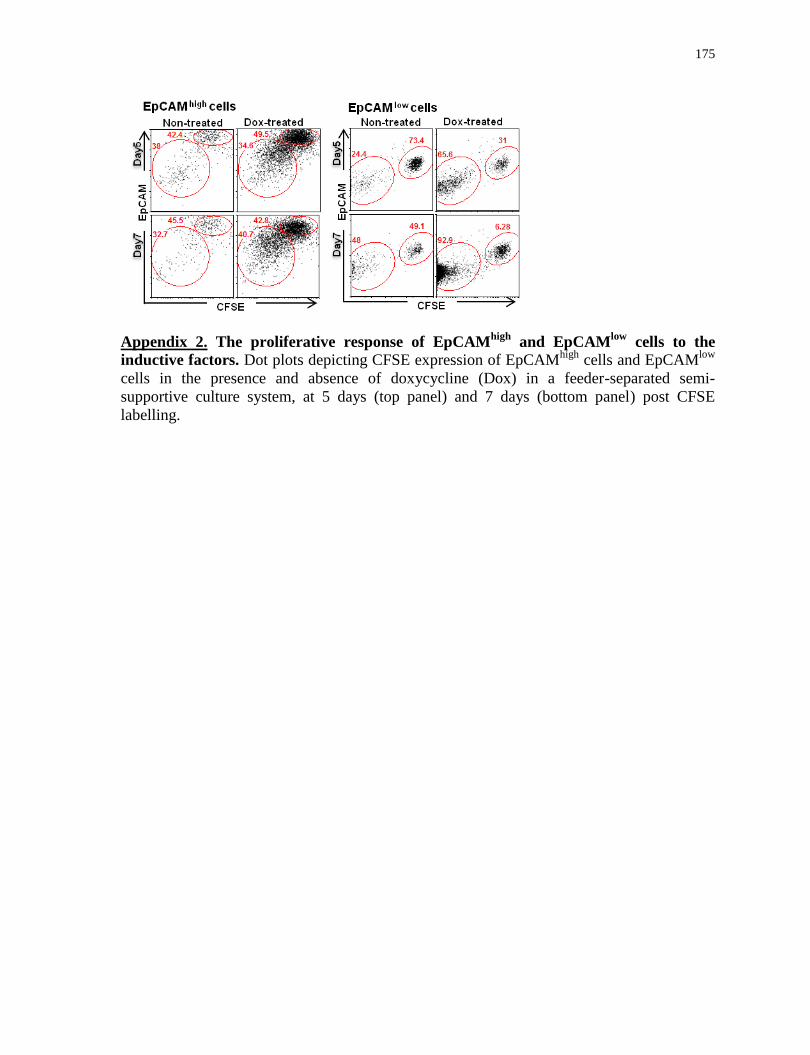
175
Appendix 2. The proliferative response of EpCAMhigh
and EpCAMlow
cells to the
inductive factors. Dot plots depicting CFSE expression of EpCAMhigh
cells and EpCAMlow
cells in the presence and absence of doxycycline (Dox) in a feeder-separated semi-
supportive culture system, at 5 days (top panel) and 7 days (bottom panel) post CFSE
labelling.
176
a. EpCAMhigh
population
177
b. EpCAMlow
population

178
Appendix 3. Characterization of induced colonies derived from EpCAMhigh
and
EpCAMlow
populations in 2D conditions. Light microscopy bright-field images showing
the generation of induced colonies and alkaline phosphatise staining. Confocal microscopy
images depicting induced colonies stained with nuclear stain DAPI (blue), Oct4/SSEA-1
(green) and Nanog/E-cadherin (red).
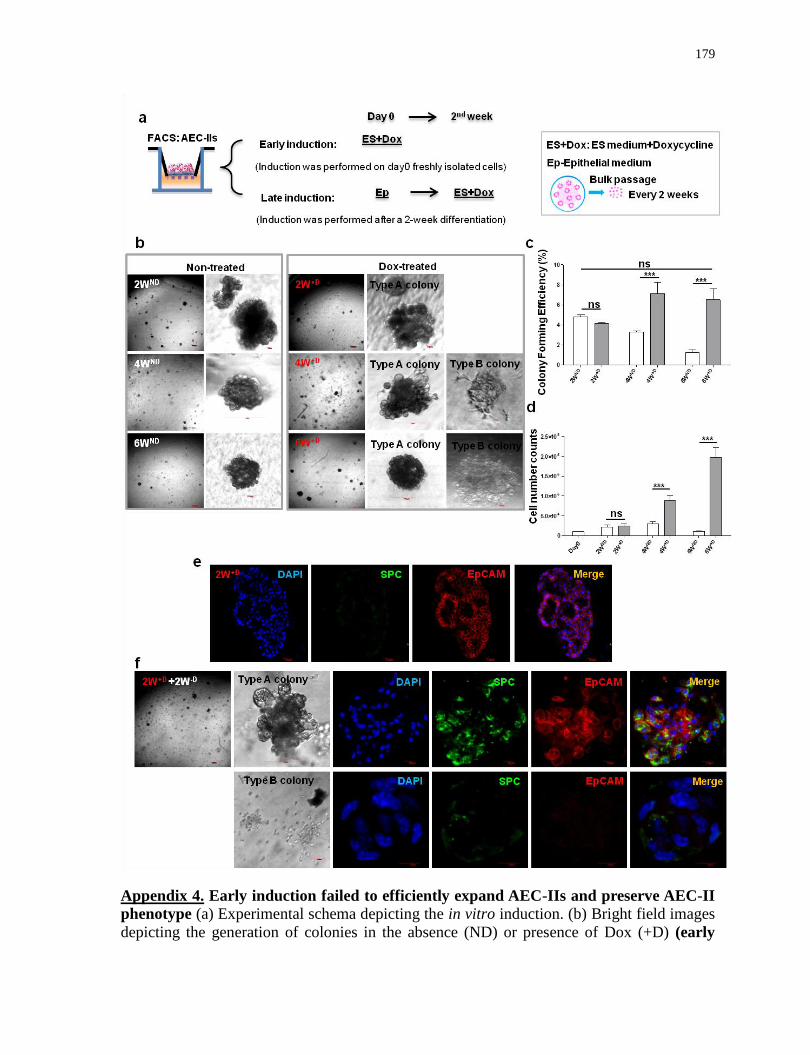
179
Appendix 4. Early induction failed to efficiently expand AEC-IIs and preserve AEC-II
phenotype (a) Experimental schema depicting the in vitro induction. (b) Bright field images
depicting the generation of colonies in the absence (ND) or presence of Dox (+D) (early

180
induction) in Matrigel-based 3D conditions. AEC-II cells treated with Dox for 4weeks
(4W+D
, 6W+D
) resulted in generation of type A (epithelial-like) and type B (mesenchymal-
like) colonies with distinct morphologies. The colony forming efficiency (c) and the fold
changes in total cell number (d) shown relative to day0 seeded cells (5,000 cells/ well).
Confocal microscopy images of 2-week Dox-treated cells (2W+D
) (e) and 2W+D
cells with
subsequent 2-week culture in Dox-free media (2W+D
+2W-D
) (f) showing nuclear stain DAPI
(blue), SPC (green) and EpCAM (red). For c and d, values are mean S.D. of three
independent biological replicates. *, p<0.05; **, p<0.001; ***, p<0.0001.
Appendix 5. Transient activation of inductive factors results an enhanced clonal
proliferation of EpCAMlow
population. Schematic graph (top panel) representing potential
outcomes of mix-culture experiment. Green, red and overlay (bottom panel) of fluorescence
from coculture of GFPpos
inducible and RFP non-inducible EpCAMlow
cells. Scale bar, 100
µm.
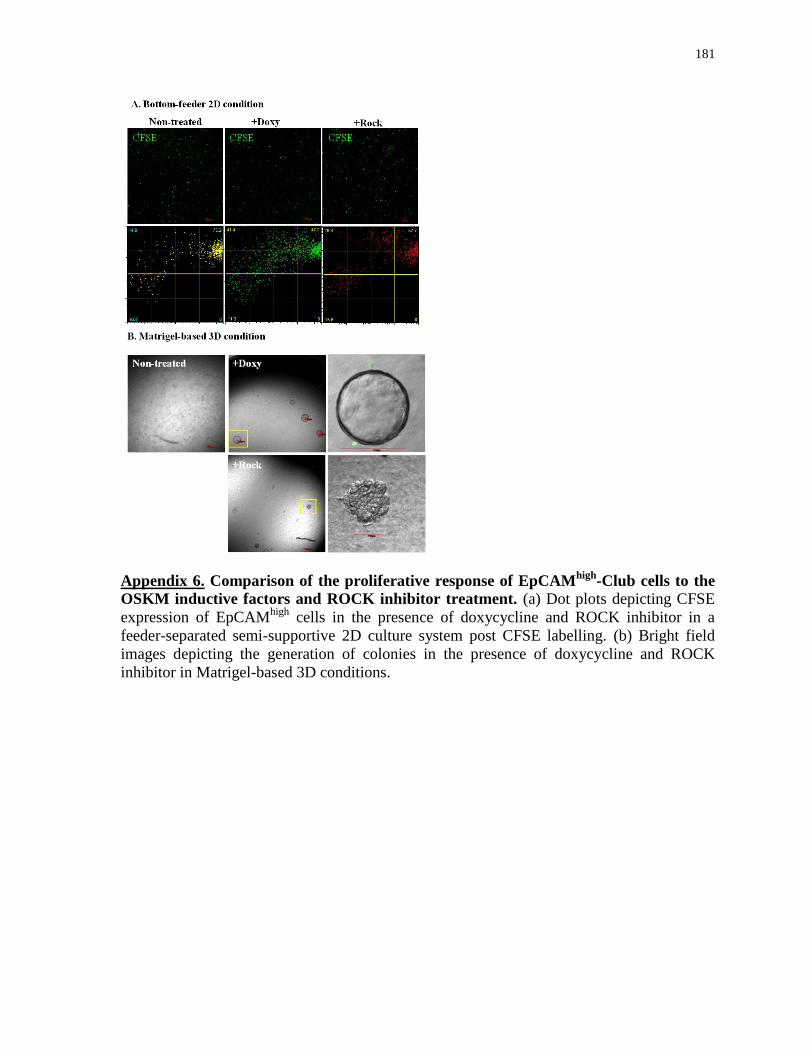
181
Appendix 6. Comparison of the proliferative response of EpCAMhigh
-Club cells to the
OSKM inductive factors and ROCK inhibitor treatment. (a) Dot plots depicting CFSE
expression of EpCAMhigh
cells in the presence of doxycycline and ROCK inhibitor in a
feeder-separated semi-supportive 2D culture system post CFSE labelling. (b) Bright field
images depicting the generation of colonies in the presence of doxycycline and ROCK
inhibitor in Matrigel-based 3D conditions.

182
References
Aasen, T., Raya, A., Barrero, M.J., Garreta, E., Consiglio, A., Gonzalez, F., Vassena, R.,
Bilić, J., Pekarik, V., Tiscornia, G., et al. (2008). Efficient and rapid generation of induced
pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 26, 1276–1284.
Abyzov, A., Mariani, J., Palejev, D., Zhang, Y., Haney, M.S., Tomasini, L., Ferrandino,
A.F., Rosenberg Belmaker, L.A., Szekely, A., Wilson, M., et al. (2012). Somatic copy
number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 492,
438–442.
Ahmad, T., Sundar, I.K., Lerner, C.A., Gerloff, J., Tormos, A.M., Yao, H., and Rahman, I.
(2015). Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence:
implications for chronic obstructive pulmonary disease. The FASEB Journal 29, 2912–2929.
Akram, K.M., Lomas, N.J., Forsyth, N.R., and Spiteri, M.A. (2014). Alveolar epithelial cells
in idiopathic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression
with simultaneous preferential over-expression of pro-apoptotic marker p53. Int J Clin Exp
Pathol 7, 552–564.
Alder, J.K., Chen, J.J.-L., Lancaster, L., Danoff, S., Su, S., Cogan, J.D., Vulto, I., Xie, M.,
Qi, X., Tuder, R.M., et al. (2008). Short telomeres are a risk factor for idiopathic pulmonary
fibrosis. Proc. Natl. Acad. Sci. U.S.A. 105, 13051–13056.
Alder, J.K., Guo, N., Kembou, F., Parry, E.M., Anderson, C.J., Gorgy, A.I., Walsh, M.F.,
Sussan, T., Biswal, S., Mitzner, W., et al. (2011). Telomere Length Is a Determinant of
Emphysema Susceptibility. American Journal of Respiratory and Critical Care Medicine
184, 904–912.
Alder, J.K., Barkauskas, C.E., Limjunyawong, N., Stanley, S.E., Kembou, F., Tuder, R.M.,
Hogan, B.L., Mitzner, W., and Armanios, M. (2015). Telomere dysfunction causes alveolar
stem cell failure. Proceedings of the National Academy of Sciences 112, 5099–5104.
Aldridge, J.R., Moseley, C.E., Boltz, D.A., Negovetich, N.J., Reynolds, C., Franks, J.,
Brown, S.A., Doherty, P.C., Webster, R.G., and Thomas, P.G. (2009). TNF/iNOS-producing
dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci.
U.S.A. 106, 5306–5311.
Ambasudhan, R., Talantova, M., Coleman, R., Yuan, X., Zhu, S., Lipton, S.A., and Ding, S.
(2011). Direct reprogramming of adult human fibroblasts to functional neurons under
defined conditions. Cell Stem Cell 9, 113–118.
Aoi, T., Yae, K., Nakagawa, M., Ichisaka, T., Okita, K., Takahashi, K., Chiba, T., and
Yamanaka, S. (2008). Generation of pluripotent stem cells from adult mouse liver and
stomach cells. Science 321, 699–702.

183
Araya, J., Cambier, S., Markovics, J.A., Wolters, P., Jablons, D., Hill, A., Finkbeiner, W.,
Jones, K., Broaddus, V.C., Sheppard, D., et al. (2007). Squamous metaplasia amplifies
pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Invest. 117,
3551–3562.
Armanios, M. (2012). Telomerase and idiopathic pulmonary fibrosis. Mutation
Research/Fundamental and Molecular Mechanisms of Mutagenesis 730, 52–58.
Armanios, M. (2013). Telomeres and age-related disease: how telomere biology informs
clinical paradigms. Journal of Clinical Investigation 123, 996–1002.
Armanios, M., and Blackburn, E.H. (2012). The telomere syndromes. Nature Reviews
Genetics 13, 693–704.
Armanios, M., Chen, J.-L., Chang, Y.-P.C., Brodsky, R.A., Hawkins, A., Griffin, C.A.,
Eshleman, J.R., Cohen, A.R., Chakravarti, A., Hamosh, A., et al. (2005). Haploinsufficiency
of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis
congenita. Proceedings of the National Academy of Sciences of the United States of
America 102, 15960–15964.
Armanios, M., Alder, J.K., Parry, E.M., Karim, B., Strong, M.A., and Greider, C.W. (2009).
Short Telomeres are Sufficient to Cause the Degenerative Defects Associated with Aging.
The American Journal of Human Genetics 85, 823–832.
Armanios, M.Y., Chen, J.J.-L., Cogan, J.D., Alder, J.K., Ingersoll, R.G., Markin, C.,
Lawson, W.E., Xie, M., Vulto, I., Phillips III, J.A., et al. (2007). Telomerase mutations in
families with idiopathic pulmonary fibrosis. New England Journal of Medicine 356, 1317–
1326.
Ashbaugh, D.G., Bigelow, D.B., Petty, T.L., and Levine, B.E. (1967). Acute respiratory
distress in adults. Lancet 2, 319–323.
Atkinson, J.J., Adair-Kirk, T.L., Kelley, D.G., deMello, D., and Senior, R.M. (2008). Clara
cell adhesion and migration to extracellular matrix. Respiratory Research 9, 1.
Avrahami, D., Li, C., Zhang, J., Schug, J., Avrahami, R., Rao, S., Stadler, M.B., Burger, L.,
Schübeler, D., Glaser, B., et al. (2015). Aging-Dependent Demethylation of Regulatory
Elements Correlates with Chromatin State and Improved β Cell Function. Cell Metabolism
22, 619–632.
Ayers, M.M., and Jeffery, P.K. (1988). Proliferation and differentiation in mammalian
airway epithelium. Eur. Respir. J. 1, 58–80.
Bachofen, M., and Weibel, E.R. (1982). Structural alterations of lung parenchyma in the
adult respiratory distress syndrome. Clin. Chest Med. 3, 35–56.

184
Barkauskas, C.E., Cronce, M.J., Rackley, C.R., Bowie, E.J., Keene, D.R., Stripp, B.R.,
Randell, S.H., Noble, P.W., and Hogan, B.L.M. (2013). Type 2 alveolar cells are stem cells
in adult lung. Journal of Clinical Investigation 123, 3025–3036.
Bar-Nur, O., Russ, H.A., Efrat, S., and Benvenisty, N. (2011). Epigenetic Memory and
Preferential Lineage-Specific Differentiation in Induced Pluripotent Stem Cells Derived
from Human Pancreatic Islet Beta Cells. Cell Stem Cell 9, 17–23.
Bastacky, J., Lee, C.Y., Goerke, J., Koushafar, H., Yager, D., Kenaga, L., Speed, T.P., Chen,
Y., and Clements, J.A. (1995). Alveolar lining layer is thin and continuous: low-temperature
scanning electron microscopy of rat lung. J. Appl. Physiol. 79, 1615–1628.
Batista, L.F.Z., Pech, M.F., Zhong, F.L., Nguyen, H.N., Xie, K.T., Zaug, A.J., Crary, S.M.,
Choi, J., Sebastiano, V., Cherry, A., et al. (2011). Telomere shortening and loss of self-
renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474, 399–402.
Beers, M.F., and Morrisey, E.E. (2011). The three R’s of lung health and disease: repair,
remodeling, and regeneration. J. Clin. Invest. 121, 2065–2073.
Benayoun, B.A., Pollina, E.A., and Brunet, A. (2015). Epigenetic regulation of ageing:
linking environmental inputs to genomic stability. Nature Reviews Molecular Cell Biology
16, 593–610.
Ben-David, U., and Benvenisty, N. (2011). The tumorigenicity of human embryonic and
induced pluripotent stem cells. Nat. Rev. Cancer 11, 268–277.
Bernardes de Jesus, B., Vera, E., Schneeberger, K., Tejera, A.M., Ayuso, E., Bosch, F., and
Blasco, M.A. (2012). Telomerase gene therapy in adult and old mice delays aging and
increases longevity without increasing cancer: TERT alone extends lifespan of adult/old
mice. EMBO Molecular Medicine 4, 691–704.
Berndt-Weis, M.L., Kauri, L.M., Williams, A., White, P., Douglas, G., and Yauk, C. (2009).
Global transcriptional characterization of a mouse pulmonary epithelial cell line for use in
genetic toxicology. Toxicology in Vitro 23, 816–833.
Bertoncello, I., and McQualter, J.L. (2010). Endogenous lung stem cells: what is their
potential for use in regenerative medicine? Expert Rev Respir Med 4, 349–362.
Birch, J., Anderson, R.K., Correia-Melo, C., Jurk, D., Hewitt, G., Marques, F.M., Green,
N.J., Moisey, E., Birrell, M.A., Belvisi, M.G., et al. (2015). DNA damage response at
telomeres contributes to lung ageing and chronic obstructive pulmonary disease. American
Journal of Physiology - Lung Cellular and Molecular Physiology ajplung.00293.2015.
Blackburn, E.H., Greider, C.W., and Szostak, J.W. (2006). Telomeres and telomerase: the
path from maize, Tetrahymena and yeast to human cancer and aging. Nature Medicine 12,
1133–1138.

185
Blasco, M.A. (2007). The epigenetic regulation of mammalian telomeres. Nature Reviews
Genetics 8, 299–309.
Blelloch, R., Venere, M., Yen, J., and Ramalho-Santos, M. (2007). Generation of induced
pluripotent stem cells in the absence of drug selection. Cell Stem Cell 1, 245–247.
Bock, C., Kiskinis, E., Verstappen, G., Gu, H., Boulting, G., Smith, Z.D., Ziller, M., Croft,
G.F., Amoroso, M.W., Oakley, D.H., et al. (2011). Reference Maps of human ES and iPS
cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144,
439–452.
Boers, J.E., Ambergen, A.W., and Thunnissen, F.B. (1998). Number and proliferation of
basal and parabasal cells in normal human airway epithelium. American Journal of
Respiratory and Critical Care Medicine 157, 2000–2006.
Boland, M.L., Chourasia, A.H., and Macleod, K.F. (2013). Mitochondrial Dysfunction in
Cancer. Frontiers in Oncology 3.
Bonasio, R., Tu, S., and Reinberg, D. (2010). Molecular Signals of Epigenetic States.
Science 330, 612–616.
Bormann, F., Rodríguez-Paredes, M., Hagemann, S., Manchanda, H., Kristof, B., Gutekunst,
J., Raddatz, G., Haas, R., Terstegen, L., Wenck, H., et al. (2016). Reduced DNA methylation
patterning and transcriptional connectivity define human skin aging. Aging Cell 15, 563–
571.
Boucher, R.C. (2003). Regulation of airway surface liquid volume by human airway
epithelia. Pflugers Arch. 445, 495–498.
Bove, P.F., Dang, H., Cheluvaraju, C., Jones, L.C., Liu, X., O’Neal, W.K., Randell, S.H.,
Schlegel, R., and Boucher, R.C. (2014). Breaking the In Vitro Alveolar Type II Cell
Proliferation Barrier while Retaining Ion Transport Properties. American Journal of
Respiratory Cell and Molecular Biology 50, 767–776.
Brambrink, T., Foreman, R., Welstead, G.G., Lengner, C.J., Wernig, M., Suh, H., and
Jaenisch, R. (2008). Sequential expression of pluripotency markers during direct
reprogramming of mouse somatic cells. Cell Stem Cell 2, 151–159.
Bratic, A., and Larsson, N.-G. (2013). The role of mitochondria in aging. Journal of Clinical
Investigation 123, 951–957.
Brock, T.G., Lee, Y.-J., Maydanski, E., Marburger, T.L., Luo, M., Paine, R., and Peters-
Golden, M. (2005). Nuclear localization of leukotriene A4 hydrolase in type II alveolar
epithelial cells in normal and fibrotic lung. Am. J. Physiol. Lung Cell Mol. Physiol. 289,
L224–L232.

186
Brody, S.L., Yan, X.H., Wuerffel, M.K., Song, S.-K., and Shapiro, S.D. (2000). Ciliogenesis
and left–right axis defects in Forkhead factor HFH-4–null mice. American Journal of
Respiratory Cell and Molecular Biology 23, 45–51.
Brune, K., Frank, J., Schwingshackl, A., Finigan, J., and Sidhaye, V.K. (2015). Pulmonary
epithelial barrier function: some new players and mechanisms. Am. J. Physiol. Lung Cell
Mol. Physiol. 308, L731–L745.
Brunet, A., and Berger, S.L. (2014). Epigenetics of Aging and Aging-related Disease. The
Journals of Gerontology Series A: Biological Sciences and Medical Sciences 69, S17–S20.
Budinger, G.R.S., Mutlu, G.M., Urich, D., Soberanes, S., Buccellato, L.J., Hawkins, K.,
Chiarella, S.E., Radigan, K.A., Eisenbart, J., Agrawal, H., et al. (2011). Epithelial cell death
is an important contributor to oxidant-mediated acute lung injury. Am. J. Respir. Crit. Care
Med. 183, 1043–1054.
Buganim, Y., Faddah, D.A., Cheng, A.W., Itskovich, E., Markoulaki, S., Ganz, K., Klemm,
S.L., van Oudenaarden, A., and Jaenisch, R. (2012). Single-cell expression analyses during
cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150,
1209–1222.
Caiazzo, M., Dell’Anno, M.T., Dvoretskova, E., Lazarevic, D., Taverna, S., Leo, D.,
Sotnikova, T.D., Menegon, A., Roncaglia, P., Colciago, G., et al. (2011). Direct generation
of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–
227.
Caiazzo, M., Okawa, Y., Ranga, A., Piersigilli, A., Tabata, Y., and Lutolf, M.P. (2016).
Defined three-dimensional microenvironments boost induction of pluripotency. Nature
Materials 15, 344–352.
Calado, R.T., Regal, J.A., Kleiner, D.E., Schrump, D.S., Peterson, N.R., Pons, V., Chanock,
S.J., Lansdorp, P.M., and Young, N.S. (2009). A Spectrum of Severe Familial Liver
Disorders Associate with Telomerase Mutations. PLoS ONE 4, e7926.
Cantin, A.M., Hanrahan, J.W., Bilodeau, G., Ellis, L., Dupuis, A., Liao, J., Zielenski, J., and
Durie, P. (2006). Cystic fibrosis transmembrane conductance regulator function is
suppressed in cigarette smokers. Am. J. Respir. Crit. Care Med. 173, 1139–1144.
Carey, B.W., Markoulaki, S., Beard, C., Hanna, J., and Jaenisch, R. (2009). Single-gene
transgenic mouse strains for reprogramming adult somatic cells. Nature Methods.
Carey, B.W., Markoulaki, S., Beard, C., Hanna, J., and Jaenisch, R. (2010). Single-gene
transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods 7, 56–59.
Cargnoni, A., Gibelli, L., Tosini, A., Signoroni, P.B., Nassuato, C., Arienti, D., Lombardi,
G., Albertini, A., Wengler, G.S., and Parolini, O. (2009). Transplantation of allogeneic and
xenogeneic placenta-derived cells reduces bleomycin-induced lung fibrosis. Cell
Transplantation 18, 405–422.

187
Castellani, C., Cuppens, H., Macek, M., Cassiman, J.J., Kerem, E., Durie, P., Tullis, E.,
Assael, B.M., Bombieri, C., Brown, A., et al. (2008). Consensus on the use and
interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 7, 179–
196.
Cerletti, M., Jang, Y.C., Finley, L.W.S., Haigis, M.C., and Wagers, A.J. (2012). Short-Term
Calorie Restriction Enhances Skeletal Muscle Stem Cell Function. Cell Stem Cell 10, 515–
519.
Chapman, H.A., Li, X., Alexander, J.P., Brumwell, A., Lorizio, W., Tan, K., Sonnenberg,
A., Wei, Y., and Vu, T.H. (2011). Integrin α6β4 identifies an adult distal lung epithelial
population with regenerative potential in mice. Journal of Clinical Investigation 121, 2855–
2862.
Chapman, S., Liu, X., Meyers, C., Schlegel, R., and McBride, A.A. (2010). Human
keratinocytes are efficiently immortalized by a Rho kinase inhibitor. Journal of Clinical
Investigation 120, 2619–2626.
Charville, G.W., and Rando, T.A. (2011). Stem cell ageing and non-random chromosome
segregation. Philosophical Transactions of the Royal Society B: Biological Sciences 366,
85–93.
Chen, R., Zhang, K., Chen, H., Zhao, X., Wang, J., Li, L., Cong, Y., Ju, Z., Xu, D.,
Williams, B.R.G., et al. (2015). Telomerase Deficiency Causes Alveolar Stem Cell
Senescence-associated Low-grade Inflammation in Lungs. Journal of Biological Chemistry
290, 30813–30829.
Cheng, L., Hansen, N.F., Zhao, L., Du, Y., Zou, C., Donovan, F.X., Chou, B.-K., Zhou, G.,
Li, S., Dowey, S.N., et al. (2012). Low incidence of DNA sequence variation in human
induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem
Cell 10, 337–344.
Chilosi, M., Carloni, A., Rossi, A., and Poletti, V. (2013). Premature lung aging and cellular
senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema.
Translational Research 162, 156–173.
Chin, M.H., Mason, M.J., Xie, W., Volinia, S., Singer, M., Peterson, C., Ambartsumyan, G.,
Aimiuwu, O., Richter, L., Zhang, J., et al. (2009). Induced pluripotent stem cells and
embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell 5,
111–123.
Chin, M.H., Pellegrini, M., Plath, K., and Lowry, W.E. (2010). Molecular analyses of
human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell 7, 263–269.
Christodoulou, C., Longmire, T.A., Shen, S.S., Bourdon, A., Sommer, C.A., Gadue, P.,
Spira, A., Gouon-Evans, V., Murphy, G.J., Mostoslavsky, G., et al. (2011). Mouse ES and
iPS cells can form similar definitive endoderm despite differences in imprinted genes. J.
Clin. Invest. 121, 2313–2325.

188
Clancy, J.L., Patel, H.R., Hussein, S.M.I., Tonge, P.D., Cloonan, N., Corso, A.J., Li, M.,
Lee, D.-S., Shin, J.-Y., Wong, J.J.L., et al. (2014). Small RNA changes en route to distinct
cellular states of induced pluripotency. Nature Communications 5, 5522.
Cole, B.B., Smith, R.W., Jenkins, K.M., Graham, B.B., Reynolds, P.R., and Reynolds, S.D.
(2010). Tracheal Basal cells: a facultative progenitor cell pool. Am. J. Pathol. 177, 362–376.
Cronkhite, J.T., Xing, C., Raghu, G., Chin, K.M., Torres, F., Rosenblatt, R.L., and Garcia,
C.K. (2008). Telomere Shortening in Familial and Sporadic Pulmonary Fibrosis. American
Journal of Respiratory and Critical Care Medicine 178, 729–737.
Crosby, L.M., and Waters, C.M. (2010). Epithelial repair mechanisms in the lung. Am. J.
Physiol. Lung Cell Mol. Physiol. 298, L715–L731.
Crosby, L.M., Luellen, C., Zhang, Z., Tague, L.L., Sinclair, S.E., and Waters, C.M. (2011).
Balance of life and death in alveolar epithelial type II cells: proliferation, apoptosis, and the
effects of cyclic stretch on wound healing. Am. J. Physiol. Lung Cell Mol. Physiol. 301,
L536–L546.
Crystal, R.G., Randell, S.H., Engelhardt, J.F., Voynow, J., and Sunday, M.E. (2008). Airway
Epithelial Cells: Current Concepts and Challenges. Proceedings of the American Thoracic
Society 5, 772–777.
Cutz, E., Yeger, H., and Pan, J. (2007). Pulmonary Neuroendocrine Cell System in Pediatric
Lung Disease?Recent Advances. Pediatric and Developmental Pathology 10, 419–435.
Davis, P.B. (2006). Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 173, 475–482.
De Los Angeles, A., Ferrari, F., Xi, R., Fujiwara, Y., Benvenisty, N., Deng, H.,
Hochedlinger, K., Jaenisch, R., Lee, S., Leitch, H.G., et al. (2015). Hallmarks of
pluripotency. Nature 525, 469–478.
Desai, T.J., Brownfield, D.G., and Krasnow, M.A. (2014). Alveolar progenitor and stem
cells in lung development, renewal and cancer. Nature 507, 190–194.
De Water, R., Willems, L.N., Van Muijen, G.N., Franken, C., Fransen, J.A., Dijkman, J.H.,
and Kramps, J.A. (1986). Ultrastructural localization of bronchial antileukoprotease in
central and peripheral human airways by a gold-labeling technique using monoclonal
antibodies. Am. Rev. Respir. Dis. 133, 882–890.
Dobbs, L.G., and Gutierrez, J.A. (2001). Mechanical forces modulate alveolar epithelial
phenotypic expression. Comp. Biochem. Physiol., Part A Mol. Integr. Physiol. 129, 261–
266.
Dobbs, L.G., and Johnson, M.D. (2007). Alveolar epithelial transport in the adult lung.
Respir Physiol Neurobiol 159, 283–300.

189
Doi, A., Park, I.-H., Wen, B., Murakami, P., Aryee, M.J., Irizarry, R., Herb, B., Ladd-
Acosta, C., Rho, J., Loewer, S., et al. (2009). Differential methylation of tissue- and cancer-
specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic
stem cells and fibroblasts. Nat. Genet. 41, 1350–1353.
Du, H.-Y., Pumbo, E., Ivanovich, J., An, P., Maziarz, R.T., Reiss, U.M., Chirnomas, D.,
Shimamura, A., Vlachos, A., Lipton, J.M., et al. (2008). TERC and TERT gene mutations in
patients with bone marrow failure and the significance of telomere length measurements.
Blood 113, 309–316.
Duchesneau, P., Wong, A.P., and Waddell, T.K. (2010). Optimization of targeted cell
replacement therapy: a new approach for lung disease. Mol. Ther. 18, 1830–1836.
Duchesneau, P., Besla, R., Derouet, M.F., Guo, L., Karoubi, G., Silberberg, A., Wong, A.P.,
and Waddell, T.K. (2017). Partial Restoration of CFTR Function in cftr-Null Mice following
Targeted Cell Replacement Therapy. Molecular Therapy.
Efe, J.A., Hilcove, S., Kim, J., Zhou, H., Ouyang, K., Wang, G., Chen, J., and Ding, S.
(2011). Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming
strategy. Nat. Cell Biol. 13, 215–222.
Egan, J.J., Stewart, J.P., Hasleton, P.S., Arrand, J.R., Carroll, K.B., and Woodcock, A.A.
(1995). Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic
fibrosing alveolitis. Thorax 50, 1234–1239.
Eisner, M.D., Anthonisen, N., Coultas, D., Kuenzli, N., Perez-Padilla, R., Postma, D.,
Romieu, I., Silverman, E.K., Balmes, J.R., and Committee on Nonsmoking COPD,
Environmental and Occupational Health Assembly (2010). An official American Thoracic
Society public policy statement: Novel risk factors and the global burden of chronic
obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 182, 693–718.
Elborn, J.S. (2016). Cystic fibrosis. Lancet 388, 2519–2531.
Eminli, S., Utikal, J., Arnold, K., Jaenisch, R., and Hochedlinger, K. (2008).
Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence
of exogenous Sox2 expression. Stem Cells 26, 2467–2474.
Epperly, M.W., Guo, H., Gretton, J.E., and Greenberger, J.S. (2003). Bone Marrow Origin
of Myofibroblasts in Irradiation Pulmonary Fibrosis. American Journal of Respiratory Cell
and Molecular Biology 29, 213–224.
Evans, C.M., and Koo, J.S. (2009). Airway mucus: the good, the bad, the sticky. Pharmacol.
Ther. 121, 332–348.
Evans, M.J., and Plopper, C.G. (1988). The Role of Basal Cells in Adhesion of Columnar
Epithelium to Airway Basement Membrane1-3. Am Rev Respir Dis 138, 481–483.

190
Evans, C.M., Williams, O.W., Tuvim, M.J., Nigam, R., Mixides, G.P., Blackburn, M.R.,
DeMayo, F.J., Burns, A.R., Smith, C., Reynolds, S.D., et al. (2004). Mucin is produced by
clara cells in the proximal airways of antigen-challenged mice. Am. J. Respir. Cell Mol.
Biol. 31, 382–394.
Evans, M.J., Van Winkle, L.S., Fanucchi, M.V., and Plopper, C.G. (2001). Cellular and
molecular characteristics of basal cells in airway epithelium. Experimental Lung Research
27, 401–415.
Faner, R., Rojas, M., MacNee, W., and Agustí, A. (2012). Abnormal Lung Aging in Chronic
Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. American Journal of
Respiratory and Critical Care Medicine 186, 306–313.
Fedorov, I.A., Wilson, S.J., Davies, D.E., and Holgate, S.T. (2005). Epithelial stress and
structural remodelling in childhood asthma. Thorax 60, 389–394.
Fehrenbach, H. (2001). Alveolar epithelial type II cell: defender of the alveolus revisited.
Respiratory Research 2, 33.
Fehrenbach, H., Kasper, M., Tschernig, T., Pan, T., Schuh, D., Shannon, J.M., Müller, M.,
and Mason, R.J. (1999). Keratinocyte growth factor-induced hyperplasia of rat alveolar type
II cells in vivo is resolved by differentiation into type I cells and by apoptosis. Eur. Respir. J.
14, 534–544.
Feser, J., and Tyler, J. (2011). Chromatin structure as a mediator of aging. FEBS Letters
585, 2041–2048.
Firth, A.L., Dargitz, C.T., Qualls, S.J., Menon, T., Wright, R., Singer, O., Gage, F.H.,
Khanna, A., and Verma, I.M. (2014). Generation of multiciliated cells in functional airway
epithelia from human induced pluripotent stem cells. Proceedings of the National Academy
of Sciences 111, E1723–E1730.
Fisher, J.T., Zhang, Y., and Engelhardt, J.F. (2011). Comparative biology of cystic fibrosis
animal models. Methods Mol. Biol. 742, 311–334.
Flores, I. (2005). Effects of Telomerase and Telomere Length on Epidermal Stem Cell
Behavior. Science 309, 1253–1256.
Flores, I., and Blasco, M.A. (2010). The role of telomeres and telomerase in stem cell aging.
FEBS Letters 584, 3826–3830.
Fraga, M.F., and Esteller, M. (2007). Epigenetics and aging: the targets and the marks.
Trends in Genetics 23, 413–418.
Fulcher, M.L., and Randell, S.H. (2013). Human nasal and tracheo-bronchial respiratory
epithelial cell culture. Methods Mol. Biol. 945, 109–121.

191
Fussner, E., Djuric, U., Strauss, M., Hotta, A., Perez-Iratxeta, C., Lanner, F., Dilworth, F.J.,
Ellis, J., and Bazett-Jones, D.P. (2011). Constitutive heterochromatin reorganization during
somatic cell reprogramming. EMBO J. 30, 1778–1789.
Ganesan, S., Comstock, A.T., and Sajjan, U.S. (2013). Barrier function of airway tract
epithelium. Tissue Barriers 1, e24997.
Gangl, K., Reininger, R., Bernhard, D., Campana, R., Pree, I., Reisinger, J., Kneidinger, M.,
Kundi, M., Dolznig, H., Thurnher, D., et al. (2009). Cigarette smoke facilitates allergen
penetration across respiratory epithelium. Allergy 64, 398–405.
Gems, D., and Partridge, L. (2013). Genetics of Longevity in Model Organisms: Debates
and Paradigm Shifts. Annual Review of Physiology 75, 621–644.
Ghadiri, M., Young, P.M., and Traini, D. (2016). Cell-based therapies for the treatment of
idiopathic pulmonary fibrosis (IPF) disease. Expert Opinion on Biological Therapy 16, 375–
387.
Ghaedi, M., Calle, E.A., Mendez, J.J., Gard, A.L., Balestrini, J., Booth, A., Bove, P.F., Gui,
L., White, E.S., and Niklason, L.E. (2013). Human iPS cell–derived alveolar epithelium
repopulates lung extracellular matrix. Journal of Clinical Investigation 123, 4950–4962.
Ghosh, M., Helm, K.M., Smith, R.W., Giordanengo, M.S., Li, B., Shen, H., and Reynolds,
S.D. (2011). A single cell functions as a tissue-specific stem cell and the in vitro niche-
forming cell. Am. J. Respir. Cell Mol. Biol. 45, 459–469.
Ghosh, Z., Wilson, K.D., Wu, Y., Hu, S., Quertermous, T., and Wu, J.C. (2010). Persistent
donor cell gene expression among human induced pluripotent stem cells contributes to
differences with human embryonic stem cells. PLoS ONE 5, e8975.
Giangreco, A., Arwert, E.N., Rosewell, I.R., Snyder, J., Watt, F.M., and Stripp, B.R. (2009).
Stem cells are dispensable for lung homeostasis but restore airways after injury. Proc. Natl.
Acad. Sci. U.S.A. 106, 9286–9291.
Gifford, A.M., and Chalmers, J.D. (2014). The role of neutrophils in cystic fibrosis. Curr.
Opin. Hematol. 21, 16–22.
Gohy, S.T., Hupin, C., Fregimilicka, C., Detry, B.R., Bouzin, C., Gaide Chevronay, H.,
Lecocq, M., Weynand, B., Ladjemi, M.Z., Pierreux, C.E., et al. (2015). Imprinting of the
COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur. Respir. J.
45, 1258–1272.
Gohy, S.T., Hupin, C., Pilette, C., and Ladjemi, M.Z. (2016). Chronic inflammatory airway
diseases: the central role of the epithelium revisited. Clin. Exp. Allergy 46, 529–542.
González, F., Georgieva, D., Vanoli, F., Shi, Z.-D., Stadtfeld, M., Ludwig, T., Jasin, M., and
Huangfu, D. (2013). Homologous recombination DNA repair genes play a critical role in
reprogramming to a pluripotent state. Cell Rep 3, 651–660.

192
Gotoh, S., Ito, I., Nagasaki, T., Yamamoto, Y., Konishi, S., Korogi, Y., Matsumoto, H.,
Muro, S., Hirai, T., Funato, M., et al. (2014). Generation of Alveolar Epithelial Spheroids
via Isolated Progenitor Cells from Human Pluripotent Stem Cells. Stem Cell Reports 3, 394–
403.
Grant, M.R., Mostov, K.E., Tlsty, T.D., and Hunt, C.A. (2006). Simulating properties of in
vitro epithelial cell morphogenesis. PLoS Computational Biology 2, e129.
Green, D.R., Galluzzi, L., and Kroemer, G. (2011a). Mitochondria and the Autophagy-
Inflammation-Cell Death Axis in Organismal Aging. Science 333, 1109–1112.
Green, M.D., Chen, A., Nostro, M.-C., d’Souza, S.L., Schaniel, C., Lemischka, I.R., Gouon-
Evans, V., Keller, G., and Snoeck, H.-W. (2011b). Generation of anterior foregut endoderm
from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 29, 267–272.
Gregory, T.J., Longmore, W.J., Moxley, M.A., Whitsett, J.A., Reed, C.R., Fowler, A.A.,
Hudson, L.D., Maunder, R.J., Crim, C., and Hyers, T.M. (1991). Surfactant chemical
composition and biophysical activity in acute respiratory distress syndrome. J. Clin. Invest.
88, 1976–1981.
Gribbin, J., Hubbard, R.B., Le Jeune, I., Smith, C.J.P., West, J., and Tata, L.J. (2006).
Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax
61, 980–985.
Gumbleton, M. (2001). Caveolae as potential macromolecule trafficking compartments
within alveolar epithelium. Adv. Drug Deliv. Rev. 49, 281–300.
Guseh, J.S., Bores, S.A., Stanger, B.Z., Zhou, Q., Anderson, W.J., Melton, D.A., and
Rajagopal, J. (2009). Notch signaling promotes airway mucous metaplasia and inhibits
alveolar development. Development 136, 1751–1759.
Gustafsson, B.I., Kidd, M., Chan, A., Malfertheiner, M.V., and Modlin, I.M. (2008).
Bronchopulmonary neuroendocrine tumors. Cancer 113, 5–21.
Hackett, B.P., and Gitlin, J.D. (1992). Cell-specific expression of a Clara cell secretory
protein-human growth hormone gene in the bronchiolar epithelium of transgenic mice. Proc.
Natl. Acad. Sci. U.S.A. 89, 9079–9083.
Hackett, T.-L., Shaheen, F., Johnson, A., Wadsworth, S., Pechkovsky, D.V., Jacoby, D.B.,
Kicic, A., Stick, S.M., and Knight, D.A. (2008). Characterization of side population cells
from human airway epithelium. Stem Cells 26, 2576–2585.
Hackett, T.-L., de Bruin, H.G., Shaheen, F., van den Berge, M., van Oosterhout, A.J.,
Postma, D.S., and Heijink, I.H. (2013). Caveolin-1 controls airway epithelial barrier
function. Implications for asthma. Am. J. Respir. Cell Mol. Biol. 49, 662–671.

193
Halliwell, B., and Whiteman, M. (2004). Measuring reactive species and oxidative damage
in vivo and in cell culture: how should you do it and what do the results mean? British
Journal of Pharmacology 142, 231–255.
Han, S., and Brunet, A. (2012). Histone methylation makes its mark on longevity. Trends in
Cell Biology 22, 42–49.
Hanna, J., Markoulaki, S., Schorderet, P., Carey, B.W., Beard, C., Wernig, M., Creyghton,
M.P., Steine, E.J., Cassady, J.P., Foreman, R., et al. (2008). Direct reprogramming of
terminally differentiated mature B lymphocytes to pluripotency. Cell 133, 250–264.
Hanna, J., Saha, K., Pando, B., van Zon, J., Lengner, C.J., Creyghton, M.P., van
Oudenaarden, A., and Jaenisch, R. (2009). Direct cell reprogramming is a stochastic process
amenable to acceleration. Nature 462, 595–601.
Hansson, J., Rafiee, M.R., Reiland, S., Polo, J.M., Gehring, J., Okawa, S., Huber, W.,
Hochedlinger, K., and Krijgsveld, J. (2012). Highly Coordinated Proteome Dynamics during
Reprogramming of Somatic Cells to Pluripotency. Cell Reports 2, 1579–1592.
Hartsock, A., and Nelson, W.J. (2008). Adherens and tight junctions: structure, function and
connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669.
Hashimoto, Y., Moki, T., Takizawa, T., Shiratsuchi, A., and Nakanishi, Y. (2007). Evidence
for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages
in mice. J. Immunol. 178, 2448–2457.
Hawkins, R.D., Hon, G.C., Lee, L.K., Ngo, Q., Lister, R., Pelizzola, M., Edsall, L.E., Kuan,
S., Luu, Y., Klugman, S., et al. (2010). Distinct epigenomic landscapes of pluripotent and
lineage-committed human cells. Cell Stem Cell 6, 479–491.
Hayflick, L. (2007). Biological Aging Is No Longer an Unsolved Problem. Annals of the
New York Academy of Sciences 1100, 1–13.
Hayflick, L., and MOORHEAD, P. (1973). The Serial Cultivation of Human Diploid. Read.
Mamm. Cell Cult 25, 66.
Hecker, L., Logsdon, N.J., Kurundkar, D., Kurundkar, A., Bernard, K., Hock, T., Meldrum,
E., Sanders, Y.Y., and Thannickal, V.J. (2014). Reversal of Persistent Fibrosis in Aging by
Targeting Nox4-Nrf2 Redox Imbalance. Science Translational Medicine 6, 231ra47–
ra231ra47.
Heijink, I.H., Brandenburg, S.M., Postma, D.S., and van Oosterhout, A.J.M. (2012).
Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery.
Eur. Respir. J. 39, 419–428.
Held, N.M., and Houtkooper, R.H. (2015). Mitochondrial quality control pathways as
determinants of metabolic health. BioEssays 37, 867–876.

194
Herold, S., Tabar, T.S., Janssen, H., Hoegner, K., Cabanski, M., Lewe-Schlosser, P.,
Albrecht, J., Driever, F., Vadasz, I., Seeger, W., et al. (2011). Exudate macrophages
attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia.
Am. J. Respir. Crit. Care Med. 183, 1380–1390.
Herold, S., Becker, C., Ridge, K.M., and Budinger, G.R.S. (2015). Influenza virus-induced
lung injury: pathogenesis and implications for treatment. Eur. Respir. J. 45, 1463–1478.
Hiemstra, P.S., McCray, P.B., and Bals, R. (2015). The innate immune function of airway
epithelial cells in inflammatory lung disease. Eur. Respir. J. 45, 1150–1162.
Hochberg, C.H., and Sidhaye, V.K. (2017). The Respiratory Epithelium in COPD. In Lung
Epithelial Biology in the Pathogenesis of Pulmonary Disease, (Elsevier), pp. 165–184.
Hockemeyer, D., Soldner, F., Cook, E.G., Gao, Q., Mitalipova, M., and Jaenisch, R. (2008).
A drug-inducible system for direct reprogramming of human somatic cells to pluripotency.
Cell Stem Cell 3, 346–353.
Hoegger, M.J., Fischer, A.J., McMenimen, J.D., Ostedgaard, L.S., Tucker, A.J., Awadalla,
M.A., Moninger, T.O., Michalski, A.S., Hoffman, E.A., Zabner, J., et al. (2014). Impaired
mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis.
Science 345, 818–822.
Hogan, B.L.M., Barkauskas, C.E., Chapman, H.A., Epstein, J.A., Jain, R., Hsia, C.C.W.,
Niklason, L., Calle, E., Le, A., Randell, S.H., et al. (2014). Repair and Regeneration of the
Respiratory System: Complexity, Plasticity, and Mechanisms of Lung Stem Cell Function.
Cell Stem Cell 15, 123–138.
Hogg, J.C., and Timens, W. (2009). The pathology of chronic obstructive pulmonary
disease. Annu Rev Pathol 4, 435–459.
Hong, K.U. (2003). In vivo differentiation potential of tracheal basal cells: evidence for
multipotent and unipotent subpopulations. AJP: Lung Cellular and Molecular Physiology
286, 643L – 649.
Hong, H., Takahashi, K., Ichisaka, T., Aoi, T., Kanagawa, O., Nakagawa, M., Okita, K., and
Yamanaka, S. (2009). Suppression of induced pluripotent stem cell generation by the p53-
p21 pathway. Nature 460, 1132–1135.
Hong, K.U., Reynolds, S.D., Giangreco, A., Hurley, C.M., and Stripp, B.R. (2001). Clara
cell secretory protein–expressing cells of the airway neuroepithelial body microenvironment
include a label-retaining subset and are critical for epithelial renewal after progenitor cell
depletion. American Journal of Respiratory Cell and Molecular Biology 24, 671–681.
Hong, K.U., Reynolds, S.D., Watkins, S., Fuchs, E., and Stripp, B.R. (2004). Basal cells are
a multipotent progenitor capable of renewing the bronchial epithelium. The American
Journal of Pathology 164, 577–588.

195
Horowitz, J.C., Cui, Z., Moore, T.A., Meier, T.R., Reddy, R.C., Toews, G.B., Standiford,
T.J., and Thannickal, V.J. (2006). Constitutive activation of prosurvival signaling in alveolar
mesenchymal cells isolated from patients with nonresolving acute respiratory distress
syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L415–L425.
Hou, P., Li, Y., Zhang, X., Liu, C., Guan, J., Li, H., Zhao, T., Ye, J., Yang, W., Liu, K., et
al. (2013). Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule
Compounds. Science 341, 651–654.
Huang, S.X.L., Islam, M.N., O’Neill, J., Hu, Z., Yang, Y.-G., Chen, Y.-W., Mumau, M.,
Green, M.D., Vunjak-Novakovic, G., Bhattacharya, J., et al. (2013). Efficient generation of
lung and airway epithelial cells from human pluripotent stem cells. Nature Biotechnology.
Huangfu, D., Maehr, R., Guo, W., Eijkelenboom, A., Snitow, M., Chen, A.E., and Melton,
D.A. (2008). Induction of pluripotent stem cells by defined factors is greatly improved by
small-molecule compounds. Nat. Biotechnol. 26, 795–797.
Hussein, S.M.I., Puri, M.C., Tonge, P.D., Benevento, M., Corso, A.J., Clancy, J.L.,
Mosbergen, R., Li, M., Lee, D.-S., Cloonan, N., et al. (2014). Genome-wide characterization
of the routes to pluripotency. Nature 516, 198–206.
Imai, Y., Parodo, J., Kajikawa, O., de Perrot, M., Fischer, S., Edwards, V., Cutz, E., Liu, M.,
Keshavjee, S., Martin, T.R., et al. (2003). Injurious mechanical ventilation and end-organ
epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory
distress syndrome. JAMA 289, 2104–2112.
Ingenito, E.P., Mora, R., Cullivan, M., Marzan, Y., Haley, K., Mark, L., and Sonna, L.A.
(2001). Decreased surfactant protein-B expression and surfactant dysfunction in a murine
model of acute lung injury. Am. J. Respir. Cell Mol. Biol. 25, 35–44.
Issa, J.-P. (2014). Aging and epigenetic drift: a vicious cycle. Journal of Clinical
Investigation 124, 24–29.
Ito, T., Udaka, N., Yazawa, T., Okudela, K., Hayashi, H., Sudo, T., Guillemot, F.,
Kageyama, R., and Kitamura, H. (2000). Basic helix-loop-helix transcription factors regulate
the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development 127,
3913–3921.
Ito, T., Udaka, N., Okudela, K., Yazawa, T., and Kitamura, H. (2003). Mechanisms of
neuroendocrine differentiation in pulmonary neuroendocrine cells and small cell carcinoma.
Endocrine Pathology 14, 133–139.
Jain, R., Barkauskas, C.E., Takeda, N., Bowie, E.J., Aghajanian, H., Wang, Q.,
Padmanabhan, A., Manderfield, L.J., Gupta, M., Li, D., et al. (2015). Plasticity of Hopx+
type I alveolar cells to regenerate type II cells in the lung. Nature Communications 6, 6727.

196
Jaskelioff, M., Muller, F.L., Paik, J.-H., Thomas, E., Jiang, S., Adams, A.C., Sahin, E., Kost-
Alimova, M., Protopopov, A., Cadiñanos, J., et al. (2011). Telomerase reactivation reverses
tissue degeneration in aged telomerase-deficient mice. Nature 469, 102–106.
Jeffery, P.K. (1983). Morphologic features of airway surface epithelial cells and glands. Am.
Rev. Respir. Dis. 128, S14–S20.
Jia, F., Wilson, K.D., Sun, N., Gupta, D.M., Huang, M., Li, Z., Panetta, N.J., Chen, Z.Y.,
Robbins, R.C., Kay, M.A., et al. (2010). A nonviral minicircle vector for deriving human
iPS cells. Nat. Methods 7, 197–199.
Jones, J.G., Minty, B.D., Lawler, P., Hulands, G., Crawley, J.C., and Veall, N. (1980).
Increased alveolar epithelial permeability in cigarette smokers. Lancet 1, 66–68.
José, R.J., Williams, A.E., Mercer, P.F., Sulikowski, M.G., Brown, J.S., and Chambers, R.C.
(2015). Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during
bacterial pulmonary infection. J. Immunol. 194, 6024–6034.
Jung, M., and Pfeifer, G.P. (2015). Aging and DNA methylation. BMC Biology 13, 7.
Kalluri, R., and Weinberg, R.A. (2009). The basics of epithelial-mesenchymal transition. J.
Clin. Invest. 119, 1420–1428.
Kastenberg, Z.J., and Odorico, J.S. (2008). Alternative sources of pluripotency: science,
ethics, and stem cells. Transplant Rev (Orlando) 22, 215–222.
Kim, C.F.B., Jackson, E.L., Woolfenden, A.E., Lawrence, S., Babar, I., Vogel, S., Crowley,
D., Bronson, R.T., and Jacks, T. (2005). Identification of Bronchioalveolar Stem Cells in
Normal Lung and Lung Cancer. Cell 121, 823–835.
Kim, D., Kim, C.-H., Moon, J.-I., Chung, Y.-G., Chang, M.-Y., Han, B.-S., Ko, S., Yang, E.,
Cha, K.Y., Lanza, R., et al. (2009). Generation of human induced pluripotent stem cells by
direct delivery of reprogramming proteins. Cell Stem Cell 4, 472–476.
Kim, J., Efe, J.A., Zhu, S., Talantova, M., Yuan, X., Wang, S., Lipton, S.A., Zhang, K., and
Ding, S. (2011). Direct reprogramming of mouse fibroblasts to neural progenitors. Proc.
Natl. Acad. Sci. U.S.A. 108, 7838–7843.
Kim, J.B., Zaehres, H., Wu, G., Gentile, L., Ko, K., Sebastiano, V., Araúzo-Bravo, M.J.,
Ruau, D., Han, D.W., Zenke, M., et al. (2008). Pluripotent stem cells induced from adult
neural stem cells by reprogramming with two factors. Nature 454, 646–650.
Kim, K., Doi, A., Wen, B., Ng, K., Zhao, R., Cahan, P., Kim, J., Aryee, M.J., Ji, H., Ehrlich,
L.I.R., et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–
290.

197
Kim, S., Ahn, S.E., Lee, J.H., Lim, D.-S., Kim, K.-S., Chung, H.-M., and Lee, S.-H. (2007).
A novel culture technique for human embryonic stem cells using porous membranes. Stem
Cells 25, 2601–2609.
Kimbrel, E.A., and Lanza, R. (2015). Current status of pluripotent stem cells: moving the
first therapies to the clinic. Nature Reviews Drug Discovery 14, 681–692.
King, T.E., Pardo, A., and Selman, M. (2011). Idiopathic pulmonary fibrosis. The Lancet
378, 1949–1961.
King, T.E., Bradford, W.Z., Castro-Bernardini, S., Fagan, E.A., Glaspole, I., Glassberg,
M.K., Gorina, E., Hopkins, P.M., Kardatzke, D., Lancaster, L., et al. (2014). A Phase 3 Trial
of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. New England Journal of
Medicine 370, 2083–2092.
Kirkwood, T.B.L. (2005). Understanding the Odd Science of Aging. Cell 120, 437–447.
Kirkwood, T.B.L., and Shanley, D.P. (2010). The connections between general and
reproductive senescence and the evolutionary basis of menopause: Kirkwood &
Shanley. Annals of the New York Academy of Sciences 1204, 21–29.
Knight, D.A., and Holgate, S.T. (2003). The airway epithelium: structural and functional
properties in health and disease. Respirology 8, 432–446.
Knowles, M.R., Stutts, M.J., Spock, A., Fischer, N., Gatzy, J.T., and Boucher, R.C. (1983).
Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science 221, 1067–
1070.
Koche, R.P., Smith, Z.D., Adli, M., Gu, H., Ku, M., Gnirke, A., Bernstein, B.E., and
Meissner, A. (2011). Reprogramming factor expression initiates widespread targeted
chromatin remodeling. Cell Stem Cell 8, 96–105.
Koga, H., Kaushik, S., and Cuervo, A.M. (2011). Protein homeostasis and aging: The
importance of exquisite quality control. Ageing Research Reviews 10, 205–215.
Koma, T., Yoshimatsu, K., Nagata, N., Sato, Y., Shimizu, K., Yasuda, S.P., Amada, T.,
Nishio, S., Hasegawa, H., and Arikawa, J. (2014). Neutrophil depletion suppresses
pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by
hantavirus infection in C.B-17 SCID mice. J. Virol. 88, 7178–7188.
Kosmider, B., Messier, E.M., Chu, H.W., and Mason, R.J. (2011). Human Alveolar
Epithelial Cell Injury Induced by Cigarette Smoke. PLoS ONE 6, e26059.
Kotton, D.N., and Morrisey, E.E. (2014). Lung regeneration: mechanisms, applications and
emerging stem cell populations. Nat. Med. 20, 822–832.

198
Kotton, D.N., Fabian, A.J., and Mulligan, R.C. (2005). Failure of bone marrow to
reconstitute lung epithelium. American Journal of Respiratory Cell and Molecular Biology
33, 328–334.
Kroegel, C., Bergmann, N., Heider, C., Moeser, A., Happe, J., Schlenker, Y., Bartuschka,
B., Henzgen, M., Walther, R., Reissig, A., et al. (2009). [Interferon-alpha as treatment
option in severe persistent uncontrolled bronchial asthma: an open label study].
Pneumologie 63, 307–313.
Kumamoto, M., Nishiwaki, T., Matsuo, N., Kimura, H., and Matsushima, K. (2009).
Minimally cultured bone marrow mesenchymal stem cells ameliorate fibrotic lung injury.
European Respiratory Journal 34, 740–748.
Kumar, P.A., Hu, Y., Yamamoto, Y., Hoe, N.B., Wei, T.S., Mu, D., Sun, Y., Joo, L.S.,
Dagher, R., Zielonka, E.M., et al. (2011). Distal Airway Stem Cells Yield Alveoli In Vitro
and during Lung Regeneration following H1N1 Influenza Infection. Cell 147, 525–538.
Lam, H.C., Cloonan, S.M., Bhashyam, A.R., Haspel, J.A., Singh, A., Sathirapongsasuti, J.F.,
Cervo, M., Yao, H., Chung, A.L., Mizumura, K., et al. (2013). Histone deacetylase 6-
mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Invest.
123, 5212–5230.
Lama, V., Moore, B.B., Christensen, P., Toews, G.B., and Peters-Golden, M. (2002).
Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial
cells is cyclooxygenase-2-dependent. Am. J. Respir. Cell Mol. Biol. 27, 752–758.
de Lange, T. (2009). How telomeres solve the end-protection problem. Science 326, 948–
952.
Lapasset, L., Milhavet, O., Prieur, A., Besnard, E., Babled, A., Ait-Hamou, N., Leschik, J.,
Pellestor, F., Ramirez, J.-M., De Vos, J., et al. (2011). Rejuvenating senescent and
centenarian human cells by reprogramming through the pluripotent state. Genes &
Development 25, 2248–2253.
Lauweryns, J.M., Van Lommel, A.T., and Dom, R.J. (1985). Innervation of rabbit
intrapulmonary neuroepithelial bodies. Quantitative and qualitative ultrastructural study
after vagotomy. J. Neurol. Sci. 67, 81–92.
Lavelle, G.M., White, M.M., Browne, N., McElvaney, N.G., and Reeves, E.P. (2016).
Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences.
Biomed Res Int 2016, 5258727.
Lawson, W.E., Cheng, D.-S., Degryse, A.L., Tanjore, H., Polosukhin, V.V., Xu, X.C.,
Newcomb, D.C., Jones, B.R., Roldan, J., Lane, K.B., et al. (2011). Endoplasmic reticulum
stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. U.S.A. 108, 10562–
10567.

199
Lee, C., Mitsialis, S.A., Aslam, M., Vitali, S.H., Vergadi, E., Konstantinou, G., Sdrimas, K.,
Fernandez-Gonzalez, A., and Kourembanas, S. (2012). Exosomes Mediate the
Cytoprotective Action of Mesenchymal Stromal Cells on Hypoxia-Induced Pulmonary
Hypertension. Circulation 126, 2601–2611.
Lee, J.-H., Bhang, D.H., Beede, A., Huang, T.L., Stripp, B.R., Bloch, K.D., Wagers, A.J.,
Tseng, Y.-H., Ryeom, S., and Kim, C.F. (2014). Lung Stem Cell Differentiation in Mice
Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 156,
440–455.
Lee, J.-H., Bhang, D.H., Beede, A., Huang, T.L., Stripp, B.R., Bloch, K.D., Wagers, A.J.,
Tseng, Y.-H., Ryeom, S., and Kim, C.F. (2014c). Lung Stem Cell Differentiation in Mice
Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 156,
440–455.
Leeman, K.T., Fillmore, C.M., and Kim, C.F. (2014). Lung stem and progenitor cells in
tissue homeostasis and disease. Curr. Top. Dev. Biol. 107, 207–233.
Leonardo, T.R., Schultheisz, H.L., Loring, J.F., and Laurent, L.C. (2012). The functions of
microRNAs in pluripotency and reprogramming. Nature Cell Biology 14, 1114–1121.
Lerou, P.H., Yabuuchi, A., Huo, H., Miller, J.D., Boyer, L.F., Schlaeger, T.M., and Daley,
G.Q. (2008). Derivation and maintenance of human embryonic stem cells from poor-quality
in vitro fertilization embryos. Nat Protoc 3, 923–933.
Levenberg, S., Huang, N.F., Lavik, E., Rogers, A.B., Itskovitz-Eldor, J., and Langer, R.
(2003). Differentiation of human embryonic stem cells on three-dimensional polymer
scaffolds. Proc. Natl. Acad. Sci. U.S.A. 100, 12741–12746.
Li, R., Liang, J., Ni, S., Zhou, T., Qing, X., Li, H., He, W., Chen, J., Li, F., Zhuang, Q., et al.
(2010). A mesenchymal-to-epithelial transition initiates and is required for the nuclear
reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63.
Liang, J., Zhang, Y., Xie, T., Liu, N., Chen, H., Geng, Y., Kurkciyan, A., Mena, J.M.,
Stripp, B.R., Jiang, D., et al. (2016). Hyaluronan and TLR4 promote surfactant-protein-C-
positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice.
Nature Medicine 22, 1285–1293.
Liao, B., Bao, X., Liu, L., Feng, S., Zovoilis, A., Liu, W., Xue, Y., Cai, J., Guo, X., Qin, B.,
et al. (2011). MicroRNA cluster 302-367 enhances somatic cell reprogramming by
accelerating a mesenchymal-to-epithelial transition. J. Biol. Chem. 286, 17359–17364.
Liebler, J.M., Marconett, C.N., Juul, N., Wang, H., Liu, Y., Flodby, P., Laird-Offringa, I.A.,
Minoo, P., and Zhou, B. (2016). Combinations of differentiation markers distinguish
subpopulations of alveolar epithelial cells in adult lung. American Journal of Physiology -
Lung Cellular and Molecular Physiology 310, L114–L120.

200
Lin, C.Y., Lovén, J., Rahl, P.B., Paranal, R.M., Burge, C.B., Bradner, J.E., Lee, T.I., and
Young, R.A. (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell
151, 56–67.
Lin, K.L., Sweeney, S., Kang, B.D., Ramsburg, E., and Gunn, M.D. (2011). CCR2-
antagonist prophylaxis reduces pulmonary immune pathology and markedly improves
survival during influenza infection. J. Immunol. 186, 508–515.
Lister, R., Pelizzola, M., Kida, Y.S., Hawkins, R.D., Nery, J.R., Hon, G., Antosiewicz-
Bourget, J., O’Malley, R., Castanon, R., Klugman, S., et al. (2011). Hotspots of aberrant
epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73.
Liu, L., and Rando, T.A. (2011). Manifestations and mechanisms of stem cell aging. The
Journal of Cell Biology 193, 257–266.
Liu, G., Cheresh, P., and Kamp, D.W. (2013). Molecular Basis of Asbestos-Induced Lung
Disease. Annual Review of Pathology: Mechanisms of Disease 8, 161–187.
Liu, G.-H., Barkho, B.Z., Ruiz, S., Diep, D., Qu, J., Yang, S.-L., Panopoulos, A.D., Suzuki,
K., Kurian, L., Walsh, C., et al. (2011a). Recapitulation of premature ageing with iPSCs
from Hutchinson–Gilford progeria syndrome. Nature 472, 221–225.
Liu, L., van Groen, T., Kadish, I., Li, Y., Wang, D., James, S.R., Karpf, A.R., and
Tollefsbol, T.O. (2011b). Insufficient DNA methylation affects healthy aging and promotes
age-related health problems. Clinical Epigenetics 2, 349–360.
Liu, X., Ory, V., Chapman, S., Yuan, H., Albanese, C., Kallakury, B., Timofeeva, O.A.,
Nealon, C., Dakic, A., Simic, V., et al. (2012). ROCK Inhibitor and Feeder Cells Induce the
Conditional Reprogramming of Epithelial Cells. The American Journal of Pathology 180,
599–607.
Liu, Y., Kumar, V.S., Zhang, W., Rehman, J., and Malik, A.B. (2015). Activation of type II
cells into regenerative stem cell antigen-1(+) cells during alveolar repair. Am. J. Respir. Cell
Mol. Biol. 53, 113–124.
Loewer, S., Cabili, M.N., Guttman, M., Loh, Y.-H., Thomas, K., Park, I.H., Garber, M.,
Curran, M., Onder, T., Agarwal, S., et al. (2010). Large intergenic non-coding RNA-RoR
modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 42, 1113–
1117.
Lok, S.S., Haider, Y., Howell, D., Stewart, J.P., Hasleton, P.S., and Egan, J.J. (2002).
Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in
bleomycin resistant mice. Eur. Respir. J. 20, 1228–1232.
Lomas, N.J., Watts, K.L., Akram, K.M., Forsyth, N.R., and Spiteri, M.A. (2012). Idiopathic
pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue
remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol 5, 58–71.

201
Longmire, T.A., Ikonomou, L., Hawkins, F., Christodoulou, C., Cao, Y., Jean, J.C., Kwok,
L.W., Mou, H., Rajagopal, J., Shen, S.S., et al. (2012). Efficient derivation of purified lung
and thyroid progenitors from embryonic stem cells. Cell Stem Cell 10, 398–411.
López-Otín, C., Blasco, M.A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The
Hallmarks of Aging. Cell 153, 1194–1217.
Love, D., Li, F.-Q., Burke, M.C., Cyge, B., Ohmitsu, M., Cabello, J., Larson, J.E., Brody,
S.L., Cohen, J.C., and Takemaru, K.-I. (2010). Altered Lung Morphogenesis, Epithelial Cell
Differentiation and Mechanics in Mice Deficient in the Wnt/β-Catenin Antagonist Chibby.
PLoS ONE 5, e13600.
Luger, K., Mäder, A.W., Richmond, R.K., Sargent, D.F., and Richmond, T.J. (1997). core
particle at 2.8 A resolution. Nature 389, 18.
Lujan, E., Zunder, E.R., Ng, Y.H., Goronzy, I.N., Nolan, G.P., and Wernig, M. (2015). Early
reprogramming regulators identified by prospective isolation and mass cytometry. Nature
521, 352–356.
Lumsden, A.B., McLean, A., and Lamb, D. (1984). Goblet and Clara cells of human distal
airways: evidence for smoking induced changes in their numbers. Thorax 39, 844–849.
Maherali, N., Sridharan, R., Xie, W., Utikal, J., Eminli, S., Arnold, K., Stadtfeld, M.,
Yachechko, R., Tchieu, J., Jaenisch, R., et al. (2007). Directly reprogrammed fibroblasts
show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1,
55–70.
Maherali, N., Ahfeldt, T., Rigamonti, A., Utikal, J., Cowan, C., and Hochedlinger, K.
(2008). A high-efficiency system for the generation and study of human induced pluripotent
stem cells. Cell Stem Cell 3, 340–345.
Marchetto, M.C.N., Yeo, G.W., Kainohana, O., Marsala, M., Gage, F.H., and Muotri, A.R.
(2009). Transcriptional signature and memory retention of human-induced pluripotent stem
cells. PLoS ONE 4, e7076.
Marión, R.M., and Blasco, M.A. (2010). Telomere rejuvenation during nuclear
reprogramming. Current Opinion in Genetics & Development 20, 190–196.
Marion, R.M., Strati, K., Li, H., Tejera, A., Schoeftner, S., Ortega, S., Serrano, M., and
Blasco, M.A. (2009). Telomeres Acquire Embryonic Stem Cell Characteristics in Induced
Pluripotent Stem Cells. Cell Stem Cell 4, 141–154.
Marsh, L.M., Cakarova, L., Kwapiszewska, G., von Wulffen, W., Herold, S., Seeger, W.,
and Lohmeyer, J. (2009). Surface expression of CD74 by type II alveolar epithelial cells: a
potential mechanism for macrophage migration inhibitory factor-induced epithelial repair.
AJP: Lung Cellular and Molecular Physiology 296, L442–L452.

202
Marshall, R.P., Bellingan, G., Webb, S., Puddicombe, A., Goldsack, N., McAnulty, R.J., and
Laurent, G.J. (2000). Fibroproliferation occurs early in the acute respiratory distress
syndrome and impacts on outcome. Am. J. Respir. Crit. Care Med. 162, 1783–1788.
Marson, A., Levine, S.S., Cole, M.F., Frampton, G.M., Brambrink, T., Johnstone, S.,
Guenther, M.G., Johnston, W.K., Wernig, M., Newman, J., et al. (2008). Connecting
microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells.
Cell 134, 521–533.
Martin-Belmonte, F., Yu, W., Rodr?guez-Fraticelli, A.E., Ewald, A., Werb, Z., Alonso,
M.A., and Mostov, K. (2008). Cell-Polarity Dynamics Controls the Mechanism of Lumen
Formation in Epithelial Morphogenesis. Current Biology 18, 507–513.
Masui, S., Nakatake, Y., Toyooka, Y., Shimosato, D., Yagi, R., Takahashi, K., Okochi, H.,
Okuda, A., Matoba, R., Sharov, A.A., et al. (2007). Pluripotency governed by Sox2 via
regulation of Oct3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 9, 625–635.
Matthay, M.A., Ware, L.B., and Zimmerman, G.A. (2012). The acute respiratory distress
syndrome. J. Clin. Invest. 122, 2731–2740.
McQualter, J.L., Yuen, K., Williams, B., and Bertoncello, I. (2010). Evidence of an
epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proceedings of the
National Academy of Sciences 107, 1414–1419.
Meiners, S., Eickelberg, O., and Königshoff, M. (2015). Hallmarks of the ageing lung.
European Respiratory Journal 45, 807–827.
Mercer, R.R., Russell, M.L., Roggli, V.L., and Crapo, J.D. (1994). Cell number and
distribution in human and rat airways. Am. J. Respir. Cell Mol. Biol. 10, 613–624.
Metzger, R.J., Klein, O.D., Martin, G.R., and Krasnow, M.A. (2008). The branching
programme of mouse lung development. Nature 453, 745–750.
Mihai, C., Bao, S., Lai, J.-P., Ghadiali, S.N., and Knoell, D.L. (2012). PTEN inhibition
improves wound healing in lung epithelia through changes in cellular mechanics that
enhance migration. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L287–L299.
Mikkelsen, T.S., Hanna, J., Zhang, X., Ku, M., Wernig, M., Schorderet, P., Bernstein, B.E.,
Jaenisch, R., Lander, E.S., and Meissner, A. (2008). Dissecting direct reprogramming
through integrative genomic analysis. Nature 454, 49–55.
Milara, J., Peiró, T., Serrano, A., and Cortijo, J. (2013). Epithelial to mesenchymal transition
is increased in patients with COPD and induced by cigarette smoke. Thorax 68, 410–420.
Miller, B.E., and Hook, G.E. (1990). Hypertrophy and hyperplasia of alveolar type II cells in
response to silica and other pulmonary toxicants. Environ. Health Perspect. 85, 15–23.

203
Miyoshi, K., Yanagi, S., Kawahara, K., Nishio, M., Tsubouchi, H., Imazu, Y., Koshida, R.,
Matsumoto, N., Taguchi, A., Yamashita, S., et al. (2013). Epithelial Pten controls acute lung
injury and fibrosis by regulating alveolar epithelial cell integrity. Am. J. Respir. Crit. Care
Med. 187, 262–275.
Molenda, N., Urbanova, K., Weiser, N., Kusche-Vihrog, K., Günzel, D., and Schillers, H.
(2014). Paracellular transport through healthy and cystic fibrosis bronchial epithelial cell
lines--do we have a proper model? PLoS ONE 9, e100621.
Molina, S.A., Stauffer, B., Moriarty, H.K., Kim, A.H., McCarty, N.A., and Koval, M.
(2015). Junctional abnormalities in human airway epithelial cells expressing F508del CFTR.
Am. J. Physiol. Lung Cell Mol. Physiol. 309, L475–L487.
Molofsky, A.V., Slutsky, S.G., Joseph, N.M., He, S., Pardal, R., Krishnamurthy, J.,
Sharpless, N.E., and Morrison, S.J. (2006). Increasing p16INK4a expression decreases
forebrain progenitors and neurogenesis during ageing. Nature 443, 448–452.
Montgomery, M.K., and Turner, N. (2014). Mitochondrial dysfunction and insulin
resistance: an update. Endocrine Connections 4, R1–R15.
Moodley, Y., Atienza, D., Manuelpillai, U., Samuel, C.S., Tchongue, J., Ilancheran, S.,
Boyd, R., and Trounson, A. (2009). Human Umbilical Cord Mesenchymal Stem Cells
Reduce Fibrosis of Bleomycin-Induced Lung Injury. The American Journal of Pathology
175, 303–313.
Mori, M., Mahoney, J.E., Stupnikov, M.R., Paez-Cortez, J.R., Szymaniak, A.D., Varelas, X.,
Herrick, D.B., Schwob, J., Zhang, H., and Cardoso, W.V. (2015). Notch3-Jagged signaling
controls the pool of undifferentiated airway progenitors. Development 142, 258–267.
Moss, M., Bucher, B., Moore, F.A., Moore, E.E., and Parsons, P.E. (1996). The role of
chronic alcohol abuse in the development of acute respiratory distress syndrome in adults.
JAMA 275, 50–54.
Mossman, B.T., Lippmann, M., Hesterberg, T.W., Kelsey, K.T., Barchowsky, A., and
Bonner, J.C. (2011). Pulmonary Endpoints (Lung Carcinomas and Asbestosis) Following
Inhalation Exposure to Asbestos. Journal of Toxicology and Environmental Health, Part B
14, 76–121.
Mou, H., Zhao, R., Sherwood, R., Ahfeldt, T., Lapey, A., Wain, J., Sicilian, L., Izvolsky, K.,
Lau, F.H., Musunuru, K., et al. (2012). Generation of Multipotent Lung and Airway
Progenitors from Mouse ESCs and Patient-Specific Cystic Fibrosis iPSCs. Cell Stem Cell
10, 385–397.
Mutze, K., Vierkotten, S., Milosevic, J., Eickelberg, O., and Königshoff, M. (2015). Enolase
1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/β-catenin-
driven trans-differentiation of murine alveolar epithelial cells. Dis Model Mech 8, 877–890.

204
Nagy, A., and Nagy, K. (2010). The mysteries of induced pluripotency: where will they
lead? Nat. Methods 7, 22–24.
Nagy, A., Gócza, E., Diaz, E.M., Prideaux, V.R., Iványi, E., Markkula, M., and Rossant, J.
(1990). Embryonic stem cells alone are able to support fetal development in the mouse.
Development 110, 815–821.
Nagy, A., Rossant, J., Nagy, R., Abramow-Newerly, W., and Roder, J.C. (1993). Derivation
of completely cell culture-derived mice from early-passage embryonic stem cells.
Proceedings of the National Academy of Sciences 90, 8424–8428.
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., Okita, K.,
Mochiduki, Y., Takizawa, N., and Yamanaka, S. (2008). Generation of induced pluripotent
stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106.
Nakajima, Y., Kawanami, O., Jin, E., Ghazizadeh, M., Honda, M., Asano, G., Horiba, K.,
and Ferrans, V.J. (1998). Immunohistochemical and ultrastructural studies of basal cells,
Clara cells and bronchiolar cuboidal cells in normal human airways. Pathology International
48, 944–953.
Nakamura, T., Sato, E., Fujiwara, N., Kawagoe, Y., Maeda, S., and Yamagishi, S. (2011).
Increased levels of soluble receptor for advanced glycation end products (sRAGE) and high
mobility group box 1 (HMGB1) are associated with death in patients with acute respiratory
distress syndrome. Clin. Biochem. 44, 601–604.
Narasaraju, T.A., Chen, H., Weng, T., Bhaskaran, M., Jin, N., Chen, J., Chen, Z., Chinoy,
M.R., and Liu, L. (2006). Expression profile of IGF system during lung injury and recovery
in rats exposed to hyperoxia: a possible role of IGF-1 in alveolar epithelial cell proliferation
and differentiation. J. Cell. Biochem. 97, 984–998.
National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS)
Clinical Trials Network, Wiedemann, H.P., Wheeler, A.P., Bernard, G.R., Thompson, B.T.,
Hayden, D., deBoisblanc, B., Connors, A.F., Hite, R.D., and Harabin, A.L. (2006).
Comparison of two fluid-management strategies in acute lung injury. N. Engl. J. Med. 354,
2564–2575.
Navarro, S., and Driscoll, B. (2017). Regeneration of the Aging Lung: A Mini-Review.
Gerontology 63, 270–280.
Noble, P.W., Barkauskas, C.E., and Jiang, D. (2012). Pulmonary fibrosis: patterns and
perpetrators. Journal of Clinical Investigation 122, 2756–2762.
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: In Sickness and in Health. Cell 148,
1145–1159.
Ocampo, A., Reddy, P., and Belmonte, J.C.I. (2016a). Anti-Aging Strategies Based on
Cellular Reprogramming. Trends in Molecular Medicine 22, 725–738.

205
Ocampo, A., Reddy, P., Martinez-Redondo, P., Platero-Luengo, A., Hatanaka, F., Hishida,
T., Li, M., Lam, D., Kurita, M., Beyret, E., et al. (2016b). In Vivo Amelioration of Age-
Associated Hallmarks by Partial Reprogramming. Cell 167, 1719–1733.e12.
Odorico, J.S., Kaufman, D.S., and Thomson, J.A. (2001). Multilineage differentiation from
human embryonic stem cell lines. Stem Cells 19, 193–204.
Ohta, H., Chiba, S., Ebina, M., Furuse, M., and Nukiwa, T. (2012). Altered expression of
tight junction molecules in alveolar septa in lung injury and fibrosis. Am. J. Physiol. Lung
Cell Mol. Physiol. 302, L193–L205.
Okita, K., Nakagawa, M., Hyenjong, H., Ichisaka, T., and Yamanaka, S. (2008). Generation
of mouse induced pluripotent stem cells without viral vectors. Science 322, 949–953.
Okubo, T. (2005). Nmyc plays an essential role during lung development as a dosage-
sensitive regulator of progenitor cell proliferation and differentiation. Development 132,
1363–1374.
Olivera, W., Ridge, K., Wood, L.D., and Sznajder, J.I. (1994). Active sodium transport and
alveolar epithelial Na-K-ATPase increase during subacute hyperoxia in rats. Am. J. Physiol.
266, L577–L584.
Olsen, C.O., Isakson, B.E., Seedorf, G.J., Lubman, R.L., and Boitano, S. (2005).
Extracellular matrix-driven alveolar epithelial cell differentiation in vitro. Exp. Lung Res.
31, 461–482.
Olson, A.L., Swigris, J.J., Lezotte, D.C., Norris, J.M., Wilson, C.G., and Brown, K.K.
(2007). Mortality from Pulmonary Fibrosis Increased in the United States from 1992 to
2003. American Journal of Respiratory and Critical Care Medicine 176, 277–284.
Ordoñez, C.L., Khashayar, R., Wong, H.H., Ferrando, R., Wu, R., Hyde, D.M., Hotchkiss,
J.A., Zhang, Y., Novikov, A., Dolganov, G., et al. (2001). Mild and moderate asthma is
associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression.
Am. J. Respir. Crit. Care Med. 163, 517–523.
Ortiz, L.A., Gambelli, F., McBride, C., Gaupp, D., Baddoo, M., Kaminski, N., and Phinney,
D.G. (2003). Mesenchymal stem cell engraftment in lung is enhanced in response to
bleomycin exposure and ameliorates its fibrotic effects. Proceedings of the National
Academy of Sciences 100, 8407–8411.
O’Sullivan, R.J., and Karlseder, J. (2012). The great unravelling: chromatin as a modulator
of the aging process. Trends in Biochemical Sciences 37, 466–476.
Pal, S., and Tyler, J.K. (2016). Epigenetics and aging. Science Advances 2, e1600584–
e1600584.

206
Pardo, A., Gibson, K., Cisneros, J., Richards, T.J., Yang, Y., Becerril, C., Yousem, S.,
Herrera, I., Ruiz, V., Selman, M., et al. (2005). Up-regulation and profibrotic role of
osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2, e251.
Pardo-Saganta, A., Law, B.M., Gonzalez-Celeiro, M., Vinarsky, V., and Rajagopal, J.
(2013). Ciliated cells of pseudostratified airway epithelium do not become mucous cells
after ovalbumin challenge. American Journal of Respiratory Cell and Molecular Biology 48,
364–373.
Pardo-Saganta, A., Law, B.M., Tata, P.R., Villoria, J., Saez, B., Mou, H., Zhao, R., and
Rajagopal, J. (2015). Injury induces direct lineage segregation of functionally distinct airway
basal stem/progenitor cell subpopulations. Cell Stem Cell 16, 184–197.
Pereira, C.F., Piccolo, F.M., Tsubouchi, T., Sauer, S., Ryan, N.K., Bruno, L., Landeira, D.,
Santos, J., Banito, A., Gil, J., et al. (2010). ESCs require PRC2 to direct the successful
reprogramming of differentiated cells toward pluripotency. Cell Stem Cell 6, 547–556.
Perl, A.-K.T., Kist, R., Shan, Z., Scherer, G., and Whitsett, J.A. (2005). Normal lung
development and function after Sox9 inactivation in the respiratory epithelium. Genesis 41,
23–32.
Petecchia, L., Sabatini, F., Varesio, L., Camoirano, A., Usai, C., Pezzolo, A., and Rossi,
G.A. (2009). Bronchial airway epithelial cell damage following exposure to cigarette smoke
includes disassembly of tight junction components mediated by the extracellular signal-
regulated kinase 1/2 pathway. Chest 135, 1502–1512.
Peterson, M.W., Walter, M.E., and Nygaard, S.D. (1995). Effect of neutrophil mediators on
epithelial permeability. Am. J. Respir. Cell Mol. Biol. 13, 719–727.
Petkova, D.K., Clelland, C.A., Ronan, J.E., Lewis, S., and Knox, A.J. (2003). Reduced
expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis.
Histopathology 43, 381–386.
Pfisterer, U., Kirkeby, A., Torper, O., Wood, J., Nelander, J., Dufour, A., Björklund, A.,
Lindvall, O., Jakobsson, J., and Parmar, M. (2011a). Direct conversion of human fibroblasts
to dopaminergic neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 10343–10348.
Pfisterer, U., Kirkeby, A., Torper, O., Wood, J., Nelander, J., Dufour, A., Björklund, A.,
Lindvall, O., Jakobsson, J., and Parmar, M. (2011b). Direct conversion of human fibroblasts
to dopaminergic neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 10343–10348.
Plath, K., and Lowry, W.E. (2011). Progress in understanding reprogramming to the induced
pluripotent state. Nat. Rev. Genet. 12, 253–265.
Pollina, E.A., and Brunet, A. (2011). Epigenetic regulation of aging stem cells. Oncogene
30, 3105–3126.

207
Polo, J.M., Liu, S., Figueroa, M.E., Kulalert, W., Eminli, S., Tan, K.Y., Apostolou, E.,
Stadtfeld, M., Li, Y., Shioda, T., et al. (2010). Cell type of origin influences the molecular
and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 28, 848–
855.
Polo, J.M., Anderssen, E., Walsh, R.M., Schwarz, B.A., Nefzger, C.M., Lim, S.M., Borkent,
M., Apostolou, E., Alaei, S., Cloutier, J., et al. (2012). A molecular roadmap of
reprogramming somatic cells into iPS cells. Cell 151, 1617–1632.
Powers, E.T., Morimoto, R.I., Dillin, A., Kelly, J.W., and Balch, W.E. (2009). Biological
and Chemical Approaches to Diseases of Proteostasis Deficiency. Annual Review of
Biochemistry 78, 959–991.
Prigione, A., Hossini, A.M., Lichtner, B., Serin, A., Fauler, B., Megges, M., Lurz, R.,
Lehrach, H., Makrantonaki, E., Zouboulis, C.C., et al. (2011). Mitochondrial-Associated
Cell Death Mechanisms Are Reset to an Embryonic-Like State in Aged Donor-Derived iPS
Cells Harboring Chromosomal Aberrations. PLoS ONE 6, e27352.
Puchelle, E., Zahm, J.-M., Tournier, J.-M., and Coraux, C. (2006). Airway epithelial repair,
regeneration, and remodeling after injury in chronic obstructive pulmonary disease.
Proceedings of the American Thoracic Society 3, 726–733.
Puddicombe, S.M., Torres-Lozano, C., Richter, A., Bucchieri, F., Lordan, J.L., Howarth,
P.H., Vrugt, B., Albers, R., Djukanovic, R., Holgate, S.T., et al. (2003). Increased
expression of p21(waf) cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium.
Am. J. Respir. Cell Mol. Biol. 28, 61–68.
Que, J., Luo, X., Schwartz, R.J., and Hogan, B.L.M. (2009). Multiple roles for Sox2 in the
developing and adult mouse trachea. Development 136, 1899–1907.
Quinton, P.M. (1983). Chloride impermeability in cystic fibrosis. Nature 301, 421–422.
Raddatz, G., Hagemann, S., Aran, D., Söhle, J., Kulkarni, P.P., Kaderali, L., Hellman, A.,
Winnefeld, M., and Lyko, F. (2013). Aging is associated with highly defined epigenetic
changes in the human epidermis. Epigenetics & Chromatin 6, 36.
Raghu, G., Weycker, D., Edelsberg, J., Bradford, W.Z., and Oster, G. (2006). Incidence and
prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 174, 810–816.
Rahl, P.B., Lin, C.Y., Seila, A.C., Flynn, R.A., McCuine, S., Burge, C.B., Sharp, P.A., and
Young, R.A. (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445.
Rai, P., Parrish, M., Tay, I.J.J., Li, N., Ackerman, S., He, F., Kwang, J., Chow, V.T., and
Engelward, B.P. (2015). Streptococcus pneumoniae secretes hydrogen peroxide leading to
DNA damage and apoptosis in lung cells. Proc. Natl. Acad. Sci. U.S.A. 112, E3421–E3430.
Raiser, D.M., and Kim, C.F. (2009). Commentary: Sca-1 and Cells of the Lung: A matter of
Different Sorts. Stem Cells 27, 606–611.

208
Randell, S.H. (2006). Airway Epithelial Stem Cells and the Pathophysiology of Chronic
Obstructive Pulmonary Disease. Proceedings of the American Thoracic Society 3, 718–725.
Rando, T.A. (2006). Stem cells, ageing and the quest for immortality. Nature 441, 1080–
1086.
Rando, O.J., and Chang, H.Y. (2009). Genome-Wide Views of Chromatin Structure. Annual
Review of Biochemistry 78, 245–271.
Rando, T.A., and Chang, H.Y. (2012). Aging, Rejuvenation, and Epigenetic
Reprogramming: Resetting the Aging Clock. Cell 148, 46–57.
Rao, R. (2008). Oxidative stress-induced disruption of epithelial and endothelial tight
junctions. Front. Biosci. 13, 7210–7226.
Rawlins, E.L., and Hogan, B.L.M. (2006). Epithelial stem cells of the lung: privileged few
or opportunities for many? Development 133, 2455–2465.
Rawlins, E.L., Okubo, T., Xue, Y., Brass, D.M., Auten, R.L., Hasegawa, H., Wang, F., and
Hogan, B.L.M. (2009). The role of Scgb1a1+ Clara cells in the long-term maintenance and
repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4, 525–534.
Reinert, T., Baldotto, C.S. da R., Nunes, F.A.P., and Scheliga, A.A. de S. (2013).
Bleomycin-Induced Lung Injury. Journal of Cancer Research 2013, 1–9.
Rera, M., Clark, R.I., and Walker, D.W. (2012). Intestinal barrier dysfunction links
metabolic and inflammatory markers of aging to death in Drosophila. Proceedings of the
National Academy of Sciences 109, 21528–21533.
Reynolds, S.D., and Malkinson, A.M. (2010). Clara cell: Progenitor for the bronchiolar
epithelium. The International Journal of Biochemistry & Cell Biology 42, 1–4.
Reynolds, S.D., Hong, K.U., Giangreco, A., Mango, G.W., Guron, C., Morimoto, Y., and
Stripp, B.R. (2000a). Conditional clara cell ablation reveals a self-renewing progenitor
function of pulmonary neuroendocrine cells. American Journal of Physiology-Lung Cellular
and Molecular Physiology 278, L1256–L1263.
Reynolds, S.D., Giangreco, A., Power, J.H., and Stripp, B.R. (2000b). Neuroepithelial
bodies of pulmonary airways serve as a reservoir of progenitor cells capable of epithelial
regeneration. The American Journal of Pathology 156, 269–278.
Reynolds, S.D., Reynolds, P.R., Pryhuber, G.S., Finder, J.D., and Stripp, B.R. (2002).
Secretoglobins SCGB3A1 and SCGB3A2 Define Secretory Cell Subsets in Mouse and
Human Airways. American Journal of Respiratory and Critical Care Medicine 166, 1498–
1509.
Reynolds, S.D., Rios, C., Wesolowska-Andersen, A., Zhuang, Y., Pinter, M., Happoldt, C.,
Hill, C.L., Lallier, S.W., Cosgrove, G.P., Solomon, G.M., et al. (2016). Airway Progenitor

209
Clone Formation Is Enhanced by Y-27632-Dependent Changes in the Transcriptome. Am. J.
Respir. Cell Mol. Biol. 55, 323–336.
Richeldi, L., du Bois, R.M., Raghu, G., Azuma, A., Brown, K.K., Costabel, U., Cottin, V.,
Flaherty, K.R., Hansell, D.M., Inoue, Y., et al. (2014). Efficacy and Safety of Nintedanib in
Idiopathic Pulmonary Fibrosis. New England Journal of Medicine 370, 2071–2082.
Rieger, M.E., Zhou, B., Solomon, N., Sunohara, M., Li, C., Nguyen, C., Liu, Y., Pan, J.,
Minoo, P., Crandall, E.D., et al. (2016). p300/β-Catenin Interactions Regulate Adult
Progenitor Cell Differentiation Downstream of WNT5a/Protein Kinase C (PKC). J. Biol.
Chem. 291, 6569–6582.
Rock, J.R., and Hogan, B.L.M. (2011). Epithelial Progenitor Cells in Lung Development,
Maintenance, Repair, and Disease. Annual Review of Cell and Developmental Biology 27,
493–512.
Rock, J.R., Onaitis, M.W., Rawlins, E.L., Lu, Y., Clark, C.P., Xue, Y., Randell, S.H., and
Hogan, B.L.M. (2009). Basal cells as stem cells of the mouse trachea and human airway
epithelium. Proc. Natl. Acad. Sci. U.S.A. 106, 12771–12775.
Rock, J.R., Randell, S.H., and Hogan, B.L.M. (2010). Airway basal stem cells: a perspective
on their roles in epithelial homeostasis and remodeling. Disease Models & Mechanisms 3,
545–556.
Rogers, D.F. (2005). The role of airway secretions in COPD: pathophysiology,
epidemiology and pharmacotherapeutic options. COPD 2, 341–353.
Rogers, C.S., Stoltz, D.A., Meyerholz, D.K., Ostedgaard, L.S., Rokhlina, T., Taft, P.J.,
Rogan, M.P., Pezzulo, A.A., Karp, P.H., Itani, O.A., et al. (2008). Disruption of the CFTR
gene produces a model of cystic fibrosis in newborn pigs. Science 321, 1837–1841.
Rohlfs, E.M., Zhou, Z., Heim, R.A., Nagan, N., Rosenblum, L.S., Flynn, K., Scholl, T.,
Akmaev, V.R., Sirko-Osadsa, D.A., Allitto, B.A., et al. (2011). Cystic fibrosis carrier testing
in an ethnically diverse US population. Clin. Chem. 57, 841–848.
Rojas, M., Levine, M., and Alvarez, D. (2015). Regenerative medicine in the treatment of
idiopathic pulmonary fibrosis: current position. Stem Cells and Cloning: Advances and
Applications 61.
Rose, M.C., and Voynow, J.A. (2006). Respiratory tract mucin genes and mucin
glycoproteins in health and disease. Physiol. Rev. 86, 245–278.
Rubins, J.B., and Janoff, E.N. (1998). Pneumolysin: a multifunctional pneumococcal
virulence factor. J. Lab. Clin. Med. 131, 21–27.
Ruiz, S., Panopoulos, A.D., Herrerías, A., Bissig, K.-D., Lutz, M., Berggren, W.T., Verma,
I.M., and Izpisua Belmonte, J.C. (2011). A high proliferation rate is required for cell

210
reprogramming and maintenance of human embryonic stem cell identity. Curr. Biol. 21, 45–
52.
Sahin, E., Colla, S., Liesa, M., Moslehi, J., Müller, F.L., Guo, M., Cooper, M., Kotton, D.,
Fabian, A.J., Walkey, C., et al. (2011). Telomere dysfunction induces metabolic and
mitochondrial compromise. Nature 470, 359–365.
Samavarchi-Tehrani, P., Golipour, A., David, L., Sung, H., Beyer, T.A., Datti, A., Woltjen,
K., Nagy, A., and Wrana, J.L. (2010). Functional Genomics Reveals a BMP-Driven
Mesenchymal-to-Epithelial Transition in the Initiation of Somatic Cell Reprogramming. Cell
Stem Cell 7, 64–77.
Sancho-Martinez, I., and Izpisua Belmonte, J.C. (2013). Stem cells: Surf the waves of
reprogramming. Nature 493, 310–311.
Sancho-Martinez, I., Baek, S.H., and Belmonte, J.C.I. (2012). Lineage conversion
methodologies meet the reprogramming toolbox. Nature Cell Biology 14, 892–899.
Scholte, B.J., Davidson, D.J., Wilke, M., and De Jonge, H.R. (2004). Animal models of
cystic fibrosis. J. Cyst. Fibros. 3 Suppl 2, 183–190.
Schumacker, P.T., Gillespie, M.N., Nakahira, K., Choi, A.M.K., Crouser, E.D., Piantadosi,
C.A., and Bhattacharya, J. (2014). Mitochondria in lung biology and pathology: more than
just a powerhouse. AJP: Lung Cellular and Molecular Physiology 306, L962–L974.
Schwartz, R.E., Fleming, H.E., Khetani, S.R., and Bhatia, S.N. (2014). Pluripotent stem cell-
derived hepatocyte-like cells. Biotechnology Advances 32, 504–513.
Schwiebert, E.M., Benos, D.J., Egan, M.E., Stutts, M.J., and Guggino, W.B. (1999). CFTR
is a conductance regulator as well as a chloride channel. Physiol. Rev. 79, S145–S166.
Selman, M., and Pardo, A. (2003). The epithelial/fibroblastic pathway in the pathogenesis of
idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 29, S93–S97.
Selman, M., and Pardo, A. (2014). Revealing the Pathogenic and Aging-related Mechanisms
of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. American Journal of
Respiratory and Critical Care Medicine 189, 1161–1172.
Serrano-Mollar, A., Nacher, M., Gay-Jordi, G., Closa, D., Xaubet, A., and Bulbena, O.
(2007). Intratracheal Transplantation of Alveolar Type II Cells Reverses Bleomycin-induced
Lung Fibrosis. American Journal of Respiratory and Critical Care Medicine 176, 1261–
1268.
Serrano-Mollar, A., Gay-Jordi, G., Guillamat-Prats, R., Closa, D., Hernandez-Gonzalez, F.,
Marin, P., Burgos, F., Martorell, J., Sánchez, M., Arguis, P., et al. (2016). Safety and
tolerability of alveolar type II cell transplantation in idiopathic pulmonary fibrosis. CHEST
Journal 150, 533–543.

211
Shah, V.S., Meyerholz, D.K., Tang, X.X., Reznikov, L., Abou Alaiwa, M., Ernst, S.E.,
Karp, P.H., Wohlford-Lenane, C.L., Heilmann, K.P., Leidinger, M.R., et al. (2016). Airway
acidification initiates host defense abnormalities in cystic fibrosis mice. Science 351, 503–
507.
Shakiba, N., White, C.A., Lipsitz, Y.Y., Yachie-Kinoshita, A., Tonge, P.D., Hussein, S.M.I.,
Puri, M.C., Elbaz, J., Morrissey-Scoot, J., Li, M., et al. (2015). CD24 tracks divergent
pluripotent states in mouse and human cells. Nature Communications 6, 7329.
Shan, L., Aster, J.C., Sklar, J., and Sunday, M.E. (2006). Notch-1 regulates pulmonary
neuroendocrine cell differentiation in cell lines and in transgenic mice. AJP: Lung Cellular
and Molecular Physiology 292, L500–L509.
Sharpless, N.E., and DePinho, R.A. (2007). How stem cells age and why this makes us grow
old. Nature Reviews Molecular Cell Biology 8, 703–713.
Shaykhiev, R., Zuo, W.-L., Chao, I., Fukui, T., Witover, B., Brekman, A., and Crystal, R.G.
(2013). EGF shifts human airway basal cell fate toward a smoking-associated airway
epithelial phenotype. Proc. Natl. Acad. Sci. U.S.A. 110, 12102–12107.
Shi, Y., Desponts, C., Do, J.T., Hahm, H.S., Schöler, H.R., and Ding, S. (2008). Induction of
pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-
molecule compounds. Cell Stem Cell 3, 568–574.
Shi, Y., Inoue, H., Wu, J.C., and Yamanaka, S. (2016). Induced pluripotent stem cell
technology: a decade of progress. Nature Reviews Drug Discovery 16, 115–130.
Shipony, Z., Mukamel, Z., Cohen, N.M., Landan, G., Chomsky, E., Zeliger, S.R., Fried,
Y.C., Ainbinder, E., Friedman, N., and Tanay, A. (2014). Dynamic and static maintenance
of epigenetic memory in pluripotent and somatic cells. Nature 513, 115–119.
Silva, J., Barrandon, O., Nichols, J., Kawaguchi, J., Theunissen, T.W., and Smith, A. (2008).
Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol.
6, e253.
Silva, J., Nichols, J., Theunissen, T.W., Guo, G., van Oosten, A.L., Barrandon, O., Wray, J.,
Yamanaka, S., Chambers, I., and Smith, A. (2009). Nanog is the gateway to the pluripotent
ground state. Cell 138, 722–737.
Singovski, G., Bernal, C., Kuciak, M., Siegl-Cachedenier, I., Conod, A., and Ruiz i Altaba,
A. (2016). In vivo epigenetic reprogramming of primary human colon cancer cells enhances
metastases. Journal of Molecular Cell Biology 8, 157–173.
Sinha, A., Nightingale, J., West, K.P., Berlanga-Acosta, J., and Playford, R.J. (2003).
Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided
ulcerative colitis or proctitis. N. Engl. J. Med. 349, 350–357.

212
Slutsky, A.S., and Ranieri, V.M. (2013). Ventilator-induced lung injury. N. Engl. J. Med.
369, 2126–2136.
Smith, A. (2006). A glossary for stem-cell biology. Nature 441, 1060–1060.
Smith, Z.D., Nachman, I., Regev, A., and Meissner, A. (2010). Dynamic single-cell imaging
of direct reprogramming reveals an early specifying event. Nat. Biotechnol. 28, 521–526.
Sohal, S.S., Reid, D., Soltani, A., Ward, C., Weston, S., Muller, H.K., Wood-Baker, R., and
Walters, E.H. (2010). Reticular basement membrane fragmentation and potential epithelial
mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive
pulmonary disease. Respirology 15, 930–938.
Sokocevic, D., Bonenfant, N.R., Wagner, D.E., Borg, Z.D., Lathrop, M.J., Lam, Y.W.,
Deng, B., DeSarno, M.J., Ashikaga, T., Loi, R., et al. (2013). The effect of age and
emphysematous and fibrotic injury on the re-cellularization of de-cellularized lungs.
Biomaterials 34, 3256–3269.
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G.W., Cook, E.G., Hargus, G., Blak,
A., Cooper, O., Mitalipova, M., et al. (2009). Parkinson’s disease patient-derived induced
pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977.
Song, H., Yao, E., Lin, C., Gacayan, R., Chen, M.-H., and Chuang, P.-T. (2012). Functional
characterization of pulmonary neuroendocrine cells in lung development, injury, and
tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 17531–17536.
Sousa-Victor, P., Gutarra, S., García-Prat, L., Rodriguez-Ubreva, J., Ortet, L., Ruiz-Bonilla,
V., Jardí, M., Ballestar, E., González, S., Serrano, A.L., et al. (2014). Geriatric muscle stem
cells switch reversible quiescence into senescence. Nature 506, 316–321.
Spina, D. (1998). Epithelium smooth muscle regulation and interactions. American Journal
of Respiratory and Critical Care Medicine 158, S141–S145.
Spragg, R.G., Bernard, G.R., Checkley, W., Curtis, J.R., Gajic, O., Guyatt, G., Hall, J.,
Israel, E., Jain, M., Needham, D.M., et al. (2010). Beyond mortality: future clinical research
in acute lung injury. Am. J. Respir. Crit. Care Med. 181, 1121–1127.
Sridharan, R., Tchieu, J., Mason, M.J., Yachechko, R., Kuoy, E., Horvath, S., Zhou, Q., and
Plath, K. (2009). Role of the murine reprogramming factors in the induction of pluripotency.
Cell 136, 364–377.
Stadtfeld, M., Brennand, K., and Hochedlinger, K. (2008a). Reprogramming of pancreatic
beta cells into induced pluripotent stem cells. Curr. Biol. 18, 890–894.
Stadtfeld, M., Nagaya, M., Utikal, J., Weir, G., and Hochedlinger, K. (2008b). Induced
pluripotent stem cells generated without viral integration. Science 322, 945–949.

213
Stadtfeld, M., Maherali, N., Breault, D.T., and Hochedlinger, K. (2008c). Defining
molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem
Cell 2, 230–240.
Stadtfeld, M., Apostolou, E., Akutsu, H., Fukuda, A., Follett, P., Natesan, S., Kono, T.,
Shioda, T., and Hochedlinger, K. (2010). Aberrant silencing of imprinted genes on
chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465, 175–181.
Stripp, B.R., Maxson, K., Mera, R., and Singh, G. (1995). Plasticity of airway cell
proliferation and gene expression after acute naphthalene injury. Am. J. Physiol. 269, L791–
L799.
Suhr, S.T., Chang, E.A., Tjong, J., Alcasid, N., Perkins, G.A., Goissis, M.D., Ellisman,
M.H., Perez, G.I., and Cibelli, J.B. (2010). Mitochondrial Rejuvenation After Induced
Pluripotency. PLoS ONE 5, e14095.
Suprynowicz, F.A., Upadhyay, G., Krawczyk, E., Kramer, S.C., Hebert, J.D., Liu, X., Yuan,
H., Cheluvaraju, C., Clapp, P.W., Boucher, R.C., et al. (2012). Conditionally reprogrammed
cells represent a stem-like state of adult epithelial cells. Proceedings of the National
Academy of Sciences 109, 20035–20040.
Sweerus, K., Lachowicz-Scroggins, M., Gordon, E., LaFemina, M., Huang, X., Parikh, M.,
Kanegai, C., Fahy, J.V., and Frank, J.A. (2017). Claudin-18 deficiency is associated with
airway epithelial barrier dysfunction and asthma. J. Allergy Clin. Immunol. 139, 72–81.e1.
Swindle, E.J., Collins, J.E., and Davies, D.E. (2009). Breakdown in epithelial barrier
function in patients with asthma: identification of novel therapeutic approaches. J. Allergy
Clin. Immunol. 124, 23–34; quiz 35–36.
Taggart, C., Coakley, R.J., Greally, P., Canny, G., O’Neill, S.J., and McElvaney, N.G.
(2000). Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-
alpha and interleukin-8. Am. J. Physiol. Lung Cell Mol. Physiol. 278, L33–L41.
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse
embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., and
Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by
defined factors. Cell 131, 861–872.
Talens, R.P., Christensen, K., Putter, H., Willemsen, G., Christiansen, L., Kremer, D.,
Suchiman, H.E.D., Slagboom, P.E., Boomsma, D.I., and Heijmans, B.T. (2012). Epigenetic
variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic
twin pairs: Talens_Increasing epigenetic variation. Aging Cell 11, 694–703.
Tang, Y.-W., Johnson, J.E., Browning, P.J., Cruz-Gervis, R.A., Davis, A., Graham, B.S.,
Brigham, K.L., Oates, J.A., Loyd, J.E., and Stecenko, A.A. (2003). Herpesvirus DNA is

214
consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J. Clin.
Microbiol. 41, 2633–2640.
Tapia, N., and Schöler, H.R. (2016). Molecular Obstacles to Clinical Translation of iPSCs.
Cell Stem Cell 19, 298–309.
Tashiro, J., Elliot, S.J., Gerth, D.J., Xia, X., Pereira-Simon, S., Choi, R., Catanuto, P.,
Shahzeidi, S., Toonkel, R.L., Shah, R.H., et al. (2015). Therapeutic benefits of young, but
not old, adipose-derived mesenchymal stem cells in a chronic mouse model of bleomycin-
induced pulmonary fibrosis. Translational Research 166, 554–567.
Tata, P.R., Mou, H., Pardo-Saganta, A., Zhao, R., Prabhu, M., Law, B.M., Vinarsky, V.,
Cho, J.L., Breton, S., Sahay, A., et al. (2013). Dedifferentiation of committed epithelial cells
into stem cells in vivo. Nature 503, 218–223.
Taylor, P.R., Bonfield, T.L., Chmiel, J.F., and Pearlman, E. (2016). Neutrophils from
F508del cystic fibrosis patients produce IL-17A and express IL-23 - dependent IL-17RC.
Clin. Immunol. 170, 53–60.
Teisanu, R.M., Chen, H., Matsumoto, K., McQualter, J.L., Potts, E., Foster, W.M.,
Bertoncello, I., and Stripp, B.R. (2011). Functional Analysis of Two Distinct Bronchiolar
Progenitors during Lung Injury and Repair. American Journal of Respiratory Cell and
Molecular Biology 44, 794–803.
Thannickal, V.J. (2013). Mechanistic links between aging and lung fibrosis. Biogerontology
14, 609–615.
Theunissen, T.W., van Oosten, A.L., Castelo-Branco, G., Hall, J., Smith, A., and Silva,
J.C.R. (2011). Nanog overcomes reprogramming barriers and induces pluripotency in
minimal conditions. Curr. Biol. 21, 65–71.
Thompson, A.B., Robbins, R.A., Romberger, D.J., Sisson, J.H., Spurzem, J.R., Teschler, H.,
and Rennard, S.I. (1995). Immunological functions of the pulmonary epithelium. European
Respiratory Journal 8, 127–149.
Tomás-Loba, A., Flores, I., Fernández-Marcos, P.J., Cayuela, M.L., Maraver, A., Tejera, A.,
Borrás, C., Matheu, A., Klatt, P., Flores, J.M., et al. (2008). Telomerase Reverse
Transcriptase Delays Aging in Cancer-Resistant Mice. Cell 135, 609–622.
Tonge, P.D., Corso, A.J., Monetti, C., Hussein, S.M.I., Puri, M.C., Michael, I.P., Li, M.,
Lee, D.-S., Mar, J.C., Cloonan, N., et al. (2014). Divergent reprogramming routes lead to
alternative stem-cell states. Nature 516, 192–197.
Toonkel, R.L., Hare, J.M., Matthay, M.A., and Glassberg, M.K. (2013). Mesenchymal Stem
Cells and Idiopathic Pulmonary Fibrosis. Potential for Clinical Testing. American Journal of
Respiratory and Critical Care Medicine 188, 133–140.

215
Treutlein, B., Brownfield, D.G., Wu, A.R., Neff, N.F., Mantalas, G.L., Espinoza, F.H.,
Desai, T.J., Krasnow, M.A., and Quake, S.R. (2014). Reconstructing lineage hierarchies of
the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375.
Treutlein, B., Lee, Q.Y., Camp, J.G., Mall, M., Koh, W., Shariati, S.A.M., Sim, S., Neff,
N.F., Skotheim, J.M., Wernig, M., et al. (2016). Dissecting direct reprogramming from
fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395.
Tsakiri, K.D., Cronkhite, J.T., Kuan, P.J., Xing, C., Raghu, G., Weissler, J.C., Rosenblatt,
R.L., Shay, J.W., and Garcia, C.K. (2007). Adult-onset pulmonary fibrosis caused by
mutations in telomerase. Proceedings of the National Academy of Sciences 104, 7552–7557.
Tsao, P.-N., Vasconcelos, M., Izvolsky, K.I., Qian, J., Lu, J., and Cardoso, W.V. (2009).
Notch signaling controls the balance of ciliated and secretory cell fates in developing
airways. Development 136, 2297–2307.
Tsuji, T., Aoshiba, K., and Nagai, A. (2006). Alveolar Cell Senescence in Patients with
Pulmonary Emphysema. American Journal of Respiratory and Critical Care Medicine 174,
886–893.
Twigg, M.S., Brockbank, S., Lowry, P., FitzGerald, S.P., Taggart, C., and Weldon, S.
(2015). The Role of Serine Proteases and Antiproteases in the Cystic Fibrosis Lung.
Mediators Inflamm. 2015, 293053.
Uhal, B.D., and Nguyen, H. (2013). The Witschi Hypothesis revisited after 35 years: genetic
proof from SP-C BRICHOS domain mutations. AJP: Lung Cellular and Molecular
Physiology 305, L906–L911.
Utikal, J., Polo, J.M., Stadtfeld, M., Maherali, N., Kulalert, W., Walsh, R.M., Khalil, A.,
Rheinwald, J.G., and Hochedlinger, K. (2009). Immortalization eliminates a roadblock
during cellular reprogramming into iPS cells. Nature 460, 1145–1148.
Van Haute, L., De Block, G., Liebaers, I., Sermon, K., and De Rycke, M. (2009). Generation
of lung epithelial-like tissue from human embryonic stem cells. Respir. Res. 10, 105.
Van Houten, B., Woshner, V., and Santos, J.H. (2006). Role of mitochondrial DNA in toxic
responses to oxidative stress. DNA Repair 5, 145–152.
Van Lommel, A. (2001). Pulmonary neuroendocrine cells (PNEC) and neuroepithelial
bodies (NEB): chemoreceptors and regulators of lung development. Paediatr Respir Rev 2,
171–176.
Van Lommel, A., Lauweryns, J.M., and Berthoud, H.R. (1998). Pulmonary neuroepithelial
bodies are innervated by vagal afferent nerves: an investigation with in vivo anterograde DiI
tracing and confocal microscopy. Anatomy and Embryology 197, 325–330.

216
Vaughan, A.E., Brumwell, A.N., Xi, Y., Gotts, J.E., Brownfield, D.G., Treutlein, B., Tan,
K., Tan, V., Liu, F.C., Looney, M.R., et al. (2014). Lineage-negative progenitors mobilize to
regenerate lung epithelium after major injury. Nature 517, 621–625.
Vermulst, M., Wanagat, J., Kujoth, G.C., Bielas, J.H., Rabinovitch, P.S., Prolla, T.A., and
Loeb, L.A. (2008). DNA deletions and clonal mutations drive premature aging in
mitochondrial mutator mice. Nature Genetics 40, 392–394.
Vijg, J., and Campisi, J. (2008). Puzzles, promises and a cure for ageing. Nature 454, 1065–
1071.
Voltz, J.W., Card, J.W., Carey, M.A., DeGraff, L.M., Ferguson, C.D., Flake, G.P., Bonner,
J.C., Korach, K.S., and Zeldin, D.C. (2008). Male Sex Hormones Exacerbate Lung Function
Impairment after Bleomycin-Induced Pulmonary Fibrosis. American Journal of Respiratory
Cell and Molecular Biology 39, 45–52.
Waghray, M., Cui, Z., Horowitz, J.C., Subramanian, I.M., Martinez, F.J., Toews, G.B., and
Thannickal, V.J. (2005). Hydrogen peroxide is a diffusible paracrine signal for the induction
of epithelial cell death by activated myofibroblasts. FASEB J. 19, 854–856.
Wallace, D.C. (2013). A mitochondrial bioenergetic etiology of disease. Journal of Clinical
Investigation 123, 1405–1412.
Wang, J., Edeen, K., Manzer, R., Chang, Y., Wang, S., Chen, X., Funk, C.J., Cosgrove,
G.P., Fang, X., and Mason, R.J. (2007). Differentiated Human Alveolar Epithelial Cells and
Reversibility of their Phenotype In Vitro. American Journal of Respiratory Cell and
Molecular Biology 36, 661–668.
Wang, Y., Minshall, R.D., Schwartz, D.E., and Hu, G. (2011). Cyclic stretch induces
alveolar epithelial barrier dysfunction via calpain-mediated degradation of p120-catenin.
Am. J. Physiol. Lung Cell Mol. Physiol. 301, L197–L206.
Wang, Y., Wang, G.Z., Rabinovitch, P.S., and Tabas, I. (2014). Macrophage mitochondrial
oxidative stress promotes atherosclerosis and nuclear factor-κB–mediated inflammation in
macrophages. Circulation Research 114, 421–433.
Wang, Z., Zang, C., Rosenfeld, J.A., Schones, D.E., Barski, A., Cuddapah, S., Cui, K., Roh,
T.-Y., Peng, W., Zhang, M.Q., et al. (2008). Combinatorial patterns of histone acetylations
and methylations in the human genome. Nature Genetics 40, 897–903.
Wang, Z., Oron, E., Nelson, B., Razis, S., and Ivanova, N. (2012). Distinct lineage
specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell
Stem Cell 10, 440–454.
Wansleeben, C., Barkauskas, C.E., Rock, J.R., and Hogan, B.L.M. (2013). Stem cells of the
adult lung: their development and role in homeostasis, regeneration, and disease. Wiley
Interdiscip Rev Dev Biol 2, 131–148.

217
Wansleeben, C., Bowie, E., Hotten, D.F., Yu, Y.-R.A., and Hogan, B.L.M. (2014). Age-
Related Changes in the Cellular Composition and Epithelial Organization of the Mouse
Trachea. PLoS ONE 9, e93496.
Ware, L.B., and Matthay, M.A. (2001). Alveolar fluid clearance is impaired in the majority
of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir.
Crit. Care Med. 163, 1376–1383.
Watanabe, K., Ueno, M., Kamiya, D., Nishiyama, A., Matsumura, M., Wataya, T.,
Takahashi, J.B., Nishikawa, S., Nishikawa, S., Muguruma, K., et al. (2007). A ROCK
inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25,
681–686.
Waters, C.M., and Savla, U. (1999). Keratinocyte growth factor accelerates wound closure
in airway epithelium during cyclic mechanical strain. J. Cell. Physiol. 181, 424–432.
Watson, J.K., Rulands, S., Wilkinson, A.C., Wuidart, A., Ousset, M., Van Keymeulen, A.,
Göttgens, B., Blanpain, C., Simons, B.D., and Rawlins, E.L. (2015). Clonal Dynamics
Reveal Two Distinct Populations of Basal Cells in Slow-Turnover Airway Epithelium. Cell
Rep 12, 90–101.
Weidner, C.I., and Wagner, W. (2014). The epigenetic tracks of aging. Biological Chemistry
395.
Weidner, C.I., Lin, Q., Koch, C.M., Eisele, L., Beier, F., Ziegler, P., Bauerschlag, D.O.,
Jöckel, K.-H., Erbel, R., Mühleisen, T.W., et al. (2014). Aging of blood can be tracked by
DNA methylation changes at just three CpG sites. Genome Biology 15, R24.
Weiss, D.J. (2014). Concise Review: Current Status of Stem Cells and Regenerative
Medicine in Lung Biology and Diseases: Advances in Lung Regenerative Medicine. STEM
CELLS 32, 16–25.
Weiss, D.J., and Ortiz, L.A. (2013). Cell therapy trials for lung diseases: progress and
cautions. American Journal of Respiratory and Critical Care Medicine 188, 123–125.
Weiss, C.H., Budinger, G.R.S., Mutlu, G.M., and Jain, M. (2010). Proteasomal Regulation
of Pulmonary Fibrosis. Proceedings of the American Thoracic Society 7, 77–83.
Welsh, M.J., and Liedtke, C.M. (1986). Chloride and potassium channels in cystic fibrosis
airway epithelia. Nature 322, 467–470.
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K.,
Bernstein, B.E., and Jaenisch, R. (2007). In vitro reprogramming of fibroblasts into a
pluripotent ES-cell-like state. Nature 448, 318–324.
Wernig, M., Lengner, C.J., Hanna, J., Lodato, M.A., Steine, E., Foreman, R., Staerk, J.,
Markoulaki, S., and Jaenisch, R. (2008). A drug-inducible transgenic system for direct
reprogramming of multiple somatic cell types. Nat. Biotechnol. 26, 916–924.

218
Whitsett, J.A., and Alenghat, T. (2014). Respiratory epithelial cells orchestrate pulmonary
innate immunity. Nature Immunology 16, 27–35.
Whitsett, J.A., and Alenghat, T. (2015). Respiratory epithelial cells orchestrate pulmonary
innate immunity. Nat. Immunol. 16, 27–35.
Whitsett, J.A., Wert, S.E., and Weaver, T.E. (2010). Alveolar Surfactant Homeostasis and
the Pathogenesis of Pulmonary Disease. Annual Review of Medicine 61, 105–119.
Wiener-Kronish, J.P., Albertine, K.H., and Matthay, M.A. (1991). Differential responses of
the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J.
Clin. Invest. 88, 864–875.
Williams, O.W., Sharafkhaneh, A., Kim, V., Dickey, B.F., and Evans, C.M. (2006). Airway
mucus: From production to secretion. Am. J. Respir. Cell Mol. Biol. 34, 527–536.
Woltjen, K., Michael, I.P., Mohseni, P., Desai, R., Mileikovsky, M., Hämäläinen, R.,
Cowling, R., Wang, W., Liu, P., Gertsenstein, M., et al. (2009). piggyBac transposition
reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770.
Wong, A.P., Bear, C.E., Chin, S., Pasceri, P., Thompson, T.O., Huan, L.-J., Ratjen, F., Ellis,
J., and Rossant, J. (2012). Directed differentiation of human pluripotent stem cells into
mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 30, 876–882.
Wood, S.J., Goldufsky, J.W., Bello, D., Masood, S., and Shafikhani, S.H. (2015).
Pseudomonas aeruginosa ExoT Induces Mitochondrial Apoptosis in Target Host Cells in a
Manner That Depends on Its GTPase-activating Protein (GAP) Domain Activity. J. Biol.
Chem. 290, 29063–29073.
Xiao, C., Puddicombe, S.M., Field, S., Haywood, J., Broughton-Head, V., Puxeddu, I.,
Haitchi, H.M., Vernon-Wilson, E., Sammut, D., Bedke, N., et al. (2011). Defective epithelial
barrier function in asthma. J. Allergy Clin. Immunol. 128, 549–556.e1–e12.
Xiao, H., Li, D.X., and Liu, M. (2012). Knowledge translation: airway epithelial cell
migration and respiratory diseases. Cell. Mol. Life Sci. 69, 4149–4162.
Yamaguchi, H., Calado, R.T., Ly, H., Kajigaya, S., Baerlocher, G.M., Chanock, S.J.,
Lansdorp, P.M., and Young, N.S. (2005). Mutations in TERT, the gene for telomerase
reverse transcriptase, in aplastic anemia. New England Journal of Medicine 352, 1413–1424.
Yanagi, S., Tsubouchi, H., Miura, A., Matsumoto, N., and Nakazato, M. (2015). Breakdown
of Epithelial Barrier Integrity and Overdrive Activation of Alveolar Epithelial Cells in the
Pathogenesis of Acute Respiratory Distress Syndrome and Lung Fibrosis. BioMed Research
International 2015, 1–12.
Yang, Y., Jiao, J., Gao, R., Le, R., Kou, X., Zhao, Y., Wang, H., Gao, S., and Wang, Y.
(2015). Enhanced Rejuvenation in Induced Pluripotent Stem Cell-Derived Neurons

219
Compared with Directly Converted Neurons from an Aged Mouse. Stem Cells and
Development 24, 2767–2777.
Yokohori, N., Aoshiba, K., and Nagai, A. (2004). Increased levels of cell death and
proliferation in alveolar wall cells in patients with pulmonary emphysema. CHEST Journal
125, 626–632.
Yu, J., Vodyanik, M.A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J.L., Tian, S., Nie,
J., Jonsdottir, G.A., Ruotti, V., Stewart, R., et al. (2007). Induced pluripotent stem cell lines
derived from human somatic cells. Science 318, 1917–1920.
Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., Slukvin, I.I., and Thomson, J.A.
(2009). Human induced pluripotent stem cells free of vector and transgene sequences.
Science 324, 797–801.
Yue, R., Xia, X., Jiang, J., Yang, D., Han, Y., Chen, X., Cai, Y., Li, L., Wang, W.E., and
Zeng, C. (2015). Mitochondrial DNA Oxidative Damage Contributes to Cardiomyocyte
Ischemia/Reperfusion-Injury in Rats: Cardioprotective Role of Lycopene:
MITOCHONDRIAL DNA OXIDATIVE DAMAGE AND I/R-INJURY. Journal of Cellular
Physiology 230, 2128–2141.
Zampieri, M., Ciccarone, F., Calabrese, R., Franceschi, C., Bürkle, A., and Caiafa, P.
(2015). Reconfiguration of DNA methylation in aging. Mechanisms of Ageing and
Development 151, 60–70.
Zane, L., Sharma, V., and Misteli, T. (2014). Common features of chromatin in aging and
cancer: cause or coincidence? Trends in Cell Biology 24, 686–694.
Zemans, R.L., Colgan, S.P., and Downey, G.P. (2009). Transepithelial migration of
neutrophils: mechanisms and implications for acute lung injury. Am. J. Respir. Cell Mol.
Biol. 40, 519–535.
Zemans, R.L., McClendon, J., Aschner, Y., Briones, N., Young, S.K., Lau, L.F., Kahn, M.,
and Downey, G.P. (2013). Role of β-catenin-regulated CCN matricellular proteins in
epithelial repair after inflammatory lung injury. Am. J. Physiol. Lung Cell Mol. Physiol.
304, L415–L427.
Zemke, A.C., Snyder, J.C., Brockway, B.L., Drake, J.A., Reynolds, S.D., Kaminski, N., and
Stripp, B.R. (2008). Molecular Staging of Epithelial Maturation Using Secretory Cell-
Specific Genes as Markers. American Journal of Respiratory Cell and Molecular Biology
40, 340–348.
Zhang, J., Nuebel, E., Daley, G.Q., Koehler, C.M., and Teitell, M.A. (2012). Metabolic
regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell
11, 589–595.

220
Zhao, Y., Yin, X., Qin, H., Zhu, F., Liu, H., Yang, W., Zhang, Q., Xiang, C., Hou, P., Song,
Z., et al. (2008). Two supporting factors greatly improve the efficiency of human iPSC
generation. Cell Stem Cell 3, 475–479.
Zheng, D., Limmon, G.V., Yin, L., Leung, N.H.N., Yu, H., Chow, V.T.K., and Chen, J.
(2012). Regeneration of alveolar type I and II cells from Scgb1a1-expressing cells following
severe pulmonary damage induced by bleomycin and influenza. PLoS ONE 7, e48451.
Zhou, H., Wu, S., Joo, J.Y., Zhu, S., Han, D.W., Lin, T., Trauger, S., Bien, G., Yao, S., Zhu,
Y., et al. (2009). Generation of induced pluripotent stem cells using recombinant proteins.
Cell Stem Cell 4, 381–384.
Zhou, L., Dey, C.R., Wert, S.E., DuVall, M.D., Frizzell, R.A., and Whitsett, J.A. (1994).
Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR.
Science 266, 1705–1708.
Zhou, Q., Ye, X., Sun, R., Matsumoto, Y., Moriyama, M., Asano, Y., Ajioka, Y., and Saijo,
Y. (2014). Differentiation of Mouse Induced Pluripotent Stem Cells Into Alveolar Epithelial
Cells In Vitro for Use In Vivo. STEM CELLS Translational Medicine 3, 675–685.
Zoz, D.F., Lawson, W.E., and Blackwell, T.S. (2011). Idiopathic Pulmonary Fibrosis: A
Disorder of Epithelial Cell Dysfunction. The American Journal of the Medical Sciences 341,
435–438.
Zunder, E.R., Lujan, E., Goltsev, Y., Wernig, M., and Nolan, G.P. (2015). A Continuous
Molecular Roadmap to iPSC Reprogramming through Progression Analysis of Single-Cell
Mass Cytometry. Cell Stem Cell 16, 323–337.
Zuo, W., Zhang, T., Wu, D.Z., Guan, S.P., Liew, A.-A., Yamamoto, Y., Wang, X., Lim,
S.J., Vincent, M., Lessard, M., et al. (2014). p63+Krt5+ distal airway stem cells are essential
for lung regeneration. Nature 517, 616–620.
(1985). The definition of emphysema. Report of a National Heart, Lung, and Blood Institute,
Division of Lung Diseases workshop. Am. Rev. Respir. Dis. 132, 182–185.

221
Related Documents











