Shawn Weng Ke Gu Philip W. Hammond Peter Lohse Cecil Rise Richard W. Wagner Martin C. Wright Robert G. Kuimelis Phylos, Lexington, MA, USA Generating addressable protein microarrays with PROfusion covalent mRNA-protein fusion technology An mRNA-protein fusion consists of a polypeptide covalently linked to its correspond- ing mRNA. These species, prepared individually or en masse by in vitro translation with a modified mRNA conjugate (the PROfusion process), link phenotype to genotype and enable powerful directed evolution schemes. We have exploited the informational content of the nucleic acid component of the mRNA-protein fusion to create an addressable protein microarray that self-assembles via hybridization to surface-bound DNA capture probes. The nucleic acid component not only directs the mRNA-protein fusion to the proper coordinate of the microarray, but also positions the protein in a uniform orientation. We demonstrate the feasibility of this protein chip concept with several mRNA-protein fusions, each possessing a unique peptide epitope sequence. These addressable proteins could be visualized on the microarray both by autoradi- ography and highly specific monoclonal antibody binding. The anchoring of the protein to the chip surface is surprisingly robust, and the system is sensitive enough to detect sub-attomole quantities of displayed protein without signal amplification. Such protein arrays should be useful for functional screening in massively parallel formats, as well as other applications involving immobilized peptides and proteins. Keywords: Protein chip / Biochip / Microarray / mRNA display PRO 0142 1 Introduction The large number of novel genes identified by various ongoing genome sequencing projects will require novel platforms to study protein function in a high-throughput fashion. Conventional assay systems for screening pro- teins usually involve highly repetitive binding measure- ments in a microtitre plate format. This requires prepar- ation of large quantities of reagents, followed by deposi- tion into individual wells. To reduce screening time and cost, both academic and industrial laboratories have put substantial effort into miniaturizing and multiplexing such assays [1, 2]. New methodologies utilizing assays on planar solid supports have been developed recently to achieve an increasing level of miniaturization and to allow for functional protein analysis in a massively parallel fash- ion [3–14]. These new methodologies will eventually allow the simultaneous measurement of the concentrations of thousands of different proteins in a sample. The ability to rapidly generate a qualitative or quantitative snapshot of the protein makeup of a complex biological sample, in a routine and reproducible manner, will open up many new applications in both research and diagnostics [10, 11]. We envisage that these new approaches will be com- plementary to traditional two-dimensional gel electro- phoresis techniques. In this paper, we report a new approach for the prepar- ation of self-assembling protein microarrays that com- bines aspects of DNA chip technology [15] with covalent mRNA-protein fusion (PROfusion ) technology [16]. The transformation of a high-density DNA array into an addressable protein array is achieved by Watson-Crick hybridization of mRNA-protein fusions to surface-immo- bilized DNA capture probes. mRNA-protein fusion mole- cules consist of a protein sequence covalently linked via the C-terminus to the 3’-end of their encoding mRNA [16]. Because the coding sequence and the protein are cova- lently linked, mRNA-protein fusions provide a means for immobilizing a protein by nucleic acid hybridization of the attached RNA to complementary, surface-immobi- lized DNA sequences. Moreover, when immobilized in this fashion, the displayed protein is presented at the sur- face in a productive and uniform orientation, thus enabling efficient interaction with the solution-phase. Since conventional DNA microarrays routinely achieve densities on the order of 10 3 –10 4 features per square cen- timeter, it is possible to imagine a similar number of immo- bilized proteins in a microarray format, arising from a standard DNA microarray. Correspondence: Dr. Robert G. Kuimelis, Phylos, Inc., 128 Spring Street, Lexington, MA 02421, USA E-mail: [email protected] Fax: +1-781402-8800 Abbreviations: Cy , cysteine; SSC, saline sodium citrate; TMV , tobacco mosaic virus 48 Proteomics 2002, 2, 48–57 ª WILEY-VCH Verlag GmbH, 69451 Weinheim, 2002 1615-9853/02/0101–48 $17.50+.50/0

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Shawn WengKe GuPhilip W. HammondPeter LohseCecil RiseRichard W. WagnerMartin C. WrightRobert G. Kuimelis
Phylos,Lexington, MA, USA
Generating addressable protein microarrayswith PROfusion covalent mRNA-protein fusiontechnology
An mRNA-protein fusion consists of a polypeptide covalently linked to its correspond-ing mRNA. These species, prepared individually or en masse by in vitro translation witha modified mRNA conjugate (the PROfusion process), link phenotype to genotypeand enable powerful directed evolution schemes. We have exploited the informationalcontent of the nucleic acid component of the mRNA-protein fusion to create anaddressable protein microarray that self-assembles via hybridization to surface-boundDNA capture probes. The nucleic acid component not only directs the mRNA-proteinfusion to the proper coordinate of the microarray, but also positions the protein in auniform orientation. We demonstrate the feasibility of this protein chip concept withseveral mRNA-protein fusions, each possessing a unique peptide epitope sequence.These addressable proteins could be visualized on the microarray both by autoradi-ography and highly specific monoclonal antibody binding. The anchoring of the proteinto the chip surface is surprisingly robust, and the system is sensitive enough to detectsub-attomole quantities of displayed protein without signal amplification. Such proteinarrays should be useful for functional screening in massively parallel formats, as well asother applications involving immobilized peptides and proteins.
Keywords: Protein chip / Biochip / Microarray / mRNA display PRO 0142
1 Introduction
The large number of novel genes identified by variousongoing genome sequencing projects will require novelplatforms to study protein function in a high-throughputfashion. Conventional assay systems for screening pro-teins usually involve highly repetitive binding measure-ments in a microtitre plate format. This requires prepar-ation of large quantities of reagents, followed by deposi-tion into individual wells. To reduce screening time andcost, both academic and industrial laboratories have putsubstantial effort into miniaturizing and multiplexing suchassays [1, 2]. New methodologies utilizing assays onplanar solid supports have been developed recently toachieve an increasing level of miniaturization and to allowfor functional protein analysis in a massively parallel fash-ion [3–14]. These new methodologies will eventually allowthe simultaneous measurement of the concentrations ofthousands of different proteins in a sample. The ability torapidly generate a qualitative or quantitative snapshot ofthe protein makeup of a complex biological sample, in
a routine and reproducible manner, will open up manynew applications in both research and diagnostics [10,11]. We envisage that these new approaches will be com-plementary to traditional two-dimensional gel electro-phoresis techniques.
In this paper, we report a new approach for the prepar-ation of self-assembling protein microarrays that com-bines aspects of DNA chip technology [15] with covalentmRNA-protein fusion (PROfusion ) technology [16]. Thetransformation of a high-density DNA array into anaddressable protein array is achieved by Watson-Crickhybridization of mRNA-protein fusions to surface-immo-bilized DNA capture probes. mRNA-protein fusion mole-cules consist of a protein sequence covalently linked viathe C-terminus to the 3’-end of their encoding mRNA [16].Because the coding sequence and the protein are cova-lently linked, mRNA-protein fusions provide a means forimmobilizing a protein by nucleic acid hybridization ofthe attached RNA to complementary, surface-immobi-lized DNA sequences. Moreover, when immobilized inthis fashion, the displayed protein is presented at the sur-face in a productive and uniform orientation, thusenabling efficient interaction with the solution-phase.Since conventional DNA microarrays routinely achievedensities on the order of 103–104 features per square cen-timeter, it is possible to imagine a similar number of immo-bilized proteins in a microarray format, arising from astandard DNA microarray.
Correspondence: Dr. Robert G. Kuimelis, Phylos, Inc., 128Spring Street, Lexington, MA 02421, USAE-mail: [email protected]: +1-781402-8800
Abbreviations: Cy, cysteine; SSC, saline sodium citrate; TMV,tobacco mosaic virus
48 Proteomics 2002, 2, 48–57
ª WILEY-VCH Verlag GmbH, 69451 Weinheim, 2002 1615-9853/02/0101–48 $17.50+.50/0

We have enabled the concept of converting a DNA arrayinto a protein array and demonstrate here the sequence-directed self-assembly of a protein microarray on a con-ventional DNA chip bearing oligonucleotide captureprobes. The correct location and identity of the immobi-lized proteins could be confirmed by autoradiography andhighly specific monoclonal antibody binding experiments.The methods we describe here represent an efficientmeans to convert readily available high-density DNAmicroarrays into high-density protein microarrays forpotential parallel functional screening, biosensor, diag-nostic, and proteomics applications.
2 Materials and methods
2.1 mRNA-protein synthesis
mRNA-protein fusions were prepared essentially asdescribed [16, 17]. PCR was first used to individuallyamplify each of the following sequences with the indi-cated common primers:
[FLAG] 5’-TAA TAC GAC TCA CTA TAG GGA CAA TTACTATTTACA ATTACA ATG GAC TAC AAG GAC GAT GACGAT AAG GGC GGC TGG TCC CAC CCC CAG TTC GAGAAG-3’
[HA11] 5’-TAA TAC GAC TCA CTA TAG GGA CAA TTACTA TTT ACA ATT ACA ATG TAC CCC TAC GAC GTGCCC GAC TAC GCC GGC GGC TGG TCC CAC CCCCAG TTC GAG AAG-3’
[MYC] 5’-TAA TAC GAC TCA CTA TAG GGA CAA TTACTA TTT ACA ATT ACA ATG AGC GCA AGA GTT ACGCAG CTG TTC CAG TTT GTG TTT CAG CTG TTC ACGACG TTT ACG CAG CAG GTC TTC TTC AGA GAT CAGTTT CTG TTC TTC AGC CAT GGC GGC TGG TCC CACCCC CAG TTC GAG AAG-3’
5’-primer = TAA TAC GAC TCA CTA TAG GGA CAA TTACTA TTT ACA ATT
3’-primer = AGC GGA TGC CTT CTC GAA CTG GGGGTG GGA
The resulting PCR products were transcribed in vitrousing T7 RNA polymerase to produce an mRNA contain-ing the coding region for the polypeptide and a tobaccomosaic virus (TMV) ribosome binding site. The RNA wassplint-ligated to the sequence 5’-d(AAA AAA AAA AAAAAA AAA AAA AAA AAA CC-P) possessing a 5’-phos-phate and a 3’-puromycin by the action of T4 DNA ligase(puromycin is indicated by P in the above sequence).The individual gel purified ligation products (200 pmol)were in vitro translated in rabbit reticulocyte lysate fromAmbion (Austin, TX, USA) (300 �L) containing [35S]
methionine according to the manufacturer’s protocolexcept that the solution was adjusted to 150 mM MgCl2and 425 mM KCl after 30 min to promote the formation ofthe puromycin-peptide bond. The mRNA-protein fusionswere isolated directly from lysate either by oligo dT-cellu-lose affinity chromatography or Strep-TagII affinity chro-matography. Quantitation was performed by scintillationcounting. Analysis of the mRNA-protein fusion reactionswas carried out by gel electrophoresis with NuPAGE(Carlsbad, CA, USA) 4–12% polyacrylamide gels employ-ing NuPAGE MOPS SDS running buffer.
2.2 Microarray fabrication
Precleaned 1�3 inch glass microscope slides (Goldseal,3010, Portsmouth, NH, USA ) were treated with Nanostrip(Cyantek, Fremont, CA, USA) for 15 min, 10% aqueousNaOH at 70�C for 3 min, and 1% aqueous HCl for 1 min,thoroughly rinsing with deionized water after each reagent.The slides were then dried in a vacuum desiccator overanhydrous calcium sulfate for several hours. A 1% solutionof aminopropytrimethoxysilane in 95% acetone/5% waterwas prepared and allowed to hydrolyze for 20 min. Theglass slides were immersed in the hydrolyzed silane solu-tion for 5 min with gentle agitation. Excess silane wasremoved by subjecting the slides to 10�5 min washes,using fresh portions of 95% acetone/5% water for eachwash, with gentle agitation. The slides were then cured byheating them at 110�C for 20 min. The cooled, silane trea-ted slides were immersed in a freshly prepared 0.2% solu-tion of phenylene 1,4-diisothiocyanate in 90% DMF/10%pyridine for two hours, with gentle agitation. The slideswere washed sequentially with 90% DMF/10% pyridine,methanol and acetone. After air drying, the functionalizedslides were stored at 0�C in a vacuum desiccator overanhydrous calcium sulfate.
Specific, non interacting, and thermodynamically isoener-getic sequences along the target RNAs were identified toserve as capture points. The software program HybSimu-lator V4.0 (Advanced Gene Computing Technology, Irvine,CA, USA) facilitated the identification and analysis ofpotential capture probes. A single specific capture probe(cp_flag, cp_ha11 and cp_myc) was ultimately identifiedfor each target RNA. In addition, two sequences commonto each RNA (cp_5 and cp_3) were also identified toserve as positive controls. One non-sense sequence(cp_ au1) was generated as well to serve as a hybridizationnegative control. The following is a list of the capture probesequences that were employed (written 5’� 3’):
cp_5: TGTAAATAGTAATTGTCCCcp_3: TTTTTTTTTTTTTTTTTTTTcp_ au1: CCTGTAGGTGTCCAT
Proteomics 2002, 2, 48–57 Addressable protein microarrays 49

cp_flag: CATCGTCCTTGTAGTCcp_ha11: CGTCGTAGGGGTAcp_myc: CAGGTCTTCTTCAGAGA
Oligonucleotide capture probes were prepared with anautomated DNA synthesizer (PE Biosystems, Foster City,CA, USA; Expedite 8909) using conventional phosphor-amidite chemistry. All reagents were from Glen Research(Sterling, VA, USA). Synthesis was initiated with a solidsupport bearing an orthogonally protected amino func-tionality, whereby the 3’-terminal amine is not unmaskeduntil the final deprotection step. The first four monomersto be added were hexaethylene oxide units, followed bythe standard A, G, C and T monomers. All capture oligosequences were cleaved from the solid support anddeprotected with ammonium hydroxide, concentrated todryness, precipitated in ethanol, and purified by reverse-phase HPLC using an acetonitrile gradient in triethyl-ammonium acetate buffer. Appropriate fractions from theHPLC were collected, evaporated to dryness in a vacuumcentrifuge, and then coevaporated with a portion of water.
The purified, amine-labeled capture oligos were adjustedto a concentration of 250 �M in 50 mM sodium carbonatebuffer (pH 9.0) containing 10% glycerol. The probes werespotted onto the amine-reactive substrate at definedpositions in a 5�5�6 array pattern with a 3-axis robot(MicroGrid, BioRobotics, Cambridge, UK). A 16-pin toolwas used to transfer the liquid from 384-well microtiterplates, producing 200 �m features with a 400 or 600 �mpitch. For the typical format, each sub-grid of 24 featuresrepresents a single capture oligo (i.e., 24 duplicate spots).For the 384-feature array, a single-pin tool was used toprint a 16�24 format. The arrays were incubated atroom temperature in a moisture-saturated environmentfor 12–18 h. The attachment reaction was terminated byimmersing the chips in 2% aqueous ammonium hydrox-ide for five minutes with gentle agitation, followed by rin-sing with distilled water (3�5 min). The array was finallysoaked in 10X PBS solution for 30 min at room tempera-ture, and then rinsed again for 5 min in distilled water.Arrays were stored at –20�C for up to three months.
2.3 mRNA-protein hybridization to DNAmicroarray
Individual or multiple mRNA-protein fusions, typically0.2 pmol each, were adjusted to 5X saline sodium citrate(SSC) containing 0.02% Tween-20 and 2 mM vanadylribonucleotide complex in a total volume of 200 �L. Theentire volume was applied to the microarray under agasket device and the assembly was continuously rotatedfor 2–12 h at room temperature. Similar results wereobtained with a static hybridization of 10 �L volumes
under a glass coverslip. After hybridization the slide waswashed sequentially with stirred 500 mL portions of 5XSSC � 0.02% Tween-20, 1X SSC � 0.02% Tween-20,and 1X SSC for 5 min each, followed by a brief rinse with0.2X SSC. Traces of liquid were removed by centrifu-gation and the slide was allowed to air-dry.
2.4 Antibody binding
Each of the following processes was conducted at 4�Cwith continuous rotation or mixing. The protein microarraysurface was passivated by treatment with 1X TBS con-taining 0.02% Tween-20 and 0.2% BSA (200 �L) for60 min. Affinity purified monoclonal antibodies (Covance,Princeton, NJ, USA) were adjusted to 10 �g/mL (100:1dilution) in the same buffer and contacted with the micro-array for 120 min. Non-bound antibody was removed bywashing with 1X TBS containing 0.02% Tween-20 (3X 50mL, 2 min each wash). Cy-3 labeled goat antimouse IgGadjusted to 2.5 �g/mL (400:1 dilution) in 1X TBS contain-ing 0.02% Tween-20 and 0.2% BSA was contacted withthe microarray for 60 min. Non-bound IgG was removedby washing with 1X TBS containing 0.02% Tween-20(2X 50 mL, 5 min each wash) followed by a 3 min rinsewith 1X TBS. Traces of liquid were removed by centrifu-gation and the slide was allowed to air-dry at room tem-perature.
For the 384-feature microarray, primary monoclonal anti-bodies were labeled with monofunctional Cy-3 or Cy-5dyes according to the protocol supplied with the kit(Amersham-Pharmacia, Uppsala, Sweden). A mixture ofthe primary-labeled monoclonal antibodies (2.5 �g/mL)was contacted with the passivated microarray surface asdescribed above. The remaining processing steps wereperformed as described above.
2.5 Detection
Phosphorimage analysis was carried out with a MolecularDynamics (Sunnyvale, CA, USA) Storm system. Typicalexposure times were 1–3 d with direct contact betweenthe microarray and the phosphor storage screen. Scan-ning was performed at the highest (50 �m) resolution set-ting. Phosphorimage data was extracted and quantitatedwith ImageQuant software (Molecular Dynamics). Fluor-escence laser scanning was carried out with a GSILumonics (Watertown, MA, USA) ScanArray 5000 systemusing preset excitation and emission wavelengths foreach fluorophore and 10 �m pixel resolution. Fluores-cence data was extracted and quantitated with ImaGeneV.2 software (BioDiscovery, Marina Del Ray, CA, USA).
50 S. Weng et al. Proteomics 2002, 2, 48–57

Figure 1. (A) PROfusion mRNA-protein fusion formation showing (a) polypeptide synthesis on theribosome, the RNA/DNA junction and the terminal puromycin moiety, (b) ribosome catalyzed bondformation between the puromycin moiety and the C-terminus of the polypeptide, (c) isolated mRNA-protein fusion. (B) Schematic of the chip surface illustrating an mRNA-protein fusion hybridized to acovalently immobilized DNA capture probe.
3 Results and discussion
3.1 Preparing the mRNA-protein fusions
The mRNA-protein fusion process originally described byRoberts and Szostak covalently links a polypeptide to itsown encoding mRNA during in vitro translation in rabbitreticulocyte lysate (Fig. 1a) [16, 18]. In this approach, theribosome binds and transits an mRNA-puromycin con-jugate, producing a polypeptide. When the ribosomereaches the engineered RNA/DNA junction, it pauses andallows entry of the puromycin moiety into the peptidyltransferase center to form an mRNA-protein fusion pro-duct (i.e., the PROfusion product). Metal chelation dis-sociates the ribosome complex and allows recovery ofisolated mRNA-protein fusions. The efficiency of the fusionreaction can be as high as 20% relative to the input mRNAwhen the fusion reaction is conducted at the subnanomolescale. When expressed in terms of the total protein pro-duced, the efficiency is normally at least 80%. The generalprocess is amenable both to single mRNA sequences andvery complex libraries of mRNA containing up to 1015 dis-tinct sequences [19, 20]. The importance of large librariesfor directed evolution applications is well known [18, 21].There does not appear to be a significant limitation on thelength of mRNA used, and therefore the size of the proteinfused, although there are obvious potential complicationswith proper protein folding. To maintain focus on the manyissues relating to the conversion of a DNA microarray intoa protein microarray, this study has employed relativelysmall proteins (up to 44 amino acids) that are not expectedto have significant secondary structures.
The mRNA constructs utilized here contain a T7 promotersite, a 25-nucleotide (nt) TMV ribosome binding site, acoding region that ranges between 57-nt and 132-nt,and a 29-nt DNA sequence terminating with a 3’-puro-mycin moiety. The mRNA coding region contains a com-mon sequence of 30-nt (coding for StrepTagII peptide)and also a unique sequence for one of three epitopes,comprising 27-nt, 30-nt, or 102-nt. The three peptide epi-topes (FLAG, HA11 and MYC, respectively) were chosenbased on the commercial availability of monoclonal anti-bodies raised against them, as we intended to use theseantibodies as specific binding agents to probe the immo-bilized polypeptide’s identity and accessability. mRNA-protein fusions were prepared individually and separatedfrom other reaction components by either oligo dT-cellu-lose affinity chromatography (targeting the DNA linker) orStrepTagII affinity chromatography (a C-terminal peptidesequence common to each of the constructs). The pep-tide-based separation scheme was especially useful forremoving excess “unfused” mRNA from the reaction mix-ture, which accounts for as much as 80% of the nucleicacid and can occupy DNA capture probe sites on themicroarray, potentially diminishing the immobilized pro-tein density and therefore the subsequent signal intensity.However, we routinely obtained very satisfactory resultswith only an oligo-dT separation step, despite the pre-sence of excess unfused mRNA. As a precaution due tothe relatively long hybridization times (vide infra), we rou-tinely added the broad spectrum ribonuclease inhibitorvanadyl ribonucleoside complex. mRNA-protein fusionswere either used individually, to test hybridization specifi-city, or combined together in a multiplex format.
Proteomics 2002, 2, 48–57 Addressable protein microarrays 51

3.2 Designing and fabricating the array
Oligonucleotide capture probes were prepared with a3’-terminal amine to serve as an attachment point to thesolid phase. Attachment at this terminus ultimately orientsthe polypeptide portion of the hybridized mRNA-proteinfusion toward the solution and away from the solid sur-face, where it may be less accessible for interaction withbinding partners (Fig. 1b). In comparison experiments, wehave generally had better results with 3’-vs. 5’-terminalattachment, and we attribute these benefits to an orien-tation effect. Similar orientation effects have beenobserved in the field of protein immobilization [22, 23].Although the 3’-terminal amine moiety possesses a six-carbon linker of its own, we have incorporated an addi-tional four hexaethylene oxide units, spaced by phospho-diester moieties, between the 3’-terminal amine and theDNA capture probe sequence. These flexible, hydrophilicspacing elements help to move the capture probe awayfrom the solid surface and, therefore, help to make theprotein more accessible to the analyte-containing solu-tion phase. We also speculate that the hexaethyleneoxide spacers aid in minimizing unwanted nonspecificprotein interactions with the surface, as has been reportedfor poly(ethylene oxide) coated surfaces [24].
The DNA capture probe sequences were designed to givea predicted melting temperature (Tm) of ca. 55�C andwere also selected to be maximally specific in the contextof all the anticipated nucleic acid sequence(s) presentduring hybridization. Computer hybridization simulations,based on thermodynamic nearest neighbor interactions,were carried out to help identify promising capture probesequences. Due to the relatively unstable nature of manyprotein secondary and tertiary structures, we designedthe capture probes so that hybridization with a mixture ofmRNA-protein fusions could be conducted at a relativelylow temperature, while still maintaining reasonable hy-bridization specificity. The capture probes employedhere range in length from 13 to 20 nucleotides. In someinstances it was possible to identify a number of differentcandidate capture probes along the length of the fusion’smRNA. With the relatively small mRNA-protein fusionsused in this study (up to 44 amino acids), there did notappear to be a significant benefit to targeting either the3’- or 5’-region of the mRNA, or the middle region, forcapture; all areas worked well.
The solid phase of the microarray was prepared as anamine-reactive self-assembled monolayer using silanechemistry, similar to a process previously reported [25].Standard glass microscope slides were carefully cleaned,treated with an aminosilane, and finally functionalizedwith the homobifunctional coupling agent phenylene 1,4-diisothiocyanate. A commercial three-axis motion control
Figure 2. Layout of epitope chip showing location ofcommon 5’-capture probe (5’-(�)), common 3’-captureprobe (3’-(�)), non-sense negative control capture probe((�)), capture probe specific for the FLAG epitope [FLAG],capture probe specific for the HA11 epitope [HA11], andcapture probe specific for the MYC epitope [MYC]. Eachmeta grid contains 24 replicate spots in a 5�5 grid pat-tern (200 �m diameter feature size).
system was used to transfer small volumes of oligo-nucleotide capture probes (about 1 nL) from microtitretrays to the microarray surface via solid pins 150 micronsin diameter. After incubation, blocking and washing themicroarrays are ready for hybridization with mRNA-pro-tein fusions (vide infra).
The layout of the microarray normally consisted of sixmeta grids, each containing 24 replicate spots with afeature size of ca. 200 �m on a 400 or 600 �m pitch(Fig. 2). Three of the six meta grids contain captureprobes specific for one of the three mRNA-protein fusions(FLAG, HA11 or MYC). The remaining three meta grids arecontrol areas: a non-sense capture probe to function as anegative hybridization control, a sequence residing at the5’-terminus that is common to each of the three mRNA-protein fusions and functions a positive control, andanother common sequence residing at the 3’-terminusthat also serves as a positive control. The 3’-terminalcommon sequence targets the DNA linker of the mRNA-protein fusion and can be helpful to diagnose unexpectedmRNA degradation, since DNA is not degraded by ribo-nucleases; large differences in signal between the 5’- and3’-capture probe suggest RNA degradation. Althoughthe microarrays utilized in this validation study are fairlylow density, there are very few practical reasons whythe density (features per square centimeter), or the totalnumber of features, could not be substantially higher. Infact, decreasing the feature’s diameter can give rise toimproved sensitivities and performance in conventionalimmunoassays [1, 26].
3.3 Generating the protein array
Protein arrays were generated by simply contacting theDNA microarray with a solution of mRNA-protein fusionat room temperature, followed by removal (rinsing) of
52 S. Weng et al. Proteomics 2002, 2, 48–57

the unhybridized material. The mRNA-protein moleculesself-assemble according to standard Watson-Crickbase-pairing rules, and one is left with an addressableprotein array that is immediately ready for use. In practice,the mRNA-protein solution is made up in a suitablehybridization buffer, such as SSC, and a nondenaturingsurfactant, such as Tween-20, is included to minimizebackground binding. Also, we have found it helpful tocontinuously mix the mRNA-protein solution over the sur-face of the microarray during hybridization. We generallyuse between 50 and 200 femtomoles of each mRNA-pro-tein fusion for the hybridization, but higher protein densi-ties can be achieved if picomole quantities are used. Thedried protein arrays appear to be quite robust and havebeen successfully used for antibody binding experimentsafter several weeks of storage at room temperature.
The distribution of protein on the self-assembled micro-array following hybridization can be readily confirmed byautoradiography. The nucleic acid component of thefusion is not radiolabeled, whereas the polypeptide com-ponent contains a [35S]methionine incorporated duringtranslation. Figure 3A shows a panel of three microarraysthat have been hybridized with MYC, HA11, or FLAGfusion and then visualized by phosphorimaging. Auto-radiography has been used here solely as a validationand development tool. Unfortunately the phosphorimagerused here is only capable of 50 �m pixel resolution, so themagnified image of the 200 �m features is not wellresolved, but the hybridization specificity is still clearlydemonstrated in these images. Figure 4A shows a panelof three replicate microarrays that have been hybridizedwith a mixture of all three mRNA-protein fusions and thenvisualized by phosphorimaging; the polypeptide is dis-tributed as expected amongst the two common captureprobes and the three specific capture probes.
The preferred method for generating protein arraysemploys partially purified mRNA-protein fusion mixturesthat have been adjusted to a suitable salt concentrationto allow for specific hybridization at a given temperature.Alternatively, we have successfully generated useful pro-tein arrays by simply diluting the crude rabbit reticulocytelysate in the standard hybridization buffer, followed byapplication directly onto the DNA microarray and sub-sequent removal of unhybridized components by rinsing(data not shown). In these cases the background signalsare somewhat higher, and the surface protein concentra-tions are diminished due to the excess mRNA, but thisapproach provides a very rapid means to generate a pro-tein array. The use of crude lysates fully exploits thetremendous resolving power of the biochip format, asthe crude lysate mixture is a highly complex biologicalfluid with numerous potential interferents.
Figure 3. Individual hybridizations. Replicate DNA chipstreated with either MYC (top) HA11 (middle) or FLAG (bot-tom) mRNA-protein fusion followed by binding with fluor-escently labeled mAb as described in Section 2. Visual-ization performed by phosphorimaging (A) and by fluores-cence laser scanning (B).
3.4 Probing the protein array with labeledantibodies
Autoradiography clearly established the presence ofimmobilized protein on the microarray at the expectedlocations, demonstrating the ability of the system to self-assemble according to predictable Watson-Crick basepairing rules. However, an equally important question isthe functionality and accessibility of the protein immobil-ized on the solid phase via hybridization. To begin toaddress this point we treated the single-protein micro-arrays from Fig. 3 with individual monoclonal antibodies(mAbs) specific for an epitope sequence of the mRNA-protein fusion (MYC, FLAG or HA11). A fluorescentlylabeled antimouse secondary antibody was used fordetection. Figure 3B shows that each of the individualmAbs could bind to their respective epitope sequence.
Proteomics 2002, 2, 48–57 Addressable protein microarrays 53

Figure 4. Multiplex hybridization. Replicate DNA chipstreated with mixtures of MYC, HA11 and FLAG mRNA-protein fusion followed by binding with fluorescentlylabeled mAb as described in Section 2. Visualization wasaccomplished both by phosphorimaging (A) and by fluor-escence laser scanning (B).
Moreover, the fluorescence signal is comparable forcapture probes targeting 3’-, 5’- or internal (specific)regions of the mRNA. Only the anti-FLAG mAb showsmeasurable nonspecific interaction with nonprotein con-taining capture probes, likely resulting from electrostaticinteractions between the nucleic acid and this particularmAb. These results demonstrate that these small pro-teins, when immobilized to a solid phase via hybridizationand in the context of a mRNA-protein fusion molecule, arecapable of productively interacting with solution-phasebinding partners, in this case rather large immunoglobu-lins. Typical signal-to-noise values for the experimentsdescribed above are 200:1. It is likely that alternative sur-face chemistries, or optimization of passivation routinesand blocking agents, could significantly reduce the back-
ground levels and therefore substantially increase theobserved signal-to-noise ratio. Improved surface chemis-tries and materials may also enhance the functionalimmobilization of proteins to the surface, thereby increas-ing signal.
To explore the specificity of the mAb-epitope interaction,we generated protein microarrays displaying all of theepitopes (Fig. 4). Each of these microarrays was thentreated with one of the three individual mAbs as describedabove. However, instead of having only one epitope bind-ing site to choose from, the mAb is presented with threeproteins which are immobilized to the solid phase. AsFig. 4B illustrates, each of the three mAbs interact speci-fically only with the expected epitope, and each exhibitsvery little cross-talk with the other members. The anti-HA11 mAb showed the most nonspecific interaction(with MYC mRNA-protein fusion), but even in this casethe average integrated fluorescence signal for HA11:MYCwas � 20:1.
3.5 Quantitating the protein array
A sequence of experiments was performed to measurethe quantity of protein at each feature and also to corre-late the fluorescence signal from the bound antibody withthe amount of displayed protein. First, a microarray wasprinted with an increasingly dilute capture probe sequenceto create an arbitrary decreasing gradient of attachedcapture probe concentration on the chip surface. Thecapture probe printing concentration was varied from250 �M to 0.12 �M in two-fold dilutions. Next, MYCmRNA-protein fusion was hybridized in the usual fashion,followed by primary and secondary antibody bindingaccording to the techniques described in Section 2. Themicroarray was subsequently quantitatively analyzed bothby fluorescence scanning (antibody detection, Fig. 5B)and phosphorimaging (fusion detection, Fig. 5A). Astandard curve was independently created to convertthe integrated phosphorimage signal from an arbitraryvalue to the actual number of mRNA-protein fusion mole-cules. This standard curve was generated by applyingknown quantities of mRNA-protein to a microscope slide,which was then exposed to the phosphor storage screenalongside the hybridized protein microarray. The standardcurve produced a linear fit between 8–125 attomoles ofmRNA-protein fusion (R2 = 0.991) and allowed the directdetermination of the amount of hybridized mRNA-proteinfusion at each 200 �m diameter feature, which varied from97–1 attomole depending on the capture probe density.These data, when plotted against the integrated fluores-cence signals, reveal the expected saturation behavior(Fig. 5C). Using a similar standard curve approach withan aim to measuring the maximum amount of mRNA-
54 S. Weng et al. Proteomics 2002, 2, 48–57
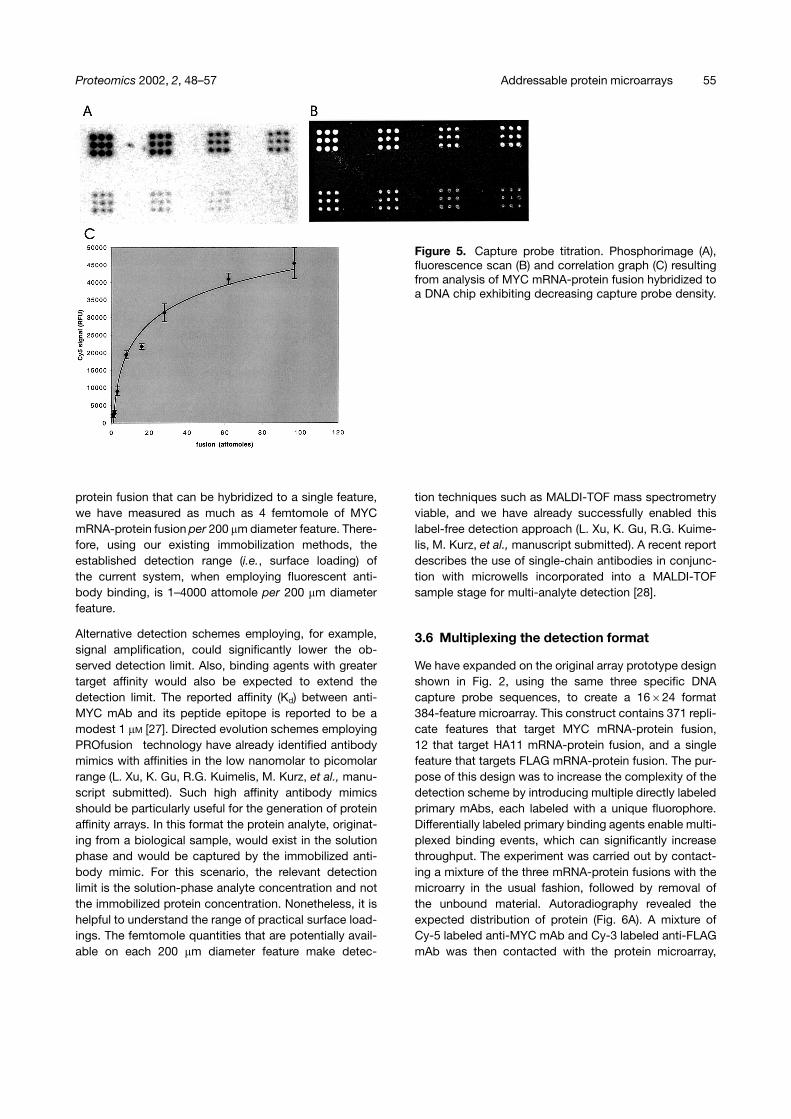
Figure 5. Capture probe titration. Phosphorimage (A),fluorescence scan (B) and correlation graph (C) resultingfrom analysis of MYC mRNA-protein fusion hybridized toa DNA chip exhibiting decreasing capture probe density.
protein fusion that can be hybridized to a single feature,we have measured as much as 4 femtomole of MYCmRNA-protein fusion per 200 �m diameter feature. There-fore, using our existing immobilization methods, theestablished detection range (i.e., surface loading) ofthe current system, when employing fluorescent anti-body binding, is 1–4000 attomole per 200 �m diameterfeature.
Alternative detection schemes employing, for example,signal amplification, could significantly lower the ob-served detection limit. Also, binding agents with greatertarget affinity would also be expected to extend thedetection limit. The reported affinity (Kd) between anti-MYC mAb and its peptide epitope is reported to be amodest 1 �M [27]. Directed evolution schemes employingPROfusion technology have already identified antibodymimics with affinities in the low nanomolar to picomolarrange (L. Xu, K. Gu, R.G. Kuimelis, M. Kurz, et al., manu-script submitted). Such high affinity antibody mimicsshould be particularly useful for the generation of proteinaffinity arrays. In this format the protein analyte, originat-ing from a biological sample, would exist in the solutionphase and would be captured by the immobilized anti-body mimic. For this scenario, the relevant detectionlimit is the solution-phase analyte concentration and notthe immobilized protein concentration. Nonetheless, it ishelpful to understand the range of practical surface load-ings. The femtomole quantities that are potentially avail-able on each 200 �m diameter feature make detec-
tion techniques such as MALDI-TOF mass spectrometryviable, and we have already successfully enabled thislabel-free detection approach (L. Xu, K. Gu, R.G. Kuime-lis, M. Kurz, et al., manuscript submitted). A recent reportdescribes the use of single-chain antibodies in conjunc-tion with microwells incorporated into a MALDI-TOFsample stage for multi-analyte detection [28].
3.6 Multiplexing the detection format
We have expanded on the original array prototype designshown in Fig. 2, using the same three specific DNAcapture probe sequences, to create a 16�24 format384-feature microarray. This construct contains 371 repli-cate features that target MYC mRNA-protein fusion,12 that target HA11 mRNA-protein fusion, and a singlefeature that targets FLAG mRNA-protein fusion. The pur-pose of this design was to increase the complexity of thedetection scheme by introducing multiple directly labeledprimary mAbs, each labeled with a unique fluorophore.Differentially labeled primary binding agents enable multi-plexed binding events, which can significantly increasethroughput. The experiment was carried out by contact-ing a mixture of the three mRNA-protein fusions with themicroarry in the usual fashion, followed by removal ofthe unbound material. Autoradiography revealed theexpected distribution of protein (Fig. 6A). A mixture ofCy-5 labeled anti-MYC mAb and Cy-3 labeled anti-FLAGmAb was then contacted with the protein microarray,
Proteomics 2002, 2, 48–57 Addressable protein microarrays 55

Figure 6. Multiplex, multicolor experiment. Phosphorimage (A), Channel A fluorescence scan (B),and Channel B fluorescence scan (C) of a 16�24 microarray created by hybridizing a mixture ofMYC, HA11 and FLAG mRNA-protein fusion to a DNA microarray followed by binding with a mixtureof two mAbs, each labeled with a different fluorophore.
unbound material was washed away, and the fluores-cence was measured separately in both the Cy-5 andCy-3 channels (Fig. 6B and 6C). The images demonstratethat labeled primary binding agents, even when multi-plexed, are very effective.
4 Concluding remarks
The data presented here demonstrate that protein micro-arrays can be effectively self-assembled from conven-tional DNA microarrays using covalent mRNA-proteinfusion technology. Moreover, proteins immobilized to thesolid-phase through Watson-Crick base pairing retainbinding functionality and are extremely robust. Althoughthis work has employed fairly low density protein micro-arrays and small, unstructured proteins, it should be fea-sible to both create significantly more dense proteinmicroarrays as well as utilize larger proteins. Future workwill focus primarily on these two aspects. In addition toplanar, two-dimensional constructs, this general approachwould also be suitable for microbead systems [29] andshould also find potential application in lab-on-a-chip[30] formats. We have demonstrated the feasibility of thisapproach with mRNA-protein fusions, but these methodsshould also be applicable to proteins that have beenchemically derivatized with nucleic acids. Opportunitiesexist in both approaches to utilize nucleic acid analougeswith improved biophysical properties, especially withregard to hybridization temperature and hybridizationspecificity. The emphasis of future work will exploit the invitro selection capabilities of the PROfusion process togenerate numerous specific and high affinity proteinsagainst highly relevant proteins associated with variousdisease states. These novel high affinity proteins (i.e.,antibody mimics based on a common scaffold) will serveas the capture agents for affinity protein microarrays thatmay find application in, for example, cytokine/chemokine
profiling or cancer diagnostics. The common scaffoldshould allow for more uniform and predictable proteinproperties, as well as consistent chemistries (e.g., liga-tion, fusion formation). A recent report describes thegeneration of comprehensive PROfusion libraries fromhighly complex, tissue-specific mRNA samples [31]. Al-though protein microarray technology is still in its infancy,and its limits have yet to be established, we ultimatelyenvisage a single protein microarray capable of directlyand simultaneously measuring the protein levels of anentire proteome, thereby providing a snapshot of the pro-teome under a given set of conditions at a specific pointin time. The protein microarray approach should comple-ment traditional 2-D gel electrophoresis techniques, andthe approach additionally holds promise for improvingthe reproducibility, repeatability and scalability of pro-teome-scale protein analysis.
The authors wish to thank Jack W. Szostak for his scien-tific support and also for helpful comments on the manu-script.
Received May 29, 2001
5 References
[1] Ekins, R. P., Chu, F., Biggart, E., Anal. Chim. Acta 1989, 227,73–96.
[2] Mendoza, L. G., McQuary, P., Mongan, A., Gangadharan, R.,et al., BioTechniques, 1999, 27, 778–788.
[3] Silzel, J. W., Cerek, B., Dodson, C., Tsay, T., Obremski, R. J.,Clin. Chem. 1998, 44, 2036–2043.
[4] MacBeath, G., Schreiber, S. L., Science 2000, 289, 1760–1763.
[5] Paweletz, C. P., Charboneau, L., Bichsel, V. E., Simone, N. L.,et al., Oncogene 2001, 20, 1981–1989.
[6] DeWildt, R. M., Mundy, C. R., Gorick, B. D., Tomlinson, I. M.,Nat. Biotechnol. 2000, 18, 989–994.
[7] Holt, L. J., Bussow, K., Walter, G., Tomlinson, I. M., NucleicAcids Res. 2000, 28, E72.
56 S. Weng et al. Proteomics 2002, 2, 48–57

[8] Emili, A. Q., Cagney, G., Nat. Biotechnol. 2000, 18, 393–397.[9] Haab, B. B., Dunham, M. J., Brown, P. O., Genome Biol.
2001, 2, 0004.1.[10] Abbott, A., Nature 1999, 402, 715–720.[11] Humphery-Smith, I., Blackstock, W., J. Protein Chem. 1997,
16, 537–544.[12] Lueking, A., Horn, M., Eichoff, H., Buessow, K., et al., Anal.
Biochem. 1999, 270, 103–111.[13] Ge, H., Nucleic Acids Res. 2000, 28, e3.[14] Arenkov, P., Kukhtin, A., Gemmell, A., Voloshuk, S., et al.,
Anal. Biochem. 2000, 278, 123–131.[15] Eisen, M. B., Brown, P. O., Methods Enzymol. 1999, 303,
179–205.[16] Roberts, R. W., Szostak, J. W., Proc. Natl. Acad. Sci. USA
1997, 94, 12297–12302.[17] Liu, R., Barrick, J. E., Szostak, J. W., Roberts, R. W., Meth-
ods Enzymol. 2000, 318, 268–293.[18] Roberts, R. W., Curr. Opin. Chem. Biol. 1999, 3, 268–273.[19] Keefe, A. D., Szostak, J. W., Nature 2001, 410, 715–718.[20] Wilson, D. S., Keefe, A. D., Szostak, J. W., Proc. Natl. Acad.
Sci. USA 2001, 98, 3750–3755.
[21] Gold, L., Proc. Natl. Acad. Sci. USA 2001, 98, 4825–4826.[22] O’Shannessy, D. J., Wilchek, M., Anal. Biochem. 1990, 191,
1–8.[23] Turkova, J., J. Chromatogr. B. 1999, 722, 11–31.[24] Kishida, A., Mishima, K., Corretge, E., Konishi, H., Ikada, Y.,
Biomaterials 1992, 13, 113–118.[25] Guo, Z., Guilfoyle, R. A., Thiel, A. J., Wang, R., Smith, L. M.,
Nucleic Acid Res. 1994, 22, 5456–5465.[26] Ekins, R. P., Chu, F. W., Clin. Chem. 1991, 37, 1955–1967.[27] Evan, G., Lewis, G., Ramsey, G., Bishop, J. M., Mol. Cell.
Biol. 1985, 5, 3610–3616.[28] Borrebaeck, C. A. K., Ekstrom, S., Malmborg Hager, A. C.,
Nilsson, J., et al., Biotechniques 2001, 30, 1126–1132.[29] Szurdoki, F., Michael, K. L., Walt, D. R., Anal. Biochem.
2001, 291, 219–228.[30] Sanders, G. H. W., Manz, A., Trends Anal. Chem. 2000, 19,
364–378.[31] Hammond, P. W., Alpin, J., Rise, C. E., Wright, M., Kreider,
B. L., J. Biol. Chem. 2001, 15, 20898–20906.
Proteomics 2002, 2, 48–57 Addressable protein microarrays 57
Related Documents











